 Grant Pfundstein
Grant Pfundstein Alexander G. Nikonenko
Alexander G. Nikonenko Vladimir Sytnyk
Vladimir Sytnyk
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 26 July 2022
Sec. Cell Adhesion and Migration
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.969547
This article is part of the Research Topic Editors' Showcase 2021/2022: Insights in Cell Adhesion and Migration View all 6 articles
Alzheimer’s disease (AD) is an incurable neurodegenerative disorder in which dysfunction and loss of synapses and neurons lead to cognitive impairment and death. Accumulation and aggregation of neurotoxic amyloid-β (Aβ) peptides generated via amyloidogenic processing of amyloid precursor protein (APP) is considered to play a central role in the disease etiology. APP interacts with cell adhesion molecules, which influence the normal physiological functions of APP, its amyloidogenic and non-amyloidogenic processing, and formation of Aβ aggregates. These cell surface glycoproteins also mediate attachment of Aβ to the neuronal cell surface and induce intracellular signaling contributing to Aβ toxicity. In this review, we discuss the current knowledge surrounding the interactions of cell adhesion molecules with APP and Aβ and analyze the evidence of the critical role these proteins play in regulating the processing and physiological function of APP as well as Aβ toxicity. This is a necessary piece of the complex AD puzzle, which we should understand in order to develop safe and effective therapeutic interventions for AD.
Alzheimer’s disease (AD) is an incurable neurodegenerative disorder in which progressive synapse loss and neuronal dysfunction are followed by neuronal death in the brain. People with AD suffer from severe cognitive impairments and eventually die as a result of the disease. AD is the seventh leading cause of death in the United States (Alzheimer’s Association, 2022). Its global economic burden, together with other dementias, is estimated to be nearly $1 trillion (USD) per annum (Prince et al., 2015). Despite decades of research, effective therapeutic interventions for AD remain elusive, presenting a considerable problem given the increasingly aging population of our world today.
The amyloid hypothesis has been central to our understanding of AD for the past three decades. It proposes that aggregates of the amyloid-β (Aβ) peptide, the primary component of senile plaques formed in brains of people with AD, are the main causative agents in AD pathogenesis (Hardy and Allsop, 1991; Selkoe, 1991; Hardy and Higgins, 1992; Selkoe and Hardy, 2016; Karran and De Strooper, 2022). These Aβ aggregates bind to cell surface receptors on neurons and glial cells, inducing synaptic dysfunction and neuroinflammation, triggering aberrant intracellular signaling cascades that lead to hyperphosphorylation of the microtubule associated protein tau, causing disruption of axonal transport, oxidative damage, breakdown of homeostasis, and resulting in neurotoxicity (LaFerla et al., 1995; Sakono and Zako, 2010; Xia et al., 2016; Yin et al., 2017; Sushma and Mondal, 2019). The gradual loss of synapses and neurons is responsible for cognitive impairment, memory loss, and, eventually, death.
Aβ peptides are generated via the proteolysis of amyloid precursor protein (APP). APP is a type I transmembrane glycoprotein with a large N-terminal extracellular domain, transmembrane region, and short intracellular tail (Figure 1A). APP is encoded by a single gene on chromosome 21, and alternative splicing generates three main isoforms, APP695, APP751, and APP770, denoted by their amino acid length. The extracellular part of all isoforms is comprised of two rigidly folded domains, E1 and E2, joined by a flexible acidic domain (AcD) and connected to the transmembrane region by a mostly unstructured juxtamembrane linker (Coburger et al., 2013; Coburger et al., 2014) (Figure 1A). E1 contains a heparin-binding domain (HBD) within a larger growth factor-like domain (GFLD), as well as a copper/zinc-binding domain (CuBD). E2 contains the second HBD and CuBD (Dahms et al., 2012). The extracellular domain mediates homophilic trans-interactions, where an APP molecule on one cell binds to an APP molecule on an adjacent cell, allowing APP to function as a cell adhesion molecule (Soba et al., 2005; Stahl et al., 2014). The extracellular domain also binds other ligands such as epidermal growth factor (EGF) (da Rocha et al., 2021) and reelin (Hoe et al., 2009), and acts as a ligand itself after being shed into the extracellular space where it interacts with other receptors (Caillé et al., 2004). The single-pass transmembrane domain is involved in cholesterol binding (Barrett et al., 2012), while the short intracellular tail contains a YENPTY motif that binds cytoplasmic adaptor proteins such as Dab1 and Mint, which mediate APP-dependent signaling (Borg et al., 1996; King et al., 2003; Schettini et al., 2010). The Kunitz protease inhibitor (KPI) domain is present in the two longer isoforms, APP751 and APP770, and the OX-2 sequence is contained in APP770 (Tanzi et al., 1987; Sandbrink et al., 1996). APP is widely expressed in many tissues. APP695 is the major neuronal isoform, while APP751 and APP770 are highly expressed in non-neuronal cells (Rohan de Silva et al., 1997).
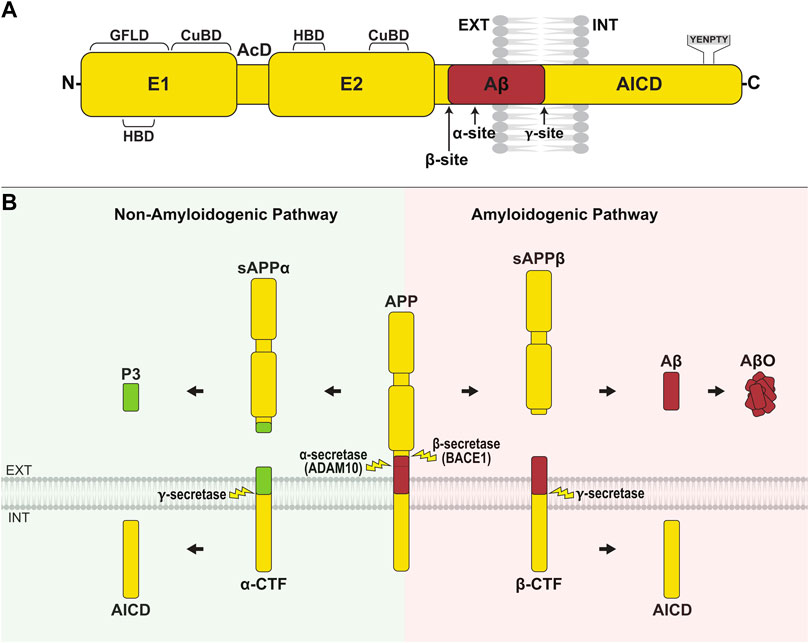
FIGURE 1. APP structure and processing. (A) APP is composed of a large N-terminal extracellular domain, transmembrane region, and short cytoplasmic tail. The extracellular domain comprises two rigidly folded regions, E1 and E2, joined by an acidic domain (AcD). E1 contains a heparin-binding domain (HBD) within a larger growth factor-like domain (GFLD), and a copper/zinc-binding domain (CuBD). E2 comprises the second HBD and CuBD. The juxtamembrane region contains the α- and β-cleavage sites, while the γ-cleavage site is located within the transmembrane domain. The C-terminal intracellular domain (AICD) contains the YENPTY sequence, which binds cytosolic adaptor proteins. (B) APP is primarily processed along two opposing pathways. In amyloidogenic processing, APP is cleaved by β-secretase (BACE1) at the N-terminus of Aβ, generating sAPPβ and the membrane-bound β-CTF. Subsequent γ-secretase cleavage of β-CTF releases the Aβ peptide into the extracellular/lumenal space and AICD into the cytosol. Aβ peptides aggregate and form oligomers (AβO). In non-amyloidogenic processing, APP is cleaved by α-secretase (ADAM10) within the Aβ region, producing sAPPα and α-CTF. Ensuing cleavage of α-CTF by γ-secretase liberates P3 into the extracellular/lumenal space and AICD into the cytosol.
APP undergoes complex proteolytic processing, yielding a number of biologically active fragments. Processing along the amyloidogenic pathway is initiated by β-site APP cleaving enzyme 1 (BACE1), which cleaves APP at the amino terminus of the Aβ region, producing sAPPβ and the membrane-bound β-C-terminal fragment, β-CTF (Vassar et al., 1999; Yan et al., 1999; Zhang et al., 2011) (Figure 1B). In the non-amyloidogenic pathway, α-secretase (ADAM10; a disintegrin and metalloproteinase 10) cleaves APP between Lys 16 and Leu 17 of the Aβ region, preventing the formation of Aβ and releasing sAPPα and α-CTF instead (Lammich et al., 1999; Asai et al., 2003; Kuhn et al., 2010). The cell surface or trans-Golgi network accumulation of APP favors non-amyloidogenic processing (Parvathy et al., 1999; Tan and Gleeson, 2019), whereas the retention of APP in endocytic compartments promotes amyloidogenic processing (Das et al., 2016). In both pathways, γ-secretase, a complex composed of presenilin 1 (PS1), nicastrin, anterior pharynx-defective 1 (APH-1), and presenilin enhancer 2 (PEN-2), cleaves the membrane-tethered CTFs yielding Aβ from β-CTF and P3 from α-CTF, as well as the APP intracellular domain (AICD) (Figure 1B). In the amyloidogenic pathway, γ-secretase produces peptides of varying length with Aβ40, denoted by amino acid length, being most abundant and Aβ42 being most aggregation prone (Jarrett et al., 1993; Hamley, 2012; Chen et al., 2017). A shift towards the generation of Aβ42 over Aβ40, or an imbalance in the overall production and clearance of Aβ peptides predisposes to the formation of neurotoxic oligomers (AβO), fibrils (AβF) and plaques (Masters et al., 1985; Bharadwaj et al., 2009; Mawuenyega et al., 2010). In addition, there are several non-canonical pathways through which APP can be processed, including proteolysis by η-secretase, δ-secretase, meprin, and caspases (Andrew et al., 2016).
APP is a member of a small family of proteins consisting of APP and APP-like proteins 1 and 2 (APLP1 and APLP2) (Heber et al., 2000; Walsh et al., 2007). Although structurally very similar to APP, APLPs lack the Aβ region and are thus non-amyloidogenic. As a whole, members of the APP family share poorly understood roles in synaptic plasticity, synaptogenesis, neurite outgrowth, learning, and memory (Small et al., 1994; Müller and Zheng, 2012; Klevanski et al., 2015).
Many therapeutic interventions have targeted Aβ and the proteases responsible for its generation, however thus far, all attempts have failed to demonstrate reasonable efficacy and are associated with worsening cognition and other side effects (Kumar et al., 2018; Hampel et al., 2021; Karran and De Strooper, 2022). These failures can be partly attributed to an incomplete understanding of the complex cell biology underlying: 1) the processing of APP, 2) the normal physiological function of APP and its proteases, and 3) the mechanisms of Aβ-induced toxicity. A class of proteins intimately involved in these poorly understood processes are cell adhesion molecules (CAMs), a vast category of cell surface proteins that mediate adhesion of cells to one another and to the extracellular matrix (ECM). These broadly include CAMs of the immunoglobulin superfamily (IgSF), integrins, cadherins, selectins, and other uncategorized proteins that possess adhesive function, including APP itself. CAMs are of particular interest in amyloid-dependent AD pathology as their structure and cell surface localization make them well-suited for interactions with both APP and Aβ, many of which have been found to date. Multiple gene ontological analyses have also described cell adhesion as an affected pathway in AD (Blalock et al., 2004; Cui et al., 2018; Wang and Li, 2021; Shu et al., 2022).
Thus, in this review, we summarize the current knowledge surrounding the interactions of CAMs with APP and Aβ and how CAMs influence the processing and physiological function of APP as well as their role in Aβ toxicity. This breadth of knowledge is a necessary piece of the complex AD puzzle, which we should better understand to enable the development of safe and effective therapeutics for the treatment and prevention of AD.
As APP family proteins are themselves CAMs, we first discuss the role that homo- and hetero-dimerization of the members of this family plays in APP processing, functioning, and Aβ toxicity.
APP interacts with itself forming homodimers in the mouse brain (Schmidt et al., 2012; Herr et al., 2017) and numerous cell lines, with at least four regions of this molecule, namely the GFLD of E1 (Kaden et al., 2009), CuBD of E1 (Noda et al., 2013), E2 (Wang and Ha, 2004), and TMD (Munter et al., 2007; Sato et al., 2009), being engaged in dimer formation (Figure 2A and Table 1). The impact of dimerization on APP processing is complex and remains controversial. Several reports suggest that APP dimerization modulates the balance between its α- and β-cleavage. For example, inhibition of APP dimerization using an APP-E1-derived peptide leads to an increase in sAPPα levels and reduction in sAPPβ and Aβ levels in SH-SY5Y cells (Kaden et al., 2008). Similarly, small molecule inhibitors of APP dimerization reduce sAPPβ and Aβ levels in HEK293 cells without affecting Aβ42/40 ratio (So et al., 2012), as well as favoring sAPPα over sAPPβ production in CHO and B103 cell lines (Libeu et al., 2012). Together, these findings suggest that APP dimerization promotes BACE1-mediated cleavage of APP. However, other studies report that APP dimerization in CHO cells leads to an increase in sAPPα levels and decrease in sAPPβ levels (Decock et al., 2015), while statin-induced TMD dimerization in iPSC-derived neurons reduces both sAPPα and sAPPβ levels, and decreases production of Aβ (Langness et al., 2021). In addition, a number of studies suggest that APP dimerization influences its γ-secretase-mediated cleavage. For example, mutationally induced dimerization of the juxtamembrane region of APP leads to an increase in Aβ levels without affecting sAPPα or sAPPβ levels, suggesting that the γ-secretase cleavage efficiency of APP-CTFs may be influenced by APP dimerization (Scheuermann et al., 2001). The preferred γ-cleavage site within APP may also be affected by the mode of dimerization, as copper-induced APP dimerization via the CuBD of E1 favors production of Aβ40 over Aβ42 while TMD dimerization promotes generation of Aβ42 over Aβ40 (Munter et al., 2007; Noda et al., 2013). It has also been suggested that juxtamembrane domain/TMD dimerization may predispose Aβ to form dimers and oligomers, which may in turn affect the levels and toxicity of Aβ (Scheuermann et al., 2001). A possible explanation for these controversies may lie in the fact that various regions of APP contribute to dimerization, and it is quite probable that variations in the contact sites involved may influence APP processing in different ways.
