 Georg A. Petroianu
Georg A. Petroianu Lujain Aloum
Lujain Aloum Abdu Adem
Abdu Adem
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 16 January 2023
Sec. Signaling
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1072629
This article is part of the Research Topic Emerging Insights in Glutamate Receptor Signaling in Psychiatric, Neurodevelopmental, and Neurodegenerative Diseases View all 5 articles
The physiopathology and neurotransmission of pain are of an owe inspiring complexity. Our ability to satisfactorily suppress neuropathic or other forms of chronic pain is limited. The number of pharmacodynamically distinct and clinically available medications is low and the successes achieved modest. Pain Medicine practitioners are confronted with the ethical dichotomy imposed by Hippocrates: On one hand the mandate of primum non nocere, on the other hand, the promise of heavenly joys if successful divinum est opus sedare dolorem. We briefly summarize the concepts associated with nociceptive pain from nociceptive input (afferents from periphery), modulatory output [descending noradrenergic (NE) and serotoninergic (5-HT) fibers] to local control. The local control is comprised of the “inflammatory soup” at the site of pain origin and synaptic relay stations, with an ATP-rich environment promoting inflammation and nociception while an adenosine-rich environment having the opposite effect. Subsequently, we address the transition from nociceptor pain to neuropathic pain (independent of nociceptor activation) and the process of sensitization and pain chronification (transient pain progressing into persistent pain). Having sketched a model of pain perception and processing we attempt to identify the sites and modes of action of clinically available drugs used in chronic pain treatment, focusing on adjuvant (co-analgesic) medication.
The neurotransmission of pain is of an owe inspiring complexity. In contrast, our ability to satisfactorily suppress neuropathic and/or chronic pain is limited. The number of pharmacodynamically distinct available medications is low, and the therapeutical successes achieved modest. In the words of Carl Edward Noe ….when pain leads to suffering, it ceases to be a teacher and becomes the oldest medical malady (Noe, 2020). Pain Medicine practitioners are confronted with the ethical dilemma imposed by Hippocrates, the delicate balance act between the mandate of primum non nocere, and the promise of heavenly joys if successful in delivering relief divinum est opus sedare dolorem.
Upon reviewing the current understanding of pain perception and processing we highlight the possible sites and modes of action of clinically available drugs used in pain treatment, focusing on adjuvant (co-analgesic) medication.
Noxious stimuli in the periphery are perceived via nociceptors and the information is relayed to the dorsal horn where the first synapse of the ascending pain pathway is localized. Post-synaptically the nociceptive input is relayed to higher (supra-spinal) centers. Modulating descending output from supra-spinal centers is received at dorsal horn level. The micro-environment (inflammatory soup) at the site of origin of noxious stimuli (injury) and around the dorsal horn have a significant influence on the intensity and duration of the signal (Figure 1).
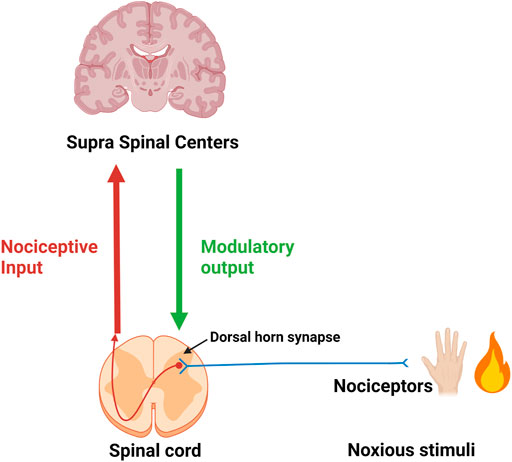
FIGURE 1. First neuron (in blue) of the ascending pain pathway. Red-brown arrow: Second ascending neuron leading to supra-spinal centers. Green arrow: Descending modulatory output from supra-spinal centers.
Injury leads to activation of Transient Receptor Potential (TRP) nociceptors in the periphery; subsequent opening of cation channels results in depolarization and action potential propagation along afferent sensory fibers to the dorsal horn synapse. Presynaptic vesicles release excitatory non-peptide transmitter (glutamate, AMPA and NMDA receptor agonist), and peptide transmitters [Substance P (SP) (NK1 neurokinin receptor 1 agonist) and Calcitonin Gene-Related Peptide (CGRP) (Calcitonin like Receptor and Receptor Activity Modifying Protein complex agonist)]. Neuropeptide Y (NPY) and CGRP receptors co-localize extensively; the two neurotransmitters have mostly opposite effects (Nelson and Taylor, 2021). NPY acts at Y2 receptors on the central terminals of primary afferents to inhibit SP release (Duggan et al., 1991).
In blood vessels, CGRP acts as a potent vasodilator when compared to several known vasodilators such as histamine, prostaglandin E2 and SP. Inhibitors of the CGRP receptor are identified by the suffix–gepant (ubrogepant; atogepant). AMPA receptor antagonists are identified by the suffix–ampanel (perampanel) (De Caro et al., 2020). Kynurenic acid is one of the endogenous antagonists at ionotropic AMPA, NMDA (glycine-site ligand), and kainate glutamate receptors.
Second neuron: After crossing to the contralateral side, the signal travels in the ascending lateral spino-thalamic tract (TST) to the thalamus.
Third neuron: From the thalamus to the sensory cortex in the parietal lobe in the thalamo-cortical tract TTC (Brodmann areas 1, 2, and 3) allowing pain localization (Figure 2).
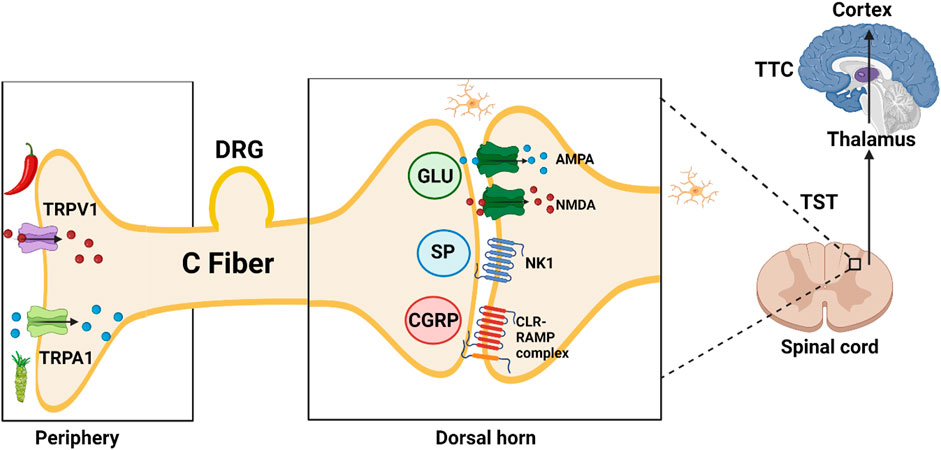
FIGURE 2. TRPV1, transient receptor potential vanilloid subfamily, member 1, capsaicin or hot chili pepper receptor; TRPA1, transient receptor potential ankyrin subfamily, member 1, allyl isothiocyanate or wasabi receptor; DRG, dorsal root ganglion; GLU, glutamate; SP, substance P (11 AA); AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA, N-methyl-d-aspartate; NK1, Neurokinin 1 receptor; CGRP, calcitonin gene-related peptide (37 AA); CLR-RAMP, calcitonin like receptor—receptor activity modifying protein complex; TST, Tractus spino-thalamicus; TTC, tractus thalamo-corticalis;




Descending noradrenergic (norepinephrine; NE) and serotoninergic (5-HT) fibers influence pain perception. Most noradrenergic fibers originate from the pontine locus coeruleus (LC; blue spot) while descending serotonergic pathways originate from the floor of the medulla oblongata from the nucleus raphe magnus (NRM). Both NE and 5-HT induce membrane hyperpolarization while decreasing the excitatory transmitter release from primary Aδ and C afferent fibers pre-synaptically and increasing the release of inhibitory GABA and glycine from interneurons (Yoshimura and Furue, 2006). The administration of 5-HT produces membrane hyperpolarization in about 50% of dorsal horn neurons, while NE hyperpolarizes more than 80% of them, suggesting the need to augment both NE and 5-HT concentrations in order to suppress algesia. Neither atomoxetine (selective noradrenaline reuptake inhibitor) nor an SSRI alone have such a marked effect (Miyahara et al., 2019).
Norepinephrine exerts via α2-adrenoceptors an inhibitory influence on neuropathic pain while α1-adrenoceptors exacerbate it (Kim et al., 2005).
As to the receptors involved in serotoninergic pain modulation findings indicate a role for 5-HT7 receptors in antinociception, and a role for 5-HT3 in pro-nociceptive facilitation (Dogrul et al., 2009). Activation of 5-HT7 receptors does not directly inhibit nociceptive dorsal horn neurons because these receptors are positively coupled to adenylate cyclase and their stimulation is excitatory. However, activation of 5-HT7 receptors localized on spinal inhibitory enkephalinergic or GABAergic interneurons, to evoke the release of enkephalins or GABA, produces an inhibition of nociceptive transmission (Brenchat et al., 2009; Liu et al., 2020) (Figure 3).
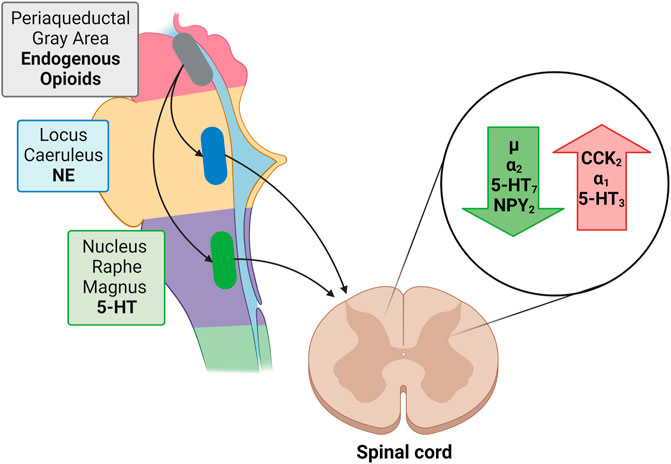
FIGURE 3. Green arrow: Receptors reducing nociception: µ opioid receptor; α2, adrenergic receptor; 5-HT7, serotonin receptor; NPY2, neuropeptide Y receptor type 2. Red arrow: Receptors enhancing nociception: CCK2, cholecystokinin 2(B) receptor; α1, adrenergic receptor; 5-HT3, ionotropic serotonin receptor;



The activity of the feed-back loop consisting of (afferent) nociceptive input, and modulatory (efferent) output is markedly influenced by the composition of the “cytokine soup” at the site of origin and synaptic relay stations, with an ATP-rich environment promoting inflammation and nociception while an adenosine-rich environment having the opposite effect.
In an injured environment glia cells release intracellular ATP via the vesicular nucleotide transporter (VNUT) into the extracellular space. ATP activates purinergic P2X ionotropic and metabotropic P2Y receptors that increase the sodium and calcium conductance of various TRP receptors. Cell surface (ecto)nucleotidases by degrading nucleotides serve to terminate purinergic signaling; their function is analogous to the activity of cholinesterase at cholinergic synapses (Liu et al., 2017; Stokes et al., 2017).
Extracellular adenosine generated by metabolic break-down of nucleotides activates adenosine receptors (A1, A2A, A2B, and A3) that decrease the sodium and calcium permeability of TRP receptors (Figure 4) (Battastini et al., 2021). The antinociceptive activity of adenosine is mediated mainly via activation of adenosine A1 receptors; activation of A3A receptors seems to play a role as well. However, the function of adenosine A2A and A2B receptors is more debatable as their activation causes both nociception and anti-nociception effects (Vincenzi et al., 2020).
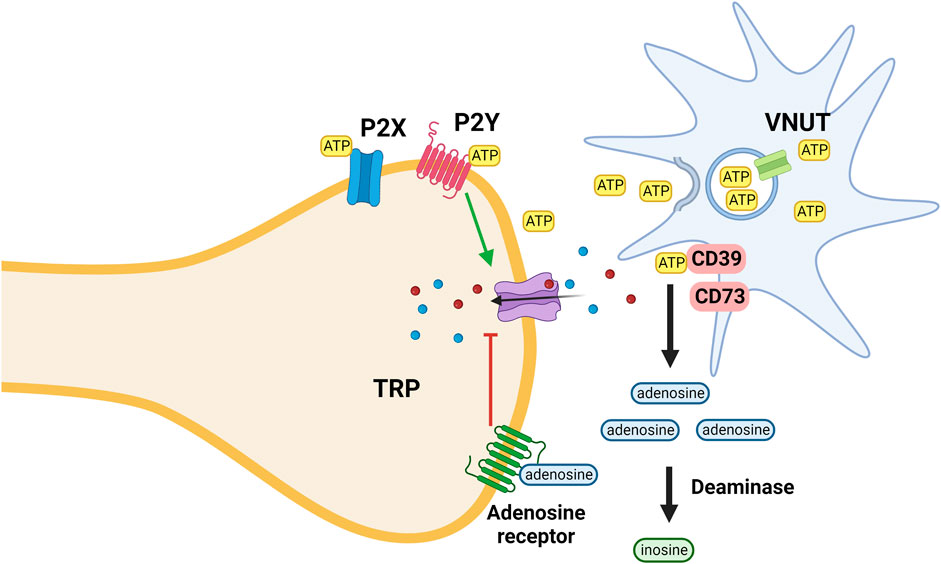
FIGURE 4. ATP is released from microglia, keratinocytes and other cells via the activity of the VNUT; inhibition of the transporter reduces the ATP concentration and thus its pro-inflammatory effect mediated via interaction with both ionotropic (P2X) and metabotropic (P2Y) purinergic receptors. ATP is metabolized by ectoenzymes (CD; cluster of differentiation 73 and 39). Extracellular adenosine generated by metabolic break-down of nucleotides activates adenosine receptors that decrease the sodium and calcium permeability of TRP receptors. Adenosine is metabolized by deaminases.
