 Xiaolei Zhao
Xiaolei Zhao Shannon Erhardt
Shannon Erhardt Kihan Sung
Kihan Sung Jun Wang
Jun Wang- 1Department of Pediatrics, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX, United States
- 2MD Anderson Cancer Center and UT Health Graduate School of Biomedical Sciences, The University of Texas, Houston, TX, United States
- 3Department of BioSciences, Rice University, Houston, TX, United States
Suture mesenchymal stem cells (SMSCs) are a heterogeneous stem cell population with the ability to self-renew and differentiate into multiple cell lineages. The cranial suture provides a niche for SMSCs to maintain suture patency, allowing for cranial bone repair and regeneration. In addition, the cranial suture functions as an intramembranous bone growth site during craniofacial bone development. Defects in suture development have been implicated in various congenital diseases, such as sutural agenesis and craniosynostosis. However, it remains largely unknown how intricate signaling pathways orchestrate suture and SMSC function in craniofacial bone development, homeostasis, repair and diseases. Studies in patients with syndromic craniosynostosis identified fibroblast growth factor (FGF) signaling as an important signaling pathway that regulates cranial vault development. A series of in vitro and in vivo studies have since revealed the critical roles of FGF signaling in SMSCs, cranial suture and cranial skeleton development, and the pathogenesis of related diseases. Here, we summarize the characteristics of cranial sutures and SMSCs, and the important functions of the FGF signaling pathway in SMSC and cranial suture development as well as diseases caused by suture dysfunction. We also discuss emerging current and future studies of signaling regulation in SMSCs.
1 Introduction
Different from long bones, which are formed through endochondral ossification, most cranial bones are formed through intramembranous ossification directly from mesenchymal cells without a cartilaginous template (Ishii et al., 2015). As shown in Figure 1, these cranial bones are connected by fibrous joints, known as cranial sutures, consisting of fibrous tissues with mesenchyme, two osteogenic fronts (OFs) of the approximating bone plates, underlying dura mater, and overlying periosteum. Notably, the OFs of the bone plates of the coronal and lambdoid sutures partially overlap, whereas the OFs of the metopic and sagittal sutures abut from end to end (Lenton et al., 2005). Cranial sutures provide postnatal locomotive shock absorption and allow joint mobility during feeding (White et al., 2021). They also function as an intramembranous bone growth site for cranial bone expansion during embryogenesis and postnatal craniofacial growth (Opperman, 2000). Furthermore, the cranial suture provides a niche for mesenchymal stem cells (Zhao et al., 2015), called suture mesenchymal stem cells (SMSCs), which maintain suture patency during craniofacial development and craniofacial bones homeostasis, repair and regeneration. In mice, most sutures remain patent throughout the lifetime except for the posterior frontal suture (PFS) located between the frontal bones (Sahar et al., 2005). In humans, the metopic suture (also known as the frontal suture) fuses between 3 and 8 months of age, whereas other cranial sutures fuse between 20 and 30 years, and facial sutures fuse after 50 years (Vu et al., 2001; Weinzweig et al., 2003; White et al., 2021). Cranial suture patency is important for allowing the skull to grow in concert with the development of the brain during childhood. Aberrant development of cranial sutures leads to various congenital diseases such as sutural agenesis and craniosynostosis (Cohen, 1993; Barnes, 2012; Ishii et al., 2015). Despite the considerable significance of cranial sutures and SMSCs, they have remained poorly understood. Recently, however, multiple single cell RNA-sequencing (scRNA-seq) studies of frontal and coronal suture tissues have characterized SMSC populations to a certain extent (Holmes et al., 2020; Farmer et al., 2021; Holmes et al., 2021).
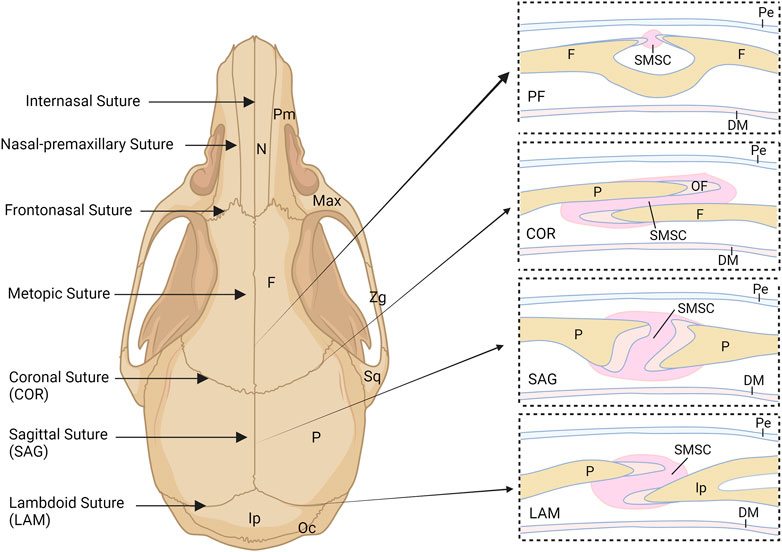
FIGURE 1. Schematic illustration of the murine skull and cranial sutures. COR, coronal suture; DM, dural mater; F, frontal bone; Ip, interparietal bone; LAM, lambdoid suture; Max, maxilla; N, nasal bone; OF, osteogenic front; Oc, occipital bone; P, parietal bone; Pe, periosteum; PF, posterior frontal suture; Pm, premaxilla; SAG, sagittal suture; SMSC, suture mesenchymal stem cell; Sq, squamosal; Zg, zygomatic.
The development of cranial sutures and SMSCs involves multiple factors including Twist (Carver et al., 2002; Yoshida et al., 2005; Ting et al., 2009), fibroblast growth factor (FGF) ligands and receptors (FGFRs) (Johnson et al., 2000; Rice et al., 2000; Greenwald et al., 2001; Marie et al., 2005; Wang et al., 2005; Robin et al., 2011), Msx1/2 (Liu et al., 1999; Merrill, 2005), TCF12 (Ting et al., 2022), Axin2 (Yu et al., 2005; Behr et al., 2013) and Gli1 (Zhao et al., 2015; Yu et al., 2021; Jing et al., 2022), as well as signaling such as Hedgehog (Kim et al., 1998; Jenkins et al., 2007; Rice et al., 2010), wingless-related integration site (Wnt) (Behr et al., 2010), Notch (Liu et al., 2017), transforming growth factor/bone morphogenetic protein (TGF/BMP) (Opperman et al., 1997; Clendenning and Mortlock, 2012), Hippo-Yap (Dong et al., 2021; Zhao et al., 2022), and mechanical signaling (Herring, 2008; Wang et al., 2014). Among them, the fundamental FGF signaling has been shown to play a pivotal role in maintaining cranial suture patency and SMSC development. In humans, FGFR mutations have been associated with craniosynostosis in patients with Apert and Crouzon syndromes (FGFR2 gain-of-function mutation) and Muenke syndrome (FGFR3 gain-of-function mutation) (Wilkie et al., 1995; Doherty et al., 2007). In addition, ectopic FGF2 expression in mouse embryos was shown to lead to coronal suture synostosis (Mathijssen et al., 2000). In humans, the FGF family includes 22 ligands, 4 of which are not secreted and act intracellularly (Olsen et al., 2003). The remaining 18 ligands (FGF1-10 and FGF16-23) act through 4 transmembrane tyrosine kinase receptors (FGFR1-4) and are involved in multiple cell functions, such as cellular stemness, proliferation, differentiation and regeneration (Sasaki et al., 2006; Gotoh, 2009; Li et al., 2010; Mossahebi-Mohammadi et al., 2020; Farooq et al., 2021; Kumar et al., 2021). However, our understanding of the precise role of FGF-mediated signaling in cranial suture development and related diseases is limited. In this review, we summarize the most up-to-date advances in the cranial suture and SMSC research. We provide an overview of the FGF pathway and its crosstalk with other signals in cranial suture development in different experimental models to provide deeper insight into the mechanisms of the FGF pathway in cranial suture development and related diseases. Finally, we discuss areas for future studies of the regulation of FGF signaling in cranial suture development and diseases, such as craniosynostosis.
2 Cranial sutures and SMSCs in cranial bone formation and repair
Most cranial bones, such as the nasal bone, frontal bone, and part of the interparietal bone, are derived from neural crest (NC) cells, whereas the parietal bone and most of the occipital bones originate from paraxial mesoderm cells (Jiang et al., 2002; Yoshida et al., 2008; Doro et al., 2019). Reports have shown that the intrinsic proliferation and osteogenic abilities of NC-derived mesenchyme are higher than those of mesoderm-derived (Jiang et al., 2002; Doro et al., 2019; Siismets and Hatch, 2020; Srinivasan et al., 2020). The cranial sutures connect the separate cranial bones as a rigid entity to support the craniofacial structures and to provide a protective cavity for the brain (Li et al., 2021). The major sutures of the skull vault include the metopic (frontal/interfrontal) suture located between the two frontal bone plates, the sagittal suture located between the two parietal bone plates, the coronal suture located between the frontal bone and parietal bone, and the lambdoid suture located between the parietal bone and occipital bone (Li et al., 2021) (Figure 1). The sutures between cranial bones are also populated by mesenchymal cell populations from different embryological origins. For example, the metopic and predominant sagittal sutures are derived from NC cells and the coronal suture is derived from paraxial mesoderm, confirmed by Jiang et al. and Lenton et al. (Jiang et al., 2002; Lenton et al., 2005). However, Doro et al. recently found that both the NC and mesoderm contribute to the coronal suture (Doro et al., 2019). The origin of the lambdoid suture remains unclear. The results of lineage tracking experiments have indicated that the underlying dura mater surrounding the cerebral hemispheres but not the midbrain or hindbrain originates from NC cells (Jiang et al., 2002). Different embryonic origins may result in distinct properties of SMSCs and their derivatives within various sutures.
