 Charlotte Roth
Charlotte Roth Helena Kilpinen2,3
Helena Kilpinen2,3 Manju A. Kurian
Manju A. Kurian Serena Barral
Serena Barral
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 27 February 2023
Sec. Epigenomics and Epigenetics
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1090046
This article is part of the Research TopicEpigenetic Factors in Neurodevelopment and Neurodegenerative DiseasesView all 6 articles
Neurodevelopmental disorders encompass a group of debilitating diseases presenting with motor and cognitive dysfunction, with variable age of onset and disease severity. Advances in genetic diagnostic tools have facilitated the identification of several monogenic chromatin remodeling diseases that cause Neurodevelopmental disorders. Chromatin remodelers play a key role in the neuro-epigenetic landscape and regulation of brain development; it is therefore not surprising that mutations, leading to loss of protein function, result in aberrant neurodevelopment. Heterozygous, usually de novo mutations in histone lysine methyltransferases have been described in patients leading to haploinsufficiency, dysregulated protein levels and impaired protein function. Studies in animal models and patient-derived cell lines, have highlighted the role of histone lysine methyltransferases in the regulation of cell self-renewal, cell fate specification and apoptosis. To date, in depth studies of histone lysine methyltransferases in oncology have provided strong evidence of histone lysine methyltransferase dysregulation as a determinant of cancer progression and drug resistance. As a result, histone lysine methyltransferases have become an important therapeutic target for the treatment of different cancer forms. Despite recent advances, we still lack knowledge about the role of histone lysine methyltransferases in neuronal development. This has hampered both the study and development of precision therapies for histone lysine methyltransferases-related Neurodevelopmental disorders. In this review, we will discuss the current knowledge of the role of histone lysine methyltransferases in neuronal development and disease progression. We will also discuss how RNA-based technologies using small-activating RNAs could potentially provide a novel therapeutic approach for the future treatment of histone lysine methyltransferase haploinsufficiency in these Neurodevelopmental disorders, and how they could be first tested in state-of-the-art patient-derived neuronal models.
Histone lysine methyltransferases (HKMTs) are part of the epigenetic system that regulates chromatin remodeling and ultimately gene expression. HKMTs are chromatin modifying enzymes that catalyze the transfer of a methyl group (CH3) from a donor molecule S-Adenosyl methionine (SAM) on the N-terminal of histone proteins (Murray, 1964; Cantoni, 1975; Finkelstein, 1990). This process, which was initially thought to be irreversible, has been now recognized to be part of a highly dynamic system involved in cellular renewal, specification of cell identity, cell differentiation, maintenance of DNA integrity, and DNA replication (Desrosiers and Tanguay, 1988; Benveniste et al., 1995; Aletta et al., 1998; Chen et al., 1999; Fuks et al., 2000; Sun et al., 2005; Pfister et al., 2014; Pai et al., 2017). Depending on tissue localization and biological context, histone methylation can result in transcriptional activation or repression (Strahl et al., 1999; Aymard et al., 2014; Salinas et al., 2020). Mutations in HKMT genes have been reported to cause childhood neurodevelopmental disorders (NDDs) (Kuroki et al., 1981; Jones et al., 2012; Koemans et al., 2017) and several form of cancers in childhood (Attarbaschi et al., 2022) and adulthood (Turkmen et al., 2012; Gala et al., 2018; Alam et al., 2020).
Studies in cancer have improved our understanding of the role of HKMTs in the regulation of cellular processes. It is hypothesized that epigenetic dysregulation could precede or initiate the neoplastic process (Fraga et al., 2005; Kuo et al., 2011). HKMTs are commonly overexpressed in cancers, leading to an imbalance of epigenetic regulation, either by inadequate activating or repressing gene expression (Barlesi et al., 2007; Bianco-Miotto et al., 2010). For some HKMTs, downregulation of chromatin remodelers has been related to poor prognosis, higher metastatic risks and drug resistance (Cheng et al., 2001; Ruault et al., 2002; Lee et al., 2009; Morin et al., 2011; Watanabe et al., 2011; Chen et al., 2014; Will and Steidl, 2014). Importantly, the role of HKMT in cancer has also been attributed to their capacity to methylate non-histone targets, as described for tumor suppressor p53 which requires HKMT-mediated methylation to be activated (Lee et al., 2009; Li et al., 2020). Not surprisingly, the role of HKMT in cancer has accelerated the design of novel therapies aiming to modulate their expression and restore physiological levels (Gilbert et al., 2004; Barlesi et al., 2007), for example, by using HKMT inhibitors (Cameron et al., 1999; Baylin and Jones, 2011).
Due to their key role in regulating early stages of human development (Xu et al., 2019), it is not surprising that germline mutation in HKMT genes are frequently reported in NDDs. It is now well recognized that de novo mutations in HKMT genes can lead to haploinsufficiency with reduced protein expression and impaired protein function (Faundes et al., 2018). Here, we will provide a brief overview of the role of HKMTs, both in normal cellular function and NDD pathophysiology. We will focus on the KMT2 family, for which several monogenic diseases have been described. We will discuss how advances in small-activating RNA (saRNA) technologies could potentially be exploited as a novel precision therapy approach for KMT2 haploinsufficiency. Finally, we will discuss how these therapeutic strategies could be further investigated in patient-derived neuronal models, enabling preclinical development of saRNA-based novel therapies for this group of diseases.
NDDs encompass a broad heterogeneous spectrum of disabling neurological diseases characterized by impairment of one or more aspects of human development (Pichot, 1986). By definition, they present in childhood, usually during the early years of infancy or childhood (Howlin et al., 2004; Morris-Rosendahl and Crocq, 2020). Neurological features such as intellectual disability are a core component of NDDs; however, NDDs are often multisystemic disorders with a complex pathophysiological phenotype (Kim and Leventhal, 2015; Niemi et al., 2018). Recent epidemiological studies have highlighted the co-occurrence of multiple neurological phenotypes in 70% of people with NDDs, complicating their classification and diagnosis (Fombonne, 2003; Straub et al., 2022). Despite advances in diagnostic tools such as next-generation sequencing technologies, many NDDs still remain undiagnosed. Therefore, the overall disease incidence is estimated to be higher than currently reported (Deciphering Developmental Disorders Study, 2017; Frances et al., 2022; Straub et al., 2022). Collectively, the social, economic and personal costs of NDDs present significant burden to both families and society. For the great majority, there are no disease-modifying treatments and care consists solely of symptomatic and palliative management (Thapar et al., 2017; Ismail and Shapiro, 2019). Etiologically, NDDs may be broadly divided into those predominantly caused by environmental factors (including birth injury, nutritional defects, infections) and genetic factors. Epigenetic factors are also likely to play a role in some NDDs (Lister et al., 2013; Salinas et al., 2020). An increasing number of NDDs have recently been reported to be caused by de novo mutations in HKMT genes (Ciptasari and van Bokhoven, 2020; Reichard and Zimmer-Bensch, 2021). Different forms of NDDs have been associated with loss-of-function mutations leading to haploinsufficiency (Singh et al., 2016; Zech et al., 2016; Deciphering Developmental Disorders Study, 2017; Yu et al., 2019; Kummeling et al., 2021).
HKMTs are ubiquitously expressed, highly conserved enzymes responsible for the methylation of lysine residues positioned along histone tails emerging from the nucleosome. Nucleosomes are made of four pairs of histone proteins: H2.A, H2.B, H3 and H4 around which the DNA is wrapped, allowing close interaction between DNA and histone proteins (Richmond et al., 1984; Arents and Moudrianakis, 1993). Methylation of the lysine residues of histones is a post-translational modification (PTM) which acts in synergy with other PTMs such as acetylation, ubiquitylation and phosphorylation, allowing interaction with a range of cofactors involved in the regulation of gene transcription (Fuks et al., 2000; Strahl and Allis, 2000; Ballestar and Esteller, 2002; Wang et al., 2004; Chaumeil et al., 2006). Notably, PTMs such as acetylation and phosphorylation are responsible for introducing a positive charge to histone proteins and directly affect the position of the histone tails (Bradbury, 1992). Histone methylation shapes the conformation of the chromatin through recruitment and interaction with complexes regulating the state of chromatin compaction and thus accessibility to DNA (Chen et al., 1999; Greer and Shi, 2012). Overall, the chromatin state is crucial to spatially control the access of transcription machinery to specific genes responsible for cellular physiology and cell identity. Although there are exceptions, methylation of the lysine 4 and 36 of the histone protein 3 (H3K4 and H3K36, respectively) activate gene transcription (Strahl et al., 1999; Heintzman et al., 2007; Aymard et al., 2014), whereas methylation in histone three lysine residues 9, 27, 79 (H3K9, H3K27, H3K79) and in histone four lysine 20 (H4K20) are more likely to be found in transcriptionally inactive heterochromatin domains (Tachibana et al., 2001; Lachner and Jenuwein, 2002; Shi et al., 2003; Peinado et al., 2004; Yang et al., 2008). Adding to the intricacy of this regulatory system, lysine residues can be mono-, di- or tri-methylated (Murray, 1964; Paik and Kim, 1967; Hempel et al., 1968). Each HKMT-related PTM at specific amino acid residues has been associated with different functions (Table 1).
