 Xian-Tao Li1,2*
Xian-Tao Li1,2*- 1School of Medicine, Guizhou University, Guiyang, China
- 2Department of Neuroscience, South-Central University for Nationalities, Wuhan, China
Estrogens, the sex hormones, have the potential to govern multiple cellular functions, such as proliferation, apoptosis, differentiation, and homeostasis, and to exert numerous beneficial influences for the cardiovascular system, nervous system, and bones in genomic and/or non-genomic ways. Converging evidence indicates that estrogens serve a crucial role in counteracting neurodegeneration and ischemic injury; they are thereby being considered as a potent neuroprotectant for preventing neurological diseases such as Alzheimer’s disease and stroke. The underlying mechanism of neuroprotective effects conferred by estrogens is thought to be complex and multifactorial, and it remains obscure. It is well established that the K+ channels broadly expressed in a variety of neural subtypes determine the essential physiological features of neuronal excitability, and dysfunction of these channels is closely associated with diverse brain deficits, such as ataxia and epilepsy. A growing body of evidence supports a neuroprotective role of K+ channels in malfunctions of nervous tissues, with the channels even being a therapeutic target in clinical trials. As multitarget steroid hormones, estrogens also regulate the activity of distinct K+ channels to generate varying biological actions, and accumulated data delineate that some aspects of estrogen-mediated neuroprotection may arise from the impact on multiple K+ channels, including Kv, BK, KATP, and K2P channels. The response of these K+ channels after acute or chronic exposure to estrogens may oppose pathological abnormality in nervous cells, which serves to extend our understanding of these phenomena.
Introduction
Estrogens, a group of sex steroid hormones that are majorly synthesized in the ovary, serve an important role in tuning physiological and pathophysiological functions of multiple tissues, such as the heart and brain. In the clinic, therefore, hormone replacement therapy (HRT) with estrogens exhibits numerous beneficial influences on postmenopausal women, including reducing cardiovascular incidence (Mendelsohn and Karas, 2007), protecting cognitive functions (Brinton, 2001), and preventing the occurrence of osteoporosis (Ikeda et al., 2019). Nevertheless, clinical studies reveal that the administration of estrogens is capable of enhancing the risk of breast, ovarian, and endometrial cancer (Cheskis et al., 2007), with several of the biggest negative effects of this therapy substantially threatening woman’s health. In order to avoid the adverse effects of HRT on the human body, selective estrogen receptor modulators (SERMs), such as tamoxifen and raloxifene, have been developed to impact selective target cells or tissues, exerting the therapeutic benefits without the proliferative action (Cheskis et al., 2007).
It is well established that two identified subtypes of nuclear estrogen receptors (ERs), ERα and ERβ, are responsible for estrogen-induced biological effects on distinct tissues. In the classical model, ERs bind to estrogen response elements (EREs), which reside in the promoters of specific genes, and subsequently regulate the related gene expression (Björnström and Sjöberg, 2005). Apart from the genomic actions, membrane-associated ERs enable rapid non-genomic actions, thereby activating protein-kinase cascades (Lösel and Wehling, 2003) or tuning other targets such as K+ channels (Tanabe et al., 1999).
A great deal of evidence indicates that the normal functions of the brain, such as emotion, memory, and cognition, are noticeably influenced by sex steroid estrogens throughout life (Taxier et al., 2020). Interestingly, these ovarian hormones impact not only female but also male sexual behaviors by modulating neuronal functions (Taziaux et al., 2007; Taxier et al., 2020). Clinical data have revealed that treatment with estrogens leads to the improvement of menopause-related depressive symptoms (Toffol et al., 2015); moreover, neuroprotective effects are substantially evident after estrogen-containing hormone therapy by researchers, which exhibits a positive action in the broad spectrum of nervous malfunctions including cognitive aging and brain injury (Engler-Chiurazzi et al., 2017). Estrogens conduct multiple actions on the structure and function of the hippocampus, a key brain region for learning and memory. These hormones are able to generate enhanced long-term potentiation by increasing dendritic spine density and the ratio of NMDA transmission to AMPA transmission in the hippocampal neurons (Smith and McMahon, 2005). ERβ activation can boost the dendritic branching and density of mushroom-type spines in these neurons, resulting in improving hippocampus-dependent cognition (Liu et al., 2008). The critical role of the hippocampus in mediating the memory-enhancing effects of estrogen is well established (Tuscher et al., 2019). Estrogen deficiency leads to enhanced hippocampal autophagy and promotes apoptosis in the hippocampus, consequently inducing memory loss (Pandey et al., 2020). Modulation of the hippocampal function by estrogens contributes to the estrogenic facilitation of learning and memory through diverse aspects, such as intracellular cascade, dendritic spinogenesis, and synaptic function and plasticity, and it protects hippocampal cells against excitotoxicity arising from drugs (Taxier et al., 2020). 17β-Estradiol (E2), an isomer of estradiol that is the most potent estrogen, is able to induce upregulation of the expression of neuroprotectant neuroglobin (NGB) to counteract H2O2-induced apoptosis (De Marinis et al., 2013; Barreto et al., 2021). Exposure to 17β-estradiol also protects the brain against reperfusion injury after acute ischemic insults by reducing lesion formation and brain atrophy, as well as improving cerebral blood flow in rat ischemic stroke (Carpenter et al., 2016). In global ischemia, treatment with 17β-estradiol prevents the loss of CA1 hippocampal neurons in response to ischemic injury (Shughrue and Merchenthaler, 2003), and neuroprotection of estrogen on hippocampal neurons can be achieved by enabling ERα and ERβ (Miller et al., 2005). On the other hand, neuron-derived 17β-estradiol (E2), which is produced locally by the neurons and which is essential for governing synaptic plasticity and cognitive function (Lu et al., 2019), also displays neuroprotective effects through induction of astrocyte activation, which in turn facilitates the release of the neurotrophic factors, BDNF and IGF-1, and the clearance of neurotoxic glutamate after ischemic brain injury (Lu et al., 2020). Collectively, the accrued evidence provides insight that the neuroprotective actions conferred by estrogens are mediated in multiple ways, including antagonizing inflammatory injuries, Aβ toxicity, and oxidative insults, as well as maintaining mitochondrial membrane integrity and calcium homeostasis (Simpkins et al., 2009). Apparently, the interaction of estrogens with multiple targets is needed to support the profound role of this female hormone in protecting the nervous system, and a growing body of evidence has suggested that the K+ channel is as a critical mediator involved in the neuroprotective benefits induced by estrogens, providing an alternative mechanistic explanation.
K+ channels that selectively conduct K+ ions are ubiquitously distributed in non-excitable and excitable tissues, with the latter including cardiomyocytes, neurons, and pancreatic β-cells, and they serve crucial roles in regulating a variety of biological processes, such as proliferation, apoptosis, cellular volume, and secretion (Shieh et al., 2000). In the central and peripheral nervous system, K+ channels, especially voltage-gated K+ (Kv) channels, are important to set the resting membrane potential, shape the action potential, and tune the firing frequency, contributing to neuronal excitability, synaptic transmission, and neurotransmitter release (Shah and Aizenman, 2014). It is well established that K+ channels are closely associated with varying neurological disorders, and dysfunction of these channels can participate in the development of human neurological disease, such as Alzheimer’s disease (AD) and epilepsy (Djillani et al., 2019; Villa et al., 2020). For instance, the mutated KCNQ2 and KCNQ3 genes, which encode Kv7.2 and Kv7.3, respectively, enable benign familial neonatal convulsions (Maljevic and Lerche, 2014), and genetic suppression of small-conductance Ca2+-activated K+ channels (SK) in deep cerebellar neurons can result in ataxia (Lam et al., 2013). Investigators consider K+ channels as a therapeutic drug target for these neurological abnormalities (Humphries and Dart, 2015), and there are even existing clinical studies of treating epilepsy with a drug targeting K+ channels (Shieh et al., 2000).
Indeed, experimental results have indicated that K+ channels are a key protein for participation in the development of distinct neurological pathologies, indicating a possible role of these channels for neuroprotection. Oxidation of Kv2.1 channels, a major component of the delayed rectifier K+ current in neurons (Murakoshi and Trimmer, 1999), leads to enhanced channel activity, and more effluxes of K+ ions can in turn promote caspase activation and apoptosis (Shah and Aizenman, 2014). In Alzheimer’s disease (AD), the most prevalent age-related neurodegenerative disorder with progressive impairment of cognitive function and memory, the malfunction of Kv channels is referred to as an essential factor that is relevant to the pathogenesis of AD. Neurotoxic Aβ peptides enable the elevated expression of Kv3.4 and Kv4.2 channels, accounting for A-type K+ currents (IK(A)) and accordingly triggering neuronal apoptosis (Pan et al., 2004; Pannaccione et al., 2007). Hence, researchers may assume that a promising neuroprotective manner against proapoptotic stimuli serves to prevent K+ efflux in nerve tissues (Yeh et al., 2020). On the other hand, excitotoxicity mediated by ischemic or epileptic insults results in the boost of K+ efflux by activating Kv channels such as Kv2.1 channels, with a hyperpolarizing shift of voltage-gated activation (Aras et al., 2009), to mitigate the excitability in cultured rat cortical neurons. As a clinical medicine, the Kv7 channel agonist retigabine, which can limit the neuronal excitability, is approved by the FDA to treat epilepsy (Amabile and Vasudevan, 2013). Considerable evidence also reveals that K+ channels are emerging as one of the multitargets of estrogens known as potent neuroprotectants, which is indicative of a possibility in which modulation of K+ channels may contribute to the neuroprotective actions of these steroid hormones in the nervous system. This review summarizes the reported observations whereby the application of estrogens generates an impact on varying K+ channels, which may allow an alternative delineation concerning the involvement of K+ channels in estrogen-mediated neuroprotective effects.
Kv channels
Voltage-gated K+ (Kv) channels, consisting of six transmembrane domains and cytosolic N- and C-terminal domains, are the largest and most complex family. Kv channels widely expressed in many types of neural tissues serve to participate in the repolarization of action potentials and govern the firing rates, thereby tuning neuronal excitability and synaptic transmission (Norris et al., 2010). Emerging evidence implies that exposure to estrogens could have an impact on diverse subtypes of Kv channels, such as Kv1.5, Kv2.1, Kv2.2, Kv4.2, and Kv4.3, and corresponding native currents, to produce genomic and non-genomic effects.
Kv1.5 channels, underlying the ultra-rapid rectifier K+ current (IKur) in cardiac atria, are located in nervous tissues such as the hippocampal and cortical neurons, and they also conduct the delayed rectifying K+ current (IK(DR)) in the brain (Maletic-Savatic et al., 1995). Lowering the expression of Kv1.5 channels confers a decrease in neuronal injury and death in ischemic stroke (Stenzel-Poore et al., 2003). Treatment with estrogens results in the downregulation of Kv1.5 transcripts (Tsang et al., 2004; Saito et al., 2009), but no direct inhibition on this channel is observed (Wong et al., 2008) that is indicative of a genomic action of the sex hormones. In line with the data, tamoxifen, a selective estrogen receptor modulator (SERM), conducts an enhanced expression of Kv1.5 channels through blocking the estrogen receptor (ER) (El Gebeily and Fiset, 2011).
As slowly inactivating K+ channels, Kv2.1 channels conduct the major delayed rectifier K+ current in the cortical and hippocampal neurons (Murakoshi and Trimmer, 1999), holding a fundamental role in shaping intrinsic neuronal excitability. Apart from those expressed in nervous tissues, the distribution of Kv2.1 channels is evident in diverse cell subtypes, such as pancreatic β-cells (MacDonald and Wheeler, 2003), cardiomyocytes (Nerbonne and Kass, 2005), and smooth muscle cells (Schmalz et al., 1998), exhibiting varying effects in regulating physiological functions. Extensive studies have established a clear role of Kv2.1 for modulating neuronal excitability (Speca et al., 2014); furthermore, this channel is also thought to be a crucial regulator of neuronal apoptosis in a number of neural cells in response to diverse apoptotic stimuli, including oxidative stress, ischemic injury, and Aβ toxins (Pannaccione et al., 2007; Sesti et al., 2014; Shah and Aizenman, 2014). Hence, Kv2.1 channels are closely associated with neuroprotective effects owing to their ability to regulate the excitability and apoptosis of neurons. Acute exposure to 17β-estradiol significantly blocks Kv2.1-dominated outward Kv currents in human osteoblast-like MG63 cells and Kv2.1 channels reconstituted in COS-7 cells, with an IC50 of 0.3 μM (Li et al., 2013). Corresponding with this report, Kv2.1- and Kv2.2-mediated Kv currents are also inhibited by acute application of 17β-estradiol in the medial preoptic nucleus (MPN), with an EC50 value of 9.7 μM (Druzin et al., 2011). Although the normal levels of estrogens are less than 10 nM in mammals (Naftolin et al., 1990), the concentrations of estrogens in target tissues could reach the range of micromolar as the result of an accumulation effect (Noe et al., 1992; Clarke et al., 2001). In addition to rapid non-genomic effects, the genomic regulation induced by estrogens is manifested in a reverse way in that expression of Kv2.1 channels is increased because of blockage of estrogen receptors by tamoxifen (El Gebeily and Fiset, 2011). Consistent with the notion of Kv2.1 inhibition in favor of anti-apoptotic effects (Sesti et al., 2014), therefore, the acute and chronic inhibitory actions of Kv2.1 channels conferred by estrogens may contribute to their neuroprotective influence. Moreover, the decrease in expression of Kv2.1 channels induced by tacrine, an acetylcholinesterase (AChE) inhibitor, leads to a boost in the proliferation of N2A cells, an alternative way to protect neuronal cells (Hu et al., 2020). Another member of the Kv2 family, the Kv2.2 channel, is also abundantly expressed in a variety of neuronal subtypes, and resveratrol, a naturally occurring phytoalexin, produces a blocking effect on Kv2.2 channels through the estrogen receptor GPR30-mediated PKC pathway in cerebellar granule cells (Dong et al., 2013).
Neuronal A-type K+ current (I(A)) is activated at potentials below the firing threshold of action potentials, with rapidly activating and inactivating kinetics (Johnston et al., 2010). Generally, it is believed that Kv4.2 and Kv4.3 channels are responsible for I(A) in broad areas of the brain, including hippocampal and cortical neurons (Norris and Nerbonne, 2010). The downregulation of Kv4.2, but not Kv4.3, mRNA levels by 17β-estradiol (E2) replacement is observed in the hypothalamic paraventricular nucleus (PVN) neurons (Lee et al., 2013). Nevertheless, Saito et al. (2009)revealed that downregulation of Kv4.3 expression is also conducted by application of estrogens in ventricular myocytes. To date, it appears that estrogens fail to exert an acute non-genomic effect on I(A) (Knock et al., 2001). As mentioned previously, injection of Aβ peptides is able to generate an upregulation of Kv4.2 channel expression in the hippocampus and cerebral cortex, and this event participates in pathogenesis of AD (Pan et al., 2004). As such, the estrogen-mediated inhibition for Kv4.2 and/or Kv4.3 may counteract the Aβ neurotoxic effects on neuronal cells. The involvement of estrogen in regulating Kv channels in neuroprotection can be seen in Figure 1.
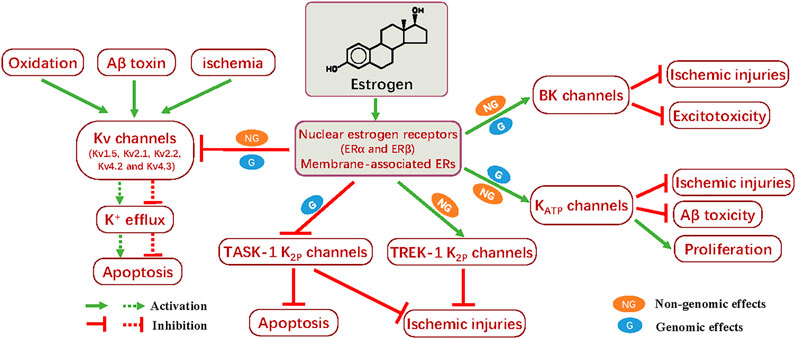
FIGURE 1. Summary of the neuroprotective effects of estrogens through modulation of potassium channels. Treatment with estrogen generates neuroprotective actions by opposing apoptosis and ischemic injuries; this effect is mediated by the inhibitory impact on the Kv, including Kv1.5, Kv2.1, Kv2.2, Kv4.2, and Kv4.3, and TASK-1 K2P channels. On the other hand, enhanced activity of BK, KATP, and TREK-1 K2P channels induced by estrogen also facilitates the mitigation of ischemic injuries, as well as Aβ toxicity or excitotocity, even promoting cell proliferation. The nuclear estrogen receptors (ERα and ERβ) and membrane-associated ERs participate in the genomic and non-genomic effects, respectively. These distinct approaches could, at least in part, underlie the neuroprotective mechanism of estrogen in modulating K+ channels.
BK channels
Large-conductance Ca2+- and voltage-activated K+ (BK) channels, encoded by the Kcnma1 gene, are distributed in diverse tissues, such as neuronal and non-neuronal cells. Normally, they can be activated in response to elevation in intracellular Ca2+concentrations and membrane depolarization; in turn, they oppose Ca2+ influx and neurotransmitter release, a mechanism for reducing cell excitation (Griguoli et al., 2016). Collected evidence has established a clear neuroprotective role for the opening of BK channels in ischemic brain damage. In acute focal cerebral ischemia, BK channels conduct a beneficial action to limit brain infarction and promote survival, which is also evident in hippocampal slice cultures (Liao et al., 2010), and activation of these channels can contribute to chlorpromazine (CPZ)- and vitamin C-mediated reduction in ischemic brain injury (Li et al., 2014; Li et al., 2019). In hippocampal pyramidal neurons, blockade of BK channels aggravates the cell damage arising from oxygen and glucose deprivation (OGD) (Rundén-Pran et al., 2002). In addition, BK channels predominantly expressed in the spinal cord could generate a protective effect on acute and chronic spinal cord injury (SCI) by improving neuronal functions (Ye et al., 2012; Jacobsen et al., 2018), and the BK channel agonist, isopimaric acid (ISO), mitigates cognitive deficits in 3xTg Alzheimer’s disease (AD) model mice (Wang et al., 2015), indicating obvious and various manners of neuroprotection.
It is well known that BK channels are tetramers of four pore-forming α subunits and four β regulatory subunits, which can be directly activated by estradiol binding to the β subunits (Valverde et al., 1999). Furthermore, several lines of evidence show that estrogens are capable of increasing the expression of BK channels in the different subtypes of neural cells via the estrogen receptor β (Nishimura et al., 2008; Li and Qiu, 2015). As a result, elevated activity of BK channels arising from the genomic and/or non-genomic effects of estrogens facilitates their positive influence in cellular survival and proliferation (Coiret et al., 2005). Additionally, 17β-estradiol can activate estrogen receptors to lower the excitotoxicity in ischemia by potentiating the activity of BK channels and decreasing the AMPA/NMDA receptor-mediated spontaneous excitatory postsynaptic currents (sEPSCs), a process that is indicative of the interplay between BK and other ion channels in estrogen-induced neuroprotection (Zhang et al., 2009). The activation of neuroprotective BK channels by 17β-estradiol also leads to an attenuation of neuronal excitability impairment in the ischemia model of oxygen and glucose deprivation (OGD) in rat hippocampal slices (Zhang et al., 2009), which is suggestive of the protective action on memory conferred by estrogen. Taken together, the estrogen-enhanced activity of BK channels facilitates neuroprotection (Figure 1).
KATP channels
In addition to regulating the release of pancreatic β-cells, KATP channels expressed in a number of different brain areas, especially with high levels in the hippocampus and cortex, are thought to be a potential regulator of excitability and neurotransmitter release (Szeto et al., 2018). Neuronal KATP channels are hetero-octameric complexes consisting of four regulatory subunits SURx and four pore-forming subunits Kir6.x. Activation of KATP channels conducts the potential effects on mitigating hypoxic-ischemic brain injury, considered as a promising therapeutic target for ischemic stroke (Szeto et al., 2018; Zhao et al., 2021). Remote ischemic preconditioning (RIPC) is capable of lowering the hippocampus damage induced by global cerebral ischemia, which is mediated by the activation of KATP channels (Mehrjerdi et al., 2015). The elevated neuronal damage after ischemic insults in the hippocampus and neocortex is also observed in the mice lacking Kir6.2-containing KATP channels (Sun et al., 2006). Overexpression of the SUR1 subunit of KATP channels in the forebrain opposes seizure induction and excitotoxic damage of hippocampal pyramidal neurons (Hernández-Sánchez et al., 2001). Ketones generate hippocampal synaptic protection in part through increasing the activity of KATP channels (Kim et al., 2015). Moreover, opening of KATP channels after application of diazoxide may protect cortical and hippocampal cholinergic neurons against Aβ1-42-induced neurotoxicity (Tan et al., 2012). Likewise, sildenafil and diazoxide could reduce the Aβ25-35-mediated mitochondrial toxicity through KATP channels (Son et al., 2018), which is indicative of another protective process arising from this channel. Therefore, KATP channels in different brain regions, including the hippocampus, may participate in the process of neuroprotection through counteracting ischemia and Aβ neurotoxicity.
Intriguingly, increasing evidence indicates that estrogens also produce a regulatory action in KATP channels. In GnRH neurons important to the control the female reproductive cycle, 17β-estradiol enhances the activity of KATP channels via a membrane ER-activated PKC-PKA signaling pathway, without affecting the mRNA levels of the Kir6.2 and SUR1 subunits (Zhang et al., 2007; Zhang et al., 2010). However, estrogen-induced upregulation of the Kir6.2 and SUR1 subunits is also observed in brain cortices, opposing focal cerebral ischemia (Zhang et al., 2012). Enhanced expression of the SUR2A subunit of KATP channels underlies the protective action of estrogen against hypoxia-reoxygenation (Ranki et al., 2002), and estrogen-mediated protection during ischemia-reperfusion stress can be attributed to the elevated expression of the SUR2 subunit (Gao et al., 2014). Moreover, increased expression of KATP channels after treatment with estrogens exhibits a positive effect on cell proliferation (Park et al., 2008), favoring the neuroprotection of estrogens. Collectively, upregulation of the activity of KATP channels can contribute to the beneficial effects of estrogens on the nervous system (Figure 1).
K2P channels
The two-pore-domain background potassium (K2P) channels possess a distinct structure known as dimers, each subunit of which comprise two pore-forming loops and four transmembrane (TM) domains (Feliciangeli et al., 2015). These channels remain active over the entire range of membrane potentials owing to the lack of a voltage sensor; as such, they are regarded as “leak” channels (Bayliss and Barrett, 2008). The existence of K2P channels is evident in a variety of cell subtypes, and their function is to stabilize the membrane potential and regulate the excitability and cell volume (Feliciangeli et al., 2015). Of 15 members of the K2P channel family, the stretch- and lipid-activated TREK-1 channels are referred to as the mediator implicated in neuroprotection because of activation by polyunsaturated fatty acids (PUFAs), which occurs against cerebral ischemic insult (Heurteaux et al., 2004). Several K2P channels such as the TREK and TASK channels are located in astrocytes important to brain volume homeostasis, and the ethacrynic acid derivative 4-(2-butyl-6,7-dichloro-2-cyclopentylindan-1-on-5-yl)oxobutyric acid (DCPIB) can inhibit the astroglial swelling and reduce the size of infarct volume during ischemic damage; the neuroprotective effects are thought to arise from activation of TREK-1 channels (Minieri et al., 2013). Choudhury and Sikdar (2018)revealed that 17β-estradiol is able to activate the TREK-1 channels by binding to the G protein-coupled estrogen receptor (GPER), contributing to the neuroprotective effect of this hormone.
The acid- and oxygen-sensitive TASK channel is thought to exert a protective effect on stroke development (Ehling et al., 2015). TASK-1−/− mice show larger infarct volumes in a model of focal cerebral ischemia, and TASK channel inhibition by antagonists increases stroke development (Meuth et al., 2009; Muhammad et al., 2010). Moreover, inactivation of the TASK-1 and TASK-3 channels results in a significant attenuation in the death of cerebellar granule neurons (Lauritzen et al., 2003). These data have established a clear role for TASK channels in counteracting ischemic and apoptotic neuronal death. Chronic applications of 17β-estradiol generate a downregulation of TASK-1 channel expression and improve the proliferation of neuroblastoma N2A cells (Hao and Li, 2014), which is suggestive of the involvement of K2P channel inhibition in estrogen-mediated neuroprotection. Interestingly, both TASK-1 inhibition and TREK-1 activation are capable of participating in the neuroprotective effects conducted by estrogens (Figure 1). More studies are needed to explore the action of estrogens on these background K+ channels for the exact underlying mechanism.
Summary
Collectively, earlier and current data have indicated that estrogens are capable of generating diverse biological actions through multiple approaches and accordingly producing beneficial effects for the symptoms of menopause in women. Many women take estrogen-containing therapies for lots of health-related problems, and clinical trials are also being undertaken to evaluate the outcomes and search for better ways with less adverse actions (Rossouw et al., 2002). The potent neuroprotection conferred by estrogens shows many positive influences to attenuate cognitive aging and brain damage, and extensive research is being conducted to clarify the exact causes for these issues. Clearly, the collected data indicate that many kinds of K+ channels, such as Kv, BK, KATP, and K2P channels, also participate in estrogen-mediated actions in nervous tissues to protect against apoptosis, ischemic insult, and Aβ toxicity (Figure 1; Table 1). Interestingly, administration of estrogens produces an inhibitory effect on the Kv and TASK-1 K2P channels but activates the BK, KATP, and TREK-1 K2P channels for a common protective goal. The virtual causes for these phenomena are unknown, and the varying regulation of these K+ channels by the genomic or non-genomic actions of estrogens alone or in combination may provide a possible answer. Definitely, additional studies are needed to address these issues. Currently, although the clear role of K+ channels in the neuroprotective actions of estrogens is established, experimental data are relatively limited. Accordingly, intensive research needs to be performed to explore the more detailed events of this subject in the future.