Several populations of SMSCs in cranial sutures, including Gli1+ (Zhao et al., 2015; Yu et al., 2021), Axin2+ (Maruyama et al., 2016), Prrx1+ (Wilk et al., 2017), and Ctsk+ (Debnath et al., 2018; Otaify et al., 2018) mesenchymal cells, have been identified and proposed as major populations of SMSCs. The characteristics of these four populations of SMSCs have been well summarized in reviews by Doro et al. (Doro et al., 2017) and Li et al. (Li et al., 2021). In general, these four subpopulations of SMSCs have similar but not identical characteristics. They all possess self-renewal and multi-lineage differentiation abilities in mice (except for Prrx1+ SMSCs which were tested only for osteogenic differentiation) and are maintained abundantly in the cranial suture for more than 1 year in mice (excluding the Prrx1+ SMSCs population, which significantly and continuously decreased with age from 8 to 32 weeks of age), and they contribute to calvarial bone injury repair. Additionally, in lineage tracking studies in mice, the subpopulations of SMSCs showed different functions and different abilities to generate calvaria tissues in vivo. Gli1+ SMSCs and their derivatives were detectable in cranial suture mesenchyme, periosteum, dura mater, and quite a few osteocytes in the calvaria bones (Zhao et al., 2015). Notably, the Axin2-expressing cells and their derivatives remained detectable in the middle of the suture, and the population continued to increase in all sutures, except for the PFS (Maruyama et al., 2016). Ctsk + SMSCs and their derivatives were detectable in the cranial suture, periosteum, dura mater and bone marrow cavity of the calvarium, and also contribute to the intramembranous bone formation (Debnath et al., 2018).
In addition to the above SMSC populations, Holmes et al. recently found that Hhip, an inhibitor of Hedgehog signaling, marks a new mesenchymal population that persists only in the coronal suture, although it is also enriched in the OFs of other skull sutures (Holmes et al., 2021). Hhip distinguishes the coronal suture mesenchyme from other skull sutures. Hhip+ populations are highly enriched in the suture and can not differentiate rapidly to osteoblasts during early postnatal periods. After 90 days of tracking in mouse, Hhip-labeled cells were incorporated as osteoblasts and osteocytes in the frontal and parietal bones, but most of them remained in the coronal suture mesenchyme. Loss of Hhip population in the coronal suture resulted in apposed osteogenic fronts and depleted suture mesenchyme at E18.5 mouse embryos (Holmes et al., 2021). Farmer et al. identified a Six2+ osteoprogenitor population in the coronal suture by performing scRNA-seq of coronal suture tissues (Farmer et al., 2021). The Six2 population contributed extensively to the mesenchyme of coronal sutures and to the osteocytes of frontal and parietal bones that are close to the suture. However, it remains largely unknown whether these 6 subpopulations overlap in identity and function, how they interact among different subpopulations of SMSCs, and how these interactions contribute to craniofacial bone development, homeostasis, repair, regeneration and diseases.
Consistent with the self-renewal and multi-lineage differentiation abilities of SMSCs, they have been shown to play indispensable roles in suture patency, injury repair and tissue regeneration. Using Gli1-CreERT2; R26-tdTomatoflox mice in which Gli1+ cells and their derivatives could be labelled with tdTomato, Zhao et al. found that Gli1 Lineage cells in the suture mesenchyme of the sagittal suture can be promptly activated to proliferate within 24 h after injury by drilling a 1 mm diameter hole 2 mm away from the sagittal suture in the parietal bone, and the majority of the cells within the injured area were Gli1 lineage after 2 weeks post-injury (Zhao et al., 2015). One month post-injury, the dura mater, periosteum and many osteocytes in the repair site were robustly labelled with tdTomato indicating that Gli1 Lineage contribute to injury repair of the calvarial bone (Zhao et al., 2015). Their further study displayed that a piece of transplanted parietal bone containing the sagittal suture can generate new dura mater and periosteum in the 4 mm2 defect region of the parietal bone of recipient nude mice, and can merge with the host bone after 1 month of transplantation, while the parietal bones without a portion of the suture fail to do so (Zhao et al., 2015). Deleting Gli1 Lineage using cre-inducible diphtheria toxin A (DTA) in one-month-old Gli1-CreERT2;DTAflox/flox mice led to coronal and frontal-premaxilla suture fusion after 1-month induction and all craniofacial sutures fusion after 2-month induction with skull growth arrest and osteoporosis. This further revealed the indispensable roles of the Gli1+ population of SMSCs in suture patency maintenance, efficient cranial bone repair and regeneration (Zhao et al., 2015).
Similar to the Gli1+ SMSCs, Axin2+ SMSCs also rapidly respond to calvarial bone injury and directly contribute to calvarial bone regeneration in response to injury in mice (Maruyama et al., 2016). Additionally, Axin2 plays an important role in maintaining suture patency (Di Pietro et al., 2020), and the targeted disruption of Axin2 in mice induces malformations of skull structures, a phenotype resembling craniosynostosis in humans (Yu et al., 2005). Similarly, Wilk et al. found that Prrx1+ SMSCs contributed to calvarial bone repair and regeneration in both NC-derived (frontal) and mesoderm-derived (parietal) bones in mice (Wilk et al., 2017). Unlike Gli1+ and Axin2+ SMSCs, the global deletion of postnatal Prrx1+ cells in mice did not lead to craniosynostosis or any other craniofacial phenotype (Wilk et al., 2017). However, ablation of Prrx1+ cells in the embryonic stage of gestation resulted in incomplete calvarial bone formation, indicating that Prrx1+ SMSCs mainly function in the earlier stage of calvarial bone development (Wilk et al., 2017). Ctsk+ mesenchyme has been shown to contribute to long bone fracture healing. Patients with CTSK mutation display abnormal suture and craniofacial bone development including delayed closure of fontanels, hypoplastic premaxilla and obtuse mandibular angle (Debnath et al., 2018; Otaify et al., 2018), yet Ctsk+ SMSCs functions in calvarial bone repair have not been tested. Hhip+ SMSCs were also required for normal coronal suture development. Hhip knockout (KO) mice displayed coronal suture dysgenesis, characterized by the reduced or absent overlap of frontal and parietal bones seen in wildtype mice with little or no intervening suture mesenchyme, resulting in more closely apposed OFs in the coronal suture (Holmes et al., 2021). Additionally, Six2+ SMSCs were reduced in the coronal suture of E14.5 and E15.5 embryos from a mouse model of Saethre-Chotzen syndrome (Twist+/−; Tcf12+/−) with coronal synostosis, suggesting the potential functions of Six2+ SMSCs in suture patency (Farmer et al., 2021).
3 FGF signaling
FGF signaling is a conserved, fundamental pathway that plays distinguished roles in embryonic development and organogenesis, metabolism homeostasis, tissue repair and regeneration, and tumor angiogenesis through the regulation of numerous cellular functions such as cell proliferation, pluripotency, migration, survival and differentiation (Boilly et al., 2000; Cao et al., 2013; Moosa and Wollnik, 2016; Mossahebi-Mohammadi et al., 2020). The FGF family includes multiple FGF ligands and receptors (FGFRs) as mentioned above. FGFRs share a highly conserved structure (Figure 2A) consisting of an extracellular domain that contains 3 immunoglobulin (Ig)-like domains (D1, D2 and D3), an acid box linker region (AB/linker) between D1 and D2, a single transmembrane domain, and a split cytoplasmic tyrosine kinase domain (Wang et al., 1995; Gong, 2014). The FGF binding sites are primarily regulated by the D2 domain, the linker region of D2/D3, and the N-terminus of D3 (Hunter, 2000; Schlessinger, 2000; Gong, 2014; Moosa and Wollnik, 2016; Farrell and Breeze, 2018). Among them, the linker region of D2/D3 is associated with regulating the affinity regulation of both FGFs and heparin/heparan sulfate (HS) (Johnson and Williams, 1992; Mohammadi et al., 2005). Additionally, the specificity of FGF binding is primarily modulated by the alternative mRNA splicing of the C-terminal half of the D3 domain in FGFRs, which generates different FGFR isoforms (McKeehan and Kan, 1994; Wang et al., 1999). For FGFR1-3, the D3 domain includes 3a and 3b or 3c domains and is encoded by exons 7 to 9. The N-terminal half of D3, named 3a, is encoded by exon 7, whereas the C-terminal half containing 3b or 3c is encoded by the alternative use of either exon 8 or 9, which generates the 3b and 3c isoforms of FGFRs, respectively (Johnson and Williams, 1992; Werner et al., 1992; Orr-Urtreger et al., 1993; Cheon et al., 1994). These two different isoforms endow FGFRs with different tissue-expression specificity and ligand-binding affinity. For example, the 3b isoform is predominantly expressed in epithelia tissues, whereas the 3c isoform is mainly expressed in mesenchymal tissues. Ligands activate either the epithelial or mesenchymal FGFR isoforms, with the exception of FGF1, which activates both isoforms (Johnson et al., 1991; Beenken and Mohammadi, 2009; Gong, 2014). Unlike FGFR1-3, FGFR4 has only one isoform (3b) because it contains only one exon encoding the C-terminal half of D3 (Kostrzewa and Müller, 1998). The other alternatively spliced FGFR isoforms are lacking the D1 and/or AB/linker domains (Johnson et al., 1990; Eisemann et al., 1991). The presence or absence of D1 is associated with FGFR autoinhibition rather than their ligand binding activity (Johnson et al., 1990; Chellaiah et al., 1999; Olsen et al., 2004; Kalinina et al., 2012). The FGFR isoforms lacking D1 or AB/linker domains promote the affinity of FGFR for FGFs and enhance the capacity of FGF signaling (Xu et al., 1992; Shi et al., 1993; Wang et al., 1995; Roghani and Moscatelli, 2007).
