 Niki Esfahanian†‡
Niki Esfahanian†‡ Khosrow Rezvani
Khosrow Rezvani
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 02 February 2023
Sec. Cellular Biochemistry
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1028519
This article is part of the Research TopicThe Role of Mortalin in Biology and DiseaseView all 8 articles
Mortalin (GRP75, HSPA9A), a heat shock protein (HSP), regulates a wide range of cellular processes, including cell survival, growth, and metabolism. The regulatory functions of mortalin are mediated through a diverse set of protein partners associated with different cellular compartments, which allows mortalin to perform critical functions under physiological conditions, including mitochondrial protein quality control. However, alteration of mortalin’s activities, its abnormal subcellular compartmentalization, and its protein partners turn mortalin into a disease-driving protein in different pathological conditions, including cancers. Here, mortalin’s contributions to tumorigenic pathways are explained. Pathology information based on mortalin’s RNA expression extracted from The Cancer Genome Atlas (TCGA) transcriptomic database indicates that mortalin has an independent prognostic value in common tumors, including lung, breast, and colorectal cancer (CRC). Subsequently, the binding partners of mortalin reported in different cellular models, from yeast to mammalian cells, and its regulation by post-translational modifications are discussed. Finally, we focus on colorectal cancer and discuss how mortalin and its tumorigenic downstream protein targets are regulated by a ubiquitin-like protein through the 26S proteasomal degradation machinery. A broader understanding of the function of mortalin and its positive and negative regulation in the formation and progression of human diseases, particularly cancer, is essential for developing new strategies to treat a diverse set of human diseases critically associated with dysregulated mortalin.
In the 1960s, Italian scientist Ferruccio Ritossa observed that cells could induce strong transcriptional activity in response to elevated temperatures (Ritossa, 1962; De Maio et al., 2012). This initial report led to the discovery of heat shock proteins (HSPs) several years later (Tissières et al., 1974). Besides stress-inducible HSPs, there are HSP genes that encode constitutively expressed members (Brown et al., 1993). HSPs have two fundamental roles in cells: Maintaining proper protein synthesis, and restoring cellular homeostasis in response to internal and external environmental stresses (Kumar et al., 2022). Constitutive and stress-responsive proteins are essential for properly folding proteins and preventing the aggregation of misfolded proteins, particularly as part of endoplasmic reticulum-associated degradation (ERAD) (Brodsky, 2007). Meanwhile, damaged proteins generated as a result of cellular physiological and pathological stressors are targeted by HSPs for a recovery process to return to their normal functions. Aside from protein folding, HSPs are necessary for cytoskeletal recognition and organization (Muranova et al., 2022) and play essential roles in the protection, integrity, and function of several intracellular organelles, including the mitochondria (Craig, 2018), Golgi apparatus (Wu et al., 2019), endoplasmic reticulum (Matsumura et al., 2013), lysosomes (Ingemann and Kirkegaard, 2014), and cell membrane (De Maio and Hightower, 2021). Alongside intracellular functions, HSPs are present in extracellular compartments covering several different roles (De Maio and Vazquez, 2013). Extracellular vesicles (EVs), including exosomes, are examples of extracellular compartments where HSPs demonstrate different biological impacts on cellular events. For example, extracellular HSPs play a critical role in cell/tissue homeostasis under normal conditions (Calderwood et al., 2021). Additionally, these extracellular HSPs (eHSPs) contribute to various diseases, including human cancers, by mediating inflammation and immunity (Lancaster and Febbraio, 2005; Wyciszkiewicz et al., 2019; Komarova et al., 2021; Li et al., 2022).
HSPs are historically divided into subfamilies according to their molecular weight, ranging from 10 to more than 100 kDa in molecular size, and are located in various cellular compartments (Jee, 2016). The 70-kDa subfamily (HSP70) is a family of proteins that function as molecular chaperones and are responsible for regulating the necessary housekeeping activities of the cell. They are essential for protein folding and refolding, protein modification, translocation, prevention of protein aggregation, protein degradation, formation and disassembly of protein complexes, regulation of apoptosis, and cell survival (Radons, 2016; Albakova et al., 2020). In addition to intracellular HSPs, extracellular HSP70 released from necrotic or stressed cells is immunogenic and contributes to the autoimmune reaction (Tukaj, 2020). Typically, HSP70 proteins require additional co-chaperones to aid their functions. These co-chaperones can positively or negatively interfere with the function of HSP70s (Laufen et al., 1999; Bhattacharya et al., 2020). A diverse set of HSP70 subfamily members are found in human cells, comprising the following 13 gene products: HSPA1A (HSP70-1), HSPA1B (HSP70-2), HSPA1L (hum70t), HSPA2 (heat-shock 70 kD protein-2), HSPA5 (Grp78, Bip, MIF2), HSPA6 (heat-shock 70 kD protein 6, or HSP70B′), HSPA7 (heat-shock 70 kD protein 7), HSPA8 (HSC70, HSC71, HSP71, HSP73), HSPA9 (Grp75, MOT, MOT2, PBP74, mot-2), HSPA12A (FLJ13874, KIAA0417), HSPA12B (RP23-32L15.1), HSPA13b (Stch), and HSPA14 (HSP70-4, HSP70L1; (Kampinga et al., 2009). HSP70 subfamily members are found in various cellular compartments such as the cytoplasm, nucleus, lysosomes, endoplasmic reticulum, and mitochondria, indicating their essential biological functions in these organelles. Isoforms of HSP70 proteins have two major domains: An N-terminal ATPase domain, and a C-terminal substrate binding domain (SBD) connected by a hydrophobic linker domain (Zhuravleva and Gierasch, 2011). The C-terminal SBD domain consists of two subdomains, α and β, which act as a flexible lid and sandwich subdomain, respectively. However, structural differences determine their respective substrates, such as DnaK (prokaryotic HSP70) versus eukaryotic HSP70 (Zhang et al., 2014). While HSP70 subfamily proteins share the majority of structural features, mutations at their coding regions have allowed them to be targeted and retained to particular organelles (McCallister et al., 2015). One example is GRP75, referred to as mortalin hereafter, which has a 46-amino acid mitochondrial-targeting signal peptide at its N terminus (Dahlseid et al., 1994).
In addition to the basal function of HSPs in normal cells, HSPs have protective roles in cancerous cells. Intrinsic (elevation of oncoproteins during carcinogenesis) and extrinsic (low glucose, pH, and oxygen) stresses developed in cancer cells lead to overexpression of HSPs in diverse malignant tumors. High levels of HSPs correlate with more aggressive characteristics and chemoresistance (Ciocca and Calderwood, 2005). However, patterns of HSP expression are selectively determined by the stages of cancer as well as tissue types. Some examples are the biological impacts and prognostic values of HSPs (HSP27, HSP60, and HSP70) in prostate, breast, and colorectal cancers in both early and advanced cancers (Glaessgen et al., 2008; Javid et al., 2022; Sun et al., 2022; Tsai et al., 2022).
Human mitochondrial Hsp70 (mortalin/mot-2/GRP75) is a heat uninducible HSP70 protein (Londono et al., 2012). Mortalin is similar to other HSPs, containing an N-terminus ATPase and SBD located at the C-terminus (Luo et al., 2010). Similar to other HSP70s, mortalin is shown to be highly expressed in several types of tumors. As a mitochondrial chaperone protein, mortalin plays a critical role in mitochondrial biogenesis. Mitochondrial mortalin mediates polypeptide chain translocation through the TOM/TIM translocase complexes, unfolds/folds nuclear proteins, and stabilizes translocated proteins. Additionally, mitochondrial mortalin is involved in biogenesis of iron-sulfur clusters in the matrix (Gambill et al., 1993; Voisine et al., 1999; Dutkiewicz et al., 2003), responds to glucose deprivation (Merrick et al., 1997), and protects against reactive oxygen species (ROS) (Tai-Nagara et al., 2014). While mortalin is a crucial mitochondrial protein, around 30% of mortalin in the cell is located in other cellular compartments such as the cytoplasm, endoplasmic reticulum (ER) (Szabadkai et al., 2006; D'Eletto et al., 2018; Liu et al., 2019b), and nuclear (Ryu et al., 2014) compartments and is circulated in blood (Priyanka and Seth, 2022). It has been shown that extracellular mortalin can have a vital survival role in cells attacked by complement-dependent cytotoxicity (CDC). Translocation of mitochondrial mortalin from mitochondria to the plasma membrane allows it to bind to the C8 and C9 complement components and inhibit C5b-9 assembly and stability, thus preventing induction of cell death mediated by complement-mediated lysis (Pilzer and Fishelson, 2005; Saar Ray et al., 2014; Tai-Nagara et al., 2014; Mazkereth et al., 2016). These locations, combined with a diverse set of protein partners, allow mortalin to positively and negatively regulate several pathways in both normal and pathological status.
Overexpression of mortalin correlates with diminished patient survival in several forms of cancer, including early-stage non-small cell lung cancer (Sun et al., 2017), breast cancer (Zhang et al., 2021), hepatocellular carcinoma (Cheng et al., 2019), ovarian and cervical cancers (Putri et al., 2019; Xu et al., 2019), gastric cancer (Dai et al., 2021), and CRC (Rozenberg et al., 2013; Javid et al., 2022). In the context of CRC, overexpressed mortalin was observed in adenoma and colorectal adenocarcinomas (Dundas et al., 2005). In addition to the intracellular tumorigenic role of mortalin in above malignant tumors, exosomes derived from tumor cells of breast, ovarian, prostate, hepatic, gastric, colon, but not pancreatic carcinoma have high levels of mortalin expression compared to normal tissues (Huang et al., 2019).