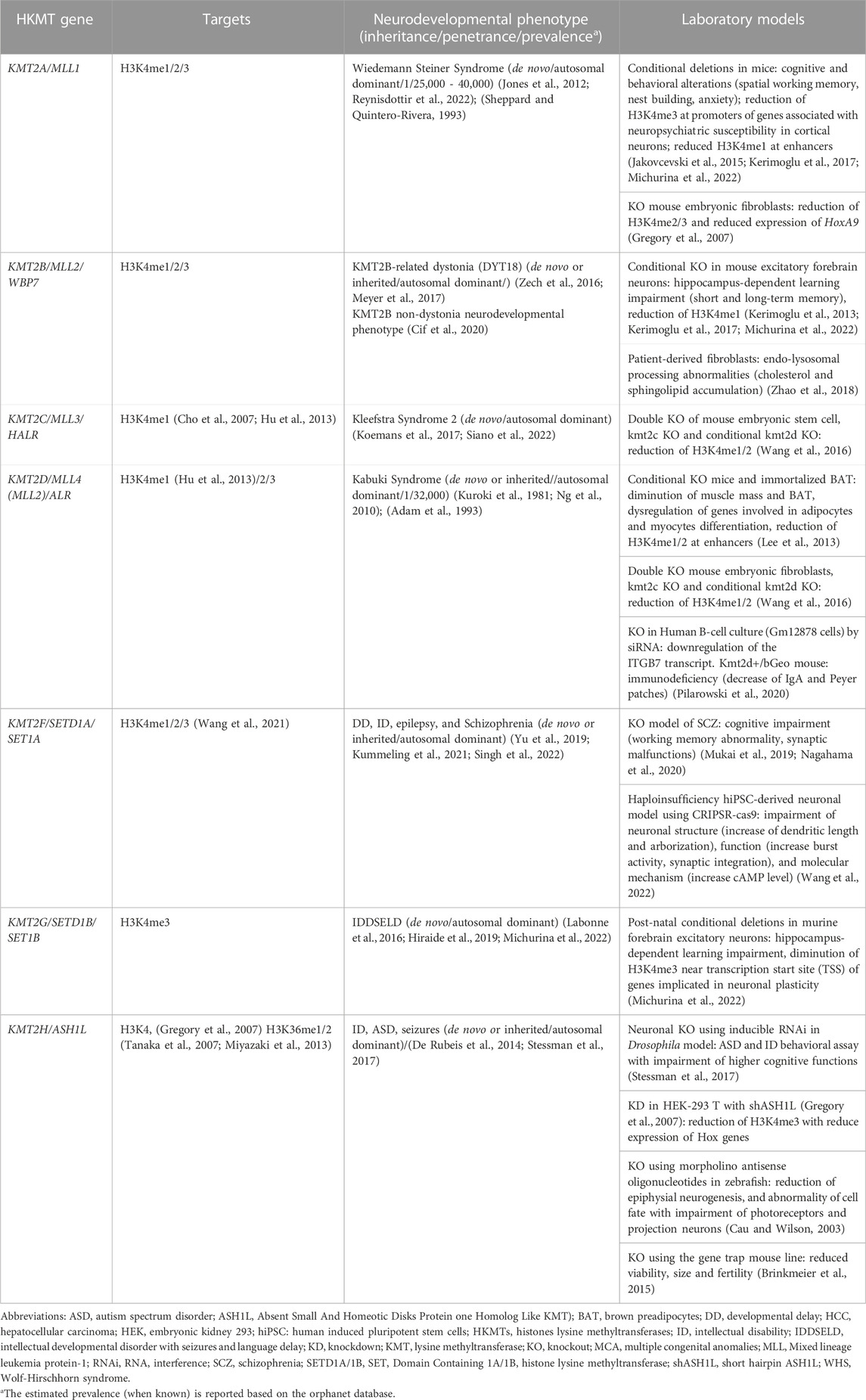
TABLE 1. Histone lysine methyltransferases causing neurodevelopmental disorders and their relevant cell/animal models.
One of the most studied subgroups of HKMTs is the lysine methyltransferase 2 (KMT2) family, which methylates the lysine four of the histone 3 (H3K4) via its catalytic SET (Su(var) 3-9, Enhancer-of-zeste, Trithorax) domain (Rea et al., 2000). Mono-methylation of H3K4 (H3K4me) is mostly present in enhancers (Heintzman et al., 2009), di-methylation (H3K4me2) is found throughout active genes and tri-methylation (H3K4me3) is enriched at active promoters (Guenther et al., 2005; Heintzman et al., 2007). Crosstalk between the different methylation states, other histones PTMs and single proteins or protein complexes is essential for genomic transcription.
Six members of the KMT2 family interact within the Complex of Proteins Associated with Set1 (COMPASS) and COMPASS-like complexes (also called MLL complexes), composed of ‘writers’ (methyltransferase), ‘erasers’ (demethylase) and ‘readers’ (Park et al., 2019; Lavery et al., 2020). Importantly, the subunit ‘WRAD’, composed of WD repeat domains 5 (WDR5), retinoblastoma binding protein 5 (RrBP5), absent small or homeotic 2-like (ASH2L), and dumpy-30 (DPY-30), is a subunit present across each of the MLL complexes and which enhances the methylation capability as well as substrate specificity (Steward et al., 2006; Cho et al., 2007; Cao et al., 2010; Dharmarajan et al., 2012). Although KMT2A and B, KMT2C and D, KMT2G and H are respectively paralogues, they have been shown to have stringent substrate specificity due to their association with distinct co-factors (Figure 1) (Hughes et al., 2004; Cho et al., 2007). As a result, mutations in each of the KMT2 genes have been linked to different NDDs, with distinct but overlapping features.
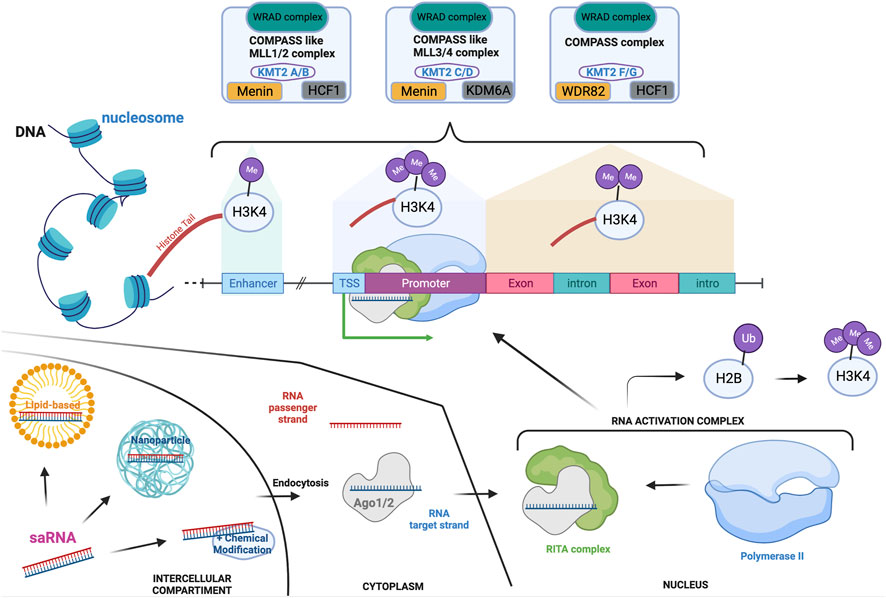
FIGURE 1. KMT2-associated complexes and saRNA mode of action in the transcription machinery. Transcription activation is a unique mechanism that is in part controlled by the addition of mono-(at enhancer), di- (at actively transcribe gene locus) or tri-(at promoter) methylation on the lysine four of the histone 3. These methylations are mediated by the COMPASS and COMPASS-like complexes which are formed by the association of specific subunits. Each of the complexes hold a common subunit called WRAD to which associate specific subunits: Menin and HCF1 to generate the COMPASS like/MLL1-2 complexes; Menin and KDM6A for the COMPASS like/MLL3-4 complex; HCF-1 and WDR82 for the COMPASS complex. saRNAs are synthetic RNA probes which enable transcription activation. Double-stranded saRNA can be delivered by various vehicles to the target cell, such as lipid-based or nanoparticle technology. After endocytosis, the single RNA target strand is loaded on the Ago protein before translocating to the nucleus. The loaded Ago protein associates with the RITA complex to recruit polymerase II and form the RNA activation complex that is guided to the promoter of the target gene. This is associated to ubiquitination of the histone 2 b which then triggers H3K4me3, enhancing the activation of transcription. If targeting HKMTs, this will increment their transcription and transduction enabling mono-, di- or tri-methylation of specific lysine (K) on histone (H) tails. Depending on the lysine localization, these post-translational modifications will repress or activate specific genes. Created with BioRender.com.
Mutations in KMT2A have been linked to Wiedemann Steiner Syndrome (WSS), an autosomal-dominant disorder that presents with intellectual disability (ID), facial dysmorphism, hypertrichosis, motor delay and hypotonia (Steiner and Marques, 2000; Jones et al., 2012). Most WSS patients have premature stop-codon mutations in the enzymatic SET domain, leading to loss-of-function and haploinsufficiency (Jones et al., 2012). However, more recently, some variants have been identified in the CXXC domain which result in increased binding at unmethylated CpG islands of DNA, leading to KTM2A overexpression and abnormal nuclear localization (Lebrun et al., 2018). Artificial intelligence (AI) systems are now facilitating the analysis of variant pathogenicity; AlphaFold2 can predict the effect of different variants of the KMT1A-CXXC domain on the 3D conformation of methyltransferases (Tunyasuvunakool et al., 2021; Reynisdottir et al., 2022).
Haploinsufficiency of KMT2B causes KMT2B-related dystonia as well as a neurodevelopmental (non-dystonic) phenotype (Zech et al., 2016; Meyer et al., 2017; Faundes et al., 2018). Since its discovery as a disease-causing gene, over a hundred pathogenic variants have been identified, making it one of the most common genetic etiologies of complex childhood onset dystonia (Gorman et al., 2018; Cif et al., 2020; Damasio et al., 2021; Grosz et al., 2022). KMT2B-related dystonia usually presents early in childhood, with lower limb dystonia progressing into a generalized dystonia with prominent cervical, cranial, and laryngeal involvement (Zech et al., 2016; Meyer et al., 2017). DNA methylation profiling reveals a unique methylation signature in KMT2B-related disease, which not only aids in determining the pathogenicity of KMT2B variants of uncertain significance (VUS) but could also be predictive of the onset and severity of disease (Ciolfi et al., 2021). Such methylation signatures also differentiate KMT2B-related disease from Kabuki syndrome, which has a different methylation (Lee et al., 2022).
Although KMT2A and KMT2B are phylogenetically closely related, and both are responsible for the methylation of the H3K4 residue, systematic studies have highlighted unique roles for each (Kerimoglu et al., 2017). For instance, a recent study of knock-out (KO) mouse models of these two methyl transferases showed that both KMT2A and KMT2B are associated with memory consolidation (Kerimoglu et al., 2013; Kerimoglu et al., 2017). However, RNA sequencing and Chromatin Immunoprecipitation Sequencing (ChIP-seq) analysis have shown limited overlap of the gene expression profiles between KMT2A and KMT2B-KO mice (Michurina et al., 2022). Moreover, these studies have highlighted that the observed reduction of H3K4me3 at specific promoters was dependent on the specific KMT2-KO (Kerimoglu et al., 2013; Kerimoglu et al., 2017). KMT2B, and not KMT2A, seems to have a pivotal role at bivalent promoters (Tomizawa et al., 2018). Bivalency is a chromatin state where repressive (H3K27) and active (H3K4) marks co-occur at specific promoters, which are hypothesized to control bivalent genes that arekey regulators of either retaining stem cell identity or initiating cellular differentiation processes (Macrae et al., 2022).