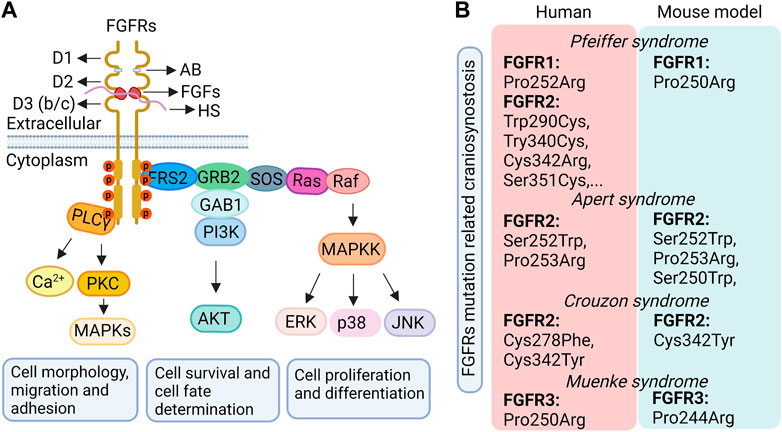
FIGURE 2. The classical FGF pathway (A) and the craniosynostosis-related syndromes caused by FGFR mutations in humans and related mouse models (B). D1, D2, D3, immunoglobulin (Ig)-like domains 1, 2, 3; AB, acid box; HS, heparin/heparan sulfate; FGFs, fibroblast growth factor ligands; FGFRs, fibroblast growth factor receptors.
FGF ligands can induce FGFR dimerization by binding to the extracellular domain of the inactive FGFR monomer. This dimerization subsequently results in the two intracellular kinase domains of the paired FGFRs phosphorylating each other on specific tyrosine residues to activate the FGFR. The activated FGFR then further activates a complex cascade of intracellular signaling events through several downstream pathways, including the Ras-MAP kinase pathway (ERK1/2, p38 and JNK kinase), the PI3 kinase/AKT pathway, and the phospholipase Cγ (PLCγ) kinase pathway (Figure 2A). The activity of these different downstream pathways depends on the cell type with the exception of the Ras-MAP kinase pathway which is activated in almost all cell types (Moosa and Wollnik, 2016). Generally, the Ras-MAP kinase pathway, the main downstream pathway of FGF signaling, is associated with cellular proliferation and differentiation; the PI3 kinase/AKT pathway is associated with cellular survival and cell fate determination and, occasionally, cell polarity; and the PLCγ kinase pathway impacts cell morphology, migration, and adhesion (Teven et al., 2014). Most of these downstream phosphorylation transduction pathways target transcription factors within the nuclei to influence cell proliferation, stemness, migration, survival, and differentiation by regulating gene expression (Moosa and Wollnik, 2016).
FGF signaling contributes to the development of most craniofacial structures, such as the development and outgrowth of the facial primordia, craniofacial skeletogenesis, palatogenesis, as well as development of submandibular salivary gland, teeth, eye lids, craniofacial muscles, and muscular tongue (Nie et al., 2006; Prochazkova et al., 2018; Weng et al., 2018). Perturbation of FGF signaling is involved in various craniofacial abnormalities, including facial or palatal cleft, midface agenesis, mandibular hypoplasia, open eyelids at an early postnatal stage, and craniosynostosis (Ibrahimi et al., 2004; Rice et al., 2004; Wang et al., 2013; Prochazkova et al., 2018; Ray et al., 2020; Xu et al., 2020).
4 FGF signaling in cranial sutures
Throughout cranial suture, SMSCs participate in cranial bone growth and development, homeostatic maintenance, injury repair, and cranial suture patency or fusion, which are precisely orchestrated by fine-tuned signals. Studies on patients with syndromic craniosynostosis and a series of mouse studies have indicated a pivotal role of FGF signaling in the development of cranial sutures.
FGF ligands and FGFRs of FGF signaling have distinct spatiotemporal expression patterns in the cranial sutures and SMSCs, depending on their specific functions. FGF ligand family, including FGF2, FGF4, and FGF18, plays important roles in embryonic or postnatal cranial suture development (Moosa and Wollnik, 2016). Among these, FGF18 is the first to be detected in calvarial mesenchymal cells and is later expressed in the osteogenic mesenchyme and differentiated osteoblasts on the endosteal and periosteal surface of skull bones (Moosa and Wollnik, 2016). Fgf18-deficient mice exhibited delayed suture closure with decreased proliferation of osteogenic mesenchymal cells and delayed terminal differentiation of osteoblasts (Ohbayashi et al., 2002). The spatiotemporal distribution of FGF2 was distinct among different cranial sutures. For example, FGF2 expression was significantly higher in posterior frontal SMSCs and the underlying dura than in sagittal SMSCs and the underlying dura during the onset of PFS fusion in mice (Gosain et al., 2004). Of note, Mehrara et al. observed that, FGF2 expression dramatically increased in PFS tissues throughout the process of PFS fusion and reduced after PFS fusion, suggesting that FGF2 benefits PFS fusion (Mehrara et al., 1998). However, FGF2 expression in the sagittal suture tissues was minimal all times (Mehrara et al., 1998). In rat organ culture studies, PFS treated with FGF2 showed significantly increased fusion on the dura side of the suture compared with the non-treated controls (Moursi et al., 2002). In addition, increasing FGF2 activity also induced coronal suture fusion in rats and mice (Iseki et al., 1999; Greenwald et al., 2001).
In addition to FGF2, FGF3 and FGF4 also play crucial roles during cranial suture development. When the ectopic expression of FGF3 and FGF4 were induced by retroviral insertion in the cranial suture region of mice, extensive premature closure was observed in the cranial sutures, including the metopic, sagittal, coronal, interparietal/occipital and intermaxillary sutures (Carlton et al., 1998). The ex vivo culture of E15 mouse calvarial explants with FGF4 bonded beads showed that FGF4 accelerated sagittal sutural closure when beads were inserted in the osteogenic fronts but not when the beads were inserted in the mid-sutural mesenchyme (Kim et al., 1998). Additionally, in humans, an FGF9 missense mutation led to craniosynostosis with multiple synostosis. This phenotype was mimicked in mice with a spontaneous heterozygous FGF9 mutation, suggesting that FGF9 plays an important role during cranial suture development (Murakami et al., 2002; Rodriguez-Zabala et al., 2017). Numerous studies have shown that FGFRs, mainly FGFR1-3, are also indispensable in the regulation of cranial suture development. FGFR1 is primarily expressed in the osteoblast and mesenchyme of the calvarium and is associated with osteoprogenitor differentiation. FGFR2 is mainly expressed in proliferating osteogenic stem cells and is involved in regulating cell proliferation. Accordingly, in mice, the onset of osteoprogenitor differentiation in the coronal suture is preceded by the downregulation of FGFR2 expression and the upregulation of FGFR1 expression (Iseki et al., 1999). Consistently, the expression of a dominant-negative FGFR1 gene in rat calvaria inhibits suture fusion (Greenwald et al., 2001). These data indicated that the gradients of FGFR1 and FGFR2 expression may play important roles in balancing the proliferation and differentiation of osteoprogenitor cells in the cranial suture (Ornitz and Marie, 2002). Iseki et al. also found that FGFR1 expression was downregulated following the upregulation of osteoblast differentiation markers in mice, indicating that FGFR1 is related to the osteogenic differentiation process but is not involved in maintaining the differentiation stage (Iseki et al., 1999). However, the detailed mechanism needs to be further studied. FGFR3, which is expressed at a later stage than FGFR1 and FGFR2 in mice, is expressed at low levels in the OFs of suture, and is also expressed in the chondrogenic regions of the skeletogenic membrane, including a thin plate of cartilage underlying the coronal suture (Iseki et al., 1999), suggesting a dual role of FGFR3 in both osteoblasts and chondrocytes during mouse skull development. The FGFR4 expression has been confined to the cranial musculature (Iseki et al., 1999), while its specific role in cranial suture development remains unknown.
To dissect the mechanisms underlying FGF-FGFR-mediated cranial suture development, the downstream pathways of FGF-FGFR-mediated signaling including Ras-MAP kinase, PI3 kinase-AKT and PLCγ-PKC pathways were studied. Blocking of the ERK pathway using an inhibitor (PD98059) repressed FGF2-induced cranial suture closure in cultured mouse calvaria, and decreased osteoblast differentiation (Kim et al., 2003). Repression of p-ERK1/2 activity in FGFR2+/S252W mutant mice using U0126 significantly inhibited craniosynostosis (Shukla et al., 2007). A study from Holmes et al. showed that p-AKT and p-p38 were increased in the calvarial tissues of newborn FGFR2+/S252W mutant mice (Holmes et al., 2009). Wang et al. discovered that compared with controls, FGFR2+/P253R mutant mice had increased levels of p-p38 and p-ERK1/2 in the neurocranium, together with enhanced osteogenic differentiation and reduced proliferation but without apoptosis changes in the coronal suture (Wang et al., 2010). However, p-AKT and PKCα were not obviously changed in these mutant mice (Wang et al., 2010). Additionally, increased p-ERK1/2 were found in the prematurely fused coronal suture of FGFR2c+/C342γ gain-of-function mutant mice, along with enhanced cellularity and dysregulated differentiation of osteoblasts (Pfaff et al., 2016). Together, these results suggest that the downstream pathways of FGF-FGFR-mediated signaling, especially the Ras-MAP kinase pathway, play important roles in FGF-FGFR-mediated cranial suture development and are context-dependent. However, further studies are needed.
