
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 10 August 2022
Sec. Signaling
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.976953
This article is part of the Research Topic Neuroinflammation in Acquired Epilepsy View all 10 articles
 Timothy Fabisiak
Timothy Fabisiak Manisha Patel*
Manisha Patel*The roles of both neuroinflammation and oxidative stress in the pathophysiology of epilepsy have begun to receive considerable attention in recent years. However, these concepts are predominantly studied as separate entities despite the evidence that neuroinflammatory and redox-based signaling cascades have significant crosstalk. Oxidative post-translational modifications have been demonstrated to directly influence the function of key neuroinflammatory mediators. Neuroinflammation can further be controlled on the transcriptional level as the transcriptional regulators NF-KB and nrf2 are activated by reactive oxygen species. Further, neuroinflammation can induce the increased expression and activity of NADPH oxidase, leading to a highly oxidative environment. These factors additionally influence mitochondria function and the metabolic status of neurons and glia, which are already metabolically stressed in epilepsy. Given the implication of this relationship to disease pathology, this review explores the numerous mechanisms by which neuroinflammation and oxidative stress influence one another in the context of epilepsy. We further examine the efficacy of treatments targeting oxidative stress and redox regulation in animal and human epilepsies in the literature that warrant further investigation. Treatment approaches aimed at rectifying oxidative stress and aberrant redox signaling may enable control of neuroinflammation and improve patient outcomes.
There are 31 types of epilepsy syndromes classified by the International League Against Epilepsy (Epilepsy Syndromes, 2022) based on numerous features such as seizure type, epilepsy presentation, and etiology (Scheffer et al., 2017). This disease complexity presents a challenge in the treatment of epilepsy, where available anti-seizure medications (ASMs) that are tailored to the epilepsy syndrome generally only achieve a 30–50% responder rate (Löscher and Klein, 2021). The prevalence of drug-resistant epilepsy highlights a need to further understand underlying mechanisms in epilepsy such as neuroinflammation and consequential signaling cascades, as reviewed further elsewhere (Vezzani et al., 2019). Yet more recently, oxidative stress and redox dysregulation have also emerged as hallmarks in the epilepsy literature (Patel, 2004; Pearson-Smith & Patel, 2017; Geronzi et al., 2018).
Interestingly, in human epilepsies and animal epilepsy models, various biomarkers of both oxidative stress and neuroinflammation are elevated in the brain and periphery. Neuroinflammation and reactive oxygen species (ROS) production are both prominent effectors of signaling and are therefore heavily regulated. It is not unexpected for these prominent cell signaling processes to have influence on the regulation of one another. As an evolutionarily conserved defense mechanism, inflammation in response to infection or injury promotes and utilizes ROS to kill pathogens or enact rapid, local signaling. At physiological levels, ROS is an important second messenger that modulates neuroinflammation at numerous stages through redox-sensitive mechanisms. Further, excessive ROS production or redox dysregulation under oxidative stress damages cells and produces danger signals that incite neuroinflammation. As oxidative stress and neuroinflammatory pathways have considerable crosstalk (See Scheme 1), there is an unmet need for further research exploring this relationship in the pathophysiology of epilepsy. This can enable the development of novel redox-based therapeutics, beyond traditional ASMs which primarily aim to rectify neural excitatory/inhibitor imbalances at the synapse, to control neuroinflammation and in turn, epilepsy and/or its comorbidities.
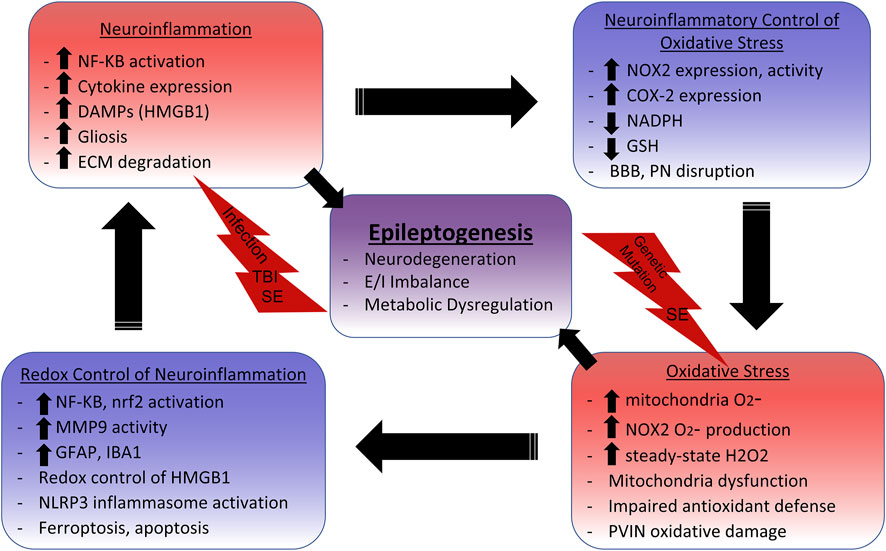
SCHEME 1. Neuroinflammation and Oxidative Stress Cycle in Epilepsy.Initial insults such as systemic infection, traumatic brain injury (TBI), status epilepticus (SE), and genetic mutations initiate inflammatory and oxidative signaling cascades. NF-KB activation leads to the production of proinflammatory cytokines such as IL-1β, TNFα, and IL-6, which have been shown to i nduce gliosis, mitochondrial dysfunction, and glutathione (GSH) depletion. GSH depletion impairs cellular and mitochondrial antioxidant defenses, enabling aberrant oxidative signaling and damage. NAPDH Oxidase 2 (NOX2) upregulation also commonly occurs with neuroinflammation. NOX2 utilizes NADPH, which is needed for the reductive abilities of antioxidant systems like thioredoxin reductase and glutathione peroxidase, to produce superoxide (O2-). O2-is primarily enzymatically converted to the redox-signaling reactive H2O2. The increased levels of steady-state H2O2 leads to the activation of numerous redox-sensitive pathways that influence neuroinflammation. For example, NF-KB activation and subsequent microglia activation relies on NOX2 activity, indicating that reactive oxygen species (ROS) production perpetuates neuroinflammation. Nrf2 is also activated by ROS to increase expression of the antioxidant response element (ARE) to combat oxidative damage and neuroinflammation, making it an attractive therapeutic target. The danger-associated molecular pattern (DAMP) HMGB1 is released in response to neuronal damage, which signals through TLR4 to activate NF-KB, induce gliosis, and increase NOX2 and COX-2 expression. Reduced HMGB1 has chemoattractant properties, but adjacent cysteine residues act as a redox switch where oxidation to a disulfide form increases affinity for TLR4 and induces cytokine like properties. The NLRP3 inflammasome, which activates IL-1β, can also by activated by ROS from the mitochondria and NOX. Extracellular matrix (ECM) digesting proteinases such as matrix metalloproteinases (MMPs) are also upregulated in the neuroinflammatory environment. This can lead to the disruption of the blood brain barrier (BBB) and perineuronal nets (PNs) surrounding inhibitory parvalbumin interneurons (PVINs). MMP9 has demonstrated redox-sensitive activation, which is associated with the loss of PNs and death of PVINs. Mitochondria dysfunction can result from excessive mitochondrial ROS (mtROS) which not only accounts for metabolic alterations, but can lead to inflammation-inducing events such as astrogliosis, apoptosis, and lipid peroxidase mediated death (ferroptosis). Metabolic dysfunction, excitatory/inhibitory (E/I) imbalance, and neurodegeneration can all result from these oxidative and inflammatory processes which ultimately contribute to epileptogenesis.
The brain is particularly susceptible to oxidative damage due to its high metabolic demand and per weight rate of oxygen consumption. It has long been recognized that ictal cerebral hypermetabolism is characteristic of seizure disorders, where increased cerebral oxygen (Meyer et al., 1966) and glucose consumption (Ingvar and Siesjö, 1983) are observed during seizure activity in experimental epilepsy models and in humans (During et al., 1994). These metabolic stresses lead to interictal metabolic dysfunction which is evidenced by glucose hypometabolism in chronic epilepsy (Guo et al., 2009) and mitochondrial metabolic deficits resulting from inhibition of mitochondrial enzyme complexes in animal and human epilepsies (Kunz et al., 2000, 1999). It has been demonstrated that mitochondrial reactive oxygen species (mtROS) damage mitochondrial electron transport chain complex I, subsequently decreasing mitochondrial oxidative phosphorylation (Ryan et al., 2012; Rowley et al., 2015). Further, pharmacological inhibition of mitochondrial enzyme complexes results in decreased oxidative phosphorylation and seizure activity (Fleck et al., 2004).