Both KMT2C and KMT2D are histone methyltransferases of H3K4 and primarily target gene enhancers (Hu et al., 2013). Monoallelic KMT2C variants have recently been described in Kleefstra syndrome type 2 (Cheema et al., 2022; Siano et al., 2022). Notably, Kleefstra syndrome type 1 is caused by deletions in the sub-telomeric region of chromosome 9q which contains the KMT1D gene, a HKMT protein that mono- and di-methylates H3K9 (Kleefstra et al., 2006; Kleefstra et al., 2009). Patients with Kleefstra syndrome present with NDD, microcephaly, ID, autism spectrum disorder (ASD), and characteristic facial features, which are associated with hypotonia and heart defects in half of the cases, and epilepsy in a fifth of the patients. In contrast, mutations in KMT2D lead to a NDD called Kabuki Syndrome (KS) type 1 (Kuroki et al., 1981), a multisystemic disorder associated with developmental delay, short stature, variable ID, increased susceptibility to infection, risk of autoimmune disorders and characteristic facial dysmorphism (Stagi et al., 2016; Adam et al., 2019; Pilarowski et al., 2020). Most of the reported cases of KS are due to loss-of-function monoallelic mutations in KMT2D, but surprisingly, the second most frequent mutated gene is the X-linked Lysine-Demethylase-6A (KDM6A), an enzyme with quite different functions to KMT2D (Lederer et al., 2012; Micale et al., 2014). Both KMT2D and KDM6A are subunits of the MLL4/COMPASS-like complex (Figure Figure1), which could explain why they lead to phenotypically similar syndromes in the disease state.
Monoallelic variants in KMT2F, have been linked to schizophrenia (SCZ) as well as NDD, ID, and early onset epilepsy (Singh et al., 2016; Yu et al., 2019; Kummeling et al., 2021). Kummeling et al. described a neurodevelopmental syndrome caused by heterozygous loss-of-function mutations in KMT2F, presenting with ID, NDD, visual and/or hearing defects and psychiatric symptoms (Kummeling et al., 2021). Recently, using a humanised induced pluripotent stem cell (iPSC)-derived neuronal model of SCZ, Wang and others have elegantly demonstrated that haploinsufficiency of KMT2F leads to major neuronal network reorganization and modification of neuronal morphology (Table 1) (Wang et al., 2022).
KMT2G dysfunction has been linked to autosomal dominant NDD that typically presents with DD, ID, ASD, and drug-resistant early onset epilepsy (Labonne et al., 2016; Hiraide et al., 2019; Roston et al., 2021). Dysregulation of methylation has been shown in patients with characteristic hypermethylation signatures (Krzyzewska et al., 2019). Recently, the postnatal effect of loss-of-function of KMT2G have been explored in a conditional KO mice model and highlighted defects in H3K4me3 deposition in genes involved in neuronal plasticity, which affected hippocampus-dependent learning (Michurina et al., 2022).
KMT2H differs from the rest of the KMT2 family with regard to substrate. It has been reported that KMT2H methylates H3K4 (Gregory et al., 2007) as well as H3K36 (Tanaka et al., 2007; An et al., 2011; Miyazaki et al., 2013; Okamoto et al., 2017). The crystal structure suggests the existence of an auto-inhibitory loop (An et al., 2011), which is potentially inhibited upon interaction with the nucleosome. Monoallelic variants have been identified in patients with ID, ASD and seizures.
To date, treatment of NDDs related to defective HKMTs has been limited to symptomatic management (Thapar et al., 2017; Ismail and Shapiro, 2019). NDDs caused by monoallelic loss-of-function mutations represent an attractive group of disorders that could benefit from a strategy aiming to restore baseline gene expression by increasing the transcription of the healthy non-mutated allele.
SaRNAs are double stranded, small non-coding activating RNAs made of 21 nucleotides with a two-base overhang on each end (Li et al., 2006). Despite being structurally identical to small-inhibiting RNA (siRNA), they have the opposite effect of increasing target gene expression by a mechanism called RNA activation (RNAa) (Huang et al., 2010; Voutila et al., 2017). RNAa is mediated by an oligonucleotide sequence complementary to the promoter of the gene of interest. Binding of the saRNA to its complementary promoter sequence mediates target gene upregulation (Janowski et al., 2007). RNAa was first reported in 2006 using synthetically designed saRNAs, which were able to increase the transcription of a specific gene with high specificity and efficacy (Li et al., 2006). After entering a cell via endocytosis, one strand (either the sense or antisense, depending on cell type) of the saRNA is loaded to a protein of the Argonaut family (Ago1 or Ago2) and guided to its targeted promoter in the nucleus (Janowski et al., 2006; Portnoy et al., 2016) (Figure Figure1). This is another major difference with siRNA, which are loaded on Ago1-4 (Schwartz et al., 2008; Fimiani et al., 2016). Once in the nucleus, the Ago carrying the saRNA strand associates with the RNA-induced transcriptional activation (RITA) complex which recruits RNA polymerase II and promotes transcription (Portnoy et al., 2016). Additionally, it has been proposed that PTMs -such as ubiquitination of H2B that promotes methylation of H3K4 - participate in saRNA/Ago2-mediated transcriptional activation (Portnoy et al., 2016). Interestingly, although Ago1 has been primarily implicated in gene silencing, in some cases, Ago1 is key for the function of saRNAs. Indeed, the use of an Ago1 inhibitor leads to a complete loss of saRNA activity (Fimiani et al., 2016). Notably, mutations in Ago2 and Ago1 have also recently been associated with NDDs (Lessel et al., 2020; Schalk et al., 2022).
While several RNA-based strategies [such as antisense oligonucleotide (ASO), micro-RNA (miRNA), small interfering RNA (siRNA) short hairpin RNAs (shRNAs)] aim to inhibit expression of disease-causing genes (Matzke and Birchler, 2005; Lanford et al., 2010; Shen et al., 2012), Li and others’ study first allowed the exploration of RNA-based therapeutic strategies to increase expression of the target gene (Li et al., 2006). Moreover, their strategy showed that the saRNA-mediated transcriptional activation increased the protein production only in cells where the target gene was in a non-condensed accessible state, with limited if not null binding to genes in the euchromatin state. Therefore, saRNA are mostly functional in cells that physiologically express the target gene. This characteristic could potentially avoid off-target effects by only targeting cells in a specific cellular state where the gene of interest is expressed.
Soon after saRNA discovery, increased expression of the progesterone receptor (PR) was successfully achieved in two breast cancer cell lines (MCF7 and T47D) using a saRNA construct targeting the PR promoter (Janowski et al., 2007). The saRNA increased PR expression up to 18-fold compared to untreated cells. The addition of a deacetylase inhibitor and a methyltransferase inhibitor reduced the saRNA effect, indicating the potentially important role of PTMs in fine tuning gene expression via RNA activation. The efficacy of the saRNA construct was significantly higher when the sequence was designed to have 100% complementary with the target sequence, while saRNA sequences containing mismatched or scrambled saRNAs showed less or no efficacy, respectively (Janowski et al., 2007). The specificity of the saRNA resides in its seed region, which span from the second to the eighth nucleotide commencing at the 5′end, where a single nucleotide mismatched within this region could lead to decreased or null activity. Variants located outside of the seed region are more tolerated but may hamper saRNA functionality (Meng et al., 2016; Voutila et al., 2017).
In a very recent study, Andrikakou and others tested saRNA technology to increase the expression of a histone deacetylase, Sirtuin 1 (encoded by SIRT1) (Andrikakou et al., 2022). SIRT1 is known for its protective effect on age-related disorders such as metabolic syndrome (Pfluger et al., 2008). Using a high fat diet rat model, they injected a saRNA against SIRT1 conjugated with an aptamer to enable systemic delivery and showed a reduction of 0.6-fold decrease of serum cholesterol, 0.7-fold decrease of triglycerides and an increase of 1.6-fold increase of HDL/LDL ratio. They also observed a significant decrease in intracellular lipid accumulation and a reduction of rat weight (Andrikakou et al., 2022). This study therefore demonstrated well-tolerated systemic delivery of saRNA in vivo, without noticeable toxicity.
SaRNAs have also been tested in vivo in a mouse model of a Rett-like condition to correct haploinsufficiency of Foxg1 (Fimiani et al., 2016). FOXG1 is a transcription factor implicated in cortical development with a role in the maintenance of neural cells in the precursor state (Hanashima et al., 2004; Martynoga et al., 2005). Lentiviral delivery of eight sense and antisense saRNAs to a primary culture of E12.5 murine neocortical precursors showed an increase of Foxg1 expression with all constructs tested (Fimiani et al., 2016). Moreover, saRNAs were able to induce the same results in mouse embryonic fibroblast (NIH3T3) and human embryonic kidney 293 (HEK) cell lines (Fimiani et al., 2016). To further analyze the potency of the saRNA on differentiating cells and their effect on the differentiation process, an inducible construct was delivered to E16.5 murine neocortical precursors. Conditional activation in cultured neocortical precursors over 5 days induced Foxg1 expression and a significant reduction in Tubulin beta class III (TUBB3) in postmitotic neurons (Fimiani et al., 2016). Thus, this saRNA strategy was able to decrease neuronal maturation downstream of increased Foxg1 expression. Interestingly, only one of the selected saRNAs was effective in E16.5 mouse primary neuronal cultures. This highlights the limited efficacy of saRNAs on cells where the baseline expression of the target gene is constitutively low. Intraventricular injection into neonate mouse pups with AAV-saRNA lead to a 1.66-fold increase in the mRNA expression of Foxg1, despite low infection efficiency (Fimiani et al., 2016).
Improvements in saRNA technology have led to the first clinical trial for the treatment of hepatocellular carcinoma using saRNA and nanoparticle-mediated delivery-NOV340-SMARTICLES (NCT02716012) (Sarker et al., 2020; Hashimoto et al., 2021). The therapeutic strategy is based on the MTL-CEBPA (MiNA therapeutics) - an saRNA targeting the CCAAT/enhancer-binding protein alpha (CEBPA) that codes the transcription factor C/EBP (Voutila et al., 2017; Reebye et al., 2018). MTL-CEBPA was first tested on a cirrhotic rat model (Reebye et al., 2014), then on human hepatocellular carcinoma cells HepG2 (Voutila et al., 2017) and finally in a rodent model (Reebye et al., 2018). MTL-CEBPA successfully increases CEBPA protein expression, leading to an increase in albumin and a reduction in tumor mass (Reebye et al., 2018). Interestingly, another group demonstrated the anti-inflammatory effect of MTL-CEBPA in an E. coli-derived lipopolysaccharide-challenged humanized mouse model (Zhou et al., 2019), demonstrating the potential versatility of saRNA.