5 FGF signaling in craniosynostosis
Given the complicated functions of FGF signaling in cranial sutures, it is no surprise that its dysfunction gives rise to various craniofacial related diseases. Familial studies have revealed that patients with craniosynostosis primarily show a gain-of-function mutation within the gene region of FGFRs responsible for the linker between the D2 and D3 extracellular domains (Figure 2B). This type of mutation may activate FGF signaling either in a ligand-dependent manner by changing the affinity and specificity of FGFRs to their corresponding FGF ligands (Ibrahimi et al., 2001; Ibrahimi et al., 2004; Moosa and Wollnik, 2016), or in a ligand-independent manner by enhancing FGFR dimerization (Kan et al., 2002; Moosa and Wollnik, 2016). As a result, the proliferation, differentiation and/or apoptosis of cells in the cranial suture are changed resulting in craniosynostosis (Passos-Bueno et al., 1999; Teven et al., 2014). For instance, in humans, Apert syndrome, characterized by premature fusion of the bilateral coronal sutures and severe syndactyly of the feet and hands, is caused by Ser252Trp and Pro253Arg mutations of the FGFR2 gene in the D2-D3 linker region, which leads to FGFR2 gain-of-function in a ligand-dependent manner (Slaney et al., 1996; Ferreira et al., 1999; Ibrahimi et al., 2001; Andreou et al., 2006; Ko, 2016; Kunwar et al., 2017). Pfeiffer syndrome in humans, which shows similar craniofacial anomalies to those seen in Apert syndrome along with big toes and broad radially deviated thumbs (Giancotti et al., 2017), is due to a mutation in either FGFR1 (Pro252Arg) or FGFR2 (Trp290Cys, Try340Cys, Cys342Arg, or Ser351Cys, etc.) (Azoury et al., 2017). The FGFR1 (Pro252Arg) mutation leads to a bulkier residue that enhances the binding affinity of the receptor to the ligand to increase receptor activation (Ibrahimi et al., 2004). The FGFR2 mutation mainly causes the ligand-independent activation of the receptor by leading to an unpaired cysteine residue that forms an intermolecular disulfide bond (Cornejo-Roldan et al., 1999; Lajeunie et al., 2006). Muenke syndrome in humans, characterized by craniosynostosis with uni- or bicoronal synostosis, comes from an FGFR3 Pro250Arg mutation resulting in the increased binding affinity of FGFR3 to its ligand (Muenke et al., 1997; Ibrahimi et al., 2004), such as FGF9, by the substitution of a bulkier residue. In addition, other craniosynostosis syndromes, including Jackson-Weiss syndrome and Crouzon syndrome, are also caused by gain-of-function mutations in the D2-D3 linker region of FGFR1 or FGFR2 in a ligand-dependent or independent manner (Moosa and Wollnik, 2016). However, FGFR2 mutations contribute to the majority of craniosynostosis syndromes in humans (Ornitz and Marie, 2002).
Results from animal studies have further supported the critical roles of FGF signaling in cranial suture development. As mentioned above, in mice, FGF2, FGF3, and FGF4 overexpression lead to suture synostosis, and FGF18 loss-of-function results in delayed suture closure. Gain-of-function mutations of FGFR1 and FGFR2 in mice also impact cranial suture development (Figure 2B). FGFR1 P250R mutation in mice, which is orthologous to the Pfeiffer syndrome mutation (FGFR1 P252R) in humans, leads to the premature fusion of calvarial sutures including frontal, sagittal, and coronal sutures (Zhou et al., 2000). FGFR2+/S250W transgenic mouse, an Apert syndrome mouse model, showed premature closure of the coronal suture (Chen et al., 2003). Additionally, Wang et al. observed that FGFR2+/S252W mutant mice, another Apert syndrome mouse model with FGFR2 gain-of-function mutation, showed proximate OFs of two parietal bones and abnormal osteoid deposited between them when compared with controls; while the interfrontal suture of mutant mice exhibited a broad gap between the OFs of frontal bones when compared with control ones (Wang et al., 2005). Concomitantly, they found that FGFR2+/P253R mutant mice, another FGFR2 gain-of-function mutation that commonly occurs in patients with Apert syndrome, had cranial features that resembled those shown in FGFR2 +/S252W mutant mice (Wang et al., 2010). Additionally, FGFR3Y367C/+ (FGFR3 gain-of-function) mutant mice also showed partial premature fusion of coronal sutures and impaired frontal bones, suggesting important roles of FGFR3 in suture patency and membranous ossification (Di Rocco et al., 2014). Nevertheless, FGFR3P244R/+ mutant mice, a model of Muenke syndrome with FGFR3 gain-of-function, displayed mild skull deformities and rarely showed premature fusion of the coronal suture (Twigg et al., 2009). FGFR3 KO mice did not show obvious calvarial bone defects (Valverde-Franco et al., 2004). Furthermore, mice with FGFR3 P244R mutation (equivalent to the human P250R mutation), a genetic model for Muenke syndrome, show a rounded skull and shortened snout with dental malocclusion which are similar to Muenke syndrome features in humans. However, coronal craniosynostosis in human patients is not reliably reproduced in this mouse model (Twigg et al., 2009). This suggests different functions of FGFR3 between mice and humans. Whereas the detailed pathological mechanism underlying FGF/FGFR related craniosynostosis is still poorly understood.
6 FGF signaling crosstalks with other signals to regulate cranial suture development
As a pivotal regulatory signaling that functions during cranial suture development, FGF signaling broadly crosstalks with many other transcription factors and signals to orchestrate complicated processes. For example, Twist, a basic helix-loop-helix transcription factor, is expressed in SMSCs and regulates osteoblast differentiation and cells apoptosis (Howard et al., 1997; Yousfi et al., 2002). The haploinsufficiency of Twist leads to premature fusion of the cranial suture (Yousfi et al., 2002). In contrast, trisomy at the human TWIST locus results in delayed suture closure (Stankiewicz et al., 2001). In addition, the distribution pattern of FGFR2 was changed in the sagittal suture of Twist+/− mice when compared with wildtype mice (Rice et al., 2000). In wildtype mice, FGFR2 was mainly expressed in osteoblasts of the OFs and weakly and diffusely expressed in SMSCs in the sagittal suture. However, in Twist+/− mice, FGFR2 localized more in the mid-sutural mesenchyme. Additionally, their study also displayed that exogenous FGF2 in the mid-suture mesenchyme stimulated Twist expression in ex vivo cultured sagittal sutures to inhibit osteoblast differentiation of suture mesenchyme (Rice et al., 2000). Accordingly, they brought the point that Twist could be a potential transcriptional regulator that modulates the inhibitory effects of FGF2 on osteoblast differentiation (Rice et al., 2000). MSX1 and MSX2, which are homeobox-containing transcription factors, are expressed in the mesenchyme and are involved in the differentiation of NC-derived calvarial bones (Ornitz and Marie, 2002). MSX2 overexpression in mice or mutation in humans leads to craniosynostosis with an increased osteoprogenitor population. Conversely, MSX2 haploinsufficiency in mice or humans results in reduced cell proliferation and delays suture closure, together with defective skull bone ossification (Ornitz and Marie, 2002). In mouse and rat calvarial cells, MSX2 was identified as an upstream factor to inhibit the osteogenic activity of FGF2. In addition, FGF4 could enhance MSX1 expression and cell proliferation. Runx2/Cbfa1 is a key transcription factor to initiate mesenchymal stem cells to differentiate into osteoblasts. Heterozygous loss-of-function mutation of RUNX2 in humans is associated with cleidocranial dysplasia (CCD) with open fontanelles. Similarly, open fontanelles were also observed in Runx2+/− mutant mice with disturbed sagittal suture formation (Qin et al., 2019). Interestingly, Qin et al. found that Runx2 loss-of-function in mice led to reduced proliferation and condensation of SMSCs (Qin et al., 2019). They further discovered that the expression of FGF signaling related genes, including FGFR1, FGFR2 and FGFR3, was significantly reduced in the suture regions but not in the calvarial bone tissues of Runx2+/− mutant mice. In addition, the expression of several other signaling factors was also decreased, such as Gli1, Ptch1 and Ihh in Hedgehog signaling, and Tcf7, Wnt10b and Wnt1 in Wnt signaling, suggesting the important role of coordinated signaling in SMSCs during cranial suture development (Qin et al., 2019). Additionally, TGF-β1, similar to FGF2, is upregulated in the PFS mesenchyme and dura during the closure of the PFS (Most et al., 1998; Gosain et al., 2004). Sasaki et al. found that FGF acts downstream of TGF-β signaling to promote cranial NC cell proliferation during frontal bone development, and FGF2 could rescue the proliferation defect caused by Tgfbr2 mutation (Sasaki et al., 2006). BMP signaling is required for osteoblast differentiation and may function in concert with FGFs to control calvarial bone development (Schliermann and Nickel, 2018). Moreover, Jiang et al. revealed that BMP2 was crucial for the FGF2-dependent later-stage osteoblastic differentiation of cranial suture cells that were isolated from bone fragments around the coronal and sagittal sutures of newborn rats. They found that the expression of BMP2 could be initiated by FGF2 in a time and dose-dependent manner (Jiang et al., 2015). FGF2 treatment may reduce the early osteoblast differentiation marker, Col1a1, expression, while enhancing the late markers (Alp, Ocn and Bsp) expression to promote mineralization. BMP2 inhibition could reduce the induction of FGF2 to later-stage osteoblast differentiation of cranial suture cells (Jiang et al., 2015). Recently, Min Swe et al. found that Lrp5 and Lrp6, co-receptors of Wnt/β-catenin signaling, were aberrantly activated in the developing coronal sutures of Apert syndrome (FGFR2+/S252W) mouse models (Min Swe et al., 2021). Lrp5 and Lrp6 knockdown dramatically decreased osteoblast differentiation markers (Runx2, Col1a1, Ocn and Alp) expression in cultured cells isolated from coronal sutures of FGFR2+/S252W mice, indicating an interaction between FGFR2 and Wnt/β-catenin signaling (Min Swe et al., 2021). The FGF signaling pathway has also been found to interact with other signaling pathways, such as Notch, Hedgehog, Hippo, and mechanical signaling pathways, which also play important roles in the proliferation, differentiation, and apoptosis of osteoprogenitors and osteoblasts (Byun et al., 2014; Li et al., 2020; Zhao et al., 2021; Zhao et al., 2022). However, whether these interactions play a role during cranial suture development and how they function in SMSC proliferation and differentiation remain largely unknown.
7 Conclusion and future prospectives
SMSCs are located in the cranial suture and are characterized as a heterogeneous stem cell population. SMSCs have a distinct ability to self-renew and differentiate into multiple cell lineages, including osteoblasts and chondrocytes, in a tempo-spatial dependent manner. SMSCs make significant contributions to craniofacial development, suture patency maintenance, and cranial bone repair and regeneration. It has been established that the proliferation, differentiation, and apoptosis of SMSCs and their derivatives are associated with multiple factors and signaling pathways, including Twist, Msx1/2, Gli1, Axin2, as well as FGF, Wnt, Hedgehog, NOTCH, Hippo, and mechanical signaling to orchestrate SMSCs and cranial suture development. Any perturbation of these factors and pathways may open a window for an array of diseases caused by abnormal development of SMSCs and cranial sutures, especially those characterized by craniosynostosis.