In transformed cells, nuclear localization of p53 is essential for its tumor suppressor activity in terms of growth arrest and apoptosis (Shaulsky et al., 1991). Overexpression and enriched cytoplasmic mortalin in transformed cells allow mortalin’s SBD to bind to and interfere with the normal function of several proteins, including p53. One of the most well-studied oncogenic roles of mortalin is its ability to bind cytoplasmic p53, thereby sequestering it in the cytoplasm, inhibiting its nuclear localization and tumor suppressive properties (Gestl and Anne Bottger, 2012; Elwakeel, 2022). Meanwhile, it has been shown that mortalin-p53 interactions can be regulated with physiological or chemical stresses in a cancer-dependent manner (Lu et al., 2011). Delivery of mortalin-specific shRNA-expressing by adenoviruses can successfully supress the growth of breast cancer MCF7 xenograft tumors (Yoo et al., 2010). Supressing mortalin-p53 interaction by siRNA pharmacological tools such as MKT-077, Withaferin A, and mortalin protein suppressors leads to growth arrest and apoptosis, supporting the dominant p53 inhibitory role of mortalin in malignant tumors (Wadhwa et al., 2000; Wadhwa et al., 2002; Wadhwa et al., 2003a; Kaul et al., 2005; Yoo et al., 2010; Sane et al., 2014; Nigam et al., 2015).
Another protein that may explain the role of mortalin in tumorigenesis is CD151. CD151 is overexpressed in malignant tumor tissues and is found to be a driver of tumor progression and metastasis in several cancer types through the formation of tetraspanin CD151-enriched microdomains (Sadej et al., 2014). In hepatocellular carcinoma (HCC), mortalin was found to stabilize CD151-dependent tetraspanin-enriched microdomains and contribute to the progression of HCC (Liu et al., 2019a). Furthermore, altered centrosome amplification can lead to chromosomal instability, a highlighted feature of cancer cells. Ma et al. (2006) reported that overexpression of mortalin resulted in its localization to the centrosome and inhibition of p53-mediated suppression of centrosome duplication. Interestingly, a dominant mortalin inhibitor, UBXN2A, which interferes with mortalin-p53 interaction (Sane et al., 2016), is enriched in centrosomes [https://www.proteinatlas.org, UBXN2A’s subcellular section; (Thul et al., 2017)]. Another critical event in tumor progression is uncontrolled cell division. The Raf/MEK/ERK pathway is a signaling pathway responsible for cell survival. Studies in ovarian cancer demonstrated the expression of mortalin-induced Raf/MEK/ERK activation, highlighting mortalin’s regulatory role in the MAPK/ERK pathway, and tumor progression (Dundas et al., 2005; Hu et al., 2016). Further studies on the effect of mortalin on the Raf/MEK/ERK signaling pathway revealed that reduction of mortalin in MEK/ERK activated cells leads to elevation in the level of p21CIP1. The cyclin-dependent kinase inhibitor p21CIP1 triggers innate tumor suppressive mechanisms by the inhibition of cell growth. Interestingly, the mortalin-dependent regulation of p21CIP1 depends on MEK/ERK activity. The regulatory mechanism of mortalin on p21CIP1 is uniquely mediated by mortalin independent of p53 (Wu et al., 2013). Finally, a set of experiments in a rat adrenal pheochromocytoma (PC12) cell line under glucose deprivation revealed that mortalin can activate AKT in a PI3K-independent manner through the Raf/MEK/ERK signaling pathway. Activation of AKT and ERK by mortalin suppress conformational change in Bax, decrease cytochrome c release, and reduce apoptosis (Yang et al., 2011).
Besides tumorigenic functions in the cytoplasm, nuclear mortalin without the mitochondrial localization peptide was shown to increase the malignant properties of cancer cells, including cell proliferation, colony formation, motility, and tumor forming capacity in both in vitro and in vivo models. The nuclear mot-N mechanistically binds and supresses p53 function while functionally activating telomerase and heterogeneous ribonucleoprotein K (hnRNP-K) proteins to promote carcinogenesis (Ryu et al., 2014).
Lastly, overexpression of mortalin is associated with the enrichment of several cancer cells stemness markers, such as ABCG2, OCT-4, CD133, ALDH1, CD9, MRP1, and connexin. By increasing the population of stem cells, mortalin promotes the formation of spheroids (Yun et al., 2017). Elevation of cancer stem cells (CSCs) may explain the higher migration and drug resistance seen in the presence of overexpressed mortalin in cancer cells. Using breast cancer cells, Na et al. showed that overexpressed mortalin can downregulate epithelial markers E-cadherin (CDH1), CK8 (KRT8), and CK18 (KRT18). Simultaneously, overexpressed mortalin enhances mesenchymal markers, including vimentin, fibronectin, β-catenin, CK14, and hnRNP-K (Na et al., 2016). Mortalin’s multi-functional roles in tumorigenic pathways summarized in this section explain mortalin’s contributions to epithelial-to-mesenchymal transition (EMT), early tumor recurrence, tumor migration/invasion, stemness, angiogenesis, and tumor metastasis (Wadhwa et al., 2006; Yi et al., 2008; Chen et al., 2014; Na et al., 2016; Xu et al., 2020). Finally, a recent study in human breast cancer cells has shown that mortalin supports breast cancer stem cells and enhances EMT through activation of the Wnt/GSK3β/β-catenin signaling pathway (Wei et al., 2021).
Based on the tumorigenic activities of mortalin in malignant tumors discussed above, we decided to investigate the survival curves of cancer patients by assessing the RNA expression level of HSPA9/mortalin using The Cancer Genome Atlas (TCGA) available in the human protein atlas database (Uhlen et al., 2017). Figure 1 shows that mortalin is a prognostic factor in breast and colorectal cancers, the second and third most common cancers, respectively (Bray et al., 2018). However, in colorectal cancer (carcinoma or adenocarcinoma), low expression of mortalin is an adverse prognostic factor (Figures 1A–F), whereas in breast cancer low expression of mortalin is a favorable prognostic factor (Figure 1G). The adverse effect of low RNA expression of mortalin in colon and rectal adenocarcinoma has no correlation with expression of mortalin protein previously reported (Dundas et al., 2005). A part of this discrepancy is that the RNA and protein levels of specific genes are not correlated in tumor tissues. The dysregulated transcription machinery, microRNAs and post-translational modifications in transforming cells can cause uncorrelated RNA-protein relations for specific genes as previously described (Kosti et al., 2016). Interestingly, mortalin’s impact on prognosis differs between rectal and colon adenocarcinoma (Figures 1C, D versus Figures 1E, F), as these two tissues are different in terms of pathological features (Hemminki et al., 2010) and survival rate (Lee et al., 2013).
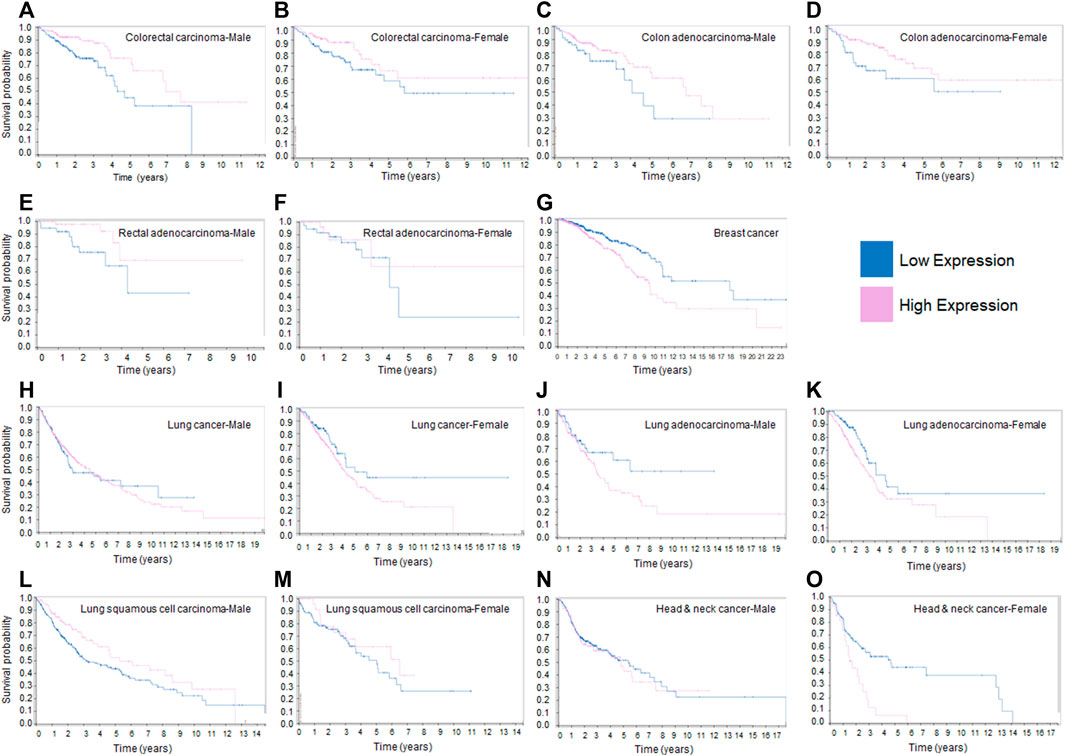
FIGURE 1. Kaplan-Meier survival curves of patients with different human cancers sorted according to expression levels of HSPA9/mortalin. (A). In male patients with colorectal carcinoma, low expression of HSPA9 has a negative effect on survival. (B). In female patients with colorectal carcinoma, low expression of HSPA9 has a negative effect on survival. (C). In male patients with colon adenocarcinoma, low expression of HSPA9 has a negative effect on survival. (D). In female patients with colon adenocarcinoma, low expression of HSPA9 has a negative effect on survival. (E). In male patients with rectal adenocarcinoma, low expression of HSPA9 has a negative effect on survival. (F). In female patients with rectal adenocarcinoma, low expression of HSPA9 has a negative effect on survival. (G). In female patients with breast cancer, high expression of HSPA9 has a negative effect on survival. (H). In male patients with lung cancer, HSPA9 expression levels have no effect on survival. (I). In female patients with lung cancer, high expression of HSPA9 has a non-significant negative effect on survival. (J). In male patients with lung adenocarcinoma, high expression of HSPA9 has a non-significant negative effect on survival. (K). In female patients with lung adenocarcinoma, high expression of HSPA9 has a non-significant negative effect on survival. (L). In male patients with lung squamous cell carcinoma, HSPA9 expression levels have no effect on survival. (M). In female patients with lung squamous cell carcinoma, HSPA9 expression levels have no effect on survival. (N). In male patients with head and neck cancer, HSPA9 expression levels have no effect on survival. (O). In female patients with head and neck cancer, high expression of HSPA9 has a non-significant negative effect on survival. The prognostic value of mortalin RNA expression in different cancers illustrated in this figure were downloaded from the Human Protein Atlas: https://www.proteinatlas.org/ENSG00000113013-HSPA9/pathology.