There are numerous indications that increased ROS production occurs across epilepsy models, as evidenced by oxidative damage and impaired redox status. In addition to Complex I inhibition in humans and animal models of temporal lobe epilepsy (TLE), the redox regulated TCA cycle enzymes aconitase and α-ketoglutarate dehydrogenase have been shown to be inactivated following status epilepticus (SE) in rats (Cock et al., 2002). Increased mitochondria oxidative phosphorylation is also shown to increase complex III mediated mtROS production (Malinska et al., 2010). Mitochondrial DNA (mtDNA) oxidative damage has also been found in the kainate model of TLE, along with increases in mitochondrial H2O2 (Jarrett et al., 2008a). Further, numerous other animal models and human epilepsies revealed depleted glutathione (GSH) levels, paired with increased oxidized glutathione (glutathione disulfide or GSSG) in the hippocampus or neocortex, indicative of an oxidative environment (Rumià et al., 2013; Cárdenas-Rodríguez et al., 2014; Pearson-Smith and Patel, 2017). As GSH acts as a major antioxidant in the brain, this impaired GSH redox status can lead to further oxidative damage and redox dysregulation, which is associated with cognitive deficits, neuronal death, and mortality in animal epilepsy models. Indeed, loss of glutathione peroxidase 4, a oxidation-resistant selenoprotein that utilizes GSH, leads to lipid peroxidation and subsequent ferroptosis, which is also found in epilepsy models (Ingold et al., 2018; Cai and Yang, 2021).
The NADPH oxidase (NOX) family of enzymes is another relevant major source of ROS in the context of epilepsy as evidenced by activation of NOX2 by kainate-induced SE (Patel et al., 2005). NOX enzymes utilize NADPH to produce superoxide (O2−), which then undergoes primarily enzymatic dismutation to H2O2. Interestingly, it has been demonstrated that NMDA receptor activation, which is increased due to excessive extracellular glutamate in epilepsy, increases O2− and subsequent H2O2 production via NOX induction (Brennan et al., 2009; Kovac et al., 2014). Further, NOX activation has been shown to be a sufficient trigger of seizure activity, while NOX inhibition reduced hyperactivity in multiple animal models of epilepsy (Malkov et al., 2019). It is argued that NOX2, the primary isoform in microglia, is thought to be the major contributor of SE-induced ROS production in acquired epilepsies, which has also been linked to glucose hypometabolism in epileptogenesis (Zilberter et al., 2022).
Biomarkers of oxidative stress in human epilepsies have recently received further attention. In the blood of patients with SE, lower levels of SOD, catalase, GSH, and total antioxidant capacity were found (Kalita et al., 2019). In drug-resistant complex partial seizure patient blood, there was an inverse correlation of vitamin C and positive correlation of the oxidative damage marker 3-nitrotyrosine to seizure frequency (Lorigados Pedre et al., 2018). Justifiably, therapies targeting oxidative stress are currently under investigation. In rat electrical status epilepticus, GSH increasing drug treatment was neuroprotective and reduced seizure frequency (Pauletti et al., 2019). Inhibition of KEAP1 by RTA 408 disinhibits the antioxidant transcription factor nrf2 and has neuroprotective and seizure reducing effects in the kainic model of TLE (Shekh-Ahmad et al., 2018). Treatment with a NOX inhibitor, a catalytic antioxidant, and a scavenger of reactive oxidized compounds gamma-ketoaldehydes, have all been shown to prevent experimental TLE seizure-induced neuronal death (Kim et al., 2013; Pearson et al., 2017; Pearson-Smith et al., 2017). In humans, Vitamin E in conjunction with ASMs has been shown to reduce seizure frequency and oxidative stress (Mehvari et al., 2016). Further, the high-fat low-carbohydrate ketogenic diet (KD), which shifts metabolism towards fatty acid oxidation and away from glycolysis, also has antioxidant properties which may contribute to its clinical efficacy in Dravet Syndrome (Caraballo et al., 2005). As a treatment for experimental or genetic epilepsies in rodents, this diet can reduce ROS production, activate nrf2, and increase the synthesis of GSH (Jarrett et al., 2008b; Milder and Patel, 2012).
Clearly, oxidative stress is associated with epilepsy which may be a consequence of the disease. Interestingly, there is further evidence indicating that mitochondrial dysfunction and oxidative stress contribute to epilepsy pathogenesis (Patel, 2004). The most robust links between oxidative stress or mitochondrial dysfunction and epilepsy are clear in mitochondrial encephalopathies such as myclonic epilepsy with ragged-red fibers (MERFF) and Leigh syndrome. These disorders are characterized by mtDNA mutations that impair mitochondria electron transport chain enzyme complexes, which can result in increased ROS production, oxidative damage, and decreased ATP production (Quintana et al., 2010; Wu et al., 2010; Wojtala et al., 2017). Other genetic epilepsies such as Dravet Syndrome are further associated with mitochondrial dysfunction (Kumar et al., 2016; Banerji et al., 2021). Genetic knockouts in mice of mitochondrial superoxide dismutase (SOD2), which detoxifies O2− to H2O2, increases susceptibility to spontaneous and induced seizures (Liang and Patel, 2004; Liang et al., 2012). Further, the forebrain neuron-specific conditional knockout of SOD2 also leads to the presentation of epilepsy in mice (Rowley et al., 2015). Together, this body of work demonstrates that oxidative stress is not only a consequence of epilepsy, but also a cause.
Seizure inciting events such as infection or traumatic brain injury (TBI) that initiate proinflammatory cascades are also linked to oxidative stress. Post-traumatic epilepsy (PTE) develops in as many as 50% of TBI patients, and TBI and chronic epilepsy brain tissue both display elevated markers of oxidative stress in addition to cytokines and neurodegeneration (Vezzani et al., 2008; Helmy et al., 2011; Terrone et al., 2019). NOX2 expression is additionally elevated in the brain following TBI and in epilepsy as reviewed by Ma et al., 2018. In experimental models of acquired epilepsy, such as chemically induced status epilepticus, there are clear signs of inflammation, reactive gliosis, and neurodegeneration (Loewen et al., 2016) as well as metabolic and redox dysregulation as reviewed by Pearson-Smith & Patel, 2017. In a zebrafish model of Dravet syndrome, there are metabolic irregularities (Kumar et al., 2016; Banerji et al., 2021) as well as increased astrogliosis associated gene expression (Tiraboschi et al., 2020), which is also apparent in a mouse DS model (Valassina et al., 2022). In pediatric drug-resistant epilepsy, levels of IL-1β in peripheral monocytes correlate to seizure frequency (Yamanaka et al., 2021). Further, an LPS administration model of systemic inflammation in sepsis in conjunction with convulsant PTZ treatment in mice increased seizure susceptibility, which was attenuated with NOX2 genetic ablation and inhibition (Huang et al., 2018). Theiler’s murine encephalomyelitis virus (TMEV) model of temporal lope epilepsy is known to induce neuroinflammation and seizures, but also leads to oxidative stress as indicated by elevated 3-nitrotyrasine levels and reduced GSH/GSSG ratios in the hippocampus (Bhuyan et al., 2015).
Epileptic encephalopathies and genetic epilepsies such as West, Lennox-Gastaut, Dravet, Lafora, and Leigh syndromes have additionally shown evidence of gliosis and neuroinflammation in humans (Kawashima et al., 1999; You et al., 2009; Salar and Galanopoulou, 2018; Lahuerta et al., 2020; Valassina et al., 2022). There is also evidence that most of these types of epilepsies also have a metabolic dysfunction component given the efficacy of the KD in patients (Wijburg et al., 1992; Caraballo et al., 2005, 2014; You et al., 2009). As both neuroinflammation and oxidative stress or metabolic dysregulation are prevalent across the epilepsies, which are both argued to be causal and consequential to the disease, the direct interactions between neuroinflammation and oxidative stress are key components of disease pathology which deserve further evaluation.
The role of NADPH oxidases in epilepsy has drawn considerable attention, with findings highlighting a link between oxidative stress production and inflammation. NOX expression of the brain isoforms 1, 2 and 4 are highly inducible, with signs of NOX2 upregulation prevalent in human and animal epilepsies. As described previously, SE can induce ROS production through NOX via NMDA receptor activation. However, NOX expression and activity is also closely linked to neuroinflammation. Pattern recognition receptor (PRR) CR3 and TLR4 signaling by damage associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1) released by damaged neurons, is linked to NOX2 expression and activation in microglia (Pei et al., 2007; Gao et al., 2011; Bell et al., 2013; Hou et al., 2018). The inhibition of NF-KB signaling has been shown to reduce LPS induced NOX and inducible nitric oxide synthase (iNOS) expression in various peripheral cell types (Kim B et al., 2008; Al-Harbi et al., 2020; Sul and Ra, 2021). NOX-mediated aberrant ROS production in response to proinflammatory signaling is a major source of oxidative stress in epilepsy, and is further linked to interictal glucose hypometabolism (Malkov et al., 2018). It is suggested that increased NOX activity induced by SE or trauma increases steady-state levels of H2O2 directly and indirectly by utilizing NADPH needed for glutathione reduction, leading to the inhibition of glycolysis and increased cell susceptibility to oxidative stress. Further, other NOX2-mediated H2O2 redox signaling is shown to directly increase mtROS production in one of the forms of ROS-induced ROS release (Kim et al., 2017).
DAMPs, particularly HMGB1, have been implicated in epilepsy pathogenesis. HMGB1 and the PRR it can signal through, TLR4, have increased expression in human and animal epilepsies. HMGB1 released from neurons and proinflammatory microglia and has been attributed to both TBI and epilepsy pathogenesis in experimental models as reviewed by Paudel et al., 2018. In animal TLE models, HMGB1 acts through TLR-4 to increase production of cytokines such as IL-1β, TNFα, and IL-6 (Maroso et al., 2010). This TLR-4 mediated signaling cascade can activate NF-KB, leading to the production of these pro-convulsant cytokines. Cytokines like TNFα can, in turn, induce COX-2 and NOX2 gene expression (Newton et al., 1997; Li et al., 2009). Interestingly, peripheral inflammation induced by TLR-4 activation by systemic LPS exacerbated kainic-acid induced seizures and elevated hippocampal reactive microglia, levels of IL-1β, TNFα, and IL-6, and NOX subunit expression (Ho et al., 2015). The ability of PRR-mediated inflammatory signaling to increase NOX expression in epilepsy suggests that enabling ROS production may act as a second messenger to neuroinflammation.
Tumor necrosis factor alpha (TNFα) further impacts the oxidative environment and metabolism in the brain by targeting mitochondria. In neuronal cultures, direct treatment with TNFα resulted in a time dependent decrease in mitochondrial respiration, followed by a reduction in cell viability correlated with increase cytosolic cytochrome c levels (Doll et al., 2015). In mice, systemic LPS increased brain region specific expression of IL-1β as well as mitochondrial complex II/III activity, while decreasing GSH (Noh et al., 2014). Local brain LPS administration, as well as astrocyte exposure to TNFα and IL-1β, depleted GSH (Gavillet et al., 2008; Ariza et al., 2010). These studies indicate that neuroinflammation can influence mitochondrial function, impair antioxidant defenses, and cause oxidative stress to contribute to neurodegeneration.
Neuroinflammation in epilepsy is commonly associated with blood brain barrier (BBB) disruption, allowing infiltration of the brain parenchyma by peripheral immune cells and serum albumin as reviewed in detail by Van Vliet et al. (2015). In addition to exacerbating gliosis and inflammation, BBB disruption also leads to oxidative damage such as in ischemia-reperfusion injury where SOD1 deficiency causes exacerbated neuronal damage and infarct volume (Gasche et al., 2001). Inhibition of matrix metalloproteinase 9 (MMP9), which degrades extracellular matrix around the vasculature, rescues this effect. MMP9 also contributes to parvalbumin positive inhibitory interneuron (PVIN) cell loss that is found in epilepsy. MMP9 is upregulated in epilepsy models, and this ECM proteinase has been shown to degrade the specialized ECM called perineuronal nets (PN) surrounding PVINs (Kim et al., 2009; Acar et al., 2015; Rankin-Gee et al., 2015; Dubey et al., 2017). Interestingly, PVINs are shown to be particularly susceptible to oxidative stress, and the neuroinflammatory based degradation of their neuroprotective PNs leads to oxidative damage and death of these cells (Cabungcal et al., 2013). In fact, neuronal NOX2 expression which is induced by neuroinflammation colocalizes with parvalbumin immunoreactivity in areas of PVIN loss in human TBI (Schiavone et al., 2017).
NOX activity may be induced by proinflammatory signaling cascades, but it is also necessary for inducing neuroinflammation. In the kainate and pilocarpine rat experimental models of TLE, ROS production through NOX activity is associated with microglial activation and is responsible for neurodegeneration in these models (Patel et al., 2005; Pestana et al., 2010). SE induced ROS production and neural cell death were attenuated by NOX inhibitors (Pestana et al., 2010; Williams et al., 2015) and overexpression of extracellular SOD (Patel et al., 2005). Interestingly, the deletion of NOX2 following mouse TBI (Dohi et al., 2010; Wang et al., 2018) or PTZ-induced SE (Huang et al., 2018) enhances neurogenesis and reduces cytokine production, indicating that the ROS production occurring following trauma is deleterious to neural recovery. A direct link between NOX activity and inflammation is apparent, as NOX2 mediated production of O2− and resulting increased steady-state H2O2 levels is necessary for NF-KB activation in macrophages (Li et al., 2018) and for microglia proliferation (Mander et al., 2006). The NOX2 inhibitor celastrol attenuated kainate-induced epileptiform activity, indicating that NOX2 is important for seizure initiation (Malkov et al., 2019). As it has been suggested that NOX2 is important for the initial oxidative burst following SE that contributes to seizures in trauma and chemically induced epilepsy models (Zilberter et al., 2022), the efficacy of NOX inhibition in chronic epilepsy and in a clinically relevant window following a seizure inciting event needs further evaluation. Chronic administration of apocynin, a NOX inhibitor antioxidant, following pilocarpine-induced SE did significantly increase neural survival (Lee et al., 2018). Despite the promise of NOX2 inhibition in epilepsy models, further efforts to develop and evaluate clinically translatable BBB permeable compounds such as GSK2795039 are needed (Hirano et al., 2015; Malkov et al., 2019).
Excessive ROS levels are toxic, but at physiological levels ROS such as H2O2 are important second messengers acting through redox signaling. H2O2 is an efficient activator of the redox-regulated proinflammatory transcription factor NF-KB, where redox-regulation activates upstream kinases P13K and NIK leading to NF-KB translocation and promotor binding. Further, NOX2 depletion and the NOX inhibitor apocynin were able to reduce NF-KB activation, demonstrating how ROS can lead to the increased expression of proinflammatory cytokines (Kim J et al., 2008). Nrf2, the redox-sensitive transcription factor controlling expression of the antioxidant response element (ARE), is an important modulator of the proinflammatory effects of ROS. For example, nrf2 depletion has been shown to increase NF-ΚB activation, as well as the activation of MMP3/9 and TGF-β following mouse TBI leading to neurodegeneration (Bhowmick et al., 2019). The nrf2 targets heme oxygenase 1 (HO-1), NQO1, and the GSH producing enzymes GCLC and GCLM have all been shown to exert anti-inflammatory effects as reviewed in detail by Ahmed et al., 2017. Not surprisingly, the inhibition of the nrf2 inhibitor KEAP1 with RTA 408, particularly in combination with a NOX inhibitor, prevented cell death and dramatically reduced kainate-induced seizures (Shekh-Ahmad et al., 2019, 2018). The KD’s clinical efficacy in the treatment of various epilepsies may be mediated in part due to its apparent ability to activate nrf2 (Milder et al., 2010). These inflammation modifying roles of nrf2 demonstrate redox-neuroinflammation crosstalk at the transcriptional level which highlights nrf2 as an important therapeutic target for controlling both ROS-mediated damage and activation of proinflammatory cascades.
Certain proinflammatory molecules are also directly regulated by ROS such as IL-1β, HMGB1, and MMP9. IL-1β activation is induced by caspase-1 which is activated by the DAMP-sensitive NLRP3 and NLRC4 inflammasomes (Poyet et al., 2001; Martinon et al., 2002). NLRP3 has been extensively linked to epilepsy, where heightened expression and activation is found in animal and human epilepsies as reviewed by Mohseni-Moghaddam et al., 2021. ROS can lead to NLRP3 activation through binding to thioredoxin-interacting protein (TXNIP) binding following thioredoxin oxidation (Zhou et al., 2011). Further, the crystal structure of NLRP3 revealed a disulfide bond between conserved cysteine residues in the inflammasome active site, suggesting redox-sensitive activity (Bae and Park, 2011). NLRP3 has indeed been shown to be activated by NOX2 mediated ROS production (Abais et al., 2014, 2013). Further, mtROS acts as a second messenger stimulating NLRP3 activation and localization to the mitochondria to promote IL-1β activation, thus inciting a proinflammatory signal in response to mitochondria dysfunction (Zhou et al., 2011; Subramanian et al., 2013). Interestingly, sulforaphane, an nrf2 activator, has been shown to limit mtROS generation and prevent both NLRP3 and NLRC4 activation in murine macrophages (Lee et al., 2016). Although implicated in neurodegenerative disorders but not currently in epilepsy, NLRC4 activation has also been found to be sensitive to mtROS production in astrocytes and contributes to gliosis (Freeman et al., 2017; Samidurai et al., 2020). NLRP3 activation further appears to be inhibited by the activity of the ketone body βHB produced by the KD, suggesting that the modulation of neuroinflammation contributes to KD efficacy (Youm et al., 2015). ROS-mediated inflammasome activation could explain the increased prevalence of IL-1β found in epilepsy as described above, which may in turn perpetuate cycle of oxidative stress and neuroinflammation.
HMGB1 contains three redox sensitive cysteine residues and carries out different immune system roles depending on its redox state. In the reduced state, HMGB1 acts as a chemoattractant. In the oxidized state with two nearby cysteines forming a disulfide bond and the third cysteine unmodified, HMGB1 acts as a proinflammatory cytokine (Yang et al., 2021) which can interact with TLR4 (Balosso et al., 2014). Overoxidation inhibits all HMGB1 functions. Interestingly, combinational drug treatment with sulforaphane and N-actylcysteine (NAC), a compound used to enhance GSH levels, also reduced the production of HMGB1 in the brain and blood in an electrical SE model (Pauletti et al., 2019).
Finally, MMP9 has also been found to have redox-sensitive activation, as oxidants have been shown to directly increase MMP function, contributing to BBB dysfunction (Haorah et al., 2007). IL-1β-induction of MMP9 was dependent on NOX2 activity leading to NF-KB and AP-1 transcription factor activation. Further, this enabled MMP9-dependent astrocyte migration (Yang et al., 2015). This relationship was found to be inhibited by the nrf2 activator RTA 408 (Yang et al., 2019). Incredibly, this redox-sensitive activity of MMP9 is linked to PVIN/PNN irregularities in neurons with impaired GSH synthesis (Gclm−/−) in vivo. MMP9 inhibition prevented NF-KB activation and restored PVIN/PNN integrity, linking redox-controlled MMP9 to excitatory/inhibitory imbalance (Dwir et al., 2020). Rectifying redox dysregulation in epilepsy may enable the control of these proinflammatory pathways.
In the brain, the major regulator of redox status is GSH, which has also been shown to regulate inflammation. This antioxidant thiol is utilized as the electron donor by glutathione peroxidase (GPx) to convert H2O2 to H2O, so the GSH/GSSG ratio is indicative of the oxidative environment. The expression of enzymes involved in this process such as GCL and GCLM is controlled by nrf2 and NF-KB. Post-translational activation of GCL by dimercaprol in cultured BV-2 microglia prevents LPS-induced production of proinflammatory cytokines and iNOS induction (McElroy et al., 2017a). Depletion of GSH with the GCL inhibitor BSO leads to gliosis and neuroinflammation in the rat brain, with a particular increase in levels of TNFα (González-Fraguela et al., 2018). In this study, this is associated with mild cognitive impairment, suggesting that impaired redox status could contribute to associated comorbidities in epilepsy. GSH in the mitochondria is protective against TNFα-induced neurotoxicity, indicating that GSH depletion in epilepsy may contribute to a vicious cycle of oxidative stress-neuroinflammation-metabolic stress (Fernandez-Checa et al., 1997). Impaired glutathione redox status also enables aberrant oxidative signaling to occur, which can influence redox sensitive ion channels, neurotransmitter receptors, and glutamate reuptake by astrocytes. Restoring GSH redox status may not only control neuroinflammation, but rectify redox based post-translational modifications that can directly influence neural excitability. The therapeutic potential of GSH increasing treatments such as NAC, Coenzyme Q10, sulforaphane, and Vitamin E has begun to be explored in epilepsy models.
Oxidative stress involves the damage to mtDNA, lipids, and proteins in the cell which can lead to neuroinflammation. Astrogliosis is prevalent in the epilepsies, and interestingly a mouse forebrain neuron-specific knockout of SOD2 resulted in seizures, oxidative stress, and marked GFAP and vimentin gene upregulation, indicative of astrogliosis resulting from increased neuronal mtROS (Fulton et al., 2021). Oxidative stress resulting from increased mtROS has significant impacts on redox signaling within neurons (Bao et al., 2009), and can lead to neuronal apoptosis as described in detail by Méndez-Armenta et al., 2014. This oxidative damage originating from neurons can induce neuroinflammation and gliosis through the release of DAMPs. Further, oxidative damage to phospholipids can induce ferroptosis, as described earlier, which has now been directly linked to the induction of neuroinflammation (Cao Y et al., 2021; Cui et al., 2021).
Together, these studies examining oxidative stress, redox dysregulation, metabolic alterations, and neuroinflammation suggest that these processes are all not only involved in the pathogenesis of epilepsy, but fundamentally linked (See Scheme 1). An infection or traumatic brain injury initiates proinflammatory cascades that involve the upregulation and increased activity of NOX. Excessive excitatory signaling induced by status epilepticus or genetic mutations also increase NOX activity. Seizure activity increases metabolic demand in neuronal mitochondria, which increases O2− production. The increased oxidative environment resulting from NOX activity and mtROS production leads to GSH depletion. Importantly, NOX utilizes NADPH to produce superoxide, thus depleting NADPH needed by antioxidant systems such as thioredoxin reductase and glutathione peroxidase. Together, these processes exacerbate neuroinflammation through oxidative damage and redox based activation of NF-KB, as well as of HMGB1, inflammasomes and IL-1β, and MMP9. NF-KB activation also increases TNFα, NOX, and COX-2 expression. Neuroinflammatory cytokines such as TNFα cause mitochondrial dysfunction when GSH is depleted, as well as induce reactive astrocytes. This can impair astrocyte regulation of the tripartite synapse, BBB, and neuronal metabolism. Damaged neurons and reactive microglia can increase NOX expression, as well as release MMP9 which degrades ECM around the BBB and PVIN neurons, leaving them susceptible to oxidative damage. Regardless of the initial insult, oxidative bursts through NOX and increased oxidative phosphorylation in the mitochondria induce metabolic alterations responsible for observed interictal glucose hypometabolism. Glycolysis further fuels the pentose phosphate pathway (PPP) which produces NADPH. Together, oxidative stress and redox dysregulation form a vicious cycle with neuroinflammation that likely underlies epileptogenesis and undermines current treatment strategies.
Developing combinational therapies that address neuronal hyperexcitability as well as oxidative stress may prevent the deleterious cycle of neuroinflammation and oxidative stress that contribute to epileptogenesis. Recent advances have identified redox-based treatment strategies that warrant further investigation (Yang et al., 2020; Parsons et al., 2022). NOX-inhibitor therapy has shown neuroprotective effects in numerous epilepsy models. However, research suggesting that NOX2 is primarily responsible for the oxidative burst immediately following SE may not allow for a wide enough treatment window in patients following their first SE event. Inhibited interictal glycolysis may contribute to a more oxidative environment due to the decreased substrate pool for the PPP. Interestingly, the KD pushes metabolism away from glycolysis, potentially removing fuel needed to trigger seizures while introducing ketone bodies. This can diminish neuroinflammation and induce Nrf2 activation to express the ARE, which has proven efficacy in treating epilepsy patients. Indeed, Nrf2 activators such as sulforaphane and hydroxylated fullerene have restored GSH levels and shown antiepileptic and neuroprotective properties in numerous epilepsy models (Wang et al., 2014; Carrasco-Pozo et al., 2015; Pauletti et al., 2019; Uddin et al., 2020; Cao H et al., 2021; Sandouka and Shekh-Ahmad, 2021). An antioxidant and metal chelator, curcumin, was able to reduce gliosis and cytokine levels in PTZ-induced epilepsy (Kaur et al., 2015). Other redox-based therapies such as AEOL10150, salicylamine, Coenzyme Q10, and NAC have also been shown to restore GSH homeostasis, which is critical to control inflammation, leading to reduction of seizure burden or comorbidities in animal models (Shin et al., 2005; Tawfik, 2011; Pearson et al., 2015; McElroy et al., 2017b). Further, in a small study of patients with refractory epilepsy, Vitamin E in combination with ASMs increased total antioxidant capacity, catalase, and glutathione and reduced seizure frequency (Mehvari et al., 2016). It is herein proposed that tempering excessive ROS production and reducing oxidative stress could thereby control neuroinflammatory processes involved in epilepsy. A comprehensive treatment strategy broadly effective in epilepsy may therefore involve a combinational approach of ASMs coupled with GSH inducing and nrf2 activating compounds.
TF authored the review and generated the schematic. MP provided content ideas as well as reviewing and editing of the manuscript.
The authors are grateful for the support of NIH grants R01NS039587, R01NS086423 and R01HD102071.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abais, J. M., Xia, M., Li, G., Chen, Y., Conley, S. M., Gehr, T. W. B., et al. (2014). Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J. Biol. Chem. 289, 27159–27168. doi:10.1074/jbc.M114.567537
Abais, J. M., Zhang, C., Xia, M., Liu, Q., Gehr, T. W. B., Boini, K. M., et al. (2013). NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid. Redox Signal. 18, 1537–1548. doi:10.1089/ars.2012.4666
Acar, G., Tanriover, G., Acar, F., and Demir, R. (2015). Increased expression of matrix metalloproteinase-9 in patients with temporal lobe epilepsy. Turk Neurosurg. 25 (5), 749–756. doi:10.5137/1019-5149.JTN.10738-14.0
Ahmed, S. M. U., Luo, L., Namani, A., Wang, X. J., and Tang, X. (2017). Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta. Mol. Basis Dis. 1863, 585–597. doi:10.1016/j.bbadis.2016.11.005
Al-Harbi, N. O., Nadeem, A., Ahmad, S. F., AL-Ayadhi, L. Y., Al-Harbi, M. M., As Sobeai, H. M., et al. (2020). Elevated expression of toll-like receptor 4 is associated with NADPH oxidase-induced oxidative stress in B cells of children with autism. Int. Immunopharmacol. 84, 106555. doi:10.1016/j.intimp.2020.106555
Ariza, D., Lima, M. M. S., Moreira, C. G., Dombrowski, P. A., Avila, T. V., Allemand, A., et al. (2010). Intranigral LPS administration produces dopamine, glutathione but not behavioral impairment in comparison to MPTP and 6-OHDA neurotoxin models of Parkinson’s disease. Neurochem. Res. 35, 1620–1627. doi:10.1007/s11064-010-0222-3
Bae, J. Y., and Park, H. H. (2011). Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 286, 39528–39536. doi:10.1074/jbc.M111.278812
Balosso, S., Liu, J., Bianchi, M. E., and Vezzani, A. (2014). Disulfide-containing high mobility group box-1 promotes N-Methyl-d-Aspartate receptor function and excitotoxicity by activating toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid. Redox Signal. 21, 1726–1740. doi:10.1089/ars.2013.5349
Banerji, R., Huynh, C., Figueroa, F., Dinday, M. T., Baraban, S. C., and Patel, M. (2021). Enhancing glucose metabolism via gluconeogenesis is therapeutic in a zebrafish model of Dravet syndrome. Brain Commun. 3, fcab004. doi:10.1093/braincomms/fcab004
Bao, L., Avshalumov, M. V., Patel, J. C., Lee, C. R., Miller, E. W., Chang, C. J., et al. (2009). Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 29, 9002–9010. doi:10.1523/JNEUROSCI.1706-09.2009
Bell, M. T., Puskas, F., Agoston, V. A., Cleveland, J. C., Freeman, K. A., Gamboni, F., et al. (2013). Toll-like receptor 4–dependent microglial activation mediates spinal cord ischemia–reperfusion injury. Circulation 128, S152–S156. doi:10.1161/CIRCULATIONAHA.112.000024
Bhowmick, S., D’Mello, V., Caruso, D., and Abdul-Muneer, P. M. (2019). Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death. J. Mol. Med. 97, 1627–1641. doi:10.1007/s00109-019-01851-4
Bhuyan, P., Patel, D. C., Wilcox, K. S., and Patel, M. (2015). Oxidative stress in murine Theiler’s virus-induced temporal lobe epilepsy. Exp. Neurol. 271, 329–334. doi:10.1016/j.expneurol.2015.06.012
Brennan, A. M., Won Suh, S., Joon Won, S., Narasimhan, P., Kauppinen, T. M., Lee, H., et al. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. doi:10.1038/nn.2334
Cabungcal, J.-H., Steullet, P., Morishita, H., Kraftsik, R., Cuenod, M., Hensch, T. K., et al. (2013). Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 110, 9130–9135. doi:10.1073/pnas.1300454110
Cai, Y., and Yang, Z. (2021). Ferroptosis and its role in epilepsy. Front. Cell. Neurosci. 15, 696889. doi:10.3389/fncel.2021.696889
Cao, H., Zhang, L., Qu, Z., Tian, S., Wang, Z., Jiang, Y., et al. (2021). The protective effect of hydroxylated fullerene pretreatment on pilocarpine-induced status epilepticus. Brain Res. 1764, 147468. doi:10.1016/j.brainres.2021.147468
Cao, Y., Li, Y., He, C., Yan, F., Li, J.-R., Xu, H.-Z., et al. (2021). Selective ferroptosis inhibitor liproxstatin-1 attenuates neurological deficits and neuroinflammation after subarachnoid hemorrhage. Neurosci. Bull. 37, 535–549. doi:10.1007/s12264-020-00620-5
Caraballo, R. H., Cersósimo, R. O., Sakr, D., Cresta, A., Escobal, N., and Fejerman, N. (2005). Ketogenic diet in patients with Dravet syndrome. Epilepsia 46, 1539–1544. doi:10.1111/j.1528-1167.2005.05705.x
Caraballo, R. H., Fortini, S., Fresler, S., Armeno, M., Ariela, A., Cresta, A., et al. (2014). Ketogenic diet in patients with Lennox–Gastaut syndrome. Seizure 23, 751–755. doi:10.1016/j.seizure.2014.06.005
Cárdenas-Rodríguez, N., Coballase-Urrutia, E., Pérez-Cruz, C., Montesinos-Correa, H., Rivera-Espinosa, L., Sampieri, A., et al. (2014). Relevance of the glutathione system in temporal lobe epilepsy: Evidence in human and experimental models. Oxid. Med. Cell. Longev. 2014, e759293. doi:10.1155/2014/759293
Carrasco-Pozo, C., Tan, K. N., and Borges, K. (2015). Sulforaphane is anticonvulsant and improves mitochondrial function. J. Neurochem. 135, 932–942. doi:10.1111/jnc.13361
Cock, H. R., Tong, X., Hargreaves, I. P., Heales, S. J. R., Clark, J. B., Patsalos, P. N., et al. (2002). Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 48, 157–168. doi:10.1016/S0920-1211(01)00334-5
Cui, Y., Zhang, Y., Zhao, X., Shao, L., Liu, G., Sun, C., et al. (2021). ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 93, 312–321. doi:10.1016/j.bbi.2021.01.003
Dohi, K., Ohtaki, H., Nakamachi, T., Yofu, S., Satoh, K., Miyamoto, K., et al. (2010). Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J. Neuroinflammation 7, 41. doi:10.1186/1742-2094-7-41
Doll, D. N., Rellick, S. L., Barr, T. L., Ren, X., and Simpkins, J. W. (2015). Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 132, 443–451. doi:10.1111/jnc.13008
Dubey, D., McRae, P. A., Rankin-Gee, E. K., Baranov, E., Wandrey, L., Rogers, S., et al. (2017). Increased metalloproteinase activity in the hippocampus following status epilepticus. Epilepsy Res. 132, 50–58. doi:10.1016/j.eplepsyres.2017.02.021
During, M. J., Fried, I., Leone, P., Katz, A., and Spencer, D. D. (1994). Direct measurement of extracellular lactate in the human hippocampus during spontaneous seizures. J. Neurochem. 62, 2356–2361. doi:10.1046/j.1471-4159.1994.62062356.x
Dwir, D., Giangreco, B., Xin, L., Tenenbaum, L., Cabungcal, J.-H., Steullet, P., et al. (2020). MMP9/RAGE pathway overactivation mediates redox dysregulation and neuroinflammation, leading to inhibitory/excitatory imbalance: A reverse translation study in schizophrenia patients. Mol. Psychiatry 25, 2889–2904. doi:10.1038/s41380-019-0393-5
Epilepsy Syndromes (2022). EPILEPSY SYNDROMES [WWW document]. URL: https://www.epilepsydiagnosis.org/syndrome/epilepsy-syndrome-groupoverview.html (accessed 524, 22).
Fernandez-Checa, J. C., Kaplowitz, N., Garcia-Ruiz, C., Colell, A., Miranda, M., Mari, M., et al. (1997). GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 273, G7–G17. doi:10.1152/ajpgi.1997.273.1.G7
Fleck, J., Ribeiro, M. C. P., Schneider, C. M., Sinhorin, V. D. G., Rubin, M. A., and Mello, C. F. (2004). Intrastriatal malonate administration induces convulsive behaviour in rats. J. Inherit. Metab. Dis. 27, 211–219. doi:10.1023/B:BOLI.0000028769.15474.7e
Freeman, L., Guo, H., David, C. N., Brickey, W. J., Jha, S., and Ting, J. P.-Y. (2017). NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 214, 1351–1370. doi:10.1084/jem.20150237
Fulton, R. E., Pearson-Smith, J. N., Huynh, C. Q., Fabisiak, T., Liang, L.-P., Aivazidis, S., et al. (2021). Neuron-specific mitochondrial oxidative stress results in epilepsy, glucose dysregulation and a striking astrocyte response. Neurobiol. Dis. 158, 105470. doi:10.1016/j.nbd.2021.105470
Gao, H.-M., Zhou, H., Zhang, F., Wilson, B. C., Kam, W., and Hong, J.-S. (2011). HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 31, 1081–1092. doi:10.1523/JNEUROSCI.3732-10.2011
Gasche, Y., Copin, J.-C., Sugawara, T., Fujimura, M., and Chan, P. H. (2001). Matrix metalloproteinase inhibition prevents oxidative stress-associated blood–brain barrier disruption after transient focal cerebral ischemia. J. Cereb. Blood Flow. Metab. 21, 1393–1400. doi:10.1097/00004647-200112000-00003
Gavillet, M., Allaman, I., and Magistretti, P. J. (2008). Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia 56, 975–989. doi:10.1002/glia.20671
Geronzi, U., Lotti, F., and Grosso, S. (2018). Oxidative stress in epilepsy. Expert Rev. Neurother. 18, 427–434. doi:10.1080/14737175.2018.1465410
González-Fraguela, M. E., Blanco, L., Fernández, C. I., Lorigados, L., Serrano, T., and Fernández, J. L. (2018). Glutathione depletion: Starting point of brain metabolic stress, neuroinflammation and cognitive impairment in rats. Brain Res. Bull. 137, 120–131. doi:10.1016/j.brainresbull.2017.11.015
Guo, Y., Gao, F., Wang, S., Ding, Y., Zhang, H., Wang, J., et al. (2009). In vivo mapping of temporospatial changes in glucose utilization in rat brain during epileptogenesis: An 18F-fluorodeoxyglucose–small animal positron emission tomography study. Neuroscience 162, 972–979. doi:10.1016/j.neuroscience.2009.05.041
Haorah, J., Ramirez, S. H., Schall, K., Smith, D., Pandya, R., and Persidsky, Y. (2007). Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood–brain barrier dysfunction. J. Neurochem. 101, 566–576. doi:10.1111/j.1471-4159.2006.04393.x
Helmy, A., De Simoni, M.-G., Guilfoyle, M. R., Carpenter, K. L. H., and Hutchinson, P. J. (2011). Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog. Neurobiol. 95, 352–372. doi:10.1016/j.pneurobio.2011.09.003
Hirano, K., Chen, W. S., Chueng, A. L. W., Dunne, A. A., Seredenina, T., Filippova, A., et al. (2015). Discovery of GSK2795039, a novel small molecule NADPH oxidase 2 inhibitor. Antioxid. Redox Signal. 23, 358–374. doi:10.1089/ars.2014.6202
Ho, Y.-H., Lin, Y.-T., Wu, C.-W. J., Chao, Y.-M., Chang, A. Y. W., and Chan, J. Y. H. (2015). Peripheral inflammation increases seizure susceptibility via the induction of neuroinflammation and oxidative stress in the hippocampus. J. Biomed. Sci. 22, 46. doi:10.1186/s12929-015-0157-8
Hou, L., Wang, K., Zhang, C., Sun, F., Che, Y., Zhao, X., et al. (2018). Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol. 14, 250–260. doi:10.1016/j.redox.2017.09.017
Huang, W.-Y., Lin, S., Chen, H.-Y., Chen, Y.-P., Chen, T.-Y., Hsu, K.-S., et al. (2018). NADPH oxidases as potential pharmacological targets against increased seizure susceptibility after systemic inflammation. J. Neuroinflammation 15, 140. doi:10.1186/s12974-018-1186-5
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422. e21. doi:10.1016/j.cell.2017.11.048
Ingvar, M., and Siesjö, B. K. (1983). Local blood flow and glucose consumption in the rat brain during sustained bicuculline-induced seizures. Acta Neurol. Scand. 68, 129–144. doi:10.1111/j.1600-0404.1983.tb05339.x
Jarrett, S. G., Liang, L.-P., Hellier, J. L., Staley, K. J., and Patel, M. (2008a). Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol. Dis. 30, 130–138. doi:10.1016/j.nbd.2007.12.009
Jarrett, S. G., Milder, J. B., Liang, L.-P., and Patel, M. (2008b). The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 106, 1044–1051. doi:10.1111/j.1471-4159.2008.05460.x
Kalita, J., Misra, U. K., Singh, L. S., and Tiwari, A. (2019). Oxidative stress in status epilepticus: A clinical-radiological correlation. Brain Res. 1704, 85–93. doi:10.1016/j.brainres.2018.09.038
Kaur, H., Patro, I., Tikoo, K., and Sandhir, R. (2015). Curcumin attenuates inflammatory response and cognitive deficits in experimental model of chronic epilepsy. Neurochem. Int. 89, 40–50. doi:10.1016/j.neuint.2015.07.009
Kawashima, T., Adachi, T., Tokunaga, Y., Furuta, A., Suzuki, S. O., Doh-ura, K., et al. (1999). Immunohistochemical analysis in a case of idiopathic Lennox-Gastaut syndrome. Clin. Neuropathol. 18, 286–292.
Kim, B. H., Roh, E., Lee, H. Y., Lee, I.-J., Ahn, B., Jung, S.-H., et al. (2008). Benzoxathiole derivative blocks lipopolysaccharide-induced nuclear factor-kappaB activation and nuclear factor-kappaB-regulated gene transcription through inactivating inhibitory kappaB kinase beta. Mol. Pharmacol. 73, 1309–1318. doi:10.1124/mol.107.041251
Kim, G. W., Kim, H.-J., Cho, K.-J., Kim, H.-W., Cho, Y.-J., and Lee, B. I. (2009). The role of MMP-9 in integrin-mediated hippocampal cell death after pilocarpine-induced status epilepticus. Neurobiol. Dis. 36, 169–180. doi:10.1016/j.nbd.2009.07.008
Kim, J.-H., Na, H.-J., Kim, C.-K., Kim, J.-Y., Ha, K.-S., Lee, H., et al. (2008). The non-provitamin A carotenoid, lutein, inhibits NF-kappaB-dependent gene expression through redox-based regulation of the phosphatidylinositol 3-kinase/PTEN/akt and NF-kappaB-inducing kinase pathways: Role of H(2)O(2) in NF-kappaB activation. Free Radic. Biol. Med. 45, 885–896. doi:10.1016/j.freeradbiomed.2008.06.019
Kim, J. H., Jang, B. G., Choi, B. Y., Kim, H. S., Sohn, M., Chung, T. N., et al. (2013). Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 1499, 163–172. doi:10.1016/j.brainres.2013.01.007
Kim, Y.-M., Kim, S.-J., Tatsunami, R., Yamamura, H., Fukai, T., and Ushio-Fukai, M. (2017). ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 312, C749–C764. doi:10.1152/ajpcell.00346.2016
Kovac, S., Domijan, A.-M., Walker, M. C., and Abramov, A. Y. (2014). Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 5, e1442. doi:10.1038/cddis.2014.390
Kumar, M. G., Rowley, S., Fulton, R., Dinday, M. T., Baraban, S. C., and Patel, M. (2016). Altered glycolysis and mitochondrial respiration in a zebrafish model of Dravet syndrome. eNeuro 3, 1. doi:10.1523/ENEURO.0008-16.2016
Kunz, W. S., Goussakov, I. V., Beck, H., and Elger, C. E. (1999). Altered mitochondrial oxidative phosphorylation in hippocampal slices of kainate-treated rats. Brain Res. 826, 236–242. doi:10.1016/s0006-8993(99)01279-2
Kunz, W. S., Kudin, A. P., Vielhaber, S., Blümcke, I., Zuschratter, W., Schramm, J., et al. (2000). Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurology 48, 766–773. doi:10.1002/1531-8249(200011)48:5<766:AID-ANA10>3.0.CO;2-M
Lahuerta, M., Gonzalez, D., Aguado, C., Fathinajafabadi, A., García-Giménez, J. L., Moreno-Estellés, M., et al. (2020). Reactive glia-derived neuroinflammation: A novel hallmark in Lafora progressive myoclonus epilepsy that progresses with age. Mol. Neurobiol. 57, 1607–1621. doi:10.1007/s12035-019-01842-z
Lee, J., Ahn, H., Hong, E.-J., An, B.-S., Jeung, E.-B., and Lee, G.-S. (2016). Sulforaphane attenuates activation of NLRP3 and NLRC4 inflammasomes but not AIM2 inflammasome. Cell. Immunol. 306 (307), 53–60. doi:10.1016/j.cellimm.2016.07.007
Lee, S. H., Choi, B. Y., Kho, A. R., Jeong, J. H., Hong, D. K., Kang, D. H., et al. (2018). Inhibition of NADPH oxidase activation by apocynin rescues seizure-induced reduction of adult hippocampal neurogenesis. Int. J. Mol. Sci. 19, 3087. doi:10.3390/ijms19103087
Li, H., Luo, Y.-F., Wang, Y.-S., Yang, Q., Xiao, Y.-L., Cai, H.-R., et al. (2018). Using ROS as a second messenger, NADPH oxidase 2 mediates macrophage senescence via interaction with NF-κB during Pseudomonas aeruginosa infection. Oxid. Med. Cell. Longev. 2018, e9741838. doi:10.1155/2018/9741838
Li, Q., Spencer, N. Y., Oakley, F. D., Buettner, G. R., and Engelhardt, J. F. (2009). Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid. Redox Signal. 11, 1249–1263. doi:10.1089/ars.2008.2407
Liang, L.-P., and Patel, M. (2004). Mitochondrial oxidative stress and increased seizure susceptibility in Sod2−/+ mice. Free Radic. Biol. Med. 36, 542–554. doi:10.1016/j.freeradbiomed.2003.11.029
Liang, L.-P., Waldbaum, S., Rowley, S., Huang, T.-T., Day, B. J., and Patel, M. (2012). Mitochondrial oxidative stress and epilepsy in SOD2 deficient mice: Attenuation by a lipophilic metalloporphyrin. Neurobiol. Dis. 45, 1068–1076. doi:10.1016/j.nbd.2011.12.025
Loewen, J. L., Barker-Haliski, M. L., Dahle, E. J., White, H. S., and Wilcox, K. S. (2016). Neuronal injury, gliosis, and glial proliferation in two models of temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 75, 366–378. doi:10.1093/jnen/nlw008
Lorigados Pedre, L., Gallardo, J. M., Morales Chacón, L. M., Vega García, A., Flores-Mendoza, M., Neri-Gómez, T., et al. (2018). Oxidative stress in patients with drug resistant partial complex seizure. Behav. Sci. 8, 59. doi:10.3390/bs8060059
Löscher, W., and Klein, P. (2021). The pharmacology and clinical efficacy of antiseizure medications: From bromide salts to cenobamate and beyond. CNS Drugs 35, 935–963. doi:10.1007/s40263-021-00827-8
Ma, M. W., Wang, J., Dhandapani, K. M., Wang, R., and Brann, D. W. (2018). NADPH oxidases in traumatic brain injury – promising therapeutic targets? Redox Biol. 16, 285–293. doi:10.1016/j.redox.2018.03.005
Malinska, D., Kulawiak, B., Kudin, A. P., Kovacs, R., Huchzermeyer, C., Kann, O., et al. (2010). Complex III-dependent superoxide production of brain mitochondria contributes to seizure-related ROS formation. Biochim. Biophys. Acta 2010, 1163–1170. doi:10.1016/j.bbabio.2010.03.001
Malkov, A., Ivanov, A. I., Buldakova, S., Waseem, T., Popova, I., Zilberter, M., et al. (2018). Seizure-induced reduction in glucose utilization promotes brain hypometabolism during epileptogenesis. Neurobiol. Dis. 116, 28–38. doi:10.1016/j.nbd.2018.04.016
Malkov, A., Ivanov, A. I., Latyshkova, A., Bregestovski, P., Zilberter, M., and Zilberter, Y. (2019). Activation of nicotinamide adenine dinucleotide phosphate oxidase is the primary trigger of epileptic seizures in rodent models. Ann. Neurol. 85, 907–920. doi:10.1002/ana.25474
Mander, P. K., Jekabsone, A., and Brown, G. C. (2006). Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 176, 1046–1052. doi:10.4049/jimmunol.176.2.1046
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., et al. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419. doi:10.1038/nm.2127
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi:10.1016/S1097-2765(02)00599-3
McElroy, P. B., Hari, A. S., Day, B. J., and Patel, M. (2017a). Post-translational activation of glutamate cysteine ligase with dimercaprol: A novel mechanism of inhibiting neuroinflammation in vitro. J. Biol. Chem. 292, 5532–5545. doi:10.1074/jbc.M116.723700
McElroy, P. B., Liang, L.-P., Day, B. J., and Patel, M. (2017b). Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 298, 13–22. doi:10.1016/j.expneurol.2017.08.009
Mehvari, J., Motlagh, F. G., Najafi, M., Ghazvini, M. R. A., Naeini, A. A., and Zare, M. (2016). Effects of Vitamin E on seizure frequency, electroencephalogram findings, and oxidative stress status of refractory epileptic patients. Adv. Biomed. Res. 5, 36. doi:10.4103/2277-9175.178780
Méndez-Armenta, M., Nava-Ruíz, C., Juárez-Rebollar, D., Rodríguez-Martínez, E., and Yescas Gómez, P. (2014). Oxidative stress associated with neuronal apoptosis in experimental models of epilepsy. Oxid. Med. Cell. Longev. 2014, e293689. doi:10.1155/2014/293689
Meyer, J. S., Gotoh, F., and Favale, E. (1966). Cerebral metabolism during epileptic seizures in man. Electroencephalogr. Clin. Neurophysiol. 21, 10–22. doi:10.1016/0013-4694(66)90054-X
Milder, J. B., Liang, L.-P., and Patel, M. (2010). Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 40, 238–244. doi:10.1016/j.nbd.2010.05.030
Milder, J., and Patel, M. (2012). Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 100, 295–303. doi:10.1016/j.eplepsyres.2011.09.021
Mohseni-Moghaddam, P., Roghani, M., Khaleghzadeh-Ahangar, H., Sadr, S. S., and Sala, C. (2021). A literature overview on epilepsy and inflammasome activation. Brain Res. Bull. 172, 229–235. doi:10.1016/j.brainresbull.2021.05.001
Newton, R., Kuitert, L. M. E., Bergmann, M., Adcock, I. M., and Barnes, P. J. (1997). Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. Biochem. Biophys. Res. Commun. 237, 28–32. doi:10.1006/bbrc.1997.7064
Noh, H., Jeon, J., and Seo, H. (2014). Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 69, 35–40. doi:10.1016/j.neuint.2014.02.008
Parsons, A. L. M., Bucknor, E. M. V., Castroflorio, E., Soares, T. R., Oliver, P. L., and Rial, D. (2022). The interconnected mechanisms of oxidative stress and neuroinflammation in epilepsy. Antioxidants 11, 157. doi:10.3390/antiox11010157
Patel, M., Li, Q.-Y., Chang, L.-Y., Crapo, J., and Liang, L.-P. (2005). Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J. Neurochem. 92, 123–131. doi:10.1111/j.1471-4159.2004.02838.x
Patel, M. (2004). Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med. 37, 1951–1962. doi:10.1016/j.freeradbiomed.2004.08.021
Paudel, Y. N., Shaikh, Mohd.F., Chakraborti, A., Kumari, Y., Aledo-Serrano, Á., Aleksovska, K., et al. (2018). HMGB1: A common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front. Neurosci. 12, 628. doi:10.3389/fnins.2018.00628
Pauletti, A., Terrone, G., Shekh-Ahmad, T., Salamone, A., Ravizza, T., Rizzi, M., et al. (2019). Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 142, e39. doi:10.1093/brain/awz130
Pearson, J. N., Rowley, S., Liang, L.-P., White, A. M., Day, B. J., and Patel, M. (2015). Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol. Dis. 82, 289–297. doi:10.1016/j.nbd.2015.07.005
Pearson, J. N., Warren, E., Liang, L.-P., Roberts, L. J., and Patel, M. (2017). Scavenging of highly reactive gamma-ketoaldehydes attenuates cognitive dysfunction associated with epileptogenesis. Neurobiol. Dis. 98, 88–99. doi:10.1016/j.nbd.2016.11.011
Pearson-Smith, J. N., Liang, L.-P., Rowley, S. D., Day, B. J., and Patel, M. (2017). Oxidative stress contributes to status epilepticus associated mortality. Neurochem. Res. 42, 2024–2032. doi:10.1007/s11064-017-2273-1
Pearson-Smith, J., and Patel, M. (2017). Metabolic dysfunction and oxidative stress in epilepsy. Int. J. Mol. Sci. 18, 2365. doi:10.3390/ijms18112365
Pei, Z., Pang, H., Qian, L., Yang, S., Wang, T., Zhang, W., et al. (2007). MAC1 mediates LPS-induced production of superoxide by microglia: The role of pattern recognition receptors in dopaminergic neurotoxicity. Glia 55, 1362–1373. doi:10.1002/glia.20545
Pestana, R. R. F., Kinjo, E. R., Hernandes, M. S., and Britto, L. R. G. (2010). Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 484, 187–191. doi:10.1016/j.neulet.2010.08.049
Poyet, J. L., Srinivasula, S. M., Tnani, M., Razmara, M., Fernandes-Alnemri, T., and Alnemri, E. S. (2001). Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 276, 28309–28313. doi:10.1074/jbc.C100250200
Quintana, A., Kruse, S. E., Kapur, R. P., Sanz, E., and Palmiter, R. D. (2010). Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. U. S. A. 107, 10996–11001. doi:10.1073/pnas.1006214107
Rankin-Gee, E. K., McRae, P. A., Baranov, E., Rogers, S., Wandrey, L., and Porter, B. E. (2015). Perineuronal net degradation in epilepsy. Epilepsia 56, 1124–1133. doi:10.1111/epi.13026
Rowley, S., Liang, L.-P., Fulton, R., Shimizu, T., Day, B., and Patel, M. (2015). Mitochondrial respiration deficits driven by reactive oxygen species in experimental temporal lobe epilepsy. Neurobiol. Dis. 75, 151–158. doi:10.1016/j.nbd.2014.12.025
Rumià, J., Marmol, F., Sanchez, J., Giménez-Crouseilles, J., Carreño, M., Bargalló, N., et al. (2013). Oxidative stress markers in the neocortex of drug-resistant epilepsy patients submitted to epilepsy surgery. Epilepsy Res. 107, 75–81. doi:10.1016/j.eplepsyres.2013.08.020