The FGF signaling pathway is a highly conserved, fundamental pathway that regulates numerous processes, ranging from embryonic development and organogenesis to adult tissue repair and regeneration. Dysfunction of FGF signaling has been linked to multiple human diseases (Xie et al., 2020), such as dwarfism syndrome, chronic kidney diseases (CKD), various tumors, and craniosynostosis, Clinical and experimental evidence showed that FGF signaling controls cranial suture development likely through modulating a balance among the proliferation, differentiation, and apoptosis of cranial sutural cells in a tissue- and stage-specific manner, but this still needs further study. Notably, most of the FGF signaling related craniosynostosis diseases are thought to be FGFR gain-of-function mutations, either in a ligand-dependent manner by altering the ligand-binding affinity or specificity, or in a ligand-independent manner through stabilizing intermolecular disulfide bonds to constitutively activate the receptor and signaling. Reports have shown that the majority of craniosynostosis syndromes were related to FGFR2 gain-of-function mutations. However, it is worth noting that different sutures respond to FGF-FGFR signaling differently. Compared with other sutures, craniosynostosis mainly occurs in the coronal sutures in FGFR gain-of-function mutant animal models, such as FGFR2+/S250W and FGFR2+/S252W transgenic mice, and FGFR3Y367C/+ mutant murine models (Wang et al., 2005). This may be due to the spatiotemporal- and tissue-specific expression pattern of FGFs and FGFRs as well as the different embryogenic origins of suture cells that have different responses to FGF signaling. As mentioned above, SMSCs of the coronal suture are mostly derived from mesodermal cells while the frontal and sagittal sutures are mainly derived from NC cells. NC-derived mesenchyme showed higher intrinsic proliferation and osteogenic abilities than mesoderm-derived mesenchyme, and expression of FGF18 and FGFR3 was higher in NC-derived MSCs than in mesoderm-derived MSCs. This leads to varying responses by cells of different embryonic origins to FGF signaling that is associated with cell proliferation, differentiation and apoptosis, representing an interesting field for further studies. Robust studies have been performed to explore the pathological processes of craniosynostosis and significant progress has been made. However, the detailed molecular mechanism of how FGFR mutations impact downstream molecules and signaling pathways leading to various diseases and how such molecules and pathways provide feedback to regulate FGF signaling is still poorly understood due to the intricate nature of FGFs and FGFRs and their multiple downstream pathways, as well as complicated SMSCs. Taken together, these findings described the populations and characteristics of SMSCs and indicated the complicated and critical roles of the FGF pathway in the development of the cranial suture and SMSCs, meanwhile highlighting the significance of studying the FGF pathway in cranial suture development and related diseases, especially craniosynostosis. We also summarized the broad crosstalk between the FGF pathway and other factors and pathways during cranial suture development and related diseases, which sheds light on the mechanistic studies of FGF-related craniofacial diseases. However, further investigations of the interactions and functions of the SMSC population, and the detailed mechanism underlying how environment transcription factors and signaling pathways coordinate with FGF signaling to orchestrate cranial suture and SMSCs development or cause suture-related diseases are urgently demanded. These may contribute to the development of therapeutic interventions with SMSCs for cranial diseases. In summary, the FGF signaling pathway has pivotal functions in cranial suture and SMSCs development and warrants further investigation in the mechanisms underlying cranial suture development and related diseases with the hopes of improving current diagnostic and therapeutic options.
Author contributions
XZ wrote the initial manuscript. XZ, SE, KS and JW revised the manuscript. XZ made the figure. All authors contributed to the article and approved the submitted version.
Funding
The National Institutes of Health (K01DE026561, and R01DE029014to JW).
Acknowledgments
The authors thank Nicole Stancel, PhD, ELS (D) of the Department of Scientific Publications at The Texas Heart Institute, for providing editorial assistance with the manuscript, as well as members of the JW laboratory at The University of Texas Health Science Center at Houston, for their contributions. The authors also apologize to colleagues whose work was not cited due to space limitations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
SMSC, Suture mesenchymal stem cell; NC, neural crest; OFs, osteogenic fronts; CCD, Cleidocranial dysplasia; FGFRs, fibroblast growth factor receptors; MSCs, mesenchymal stem cells; PFS, posterior frontal suture; MAPK, mitogen-activated protein kinases.
References
Andreou, A., Lamy, A., Layet, V., Cailliez, D., Gobet, F., Pfister, C., et al. (2006). Early-onset low-grade papillary carcinoma of the bladder associated with Apert syndrome and a germline FGFR2 mutation (Pro253Arg). Am. J. Med. Genet. Part A 140, 2245–2247. doi:10.1002/ajmg.a.31430
Azoury, S. C., Reddy, S., Shukla, V., and Deng, C.-X. (2017). Fibroblast growth factor receptor 2 (FGFR2) mutation related syndromic craniosynostosis. Int. J. Biol. Sci. 13, 1479–1488. doi:10.7150/ijbs.22373
Barnes, E. (2012). Atlas of developmental field anomalies of the human skeleton: A paleopathology perspective. United States: John Wiley & Sons.
Beenken, A., and Mohammadi, M. (2009). The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8, 235–253. doi:10.1038/nrd2792
Behr, B., Longaker, M. T., and Quarto, N. (2013). Absence of endochondral ossification and craniosynostosis in posterior frontal cranial sutures of Axin2−/− mice. PLoS One 8, e70240. doi:10.1371/journal.pone.0070240
Behr, B., Longaker, M. T., and Quarto, N. (2010). Differential activation of canonical Wnt signaling determines cranial sutures fate: A novel mechanism for sagittal suture craniosynostosis. Dev. Biol. 344, 922–940. doi:10.1016/j.ydbio.2010.06.009
Boilly, B., Vercoutter-Edouart, A., Hondermarck, H., Nurcombe, V., and Le Bourhis, X. (2000). FGF signals for cell proliferation and migration through different pathways. Cytokine & growth factor Rev. 11, 295–302. doi:10.1016/s1359-6101(00)00014-9
Byun, M. R., Kim, A. R., Hwang, J.-H., Kim, K. M., Hwang, E. S., and Hong, J.-H. (2014). FGF2 stimulates osteogenic differentiation through ERK induced TAZ expression. Bone 58, 72–80. doi:10.1016/j.bone.2013.09.024
Cao, X.-C., Zhang, W.-R., Cao, W.-F., Liu, B.-W., Zhang, F., Zhao, H.-M., et al. (2013). Aquaporin3 is required for FGF-2-induced migration of human breast cancers. PloS one 8, e56735. doi:10.1371/journal.pone.0056735
Carlton, M. B., Colledge, W. H., and Evans, M. J. (1998). Crouzon-like craniofacial dysmorphology in the mouse is caused by an insertional mutation at the Fgf3/Fgf4 locus. Dev. Dyn. official Publ. Am. Assoc. Anatomists 212, 242–249.
Carver, E. A., Oram, K. F., and Gridley, T. (2002). Craniosynostosis in twist heterozygous mice: A model for saethre-chotzen syndrome. Anatomical Rec. 268, 90–92.
Chellaiah, A., Yuan, W., Chellaiah, M., and Ornitz, D. M. (1999). Mapping ligand binding domains in chimeric fibroblast growth factor receptor molecules: Multiple regions determine ligand binding specificity. J. Biol. Chem. 274, 34785–34794. doi:10.1074/jbc.274.49.34785
Chen, L., Li, D., Li, C., Engel, A., and Deng, C.-X. (2003). A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone 33, 169–178. doi:10.1016/s8756-3282(03)00222-9
Cheon, H.-G., LaRochelle, W. J., Bottaro, D. P., Burgess, W. H., and Aaronson, S. A. (1994). High-affinity binding sites for related fibroblast growth factor ligands reside within different receptor immunoglobulin-like domains. Proc. Natl. Acad. Sci. 91, 989–993. doi:10.1073/pnas.91.3.989
Clendenning, D. E., and Mortlock, D. P. (2012). The BMP ligand Gdf6 prevents differentiation of coronal suture mesenchyme in early cranial development. PloS one 7, e36789. doi:10.1371/journal.pone.0036789
Cohen, M. M. (1993). Sutural biology and the correlates of craniosynostosis. Am. J. Med. Genet. 47, 581–616. doi:10.1002/ajmg.1320470507
Cornejo-Roldan, L. R., Roessler, E., and Muenke, M. (1999). Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum. Genet. 104, 425–431. doi:10.1007/s004390050979
Debnath, S., Yallowitz, A. R., McCormick, J., Lalani, S., Zhang, T., Xu, R., et al. (2018). Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139. doi:10.1038/s41586-018-0554-8
Di Pietro, L., Barba, M., Prampolini, C., Ceccariglia, S., Frassanito, P., Vita, A., et al. (2020). GLI1 and AXIN2 are distinctive markers of human calvarial mesenchymal stromal cells in nonsyndromic craniosynostosis. Int. J. Mol. Sci. 21, 4356. doi:10.3390/ijms21124356
Di Rocco, F., Biosse Duplan, M., Heuzé, Y., Kaci, N., Komla-Ebri, D., Munnich, A., et al. (2014). FGFR3 mutation causes abnormal membranous ossification in achondroplasia. Hum. Mol. Genet. 23, 2914–2925. doi:10.1093/hmg/ddu004
Doherty, E. S., Lacbawan, F., Hadley, D. W., Brewer, C., Zalewski, C., Kim, H. J., et al. (2007). Muenke syndrome (FGFR3-related craniosynostosis): Expansion of the phenotype and review of the literature. Am. J. Med. Genet. Part A 143, 3204–3215. doi:10.1002/ajmg.a.32078
Dong, X.-H., Zhang, M.-Z., Lai, C.-Z., Li, C.-C., Du, L., Song, G.-D., et al. (2021). Dura cells in the etiopathogenesis of Crouzon syndrome: The effects of FGFR2 mutations in the dura cells on the proliferation of osteoblasts through the hippo/YAP mediated transcriptional regulation pathway. Am. J. Transl. Res. 13, 11255–11270.
Doro, D. H., Grigoriadis, A. E., and Liu, K. J. (2017). Calvarial suture-derived stem cells and their contribution to cranial bone repair. Front. Physiology 8, 956. doi:10.3389/fphys.2017.00956
Doro, D., Liu, A., Grigoriadis, A. E., and Liu, K. J. (2019). The osteogenic potential of the neural crest lineage may contribute to craniosynostosis. Mol. Syndromol. 10, 48–57. doi:10.1159/000493106
Eisemann, A., Ahn, J., Graziani, G., Tronick, S., and Ron, D. (1991). Alternative splicing generates at least five different isoforms of the human basic-FGF receptor. Oncogene 6, 1195–1202.