The purinergic influence is complemented by a plethora of pro- and anti-nociceptive substances, both oligopeptide [endogenous opioid peptides, the anti-opioid cholecystokinin (CCK), bradykinin, and cytokines] and non-peptide (histamine, prostanoids, leukotrienes).
Non-steroidal anti-inflammatory drugs (NSAIDs) are effective in nociceptive pain and controlling pain associated with inflammation via inhibition of cyclooxygenase-2. They are commonly prescribed for neuropathic pain however; there is not sufficient evidence to support the usage of NSAIDs for such pain and thus are not listed in the major guidelines addressing the treatment of neuropathic pain (Vane and Botting, 1998; Cohen and Mao, 2014; Moore et al., 2015; Kappelmann et al., 2018). One has to consider however that pain is rarely exclusively neuropathic or nociceptive.
Nociceptive pain is the physiologic response to nociceptor activation, and -as it is conducive to protective responses-it serves a useful purpose. Pain independent of nociceptor activation is called neuropathic; it is -most likely-not beneficial and it is the result of sensitization and autonomous ectopic activity of various pro-nociceptive entities, at the most basic level sodium and calcium channels. Dull nociceptive pain is contrasted by the sharp neuropathic one. There is overlap between chronic and neuropathic pain: neuropathic pain is chronic but not all chronic pain is neuropathic.
Most likely as a result of increased activity of the endogenous pro-nociceptive neuropeptide CCK, neuropathic pain does not respond well to opioids (Wiesenfeld-Hallin et al., 2002). The predominant form of CCK in the central nervous system is an octapeptide (Bowers et al., 2012). Opioid-induced CCK release is one of the postulated mechanisms explaining opioid tolerance and hyperalgesia (Smith, 2012). Recently, heterodimerization of opioid and CCK receptors subsequent to CCK octapeptide binding was demonstrated. This interaction was identified as the basis of the inhibition of opioid signal transduction and the antagonism of morphine analgesia (Yang et al., 2018; Bernard et al., 2021). CCK antagonists enhance the analgesic efficacy of endogenous opioids in animal models of pain (Inoue, 2017). NPY is pro-analgesic and naloxone administration reduces NPY analgesia (Upadhya et al., 2009). Even though opioids have been very effective in the treatment of pain but in recent years opioid overdose has been a major challenge for the clinical management of pain. Indeed, the most tragic reported drug overdose epidemic in US history was due to opioids. The introduction of a slow-release formulation of oxycodone, in 1996 was found to play a major role in this overdose death or so-called US opioid epidemic (Egilman et al., 2019; Dyer, 2020; Alpert et al., 2021).
The need to estimate and compare drug efficacy for a particular disease is partially satisfied by using NNT (number needed to treat) and NNH (number needed to harm). NNT is defined as the number of patients needed to treat with a certain drug to obtain one patient with a defined degree of pain relief. NNH indicates the number of patients that need to be treated for one patient to drop out due to adverse effects. While the approach works reasonably well for a defined condition, it becomes fuzzy when a multitude of pain causes are amalgamated. Figure 5 shows NNTs for the main drugs used in chronic pain treatment across the different conditions (Finnerup et al., 2005). Table 1 summarizes the sites, modes of action, mechanisms, and examples of drugs used in the relief of pain.
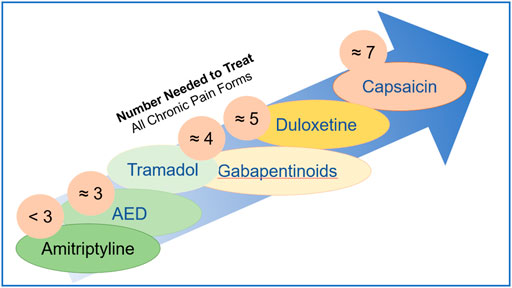
FIGURE 5. Number needed to treat is defined as the number of patients needed to treat with a certain drug to obtain one patient with a defined degree of pain relief.
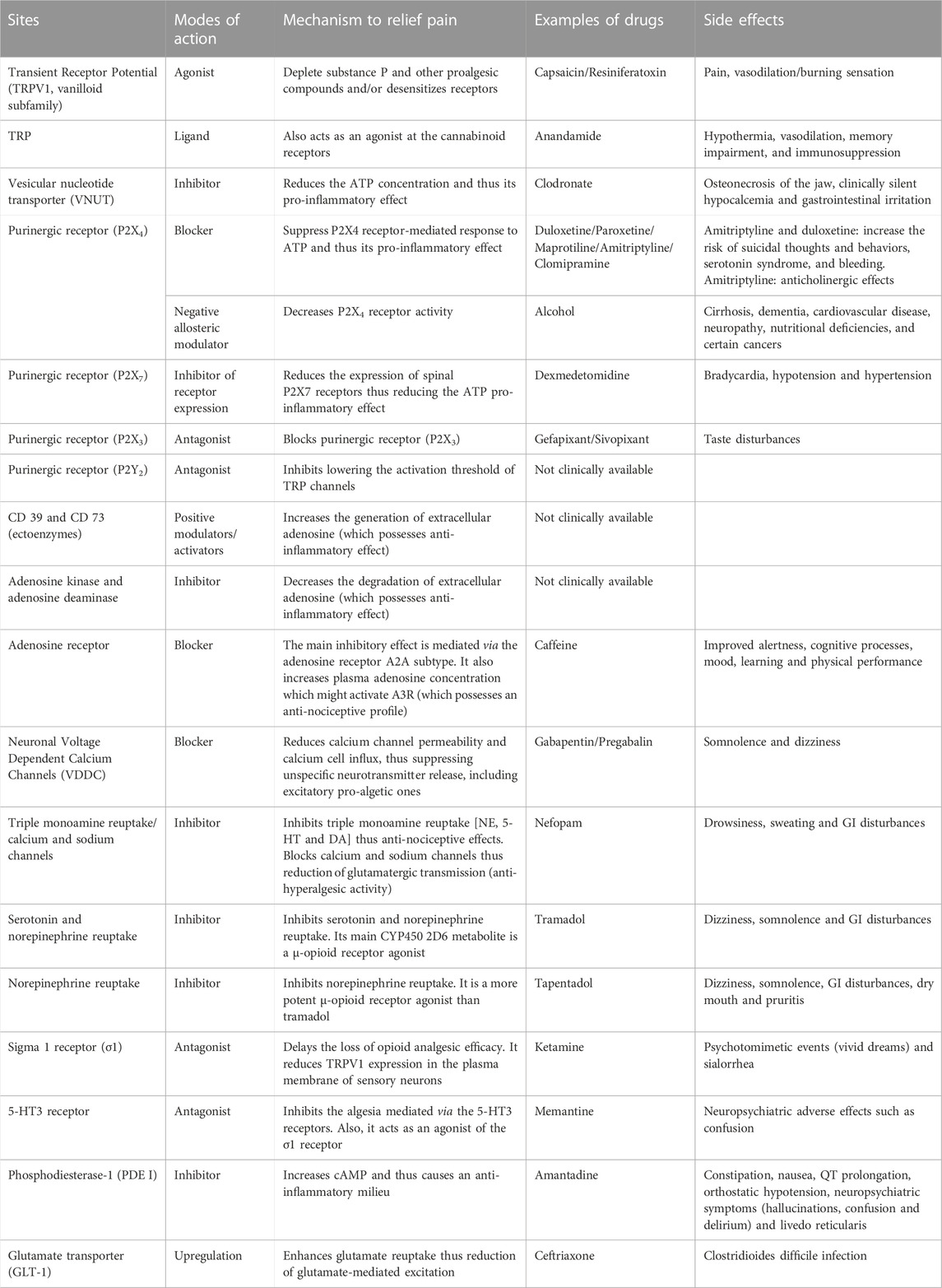
TABLE 1. The sites, modes of action, mechanism and examples of drugs used in the relief of pain.
The TRP channel superfamily consists of many cation channels divided into six subfamilies. The TRPA1 (ankyrin subfamily, member 1) is known as the wasabi receptor (typical ligand is allyl isothiocyanate, the main pungent compound of wasabi), while the TRPV1 (vanilloid subfamily, member 1) is known as the hot chili pepper receptor [typical ligand is capsaicin (Figure 6), the pungent compound of hot chili pepper]. TRPA1 and TRPV1 have been associated with pain perception (Gouin et al., 2017). Remarkably, it has been reported that almost all sensory neurons expressing TRPA1 (approximately 97%) also express TRPV1 (Story et al., 2003). Functional cross-desensitization has also been reported between the typical agonists of TRPA1 (allyl isothiocyanate; wasabi) and TRPV1 (capsaicin) (Mihara and Shibamoto, 2015). Furthermore, studies have shown that TRPA1 and TRPV1 can form a complex in the plasma membrane, and therefore influence each other’s characteristics (Staruschenko et al., 2010). Thus, Fernandes et al. (2012) described TRPA1 and TRPV1 channels as “partners in crime.”
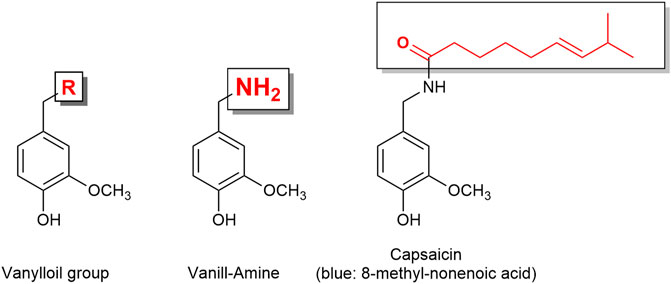
FIGURE 6. Capsaicin is a vanilloid derivative. The capsaicin receptor was therefore named vanilloid receptor.
Ligands at these receptors act from the intracellular side. The best-known activators of TRPV1 are temperature greater than 43°C (109°F), hydrogen ions, capsaicin, allyl isothiocyanate (mustard oil) and resiniferatoxin (Aloum et al., 2021).
Resiniferatoxin (RTX) is a chemical found in some Euphorbia species from Africa. Pure RTX is two-to three orders of magnitude hotter than pure capsaicin on the eponymous scale devised by Wilbur Scoville (1865–1942).
Capsaicin has been available as transdermal therapeutic system for many years. On dermal application (8%), it causes pain (dolor) and vasodilation (rubor); many pain specialists advise for use of lidocaine gel/cream prior (1 h) to capsaicin application. Tramadol given well before patch application is considered as an alternative pretreatment option in patients receiving capsaicin patch treatment (Jensen et al., 2014). Resiniferatoxin also causes burning sensation, thus anesthesia or nerve block can be used prior to resiniferatoxin injection (Salas et al., 2017).
Subsequently chronic pain relief sets in with various degrees of efficacy and duration. The assumed mechanism of action is depletion of SP and other proalgesic compounds and/or desensitization of receptors. Overall, the therapeutic approach is less successful than initially hoped for.
Anandamide is an endocannabinoid with anxiolytic and hypoalgesic properties (Habib et al., 2019). The name “anandamide” is derived from the Sanskrit word for bliss or delight ananda, and amide (Fride and Mechoulam, 1993). In addition to effects mediated via cannabinoid receptors, anandamide is also an endogenous ligand (agonist?) for TRP receptors, contributing i.e. to the vasodilation component of inflammation (Russell et al., 2014). Hypothermia, vasodilation, memory impairment, and immunosuppression are side effects of anandamide (Greenberg, 2003) Many other candidates that belong to the cannabinoids family has been used for the management of neuropathic pain as reviewed in (Aviram and Samuelly-Leichtag, 2017; Romero-Sandoval et al., 2017; Bruni et al., 2018; Lee et al., 2018; Mücke et al., 2018; Romero-Sandoval et al., 2018; Boyaji et al., 2020; Meng et al., 2020; Mlost et al., 2020; Urits et al., 2020; Campos et al., 2021). Recently, after the opioid epidemic, attention was diverted to cannabinoids as a therapeutic option for the treatment of pain (Urits et al., 2020).
TRPV1 activity is subject to regulation by a host of intracellular signaling cascades and extracellular events. As such, modulation of these is a possible and attractive avenue for chronic pain treatment. The fundamental premise as stated by (De Marchi et al., 2019) and other groups is that ATP-induces a pro-inflammatory milieu while an anti-inflammatory environment is adenosine-driven. ATP is released from microglia, keratinocytes, and other cells via the activity of the VNUT; inhibition of the transporter reduces the ATP concentration and thus its pro-inflammatory effect mediated via interaction with both ionotropic (P2X) and metabotropic (P2Y) purinergic receptors.
Clodronate (Figure 7), a first-generation non-nitrogen-containing bisphosphonate, is a potent and selective inhibitor of the VNUT and thus an inhibitor of ATP release. In vitro clodronate inhibits VNUT at an IC50 ≈ 16 nM without affecting other vesicular neurotransmitter transporters. Clodronate has the potential to suppress inflammatory and neuropathic pain by shifting the balance ATP/adenosine from pro-inflammatory ATP to anti-inflammatory adenosine (Kato et al., 2017; De Marchi et al., 2019; Miras-Portugal et al., 2019; Hasuzawa et al., 2020). Osteonecrosis of the jaw and clinically silent hypocalcemia are side effects associated with the usage of clodronate (Frediani et al., 2018). The gastrointestinal irritation (such as nausea, dyspepsia) is less severe compared to nitrogen-containing bisphosphonates (Suri et al., 2001).
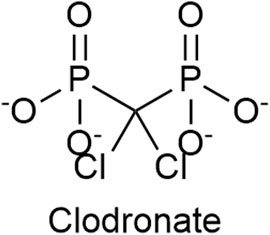
FIGURE 7. The structure of Clodronate, a first generation non-nitrogen-containing bisphosphonate, which is an inhibitor of the vesicular nucleotide transporter (VNUT).
P2X ionotropic purinergic receptors mediate excitatory postsynaptic responses; the main pharmacological distinction between the members of the P2X purinoceptor family is their relative sensitivity to the antagonist suramin (the drug used for the treatment of trypanosomial-induced human sleeping sickness). Seven different P2X receptor subtypes (P2X1-7) have been identified to date; trimeric assembly creates various ionic channels. P2X4 receptors have the highest permeability for calcium ions and are thus viewed as most relevant to pain.
P2X4 activation in spinal microglia results in pain hypersensitivity (Inoue, 2017; Stokes et al., 2017; Tozaki-Saitoh et al., 2022).
P2X4 receptor activity is positively modulated by ivermectin, a broad-spectrum anti-parasitic agent included in the WHO essential medicines list for several parasitic diseases (Nörenberg et al., 2012).
The selective serotonin and norepinephrine reuptake inhibitor (SSNeRI) duloxetine (Figure 8) reduces pain via augmenting serotonin and norepinephrine mediated inhibitory pain pathways (Smith and Nicholson, 2007). It might alleviate chronic pain also through P2X4 receptor blockade Duloxetine was able to almost completely suppress P2X4 receptor-mediated response to micromolar ATP (Yamashita et al., 2016). The same applies for the selective serotonin reuptake inhibitor (SSRI) paroxetine, and to a lesser extent (≈30% of control) to maprotiline (secondary amine), classical (tertiary amine) tricyclic antidepressants (TCA) (≈50% of control) (amitriptyline and clomipramine) and possibly to most antidepressants used in chronic pain treatment. No effect on P2X4 receptor activity was noticed for mirtazapine and bupropion (Yamashita et al., 2016; Kohno and Tsuda, 2021).
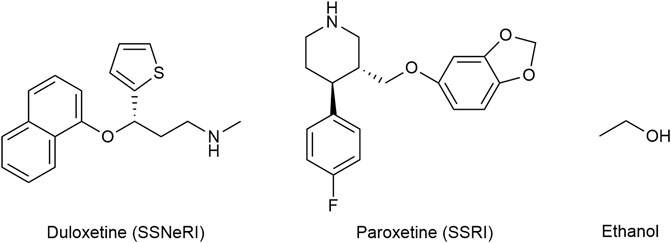
FIGURE 8. The structures of duloxetine (selective serotonin and norepinephrine reuptake inhibitor—SSNeRI), paroxetine (selective serotonin reuptake inhibitor—SSRI), and ethanol.
Amitriptyline and duloxetine are recommended first-line treatment for neuropathic pain. Amitriptyline and duloxetine both increase the risk of suicidal thoughts and behaviors, serotonin syndrome, and bleeding; only the former precipitates anticholinergic effects (Bryson and Wilde, 1996; Dougherty et al., 2002; Friedman and Leon, 2007; Leon, 2007; Bartlett, 2017; Bixby et al., 2019; Dhaliwal et al., 2022). The NNT and NNH values are 3.6 (3.0–4.4) and 13.4 (9.3–24.4) for amitriptyline and 6.4 (5.2–8.4) and 11.8 (9.5–15.2) for duloxetine, respectively. Amitriptyline has a selectivity ratio serotonin-to-norepinephrine transporters of approximately 3:1, whereas duloxetine has a 10-fold selectivity for serotonin transporters (Bymaster et al., 2001; Finnerup et al., 2015; Patel and Dickenson, 2022).
The superior NNT for amitriptyline vs. duloxetine contrasts their blocking ability at P2X4 receptor (50% vs. >90%). Several explanations are possible, such as that efficacy for neuropathic pain treatment requires additional mechanisms beyond P2X4 antagonism and that a balanced reuptake inhibition (serotonin to norepinephrine) is of equal, if not greater importance.
While such a line of thinking is perfectly plausible, an alternative explanation is the fact that NNTs are estimated against placebo; a highly sedating tertiary amine such as amitriptyline (IC50 = 1 nM for the histamine H1 receptor) would have a perceived “beneficiary” effect of facilitating sleep that is difficult to separate from pain reduction. Duloxetine, a non-sedating agent with an IC50 = 2,300 nM for the histamine H1 receptor would not have such a benefit (Bymaster et al., 2001). Milnacipran, a non-sedating agent with a selectivity ratio serotonin-to-norepinephrine transporters of approximately 1:3 (mirror values of amitriptyline) has a modest effect in chronic pain with a NNT of only 6 to 10.
The mechanism of action of antidepressants in chronic pain remains highly controversial (Stokes et al., 2017).
Historically ethanol (Figure 8) is one of the oldest analgesics and sedative-hypnotics known to mankind. The molecule being quite promiscuous, the mechanism of action of alcohol is certainly complex. P2X4 are the most ethanol sensitive P2X receptors in the central nervous system and likely mediate some if not the main effect of ethanol on the brain. As the ethanol concentration increases, P2X4 activity decreases (Zhang et al., 2020). Alcohol acts as a negative allosteric modulator that blocks the open channel; ivermectin can antagonize the inhibitory effect of ethanol on P2X4 and is thought to interfere with ethanol binding to the channel (Kanellopoulos et al., 2021). Chronic alcohol consumption is associated with cirrhosis, dementia, cardiovascular disease (such as hypertension, heart failure, atrial fibrillation), neuropathy, nutritional deficiencies, and certain cancers (Bielinska-Kwapisz and Mielecka-Kubien, 2011; Obad et al., 2018).
Antagonism at P2X7 receptors is assumed to provide multiple benefits (Mishra et al., 2021). Oxidized ATP irreversibly antagonizes P2X7 receptor activation, but lacks selectivity (Murgia et al., 1993; Savio et al., 2018; Rotondo et al., 2022). Brilliant Blue G produced a non-competitive inhibition of human P2X7 receptors with IC50 values of 200 nM.
While no clinically used compounds interacting selectively/exclusively with P2X7 receptor are available, dexmedetomidine, a centrally acting α2-adrenoceptor agonist used for sedation, was proposed to attenuate neuropathic pain i.a. through inhibition of spinal P2X7 receptor expression (Lin et al., 2018; Zhao et al., 2020) (Figure 9). Bradycardia, hypotension, and hypertension are the most commonly reported side effects (Kaye et al., 2020).
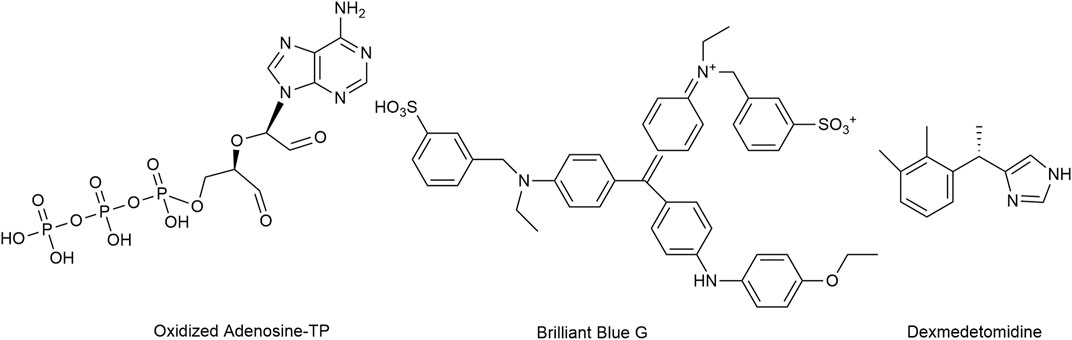
FIGURE 9. The structures of oxidized Adenosine-TP, Brilliant Blue G and dexmedetomidine, which mediate the inhibition of P2X7 receptors.
Gefapixant (Merck-7264), structurally similar to trimethoprim, is a P2X3 receptor antagonist named in honour of Geoffrey Burnstock (1929–2020), the father of purinergic pharmacology. Japan authorities approved gefapixant for adults with refractory or unexplained chronic cough. The suffix -pixant was adopted for P2X antagonists. Gefapixant has however a low selectivity for P2X3 receptors, whereas the newly developed sivopixant has a much higher affinity and selectivity (P2X3 IC50 ≈ 5 nM). Taste disturbances such as loss or reduced sense of taste are the most reported side effects with gefapixant since P2X3 receptors plays an important role in taste perception. Other common side effects include nausea and upper respiratory tract infection. However, lower incidence of taste disturbances is conveyed with sivopixant administration (Abu-Zaid et al., 2021; Niimi et al., 2022).
P2Y metabotropic receptors are G-protein-coupled receptors that are stimulated by purine and pyrimidine nucleotides. P2Y1 and/or P2Y2 receptor activation modulates TRP channels by lowering their activation threshold and thus favoring nociception (Tozaki-Saitoh et al., 2022).
Diquafosol, a P2Y2 receptor agonist that promotes fluid transfer and mucin secretion by activating receptors expressed on the ocular surface, has been approved in Japan and South Korea to treat dry eye disease (Murakami et al., 2004; Keating, 2015; Nam et al., 2019).
In contrast, an experimental P2Y2 receptor-selective antagonist, reversed allodynia in a chronic pain model (Magni et al., 2015). No P2Y2 antagonists are clinically available.
CD 39 (EC 3.6.1.5) and CD 73 (EC 3.1.3.5) are ectoenzymes involved in the production of AMP from phosphorylated precursors (CD 39) and adenosine from AMP (CD 73) (Liu et al., 2017). They participate in regulating inflammatory processes and in nociceptive modulation by affecting extracellular adenosine generation.
Cell surface (ecto)nucleotidases serving to terminate purinergic signaling are seen as analogous to the activity of cholinesterases at cholinergic synapses (Stokes et al., 2017). Manipulating extracellular AMP hydrolysis could provide an alternative mechanism to control pain (Zylka, 2011; Liu et al., 2017).
Inhibiting overexpressed ectoenzymes involved in the production of adenosine in cancer cells may improve outcomes of conventional cancer therapy by decreasing adenosine levels and, consequently, promoting antitumor immune cells (Azambuja et al., 2019; Battastini et al., 2021).
Conversely, positive modulators/activators of ectoenzymes, are of potential interest in reducing/modulating pain, contingent on our ability to control off-target effects (Sowa et al., 2010). No positive modulators or antagonists are to date clinically available.
Adenosine is rapidly removed from the extracellular space by nucleoside transporters and metabolic enzymes, including adenosine kinase (intracellular conversion to AMP and trapping) and adenosine deaminase (conversion to inosine). Indeed, the extracellular concentration of adenosine and the antinociceptive effects of adenosine can be increased by pharmacologically inhibiting these metabolic enzymes (Sowa et al., 2010; Pastor-Anglada and Pérez-Torras, 2018).
Clinical development of adenosine kinase inhibitors was stopped due to toxicity (Boison and Jarvis, 2021).
Caffeine (Figure 10), a tri-methylated (1; 3;7) xanthine derivative and the most consumed psychoactive drug worldwide, has putative analgesic and/or anti-nociceptive effects. Its inclusion as an adjuvant in a number of double combination preparations, mostly with an NSAID (ibuprofen, naproxen, acetylsalicylic acid) or acetaminophen and triple combinations with an NSAID and acetaminophen modestly lowers the NNT (the lower the NNT the more efficacious the drug is) (Petersen, 2013; Yancey and Dattoli, 2013). The usual caffeine dose in combination preparations is 65 mg with acetaminophen and 100 mg with ibuprofen. For the general population of healthy adults, Health Canada advises a daily intake of no more than 400 mg.
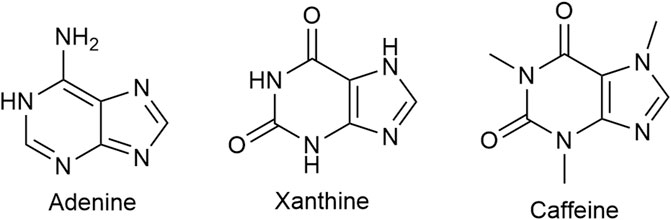
FIGURE 10. Caffeine, a xanthine derivative, is a non-selective adenosine receptor blocker. Adenine is a precursor of xanthine and adenine nucleotide is a precursor of adenosine.
The exact mechanism(s) of analgesic and/or anti-nociceptive effect(s) of caffeine is/are not known.
Caffeine is a non-selective adenosine receptor (AR) blocker. The affinity for the AR subtypes varies. Researchers from the Swiss Federal Institute of Technology in Lausanne report affinity values of caffeine for the human adenosine receptors as 12 at A1, 2.4 at A2A, 13 at A2B, and 80 μM at A3 (Froestl et al., 2012).
Administration of 160 mg caffeine (≈800 μM), the rough equivalent of one cup of coffee, generates mean maximal plasma concentrations of 18 μM (White et al., 2016). Administration of 100 mg caffeine (≈500 μM) generates mean maximal plasma concentrations of 15 μM (Grzegorzewski et al., 2021). Caffeine concentration in brain structures is about 50% of caffeine level in plasma (Orrego et al., 2016).
With estimated maximal CNS concentrations of caffeine at 10 μM, one can assume that the main inhibitory effect is mediated via A2A receptors; some inhibition of A1 and A2B receptors is possible, while a direct effect at A3 receptors appears unlikely.
A2A have anti-inflammatory effects and modify pain by direct and indirect actions (Sawynok, 2016). A2AR agonists however show some peripheral pro-nociceptive effects (Jung et al., 2022).
Furthermore, caffeine significantly increases plasma adenosine concentration (Conlay et al., 1997). As such, a shift of adenosine effect from caffeine blocked receptors to the unaffected A3R appears possible. A3R have an anti-nociceptive profile via actions on spinal microglia (Sawynok, 2016). Several experimental A3R agonists are being considered as promising analgesic agents (Jung et al., 2022).
Adding further complexity is the propensity of adenosine receptors to form homo-and hetero-dimers such as A1-A2A. In a must-read review titled “Pharmacology of Adenosine Receptors: The State of the Art” the authors point out that: “Low adenosine concentration preferentially stimulates the A1 protomer of the heteromer, which would inhibit glutamatergic transmission. On the other hand, high adenosine concentration activates adenosine A2A protomer of the heteromer that blocks adenosine A1-mediated effects and results in potentiation of glutamate release” (Borea et al., 2018). No information concerning the interaction between caffeine and A1-A2A adenosine receptor dimers is available to us.
In addition, caffeine is also an antagonist at the inositol trisphosphate (IP3)- receptors subtype 1. High concentrations of caffeine (10–70 mM) selectively inhibit IP3-R1 without affecting IP3 binding and oppose the intracellular Ca2+ increase (Saleem et al., 2014). The caffeine concentrations required for IP3-R1 inhibition are in the toxic range and therefore clinically not relevant.
Same applies to the ryanodine receptor where low mM (5 mM) concentrations of caffeine activate the receptor.
Caffeine is also a weak blocker of the ionotropic-glycine receptor (GlyR). Docking simulations indicate that caffeine and strychnine may bind to similar sites at the GlyR. Tested against the EC50 of each GlyR subtype, the inhibitory caffeine concentration (IC50) is in the range of 200–1,000 μM. GlyR produces its inhibitory effects through hyperpolarizing chloride currents (Duan et al., 2009). The caffeine concentrations required for GlyR inhibition are in the toxic range and therefore clinically not relevant for analgesia.
In conclusion, the analgesic/anti-nociceptive mechanism of action of caffeine is unclear. However, the effects are more established including improved alertness, cognitive processes, mood, learning and physical performance (Wikoff et al., 2017; Soós et al., 2021).
Gabapentin and pregabalin are structurally related to the endogenous inhibitory neurotransmitter GABA and to the amino acids leucine and isoleucine; in fact, both gabapentin and pregabalin contain GABA in both name and structure (Figure 11). Despite the similarity, their affinity for GABA receptors is orders of magnitude lower (high Ki) than for the α2δ auxiliary subunit of neuronal Voltage Dependent Calcium Channels (VDDC), which they bind to and block. By doing so, calcium channel permeability and calcium cell influx are reduced, leading subsequently to an unspecific neurotransmitter release reduction, including excitatory pro-algetic ones (Lanzetti and Di Biase, 2022). It is assumed that the endogenous agonists at the α2δ auxiliary subunit are the essential, branched-chain amino acids leucine and isoleucine, and thus gabapentin and pregabalin function as leucine/isoleucine competitive antagonists. While both drugs are recommended as possible first-line treatment for neuropathic pain by various medical authorities, the NNTs are high. Generally, the consensus is that tertiary amine tricyclic antidepressants (TA-TCA; amitriptyline) have superior (low) NNT values, but gabapentinoids are safer drugs (Finnerup et al., 2005). Stated more bluntly, it is easy and often infaust to overdose on TA-TCA, while a similar outcome would require enthusiastic use of gabapentinoids and still not offer any guarantee (Montgomery et al., 1989; Fischer et al., 1994). However, both medications share the most frequently reported adverse effects, which are somnolence and dizziness (Baidya et al., 2011; Chang et al., 2014).
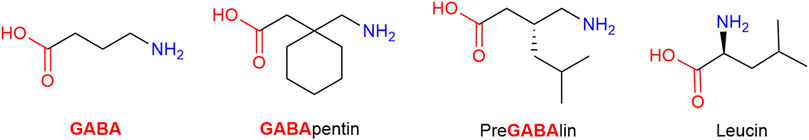
FIGURE 11. Structurally gabapentin and pregabalin are related to the endogenous inhibitory neurotransmitter GABA and to the amino acid leucine.
Nefopam (NFP) is a non-opioid, non-steroidal, centrally acting analgesic drug that does not inhibit PG synthesis. Structurally a benz-oxazocine, NFP is related to orphenadrine and diphen-hydramine. It has anti-cholinergic but no anti-histaminergic side-effects (Figure 12) (Heel et al., 1980).
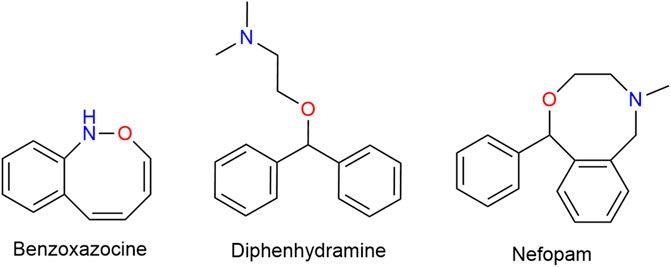
FIGURE 12. Structurally a benzoxazocine, nefopam is related to diphenhydramine.
NFP acts at spinal and supra spinal sites and exerts anti-nociceptive effects through triple monoamine reuptake inhibition [NE, 5-HT and DA], and anti-hyperalgesic activity through blockade of calcium and sodium channels and subsequent reduction of glutamatergic transmission (Alfonsi et al., 2004; Kim and Abdi, 2014; Girard et al., 2016). NFP is also effective for prevention of intra- and post-anesthetic shivering (meperidine/pethidine-like effect). The substance has approximately half of the analgesic potency of morphine but does not produce respiratory depression (Piper et al., 1998; Piper et al., 2004). NFP overdose causes neurological impairment such as disorientation, seizure, and cardiovascular and anticholinergic effects. However, NFP is generally well-tolerated, in which the reported side effects are minor for example drowsiness, sweating and GI disturbances (Park et al., 2014; Jeon et al., 2019). Based on NFP’s mechanisms of analgesic action, it is more suitable for the treatment of neuropathic pain (Kim and Abdi, 2014). The 5-HT7 receptor (but not 5-HT3) is involved in the antiallodynic action of NFP in the spinal cord (Dam et al., 2014; Lee et al., 2015).
Tramadol (Figure 13) is a familiar drug to physicians, having been on the European market for over 50 years and its pharmacodynamic actions are well understood. The (+) enantiomer preferentially inhibits serotonin reuptake and enhances serotonin efflux in the brain, whereas the (−) enantiomer mainly inhibits noradrenaline reuptake thus the racemic mixture itself is a balanced (equal serotonin and norepinephrine) reuptake inhibitor. Its main CYP450 2D6 metabolite (M1: ODM-tramadol) is an µ-opioid receptor agonist with poor blood-brain barrier permeability (Fudin and Boglish, 2016). The opioid mediated analgesic efficacy of tramadol (after metabolic conversion) is about 1/10 of that of morphine (comparable with codeine) and dependent on the activity of the highly polymorphic cytochrome P450 enzyme 2D6 (Gan et al., 2007).
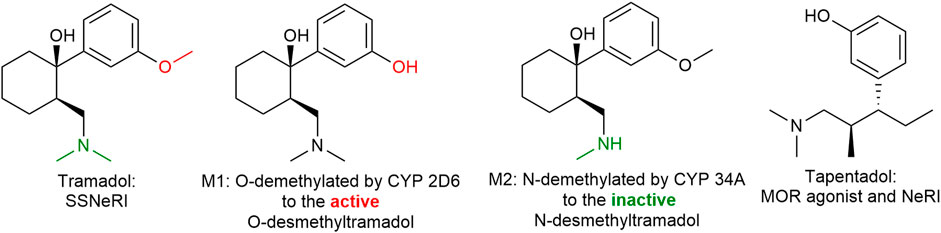
FIGURE 13. Tramadol and tapentadol are similar in their inhibition of norepinephrine reuptake. Tapentadol is also a more potent µ-opioid receptor agonist than tramadol. SSNeRI: selective serotonin and norepinephrine reuptake inhibitor, MOR: µ-opioid receptor, and NeRI: norepinephrine reuptake inhibitor.
As an intravenous drug it was (and still is) used for acute pain on ambulances and in hospital with various degrees of success due to the slow onset of action and variable efficacy. More established is tramadol’s use orally for chronic pain (Fudin and Boglish, 2016; Subedi et al., 2019). In addition, it is preferred over NSAIDs due to the better tolerability as tramadol common adverse effects include dizziness, somnolence and GI disturbances while NSAIDs can cause renal and GI impairment. The low addiction rate also makes tramadol a preferable choice for analgesia in comparison to other opioid medications (Vazzana et al., 2015).
Tapentadol (Figure 13), is similar to (−)tramadol in as much as it inhibits mainly norepinephrine reuptake. It is a more potent µ-opioid receptor agonist than tramadol (in fact tramadol is only a partial agonist) and does not require hepatic activation, thus having a more predictable and faster onset of action. The side effect profile is similar to tramadol and other opioids such as GI disturbances, somnolence, dizziness, dry mouth, and pruritis (Singh et al., 2013). Tapentadol might possess a lower risk of nausea, vomiting, and hypoglycemia however a greater risk of respiratory depression (yet rare), constipation, and overdose complications than tramadol (Roulet et al., 2021).
Ketamine is a familiar drug to most emergency and trauma physicians, having been clinically available for over half a century. Since the Vietnam War ketamine is the standard analgesic/anesthetic for mass casualties and out-of-hospital trauma emergencies. Chemically an arylcyclohexylamine derivative related to phencyclidine (phenyl-cyclohexyl-piperidine; PCP), ketamine (Figure 14) is a promiscuous drug interacting with a large number of receptors (Diwo and Petroianu, 2002). The main mechanism of anesthetic action is NMDA receptor antagonism (Ki ≈ 200–500 nM) translating in a unique state of dissociation (dissociative anesthesia) assumed to be due to interruption of the thalamo-cortical pathway. The major advantage of ketamine anesthesia is maintenance of respiratory drive and cardiac output, associated with sympathetic nervous system activation. On the negative side there is a relatively high incidence of psychotomimetic events (vivid dreams) and sialorrhea, the later likely due to muscarinic M3 receptor activation. Co-administration of a benzodiazepine and a muscarinic antagonist reduces these side-effects.
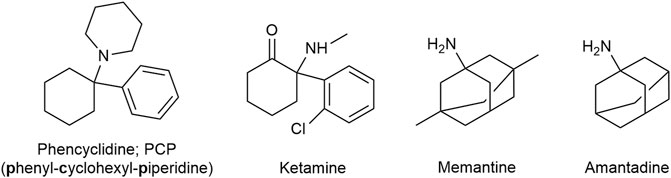
FIGURE 14. Ketamine is related to Phencyclidine. Memantine is an amantadine derivative.
The role of subanesthetic doses of ketamine in the treatment of neuropathic and chronic pain is less well defined. Also less understood is the site and mechanism of action of ketamine in neuropathic/chronic pain, most likely involving actions at additional receptors [sigma 1 (σ1) receptor] and efferent inhibitory pain pathways (Niesters et al., 2014). The σ1 ligands that enhance µ-opioid receptor (MOR) analgesia are referred to as antagonists, and those that reduce opioid analgesia and/or oppose the effects of antagonists are classified as agonists. The antagonistic activity of ketamine at σ1 receptors translates clinically in delayed loss of opioid analgesic efficacy; the σ1 receptor and the MOR co-regulate the activity of many TRP calcium channels (Cortés-Montero et al., 2019). This is the theoretical basis for co-administration of ketamine with an opiate. In addition, σ1 antagonists, alter or reduce TRPV1 expression in the plasma membrane of sensory neurons (Ortíz-Rentería et al., 2018).
Sanacora and Schatzberg (2015) in their review speak of a web of glutamatergic confusion.
Memantine (Figure 14), an amantadine derivative marketed for Alzheimer dementia as “the better magnesium,” is another promiscuous NMDA antagonist. The Ki-value of memantine at the phencyclidine binding site of the NMDA receptor is similar to that of ketamine (Kornhuber et al., 1989). In contrast to ketamine however, memantine is not trapped in the cationic channel and can rapidly dissociate out of the protein (rapid roll in - rapid roll off). Memantine also shows antagonist effects at the 5-HT3 receptor, with an affinity similar to that for the NMDA receptor. 5-HT3 receptor activation is assumed to favor algesia. Memantine (and amantadine; see below) also bind to and act as agonists (with comparable affinities) of the σ1 receptor. The drug was used for neuropathic/chronic pain treatment, the data is however very limited (Sanacora and Schatzberg, 2015; Nair and Sahoo, 2019). Memantine is generally well-tolerated with no serious side effects however high dosages can precipitate neuropsychiatric adverse effects such as confusion (Kornhuber et al., 2007; Nair and Sahoo, 2019).
Eisenberg and Pud (1998) asked Can patients with chronic neuropathic pain be cured by acute administration of the NMDA receptor antagonist amantadine?
Amantadine has been suggested to inhibit NMDA receptors by accelerating the channel closing, in turn leading to stabilization of the channel in the closed state (Blanpied et al., 2005). Recently it was questioned whether NMDA receptors are major targets for the therapeutic activity of amantadine. (Danysz et al., 2021). The authors indicate that at therapeutically achievable concentrations the most likely targets are intra-cellular proteins: phosphodiesterase-1 (PDE I), aromatic amino acids decarboxylase (AADC) and the sigma 1 receptor (σ1R).
PDE I inhibition translates in a cAMP increase and thus possibly in an anti-inflammatory milieu (O'Brien et al., 2020). Regardless of the actual mechanism of action therapeutic success in pain control appears to be limited (Aiyer et al., 2018). A range of side effects have been reported with the usage of amantadine including constipation, nausea, QT prolongation, orthostatic hypotension, neuropsychiatric symptoms (hallucinations, confusion and delirium) and livedo reticularis (Perez-Lloret and Rascol, 2018).
Magnesium, the endogenous NMDA antagonist, is assumed to play an important role in the prevention of central sensitization and in the attenuation of established pain hypersensitivity. The drug was used for neuropathic/chronic pain treatment, the supportive data is however limited (Na et al., 2011; Shin et al., 2020; Morel et al., 2021).
Several lines of evidence suggest analgesic effects of the third-generation parenteral cephalosporin antibiotic ceftriaxone in preclinical pain models. The assumed mechanism of action is enhancement of the glutamate reuptake by upregulation of the glutamate transporter GLT-1 in the spinal cord and elsewhere (Sisignano et al., 2022). GLT-1 is a sodium-dependent transporter that plays a key role in glutamate homeostasis by removing excess glutamate in the central nervous system (Peterson and Binder, 2019). Due to GLT-1 upregulation, glutamate concentration and glutamate mediated excitation decrease. The glial glutamate transporter is referred to as GLT-1 in the rodent literature and excitatory amino acid transporter 2 (EAAT2) in the human literature (Wikipedia, 2022). Ceftriaxone share similar side effect to other cephalosporins including clostridioides difficile infection (Hensgens et al., 2012).
While CGRP receptor inhibitors (-gepant) are mainly used for the treatment of migraine attacks, monoclonal antibodies targeting either CGRP receptor or ligand are effective in the prevention of migraine attacks (Edvinsson, 2021). FDA approved anti-CGRP antibodies erenumab, fremanezumab, and galcanezumab in 2018 and eptinezumab in 2020 for the prophylactic treatment in chronic migraine (Schwedt and Garza, 2022). The stimulation of trigeminal ganglion causes CGRP release; CGRP concentrations in jugular blood were found to be elevated during migraine attacks (Goadsby and Edvinsson, 1993). The monoclonal antibodies against the neuropeptide CGRP act via inhibition of neurogenic vasodilation in the dura and skin. Anti-CGRP antibodies are superior to other treatments due to their high specificity for the target, limited side effects and toxicities and long duration of action (Charles and Pozo-Rosich, 2019; Edvinsson, 2021).
Nerve growth factor (NGF), secreted following nerve injury, binds to its receptor and activates MAPKs and nuclear factor-kappa B (NF-κB) signaling pathways. This mediates the increase and sensitization of several substances such as substance P, CGRP, brain-derived neurotrophic factor (BDNF) and TRPV1 thus modulating neuropathic pain (Dai et al., 2020). Several studies showed that NGF concentration is elevated in chronic pain such as lumbar disc herniation, osteoarthritis, and low back pain (Onda et al., 2005; Orita et al., 2011; Shi et al., 2018). Thus, antibodies targeting NGF including tanezumab, fulranumab, and fasinumab have been developed and tested in clinical trials, however unfortunately FDA stopped all trials related to anti-NGF antibodies due to reported fast joint destruction (Bannwarth and Kostine, 2014).
Neuropathic pain is a major clinical problem; unfortunately, we are still not able to adequately relief pain. Several other pathways have been found to be involved in pain mechanism and thus are potential future drug targets. The α-Subunit of the voltage-gated sodium channel, Nav1.7, is a key player in the transmission of pain signals; it evokes an action potential following depolarization of neurons by harmful stimuli (Minett et al., 2012). Mutations in SCN9A gene encoding for Nav1.7 are associated with pain disorders such primary hereditary erythromelalgia and paroxysmal extreme pain disorder (Yang et al., 2004; Drenth et al., 2005; Fertleman et al., 2006; Sheets et al., 2007). On the contrary, mutations involving loss of function of Nav1.7 causes severe impairment of pain perception known as congenital indifference to pain (Cox et al., 2006; Goldberg et al., 2007). Hence, Nav1.7-targeted blockers are potential analgesics; the combination with opioids produced analgesia in animals (Emery et al., 2016).
In neuropathic pain, loss of Potassium-Chloride Cotransporter (KCC2) functional expression in spinal cord dorsal horn neurons contributes to GABAergic disinhibition. KCC2 expels chloride ions out of the neurons and hence guarantees a low intracellular chloride concentration, which is needed for proper GABAergic transmission. The activation of GABA-A receptors causes an influx of chloride and ultimately hyperpolarization. When KCC2 activity is reduced, chloride will accumulate in the neuron and thus less hyperpolarization will occur upon GABA-A receptor activation, causing neuropathic pain. Hence, KCC2 enhancers constitute another potential new therapeutic strategy to reduce neuropathic pain (Kahle et al., 2014). Gagnon et al. (2013) were successful in finding a KCC2 analogue (CLP257); this agonist enhanced the extrusion of chloride and thus caused hyperpolarization in spinal neurons of a rat model of neuropathic pain. In addition, kenpaullone is another successful example of KCC2 modulator. It improved Kcc2/KCC2 gene expression by negatively affecting the function of glycogen synthase kinase-3 (GSK3ß). Kenpaullone was found to be an effective analgesic in mouse models of pathologic pain (Yeo et al., 2021). Therefore, many future directions are yet to be explored from classic receptor modulation to novel genetic reprogramming.
Acute pain is a protective physiological reaction to noxious stimuli; in contrast chronic pain is pain that lost most or any usefulness. Treatment of chronic pain is difficult and often frustrating -both for patient and physician-as symptomatic treatment so often is. The symptom of chronic pain is the common final pathway for many conditions of most various etiology. It is not surprising that therapeutic successes are limited. Understanding the multitude of possible molecular mechanisms generating the common symptom pain is a highly desirable but a faraway goal. Familiarity with the pharmacodynamic action of the available analgesic drugs allows drug combinations with complementary, possibly synergistic actions, and non-additive adverse reactions.
GP wrote the manuscript. LA and AA reviewed the manuscript. LA created most figures and the table.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abu-Zaid, A., Aljaili, A. K., Althaqib, A., Adem, F., Alhalal, D. A., Almubarak, A. F., et al. (2021). Safety and efficacy of gefapixant, a novel drug for the treatment of chronic cough: A systematic review and meta-analysis of randomized controlled trials. Ann. Thorac. Med. 16 (2), 127–140. doi:10.4103/atm.ATM_417_20
Aiyer, R., Mehta, N., Gungor, S., and Gulati, A. (2018). A systematic review of NMDA receptor antagonists for treatment of neuropathic pain in clinical practice. Clin. J. Pain 34 (5), 450–467. doi:10.1097/ajp.0000000000000547
Alfonsi, P., Adam, F., Passard, A., Guignard, B., Sessler, D. I., and Chauvin, M. (2004). Nefopam, a nonsedative benzoxazocine analgesic, selectively reduces the shivering threshold in unanesthetized subjects. Anesthesiology 100 (1), 37–43. doi:10.1097/00000542-200401000-00010
Aloum, L., Alefishat, E., Shaya, J., and Petroianu, G. A. (2021). Remedia sternutatoria over the centuries: TRP mediation. Molecules 26 (6), 1627. doi:10.3390/molecules26061627
Alpert, A., Evans, W. N., Lieber, E. M. J., and Powell, D. (2021). Origins of the opioid crisis and its enduring impacts. Q. J. Econ. 137 (2), 1139–1179. doi:10.1093/qje/qjab043
Aviram, J., and Samuelly-Leichtag, G. (2017). Efficacy of cannabis-based medicines for pain management: A systematic review and meta-analysis of randomized controlled trials. Pain Physician 20 (6), E755–E796. doi:10.36076/ppj.20.5.e755
Azambuja, J. H., Ludwig, N., Braganhol, E., and Whiteside, T. L. (2019). Inhibition of the adenosinergic pathway in cancer rejuvenates innate and adaptive immunity. Int. J. Mol. Sci. 20 (22), 5698. doi:10.3390/ijms20225698
Baidya, D. K., Agarwal, A., Khanna, P., and Arora, M. K. (2011). Pregabalin in acute and chronic pain. J. Anaesthesiol. Clin. Pharmacol. 27 (3), 307–314. doi:10.4103/0970-9185.83672
Bannwarth, B., and Kostine, M. (2014). Targeting nerve growth factor (NGF) for pain management: What does the future hold for NGF antagonists? Drugs 74 (6), 619–626. doi:10.1007/s40265-014-0208-6
Bartlett, D. (2017). Drug-induced serotonin syndrome. Crit. Care Nurse 37 (1), 49–54. doi:10.4037/ccn2017169
Battastini, A. M. O., Figueiró, F., Leal, D. B. R., Doleski, P. H., and Schetinger, M. R. C. (2021). CD39 and CD73 as promising therapeutic targets: What could Be the limitations? Front. Pharmacol. 12, 633603. doi:10.3389/fphar.2021.633603
Bernard, A., Danigo, A., Bourthoumieu, S., Mroué, M., Desmoulière, A., Sturtz, F., et al. (2021). The cholecystokinin type 2 receptor, a pharmacological target for pain management. Pharm. (Basel) 14 (11), 1185. doi:10.3390/ph14111185
Bielinska-Kwapisz, A., and Mielecka-Kubien, Z. (2011). Alcohol consumption and its adverse effects in Poland in years 1950–2005. Econ. Res. Int. 2011, 1–13. doi:10.1155/2011/870714
Bixby, A. L., VandenBerg, A., and Bostwick, J. R. (2019). Clinical management of bleeding risk with antidepressants. Ann. Pharmacother. 53 (2), 186–194. doi:10.1177/1060028018794005
Blanpied, T. A., Clarke, R. J., and Johnson, J. W. (2005). Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 25 (13), 3312–3322. doi:10.1523/jneurosci.4262-04.2005
Boison, D., and Jarvis, M. F. (2021). Adenosine kinase: A key regulator of purinergic physiology. Biochem. Pharmacol. 187, 114321. doi:10.1016/j.bcp.2020.114321
Borea, P. A., Gessi, S., Merighi, S., Vincenzi, F., and Varani, K. (2018). Pharmacology of adenosine receptors: The state of the Art. Physiol. Rev. 98 (3), 1591–1625. doi:10.1152/physrev.00049.2017
Bowers, M. E., Choi, D. C., and Ressler, K. J. (2012). Neuropeptide regulation of fear and anxiety: Implications of cholecystokinin, endogenous opioids, and neuropeptide Y. Physiol. Behav. 107 (5), 699–710. doi:10.1016/j.physbeh.2012.03.004
Boyaji, S., Merkow, J., Elman, R. N. M., Kaye, A. D., Yong, R. J., and Urman, R. D. (2020). The role of cannabidiol (CBD) in chronic pain management: An assessment of current evidence. Curr. Pain Headache Rep. 24 (2), 4. doi:10.1007/s11916-020-0835-4
Brenchat, A., Romero, L., García, M., Pujol, M., Burgueño, J., Torrens, A., et al. (2009). 5-HT7 receptor activation inhibits mechanical hypersensitivity secondary to capsaicin sensitization in mice. Pain 141 (3), 239–247. doi:10.1016/j.pain.2008.11.009
Bruni, N., Della Pepa, C., Oliaro-Bosso, S., Pessione, E., Gastaldi, D., and Dosio, F. (2018). Cannabinoid delivery systems for pain and inflammation treatment. Molecules 23 (10), 2478. doi:10.3390/molecules23102478
Bryson, H. M., and Wilde, M. I. (1996). Amitriptyline. A review of its pharmacological properties and therapeutic use in chronic pain states. Amitriptyline. Drugs & Aging 8 (6), 459–476. doi:10.2165/00002512-199608060-00008
Bymaster, F. P., Dreshfield-Ahmad, L. J., Threlkeld, P. G., Shaw, J. L., Thompson, L., Nelson, D. L., et al. (2001). Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology 25 (6), 871–880. doi:10.1016/s0893-133x(01)00298-6
Campos, R. M. P., Aguiar, A. F. L., Paes-Colli, Y., Trindade, P. M. P., Ferreira, B. K., de Melo Reis, R. A., et al. (2021). Cannabinoid therapeutics in chronic neuropathic pain: From animal research to human treatment. Front. Physiol. 12, 785176. doi:10.3389/fphys.2021.785176
Chang, C. Y., Challa, C. K., Shah, J., and Eloy, J. D. (2014). Gabapentin in acute postoperative pain management. BioMed Res. Int. 2014, 631756. doi:10.1155/2014/631756
Charles, A., and Pozo-Rosich, P. (2019). Targeting calcitonin gene-related peptide: A new era in migraine therapy. Lancet 394 (10210), 1765–1774. doi:10.1016/s0140-6736(19)32504-8
Cohen, S. P., and Mao, J. (2014). Neuropathic pain: Mechanisms and their clinical implications. Bmj 348, f7656. doi:10.1136/bmj.f7656
Conlay, L. A., Conant, J. A., deBros, F., and Wurtman, R. (1997). Caffeine alters plasma adenosine levels. Nature 389 (6647), 136. doi:10.1038/38160
Cortés-Montero, E., Sánchez-Blázquez, P., Onetti, Y., Merlos, M., and Garzón, J. (2019). Ligands exert biased activity to regulate sigma 1 receptor interactions with cationic TRPA1, TRPV1, and TRPM8 channels. Front. Pharmacol. 10, 634. doi:10.3389/fphar.2019.00634
Cox, J. J., Reimann, F., Nicholas, A. K., Thornton, G., Roberts, E., Springell, K., et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444 (7121), 894–898. doi:10.1038/nature05413
Dai, W.-L., Yan, B., Bao, Y.-N., Fan, J.-F., and Liu, J.-H. (2020). Suppression of peripheral NGF attenuates neuropathic pain induced by chronic constriction injury through the TAK1-MAPK/NF-κB signaling pathways. Cell Commun. Signal. 18 (1), 66. doi:10.1186/s12964-020-00556-3
Dam, L. J., Hai, L., and Ha, Y. M. (2014). Role of the 5-HT(7) receptor in the effects of intrathecal nefopam in neuropathic pain in rats. Neurosci. Lett. 566, 50–54. doi:10.1016/j.neulet.2014.02.021
Danysz, W., Dekundy, A., Scheschonka, A., and Riederer, P. (2021). Amantadine: Reappraisal of the timeless diamond-target updates and novel therapeutic potentials. J. Neural Transm. (Vienna) 128 (2), 127–169. doi:10.1007/s00702-021-02306-2
De Caro, C., Cristiano, C., Avagliano, C., Cuozzo, M., La Rana, G., Aviello, G., et al. (2020). Analgesic and anti-inflammatory effects of perampanel in acute and chronic pain models in mice: Interaction with the cannabinergic system. Front. Pharmacol. 11, 620221. doi:10.3389/fphar.2020.620221
De Marchi, E., Orioli, E., Pegoraro, A., Sangaletti, S., Portararo, P., Curti, A., et al. (2019). The P2X7 receptor modulates immune cells infiltration, ectonucleotidases expression and extracellular ATP levels in the tumor microenvironment. Oncogene 38 (19), 3636–3650. doi:10.1038/s41388-019-0684-y
Dhaliwal, J. S., Spurling, B. C., and Molla, M. (2022). Duloxetine. Treasure Island (FL): StatPearls Publishing.
Diwo, S., and Petroianu, G. (2002). “Die Pharmakologie von Ketamin: Enantiomere, Distomere, Eutomere und Razemat,” in S)-Ketamin: Aktuelle interdisziplinäre AspekteR. Klose & U. Hoppe. (Berlin, Heidelberg: Springer Berlin Heidelberg), 1–16.
Dogrul, A., Ossipov, M. H., and Porreca, F. (2009). Differential mediation of descending pain facilitation and inhibition by spinal 5HT-3 and 5HT-7 receptors. Brain Res. 1280, 52–59. doi:10.1016/j.brainres.2009.05.001
Dougherty, J. A., Young, H., and Shafi, T. (2002). Serotonin syndrome induced by amitriptyline, meperidine, and venlafaxine. Ann. Pharmacother. 36 (10), 1647–1648. doi:10.1345/aph.1C091
Drenth, J. P., te Morsche, R. H., Guillet, G., Taieb, A., Kirby, R. L., and Jansen, J. B. (2005). SCN9A mutations define primary erythermalgia as a neuropathic disorder of voltage gated sodium channels. J. Invest. Dermatol 124 (6), 1333–1338. doi:10.1111/j.0022-202X.2005.23737.x
Duan, L., Yang, J., and Slaughter, M. M. (2009). Caffeine inhibition of ionotropic glycine receptors. J. Physiol. 587 (16), 4063–4075. doi:10.1113/jphysiol.2009.174797
Duggan, A. W., Hope, P. J., and Lang, C. W. (1991). Microinjection of neuropeptide Y into the superficial dorsal horn reduces stimulus-evoked release of immunoreactive substance P in the anaesthetized cat. Neuroscience 44 (3), 733–740. doi:10.1016/0306-4522(91)90092-3
Dyer, O. (2020). Purdue Pharma to plead guilty and pay $8.3bn over opioid marketing. Bmj 371, m4103. doi:10.1136/bmj.m4103
Edvinsson, L. (2021). CGRP and migraine: From bench to bedside. Rev. Neurol. 177 (7), 785–790. doi:10.1016/j.neurol.2021.06.003
Egilman, D., Collins, G., Falender, J., Shembo, N., Keegan, C., and Tohan, S. (2019). The marketing of OxyContin®: A cautionary tale. Indian J Med Ethics 4 (3), 183–193. doi:10.20529/ijme.2019.043
Eisenberg, E., and Pud, D. (1998). Can patients with chronic neuropathic pain be cured by acute administration of the NMDA receptor antagonist amantadine? Pain 74 (2-3), 337–339. doi:10.1016/s0304-3959(97)00198-x
Emery, E. C., Luiz, A. P., and Wood, J. N. (2016). Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin. Ther. Targets 20 (8), 975–983. doi:10.1517/14728222.2016.1162295
Fernandes, E. S., Fernandes, M. A., and Keeble, J. E. (2012). The functions of TRPA1 and TRPV1: Moving away from sensory nerves. Br. J. Pharmacol. 166 (2), 510–521. doi:10.1111/j.1476-5381.2012.01851.x
Fertleman, C. R., Baker, M. D., Parker, K. A., Moffatt, S., Elmslie, F. V., Abrahamsen, B., et al. (2006). SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 52 (5), 767–774. doi:10.1016/j.neuron.2006.10.006
Finnerup, N. B., Attal, N., Haroutounian, S., McNicol, E., Baron, R., Dworkin, R. H., et al. (2015). Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 14 (2), 162–173. doi:10.1016/s1474-4422(14)70251-0
Finnerup, N. B., Otto, M., McQuay, H. J., Jensen, T. S., and Sindrup, S. H. (2005). Algorithm for neuropathic pain treatment: An evidence based proposal. Pain 118 (3), 289–305. doi:10.1016/j.pain.2005.08.013
Fischer, J. H., Barr, A. N., Rogers, S. L., Fischer, P. A., and Trudeau, V. L. (1994). Lack of serious toxicity following gabapentin overdose. Neurology 44 (5), 982–983. doi:10.1212/wnl.44.5.982
Frediani, B., Giusti, A., Bianchi, G., Dalle Carbonare, L., Malavolta, N., Cantarini, L., et al. (2018). Clodronate in the management of different musculoskeletal conditions. Minerva Med. 109 (4), 300–325. doi:10.23736/s0026-4806.18.05688-4
Fride, E., and Mechoulam, R. (1993). Pharmacological activity of the cannabinoid receptor agonist, anandamide, a brain constituent. Eur. J. Pharmacol. 231 (2), 313–314. doi:10.1016/0014-2999(93)90468-W
Friedman, R. A., and Leon, A. C. (2007). Expanding the black box - depression, antidepressants, and the risk of suicide. N. Engl. J. Med. 356 (23), 2343–2346. doi:10.1056/NEJMp078015
Froestl, W., Muhs, A., and Pfeifer, A. (2012). Cognitive enhancers (nootropics). Part 1: Drugs interacting with receptors. J. Alzheimers Dis. 32 (4), 793–887. doi:10.3233/jad-2012-121186
Fudin, J., and Boglish, P. (2016). Ask The Expert: Is Tapentadol a Glorified Tramadol? Pract. Pain Manag 16 (1).
Gagnon, M., Bergeron, M. J., Lavertu, G., Castonguay, A., Tripathy, S., Bonin, R. P., et al. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19 (11), 1524–1528. doi:10.1038/nm.3356
Gan, S. H., Ismail, R., Wan Adnan, W. A., and Zulmi, W. (2007). Impact of CYP2D6 genetic polymorphism on tramadol pharmacokinetics and pharmacodynamics. Mol. Diagn Ther. 11 (3), 171–181. doi:10.1007/bf03256239
Girard, P., Chauvin, M., and Verleye, M. (2016). Nefopam analgesia and its role in multimodal analgesia: A review of preclinical and clinical studies. Clin. Exp. Pharmacol. Physiol. 43 (1), 3–12. doi:10.1111/1440-1681.12506
Goadsby, P. J., and Edvinsson, L. (1993). The trigeminovascular system and migraine: Studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann. Neurol. 33 (1), 48–56. doi:10.1002/ana.410330109
Goldberg, Y. P., MacFarlane, J., MacDonald, M. L., Thompson, J., Dube, M. P., Mattice, M., et al. (2007). Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 71 (4), 311–319. doi:10.1111/j.1399-0004.2007.00790.x
Gouin, O., L'Herondelle, K., Lebonvallet, N., Le Gall-Ianotto, C., Sakka, M., Buhé, V., et al. (2017). TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: Pro-inflammatory response induced by their activation and their sensitization. Protein Cell 8 (9), 644–661. doi:10.1007/s13238-017-0395-5
Greenberg, D. A. (2003). “Endocannabinoids,” in Encyclopedia of the neurological sciences. Editors M. J. Aminoff, and R. B. Daroff (New York: Academic Press), 143–144.
Grzegorzewski, J., Bartsch, F., Köller, A., and König, M. (2021). Pharmacokinetics of caffeine: A systematic analysis of reported data for application in metabolic phenotyping and liver function testing. Front. Pharmacol. 12, 752826. doi:10.3389/fphar.2021.752826
Habib, A. M., Okorokov, A. L., Hill, M. N., Bras, J. T., Lee, M.-C., Li, S., et al. (2019). Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br. J. Anaesth. 123 (2), e249–e253. doi:10.1016/j.bja.2019.02.019
Hasuzawa, N., Moriyama, S., Moriyama, Y., and Nomura, M. (2020). Physiopathological roles of vesicular nucleotide transporter (VNUT), an essential component for vesicular ATP release. Biochim. Biophys. Acta Biomembr. 1862 (12), 183408. doi:10.1016/j.bbamem.2020.183408
Heel, R. C., Brogden, R. N., Pakes, G. E., Speight, T. M., and Avery, G. S. (1980). Nefopam: A review of its pharmacological properties and therapeutic efficacy. Drugs 19 (4), 249–267. doi:10.2165/00003495-198019040-00001
Hensgens, M. P., Goorhuis, A., Dekkers, O. M., and Kuijper, E. J. (2012). Time interval of increased risk for Clostridium difficile infection after exposure to antibiotics. J. Antimicrob. Chemother. 67 (3), 742–748. doi:10.1093/jac/dkr508
Inoue, K. (2017). Purinergic signaling in microglia in the pathogenesis of neuropathic pain. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 93 (4), 174–182. doi:10.2183/pjab.93.011
Jensen, T. S., Høye, K., Fricová, J., Vanelderen, P., Ernault, E., Siciliano, T., et al. (2014). Tolerability of the capsaicin 8% patch following pretreatment with lidocaine or tramadol in patients with peripheral neuropathic pain: A multicentre, randomized, assessor-blinded study. Eur. J. Pain 18 (9), 1240–1247. doi:10.1002/j.1532-2149.2014.00479.x
Jeon, Y. T., Oh, A. Y., Jin, S. J., Park, B. S., and Choi, E. S. (2019). Effects of intraoperative nefopam on catheter-related bladder discomfort in patients undergoing robotic nephrectomy: A randomized double-blind study. J. Clin. Med. 8 (4), 519. doi:10.3390/jcm8040519
Jung, S. M., Peyton, L., Essa, H., and Choi, D. S. (2022). Adenosine receptors: Emerging non-opioids targets for pain medications. Neurobiol. Pain 11, 100087. doi:10.1016/j.ynpai.2022.100087
Kahle, K. T., Khanna, A., Clapham, D. E., and Woolf, C. J. (2014). Therapeutic restoration of spinal inhibition via druggable enhancement of potassium-chloride cotransporter KCC2-mediated chloride extrusion in peripheral neuropathic pain. JAMA Neurol. 71 (5), 640–645. doi:10.1001/jamaneurol.2014.21
Kanellopoulos, J. M., Almeida-da-Silva, C. L. C., Rüütel Boudinot, S., and Ojcius, D. M. (2021). Structural and functional features of the P2X4 receptor: An immunological perspective. Front. Immunol. 12, 645834. doi:10.3389/fimmu.2021.645834
Kappelmann, N., Lewis, G., Dantzer, R., Jones, P. B., and Khandaker, G. M. (2018). Antidepressant activity of anti-cytokine treatment: A systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 23 (2), 335–343. doi:10.1038/mp.2016.167
Kato, Y., Hiasa, M., Ichikawa, R., Hasuzawa, N., Kadowaki, A., Iwatsuki, K., et al. (2017). Identification of a vesicular ATP release inhibitor for the treatment of neuropathic and inflammatory pain. Proc. Natl. Acad. Sci. U. S. A. 114 (31), E6297–e6305. doi:10.1073/pnas.1704847114
Kaye, A. D., Chernobylsky, D. J., Thakur, P., Siddaiah, H., Kaye, R. J., Eng, L. K., et al. (2020). Dexmedetomidine in enhanced recovery after surgery (ERAS) protocols for postoperative pain. Curr. Pain Headache Rep. 24 (5), 21. doi:10.1007/s11916-020-00853-z
Keating, G. M. (2015). Diquafosol ophthalmic solution 3 %: a review of its use in dry eye. Drugs 75 (8), 911–922. doi:10.1007/s40265-015-0409-7
Kim, K. H., and Abdi, S. (2014). Rediscovery of nefopam for the treatment of neuropathic pain. Korean J. Pain 27 (2), 103–111. doi:10.3344/kjp.2014.27.2.103
Kim, S. K., Min, B.-I., Kim, J. H., Hwang, B. G., Yoo, G. Y., Park, D. S., et al. (2005). Effects of alpha1-and alpha2-adrenoreceptor antagonists on cold allodynia in a rat tail model of neuropathic pain. Brain Res. 1039 (1), 207–210. doi:10.1016/j.brainres.2005.01.051
Kohno, K., and Tsuda, M. (2021). Role of microglia and P2X4 receptors in chronic pain. Pain Rep. 6 (1), e864. doi:10.1097/pr9.0000000000000864
Kornhuber, J., Kennepohl, E. M., Bleich, S., Wiltfang, J., Kraus, T., Reulbach, U., et al. (2007). Memantine pharmacotherapy: A naturalistic study using a population pharmacokinetic approach. Clin. Pharmacokinet. 46 (7), 599–612. doi:10.2165/00003088-200746070-00005
Kornhuber, J., Mack-Burkhardt, F., Riederer, P., Hebenstreit, G. F., Reynolds, G. P., Andrews, H. B., et al. (1989). [3H]MK-801 binding sites in postmortem brain regions of schizophrenic patients. J. Neural Transm. 77 (2-3), 231–236. doi:10.1007/bf01248936
Lanzetti, S., and Di Biase, V. (2022). Small molecules as modulators of voltage-gated calcium channels in neurological disorders: State of the Art and perspectives. Molecules 27 (4), 1312. doi:10.3390/molecules27041312
Lee, G., Grovey, B., Furnish, T., and Wallace, M. (2018). Medical cannabis for neuropathic pain. Curr. Pain Headache Rep. 22 (1), 8. doi:10.1007/s11916-018-0658-8
Lee, H. G., Kim, W. M., Kim, J. M., Bae, H. B., and Choi, J. I. (2015). Intrathecal nefopam-induced antinociception through activation of descending serotonergic projections involving spinal 5-HT7 but not 5-HT3 receptors. Neurosci. Lett. 587, 120–125. doi:10.1016/j.neulet.2014.12.040
Leon, A. C. (2007). The revised warning for antidepressants and suicidality: Unveiling the black box of statistical analyses. Am. J. Psychiatry 164 (12), 1786–1789. doi:10.1176/appi.ajp.2007.07050775
Lin, J. P., Chen, C. Q., Huang, L. E., Li, N. N., Yang, Y., Zhu, S. M., et al. (2018). Dexmedetomidine attenuates neuropathic pain by inhibiting P2X7R expression and ERK phosphorylation in rats. Exp. Neurobiol. 27 (4), 267–276. doi:10.5607/en.2018.27.4.267
Liu, Q. Q., Yao, X. X., Gao, S. H., Li, R., Li, B. J., Yang, W., et al. (2020). Role of 5-HT receptors in neuropathic pain: Potential therapeutic implications. Pharmacol. Res. 159, 104949. doi:10.1016/j.phrs.2020.104949
Liu, X., Ma, L., Zhang, S., Ren, Y., and Dirksen, R. T. (2017). CD73 controls extracellular adenosine generation in the trigeminal nociceptive nerves. J. Dent. Res. 96 (6), 671–677. doi:10.1177/0022034517692953
Magni, G., Merli, D., Verderio, C., Abbracchio, M. P., and Ceruti, S. (2015). P2Y2 receptor antagonists as anti-allodynic agents in acute and sub-chronic trigeminal sensitization: Role of satellite glial cells. Glia 63 (7), 1256–1269. doi:10.1002/glia.22819
Meng, H., Dai, T., Hanlon, J. G., Downar, J., Alibhai, S. M. H., and Clarke, H. (2020). Cannabis and cannabinoids in cancer pain management. Curr. Opin. Support Palliat. Care 14 (2), 87–93. doi:10.1097/spc.0000000000000493
Mihara, S., and Shibamoto, T. (2015). The role of flavor and fragrance chemicals in TRPA1 (transient receptor potential cation channel, member A1) activity associated with allergies. Allergy Asthma Clin. Immunol. 11 (1), 11. doi:10.1186/s13223-015-0074-0
Minett, M. S., Nassar, M. A., Clark, A. K., Passmore, G., Dickenson, A. H., Wang, F., et al. (2012). Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 3, 791. doi:10.1038/ncomms1795
Miras-Portugal, M. T., Menéndez-Méndez, A., Gómez-Villafuertes, R., Ortega, F., Delicado, E. G., Pérez-Sen, R., et al. (2019). Physiopathological role of the vesicular nucleotide transporter (VNUT) in the central nervous system: Relevance of the vesicular nucleotide release as a potential therapeutic target. Front. Cell Neurosci. 13, 224. doi:10.3389/fncel.2019.00224
Mishra, A., Behura, A., Kumar, A., Naik, L., Swain, A., Das, M., et al. (2021). P2X7 receptor in multifaceted cellular signalling and its relevance as a potential therapeutic target in different diseases. Eur. J. Pharmacol. 906, 174235. doi:10.1016/j.ejphar.2021.174235
Miyahara, Y., Funahashi, H., Naono-Nakayama, R., Haruta-Tsukamoto, A., Nishimori, T., and Ishida, Y. (2019). Role of serotonin and noradrenaline in the acute itch processing in mice. Eur. J. Pharmacol. 850, 118–125. doi:10.1016/j.ejphar.2019.02.013
Mlost, J., Bryk, M., and Starowicz, K. (2020). Cannabidiol for pain treatment: Focus on pharmacology and mechanism of action. Int. J. Mol. Sci. 21 (22), 8870. doi:10.3390/ijms21228870
Montgomery, S. A., Baldwin, D., and Green, M. (1989). Why do amitriptyline and dothiepin appear to be so dangerous in overdose? Acta Psychiatr. Scand. Suppl. 354, 47–53. doi:10.1111/j.1600-0447.1989.tb03046.x
Moore, R. A., Chi, C. C., Wiffen, P. J., Derry, S., and Rice, A. S. (2015). Oral nonsteroidal anti-inflammatory drugs for neuropathic pain. Cochrane Database Syst. Rev. 2015 (10), Cd010902. doi:10.1002/14651858.CD010902.pub2
Morel, V., Pickering, M. E., Goubayon, J., Djobo, M., Macian, N., and Pickering, G. (2021). Magnesium for pain treatment in 2021? State of the Art. Nutrients 13 (5), 1397. doi:10.3390/nu13051397
Mücke, M., Phillips, T., Radbruch, L., Petzke, F., and Häuser, W. (2018). Cannabis-based medicines for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 3 (3), Cd012182. doi:10.1002/14651858.CD012182.pub2
Murakami, T., Fujihara, T., Horibe, Y., and Nakamura, M. (2004). Diquafosol elicits increases in net Cl-transport through P2Y2 receptor stimulation in rabbit conjunctiva. Ophthalmic Res. 36 (2), 89–93. doi:10.1159/000076887
Murgia, M., Hanau, S., Pizzo, P., Rippa, M., and Di Virgilio, F. (1993). Oxidized ATP. An irreversible inhibitor of the macrophage purinergic P2Z receptor. J. Biol. Chem. 268 (11), 8199–8203. doi:10.1016/s0021-9258(18)53082-9
Na, H. S., Ryu, J. H., and Do, S. H. (2011). “The role of magnesium in pain,” in Magnesium in the central nervous system. Editors R. Vink, and M. Nechifor (Adelaide (AU): University of Adelaide Press).
Nair, A. S., and Sahoo, R. K. (2019). Efficacy of memantine hydrochloride in neuropathic pain. Indian J. Palliat. Care 25 (1), 161–162. doi:10.4103/ijpc.ijpc_189_18
Nam, K., Kim, H. J., and Yoo, A. (2019). Efficacy and safety of topical 3% diquafosol ophthalmic solution for the treatment of multifactorial dry eye disease: Meta-analysis of randomized clinical trials. Ophthalmic Res. 61 (4), 188–198. doi:10.1159/000492896
Nelson, T. S., and Taylor, B. K. (2021). Targeting spinal neuropeptide Y1 receptor-expressing interneurons to alleviate chronic pain and itch. Prog. Neurobiol. 196, 101894. doi:10.1016/j.pneurobio.2020.101894
Niesters, M., Martini, C., and Dahan, A. (2014). Ketamine for chronic pain: Risks and benefits. Br. J. Clin. Pharmacol. 77 (2), 357–367. doi:10.1111/bcp.12094
Niimi, A., Saito, J., Kamei, T., Shinkai, M., Ishihara, H., Machida, M., et al. (2022). Randomised trial of the P2X(3) receptor antagonist sivopixant for refractory chronic cough. Eur. Respir. J. 59 (6), 2100725. doi:10.1183/13993003.00725-2021
Noe, C. E. (2020). Pain management for clinicians A guide to assessment and treatment. Switzerland: Springer Nature.
Nörenberg, W., Sobottka, H., Hempel, C., Plötz, T., Fischer, W., Schmalzing, G., et al. (2012). Positive allosteric modulation by ivermectin of human but not murine P2X7 receptors. Br. J. Pharmacol. 167 (1), 48–66. doi:10.1111/j.1476-5381.2012.01987.x
O'Brien, J. J., O'Callaghan, J. P., Miller, D. B., Chalgeri, S., Wennogle, L. P., Davis, R. E., et al. (2020). Inhibition of calcium-calmodulin-dependent phosphodiesterase (PDE1) suppresses inflammatory responses. Mol. Cell Neurosci. 102, 103449. doi:10.1016/j.mcn.2019.103449
Obad, A., Peeran, A., Little, J. I., Haddad, G. E., and Tarzami, S. T. (2018). Alcohol-mediated organ damages: Heart and brain. Front. Pharmacol. 9, 81. doi:10.3389/fphar.2018.00081
Onda, A., Murata, Y., Rydevik, B., Larsson, K., Kikuchi, S., and Olmarker, K. (2005). Nerve growth factor content in dorsal root ganglion as related to changes in pain behavior in a rat model of experimental lumbar disc herniation. Spine (Phila Pa 1976) 30 (2), 188–193. doi:10.1097/01.brs.0000150830.12518.26
Orita, S., Ishikawa, T., Miyagi, M., Ochiai, N., Inoue, G., Eguchi, Y., et al. (2011). Pain-related sensory innervation in monoiodoacetate-induced osteoarthritis in rat knees that gradually develops neuronal injury in addition to inflammatory pain. BMC Musculoskelet. Disord. 12, 134. doi:10.1186/1471-2474-12-134
Orrego, M., Jiménez-Rodriguez, A., Osorio-Forero, A., Restrepo, F., Arias, J., Tamayo, L., et al. (2016). Different brain structures exhibit the same caffeine levels after the administration of A single dose of caffeine. Biosalud 15, 20–27. doi:10.17151/biosa.2016.15.2.3
Ortíz-Rentería, M., Juárez-Contreras, R., González-Ramírez, R., Islas, L. D., Sierra-Ramírez, F., Llorente, I., et al. (2018). TRPV1 channels and the progesterone receptor Sig-1R interact to regulate pain. Proc. Natl. Acad. Sci. U. S. A. 115 (7), E1657–e1666. doi:10.1073/pnas.1715972115
Park, Y. S., Kim, Y. B., and Kim, J. M. (2014). Status epilepticus caused by nefopam. J. Korean Neurosurg. Soc. 56 (5), 448–450. doi:10.3340/jkns.2014.56.5.448
Pastor-Anglada, M., and Pérez-Torras, S. (2018). Who is who in adenosine transport. Front. Pharmacol. 9, 627. doi:10.3389/fphar.2018.00627
Patel, R., and Dickenson, A. H. (2022). Neuropharmacological basis for multimodal analgesia in chronic pain. Postgrad. Med. 134 (3), 245–259. doi:10.1080/00325481.2021.1985351
Perez-Lloret, S., and Rascol, O. (2018). Efficacy and safety of amantadine for the treatment of L-DOPA-induced dyskinesia. J. Neural Transm. (Vienna) 125 (8), 1237–1250. doi:10.1007/s00702-018-1869-1
Petersen, K. U. (2013). Caffeine in analgesics--myth or medicine? MMW Fortschr Med. 155 (4), 109–114. doi:10.1007/s15006-013-2541-1
Peterson, A. R., and Binder, D. K. (2019). Post-translational regulation of GLT-1 in neurological diseases and its potential as an effective therapeutic target. Front. Mol. Neurosci. 12, 164. doi:10.3389/fnmol.2019.00164
Piper, S. N., Röhm, K. D., Suttner, S. W., Maleck, W. H., Kranke, P., and Boldt, J. (2004). A comparison of nefopam and clonidine for the prevention of postanaesthetic shivering: A comparative, double-blind and placebo-controlled dose-ranging study. Anaesthesia 59 (6), 559–564. doi:10.1111/j.1365-2044.2004.03734.x
Piper, S. N., Schmidt, C. C., Suttner, S. W., Kumle, B., Triem, J. G., Maleck, W. H., et al. (1998). Prophylactic nefopam administration for post-anesthetic shivering. Anasthesiol Intensivmed. Notfallmed Schmerzther 33 (12), 786–789. doi:10.1055/s-2007-994854
Romero-Sandoval, E. A., Fincham, J. E., Kolano, A. L., Sharpe, B. N., and Alvarado-Vázquez, P. A. (2018). Cannabis for chronic pain: Challenges and considerations. Pharmacotherapy 38 (6), 651–662. doi:10.1002/phar.2115
Romero-Sandoval, E. A., Kolano, A. L., and Alvarado-Vázquez, P. A. (2017). Cannabis and cannabinoids for chronic pain. Curr. Rheumatol. Rep. 19 (11), 67. doi:10.1007/s11926-017-0693-1
Rotondo, J. C., Mazziotta, C., Lanzillotti, C., Stefani, C., Badiale, G., Campione, G., et al. (2022). The role of purinergic P2X7 receptor in inflammation and cancer: Novel molecular insights and clinical applications. Cancers (Basel) 14 (5), 1116. doi:10.3390/cancers14051116
Roulet, L., Rollason, V., Desmeules, J., and Piguet, V. (2021). Tapentadol versus tramadol: A narrative and comparative review of their pharmacological, efficacy and safety profiles in adult patients. Drugs 81 (11), 1257–1272. doi:10.1007/s40265-021-01515-z
Russell, F. A., King, R., Smillie, S.-J., Kodji, X., and Brain, S. D. (2014). Calcitonin gene-related peptide: Physiology and pathophysiology. Physiol. Rev. 94 (4), 1099–1142. doi:10.1152/physrev.00034.2013
Salas, M. M., Clifford, J. L., Hayden, J. R., Iadarola, M. J., and Averitt, D. L. (2017). Local resiniferatoxin induces long-lasting analgesia in a rat model of full thickness thermal injury. Pain Med. 18 (12), 2453–2465. doi:10.1093/pm/pnw260
Saleem, H., Tovey, S. C., Molinski, T. F., and Taylor, C. W. (2014). Interactions of antagonists with subtypes of inositol 1, 4, 5-trisphosphate (IP3) receptor. Br. J. Pharmacol. 171 (13), 3298–3312. doi:10.1111/bph.12685
Sanacora, G., and Schatzberg, A. F. (2015). Ketamine: Promising path or false prophecy in the development of novel therapeutics for mood disorders? Neuropsychopharmacology 40 (2), 259–267. doi:10.1038/npp.2014.261
Savio, L. E. B., de Andrade Mello, P., da Silva, C. G., and Coutinho-Silva, R. (2018). The P2X7 receptor in inflammatory diseases: Angel or demon? Front. Pharmacol. 9, 52. doi:10.3389/fphar.2018.00052
Sawynok, J. (2016). Adenosine receptor targets for pain. Neuroscience 338, 1–18. doi:10.1016/j.neuroscience.2015.10.031
Schwedt, T. J., and Garza, M. (2022). Preventive treatment of episodic migraine in adults. Available at: https://www-uptodate-com.libconnect.ku.ac.ae/contents/preventive-treatment-of-episodic-migraine-in-adults?search=erenumab&source=search_result&selectedTitle=2∼5&usage_type=default&display_rank=1#H2010410496 (Accessed 05 12, 2022).
Sheets, P. L., Jackson, J. O., Waxman, S. G., Dib-Hajj, S. D., and Cummins, T. R. (2007). A Nav1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J. Physiol. 581 (3), 1019–1031. doi:10.1113/jphysiol.2006.127027
Shi, C., Das, V., Li, X., Kc, R., Qiu, S., I, O. S., et al. (2018). Development of an in vivo mouse model of discogenic low back pain. J. Cell Physiol. 233 (10), 6589–6602. doi:10.1002/jcp.26280
Shin, H. J., Na, H. S., and Do, S. H. (2020). Magnesium and pain. Nutrients 12 (8), 2184. doi:10.3390/nu12082184
Singh, D., Nag, K., Shetti, A., and Krishnaveni, N. (2013). Tapentadol hydrochloride: A novel analgesic. Saudi J. Anaesth. 7 (3), 322–326. doi:10.4103/1658-354x.115319
Sisignano, M., Gribbon, P., and Geisslinger, G. (2022). Drug repurposing to target neuroinflammation and sensory neuron-dependent pain. Drugs 82 (4), 357–373. doi:10.1007/s40265-022-01689-0
Smith, H. S. (2012). Opioids and neuropathic pain. Pain Physician 15, Es93–110. doi:10.36076/ppj.2012/15/es93
Smith, T., and Nicholson, R. A. (2007). Review of duloxetine in the management of diabetic peripheral neuropathic pain. Vasc. Health Risk Manag. 3 (6), 833–844.
Soós, R., Gyebrovszki, Á., Tóth, Á., Jeges, S., and Wilhelm, M. (2021). Effects of caffeine and caffeinated beverages in children, adolescents and young adults: Short review. Int. J. Environ. Res. Public Health 18 (23), 12389. doi:10.3390/ijerph182312389
Sowa, N. A., Taylor-Blake, B., and Zylka, M. J. (2010). Ecto-5'-nucleotidase (CD73) inhibits nociception by hydrolyzing AMP to adenosine in nociceptive circuits. J. Neurosci. 30 (6), 2235–2244. doi:10.1523/jneurosci.5324-09.2010
Staruschenko, A., Jeske, N. A., and Akopian, A. N. (2010). Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J. Biol. Chem. 285 (20), 15167–15177. doi:10.1074/jbc.M110.106153
Stokes, L., Layhadi, J. A., Bibic, L., Dhuna, K., and Fountain, S. J. (2017). P2X4 receptor function in the nervous system and current breakthroughs in pharmacology. Front. Pharmacol. 8, 291. doi:10.3389/fphar.2017.00291
Story, G. M., Peier, A. M., Reeve, A. J., Eid, S. R., Mosbacher, J., Hricik, T. R., et al. (2003). ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112 (6), 819–829. doi:10.1016/s0092-8674(03)00158-2
Subedi, M., Bajaj, S., Kumar, M. S., and Yc, M. (2019). An overview of tramadol and its usage in pain management and future perspective. Biomed. Pharmacother. 111, 443–451. doi:10.1016/j.biopha.2018.12.085
Suri, S., Mönkkönen, J., Taskinen, M., Pesonen, J., Blank, M. A., Phipps, R. J., et al. (2001). Nitrogen-containing bisphosphonates induce apoptosis of caco-2 cells in vitro by inhibiting the mevalonate pathway: A model of bisphosphonate-induced gastrointestinal toxicity. Bone 29 (4), 336–343. doi:10.1016/S8756-3282(01)00589-0
Tozaki-Saitoh, H., Takeda, H., and Inoue, K. (2022). The role of microglial purinergic receptors in pain signaling. Molecules 27 (6), 1919. doi:10.3390/molecules27061919
Upadhya, M. A., Dandekar, M. P., Kokare, D. M., Singru, P. S., and Subhedar, N. K. (2009). Involvement of neuropeptide Y in the acute, chronic and withdrawal responses of morphine in nociception in neuropathic rats: Behavioral and neuroanatomical correlates. Neuropeptides 43 (4), 303–314. doi:10.1016/j.npep.2009.05.003
Urits, I., Gress, K., Charipova, K., Habib, K., Lee, D., Lee, C., et al. (2020). Use of cannabidiol (CBD) for the treatment of chronic pain. Best. Pract. Res. Clin. Anaesthesiol. 34 (3), 463–477. doi:10.1016/j.bpa.2020.06.004
Vane, J. R., and Botting, R. M. (1998). Anti-inflammatory drugs and their mechanism of action. Inflamm. Res. 47 (2), S78–S87. doi:10.1007/s000110050284
Vazzana, M., Andreani, T., Fangueiro, J., Faggio, C., Silva, C., Santini, A., et al. (2015). Tramadol hydrochloride: Pharmacokinetics, pharmacodynamics, adverse side effects, co-administration of drugs and new drug delivery systems. Biomed. Pharmacother. 70, 234–238. doi:10.1016/j.biopha.2015.01.022
Vincenzi, F., Pasquini, S., Borea, P. A., and Varani, K. (2020). Targeting adenosine receptors: A potential pharmacological avenue for acute and chronic pain. Int. J. Mol. Sci. 21 (22), 8710. doi:10.3390/ijms21228710
White, J. R., Padowski, J. M., Zhong, Y., Chen, G., Luo, S., Lazarus, P., et al. (2016). Pharmacokinetic analysis and comparison of caffeine administered rapidly or slowly in coffee chilled or hot versus chilled energy drink in healthy young adults. Clin. Toxicol. (Phila) 54 (4), 308–312. doi:10.3109/15563650.2016.1146740
Wiesenfeld-Hallin, Z., Xu, X. J., and Hökfelt, T. (2002). The role of spinal cholecystokinin in chronic pain states. Pharmacol. Toxicol. 91 (6), 398–403. doi:10.1034/j.1600-0773.2002.910619.x
Wikipedia (2022). Glutamate transporter. Available at: https://en.wikipedia.org/wiki/Glutamate_transporter (Accessed October 1, 2022).
Wikoff, D., Welsh, B. T., Henderson, R., Brorby, G. P., Britt, J., Myers, E., et al. (2017). Systematic review of the potential adverse effects of caffeine consumption in healthy adults, pregnant women, adolescents, and children. Food Chem. Toxicol. 109, 585–648. doi:10.1016/j.fct.2017.04.002
Yamashita, T., Yamamoto, S., Zhang, J., Kometani, M., Tomiyama, D., Kohno, K., et al. (2016). Duloxetine inhibits microglial P2X4 receptor function and alleviates neuropathic pain after peripheral nerve injury. PLoS One 11 (10), e0165189. doi:10.1371/journal.pone.0165189
Yancey, J. R., and Dattoli, G. (2013). Caffeine as an analgesic adjuvant for acute pain in adults. Am. Fam. Physician 87 (1), 11.
Yang, Y., Li, Q., He, Q. H., Han, J. S., Su, L., and Wan, Y. (2018). Heteromerization of μ-opioid receptor and cholecystokinin B receptor through the third transmembrane domain of the μ-opioid receptor contributes to the anti-opioid effects of cholecystokinin octapeptide. Exp. Mol. Med. 50 (5), 1–16. doi:10.1038/s12276-018-0090-5
Yang, Y., Wang, Y., Li, S., Xu, Z., Li, H., Ma, L., et al. (2004). Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 41 (3), 171–174. doi:10.1136/jmg.2003.012153
Yeo, M., Chen, Y., Jiang, C., Chen, G., Wang, K., Chandra, S., et al. (2021). Repurposing cancer drugs identifies kenpaullone which ameliorates pathologic pain in preclinical models via normalization of inhibitory neurotransmission. Nat. Commun. 12 (1), 6208. doi:10.1038/s41467-021-26270-3
Yoshimura, M., and Furue, H. (2006). Mechanisms for the anti-nociceptive actions of the descending noradrenergic and serotonergic systems in the spinal cord. J. Pharmacol. Sci. 101 (2), 107–117. doi:10.1254/jphs.crj06008x
Zhang, W. J., Luo, H. L., and Zhu, Z. M. (2020). The role of P2X4 receptors in chronic pain: A potential pharmacological target. Biomed. Pharmacother. 129, 110447. doi:10.1016/j.biopha.2020.110447
Zhao, Y., He, J., Yu, N., Jia, C., and Wang, S. (2020). Mechanisms of dexmedetomidine in neuropathic pain. Front. Neurosci. 14, 330. doi:10.3389/fnins.2020.00330
Keywords: chronic pain, nociceptive pain, neuropathic pain, ATP, adenosine, capsaicin, clodronate, pregabalin
Citation: Petroianu GA, Aloum L and Adem A (2023) Neuropathic pain: Mechanisms and therapeutic strategies. Front. Cell Dev. Biol. 11:1072629. doi: 10.3389/fcell.2023.1072629
Received: 17 October 2022; Accepted: 06 January 2023;
Published: 16 January 2023.
Edited by:
Jean-Martin Beaulieu, University of Toronto, CanadaReviewed by:
Louis-Etienne Lorenzo, Laval University, CanadaCopyright © 2023 Petroianu, Aloum and Adem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georg A. Petroianu, Z2VvcmcucGV0cm9pYW51QGt1LmFjLmFl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.