In lung cancer, mortalin is not found to be a significant prognostic factor. However, in women, a high expression of mortalin appears to be an adverse prognostic factor (Figures 1H, I). When categorizing lung cancer into adenocarcinoma and squamous cell carcinoma, a high expression of mortalin is found to have an adverse effect on survival in the former (Figures 1J, K) but no significant impact in the latter (Figures 1L, M). Finally, in head and neck cancer, while mortalin is not a prognostic factor in male patients, there appears to be a trend towards higher expression being an adverse prognostic factor in female patients (Figures 1N, O). The RNA-sequencing data in Figure 1, generated from TCGA transcriptome database, suggests mortalin’s tumorigenic or tumor suppressive function depends on the specific cancer tissues. Furthermore, this data suggests sex is another factor that can determine the function of mortalin in malignant cells, as previously described (Li et al., 2018). However, it has been shown that the gene-expression values derived by TCGA pipeline can vary considerably across biological replicates (Rahman et al., 2015). Further studies are necessary to determine the role of mortalin, particularly at the protein level, in a tissue-specific, cell-type-specific, and stage-dependent manner in tumorigenesis and the organization of oncogenic signaling pathways. Western blot experiments revealed that human CRC and breast tumors have significantly elevated levels of mortalin and showed that patients with overexpressed mortalin are commonly diagnosed with a higher grade and stage of tumors, indicating lower chance of survival (Abdullah et al., 2015a). Beyond the expression levels of mortalin, its binding partners and post-translational modifications are other critical factors in mortalin’s functions in malignant tumors (Schneider et al., 2017).
Published research articles indicate mortalin crosstalks with a diverse protein network. A variety of biochemical, biological, and computational methods with different levels of selectivity and specificity have been used to investigate mortalin’s interaction partners and their biological mechanisms in diverse types of in vitro and in vivo models. Based on the interaction dynamics, interactions between mortalin and its partners can be weak or strong, transient or stable, non-permanent or permanent. Several molecular and cellular factors such as kinetics, thermodynamics, stoichiometry, and cofactors can affect mortalin’s interaction with its partners in a spatiotemporal manner (Lobingier et al., 2017). We decided to list several of mortalin’s partners reported in the last decade in the literature. While discussion of mortalin’s protein partners can provide a valuable resource for future studies, some of these protein-protein interactions and their roles at the molecular and cellular levels needs further confirmation.
Mortalin has several different protein partners crucial for its function in the mitochondria. The main binding partners of the HSP70 subfamiliy is comprised of two co-chaperones: J-domain proteins, and nucleotide exchange factors (NEFs). These two co-chaperones are essential for the protein-folding function of HSP70 proteins, as J-protein stimulates ATPase activity, and NEFs promote the release of nucleotides. In eukaryotic cells, NEFs have a vital role in protein folding and import as well as protein quality control (Bracher and Verghese, 2015). Different cellular compartments have various combinations of these two proteins, allowing for specialized functions of HSP70 proteins (in this case, mortalin) in the mitochondria (Moro and Muga, 2006; Marszalek, 2016).
P66shc protein mediates insulin resistance, promotes apoptosis, and controls the intracellular redox balance (Biondi et al., 2022; Haslem et al., 2022). In HCC, downregulated p66shc is associated with better survival (Fasolato et al., 2021). Under normal conditions, p66Shc is tightly controlled to avoid unnecessary induction of apoptotic signals originated in the mitochondria (Orsini et al., 2006). In the absence of cell stressors, a fraction of cytosolic p66Shc localizes within the mitochondrial matrix to form a complex with mortalin. Apoptotic events such as ultraviolet radiation induce the dissociation of p66Shc from mortalin and the release of free p66Shc. The dissociated p66Shc can enhance the mitochondrial pathway of apoptosis by inducing mitochondrial damage (Orsini et al., 2004).
A mitochondrial zinc finger motif heat shock protein, Zim17, binds to mortalin inside the mitochondria and is involved in several critical mitochondrial functions. The TimI5/Ziml7 complex cooperates with mortalin to facilitate the transfer of unfolded proteins into the mitochondrial matrix (Yamamoto et al., 2005). Studies in yeast have shown that the depletion of Zim17 suppresses mitochondrial protein import, interferes with Fe/S protein biogenesis, and leads to aggregation of mortalin in the matrix (Sanjuán Szklarz et al., 2005). Human Hsp70 escort protein (Hep) is a human Zim17 ortholog. Uniquely, mammalian Zim17 (Hep) can facilitate the ATPase activity of human mortalin, and it prevents the aggregation of unfolded target proteins (Goswami et al., 2010).
In addition to mortalin, HSP60 is a vital chaperone for unfolded proteins in the mitochondria (Cheng et al., 1989). The two proteins work similarly to assist protein folding in the mitochondrial matrix but are also found in other subcellular locations with similar functions (Deocaris et al., 2006). An interaction between the N-terminal domain of mortalin and HSP60 has also been demonstrated in both in vitro and in vivo conditions (Wadhwa et al., 2005). Malfunction of mortalin and its HSP60 partner leads to the accumulation of unfolded aggregated proteins in response to stress, aging, or disease (Iosefson et al., 2012). A more recent study revealed that proteasomal degradation of mortalin in the cytoplasmic compartment leads to reduction of mortalin’s partner, HSP60, in mitochondria. This finding indicates that the presence of mortalin is essential for mortalin-HSP60 protein quality machinery in the mitochondria (Sane et al., 2018). Another functional binding partner for mortalin’s disaggregating mechanism in the mitochondria is Tid1, a known HSP40 protein. Therefore, the mortalin-TID1 complex can potentially respond to aggregation during cellular stress. More importantly, the efficient function of mortalin-Tid1 can be a therapeutic target to slow or reverse the aggregation of toxic proteins in pathological conditions such as neurodegenerative diseases (Khan et al., 2020).
Accumulating evidence indicates that mitochondria play critical roles in the cancer cell metastatic cascade by promoting invasion, intravasation, and extravasation. A dysregulated mitochondrial bioenergetic state due to the abnormal function of mitochondrial protein quality control supports cancer cells in their divergent microenvironments during tumor cell migration and invasion (Guha et al., 2014; Cui et al., 2017; Han et al., 2018; Lin et al., 2018; Porporato et al., 2018; Emmings et al., 2019). The lack of knowledge regarding regulatory pathways abused by mitochondria in cancer and the unexamined protein complexes within in vivo models limit the number of therapeutic approaches that specifically target mitochondria in cancer cells. Finding novel pathways such as the mortalin-HSP60 axis and its regulatory elements can be a new platform for a therapeutic approach that not only effectively represses primary tumor progression but also suppresses the development of metastatic characteristics interfering with overactive mitochondria.
Interactions between the mitochondria and the ER allow for resident ER proteins to interact with mortalin. Two important proteins that cooperate with mortalin are the inositol 1,4,5 triphosphate receptor (IP3R) and voltage-dependent anion channel 1 (VDAC1) (Xu et al., 2018). Together, these three proteins assist with Ca2+ transfer from the ER to mitochondria, with mortalin acting as the bridge between IP3R and VDAC1 (Xu et al., 2018). Transglutaminase type 2 (TG2) is a mortalin’s partner in mitochondria-associated membranes (MAMs) that regulate ER-mitochondria contact sites. Reduction of TG2-mortalin increases interaction between IP3R3 and mortalin. TG2 and its binding to mortalin can control ER-mitochondrial Ca2+ flux and protein expression (D'Eletto et al., 2018). A study focused on early onset Parkinson’s disease (PD) has shown that DJ-1 interacts with the IP3R-mortalin-VDAC complex and is essential for proper communication between the mitochondria and ER (Liu et al., 2019b).
Another key partner of mortalin is apoptosis-inducing factor (AIF), which plays a major role in the caspase-independent apoptotic pathway (Liu et al., 2006). It has been shown that mortalin interacts with AIF to mediate its binding to the outer mitochondrial membrane (OMM). In addition, chemotherapeutic agents decrease AIF-mortalin interaction, resulting in the dissociation of AIF from the OMM and, consequently, the induction of apoptosis (Fadeeva et al., 2018). Further studies are needed to determine whether inhibition of AIF-mortalin interaction, dissociation of AIF from the OMM, and subsequent apoptosis can potentially turn into a therapeutic strategy in cancer cells with high levels of mortalin, such as glioma patients.
Outside of the mitochondria, mortalin’s binding partners are varied. In vivo studies demonstrated that GRP94, an HSP90-like chaperone of the endoplasmic reticulum, is a binding partner (Takano et al., 2001; Marzec et al., 2012). Similar to its role in the mitochondrial import of unfolded proteins, mortalin mediates a similar role in intracellular import in the cytoplasm (Mizukoshi et al., 1999). Mortalin binds fibroblast growth factor 1 (FGF1), allowing for its uptake and appropriate path to its target organelle in the cell (Mizukoshi et al., 1999). The IL-1 receptor is similarly internalized through ATP hydrolysis by mortalin, leading to downstream cytokine pathways in the cell (Sacht et al., 1999). Additionally, a set of cytoplasmic proteins interact with mortalin and interfere with the formation of a mortalin-p53 complex in the cytoplasm. One such example is DAB2IP, which competitively binds mortalin, effectively preventing the degradation of p53 (Feng et al., 2022).
A set of studies reported by Wadhwa et al. (2003b) has shown that interaction between mortalin and mevalonate pyrophosphate decarboxylase (MPD) can result in reduced levels of Ras and phosphorylated ERK2. Since oncogenic Ras facilitates cancer cell proliferation, inhibition by mortalin suggests a safeguard mechanism. This study indicates that mortalin affects cell proliferation by more than one mechanism, which can be exchanged during cancer cell transformation in a cellular context matter. Current evidence strongly supports mortalin’s functions as a proliferation-controlling complex under normal and pathological conditions. Future mechanistic studies will determine whether mortalin is a promising therapeutic target in selected malignant tumors.
Using a set of protein-protein assays, Hansen et al. (2015) reported that mortalin binds to the CREC family, which is a set of multiple EF-hand Ca2+-binding proteins. Reticulocalbin, ERC-55 and its splice variants, reticulocalbin-3, Cab45 and its splice variants, and calumenin are all in the CREC family. Members of the CREC family bind and regulate a number of proteins in the ER and sarcoplasmic reticulum (SR). By binding to mortalin, calumenin and reticulocalbin allow CREC proteins to contribute to diverse pathways, including chaperone activity, cell proliferation, transformation, and cellular aging (Hansen et al., 2015).
It has been shown that mitochondrial proteolytic stress induced by loss of mortalin function can be rescued by Parkin and PINK1 protein. To rescue loss of mortalin phenotypes, Parkin and PINK1 increase lysosomal-mediated mitochondrial clearance and facilitate autophagic machinery (Burbulla et al., 2014). The current evidence indicates that PINK1 can function as either a pro- or an anti-tumorigenic protein in different tumors, showing a duality dependent on context (Wang et al., 2022). PINK1 regulates cell survival, stress resistance, mitochondrial homeostasis, and the cell cycle, all of which are key signaling pathways misused by cancer cells during tumor initiation and progression. Mechanistically, PINK1 interferes with the PI3-kinase/Akt/mTOR signaling pathway, resulting in critical changes in mitochondrial and metabolic functions. These are essential for cancer survival, growth, stress resistance, and the cell cycle (Rakovic et al., 2011). Mechanistic crosstalk between PINK1 and mortalin requires further study, as PINK1-mortalin interaction could be a potential target for new therapeutic approaches in certain diseases, including Parkinson’s disease and malignant tumors.
Cancer cells express a diverse set of regulatory proteins to block the C5b-9 membrane attack complex (MAC) which induces cell death (Fishelson et al., 2003). By shedding of membrane vesicles loaded with complement MAC, mortalin can protects cancer cells from complement-mediated lysis (Fishelson et al., 2003). Rozenberg et al. has shown that Hsp90 can additionally protect cells from the complement-dependent cytotoxicity (CDC) by inhibiting C5b-9 assembly and/or stability at the plasma membrane. Interestingly, the complement activation can lead to direct binding of Hsp90 to mortalin. Formation of Hsp90–mortalin heterocomplexes in the presence of activated C5b-9 can generate an efficient protection against C5b-9 (Rozenberg et al., 2018).
Post-translational modifications (PTMs) of mortalin can affect its binding partners and specific function. For instance, phosphorylation of tyrosine residues augmented the interaction between mortalin and fibroblast growth factor 1 in the late G1 phase, emphasizing the importance of mortalin post-translational modifications in the regulation of the cell cycle (Mizukoshi et al., 1999). Additional PTMs include acetylation, oxidation, and ubiquitination (Deocaris et al., 2008) but further studies are needed to understand these PTMs and their function at the cellular level. Subsequently, selective activation or inhibition of mortalin’s PTMs by therapeutic interventions may be a possible avenue of therapy, preventing the downstream cancer-promoting signaling cascades.
Normal evolutionary mammalian cells have developed several regulatory mechanisms to avoid malignant transformation of normal cells in a tissue-dependent manner. Anti-tumorigenic mechanisms mediated by these tumor suppressor proteins and their associated proteins can target key proteins involved in tumorigenic pathways and stop or slow tumor progression. One protein that promotes the degradation of mortalin is UBXN2A (Sane et al., 2018). Similar to other ubiquitin-like proteins, UBXN2A targets a specific set of substrates in a cell-dependent manner. UBXN2A has a dual effect on the mortalin tumorigenic pathway. By binding to mortalin’s binding pocket (Sane et al., 2016), UBXN2A blocks mortalin’s interaction with its substrates, including the p53 tumor suppressor protein (Abdullah et al., 2015b). Consequently, UBXN2A recruits the CHIP E3 ubiquitin ligase and facilitates the ubiquitination of mortalin. Ubiquitination of the carboxy-terminal of mortalin leads to its proteasomal degradation (Sane et al., 2018). Future studies, including ongoing projects in our group, will determine whether UBXN2A-dependent degradation of mortalin alters the fate of mortalin protein partners and improves cancer cell response to traditional anti-cancer therapies. Besides its tumor suppressor function, UBXN2A plays a regulatory role in the nervous system (Rezvani et al., 2009; Teng et al., 2015). The mechanistic relation between UBXN2A and mortalin in the nervous system is another potential future study that can uncover the function of mortalin in neurodegenerative diseases.
The ubiquitin and ubiquitin-like proteins are involved in various pathologies such as inflammatory and neurodegenerative diseases as well as cancer (Hwang et al., 2022). It has been shown that many of these disease-related pathways can be modified and regulated by different types of modifications. The ubiquitin-like proteins can determine the fate of selected proteins using a single or combination of mono-ubiquitination, poly-ubiquitination, sumoylation, and neddylation. Therefore, understanding the mechanisms behind the upregulation and downregulation of mortalin by the ubiquitin-proteasome pathway can potentially lead to more specific anti-mortalin drugs.
Mortalin, as a mitochondrial HSP, controls the protein quality process in the mitochondrial matrix under physiological conditions. However, overexpression and abnormal cellular compartmentalization turn this HSP into a driver of tumor progression in diverse types of malignant tumors. Mortalin’s diverse network of interaction partners enable it to positively or negatively regulate several pathways involved in cancer cell initiation and progression. In addition, mortalin promotes cancer cell migration and invasion, two key elements during tumor metastasis. As an element of the hallmarks of cancer, metastasis is the most significant factor in cancer-related deaths. Understanding the biological impact of mortalin during cancer progression and its tumorigenic mechanisms will introduce new paradigms to the study of tumor growth and metastasis. Certainly, mortalin’s partners and subcellular localizations are two key elements in mortalin’s tumorigenic functions. Additionally, the tumorigenic function of extracellular mortalin and its interaction with the tumor microenvironment is another unstudied area. Investigating the spatial and temporal function of mortalin in a cancer-dependent manner will improve the understanding of the pathological principle of mortalin, resulting in more effective and safer targeted therapies. Figure 2 and Supplementary Table S1 in this review list a set of mortalin’s partners reported in different in vitro and in vivo models using diverse methods and analytical approaches. These partners have different binding affinities to mortalin, and their interactions are conditional on the cellular context. Summarization of mortalin partners, and furthermore their direct and indirect relation with mortalin, illustrated in Figure 2, provides a helpful guideline for researchers who wish to conduct further studies on mortalin or mortalin partners. Understanding the biological mechanisms of pro-cancer proteins such as mortalin is crucial to opening therapeutic windows for successful novel interventions. The background and recent advancements covered in this review article indicate a safe anti-mortalin treatment can become an effective targeted therapy in a set of cancers with abnormal mortalin.
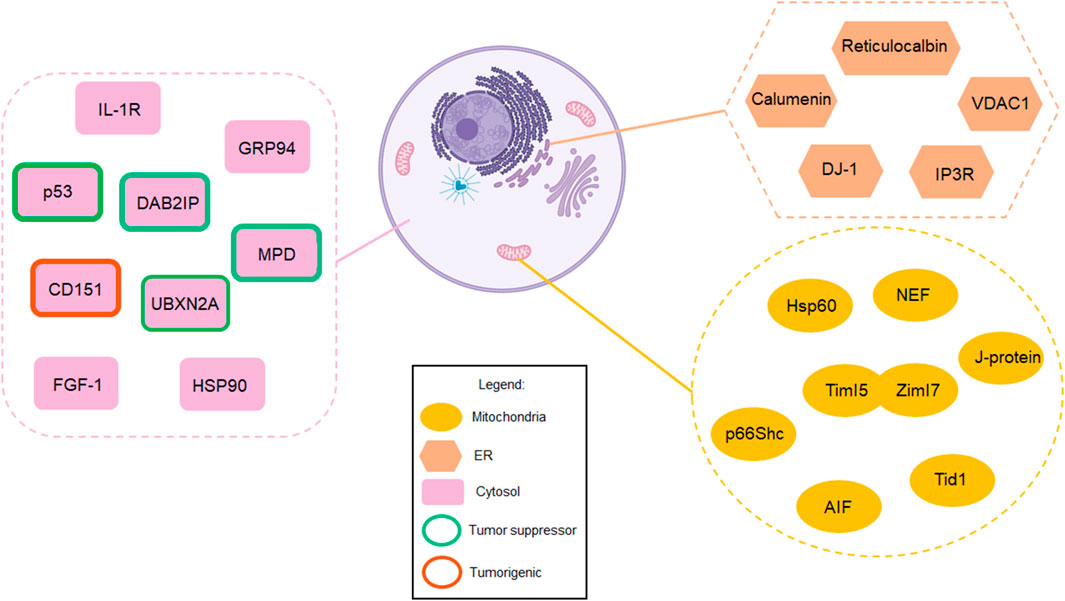
FIGURE 2. Mortalin acts as a “double agent,” playing contrasting roles in normal cells versus cancer cells. This figure summarizes mortalin’s partners in different subcellular compartments, including mitochondria, the cytosol, and the endoplasmic reticulum (created with BioRender.com). While mortalin’s functions are essential for normal cells, its interaction partners allow mortalin to become a poor prognostic factor in human cancer. Understanding these two different faces of mortalin will enable the design of anti-mortalin drugs that regulate its tumorigenic pathways in cancer cells while its physiological functions in normal cells remain intact. These partners have different binding affinities to mortalin, and their interactions are conditional on the cellular context. Therefore, future studies will determine the biological and pathological impact of these interactions in a tissue-dependent manner.
NE and CK wrote the manuscript with support from GB; KR supervised the project.
The support for this work was provided by the University of South Dakota Division of Basic Biomedical Sciences (READ award) and CBBRe (Center for Brain and Behavior Research, University of South Dakota). The DaCCoTA funding is supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number U54GM128729 and the National Cancer Institute of the National Institutes of Health under award number 1R03CA223935-01. Additionally, this research was supported, in part, by a grant from the National Science Foundation (DGE-1633213).
We are grateful for the insightful comments offered by Rosha Vahadi. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1028519/full#supplementary-material
Abdullah, A., Sane, S., Branick, K. A., Freeling, J. L., Wang, H., Zhang, D., et al. (2015a). A plant alkaloid, veratridine, potentiates cancer chemosensitivity by UBXN2A-dependent inhibition of an oncoprotein, mortalin-2. Oncotarget 16, 23561–23581. doi:10.18632/oncotarget.4452
Abdullah, A., Sane, S., Freeling, J. L., Wang, H., Zhang, D., and Rezvani, K. (2015b). Nucleocytoplasmic translocation of UBXN2A is required for apoptosis during DNA damage stresses in colon cancer cells. J. Cancer 6, 1066–1078. doi:10.7150/jca.12134
Albakova, Z., Armeev, G. A., Kanevskiy, L. M., Kovalenko, E. I., and Sapozhnikov, A. M. (2020). HSP70 multi-functionality in cancer. Cells 9, 587. doi:10.3390/cells9030587
Bhattacharya, K., Weidenauer, L., Luengo, T. M., Pieters, E. C., Echeverría, P. C., Bernasconi, L., et al. (2020). The Hsp70-Hsp90 co-chaperone Hop/Stip1 shifts the proteostatic balance from folding towards degradation. Nat. Commun. 11, 5975. doi:10.1038/s41467-020-19783-w
Biondi, G., Marrano, N., Dipaola, L., Borrelli, A., Rella, M., D'Oria, R., et al. (2022). The p66Shc protein mediates insulin resistance and secretory dysfunction in pancreatic β-cells under lipotoxic conditions. Diabetes 71, 1763–1771. doi:10.2337/db21-1066
Bracher, A., and Verghese, J. (2015). The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2. doi:10.3389/fmolb.2015.00010
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 68, 394–424. doi:10.3322/caac.21492
Brodsky, J. L. (2007). The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem. J. 404, 353–363. doi:10.1042/BJ20061890
Brown, C. R., Martin, R. L., Hansen, W. J., Beckmann, R. P., and Welch, W. J. (1993). The constitutive and stress inducible forms of hsp 70 exhibit functional similarities and interact with one another in an ATP-dependent fashion. J. Cell Biol. 120, 1101–1112. doi:10.1083/jcb.120.5.1101
Burbulla, L. F., Fitzgerald, J. C., Stegen, K., Westermeier, J., Thost, A. K., Kato, H., et al. (2014). Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis. 5, e1180. doi:10.1038/cddis.2014.103
Calderwood, S. K., Borges, T. J., Eguchi, T., Lang, B. J., Murshid, A., Okusha, Y., et al. (2021). Extracellular Hsp90 and protection of neuronal cells through Nrf2. Biochem. Soc. Trans. 49, 2299–2306. doi:10.1042/BST20210370
Chen, J., Liu, W. B., Jia, W. D., Xu, G. L., Ma, J. L., Huang, M., et al. (2014). Overexpression of Mortalin in hepatocellular carcinoma and its relationship with angiogenesis and epithelial to mesenchymal transition. Int. J. Oncol. 44, 247–255. doi:10.3892/ijo.2013.2161
Cheng, M. Y., Hartl, F. U., Martin, J., Pollock, R. A., Kalousek, F., Neupert, W., et al. (1989). Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature 337, 620–625. doi:10.1038/337620a0
Cheng, W., Zhang, B., Zikeliyar, M., Wang, J., Jian, H., Wu, K., et al. (2019). Elevated Mortalin correlates with poor outcome in hepatocellular carcinoma. Ann. Diagn Pathol. 42, 59–63. doi:10.1016/j.anndiagpath.2019.06.011
Ciocca, D. R., and Calderwood, S. K. (2005). Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10, 86–103. doi:10.1379/csc-99r.1
Craig, E. A. (2018). Hsp70 at the membrane: Driving protein translocation. BMC Biol. 16, 11. doi:10.1186/s12915-017-0474-3
Cui, X., Li, Z., Piao, J., Li, J., Li, L., Lin, Z., et al. (2017). Mortalin expression in pancreatic cancer and its clinical and prognostic significance. Hum. Pathol. 64, 171–178. doi:10.1016/j.humpath.2017.03.015
D'Eletto, M., Rossin, F., Occhigrossi, L., Farrace, M. G., Faccenda, D., Desai, R., et al. (2018). Transglutaminase type 2 regulates ER-mitochondria contact sites by interacting with GRP75. Cell Rep. 25, 3573–3581. doi:10.1016/j.celrep.2018.11.094
Dahlseid, J. N., Lill, R., Green, J. M., Xu, X., Qiu, Y., and Pierce, S. K. (1994). PBP74, a new member of the mammalian 70-kDa heat shock protein family, is a mitochondrial protein. Mol. Biol. Cell 5, 1265–1275. doi:10.1091/mbc.5.11.1265
Dai, Y., Li, F., Jiao, Y., Wang, G., Zhan, T., Xia, Y., et al. (2021). Mortalin/glucose-regulated protein 75 promotes the cisplatin-resistance of gastric cancer via regulating anti-oxidation/apoptosis and metabolic reprogramming. Cell Death Discov. 7, 140. doi:10.1038/s41420-021-00517-w
De Maio, A., and Hightower, L. (2021). The interaction of heat shock proteins with cellular membranes: A historical perspective. Cell Stress Chaperones 26, 769–783. doi:10.1007/s12192-021-01228-y
De Maio, A., Santoro, M. G., Tanguay, R. M., and Hightower, L. E. (2012). Ferruccio ritossa's scientific legacy 50 years after his discovery of the heat shock response: A new view of biology, a new society, and a new journal. Cell Stress Chaperones 17, 139–143. doi:10.1007/s12192-012-0320-z
De Maio, A., and Vazquez, D. (2013). Extracellular heat shock proteins: A new location, a new function. Shock 40, 239–246. doi:10.1097/SHK.0b013e3182a185ab
Deocaris, C. C., Kaul, S. C., and Wadhwa, R. (2008). From proliferative to neurological role of an hsp70 stress chaperone, mortalin. Biogerontology 9, 391–403. doi:10.1007/s10522-008-9174-2
Deocaris, C. C., Kaul, S. C., and Wadhwa, R. (2006). On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress & Chaperones 11, 116–128. doi:10.1379/csc-144r.1
Dundas, S. R., Lawrie, L. C., Rooney, P. H., and Murray, G. I. (2005). Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J. Pathol. 205, 74–81. doi:10.1002/path.1672
Dutkiewicz, R., Schilke, B., Knieszner, H., Walter, W., Craig, E. A., and Marszalek, J. (2003). Ssq1, a mitochondrial Hsp70 involved in iron-sulfur (Fe/S) center biogenesis. Similarities to and differences from its bacterial counterpart. J. Biol. Chem. 278, 29719–29727. doi:10.1074/jbc.M303527200
Elwakeel, A. (2022). Abrogating the interaction between p53 and mortalin (Grp75/HSPA9/mtHsp70) for cancer therapy: The story so far. Front. Cell Dev. Biol. 10, 879632. doi:10.3389/fcell.2022.879632
Emmings, E., Mullany, S., Chang, Z., Landen, C. N., Linder, S., and Bazzaro, M. (2019). Targeting mitochondria for treatment of chemoresistant ovarian cancer. Int. J. Mol. Sci. 20, 229. doi:10.3390/ijms20010229
Fadeeva, N. P., Antipova, N. V., Shender, V. O., Anufrieva, K. S., Stepanov, G. A., Bastola, S., et al. (2018). Identification of novel interaction partners of AIF protein on the outer mitochondrial membrane. Acta Naturae 10, 100–109. doi:10.32607/20758251-2018-10-4-100-109
Fasolato, S., Ruvoletto, M., Nardo, G., Rasola, A., Sciacovelli, M., Zanus, G., et al. (2021). Low P66shc with high SerpinB3 levels favors necroptosis and better survival in hepatocellular carcinoma. Biol. (Basel) 10, 363. doi:10.3390/biology10050363
Feng, S., Huang, Q., Deng, J., Jia, W., Gong, J., Xie, D., et al. (2022). DAB2IP suppresses tumor malignancy by inhibiting GRP75-driven p53 ubiquitination in colon cancer. Cancer Lett. 532, 215588. doi:10.1016/j.canlet.2022.215588
Fishelson, Z., Donin, N., Zell, S., Schultz, S., and Kirschfink, M. (2003). Obstacles to cancer immunotherapy: Expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol. Immunol. 40, 109–123. doi:10.1016/s0161-5890(03)00112-3
Gambill, B. D., Voos, W., Kang, P. J., Miao, B., Langer, T., Craig, E. A., et al. (1993). A dual role for mitochondrial heat shock protein 70 in membrane translocation of preproteins. J. Cell Biol. 123, 109–117. doi:10.1083/jcb.123.1.109
Gestl, E. E., and Anne Bottger, S. (2012). Cytoplasmic sequestration of the tumor suppressor p53 by a heat shock protein 70 family member, mortalin, in human colorectal adenocarcinoma cell lines. Biochem. Biophys. Res. Commun. 423, 411–416. doi:10.1016/j.bbrc.2012.05.139
Glaessgen, A., Jonmarker, S., Lindberg, A., Nilsson, B., Lewensohn, R., Ekman, P., et al. (2008). Heat shock proteins 27, 60 and 70 as prognostic markers of prostate cancer. Apmis 116, 888–895. doi:10.1111/j.1600-0463.2008.01051.x
Goswami, A. V., Chittoor, B., and D'Silva, P. (2010). Understanding the functional interplay between mammalian mitochondrial Hsp70 chaperone machine components. J. Biol. Chem. 285, 19472–19482. doi:10.1074/jbc.M110.105957
Guha, M., Srinivasan, S., Ruthel, G., Kashina, A. K., Carstens, R. P., Mendoza, A., et al. (2014). Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene 33, 5238–5250. doi:10.1038/onc.2013.467
Han, S. Y., Jeong, Y. J., Choi, Y., Hwang, S. K., Bae, Y. S., and Chang, Y. C. (2018). Mitochondrial dysfunction induces the invasive phenotype, and cell migration and invasion, through the induction of AKT and AMPK pathways in lung cancer cells. Int. J. Mol. Med. 42, 1644–1652. doi:10.3892/ijmm.2018.3733
Hansen, G. A., Ludvigsen, M., Jacobsen, C., Cangemi, C., Rasmussen, L. M., Vorum, H., et al. (2015). Fibulin-1C, C1 esterase inhibitor and glucose regulated protein 75 interact with the CREC proteins, calumenin and reticulocalbin. PLoS One 10, e0132283. doi:10.1371/journal.pone.0132283
Haslem, L., Hays, J. M., and Hays, F. A. (2022). p66Shc in cardiovascular pathology. Cells 11, 1855. doi:10.3390/cells11111855
Hemminki, K., Santi, I., Weires, M., Thomsen, H., Sundquist, J., and Bermejo, J. L. (2010). Tumor location and patient characteristics of colon and rectal adenocarcinomas in relation to survival and TNM classes. BMC Cancer 10, 688. doi:10.1186/1471-2407-10-688
Hu, Y., Yang, L., Yang, Y., Han, Y., Wang, Y., Liu, W., et al. (2016). Oncogenic role of mortalin contributes to ovarian tumorigenesis by activating the MAPK-ERK pathway. J. Cell Mol. Med. 20, 2111–2121. doi:10.1111/jcmm.12905
Huang, M. B., Xia, M., Gao, Z., Zhou, H., Liu, M., Huang, S., et al. (2019). Characterization of exosomes in plasma of patients with breast, ovarian, prostate, hepatic, gastric, colon, and pancreatic cancers. J. Cancer Ther. 10, 382–399. doi:10.4236/jct.2019.105032
Hwang, J. T., Lee, A., and Kho, C. (2022). Ubiquitin and ubiquitin-like proteins in cancer, neurodegenerative disorders, and heart diseases. Int. J. Mol. Sci. 23, 5053. doi:10.3390/ijms23095053
Ingemann, L., and Kirkegaard, T. (2014). Lysosomal storage diseases and the heat shock response: Convergences and therapeutic opportunities. J. Lipid Res. 55, 2198–2210. doi:10.1194/jlr.R048090
Iosefson, O., Sharon, S., Goloubinoff, P., and Azem, A. (2012). Reactivation of protein aggregates by mortalin and Tid1-the human mitochondrial Hsp70 chaperone system. Cell Stress Chaperones 17, 57–66. doi:10.1007/s12192-011-0285-3
Javid, H., Hashemian, P., Yazdani, S., Sharbaf Mashhad, A., and Karimi-Shahri, M. (2022). The role of heat shock proteins in metastatic colorectal cancer: A review. J. Cell Biochem. 123, 1704–1735. doi:10.1002/jcb.30326
Jee, H. (2016). Size dependent classification of heat shock proteins: A mini-review. J. Exerc Rehabil. 12, 255–259. doi:10.12965/jer.1632642.321
Kampinga, H. H., Hageman, J., Vos, M. J., Kubota, H., Tanguay, R. M., Bruford, E. A., et al. (2009). Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105–111. doi:10.1007/s12192-008-0068-7
Kaul, S. C., Aida, S., Yaguchi, T., Kaur, K., and Wadhwa, R. (2005). Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J. Biol. Chem. 280, 39373–39379. doi:10.1074/jbc.M500022200
Khan, M. R., Khan, M. S., Ahmed, A., Malik, A., and Qamar, W. (2020). Optimization of expression and purification of mitochondrial HSP 40 (Tid1-L) chaperone: Role of mortalin and tid1 in the reactivation and amyloid inhibition of proteins. Saudi J. Biol. Sci. 27, 3099–3105. doi:10.1016/j.sjbs.2020.09.006
Komarova, E. Y., Suezov, R. V., Nikotina, A. D., Aksenov, N. D., Garaeva, L. A., Shtam, T. A., et al. (2021). Hsp70-containing extracellular vesicles are capable of activating of adaptive immunity in models of mouse melanoma and colon carcinoma. Sci. Rep. 11, 21314. doi:10.1038/s41598-021-00734-4
Kosti, I., Jain, N., Aran, D., Butte, A. J., and Sirota, M. (2016). Cross-tissue analysis of gene and protein expression in normal and cancer tissues. Sci. Rep. 6, 24799. doi:10.1038/srep24799
Kumar, V., Roy, S., Behera, B. K., and Das, B. K. (2022). Heat shock proteins (hsps) in cellular homeostasis: A promising tool for Health management in Crustacean aquaculture. Life (Basel) 12, 1777. doi:10.3390/life12111777
Lancaster, G. I., and Febbraio, M. A. (2005). Exosome-dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 280, 23349–23355. doi:10.1074/jbc.M502017200
Laufen, T., Mayer, M. P., Beisel, C., Klostermeier, D., Mogk, A., Reinstein, J., et al. (1999). Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. U. S. A. 96, 5452–5457. doi:10.1073/pnas.96.10.5452
Lee, Y. C., Lee, Y. L., Chuang, J. P., and Lee, J. C. (2013). Differences in survival between colon and rectal cancer from SEER data. PLoS One 8, e78709. doi:10.1371/journal.pone.0078709
Li, C. H., Haider, S., Shiah, Y. J., Thai, K., and Boutros, P. C. (2018). Sex differences in cancer driver genes and biomarkers. Cancer Res. 78, 5527–5537. doi:10.1158/0008-5472.CAN-18-0362
Li, D. Y., Liang, S., Wen, J. H., Tang, J. X., Deng, S. L., and Liu, Y. X. (2022). Extracellular HSPs: The potential target for human disease therapy. Molecules 27, 2361. doi:10.3390/molecules27072361
Lin, C. S., Liu, L. T., Ou, L. H., Pan, S. C., Lin, C. I., and Wei, Y. H. (2018). Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol. Rep. 39, 316–330. doi:10.3892/or.2017.6087
Liu, L. X., Lu, J. C., Zeng, H. Y., Cai, J. B., Zhang, P. F., Guo, X. J., et al. (2019a). Mortalin stabilizes CD151-depedent tetraspanin-enriched microdomains and implicates in the progression of hepatocellular carcinoma. J. Cancer 10, 6199–6206. doi:10.7150/jca.36301
Liu, T., Biddle, D., Hanks, A. N., Brouha, B., Yan, H., Lee, R. M., et al. (2006). Activation of dual apoptotic pathways in human melanocytes and protection by survivin. J. Invest. Dermatol 126, 2247–2256. doi:10.1038/sj.jid.5700381
Liu, Y., Ma, X., Fujioka, H., Liu, J., Chen, S., and Zhu, X. (2019b). DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. U. S. A. 116, 25322–25328. doi:10.1073/pnas.1906565116
Lobingier, B. T., Hüttenhain, R., Eichel, K., Miller, K. B., Ting, A. Y., Von Zastrow, M., et al. (2017). An approach to spatiotemporally resolve protein interaction networks in living cells. Cell 169, 350–360. doi:10.1016/j.cell.2017.03.022
Londono, C., Osorio, C., Gama, V., and Alzate, O. (2012). Mortalin, apoptosis, and neurodegeneration. Biomolecules 2, 143–164. doi:10.3390/biom2010143
Lu, W. J., Lee, N. P., Kaul, S. C., Lan, F., Poon, R. T., Wadhwa, R., et al. (2011). Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 18, 1046–1056. doi:10.1038/cdd.2010.177
Luo, W. I., Dizin, E., Yoon, T., and Cowan, J. A. (2010). Kinetic and structural characterization of human mortalin. Protein Expr. Purif. 72, 75–81. doi:10.1016/j.pep.2010.02.003
Ma, Z., Izumi, H., Kanai, M., Kabuyama, Y., Ahn, N. G., and Fukasawa, K. (2006). Mortalin controls centrosome duplication via modulating centrosomal localization of p53. Oncogene 25, 5377–5390. doi:10.1038/sj.onc.1209543
Marzec, M., Eletto, D., and Argon, Y. (2012). GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 1823, 774–787. doi:10.1016/j.bbamcr.2011.10.013
Matsumura, Y., Sakai, J., and Skach, W. R. (2013). Endoplasmic reticulum protein quality control is determined by cooperative interactions between hsp/c70 protein and the CHIP E3 ligase. J. Biol. Chem. 288, 31069–31079. doi:10.1074/jbc.M113.479345
Mazkereth, N., Rocca, F., Schubert, J. R., Geisler, C., Hillman, Y., Egner, A., et al. (2016). Complement triggers relocation of Mortalin/GRP75 from mitochondria to the plasma membrane. Immunobiology 221, 1395–1406. doi:10.1016/j.imbio.2016.07.005
Mccallister, C., Siracusa, M. C., Shirazi, F., Chalkia, D., and Nikolaidis, N. (2015). Functional diversification and specialization of cytosolic 70-kDa heat shock proteins. Sci. Rep. 5, 9363. doi:10.1038/srep09363
Merrick, B. A., Walker, V. R., He, C., Patterson, R. M., and Selkirk, J. K. (1997). Induction of novel Grp75 isoforms by 2-deoxyglucose in human and murine fibroblasts. Cancer Lett. 119, 185–190. doi:10.1016/s0304-3835(97)00270-x
Mizukoshi, E., Suzuki, M., Loupatov, A., Uruno, T., Hayashi, H., Misono, T., et al. (1999). Fibroblast growth factor-1 interacts with the glucose-regulated protein GRP75/mortalin. Biochem. J. 343 (2), 461–466. doi:10.1042/bj3430461
Moro, F., and Muga, A. (2006). Thermal adaptation of the yeast mitochondrial Hsp70 system is regulated by the reversible unfolding of its nucleotide exchange factor. J. Mol. Biol. 358, 1367–1377. doi:10.1016/j.jmb.2006.03.027
Muranova, L. K., Shatov, V. M., and Gusev, N. B. (2022). Role of small heat shock proteins in the remodeling of actin microfilaments. Biochem. (Mosc) 87, 800–811. doi:10.1134/S0006297922080119
Na, Y., Kaul, S. C., Ryu, J., Lee, J. S., Ahn, H. M., Kaul, Z., et al. (2016). Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 76, 2754–2765. doi:10.1158/0008-5472.CAN-15-2704
Nigam, N., Grover, A., Goyal, S., Katiyar, S. P., Bhargava, P., Wang, P. C., et al. (2015). Targeting mortalin by embelin causes activation of tumor suppressor p53 and deactivation of metastatic signaling in human breast cancer cells. PLoS One 10, e0138192. doi:10.1371/journal.pone.0138192
Orsini, F., Migliaccio, E., Moroni, M., Contursi, C., Raker, V. A., Piccini, D., et al. (2004). The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem. 279, 25689–25695. doi:10.1074/jbc.M401844200
Orsini, F., Moroni, M., Contursi, C., Yano, M., Pelicci, P., Giorgio, M., et al. (2006). Regulatory effects of the mitochondrial energetic status on mitochondrial p66Shc. Biol. Chem. 387, 1405–1410. doi:10.1515/BC.2006.176
Pilzer, D., and Fishelson, Z. (2005). Mortalin/GRP75 promotes release of membrane vesicles from immune attacked cells and protection from complement-mediated lysis. Int. Immunol. 17, 1239–1248. doi:10.1093/intimm/dxh300
Porporato, P. E., Filigheddu, N., Pedro, J. M. B., Kroemer, G., and Galluzzi, L. (2018). Mitochondrial metabolism and cancer. Cell Res. 28, 265–280. doi:10.1038/cr.2017.155
Priyanka, , , and Seth, P. (2022). Insights into the role of mortalin in alzheimer's disease, Parkinson's disease, and HIV-1-Associated neurocognitive disorders. Front. Cell Dev. Biol. 10, 903031. doi:10.3389/fcell.2022.903031
Putri, J. F., Bhargava, P., Dhanjal, J. K., Yaguchi, T., Sundar, D., Kaul, S. C., et al. (2019). Mortaparib, a novel dual inhibitor of mortalin and PARP1, is a potential drug candidate for ovarian and cervical cancers. J. Exp. Clin. Cancer Res. 38, 499. doi:10.1186/s13046-019-1500-9
Radons, J. (2016). The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 21, 379–404. doi:10.1007/s12192-016-0676-6
Rahman, M., Jackson, L. K., Johnson, W. E., Li, D. Y., Bild, A. H., and Piccolo, S. R. (2015). Alternative preprocessing of RNA-Sequencing data in the Cancer Genome Atlas leads to improved analysis results. Bioinformatics 31, 3666–3672. doi:10.1093/bioinformatics/btv377
Rakovic, A., Grünewald, A., Voges, L., Hofmann, S., Orolicki, S., Lohmann, K., et al. (2011). PINK1-Interacting proteins: Proteomic analysis of overexpressed PINK1. Park. Dis. 2011, 153979. doi:10.4061/2011/153979
Rezvani, K., Teng, Y., Pan, Y., Dani, J. A., Lindstrom, J., Garcia Gras, E. A., et al. (2009). UBXD4, a UBX-containing protein, regulates the cell surface number and stability of alpha3-containing nicotinic acetylcholine receptors. J. Neurosci. 29, 6883–6896. doi:10.1523/JNEUROSCI.4723-08.2009
Ritossa, F. (1962). A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 18, 571–573. doi:10.1007/bf02172188
Rozenberg, P., Kocsis, J., Saar, M., Prohaszka, Z., Fust, G., and Fishelson, Z. (2013). Elevated levels of mitochondrial mortalin and cytosolic HSP70 in blood as risk factors in patients with colorectal cancer. Int. J. Cancer 133, 514–518. doi:10.1002/ijc.28029
Rozenberg, P., Ziporen, L., Gancz, D., Saar-Ray, M., and Fishelson, Z. (2018). Cooperation between Hsp90 and mortalin/GRP75 in resistance to cell death induced by complement C5b-9. Cell Death Dis. 9, 150. doi:10.1038/s41419-017-0240-z
Ryu, J., Kaul, Z., Yoon, A. R., Liu, Y., Yaguchi, T., Na, Y., et al. (2014). Identification and functional characterization of nuclear mortalin in human carcinogenesis. J. Biol. Chem. 289, 24832–24844. doi:10.1074/jbc.M114.565929
Saar Ray, M., Moskovich, O., Iosefson, O., and Fishelson, Z. (2014). Mortalin/Grp75 binds to complement C9 and plays a role in resistance to complement-dependent cytotoxicity. J. Biol. Chem. 289, 15014–15022. doi:10.1074/jbc.M114.552406
Sacht, G., Brigelius-Flohé, R., Kiess, M., Sztajer, H., and Flohé, L. (1999). ATP-sensitive association of mortalin with the IL-1 receptor type I. Biofactors 9, 49–60. doi:10.1002/biof.5520090107
Sadej, R., Grudowska, A., Turczyk, L., Kordek, R., and Romanska, H. M. (2014). CD151 in cancer progression and metastasis: A complex scenario. Lab. Invest. 94, 41–51. doi:10.1038/labinvest.2013.136
Sane, S., Abdullah, A., Boudreau, D. A., Autenried, R. K., Gupta, B. K., Wang, X., et al. (2014). Ubiquitin-like (UBX)-domain-containing protein, UBXN2A, promotes cell death by interfering with the p53-Mortalin interactions in colon cancer cells. Cell Death Dis. 5, e1118. doi:10.1038/cddis.2014.100
Sane, S., Abdullah, A., Nelson, M. E., Wang, H., Chauhan, S. C., Newton, S. S., et al. (2016). Structural studies of UBXN2A and mortalin interaction and the putative role of silenced UBXN2A in preventing response to chemotherapy. Cell Stress Chaperones 2, 313–326. doi:10.1007/s12192-015-0661-5
Sane, S., Hafner, A., Srinivasan, R., Masood, D., Slunecka, J. L., Noldner, C. J., et al. (2018). UBXN2A enhances CHIP-mediated proteasomal degradation of oncoprotein mortalin-2 in cancer cells. Mol. Oncol. 12, 1753–1777. doi:10.1002/1878-0261.12372
Sanjuán Szklarz, L. K., Guiard, B., Rissler, M., Wiedemann, N., Kozjak, V., Van der Laan, M., et al. (2005). Inactivation of the mitochondrial heat shock protein zim17 leads to aggregation of matrix hsp70s followed by pleiotropic effects on morphology and protein biogenesis. J. Mol. Biol. 351, 206–218. doi:10.1016/j.jmb.2005.05.068
Schneider, G., Schmidt-Supprian, M., Rad, R., and Saur, D. (2017). Tissue-specific tumorigenesis: Context matters. Nat. Rev. Cancer 17, 239–253. doi:10.1038/nrc.2017.5
Shaulsky, G., Goldfinger, N., Tosky, M. S., Levine, A. J., and Rotter, V. (1991). Nuclear localization is essential for the activity of p53 protein. Oncogene 6, 2055–2065.
Sun, B., Li, G., Yu, Q., Liu, D., and Tang, X. (2022). HSP60 in cancer: A promising biomarker for diagnosis and a potentially useful target for treatment. J. Drug Target 30, 31–45. doi:10.1080/1061186X.2021.1920025
Sun, J., Che, S. L., Piao, J. J., Xu, M., Chen, L. Y., and Lin, Z. H. (2017). Mortalin overexpression predicts poor prognosis in early stage of non-small cell lung cancer. Tumour Biol. 39, 1010428317695918. doi:10.1177/1010428317695918
Szabadkai, G., Bianchi, K., Várnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi:10.1083/jcb.200608073
Tai-Nagara, I., Matsuoka, S., Ariga, H., and Suda, T. (2014). Mortalin and DJ-1 coordinately regulate hematopoietic stem cell function through the control of oxidative stress. Blood 123, 41–50. doi:10.1182/blood-2013-06-508333
Takano, S., Wadhwa, R., Mitsui, Y., and Kaul, S. C. (2001). Identification and characterization of molecular interactions between glucose-regulated proteins (GRPs) mortalin/GRP75/peptide-binding protein 74 (PBP74) and GRP94. Biochem. J. 357, 393–398. doi:10.1042/0264-6021:3570393
Teng, Y., Rezvani, K., and De Biasi, M. (2015). UBXN2A regulates nicotinic receptor degradation by modulating the E3 ligase activity of CHIP. Biochem. Pharmacol. 97, 518–530. doi:10.1016/j.bcp.2015.08.084
Thul, P. J., Åkesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal, H., et al. (2017). A subcellular map of the human proteome. Science 356, eaal3321. doi:10.1126/science.aal3321
Tissières, A., Mitchell, H. K., Tracy, U. M., and Tissieres, A. (1974). Protein synthesis in salivary glands of Drosophila melanogaster: Relation to chromosome puffs. J. Mol. Biol. 84, 389–398. doi:10.1016/0022-2836(74)90447-1
Tsai, C. H., Weng, J. R., Lin, H. W., Lu, M. T., Liu, Y. C., and Chu, P. C. (2022). Targeting triple negative breast cancer stem cells by heat shock protein 70 inhibitors. Cancers (Basel) 14. doi:10.3390/cancers14194898
Tukaj, S. (2020). Heat shock protein 70 as a double agent acting inside and outside the cell: Insights into autoimmunity. Int. J. Mol. Sci. 21, 5298. doi:10.3390/ijms21155298
Uhlen, M., Zhang, C., Lee, S., Sjöstedt, E., Fagerberg, L., Bidkhori, G., et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357, eaan2507. doi:10.1126/science.aan2507
Voisine, C., Craig, E. A., Zufall, N., Von Ahsen, O., Pfanner, N., and Voos, W. (1999). The protein import motor of mitochondria: Unfolding and trapping of preproteins are distinct and separable functions of matrix Hsp70. Cell 97, 565–574. doi:10.1016/s0092-8674(00)80768-0
Wadhwa, R., Ando, H., Kawasaki, H., Taira, K., and Kaul, S. C. (2003a). Targeting mortalin using conventional and RNA-helicase-coupled hammerhead ribozymes. EMBO Rep. 4, 595–601. doi:10.1038/sj.embor.embor855
Wadhwa, R., Colgin, L., Yaguchi, T., Taira, K., Reddel, R. R., and Kaul, S. C. (2002). Rhodacyanine dye MKT-077 inhibits in vitro telomerase assay but has no detectable effects on telomerase activity in vivo. Cancer Res. 62, 4434–4438.
Wadhwa, R., Sugihara, T., Yoshida, A., Nomura, H., Reddel, R. R., Simpson, R., et al. (2000). Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 60, 6818–6821.
Wadhwa, R., Takano, S., Kaur, K., Aida, S., Yaguchi, T., Kaul, Z., et al. (2005). Identification and characterization of molecular interactions between mortalin/mtHsp70 and HSP60. Biochem. J. 391, 185–190. doi:10.1042/BJ20050861
Wadhwa, R., Takano, S., Kaur, K., Deocaris, C. C., Pereira-Smith, O. M., Reddel, R. R., et al. (2006). Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 118, 2973–2980. doi:10.1002/ijc.21773
Wadhwa, R., Yaguchi, T., Hasan, M. K., Taira, K., and Kaul, S. C. (2003b). Mortalin-MPD (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem. Biophys. Res. Commun. 302, 735–742. doi:10.1016/s0006-291x(03)00226-2
Wang, M., Luan, S., Fan, X., Wang, J., Huang, J., Gao, X., et al. (2022). The emerging multifaceted role of PINK1 in cancer biology. Cancer Sci. 113, 4037–4047. doi:10.1111/cas.15568
Wei, B., Cao, J., Tian, J. H., Yu, C. Y., Huang, Q., Yu, J. J., et al. (2021). Mortalin maintains breast cancer stem cells stemness via activation of Wnt/GSK3β/β-catenin signaling pathway. Am. J. Cancer Res. 11, 2696–2716.
Wu, P. K., Hong, S. K., Veeranki, S., Karkhanis, M., Starenki, D., Plaza, J. A., et al. (2013). A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol. Cell Biol. 33, 4051–4067. doi:10.1128/MCB.00021-13
Wu, Y., Ding, Y., Zheng, X., and Liao, K. (2019). The molecular chaperone Hsp90 maintains Golgi organization and vesicular trafficking by regulating microtubule stability. J. Mol. Cell Biol. 12, 448–461. doi:10.1093/jmcb/mjz093
Wyciszkiewicz, A., Kalinowska-Łyszczarz, A., Nowakowski, B., Kaźmierczak, K., Osztynowicz, K., and Michalak, S. (2019). Expression of small heat shock proteins in exosomes from patients with gynecologic cancers. Sci. Rep. 9, 9817. doi:10.1038/s41598-019-46221-9
Xu, H., Guan, N., Ren, Y. L., Wei, Q. J., Tao, Y. H., Yang, G. S., et al. (2018). IP(3)R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 19, 140. doi:10.1186/s12882-018-0940-3
Xu, M., Jin, T., Chen, L., Zhang, X., Zhu, G., Wang, Q., et al. (2019). Mortalin is a distinct bio-marker and prognostic factor in serous ovarian carcinoma. Gene 696, 63–71. doi:10.1016/j.gene.2019.02.033
Xu, M., Zhang, Y., Cui, M., Wang, X., and Lin, Z. (2020). Mortalin contributes to colorectal cancer by promoting proliferation and epithelial-mesenchymal transition. IUBMB Life 72, 771–781. doi:10.1002/iub.2176
Yamamoto, H., Momose, T., Yatsukawa, Y., Ohshima, C., Ishikawa, D., Sato, T., et al. (2005). Identification of a novel member of yeast mitochondrial Hsp70-associated motor and chaperone proteins that facilitates protein translocation across the inner membrane. FEBS Lett. 579, 507–511. doi:10.1016/j.febslet.2004.12.018
Yang, L., Guo, W., Zhang, Q., Li, H., Liu, X., Yang, Y., et al. (2011). Crosstalk between Raf/MEK/ERK and PI3K/AKT in suppression of Bax conformational change by Grp75 under glucose deprivation conditions. J. Mol. Biol. 414, 654–666. doi:10.1016/j.jmb.2011.09.009
Yi, X., Luk, J. M., Lee, N. P., Peng, J., Leng, X., Guan, X. Y., et al. (2008). Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol. Cell Proteomics 7, 315–325. doi:10.1074/mcp.M700116-MCP200
Yoo, J. Y., Ryu, J., Gao, R., Yaguchi, T., Kaul, S. C., Wadhwa, R., et al. (2010). Tumor suppression by apoptotic and anti-angiogenic effects of mortalin-targeting adeno-oncolytic virus. J. Gene Med. 12, 586–595. doi:10.1002/jgm.1471
Yun, C. O., Bhargava, P., Na, Y., Lee, J. S., Ryu, J., Kaul, S. C., et al. (2017). Relevance of mortalin to cancer cell stemness and cancer therapy. Sci. Rep. 7, 42016. doi:10.1038/srep42016
Zhang, P., Leu, J. I., Murphy, M. E., George, D. L., and Marmorstein, R. (2014). Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PLoS One 9, e103518. doi:10.1371/journal.pone.0103518
Zhang, R., Meng, Z., Wu, X., Zhang, M., Zhang, S., and Jin, T. (2021). Mortalin promotes breast cancer malignancy. Exp. Mol. Pathol. 118, 104593. doi:10.1016/j.yexmp.2020.104593
Keywords: mortalin (HSPA9), cancer, protein partners, cellular localization, post-translational modification (PTM)
Citation: Esfahanian N, Knoblich CD, Bowman GA and Rezvani K (2023) Mortalin: Protein partners, biological impacts, pathological roles, and therapeutic opportunities. Front. Cell Dev. Biol. 11:1028519. doi: 10.3389/fcell.2023.1028519
Received: 26 August 2022; Accepted: 23 January 2023;
Published: 02 February 2023.
Edited by:
Venkaiah Betapudi, United States Department of Health and Human Services, United StatesReviewed by:
Zvi Fishelson, Tel Aviv University, IsraelCopyright © 2023 Esfahanian, Knoblich, Bowman and Rezvani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khosrow Rezvani, a2hvc3Jvdy5yZXp2YW5pQHVzZC5lZHU=
†These authors have contributed equally to this work
‡Present address: Niki Esfahanian, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.