Farmer, D. J. T., Mlcochova, H., Zhou, Y., Koelling, N., Wang, G., Ashley, N., et al. (2021). The developing mouse coronal suture at single-cell resolution. Nat. Commun. 12, 4797–4814. doi:10.1038/s41467-021-24917-9
Farooq, M., Khan, A. W., Kim, M. S., and Choi, S. (2021). The role of fibroblast growth factor (FGF) signaling in tissue repair and regeneration. Cells 10, 3242. doi:10.3390/cells10113242
Farrell, B., and Breeze, A. L. (2018). Structure, activation and dysregulation of fibroblast growth factor receptor kinases: Perspectives for clinical targeting. Biochem. Soc. Trans. 46, 1753–1770. doi:10.1042/BST20180004
Ferreira, J., Carter, S., Bernstein, P., Jabs, E., Glickstein, J., Marion, R., et al. (1999). Second-trimester molecular prenatal diagnosis of sporadic Apert syndrome following suspicious ultrasound findings. Ultrasound Obstetrics Gynecol. 14, 426–430. doi:10.1046/j.1469-0705.1999.14060426.x
Giancotti, A., D’Ambrosio, V., Marchionni, E., Squarcella, A., Aliberti, C., La Torre, R., et al. (2017). Pfeiffer syndrome: Literature review of prenatal sonographic findings and genetic diagnosis. J. Maternal-Fetal Neonatal Med. 30, 2225–2231. doi:10.1080/14767058.2016.1243099
Gong, S. G. (2014). Isoforms of receptors of fibroblast growth factors. J. Cell. physiology 229, 1887–1895. doi:10.1002/jcp.24649
Gosain, A. K., Recinos, R. F., Agresti, M., and Khanna, A. K. (2004). TGF-beta1, FGF-2, and receptor mRNA expression in suture mesenchyme and dura versus underlying brain in fusing and nonfusing mouse cranial sutures. Plastic Reconstr. Surg. 113, 1675–1684. doi:10.1097/01.prs.0000117362.33347.43
Gotoh, N. (2009). Control of stemness by fibroblast growth factor signaling in stem cells and cancer stem cells. Curr. stem Cell Res. Ther. 4, 9–15. doi:10.2174/157488809787169048
Greenwald, J. A., Mehrara, B. J., Spector, J. A., Warren, S. M., Fagenholz, P. J., Smith, L. P., et al. (2001). In vivo modulation of FGF biological activity alters cranial suture fate. Am. J. pathology 158, 441–452. doi:10.1016/s0002-9440(10)63987-9
Herring, S. W. (2008). Mechanical influences on suture development and patency. Craniofacial sutures 12, 41–56. doi:10.1159/0000115031
Holmes, G., Gonzalez-Reiche, A. S., Lu, N., Zhou, X., Rivera, J., Kriti, D., et al. (2020). Integrated transcriptome and network analysis reveals spatiotemporal dynamics of calvarial suturogenesis. Cell. Rep. 32, 107871. doi:10.1016/j.celrep.2020.107871
Holmes, G., Gonzalez-Reiche, A. S., Saturne, M., Motch Perrine, S. M., Zhou, X., Borges, A. C., et al. (2021). Single-cell analysis identifies a key role for Hhip in murine coronal suture development. Nat. Commun. 12, 7132–7216. doi:10.1038/s41467-021-27402-5
Holmes, G., Rothschild, G., Roy, U. B., Deng, C.-X., Mansukhani, A., and Basilico, C. (2009). Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev. Biol. 328, 273–284. doi:10.1016/j.ydbio.2009.01.026
Howard, T. D., Paznekas, W. A., Green, E. D., Chiang, L. C., Ma, N., Luna, R. I. O. D., et al. (1997). Mutations in TWIST, a basic helix–loop–helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 15, 36–41. doi:10.1038/ng0197-36
Ibrahimi, O. A., Eliseenkova, A. V., Plotnikov, A. N., Yu, K., Ornitz, D. M., and Mohammadi, M. (2001). Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. 98, 7182–7187. doi:10.1073/pnas.121183798
Ibrahimi, O. A., Zhang, F., Eliseenkova, A. V., Linhardt, R. J., and Mohammadi, M. (2004). Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum. Mol. Genet. 13, 69–78. doi:10.1093/hmg/ddh011
Iseki, S., Wilkie, A., and Morriss-Kay, G. (1999). Fgfr1 and Fgfr2 have distinct differentiation-and proliferation-related roles in the developing mouse skull vault. Development 126, 5611–5620. doi:10.1242/dev.126.24.5611
Ishii, M., Sun, J., Ting, M.-C., and Maxson, R. E. (2015). The development of the calvarial bones and sutures and the pathophysiology of craniosynostosis. Curr. Top. Dev. Biol. 115, 131–156. doi:10.1016/bs.ctdb.2015.07.004
Jenkins, D., Seelow, D., Jehee, F. S., Perlyn, C. A., Alonso, L. G., Bueno, D. F., et al. (2007). RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 80, 1162–1170. doi:10.1086/518047
Jiang, T., Ge, S., Shim, Y. H., Zhang, C., and Cao, D. (2015). Bone morphogenetic protein is required for fibroblast growth factor 2-dependent later-stage osteoblastic differentiation in cranial suture cells. Int. J. Clin. Exp. pathology 8, 2946–2954.
Jiang, X., Iseki, S., Maxson, R. E., Sucov, H. M., and Morriss-Kay, G. M. (2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241, 106–116. doi:10.1006/dbio.2001.0487
Jing, D., Chen, Z., Men, Y., Yi, Y., Wang, Y., Wang, J., et al. (2022). Response of Gli1+ suture stem cells to mechanical force upon suture expansion. J. Bone Mineral Res. 37, 1307–1320. doi:10.1002/jbmr.4561
Johnson, D. E., Lee, P. L., Lu, J., and Williams, L. T. (1990). Diverse forms of a receptor for acidic and basic fibroblast growth factors. Mol. Cell. Biol. 10, 4728–4736. doi:10.1128/mcb.10.9.4728
Johnson, D. E., Lu, J., Chen, H., Werner, S., and Williams, L. T. (1991). The human fibroblast growth factor receptor genes: A common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol. 11, 4627–4634. doi:10.1128/mcb.11.9.4627
Johnson, D. E., and Williams, L. T. (1992). Structural and functional diversity in the FGF receptor multigene family. Adv. cancer Res. 60, 1–41. doi:10.1016/s0065-230x(08)60821-0
Johnson, D., Iseki, S., Wilkie, A., and Morriss-Kay, G. (2000). Expression patterns of Twist and Fgfr1,-2 and-3 in the developing mouse coronal suture suggest a key role for twist in suture initiation and biogenesis. Mech. Dev. 91, 341–345. doi:10.1016/s0925-4773(99)00278-6
Kalinina, J., Dutta, K., Ilghari, D., Beenken, A., Goetz, R., Eliseenkova, A. V., et al. (2012). The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 20, 77–88. doi:10.1016/j.str.2011.10.022
Kan, S.-h., Elanko, N., Johnson, D., Cornejo-Roldan, L., Cook, J., Reich, E. W., et al. (2002). Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am. J. Hum. Genet. 70, 472–486. doi:10.1086/338758
Kim, H.-J., Rice, D., Kettunen, P. J., and Thesleff, I. (1998). FGF-BMP-and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 125, 1241–1251. doi:10.1242/dev.125.7.1241
Kim, H. J., Lee, M. H., Park, H. S., Park, M. H., Lee, S. W., Kim, S. Y., et al. (2003). Erk pathway and activator protein 1 play crucial roles in FGF2-stimulated premature cranial suture closure. Dev. Dyn. 227, 335–346.
Ko, J. M. (2016). Genetic syndromes associated with craniosynostosis. J. Korean Neurosurg. Soc. 59, 187–191. doi:10.3340/jkns.2016.59.3.187
Kostrzewa, M., and Müller, U. (1998). Genomic structure and complete sequence of the human FGFR4 gene. Mamm. genome 9, 131–135. doi:10.1007/s003359900703
Kumar, V., Goutam, R. S., Park, S., Lee, U., and Kim, J. (2021). Functional roles of FGF signaling in early development of vertebrate embryos. Cells 10, 2148. doi:10.3390/cells10082148
Kunwar, F., Tewari, S., and Bakshi, S. R. (2017). Apert syndrome with S252W FGFR2 mutation and characterization using phenomizer: An Indian case report. J. Oral Biol. Craniofacial Res. 7, 67–71. doi:10.1016/j.jobcr.2016.07.002
Lajeunie, E., Heuertz, S., El Ghouzzi, V., Martinovic, J., Renier, D., Le Merrer, M., et al. (2006). Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. Eur. J. Hum. Genet. 14, 289–298. doi:10.1038/sj.ejhg.5201558
Lenton, K. A., Nacamuli, R. P., Wan, D. C., Helms, J. A., and Longaker, M. T. (2005). Cranial suture biology. Curr. Top. Dev. Biol. 66, 287–328. doi:10.1016/S0070-2153(05)66009-7
Li, B., Wang, Y., Fan, Y., Ouchi, T., Zhao, Z., and Li, L. (2021). Cranial suture mesenchymal stem cells: Insights and advances. Biomolecules 11, 1129. doi:10.3390/biom11081129
Li, S., Quarto, N., and Longaker, M. T. (2010). Activation of FGF signaling mediates proliferative and osteogenic differences between neural crest derived frontal and mesoderm parietal derived bone. PloS one 5, e14033. doi:10.1371/journal.pone.0014033
Li, W., Zhao, J., Wang, J., Sun, L., Xu, H., Sun, W., et al. (2020). ROCK-TAZ signaling axis regulates mechanical tension-induced osteogenic differentiation of rat cranial sagittal suture mesenchymal stem cells. J. Cell. Physiology 235, 5972–5984. doi:10.1002/jcp.29522
Liu, X., Zhang, C., Jing, J., Peng, W., Zhu, S., and Zou, S. (2017). Role of notch signaling in the physiological patterning of Posterofrontal and sagittal cranial sutures. J. Craniofacial Surg. 28, 1620–1625. doi:10.1097/SCS.0000000000003721
Liu, Y.-H., Tang, Z., Kundu, R. K., Wu, L., Luo, W., Zhu, D., et al. (1999). Msx2Gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: A possible mechanism forMSX2-mediated craniosynostosis in humans. Dev. Biol. 205, 260–274. doi:10.1006/dbio.1998.9114
Marie, P., Coffin, J., and Hurley, M. (2005). FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J. Cell. Biochem. 96, 888–896. doi:10.1002/jcb.20582
Maruyama, T., Jeong, J., Sheu, T.-J., and Hsu, W. (2016). Stem cells of the suture mesenchyme in craniofacial bone development, repair and regeneration. Nat. Commun. 7, 10526–10611. doi:10.1038/ncomms10526
Mathijssen, I., van Leeuwen, J., and Vermeij-Keers, C. (2000). Simultaneous induction of apoptosis, collagen type I expression and mineralization in the developing coronal suture following FGF4 and FGF2 application. J. Craniofacial Genet. Dev. Biol. 20, 127–136.
McKeehan, W. L., and Kan, M. (1994). Heparan sulfate fibroblast growth factor receptor complex: Structure-function relationships. Mol. reproduction Dev. 39, 69–81. doi:10.1002/mrd.1080390112
Mehrara, B. J., Mackool, R. J., McCarthy, J. G., Gittes, G. K., and Longaker, M. T. (1998). Immunolocalization of basic fibroblast growth factor and fibroblast growth factor receptor-1 and receptor-2 in rat cranial sutures. Plastic Reconstr. Surg. 102, 1805–1817. doi:10.1097/00006534-199811000-00001
Merrill, A. E. (2005). Opposing activities of Twist and Msx2 regulate the development of the coronal suture and the neural crest-mesoderm boundary in the murine skull vault. United States: University of Southern California.
Min Swe, N. M., Kobayashi, Y., Kamimoto, H., and Moriyama, K. (2021). Aberrantly activated Wnt/β-catenin pathway co-receptors LRP5 and LRP6 regulate osteoblast differentiation in the developing coronal sutures of an Apert syndrome (Fgfr2 S252W/+) mouse model. Dev. Dyn. 250, 465–476. doi:10.1002/dvdy.239
Mohammadi, M., Olsen, S. K., and Ibrahimi, O. A. (2005). Structural basis for fibroblast growth factor receptor activation. Cytokine & growth factor Rev. 16, 107–137. doi:10.1016/j.cytogfr.2005.01.008
Moosa, S., and Wollnik, B. (2016). Altered FGF signalling in congenital craniofacial and skeletal disorders. Seminars Cell & Dev. Biol. 53, 115–125. doi:10.1016/j.semcdb.2015.12.005
Mossahebi-Mohammadi, M., Quan, M., Zhang, J.-S., and Li, X. (2020). FGF signaling pathway: A key regulator of stem cell pluripotency. Front. Cell Dev. Biol. 8, 79. doi:10.3389/fcell.2020.00079
Most, D., Levine, J. P., Chang, J., Sung, J., McCarthy, J. G., Schendel, S. A., et al. (1998). Studies in cranial suture biology: Up-regulation of transforming growth factor-beta1 and basic fibroblast growth factor mRNA correlates with posterior frontal cranial suture fusion in the rat. Plastic Reconstr. Surg. 101, 1431–1440. doi:10.1097/00006534-199805000-00001
Moursi, A. M., Winnard, P. L., Winnard, A. V., Rubenstrunk, J. M., and Mooney, M. P. (2002). Fibroblast growth factor 2 induces increased calvarial osteoblast proliferation and cranial suture fusion. Cleft palate-craniofacial J. 39, 487–496. doi:10.1597/1545-1569_2002_039_0487_fgfiic_2.0.co_2
Muenke, M., Gripp, K., McDonald-McGinn, D., Gaudenz, K., Whitaker, L., Bartlett, S., et al. (1997). A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am. J. Hum. Genet. 60, 555–564.
Murakami, H., Okawa, A., Yoshida, H., Nishikawa, S.-i., Moriya, H., and Koseki, H. (2002). Elbow knee synostosis (eks): A new mutation on mouse chromosome 14. Mamm. genome 13, 341–344. doi:10.1007/s00335-001-2143-6
Nie, X., Luukko, K., and Kettunen, P. (2006). FGF signalling in craniofacial development and developmental disorders. Oral Dis. 12, 102–111. doi:10.1111/j.1601-0825.2005.01176.x
Ohbayashi, N., Shibayama, M., Kurotaki, Y., Imanishi, M., Fujimori, T., Itoh, N., et al. (2002). FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes & Dev. 16, 870–879. doi:10.1101/gad.965702
Olsen, S. K., Garbi, M., Zampieri, N., Eliseenkova, A. V., Ornitz, D. M., Goldfarb, M., et al. (2003). Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 278, 34226–34236. doi:10.1074/jbc.M303183200
Olsen, S. K., Ibrahimi, O. A., Raucci, A., Zhang, F., Eliseenkova, A. V., Yayon, A., et al. (2004). Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. 101, 935–940. doi:10.1073/pnas.0307287101
Opperman, L. A. (2000). Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 219, 472–485.
Opperman, L. A., Nolen, A. A., and Ogle, R. C. (1997). TGF-β1, TGF-β2, and TGF-β3 exhibit distinct patterns of expression during cranial suture formation and obliteration in vivo and in vitro. J. Bone Mineral Res. 12, 301–310. doi:10.1359/jbmr.1997.12.3.301
Ornitz, D. M., and Marie, P. J. (2002). FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes & Dev. 16, 1446–1465. doi:10.1101/gad.990702
Orr-Urtreger, A., Bedford, M. T., Burakova, T., Arman, E., Zimmer, Y., Yayon, A., et al. (1993). Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev. Biol. 158, 475–486. doi:10.1006/dbio.1993.1205
Otaify, G., Abdel-Hamid, M., Mehrez, M., Aboul-Ezz, E., Zaki, M., Aglan, M., et al. (2018). Genetic study of eight Egyptian patients with pycnodysostosis: Identification of novel CTSK mutations and founder effect. Osteoporos. Int. 29, 1833–1841. doi:10.1007/s00198-018-4555-0
Passos-Bueno, M., Wilcox, W., Jabs, E., Sertie, A., Alonso, L. G., and Kitoh, H. (1999). Clinical spectrum of fibroblast growth factor receptor mutations. Hum. Mutat. 14, 115–125. doi:10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2
Pfaff, M. J., Xue, K., Li, L., Horowitz, M. C., Steinbacher, D. M., and Eswarakumar, J. V. (2016). FGFR2c-mediated ERK–MAPK activity regulates coronal suture development. Dev. Biol. 415, 242–250. doi:10.1016/j.ydbio.2016.03.026
Prochazkova, M., Prochazka, J., Marangoni, P., and Klein, O. D. (2018). Bones, glands, ears and more: The multiple roles of FGF10 in craniofacial development. Front. Genet. 9, 542. doi:10.3389/fgene.2018.00542
Qin, X., Jiang, Q., Miyazaki, T., and Komori, T. (2019). Runx2 regulates cranial suture closure by inducing hedgehog, Fgf, Wnt and Pthlh signaling pathway gene expressions in suture mesenchymal cells. Hum. Mol. Genet. 28, 896–911. doi:10.1093/hmg/ddy386
Ray, A. T., Mazot, P., Brewer, J. R., Catela, C., Dinsmore, C. J., and Soriano, P. (2020). FGF signaling regulates development by processes beyond canonical pathways. Genes & Dev. 34, 1735–1752. doi:10.1101/gad.342956.120
Rice, D., Aberg, T., Chan, Y., Tang, Z., Kettunen, P. J., Pakarinen, L., et al. (2000). Integration of FGF and TWIST in calvarial bone and suture development. Development 127, 1845–1855. doi:10.1242/dev.127.9.1845
Rice, D. P., Connor, E. C., Veltmaat, J. M., Lana-Elola, E., Veistinen, L., Tanimoto, Y., et al. (2010). Gli3 Xt− J/Xt− J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum. Mol. Genet. 19, 3457–3467. doi:10.1093/hmg/ddq258
Rice, R., Spencer-Dene, B., Connor, E. C., Gritli-Linde, A., McMahon, A. P., Dickson, C., et al. (2004). Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. investigation 113, 1692–1700. doi:10.1172/JCI20384
Robin, N. H., Falk, M. J., and Haldeman-Englert, C. R. (2011). FGFR-related craniosynostosis syndromes. Am. J. Med. Genet. A 170, 3215–3221. doi:10.1002/ajmg.a.37862
Rodriguez-Zabala, M., Aza-Carmona, M., Rivera-Pedroza, C. I., Belinchón, A., Guerrero-Zapata, I., Barraza-García, J., et al. (2017). FGF9 mutation causes craniosynostosis along with multiple synostoses. Hum. Mutat. 38, 1471–1476. doi:10.1002/humu.23292
Roghani, M., and Moscatelli, D. (2007). Prostate cells express two isoforms of fibroblast growth factor receptor 1 with different affinities for fibroblast growth factor-2. Prostate 67, 115–124. doi:10.1002/pros.20448
Sahar, D. E., Longaker, M. T., and Quarto, N. (2005). Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Dev. Biol. 280, 344–361. doi:10.1016/j.ydbio.2005.01.022
Sasaki, T., Ito, Y., Bringas, P., Chou, S., Urata, M. M., Slavkin, H., et al. (2006). TGFβ-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Development 133, 371–381. doi:10.1242/dev.02200
Schlessinger, J. (2000). Cell signaling by receptor tyrosine kinases. Cell. 103, 211–225. doi:10.1016/s0092-8674(00)00114-8
Schliermann, A., and Nickel, J. (2018). Unraveling the connection between fibroblast growth factor and bone morphogenetic protein signaling. Int. J. Mol. Sci. 19, 3220. doi:10.3390/ijms19103220
Shi, E., Kan, M., Xu, J., Wang, F., Hou, J., and McKeehan, W. (1993). Control of fibroblast growth factor receptor kinase signal transduction by heterodimerization of combinatorial splice variants. Mol. Cell. Biol. 13, 3907–3918. doi:10.1128/mcb.13.7.3907
Shukla, V., Coumoul, X., Wang, R.-H., Kim, H.-S., and Deng, C.-X. (2007). RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 39, 1145–1150. doi:10.1038/ng2096
Siismets, E. M., and Hatch, N. E. (2020). Cranial neural crest cells and their role in the pathogenesis of craniofacial anomalies and coronal craniosynostosis. J. Dev. Biol. 8, 18. doi:10.3390/jdb8030018
Slaney, S. F., Oldridge, M., Hurst, J. A., Moriss-Kay, G., Hall, C. M., Poole, M. D., et al. (1996). Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am. J. Hum. Genet. 58, 923–932.
Srinivasan, A., Teo, N., Poon, K. J., Tiwari, P., Ravichandran, A., Wen, F., et al. (2020). Comparative craniofacial bone regeneration capacities of mesenchymal stem cells derived from human neural crest stem cells and bone marrow. ACS Biomaterials Sci. Eng. 7, 207–221. doi:10.1021/acsbiomaterials.0c00878
Stankiewicz, P., Thiele, H., Baldermann, C., Krüger, A., Giannakudis, I., Dörr, S., et al. (2001). Phenotypic findings due to trisomy 7p15, 3-pter including the TWIST locus. Am. J. Med. Genet. 103, 56–62. doi:10.1002/ajmg.1512
Teven, C. M., Farina, E. M., Rivas, J., and Reid, R. R. (2014). Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes & Dis. 1, 199–213. doi:10.1016/j.gendis.2014.09.005
Ting, M.-c., Farmer, D. J. T., Teng, C. S., He, J., Chai, Y., Crump, J. G., et al. (2022). Embryonic requirements for Tcf12 in the development of the mouse coronal suture. Development 149, dev199575. doi:10.1242/dev.199575
Ting, M.-C., Wu, N. L., Roybal, P. G., Sun, J., Liu, L., Yen, Y., et al. (2009). EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development 136, 855–864. doi:10.1242/dev.028605
Twigg, S. R., Healy, C., Babbs, C., Sharpe, J. A., Wood, W. G., Sharpe, P. T., et al. (2009). Skeletal analysis of the Fgfr3P244R mouse, a genetic model for the Muenke craniosynostosis syndrome. Dev. Dyn. 238, 331–342. doi:10.1002/dvdy.21790
Valverde-Franco, G., Liu, H., Davidson, D., Chai, S., Valderrama-Carvajal, H., Goltzman, D., et al. (2004). Defective bone mineralization and osteopenia in young adult FGFR3−/− mice. Hum. Mol. Genet. 13, 271–284. doi:10.1093/hmg/ddh034
Vu, H. L., Panchal, J., Parker, E. E., Levine, N. S., and Francel, P. (2001). The timing of physiologic closure of the metopic suture: A review of 159 patients using reconstructed 3D CT scans of the craniofacial region. J. Craniofacial Surg. 12, 527–532. doi:10.1097/00001665-200111000-00005
Wang, C., Chang, J. Y. F., Yang, C., Huang, Y., Liu, J., You, P., et al. (2013). Type 1 fibroblast growth factor receptor in cranial neural crest cell-derived mesenchyme is required for palatogenesis. J. Biol. Chem. 288, 22174–22183. doi:10.1074/jbc.M113.463620
Wang, F., Kan, M., Xu, J., Yan, G., and McKeehan, W. L. (1995). Ligand-specific structural domains in the fibroblast growth factor receptor (∗). J. Biol. Chem. 270, 10222–10230. doi:10.1074/jbc.270.17.10222
Wang, F., Lu, W., McKeehan, K., Mohamedali, K., Gabriel, J. L., Kan, M., et al. (1999). Common and specific determinants for fibroblast growth factors in the ectodomain of the receptor kinase complex. Biochemistry 38, 160–171. doi:10.1021/bi981758m
Wang, J., Zou, D., Li, Z., Huang, P., Li, D., Shao, Y., et al. (2014). Mechanical properties of cranial bones and sutures in 1–2-year-old infants. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 20, 1808–1813. doi:10.12659/MSM.892278
Wang, Y., Sun, M., Uhlhorn, V. L., Zhou, X., Peter, I., Martinez-Abadias, N., et al. (2010). Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2+ P253R mice. BMC Dev. Biol. 10, 22–20. doi:10.1186/1471-213X-10-22
Wang, Y., Xiao, R., Yang, F., Karim, B. O., Iacovelli, A. J., Cai, J., et al. (2005). Abnormalities in cartilage and bone development in the Apert syndrome FGFR2+/S252W mouse. Development 132, 3537–3548. doi:10.1242/dev.01914
Weinzweig, J., Kirschner, R. E., Farley, A., Reiss, P., Hunter, J., Whitaker, L. A., et al. (2003). Metopic synostosis: Defining the temporal sequence of normal suture fusion and differentiating it from synostosis on the basis of computed tomography images. Plastic Reconstr. Surg. 112, 1211–1218. doi:10.1097/01.PRS.0000080729.28749.A3
Weng, M., Chen, Z., Xiao, Q., Li, R., and Chen, Z. (2018). A review of FGF signaling in palate development. Biomed. Pharmacother. 103, 240–247. doi:10.1016/j.biopha.2018.04.026
Werner, S., Duan, D., de Vries, C., Peters, K., Johnson, D., and Williams, L. (1992). Differential splicing in the extracellular region of fibroblast growth factor receptor 1 generates receptor variants with different ligand-binding specificities. Mol. Cell. Biol. 12, 82–88. doi:10.1128/mcb.12.1.82
White, H. E., Goswami, A., and Tucker, A. S. (2021). The intertwined evolution and development of sutures and cranial morphology. Front. Cell. Dev. Biol. 690, 653579. doi:10.3389/fcell.2021.653579
Wilk, K., Yeh, S.-C. A., Mortensen, L. J., Ghaffarigarakani, S., Lombardo, C. M., Bassir, S. H., et al. (2017). Postnatal calvarial skeletal stem cells expressing PRX1 reside exclusively in the calvarial sutures and are required for bone regeneration. Stem Cell Rep. 8, 933–946. doi:10.1016/j.stemcr.2017.03.002
Wilkie, A. O., Slaney, S. F., Oldridge, M., Poole, M. D., Ashworth, G. J., Hockley, A. D., et al. (1995). Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat. Genet. 9, 165–172. doi:10.1038/ng0295-165
Xie, Y., Su, N., Yang, J., Tan, Q., Huang, S., Jin, M., et al. (2020). FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 5, 181–238. doi:10.1038/s41392-020-00222-7
Xu, J., Nakahara, M., Crabb, J., Shi, E., Matuo, Y., Fraser, M., et al. (1992). Expression and immunochemical analysis of rat and human fibroblast growth factor receptor (flg) isoforms. J. Biol. Chem. 267, 17792–17803. doi:10.1016/s0021-9258(19)37114-5
Xu, J., Wang, L., Huang, Z., Chen, Y., and Shao, M. (2020). Exogenous FGF8 signaling in osteocytes leads to mandibular hypoplasia in mice. Oral Dis. 26, 590–596. doi:10.1111/odi.13262
Yoshida, T., Phylactou, L. A., Uney, J. B., Ishikawa, I., Eto, K., and Iseki, S. (2005). Twist is required for establishment of the mouse coronal suture. J. Anat. 206, 437–444. doi:10.1111/j.1469-7580.2005.00411.x
Yoshida, T., Vivatbutsiri, P., Morriss-Kay, G., Saga, Y., and Iseki, S. (2008). Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev. 125, 797–808. doi:10.1016/j.mod.2008.06.007
Yousfi, M., Lasmoles, F., El Ghouzzi, V., and Marie, P. J. (2002). Twist haploinsufficiency in Saethre-Chotzen syndrome induces calvarial osteoblast apoptosis due to increased TNFalpha expression and caspase-2 activation. Hum. Mol. Genet. 11, 359–369. doi:10.1093/hmg/11.4.359
Yu, H.-M. I., Jerchow, B., Sheu, T.-J., Liu, B., Costantini, F., Puzas, J. E., et al. (2005). The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development 132, 1995–2005. doi:10.1242/dev.01786
Yu, M., Ma, L., Yuan, Y., Ye, X., Montagne, A., He, J., et al. (2021). Cranial suture regeneration mitigates skull and neurocognitive defects in craniosynostosis. Cell. 184, 243–256.e18. doi:10.1016/j.cell.2020.11.037
Zhao, H., Feng, J., Ho, T.-V., Grimes, W., Urata, M., and Chai, Y. (2015). The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat. Cell Biol. 17, 386–396. doi:10.1038/ncb3139
Zhao, X., Le, T. P., Erhardt, S., Findley, T. O., and Wang, J. (2021). Hippo-yap pathway orchestrates neural crest ontogenesis. Front. Cell. Dev. Biol. 9, 706623. doi:10.3389/fcell.2021.706623
Zhao, X., Tang, L., Le, T. P., Nguyen, B. H., Chen, W., Zheng, M., et al. (2022). Yap and Taz promote osteogenesis and prevent chondrogenesis in neural crest cells in vitro and in vivo. Sci. Signal. 15, eabn9009. doi:10.1126/scisignal.abn9009
Keywords: suture mesenchymal stem cell, neural crest, cranial suture, repair, craniosynostosis, FGF signaling
Citation: Zhao X, Erhardt S, Sung K and Wang J (2023) FGF signaling in cranial suture development and related diseases. Front. Cell Dev. Biol. 11:1112890. doi: 10.3389/fcell.2023.1112890
Received: 30 November 2022; Accepted: 22 May 2023;
Published: 01 June 2023.
Edited by:
Rulang Jiang, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Sachiko Iseki, Tokyo Medical and Dental University, JapanKatherine Ann Fantauzzo, University of Colorado Anschutz Medical Campus, United States
Copyright © 2023 Zhao, Erhardt, Sung and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolei Zhao, WGlhb2xlaS56aGFvQHV0aC50bWMuZWR1