 Zhanfang Kang
Zhanfang Kang Feng Chen2†
Feng Chen2† Tianda Chen
Tianda Chen Fang Xu
Fang Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 16 September 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.974083
This article is part of the Research Topic Mitochondrial Dysfunction in Metabolic Diseases View all 6 articles
The mitochondrial unfolded protein response (UPRmt) is a molecular mechanism that maintains mitochondrial proteostasis under stress and is closely related to various metabolic diseases, such as type 2 diabetes (T2D). Similarly, the unfolded protein response of the endoplasmic reticulum (UPRER) is responsible for maintaining proteomic stability in the endoplasmic reticulum (ER). Since the mitochondria and endoplasmic reticulum are the primary centers of energy metabolism and protein synthesis in cells, respectively, a synergistic mechanism must exist between UPRmt and UPRER to cooperatively resist stresses such as hyperglycemia in T2D. Increasing evidence suggests that the protein kinase RNA (PKR)-like endoplasmic reticulum kinase (PERK) signaling pathway is likely an important node for coordinating UPRmt and UPRER. The PERK pathway is activated in both UPRmt and UPRER, and its downstream molecules perform important functions. In this review, we discuss the mechanisms of UPRmt, UPRER and their crosstalk in T2D.
There are many internal and external challenges for cells that living systems must manage to sustain normal and healthy living activities. Therefore, some organelle-specific stress responses have evolved to deal with these external factors, such as the unfolded protein response in endoplasmic reticulum and mitochondria (UPRER and UPRmt, respectively) (Wang and Kaufman, 2012; Jovaisaite et al., 2014; Lee and Ozcan, 2014). The endoplasmic reticulum (ER) is a compartment that facilitates the synthesis, folding, modification and transportation of proteins. Therefore, the proteostasis in ER is so critical that cells have evolved relevant stress response signals to manage the homeostasis disruption. For example, UPRER is a regulation mechanism that disposes of the proteostasis imbalance in the ER (Kaufman, 2002; Ron, 2002; Rutkowski and Kaufman, 2004; Ron and Walter, 2007). Furthermore, the mitochondrion can code proteins related to oxidative phosphorylation (OXPHOS), which has an intimate connection with energy metabolism. When mitochondria malfunction and there is no remedial measure, cells die. Hence, a remedial measure or surveillance program is important for maintaining living systems (Wouters and Koritzinsky, 2008; Pellegrino et al., 2013; Nagelkerke et al., 2014; Münch and Harper, 2016; Qureshi et al., 2017). One such a measure, UPRmt, consistently monitors the environment in mitochondria, which is vital to the organelle’s normal functioning (Yoneda et al., 2004; Benedetti et al., 2006; Houtkooper et al., 2013). Recent evidence suggests that if cells cannot dispose of various stresses and resolve the disruption of homeostasis rapidly, multiple disorders such as metabolic and neurodegenerative diseases occur (Wang and Kaufman, 2012; Jovaisaite et al., 2014; Lee and Ozcan, 2014). In addition, the cell is a highly dynamic but orderly system wherein the organelles coordinate to ensure its healthy and orderly activities (Csordás et al., 2006; West et al., 2011; Grimm, 2012; Allison et al., 2017). Mitochondria ER contacts (MERCs), also called mitochondria associated membranes (MAMs) are composed of ER and mitochondrion membranes, house many molecules that are situated at contact sites, and facilitate signal exchange between two compartments (Vance, 2014; Ilacqua et al., 2017; Zhang et al., 2021a; Sukhorukov et al., 2022). Therefore, organelle-specific stress responses, such as UPRER and UPRmt, can also coordinate to manage stresses so as to maintain cellular homeostasis and prevent the development of multiple disorders. In this study, we evaluate UPRmt and UPRER, as well as the crosstalk between them, and highlight the coordinative impact of them in T2D.
Mitochondria are organelles with double membranes, which contain proteins and RNAs encoded by the nuclear genome and are also capable of encoding proteins and RNAs related to OXPHOS (Sloan et al., 2018). Mitochondria that do not work normally cannot provide ATP for the efficient functioning of cells with their highly dynamic and interconnected mitochondrial network (Wilkins et al., 2017; Sloan et al., 2018; Zhang et al., 2018). In addition to producing energy, mitochondria also play an indispensable role in other signaling cascades, such as the signaling axis that regulates apoptosis (Wilkins et al., 2017; Zhang et al., 2018; Dall and Færgeman, 2019). Simultaneously, mitochondria have waste removal and utilization systems (Vakifahmetoglu-Norberg et al., 2017). Therefore, mitochondrial proteostasis is indispensable, and without it, mitochondria cannot maintain their homeostasis and perform their normal tasks (Roger et al., 2017). The bulk of proteins in mitochondria are encoded by nuclear genes, which are then translated through free ribosomes (not through co-translation), and can finally be transported to the mitochondria to produce their marked effect. However, mitochondrial DNA that codes for 13 OXPHOS reaction center proteins are translated into the mitochondrial matrix (Hansen and Herrmann, 2019). Proteins encoded by nuclear genes or proteins encoded by mitochondrial DNA must be correctly folded and subjected to quality control to sustain mitochondrial proteostasis. Misfolded proteins must be refolded or degraded to rebuild mitochondrial proteostasis (Schmidt et al., 2010; Ott et al., 2016).
The UPRmt is a mitochondrial stress response that maintains proteostasis in the mitochondria (Haynes and Ron, 2010; Schmidt et al., 2010; Durieux et al., 2011; Ott et al., 2016; Xie et al., 2017; Smyrnias et al., 2019). When proteins that need to be processed exceed the protein-handling capacity of the mitochondria, UPRmt initiates an increase in protein degradation and expansion of the mitochondrial matrix folding capacity to restore protein homeostasis (Ryan and Hoogenraad, 2007). Restoring protein homeostasis in mitochondria relies on chaperones that increase protein folding and block protein aggregation and are imperative to all cells (Bukau et al., 2006). Different stressors, such as oxidative stress, infections, and alterations in metabolism, are not beneficial for protein folding (Aldridge et al., 2007; Ron and Walter, 2007). In addition, because the influx of mitochondrial precursor proteins and the correct assembly of nuclear and mitochondrial DNA-encoded proteins are essential for the healthy survival of cells, restoration of mitochondrial protein homeostasis encounters unique challenges (Ryan and Hoogenraad, 2007). A series of molecular chaperones and proteases are indispensable for protein proteostasis in mitochondria, participating in the transduction pathway of UPRmt as mediators and targets (Zhao et al., 2002; Aldridge et al., 2007).
In addition to proteostasis disturbances, the disruption of mitochondrial metabolic processes also activates UPRmt. Multiple drugs suppress OXPHOS and activate UPRmt (Yoneda et al., 2004; Nargund et al., 2012; Rossmanith et al., 2012; Houtkooper et al., 2013; Zhang et al., 2021b), including inhibitors of complex I, such as rotenone and petasin (Heishima et al., 2021), an inhibitor of complex III, antimycin, and inhibitors of complex V, oligomycin and paraquat. Fumarate hydratase, an enzyme that converts fumarate to malate during the tricarboxylic acid cycle (TCA), can be inhibited to activate UPRmt, demonstrating that the mitochondrial stress response can be activated not only by mitochondrial proteostasis disruption but also by metabolic disturbances (Wang et al., 2016).
UPRmt has been associated with multiple biological processes, including development (Sukhorukov et al., 2022), innate immune signaling (Zhu et al., 2021), aging (Mohrin et al., 2015; Lin et al., 2016; Wang et al., 2016; Zhu et al., 2021) and cardioprotection (Qureshi et al., 2017). Once the UPRmt signal is prolonged to a chronic state potentially maladaptive to organelles, it presages a therapeutic target for a broad spectrum of illnesses. Rapid activation of UPRmt facilitates the adaptability of cells to environmental stresses and physiological stimuli. The transduction signal of UPRmt promotes the proliferation of defective mitochondrial genomes, which ultimately leads to congenital metabolic abnormalities (Haynes et al., 2007; Yan et al., 2020). In addition, dysregulated UPRmt signaling leads to the development of diseases such as cancer (Martinus et al., 1996; Shpilka et al., 2021), neurodegenerative disorders (Roger et al., 2017; Hansen and Herrmann, 2019), fatty liver, and diabetes (Haynes et al., 2007; Yan et al., 2020). Also, UPRmt promotes host tolerance and protects against pathogenic infections (Mehrbod et al., 2019).
Although UPRmt was initially discovered and characterized in mammalian cells (Martinus et al., 1996; Zhao et al., 2002), research on UPRmt has been more thorough in Caenorhabditis elegans (C. elegans). In C. elegans, mitochondrial matrix proteins can be hydrolyzed to generate short peptides by CLPP-1 when the processing capability of chaperone proteins cannot afford the aggregation of misfolded and unassembled proteins (Nicolas et al., 2019). HAF-1, a mitochondria-localized ATP-binding cassette protein, causes an influx of these peptides in the mitochondrial matrix into the cytoplasm in an ATP-dependent manner, thereby inhibiting the import of mitochondrial proteins (Haynes et al., 2010). In addition, mitochondrial stressors diminish mitochondrial membrane potential, which hinders the import of mitochondrial proteins (Rolland et al., 2019). The basic leucine zipper (bZIP) transcription factor ATFS-1, which has two special sequences, the mitochondrial targeting sequence (MTS) and the nuclear localization sequence (NLS), plays a crucial role in the regulation of the UPRmt (Haynes et al., 2010; Nargund et al., 2012). The mitochondrial-to-nuclear communication is mediated by dual subcellular localization sequences (Lin et al., 2016; Sorrentino et al., 2017). Thus, the import efficiency of mitochondrial proteins is a possible indicator of the general mitochondrial function in cells where ATFS-1 is used as a sensor. Under normal circumstances, ATFS-1 is transported into the mitochondrial matrix, where the protein is degraded. However, in times of stress, the influx of ATFS-1 is inhibited, leading to its accumulation in the nucleus; this activates the transcription of a set of genes related to encoding chaperones and proteases participating in UPRmt, suggesting that mitochondrial import efficiency is a key regulator of UPRmt activation (Nargund et al., 2012; ; Nargund et al., 2015). DVE-1, a UPRmt regulator capable of binding to the promoter of heat shock protein (HSP60), has recently been found downstream of mitochondrial dysfunction-driven chromatin reorganization. Histone methyltransferases, MET-2 and LIN-65, are involved in chromatin reorganization, and regardless of which is inhibited, they would reduce the methylation of histone H3K9 and block the induction of UPRmt downstream of CLPP-1. In addition, it was found that MET-2 was necessary for the accumulation of LIN-65 in the nucleus, which could promote the opening of a portion of chromatin, facilitating the binding to open regions and the transcriptional activity of DVE-1 (Tian et al., 2016; Wang et al., 2019). Furthermore, demethylation of JMJD-1.2 and JMJD-3.1 in histone H3K27 is also required for UPRmt induction. Importantly, overexpression of any of these demethylases leads to a transcriptional response resembling that observed during OXPHOS dysfunction (Merkwirth et al., 2016). These studies highlight that the DVE-1-mediated transcriptional response in times of mitochondrial stress is an important precursor for chromatin remodeling. Both ATFS-1 and DVE-1 are regulated by small ubiquitin-like modifier (SUMO) proteins, and the SUMO moieties of the peptidase have been previously identified to be required for ULP-4 cleavage to UPRmt induction (Liu et al., 2014a). AVE-1 and ATFS-1 were SUMOylated at K327 and K326, respectively, leading to accelerated proteolysis of ATFS-1 and import of DVE-1. In the absence of ulp-4, mutations that eliminate SUMOylation of these transcription factors also activate UPRmt (Gao et al., 2019).
Although there is considerable overlap in the regulatory mechanisms between mammalian UPRmt and C. elegans UPRmt, mammalian UPRmt regulatory mechanisms are more sophisticated (Fiorese et al., 2016; Münch and Harper, 2016; Quirós et al., 2017). In mammalian cells facing mitochondrial stress, eIF2α-specific kinases such as GCN2, PERK, PKR, and HRI are activated for the phosphorylation of the translation initiation factor eIF2α, which restrains the synthesis of general proteins while increasing the translation of some specific proteins (Barbosa et al., 2013; Pakos-Zebrucka et al., 2016). For example, CHOP, ATF4, and ATF5 bZIP transcription factors were increased in UPRmt. ATF5, a functional mammalian homolog of the transcription factor ATFS-1, which has been shown in C. elegans to regulate UPRmt, elicits a transcriptional response (; Persengiev et al., 2002). Like ATFS-1 in C. elegans, ATF5 harbors a sequence similar to MTS, which allows it to be degraded through the mitochondrial membrane. However, during UPRmt, it could not efficiently import into the mitochondria to accumulate in the nucleus and induce the expression of genes that have a bearing on several mitochondrial chaperones and proteases, a process that promotes OXPHOS and cell growth during mitochondrial homeostasis disorder to help cells adapt and survive (Silva et al., 2009; Tyynismaa et al., 2010; Martínez-Reyes et al., 2012; Michel et al., 2015; Münch and Harper, 2016; Quirós et al., 2017). In addition to ATF5, two other bZIP transcription factors, ATF4 and CHOP, also participate in UPRmt activation, and an intimate connection exists between ATF4, CHOP, and ATF5 during times of mitochondrial dysfunction. Both CHOP and ATF4 regulate mitochondrial proteins by inducing the transcription of ATF5 (Hatano et al., 2013; Zhu et al., 2021). Furthermore, the transcription factor c-Jun is activated by c-Jun N-terminal kinase (JNK)2 and dsRNA-activated protein kinase (PKR) during stress, which subsequently attaches to AP-1, triggering the translation of CHOP and C/EBP. CHOP combines with C/EBP to form a dimer that is bound to the promoters of UPRmt genes, which encode chaperones HSP60, CLPP-1, and mitochondrial import components (Jovaisaite et al., 2014; Münch and Harper, 2016). In addition, the kinases of eIF2, including GCN2 and PERK, promote the phosphorylation of elF2, thereby suppressing the synthesis of proteins while facilitating the output of CHOP, ATF4, and ATF5 (Teske et al., 2013; Pakos-Zebrucka et al., 2016). Additionally, heat shock transcription factor 1(HSF1) combines with single-stranded DNA binding protein 1(SSBP1) to generate a complex that increases the yield of heat shock proteins HSP60, HSP10, and mtHSP70 during times of stress (Tan et al., 2015). Members of the sirtuin family, including SIRT3 and SIRT7, may also contribute to the regulation of UPRmt (Papa and Germain, 2014). The buildup of proteins in the intermembrane space (IMS) of the mitochondria generates excessive reactive oxygen species (ROS). In turn, AKT and protein kinase B can be stimulated by excessive ROS, activating the phosphorylated estrogen receptor so that it enhances the activities of proteasome 26S and stimulates the translation of intermembrane space proteases HTRA2, OMI, and the mitochondrial regulator NRF1, hence preserving quality control within the intermembrane space protein (Papa and Germain, 2011).
Moreover, the substrate EPS-8/EPS8 of the epidermal growth factor receptor pathway, as a signaling protein adaptor, plays a role in general homeostasis in mitochondria and the regulation of UPRmt by reorganizing the actin cytoskeleton mediated by integrin (Kaspar et al., 2021; Moehle et al., 2021). Rox1 binds mtDNA and performs a TFAM-like function. By inducing UPRmt, Rox1 serves as the initial line of defense (Poveda-Huertes et al., 2020).
The UPRER is a set of evolutionarily conserved signaling axes that continuously monitor the quality of proteins that have intimate connections with the protein-folding capacity within the ER lumen (Cox et al., 1993; Cox and Walter, 1996; Sidrauski and Walter, 1997; Yoshida et al., 1998; Niwa et al., 1999; Harding et al., 2000; Tirasophon et al., 2000; Yoshida et al., 2001). When the ER protein-folding capacity is so limited that it cannot satisfy the demand for ER function owing to ER stress, UPRER is activated, assisting the cell in modifying the organelle’s folding function to restore equilibrium. For example, it can expand the size of the ER lumen and increase the expression of genes encoding proteins related to ER folding capacity. Complementary UPRER actions coordinate protein homeostasis within the ER lumen. If proteostasis cannot be restored, UPRER initiates cell-apoptosis signaling pathways that destroy defective cells in an effort to benefit the organism (Lin et al., 2007; Lu et al., 2014a). In mammalian species, three distinct transmembrane UPRER sensors of unfolded proteins accumulate in tandem to resolve homeostatic disruption (Cox et al., 1993; Cox and Walter, 1996; Sidrauski and Walter, 1997; Yoshida et al., 1998; Niwa et al., 1999; Harding et al., 2000; Tirasophon et al., 2000; Yoshida et al., 2001). These three transmembrane proteins composed of sensors are PERK, membrane-tethered activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1α (IRE1α) (Cox et al., 1993; Cox and Walter, 1996; Sidrauski and Walter, 1997; Yoshida et al., 1998; Yoshida et al., 2001; Calfon et al., 2002; Aragón et al., 2009; Korennykh et al., 2009; Li et al., 2010). In times of ER stress, IRE1, as the most evolutionarily conserved arm of UPRER, oligomerizes in the plane of the ER membrane, which promotes the dissociation of Bip from the luminal domain of IRE1, causing subsequent autophosphorylation and allosteric activation of its cytosolic RNase domain, initiating the non-canonical splicing of the 26-nt intron from X-box binding protein 1 (XBP1) as mRNA, producing XBP1s, which is an active transcription factor. Once XBP1s are encoded and translated, they can enter the nucleus to facilitate the production of chaperones, components of ER-associated degradation (ERAD), and foldases, which participate in protein trafficking, degradation, and folding by driving the activation of UPRER-related genes (Bertolotti et al., 2001; Calfon et al., 2002; Lee et al., 2003; Hollien and Weissman, 2006; Acosta-Alvear et al., 2007; Aragón et al., 2009; Hollien et al., 2009; Korennykh et al., 2009; Silva et al., 2009; Li et al., 2010; Hatano et al., 2013; Michel et al., 2015; Sundaram et al., 2017). In addition to XBP1s activation, active IRE1 can also be degraded. ER-localized mRNA transcripts are regulated by IRE1-dependent decay (RIDD) to limit the influx of client proteins into the ER, thereby reducing the protein-folding load in the organelle (Hollien and Weissman, 2006; Acosta-Alvear et al., 2007; Hollien et al., 2009).
PERK, an ER-resident transmembrane serine/threonine kinase, mediates the second branch of UPRER (Acosta-Alvear et al., 2007). Similar to IRE1, PERK dissociates with Bip to unlock the dormant status and then homomultimerizes as a transmembrane protein of the ER upon sensing ER stress, leading to its autophosphorylation and activation (Harding et al., 1999; Bertolotti et al., 2000; Harding et al., 2002; Marciniak et al., 2006; Hollien et al., 2009). Activation of PERK phosphorylates eIF2, a critical regulator of protein synthesis (Shi et al., 1998; Harding et al., 1999; Harding et al., 2002). Phosphorylation of eIF2 suppresses general translation, cutting down the expression of proteins into the endoplasmic reticulum, thereby mitigating ER stress in a manner analogous to the attenuation of the protein-folding burden offered by RIDD. Paradoxically, eIF2 phosphorylation promotes preferential translation of a select subset of mRNAs with short open reading frames (uORFs) in their upstream untranslated regions. One of these mRNAs transcripts is ATF4, which, like XBP1s, facilitates the expression of various genes that have something to do with combatting ER dysfunction by improving the cell’s capacity to process native and functional bioproducts, such as the redox homeostasis regulators and importers encoding amino acid (Harding et al., 2003). Recent studies have indicated that PERK has an intimate connection with the formation of ER whorls and is categorized into ER whorls in times of ER stress, and the autophosphorylation activating response of PERK as a result of ER stress and the inhibition of translation cannot be maintained for a long time without the formation of ER whorl precursors. Furthermore, most translocons are isolated into ER whorls during prolonged ER stress, taking translocons away from ribosomes, which augments the interruption of translation. ER whorls are a new kind of ER stress response that attenuates ER perturbations by promoting PERK activation and modulating the co-translation and influx of nascent proteins into the ER (Xu et al., 2021).
ATF6 regulates the third branch of the UPR (Figure 1). When the site of two Golgi-localization signals is unmasked via the dissociation of Bip during ER stress, ATF6 is exported to the Golgi apparatus with an accumulation of unfolded proteins, where the entire protein is sequentially and successively digested by site-1 protease (S1P) and site 2 protease (S2P) upon reaching the Golgi apparatus by removing their lumenal domain and transmembrane anchor, respectively, releasing the fragment facing cytosol that is translocated to the nucleus to initiate transcription related to degradation of abnormal proteins (Haze et al., 1999; Wang et al., 2000; Ye et al., 2000). However, ATF6 differs from PERK and IRE1 in that it does not inhibit the influx of client proteins into the ER. Rather, it boosts the ER’s protein processing and degradation capacity, as well as its volume (Wang et al., 2000; Nadanaka et al., 2006; Adachi et al., 2008; Bommiasamy et al., 2009). Therefore, it is suspected that the parallel activation and action of IRE1, PERK, and ATF6 combine to form an adaptive UPRER phase so that organelles can rapidly respond to ER stress and take timely measures simultaneously to maintain ER-folding equilibrium.
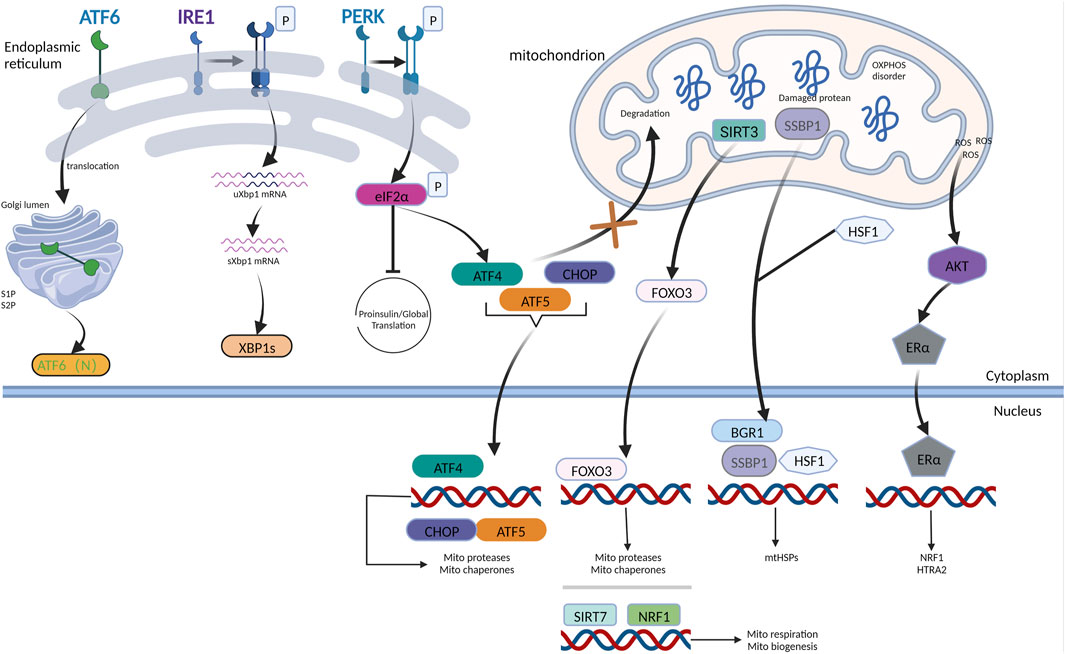
FIGURE 1. UPRmt mechanism in mammals. The AKT-ER axis is induced by the accumulation of ROS in IMS to trigger the transcription of NRF1 and HTRA2 upon responding to IMS damage. When ATF5 receives the signal derived from the dysfunctional mitochondria, it influxes into the nucleus, accompanied by ATF4 and CHOP, facilitating the production of chaperones and proteases belonging to mitochondria. HSF1 combines with SSBP1, forming a complex bound to the chromatin remodeling protein BRG1, prompting the expression of mtHSPs. Mitophagy and oxidative stress can trigger the transduction of SIRT3-FOXO3 signaling pathways. NRF1 coordinating with SIRT7 reduces the burden of damaged proteins via inhibition of mitochondrial respiration and synthesis. In conclusion, the mammalian UPRmt facilitates mitochondrial recovery and maintains proteostasis via multiple mechanisms. IRE1, PERK, and ATF6 are three ER stress sensors that monitor protein-folding conditions in the ER lumen. PERK-ATF4 axis is the main hub coordinating UPRER and UPRmt.
Several studies have demonstrated that UPRmt and UPRER have intersecting crossroads upon facing disruption of homeostasis, which can integrate their signaling and co-activate in some conditions, thereby enhancing ER and mitochondrial function in response to stimuli that dysregulate proteostasis in these two organelles (Liu et al., 2014b; Lu et al., 2014b; Rainbolt et al., 2014). PERK is a putative signal-transducing crossroad connecting UPRmt and UPRER, which has been intensively explored. PERK is located at the ER-mitochondria contact sites, providing a PERK-mitochondria signaling pathway that enables PERK to detect stress derived from the proteostasis of both organelles. The PERK/ATF4 signaling axis, which is crucial for UPRER (Liu et al., 2014b), is also implicated in UPRmt (Rainbolt et al., 2014). Activation of PERK/eIF2 signaling lowers the production of reactive oxygen species from the mitochondrial complex. Similar to ATF4, the ATF5 and CHOP transcripts also contain ORFs in their upstream untranslated region. Hence, ATF5 and CHOP levels would increase upon PERK activation by UPRER or UPRmt (Harding et al., 2003). PERK is necessary for ATF5 expression, as genetic deletion of PERK prevents the increase in ATF5 levels in ER-stressed cells (Zhou et al., 2008; Teske et al., 2013). During ER stress, the levels of the UPRmt transcripts, Lon protease, and mitochondrial HSP70 (GRP75) increased in PERK- eIF2- ATF4 (Lebeau et al., 2018). These findings imply that following ER stress, the PERK- eIF2- ATF4 pathway can promote collaboration between ATF4 and CHOP, consequently increasing the expression of ATF5 and enhancing the production of mitochondrial chaperones, facilitating the correct formation of proteins and proteases that degrade abnormal proteins (Figure 1).
Mitochondria are known as vital organelles that are involved in metabolism (especially energy metabolism), and maintaining homeostasis is not only important for the organelle itself, but also for the whole cell. As an essential mechanism to cope with homeostasis disruption, UPRmt and UPRER participate in the development of various metabolic diseases, such as diabetes, obesity, and fatty liver (Qureshi et al., 2017; Yan et al., 2020). For instance, evidence has indicated that both UPRmt and UPRER participates in obesity development. Obesity is a metabolic disease caused by the fact that fat cannot be consumed efficiently and is instead produced excessively and accumulates for a long time. Fat can be oxidized in mitochondria as a way of consumption, and activating UPRmt facilitates efficient fat utilization, therefore UPRmt alleviates obesity (Bhaskaran et al., 2017; Chung et al., 2017). The genes of HSP60, SIRT1, SIRT3, LONP1 and CLPP related to UPRmt were significantly downregulated in the individuals with larger body mass index. Furthermore, the activation of UPRmt efficiently facilitates fat utilization in an unsaturated fat diet, whereas inhibiting UPRmt promotes the accumulation of fat in a saturated fat diet. In addition, UPRmt promotes systemic energy expenditure via GDF15 that is capable of enhancing lipid consumption in muscle and adipose tissues, which in turn contributes to protecting the body from obesity. To summarize, in terms of stress, UPRmt mainly regulates metabolic alterations through augmenting glycolysis and restraining OXPHOS (Jukarainen et al., 2016; Bhaskaran et al., 2017; Chung et al., 2017). In addition, the expression of ER stress markers are enhanced in obesity models and ER stress is related to the development of leptin resistance, which is an obesity inducer. The leptin hormone increases energy expenditure, which plays a protective role against obesity. Leptin-induced STAT3 phosphorylation can be inhibited by ER stress inducers, which are associated with leptin-resistance and body weight gain. XBPs and ATF6 overexpression associated with UPRER alleviate ER stress and promote the transduction of leptin signaling. Additionally, mice with a XBPs deletion in neurons consequently reduced ER folding capacity, and displayed an obesogenic phenotype upon high fat diet feeding accompanied with decreased energy expenditure and hyperphagia. Furthermore, PERK phosphorylation levels are significantly increased and the STAT3 phosphorylation induced by leptin was markedly blunted in such mice (Ozcan et al., 2004; Hosoi et al., 2008; Zhang et al., 2008; Ozcan et al., 2009; Won et al., 2009; Cakir et al., 2013; Schneeberger et al., 2013; Lee and Ozcan, 2014; Williams et al., 2014). Notably, UPRmt coordinating with UPRER participates in the development of metabolic diseases. For example, oxidative stressors such as ROS in mitochondria, associated with the occurrence and development of metabolic diseases, can initiate UPRER before activating UPRmt. UPRER can touch off UPRmt via a ATF4-dependent manner. Moreover, LONP1, as a vital protease in UPRmt, can also participate in UPRER. Therefore, the coordination between UPRmt and UPRER must be remarkable to manage stress and maintain cellular homeostasis. However, the detailed mechanism related to the crosstalk between UPRmt and UPRER in metabolism are lacking, and requires additional studies (Iqbal and Hood, 2014; Yang et al., 2018; Waldherr et al., 2019; Jiang et al., 2020)
T2D is a global health concern. It is the leading cause of morbidity and mortality worldwide. T2D is characterized by insulin resistance in peripheral tissues (skeletal muscles, adipose tissues, etc.) and pancreatic beta cell dysfunction. Initially, factors such as obesity led to insulin resistance, forcing pancreatic beta cells to exert excessive normal action for more insulin to maintain normal glucose levels. However, prolonged overstimulation results in beta cell dysfunction and death, ultimately leading to hyperglycemia and gradually leading to diabetes. UPRmt plays a crucial role in T2D, from insulin resistance to beta cell dysfunction. Increased glucose and fatty acid metabolism in cells in hyperglycemia and hyperlipidemia result in increased ROS production and protein aggregation (Fernandes et al., 2020; Kos, 2020). Consequently, UPRmt is activated to increase glucose metabolism and decrease ROS generation and protein aggregation (Fernandes et al., 2020). UPRmt regulates OXPHOS and glycolysis to increase glucose metabolism. Chronic hyperglycemia impairs the UPRmt in people with diabetes. UPRmt has been observed to be decreased in the brains of insulin-resistant mice, mice lacking insulin signaling, and people with T2D (Fernandes et al., 2020). Moreover, it has been demonstrated that insulin working in the brain promotes UPRmt and attenuates diet-induced weight gain (Wardelmann et al., 2019).
UPRER coordinates with UPRmt in T2D by activating the PERK pathway. Downstream PERK has been reported to be involved in T2D. The homeodomain transcription factor of pancreas/duodenum homeobox protein 1 (Pdx1) and human diabetes genes partially regulate beta cell survival via direct regulation of the activating transcription factor ATF5. Significantly, the absence of ATF5 decreased the survival rate under stressful conditions. ATF5 is parallel to ATF4 downstream under the control of eIF4E-binding protein 1 (4ebp1), a component of the mammalian target of rapamycin (mTOR) pathway that slows the translation of proteins, based on loss-of-function and chromatin occupancy tests. Consequently, the absence of ATF5 decreased the stress-induced inhibition of almost the whole translation, thereby increasing the vulnerability of cells to stress-induced death (Pakos-Zebrucka et al., 2016). CHOP depletion has been demonstrated as a therapeutic method for reducing dysregulated insulin secretion and fatty liver pathology in T2D (Wardelmann et al., 2019).
PERK, a crucial sensor for responses to ER stress, is located in the MAMs, which is composed of membrane compositions from mitochondria and ER, has a closely related impact on the ER–mitochondrial interaction and mitochondrial apoptosis mediated by ROS (Verfaillie et al., 2012; Verfaillie et al., 2013). For example knocking out PERK in murine embryonic fibroblasts (MEFs) causes a disturbance to the ER–mitochondria association, consequentially causing disruption to ER morphology, decreasing the transduction of ROS signaling from ER to mitochondria, reducing the flux of Ca2+ from ER to mitochondria, and bringing about decline in mitochondrial apoptosis induced by ROS (Liu et al., 2013). The leucine-rich repeat kinase 2 (LRRK2) enzyme suppresses PERK-induced phosphorylation of the E3 ubiquitin ligases, parkin. In LRRK2 KO MEFs, parkin is phosphorylated by PERK, and mitofusin-2 (Mfn2) (located at the MAMs) is ubiquitinated and degraded, which causes the ER–mitochondria association to decrease (Toyofuku et al., 2020). Furthermore, PERK can form the PERK–Mfn2 complex and PERK–S1R complex contacting with Mfn2 and sigma-1 receptor (S1R), which are also involved in the composition of MAMs and sustain the stabilization of ER-mitochondrial contacts (Liu et al., 2013; Cao et al., 2021).
Disruption of Ca2+ homeostasis is to some extent related to metabolic disorders such as insulin resistance, obesity, and T2D in both humans and animals. For example, primary adipocytes isolated from humans with obesity and insulin resistance (Draznin et al., 1988) or obtained from rats with diabetes (Draznin et al., 1989) exhibited higher Ca2+ levels within the cell compared to ordinary individuals. Simultaneously, MAMs are the contact interfaces linking the outer membrane of mitochondria and ER membrane where the molecules between two organelles communicating with each other, also has a definition of structural membranes between the two compartments (Phillips and Voeltz, 2016). Notably, mitochondria and ER, shown as the two largest compartments storing Ca2+ in human cells, have Ca2+ ion transportation and the efficient and precise communication between the two organelles, relies on such a special membrane structure. Therefore, MAMs shoulder the responsibility of dynamically and efficiently transmitting Ca2+ signals between the ER and the mitochondria in physiology and pathology. Owing to Ca2+ handling proteins that are enriched in the sites of MAMs, regulating intracellular Ca2+ homeostasis is dependent on the functional coupling at the contact sites of ER-mitochondria when the cells cope with various pathophysiological and environmental stimuli and go through metabolic reprogramming (Theurey and Rieusset, 2017). In addition, there are abundant functional enzymes and proteins that participate in the metabolism of fatty acids and glucose locating at MAMs, thus MAMs are referred as an integrator of energy metabolism (Sala-Vila et al., 2016; Rieusset, 2017). In conclusion, we believe that MAMs has an intimate relationship with the occurrence of T2D.
In addition, other UPRmt components, such as SIRT1, SIRT3, and SIRT5, have positive regulatory effects on insulin sensitivity (Owusu-Ansah et al., 2013; Jukarainen et al., 2016; Yong et al., 2021). HSP60, HSP72, and HSP70, which are mitochondrial chaperone proteins, substantially impact T2D (Lee et al., 2011; Kim et al., 2013; Kleinridders et al., 2013; Henstridge et al., 2014; Mottis et al., 2014; Lee et al., 2015; Aluksanasuwan et al., 2017). Moreover, high levels of mitochondrial proteases, including LONP1, CLPP, and OMA1, are also involved in protective action in T2D (Lee et al., 2011; Lee et al., 2015; Wu et al., 2019; Kalvala et al., 2020). (Figure 2).
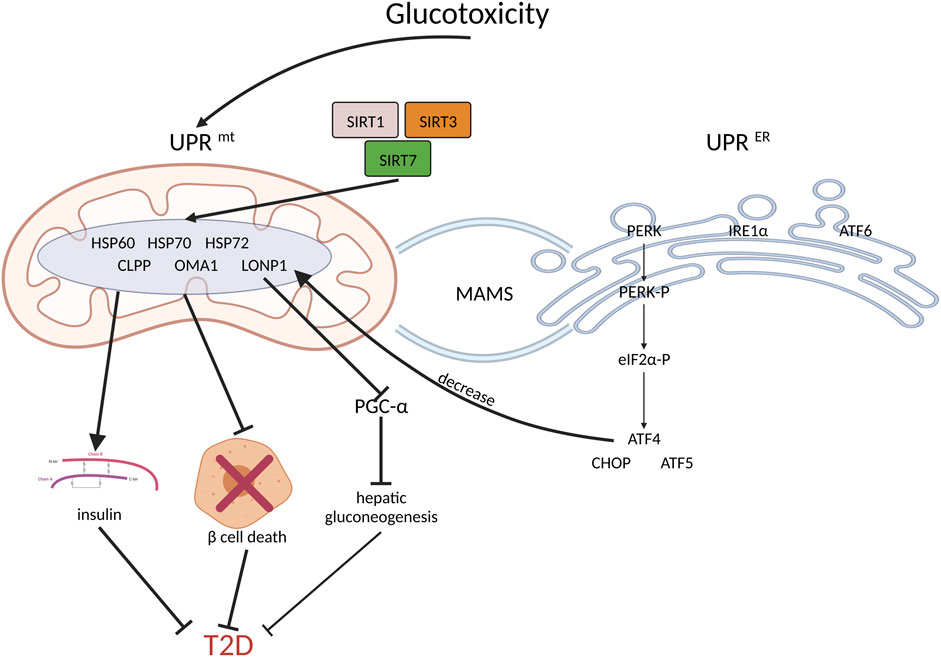
FIGURE 2. UPRmt and coordinated UPRER increase insulin sensitivity, inhibit beta cell death and benefit T2D. Firstly, hyperglycemia induces UPRmt and UPRER. As the downstream of PERK, ATF4, CHOP and ATF5 will inhibit beta cell death; thus, they have beneficial effects on T2D management. Secondly, SIRT1, SIRT3 and SIRT5 activate UPRmt, which improves insulin sensitivity. In addition, the increased expression of mitochondrial chaperone proteins and proteases, such as HSP60, HSP70, HSP72, LONP1, CLPP, and OMA1, may reduce insulin resistance. Moreover, by inhibiting PGC-1a, LONP1 suppresses hepatic gluconeogenesis, thereby ameliorating hepatic insulin resistance.
A germline deletion of the CHOP gene, which encodes the C/EBP homologous protein as a transcription factor bound to enhancer, can prevent β cell failure in diabetes models (Song et al., 2008; Satoh et al., 2011; Maris et al., 2012). However, whether the CHOP deletion gives protection to β cells by the means of a cell-autonomous manner, remains unclear (Kempf et al., 2012).
Currently, researchers investigated whether there are therapeutic benefits through targeting CHOP specifically in β cells of mice. First, CHOP deletions in ß cells alleviated ER stress and the glucose-stimulated insulin secretion is delayed in the mice that are fed a high fat diet (HFD-fed mice). Second, specific CHOP deletion in ß cell can protect aged HFD-fed mice against liver hepatomegaly and steatosis and not influence basal glucose homeostasis. Third, the Ca2+ buffering capacity within ER is reduced and glucose-induced Ca2+ oscillations is regulated in terms of CHOP depletion in cells, causing a result of changes in transcriptional profile of the ER chaperone. Finally, there is decline of pancreatic insulin content and liver triglycerides under the operation of exerting CHOP antisense oligonucleotide conjugated with GLP1. In conclusion, these results suggest a novel therapeutic strategy via targeting CHOP in β cells to mitigate the development of T2D, while regulating insulin secretion and alleviating syndromes of consequent fatty liver disease (Yong et al., 2021).
As a member of TGF-β superfamily, the growth differentiation factor-15 (GDF-15) cytokine has an impact on the development of several metabolic disorders via plasmatic level regulation, including T2D (Kempf et al., 2012; Bao et al., 2019), cardiovascular diseases (Brown et al., 2002), obesity (Dostálová et al., 2009; Vila et al., 2011), non-alcoholic steatohepatitis (NASH) (Kempf et al., 2012), and various cancers (Brown et al., 2003; Koopmann et al., 2004). Moreover, there is some connection between the CHOP and GDF-15. The transcription factor CHOP, which is directly bound to the promoter region of GDF15, modulates the expression of GDF15. In addition, saturated fatty acids (SFAs) can significantly induce the expression and secretion of GDF15 (L'Homme et al., 2020). Therefore, GDF-15 may have a relationship with the development of several metabolic disorders, which could be a potential target of T2D.
The study of UPRmt is still in its infancy. Despite significant progress in the past few years, the crucial action of UPRmt in metabolism, such as T2D, is gradually being understood. Here, we describe our current understanding of UPRmt and its crosstalk with UPRER, mainly through the PERK signaling pathway. Furthermore, we discuss the functions and molecular mechanisms of UPRmt and coordinated UPRER in the context of T2D. Although our knowledge of the topics in this review has largely improved, many fundamental questions remain unanswered: Do UPRER components other than PERK also affect UPRmt? Does UPRmt also regulate UPRER? Which components of UPRmt respond to insulin resistance development in T2D? Answers to these questions will help identify new candidate interventions for T2D.
As the first authors, ZK and FC make the same important contributions to design concept, collected and analyzed materials, wrote articles and drew. FX make the final review and finalization of the articles to be published as the corresponding authors. All authors contributed to the article and approved the submitted version.
This work was funded by National Natural Science Foundation of China (No. 32170705); Natural Science Foundation of Guangdong Province (No. 2022A1515012303) and Guangzhou key medical discipline construction project fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., et al. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell. 27 (1), 53–66. doi:10.1016/j.molcel.2007.06.011
Adachi, Y., Yamamoto, K., Okada, T., Yoshida, H., Harada, A., and Mori, K. (2008). ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell. Struct. Funct. 33 (1), 75–89. doi:10.1247/csf.07044
Aldridge, J. E., Horibe, T., and Hoogenraad, N. J. (2007). Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2 (9), e874. doi:10.1371/journal.pone.0000874
Allison, R., Edgar, J. R., Pearson, G., Rizo, T., Newton, T., Gunther, S., et al. (2017). Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell. Biol. 216 (5), 1337–1355. doi:10.1083/jcb.201609033
Aluksanasuwan, S., Sueksakit, K., Fong-Ngern, K., and Thongboonkerd, V. (2017). Role of HSP60 (HSPD1) in diabetes-induced renal tubular dysfunction: Regulation of intracellular protein aggregation, ATP production, and oxidative stress. FASEB J. 31 (5), 2157–2167. doi:10.1096/fj.201600910RR
Aragón, T., van Anken, E., Pincus, D., Serafimova, I. M., Korennykh, A. V., Rubio, C. A., et al. (2009). Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 457 (7230), 736–740. doi:10.1038/nature07641
Bao, X., Borne, Y., Muhammad, I. F., Nilsson, J., Lind, L., Melander, O., et al. (2019). Growth differentiation factor 15 is positively associated with incidence of diabetes mellitus: The malmö diet and cancer-cardiovascular cohort. Diabetologia 62 (1), 78–86. doi:10.1007/s00125-018-4751-7
Barbosa, C., Peixeiro, I., and Romão, L. (2013). Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 9 (8), e1003529. doi:10.1371/journal.pgen.1003529
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P., and Ron, D. (2006). Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174 (1), 229–239. doi:10.1534/genetics.106.061580
Bertolotti, A., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell. Biol. 2 (6), 326–332. doi:10.1038/35014014
Bertolotti, A., Wang, X., Novoa, I., Jungreis, R., Schlessinger, K., Cho, J. H., et al. (2001). Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J. Clin. Invest. 107 (5), 585–593. doi:10.1172/JCI11476
Bhaskaran, S., Unnikrishnan, A., Ranjit, R., Qaisar, R., Pharaoh, G., Matyi, S., et al. (2017). A fish oil diet induces mitochondrial uncoupling and mitochondrial unfolded protein response in epididymal white adipose tissue of mice. Free Radic. Biol. Med. 108, 704–714. doi:10.1016/j.freeradbiomed.2017.04.028
Bommiasamy, H., Back, S. H., Fagone, P., Lee, K., Meshinchi, S., Vink, E., et al. (2009). ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell. Sci. 122, 1626–1636. doi:10.1242/jcs.045625
Brown, D. A., Breit, S. N., Buring, J., Fairlie, W. D., Bauskin, A. R., Liu, T., et al. (2002). Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: A nested case-control study. Lancet 359 (9324), 2159–2163. doi:10.1016/S0140-6736(02)09093-1
Brown, D. A., Ward, R. L., Buckhaults, P., Liu, T., Romans, K. E., Hawkins, N. J., et al. (2003). MIC-1 serum level and genotype: Associations with progress and prognosis of colorectal carcinoma. Clin. Cancer Res. 9 (7), 2642–2650.
Bukau, B., Weissman, J., and Horwich, A. (2006). Molecular chaperones and protein quality control. Cell. 125 (3), 443–451. doi:10.1016/j.cell.2006.04.014
Cakir, I., Cyr, N. E., Perello, M., Litvinov, B. P., Romero, A., Stuart, R. C., et al. (2013). Obesity induces hypothalamic endoplasmic reticulum stress and impairs proopiomelanocortin (POMC) post-translational processing. J. Biol. Chem. 288 (24), 17675–17688. doi:10.1074/jbc.M113.475343
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415 (6867), 92–96. doi:10.1038/415092a
Cao, Y., Chen, Z., Hu, J., Feng, J., Zhu, Z., Fan, Y., et al. (2021). Mfn2 regulates high glucose-induced MAMs dysfunction and apoptosis in podocytes via PERK pathway. Front. Cell. Dev. Biol. 9, 769213. doi:10.3389/fcell.2021.769213
Chung, H. K., Ryu, D., Kim, K. S., Chang, J. Y., Kim, Y. K., Yi, H. S., et al. (2017). Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell. Biol. 216 (1), 149–165. doi:10.1083/jcb.201607110
Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 73 (6), 1197–1206. doi:10.1016/0092-8674(93)90648-a
Cox, J. S., and Walter, P. (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 87 (3), 391–404. doi:10.1016/s0092-8674(00)81360-4
Csordás, G., Renken, C., Varnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell. Biol. 174 (7), 915–921. doi:10.1083/jcb.200604016
Dall, K. B., and Færgeman, N. J. (2019). Metabolic regulation of lifespan from a C. elegans perspective. Genes. Nutr. 14, 25. doi:10.1186/s12263-019-0650-x
Dostálová, I., Roubicek, T., Bartlova, M., Mraz, M., Lacinova, Z., Haluzikova, D., et al. (2009). Increased serum concentrations of macrophage inhibitory cytokine-1 in patients with obesity and type 2 diabetes mellitus: The influence of very low calorie diet. Eur. J. Endocrinol. 161 (3), 397–404. doi:10.1530/EJE-09-0417
Draznin, B., Lewis, D., HoulderN., , ShermaNN., , AdaMoM., , Garvey, W. T., et al. (1989). Mechanism of insulin resistance induced by sustained levels of cytosolic free calcium in rat adipocytes. Endocrinology 125 (5), 2341–2349. doi:10.1210/endo-125-5-2341
Draznin, B., Sussman, K. E., Eckel, R. H., KaoM., , YosT, T., and Sherman, N. A. (1988). Possible role of cytosolic free calcium concentrations in mediating insulin resistance of obesity and hyperinsulinemia. J. Clin. Invest. 82 (6), 1848–1852. doi:10.1172/JCI113801
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 144 (1), 79–91. doi:10.1016/j.cell.2010.12.016
Fernandes, V., Choudhary, M., Kumar, A., and Singh, S. B. (2020). Proteotoxicity and mitochondrial dynamics in aging diabetic brain. Pharmacol. Res. 159, 104948. doi:10.1016/j.phrs.2020.104948
Fiorese, C. J., Schulz, A. M., Lin, Y. F., Rosin, N., Pellegrino, M. W., and Haynes, C. M. (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26 (15), 2037–2043. doi:10.1016/j.cub.2016.06.002
Gao, K., Li, Y., Hu, S., Liu, Y., et al. (2019). SUMO peptidase ULP-4 regulates mitochondrial UPR mediated innate immunity and lifespan extension. Elife 8, e41792. doi:10.7554/eLife.41792
Grimm, S. (2012). The ER-mitochondria interface: The social network of cell death. Biochim. Biophys. Acta 1823 (2), 327–334. doi:10.1016/j.bbamcr.2011.11.018
Hansen, K. G., and Herrmann, J. M. (2019). Transport of proteins into mitochondria. Protein J. 38 (3), 330–342. doi:10.1007/s10930-019-09819-6
Harding, H. P., Calfon, M., Urano, F., Novoa, I., and Ron, D. (2002). Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell. Dev. Biol. 18, 575–599. doi:10.1146/annurev.cellbio.18.011402.160624
Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., and Ron, D. (2000). Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 5 (5), 897–904. doi:10.1016/s1097-2765(00)80330-5
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397 (6716), 271–274. doi:10.1038/16729
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 11 (3), 619–633. doi:10.1016/s1097-2765(03)00105-9
Hatano, M., Umemura, M., Kimura, N., Yamazaki, T., Takeda, H., Nakano, H., et al. (2013). The 5'-untranslated region regulates ATF5 mRNA stability via nonsense-mediated mRNA decay in response to environmental stress. FEBS J. 280 (18), 4693–4707. doi:10.1111/febs.12440
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C-elegans. Dev. Cell. 13 (4), 467–480. doi:10.1016/j.devcel.2007.07.016
Haynes, C. M., and Ron, D. (2010). The mitochondrial UPR - protecting organelle protein homeostasis. J. Cell. Sci. 123 (22), 3849–3855. doi:10.1242/jcs.075119
Haynes, C. M., Yang, Y., Blais, S. P., Neubert, T. A., and Ron, D. (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell. 37 (4), 529–540. doi:10.1016/j.molcel.2010.01.015
Haze, K., YosHida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 10 (11), 3787–3799. doi:10.1091/mbc.10.11.3787
Heishima, K., Sugito, N., Soga, T., Nishikawa, M., Ito, Y., Honda, R., et al. (2021). Petasin potently inhibits mitochondrial complex I-based metabolism that supports tumor growth and metastasis. J. Clin. Invest. 131 (17), e139933. doi:10.1172/JCI139933
Henstridge, D. C., Bruce, C. R., Drew, B. G., Tory, K., Kolonics, A., Estevez, E., et al. (2014). Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 63 (6), 1881–1894. doi:10.2337/db13-0967
Hollien, J., Lin, J. H., Li, H., Stevens, N., Walter, P., and Weissman, J. S. (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell. Biol. 186 (3), 323–331. doi:10.1083/jcb.200903014
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313 (5783), 104–107. doi:10.1126/science.1129631
Hosoi, T., Sasaki, M., Miyahara, T., Hashimoto, C., Matsuo, S., Yoshii, M., et al. (2008). Endoplasmic reticulum stress induces leptin resistance. Mol. Pharmacol. 74 (6), 1610–1619. doi:10.1124/mol.108.050070
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497 (7450), 451–457. doi:10.1038/nature12188
Ilacqua, N., Sanchez-Alvarez, M., Bachmann, M., Costiniti, V., Del Pozo, M. A., and Giacomello, M. (2017). Protein localization at mitochondria-ER contact sites in basal and stress conditions. Front. Cell. Dev. Biol. 5, 107. doi:10.3389/fcell.2017.00107
Iqbal, S., and Hood, D. A. (2014). Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol. Cell. Physiol. 306 (12), C1176–C1183. doi:10.1152/ajpcell.00017.2014
Jiang, D., Cui, H., Xie, N., Banerjee, S., Liu, R. M., Dai, H., et al. (2020). ATF4 mediates mitochondrial unfolded protein response in alveolar epithelial cells. Am. J. Respir. Cell. Mol. Biol. 63 (4), 478–489. doi:10.1165/rcmb.2020-0107OC
Jovaisaite, V., Mouchiroud, L., and Auwerx, J. (2014). The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 217, 137–143. doi:10.1242/jeb.090738
Jukarainen, S., Heinonen, S., Ramo, J. T., Rinnankoski-Tuikka, R., Rappou, E., Tummers, M., et al. (2016). Obesity is associated with low NAD (+)/SIRT pathway expression in adipose tissue of BMI-discordant monozygotic twins. J. Clin. Endocrinol. Metab. 101 (1), 275–283. doi:10.1210/jc.2015-3095
Kalvala, A. K., Yerra, V. G., Sherkhane, B., Gundu, C., Arruri, V., Kumar, R., et al. (2020). Chronic hyperglycemia impairs mitochondrial unfolded protein response and precipitates proteotoxicity in experimental diabetic neuropathy: Focus on LonP1 mediated mitochondrial regulation. Pharmacol. Rep. 72 (6), 1627–1644. doi:10.1007/s43440-020-00147-6
Kaspar, S., Oertlin, C., Szczepanowska, K., Kukat, A., Senft, K., Lucas, C., et al. (2021). Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 7 (22), eabf0971. doi:10.1126/sciadv.abf0971
Kaufman, R. J. (2002). Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 110 (10), 1389–1398. doi:10.1172/JCI16886
Kempf, T., Guba-Quint, A., Torgerson, J., Magnone, M. C., Haefliger, C., Bobadilla, M., et al. (2012). Growth differentiation factor 15 predicts future insulin resistance and impaired glucose control in obese nondiabetic individuals: Results from the XENDOS trial. Eur. J. Endocrinol. 167 (5), 671–678. doi:10.1530/EJE-12-0466
Kim, K. H., Jeong, Y. T., Oh, H., Kim, S. H., Cho, J. M., Kim, Y. N., et al. (2013). Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 19 (1), 83–92. doi:10.1038/nm.3014
Kleinridders, A., Lauritzen, H. P. M. M., Ussar, S., Christensen, J. H., Mori, M. A., Bross, P., et al. (2013). Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J. Clin. Invest. 123 (11), 4667–4680. doi:10.1172/JCI67615
Koopmann, J., Buckhaults, P., Brown, D. A., Zahurak, M. L., Sato, N., Fukushima, N., et al. (2004). Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other periampullary cancers. Clin. Cancer Res. 10 (7), 2386–2392. doi:10.1158/1078-0432.ccr-03-0165
Korennykh, A. V., Egea, P. F., Korostelev, A. A., Finer-Moore, J., Zhang, C., Shokat, K. M., et al. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature 457 (7230), 687–693. doi:10.1038/nature07661
Kos, K. (2020). Cardiometabolic morbidity and mortality with smoking cessation, review of recommendations for people with diabetes and obesity. Curr. Diab. Rep. 20 (12), 82. doi:10.1007/s11892-020-01352-6
L'Homme, L., Sermikli, B. P., Staels, B., Piette, J., Legrand-Poels, S., and Dombrowicz, D. (2020). Saturated fatty acids promote GDF15 expression in human macrophages through the PERK/eIF2/CHOP signaling pathway. Nutrients 12 (12), E3771. doi:10.3390/nu12123771
Lebeau, J., Saunders, J. M., Moraes, V. W. R., Madhavan, A., Madrazo, N., Anthony, M. C., et al. (2018). The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell. Rep. 22 (11), 2827–2836. doi:10.1016/j.celrep.2018.02.055
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23 (21), 7448–7459. doi:10.1128/mcb.23.21.7448-7459.2003
Lee, C., Zeng, J., Drew, B. G., Sallam, T., Martin-Montalvo, A., Wan, J., et al. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell. Metab. 21 (3), 443–454. doi:10.1016/j.cmet.2015.02.009
Lee, H. J., Chung, K., Lee, K., Lim, J. H., and Song, J. (2011). Downregulation of mitochondrial lon protease impairs mitochondrial function and causes hepatic insulin resistance in human liver SK-HEP-1 cells. Diabetologia 54 (6), 1437–1446. doi:10.1007/s00125-011-2074-z
Lee, J., and Ozcan, U. (2014). Unfolded protein response signaling and metabolic diseases. J. Biol. Chem. 289 (3), 1203–1211. doi:10.1074/jbc.R113.534743
Li, H., Korennykh, A. V., Behrman, S. L., and Walter, P. (2010). Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. U. S. A. 107 (37), 16113–16118. doi:10.1073/pnas.1010580107
Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., et al. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318 (5852), 944–949. doi:10.1126/science.1146361
Lin, Y. F., Schulz, A. M., Pellegrino, M. W., Lu, Y., Shaham, S., and Haynes, C. M. (2016). Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533 (7603), 416–419. doi:10.1038/nature17989
Liu, X., Kwak, D., Lu, Z., Xu, X., Fassett, J., Wang, H., et al. (2014). Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension 64 (4), 738–744. doi:10.1161/HYPERTENSIONAHA.114.03811
Liu, Y., Samuel, B. S., Breen, P. C., and Ruvkun, G. (2014). Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508 (7496), 406–410. doi:10.1038/nature13204
Liu, Z. W., Zhu, H. T., Chen, K. L., Dong, X., Wei, J., Qiu, C., et al. (2013). Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc. Diabetol. 12, 158. doi:10.1186/1475-2840-12-158
Lu, M., Lawrence, D. A., Marsters, S., Acosta-Alvear, D., Kimmig, P., Mendez, A. S., et al. (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345 (6192), 98–101. doi:10.1126/science.1254312
Lu, Z., Xu, X., Fassett, J., Kwak, D., Liu, X., Hu, X., et al. (2014). Loss of the eukaryotic initiation factor 2α kinase general control nonderepressible 2 protects mice from pressure overload-induced congestive heart failure without affecting ventricular hypertrophy. Hypertension 63 (1), 128–135. doi:10.1161/HYPERTENSIONAHA.113.02313
Marciniak, S. J., Garcia-Bonilla, L., Hu, J., Harding, H. P., and Ron, D. (2006). Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell. Biol. 172 (2), 201–209. doi:10.1083/jcb.200508099
Maris, M., Overbergh, L., Gysemans, C., WAget, A., Cardozo, A. K., VErdrEngh, E., et al. (2012). Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia 55 (4), 1167–1178. doi:10.1007/s00125-011-2427-7
Martínez-Reyes, I., Sánchez-Aragó, M., and Cuezva, J. M. (2012). AMPK and GCN2-ATF4 signal the repression of mitochondria in colon cancer cells. Biochem. J. 444 (2), 249–259. doi:10.1042/BJ20111829
Martinus, R. D., Garth, G. P., Webster, T. L., Cartwright, P., Naylor, D. J., Hoj, P. B., et al. (1996). Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 240 (1), 98–103. doi:10.1111/j.1432-1033.1996.0098h.x
Mehrbod, P., Ande, S. R., Alizadeh, J., Rahimizadeh, S., Shariati, A., Malek, H., et al. (2019). The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. VIRULENCE 10 (1), 376–413. doi:10.1080/21505594.2019.1605803
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 165 (5), 1209–1223. doi:10.1016/j.cell.2016.04.012
Michel, S., Canonne, M., Arnould, T., and Renard, P. (2015). Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 21, 58–68. doi:10.1016/j.mito.2015.01.005
Moehle, E. A., Higuchi-Sanabria, R., Tsui, C. K., Homentcovschi, S., Tharp, K. M., Zhang, H., et al. (2021). Cross-species screening platforms identify EPS-8 as a critical link for mitochondrial stress and actin stabilization. Sci. Adv. 7 (44), eabj6818. doi:10.1126/sciadv.abj6818
Mohrin, M., Shin, J., Liu, Y., Brown, K., Luo, H., Xi, Y., et al. (2015). Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. SCIENCE 347 (6228), 1374–1377. doi:10.1126/science.aaa2361
Mottis, A., Jovaisaite, V., and Auwerx, J. (2014). The mitochondrial unfolded protein response in mammalian physiology. Mamm. Genome 25 (9-10), 424–433. doi:10.1007/s00335-014-9525-z
Münch, C., and Harper, J. W. (2016). Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534 (7609), 710–713. doi:10.1038/nature18302
Nadanaka, S., Yoshida, H., Sato, R., and Mori, K. (2006). Analysis of ATF6 activation in Site-2 protease-deficient Chinese hamster ovary cells. Cell. Struct. Funct. 31 (2), 109–116. doi:10.1247/csf.06015
Nagelkerke, A., Bussink, J., Sweep, F. C. G. J., and Span, P. N. (2014). The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta 1846 (2), 277–284. doi:10.1016/j.bbcan.2014.07.006
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P., and Haynes, C. M. (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR (mt). Mol. Cell. 58 (1), 123–133. doi:10.1016/j.molcel.2015.02.008
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337 (6094), 587–590. doi:10.1126/science.1223560
Nicolas, E., Tricarico, R., Savage, M., Golemis, E. A., and Hall, M. J. (2019). Disease-associated genetic variation in human mitochondrial protein import. Am. J. Hum. Genet. 104 (5), 784–801. doi:10.1016/j.ajhg.2019.03.019
Niwa, M., Sidrauski, C., Kaufman, R. J., and Walter, P. (1999). A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell. 99 (7), 691–702. doi:10.1016/s0092-8674(00)81667-0
Ott, M., Amunts, A., and Brown, A. (2016). Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 85, 77–101. doi:10.1146/annurev-biochem-060815-014334
Owusu-Ansah, E., Song, W., and Perrimon, N. (2013). Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 155 (3), 699–712. doi:10.1016/j.cell.2013.09.021
Ozcan, L., Ergin, A. S., Lu, A., Chung, J., Sarkar, S., Nie, D., et al. (2009). Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell. Metab. 9 (1), 35–51. doi:10.1016/j.cmet.2008.12.004
Ozcan, U., Cao, Q., Yilmaz, E., Lee, A. H., Iwakoshi, N. N., Ozdelen, E., et al. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306 (5695), 457–461. doi:10.1126/science.1103160
Pakos-Zebrucka, K., Koryga, I., Mnich, K., Ljujic, M., Samali, A., and Gorman, A. M. (2016). The integrated stress response. EMBO Rep. 17 (10), 1374–1395. doi:10.15252/embr.201642195
Papa, L., and Germain, D. (2011). Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell. Sci. 124 (9), 1396–1402. doi:10.1242/jcs.078220
Papa, L., and Germain, D. (2014). SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 34 (4), 699–710. doi:10.1128/MCB.01337-13
Pellegrino, M. W., Nargund, A. M., and Haynes, C. M. (2013). Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833 (2), 410–416. doi:10.1016/j.bbamcr.2012.02.019
Persengiev, S. P., Devireddy, L. R., and Green, M. R. (2002). Inhibition of apoptosis by ATFx: A novel role for a member of the ATF/CREB family of mammalian bZIP transcription factors. Genes. Dev. 16 (14), 1806–1814. doi:10.1101/gad.992202
Phillips, M. J., and Voeltz, G. K. (2016). Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell. Biol. 17 (2), 69–82. doi:10.1038/nrm.2015.8
Poveda-Huertes, D., Matic, S., Marada, A., Habernig, L., Licheva, M., Myketin, L., et al. (2020). An early mtUPR: Redistribution of the nuclear transcription factor Rox1 to mitochondria protects against intramitochondrial proteotoxic aggregates. Mol. Cell. 77 (1), 180–188. e9. doi:10.1016/j.molcel.2019.09.026
Quirós, P. M., Prado, M. A., Zamboni, N., D'Amico, D., Williams, R. W., Finley, D., et al. (2017). Multi -omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell. Biol. 216 (7), 2027–2045. doi:10.1083/jcb.201702058
Qureshi, M. A., Haynes, C. M., and Pellegrino, M. W. (2017). The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 292 (33), 13500–13506. doi:10.1074/jbc.R117.791061
Rainbolt, T. K., Saunders, J. M., and Wiseman, R. L. (2014). Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 25 (10), 528–537. doi:10.1016/j.tem.2014.06.007
Rieusset, J. (2017). Endoplasmic reticulum-mitochondria calcium signaling in hepatic metabolic diseases. Biochim. Biophys. Acta. Mol. Cell. Res. 1864 (6), 865–876. doi:10.1016/j.bbamcr.2017.01.001
Roger, A. J., Munoz-Gomez, S. A., and Kamikawa, R. (2017). The origin and diversification of mitochondria. Curr. Biol. 27 (21), R1177–R1192. doi:10.1016/j.cub.2017.09.015
Rolland, S. G., Schneid, S., Schwarz, M., Rackles, E., Fischer, C., Haeussler, S., et al. (2019). Compromised mitochondrial protein import acts as a signal for UPR (mt). Cell. Rep. 28 (7), 1659–1669. e5. doi:10.1016/j.celrep.2019.07.049
Ron, D. (2002). Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110 (10), 1383–1388. doi:10.1172/JCI16784
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 8 (7), 519–529. doi:10.1038/nrm2199
Rossmanith, W., Of, P., and and, Z. (2012). Of P and Z: Mitochondrial tRNA processing enzymes. Biochim. Biophys. Acta 1819 (9-10), 1017–1026. doi:10.1016/j.bbagrm.2011.11.003
Rutkowski, D. T., and Kaufman, R. J. (2004). A trip to the ER: Coping with stress. Trends Cell. Biol. 14 (1), 20–28. doi:10.1016/j.tcb.2003.11.001
Ryan, M. T., and Hoogenraad, N. J. (2007). Mitochondrial-nuclear communications. Annu. Rev. Biochem. 76, 701–722. doi:10.1146/annurev.biochem.76.052305.091720
Sala-Vila, A., Navarro-Lerida, I., Sanchez-Alvarez, M., Bosch, M., Calvo, C., Lopez, J. A., et al. (2016). Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 6, 27351. doi:10.1038/srep27351
Satoh, T., AbiruN., , KobayashiM., , ZHou, H., NaKamura, K., Kuriya, G., et al. (2011). CHOP deletion does not impact the development of diabetes but suppresses the early production of insulin autoantibody in the NOD mouse. Apoptosis 16 (4), 438–448. doi:10.1007/s10495-011-0576-2
Schmidt, O., Pfanner, N., and Meisinger, C. (2010). Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell. Biol. 11 (9), 655–667. doi:10.1038/nrm2959
Schneeberger, M., Dietrich, M. O., Sebastian, D., Imbernon, M., Castano, C., Garcia, A., et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 155 (1), 172–187. doi:10.1016/j.cell.2013.09.003
Shi, Y., Vattem, K. M., Sood, R., An, J., Liang, J., Stramm, L., et al. (1998). Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18 (12), 7499–7509. doi:10.1128/mcb.18.12.7499
Shpilka, T., Du, Y., Yang, Q., Melber, A., Uma Naresh, N., Lavelle, J., et al. (2021). UPR (mt) scales mitochondrial network expansion with protein synthesis via mitochondrial import in Caenorhabditis elegans. Nat. Commun. 12 (1), 479. doi:10.1038/s41467-020-20784-y
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 90 (6), 1031–1039. doi:10.1016/s0092-8674(00)80369-4
Silva, J. M., Wong, A., Carelli, V., and Cortopassi, G. A. (2009). Inhibition of mitochondrial function induces an integrated stress response in oligodendroglia. Neurobiol. Dis. 34 (2), 357–365. doi:10.1016/j.nbd.2009.02.005
Sloan, D. B., Warren, J. M., Williams, A. M., Wu, Z., Abdel-Ghany, S. E., Chicco, A. J., et al. (2018). Cytonuclear integration and co-evolution. Nat. Rev. Genet. 19 (10), 635–648. doi:10.1038/s41576-018-0035-9
Smyrnias, I., Gray, S. P., Okonko, D. O., Sawyer, G., Zoccarato, A., Catibog, N., et al. (2019). Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J. Am. Coll. Cardiol. 73 (14), 1795–1806. doi:10.1016/j.jacc.2018.12.087
Song, B., Scheuner, D., Ron, D., Pennathur, S., and Kaufman, R. J. (2008). Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 118 (10), 3378–3389. doi:10.1172/JCI34587
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D'Amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552 (7684), 187–193. doi:10.1038/nature25143
Sukhorukov, V. S., Voronkova, A. S., Baranich, T. I., Gofman, A. A., Brydun, A. V., Knyazeva, L. A., et al. (2022). Molecular mechanisms of interactions between mitochondria and the endoplasmic reticulum: A new look at how important cell functions are supported. Mol. Biol. 56 (1), 69–82. doi:10.31857/S0026898422010098
Sundaram, A., Plumb, R., Appathurai, S., and Mariappan, M. (2017). The Sec61 translocon limits IRE1α signaling during the unfolded protein response. Elife 6, e27187. doi:10.7554/eLife.27187
Tan, K., Fujimoto, M., Takii, R., Takaki, E., Hayashida, N., and Nakai, A. (2015). Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat. Commun. 6, 6580. doi:10.1038/ncomms7580
Teske, B. F., Fusakio, M. E., Zhou, D., Shan, J., McClintick, J. N., Kilberg, M. S., et al. (2013). CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell. 24 (15), 2477–2490. doi:10.1091/mbc.E13-01-0067
Theurey, P., and Rieusset, J. (2017). Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol. Metab. 28 (1), 32–45. doi:10.1016/j.tem.2016.09.002
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPR (mt). Cell. 165 (5), 1197–1208. doi:10.1016/j.cell.2016.04.011
Tirasophon, W., Lee, K., Callaghan, B., WelihindA, A., and Kaufman, R. J. (2000). The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes. Dev. 14 (21), 2725–2736. doi:10.1101/gad.839400
Toyofuku, T., Okamoto, Y., Ishikawa, T., Sasawatari, S., and Kumanogoh, A. (2020). LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 39 (2), e100875. doi:10.15252/embj.2018100875
Tyynismaa, H., Carroll, C. J., Raimundo, N., Ahola-Erkkila, S., Wenz, T., Ruhanen, H., et al. (2010). Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 19 (20), 3948–3958. doi:10.1093/hmg/ddq310
Vakifahmetoglu-Norberg, H., Ouchida, A. T., and Norberg, E. (2017). The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 482 (3), 426–431. doi:10.1016/j.bbrc.2016.11.088
Vance, J. E. (2014). MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 1841 (4), 595–609. doi:10.1016/j.bbalip.2013.11.014
Verfaillie, T., RubioN., , Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell. Death Differ. 19 (11), 1880–1891. doi:10.1038/cdd.2012.74
Verfaillie, T., van Vliet, A., Garg, A. D., Dewaele, M., Rubio, N., Gupta, S., et al. (2013). Pro-apoptotic signaling induced by photo-oxidative ER stress is amplified by Noxa, not Bim. Biochem. Biophys. Res. Commun. 438 (3), 500–506. doi:10.1016/j.bbrc.2013.07.107
Vila, G., Riedl, M., Anderwald, C., Resl, M., Handisurya, A., Clodi, M., et al. (2011). The relationship between insulin resistance and the cardiovascular biomarker growth differentiation factor-15 in obese patients. Clin. Chem. 57 (2), 309–316. doi:10.1373/clinchem.2010.153726
Waldherr, S. M., Strovas, T. J., Vadset, T. A., Liachko, N. F., and Kraemer, B. C. (2019). Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 10 (1), 4443. doi:10.1038/s41467-019-12070-3
Wang, S., and Kaufman, R. J. (2012). The impact of the unfolded protein response on human disease. J. Cell. Biol. 197 (7), 857–867. doi:10.1083/jcb.201110131
Wang, X., Pandey, A. K., Mulligan, M. K., Williams, E. G., Mozhui, K., Li, Z., et al. (2016). Joint mouse-human phenome-wide association to test gene function and disease risk. Nat. Commun. 7, 10464. doi:10.1038/ncomms10464
Wang, Y., Shen, J., AreNzaNaN., , Tirasophon, W., Kaufman, R. J., and PRywes, R. (2000). Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 275 (35), 27013–27020. doi:10.1074/jbc.M003322200
Wang, Y. T., Lim, Y., McCall, M. N., Huang, K. T., Haynes, C. M., Nehrke, K., et al. (2019). Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 317 (2), H472–H478. doi:10.1152/ajpheart.00244.2019
Wardelmann, K., Blumel, S., Rath, M., Alfine, E., Chudoba, C., Schell, M., et al. (2019). Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 21, 68–81. doi:10.1016/j.molmet.2019.01.001
West, M., Zurek, N., Hoenger, A., and Voeltz, G. K. (2011). A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell. Biol. 193 (2), 333–346. doi:10.1083/jcb.201011039
Wilkins, H. M., Weidling, I. W., Ji, Y., and Swerdlow, R. H. (2017). Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 8, 508. doi:10.3389/fimmu.2017.00508
Williams, K. W., Liu, T., Kong, X., Fukuda, M., Deng, Y., Berglund, E. D., et al. (2014). Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell. Metab. 20 (3), 471–482. doi:10.1016/j.cmet.2014.06.002
Won, J. C., Jang, P. G., Namkoong, C., Koh, E. H., Kim, S. K., Park, J. Y., et al. (2009). Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obes. (Silver Spring) 17 (10), 1861–1865. doi:10.1038/oby.2009.194
Wouters, B. G., and Koritzinsky, M. (2008). Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 8 (11), 851–864. doi:10.1038/nrc2501
Wu, G., Xiong, Q., Wei, X., Wang, Y., Hu, X., He, G., et al. (2019). Mitochondrial unfolded protein response gene CLPP changes mitochondrial dynamics and affects mitochondrial function. PeerJ 7, e7209. doi:10.7717/peerj.7209
Xie, F., Wu, D., Huang, S. F., Cao, J. G., Li, H. N., He, L., et al. (2017). The endoplasmic reticulum stress-autophagy pathway is involved in apelin-13-induced cardiomyocyte hypertrophy in vitro. Acta Pharmacol. Sin. 38 (12), 1589–1600. doi:10.1038/aps.2017.97
Xu, F., Du, W., Zou, Q., Wang, Y., Zhang, X., Xing, X., et al. (2021). COPII mitigates ER stress by promoting formation of ER whorls. Cell. Res. 31 (2), 141–156. doi:10.1038/s41422-020-00416-2
Yan, J. L., Jiang, J., He, L., and Chen, L. (2020). Mitochondrial superoxide/hydrogen peroxide: An emerging therapeutic target for metabolic diseases. Free Radic. Biol. Med. 152, 33–42. doi:10.1016/j.freeradbiomed.2020.02.029
Yang, J., Chen, W., Zhang, B., Tian, F., Zhou, Z., Liao, X., et al. (2018). Lon in maintaining mitochondrial and endoplasmic reticulum homeostasis. Arch. Toxicol. 92 (6), 1913–1923. doi:10.1007/s00204-018-2210-3
Ye, J., Rawson, R. B., KomuRo, R., Chen, X., Dave, U. P., PRywes, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 6 (6), 1355–1364. doi:10.1016/s1097-2765(00)00133-7
Yoneda, T., Benedetti, C., Urano, F., Clark, S. G., Harding, H. P., and Ron, D. (2004). Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell. Sci. 117, 4055–4066. doi:10.1242/jcs.01275
Yong, J., Parekh, V. S., Reilly, S. M., Nayak, J., Chen, Z., Lebeaupin, C., et al. (2021). Chop/Ddit3 depletion in β cells alleviates ER stress and corrects hepatic steatosis in mice. Sci. Transl. Med. 13 (604), eaba9796. doi:10.1126/scitranslmed.aba9796
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and Mori, K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273 (50), 33741–33749. doi:10.1074/jbc.273.50.33741
Yoshida, H., MaTsui, T., YAmAmoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 107 (7), 881–891. doi:10.1016/s0092-8674(01)00611-0
Zhang, P., Konja, D., Zhang, Y., and Wang, Y. (2021). Communications between mitochondria and endoplasmic reticulum in the regulation of metabolic homeostasis. Cells 10 (9), 2195. doi:10.3390/cells10092195
Zhang, Q., Wang, Z., Zhang, W., Wen, Q., Li, X., Zhou, J., et al. (2021). The memory of neuronal mitochondrial stress is inherited transgenerationally via elevated mitochondrial DNA levels. Nat. Cell. Biol. 23 (8), 870–880. doi:10.1038/s41556-021-00724-8
Zhang, Q., Wu, X., Chen, P., Liu, L., Xin, N., Tian, Y., et al. (2018). The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent wnt signaling. Cell. 174 (4), 870–883. e17. doi:10.1016/j.cell.2018.06.029
Zhang, X., Zhang, G., Zhang, H., Karin, M., Bai, H., and Cai, D. (2008). Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 135 (1), 61–73. doi:10.1016/j.cell.2008.07.043
Zhao, Q., Wang, J., Levichkin, I. V., Stasinopoulos, S., Ryan, M. T., and Hoogenraad, N. J. (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21 (17), 4411–4419. doi:10.1093/emboj/cdf445
Zhou, D., Palam, L. R., Jiang, L., Narasimhan, J., Staschke, K. A., and Wek, R. C. (2008). Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 283 (11), 7064–7073. doi:10.1074/jbc.M708530200
Keywords: UPRmt, UPR, unfolded protein response, T2D, PERK (PKR-like endoplasmic reticulum kinase), mitochondia
Citation: Kang Z, Chen F, Wu W, Liu R, Chen T and Xu F (2022) UPRmt and coordinated UPRER in type 2 diabetes. Front. Cell Dev. Biol. 10:974083. doi: 10.3389/fcell.2022.974083
Received: 20 June 2022; Accepted: 29 August 2022;
Published: 16 September 2022.
Edited by:
Kanchan Phadwal, University of Edinburgh, United KingdomReviewed by:
Felipe Cabral-Miranda, Federal University of Rio de Janeiro, BrazilCopyright © 2022 Kang, Chen, Wu, Liu, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Xu, ZmFuZ3h1QGd6aG11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.