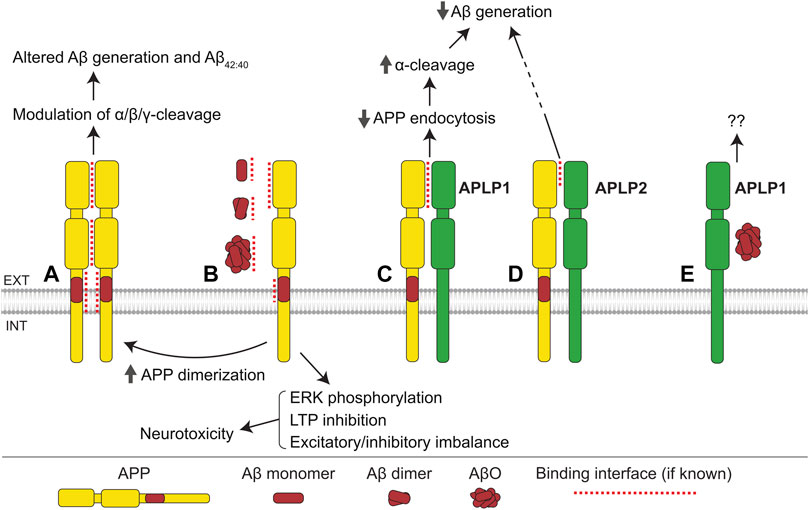
FIGURE 2. APP family interactions. (A) Interactions between E1, E2, and transmembrane domains of APP mediate formation of homodimers. Homodimerization influences the α-, β-, and γ-cleavage of APP, Aβ generation, and the ratio of Aβ42/40. (B) Monomers, dimers, and oligomers of Aβ bind to the E1 domain and cognate Aβ region of APP. The interaction with Aβ monomers and dimers promotes APP homodimerization, while the APP-AβO interaction induces ERK phosphorylation, inhibits long-term potentiation (LTP), and leads to excitatory/inhibitory imbalance, ultimately resulting in neurotoxicity. (C) APLP1 interacts with the E1 domain of APP, suppressing APP endocytosis, increasing cell surface levels of APP, and thereby facilitating the α-cleavage of APP, and consequently reducing Aβ generation. (D) APLP2 interacts with APP and reduces Aβ generation via an unknown mechanism. (E) APLP1 interacts with AβOs with unknown consequence.
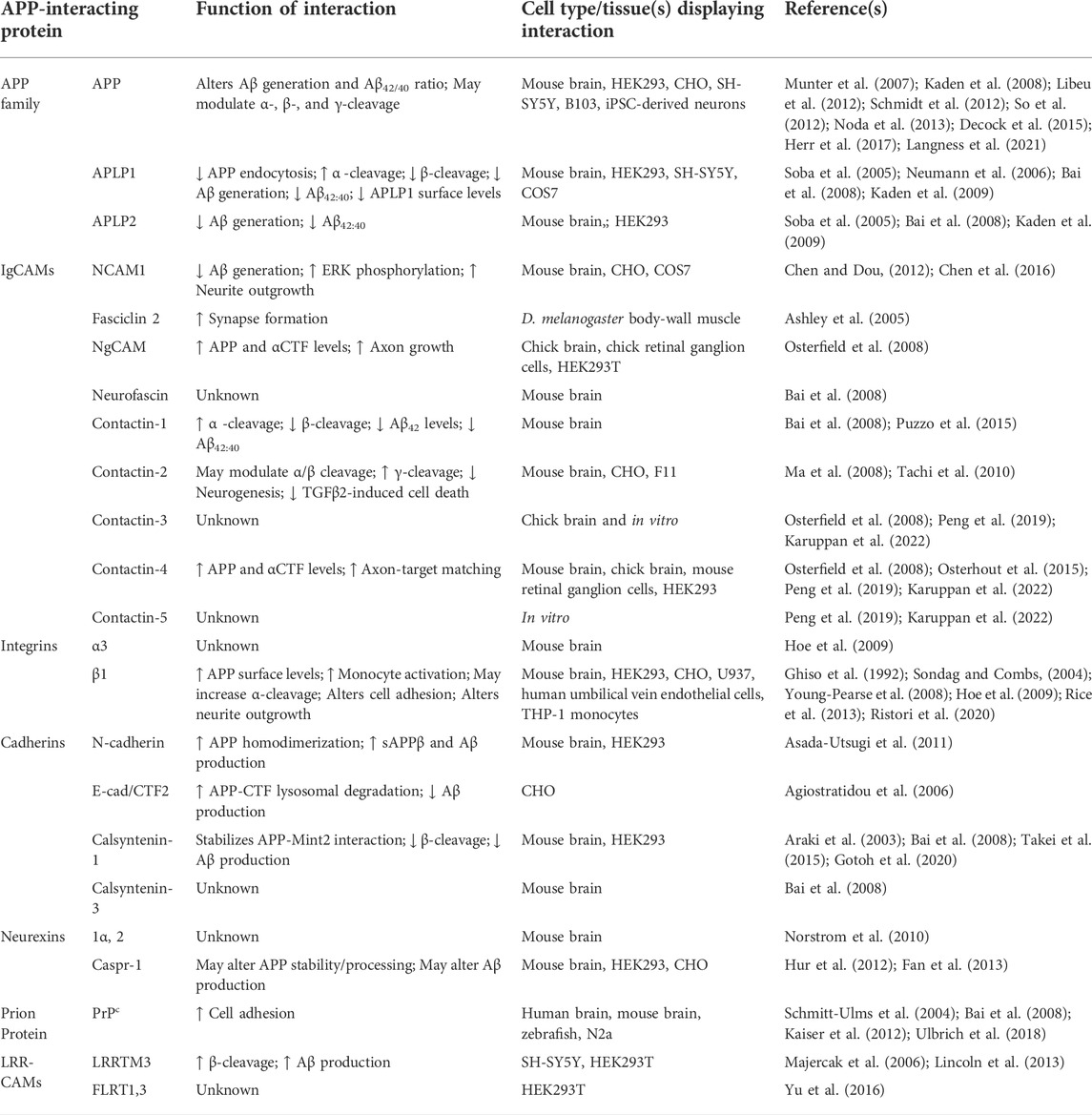
TABLE 1. APP-interacting cell adhesion molecules and their role in APP processing and physiological function.
APP interacts with APLP1 in the mouse brain (Soba et al., 2005; Bai et al., 2008). This interaction is mediated partially by the E1 domains of APP and APLP1 (Figure 2C), but other regions are suggested to be also involved (Kaden et al., 2009). While APP is mostly found intracellularly, APLP1, in contrast, is predominantly localized at the cell surface and has a much slower rate of endocytosis compared to APP (Kaden et al., 2009; Schilling et al., 2017). Accordingly, APP-APLP1 interactions are reported to suppress endocytosis of APP, increasing its α-cleavage and reducing its β-cleavage in HEK293 cells (Neumann et al., 2006). In agreement, the co-expression of APP and APLP1 in SH-SY5Y cells results in reduced Aβ generation compared to cells transfected with APP only (Kaden et al., 2009). APP also influences the subcellular distribution of APLP1, reducing its levels at the cell surface (Kaden et al., 2009). Given the emerging role of APLP1 as the primary cell adhesion molecule of the APP family (Mayer et al., 2016; Dunsing et al., 2017; Schilling et al., 2017; Onodera et al., 2021), these effects of APP on APLP1 distribution provide evidence supporting the involvement of APP in cell adhesion regulation. In the mouse brain, APP also binds to APLP2 via its GFLD of E1 (Soba et al., 2005; Kaden et al., 2009). Similarly to APLP1, APLP2 reduces Aβ generation in HEK293 cells when it is co-expressed with APP (Kaden et al., 2009) (Figure 2D). APLPs preferentially reduce Aβ42, rather than Aβ40 levels, suggesting that APP/APLP interactions affect the γ-secretase-mediated cleavage of APP-CTFs (Kaden et al., 2009).
APP interacts with Aβ monomers, oligomers, and fibrils, acting as a receptor for Aβ, which mediates Aβ-induced toxicity (Figure 2B and Table 2). Soluble Aβ was found to bind to the Aβ region of APP at the cell surface thereby inducing cell death in N2a cells (Shaked et al., 2006). This effect was dependent on the YENPTY motif in the APP intracellular domain, which interacts with a number of cytoplasmic proteins involved in intracellular signaling, suggesting that Aβ induces intracellular signaling pathways via APP. Consistent with this idea, AβO treatment of B103 cells increases Ras levels and ERK phosphorylation, both of which are dependent on APP expression (Kirouac et al., 2017). ERK induces hyperphosphorylation of tau and thereby can mediate the APP-dependent AβO toxicity (Guise et al., 2001; Siano et al., 2019). In APP−/− mice, AβO binding to synapses is reduced and AβO effects on long-term potentiation (LTP) and the balance of excitatory/inhibitory activity are attenuated (Wang et al., 2017). Cultured APP−/− neurons are less vulnerable to Aβ-induced toxicity compared to wild-type neurons (Lorenzo et al., 2000). Aβ monomers and dimers also bind to the E1 domain of APP, increasing APP homodimerization and influencing neurotransmitter release probability (Fogel et al., 2014). In addition, AβFs bind to APP leading to an increase in APP levels at the cell surface, thereby facilitating the binding of Aβ to APP (Lorenzo et al., 2000; Heredia et al., 2004). APP-Aβ interactions promote homodimerization of APP, which may in turn stimulate amyloidogenic processing of APP to a greater extent (Fogel et al., 2014). APLP1 has also been identified as a binding partner of AβO, however, the functional significance of this interaction remains to be determined (Figure 2E).
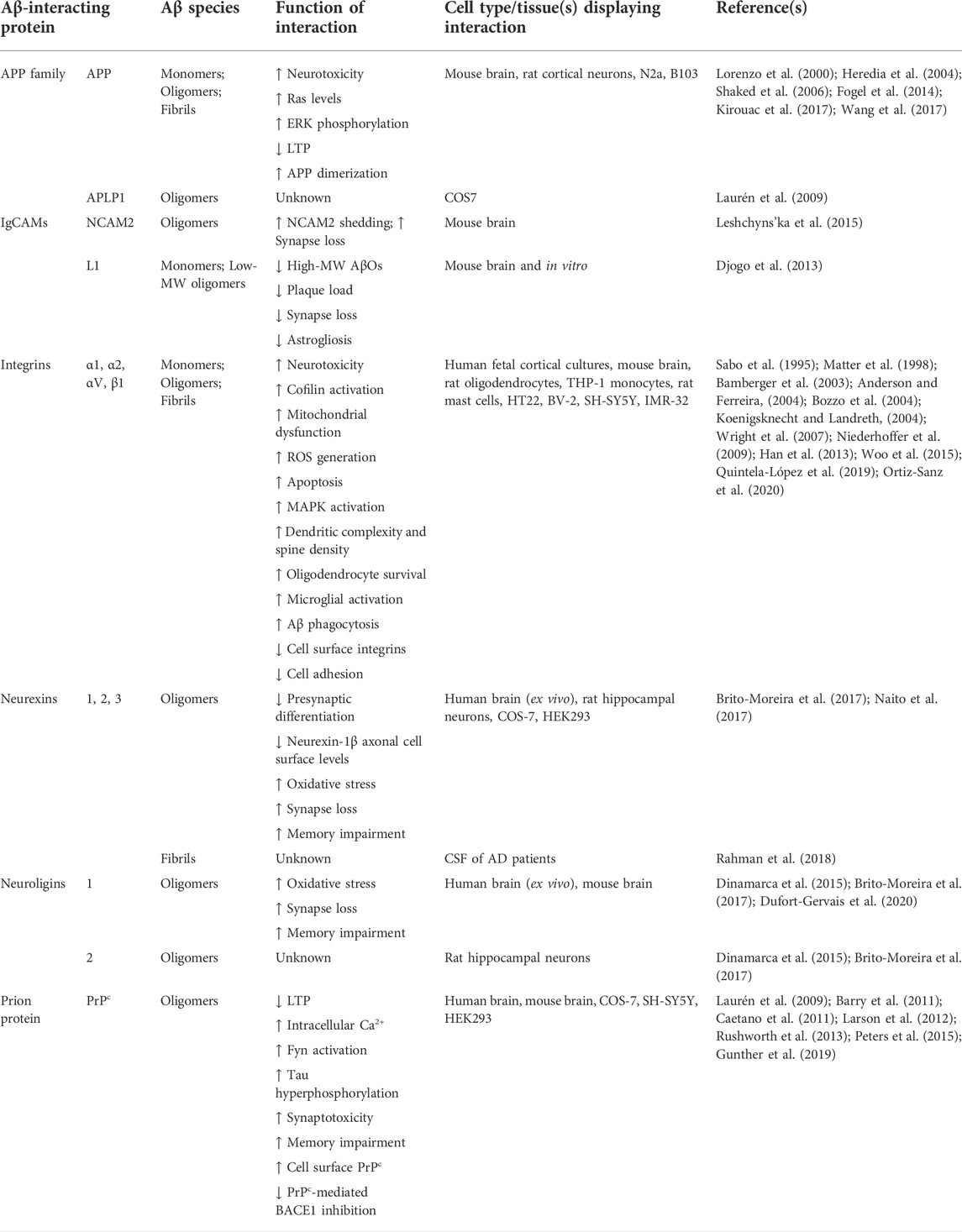
TABLE 2. Aβ-interacting cell adhesion molecules and their role in Aβ toxicity.
Members of the immunoglobulin superfamily (IgSF) of CAMs are plasma membrane-attached glycoproteins characterized by the presence of immunoglobulin-like (Ig) repeats within their extracellular domains. IgSF CAMs mediate calcium-independent homophilic adhesion between cells where identical molecules on membranes of adjacent cells bind to each other. IgSF CAMs also heterophilically interact in cis with a number of other cell surface receptors located within the same membrane or bind in trans to the cell surface receptors on membranes of other cells. These proteins play important roles in regulating neuronal development and synaptic functions (Maness and Schachner, 2007; Sytnyk et al., 2017) and have been implicated in AD (Leshchyns'ka and Sytnyk, 2016). The role that APP and Aβ play in regulating the functions of these proteins and effects of these proteins on the amyloidogenic processing of APP and Aβ toxicity are reviewed below.
NCAMs belong to a sub-family of CAMs within the IgSF. The group consists of NCAM1 (originally designated NCAM) and the lesser known NCAM2 (also designated OCAM) (Winther et al., 2012; Weledji and Assob, 2014). NCAM1 and NCAM2 are structurally similar, being composed of five N-terminal Ig domains (IgI-V) and two fibronectin type III (Fn3) repeats (Fn3I-II) (Figure 3A). Alternative splicing generates three major NCAM1 isoforms, which have identical extracellular domains and differ in the membrane attachment. Two longer isoforms of NCAM1, designated NCAM140 and NCAM180 according to their molecular weight, are transmembrane proteins with a longer intracellular tail in NCAM180. The shortest NCAM1 isoform, designated NCAM120, is a glycosylphosphatidylinositol (GPI)-anchored protein lacking the intracellular domain. Two major NCAM2 isoforms also have identical extracellular domains. The longer NCAM2 isoform is a transmembrane protein, whereas the shorter isoform is GPI-anchored to the plasma membrane. NCAMs are expressed in many tissues but are particularly enriched in the brain, where they participate in the regulation of neurite outgrowth, synaptogenesis and synaptic plasticity (Sytnyk et al., 2017; Rasmussen et al., 2018).
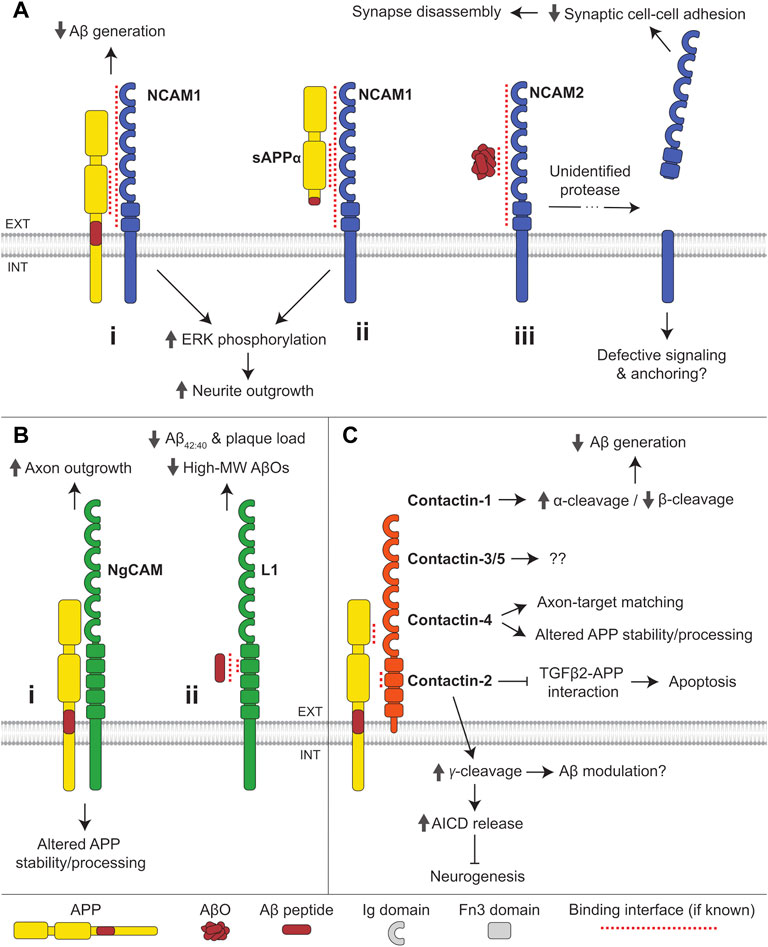
FIGURE 3. Interactions of IgSF CAMs with APP and Aβ. (A) (i) NCAM1 interacts with the E2 domain of APP and reduces Aβ generation. Binding of APP or (ii) sAPPα to NCAM1 induces ERK phosphorylation and promotes neurite outgrowth. (iii) AβOs bind to NCAM2 triggering shedding of its extracellular domain by an unidentified protease, causing synapse disassembly. (B) (i) The chicken ortholog of L1, NgCAM, interacts with APP and regulates APP stability and processing. Binding of APP to NgCAM promotes axon outgrowth in retinal ganglion cells. (ii) Aβ peptides bind to the second Fn3 domain of L1. L1 reduces Aβ42:40 ratio and the formation of high molecular-weight (MW) AβOs in favor of monomers, dimers, and tetramers. (C) APP interacts with all contactin family members except for contactin-6. The E1 CuBD of APP binds to the second Fn3 domain of contactin. Contactin-1 promotes α-cleavage and reduces β-cleavage of APP, thereby reducing Aβ generation. Contactin-2 promotes γ-cleavage of APP, thereby increasing AICD release and inhibiting neurogenesis. Contactin-2 also inhibits the binding of APP to TGFβ2 and reduces apoptosis induced by APP-TGFβ2 interactions. Contactin-4 regulates APP stability and processing and is required for APP-dependent axon-target matching. The function of APP interactions with contactin-3 and -5 remains undetermined.
NCAM1 forms a complex with APP in the mouse brain, with the E2 domain of APP and the extracellular domain of NCAM1 mediating this interaction (Chen and Dou, 2012) (Figure 3Ai). NCAM180, a splice variant of NCAM1 with a longer cytoplasmic tail, does not, however, associate with APP, suggesting that the APP/NCAM1 complex formation may also be dependent on an additional intracellular mechanism. The interaction of APP with NCAM1 is conserved in Drosophila melanogaster, where the APP homolog, APPL, binds to the NCAM ortholog Fasciclin 2 (Ashley et al., 2005; Mao and Freeman, 2009). Human APP was also found in a complex with NCAM2 in the brain of transgenic APP23 mice (Leshchyns’ka et al., 2015).
APP and NCAM1 trigger the mitogen-activated protein kinase (MAPK) pathway via phosphorylation of ERK1 and ERK2. Co-expression of NCAM1 and APP in COS7 cells promotes phosphorylation of ERK1,2 to a larger extent than found in cells expressing either protein alone (Chen and Dou, 2012) (Figure 3Ai,ii). This synergistic effect requires the extracellular, but not the intracellular domain of APP. Both APP and NCAM1 independently promote neurite outgrowth in mouse hippocampal neurons, and when present together they increase neurite outgrowth synergistically (Chen et al., 2016). The fact that the latter effect can be induced by secreted sAPPα is consistent with a non-essential role of the intracellular domain of APP, altogether indicative of a ligand-receptor interaction between APP and NCAM1 (Figure 3Aii).
The role of the interaction between NCAM and APP in synaptogenesis was shown in Drosophila, where Fasciclin 2 promotes synapse formation by interacting with APPL, which initiates signaling via the adaptor protein Mint1 (Ashley et al., 2005).
In AD, NCAM1 levels are reduced in the frontal and temporal cortex, while levels of proteolytic NCAM1 fragments in the serum are increased (Todaro et al., 2004; Aisa et al., 2010). The levels of NCAM1 carrying polysialic acid, an unusual carbohydrate predominantly found on NCAM1, are similarly diminished in the entorhinal cortex in AD-affected brains inversely correlating with hyperphosphorylated tau load (Murray et al., 2016). The levels of polysialylated NCAM1 are however increased in the AD hippocampus (Mikkonen et al., 1999).
NCAM2 is enriched in the human hippocampus, a brain region highly susceptible to Aβ-induced toxicity. In brains of people with AD, NCAM2 levels are reduced in hippocampal synapses and the levels of soluble NCAM2 are elevated suggesting increased shedding of NCAM2 from synaptic membranes (Leshchyns’ka et al., 2015). The overall levels of NCAM2 and the levels of its phosphorylation are however increased in AD brains (Leshchyns’ka et al., 2015; Sathe et al., 2020). Together these data suggest that NCAMs play a role in AD.
Co-expression of NCAM1 with APP in CHO cells reduces production of both Aβ40 and Aβ42, suggesting that NCAM1 modulates APP processing (Chen and Dou, 2012) (Figure 3Ai). The mechanism underlying this effect is currently unknown.
A single nucleotide polymorphism (SNP) in NCAM2 (rs2212624) is associated with the development of late-onset AD (Kimura et al., 2006), while another SNP in NCAM2 (rs1022442) is associated with high Aβ levels in the cerebrospinal fluid (CSF) (Han et al., 2010), together suggesting that NCAM2 may be implicated in Aβ-dependent AD pathology. The association between SNPs in NCAM2 and the levels of Aβ in CSF (Han et al., 2010) suggests that NCAM2 may influence Aβ production, presumably by interacting with APP.
A peptide derived from the fibroblast growth factor receptor (FGFR)-binding region of NCAM1 within the Fn3II domain prevents neurodegeneration and cognitive impairment in AβO-treated rats (Klementiev et al., 2007). The peptide mimics NCAM1 functions by binding to and activating FGFR (Neiiendam et al., 2004). Together these data suggest that the loss of NCAM1-FGFR interactions and attenuation of NCAM1-dependent signaling contribute to Aβ toxicity.
NCAM2 interacts with AβOs in brains of transgenic APP23 mice, and AβOs increase shedding of synaptic NCAM2 in cultured hippocampal neurons, compromising synaptic adhesion and inducing synapse disassembly (Leshchyns’ka et al., 2015) (Figure 3Aiii).
The L1 family is a group of IgSF CAMs that includes L1, close homologue of L1 (CHL1), neurofascin and NgCAM-related CAM (NrCAM). L1 is a transmembrane protein with a short cytoplasmic tail and extracellular domain composed of six Ig-like domains and five Fn3 repeats (Figure 3B). L1 is expressed in a variety of tissues, but is enriched in the nervous system, where it plays a role in synaptogenesis, neurite outgrowth, and neuromuscular junction stability (Sytnyk et al., 2017).
APP interacts with neuronal-glial CAM (NgCAM), the chicken ortholog of L1, in the chick brain, and APP and sAPPα enhance NgCAM-dependent axon growth in retinal ganglion cells (Osterfield et al., 2008) (Figure 3Bi). In the mouse brain, APP interacts with another L1 family member, neurofascin (Bai et al., 2008). The functional role of this interaction remains to be investigated.
In the CSF of AD patients, the levels of L1 proteolytic fragments are increased (Strekalova et al., 2006), while neurofascin levels are reduced (Brinkmalm et al., 2018).
The levels of full-length APP and αCTF are elevated in HEK293T cells co-expressing NgCAM, suggesting a role for the latter in modulation of APP stability and/or processing (Osterfield et al., 2008) (Figure 3Bi).
Aβ42 peptides bind to L1 in vitro via its second Fn3 domain (Djogo et al., 2013) (Figure 3Bii). This interaction reduces the formation of high-molecular weight (MW) forms of AβOs, with a corresponding increase in levels of Aβ monomers, trimers, and tetramers. Hippocampal L1 levels are reduced in aged APPswe mice, a mouse model of AD overexpressing human APP with a Swedish (KM670/671NL) mutation, which exhibits Aβ deposition with age (Hu et al., 2022). In APP/PS1 mice, a mouse AD model co-expressing mutated human APP with a Swedish mutation and mutated presenilin 1, overexpression of L1 using adeno-associated viruses results in a reduced Aβ plaque load and Aβ42/40 ratio, as well as milder hippocampal synapse loss and astrogliosis (Djogo et al., 2013). A 70 kDa fragment of L1 (L1-70) generated by proteolysis of L1 by serine proteases also reduces Aβ load in mice. This occurs via translocation of L1-70 to the nucleus inducing cytokine expression and the clearance of Aβ plaques by activated microglia (Hu et al., 2022). While microglial activation is important for Aβ clearance, in prolonged or extreme form it results in inflammation exacerbating neuronal damage in AD (Heneka et al., 2015).
Together, these findings allude to a protective role for L1 in AD via the prevention of aggregation and promotion of Aβ clearance.
Contactins are a family of CAMs within the IgSF comprising six members, including contactin-1 (also designated F3), contactin-2 (also named TAG1 or TAX1), contactin-3 (also named BIG-1 or PANG), contactin-4 (also designated BIG-2), contactin-5 (also named NB-2), and contactin-6 (also designated NB-3). Contactins are composed of six Ig domains and four Fn3 domains attached to plasma membrane via a GPI anchor (Figure 3C). They are primarily expressed in the brain where they accumulate in axons, contributing to control of axon growth and guidance, as well as to other functions distinct for each family member (Shimoda and Watanabe, 2009; Gennarini et al., 2017; Chatterjee et al., 2019).
In the mouse brain, APP was shown to interact with contactin-1 (Bai et al., 2008; Puzzo et al., 2015), contactin-2 (Ma et al., 2008), and contactin-4 (Osterhout et al., 2015). Contactin-3 and -4 were demonstrated to interact with APP in the chick brain (Osterfield et al., 2008). In vitro assays show that contactin-3 and -4 bind to APP with the highest affinity amongst the contactin family via a conserved interaction interface between the E1 CuBD of APP and second Fn3 domain of Contactin-3 and -4 (Peng et al., 2019; Karuppan et al., 2022) (Figure 3C). Contactin-5 also binds to APP via this interface, suggesting the binding site is likely to be similar for all contactins (Karuppan et al., 2022). Contactin-1 and -2 show little to no binding to APP in these in vitro assays, suggesting additional factors may mediate such interactions in the brain. Contactin-6 does not bind to APP in vitro, and this interaction has not thus far been identified in vivo, potentially making contactin-6 unique within the contactin family.
The interaction of contactin-4 with APP is required for axon-target matching in mouse retinal ganglion cells, regulating the circuitry involved in vision stabilization (Osterhout et al., 2015) (Figure 3C). The physiological role of the interactions between other contactins and APP is yet to be determined.
In mice, contactin-1 expression in the hippocampus decreases with age being associated with age-dependent cognitive decline (Shimazaki et al., 1998; Puzzo et al., 2015). Single nucleotide polymorphisms in the contactin-2 coding gene are associated with late-onset AD and contactin-2 CSF levels are reduced in AD and correlate with Aβ40 and hyperphosphorylated tau levels (Medway et al., 2010; Chatterjee et al., 2018). The levels of contactin-2 also decrease with age in mice (Tachi et al., 2010). The chromosomal region encompassing the contactin-4 coding gene was found to have suggestive linkage to late-onset AD (Blacker et al., 2003; Bamford et al., 2020).
Contactin-1 is suggested to modulate the α-/β-cleavage of APP as aged transgenic mice overexpressing contactin-1 display reduced sAPPβ and increased sAPPα levels compared to similarly aged wild-type mice (Puzzo et al., 2015) (Figure 3C). The levels of Aβ42 are decreased, while the levels of Aβ40 remain unchanged in these transgenic mice, suggesting that the preferred γ-cleavage site may also be influenced by contactin-1 overexpression. Contactin-2 may similarly influence the α-/β-cleavage of APP as mouse embryonic fibroblasts overexpressing contactin-2 show increased production of both α- and β-CTFs, with a more prominent rise in α-CTF levels (Ma et al., 2008). However, contactin-2 does not affect sAPPα or sAPPβ levels in HEK293 cells or cultured cortical neurons (Rice et al., 2013). Contactin-2 promotes the γ-cleavage of APP-CTFs as demonstrated by a decrease in AICD release in CNTN2−/− embryonic mouse brains (Ma et al., 2008). This contactin-2-dependent γ-cleavage and release of AICD initiates a signaling pathway that inhibits neurogenesis (Figure 3C). Contactin-2 is also suggested to suppress neuronal cell death, as it competitively inhibits the binding of transforming growth factor β2 (TGFβ2) to APP (Tachi et al., 2010), an interaction known to induce neuronal apoptosis (Hashimoto et al., 2005). In HEK293 cells, co-expression of contactin-4 increases the levels of full-length APP and αCTF suggesting that contactin-4 also regulates the processing of APP (Osterfield et al., 2008) (Figure 3C).
Together, these data suggest that at least some members of the contactin family regulate the function and processing of APP, and that the age-related reduction in contactin expression may contribute to the shift towards amyloidogenic processing that leads to AD.
The small GPI-anchored CAM Thy-1 has also been identified as a binding partner of APP (Bai et al., 2008), while neurotrimin and opioid-binding protein/CAM have been found to bind AβF (Verdier et al., 2005). The role these interactions play in normal physiology and amyloid-dependent pathology requires further investigation.
Integrins form a large and diverse family of ubiquitously expressed transmembrane CAMs (Barczyk et al., 2009). They are heterodimers composed of α and β subunits, combining to form at least 24 different pairs with distinct functions and expression patterns (Takada et al., 2007) (Figure 4). Integrins are the primary mediators of cell-to-ECM adhesion throughout the body and serve a wide variety of functions including regulation of cellular growth and migration (Howe et al., 1998; Huttenlocher and Horwitz, 2011). Integrins are widely expressed in cells of the nervous system, where they play important roles in neurite outgrowth, synaptic plasticity, and neural immune function (Archelos et al., 1999; Clegg et al., 2003).
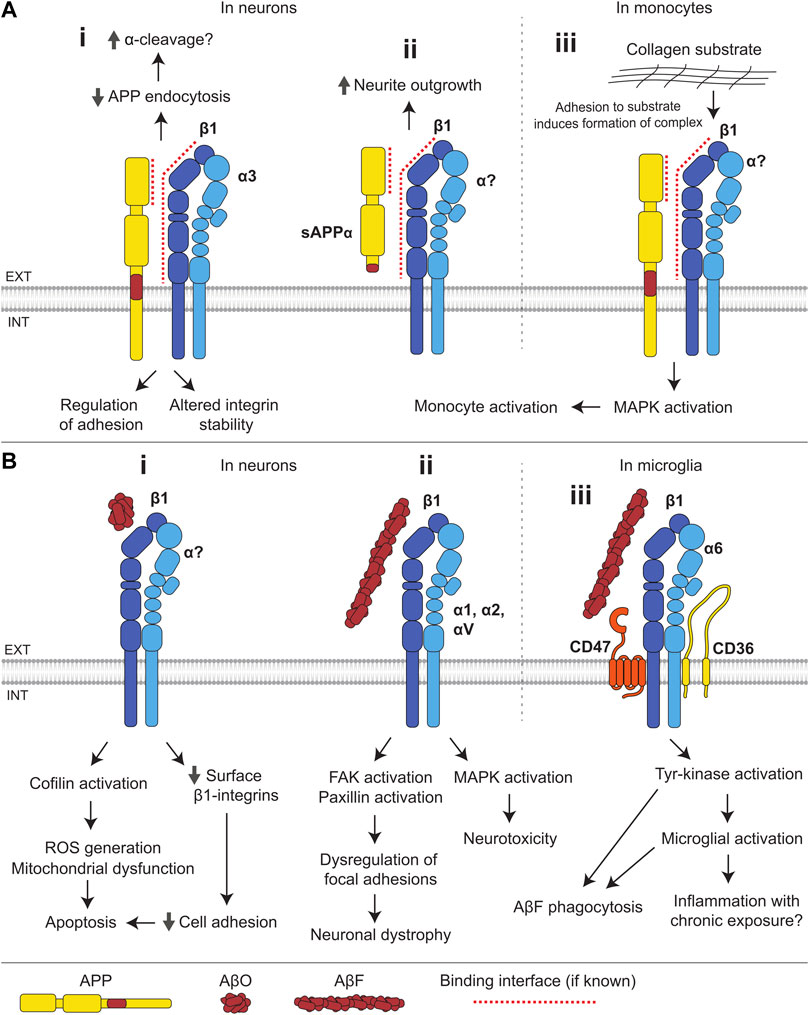
FIGURE 4. Interactions of integrins with APP and Aβ. (A) (i) The E1 domain of APP interacts with β1-and α3-integrins. Integrins suppress endocytosis of APP and thereby may facilitate its α-cleavage. APP influences integrin stability and integrin-dependent adhesion. (ii) In neurons, sAPPα binds to β1-integrins and induces neurite outgrowth. (iii) In monocytes, adhesion to a collagen substrate induces the formation of an APP-β1-integrin complex, which activates MAPK signaling resulting in monocyte activation. (B) (i) In neurons, binding of AβOs to β1-integrins leads to a reduction in the cell surface levels of β1-integrins and compromised cell adhesion, and results in activation of the f-actin severing protein, cofilin, mitochondrial dysfunction and ROS generation, leading to apoptosis. (ii) AβFs bind several integrin subunits (β1, α1, α2, and αV) and induce MAPK activation and neurotoxicity. Binding of AβF to β1-integrin also induces FAK and paxillin activation, leading to dysregulation of integrin-mediated focal adhesion and contributing to neuronal dystrophy. (iii) In microglia, AβFs bind to a receptor complex composed of α6β1-integrin, CD47, and CD36, and induce tyrosine-kinase activation, stimulating AβF phagocytosis and microglial activation.
In the mouse and rat brain, APP binds to β1-integrins and α3-integrins, but not to αM-integrins (Young-Pearse et al., 2008; Hoe et al., 2009) (Figure 4A). APP colocalizes strongly with α1β1-and α5β1-integrins in cultured neurons, and with α1β1- but not α5β1-integrins in cultured astrocytes (Yamazaki et al., 1997), suggesting cell type-specific interactions between APP and different integrin subunits. The β1-integrin-APP complex is formed via the interaction of the E1 domain of APP with the extracellular domain of β1-integrin (Young-Pearse et al., 2008). Binding sites within APP for other integrin subunits remain unknown. APP and β1-integrins may also be linked by cytoplasmic adaptor proteins, such as Dab1 and Fe65, which both APP and β1-integrins bind to (Young-Pearse et al., 2008).
Studies in multiple cell types suggest that APP regulates integrin-mediated adhesion, a function consistent with the ubiquitous expression of both proteins. The loss of APP in endothelial cells reduces expression of β1-and β3-integrins, compromising attachment of cells to collagen and fibronectin substrates (Ristori et al., 2020). Adhesion of THP-1 monocytes to collagen substrates is also dependent on APP expression, wherein adhesion induces the formation of a receptor complex containing β1-integrins and APP, initiating MAPK signaling and facilitating monocyte activation (Sondag and Combs, 2004) (Figure 4Aiii). In contrast, the loss of APP in mice leads to an increase in the levels of β1-and α3-integrins in the brain (Hoe et al., 2009), suggesting that the integrin-APP interactions in cells of the nervous system may differ from those in other tissues. In cultured neurons, blockade of β1-integrins with anti-β1-integrin antibodies leads to inhibition of neurite outgrowth induced by sAPPα, suggesting that β1-integrins function as receptors for soluble forms of APP (Young-Pearse et al., 2008) (Figure 4Aii).
In cultured hippocampal neurons overexpressing β1-integrin and APP, APP internalization is reduced and APP levels at the cell surface are increased (Hoe et al., 2009), suggesting that β1-integrins facilitate the α-cleavage of APP which is known to occur predominantly at the cell surface (Parvathy et al., 1999) (Figure 4Ai). In agreement, overexpression of β1-integrins in HEK293 cells endogenously expressing APP leads to an increase in sAPPα production, however, this effect could not be reproduced in cultured cortical neurons (Rice et al., 2013). The levels of β1-integrins are reduced in the brain of the Tg2576 mouse model of AD overexpressing a mutant form of APP (isoform 695) with the Swedish mutation (KM670/671NL). This model displays elevated Aβ levels and ultimately amyloid plaques (Hoe et al., 2009), supporting the idea that low integrin levels correlate with increased amyloidogenic APP processing. Integrin-mediated signaling was also implicated in the control of APP processing in several genome-wide siRNA screens (Camargo et al., 2015; Chapuis et al., 2017). β1-integrins interact with a 109 amino acid APP-CTF (Ghiso et al., 1992). They may therefore bind to α- and β-CTFs of APP and influence their γ-cleavage.
Integrins bind to Aβ monomers, oligomers, and fibrils. The RHDS sequence located near the N-terminus of Aβ is similar to the known integrin recognition sequence RGDS (Ghiso et al., 1992). CHO cells expressing α5β1-integrins attach to non-fibrillar Aβ-coated surfaces, and this attachment is blocked by soluble RGDS sequence-containing peptides or anti-α5-integrin antibodies (Matter et al., 1998). The binding of integrins to Aβ is subunit specific because CHO cells expressing αvβ1-integrin, but not αvβ3-integrin, also attach to Aβ-coated surfaces. Aβ40 peptides also bind to αIIbβ3-integrin (platelet integrin) via the RHDS sequence, however, other sequences in Aβ40 are also involved (Donner et al., 2016; Donner et al., 2018). The binding of α6-integrins and β1-integrins to AβF is mediated by an epitope distinct from the RHDS sequence (Bamberger et al., 2003; Venkatasubramaniam et al., 2014). β1-integrin also binds AβOs, and β1-integrin conditional knock-out mice demonstrate reduced binding of AβOs to neurons (Woo et al., 2015).
The binding of AβOs to β1-integrins in neurons results in the activation of cofilin, an f-actin severing protein, leading to the depletion of f-actin, mitochondrial dysfunction, ROS generation, and apoptosis (Woo et al., 2015) (Figure 4Bi). β1-integrin also mediates a transient increase in spine density and dendritic complexity following AβO treatment (Ortiz-Sanz et al., 2020). Changes in function of oligodendrocytes, the myelinating glial cells of the central nervous system, are associated with the onset of neurodegeneration in AD. It is noteworthy that the binding of AβOs to β1-integrin in oligodendrocytes leads to the activation of protein tyrosine kinase Fyn and serine/threonine-specific Ca2+/calmodulin-dependent protein kinase II (CaMKII), promoting differentiation, maturation, and survival of these cells (Quintela-López et al., 2019). This may represent a physiological role of Aβ which may be lost in favor of toxicity with chronic Aβ exposure.
Binding of AβF to β1-integrin in neurons results in activation of focal adhesion kinase (FAK) and the focal adhesion scaffolding protein, paxillin, leading to the formation of aberrant focal adhesion-like structures (Grace and Busciglio, 2003; Han et al., 2013). AβF-induced neuronal dystrophy is dependent on β1-integrin-induced paxillin activation, indicating that dysregulation of focal adhesions may be central to Aβ toxicity (Figure 4Bii). Furthermore, AβF-induced neurotoxicity appears to be driven by the MAPK pathway, dependent on specific integrins, with neurotoxicity mediated by α1-integrin in hippocampal neurons and α2-, αV, and β1-integrins in cortical neurons (Anderson and Ferreira, 2004; Wright et al., 2007). Binding of AβF to a receptor complex composed of α6β1-integrin, IgSF CAM CD47, and B-class scavenger receptor CD36 in microglia, immune effector cells of the central nervous system, causes Fyn activation, triggering microglial cell activation and leading to a potentially deleterious inflammatory response (Bamberger et al., 2003) (Figure 4Biii). Tyrosine kinase-dependent signaling via this receptor complex induces phagocytosis of AβF, which may facilitate Aβ clearance (Koenigsknecht and Landreth, 2004). Blockade of β1-integrins prevents AβF-induced tyrosine kinase signaling, ROS generation, and interleukin-1β production in THP-1 monocytes (Bamberger et al., 2003). AβF also stimulates histamine secretion from mast cells via binding to a β1-integrin-CD47 receptor complex (Niederhoffer et al., 2009).
While aberrant overactivation of some integrin signaling pathways appears to be a common response to Aβ, other integrin-dependent pathways are silenced in AD mouse models. For example, integrin-linked kinase (ILK) levels and activity are reduced in APP/PS1 mice expressing a chimeric mouse/human APP with a Swedish mutation and a mutant human presenilin 1 in the central nervous system neurons. Overexpression of ILK rescues hippocampal neurogenesis and memory deficits in this AD mouse model (Xu et al., 2018).
Integrin-mediated cell-to-cell and cell-to-ECM adhesion also appears to be a target of Aβ toxicity, as Aβ peptides partially block β1-integrin-mediated adhesion of SH-SY5Y cells to a fibronectin substrate (Sabo et al., 1995). AβOs induce the loss of cell surface β1-integrins in cultured neurons and α1β1-integrins in neuroblastoma cell lines (Bozzo et al., 2004; Bozzo et al., 2010; Woo et al., 2015). Overexpression of α5β1-integrins in human neuroblastoma IMR-32 cells results in inhibition of Aβ-induced apoptosis (Matter et al., 1998), suggesting that the loss of integrins contributes to AβO-induced neuronal death in AD (Figure 4Bi).
Cadherins are a family of widely expressed CAMs characterized by the presence of the extracellular cadherin (EC) domains and mediating calcium-dependent homophilic adhesion between cells. Classical cadherins such as E- or N-cadherin are transmembrane glycoproteins with a short cytoplasmic tail and extracellular domain composed of five EC domains (Figure 5B). Other cadherins contain variable numbers of EC and other domains. Cadherins play various roles in regulating cell migration, cytoskeleton organization, and are crucial components of adherens junctions in epithelia and endothelia (Angst et al., 2001; Halbleib and Nelson, 2006; Kowalczyk and Nanes, 2012; Shih and Yamada, 2012). In the brain, cadherins regulate neurite outgrowth and the formation of synaptic contacts, and are important for overall neural development (Zhu and Luo, 2004; Arikkath and Reichardt, 2008; Hansen et al., 2008).
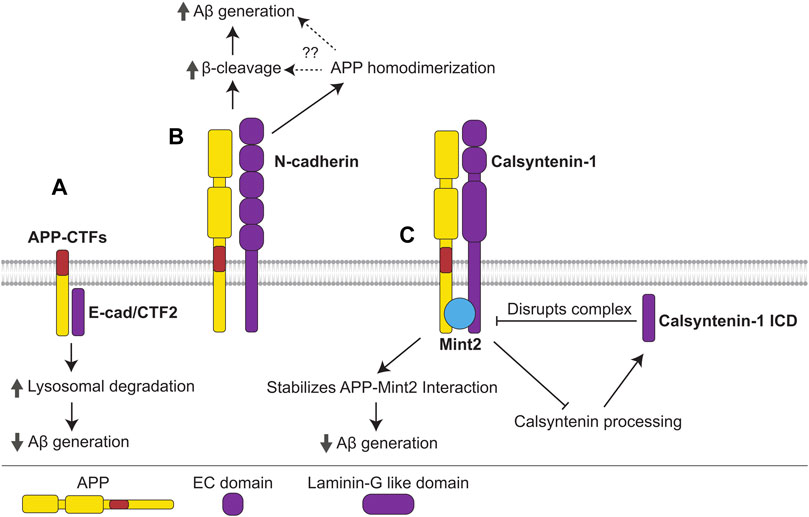
FIGURE 5. Interactions of cadherins with APP. (A) The γ-cleavage product of E-cadherin, E-cad/CTF2, interacts with APP-CTFs, promoting their lysosomal degradation and precluding Aβ generation. (B) N-cadherin interacts with APP promoting its homodimerization, β-cleavage, and Aβ generation. N-cadherin-induced APP homodimerization may promote β-cleavage and Aβ generation, however, this mechanism needs to be confirmed. (C) APP forms a complex with the non-classical cadherin, calsyntenin-1, and adaptor protein Mint2. The APP-Mint2 interaction, which may suppress Aβ generation, is stabilized in the tripartite complex with calsyntenin-1, thus enabling greater suppression of Aβ generation. Formation of the APP-Mint2-calsyntenin-1 complex also precludes calsyntenin-1 processing, reducing generation of the γ-cleavage product, calsyntenin-1 ICD, which disrupts the APP-Mint2-calsyntenin-1 complex.
The levels of N-cadherin proteolytic fragments are increased in the CSF and plasma of AD patients (Choi et al., 2020), while N-cadherin expression is upregulated in the cerebral cortex of Aβ aggregate-injected mice (Kong et al., 2005), suggesting deregulation of N-cadherin expression or cleavage of this glycoprotein in AD.
In the mouse brain, N-cadherin interacts with APP and enhances APP homodimerization as well as production of sAPPβ and Aβ (Asada-Utsugi et al., 2011) (Figure 5B). Similarly to APP, E-cadherin is processed by ADAM10 and γ-secretase (Marambaud et al., 2002; Maretzky et al., 2005). The cytoplasmic fragment of E-cadherin, termed E-cad/CTF2, which is released following γ-secretase cleavage of E-cadherin, interacts with APP-CTFs in CHO cells, promoting their lysosomal degradation and inhibiting the production of Aβ and AICD (Agiostratidou et al., 2006) (Figure 5A). E-cadherin was identified as an interacting partner of APP in silico using a protein-protein interaction tool (Dursun and Gezen-Ak, 2017). This approach also identified β-catenin, which is a cytoplasmic adaptor protein that links E-cadherin to the cytoskeleton (Tian et al., 2011), suggesting that APP modulates cadherin functions in cytoskeletal organization, signaling, and adherens junction maintenance.
In the mouse brain, APP interacts with the non-classical cadherins calsyntenin-1 and -3 (Bai et al., 2008). These cell adhesion molecules are highly expressed in neurons and accumulate in the post-synaptic membrane (Hintsch et al., 2002). Calsyntenin-1 forms a complex with APP and Mint2, stabilizing the APP-Mint2 interaction, suppressing amyloidogenic processing and reducing Aβ production (Araki et al., 2003; Gotoh et al., 2020) (Figure 5C). Accordingly, calsyntenin-1 deficiency in APP23 mice results in increased plaque deposition. On the other hand, the intracellular domain of calsyntenin-1 released following γ-secretase cleavage (calsyntenin-1 ICD) disrupts the APP-Mint2-Calsyntenin-1 complex, promoting Aβ generation (Takei et al., 2015). Since the tripartite complex additionally protects calsyntenin-1 from cleavage (Araki et al., 2004), calsyntenin-1 ICD production may promote further calsyntenin-1 cleavage and ICD production, leading to a positive feedback loop that generates increasing amounts of Aβ (Figure 5C).
Neurexins are a family of transmembrane CAMs, which includes three classical neurexins (neurexin-1 to -3) and five contactin-associated proteins (Caspr-1 to -5) named for their close association with contactins. Each neurexin gene encodes an α and β isoform, which differ in their extracellular part. The extracellular domains of α-neurexins are composed of six laminin G-like domains and three EGF-like regions, while the extracellular domains of β isoforms contain only a single laminin G-like repeat (Reissner et al., 2013). The extracellular domains of contactin-associated proteins contain four laminin G-like domains, two EGF-like regions, an F5/8 type C domain, and a fibrinogen-like part (Figure 6B).
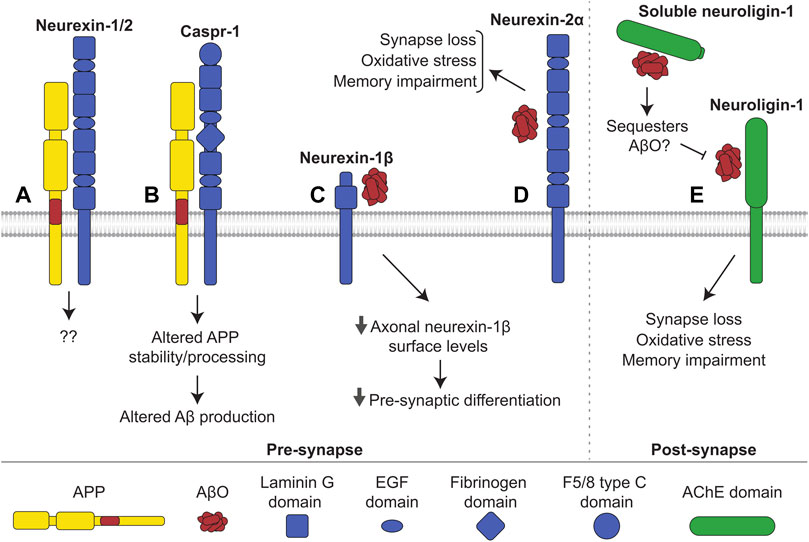
FIGURE 6. Interactions of neurexin and neuroligin with APP and Aβ. (A) Neurexin-1 and -2 interact with APP in mouse brains, with unknown consequence. (B) Caspr-1 interacts with APP. This interaction alters stability and processing of APP influencing production of Aβ. It is uncertain whether the interaction increases or decreases amyloidogenic processing. (C) AβOs bind to neurexin-1β, reducing its levels at the axonal cell surface and hindering presynaptic differentiation. (D) AβOs additionally bind to neurexin-2α triggering oxidative stress, synapse loss, and memory impairments in mice. Similarly, (E) AβOs interact with neuroligin-1 at the post-synapse, inducing oxidative stress, synapse loss, and memory impairments in mice. Shedding releases soluble neuroligin-1 which also binds to and likely sequesters AβOs, preventing interactions with membrane-bound neuroligin-1 and other receptors.
Neuroligins are a family of transmembrane CAMs, which includes five members (neuroligin-1 to -3, 4X and 4Y). The extracellular domains of neuroligins mostly consist of an acetylcholinesterase (AChE)-homologous domain, which binds in a calcium-dependent manner to the laminin G-like domains of neurexins (Bemben et al., 2015). The interaction between presynaptic neurexins and postsynaptic neuroligins plays an important role in synapse formation and maturation being critical for overall neural development (Craig and Kang, 2007; Bang and Owczarek, 2013).
Cytosolic domains of neurexins and APP interact with Mint1 and Mint2 (Biederer and Südhof, 2000), and neurexin-1 and -2 were found in a complex with APP in the brains of transgenic mice expressing affinity tagged APP (Norstrom et al., 2010) (Figure 6A). APP also interacts with Caspr-1 in the mouse brain (Fan et al., 2013) (Figure 6B). Caspr-1 interacts with and is cleaved by γ-secretase, and its silencing results in a drastic reduction of Aβ production in HEK293 cells (Hur et al., 2012). The levels of Caspr-1 are increased in APP/PS1 mice (Fan et al., 2013). Caspr-1 loss of function leads to a decrease in sAPPα production in brain endothelial cells via transcriptional regulation of the secondary α-secretase ADAM9 (Tang et al., 2020). Interestingly, Caspr-1 overexpression results in reduced levels of APP, APP-CTFs and Aβ in CHO cells transfected with APP containing an Indiana (V717F) mutation (Fan et al., 2013), which affects binding of APP to γ-secretase (De Jonghe et al., 2001).
AβOs bind to neurexin-1, -2, and -3 in transfected COS7 cells (Naito et al., 2017). The binding of AβOs to β-neurexins has no effect on neurexin-neuroligin interactions but hinders the neurexin-mediated excitatory presynaptic differentiation observed when hippocampal neurons were co-cultured together with neuroligin-expressing HEK293 cells. This occurs via a reduction in neurexin-1β levels at the axonal cell surface induced by AβOs (Figure 6C). In accordance, β-neurexins, but not α-neurexins, are downregulated in synapses of the J20 APP mouse model of AD, which overexpresses human APP with the Swedish and Indiana mutations linked to familial AD (Naito et al., 2017). AβOs also bind neurexin-2α in human brain tissue (Brito-Moreira et al., 2017) (Figure 6D). Blockade of the neurexin-2α-AβO interaction using antibodies against neurexin-2α reduces the binding of AβOs to cultured hippocampal neurons and prevents AβO-induced oxidative stress and synapse loss. AβO-induced memory impairment in mice is attenuated after injection of the aforementioned anti-neurexin-2α antibodies (Brito-Moreira et al., 2017). Neurexins-1, -2, and -3 have been reported to bind to AβF in CSF of AD patients (Rahman et al., 2018).
AβOs bind to neuroligin-1 in human brain tissue and in the rat brain (Dinamarca et al., 2015; Brito-Moreira et al., 2017). Blockade of the neuroligin-1-AβO interaction reduces binding of AβOs to neurons, inhibits AβO-induced oxidative stress and synapse loss in cultured neurons, and prevents memory impairments in mice (Brito-Moreira et al., 2017). Soluble neuroligin-1 binds to AβOs and reduces excitatory synaptotoxicity (Dinamarca et al., 2015), most likely by sequestering AβOs and inhibiting their interaction with membrane-bound neuroligin-1 (Figure 6E). On the other hand, neuroligin-1 deficient neurons are more vulnerable to AβO-induced toxicity, and AβO-induced impairments in learning are more severe in neuroligin-1 deficient mice (Dufort-Gervais et al., 2020). Neuroligin-1 levels are also reduced in the hippocampus of people with AD (Dufort-Gervais et al., 2020). These seemingly contradictory findings may suggest that although neuroligin-1 mediates toxicity via its interaction with AβOs, loss of neuroligin-1 and its associated physiological functions may weaken synapses and increase vulnerability to AβO toxicity through other receptors. AβOs also bind to neuroligin-2, but not neuroligin-3 (Brito-Moreira et al., 2017). In contrast to neuroligin-1, neuroligin-2 is localized to inhibitory synapses (Varoqueaux et al., 2004), and the effects of its interaction with AβOs remain unknown.
The cellular prion protein (PrPc) is a small glycoprotein well-known for its role in prion diseases such as Creutzfeldt-Jakob disease, where it exists in a misfolded and aggregated form termed prion protein scrapie (PrPSc) (Atkinson et al., 2016). PrPc consists of a disordered N-terminal domain and C-terminal α-helical region attached to the membrane via a GPI anchor (Watts et al., 2018). PrPc is expressed in a variety of tissues but is particularly enriched in the brain where is plays a role in regulating cell adhesion, neuronal development, synaptic plasticity, and myelin maintenance (Roucou and LeBlanc, 2005; Petit et al., 2013; Wulf et al., 2017; Watts et al., 2018).
PrPc interacts with APP in the mouse brain, zebrafish, and N2a cells (Schmitt-Ulms et al., 2004; Bai et al., 2008; Kaiser et al., 2012) (Figure 7Aii). APP was pulled down from human brain lysate with the N-terminal domain of PrPc used as bait (Ulbrich et al., 2018). In zebrafish embryos, knockdown of either APP or PrPc homologs was shown to have no effect on cell aggregation, however, knockdown of both significantly reduced the propensity for aggregation. (Kaiser et al., 2012). The loss of APP in zebrafish results in increased seizures upon exposure to low doses of convulsant, and this effect is lost in fish lacking PrPc suggesting that APP regulates neuronal excitability in a PrPc-dependent manner (Kanyo et al., 2020).
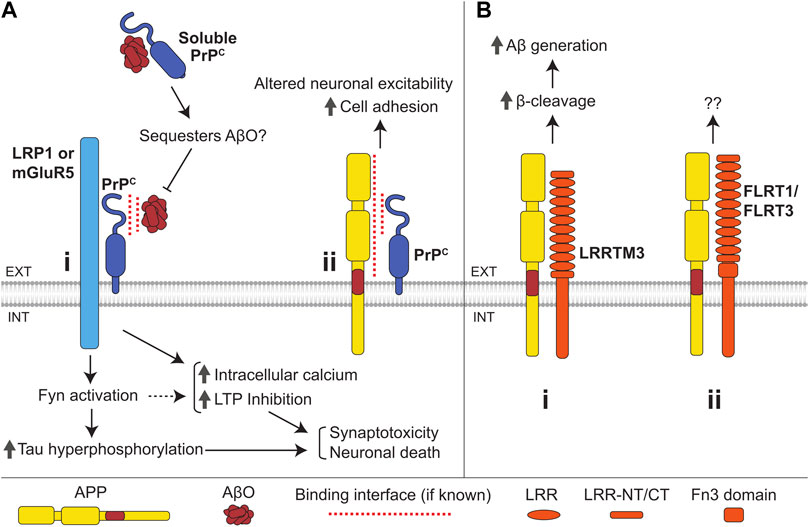
FIGURE 7. Interactions of the cellular prion protein (PrPc) and LRR superfamily CAMs with APP and Aβ. (A) (i) Binding of AβOs to PrPc induces an increase in intracellular Ca2+ levels, LTP inhibition, Fyn activation, and tau hyperphosphorylation, ultimately resulting in synaptotoxicity and neuronal death. PrPc is anchored to the outer leaflet of the plasma membrane and transmits the AβO-induced signals across the plasma membrane by interacting with the transmembrane proteins LRP1 and mGluR5. Soluble PrPc binds to and can sequester AβOs, preventing membrane-bound PrPc-mediated AβO toxicity. (ii) Interaction of APP with the N-terminal domain of PrPc enhances cell adhesion via an unknown mechanism and may also regulate neuronal excitability. (B) (i) LRRTM3 interacts with APP promoting its β-cleavage and facilitating Aβ generation. (ii) APP also interacts with FLRT1 and FLRT3, with unknown consequence.
There are some similarities between prion diseases and AD, and the gene encoding for PrPc (Prpn) has been identified as a potential susceptibility gene for AD (Riemenschneider et al., 2004; Bertram et al., 2007). PrPc levels are reduced in the temporal cortex of patients suffering from sporadic AD, and this reduction correlates with increased clinical severity of the disease (Whitehouse et al., 2013).
PrPc modulates APP processing by interacting with BACE1, where PrPc binds to immature BACE1 in the Golgi, preventing its export and maturation (Parkin et al., 2007; Griffiths et al., 2011). PrPc reduces Aβ levels in mouse brains, and PrPc levels inversely correlate with Aβ load in AD (Whitehouse et al., 2013). It is unknown whether PrPc also controls APP processing by directly interacting with APP.
AβOs bind to the N-terminal region of PrPc in AD brains but not in brains of non-demented controls (Chen et al., 2010; Dohler et al., 2014) (Figure 7Ai). The efficiency of AβO binding to the neuronal surface is strongly reduced in cultured Prpn−/− neurons, and AβOs fail to inhibit hippocampal LTP in Prpn−/− mice indicating that PrPc is one of the major neuronal receptors for AβOs (Laurén et al., 2009). The AβO-induced LTP impairment is also reduced in rats after intracerebroventricular administration of antibody fragments directed to a putative Aβ-binding site on PrPc (Barry et al., 2011). Furthermore, PrPc shedding due to cleavage by ADAM10 decreases AβO binding to iPSC-derived neurons, resulting in reduced toxicity (Jarosz-Griffiths et al., 2019), while soluble PrPc inhibits AβO-induced LTP inhibition (Calella et al., 2010; Scott-McKean et al., 2016).
Binding of AβO to PrPc leads to an increase in intracellular calcium levels, causes synaptotoxicity, and induces activation of Fyn kinase leading to tau hyperphosphorylation (Larson et al., 2012; Peters et al., 2015). PrPc transmits signals across the plasma membrane by binding to LRP1 (Rushworth et al., 2013) and mGluR5 (Um et al., 2013; Hu et al., 2014). NCAMs, which bind both PrPc and Fyn, may also be involved (Bodrikov et al., 2005; Santuccione et al., 2005; Wang et al., 2013). Binding of AβO to PrPc has been shown to hinder PrPc-dependent BACE1 inhibition, likely promoting further Aβ production and accumulation (Rushworth et al., 2013). Binding of AβO to PrPc at the cell surface leads to an increase in PrPc levels here by limiting PrPc endocytosis (Caetano et al., 2011), thereby further promoting the binding of AβO to the cell surface and enhancing AβO toxicity in a positive feedback loop.
An orally administered PrPc agonist that blocks binding of AβOs to PrPc rescues memory impairment and synaptic loss in transgenic APP/PS1 mice (Gunther et al., 2019), which overexpress human APP with a Swedish mutation and mutant PS1 and exhibit an age-dependent accumulation of AβOs and AβO-induced pathology. The effects of the PrPc agonist are seen in 12-month-old mice, when Aβ accumulation has already occurred, and memory impairment and synaptic loss are evident (Gunther et al., 2019). These data suggest that therapeutic interventions targeting the PrPc-AβO interaction may prove effective at restoring brain health in individuals already diagnosed with AD.
CAMs of the leucine-rich repeat superfamily are characterized by the presence of leucine rich-repeats (LRRs) in their extracellular domains. Leucine-rich repeat transmembrane proteins (LRRTMs) are a family within the LRR superfamily consisting of four members (LRRTM-1 to -4). They are single-pass transmembrane proteins with short cytoplasmic tails and extracellular domains comprising ten LRRs (Figure 7Bi). LRRTMs mediate synaptic adhesion by binding to neurexins (Ko, 2012). A family of fibronectin leucine-rich repeat transmembrane (FLRT) proteins also belongs to the LRR superfamily. Three members of this family (FLRT-1 to -3) are single-pass transmembrane proteins with ten extracellular LRR domains and a juxtamembrane Fn3 domain (Figure 7Bii). FLRTs mediate synaptic adhesion by binding to latrophilins (O'Sullivan et al., 2012). LRRTMs and FLRTs play an important role in synapse formation and regulation (Ko, 2012; Schroeder and de Wit, 2018).
LRRTM3 was identified as a candidate gene for AD from an siRNA screening of over 15,000 genes (Majercak et al., 2006). SNPs within the promoter and intronic regions of the gene coding for LRRTM3 are associated with AD (Reitz et al., 2012). LRRTM3 interacts with APP in HEK293 cells and colocalizes with APP in cultured neurons (Lincoln et al., 2013). siRNA-mediated knockdown of LRRTM3 leads to a reduction in sAPPβ and β-CTF levels and Aβ secretion in SH-SY5Y cells (Majercak et al., 2006; Lincoln et al., 2013), suggesting that LRRTM3 modulates β-cleavage of APP (Figure 7Bi). APP also interacts with FLRT1 and FLRT3 in HEK293T cells (Yu et al., 2016) (Figure 7Bii). The functional role of this interaction remains unclear.
The amyloid hypothesis places APP and Aβ at the center of AD etiology, however, our understanding of the normal functions of APP, regulation of its processing, and the mechanisms of Aβ-induced toxicity are still incomplete. These knowledge gaps have been showcased by the failures of therapeutics targeting APP processing and Aβ, which have been unable to demonstrate reasonable efficacy (Kumar et al., 2018; Hampel et al., 2021; Karran and De Strooper, 2022). While the physiological functions of APP are incompletely understood, its general role in regulating cell adhesion is suggested by multiple reports showing that APP not only interacts with different CAMs, but also regulates multiple functions of these proteins, including cell adhesion and neuronal growth regulation. CAMs also emerged as important regulators of the processing of APP with some CAMs promoting amyloidogenic pathway and others enhancing the non-amyloidogenic pathway. In addition to its role in AD pathogenesis, Aβ plays physiological roles in learning and memory (Puzzo et al., 2011; Kent et al., 2020). It is therefore possible that the CAM-regulated switches in the modes of APP processing are functionally important, however, the roles these switches play in the healthy brain remains poorly understood and should be analyzed in the future. Substantial evidence indicates that CAMs function as the cell surface receptors for Aβ and its oligomers. While a possible physiological role of these interactions remains also unknown, the binding of Aβ to CAMs clearly plays a role in AD by inducing aberrant signaling pathways and loss of CAM-mediated adhesion. Interestingly, some CAMs inhibit the formation of large Aβ oligomers, suggesting these proteins are involved in the regulation of Aβ turnover in the brain.
Together, the research outlined here highlights the importance of the interplay between CAMs and APP during normal physiology, a factor which should be considered when developing therapeutics that target APP as they may impact these functions. On the other hand, modulation of the interactions between APP and CAMs may represent an attractive therapeutic approach to reduce Aβ generation while limiting side effects due to overzealous obstruction of APP and its proteases. Finally, inhibition of the binding of Aβ to CAMs may be used to prevent the binding of Aβ to the neuronal surface and reduce the Aβ-induced toxic effects. There are still major gaps in our understanding of the role that CAMs play in APP and Aβ-dependent functions. Future research should focus on the mechanisms underpinning the interactions of CAMs with APP and Aβ in the brain with the hope of aiding future endeavors to develop safe and effective therapies for AD.
GP and VS contributed to conception and design of the study. GP wrote the first draft of the manuscript and prepared the figures and tables. GP, AN, and VS contributed to manuscript revisions, read, and approved the submitted version.
The study was supported by the National Health and Medical Research Council (VS, APP1129869).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agiostratidou, G., Muros, R. M., Shioi, J., Marambaud, P., and Robakis, N. K. (2006). The cytoplasmic sequence of E-cadherin promotes non-amyloidogenic degradation of Aβ precursors. J. Neurochem. 96, 1182–1188. doi:10.1111/j.1471-4159.2005.03616.x
Aisa, B., Gil-Bea, F. J., Solas, M., García-Alloza, M., Chen, C. P., Lai, M. K., et al. (2010). Altered NCAM expression associated with the cholinergic system in Alzheimer's disease. J. Alzheimers Dis. 20, 659–668. doi:10.3233/JAD-2010-1398
Alzheimer’s Association (2022). 2022 Alzheimer's disease facts and figures. Alzheimer's Dementia 18, 700–789.
Anderson, K. L., and Ferreira, A. (2004). alpha1 integrin activation: a link between beta-amyloid deposition and neuronal death in aging hippocampal neurons. J. Neurosci. Res. 75, 688–697. doi:10.1002/jnr.20018
Andrew, R. J., Kellett, K. A. B., Thinakaran, G., and Hooper, N. M. (2016). A Greek tragedy: The growing complexity of alzheimer amyloid precursor protein proteolysis. J. Biol. Chem. 291, 19235–19244. doi:10.1074/jbc.R116.746032
Angst, B. D., Marcozzi, C., and Magee, A. I. (2001). The cadherin superfamily: Diversity in form and function. J. Cell. Sci. 114, 629–641. doi:10.1242/jcs.114.4.629
Araki, Y., Miyagi, N., Kato, N., Yoshida, T., Wada, S., Nishimura, M., et al. (2004). Coordinated metabolism of alcadein and amyloid β-protein precursor regulates Fe65-dependent gene transactivation. J. Biol. Chem. 279, 24343–24354. doi:10.1074/jbc.M401925200
Araki, Y., Tomita, S., Yamaguchi, H., Miyagi, N., Sumioka, A., Kirino, Y., et al. (2003). Novel cadherin-related membrane proteins, Alcadeins, enhance the X11-like protein-mediated stabilization of amyloid beta-protein precursor metabolism. J. Biol. Chem. 278, 49448–49458. doi:10.1074/jbc.M306024200
Archelos, J. J., Previtali, S. C., and Hartung, H.-P. (1999). The role of integrins in immune-mediated diseases of the nervous system. Trends Neurosci. 22, 30–38. doi:10.1016/s0166-2236(98)01287-9
Arikkath, J., and Reichardt, L. F. (2008). Cadherins and catenins at synapses: Roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 31, 487–494. doi:10.1016/j.tins.2008.07.001
Asada-Utsugi, M., Uemura, K., Noda, Y., Kuzuya, A., Maesako, M., Ando, K., et al. (2011). N-cadherin enhances APP dimerization at the extracellular domain and modulates Aβ production. J. Neurochem. 119, 354–363. doi:10.1111/j.1471-4159.2011.07364.x
Asai, M., Hattori, C., Szabó, B., Sasagawa, N., Maruyama, K., Tanuma, S.-I., et al. (2003). Putative function of ADAM9, ADAM10, and ADAM17 as APP α-secretase. Biochem. Biophys. Res. Commun. 301, 231–235. doi:10.1016/s0006-291x(02)02999-6
Ashley, J., Packard, M., Ataman, B., and Budnik, V. (2005). Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J. Neurosci. 25, 5943–5955. doi:10.1523/JNEUROSCI.1144-05.2005
Atkinson, C. J., Zhang, K., Munn, A. L., Wiegmans, A., and Wei, M. Q. (2016). Prion protein scrapie and the normal cellular prion protein. Prion 10, 63–82. doi:10.1080/19336896.2015.1110293
Bai, Y., Markham, K., Chen, F., Weerasekera, R., Watts, J., Horne, P., et al. (2008). The in vivo brain interactome of the amyloid precursor protein. Mol. Cell. Proteomics 7, 15–34. doi:10.1074/mcp.M700077-MCP200
Bamberger, M. E., Harris, M. E., Mcdonald, D. R., Husemann, J., and Landreth, G. E. (2003). A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J. Neurosci. 23, 2665–2674. doi:10.1523/jneurosci.23-07-02665.2003
Bamford, R. A., Widagdo, J., Takamura, N., Eve, M., Anggono, V., Oguro-Ando, A., et al. (2020). The interaction between contactin and amyloid precursor protein and its role in Alzheimer's disease. Neuroscience 424, 184–202. doi:10.1016/j.neuroscience.2019.10.006
Bang, M. L., and Owczarek, S. (2013). A matter of balance: Role of neurexin and neuroligin at the synapse. Neurochem. Res. 38, 1174–1189. doi:10.1007/s11064-013-1029-9
Barczyk, M., Carracedo, S., and Gullberg, D. (2009). Integrins. Cell. Tissue Res. 339, 269–280. doi:10.1007/s00441-009-0834-6
Barrett, P. J., Song, Y., Horn, W. D. V., Hustedt, E. J., Schafer, J. M., Hadziselimovic, A., et al. (2012). The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171. doi:10.1126/science.1219988
Barry, A. E., Klyubin, I., Mc Donald, J. M., Mably, A. J., Farrell, M. A., Scott, M., et al. (2011). Alzheimer's disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263. doi:10.1523/JNEUROSCI.6500-10.2011
Bemben, M. A., Shipman, S. L., Nicoll, R. A., and Roche, K. W. (2015). The cellular and molecular landscape of neuroligins. Trends Neurosci. 38, 496–505. doi:10.1016/j.tins.2015.06.004
Bertram, L., Mcqueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 39, 17–23. doi:10.1038/ng1934
Bharadwaj, P. R., Dubey, A. K., Masters, C. L., Martins, R. N., and Macreadie, I. G. (2009). Abeta aggregation and possible implications in Alzheimer's disease pathogenesis. J. Cell. Mol. Med. 13, 412–421. doi:10.1111/j.1582-4934.2009.00609.x
Biederer, T., and Südhof, T. C. (2000). Mints as adaptors: Direct binding to neurexins and recruitment of Munc18. J. Biol. Chem. 275, 39803–39806. doi:10.1074/jbc.C000656200
Blacker, D., Bertram, L., Saunders, A. J., Moscarillo, T. J., Albert, M. S., Wiener, H., et al. (2003). Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum. Mol. Genet. 12, 23–32. doi:10.1093/hmg/ddg007
Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R., Landfield, P. W., et al. (2004). Incipient alzheimer's disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U. S. A. 101, 2173–2178. doi:10.1073/pnas.0308512100
Bodrikov, V., Leshchyns'Ka, I., Sytnyk, V., Overvoorde, J., Den Hertog, J., Schachner, M., et al. (2005). RPTPalpha is essential for NCAM-mediated p59fyn activation and neurite elongation. J. Cell. Biol. 168, 127–139. doi:10.1083/jcb.200405073
Borg, J. P., Ooi, J., Levy, E., and Margolis, B. (1996). The phosphotyrosine interaction domains of X11 and Fe65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol. Cell. Biol. 16, 6229–6241. doi:10.1128/mcb.16.11.6229
Bozzo, C., Graziola, F., Chiocchetti, A., and Canonico, P. L. (2010). Estrogen and β-amyloid toxicity: Role of integrin and PI3-K. Mol. Cell. Neurosci. 45, 85–91. doi:10.1016/j.mcn.2010.05.012
Bozzo, C., Lombardi, G., Santoro, C., and Canonico, P. L. (2004). Involvement of beta(1) integrin in betaAP-induced apoptosis in human neuroblastoma cells. Mol. Cell.Neurosci. 25, 1–8. doi:10.1016/j.mcn.2003.09.008
Brinkmalm, G., Sjödin, S., Simonsen, A. H., Hasselbalch, S. G., Zetterberg, H., Brinkmalm, A., et al. (2018). A parallel reaction monitoring mass spectrometric method for analysis of potential CSF biomarkers for Alzheimer's disease. Prot. Clin. Appl. 12, 1700131. doi:10.1002/prca.201700131
Brito-Moreira, J., Lourenco, M. V., Oliveira, M. M., Ribeiro, F. C., Ledo, J. H., Diniz, L. P., et al. (2017). Interaction of amyloid-β (Aβ) oligomers with neurexin 2α and neuroligin 1 mediates synapse damage and memory loss in mice. J. Biol. Chem. 292, 7327–7337. doi:10.1074/jbc.M116.761189
Caetano, F. A., Beraldo, F. H., Hajj, G. N. M., Guimaraes, A. L., Jürgensen, S., Wasilewska-Sampaio, A. P., et al. (2011). Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J. Neurochem. 117, 538–553. doi:10.1111/j.1471-4159.2011.07225.x
Caillé, I., Allinquant, B., Dupont, E., Bouillot, C., Langer, A., MüLler, U., et al. (2004). Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131, 2173–2181. doi:10.1242/dev.01103
Calella, A. M., Farinelli, M., Nuvolone, M., Mirante, O., Moos, R., Falsig, J., et al. (2010). Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314. doi:10.1002/emmm.201000082
Camargo, L. M., Zhang, X. D., Loerch, P., Caceres, R. M., Marine, S. D., Uva, P., et al. (2015). Pathway-based analysis of genome-wide siRNA screens reveals the regulatory landscape of APP processing. PloS one 10, e0115369. doi:10.1371/journal.pone.0115369
Chapuis, J., Flaig, A., Grenier-Boley, B., Eysert, F., Pottiez, V., Deloison, G., et al. (2017). Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol. 133, 955–966. doi:10.1007/s00401-016-1652-z
Chatterjee, M., Del Campo, M., Morrema, T. H. J., de Waal, M., van der Flier, W. M., Hoozemans, J. J. M., et al. (2018). Contactin-2, a synaptic and axonal protein, is reduced in cerebrospinal fluid and brain tissue in Alzheimer’s disease. Alzheimers Res. Ther. 10, 52. doi:10.1186/s13195-018-0383-x
Chatterjee, M., Schild, D., and Teunissen, C. E. (2019). Contactins in the central nervous system: Role in health and disease. Neural Regen. Res. 14, 206–216. doi:10.4103/1673-5374.244776
Chen, G.-F., Xu, T.-H., Yan, Y., Zhou, Y.-R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi:10.1038/aps.2017.28
Chen, K.-P., and Dou, F. (2012). Selective interaction of amyloid precursor protein with different isoforms of neural cell adhesion molecule. J. Mol. Neurosci. 46, 203–209. doi:10.1007/s12031-011-9578-3
Chen, K., Lu, H., Gao, T., Xue, X., Wang, C., Miao, F., et al. (2016). Synergic interaction between amyloid precursor protein and neural cell adhesion molecule promotes neurite outgrowth. Oncotarget 7, 14199–14206. doi:10.18632/oncotarget.7348
Chen, S., Yadav, S. P., and Surewicz, W. K. (2010). Interaction between human prion protein and amyloid-beta (abeta) oligomers: Role of N-terminal residues. J. Biol. Chem. 285, 26377–26383. doi:10.1074/jbc.M110.145516
Choi, J.-Y., Cho, S.-J., Park, J. H., Yun, S.-M., Jo, C., Kim, E.-J., et al. (2020). Elevated cerebrospinal fluid and plasma N-cadherin in Alzheimer disease. J. Neuropathol. Exp. Neurol. 79, 484–492. doi:10.1093/jnen/nlaa019
Clegg, D. O., Wingerd, K. L., Hikita, S. T., and Tolhurst, E. C. (2003). Integrins in the development, function and dysfunction of the nervous system. Front. Biosci. 8, d723–50. doi:10.2741/1020
Coburger, I., Dahms, S. O., Roeser, D., Gührs, K.-H., Hortschansky, P., Than, M. E., et al. (2013). Analysis of the overall structure of the multi-domain amyloid precursor protein (APP). PLoS one 8, e81926. doi:10.1371/journal.pone.0081926
Coburger, I., Hoefgen, S., and Than, M. E. (2014). The structural biology of the amyloid precursor protein APP - a complex puzzle reveals its multi-domain architecture. Biol. Chem. 395, 485–498. doi:10.1515/hsz-2013-0280
Craig, A. M., and Kang, Y. (2007). Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 17, 43–52. doi:10.1016/j.conb.2007.01.011
Cui, P., Ma, X., Li, H., Lang, W., and Hao, J. (2018). Shared biological pathways between Alzheimer’s disease and ischemic stroke. Front. Neurosci. 12, 605. doi:10.3389/fnins.2018.00605
Da Rocha, J. F., Bastos, L., Domingues, S. C., Bento, A. R., Konietzko, U., Da Cruz E Silva, O. A. B., et al. (2021). APP binds to the EGFR ligands HB-EGF and EGF, acting synergistically with EGF to promote ERK signaling and neuritogenesis. Mol. Neurobiol. 58, 668–688. doi:10.1007/s12035-020-02139-2
Dahms, S. O., Könnig, I., Roeser, D., Gührs, K.-H., Mayer, M. C., Kaden, D., et al. (2012). Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J. Mol. Biol. 416, 438–452. doi:10.1016/j.jmb.2011.12.057
Das, U., Wang, L., Ganguly, A., Saikia, J. M., Wagner, S. L., Koo, E. H., et al. (2016). Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 19, 55–64. doi:10.1038/nn.4188
De Jonghe, C., Esselens, C., Kumar-Singh, S., Craessaerts, K., Serneels, S., Checler, F., et al. (2001). Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 10, 1665–1671. doi:10.1093/hmg/10.16.1665
Decock, M., el Haylani, L., Stanga, S., Dewachter, I., Octave, J.-N., Smith, S. O., et al. (2015). Analysis by a highly sensitive split luciferase assay of the regions involved in APP dimerization and its impact on processing. FEBS Open Bio 5, 763–773. doi:10.1016/j.fob.2015.09.002
Dinamarca, M. C., di Luca, M., Godoy, J. A., and Inestrosa, N. C. (2015). The soluble extracellular fragment of neuroligin-1 targets Aβ oligomers to the postsynaptic region of excitatory synapses. Biochem. Biophys. Res. Commun. 466, 66–71. doi:10.1016/j.bbrc.2015.08.107
Djogo, N., Jakovcevski, I., Müller, C., Lee, H. J., Xu, J.-C., Jakovcevski, M., et al. (2013). Adhesion molecule L1 binds to amyloid beta and reduces Alzheimer's disease pathology in mice. Neurobiol. Dis. 56, 104–115. doi:10.1016/j.nbd.2013.04.014
Dohler, F., Sepulveda-Falla, D., Krasemann, S., Altmeppen, H., Schlüter, H., Hildebrand, D., et al. (2014). High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer’s disease. Brain 137, 873–886. doi:10.1093/brain/awt375
Donner, L., Fälker, K., Gremer, L., Klinker, S., Pagani, G., Ljungberg, L. U., et al. (2016). Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci. Signal. 9, ra52. ra52. doi:10.1126/scisignal.aaf6240
Donner, L., Gremer, L., Ziehm, T., Gertzen, C. G. W., Gohlke, H., Willbold, D., et al. (2018). Relevance of N-terminal residues for amyloid-β binding to platelet integrin αIIbβ3, integrin outside-in signaling and amyloid-β fibril formation. Cell. Signal. 50, 121–130. doi:10.1016/j.cellsig.2018.06.015
Dufort-Gervais, J., Provost, C., Charbonneau, L., Norris, C. M., Calon, F., Mongrain, V., et al. (2020). Neuroligin-1 is altered in the hippocampus of Alzheimer’s disease patients and mouse models, and modulates the toxicity of amyloid-beta oligomers. Sci. Rep. 10, 6956. doi:10.1038/s41598-020-63255-6
Dunsing, V., Mayer, M., Liebsch, F., Multhaup, G., and Chiantia, S. (2017). Direct evidence of amyloid precursor–like protein 1 trans interactions in cell–cell adhesion platforms investigated via fluorescence fluctuation spectroscopy. Mol. Biol. Cell. 28, 3609–3620. doi:10.1091/mbc.E17-07-0459
Dursun, E., and Gezen-Ak, D. (2017). Vitamin D receptor is present on the neuronal plasma membrane and is co-localized with amyloid precursor protein, ADAM10 or Nicastrin. PLoS one 12, e0188605. doi:10.1371/journal.pone.0188605
Fan, L.-F., Xu, D.-E., Wang, W.-H., Yan, K., Wu, H., Yao, X.-Q., et al. (2013). Caspr interaction with amyloid precursor protein reduces amyloid-β generation in vitro. Neurosci. Lett. 548, 255–260. doi:10.1016/j.neulet.2013.05.055
Fogel, H., Frere, S., Segev, O., Bharill, S., Shapira, I., Gazit, N., et al. (2014). APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell. Rep. 7, 1560–1576. doi:10.1016/j.celrep.2014.04.024
Gennarini, G., Bizzoca, A., Picocci, S., Puzzo, D., Corsi, P., Furley, A. J. W., et al. (2017). The role of GPI-anchored axonal glycoproteins in neural development and neurological disorders. Mol. Cell. Neurosci. 81, 49–63. doi:10.1016/j.mcn.2016.11.006
Ghiso, J., Rostagno, A., Gardella, J. E., Liem, L., Gorevic, P. D., and Frangione, B. (1992). A 109-amino-acid C-terminal fragment of Alzheimer's-disease amyloid precursor protein contains a sequence, -RHDS-that promotes cell adhesion. Biochem. J. 288, 1053–1059. doi:10.1042/bj2881053
Gotoh, N., Saito, Y., Hata, S., Saito, H., Ojima, D., Murayama, C., et al. (2020). Amyloidogenic processing of amyloid β protein precursor (APP) is enhanced in the brains of alcadein α-deficient mice. J. Biol. Chem. 295, 9650–9662. doi:10.1074/jbc.RA119.012386
Grace, E. A., and Busciglio, J. (2003). Aberrant activation of focal adhesion proteins mediates fibrillar amyloid β-induced neuronal dystrophy. J. Neurosci. 23, 493–502. doi:10.1523/jneurosci.23-02-00493.2003
Griffiths, H. H., Whitehouse, I. J., Baybutt, H., Brown, D., Kellett, K. A. B., Jackson, C. D., et al. (2011). Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 286, 33489–33500. doi:10.1074/jbc.M111.278556
Guise, S., Braguer, D., Carles, G., Delacourte, A., and Briand, C. (2001). Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J. Neurosci. Res. 63, 257–267. doi:10.1002/1097-4547(20010201)63:3<257::AID-JNR1019>3.0.CO;2-T
Gunther, E. C., Smith, L. M., Kostylev, M. A., Cox, T. O., Kaufman, A. C., Lee, S., et al. (2019). Rescue of transgenic Alzheimer's pathophysiology by polymeric cellular prion protein antagonists. Cell. Rep. 26, 1368. e8. doi:10.1016/j.celrep.2019.01.064
Halbleib, J. M., and Nelson, W. J. (2006). Cadherins in development: Cell adhesion, sorting, and tissue morphogenesis. Genes. Dev. 20, 3199–3214. doi:10.1101/gad.1486806
Hamley, I. W. (2012). The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 112, 5147–5192. doi:10.1021/cr3000994
Hampel, H., Vassar, R., de Strooper, B., Hardy, J., Willem, M., Singh, N., et al. (2021). The β-secretase BACE1 in Alzheimer’s disease. Biol. Psychiatry 89, 745–756. doi:10.1016/j.biopsych.2020.02.001
Han, H.-Y., Zhang, J.-P., Ji, S.-Q., Liang, Q.-M., Kang, H.-C., Tang, R.-H., et al. (2013). αν and β1 integrins mediate Aβ-induced neurotoxicity in hippocampal neurons via the FAK signaling pathway. PLoS one 8, e64839. doi:10.1371/journal.pone.0064839
Han, M.-R., Schellenberg, G. D., and Wang, L.-S.THE ALZHEIMER'S DISEASE NEUROIMAGING INIATIVE (2010). Genome-wide association reveals genetic effects on human Aβ 42 and τ protein levels in cerebrospinal fluids: A case control study. BMC Neurol. 10, 90. doi:10.1186/1471-2377-10-90
Hansen, S. M., Berezin, V., and Bock, E. (2008). Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-cadherin. Cell. Mol. Life Sci. 65, 3809–3821. doi:10.1007/s00018-008-8290-0
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: The amyloid cascade hypothesis. Science 256, 184–185. doi:10.1126/science.1566067
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388. doi:10.1016/0165-6147(91)90609-v
Hashimoto, Y., Chiba, T., Yamada, M., Nawa, M., Kanekura, K., Suzuki, H., et al. (2005). Transforming growth factor beta2 is a neuronal death-inducing ligand for amyloid-beta precursor protein. Mol. Cell. Biol. 25, 9304–9317. doi:10.1128/MCB.25.21.9304-9317.2005
Heber, S., Herms, J., Gajic, V., Hainfellner, J., Aguzzi, A., Rülicke, T., et al. (2000). Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 20, 7951–7963. doi:10.1523/jneurosci.20-21-07951.2000
Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet. Neurol. 14, 388–405. doi:10.1016/S1474-4422(15)70016-5
Heredia, L., Lin, R., Vigo, F. S., Kedikian, G., Busciglio, J., Lorenzo, A., et al. (2004). Deposition of amyloid fibrils promotes cell-surface accumulation of amyloid β precursor protein. Neurobiol. Dis. 16, 617–629. doi:10.1016/j.nbd.2004.04.015
Herr, U.-M., Strecker, P., Storck, S. E., Thomas, C., Rabiej, V., Junker, A., et al. (2017). LRP1 modulates APP intraneuronal transport and processing in its monomeric and dimeric state. Front. Mol. Neurosci. 10, 118. doi:10.3389/fnmol.2017.00118
Hintsch, G., Zurlinden, A., Meskenaite, V., Steuble, M., Fink-Widmer, K., Kinter, J., et al. (2002). The calsyntenins – A family of postsynaptic membrane proteins with distinct neuronal expression patterns. Mol. Cell. Neurosci. 21, 393–409. doi:10.1006/mcne.2002.1181
Hoe, H. S., Lee, K. J., Carney, R. S., Lee, J., Markova, A., Lee, J. Y., et al. (2009). Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 29, 7459–7473. doi:10.1523/JNEUROSCI.4872-08.2009
Howe, A., Aplin, A. E., Alahari, S. K., and Juliano, R. L. (1998). Integrin signaling and cell growth control. Curr. Opin. Cell. Biol. 10, 220–231. doi:10.1016/s0955-0674(98)80144-0
Hu, J., Lin, S. L., and Schachner, M. (2022). A fragment of cell adhesion molecule L1 reduces amyloid-β plaques in a mouse model of Alzheimer’s disease. Cell. Death Dis. 13, 48. doi:10.1038/s41419-021-04348-6
Hu, N.-W., Nicoll, A. J., Zhang, D., Mably, A. J., O’Malley, T., Purro, S. A., et al. (2014). mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 5, 3374. doi:10.1038/ncomms4374
Hur, J.-Y., Teranishi, Y., Kihara, T., Yamamoto, N. G., Inoue, M., Hosia, W., et al. (2012). Identification of novel γ-secretase-associated proteins in detergent-resistant membranes from brain. J. Biol. Chem. 287, 11991–12005. doi:10.1074/jbc.M111.246074
Huttenlocher, A., and Horwitz, A. R. (2011). Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 3, a005074. doi:10.1101/cshperspect.a005074
Jarosz-Griffiths, H. H., Corbett, N. J., Rowland, H. A., Fisher, K., Jones, A. C., Baron, J., et al. (2019). Proteolytic shedding of the prion protein via activation of metallopeptidase ADAM10 reduces cellular binding and toxicity of amyloid-β oligomers. J. Biol. Chem. 294, 7085–7097. doi:10.1074/jbc.RA118.005364
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. (1993). The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of alzheimer's disease. Biochemistry 32, 4693–4697. doi:10.1021/bi00069a001
Kaden, D., Munter, L.-M., Joshi, M., Treiber, C., Weise, C., Bethge, T., et al. (2008). Homophilic interactions of the amyloid precursor protein (APP) ectodomain are regulated by the loop region and affect β-secretase cleavage of APP. J. Biol. Chem. 283, 7271–7279. doi:10.1074/jbc.M708046200
Kaden, D., Voigt, P., Munter, L.-M., Bobowski, K. D., Schaefer, M., Multhaup, G., et al. (2009). Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J. Cell. Sci. 122, 368–377. doi:10.1242/jcs.034058
Kaiser, D. M., Acharya, M., Leighton, P. L. A., Wang, H., Daude, N., Wohlgemuth, S., et al. (2012). Amyloid beta precursor protein and prion protein have a conserved interaction affecting cell adhesion and CNS development. PLoS one 7, e51305. doi:10.1371/journal.pone.0051305
Kanyo, R., Leighton, P. L. A., Neil, G. J., Locskai, L. F., and Allison, W. T. (2020). Amyloid-β precursor protein mutant zebrafish exhibit seizure susceptibility that depends on prion protein. Exp. Neurol. 328, 113283. doi:10.1016/j.expneurol.2020.113283
Karran, E., and De Strooper, B. (2022). The amyloid hypothesis in alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 21, 306–318. doi:10.1038/s41573-022-00391-w
Karuppan, S. J., Vogt, A., Fischer, Z., Ladutska, A., Swiastyn, J., Mcgraw, H. F., et al. (2022). Members of the vertebrate contactin and amyloid precursor protein families interact through a conserved interface. J. Biol. Chem. 298, 101541. doi:10.1016/j.jbc.2021.101541
Kent, S. A., Spires-Jones, T. L., and Durrant, C. S. (2020). The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 140, 417–447. doi:10.1007/s00401-020-02196-w
Kimura, R., Kamino, K., Yamamoto, M., Nuripa, A., Kida, T., Kazui, H., et al. (2006). The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 16, 15–23. doi:10.1093/hmg/ddl437
King, G. D., Perez, R. G., Steinhilb, M. L., Gaut, J. R., and Turner, R. S. (2003). X11alpha modulates secretory and endocytic trafficking and metabolism of amyloid precursor protein: Mutational analysis of the YENPTY sequence. Neuroscience 120, 143–154. doi:10.1016/s0306-4522(03)00284-7
Kirouac, L., Rajic, A. J., Cribbs, D. H., and Padmanabhan, J. (2017). Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer’s disease. eNeuro 4, ENEURO.0149–16.2017. ENEURO.0149-16.2017. doi:10.1523/ENEURO.0149-16.2017
Klementiev, B., Novikova, T., Novitskaya, V., Walmod, P. S., Dmytriyeva, O., Pakkenberg, B., et al. (2007). A neural cell adhesion molecule-derived peptide reduces neuropathological signs and cognitive impairment induced by Abeta25-35. Neuroscience 145, 209–224. doi:10.1016/j.neuroscience.2006.11.060
Klevanski, M., Herrmann, U., Weyer, S. W., Fol, R., Cartier, N., Wolfer, D. P., et al. (2015). The APP intracellular domain is required for normal synaptic morphology, synaptic plasticity, and hippocampus-dependent behavior. J. Neurosci. 35, 16018–16033. doi:10.1523/JNEUROSCI.2009-15.2015
Ko, J. (2012). The leucine-rich repeat superfamily of synaptic adhesion molecules: LRRTMs and slitrks. Mol. Cells 34, 335–340. doi:10.1007/s10059-012-0113-3
Koenigsknecht, J., and Landreth, G. (2004). Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846. doi:10.1523/JNEUROSCI.2557-04.2004
Kong, L.-N., Zuo, P.-P., Mu, L., Liu, Y.-Y., and Yang, N. (2005). Gene expression profile of amyloid beta protein-injected mouse model for Alzheimer disease. Acta Pharmacol. Sin. 26, 666–672. doi:10.1111/j.1745-7254.2005.00129.x
Kowalczyk, A. P., and Nanes, B. A. (2012). “Adherens junction turnover: Regulating adhesion through cadherin endocytosis, degradation, and recycling,” in Adherens junctions: From molecular mechanisms to tissue development and disease. Editor T. HARRIS (Dordrecht: Springer Netherlands).
Kuhn, P.-H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., et al. (2010). ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032. doi:10.1038/emboj.2010.167
Kumar, D., Ganeshpurkar, A., Kumar, D., Modi, G., Gupta, S. K., Singh, S. K., et al. (2018). Secretase inhibitors for the treatment of Alzheimer's disease: Long road ahead. Eur. J. Med. Chem. 148, 436–452. doi:10.1016/j.ejmech.2018.02.035
Laferla, F. M., Tinkle, B. T., Bieberich, C. J., Haudenschild, C. C., and Jay, G. (1995). The Alzheimer's Aβ peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat. Genet. 9, 21–30. doi:10.1038/ng0195-21
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., et al. (1999). Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U. S. A. 96, 3922–3927. doi:10.1073/pnas.96.7.3922
Langness, V. F., Kant, R. V. D., Das, U., Wang, L., Chaves, R. D. S., Goldstein, L. S. B., et al. (2021). Cholesterol-lowering drugs reduce APP processing to Aβ by inducing APP dimerization. Mol. Biol. Cell. 32, 247–259. doi:10.1091/mbc.E20-05-0345
Larson, M., Sherman, M. A., Amar, F., Nuvolone, M., Schneider, J. A., Bennett, D. A., et al. (2012). The complex PrPc-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease. J. Neurosci. 32, 16857–1671a. doi:10.1523/JNEUROSCI.1858-12.2012
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132. doi:10.1038/nature07761
Leshchyns'ka, I., and Sytnyk, V. (2016). Synaptic cell adhesion molecules in Alzheimer's disease. Neural plasticity, 6427537. doi:10.1155/2016/6427537
Leshchyns’ka, I., Liew, H. T., Shepherd, C., Halliday, G. M., Stevens, C. H., Ke, Y. D., et al. (2015). Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease. Nat. Commun. 6, 8836. doi:10.1038/ncomms9836
Libeu, C. A. P., Descamps, O., Zhang, Q., John, V., and Bredesen, D. E. (2012). Altering APP proteolysis: Increasing sAPPalpha production by targeting dimerization of the APP ectodomain. PLoS one 7, e40027. doi:10.1371/journal.pone.0040027
Lincoln, S., Allen, M., Cox, C. L., Walker, L. P., Malphrus, K., Qiu, Y., et al. (2013). LRRTM3 interacts with APP and BACE1 and has variants associating with late-onset Alzheimer’s disease (LOAD). PLoS one 8, e64164. doi:10.1371/journal.pone.0064164
Lorenzo, A., Yuan, M., Zhang, Z., Paganetti, P. A., Sturchler-Pierrat, C., Staufenbiel, M., et al. (2000). Amyloid β interacts with the amyloid precursor protein: A potential toxic mechanism in alzheimer's disease. Nat. Neurosci. 3, 460–464. doi:10.1038/74833
Ma, Q.-H., Futagawa, T., Yang, W.-L., Jiang, X.-D., Zeng, L., Takeda, Y., et al. (2008). A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat. Cell. Biol. 10, 283–294. doi:10.1038/ncb1690
Majercak, J., Ray, W. J., Espeseth, A., Simon, A., Shi, X.-P., Wolffe, C., et al. (2006). LRRTM3 promotes processing of amyloid-precursor protein by BACE1 and is a positional candidate gene for late-onset Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 103, 17967–17972. doi:10.1073/pnas.0605461103
Maness, P. F., and Schachner, M. (2007). Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 10, 19–26. doi:10.1038/nn1827
Mao, Y., and Freeman, M. (2009). Fasciclin 2, the drosophila orthologue of neural cell-adhesion molecule, inhibits EGF receptor signalling. Development 136, 473–481. doi:10.1242/dev.026054
Marambaud, P., Shioi, J., Serban, G., Georgakopoulos, A., Sarner, S., Nagy, V., et al. (2002). A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 21, 1948–1956. doi:10.1093/emboj/21.8.1948
Maretzky, T., Reiss, K., Ludwig, A., Buchholz, J., Scholz, F., Proksch, E., et al. (2005). ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and β-catenin translocation. Proc. Natl. Acad. Sci. U. S. A. 102, 9182–9187. doi:10.1073/pnas.0500918102
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., Mcdonald, B. L., Beyreuther, K., et al. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 82, 4245–4249. doi:10.1073/pnas.82.12.4245
Matter, M. L., Zhang, Z., Nordstedt, C., and Ruoslahti, E. (1998). The alpha5beta1 integrin mediates elimination of amyloid-beta peptide and protects against apoptosis. J. Cell. Biol. 141, 1019–1030. doi:10.1083/jcb.141.4.1019
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 330, 1774. doi:10.1126/science.1197623
Mayer, M. C., Schauenburg, L., Thompson-Steckel, G., Dunsing, V., Kaden, D., Voigt, P., et al. (2016). Amyloid precursor-like protein 1 (APLP1) exhibits stronger zinc-dependent neuronal adhesion than amyloid precursor protein and APLP2. J. Neurochem. 137, 266–276. doi:10.1111/jnc.13540
Medway, C., Shi, H., Bullock, J., Black, H., Brown, K., Vafadar-Isfahani, B., et al. (2010). Using in silico LD clumping and meta-analysis of genome-wide datasets as a complementary tool to investigate and validate new candidate biomarkers in Alzheimer's disease. Int. J. Mol. Epidemiol. Genet. 1, 134–144.
Mikkonen, M., Soininen, H., Tapiola, T., Alafuzoff, I., and Miettinen, R. (1999). Hippocampal plasticity in alzheimer's disease: Changes in highly polysialylated NCAM immunoreactivity in the hippocampal formation. Eur. J. Neurosci. 11, 1754–1764. doi:10.1046/j.1460-9568.1999.00593.x
Müller, U. C., and Zheng, H. (2012). Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2, a006288. doi:10.1101/cshperspect.a006288
Munter, L.-M., Voigt, P., Harmeier, A., Kaden, D., Gottschalk, K. E., Weise, C., et al. (2007). GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 26, 1702–1712. doi:10.1038/sj.emboj.7601616
Murray, H. C., Low, V. F., Swanson, M. E. V., Dieriks, B. V., Turner, C., Faull, R. L. M., et al. (2016). Distribution of PSA-NCAM in normal, Alzheimer’s and Parkinson’s disease human brain. Neuroscience 330, 359–375. doi:10.1016/j.neuroscience.2016.06.003
Naito, Y., Tanabe, Y., Lee, A. K., Hamel, E., and Takahashi, H. (2017). Amyloid-β oligomers interact with neurexin and diminish neurexin-mediated excitatory presynaptic organization. Sci. Rep. 7, 42548. doi:10.1038/srep42548
Neiiendam, J. L., Køhler, L. B., Christensen, C., Li, S., Pedersen, M. V., Ditlevsen, D. K., et al. (2004). An NCAM-derived FGF-receptor agonist, the FGL-peptide, induces neurite outgrowth and neuronal survival in primary rat neurons. J. Neurochem. 91, 920–935. doi:10.1111/j.1471-4159.2004.02779.x
Neumann, S., Schöbel, S., Jäger, S., Trautwein, A., Haass, C., Pietrzik, C. U., et al. (2006). Amyloid precursor-like protein 1 influences endocytosis and proteolytic processing of the amyloid precursor protein. J. Biol. Chem. 281, 7583–7594. doi:10.1074/jbc.M508340200
Niederhoffer, N., Levy, R., Sick, E., Andre, P., Coupin, G., Lombard, Y., et al. (2009). Amyloid β peptides trigger CD47-dependent mast cell secretory and phagocytic responses. Int. J. Immunopathol. Pharmacol. 22, 473–483. doi:10.1177/039463200902200224
Noda, Y., Asada, M., