 Chuck T. Chen
Chuck T. Chen Zhuo Shao
Zhuo Shao Zhongjie Fu
Zhongjie Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 07 September 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.982564
This article is part of the Research Topic Molecular Mechanisms in Ocular Development and Disease View all 14 articles
Retina is rich in lipids and dyslipidemia causes retinal dysfunction and eye diseases. In retina, lipids are not only important membrane component in cells and organelles but also fuel substrates for energy production. However, our current knowledge of lipid processing in the retina are very limited. Peroxisomes play a critical role in lipid homeostasis and genetic disorders with peroxisomal dysfunction have different types of ocular complications. In this review, we focus on the role of peroxisomes in lipid metabolism, including degradation and detoxification of very-long-chain fatty acids, branched-chain fatty acids, dicarboxylic acids, reactive oxygen/nitrogen species, glyoxylate, and amino acids, as well as biosynthesis of docosahexaenoic acid, plasmalogen and bile acids. We also discuss the potential contributions of peroxisomal pathways to eye health and summarize the reported cases of ocular symptoms in patients with peroxisomal disorders, corresponding to each disrupted peroxisomal pathway. We also review the cross-talk between peroxisomes and other organelles such as lysosomes, endoplasmic reticulum and mitochondria.
Peroxisomes are membrane bound organelles found in all eukaryotic cells and heavily involved in α and β-oxidation of lipids, glyoxylate detoxification, amino acid degradation, as well as reactive oxygen species (ROS) and reactive nitrogen species (RNS) metabolism. Peroxisomes are also essential in plasmalogen, docosahexaenoic acid (DHA) and bile acid synthesis. There are abundance of enzymes and transporter proteins associated with peroxisomes that are responsible for peroxisome biogenesis, transportation across peroxisomal membrane, and metabolism. Loss of function in various proteins due to pathogenic genetic variants lead to multisystem disorders in human. As these mutations cause the potential for severe and early onset neurological, ophthalmological, craniofacial, musculoskeletal, and hepatorenal damages, they pose significant health risks and burden on patients and their families (Steinberg et al., 1993). Recent studies have also suggested that peroxisomes are involved in common neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (Lizard et al., 2012; Wojtowicz et al., 2020). Since neurodegeneration is associated with oxidative stress, lipid peroxidation, and cholesterol auto-oxidation, modifications of peroxisomal functions have been proposed as a potential mechanism for the altered metabolome. Retina is rich in lipids and highly metabolically demanding. Photoreceptors are very high in mitochondria, and nursed by retinal pigment epithelium (RPE) which uptakes nutrients from choroidal blood vessels and Müller glia which extends across the retinal layers and safeguards the blood-retinal barrier (Figure 1). This unique anatomical landscape and high metabolic demand made retina a vulnerable tissue prone to damage from metabolic insults such as peroxisomal disorders. In this review, we focus on the peroxisomal metabolism pathways that are involved in retinal health and the ocular dysfunction associated with peroxisomal disorders.
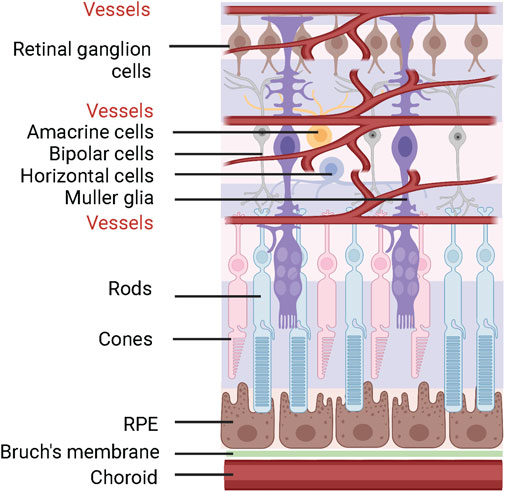
FIGURE 1. Schematics of retinal structure. The neural retina is supplied by retinal and choroidal vasculature. Photoreceptors (rods and cones) are supported by RPE and Müller glia cells. The figure was generated using Biorender (license # LX24BJEUXV).
Peroxisomes are essential for biosynthesis of DHA, plasmalogen and bile acids, as well as degradation and detoxification of very-long-chain fatty acids (VLCFAs), branched-chain fatty acids (BCFAs), dicarboxylic acids, reactive oxygen/nitrogen species (ROS/RNS), glyoxylate, and amino acids (Islinger et al., 2010; Islinger et al., 2018). In this section, we summarize the current updates on these pathways (Figure 2) and the eye diseases associated with gene mutation in these pathways (Table 1). The detailed eye symptoms are discussed in Section 3. The abbreviations are listed in Table 2.
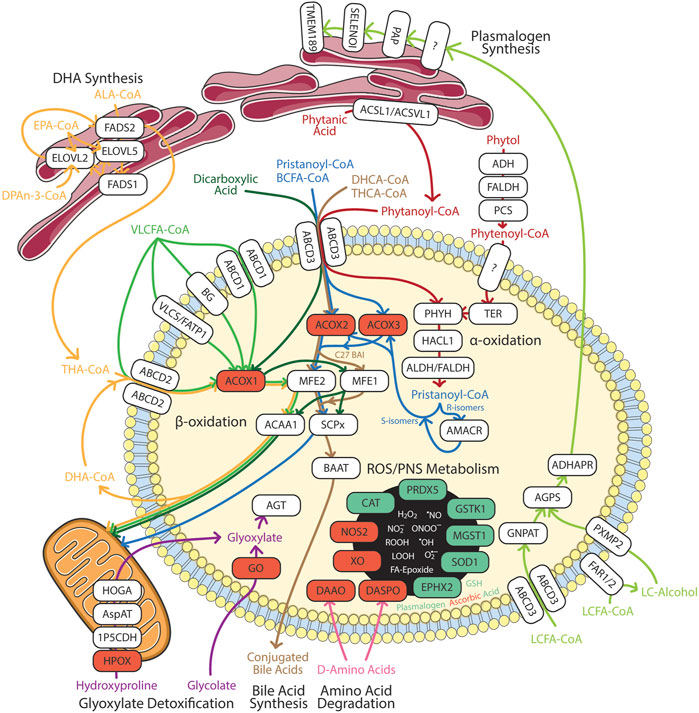
FIGURE 2. Summary of peroxisomal functions in humans. Enzymes involved in the catabolic and anabolic pathways in white boxes. Red box represents ROS/RNS producing enzymes. Green box represents anti-oxidation enzymes in redox homeostasis. 1P5CDH, Δ1-pyrroline-5-carboxylate dehydrogenase; ABCD, ATP-binding cassette sub-family D; ACAA, 3-oxoacyl thiolase; ACOX, acyl-CoA oxidase; ACSL, long chain acyl-CoA synthetase; ACSVL, very long chain acyl-CoA synthetase; ADH, alcohol dehydrogenase; ADHAPR, acyl/alkyl DHAP reductase; AGPS, alkyl-glycerone phosphate synthase; AGT, alanine:glyoxylate aminotransferase; ALA, α-linolenic acid; ALDH, aldehyde dehydrogenase; AMACR, α-methylacyl-CoA racemase; AspAT, aspartate aminotransferase; BAAT, bile acid-CoA:amino acid N-acyltransferase; BCFA, branched chain fatty acids; BG, bubblegum; CAT, catalase; DAAO, D-amino acid oxidase; DASPO, D-aspartate oxidase; DHA, docosahexaenoic acid; DHCA, 3α,7α-dihydroxy-5β-cholestanoic acid; DPAn-3, n-3 docosapentaenoic acid; ELOVL, fatty acid elongase; EPA, eicosapentaenoic acid; EPHX2, epoxide hydrolase 2; FADS, fatty acid desaturase; FALDH, fatty aldehyde dehydrogenase; FAR, fatty acyl-CoA reductase; FATP, fatty acid transport protein; GNPAT, glycerone-phosphate O-acyltransferase; GO, glycolate oxidase; GSH, glutathione; GSTK, glutathione transferase kappa; HACL, 2-hydroxyacyl-CoA lyase; HOGA, 4-hydroxy-2-oxoglutarate aldolase; HPOX, hydroxyproline oxidase; LCFA, long chain fatty acids; MFE, multifuctional enzyme; MGST, microsomal glutathione S-transferase; NOS2, inducible nitric oxide synthase; PAP, phosphatidic acid phosphatase; PCS, phytenoyl-CoA synthetase; PHYH, phytanoyl-CoA 2-hydroxylase; PRDX5, peroxiredoxin 5; PXMP2, peroxisomal membrane protein 2; SELENOI, selenoprotein 1 (ethanolamine phosphotransferase 1); SCPx, SCP-2/3-ketoacyl-CoA thiolase; SOD1, superoxide dismutase; TER, trans-2-enoyl-CoA reductase; THA, tetracosahexaenoic acid; THCA, 3α,7α, 12 α-trihydroxy-5β-cholestanoic acid; TMEM189, plasmanylethanolamine desaturase; VLCFA, very long chain fatty acids; VLCS, very long chain acyl-CoA synthetase; XO, xanthine oxidase. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
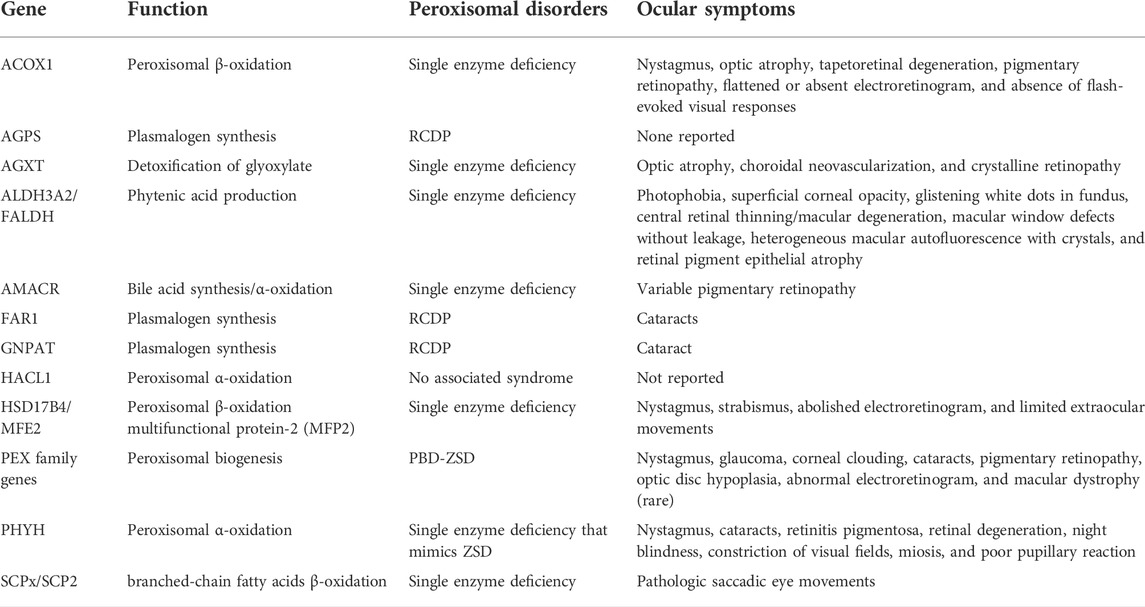
TABLE 1. Summary of common gene mutations and ocular symptoms in peroxisomal disorders.

TABLE 2. Abbreviations.
Peroxisomal β-oxidation of monocarboxylic fatty acids and dicarboxylic acids are ubiquitous within the animal kingdom (Ferdinandusse et al., 2004; Wanders, 2014). VLCFAs are a family of monocarboxylic fatty acids with greater than 22 carbons. These include saturated, monounsaturated, and polyunsaturated species of fatty acids such as behenic acid (22:0), lignoceric acid (24:0), cerotic acid (26:0), erucic acid (22:1n-9), nervonic acid (24:1n-9), adrenic acid (22:4n-6), docosapentaenoic acids (DPA; 22:5n-6 or 22:5n-3), DHA (22:6n-3), and nisinic acid/tetracosahexaenoic acid (THA; 24:6n-3). Since mitochondria lack expression of very long chain acyl-CoA synthetase (very long chain acyl-CoA synthetase/fatty acid transport protein, VLCS/FATP; bubblegum), and mitochondrial carnitine palmitoyltransferase I (CPTI) preferentially transports long-chain fatty acids (LCFA - 14–20 carbons), VLCFA are largely metabolized by the peroxisome (Gavino and Gavino, 1991; Lageweg et al., 1991; van den Bosch et al., 1992; Reddy and Mannaerts, 1994; Uchiyama et al., 1996; Coe et al., 1999; Smith et al., 1999; Steinberg et al., 1999b; Steinberg et al., 2000; Heinzer et al., 2003; Pridie et al., 2020) (Figure 3). Furthermore, peroxisomal ATP binding cassette sub family D members 1/2 (ABCD1/2) are essential for transport of VLCFA; though the exact mechanism is still debated (van Roermund et al., 2011; Kawaguchi et al., 2021). Long chain dicarboxylic acid are metabolites of monocarboxylic acid ω-oxidation by cytochrome P450 that are products of compromised β-oxidation (Grego and Mingrone, 1995). Dicarboxylic acids are transported to peroxisome via ABCD3 (van Roermund et al., 2014) (Figure 4).
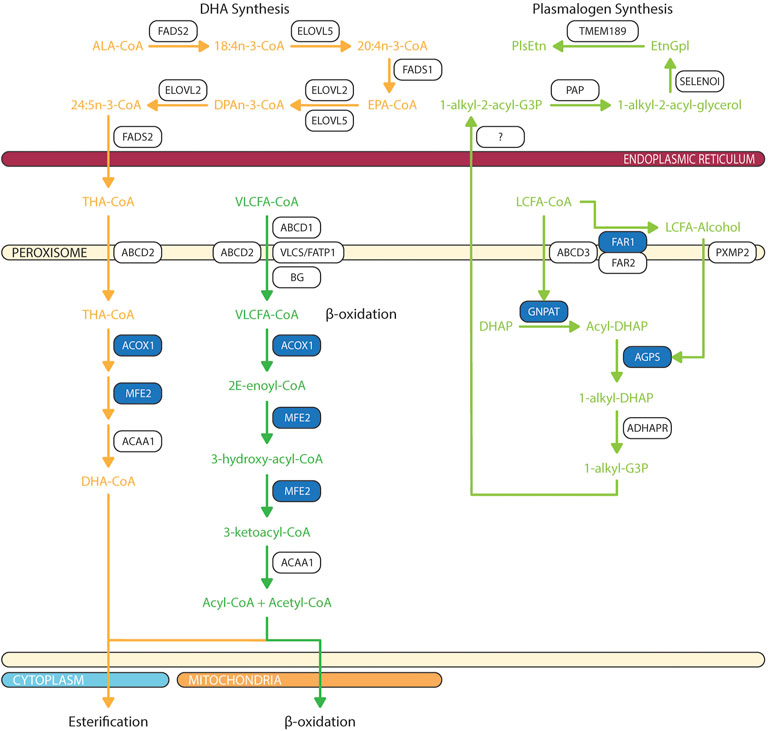
FIGURE 3. Biochemical pathways of peroxisomal lipid synthesis and degradation. Blue box represents enzymes with published pathogenic variants resulting in ocular manifestations in humans. ABCD, ATP-binding cassette sub-family D; ACAA, 3-oxoacyl thiolase; ACOX, acyl-CoA oxidase; ADHAPR, acyl/alkyl DHAP reductase; AGPS, alkyl-glycerone phosphate synthase; ALA, α-linolenic acid; BG, bubblegum; DHA, docosahexaenoic acid; DHAP, dihydroxyacetone phosphate; DPAn-3, n-3 docosapentaenoic acid; ELOVL, fatty acid elongase; EPA, eicosapentaenoic acid; EtnGpl, ethanolamine glycerophospholipids; FADS, fatty acid desaturase; FAR, fatty acyl-CoA reductase; FATP, fatty acid transport protein; G3P, glycerol-3-phosphate; GNPAT, glycerone-phosphate O-acyltransferase; LCFA, long chain fatty acids; MFE, multifuctional enzyme; PAP, phosphatidic acid phosphatase; PlsEtn, ethanolamine plasmalogens; PXMP2, peroxisomal membrane protein 2; SELENOI, selenoprotein 1 (ethanolamine phosphotransferase 1); THA, tetracosahexaenoic acid; TMEM189, plasmanylethanolamine desaturase; VLCFA, very long chain fatty acids; VLCS, very long chain acyl-CoA synthetase. The figure was partly generated using Servier Medical Art.
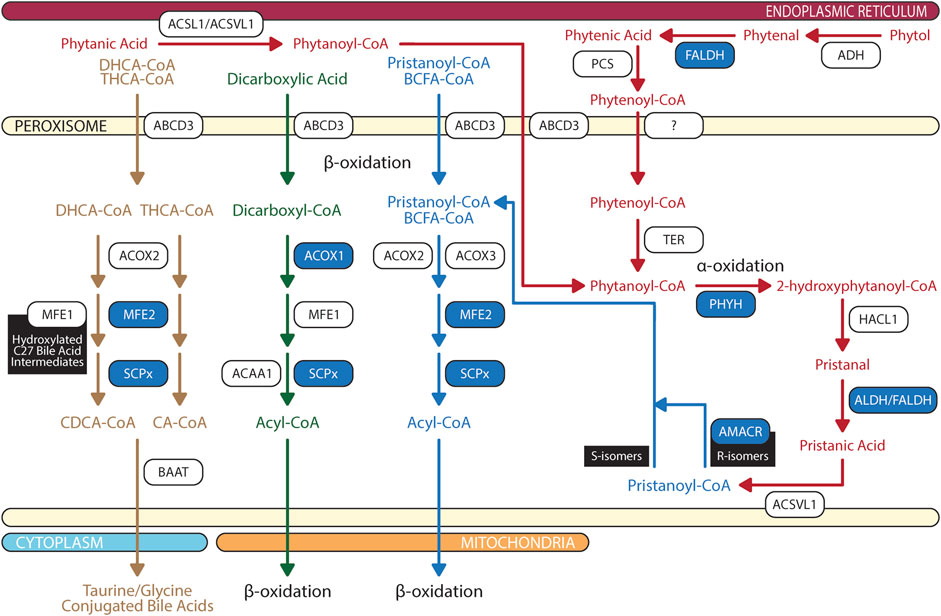
FIGURE 4. Biochemical pathways of peroxisomal branched chain fatty acid, dicarboxylic acid, and bile acid degradations. Blue box represents enzymes with published pathogenic variants resulting in ocular manifestations in humans. ABCD, ATP-binding cassette sub-family D; ACAA, 3-oxoacyl thiolase; ACOX, acyl-CoA oxidase; ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; AMACR, α-methylacyl-CoA racemase; AspAT, aspartate aminotransferase; BAAT, bile acid-CoA:amino acid N-acyltransferase; BCFA, branched chain fatty acids; CA, cholic acid; CDCA, chenodeoxycholic acid; DHCA, 3α,7α-dihydroxy-5β-cholestanoic acid; FALDH, fatty aldehyde dehydrogenase; HACL, 2-hydroxyacyl-CoA lyase; MFE, multifuctional enzyme; PCS, phytenoyl-CoA synthetase; PHYH, phytanoyl-CoA 2-hydroxylase; SCPx, SCP-2/3-ketoacyl-CoA thiolase; TER, trans-2-enoyl-CoA reductase; THCA, 3α,7α, 12 α-trihydroxy-5β-cholestanoic acid. The figure was partly generated using Servier Medical Art.
Peroxisomal β-oxidation involves four sequential reactions: 1) dehydrogenation by ACOX, 2 + 3) hydration and dehydrogenation by multifunctional enzymes (MFE), and 4) thiolytic cleavage by 3-ketoacyl-CoA thiolases. The first step of peroxisomal β-oxidation involves ACOX catalyzed acyl-CoA and oxygen to 2E-enoyl-CoA and hydrogen peroxide. In rodents, there are three ACOX with different substrate specificity. Generally, ACOX1 (straight chain-CoA oxidase) has high affinity for CoA esters of medium chain fatty acids, LCFA, and VLCFA, whereas ACOX2 (pristanoyl-CoA oxidase) and ACOX3 (cholestanoyl-CoA oxidase) prefer CoA esters of BCFA, 3α,7α-dihydroxy-5β-cholestanoic acid (DHCA), and 3α,7α, 12 α-trihydroxy-5β-cholestanoic acid (THCA), respectively (Bieber et al., 1981; Vamecq, 1987; Vanhove et al., 1993). In contrast, human ACOX2 dehydrogenate BCFA, DHCA and THCA while human ACOX3 metabolizes BCFA (Ferdinandusse et al., 2018).
Following ACOX, MFE catalyze the hydration and dehydrogenation of 2E-enoyl-CoA to chiral 3-hydroxy-acyl-CoA and 3-ketoacyl-CoA. In mammals, there are two MFE: 1) MFE-1 (L-bifunctional protein; LBP) and 2) MFE-2 (D-bifunctional protein; DBP). While MFE-1 has a broad substrate specificity which can bind CoA esters of fatty acids varying from short to long, hydroxylated C27 DHCA/THCA intermediates, and dicarboxylic acids, it predominantly catalyzes breakdown of medium and long chain dicarboxylic acids (Ferdinandusse et al., 2004; Houten et al., 2012). MFE-2 (coded by the HSD17B4 gene in human) is essential for the breakdown of VLCFA, BCFA, DHCA, and THCA (Qi et al., 1999; Su et al., 2001; Huyghe et al., 2006).
In the final step of peroxisomal β-oxidation, 3-ketoacyl-CoA thiolases catalyze the cleavage of 3-ketoacyl-CoA to acetyl-CoA and shortened acyl-CoA. In rodents, there are three 3-ketoacyl-CoA thiolases: 1) thiolase A, 2) thiolase B, and 3) SCP-2/3-ketoacyl-CoA thiolase (SCPx). Thiolase A and B have similar profile in substrate specificity, where the preference is given to saturated fatty acid in various chain length. In addition to binding of saturated 3-ketoacyl-CoA esters, SCPx also binds BCFA CoA esters (Poirier et al., 2006). In contrast to rodents, human has two 3-ketoacyl-CoA thiolases: 1) 3-oxoacyl thiolase (ACAA1) and 2) sterol carrier protein X (SCPx—ortholog of rodent SCPx). ACAA1 ligand specificity is restricted to 3-ketoacyl-CoA esters of VLCFA, whereas SCPx prefers 3-oxoacyl-CoA esters of pristanic acid, and DHCA/THCA (Seedorf et al., 1994). ACAA1 is expressed equally or more in neural retina than RPE, while reversely, SCPx expression is more abundant in RPE as compared to neural retina (Das et al., 2019).
BCFAs are a specialized group of saturated fatty acids with mono- or poly-methyl branches on the carbon chain. Monomethyl BCFA are subdivided into iso and anteiso structure. Iso-BCFA have methyl branch on the penultimate carbon which is one carbon from the methyl end; meanwhile anteiso-BCFA have methyl branch on the antepenultimate carbon that is two carbons from the methyl end. Enriched sources of BCFA include ruminant-derived and fermented foods (Taormina et al., 2020). Phytanic acid and its metabolite pristanic acid are common 3-methyl BCFA synthesized in ruminant or derived from phytol that are bioaccumulated in the marine system from phytoplankton chlorophyll (Wanders et al., 2011a; Taormina et al., 2020). In humans, tissue phytanic acid levels are strictly derived from dietary sources as endogenous synthesis are limited (Steinberg et al., 1965).
Dietary phytol is oxidized to phytenal by alcohol dehydrogenase, then converted into phytenic acid by fatty aldehyde dehydrogenase (FALDH) (Rizzo et al., 1999) (Figure 4). Phytenic acid is activated by esterification of CoA to phytenoyl-CoA by phytenoyl-CoA synthetase (PCS) after which it can bind peroxisomal trans-2-enoyl-CoA reductase (TER) and reduced to produce phytanoyl-CoA for subsequent peroxisomal phytanoyl-CoA α-oxidation and pristanoyl-CoA β-oxidation (van den Brink et al., 2005; Gloerich et al., 2006). Reduction of phytenoyl-CoA to phytanoyl-CoA is localized to peroxisome for two reasons: 1) TER is targeted to peroxisome due to attached peroxisomal targeting signal type 1 (PTS1) peptide on the enzyme and 2) mitochondrial CPTI does not catalyze carnitine esterification to phytenoyl-CoA (Wanders et al., 2011a). Though the degradation pathways are characterized, only two enzymes have been identified, FALDH and TER. Further research is required to identify enzymes catalyzing the initial conversion to phytenal and CoA esterification to phytenoyl-CoA.
Like VLCFA, phytanic acid and pristanic acid are ligands for PPARα resulting in upregulation of peroxisomal β-oxidation enzymes and cytochrome P450 4A (CYP4A) enzymes facilitated ω-oxidation of phytanic acid in endoplasmic reticulum (ER) and microsome (Zomer et al., 2000; Wanders et al., 2011b; Kersten, 2014). Under normal conditions, phytanic acid is preferentially catabolized by α-oxidation (Wanders et al., 2011a). Degradation of dietary phytanic acid begins with activation of CoA esterification by phytanoyl-CoA synthetase, ACSL1 and ACSVL1 (Watkins et al., 1994). Phytanoyl-CoA is transported into peroxisome by ABCD3 (Morita and Imanaka, 2012). Hydroxylation of phytanoyl-CoA to 2-hydroxyphytanoyl-CoA is catalyzed by peroxisomal phytanoyl-CoA 2-hydroxylase (PHYH). Similar to TER, PHYH protein sequence includes PTS2; hence after is shuttled to peroxisome (Jansen et al., 1999a). 2-hydroxyphytanoyl-CoA is then cleaved by peroxisomal 2-hydroxyacyl-CoA lyase (HACL1) to pristanal and formyl-CoA (Jansen et al., 1999b; Foulon et al., 1999). Lastly, pristanal is oxidized to pristanic acid by unknown peroxisomal aldehyde dehydrogenase (potential candidate includes FALDH). In contrast to PHYH and HACL1, which are ubiquitously expressed in RPE and neural retina, ABCD3 is more abundantly found in RPE (Das et al., 2019). This suggests that phytanoyl-CoA and pristanoyl-CoA may have alternative route of entry in neural retina as compared to RPE.
Pristanic acid is activated to pristanoyl-CoA by ACSVL1 (Steinberg et al., 1999a). α-oxidation of phytanic acid may produce pristanoyl-CoA enantiomers; therefore, R-isomers are converted to S-isomer by α-methylacyl-CoA racemase (AMACR) in order to continue peroxisomal β-oxidation cycle due to stereospecificity of ACOX2 and ACOX3 (Van Veldhoven et al., 1996). Pristanoyl-CoA is catabolized by ACOX2/3, MFE-2, and SCPx in three rounds of peroxisomal β-oxidation (Verhoeven et al., 1998; Atshaves et al., 2007). Furthermore, recent report found that PMP34 are important in the peroxisomal oxidation of pristanic acid and the export of its metabolites (Van Veldhoven et al., 2020). However, the exact mechanistic details of PMP34’s contribution to pristanic acid degradation requires further examination. AMACR deficiency results in increased plasma pristanic acid levels (Ferdinandusse et al., 2000; Thompson et al., 2009).
Glyoxylate is a highly reactive two carbon aldehyde form primarily from metabolisms of plant-source glycolate by peroxisomal glycolate oxidase (GO) and animal-source hydroxyproline by sequential breakdown via hydroxyproline oxidase (HPOX), Δ1-pyrroline-5-carboxylate dehydrogenase (1P5CDH), aspartate aminotransferase (AspAT), and 4-hydroxy-2-oxoglutarate aldolase (HOGA) (Li et al., 2016; Wang et al., 2020). Glyoxylate is readily metabolized by dehydrogenase and oxidases to oxalate which then crystalizes with calcium to form calcium oxalate stone, a common source of kidney stones (Tsujihata, 2008). Since glyoxylate accumulation is correlated with dietary sources and intake, multiple detoxification mechanisms involving peroxisome and/or mitochondria are evolved depending on the food sources of the organisms (Danpure, 1997; Birdsey et al., 2005; Martin, 2010). Three enzymes are involved in the detoxification of glyoxylate: 1) peroxisomal and mitochondrial alanine:glyoxylate aminotransferase (AGT, encoded by the gene AGXT), 2) mitochondrial and cytosolic glyoxylate reductase/hydroxypyruvate reductase (GRHPR), and 3) cytosolic lactate dehydrogenase (LDH) (Figure 5). In contrast to rat where AGT is localized to both peroxisome and mitochondria, human AGT is exclusively expressed in peroxisome (Danpure, 1997). AGT catalyzes the transamination of glyoxylate and L-alanine to form pyruvate and glycine (Cellini et al., 2007). Cellular expressions of AGT in retina and RPE has not been elucidated.
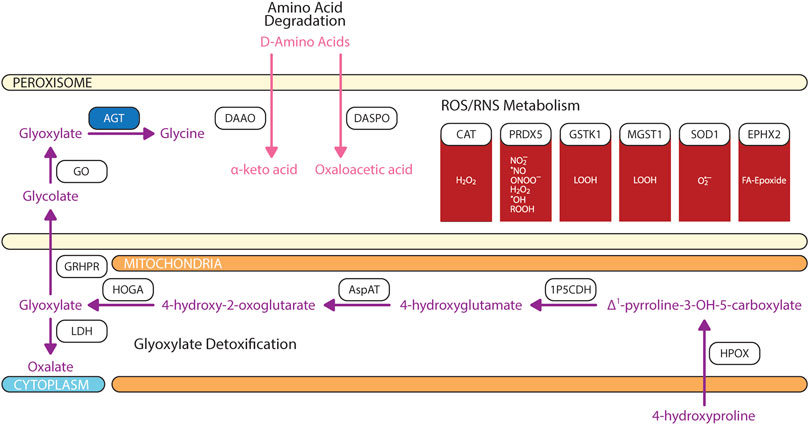
FIGURE 5. Biochemical pathways of glyoxylate, amino acids, and reactive oxygen/nitrogen species degradations. Blue box represents enzymes with published pathogenic variants resulting in ocular manifestations in humans. 1P5CDH, Δ1-pyrroline-5-carboxylate dehydrogenase; AGT, alanine:glyoxylate aminotransferase; AspAT, aspartate aminotransferase; CAT, catalase; DAAO, D-amino acid oxidase; DASPO, D-aspartate oxidase; EPHX2, epoxide hydrolase 2; GO, glycolate oxidase; GRHPR, glyoxylate and hydroxypyruvate reductase; GSH, glutathione; GSTK, glutathione transferase kappa; HOGA, 4-hydroxy-2-oxoglutarate aldolase; HPOX, hydroxyproline oxidase; LDH, lactate dehydrogenase; MGST, microsomal glutathione S-transferase; PRDX5, peroxiredoxin 5; SOD1, superoxide dismutase. The figure was partly generated using Servier Medical Art.
Peroxisomes is also engaged in the catabolism of D-amino acids (Figure 5). Two stereospecific enzymes are localized to peroxisomes: 1) D-amino acid oxidase (DAAO/DAO) and 2) D-aspartate oxidase (DASPO/DDO) (Van Veldhoven et al., 1991; Pollegioni et al., 2018). DAAO have a wide substrate specificity; however, it prefers bulky hydrophobic D-amino acids (D-DOPA, D-Tyr, D-Phe, D-Trp) while having limited catalytic activity for small uncharged D-amino acids (D-Cys, D-Ala, D-Pro, D-Ser) (Kawazoe et al., 2007; Frattini et al., 2011; Shibuya et al., 2013; Murtas et al., 2017). DAAO does not actively metabolize glycine which explain the negligible contribution of glyoxylate from DAAO-mediated glycine degradation (Thureen et al., 1995; Molla et al., 2006; Murtas et al., 2017). DASPO have high affinity for acidic D-amino acids (D-Asp, NMDA, D-Glu) followed by polar D-amino acids (D-Gln, D-Asn) (Pollegioni et al., 2021). DAAO is expressed in RPE and Müller cells (Beard et al., 1988); whereas DASPO retinal cell expression is unknown.
As its name defined, the core feature of peroxisome is the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) (De Duve and Baudhuin, 1966), which may cause biomolecular damage and cell death. As an organelle with high productions of ROS/RNS species, naturally, peroxisomes are equipped with an arsenal of antioxidant enzymes and non-enzymatic free radical scavengers (Figure 5). To-date, there is no consensus regarding peroxisome’s overall contribution as a net source of cellular ROS/RNS or sink for ROS/RNS detoxification (Fransen et al., 2013). Cellular reduction-oxidation (redox) state is dependent on several factors including the type, concentration, localization, synthetic rate, and degradation rate of oxidants formed (Trachootham et al., 2008; Forman et al., 2010). The magnitude of oxidative stress can have divergent effects on cellular fates where at high levels can induced irreversible damage to lipids, protein, and DNA resulting in apoptosis, but at low levels oxidants act as signaling messengers to promote cell survival and proliferation (Jones and Go, 2010; Holmstrom and Finkel, 2014).
The most abundant oxidants produced in peroxisomes is hydrogen peroxide (H2O2) because of flavin-containing oxidases that reduce molecular oxygen (O2) to H2O2 (Antonenkov et al., 2010). Peroxisomes account up to 35% of total hydrogen peroxide generation in the mammalian tissues and 20% of total cellular oxygen consumption (Cipolla and Lodhi, 2017). In human peroxisomes, H2O2 is degraded by catalase (CAT) and peroxiredoxin 5 (PRDX5). CAT is the most abundant peroxisomal antioxidant enzyme that is highly expressed in RPE as compared to neural retina. CAT activity exhibits a diurnal pattern where post-translational modification of CAT increases antioxidant activity in the morning during peak VLCFA β-oxidation. While PRDX5 can assist in H2O2 degradation, it is limited due to the small rate constant (Knoops et al., 2011). Superoxide anion radical (O2•–) and nitric oxide radical (•NO) are produced by xanthine oxidase and the inducible form of nitric oxide synthase. While superoxide is degraded by superoxide dismutase 1 (SOD1), •NO is thermodynamically favored to form peroxynitrite (ONOO−) with superoxide anion radical after which ONOO−is predominantly degraded by PRDX5 (Pacher et al., 2007; Trujillo et al., 2007; Antonenkov et al., 2010). Hydroxyl radical (•OH) are produced from H2O2 through Fenton reactions (Winterbourn, 1995). •OH and •NO may target protein cysteine thiol and unsaturated lipids causing cysteine oxidation and lipid peroxidation (Lismont et al., 2015). PRDX5 also degrades alkyl hydroperoxides (ROOH); whereas LOOH and fatty acid epoxides are detoxified by glutathione transferase kappa (GSTK1)/microsomal glutathione S-transferase 1 (MGST1) and epoxide hydrolase 2 (EPHX2) (Summerer et al., 2002; Sala et al., 2003; Knoops et al., 2011). Free radicals may also be non-enzymatically scavenged by glutathione (GSH), ascorbic acid (vitamin C), and plasmalogens (Circu and Aw, 2010; Ivashchenko et al., 2011). While the mechanisms by which GSH and ascorbic acid modulate oxidative stress are debated, plasmalogen quenches lipid peroxidation and protects unsaturated fatty acid from oxidation in the membrane because the vinyl-ether bond of plasmalogen interact faster with singlet oxygen and free radicals than other fatty acids (Broniec et al., 2011; Wallner and Schmitz, 2011).
Redox imbalance is a significant contributor to multiple retinal metabolic diseases (Kowluru and Chan, 2007; Bellezza, 2018; Trachsel-Moncho et al., 2018; Graziosi et al., 2020). Restoring the redox homeostasis preserves retinal neuronal and vascular stability. However, the role of peroxisomal redox contribution to retinopathy is understudied. A recent report has shown that overactivation of peroxisomal oxidation with a gain function of ACOX1 (ACOX1N237S) causes elevated ROS and retinal glial loss (Chung et al., 2020). Further exploration of peroxisomal redox balance in retinal function is needed.
Plasmalogen (Pls) are a subclass of glycerophospholipids defined by a vinyl-ether bond at sn-1 position of glycerol backbone. Dependent on the polar head group at sn-3 position, compositions of fatty acids at the sn-1 ether-linkage and sn-2 ester-linkage differs. Plasmalogen ethanolamine (PlsEtn) is the predominant subclass in photoreceptor rod inner segment and RPE (Acar et al., 2007; Bretillon et al., 2008; Acar et al., 2012). In the retina and RPE, PlsEtn accounts for 20–40 and 30% of ethanolamine glycerophospholipids, respectively (Heymans et al., 1983; Acar et al., 2007; Acar et al., 2012). PlsEtn is composed of ether-linked saturated fatty acid (16:0/18:0) or monounsaturated fatty acid (18:1n-9/18:1n-7) at sn-1 position and ester-linked PUFA (DHA/20:4n-6—arachidonic acid, ARA) at sn-2 position (Acar et al., 2012). Plasmalogen choline (PlsCho) is the predominant subclass in white matter and optic nerve where it accounts for 76 and 36% of total glycerophospholipids, respectively (Han et al., 2001; Acar et al., 2012). PlsCho is composed of ether-linked saturated fatty acid (16:0/18:0) or monounsaturated fatty acid (18:1n-9/18:1n-7) at sn-1 position and ester-linked saturated fatty acid (18:0) at sn-2 position (Han et al., 2001; Acar et al., 2012). The concentrated Pls level in RPE is likely to protect against elevated oxidative stress as discussed in previous section.
Peroxisome is critical for initiation of ether phospholipid synthesis as the initial two enzymes in a complex, glycerone-phosphate O-acyltransferase (GNPAT) and alkyl-glyceronephosphate synthase (AGPS) are targeted to inner peroxisomal membrane via PTS2 and PTS1 signal peptide, respectively (Hasan et al., 2013; Fujiki et al., 2014) (Figure 3). Dihydroxyacetone-phosphate (DHAP) is esterified to a long chain fatty acyl-CoA at sn-1 position by GNPAT, then AGPS catalyzes the exchange of 1-acyl-DHAP and long chain alcohol to produce 1-alkyl-DHAP. This exchange is essential to ensure synthesis of alk-1′-enyl ether bond by plasmanylethanolamine desaturate (encoded by TMEM189 gene) in ER (Gallego-Garcia et al., 2019; Werner et al., 2020). Long chain alcohol is synthesized by two peroxisomal acyl-CoA reductases: fatty acyl-CoA reductase 1 (FAR1) and fatty acyl-CoA reductase 2 (FAR2) (Cheng and Russell, 2004). FAR1 have high affinity for palmitoyl-CoA (16:0-CoA), stearoyl-CoA (18:0-CoA), and oleoyl-CoA (18:1n-9-CoA); thereby explaining unique composition of saturated and monounsaturated fatty acids at sn-1 ether linkage (Bishop and Hajra, 1981). In contrast, FAR2 catalyzes synthesis of long chain fatty alcohol by binding fatty acyl-CoA with >20 carbons (Otsuka et al., 2022). Finally, 1-alkyl-DHAP is reduced by peroxisomal acyl/alkyl DHAP reductase (ADHAPR—encoded by Dhrs7b) (Honsho et al., 2020). GNPAT is ubiquitously expressed in neural retina and RPE (Acar et al., 2007; Das et al., 2019). Retinal distributions of AGPS, FAR1, FAR2, and ADHAPR have not yet been characterized.
DHA is the predominant omega-3 polyunsaturated fatty acids (n-3 PUFA) in retinal phospholipid membrane and its accretion may modulate membrane fluidity which affects assemblies of protein complexes and their retinal activity (Fliesler and Anderson, 1983; Treen et al., 1992; Litman and Mitchell, 1996). In human, DHA accounts for 15.3% of total fatty acids in photoreceptor rod cell outer segments as compared to 1–4% in other tissues (Fliesler and Anderson, 1983; Acar et al., 2012). In contrast to photoreceptor rod cells, the compositions of total DHA and diDHA (unique phospholipids with two ester-linked DHA) species in phosphatidylcholine, phosphatidylethanolamine and phosphatidylserine were approximately 2-fold lower in cone-dominant retinas (Agbaga et al., 2018). The unique DHA accretion in the retina is maintained by three mechanisms: 1) dietary uptake of DHA (Young, 1976; Wang and Anderson, 1993b; Pifferi et al., 2012), 2) recycling of phospholipid DHA in rod outer segments via phagocytosis (Young, 1976; Stinson et al., 1991b; Gordon et al., 1992; Gordon and Bazan, 1993), and 3) synthesis of DHA from dietary precursors including α-linolenic acid (ALA, 18:3n-3), eicosapentaenoic acid (EPA, 20:5n-3), and DPAn-3 (22:5n-3) (Anderson et al., 1990; Wang and Anderson, 1993a; Alvarez et al., 1994; Delton-Vandenbroucke et al., 1997). Although biosynthesis of DHA from dietary n-3 PUFA precursor in retina and RPE is limited, impairment in peroxisomal β-oxidation of THA to DHA was observed in patients with X-linked retinitis pigmentosa (Wetzel et al., 1991; Hoffman et al., 2001).
DHA synthesis was first described to be solely localized to the ER where a series of alternating elongation by ELOVL5/2 and desaturation by FADS1/2 to produce DPAn-3. DPAn-3 is subsequently desaturated to DHA by Δ4-desaturase. However, the presence of Δ4-desaturase in retina is still heavily debated (Metherel and Bazinet, 2019). Since the publication of Voss et al., in 1991, the dogma of DHA synthesis shifted from a localized ER pathway to concerted efforts between ER and peroxisome which is later termed the Sprecher Pathway (Voss et al., 1991; Sprecher et al., 1995) (Figure 3). From fibroblasts of patients with Zellweger and Refsum disease, the final steps of DHA synthesis were characterized as such that DPAn-3 is elongated to tetrapentaenoic acid (TPA, 24:5n-3) by ELOVL2 and desaturated to THA by FADS2 in ER, then transported to peroxisome via ABCD2, where peroxisomal β-oxidation retroconvert THA to DHA for export (Moore et al., 1995; Morita and Imanaka, 2012). The other possibility is that peroxisomal dysfunction also affect the DHA recycling from photoreceptor outer segment shedding, which will be further discussed in Session 3.
Peroxisomal dysfunction in patients causes DHA deficiency in the brain and retina (Martinez, 1992). Lack of DHA contribute to the pathophysiology of multiple retinal disorders (Fu et al., 2019). In retinal diseases including diabetic retinopathy, retinopathy of prematurity (ROP), age-related macular degeneration (AMD) and inherited retinal degenerative diseases, DHA deficiency is observed. However, DHA supplementation shows positive impacts in some but not all studies (Fu et al., 2019).
Bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA), play an important role in homeostasis of cholesterol metabolism in the retina. Unconjugated and taurine-conjugated metabolites of CDCA, ursodeoxycholic acid and tauroursodeoxycholic acid, are neuroprotective of photoreceptors and ganglion cells (Daruich et al., 2019; Win et al., 2021). Classic bile acid synthesis commenced with conversion of cholesterol to 7α-hydroxycholesterol by 7α-hydroxylase (CYP7A1) leading to production of DHCA and THCA in mitochondria (Chiang, 2004). Then in the ER, CoA activation of DHCA and THCA is catalyzed by bile acid-CoA ligase (BACL) (Wheeler et al., 1997; Falany et al., 2002). DHCA-CoA and THCA-CoA are then transported by ABCD3 into peroxisome where β-oxidation chain shortening produce chenodeoxycholoyl-CoA and choloyl-CoA, respectively (Ferdinandusse et al., 2015) (Figure 4). In contrast to liver, retina predominantly relies on an alternative pathway for bile acid synthesis where mitochondrial CYP27A1 in rod inner segment and ER CYP46A1 in retinal ganglion cells convert cholesterol to 27-hydroxycholesterol and 24-hydroxycholesterol, respectively (Lee et al., 2006; Ramirez et al., 2008; Ishikawa et al., 2016; Mertens et al., 2017). Hydroxylated cholesterol intermediates of CYP27A1 and CYP46A1 are then converted to DHCA and THCA at which classic pathway of CDCA and CA synthesis resume (Omarova et al., 2012; Saadane et al., 2014). Chenodeoxycholoyl-CoA and choloyl-CoA are then conjugated with taurine or glycine by peroxisomal bile acid-CoA:amino acid N-acyltransferase (BAAT) to produce tauro/glycochenodeoxycholate and tauro/glycocholate (Pellicoro et al., 2007).
Based on the results of enzyme activity studies and biochemical markers, the peroxisomal disorders was historically divided into three groups: 1) peroxisomal biogenesis disorders (PBD); 2) multiple enzyme deficiencies; and 3) single enzyme/transporter deficiency disorders. With recent development in the understanding of the biochemical and molecular genetic bases of these conditions, a new system has been proposed to divide the PBD into two subtypes: Zellweger Spectrum Disorder (PBD-ZSD) and the rhizomelic chondrodysplasia punctata (RCDP) spectrum (previously known as the multiple enzyme deficiencies). Other conditions such as Heimler Syndrome (HS), which was previously classified as a distinct syndrome consists of sensorineural hearing loss, amelogenesis inperfecta (AI) and nail anomalies with or without visual defect, is now considered as part of the PBD-ZSD (Ratbi et al., 2015). Single enzyme/transporter deficiency disorders such as X-linked adrenoleukodystrophy (X-ALD) and peroxisomal acyl-CoA oxidase (ACOX) deficiency remains a separate entity.
PBD-ZSD is a group of conditions with a very broad clinical spectrum ranging from neonatal lethal conditions with severe neurological presentation to adult-onset isolated visual impairment, sensorineural hearing loss, adrenal insufficiency, or liver disease. The incidence of PBD-ZSD was estimated to be around 1 per 50,000 births (Braverman et al., 2016). PBD-ZSD is characterized by biochemical findings of elevated plasma VLCFAs (C24 and C26), elevated plasma C24:C22 and C26:C22 ratio, elevated plasma phytanic acid, pristanic acid, pipecolic acid and bile acids concentrations, reduced erythrocyte plasmalogen level, and elevated urinary excretion of dicarboxylic acid (Steinberg et al., 2006; Braverman et al., 2016) (Figure 4). Patients with PBD-ZSD typically carry biallelic (homozygous or compound heterozygous) pathogenic variants in the PEX family genes, which encode proteins involved in peroxisome biogenesis and proliferation. A total of sixteen PEX genes have been identified in humans, and PBD-ZSD have been associated with PEX1, PEX2, PEX3, PEX5, PEX6, PEX10, PEX11β, PEX12, PEX13, PEX14, PEX16, PEX19, and PEX26 (Ebberink et al., 2009; Waterham and Ebberink, 2012; Krause et al., 2013; Fedick et al., 2014; Kettelhut and Thoms, 2014; Komatsuzaki et al., 2015; Konkolova et al., 2015; Renaud et al., 2016; Waterham et al., 2016; Bjorgo et al., 2017; Rydzanicz et al., 2017; Stowe and Agarwal, 2017; Kumar et al., 2018). Patients with loss-of-function PEX gene defects and abolished peroxin activity, are most severe in their clinical presentation. In contrast, variants with residual peroxin function, such as missense variants, result in a milder cellular and clinical phenotype. However, not all missense variants have residual activity, and the clinical course of a patient cannot be predicted by the genetic variants alone.
Diagnosing patients with mild presentation of PBD-ZSD has been challenging due to the diverse and non-specific nature of the symptoms (Enns et al., 2021). Nonetheless, recent expansion in newborn screening programs have included screening of X-ALD using liquid chromatography tandem mass spectroscopy (LC-MS/MS) to detect VLCFA, C26:0-lysophosphatidyl choline (C26:0-LPC), from dried blood spots (Turk et al., 2020). First introduced in 2013, newborn screening for X-ALD is now carried out in 20 States and the District of Columbia with 2 additional states currently running pilot programs (https://adrenoleukodystrophy.info/clinical-diagnosis/ald-newborn-screening). Therefore, every newborn from these states will have C26:0-LPC testing at birth, which will identify all newborns with PBD-ZSD (Klemp et al., 2021). Concerns have been raised around the “incidental” identification of PBD-ZSD patients through the X-ALD screening due to lack of targeted treatment that modifies the course of PBD-ZSD. Therefore, development of targeted PBD-ZSD treatment is in dire need.
The central nerves system (CNS) manifestations of PBD-ZSD involves structural brain malformations, predominantly involve the neuronal migration defects such as polymicrogyria, heterotopia, agenesis/hypoplasia of the corpus callosum, pachygyria, and hypoplastic olfactory bulb. Cerebral ventricular abnormalities such as colpocephaly and progressive dilatation of the ventricles were also reported (Nakai et al., 1995). Demyelination and global cerebral atrophy have also been reported in both early and late onset cases (Renaud et al., 2016). Ocular phenotypes related to PBD-ZSD includes anterior changes of cornea clouding, cataracts, and Brushfield spots (small, white or greyish/brown spots on the periphery of the iris). Posterior ocular findings include Leber congenital amaurosis (LCA), pigmentary retinopathy, and optic nerve dysplasia (Lambert et al., 1989; Mechaussier et al., 2019). LCA is a severe form of early onset retinal dystrophy. Patients with LCA typically have visual impairment since infancy. Symptoms of LCA include photophobia, nystagmus, keratoconus, and behavior of eye poking and picking (oculo-digital signs). Retinal changes associated with PBD-ZSD involve all retinal layers from RPE to retinal ganglion cells (Haddad et al., 1976), with most severe loss in photoreceptors (Haddad et al., 1976; Kretzer et al., 1981; Garner et al., 1982). These changes are scattered from central to peripheral retina. The optic nerve of patients with PBD-ZSD can be profoundly demyelinated (Thomas et al., 1975). Fundoscopic changes in patients with PBD-ZSD can include retinal arteriolar attenuation, pale optic disc and loss of pigment epithelium most prevalent in the macular area (Folz and Trobe, 1991).
Heimler Syndrome is a condition found to be caused by pathogenic variants in PEX1 and PEX6 genes (Ratbi et al., 2015). It represents the mild end of PBD-ZSD. Long term follow-ups of index cases revealed “salt-and-pepper”-like mottling of RPE and abnormal electroretinogram (ERG) recording in young adulthood after complaints of decreased vision in a female patient. The patient also had cystoid macular edema in keeping with the diagnosis of macular dystrophy (Lima et al., 2011). After evaluating other patients with Heimler syndrome, three additional patients with PEX1 variants and one patient with PEX6 variants were also found to have retinal pigmentary changes (Heimler et al., 1991; Zaki et al., 2016). Given the rarity of these conditions, most literature is constituted with case studies describing the retinal phenotype of PBD-ZSD. Studies focusing on the natural history of retinal dystrophy in PBD-ZSD are required to understand its progressive impact on vision.
Peroxisomal acyl-CoA oxidase deficiency disorder caused by biallelic pathogenic variants in ACOX1 gene and D-bifunctional protein (DBP) deficiency disorder caused by either or both MFE 2-enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase deficiency, are disorders of peroxisomal fatty acid β oxidation (Figure 3). They can present with neonatal onset severe neurological sequela with progressive white-matter demyelination, seizures, and cortical malformations. Both conditions were associated with visual impairment, abolished electroretinogram possibly related to retinal degeneration or subretinal pigmentary retinopathy (Suzuki et al., 2002; Kurian et al., 2004; Bae et al., 2020). ACOX1 is abundantly expressed in the RPE as compared to neural retina, whereas the distribution of ACOX2 and ACOX3 in RPE and retinal layers remains unclear (Das et al., 2019). ACOX1 deficiency results in premature death in adolescence and are associated with abnormal retinal pigmentation, retinal degeneration, and optic atrophy (Das et al., 2021). Patients with ACOX1 deficiency have elevated plasma and fibroblast saturated VLCFA and comparable saturated LCFA, BCFA (phytanic acid), and DHCA/THCA to control (Ferdinandusse et al., 2007). MFE-2 is equally or more expressed in the neural retina than RPE, whereas expressions of MFE-1 has yet to be elucidated in the eyes (Das et al., 2019). Severe MFE-2 deficiency may manifest symptoms comparable to that of PBD-ZSD; however milder juvenile onset deficiency may present with abnormal retinal pigmentation with normal visual acuity (Das et al., 2021). Patients with MFE-2 deficiency have elevated levels of saturated VLCFA, BCFA (pristanic acid), and DHCA/THCA and reduced levels of plasma and retinal DHA (Martinez, 1992; Ferdinandusse et al., 2006a). Patients with peroxisomal ACOX deficiency and MFE-2 deficiency show no accumulation of pipecolic acid, with no reduction in erythrocyte plasmalogens. MFE-2 deficiency is associated with increased plasma levels of bile acid intermediates, but ACOX deficiency is not (Ferdinandusse et al., 2007).
SCPx deficiency was first reported in 2006 in a patient with normal level of VLCFA but elevated level of pristanic acid (Ferdinandusse et al., 2006b). The only ocular finding reported was pathological saccadic eye movements indicating potential pathology in the brain stem. Another patient with SCPx deficiency had mild symptoms of nyctalopia. This individual had normal visual acuity. Detailed ocular investigation revealed peripapillary atrophy, irregular autofluorescence associated with the major vascular arcades, with no peripheral pigmentary retinal changes (Horvath et al., 2015; Morarji et al., 2017). ACAA1 deficiency has yet to be associated with human disease.
FALDH deficiency leads to Sjögren-Larsson Syndrome, a condition associated with accumulation of phytol. Sjögren-Larsson Syndrome is associated with reduced visual acuity, degeneration of Müller cells in the inner retina, retinal thinning, RPE atrophy, and photoreceptor dysfunction (van den Brink et al., 2004; Nanda and Kovach, 2019). PHYH deficiency is one of the primary causes of Refsum disease (Jansen et al., 1997) (Figure 4). Patient with Refsum disease have elevated levels of phytanic acid in blood and tissues. Clinical symptoms of Refsum disease includes retinitis pigmentosa, miosis with attenuated pupillary light response, iris atrophy, and cataract (Ruether et al., 2010). Phytanic acid exposure cause morphological changes in both fetal bovine and human RPE in vitro, mimicking the RPE pathology observed in patients with Refsum’s disease (Bernstein et al., 1992). HACL1 deficiency has yet to be reported in humans; however, animal model suggests that dietary intake of phytol in Hacl1−/+ mice exhibited elevated levels of phytanic acid in blood and tissues similar to the biochemical profile of patients with Refsum disease (Mezzar et al., 2017). Interestingly, the human HACL1 is only three amino acids different from its mouse analog. Clinical phenotypes of AMACR deficiency include retinitis pigmentosa and decreased visual acuity (Thompson et al., 2009; Stewart et al., 2011).
Defects in AGXT gene results in primary hyperoxaluria type 1, which is an inborn error of metabolism characterized by accumulation of calcium oxalate (Figure 5). This process leads to calcinosis cutis metastatica of the skin, tooth mobility, vascular spasm/occlusion, and nephrocalcinosis. Retinal complications include optic atrophy, retinopathy and choroidal neovascularization (Leumann and Hoppe, 2001; Cochat et al., 2006; Lorenzo et al., 2006). Patients with primary hyperoxaluria type 1 also exhibit crystalline deposit in RPE leading to bilateral vision loss (Roth et al., 2012; Kourti et al., 2020).
GNPAT deficiency results in RCDP type 2 (Wanders et al., 1992) (Figure 3). Patients with GNPAT deficiency have low plasmalogen levels in erythrocytes. They typically have a shortened life expectancy not exceeding a decade (Itzkovitz et al., 2012). Congenital cataracts have been reported (Wanders et al., 1992). AGPS deficiency results in RCDP type 3 and patients exhibited lower levels of plasmalogen in erythrocytes (Wanders et al., 1994). FAR1 deficiency results in autosomal recessive RCDP type 4 (Buchert et al., 2014). FAR1-deficient patients have markedly lower levels of plasmalogens, and congenital cataracts is a cardinal symptom (Buchert et al., 2014). Interestingly, patients with autosomal dominant FAR1 de novo variants also shares similar clinical phenotypes including congenital and juvenile cataracts, but a divergent biochemical phenotype of elevated plasmalogens in fibroblasts (Ferdinandusse et al., 2021). FAR1 de novo variant disrupted negative feedback regulation leading to un-inhibited biosynthesis of plasmalogen (Ferdinandusse et al., 2021). Deficiency in FAR2 has yet to be associated with human diseases.
Although, ABCD1 was shown to be expressed ubiquitously between RPE and neural retina in rodent model (Das et al., 2019), patients with X-ALD do not exhibit direct retinal pathology. The visual impairment in X-ALD patients are caused by two mechanisms: 1) the loss of color vision in a subgroup of X-ALD patients are caused by continuous gene deletions in X chromosome where ABCD1 gene and red pigment gene are in close proximity (Sack et al., 1989) or by chromosome rearrangements (Alpern et al., 1993), 2) the cortical blindness associated with X-ALD is caused by visual track demyelination, which can subsequently lead to retrograde degeneration of ganglion cells (Kaplan et al., 1995; Ohkuma et al., 2014).
Patients with ABCD3 deficiency have elevated levels of DHCA and THCA, but ocular abnormalities have not yet been identified (Ferdinandusse et al., 2015). Patients with BAAT deficiency exhibited elevated levels of unconjugated bile acids (Carlton et al., 2003). BAAT expression in retinal cells are unknown and patients with BAAT deficiency has no reported ocular abnormalities.
Recently, patients with biallelic pathogenic variants in acyl-CoA binding domain containing protein 5 (ACBD5) gene have been reported (Abu-Safieh et al., 2013; Ferdinandusse et al., 2017; Bartlett et al., 2021). These patients demonstrated progressive neurological deterioration, nystagmus, optic atrophy, cone-rod dystrophy, early onset retinal dystrophy, attenuation of eye vessels, and diffuse granularity of RPE.
Currently, there is no curative or targeted disease modifying treatment for PBD-ZSD. Cholic acid (CA) therapy was approved by Food and Drug Administration (FDA) in March 2015 to treat liver disease in patients with PBD-ZSD. CA has been shown to decrease toxic bile acid intermediates, improve transaminase levels, and reduce liver inflammation, with improvement in growth parameters in pediatric populations (Anderson et al., 2021). Since PBD-ZSD patients have markedly low level of DHA (Janssen et al., 2000), trials of DHA supplementation were made as an attempt to restore the neurological, ocular and hepatic function in PBD-ZSD patients. M. Martinez conducted a cohort study in the 1990s treating 20 PBD-ZSD patients with DHA supplementation. Patients were treated with variable doses of DHA ethyl ether (depends on age and blood DHA level) between 9 months and 9 years and more than half of the patients not only had improved biochemical markers such as reduced VLCFA and increased erythrocyte plasmalogen level, but also improved brain and retinal function compared to pre-treatment (Martinez, 2001). Nonetheless, the clinical improvement was not subjectively quantified. A follow up study focusing on the visual function of this cohort demonstrated resolution of nystagmus, improvement of ERG recording, and amelioration of visual behaviors in some cases and stabilization in the rest. The improvement was better appreciated in patients who had baseline ophthalmological assessment (Noguer and Martinez, 2010). Based on this preliminary work, a randomized double-blind case control trial was conducted supplementing a fixed dose (100 mg/kg/d) of DHA triglyceride (47% DHA) and arachidonic acid (ARA; 20:4n-6) triglyceride (46% ARA) (Paker et al., 2010). This is a short-term study in 50 patients (25 in treatment group and 25 in control) with follow ups in 1 year. The study also focused exclusively on quantitative biometrics of patients such as ERG changes, weight, height and biochemical markers such as VLCFA, plasmalogen and liver enzymes. Given the short-term intervention and the broad spectrum of clinical variability of PBD-ZSD patients, it was not surprising that no statistically significant changes were obtained between the two groups. At this time, there is very limited evidence supporting the efficacy of clinical use of DHA in patients with PBD-ZSD.
Peroxisomes are highly dependent on the interaction with other organelles (ER, mitochondria and lysosomes) in order to perform proper metabolism. For example, some of the peroxisomal substrates (e.g., VLCFAs) are synthesized and elongated from shorter chain fatty acids (SCFAs) in ER. In addition, the end products of peroxisomal β-oxidation subsequently enter mitochondria for metabolism and energy production.
Most of VLCFAs are not derived from dietary source, instead, FAs are elongated in ER by cycling through a four-step process (condensation, reduction, dehydration and reduction) (Figure 6) (Kihara, 2012). Acyl-CoA is elongated to have two more carbon units in each cycle. The FA elongases ELOVL1-7 have characteristic substrate specificity (Kihara, 2012). ELOVL1,3,4,6 and 7 elongate SFAs and MUFAs, while ELOLV2 and 5 are strictly PUFA-specific. VLCPUFAs uniquely exist in mammalian retina, brain, and sperm (Kihara, 2012). ELOLV4 is critical for the synthesis of VLCFAs and expressed highest in the retina (Agbaga et al., 2010; Kihara, 2012). Mutations in the ELOVL4 gene leads to Stargardt disease type 3 that causes vision loss starting around 14 years old (Edwards et al., 1999; Agbaga et al., 2010). Loss of photoreceptor ELOLV4 in mice causes significant decrease in retinal glycero-phospholipids containing VLCPUFAs, abnormal accumulation of lipid droplets and lipofuscin-like granules, as well as decreased retinal neuronal responses by ERG (Harkewicz et al., 2012). In addition, adiponectin receptor 1 (ADIPOR1), which is predominantly expressed in photoreceptors (Fu et al., 2017), may recycle VLCPUFAs (containing DHA C22:6) from shed photoreceptor apical disk membrane back to photoreceptor inner segments and further elongate in ER, thus preserves photoreceptor cell survival (Rice et al., 2015). The VAMP-associated proteins (VAP)-ACBD5 complex acts as the primary ER-peroxisome tether, and loss of VAP or ACBD5 increases peroxisome mobility. VAP-ACBD5-mediated contact between the ER and peroxisomes is also important for peroxisome growth and lipid homeostasis (Costello et al., 2017; Hua et al., 2017).
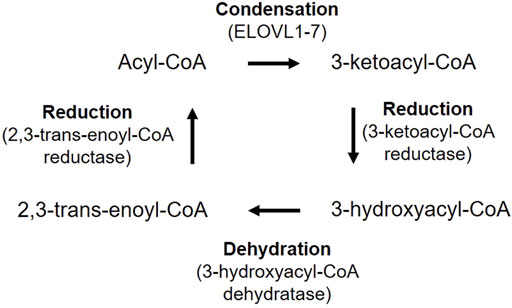
FIGURE 6. VLCFA biosynthesis in mammalians. Acyl-CoA is first condensed with malonyl-CoA to produce 3-ketoacyl-CoA, catalyzed by fatty acid elongase (ELOVL1-7). 3-ketoacyl-CoA is reduced to 3-hydroxyacyl-CoA by 3-ketoacyl-CoA reductase. 3-Hydroxyacyl-CoA is then dehydrated by 3-hydroxyacyl-CoA dehydratase and generate 2,3-trans-enoyl-CoA. Finally, 2,3-hydroxyacyl-CoA is reduced to an acyl-CoA by 2,3-trans-enoyl-CoA reductase, with two more carbon chain units than the original acyl-CoA.
In addition, plasmalogen synthesis starts in peroxisomes and completes in ER (Nagan and Zoeller, 2001; Braverman and Moser, 2012). The initial two steps are catalyzed by peroxisomal matrix enzymes dihydroxyacetone phosphate acyltransferase (DHAPAT/GNPAT) for transfer of acyl-DHAP across the enzyme active sites, and AGPS for the exchange of the acyl group (fatty acid) for an alkyl group (fatty alcohol, supplied by FAR1). Alkyl-DHAP is further reduced and plasmalogen synthesis proceeds in the ER (Figure 2).
Moreover, ER is involved in peroxisome biogenesis. Peroxisomal membrane proteins (PMP) are thought to be first inserted in ER and subsequently exit from ER to peroxisomes (Dimitrov et al., 2013). It has also been proposed that peroxisomes originate from ER and the avoidance of toxic lipid by-products is the driving force for the separation of the peroxisomes (Gabaldon, 2014). In mammalian cells, PEX16 with an appended NH2-terminal type I signal anchor sequence moves to peroxisomes after being cotranslationally synthesized in the ER (Kim et al., 2006). In mouse dendritic cells, PEX13 and PMP70 are present in the ER domain with a peroxisomal reticulum from which mature peroxisomes are derived (Geuze et al., 2003). Meanwhile, the peroxisome biogenesis factors PEX19 and PEX3 cooperate and mediates the posttranslational targeting of proteins such as UBXD8, a lipid droplet-destined membrane protein and reticulon homology domain-containing protein to ER (Schrul and Kopito, 2016; Yamamoto and Sakisaka, 2018). Therefore, the close connection between peroxisome and ER is evident.
Peroxisomal substrates such as VLCFAs can also be generated from lysosomal degradation of different lipid species. Rod photoreceptor outer segment is rich in VLCPUFA and LCPUFA (DHA and ARA), which comprise ∼60% of total phospholipids (Stinson et al., 1991a). To maintain normal retinal function, there is a continuous shedding of photoreceptor outer segment on daily basis (Fliesler & Anderson, 1983). The sheded outer segments are ingested by RPE via phagocytosis and packaged into membrane-bound phagosomes, which are then fused with lysosomes for subsequent degradation (Bosch et al., 1993; Ferrington et al., 2016). It has been reported that there is a dynamic membrane contact between peroxisomes and lysosomes mediated by lysosomal Synaptotagmin VII (Syt7) binding to the lipid PI(4,5)P2 on peroxisomal membrane, and the organelle contact facilitates the transport of cholesterol from lysosomes to peroxisomes (Chu et al., 2015). Cholesterol is accumulated in cells and animal models of peroxisomal disorders (Chu et al., 2015). These findings suggest the potential involvement of peroxisomes in VLCFAs recycling during photoreceptor outer segment shedding. Although RPE immediately recycles DHA released from the disc membrane during degradation back to photoreceptors for disc membrane synthesis (Bazan et al., 1992), the involvement of peroxisome in this process is yet to be characterized. Moreover, compromised lysosomes with impaired degradative enzyme activity leads to lipofuscin-like autofluorescence in RPE (Guha et al., 2014). Understanding of the interactions between peroxisome and lysosome will uncover the basic lipid biology and balance in retina.
Peroxisomes and mitochondria tightly interact with each other in fatty acid metabolism and undergo close communication through three major pathways: 1) there is a physical association between these two organelles, and the degree of contact changes dynamically with the cell types and microenvironment (Fransen et al., 2017). For example, the peroxisomal membrane protein PEX34 (conserved to humans) and the outer mitochondrial membrane protein FZO1 (mammalian homologue Mitofusin 2) tether the peroxisome and mitochondria in the yeast (Shai et al., 2018). 2) mitochondria-derived vesicles containing mitochondria-anchored protein ligase that can fuse with a subset of peroxisomes (Neuspiel et al., 2008); 3) peroxisomes and mitochondria communicates through releasing of biological messengers such as ROS, lipids and NAD+ etc.
VLCFAs, which cannot be degraded in mitochondria, need to be shortened by peroxisome β-oxidation. Shortened VLCFAs are further oxidized in mitochondria for energy production. Peroxisomal β-oxidation can also compensate defective mitochondrial fatty oxidation by breaking down medium-chain and long-chain fatty acids (Violante et al., 2019; Ranea-Robles et al., 2021). Studies of RPE from AMD vs control patients indicated that proliferation of peroxisomes is accompanied with mitochondrial abnormalities (Feher et al., 2006). Abnormal peroxisomal metabolism and redox status can rapidly disturb mitochondrial redox state. The antioxidant enzyme CAT is mainly localized in peroxisomes. Deficiency of CAT increases redox state of mitochondria and peroxisome-derived oxidative stress causes mitochondrial redox imbalance (Ivashchenko et al., 2011). Retinal transfection of AAV-expressing CAT and superoxide dismutases 2 (SOD2) prolonged cone photoreceptor survival in mice during retinal degeneration (Xiong et al., 2015), and AAV-CAT also exerts neuroprotective effects on retinal ganglion cells in ischemia-induced retinal injury in rats (Chen and Tang, 2011).
In addition, peroxisomal oxidation also regulates lipid composition in mitochondria membrane, which in turn regulates mitochondrial dynamics and ATP production (Petrushka et al., 1959; Hatch, 2004). Oxidative stress induces release of cytochrome c from mitochondria, and cytochrome c cleaves plasmenylcholine (plasmalogens of the phosphocholine head group class) in membrane bilayers (Jenkins et al., 2018). Loss of plasmalogens in mitochondrial membrane causes inefficient respiration (Kimura et al., 2019). As VLCPUFAs are recycled back to the retina during photoreceptor outer segment renewal (Bazan et al., 1992; Gordon et al., 1992), mitochondrial DHA and ARA content could also be potentially modulated by peroxisomal oxidation. Both dietary DHA (no ARA) and ARA (no DHA) in rats profoundly alters cardiac mitochondrial phospholipid fatty acid compositions, and suppresses Ca2+-induced opening of the mitochondrial permeability transition pore (MPTP), which causes cell death (Khairallah et al., 2010; Khairallah et al., 2012); dietary DHA (no ARA) also depletes cardiac mitochondrial ARA content (Khairallah et al., 2012). These findings suggest that peroxisomal oxidation may control mitochondrial membrane composition and activity via VLCPUFA recycling. More recently, it has been reported that adipose specific knockout of the peroxisomal biogenesis factor PEX16 or the plasmalogen synthetic enzyme GNPAT blocks cold-induced mitochondrial fission, decreases mitochondrial copy number and causes abnormal mitochondrial function. Dietary plasmalogen increases mitochondrial copy number, improves mitochondria function in mice with Pex16 deficiency (Park et al., 2019). This work provides direct evidence of peroxisome-derived ether lipid regulating mitochondrial activity.
Moreover, mitochondrial respiration is also critical to reoxidize NADH formed in peroxisomes and maintain peroxisomal β-oxidation (Wanders et al., 2015). Altered NAD (+)/NADH redox state with age-dependent increases in intracellular NADH and decreases in NAD (+) content is shown in human brain (Zhu et al., 2015), reflecting declined mitochondrial function. NAD (+) levels also decrease in mouse RPE cells with aging (Jadeja et al., 2018). Mutations of gene encoding nicotinamide mononucleotide adenylyltransferase 1 involved in NAD (+) biosynthesis reduce the enzymatic activity and causes blindness within the first year after birth (Koenekoop et al., 2012). Taken together, peroxisomal metabolic and redox homeostasis is important to maintain mitochondrial function. Further understanding of peroxisomal and mitochondrial interaction will provide more clues to understand cellular activity in healthy and diseased retinas.
Although there are limited studies regarding the disease pathogenesis of peroxisome-related retinal dysfunction, we could gain some knowledge from the brain investigations as retina is part of the CNS (London et al., 2013). Peroxisomal dysfunction leads to compromised antioxidant defense with decreased CAT expression, attenuated D-serine degradation, disrupted lipid composition in myelin sheath and cellular membranes in the CNS (Uzor et al., 2020), which in turn cause abnormalities in the myelinated axons and synaptic transmission (Berger et al., 2016). Accumulated VLCFAs and decreased plasmalogen ethanolamine levels are observed in the brain of patients with Alzheimer’s disease (Ginsberg et al., 1995; Kou et al., 2011). Reduced DHA, ARA and plasmalogen ethanolamine levels are found in the brain of patients with Parkinson’s disease, and supplementation of DHA-containing plasmalogen precursor protects against striatal dopamine loss in mice modeling Parkinson’s disease (Fabelo et al., 2011; Miville-Godbout et al., 2017). These findings suggest an essential role of peroxisomes and the related lipid metabolism in maintaining brain health.
The contribution of peroxisomes to brain function could be cell-dependent. In mice, dysfunction of peroxisome in neural cells leads to abnormal cortical neuronal migration and maturation of the cerebellum (Janssen et al., 2003; Krysko et al., 2007; Muller et al., 2011). Peroxisome deficiency in astrocytes disturbs brain-derived neurotrophic factor levels and dysregulates axogenesis of the hippocampus neurons (Abe et al., 2020), suggesting that astrocytic peroxisomes modulate neuronal integrity. Moreover, microglial peroxisomal dysfunction induces an inflammatory activated and proliferative state in the mouse brain, but does not affect neuronal health up to one-year old and the long-term impacts is yet to be evaluated (Beckers et al., 2019). Therefore, it would be important to uncover the contribution of neuronal and glial peroxisomal activity to neuronal function in the CNS.
Defective peroxisomal function causes disturbed retinal homeostasis and retinal degeneration. However, the mechanisms behind are largely unknown. We speculate that the disrupted lipid metabolism and redox balance typically regulated through the interaction between peroxisomes, lysosomes, mitochondria and ER may contribute to the disease development and progression. Further investigations in the pathophysiology and natural history of peroxisomeal disorder associated retinal dystrophy will greatly advance the potential for therapeutic development.
The retinal lipid homeostasis is essential in maintaining normal retinal neuronal and vascular function (Fu et al., 2019; Fliesler, 2021). As there are extensive lipid alterations accompanied with peroxisomal dysfunction, it is important to know that whether all or certain altered lipids are the significant contributors to the development of retinal diseases. Lipids are important membrane components of cells and organelles. Change in lipid composition alters organelle function and cell status. In addition, lipids and their downstream lipid metabolites can function as signaling molecules to induce various cellular responses in metabolism, inflammation, cell survival and apoptosis. Moreover, fatty acids such as palmitate are also possible fuel substrates for photoreceptor, but other type and source of fatty acids are understudied (Joyal et al., 2016). Supplementation of specific deficient lipids and modulating the downstream targeting pathways may be protective against retinal degeneration.
Furthermore, the interaction between RPE, Müller glia, and neuronal cells in retina is also important for the modulation of lipid availability and clearance of neighboring photoreceptors (Fu et al., 2021). For example, disrupted lipid homeostasis in RPE causes lipid droplet formation and RPE dysfunction with aging (Yako et al., 2022). Interestingly, peroxisomes and lipid droplets have physical interaction within cells. Peroxisomal β-oxidation-derived ROS regulate the levels of adipose triglyceride lipase, which control lipolysis of intracellular lipids (Ding et al., 2021). Therefore, increasing peroxisomal activity may decrease lipid droplets in RPE, preserve RPE function and in turn photoreceptor health. Fatty acid profiling focuses on the entire tissue and radiolabeled lipid tracing aids the understanding of the kinetic incorporation, turnover and loss of fatty acids in the retina (Rodriguez de Turco et al., 1990; Gordon and Bazan, 1993; Santos et al., 1995). However, the knowledge of cell-specific uses of lipids and the interactions among different lipid metabolic pathways are limited. The feasibility of single-cell transcriptomics and cell-specific lipidomics in retinal research may provide insight to the cell specific lipid and fatty acid metabolism. Furthermore, progress in characterizing dynamic processing of fatty acids in retina is often limited by the high cost of radiotracer and retinal tissue size. Recently, a cost-effective, tracer-free novel methods using compound-specific isotope analysis (CSIA) has been established to trace fatty acid metabolism by utilizing food sources differing in natural carbon isotope abundance. Briefly, because the carbon isotopic composition of a molecule is conserved following dietary intake, highly precise measurement of carbon 13 (13C) abundance can provide insights to the dietary source that contributed to the incorporation of 13C to an endogenous molecule. Therefore, by designing well-controlled dietary switch experiments that switches diets made from foods less enriched in 13C foods (C3 plants) to foods more enriched in 13C (C4 plants), fatty acid incorporation, turnover, and synthesis can be measured as effectively as radiotracer experiments (Lacombe et al., 2020; Lacombe and Bazinet, 2021). Moreover, due to the high precision of 13C measurements by gas chromatography-isotope ratio mass spectrometry (GC-IRMS), cell-specific retinal extract can be quantified.
Altered peroxisomal function has been implicated in age-related neurodegenerative diseases and diabetes-related complications (Cipolla and Lodhi, 2017). In RPE from AMD vs control patients, mitochondrial changes are associated with proliferation of peroxisomes (Feher et al., 2006). However, it is unclear that whether this mitochondrial response is protective or detrimental. Further investigation is required.
Furthermore, the knowledge of DHA metabolism and the application of dietary DHA for prevention of retinal dysfunction is translatable to a number of retinal diseases. Some (but not all) clinical trials showed that fish oil (rich in DHA and EPA) supplementation reduces risk for severe ROP (Pawlik et al., 2011; Beken et al., 2014; Pawlik et al., 2014; Molloy et al., 2016; Najm et al., 2017). Fish oil supplementation or dietary intake of food rich in DHA decreased the risk for AMD in a number of trials (Sangiovanni et al., 2009; Tan et al., 2009; Christen et al., 2011; Ho et al., 2011). However, dietary supplementation of DHA (350 mg/day) and EPA (650 mg/day) did not reduce the risk of advanced AMD in Age-Related Eye Disease Studies (AREDS/AREDS2) (Age-Related Eye Disease Study 2 Research Group, 2013). The reasons for the inconsistent observations are unclear. It has been noted that DHA and EPA supplements may lower circulating ARA levels (D’ascenzo et al., 2014; Zhao et al., 2015) and decrease the ARA/DHA ratio in preterm infants (Najm et al., 2017). Low serum ARA level in the postnatal period is strongly associated with development of clinically significant ROP (Lofqvist et al., 2018) and sufficient ARA is required for DHA to protect against severe ROP (Hellstrom et al., 2021). In addition, DHA- and ARA-oxylipins generated via cytochrome P450 oxidases exaggerates while DHA- and ARA-derived oxylipins via lipoxygenases inhibit pathological retinal neovascularization (Sapieha et al., 2011; Gong et al., 2016; Gong et al., 2017). Therefore, metabolic imbalance among these pathways may also contribute to the inconsistent observations among different trials.
In conclusion, understanding of retinal cell-specific peroxisomal function, VLCFA metabolism and natural history of retinal dysfunction in peroxisomal disorders is a key to development of disease modifying therapeutics. Given the involvement of peroxisomal dysfunction in various retinal pathophysiology, this therapeutic intervention is likely to benefit common multifactorial retinal degenerative disorders as well.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
ZF is supported by NIH R01EY032492, NIH R01EY017017, Boston Children’s Hospital (OFD/BTREC/CTREC Faculty Career Development Grant 97906, Pilot Grant 92214, and Ophthalmology Foundation 85010), Mass Lions Eye Foundation 87820.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abe, Y., Honsho, M., Kawaguchi, R., Matsuzaki, T., Ichiki, Y., Fujitani, M., et al. (2020). A peroxisome deficiency-induced reductive cytosol state up-regulates the brain-derived neurotrophic factor pathway. J. Biol. Chem. 295, 5321–5334. doi:10.1074/jbc.RA119.011989
Abu-Safieh, L., Alrashed, M., Anazi, S., Alkuraya, H., Khan, A. O., Al-Owain, M., et al. (2013). Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 23, 236–247. doi:10.1101/gr.144105.112
Acar, N., Berdeaux, O., Gregoire, S., Cabaret, S., Martine, L., Gain, P., et al. (2012). Lipid composition of the human eye: Are red blood cells a good mirror of retinal and optic nerve fatty acids? PLoS One 7, e35102. doi:10.1371/journal.pone.0035102
Acar, N., Gregoire, S., Andre, A., Juaneda, P., Joffre, C., Bron, A. M., et al. (2007). Plasmalogens in the retina: In situ hybridization of dihydroxyacetone phosphate acyltransferase (DHAP-AT)--the first enzyme involved in their biosynthesis--and comparative study of retinal and retinal pigment epithelial lipid composition. Exp. Eye Res. 84, 143–151. doi:10.1016/j.exer.2006.09.009
Agbaga, M. P., Mandal, M. N., and Anderson, R. E. (2010). Retinal very long-chain PUFAs: New insights from studies on ELOVL4 protein. J. Lipid Res. 51, 1624–1642. doi:10.1194/jlr.R005025
Agbaga, M. P., Merriman, D. K., Brush, R. S., Lydic, T. A., Conley, S. M., Naash, M. I., et al. (2018). Differential composition of DHA and very-long-chain PUFAs in rod and cone photoreceptors. J. Lipid Res. 59, 1586–1596. doi:10.1194/jlr.M082495
Age-Related Eye Disease Study 2 Research Group (2013). Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: The age-related eye disease study 2 (AREDS2) randomized clinical trial. JAMA 309, 2005–2015. doi:10.1001/jama.2013.4997
Alpern, M., Sack, G. H., Krantz, D. H., Jenness, J., Zhang, H., and Moser, H. W. (1993). Chromosomal rearrangement segregating with adrenoleukodystrophy: Associated changes in color vision. Proc. Natl. Acad. Sci. U. S. A. 90, 9494–9498. doi:10.1073/pnas.90.20.9494
Alvarez, R. A., Aguirre, G. D., Acland, G. M., and Anderson, R. E. (1994). Docosapentaenoic acid is converted to docosahexaenoic acid in the retinas of normal and prcd-affected miniature poodle dogs. Invest. Ophthalmol. Vis. Sci. 35, 402–408.
Anderson, G. J., Connor, W. E., and Corliss, J. D. (1990). Docosahexaenoic acid is the preferred dietary n-3 fatty acid for the development of the brain and retina. Pediatr. Res. 27, 89–97. doi:10.1203/00006450-199001000-00023
Anderson, J. N., Ammous, Z., Eroglu, Y., Hernandez, E., Heubi, J., Himes, R., et al. (2021). Cholbam® and zellweger spectrum disorders: Treatment implementation and management. Orphanet J. Rare Dis. 16, 388. doi:10.1186/s13023-021-01940-z
Antonenkov, V. D., Grunau, S., Ohlmeier, S., and Hiltunen, J. K. (2010). Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 13, 525–537. doi:10.1089/ars.2009.2996
Atshaves, B. P., Mcintosh, A. L., Landrock, D., Payne, H. R., Mackie, J. T., Maeda, N., et al. (2007). Effect of SCP-x gene ablation on branched-chain fatty acid metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G939–G951. doi:10.1152/ajpgi.00308.2006
Bae, E. Y., Yi, Y., Lim, H. H., Lee, J. M., Lee, B., Kim, S. Y., et al. (2020). First case of peroxisomal D-bifunctional protein deficiency with novel HSD17B4 mutations and progressive neuropathy in korea. J. Korean Med. Sci. 35, e357. doi:10.3346/jkms.2020.35.e357
Bartlett, M., Nasiri, N., Pressman, R., Bademci, G., and Forghani, I. (2021). First reported adult patient with retinal dystrophy and leukodystrophy caused by a novel ACBD5 variant: A case report and review of literature. Am. J. Med. Genet. A 185, 1236–1241. doi:10.1002/ajmg.a.62073
Bazan, N. G., Gordon, W. C., and Rodriguez De Turco, E. B. (1992). Docosahexaenoic acid uptake and metabolism in photoreceptors: Retinal conservation by an efficient retinal pigment epithelial cell-mediated recycling process. Adv. Exp. Med. Biol. 318, 295–306. doi:10.1007/978-1-4615-3426-6_26
Beard, M. E., Davies, T., Holloway, M., and Holtzman, E. (1988). Peroxisomes in pigment epithelium and Muller cells of amphibian retina possess D-amino acid oxidase as well as catalase. Exp. Eye Res. 47, 795–806. doi:10.1016/0014-4835(88)90063-2
Beckers, L., Geric, I., Stroobants, S., Beel, S., Van Damme, P., D'hooge, R., et al. (2019). Microglia lacking a peroxisomal beta-oxidation enzyme chronically alter their inflammatory profile without evoking neuronal and behavioral deficits. J. Neuroinflammation 16, 61. doi:10.1186/s12974-019-1442-3
Beken, S., Dilli, D., Fettah, N. D., Kabatas, E. U., Zenciroglu, A., and Okumus, N. (2014). The influence of fish-oil lipid emulsions on retinopathy of prematurity in very low birth weight infants: A randomized controlled trial. Early Hum. Dev. 90, 27–31. doi:10.1016/j.earlhumdev.2013.11.002
Bellezza, I. (2018). Oxidative stress in age-related macular degeneration: Nrf2 as therapeutic target. Front. Pharmacol. 9, 1280. doi:10.3389/fphar.2018.01280
Berger, J., Dorninger, F., Forss-Petter, S., and Kunze, M. (2016). Peroxisomes in brain development and function. Biochim. Biophys. Acta 1863, 934–955. doi:10.1016/j.bbamcr.2015.12.005
Bernstein, P. S., Lloyd, M. B., O'day, W. T., and Bok, D. (1992). Effect of phytanic acid on cultured retinal pigment epithelium: An in vitro model for refsum's disease. Exp. Eye Res. 55, 869–878. doi:10.1016/0014-4835(92)90013-i
Bieber, L. L., Krahling, J. B., Clarke, P. R., Valkner, K. J., and Tolbert, N. E. (1981). Carnitine acyltransferases in rat liver peroxisomes. Arch. Biochem. Biophys. 211, 599–604. doi:10.1016/0003-9861(81)90494-x
Birdsey, G. M., Lewin, J., Holbrook, J. D., Simpson, V. R., Cunningham, A. A., and Danpure, C. J. (2005). A comparative analysis of the evolutionary relationship between diet and enzyme targeting in bats, marsupials and other mammals. Proc. Biol. Sci. 272, 833–840. doi:10.1098/rspb.2004.3011
Bishop, J. E., and Hajra, A. K. (1981). Mechanism and specificity of formation of long chain alcohols by developing rat brain. J. Biol. Chem. 256, 9542–9550. doi:10.1016/s0021-9258(19)68796-x
Bjorgo, K., Fjaer, R., Mork, H. H., Ferdinandusse, S., Falkenberg, K. D., Waterham, H. R., et al. (2017). Biochemical and genetic characterization of an unusual mild PEX3-related Zellweger spectrum disorder. Mol. Genet. Metab. 121, 325–328. doi:10.1016/j.ymgme.2017.06.004
Bosch, E., Horwitz, J., and Bok, D. (1993). Phagocytosis of outer segments by retinal pigment epithelium: Phagosome-lysosome interaction. J. Histochem. Cytochem. 41, 253–263. doi:10.1177/41.2.8419462
Braverman, N. E., and Moser, A. B. (2012). Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 1822, 1442–1452. doi:10.1016/j.bbadis.2012.05.008
Braverman, N. E., Raymond, G. V., Rizzo, W. B., Moser, A. B., Wilkinson, M. E., Stone, E. M., et al. (2016). Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 117, 313–321. doi:10.1016/j.ymgme.2015.12.009
Bretillon, L., Thuret, G., Gregoire, S., Acar, N., Joffre, C., Bron, A. M., et al. (2008). Lipid and fatty acid profile of the retina, retinal pigment epithelium/choroid, and the lacrimal gland, and associations with adipose tissue fatty acids in human subjects. Exp. Eye Res. 87, 521–528. doi:10.1016/j.exer.2008.08.010
Broniec, A., Klosinski, R., Pawlak, A., Wrona-Krol, M., Thompson, D., and Sarna, T. (2011). Interactions of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free Radic. Biol. Med. 50, 892–898. doi:10.1016/j.freeradbiomed.2011.01.002
Buchert, R., Tawamie, H., Smith, C., Uebe, S., Innes, A. M., Al Hallak, B., et al. (2014). A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency. Am. J. Hum. Genet. 95, 602–610. doi:10.1016/j.ajhg.2014.10.003
Carlton, V. E., Harris, B. Z., Puffenberger, E. G., Batta, A. K., Knisely, A. S., Robinson, D. L., et al. (2003). Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat. Genet. 34, 91–96. doi:10.1038/ng1147
Cellini, B., Bertoldi, M., Montioli, R., Paiardini, A., and Borri Voltattorni, C. (2007). Human wild-type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: Functional properties and physiological implications. Biochem. J. 408, 39–50. doi:10.1042/BJ20070637
Chen, B., and Tang, L. (2011). Protective effects of catalase on retinal ischemia/reperfusion injury in rats. Exp. Eye Res. 93, 599–606. doi:10.1016/j.exer.2011.07.007
Cheng, J. B., and Russell, D. W. (2004). Mammalian wax biosynthesis. I. Identification of two fatty acyl-Coenzyme A reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 279, 37789–37797. doi:10.1074/jbc.M406225200
Chiang, J. Y. (2004). Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 40, 539–551. doi:10.1016/j.jhep.2003.11.006
Christen, W. G., Schaumberg, D. A., Glynn, R. J., and Buring, J. E. (2011). Dietary omega-3 fatty acid and fish intake and incident age-related macular degeneration in women. Arch. Ophthalmol. 129, 921–929. doi:10.1001/archophthalmol.2011.34
Chu, B. B., Liao, Y. C., Qi, W., Xie, C., Du, X., Wang, J., et al. (2015). Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306. doi:10.1016/j.cell.2015.02.019
Chung, H. L., Wangler, M. F., Marcogliese, P. C., Jo, J., Ravenscroft, T. A., Zuo, Z., et al. (2020). Loss- or gain-of-function mutations in ACOX1 cause axonal loss via different mechanisms. Neuron 106, 589–606. e586. doi:10.1016/j.neuron.2020.02.021
Cipolla, C. M., and Lodhi, I. J. (2017). Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 28, 297–308. doi:10.1016/j.tem.2016.12.003
Circu, M. L., and Aw, T. Y. (2010). Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 48, 749–762. doi:10.1016/j.freeradbiomed.2009.12.022
Cochat, P., Liutkus, A., Fargue, S., Basmaison, O., Ranchin, B., and Rolland, M. O. (2006). Primary hyperoxaluria type 1: Still challenging. Pediatr. Nephrol. 21, 1075–1081. doi:10.1007/s00467-006-0124-4
Coe, N. R., Smith, A. J., Frohnert, B. I., Watkins, P. A., and Bernlohr, D. A. (1999). The fatty acid transport protein (FATP1) is a very long chain acyl-CoA synthetase. J. Biol. Chem. 274, 36300–36304. doi:10.1074/jbc.274.51.36300
Costello, J. L., Castro, I. G., Hacker, C., Schrader, T. A., Metz, J., Zeuschner, D., et al. (2017). ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 216, 331–342. doi:10.1083/jcb.201607055
D'ascenzo, R., Savini, S., Biagetti, C., Bellagamba, M. P., Marchionni, P., Pompilio, A., et al. (2014). Higher docosahexaenoic acid, lower arachidonic acid and reduced lipid tolerance with high doses of a lipid emulsion containing 15% fish oil: A randomized clinical trial. Clin. Nutr. 33, 1002–1009. doi:10.1016/j.clnu.2014.01.009
Danpure, C. J. (1997). Variable peroxisomal and mitochondrial targeting of alanine: Glyoxylate aminotransferase in mammalian evolution and disease. Bioessays 19, 317–326. doi:10.1002/bies.950190409
Daruich, A., Picard, E., Boatright, J. H., and Behar-Cohen, F. (2019). Review: The bile acids urso- and tauroursodeoxycholic acid as neuroprotective therapies in retinal disease. Mol. Vis. 25, 610–624.
Das, Y., Roose, N., De Groef, L., Fransen, M., Moons, L., Van Veldhoven, P. P., et al. (2019). Differential distribution of peroxisomal proteins points to specific roles of peroxisomes in the murine retina. Mol. Cell. Biochem. 456, 53–62. doi:10.1007/s11010-018-3489-3
Das, Y., Swinkels, D., and Baes, M. (2021). Peroxisomal disorders and their mouse models point to essential roles of peroxisomes for retinal integrity. Int. J. Mol. Sci. 22, 4101. doi:10.3390/ijms22084101
De Duve, C., and Baudhuin, P. (1966). Peroxisomes (microbodies and related particles). Physiol. Rev. 46, 323–357. doi:10.1152/physrev.1966.46.2.323
Delton-Vandenbroucke, I., Grammas, P., and Anderson, R. E. (1997). Polyunsaturated fatty acid metabolism in retinal and cerebral microvascular endothelial cells. J. Lipid Res. 38, 147–159. doi:10.1016/s0022-2275(20)37284-9
Dimitrov, L., Lam, S. K., and Schekman, R. (2013). The role of the endoplasmic reticulum in peroxisome biogenesis. Cold Spring Harb. Perspect. Biol. 5, a013243. doi:10.1101/cshperspect.a013243
Ding, L., Sun, W., Balaz, M., He, A., Klug, M., Wieland, S., et al. (2021). Peroxisomal beta-oxidation acts as a sensor for intracellular fatty acids and regulates lipolysis. Nat. Metab. 3, 1648–1661. doi:10.1038/s42255-021-00489-2
Ebberink, M. S., Mooyer, P. A., Koster, J., Dekker, C. J., Eyskens, F. J., Dionisi-Vici, C., et al. (2009). Genotype-phenotype correlation in PEX5-deficient peroxisome biogenesis defective cell lines. Hum. Mutat. 30, 93–98. doi:10.1002/humu.20833
Edwards, A. O., Miedziak, A., Vrabec, T., Verhoeven, J., Acott, T. S., Weleber, R. G., et al. (1999). Autosomal dominant stargardt-like macular dystrophy: I. Clinical characterization, longitudinal follow-up, and evidence for a common ancestry in families linked to chromosome 6q14. Am. J. Ophthalmol. 127, 426–435. doi:10.1016/s0002-9394(98)00331-6
Enns, G. M., Ammous, Z., Himes, R. W., Nogueira, J., Palle, S., Sullivan, M., et al. (2021). Diagnostic challenges and disease management in patients with a mild Zellweger spectrum disorder phenotype. Mol. Genet. Metab. 134, 217–222. doi:10.1016/j.ymgme.2021.09.007
Fabelo, N., Martin, V., Santpere, G., Marin, R., Torrent, L., Ferrer, I., et al. (2011). Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson's disease and incidental Parkinson's disease. Mol. Med. 17, 1107–1118. doi:10.2119/molmed.2011.00119
Falany, C. N., Xie, X., Wheeler, J. B., Wang, J., Smith, M., He, D., et al. (2002). Molecular cloning and expression of rat liver bile acid CoA ligase. J. Lipid Res. 43, 2062–2071. doi:10.1194/jlr.m200260-jlr200
Fedick, A., Jalas, C., and Treff, N. R. (2014). A deleterious mutation in the PEX2 gene causes Zellweger syndrome in individuals of Ashkenazi Jewish descent. Clin. Genet. 85, 343–346. doi:10.1111/cge.12170
Feher, J., Kovacs, I., Artico, M., Cavallotti, C., Papale, A., and Balacco Gabrieli, C. (2006). Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 27, 983–993. doi:10.1016/j.neurobiolaging.2005.05.012
Ferdinandusse, S., Denis, S., Clayton, P. T., Graham, A., Rees, J. E., Allen, J. T., et al. (2000). Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nat. Genet. 24, 188–191. doi:10.1038/72861
Ferdinandusse, S., Denis, S., Hogenhout, E. M., Koster, J., Van Roermund, C. W., Moser, A. B., et al. (2007). Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Hum. Mutat. 28, 904–912. doi:10.1002/humu.20535
Ferdinandusse, S., Denis, S., Mooyer, P. A., Dekker, C., Duran, M., Soorani-Lunsing, R. J., et al. (2006a). Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann. Neurol. 59, 92–104. doi:10.1002/ana.20702
Ferdinandusse, S., Denis, S., Van Roermund, C. W. T., Preece, M. A., Koster, J., Ebberink, M. S., et al. (2018). A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta. Mol. Basis Dis. 1864, 952–958. doi:10.1016/j.bbadis.2017.12.032
Ferdinandusse, S., Denis, S., Van Roermund, C. W., Wanders, R. J., and Dacremont, G. (2004). Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of long-chain dicarboxylic acids. J. Lipid Res. 45, 1104–1111. doi:10.1194/jlr.M300512-JLR200
Ferdinandusse, S., Falkenberg, K. D., Koster, J., Mooyer, P. A., Jones, R., Van Roermund, C. W. T., et al. (2017). ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J. Med. Genet. 54, 330–337. doi:10.1136/jmedgenet-2016-104132
Ferdinandusse, S., Jimenez-Sanchez, G., Koster, J., Denis, S., Van Roermund, C. W., Silva-Zolezzi, I., et al. (2015). A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 24, 361–370. doi:10.1093/hmg/ddu448
Ferdinandusse, S., Kostopoulos, P., Denis, S., Rusch, H., Overmars, H., Dillmann, U., et al. (2006b). Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am. J. Hum. Genet. 78, 1046–1052. doi:10.1086/503921
Ferdinandusse, S., Mcwalter, K., Te Brinke, H., Mooijer, P. M., Ruiter, J. P. N., Van Lint, A. E. M., et al. (2021). An autosomal dominant neurological disorder caused by de novo variants in FAR1 resulting in uncontrolled synthesis of ether lipids. Genet. Med. 23, 740–750. doi:10.1038/s41436-020-01027-3
Ferrington, D. A., Sinha, D., and Kaarniranta, K. (2016). Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res. 51, 69–89. doi:10.1016/j.preteyeres.2015.09.002
Fliesler, S. J., and Anderson, R. E. (1983). Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res. 22, 79–131. doi:10.1016/0163-7827(83)90004-8
Fliesler, S. J. (2021). Introduction to the thematic review series: Seeing 2020: Lipids and lipid-soluble molecules in the eye. J. Lipid Res. 62, 100007. doi:10.1016/j.jlr.2020.100007
Folz, S. J., and Trobe, J. D. (1991). The peroxisome and the eye. Surv. Ophthalmol. 35, 353–368. doi:10.1016/0039-6257(91)90185-i
Forman, H. J., Maiorino, M., and Ursini, F. (2010). Signaling functions of reactive oxygen species. Biochemistry 49, 835–842. doi:10.1021/bi9020378
Foulon, V., Antonenkov, V. D., Croes, K., Waelkens, E., Mannaerts, G. P., Van Veldhoven, P. P., et al. (1999). Purification, molecular cloning, and expression of 2-hydroxyphytanoyl-CoA lyase, a peroxisomal thiamine pyrophosphate-dependent enzyme that catalyzes the carbon-carbon bond cleavage during alpha-oxidation of 3-methyl-branched fatty acids. Proc. Natl. Acad. Sci. U. S. A. 96, 10039–10044. doi:10.1073/pnas.96.18.10039
Fransen, M., Lismont, C., and Walton, P. (2017). The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 18, E1126. doi:10.3390/ijms18061126
Fransen, M., Nordgren, M., Wang, B., Apanasets, O., and Van Veldhoven, P. P. (2013). Aging, age-related diseases and peroxisomes. Subcell. Biochem. 69, 45–65. doi:10.1007/978-94-007-6889-5_3
Frattini, L. F., Piubelli, L., Sacchi, S., Molla, G., and Pollegioni, L. (2011). Is rat an appropriate animal model to study the involvement of D-serine catabolism in schizophrenia? Insights from characterization of D-amino acid oxidase. FEBS J. 278, 4362–4373. doi:10.1111/j.1742-4658.2011.08354.x
Fu, Z., Chen, C. T., Cagnone, G., Heckel, E., Sun, Y., Cakir, B., et al. (2019). Dyslipidemia in retinal metabolic disorders. EMBO Mol. Med. 11, e10473. doi:10.15252/emmm.201910473
Fu, Z., Kern, T. S., Hellstrom, A., and Smith, L. E. H. (2021). Fatty acid oxidation and photoreceptor metabolic needs. J. Lipid Res. 62, 100035. doi:10.1194/jlr.TR120000618
Fu, Z., Lofqvist, C. A., Liegl, R., Wang, Z., Sun, Y., Gong, Y., et al. (2017). Photoreceptor glucose metabolism determines normal retinal vascular growth. EMBO Mol. Med. 10, 76–90. doi:10.15252/emmm.201707966
Fujiki, Y., Okumoto, K., Mukai, S., Honsho, M., and Tamura, S. (2014). Peroxisome biogenesis in mammalian cells. Front. Physiol. 5, 307. doi:10.3389/fphys.2014.00307
Gabaldon, T. (2014). A metabolic scenario for the evolutionary origin of peroxisomes from the endomembranous system. Cell. Mol. Life Sci. 71, 2373–2376. doi:10.1007/s00018-013-1424-z
Gallego-Garcia, A., Monera-Girona, A. J., Pajares-Martinez, E., Bastida-Martinez, E., Perez-Castano, R., Iniesta, A. A., et al. (2019). A bacterial light response reveals an orphan desaturase for human plasmalogen synthesis. Science 366, 128–132. doi:10.1126/science.aay1436
Garner, A., Fielder, A. R., Primavesi, R., and Stevens, A. (1982). Tapetoretinal degeneration in the cerebro-hepato-renal (Zellweger's) syndrome. Br. J. Ophthalmol. 66, 422–431. doi:10.1136/bjo.66.7.422
Gavino, G. R., and Gavino, V. C. (1991). Rat liver outer mitochondrial carnitine palmitoyltransferase activity towards long-chain polyunsaturated fatty acids and their CoA esters. Lipids 26, 266–270. doi:10.1007/BF02537135
Geuze, H. J., Murk, J. L., Stroobants, A. K., Griffith, J. M., Kleijmeer, M. J., Koster, A. J., et al. (2003). Involvement of the endoplasmic reticulum in peroxisome formation. Mol. Biol. Cell 14, 2900–2907. doi:10.1091/mbc.e02-11-0734
Ginsberg, L., Rafique, S., Xuereb, J. H., Rapoport, S. I., and Gershfeld, N. L. (1995). Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer's disease brain. Brain Res. 698, 223–226. doi:10.1016/0006-8993(95)00931-f
Gloerich, J., Ruiter, J. P., Van Den Brink, D. M., Ofman, R., Ferdinandusse, S., and Wanders, R. J. (2006). Peroxisomal trans-2-enoyl-CoA reductase is involved in phytol degradation. FEBS Lett. 580, 2092–2096. doi:10.1016/j.febslet.2006.03.011
Gong, Y., Fu, Z., Edin, M. L., Liu, C. H., Wang, Z., Shao, Z., et al. (2016). Cytochrome P450 oxidase 2C inhibition adds to omega-3 long-chain polyunsaturated fatty acids protection against retinal and choroidal neovascularization. Arterioscler. Thromb. Vasc. Biol. 36, 1919–1927. doi:10.1161/ATVBAHA.116.307558
Gong, Y., Fu, Z., Liegl, R., Chen, J., Hellstrom, A., and Smith, L. E. (2017). ω-3 and ω-6 long-chain PUFAs and their enzymatic metabolites in neovascular eye diseases. Am. J. Clin. Nutr. 106, 16–26. doi:10.3945/ajcn.117.153825
Gordon, W. C., and Bazan, N. G. (1993). Visualization of [3H]docosahexaenoic acid trafficking through photoreceptors and retinal pigment epithelium by electron microscopic autoradiography. Invest. Ophthalmol. Vis. Sci. 34, 2402–2411.
Gordon, W. C., Rodriguez De Turco, E. B., and Bazan, N. G. (1992). Retinal pigment epithelial cells play a central role in the conservation of docosahexaenoic acid by photoreceptor cells after shedding and phagocytosis. Curr. Eye Res. 11, 73–83. doi:10.3109/02713689209069169
Graziosi, A., Perrotta, M., Russo, D., Gasparroni, G., D'egidio, C., Marinelli, B., et al. (2020). Oxidative stress markers and the retinopathy of prematurity. J. Clin. Med. 9, E2711. doi:10.3390/jcm9092711
Grego, A. V., and Mingrone, G. (1995). Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: An update. Clin. Nutr. 14, 143–148. doi:10.1016/s0261-5614(95)80011-5
Guha, S., Liu, J., Baltazar, G., Laties, A. M., and Mitchell, C. H. (2014). Rescue of compromised lysosomes enhances degradation of photoreceptor outer segments and reduces lipofuscin-like autofluorescence in retinal pigmented epithelial cells. Adv. Exp. Med. Biol. 801, 105–111. doi:10.1007/978-1-4614-3209-8_14
Haddad, R., Font, R. L., and Friendly, D. S. (1976). Cerebro-hepato-renal syndrome of Zellweger. Ocular histopathologic findings. Arch. Ophthalmol. 94, 1927–1930. doi:10.1001/archopht.1976.03910040633011
Han, X., Holtzman, D. M., and Mckeel, D. W. (2001). Plasmalogen deficiency in early alzheimer's disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 77, 1168–1180. doi:10.1046/j.1471-4159.2001.00332.x
Harkewicz, R., Du, H., Tong, Z., Alkuraya, H., Bedell, M., Sun, W., et al. (2012). Essential role of ELOVL4 protein in very long chain fatty acid synthesis and retinal function. J. Biol. Chem. 287, 11469–11480. doi:10.1074/jbc.M111.256073
Hasan, S., Platta, H. W., and Erdmann, R. (2013). Import of proteins into the peroxisomal matrix. Front. Physiol. 4, 261. doi:10.3389/fphys.2013.00261
Hatch, G. M. (2004). Cell biology of cardiac mitochondrial phospholipids. Biochem. Cell Biol. 82, 99–112. doi:10.1139/o03-074
Heimler, A., Fox, J. E., Hershey, J. E., and Crespi, P. (1991). Sensorineural hearing loss, enamel hypoplasia, and nail abnormalities in sibs. Am. J. Med. Genet. 39, 192–195. doi:10.1002/ajmg.1320390214
Heinzer, A. K., Watkins, P. A., Lu, J. F., Kemp, S., Moser, A. B., Li, Y. Y., et al. (2003). A very long-chain acyl-CoA synthetase-deficient mouse and its relevance to X-linked adrenoleukodystrophy. Hum. Mol. Genet. 12, 1145–1154. doi:10.1093/hmg/ddg126
Hellstrom, A., Pivodic, A., Granse, L., Lundgren, P., Sjobom, U., Nilsson, A. K., et al. (2021). Association of docosahexaenoic acid and arachidonic acid serum levels with retinopathy of prematurity in preterm infants. JAMA Netw. Open 4, e2128771. doi:10.1001/jamanetworkopen.2021.28771
Heymans, H. S., Schutgens, R. B., Tan, R., Van Den Bosch, H., and Borst, P. (1983). Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome). Nature 306, 69–70. doi:10.1038/306069a0
Ho, L., Van Leeuwen, R., Witteman, J. C., Van Duijn, C. M., Uitterlinden, A. G., Hofman, A., et al. (2011). Reducing the genetic risk of age-related macular degeneration with dietary antioxidants, zinc, and omega-3 fatty acids: The rotterdam study. Arch. Ophthalmol. 129, 758–766. doi:10.1001/archophthalmol.2011.141
Hoffman, D. R., Demar, J. C., Heird, W. C., Birch, D. G., and Anderson, R. E. (2001). Impaired synthesis of DHA in patients with X-linked retinitis pigmentosa. J. Lipid Res. 42, 1395–1401. doi:10.1016/s0022-2275(20)30271-6
Holmstrom, K. M., and Finkel, T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421. doi:10.1038/nrm3801
Honsho, M., Tanaka, M., Zoeller, R. A., and Fujiki, Y. (2020). Distinct functions of acyl/alkyl dihydroxyacetonephosphate reductase in peroxisomes and endoplasmic reticulum. Front. Cell Dev. Biol. 8, 855. doi:10.3389/fcell.2020.00855
Horvath, R., Lewis-Smith, D., Douroudis, K., Duff, J., Keogh, M., Pyle, A., et al. (2015). SCP2 mutations and neurodegeneration with brain iron accumulation. Neurology 85, 1909–1911. doi:10.1212/WNL.0000000000002157
Houten, S. M., Denis, S., Argmann, C. A., Jia, Y., Ferdinandusse, S., Reddy, J. K., et al. (2012). Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids. J. Lipid Res. 53, 1296–1303. doi:10.1194/jlr.M024463
Hua, R., Cheng, D., Coyaud, E., Freeman, S., Di Pietro, E., Wang, Y., et al. (2017). VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 216, 367–377. doi:10.1083/jcb.201608128
Huyghe, S., Mannaerts, G. P., Baes, M., and Van Veldhoven, P. P. (2006). Peroxisomal multifunctional protein-2: The enzyme, the patients and the knockout mouse model. Biochim. Biophys. Acta 1761, 973–994. doi:10.1016/j.bbalip.2006.04.006
Ishikawa, M., Yoshitomi, T., Zorumski, C. F., and Izumi, Y. (2016). 24(S)-Hydroxycholesterol protects the ex vivo rat retina from injury by elevated hydrostatic pressure. Sci. Rep. 6, 33886. doi:10.1038/srep33886
Islinger, M., Cardoso, M. J., and Schrader, M. (2010). Be different--the diversity of peroxisomes in the animal kingdom. Biochim. Biophys. Acta 1803, 881–897. doi:10.1016/j.bbamcr.2010.03.013
Islinger, M., Voelkl, A., Fahimi, H. D., and Schrader, M. (2018). The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 150, 443–471. doi:10.1007/s00418-018-1722-5
Itzkovitz, B., Jiralerspong, S., Nimmo, G., Loscalzo, M., Horovitz, D. D., Snowden, A., et al. (2012). Functional characterization of novel mutations in GNPAT and AGPS, causing rhizomelic chondrodysplasia punctata (RCDP) types 2 and 3. Hum. Mutat. 33, 189–197. doi:10.1002/humu.21623
Ivashchenko, O., Van Veldhoven, P. P., Brees, C., Ho, Y. S., Terlecky, S. R., and Fransen, M. (2011). Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 22, 1440–1451. doi:10.1091/mbc.E10-11-0919
Jadeja, R. N., Powell, F. L., Jones, M. A., Fuller, J., Joseph, E., Thounaojam, M. C., et al. (2018). Loss of NAMPT in aging retinal pigment epithelium reduces NAD(+) availability and promotes cellular senescence. Aging (Albany NY) 10, 1306–1323. doi:10.18632/aging.101469
Jansen, G. A., Ofman, R., Denis, S., Ferdinandusse, S., Hogenhout, E. M., Jakobs, C., et al. (1999a). Phytanoyl-CoA hydroxylase from rat liver. Protein purification and cDNA cloning with implications for the subcellular localization of phytanic acid alpha-oxidation. J. Lipid Res. 40, 2244–2254. doi:10.1016/s0022-2275(20)32099-x
Jansen, G. A., Ofman, R., Ferdinandusse, S., Ijlst, L., Muijsers, A. O., Skjeldal, O. H., et al. (1997). Refsum disease is caused by mutations in the phytanoyl-CoA hydroxylase gene. Nat. Genet. 17, 190–193. doi:10.1038/ng1097-190
Jansen, G. A., Verhoeven, N. M., Denis, S., Romeijn, G., Jakobs, C., Ten Brink, H. J., et al. (1999b). Phytanic acid alpha-oxidation: Identification of 2-hydroxyphytanoyl-CoA lyase in rat liver and its localisation in peroxisomes. Biochim. Biophys. Acta 1440, 176–182. doi:10.1016/s1388-1981(99)00126-2
Janssen, A., Baes, M., Gressens, P., Mannaerts, G. P., Declercq, P., and Van Veldhoven, P. P. (2000). Docosahexaenoic acid deficit is not a major pathogenic factor in peroxisome-deficient mice. Lab. Invest. 80, 31–35. doi:10.1038/labinvest.3780005
Janssen, A., Gressens, P., Grabenbauer, M., Baumgart, E., Schad, A., Vanhorebeek, I., et al. (2003). Neuronal migration depends on intact peroxisomal function in brain and in extraneuronal tissues. J. Neurosci. 23, 9732–9741. doi:10.1523/jneurosci.23-30-09732.2003
Jenkins, C. M., Yang, K., Liu, G., Moon, S. H., Dilthey, B. G., and Gross, R. W. (2018). Cytochrome c is an oxidative stress-activated plasmalogenase that cleaves plasmenylcholine and plasmenylethanolamine at the sn-1 vinyl ether linkage. J. Biol. Chem. 293, 8693–8709. doi:10.1074/jbc.RA117.001629
Jones, D. P., and Go, Y. M. (2010). Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 12 (2), 116–125. doi:10.1111/j.1463-1326.2010.01266.x
Joyal, J. S., Sun, Y., Gantner, M. L., Shao, Z., Evans, L. P., Saba, N., et al. (2016). Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 22, 439–445. doi:10.1038/nm.4059
Kaplan, P. W., Kruse, B., Tusa, R. J., Shankroff, J., Rignani, J., and Moser, H. W. (1995). Visual system abnormalities in adrenomyeloneuropathy. Ann. Neurol. 37, 550–552. doi:10.1002/ana.410370419
Kawaguchi, K., Mukai, E., Watanabe, S., Yamashita, A., Morita, M., So, T., et al. (2021). Acyl-CoA thioesterase activity of peroxisomal ABC protein ABCD1 is required for the transport of very long-chain acyl-CoA into peroxisomes. Sci. Rep. 11, 2192. doi:10.1038/s41598-021-81949-3
Kawazoe, T., Park, H. K., Iwana, S., Tsuge, H., and Fukui, K. (2007). Human D-amino acid oxidase: An update and review. Chem. Rec. 7, 305–315. doi:10.1002/tcr.20129
Kersten, S. (2014). Integrated physiology and systems biology of PPARα. Mol. Metab. 3, 354–371. doi:10.1016/j.molmet.2014.02.002
Kettelhut, T., and Thoms, S. (2014). Molecular machines involved in peroxisome biogenesis and maintenance. Vienna: Springer.
Khairallah, R. J., Kim, J., O'shea, K. M., O'connell, K. A., Brown, B. H., Galvao, T., et al. (2012). Improved mitochondrial function with diet-induced increase in either docosahexaenoic acid or arachidonic acid in membrane phospholipids. PLoS One 7, e34402. doi:10.1371/journal.pone.0034402
Khairallah, R. J., Sparagna, G. C., Khanna, N., O'shea, K. M., Hecker, P. A., Kristian, T., et al. (2010). Dietary supplementation with docosahexaenoic acid, but not eicosapentaenoic acid, dramatically alters cardiac mitochondrial phospholipid fatty acid composition and prevents permeability transition. Biochim. Biophys. Acta 1797, 1555–1562. doi:10.1016/j.bbabio.2010.05.007
Kihara, A. (2012). Very long-chain fatty acids: Elongation, physiology and related disorders. J. Biochem. 152, 387–395. doi:10.1093/jb/mvs105
Kim, P. K., Mullen, R. T., Schumann, U., and Lippincott-Schwartz, J. (2006). The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J. Cell Biol. 173, 521–532. doi:10.1083/jcb.200601036
Kimura, T., Kimura, A. K., Ren, M., Monteiro, V., Xu, Y., Berno, B., et al. (2019). Plasmalogen loss caused by remodeling deficiency in mitochondria. Life Sci. Alliance 2, e201900348. doi:10.26508/lsa.201900348
Klemp, H. G., Kettwig, M., Streit, F., Gartner, J., Rosewich, H., and Kratzner, R. (2021). LC-MS based platform simplifies access to metabolomics for peroxisomal disorders. Metabolites 11, 347. doi:10.3390/metabo11060347
Knoops, B., Goemaere, J., Van Der Eecken, V., and Declercq, J. P. (2011). Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-cys peroxiredoxin. Antioxid. Redox Signal. 15, 817–829. doi:10.1089/ars.2010.3584
Koenekoop, R. K., Wang, H., Majewski, J., Wang, X., Lopez, I., Ren, H., et al. (2012). Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 44, 1035–1039. doi:10.1038/ng.2356
Komatsuzaki, S., Ogawa, E., Shimozawa, N., Sakamoto, O., Haginoya, K., Uematsu, M., et al. (2015). First Japanese case of Zellweger syndrome with a mutation in PEX14. Pediatr. Int. 57, 1189–1192. doi:10.1111/ped.12713
Konkolova, J., Petrovic, R., Chandoga, J., Halasova, E., Jungova, P., and Bohmer, D. (2015). A novel mutation in the PEX12 gene causing a peroxisomal biogenesis disorder. Mol. Biol. Rep. 42, 1359–1363. doi:10.1007/s11033-015-3885-7
Kou, J., Kovacs, G. G., Hoftberger, R., Kulik, W., Brodde, A., Forss-Petter, S., et al. (2011). Peroxisomal alterations in Alzheimer's disease. Acta Neuropathol. 122, 271–283. doi:10.1007/s00401-011-0836-9
Kourti, P., Gonzalez-Martin, J., and Yeo, D. C. M. (2020). Oxalate maculopathy. Ophthalmol. Retina 4, 898. doi:10.1016/j.oret.2020.04.008
Kowluru, R. A., and Chan, P. S. (2007). Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 43603. doi:10.1155/2007/43603
Krause, C., Rosewich, H., Woehler, A., and Gartner, J. (2013). Functional analysis of PEX13 mutation in a Zellweger syndrome spectrum patient reveals novel homooligomerization of PEX13 and its role in human peroxisome biogenesis. Hum. Mol. Genet. 22, 3844–3857. doi:10.1093/hmg/ddt238
Kretzer, F. L., Hittner, H. M., and Mehta, R. (1981). Ocular manifestations of conradi and zellweger syndromes. Metab. Pediatr. Ophthalmol. 5, 1–11.
Krysko, O., Hulshagen, L., Janssen, A., Schutz, G., Klein, R., De Bruycker, M., et al. (2007). Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver. J. Neurosci. Res. 85, 58–72. doi:10.1002/jnr.21097
Kumar, K. R., Wali, G., Davis, R. L., Mallawaarachchi, A. C., Palmer, E. E., Gayevskiy, V., et al. (2018). Expanding the spectrum of PEX16 mutations and novel insights into disease mechanisms. Mol. Genet. Metab. Rep. 16, 46–51. doi:10.1016/j.ymgmr.2018.07.003
Kurian, M. A., Ryan, S., Besley, G. T., Wanders, R. J., and King, M. D. (2004). Straight-chain acyl-CoA oxidase deficiency presenting with dysmorphia, neurodevelopmental autistic-type regression and a selective pattern of leukodystrophy. J. Inherit. Metab. Dis. 27, 105–108. doi:10.1023/b:boli.0000016687.88818.6d
Lacombe, R. J. S., and Bazinet, R. P. (2021). Natural abundance carbon isotope ratio analysis and its application in the study of diet and metabolism. Nutr. Rev. 79, 869–888. doi:10.1093/nutrit/nuaa109
Lacombe, R. J. S., Lee, C. C., and Bazinet, R. P. (2020). Turnover of brain DHA in mice is accurately determined by tracer-free natural abundance carbon isotope ratio analysis. J. Lipid Res. 61, 116–126. doi:10.1194/jlr.D119000518
Lageweg, W., Sykes, J. E., Lopes-Cardozo, M., and Wanders, R. J. (1991). Oxidation of very-long-chain fatty acids in rat brain: Cerotic acid is beta-oxidized exclusively in rat brain peroxisomes. Biochim. Biophys. Acta 1085, 381–384. doi:10.1016/0005-2760(91)90144-7
Lambert, S. R., Kriss, A., Taylor, D., Coffey, R., and Pembrey, M. (1989). Follow-up and diagnostic reappraisal of 75 patients with Leber's congenital amaurosis. Am. J. Ophthalmol. 107, 624–631. doi:10.1016/0002-9394(89)90259-6
Lee, J. W., Fuda, H., Javitt, N. B., Strott, C. A., and Rodriguez, I. R. (2006). Expression and localization of sterol 27-hydroxylase (CYP27A1) in monkey retina. Exp. Eye Res. 83, 465–469. doi:10.1016/j.exer.2005.11.018
Leumann, E., and Hoppe, B. (2001). The primary hyperoxalurias. J. Am. Soc. Nephrol. 12, 1986–1993. doi:10.1681/ASN.V1291986
Li, X., Knight, J., Fargue, S., Buchalski, B., Guan, Z., Inscho, E. W., et al. (2016). Metabolism of (13)C5-hydroxyproline in mouse models of Primary Hyperoxaluria and its inhibition by RNAi therapeutics targeting liver glycolate oxidase and hydroxyproline dehydrogenase. Biochim. Biophys. Acta 1862, 233–239. doi:10.1016/j.bbadis.2015.12.001
Lima, L. H., Barbazetto, I. A., Chen, R., Yannuzzi, L. A., Tsang, S. H., and Spaide, R. F. (2011). Macular dystrophy in Heimler syndrome. Ophthalmic Genet. 32, 97–100. doi:10.3109/13816810.2010.551797
Lismont, C., Nordgren, M., Van Veldhoven, P. P., and Fransen, M. (2015). Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 3, 35. doi:10.3389/fcell.2015.00035
Litman, B. J., and Mitchell, D. C. (1996). A role for phospholipid polyunsaturation in modulating membrane protein function. Lipids 31 (1), S193–S197. doi:10.1007/BF02637075
Lizard, G., Rouaud, O., Demarquoy, J., Cherkaoui-Malki, M., and Iuliano, L. (2012). Potential roles of peroxisomes in Alzheimer's disease and in dementia of the Alzheimer's type. J. Alzheimers Dis. 29, 241–254. doi:10.3233/JAD-2011-111163
Lofqvist, C. A., Najm, S., Hellgren, G., Engstrom, E., Savman, K., Nilsson, A. K., et al. (2018). Association of retinopathy of prematurity with low levels of arachidonic acid: A secondary analysis of a randomized clinical trial. JAMA Ophthalmol. 136, 271–277. doi:10.1001/jamaophthalmol.2017.6658
London, A., Benhar, I., and Schwartz, M. (2013). The retina as a window to the brain-from eye research to CNS disorders. Nat. Rev. Neurol. 9, 44–53. doi:10.1038/nrneurol.2012.227
Lorenzo, V., Alvarez, A., Torres, A., Torregrosa, V., Hernandez, D., and Salido, E. (2006). Presentation and role of transplantation in adult patients with type 1 primary hyperoxaluria and the I244T AGXT mutation: Single-center experience. Kidney Int. 70, 1115–1119. doi:10.1038/sj.ki.5001758
Martin, W. (2010). Evolutionary origins of metabolic compartmentalization in eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 847–855. doi:10.1098/rstb.2009.0252
Martinez, M. (1992). Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders. Brain Res. 583, 171–182. doi:10.1016/s0006-8993(10)80021-6
Martinez, M. (2001). Restoring the DHA levels in the brains of Zellweger patients. J. Mol. Neurosci. 16, 309–316. doi:10.1385/JMN:16:2-3:309
Mechaussier, S., Marlin, S., Kaplan, J., Rozet, J. M., and Perrault, I. (2019). Genetic deciphering of early-onset and severe retinal dystrophy associated with sensorineural hearing loss. Adv. Exp. Med. Biol. 1185, 233–238. doi:10.1007/978-3-030-27378-1_38
Mertens, K. L., Kalsbeek, A., Soeters, M. R., and Eggink, H. M. (2017). Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front. Neurosci. 11, 617. doi:10.3389/fnins.2017.00617
Metherel, A. H., and Bazinet, R. P. (2019). Updates to the n-3 polyunsaturated fatty acid biosynthesis pathway: DHA synthesis rates, tetracosahexaenoic acid and (minimal) retroconversion. Prog. Lipid Res. 76, 101008. doi:10.1016/j.plipres.2019.101008
Mezzar, S., De Schryver, E., Asselberghs, S., Meyhi, E., Morvay, P. L., Baes, M., et al. (2017). Phytol-induced pathology in 2-hydroxyacyl-CoA lyase (HACL1) deficient mice. Evidence for a second non-HACL1-related lyase. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1862, 972–990. doi:10.1016/j.bbalip.2017.06.004
Miville-Godbout, E., Bourque, M., Morissette, M., Al-Sweidi, S., Smith, T., Jayasinghe, D., et al. (2017). Plasmalogen precursor mitigates striatal dopamine loss in MPTP mice. Brain Res. 1674, 70–76. doi:10.1016/j.brainres.2017.08.020
Molla, G., Sacchi, S., Bernasconi, M., Pilone, M. S., Fukui, K., and Polegioni, L. (2006). Characterization of human D-amino acid oxidase. FEBS Lett. 580, 2358–2364. doi:10.1016/j.febslet.2006.03.045
Molloy, C. S., Stokes, S., Makrides, M., Collins, C. T., Anderson, P. J., and Doyle, L. W. (2016). Long-term effect of high-dose supplementation with DHA on visual function at school age in children born at <33 wk gestational age: Results from a follow-up of a randomized controlled trial. Am. J. Clin. Nutr. 103, 268–275. doi:10.3945/ajcn.115.114710
Moore, S. A., Hurt, E., Yoder, E., Sprecher, H., and Spector, A. A. (1995). Docosahexaenoic acid synthesis in human skin fibroblasts involves peroxisomal retroconversion of tetracosahexaenoic acid. J. Lipid Res. 36, 2433–2443. doi:10.1016/s0022-2275(20)39724-8
Morarji, J., Gillespie, R., Sergouniotis, P. I., Horvath, R., and Black, G. C. M. (2017). An unusual retinal phenotype Associated with a mutation in sterol carrier protein SCP2. JAMA Ophthalmol. 135, 167–169. doi:10.1001/jamaophthalmol.2016.4985
Morita, M., and Imanaka, T. (2012). Peroxisomal ABC transporters: Structure, function and role in disease. Biochim. Biophys. Acta 1822, 1387–1396. doi:10.1016/j.bbadis.2012.02.009
Muller, C. C., Nguyen, T. H., Ahlemeyer, B., Meshram, M., Santrampurwala, N., Cao, S., et al. (2011). PEX13 deficiency in mouse brain as a model of zellweger syndrome: Abnormal cerebellum formation, reactive gliosis and oxidative stress. Dis. Model. Mech. 4, 104–119. doi:10.1242/dmm.004622
Murtas, G., Sacchi, S., Valentino, M., and Pollegioni, L. (2017). Biochemical properties of human D-amino acid oxidase. Front. Mol. Biosci. 4, 88. doi:10.3389/fmolb.2017.00088
Nagan, N., and Zoeller, R. A. (2001). Plasmalogens: Biosynthesis and functions. Prog. Lipid Res. 40, 199–229. doi:10.1016/s0163-7827(01)00003-0
Najm, S., Lofqvist, C., Hellgren, G., Engstrom, E., Lundgren, P., Hard, A. L., et al. (2017). Effects of a lipid emulsion containing fish oil on polyunsaturated fatty acid profiles, growth and morbidities in extremely premature infants: A randomized controlled trial. Clin. Nutr. ESPEN 20, 17–23. doi:10.1016/j.clnesp.2017.04.004
Nakai, A., Shigematsu, Y., Nishida, K., Kikawa, Y., and Konishi, Y. (1995). MRI findings of Zellweger syndrome. Pediatr. Neurol. 13, 346–348. doi:10.1016/0887-8994(95)00215-4
Nanda, T., and Kovach, J. L. (2019). Ophthalmic findings in late stage sjogren-larsson syndrome. Retin. Cases Brief. Rep. 13, 251–254. doi:10.1097/ICB.0000000000000583
Neuspiel, M., Schauss, A. C., Braschi, E., Zunino, R., Rippstein, P., Rachubinski, R. A., et al. (2008). Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108. doi:10.1016/j.cub.2007.12.038
Noguer, M. T., and Martinez, M. (2010). Visual follow-up in peroxisomal-disorder patients treated with docosahexaenoic Acid ethyl ester. Invest. Ophthalmol. Vis. Sci. 51, 2277–2285. doi:10.1167/iovs.09-4020
Ohkuma, Y., Hayashi, T., Yoshimine, S., Tsuneoka, H., Terao, Y., Akiyama, M., et al. (2014). Retinal ganglion cell loss in X-linked adrenoleukodystrophy with an ABCD1 mutation (Gly266Arg). Neuroophthalmology. 38, 331–335. doi:10.3109/01658107.2014.950430
Omarova, S., Charvet, C. D., Reem, R. E., Mast, N., Zheng, W., Huang, S., et al. (2012). Abnormal vascularization in mouse retina with dysregulated retinal cholesterol homeostasis. J. Clin. Invest. 122, 3012–3023. doi:10.1172/JCI63816
Otsuka, K., Sawai-Ogawa, M., and Kihara, A. (2022). Formation of fatty alcohols-components of meibum lipids-by the fatty acyl-CoA reductase FAR2 is essential for dry eye prevention. FASEB J. 36, e22216. doi:10.1096/fj.202101733R
Pacher, P., Beckman, J. S., and Liaudet, L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424. doi:10.1152/physrev.00029.2006
Paker, A. M., Sunness, J. S., Brereton, N. H., Speedie, L. J., Albanna, L., Dharmaraj, S., et al. (2010). Docosahexaenoic acid therapy in peroxisomal diseases: Results of a double-blind, randomized trial. Neurology 75, 826–830. doi:10.1212/WNL.0b013e3181f07061
Park, H., He, A., Tan, M., Johnson, J. M., Dean, J. M., Pietka, T. A., et al. (2019). Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J. Clin. Invest. 129, 694–711. doi:10.1172/JCI120606
Pawlik, D., Lauterbach, R., and Turyk, E. (2011). Fish-oil fat emulsion supplementation may reduce the risk of severe retinopathy in VLBW infants. Pediatrics 127, 223–228. doi:10.1542/peds.2010-2427
Pawlik, D., Lauterbach, R., Walczak, M., Hurkala, J., and Sherman, M. P. (2014). Fish-oil fat emulsion supplementation reduces the risk of retinopathy in very low birth weight infants: A prospective, randomized study. JPEN. J. Parenter. Enter. Nutr. 38, 711–716. doi:10.1177/0148607113499373
Pellicoro, A., Van Den Heuvel, F. A., Geuken, M., Moshage, H., Jansen, P. L., and Faber, K. N. (2007). Human and rat bile acid-CoA:amino acid N-acyltransferase are liver-specific peroxisomal enzymes: Implications for intracellular bile salt transport. Hepatology 45, 340–348. doi:10.1002/hep.21528
Petrushka, E., Quastel, J. H., and Scholefield, P. G. (1959). Role of phospholipids in oxidative phosphorylation and mitochondrial structure. Can. J. Biochem. Physiol. 37, 989–998. doi:10.1139/y59-108
Pifferi, F., Perret, M., Guesnet, P., Aujard, F., and Alessandri, J. M. (2012). Fatty acid composition of the brain, retina, liver and adipose tissue of the grey mouse lemur (Microcebus murinus, primate). Lipids 47, 793–801. doi:10.1007/s11745-012-3686-x
Poirier, Y., Antonenkov, V. D., Glumoff, T., and Hiltunen, J. K. (2006). Peroxisomal beta-oxidation--a metabolic pathway with multiple functions. Biochim. Biophys. Acta 1763, 1413–1426. doi:10.1016/j.bbamcr.2006.08.034
Pollegioni, L., Molla, G., Sacchi, S., and Murtas, G. (2021). Human D-aspartate oxidase: A key player in D-aspartate metabolism. Front. Mol. Biosci. 8, 689719. doi:10.3389/fmolb.2021.689719
Pollegioni, L., Sacchi, S., and Murtas, G. (2018). Human D-amino acid oxidase: Structure, function, and regulation. Front. Mol. Biosci. 5, 107. doi:10.3389/fmolb.2018.00107
Pridie, C., Ueda, K., and Simmonds, A. J. (2020). Rosy beginnings: Studying peroxisomes in Drosophila. Front. Cell Dev. Biol. 8, 835. doi:10.3389/fcell.2020.00835
Qi, C., Zhu, Y., Pan, J., Usuda, N., Maeda, N., Yeldandi, A. V., et al. (1999). Absence of spontaneous peroxisome proliferation in enoyl-CoA Hydratase/L-3-hydroxyacyl-CoA dehydrogenase-deficient mouse liver. Further support for the role of fatty acyl CoA oxidase in PPARalpha ligand metabolism. J. Biol. Chem. 274, 15775–15780. doi:10.1074/jbc.274.22.15775
Ramirez, D. M., Andersson, S., and Russell, D. W. (2008). Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J. Comp. Neurol. 507, 1676–1693. doi:10.1002/cne.21605
Ranea-Robles, P., Violante, S., Argmann, C., Dodatko, T., Bhattacharya, D., Chen, H., et al. (2021). Murine deficiency of peroxisomal L-bifunctional protein (EHHADH) causes medium-chain 3-hydroxydicarboxylic aciduria and perturbs hepatic cholesterol homeostasis. Cell. Mol. Life Sci. 78, 5631–5646. doi:10.1007/s00018-021-03869-9
Ratbi, I., Falkenberg, K. D., Sommen, M., Al-Sheqaih, N., Guaoua, S., Vandeweyer, G., et al. (2015). Heimler syndrome is caused by hypomorphic mutations in the peroxisome-biogenesis genes PEX1 and PEX6. Am. J. Hum. Genet. 97, 535–545. doi:10.1016/j.ajhg.2015.08.011
Reddy, J. K., and Mannaerts, G. P. (1994). Peroxisomal lipid metabolism. Annu. Rev. Nutr. 14, 343–370. doi:10.1146/annurev.nu.14.070194.002015
Renaud, M., Guissart, C., Mallaret, M., Ferdinandusse, S., Cheillan, D., Drouot, N., et al. (2016). Expanding the spectrum of PEX10-related peroxisomal biogenesis disorders: Slowly progressive recessive ataxia. J. Neurol. 263, 1552–1558. doi:10.1007/s00415-016-8167-3
Rice, D. S., Calandria, J. M., Gordon, W. C., Jun, B., Zhou, Y., Gelfman, C. M., et al. (2015). Corrigendum: Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell surviva.. Nat. Commun. 6, 7225. doi:10.1038/ncomms8225
Rizzo, W. B., Carney, G., and Lin, Z. (1999). The molecular basis of sjogren-larsson syndrome: Mutation analysis of the fatty aldehyde dehydrogenase gene. Am. J. Hum. Genet. 65, 1547–1560. doi:10.1086/302681
Rodriguez De Turco, E. B., Gordon, W. C., Peyman, G. A., and Bazan, N. G. (1990). Preferential uptake and metabolism of docosahexaenoic acid in membrane phospholipids from rod and cone photoreceptor cells of human and monkey retinas. J. Neurosci. Res. 27, 522–532. doi:10.1002/jnr.490270413
Roth, B. M., Yuan, A., and Ehlers, J. P. (2012). Retinal and choroidal findings in oxalate retinopathy using EDI-OCT. Ophthalmic Surg. Lasers Imaging 43, S142–S144. doi:10.3928/15428877-20121001-05
Ruether, K., Baldwin, E., Casteels, M., Feher, M. D., Horn, M., Kuranoff, S., et al. (2010). Adult Refsum disease: A form of tapetoretinal dystrophy accessible to therapy. Surv. Ophthalmol. 55, 531–538. doi:10.1016/j.survophthal.2010.03.007
Rydzanicz, M., Stradomska, T. J., Jurkiewicz, E., Jamroz, E., Gasperowicz, P., Kostrzewa, G., et al. (2017). Mild zellweger syndrome due to a novel PEX6 mutation: Correlation between clinical phenotype and in silico prediction of variant pathogenicity. J. Appl. Genet. 58, 475–480. doi:10.1007/s13353-017-0414-5
Saadane, A., Mast, N., Charvet, C. D., Omarova, S., Zheng, W., Huang, S. S., et al. (2014). Retinal and nonocular abnormalities in Cyp27a1(-/-)Cyp46a1(-/-) mice with dysfunctional metabolism of cholesterol. Am. J. Pathol. 184, 2403–2419. doi:10.1016/j.ajpath.2014.05.024
Sack, G. H., Raven, M. B., and Moser, H. W. (1989). Color vision defects in adrenomyeloneuropathy. Am. J. Hum. Genet. 44, 794–798.
Sala, A., Lanciotti, M., Valsecchi, M. G., Di Michele, P., Dufour, C., Haupt, R., et al. (2003). Genotypes of the glutathione S-transferase superfamily do not correlate with outcome of childhood acute lymphoblastic leukemia. Leukemia 17, 981–983. doi:10.1038/sj.leu.2402888
Sangiovanni, J. P., Agron, E., Meleth, A. D., Reed, G. F., Sperduto, R. D., Clemons, T. E., et al. (2009). Age-related eye disease study research, G{omega}-3 long-chain polyunsaturated fatty acid intake and 12-y incidence of neovascular age-related macular degeneration and central geographic atrophy: AREDS report 30, a prospective cohort study from the age-related eye disease study. Am. J. Clin. Nutr. 90, 1601–1607. doi:10.3945/ajcn.2009.27594
Santos, F. F., De Turco, E. B., Gordon, W. C., Peyman, G. A., and Bazan, N. G. (1995). Alterations in rabbit retina lipid metabolism induced by detachment. Decreased incorporation of [3H]DHA into phospholipids. Int. Ophthalmol. 19, 149–159. doi:10.1007/BF00133731
Sapieha, P., Stahl, A., Chen, J., Seaward, M. R., Willett, K. L., Krah, N. M., et al. (2011). 5-Lipoxygenase metabolite 4-HDHA is a mediator of the antiangiogenic effect of omega-3 polyunsaturated fatty acids. Sci. Transl. Med. 3, 69ra12. doi:10.1126/scitranslmed.3001571
Schrul, B., and Kopito, R. R. (2016). Peroxin-dependent targeting of a lipid-droplet-destined membrane protein to ER subdomains. Nat. Cell Biol. 18, 740–751. doi:10.1038/ncb3373
Seedorf, U., Brysch, P., Engel, T., Schrage, K., and Assmann, G. (1994). Sterol carrier protein X is peroxisomal 3-oxoacyl coenzyme A thiolase with intrinsic sterol carrier and lipid transfer activity. J. Biol. Chem. 269, 21277–21283. doi:10.1016/s0021-9258(17)31960-9
Shai, N., Yifrach, E., Van Roermund, C. W. T., Cohen, N., Bibi, C., Cavellini, L., et al. (2018). Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 9, 1761. doi:10.1038/s41467-018-03957-8
Shibuya, N., Koike, S., Tanaka, M., Ishigami-Yuasa, M., Kimura, Y., Ogasawara, Y., et al. (2013). A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 4, 1366. doi:10.1038/ncomms2371
Smith, K. D., Kemp, S., Braiterman, L. T., Lu, J. F., Wei, H. M., Geraghty, M., et al. (1999). X-Linked adrenoleukodystrophy: Genes, mutations, and phenotypes. Neurochem. Res. 24, 521–535. doi:10.1023/a:1022535930009
Sprecher, H., Luthria, D. L., Mohammed, B. S., and Baykousheva, S. P. (1995). Reevaluation of the pathways for the biosynthesis of polyunsaturated fatty acids. J. Lipid Res. 36, 2471–2477. doi:10.1016/s0022-2275(20)41084-3
Steinberg, D., Avigan, J., Mize, C., Eldjarn, L., Try, K., and Refsum, S. (1965). Conversion of U-C14-phytol to phytanic acid and its oxidation in heredopathia atactica polyneuritiformis. Biochem. Biophys. Res. Commun. 19, 783–789. doi:10.1016/0006-291x(65)90328-1
Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., and Moser, H. W. (2006). Peroxisome biogenesis disorders. Biochim. Biophys. Acta 1763, 1733–1748. doi:10.1016/j.bbamcr.2006.09.010
Steinberg, S. J., Morgenthaler, J., Heinzer, A. K., Smith, K. D., and Watkins, P. A. (2000). Very long-chain acyl-CoA synthetases. Human "bubblegum" represents a new family of proteins capable of activating very long-chain fatty acids. J. Biol. Chem. 275, 35162–35169. doi:10.1074/jbc.M006403200
Steinberg, S. J., Raymond, G. V., Braverman, N. E., and Moser, A. B. (1993). “Zellweger spectrum disorder,” in GeneReviews. Editors M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle (WA): University of Washington).
Steinberg, S. J., Wang, S. J., Kim, D. G., Mihalik, S. J., and Watkins, P. A. (1999a). Human very-long-chain acyl-CoA synthetase: Cloning, topography, and relevance to branched-chain fatty acid metabolism. Biochem. Biophys. Res. Commun. 257, 615–621. doi:10.1006/bbrc.1999.0510
Steinberg, S. J., Wang, S. J., Mcguinness, M. C., and Watkins, P. A. (1999b). Human liver-specific very-long-chain acyl-coenzyme A synthetase: cDNA cloning and characterization of a second enzymatically active protein. Mol. Genet. Metab. 68, 32–42. doi:10.1006/mgme.1999.2883
Stewart, M. W., Vavra, M. W., and Whaley, N. R. (2011). Fundus findings in A patient with alpha-methlyacyl-coa racemase deficiency. Retin. Cases Brief. Rep. 5, 262–266. doi:10.1097/ICB.0b013e3181f047dd
Stinson, A. M., Wiegand, R. D., and Anderson, R. E. (1991a). Fatty acid and molecular species compositions of phospholipids and diacylglycerols from rat retinal membranes. Exp. Eye Res. 52, 213–218. doi:10.1016/0014-4835(91)90261-c
Stinson, A. M., Wiegand, R. D., and Anderson, R. E. (1991b). Recycling of docosahexaenoic acid in rat retinas during n-3 fatty acid deficiency. J. Lipid Res. 32, 2009–2017. doi:10.1016/s0022-2275(20)41904-2
Stowe, R. C., and Agarwal, S. (2017). Novel PEX26 mutation causing zellweger syndrome presenting as feeding intolerance and hypotonia. Pediatr. Neurol. 75, 96–97. doi:10.1016/j.pediatrneurol.2017.06.012
Su, H. M., Moser, A. B., Moser, H. W., and Watkins, P. A. (2001). Peroxisomal straight-chain Acyl-CoA oxidase and D-bifunctional protein are essential for the retroconversion step in docosahexaenoic acid synthesis. J. Biol. Chem. 276, 38115–38120. doi:10.1074/jbc.M106326200
Summerer, S., Hanano, A., Utsumi, S., Arand, M., Schuber, F., and Blee, E. (2002). Stereochemical features of the hydrolysis of 9, 10-epoxystearic acid catalysed by plant and mammalian epoxide hydrolases. Biochem. J. 366, 471–480. doi:10.1042/BJ20011778
Suzuki, Y., Iai, M., Kamei, A., Tanabe, Y., Chida, S., Yamaguchi, S., et al. (2002). Peroxisomal acyl CoA oxidase deficiency. J. Pediatr. 140, 128–130. doi:10.1067/mpd.2002.120511
Tan, J. S., Wang, J. J., Flood, V., and Mitchell, P. (2009). Dietary fatty acids and the 10-year incidence of age-related macular degeneration: The blue mountains eye study. Arch. Ophthalmol. 127, 656–665. doi:10.1001/archophthalmol.2009.76
Taormina, V. M., Unger, A. L., Schiksnis, M. R., Torres-Gonzalez, M., and Kraft, J. (2020). Branched-chain fatty acids-an underexplored class of dairy-derived fatty acids. Nutrients 12, E2875. doi:10.3390/nu12092875
Thomas, G. H., Haslam, R. H., Batshaw, M. L., Capute, A. J., Neidengard, L., and Ransom, J. L. (1975). Hyperpipecolic acidemia associated with hepatomegaly, mental retardation, optic nerve dysplasia and progressive neurological disease. Clin. Genet. 8, 376–382. doi:10.1111/j.1399-0004.1975.tb01517.x
Thompson, S. A., Calvin, J., Hogg, S., Ferdinandusse, S., Wanders, R. J., and Barker, R. A. (2009). Relapsing encephalopathy in a patient with alpha-methylacyl-CoA racemase deficiency. BMJ Case Rep. 2009, bcr08. doi:10.1136/bcr.08.2008.0814
Thureen, P. J., Narkewicz, M. R., Battaglia, F. C., Tjoa, S., and Fennessey, P. V. (1995). Pathways of serine and glycine metabolism in primary culture of ovine fetal hepatocytes. Pediatr. Res. 38, 775–782. doi:10.1203/00006450-199511000-00023
Trachootham, D., Lu, W., Ogasawara, M. A., Nilsa, R. D., and Huang, P. (2008). Redox regulation of cell survival. Antioxid. Redox Signal. 10, 1343–1374. doi:10.1089/ars.2007.1957
Trachsel-Moncho, L., Benlloch-Navarro, S., Fernandez-Carbonell, A., Ramirez-Lamelas, D. T., Olivar, T., Silvestre, D., et al. (2018). Oxidative stress and autophagy-related changes during retinal degeneration and development. Cell Death Dis. 9, 812. doi:10.1038/s41419-018-0855-8
Treen, M., Uauy, R. D., Jameson, D. M., Thomas, V. L., and Hoffman, D. R. (1992). Effect of docosahexaenoic acid on membrane fluidity and function in intact cultured Y-79 retinoblastoma cells. Arch. Biochem. Biophys. 294, 564–570. doi:10.1016/0003-9861(92)90726-d
Trujillo, M., Clippe, A., Manta, B., Ferrer-Sueta, G., Smeets, A., Declercq, J. P., et al. (2007). Pre-steady state kinetic characterization of human peroxiredoxin 5: Taking advantage of Trp84 fluorescence increase upon oxidation. Arch. Biochem. Biophys. 467, 95–106. doi:10.1016/j.abb.2007.08.008
Tsujihata, M. (2008). Mechanism of calcium oxalate renal stone formation and renal tubular cell injury. Int. J. Urol. 15, 115–120. doi:10.1111/j.1442-2042.2007.01953.x
Turk, B. R., Theda, C., Fatemi, A., and Moser, A. B. (2020). X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int. J. Dev. Neurosci. 80, 52–72. doi:10.1002/jdn.10003
Uchiyama, A., Aoyama, T., Kamijo, K., Uchida, Y., Kondo, N., Orii, T., et al. (1996). Molecular cloning of cDNA encoding rat very long-chain acyl-CoA synthetase. J. Biol. Chem. 271, 30360–30365. doi:10.1074/jbc.271.48.30360
Uzor, N. E., Mccullough, L. D., and Tsvetkov, A. S. (2020). Peroxisomal dysfunction in neurological diseases and brain aging. Front. Cell. Neurosci. 14, 44. doi:10.3389/fncel.2020.00044
Vamecq, J. (1987). Chlorpromazine and carnitine-dependency of rat liver peroxisomal beta-oxidation of long-chain fatty acids. Biochem. J. 241, 783–791. doi:10.1042/bj2410783
van den Bosch, H., Schutgens, R. B., Wanders, R. J., and Tager, J. M. (1992). Biochemistry of peroxisomes. Annu. Rev. Biochem. 61, 157–197. doi:10.1146/annurev.bi.61.070192.001105
van den Brink, D. M., Van Miert, J. N., Dacremont, G., Rontani, J. F., Jansen, G. A., and Wanders, R. J. (2004). Identification of fatty aldehyde dehydrogenase in the breakdown of phytol to phytanic acid. Mol. Genet. Metab. 82, 33–37. doi:10.1016/j.ymgme.2004.01.019
van den Brink, D. M., Van Miert, J. N., Dacremont, G., Rontani, J. F., and Wanders, R. J. (2005). Characterization of the final step in the conversion of phytol into phytanic acid. J. Biol. Chem. 280, 26838–26844. doi:10.1074/jbc.M501861200
van Roermund, C. W., Ijlst, L., Wagemans, T., Wanders, R. J., and Waterham, H. R. (2014). A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim. Biophys. Acta 1841, 563–568. doi:10.1016/j.bbalip.2013.12.001
van Roermund, C. W., Visser, W. F., Ijlst, L., Waterham, H. R., and Wanders, R. J. (2011). Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid beta-oxidation. Biochim. Biophys. Acta 1811, 148–152. doi:10.1016/j.bbalip.2010.11.010
Van Veldhoven, P. P., Brees, C., and Mannaerts, G. P. (1991). D-aspartate oxidase, a peroxisomal enzyme in liver of rat and man. Biochim. Biophys. Acta 1073, 203–208. doi:10.1016/0304-4165(91)90203-s
Van Veldhoven, P. P., Croes, K., Asselberghs, S., Herdewijn, P., and Mannaerts, G. P. (1996). Peroxisomal beta-oxidation of 2-methyl-branched acyl-CoA esters: Stereospecific recognition of the 2S-methyl compounds by trihydroxycoprostanoyl-CoA oxidase and pristanoyl-CoA oxidase. FEBS Lett. 388, 80–84. doi:10.1016/0014-5793(96)00508-x
Van Veldhoven, P. P., De Schryver, E., Young, S. G., Zwijsen, A., Fransen, M., Espeel, M., et al. (2020). Slc25a17 gene trapped mice: PMP34 plays a role in the peroxisomal degradation of phytanic and pristanic acid. Front. Cell Dev. Biol. 8, 144. doi:10.3389/fcell.2020.00144
Vanhove, G. F., Van Veldhoven, P. P., Fransen, M., Denis, S., Eyssen, H. J., Wanders, R. J., et al. (1993). The CoA esters of 2-methyl-branched chain fatty acids and of the bile acid intermediates di- and trihydroxycoprostanic acids are oxidized by one single peroxisomal branched chain acyl-CoA oxidase in human liver and kidney. J. Biol. Chem. 268, 10335–10344. doi:10.1016/s0021-9258(18)82206-2
Verhoeven, N. M., Roe, D. S., Kok, R. M., Wanders, R. J., Jakobs, C., and Roe, C. R. (1998). Phytanic acid and pristanic acid are oxidized by sequential peroxisomal and mitochondrial reactions in cultured fibroblasts. J. Lipid Res. 39, 66–74. doi:10.1016/s0022-2275(20)34204-8
Violante, S., Achetib, N., Van Roermund, C. W. T., Hagen, J., Dodatko, T., Vaz, F. M., et al. (2019). Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 33, 4355–4364. doi:10.1096/fj.201801498R
Voss, A., Reinhart, M., Sankarappa, S., and Sprecher, H. (1991). The metabolism of 7, 10, 13, 16, 19-docosapentaenoic acid to 4, 7, 10, 13, 16, 19-docosahexaenoic acid in rat liver is independent of a 4-desaturase. J. Biol. Chem. 266, 19995–20000. doi:10.1016/s0021-9258(18)54882-1
Wallner, S., and Schmitz, G. (2011). Plasmalogens the neglected regulatory and scavenging lipid species. Chem. Phys. Lipids 164, 573–589. doi:10.1016/j.chemphyslip.2011.06.008
Wanders, R. J., Dekker, C., Hovarth, V. A., Schutgens, R. B., Tager, J. M., Van Laer, P., et al. (1994). Human alkyldihydroxyacetonephosphate synthase deficiency: A new peroxisomal disorder. J. Inherit. Metab. Dis. 17, 315–318. doi:10.1007/BF00711817
Wanders, R. J., Komen, J., and Ferdinandusse, S. (2011a). Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta 1811, 498–507. doi:10.1016/j.bbalip.2011.06.006
Wanders, R. J., Komen, J., and Kemp, S. (2011b). Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 278, 182–194. doi:10.1111/j.1742-4658.2010.07947.x
Wanders, R. J. (2014). Metabolic functions of peroxisomes in health and disease. Biochimie 98, 36–44. doi:10.1016/j.biochi.2013.08.022
Wanders, R. J., Schumacher, H., Heikoop, J., Schutgens, R. B., and Tager, J. M. (1992). Human dihydroxyacetonephosphate acyltransferase deficiency: A new peroxisomal disorder. J. Inherit. Metab. Dis. 15, 389–391. doi:10.1007/BF02435984
Wanders, R. J., Waterham, H. R., and Ferdinandusse, S. (2015). Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front. Cell Dev. Biol. 3, 83. doi:10.3389/fcell.2015.00083
Wang, B. J., Xia, J. M., Wang, Q., Yu, J. L., Song, Z., and Zhao, H. (2020). Diet and adaptive evolution of alanine-glyoxylate aminotransferase mitochondrial targeting in birds. Mol. Biol. Evol. 37, 786–798. doi:10.1093/molbev/msz266
Wang, N., and Anderson, R. E. (1993a). Synthesis of docosahexaenoic acid by retina and retinal pigment epithelium. Biochemistry 32, 13703–13709. doi:10.1021/bi00212a040
Wang, N., and Anderson, R. E. (1993b). Transport of 22:6n-3 in the plasma and uptake into retinal pigment epithelium and retina. Exp. Eye Res. 57, 225–233. doi:10.1006/exer.1993.1118
Waterham, H. R., and Ebberink, M. S. (2012). Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta 1822, 1430–1441. doi:10.1016/j.bbadis.2012.04.006
Waterham, H. R., Ferdinandusse, S., and Wanders, R. J. (2016). Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 1863, 922–933. doi:10.1016/j.bbamcr.2015.11.015
Watkins, P. A., Howard, A. E., and Mihalik, S. J. (1994). Phytanic acid must be activated to phytanoyl-CoA prior to its alpha-oxidation in rat liver peroxisomes. Biochim. Biophys. Acta 1214, 288–294. doi:10.1016/0005-2760(94)90075-2
Werner, E. R., Keller, M. A., Sailer, S., Lackner, K., Koch, J., Hermann, M., et al. (2020). The TMEM189 gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc. Natl. Acad. Sci. U. S. A. 117, 7792–7798. doi:10.1073/pnas.1917461117
Wetzel, M. G., Li, J., Alvarez, R. A., Anderson, R. E., and O'brien, P. J. (1991). Metabolism of linolenic acid and docosahexaenoic acid in rat retinas and rod outer segments. Exp. Eye Res. 53, 437–446. doi:10.1016/0014-4835(91)90161-7
Wheeler, J. B., Shaw, D. R., and Barnes, S. (1997). Purification and characterization of a rat liver bile acid coenzyme A ligase from rat liver microsomes. Arch. Biochem. Biophys. 348, 15–24. doi:10.1006/abbi.1997.0391
Win, A., Delgado, A., Jadeja, R. N., Martin, P. M., Bartoli, M., and Thounaojam, M. C. (2021). Pharmacological and metabolic significance of bile acids in retinal diseases. Biomolecules 11, 292. doi:10.3390/biom11020292
Winterbourn, C. C. (1995). Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 82-83, 969–974. doi:10.1016/0378-4274(95)03532-x
Wojtowicz, S., Strosznajder, A. K., Jezyna, M., and Strosznajder, J. B. (2020). The novel role of PPAR alpha in the brain: Promising target in therapy of alzheimer's disease and other neurodegenerative disorders. Neurochem. Res. 45, 972–988. doi:10.1007/s11064-020-02993-5
Xiong, W., Maccoll Garfinkel, A. E., Li, Y., Benowitz, L. I., and Cepko, C. L. (2015). NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Invest. 125, 1433–1445. doi:10.1172/JCI79735
Yako, T., Otsu, W., Nakamura, S., Shimazawa, M., and Hara, H. (2022). Lipid droplet accumulation promotes RPE dysfunction. Int. J. Mol. Sci. 23, 1790. doi:10.3390/ijms23031790
Yamamoto, Y., and Sakisaka, T. (2018). The peroxisome biogenesis factors posttranslationally target reticulon homology domain-containing proteins to the endoplasmic reticulum membrane. Sci. Rep. 8, 2322. doi:10.1038/s41598-018-20797-0
Young, R. W. (1976). Visual cells and the concept of renewal. Invest. Ophthalmol. Vis. Sci. 15, 700–725.
Zaki, M. S., Heller, R., Thoenes, M., Nurnberg, G., Stern-Schneider, G., Nurnberg, P., et al. (2016). PEX6 is expressed in photoreceptor cilia and mutated in deafblindness with enamel dysplasia and microcephaly. Hum. Mutat. 37, 170–174. doi:10.1002/humu.22934
Zhao, Y., Wu, Y., Pei, J., Chen, Z., Wang, Q., and Xiang, B. (2015). Safety and efficacy of parenteral fish oil-containing lipid emulsions in premature neonates. J. Pediatr. Gastroenterol. Nutr. 60, 708–716. doi:10.1097/MPG.0000000000000665
Zhu, X. H., Lu, M., Lee, B. Y., Ugurbil, K., and Chen, W. (2015). In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. U. S. A. 112, 2876–2881. doi:10.1073/pnas.1417921112
Zomer, A. W., Van Der Burg, B., Jansen, G. A., Wanders, R. J., Poll-The, B. T., and Van Der Saag, P. T. (2000). Pristanic acid and phytanic acid: Naturally occurring ligands for the nuclear receptor peroxisome proliferator-activated receptor α. J. Lipid Res. 41, 1801–1807. doi:10.1016/s0022-2275(20)31973-8
Keywords: peroxisome, retinal lipids, fatty acid oxidation, retinal dystrophy, retinopathy
Citation: Chen CT, Shao Z and Fu Z (2022) Dysfunctional peroxisomal lipid metabolisms and their ocular manifestations. Front. Cell Dev. Biol. 10:982564. doi: 10.3389/fcell.2022.982564
Received: 30 June 2022; Accepted: 17 August 2022;
Published: 07 September 2022.
Edited by:
Rajalekshmy Shyam, Indiana University Bloomington, United StatesReviewed by:
Yukio Fujiki, Kyushu University Graduate School, JapanCopyright © 2022 Chen, Shao and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongjie Fu, emhvbmdqaWUuZnVAY2hpbGRyZW5zLmhhcnZhcmQuZWR1
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.