TABLE 1. Modulation of estrogens on K+ channels for neuroprotection.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by grants from the research project of SCUN (HZY18032).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amabile, C. M., and Vasudevan, A. (2013). Ezogabine: a novel antiepileptic for adjunctive treatment of partial-onset seizures. Pharmacotherapy 33, 187–194. doi:10.1002/phar.1185
Aras, M. A., Saadi, R. A., and Aizenman, E. (2009). Zn2+ regulates Kv2.1 voltage-dependent gating and localization following ischemia. Eur. J. Neurosci. 30, 2250–2257. doi:10.1111/j.1460-9568.2009.07026.x
Barreto, G. E., McGovern, A. J., and Garcia-Segura, L. M. (2021). Role of neuroglobin in the neuroprotective actions of estradiol and estrogenic compounds. Cells 10, 1907. doi:10.3390/cells10081907
Bayliss, D. A., and Barrett, P. Q. (2008). Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol. Sci. 29, 566–575. doi:10.1016/j.tips.2008.07.013
Björnström, L., and Sjöberg, M. (2005). Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 19, 833–842. doi:10.1210/me.2004-0486
Brinton, R. D. (2001). Cellular and molecular mechanisms of estrogen regulation of memory function and neuroprotection against alzheimer's disease: recent insights and remaining challenges. Learn. Mem. 8, 121–133. doi:10.1101/lm.39601
Carpenter, R. S., Iwuchukwu, I., Hinkson, C. L., Reitz, S., Lee, W., Kukino, A., et al. (2016). High-dose estrogen treatment at reperfusion reduces lesion volume and accelerates recovery of sensorimotor function after experimental ischemic stroke. Brain Res. 15, 1639200–1639213. doi:10.1016/j.brainres.2016.01.058
Cheskis, B. J., Greger, J. G., Nagpal, S., and Freedman, L. P. (2007). Signaling by estrogens. J. Cell. Physiol. 213, 610–617. doi:10.1002/jcp.21253
Choudhury, N., and Sikdar, S. K. (2018). 17β-estradiol potentiates TREK1 channel activity through G protein-coupled estrogen receptor. J. Steroid Biochem. Mol. Biol. 183, 94–105. doi:10.1016/j.jsbmb.2018.06.001
Clarke, R., Leonessa, F., Welch, J. N., and Skaar, T. C. (2001). Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol. Rev. 53, 25–71.
Coiret, G., Matifat, F., Hague, F., and Ouadid-Ahidouch, H. (2005). 17-beta-estradiol activates maxi-K channels through a non-genomic pathway in human breast cancer cells. FEBS Lett. 6, 5792995–5793000. doi:10.1016/j.febslet.2005.02.085
De Marinis, E., Fiocchetti, M., Acconcia, F., Ascenzi, P., and Marino, M. (2013). Neuroglobin upregulation induced by 17β-estradiol sequesters cytocrome c in the mitochondria preventing H2O2-induced apoptosis of neuroblastoma cells. Cell Death Dis. 4, e508. doi:10.1038/cddis.2013.30
Djillani, A., Mazella, J., Heurteaux, C., and Borsotto, M. (2019). Role of TREK-1 in health and disease, focus on the central nervous system. Front. Pharmacol. 10, 379. doi:10.3389/fphar.2019.00379
Dong, W. H., Chen, J. C., He, Y. L., Xu, J. J., and Mei, Y. A. (2013). Resveratrol inhibits K(v)2.2 currents through the estrogen receptor GPR30-mediated PKC pathway. Am. J. Physiol. Cell Physiol. 305, C547–C557. doi:10.1152/ajpcell.00146.2013
Druzin, M., Malinina, E., Grimsholm, O., and Johansson, S. (2011). Mechanism of estradiol-induced block of voltage-gated K+ currents in rat medial preoptic neurons. PLoS One 6, e20213. doi:10.1371/journal.pone.0020213
Ehling, P., Cerina, M., Budde, T., Meuth, S. G., and Bittner, S. (2015). The CNS under pathophysiologic attack--examining the role of K₂p channels. Pflugers Arch. 467, 959–972. doi:10.1007/s00424-014-1664-2
El Gebeily, G., and Fiset, C. (2011). Upregulation of ventricular potassium channels by chronic tamoxifen treatment. Cardiovasc. Res. 1 (90), 68–76. doi:10.1093/cvr/cvq384
Engler-Chiurazzi, E. B., Brown, C. M., Povroznik, J. M., and Simpkins, J. W. (2017). Estrogens as neuroprotectants: Estrogenic actions in the context of cognitive aging and brain injury. Prog. Neurobiol. 157, 188–211. doi:10.1016/j.pneurobio.2015.12.008
Feliciangeli, S., Chatelain, F. C., Bichet, D., and Lesage, F. (2015). The family of K2P channels: salient structural and functional properties. J. Physiol. 15 (593), 2587–2603. doi:10.1113/jphysiol.2014.287268
Gao, J., Xu, D., Sabat, G., Valdivia, H., Xu, W., and Shi, N. Q. (2014). Disrupting KATP channels diminishes the estrogen-mediated protection in female mutant mice during ischemia-reperfusion. Clin. Proteomics 11, 19. doi:10.1186/1559-0275-11-19
Griguoli, M., Sgritta, M., and Cherubini, E. (2016). Presynaptic BK channels control transmitter release: physiological relevance and potential therapeutic implications. J. Physiol. 1 (594), 3489–3500. doi:10.1113/JP271841
Hao, X., and Li, X. (2014). 17β-estradiol downregulated the expression of TASK-1 channels in mouse neuroblastoma N2A cells. J. Membr. Biol. 247, 273–279. doi:10.1007/s00232-014-9632-5
Hernández-Sánchez, C., Basile, A. S., Fedorova, I., Arima, H., Stannard, B., Fernandez, A. M., et al. (2001). Mice transgenically overexpressing sulfonylurea receptor 1 in forebrain resist seizure induction and excitotoxic neuron death. Proc. Natl. Acad. Sci. U. S. A. 1398, 3549–3554. Epub 2001/03/15. doi:10.1073/pnas.051012898
Heurteaux, C., Guy, N., Laigle, C., Blondeau, N., Duprat, F., Mazzuca, M., et al. (2004). TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. Jul 7 (23), 2684–2695. Epub 2004/06/04. doi:10.1038/sj.emboj.7600234
Hu, X. M., Ren, S., Li, K., and Li, X. T. (2020). Tacrine modulates Kv2.1 channel gene expression and cell proliferation. Int. J. Neurosci. 130, 781–787. Epub 2019/12/19. doi:10.1080/00207454.2019.1705811
Humphries, E. S., and Dart, C. (2015). Neuronal and cardiovascular potassium channels as therapeutic drug targets: Promise and pitfalls. J. Biomol. Screen. 20, 1055–1073. Epub 2015/08/26. doi:10.1177/1087057115601677
Ikeda, K., Horie-Inoue, K., and Inoue, S. (2019). Functions of estrogen and estrogen receptor signaling on skeletal muscle. J. Steroid Biochem. Mol. Biol. 191, 105375. Epub 2019/05/09. doi:10.1016/j.jsbmb.2019.105375
Jacobsen, M., Lett, K., Barden, J. M., Simpson, G. L., and Buttigieg, J. (2018). Activation of the large-conductance, voltage, and Ca(2+)- activated K(+) (BK) channel in acute spinal cord injury in the wistar rat is neuroprotective. Front. Neurol. 9, 1107. Epub 2019/01/09. doi:10.3389/fneur.2018.01107
Johnston, J., Forsythe, I. D., and Kopp-Scheinpflug, C. (2010). Going native: voltage-gated potassium channels controlling neuronal excitability. J. Physiol. 1, 5883187–5883200. Epub 2010/06/04. doi:10.1113/jphysiol.2010.191973
Kim, D. Y., Abdelwahab, M. G., Lee, S. H., O'Neill, D., Thompson, R. J., Duff, H. J., et al. (2015). Ketones prevent oxidative impairment of hippocampal synaptic integrity through KATP channels. PLoS One 10, e0119316. Epub 2015/04/08. doi:10.1371/journal.pone.0119316
Knock, G. A., Tribe, R. M., Hassoni, A. A., and Aaronson, P. I. (2001). Modulation of potassium current characteristics in human myometrial smooth muscle by 17beta-estradiol and progesterone. Biol. Reprod. 64, 1526–1534. Epub 2001/04/25. doi:10.1095/biolreprod64.5.1526
Lam, J., Coleman, N., Garing, A. L., and Wulff, H. (2013). The therapeutic potential of small-conductance KCa2 channels in neurodegenerative and psychiatric diseases. Expert Opin. Ther. Targets 17, 1203–1220. Epub 2013/07/26. doi:10.1517/14728222.2013.823161
Lauritzen, I., Zanzouri, M., Honore, E., Duprat, F., Ehrengruber, M. U., Lazdunski, M., et al. (2003). K+-dependent cerebellar granule neuron apoptosis. Role of task leak K+ channels. J. Biol. Chem. 22, 32068–32076. Epub 2003/06/05. doi:10.1074/jbc.M302631200
Lee, S. K., Ryu, P. D., and Lee, S. Y. (2013). Estrogen replacement modulates voltage-gated potassium channels in rat presympathetic paraventricular nucleus neurons. BMC Neurosci. 4, 14134. Epub 2013/11/05. doi:10.1186/1471-2202-14-134
Li, H. J., Zhang, Y. J., Zhou, L., Han, F., Wang, M. Y., Xue, M. Q., et al. (2014). Chlorpromazine confers neuroprotection against brain ischemia by activating BKCa channel. Eur. J. Pharmacol. 15 (735), 38–43. doi:10.1016/j.ejphar.2014.04.017
Li, L., Li, S., Hu, C., Zhou, L., Zhang, Y., Wang, M., et al. (2019). BK(Ca) channel is a molecular target of vitamin C to protect against ischemic brain stroke. Mol. Membr. Biol. 35, 9–20. doi:10.1080/09687688.2019.1608378
Li, X., Zheng, S., Dong, X., and Xiao, J. (2013). 17β-Estradiol inhibits outward voltage-gated K⁺ currents in human osteoblast-like MG63 cells. J. Membr. Biol. 246, 39–45. doi:10.1007/s00232-012-9502-y
Li, X. T., and Qiu, X. Y. (2015). 17β-Estradiol upregulated expression of α and β subunits of larger-conductance calcium-activated K(+) channels (BK) via estrogen receptor β. J. Mol. Neurosci. 56, 799–807. Epub 2015/02/14. doi:10.1007/s12031-015-0502-0
Liao, Y., Kristiansen, A. M., Oksvold, C. P., Tuvnes, F. A., Gu, N., Rundén-Pran, E., et al. (2010). Neuronal Ca2+-activated K+ channels limit brain infarction and promote survival. PLoS One 5 (5), e15601. Epub 2011/01/07. doi:10.1371/journal.pone.0015601
Liu, F., Day, M., Muñiz, L. C., Bitran, D., Arias, R., Revilla-Sanchez, R., et al. (2008). Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 11, 334–343. Epub 2008/02/26. doi:10.1038/nn2057
Lösel, R., and Wehling, M. (2003). Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 4, 46–56. Epub 2003/01/04. doi:10.1038/nrm1009
Lu, Y., Sareddy, G. R., Wang, J., Wang, R., Li, Y., Dong, Y., et al. (2019). Neuron-derived estrogen regulates synaptic plasticity and memory. J. Neurosci. 39, 2792–2809. Epub 2019/02/08. doi:10.1523/JNEUROSCI.1970-18.2019
Lu, Y., Sareddy, G. R., Wang, J., Zhang, Q., Tang, F. L., Pratap, U. P., et al. (2020). Neuron-derived estrogen is critical for astrocyte activation and neuroprotection of the ischemic brain. J. Neurosci. 16 (40), 7355–7374. Epub 2020/08/21. doi:10.1523/JNEUROSCI.0115-20.2020
MacDonald, P. E., and Wheeler, M. B. (2003). Voltage-dependent K(+) channels in pancreatic beta cells: role, regulation and potential as therapeutic targets. Diabetologia 46, 1046–1062. Epub 2003/06/28. doi:10.1007/s00125-003-1159-8
Maletic-Savatic, M., Lenn, N. J., and Trimmer, J. S. (1995). Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J. Neurosci. 15, 3840–3851. Epub 1995/05/01. doi:10.1523/jneurosci.15-05-03840.1995
Maljevic, S., and Lerche, H. (2014). Potassium channel genes and benign familial neonatal epilepsy. Prog. Brain Res. 213, 17–53. Epub 2014/09/10. doi:10.1016/B978-0-444-63326-2.00002-8
Mehrjerdi, F. Z., Aboutaleb, N., Pazoki-Toroudi, H., Soleimani, M., Ajami, M., Khaksari, M., et al. (2015). The protective effect of remote renal preconditioning against hippocampal ischemia reperfusion injury: Role of KATP channels. J. Mol. Neurosci. 57, 554–560. Epub 2015/08/10. doi:10.1007/s12031-015-0636-0
Mendelsohn, M. E., and Karas, R. H. (2007). HRT and the young at heart. N. Engl. J. Med. 356 (356), 2639–2641. Epub 2007/06/22. doi:10.1056/NEJMe078072
Meuth, S. G., Kleinschnitz, C., Broicher, T., Austinat, M., Braeuninger, S., Bittner, S., et al. (2009). The neuroprotective impact of the leak potassium channel TASK1 on stroke development in mice. Neurobiol. Dis. 33, 1–11. Epub 2008/10/22. doi:10.1016/j.nbd.2008.09.006
Miller, N. R., Jover, T., Cohen, H. W., Zukin, R. S., and Etgen, A. M. (2005). Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology 146, 3070–3079. Epub 2005/04/09. doi:10.1210/en.2004-1515
Minieri, L., Pivonkova, H., Caprini, M., Harantova, L., Anderova, M., and Ferroni, S. (2013). The inhibitor of volume-regulated anion channels DCPIB activates TREK potassium channels in cultured astrocytes. Br. J. Pharmacol. 168, 1240–1254. Epub 2012/10/18. doi:10.1111/bph.12011
Muhammad, S., Aller, M. I., Maser-Gluth, C., Schwaninger, M., and Wisden, W. (2010). Expression of the kcnk3 potassium channel gene lessens the injury from cerebral ischemia, most likely by a general influence on blood pressure. Neuroscience 167, 758–764. Epub 2010/02/20. doi:10.1016/j.neuroscience.2010.02.024
Murakoshi, H., and Trimmer, J. S. (1999). Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J. Neurosci. 1 (19), 1728–1735. Epub 1999/02/19. doi:10.1523/jneurosci.19-05-01728.1999
Naftolin, F., Cutmann, J. N., Decherney, A. H., and Sarrel, P. M. (1990). Ovarian secretions and cardiovascular and neurological function. New York: Raven.
Nerbonne, J. M., and Kass, R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. Epub 2005/09/27. doi:10.1152/physrev.00002.2005
Nishimura, I., Ui-Tei, K., Saigo, K., Ishii, H., Sakuma, Y., and Kato, M. (2008). 17beta-estradiol at physiological concentrations augments Ca(2+) -activated K+ currents via estrogen receptor beta in the gonadotropin-releasing hormone neuronal cell line GT1-7. Endocrinology 149, 774–782. Epub 2007/10/27. doi:10.1210/en.2007-0759
Noe, G., Cheng, Y. C., Dabike, M., and Croxatto, H. B. (1992). Tissue uptake of human sex hormone-binding globulin and its influence on ligand kinetics in the adult female rat. Biol. Reprod. 47, 970–976. Epub 1992/12/01. doi:10.1095/biolreprod47.6.970
Norris, A. J., Foeger, N. C., and Nerbonne, J. M. (2010). Neuronal voltage-gated K+ (Kv) channels function in macromolecular complexes. Neurosci. Lett. 486, 48673–48677. Epub 2010/09/04. doi:10.1016/j.neulet.2010.08.067
Norris, A. J., and Nerbonne, J. M. (2010). Molecular dissection of I(A) in cortical pyramidal neurons reveals three distinct components encoded by Kv4.2, Kv4.3, and Kv1.4 alpha-subunits. J. Neurosci. 7 (30), 5092–5101. Epub 2010/04/08. doi:10.1523/JNEUROSCI.5890-09.2010
Pan, Y., Xu, X., Tong, X., and Wang, X. (2004). Messenger RNA and protein expression analysis of voltage-gated potassium channels in the brain of Abeta(25-35)-treated rats. J. Neurosci. Res. 1 (77), 94–99. Epub 2004/06/16. doi:10.1002/jnr.20134
Pandey, R., Shukla, P., Anjum, B., Gupta, H. P., Pal, S., Arjaria, N., et al. (2020). Estrogen deficiency induces memory loss via altered hippocampal HB-EGF and autophagy. J. Endocrinol. 1, 24453–24470. Epub 2019/10/28. doi:10.1530/JOE-19-0197
Pannaccione, A., Boscia, F., Scorziello, A., Adornetto, A., Castaldo, P., Sirabella, R., et al. (2007). Up-regulation and increased activity of KV3.4 channels and their accessory subunit MinK-related peptide 2 induced by amyloid peptide are involved in apoptotic neuronal death. Mol. Pharmacol. 72, 665–673. Epub 2007/05/15. doi:10.1124/mol.107.034868
Park, S. H., Ramachandran, S., Kwon, S. H., Cha, S. D., Seo, E. W., Bae, I., et al. (2008). Upregulation of ATP-sensitive potassium channels for estrogen-mediated cell proliferation in human uterine leiomyoma cells. Gynecol. Endocrinol. 24, 250–256. Epub 2008/06/24. doi:10.1080/09513590801893315
Ranki, H. J., Budas, G. R., Crawford, R. M., Davies, A. M., and Jovanović, A. (2002). 17Beta-estradiol regulates expression of K(ATP) channels in heart-derived H9c2 cells. J. Am. Coll. Cardiol. 40, 367–374. Epub 2002/07/11. doi:10.1016/s0735-1097(02)01947-2
Rossouw, J. E., Anderson, G. L., Prentice, R. L., LaCroix, A. Z., Kooperberg, C., Stefanick, M. L., et al. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women's health initiative randomized controlled trial. JAMA 288 (288), 321–333. Epub 2002/07/19. doi:10.1001/jama.288.3.321
Rundén-Pran, E., Haug, F. M., Storm, J. F., and Ottersen, O. P. (2002). BK channel activity determines the extent of cell degeneration after oxygen and glucose deprivation: a study in organotypical hippocampal slice cultures. Neuroscience 112, 277–288. Epub 2002/06/05. doi:10.1016/s0306-4522(02)00092-1
Saito, T., Ciobotaru, A., Bopassa, J. C., Toro, L., Stefani, E., and Eghbali, M. (2009). Estrogen contributes to gender differences in mouse ventricular repolarization. Circ. Res. 14 (105), 343–352. Epub 2009/07/18. doi:10.1161/CIRCRESAHA.108.190041
Schmalz, F., Kinsella, J., Koh, S. D., Vogalis, F., Schneider, A., Flynn, E. R., et al. (1998). Molecular identification of a component of delayed rectifier current in gastrointestinal smooth muscles. Am. J. Physiol. 274, G901–G911. Epub 1998/06/05. doi:10.1152/ajpgi.1998.274.5.G901
Sesti, F., Wu, X., and Liu, S. (2014). Oxidation of KCNB1 K(+) channels in central nervous system and beyond. World J. Biol. Chem. 26 (5), 85–92. Epub 2014/06/13. doi:10.4331/wjbc.v5.i2.85
Shah, N. H., and Aizenman, E. (2014). Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke Res. 5, 38–58. Epub 2013/12/11. doi:10.1007/s12975-013-0297-7
Shieh, C. C., Coghlan, M., Sullivan, J. P., and Gopalakrishnan, M. (2000). Potassium channels: molecular defects, diseases, and therapeutic opportunities. Pharmacol. Rev. 52, 557–594. Epub 2000/12/21.
Shughrue, P. J., and Merchenthaler, I. (2003). Estrogen prevents the loss of CA1 hippocampal neurons in gerbils after ischemic injury. Neuroscience 116, 851–861. Epub 2003/02/08. doi:10.1016/s0306-4522(02)00790-x
Simpkins, J. W., Perez, E., Wang, X., Yang, S., Wen, Y., and Singh, M. (2009). The potential for estrogens in preventing Alzheimer's disease and vascular dementia. Ther. Adv. Neurol. Disord. 2, 31–49. Epub 2009/11/06. doi:10.1177/1756285608100427
Smith, C. C., and McMahon, L. L. (2005). Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J. Neurosci. 25 (25), 7780–7791. Epub 2005/08/27. doi:10.1523/JNEUROSCI.0762-05.2005
Son, Y., Kim, K., and Cho, H. R. (2018). Sildenafil protects neuronal cells from mitochondrial toxicity induced by β-amyloid peptide via ATP-sensitive K(+) channels. Biochem. Biophys. Res. Commun. 500, 504–510. Epub 2018/04/22. doi:10.1016/j.bbrc.2018.04.128
Speca, D. J., Ogata, G., Mandikian, D., Bishop, H. I., Wiler, S. W., Eum, K., et al. (2014). Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Genes Brain Behav. 13, 394–408. Epub 2014/02/06. doi:10.1111/gbb.12120
Stenzel-Poore, M. P., Stevens, S. L., Xiong, Z., Lessov, N. S., Harrington, C. A., Mori, M., et al. (2003). Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 362, 1028–1037. Epub 2003/10/03. doi:10.1016/S0140-6736(03)14412-1
Sun, H. S., Feng, Z. P., Miki, T., Seino, S., and French, R. J. (2006). Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J. Neurophysiol. 95, 2590–2601. Epub 2005/12/16. doi:10.1152/jn.00970.2005
Szeto, V., Chen, N. H., Sun, H. S., and Feng, Z. P. (2018). The role of K(ATP) channels in cerebral ischemic stroke and diabetes. Acta Pharmacol. Sin. 39, 683–694. Epub 2018/04/20. doi:10.1038/aps.2018.10
Tan, S., Ma, G., Li, Y., Li, J., Yao, W., Ren, X., et al. (2012). Effects of Aβ1‐42 on the current of KATP channels in cultured cholinergic neurons. Neurol. Res. 34, 707–713. Epub 2012/08/15. doi:10.1179/1743132812Y.0000000069
Tanabe, S., Hata, T., and Hiraoka, M. (1999). Effects of estrogen on action potential and membrane currents in guinea pig ventricular myocytes. Am. J. Physiol. 277, H826–H833. Epub 1999/08/13. doi:10.1152/ajpheart.1999.277.2.H826
Taxier, L. R., Gross, K. S., and Frick, K. M. (2020). Oestradiol as a neuromodulator of learning and memory. Nat. Rev. Neurosci. 21, 535–550. Epub 2020/09/04. doi:10.1038/s41583-020-0362-7
Taziaux, M., Keller, M., Bakker, J., and Balthazart, J. (2007). Sexual behavior activity tracks rapid changes in brain estrogen concentrations. J. Neurosci. 13 (27), 6563–6572. Epub 2007/06/15. doi:10.1523/JNEUROSCI.1797-07.2007
Toffol, E., Heikinheimo, O., and Partonen, T. (2015). Hormone therapy and mood in perimenopausal and postmenopausal women: a narrative review. Menopause 22, 564–578. Epub 2014/09/10. doi:10.1097/GME.0000000000000323
Tsang, S. Y., Yao, X., Wong, C. M., Chan, F. L., Chen, Z. Y., and Huang, Y. (2004). Differential regulation of K+ and Ca2+ channel gene expression by chronic treatment with estrogen and tamoxifen in rat aorta. Eur. J. Pharmacol. 12, 483155–483162. Epub 2004/01/20. doi:10.1016/j.ejphar.2003.10.031
Tuscher, J. J., Taxier, L. R., Schalk, J. C., Haertel, J. M., and Frick, K. M. (2019). Chemogenetic suppression of medial prefrontal-dorsal hippocampal interactions prevents estrogenic enhancement of memory consolidation in female mice. eNeuro 6, ENEURO.0451-18.2019. Epub 2019/04/25. doi:10.1523/ENEURO.0451-18.2019
Valverde, M. A., Rojas, P., Amigo, J., Cosmelli, D., Orio, P., Bahamonde, M. I., et al. (1999). Acute activation of Maxi-K channels (hSlo) by estradiol binding to the beta subunit. Science 285, 1929–1931. Epub 1999/09/18. doi:10.1126/science.285.5435.1929
Villa, C., Suphesiz, H., Combi, R., and Akyuz, E. (2020). Potassium channels in the neuronal homeostasis and neurodegenerative pathways underlying Alzheimer's disease: An update. Mech. Ageing Dev. 185, 111197. Epub 2019/12/22. doi:10.1016/j.mad.2019.111197
Wang, L., Kang, H., Li, Y., Shui, Y., Yamamoto, R., Sugai, T., et al. (2015). Cognitive recovery by chronic activation of the large-conductance calcium-activated potassium channel in a mouse model of Alzheimer's disease. Neuropharmacology 92, 8–15. Epub 2015/01/13. doi:10.1016/j.neuropharm.2014.12.033
Wong, C. M., Tsang, S. Y., Yao, X., Chan, F. L., and Huang, Y. (2008). Differential effects of estrogen and progesterone on potassium channels expressed in Xenopus oocytes. Steroids 73, 272–279. Epub 2007/12/11. doi:10.1016/j.steroids.2007.10.010
Ye, H., Buttigieg, J., Wan, Y., Wang, J., Figley, S., and Fehlings, M. G. (2012). Expression and functional role of BK channels in chronically injured spinal cord white matter. Neurobiol. Dis. 47, 225–236. Epub 2012/04/27. doi:10.1016/j.nbd.2012.04.006
Yeh, C. Y., Schulien, A. J., Molyneaux, B. J., and Aizenman, E. (2020). Lessons from recent advances in ischemic stroke management and targeting Kv2.1 for neuroprotection. Int. J. Mol. Sci. 25, 6107. Epub 2020/08/29. doi:10.3390/ijms21176107
Zhang, C., Bosch, M. A., Levine, J. E., Rønnekleiv, O. K., and Kelly, M. J. (2007). Gonadotropin-releasing hormone neurons express K(ATP) channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. J. Neurosci. 27 (27), 10153–10164. Epub 2007/09/21. doi:10.1523/JNEUROSCI.1657-07.2007
Zhang, C., Kelly, M. J., and Rønnekleiv, O. K. (2010). 17 β-estradiol rapidly increases ATP-sensitive potassium channel activity in gonadotropin-releasing hormone neurons [corrected] via a protein kinase signaling pathway. Endocrinology 151, 4477–4484. Epub 2010/07/28. doi:10.1210/en.2010-0177
Zhang, D., Xia, H., Xu, L., Zhang, C., Yao, W., Wang, Y., et al. (2012). Neuroprotective effects of 17β-estradiol associate with KATP in rat brain. Neuroreport 14 (23), 952–957. Epub 2012/10/04. doi:10.1097/WNR.0b013e3283598de6
Zhang, H., Xie, M., Schools, G. P., Feustel, P. F., Wang, W., Lei, T., et al. (2009). Tamoxifen mediated estrogen receptor activation protects against early impairment of hippocampal neuron excitability in an oxygen/glucose deprivation brain slice ischemia model. Brain Res. 9, 1247196–1247211. Epub 2008/11/11. doi:10.1016/j.brainres.2008.10.015
Keywords: estrogens, neuroprotective effects, K+ channels, therapeutic benefit, apoptosis
Citation: Li X-T (2022) The modulation of potassium channels by estrogens facilitates neuroprotection. Front. Cell Dev. Biol. 10:998009. doi: 10.3389/fcell.2022.998009
Received: 19 July 2022; Accepted: 11 October 2022;
Published: 26 October 2022.
Edited by:
Bilal ÇİĞ, Ahi Evran University, TurkeyReviewed by:
Pei-Chun Chen, National Cheng Kung University, TaiwanCopyright © 2022 Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xian-Tao Li, bGl4MzU3OUBob3RtYWlsLmNvbQ==