Despite improvements and efforts to further optimize this technology, clinical translation of saRNAs still present some limitations. Indeed, as an RNA molecule, saRNAs are highly susceptible to degradation, which impacts the duration of therapeutic effect–in reality this will likely require repeated regular drug delivery to patients. Delivery of the saRNA remains one of the biggest challenges for efficient therapeutic translation of this technology. When systemic delivery is used, the saRNA are subjected to several processes, which drives their degradation, including renal clearance, nucleases, and lysosomal degradation (Alexis et al., 2008; Petros and DeSimone, 2010). Significant efforts have been made to reduce degradation of ASOs and siRNAs, and these will hopefully also translate to saRNA technology.
Another important aspect to consider is brain delivery of saRNA-based strategies for the treatment of NDDs. To date, delivery has been achieved for chemically modified RNA-based therapies via intrathecal (IT) or intracerebroventricular (ICV) injections (Hache et al., 2016; Hirunagi et al., 2021). This same administration route could potentially be used for saRNAs, although given that the effect of saRNAs is time-limited, to achieve a long-lasting effect, repetitive administration would be needed. Whilst ventricular reservoirs are also potentially another delivery route, ideally a systemic saRNA that crosses the brain blood barrier (BBB) to reach the specific brain areas would be even more desirable.
Current strategies to improve the half-life of RNA-based technologies rely on chemical modifications of the nucleobase, the backbone or ribose of the oligonucleotide (Roberts et al., 2020). Addition of phosphorothioate (PS) linkages affects the backbone, stereochemistry, and resistance to nucleases (Brown et al., 1994). However, these modifications, mainly tested in siRNA, will decrease affinity with the target gene, causing non-specific binding and global non-antisense effect, thus affecting overall efficiency and more importantly, safety (Brown et al., 1994; Guvakova et al., 1995; Giles et al., 1998; Liang et al., 2014; Liang et al., 2015). Regarding saRNAs, the 2ʹ-O-methyl or 2ʹ-Fluoro chemical modifications showed better results (Place et al., 2010; Kang et al., 2012); 2ʹ-Fluoro modified P21 targeting saRNA, when compared to its non-modified counterpart, showed an increase resistance to nuclease and reduced immunogenicity whilst maintaining efficacy. The non-immunogenicity of 2ʹ-Fluoro modification represents one of the major aspects for clinical translation of this technology. In addition, the use of lipid nanoparticles for intravesical delivery in mice bladder did not modify the saRNA measured activity (Kang et al., 2012). Indeed, other major technological advances have allowed saRNA molecules to associate with peptides, lipids, antibodies and nanocarrier liposomes to improve their stability (Kang et al., 2012; Blakney et al., 2019; Zhou et al., 2019).
When developing saRNA-based therapies for NDDs, some important aspects should be considered for preclinical proof-of-concept studies. HKMT-related NDDs present with complex developmental phenotypes. Animal models do not always recapitulate features of human disease, likely due to key differences between human and murine brain development. It would therefore be potentially advantageous to validate saRNAs in systems that more closely mimic the human brain. Patient-derived neuronal models represent an ideal platform for the development of saRNA therapies for NDDs. Since the first generation of two-dimensional (2D) iPSC-derived neuronal models, technological advances have facilitated the generation of three-dimensional (3D) neuronal systems and brain assembloids that more closely align with human development and physiology. Indeed, 3D cerebral organoid studies of key neurodevelopmental stages of the human cortex, midbrain, forebrain, hippocampus, striatum and cerebellum (Jo et al., 2016; Birey et al., 2017; Lancaster et al., 2017; Miura et al., 2020; Fear et al., 2022; Kim et al., 2023) have highlighted key differences with animal systems. Long-term maturation of neuronal organoids has allowed better recapitulation of the latter stages of fetal brain development (Giandomenico et al., 2021). Generation of patient-derived neuronal organoids has thus provided new-humanized models of disease (Lancaster et al., 2013; Bershteyn et al., 2017; Iefremova et al., 2017). which can be a unique platform for testing novel therapeutic approaches in a “first-in-human” context, providing another preclinical model for evaluating drug efficacy (Lancaster et al., 2013; Galet et al., 2020). The evaluation of saRNAs in patient-derived organoids could therefore potentially yield important proof-of-concept data with regard to drug delivery, efficacy and off-target toxicity. Moreover, the next-generation of complex organoids include the brain blood barrier, which could also inform on systemic delivery of saRNAs (Bergmann et al., 2018; Ahn et al., 2021).
Several studies have shown preclinical feasibility of saRNA-based therapies for the treatment of different disorders. Indeed, saRNAs have been shown to be effective in reversing disease-related phenotypes in cell lines and animal models, now leading to the first clinical trial (Voutila et al., 2017; Reebye et al., 2018; Zhou et al., 2019). Technological advances have allowed the development of novel delivery systems for specific targeting of cells in vivo, aiming to avoid off-targets effects. Further development and optimization of saRNA for the treatment of several diseases are ongoing (Zhou et al., 2019; Sarker et al., 2020).
In this context, HKMT-NDDs represent a promising group of diseases that could benefit from this strategy, though much work needs to be done preclinically to prove safety and efficacy. Another major point to consider is the therapeutic window for saRNA administration. We still lack knowledge on the underlying cellular and molecular mechanisms underpinning HKMT-related NDDs, and very little is known about the specific developmental stages that are most affected in disease. It is likely that some of these diseases have prenatal onset and so fetal therapy strategies may need to be considered in the future. Nevertheless, as is the case for many early onset genetic disorders (Pearson et al., 2021; Fumagalli et al., 2022; Masson et al., 2022; Strauss et al., 2022), such therapies have the potential to significantly modify disease at different stages of the disease course, which can significantly improve patient lifespan and quality of life.
In summary, saRNAs are an important and relevant therapeutic strategy to explore for HKMT-NDD. The recent clinical trial with MTL-CEBPA for treatment of HKMT-related cancer brings promise and will no doubt provide more knowledge to improve the development of future saRNA-based therapies for NDDs. If the challenges of accurate and targeted brain delivery, repeated dosing, adequate efficacy, optimum therapeutic window, safety, and off-target toxicity can be overcome, saRNA therapies could potentially be applicable to a broad range of monogenic disorders of genetic haploinsufficiency.
CR wrote the manuscript and generated the figure and table. HK and MK provided a revision of the manuscript. SB provided guidance and revisions on the original manuscript and conceptualized the manuscript with CR and MK. All authors contributed to the article and approved the submitted version.
This work was partly funded by the NIHR GOSH BRC (the views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health). SB is funded by the Great Ormond Street Hospital Children’s Charity (VS0218); MK is funded by a NIHR Professorship (NIHR-RP-2016-07-019) and Rosetrees Trust (CF2\100018); MK and CR are funded by the Jules Thorn Award for Biomedical Research (JTA-017); HK is funded by the Helsinki Institute of Life Science (HiLIFE), University of Helsinki, Finland. HK also acknowledges funding from the Wellcome Trust (207797/Z/17/Z).
We acknowledge Kimberley Reid (UCL GOS Institute of Child Health) for providing revision of the English language.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adam, M. P., Banka, S., Bjornsson, H. T., Bodamer, O., Chudley, A. E., Harris, J., et al. (2019). Kabuki syndrome: International consensus diagnostic criteria. J. Med. Genet. 56, 89–95.
Adam, M. P., Hudgins, L., and Hannibal, M. (1993). “Kabuki syndrome,” in GeneReviews((R)). Editors M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (Seattle (WA): University of Washington).
Ahn, Y., An, J. H., Yang, H. J., Lee, D. G., Kim, J., Koh, H., et al. (2021). Exendin-4 pretreatment attenuates kainic acid-induced hippocampal neuronal death. Cells 10, 2527. doi:10.3390/cells10102527
Alam, H., Tang, M., Maitituoheti, M., Dhar, S. S., Kumar, M., Han, C. Y., et al. (2020). KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. 37, 599–617 e7. doi:10.1016/j.ccell.2020.03.005
Aletta, J. M., Cimato, T. R., and Ettinger, M. J. (1998). Protein methylation: A signal event in post-translational modification. Trends Biochem. Sci. 23, 89–91. doi:10.1016/s0968-0004(98)01185-2
Alexis, F., Pridgen, E., Molnar, L. K., and Farokhzad, O. C. (2008). Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 5, 505–515. doi:10.1021/mp800051m
An, S., Yeo, K. J., Jeon, Y. H., and Song, J. J. (2011). Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. J. Biol. Chem. 286, 8369–8374. doi:10.1074/jbc.M110.203380
Andrikakou, P., Reebye, V., Vasconcelos, D., Yoon, S., Voutila, J., George, A. J. T., et al. (2022). Enhancing SIRT1 gene expression using small activating RNAs: A novel approach for reversing metabolic syndrome. Nucleic Acid. Ther. 32, 486–496. doi:10.1089/nat.2021.0115
Arents, G., and Moudrianakis, E. N. (1993). Topography of the histone octamer surface: Repeating structural motifs utilized in the docking of nucleosomal DNA. Proc. Natl. Acad. Sci. U. S. A. 90, 10489–10493. doi:10.1073/pnas.90.22.10489
Attarbaschi, A., Moricke, A., Harrison, C. J., Mann, G., Baruchel, A., De Moerloose, B., et al. (2022). Outcomes of childhood noninfant acute lymphoblastic leukemia with 11q23/kmt2a rearrangements in a modern therapy era: A retrospective international study. J. Clin. Oncol., JCO2201297. doi:10.1200/JCO.22.01297
Aymard, F., Bugler, B., Schmidt, C. K., Guillou, E., Caron, P., Briois, S., et al. (2014). Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 366–374. doi:10.1038/nsmb.2796
Ballestar, E., and Esteller, M. (2002). The impact of chromatin in human cancer: Linking DNA methylation to gene silencing. Carcinogenesis 23, 1103–1109. doi:10.1093/carcin/23.7.1103
Barlesi, F., Giaccone, G., Gallegos-Ruiz, M. I., Loundou, A., Span, S. W., Lefesvre, P., et al. (2007). Global histone modifications predict prognosis of resected non small-cell lung cancer. J. Clin. Oncol. 25, 4358–4364. doi:10.1200/JCO.2007.11.2599
Baylin, S. B., and Jones, P. A. (2011). A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer 11, 726–734. doi:10.1038/nrc3130
Benveniste, P., Zhu, W., and Cohen, A. (1995). Interference with thymocyte differentiation by an inhibitor of S-adenosylhomocysteine hydrolase. J. Immunol. 155, 536–544. doi:10.4049/jimmunol.155.2.536
Bergmann, S., Lawler, S. E., Qu, Y., Fadzen, C. M., Wolfe, J. M., Regan, M. S., et al. (2018). Blood-brain-barrier organoids for investigating the permeability of CNS therapeutics. Nat. Protoc. 13, 2827–2843. doi:10.1038/s41596-018-0066-x
Bershteyn, M., Nowakowski, T. J., Pollen, A. A., Di Lullo, E., Nene, A., Wynshaw-Boris, A., et al. (2017). Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell. Stem Cell. 20, 435–449 e4. doi:10.1016/j.stem.2016.12.007
Bianco-Miotto, T., Chiam, K., Buchanan, G., Jindal, S., Day, T. K., Thomas, M., et al. (2010). Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomarkers Prev. 19, 2611–2622. doi:10.1158/1055-9965.EPI-10-0555
Birey, F., Andersen, J., Makinson, C. D., Islam, S., Wei, W., Huber, N., et al. (2017). Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59. doi:10.1038/nature22330
Blakney, A. K., Mckay, P. F., Yus, B. I., Aldon, Y., and Shattock, R. J. (2019). Inside out: Optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 26, 363–372. doi:10.1038/s41434-019-0095-2
Bradbury, E. M. (1992). Reversible histone modifications and the chromosome cell cycle. Bioessays 14, 9–16. doi:10.1002/bies.950140103
Brinkmeier, M. L., Geister, K. A., Jones, M., Waqas, M., Maillard, I., and Camper, S. A. (2015). The histone methyltransferase gene absent, small, or homeotic discs-1 like is required for normal hox gene expression and fertility in mice. Biol. Reprod. 93, 121. doi:10.1095/biolreprod.115.131516
Brown, D. A., Kang, S. H., Gryaznov, S. M., Dedionisio, L., Heidenreich, O., Sullivan, S., et al. (1994). Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 269, 26801–26805. doi:10.1016/s0021-9258(18)47090-1
Cameron, E. E., Bachman, K. E., Myohanen, S., Herman, J. G., and Baylin, S. B. (1999). Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 21, 103–107. doi:10.1038/5047
Cantoni, G. L. (1975). Biological methylation- selected aspects. Annu. Rev. Biochem. 44, 435–451. doi:10.1146/annurev.bi.44.070175.002251
Cao, F., Chen, Y., Cierpicki, T., Liu, Y., Basrur, V., Lei, M., et al. (2010). An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS One 5, e14102. doi:10.1371/journal.pone.0014102
Cau, E., and Wilson, S. W. (2003). Ash1a and Neurogenin1 function downstream of Floating head to regulate epiphysial neurogenesis. Development 130, 2455–2466. doi:10.1242/dev.00452
Chaumeil, J., Le Baccon, P., Wutz, A., and Heard, E. (2006). A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes. Dev. 20, 2223–2237. doi:10.1101/gad.380906
Cheema, H. A., Waheed, N., and Saeed, A. (2022). Kleefstra syndrome with severe sensory neural deafness and de novo novel mutation. J. Coll. Physicians Surg. Pak 32, 236–238. doi:10.29271/jcpsp.2022.02.236
Chen, C., Liu, Y., Rappaport, A. R., Kitzing, T., Schultz, N., Zhao, Z., et al. (2014). MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 25, 652–665. doi:10.1016/j.ccr.2014.03.016
Chen, D., Ma, H., Hong, H., Koh, S. S., Huang, S. M., Schurter, B. T., et al. (1999). Regulation of transcription by a protein methyltransferase. Science 284, 2174–2177. doi:10.1126/science.284.5423.2174
Cheng, C. W., Wu, P. E., Yu, J. C., Huang, C. S., Yue, C. T., Wu, C. W., et al. (2001). Mechanisms of inactivation of E-cadherin in breast carcinoma: Modification of the two-hit hypothesis of tumor suppressor gene. Oncogene 20, 3814–3823. doi:10.1038/sj.onc.1204505
Cho, Y. W., Hong, T., Hong, S., Guo, H., Yu, H., Kim, D., et al. (2007). PTIP associates with MLL3-and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 282, 20395–20406. doi:10.1074/jbc.M701574200
Cif, L., Demailly, D., Lin, J. P., Barwick, K. E., Sa, M., Abela, L., et al. (2020). KMT2B-related disorders: Expansion of the phenotypic spectrum and long-term efficacy of deep brain stimulation. Brain 143, 3242–3261. doi:10.1093/brain/awaa304
Ciolfi, A., Foroutan, A., Capuano, A., Pedace, L., Travaglini, L., Pizzi, S., et al. (2021). Childhood-onset dystonia-causing KMT2B variants result in a distinctive genomic hypermethylation profile. Clin. Epigenetics 13, 157. doi:10.1186/s13148-021-01145-y
Ciptasari, U., and van Bokhoven, H. (2020). The phenomenal epigenome in neurodevelopmental disorders. Hum. Mol. Genet. 29, R42–R50. doi:10.1093/hmg/ddaa175
Damasio, J., Santos, M., Samoes, R., Araujo, M., Macedo, M., Sardoeira, A., et al. (2021). Novel KMT2B mutation causes cerebellar ataxia: Expanding the clinical phenotype. Clin. Genet. 100, 743–747. doi:10.1111/cge.14055
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi:10.1038/nature13772
Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi:10.1038/nature21062
Desrosiers, R., and Tanguay, R. M. (1988). Methylation of Drosophila histones at proline, lysine, and arginine residues during heat shock. J. Biol. Chem. 263, 4686–4692. doi:10.1016/s0021-9258(18)68837-4
Dharmarajan, V., Lee, J. H., Patel, A., Skalnik, D. G., and Cosgrove, M. S. (2012). Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J. Biol. Chem. 287, 27275–27289. doi:10.1074/jbc.M112.364125
Faundes, V., Newman, W. G., Bernardini, L., Canham, N., Clayton-Smith, J., Dallapiccola, B., et al. (2018). Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am. J. Hum. Genet. 102, 175–187. doi:10.1016/j.ajhg.2017.11.013
Fear, V. S., Forbes, C. A., Anderson, D., Rauschert, S., Syn, G., Shaw, N., et al. (2022). Functional validation of variants of unknown significance using CRISPR gene editing and transcriptomics: A Kleefstra syndrome case study. Gene 821, 146287. doi:10.1016/j.gene.2022.146287
Fimiani, C., Goina, E., Su, Q., Gao, G., and Mallamaci, A. (2016). RNA activation of haploinsufficient Foxg1 gene in murine neocortex. Sci. Rep. 6, 39311. doi:10.1038/srep39311
Finkelstein, J. D. (1990). Methionine metabolism in mammals. J. Nutr. Biochem. 1, 228–237. doi:10.1016/0955-2863(90)90070-2
Fombonne, E. (2003). Epidemiological surveys of autism and other pervasive developmental disorders: An update. J. Autism Dev. Disord. 33, 365–382. doi:10.1023/a:1025054610557
Fraga, M. F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., et al. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400. doi:10.1038/ng1531
Frances, L., Quintero, J., Fernandez, A., Ruiz, A., Caules, J., Fillon, G., et al. (2022). Current state of knowledge on the prevalence of neurodevelopmental disorders in childhood according to the DSM-5: A systematic review in accordance with the PRISMA criteria. Child. Adolesc. Psychiatry Ment. Health 16, 27. doi:10.1186/s13034-022-00462-1
Fuks, F., Burgers, W. A., Brehm, A., Hughes-Davies, L., and Kouzarides, T. (2000). DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 24, 88–91. doi:10.1038/71750
Fumagalli, F., Calbi, V., Natali Sora, M. G., Sessa, M., Baldoli, C., Rancoita, P. M. V., et al. (2022). Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 399, 372–383. doi:10.1016/S0140-6736(21)02017-1
Gala, K., Li, Q., Sinha, A., Razavi, P., Dorso, M., Sanchez-Vega, F., et al. (2018). KMT2C mediates the estrogen dependence of breast cancer through regulation of ERα enhancer function. Oncogene 37, 4692–4710. doi:10.1038/s41388-018-0273-5
Galet, B., Cheval, H., and Ravassard, P. (2020). Patient-derived midbrain organoids to explore the molecular basis of Parkinson's disease. Front. Neurol. 11, 1005. doi:10.3389/fneur.2020.01005
Giandomenico, S. L., Sutcliffe, M., and Lancaster, M. A. (2021). Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 16, 579–602. doi:10.1038/s41596-020-00433-w
Gilbert, J., Gore, S. D., Herman, J. G., and Carducci, M. A. (2004). The clinical application of targeting cancer through histone acetylation and hypomethylation. Clin. Cancer Res. 10, 4589–4596. doi:10.1158/1078-0432.CCR-03-0297