Ryan, K., Backos, D. S., Reigan, P., and Patel, M. (2012). Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J. Neurosci. 32, 11250–11258. doi:10.1523/JNEUROSCI.0907-12.2012
Salar, S., and Galanopoulou, A. S. (2018). “Chapter 5 - neuroinflammation in the pathogenesis of early life epileptic encephalopathies,” in Acute encephalopathy and encephalitis in infancy and its related disorders. Editors H. Yamanouchi, S. L. Moshé, and A. Okumura (Elsevier), 33–44. doi:10.1016/B978-0-323-53088-0.00005-1
Samidurai, M., Tarale, P., Janarthanam, C., Estrada, C. G., Gordon, R., Zenitsky, G., et al. (2020). Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances activation of STAT3/NLRC4 inflammasome signaling Axis through PKCδ in astrocytes: Implications for Parkinson’s disease. Cells 9, 1831. doi:10.3390/cells9081831
Sandouka, S., and Shekh-Ahmad, T. (2021). Induction of the Nrf2 pathway by sulforaphane is neuroprotective in a rat temporal lobe epilepsy model. Antioxidants 10, 1702. doi:10.3390/antiox10111702
Scheffer, I. E., Berkovic, S., Capovilla, G., Connolly, M. B., French, J., Guilhoto, L., et al. (2017). ILAE classification of the epilepsies: Position paper of the ILAE commission for classification and terminology. Epilepsia 58, 512–521. doi:10.1111/epi.13709
Schiavone, S., Neri, M., Trabace, L., and Turillazzi, E. (2017). The NADPH oxidase NOX2 mediates loss of parvalbumin interneurons in traumatic brain injury: Human autoptic immunohistochemical evidence. Sci. Rep. 7, 8752. doi:10.1038/s41598-017-09202-4
Shekh-Ahmad, T., Eckel, R., Dayalan Naidu, S., Higgins, M., Yamamoto, M., Dinkova-Kostova, A. T., et al. (2018). KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain 141, 1390–1403. doi:10.1093/brain/awy071
Shekh-Ahmad, T., Lieb, A., Kovac, S., Gola, L., Christian Wigley, W., Abramov, A. Y., et al. (2019). Combination antioxidant therapy prevents epileptogenesis and modifies chronic epilepsy. Redox Biol. 26, 101278. doi:10.1016/j.redox.2019.101278
Shin, E.-J., Suh, S. K., Lim, Y. K., Jhoo, W.-K., Hjelle, O. P., Ottersen, O. P., et al. (2005). Ascorbate attenuates trimethyltin-induced oxidative burden and neuronal degeneration in the rat hippocampus by maintaining glutathione homeostasis. Neuroscience 133, 715–727. doi:10.1016/j.neuroscience.2005.02.030
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z., and Germain, R. N. (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. doi:10.1016/j.cell.2013.02.054
Sul, O.-J., and Ra, S. W. (2021). Quercetin prevents LPS-induced oxidative stress and inflammation by modulating NOX2/ROS/NF-kB in lung epithelial cells. Molecules 26, 6949. doi:10.3390/molecules26226949
Tawfik, M. K. (2011). Coenzyme Q10 enhances the anticonvulsant effect of phenytoin in pilocarpine-induced seizures in rats and ameliorates phenytoin-induced cognitive impairment and oxidative stress. Epilepsy Behav. 22, 671–677. doi:10.1016/j.yebeh.2011.09.018
Terrone, G., Frigerio, F., Balosso, S., Ravizza, T., and Vezzani, A. (2019). Inflammation and reactive oxygen species in status epilepticus: Biomarkers and implications for therapy. Epilepsy Behav. 101, 106275. doi:10.1016/j.yebeh.2019.04.028
Tiraboschi, E., Martina, S., van der Ent, W., Grzyb, K., Gawel, K., Cordero-Maldonado, M. L., et al. (2020). New insights into the early mechanisms of epileptogenesis in a zebrafish model of Dravet syndrome. Epilepsia 61, 549–560. doi:10.1111/epi.16456
Uddin, Md.S., Mamun, A. A., Jakaria, Md., Thangapandiyan, S., Ahmad, J., Rahman, Md.A., et al. (2020). Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 707, 135624. doi:10.1016/j.scitotenv.2019.135624
Valassina, N., Brusco, S., Salamone, A., Serra, L., Luoni, M., Giannelli, S., et al. (2022). Scn1a gene reactivation after symptom onset rescues pathological phenotypes in a mouse model of Dravet syndrome. Nat. Commun. 13, 161. doi:10.1038/s41467-021-27837-w
Van Vliet, E. A., Aronica, E., and Gorter, J. A. (2015). Blood–brain barrier dysfunction, seizures and epilepsy. Semin. Cell Dev. Biol. 38, 26–34. doi:10.1016/j.semcdb.2014.10.003
Vezzani, A., Balosso, S., and Ravizza, T. (2019). Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 15, 459–472. doi:10.1038/s41582-019-0217-x
Vezzani, A., Balosso, S., and Ravizza, T. (2008). The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 22, 797–803. doi:10.1016/j.bbi.2008.03.009
Wang, J., Ma, M. W., Dhandapani, K. M., and Brann, D. W. (2018). NADPH oxidase 2 deletion enhances neurogenesis following traumatic brain injury. Free Radic. Biol. Med. 123, 62–71. doi:10.1016/j.freeradbiomed.2018.05.069
Wang, W., Wu, Y., Zhang, G., Fang, H., Wang, H., Zang, H., et al. (2014). Activation of Nrf2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 1544, 54–61. doi:10.1016/j.brainres.2013.12.004
Wijburg, F. A., Barth, P. G., Bindoff, L. A., Birch-Machin, M. A., Blij, J. F. V. D., Ruitenbeek, W., et al. (1992). Leigh syndrome associated with a deficiency of the pyruvate dehydrogenase complex: Results of treatment with a ketogenic diet. Neuropediatrics 23, 147–152. doi:10.1055/s-2008-1071331
Williams, S., Hamil, N., Abramov, A. Y., Walker, M. C., and Kovac, S. (2015). Status epilepticus results in persistent overproduction of reactive oxygen species, inhibition of which is neuroprotective. Neuroscience 303, 160–165. doi:10.1016/j.neuroscience.2015.07.005
Wojtala, A., Karkucinska-Wieckowska, A., Sardao, V. A., Szczepanowska, J., Kowalski, P., Pronicki, M., et al. (2017). Modulation of mitochondrial dysfunction-related oxidative stress in fibroblasts of patients with Leigh syndrome by inhibition of prooxidative p66Shc pathway. Mitochondrion 37, 62–79. doi:10.1016/j.mito.2017.07.002
Wu, S.-B., Ma, Y.-S., Wu, Y.-T., Chen, Y.-C., and Wei, Y.-H. (2010). Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol. Neurobiol. 41, 256–266. doi:10.1007/s12035-010-8123-7
Yamanaka, G., Takamatsu, T., Morichi, S., Yamazaki, T., Mizoguchi, I., Ohno, K., et al. (2021). Interleukin-1β in peripheral monocytes is associated with seizure frequency in pediatric drug-resistant epilepsy. J. Neuroimmunol. 352, 577475. doi:10.1016/j.jneuroim.2021.577475
Yang, C.-C., Lin, C.-C., Jou, M.-J., Hsiao, L.-D., and Yang, C.-M. (2019). RTA 408 inhibits interleukin-1β-induced MMP-9 expression via suppressing protein kinase-dependent NF-κB and AP-1 activation in rat brain astrocytes. Int. J. Mol. Sci. 20, 2826. doi:10.3390/ijms20112826
Yang, C.-M., Hsieh, H.-L., Yu, P.-H., Lin, C.-C., and Liu, S.-W. (2015). IL-1β induces MMP-9-dependent brain astrocytic migration via transactivation of PDGF receptor/NADPH oxidase 2-derived reactive oxygen species signals. Mol. Neurobiol. 52, 303–317. doi:10.1007/s12035-014-8838-y
Yang, H., Lundbäck, P., Ottosson, L., Erlandsson-Harris, H., Venereau, E., Bianchi, M. E., et al. (2021). Redox modifications of cysteine residues regulate the cytokine activity of HMGB1. Mol. Med. 27, 58. doi:10.1186/s10020-021-00307-1
Yang, N., Guan, Q.-W., Chen, F.-H., Xia, Q.-X., Yin, X.-X., Zhou, H.-H., et al. (2020). Antioxidants targeting mitochondrial oxidative stress: Promising neuroprotectants for epilepsy. Oxid. Med. Cell. Longev. 2020, e6687185. doi:10.1155/2020/6687185
You, S. J., Kim, H. D., and Kang, H.-C. (2009). Factors influencing the evolution of west syndrome to lennox-gastaut syndrome. Pediatr. Neurol. 41, 111–113. doi:10.1016/j.pediatrneurol.2009.03.006
Youm, Y.-H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 21, 263–269. doi:10.1038/nm.3804
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi:10.1038/nature09663
Keywords: neuroinflammation, oxidative stress, redox, epilepsy, mitochondria, NADPH oxidase
Citation: Fabisiak T and Patel M (2022) Crosstalk between neuroinflammation and oxidative stress in epilepsy. Front. Cell Dev. Biol. 10:976953. doi: 10.3389/fcell.2022.976953
Received: 23 June 2022; Accepted: 18 July 2022;
Published: 10 August 2022.
Edited by:
Xinjian Zhu, Southeast University, ChinaReviewed by:
Nyzil Massey, Iowa State University, United StatesCopyright © 2022 Fabisiak and Patel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manisha Patel, TWFuaXNoYS5QYXRlbEBDdUFuc2NodXR6LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.