Giles, R. V., Spiller, D. G., Grzybowski, J., Clark, R. E., Nicklin, P., and Tidd, D. M. (1998). Selecting optimal oligonucleotide composition for maximal antisense effect following streptolysin O-mediated delivery into human leukaemia cells. Nucleic Acids Res. 26, 1567–1575. doi:10.1093/nar/26.7.1567
Gorman, K. M., Meyer, E., and Kurian, M. A. (2018). Review of the phenotype of early-onset generalised progressive dystonia due to mutations in KMT2B. Eur. J. Paediatr. Neurol. 22, 245–256. doi:10.1016/j.ejpn.2017.11.009
Greer, E. L., and Shi, Y. (2012). Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357. doi:10.1038/nrg3173
Gregory, G. D., Vakoc, C. R., Rozovskaia, T., Zheng, X., Patel, S., Nakamura, T., et al. (2007). Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol. Cell. Biol. 27, 8466–8479. doi:10.1128/MCB.00993-07
Grosz, B. R., Tisch, S., Tchan, M. C., Fung, V. S. C., Darveniza, P., Fellner, A., et al. (2022). A novel synonymous KMT2B variant in a patient with dystonia causes aberrant splicing. Mol. Genet. Genomic Med. 10, e1923. doi:10.1002/mgg3.1923
Guenther, M. G., Jenner, R. G., Chevalier, B., Nakamura, T., Croce, C. M., Canaani, E., et al. (2005). Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 102, 8603–8608. doi:10.1073/pnas.0503072102
Guvakova, M. A., Yakubov, L. A., Vlodavsky, I., Tonkinson, J. L., and Stein, C. A. (1995). Phosphorothioate oligodeoxynucleotides bind to basic fibroblast growth factor, inhibit its binding to cell surface receptors, and remove it from low affinity binding sites on extracellular matrix. J. Biol. Chem. 270, 2620–2627. doi:10.1074/jbc.270.6.2620
Hache, M., Swoboda, K. J., Sethna, N., Farrow-Gillespie, A., Khandji, A., Xia, S., et al. (2016). Intrathecal injections in children with spinal muscular atrophy: Nusinersen clinical trial experience. J. Child. Neurol. 31, 899–906. doi:10.1177/0883073815627882
Hanashima, C., Li, S. C., Shen, L., Lai, E., and Fishell, G. (2004). Foxg1 suppresses early cortical cell fate. Science 303, 56–59. doi:10.1126/science.1090674
Hashimoto, A., Sarker, D., Reebye, V., Jarvis, S., Sodergren, M. H., Kossenkov, A., et al. (2021). Upregulation of C/EBPα inhibits suppressive activity of myeloid cells and potentiates antitumor response in mice and patients with cancer. Clin. Cancer Res. 27, 5961–5978. doi:10.1158/1078-0432.CCR-21-0986
Heintzman, N. D., Hon, G. C., Hawkins, R. D., Kheradpour, P., Stark, A., Harp, L. F., et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. doi:10.1038/nature07829
Heintzman, N. D., Stuart, R. K., Hon, G., Fu, Y., Ching, C. W., Hawkins, R. D., et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318. doi:10.1038/ng1966
Hempel, K., Lange, H. W., and Birkofer, L. (1968). Epsilon-N-trimethyllysine, a new amino acid in histones. Naturwissenschaften 55, 37. doi:10.1007/BF00593411
Hirunagi, T., Sahashi, K., Tachikawa, K., Leu, A. I., Nguyen, M., Mukthavaram, R., et al. (2021). Selective suppression of polyglutamine-expanded protein by lipid nanoparticle-delivered siRNA targeting CAG expansions in the mouse CNS. Mol. Ther. Nucleic Acids 24, 1–10. doi:10.1016/j.omtn.2021.02.007
Hiraide, T., Hattori, A., Ieda, D., Hori, I., Saitoh, S., Nakashima, M., et al. (2019). De novo variants in SETD1B cause intellectual disability, autism spectrum disorder, and epilepsy with myoclonic absences. Epilepsia Open 4, 476–481. doi:10.1002/epi4.12339
Howlin, P., Goode, S., Hutton, J., and Rutter, M. (2004). Adult outcome for children with autism. J. Child. Psychol. Psychiatry 45, 212–229. doi:10.1111/j.1469-7610.2004.00215.x
Hu, D., Gao, X., Morgan, M. A., Herz, H. M., Smith, E. R., and Shilatifard, A. (2013). The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 33, 4745–4754. doi:10.1128/MCB.01181-13
Huang, V., Qin, Y., Wang, J., Wang, X., Place, R. F., Lin, G., et al. (2010). RNAa is conserved in mammalian cells. PLoS One 5, e8848. doi:10.1371/journal.pone.0008848
Hughes, C. M., Rozenblatt-Rosen, O., Milne, T. A., Copeland, T. D., Levine, S. S., Lee, J. C., et al. (2004). Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell. 13, 587–597. doi:10.1016/s1097-2765(04)00081-4
Iefremova, V., Manikakis, G., Krefft, O., Jabali, A., Weynans, K., Wilkens, R., et al. (2017). An organoid-based model of cortical development identifies non-cell-autonomous defects in wnt signaling contributing to miller-dieker syndrome. Cell. Rep. 19, 50–59. doi:10.1016/j.celrep.2017.03.047
Ismail, F. Y., and Shapiro, B. K. (2019). What are neurodevelopmental disorders? Curr. Opin. Neurol. 32, 611–616. doi:10.1097/WCO.0000000000000710
Jakovcevski, M., Ruan, H., Shen, E. Y., Dincer, A., Javidfar, B., Ma, Q., et al. (2015). Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J. Neurosci. 35, 5097–5108. doi:10.1523/JNEUROSCI.3004-14.2015
Janowski, B. A., Huffman, K. E., Schwartz, J. C., Ram, R., Nordsell, R., Shames, D. S., et al. (2006). Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat. Struct. Mol. Biol. 13, 787–792. doi:10.1038/nsmb1140
Janowski, B. A., Younger, S. T., Hardy, D. B., Ram, R., Huffman, K. E., and Corey, D. R. (2007). Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol. 3, 166–173. doi:10.1038/nchembio860
Jo, J., Xiao, Y., Sun, A. X., Cukuroglu, E., Tran, H. D., Goke, J., et al. (2016). Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell. Stem Cell. 19, 248–257. doi:10.1016/j.stem.2016.07.005
Jones, W. D., Dafou, D., Mcentagart, M., Woollard, W. J., Elmslie, F. V., Holder-Espinasse, M., et al. (2012). De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 91, 358–364. doi:10.1016/j.ajhg.2012.06.008
Kang, M. R., Yang, G., Place, R. F., Charisse, K., Epstein-Barash, H., Manoharan, M., et al. (2012). Intravesical delivery of small activating RNA formulated into lipid nanoparticles inhibits orthotopic bladder tumor growth. Cancer Res. 72, 5069–5079. doi:10.1158/0008-5472.CAN-12-1871
Kerimoglu, C., Agis-Balboa, R. C., Kranz, A., Stilling, R., Bahari-Javan, S., Benito-Garagorri, E., et al. (2013). Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J. Neurosci. 33, 3452–3464. doi:10.1523/JNEUROSCI.3356-12.2013
Kerimoglu, C., Sakib, M. S., Jain, G., Benito, E., Burkhardt, S., Capece, V., et al. (2017). KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell. Rep. 20, 538–548. doi:10.1016/j.celrep.2017.06.072
Kim, W., Gwon, Y., Park, S., Kim, H., and Kim, J. (2023). Therapeutic strategies of three-dimensional stem cell spheroids and organoids for tissue repair and regeneration. Bioact. Mater 19, 50–74. doi:10.1016/j.bioactmat.2022.03.039
Kim, Y. S., and Leventhal, B. L. (2015). Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol. Psychiatry 77, 66–74. doi:10.1016/j.biopsych.2014.11.001
Kleefstra, T., Brunner, H. G., Amiel, J., Oudakker, A. R., Nillesen, W. M., Magee, A., et al. (2006). Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 79, 370–377. doi:10.1086/505693
Kleefstra, T., van Zelst-Stams, W. A., Nillesen, W. M., Cormier-Daire, V., Houge, G., Foulds, N., et al. (2009). Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J. Med. Genet. 46, 598–606. doi:10.1136/jmg.2008.062950
Koemans, T. S., Kleefstra, T., Chubak, M. C., Stone, M. H., Reijnders, M. R. F., De Munnik, S., et al. (2017). Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. 13, e1006864. doi:10.1371/journal.pgen.1006864
Krzyzewska, I. M., Maas, S. M., Henneman, P., Lip, K. V. D., Venema, A., Baranano, K., et al. (2019). A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin. Epigenetics 11, 156. doi:10.1186/s13148-019-0749-3
Kummeling, J., Stremmelaar, D. E., Raun, N., Reijnders, M. R. F., Willemsen, M. H., Ruiterkamp-Versteeg, M., et al. (2021). Characterization of SETD1A haploinsufficiency in humans and Drosophila defines a novel neurodevelopmental syndrome. Mol. Psychiatry 26, 2013–2024. doi:10.1038/s41380-020-0725-5
Kuo, A. J., Cheung, P., Chen, K., Zee, B. M., Kioi, M., Lauring, J., et al. (2011). NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell. 44, 609–620. doi:10.1016/j.molcel.2011.08.042
Kuroki, Y., Suzuki, Y., Chyo, H., Hata, A., and Matsui, I. (1981). A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 99 (4), 570–573. doi:10.1016/s0022-3476(81)80256-9
Labonne, J. D., Lee, K. H., Iwase, S., Kong, I. K., Diamond, M. P., Layman, L. C., et al. (2016). An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum. Genet. 135, 757–771. doi:10.1007/s00439-016-1668-4
Lachner, M., and Jenuwein, T. (2002). The many faces of histone lysine methylation. Curr. Opin. Cell. Biol. 14, 286–298. doi:10.1016/s0955-0674(02)00335-6
Lancaster, M. A., Corsini, N. S., Wolfinger, S., Gustafson, E. H., Phillips, A. W., Burkard, T. R., et al. (2017). Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 35, 659–666. doi:10.1038/nbt.3906
Lancaster, M. A., Renner, M., Martin, C. A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. doi:10.1038/nature12517
Lanford, R. E., Hildebrandt-Eriksen, E. S., Petri, A., Persson, R., Lindow, M., Munk, M. E., et al. (2010). Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327, 198–201. doi:10.1126/science.1178178
Lavery, W. J., Barski, A., Wiley, S., Schorry, E. K., and Lindsley, A. W. (2020). KMT2C/D COMPASS complex-associated diseases [KCDCOM-ADs]: An emerging class of congenital regulopathies. Clin. Epigenetics 12, 10. doi:10.1186/s13148-019-0802-2
Lebrun, N., Giurgea, I., Goldenberg, A., Dieux, A., Afenjar, A., Ghoumid, J., et al. (2018). Molecular and cellular issues of KMT2A variants involved in Wiedemann-Steiner syndrome. Eur. J. Hum. Genet. 26, 107–116. doi:10.1038/s41431-017-0033-y
Lederer, D., Grisart, B., Digilio, M. C., Benoit, V., Crespin, M., Ghariani, S. C., et al. (2012). Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 90, 119–124. doi:10.1016/j.ajhg.2011.11.021
Lee, J. E., Wang, C., Xu, S., Cho, Y. W., Wang, L., Feng, X., et al. (2013). H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2, e01503. doi:10.7554/eLife.01503
Lee, J., Kim, D. H., Lee, S., Yang, Q. H., Lee, D. K., Lee, S. K., et al. (2009). A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. U. S. A. 106, 8513–8518. doi:10.1073/pnas.0902873106
Lee, S., Ochoa, E., Barwick, K., Cif, L., Rodger, F., Docquier, F., et al. (2022). Comparison of methylation episignatures in KMT2B- and KMT2D-related human disorders. Child Adolesc. Psychiatry Ment. Health 16, 27.
Lessel, D., Zeitler, D. M., Reijnders, M. R. F., Kazantsev, A., Hassani Nia, F., Bartholomaus, A., et al. (2020). Germline AGO2 mutations impair RNA interference and human neurological development. Nat. Commun. 11, 5797. doi:10.1038/s41467-020-19572-5
Li, L. C., Okino, S. T., Zhao, H., Pookot, D., Place, R. F., Urakami, S., et al. (2006). Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. U. S. A. 103, 17337–17342. doi:10.1073/pnas.0607015103
Li, Y., Zhao, L., Tian, X., Peng, C., Gong, F., and Chen, Y. (2020). Crystal structure of MLL2 complex guides the identification of a methylation site on P53 catalyzed by KMT2 family methyltransferases. Structure 28, 1141–1148 e4. doi:10.1016/j.str.2020.07.002
Liang, X. H., Shen, W., Sun, H., Prakash, T. P., and Crooke, S. T. (2014). TCP1 complex proteins interact with phosphorothioate oligonucleotides and can co-localize in oligonucleotide-induced nuclear bodies in mammalian cells. Nucleic Acids Res. 42, 7819–7832. doi:10.1093/nar/gku484
Liang, X. H., Sun, H., Shen, W., and Crooke, S. T. (2015). Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 43, 2927–2945. doi:10.1093/nar/gkv143
Lister, R., Mukamel, E. A., Nery, J. R., Urich, M., Puddifoot, C. A., Johnson, N. D., et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. doi:10.1126/science.1237905
Macrae, T. A., Fothergill-Robinson, J., and Ramalho-Santos, M. (2022). Regulation, functions and transmission of bivalent chromatin during mammalian development. Nat. Rev. Mol. Cell. Biol. 24, 6–26. doi:10.1038/s41580-022-00518-2
Martynoga, B., Morrison, H., Price, D. J., and Mason, J. O. (2005). Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 283, 113–127. doi:10.1016/j.ydbio.2005.04.005
Masson, R., Mazurkiewicz-Beldzinska, M., Rose, K., Servais, L., Xiong, H., Zanoteli, E., et al. (2022). Safety and efficacy of risdiplam in patients with type 1 spinal muscular atrophy (FIREFISH part 2): Secondary analyses from an open-label trial. Lancet Neurol. 21, 1110–1119. doi:10.1016/S1474-4422(22)00339-8
Matzke, M. A., and Birchler, J. A. (2005). RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 6, 24–35. doi:10.1038/nrg1500
Meng, X., Jiang, Q., Chang, N., Wang, X., Liu, C., Xiong, J., et al. (2016). Small activating RNA binds to the genomic target site in a seed-region-dependent manner. Nucleic Acids Res. 44, 2274–2282. doi:10.1093/nar/gkw076
Meyer, E., Carss, K. J., Rankin, J., Nichols, J. M., Grozeva, D., Joseph, A. P., et al. (2017). Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat. Genet. 49, 223–237. doi:10.1038/ng.3740
Micale, L., Augello, B., Maffeo, C., Selicorni, A., Zucchetti, F., Fusco, C., et al. (2014). Molecular analysis, pathogenic mechanisms, and readthrough therapy on a large cohort of Kabuki syndrome patients. Hum. Mutat. 35, 841–850. doi:10.1002/humu.22547
Michurina, A., Sakib, M. S., Kerimoglu, C., Kruger, D. M., Kaurani, L., Islam, M. R., et al. (2022). Postnatal expression of the lysine methyltransferase SETD1B is essential for learning and the regulation of neuron-enriched genes. EMBO J. 41, e106459. doi:10.15252/embj.2020106459
Miura, Y., Li, M. Y., Birey, F., Ikeda, K., Revah, O., Thete, M. V., et al. (2020). Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 38, 1421–1430. doi:10.1038/s41587-020-00763-w
Miyazaki, H., Higashimoto, K., Yada, Y., Endo, T. A., Sharif, J., Komori, T., et al. (2013). Ash1l methylates Lys36 of histone H3 independently of transcriptional elongation to counteract polycomb silencing. PLoS Genet. 9, e1003897. doi:10.1371/journal.pgen.1003897
Morin, R. D., Mendez-Lago, M., Mungall, A. J., Goya, R., Mungall, K. L., Corbett, R. D., et al. (2011). Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476, 298–303. doi:10.1038/nature10351
Morris-Rosendahl, D. J., and Crocq, M. A. (2020). Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin. Neurosci. 22, 65–72. doi:10.31887/DCNS.2020.22.1/macrocq
Mukai, J., Cannavo, E., Crabtree, G. W., Sun, Z., Diamantopoulou, A., Thakur, P., et al. (2019). Recapitulation and reversal of schizophrenia-related phenotypes in setd1a-deficient mice. Neuron 104, 471–487 e12. doi:10.1016/j.neuron.2019.09.014
Murray, K. (1964). The occurrence of epsilon-N-methyl lysine in histones. Biochemistry 3, 10–15. doi:10.1021/bi00889a003
Nagahama, K., Sakoori, K., Watanabe, T., Kishi, Y., Kawaji, K., Koebis, M., et al. (2020). Setd1a insufficiency in mice attenuates excitatory synaptic function and recapitulates schizophrenia-related behavioral abnormalities. Cell. Rep. 32, 108126. doi:10.1016/j.celrep.2020.108126
Ng, S. B., Bigham, A. W., Buckingham, K. J., Hannibal, M. C., Mcmillin, M. J., Gildersleeve, H. I., et al. (2010). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 790–793. doi:10.1038/ng.646
Niemi, M. E. K., Martin, H. C., Rice, D. L., Gallone, G., Gordon, S., Kelemen, M., et al. (2018). Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271. doi:10.1038/s41586-018-0566-4
Okamoto, N., Miya, F., Tsunoda, T., Kato, M., Saitoh, S., Yamasaki, M., et al. (2017). Novel MCA/ID syndrome with ASH1L mutation. Am. J. Med. Genet. A 173, 1644–1648. doi:10.1002/ajmg.a.38193
Pai, C. C., Kishkevich, A., Deegan, R. S., Keszthelyi, A., Folkes, L., Kearsey, S. E., et al. (2017). Set2 methyltransferase facilitates DNA replication and promotes genotoxic stress responses through MBF-dependent transcription. Cell. Rep. 20, 2693–2705. doi:10.1016/j.celrep.2017.08.058
Paik, W. K., and Kim, S. (1967). E-N-dimethyllysine in histones. Biochem.Biophys. Res. Commun. 27, 479–483. doi:10.1016/s0006-291x(67)80010-x
Park, S. H., Ayoub, A., Lee, Y. T., Xu, J., Kim, H., Zheng, W., et al. (2019). Cryo-EM structure of the human MLL1 core complex bound to the nucleosome. Nat. Commun. 10, 5540. doi:10.1038/s41467-019-13550-2
Pearson, T. S., Gupta, N., San Sebastian, W., Imamura-Ching, J., Viehoever, A., Grijalvo-Perez, A., et al. (2021). Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat. Commun. 12, 4251. doi:10.1038/s41467-021-24524-8
Peinado, H., Ballestar, E., Esteller, M., and Cano, A. (2004). Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 24, 306–319. doi:10.1128/MCB.24.1.306-319.2004
Petros, R. A., and Desimone, J. M. (2010). Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 9, 615–627. doi:10.1038/nrd2591
Pfister, S. X., Ahrabi, S., Zalmas, L. P., Sarkar, S., Aymard, F., Bachrati, C. Z., et al. (2014). SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell. Rep. 7, 2006–2018. doi:10.1016/j.celrep.2014.05.026
Pfluger, P. T., Herranz, D., Velasco-Miguel, S., Serrano, M., and Tschop, M. H. (2008). Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. U. S. A. 105, 9793–9798. doi:10.1073/pnas.0802917105
Pichot, P. (1986). DSM-III: The 3d edition of the diagnostic and statistical manual of mental disorders from the American psychiatric association. Rev. Neurol. Paris. 142, 489–499.
Pilarowski, G. O., Cazares, T., Zhang, L., Benjamin, J. S., Liu, K., Jagannathan, S., et al. (2020). Abnormal Peyer patch development and B-cell gut homing drive IgA deficiency in Kabuki syndrome. J. Allergy Clin. Immunol. 145, 982–992. doi:10.1016/j.jaci.2019.11.034
Place, R. F., Noonan, E. J., Foldes-Papp, Z., and Li, L. C. (2010). Defining features and exploring chemical modifications to manipulate RNAa activity. Curr. Pharm. Biotechnol. 11, 518–526. doi:10.2174/138920110791591463
Portnoy, V., Lin, S. H., Li, K. H., Burlingame, A., Hu, Z. H., Li, H., et al. (2016). saRNA-guided Ago2 targets the RITA complex to promoters to stimulate transcription. Cell. Res. 26, 320–335. doi:10.1038/cr.2016.22
Rea, S., Eisenhaber, F., O'Carroll, D., Strahl, D. B., Sun, Z.-W., Schmid, M., et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. doi:10.1038/35020506
Reebye, V., Huang, K. W., Lin, V., Jarvis, S., Cutilas, P., Dorman, S., et al. (2018). Gene activation of CEBPA using saRNA: Preclinical studies of the first in human saRNA drug candidate for liver cancer. Oncogene 37, 3216–3228. doi:10.1038/s41388-018-0126-2
Reebye, V., Saetrom, P., Mintz, P. J., Huang, K. W., Swiderski, P., Peng, L., et al. (2014). Novel RNA oligonucleotide improves liver function and inhibits liver carcinogenesis in vivo. Hepatology 59, 216–227. doi:10.1002/hep.26669
Reichard, J., and Zimmer-Bensch, G. (2021). The epigenome in neurodevelopmental disorders. Front. Neurosci. 15, 776809. doi:10.3389/fnins.2021.776809
Reynisdottir, T., Anderson, K. J., Boukas, L., and Bjornsson, H. T. (2022). Missense variants causing Wiedemann-Steiner syndrome preferentially occur in the KMT2A-CXXC domain and are accurately classified using AlphaFold2. PLoS Genet. 18, e1010278. doi:10.1371/journal.pgen.1010278
Richmond, T. J., Finch, J. T., Rushton, B., Rhodes, D., and Klug, A. (1984). Structure of the nucleosome core particle at 7 A resolution. Nature 311, 532–537. doi:10.1038/311532a0
Roberts, T. C., Langer, R., and Wood, M. J. A. (2020). Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 19, 673–694. doi:10.1038/s41573-020-0075-7
Roston, A., Evans, D., Gill, H., Mckinnon, M., Isidor, B., Cogne, B., et al. (2021). SETD1B-associated neurodevelopmental disorder. J. Med. Genet. 58, 196–204. doi:10.1136/jmedgenet-2019-106756
Ruault, M., Brun, M. E., Ventura, M., Roizes, G., and De Sario, A. (2002). MLL3, a new human member of the TRX:MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene 284, 73–81. doi:10.1016/s0378-1119(02)00392-x
Salinas, R. D., Connolly, D. R., and Song, H. (2020). Invited review: Epigenetics in neurodevelopment. Neuropathol. Appl. Neurobiol. 46, 6–27. doi:10.1111/nan.12608
Sarker, D., Plummer, R., Meyer, T., Sodergren, M. H., Basu, B., Chee, C. E., et al. (2020). MTL-CEBPA, a small activating RNA therapeutic upregulating C/EBP-alpha, in patients with advanced liver cancer: A first-in-human, multicenter, open-label, phase I trial. Clin. Cancer Res. 26, 3936–3946. doi:10.1158/1078-0432.CCR-20-0414
Schalk, A., Cousin, M. A., Dsouza, N. R., Challman, T. D., Wain, K. E., Powis, Z., et al. (2022). De novo coding variants in the AGO1 gene cause a neurodevelopmental disorder with intellectual disability. J. Med. Genet. 59, 965–975. doi:10.1136/jmedgenet-2021-107751
Schwartz, J. C., Younger, S. T., Nguyen, N. B., Hardy, D. B., Monia, B. P., Corey, D. R., et al. (2008). Antisense transcripts are targets for activating small RNAs. Nat. Struct. Mol. Biol. 15, 842–848. doi:10.1038/nsmb.1444
Shen, H., Sun, T., and Ferrari, M. (2012). Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 19, 367–373. doi:10.1038/cgt.2012.22
Sheppard, S. E., and Quintero-Rivera, F. (1993). “Wiedemann-steiner syndrome,” in GeneReviews((R)). Editors M. P. ADAM, D. B. EVERMAN, G. M. MIRZAA, R. A. PAGON, S. E. WALLACE, L. J. H. BEANet al. (Seattle (WA): University of Washington).
Shi, Y., Sawada, J., Sui, G., Affar El, B., Whetstine, J. R., Lan, F., et al. (2003). Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738. doi:10.1038/nature01550
Siano, M. A., De Maggio, I., Petillo, R., Cocciadiferro, D., Agolini, E., Majolo, M., et al. (2022). De novo mutation in KMT2C manifesting as Kleefstra syndrome 2: Case report and literature review. Pediatr. Rep. 14, 131–139. doi:10.3390/pediatric14010019
Singh, T., Kurki, M. I., Curtis, D., Purcell, S. M., Crooks, L., Mcrae, J., et al. (2016). Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577. doi:10.1038/nn.4267
Singh, T., Poterba, T., Curtis, D., Akil, H., Al Eissa, M., Barchas, J. D., et al. (2022). Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 604, 509–516. doi:10.1038/s41586-022-04556-w
Stagi, S., Gulino, A. V., Lapi, E., and Rigante, D. (2016). Epigenetic control of the immune system: A lesson from Kabuki syndrome. Immunol. Res. 64, 345–359. doi:10.1007/s12026-015-8707-4
Steiner, C. E., and Marques, A. P. (2000). Growth deficiency, mental retardation and unusual facies. Clin. Dysmorphol. 9, 155–156. doi:10.1097/00019605-200009020-00021
Stessman, H. A., Xiong, B., Coe, B. P., Wang, T., Hoekzema, K., Fenckova, M., et al. (2017). Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526. doi:10.1038/ng.3792
Steward, M. M., Lee, J. S., O'Donovan, A., Wyatt, M., Bernstein, B. E., and Shilatifard, A. (2006). Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 13, 852–854. doi:10.1038/nsmb1131
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi:10.1038/47412
Strahl, B. D., Ohba, R., Cook, R. G., and Allis, C. D. (1999). Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl. Acad. Sci. U. S. A. 96, 14967–14972. doi:10.1073/pnas.96.26.14967
Straub, L., Bateman, B. T., Hernandez-Diaz, S., York, C., Lester, B., Wisner, K. L., et al. (2022). Neurodevelopmental disorders among publicly or privately insured children in the United States. JAMA Psychiatry 79, 232–242. doi:10.1001/jamapsychiatry.2021.3815
Strauss, K. A., Farrar, M. A., Muntoni, F., Saito, K., Mendell, J. R., Servais, L., et al. (2022). Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: The phase III SPR1NT trial. Nat. Med. 28, 1381–1389. doi:10.1038/s41591-022-01866-4
Sun, X. J., Wei, J., Wu, X. Y., Hu, M., Wang, L., Wang, H. H., et al. (2005). Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J. Biol. Chem. 280, 35261–35271. doi:10.1074/jbc.M504012200
Tachibana, M., Sugimoto, K., Fukushima, T., and Shinkai, Y. (2001). Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 276, 25309–25317. doi:10.1074/jbc.M101914200
Tanaka, Y., Katagiri, Z., Kawahashi, K., Kioussis, D., and Kitajima, S. (2007). Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene 397, 161–168. doi:10.1016/j.gene.2007.04.027
Thapar, A., Cooper, M., and Rutter, M. (2017). Neurodevelopmental disorders. Lancet Psychiatry 4, 339–346. doi:10.1016/S2215-0366(16)30376-5
Tomizawa, S. I., Kobayashi, Y., Shirakawa, T., Watanabe, K., Mizoguchi, K., Hoshi, I., et al. (2018). Kmt2b conveys monovalent and bivalent H3K4me3 in mouse spermatogonial stem cells at germline and embryonic promoters. Development 145 (23), dev169102. doi:10.1242/dev.169102
Turkmen, S., Timmermann, B., Bartels, G., Groger, D., Meyer, C., Schwartz, S., et al. (2012). Involvement of the MLL gene in adult T-lymphoblastic leukemia. Genes. Chromosom. Cancer 51, 1114–1124. doi:10.1002/gcc.21996
Tunyasuvunakool, K., Adler, J., Wu, Z., Green, T., Zielinski, M., Zidek, A., et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596. doi:10.1038/s41586-021-03828-1
Voutila, J., Reebye, V., Roberts, T. C., Protopapa, P., Andrikakou, P., Blakey, D. C., et al. (2017). Development and mechanism of small activating RNA targeting CEBPA, a novel therapeutic in clinical trials for liver cancer. Mol. Ther. 25, 2705–2714. doi:10.1016/j.ymthe.2017.07.018
Wang, C., Lee, J. E., Lai, B., Macfarlan, T. S., Xu, S., Zhuang, L., et al. (2016). Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. U. S. A. 113, 11871–11876. doi:10.1073/pnas.1606857113
Wang, H., Wang, L., Erdjument-Bromage, H., Vidal, M., Tempst, P., Jones, R. S., et al. (2004). Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878. doi:10.1038/nature02985
Wang, S., Bleeck, A., Nadif Kasri, N., Kleefstra, T., van Rhijn, J. R., and Schubert, D. (2021). SETD1A mediated H3K4 methylation and its role in neurodevelopmental and neuropsychiatric disorders. Front. Mol. Neurosci. 14, 772000. doi:10.3389/fnmol.2021.772000
Wang, S., Rhijn, J. V., Akkouh, I., Kogo, N., Maas, N., Bleeck, A., et al. (2022). Loss-of-function variants in the schizophrenia risk gene SETD1A alter neuronal network activity in human neurons through the cAMP/PKA pathway. Cell. Rep. 39, 110790. doi:10.1016/j.celrep.2022.110790
Watanabe, Y., Castoro, R. J., Kim, H. S., North, B., Oikawa, R., Hiraishi, T., et al. (2011). Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. PLoS One 6, e23320. doi:10.1371/journal.pone.0023320
Will, B., and Steidl, U. (2014). Combinatorial haplo-deficient tumor suppression in 7q-deficient myelodysplastic syndrome and acute myeloid leukemia. Cancer Cell. 25, 555–557. doi:10.1016/j.ccr.2014.04.018
Xu, Q., Xiang, Y., Wang, Q., Wang, L., Brind'Amour, J., Bogutz, A. B., et al. (2019). SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat. Genet. 51, 844–856. doi:10.1038/s41588-019-0398-7
Yang, H., Pesavento, J. J., Starnes, T. W., Cryderman, D. E., Wallrath, L. L., Kelleher, N. L., et al. (2008). Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J. Biol. Chem. 283, 12085–12092. doi:10.1074/jbc.M707974200
Yu, X., Yang, L., Li, J., Li, W., Li, D., Wang, R., et al. (2019). De novo and inherited SETD1A variants in early-onset epilepsy. Neurosci. Bull. 35, 1045–1057. doi:10.1007/s12264-019-00400-w
Zech, M., Boesch, S., Maier, E. M., Borggraefe, I., Vill, K., Laccone, F., et al. (2016). Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2B, results in early-onset generalized dystonia. Am. J. Hum. Genet. 99, 1377–1387. doi:10.1016/j.ajhg.2016.10.010
Zhao, K., van der Spoel, A., Castiglioni, C., Gale, S., Fujiwara, H., Ory, D. S., et al. (2018). 19q13.12 microdeletion syndrome fibroblasts display abnormal storage of cholesterol and sphingolipids in the endo-lysosomal system. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2108–2118. doi:10.1016/j.bbadis.2018.03.020
Keywords: neurodevelopmental disorders (NDDs), small-activating RNA (saRNA), histone lysine methyltransferases (HKMTs), epigenetics (chromatin remodeling), brain organoids
Citation: Roth C, Kilpinen H, Kurian MA and Barral S (2023) Histone lysine methyltransferase-related neurodevelopmental disorders: current knowledge and saRNA future therapies. Front. Cell Dev. Biol. 11:1090046. doi: 10.3389/fcell.2023.1090046
Received: 07 December 2022; Accepted: 06 February 2023;
Published: 27 February 2023.
Edited by:
Katherine Athayde Teixeira De Carvalho, Pelé Pequeno Príncipe Research Institute, BrazilReviewed by:
Luciane R. Cavalli, Pelé Pequeno Príncipe Research Institute, BrazilCopyright © 2023 Roth, Kilpinen, Kurian and Barral. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serena Barral, cy5iYXJyYWxAdWNsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.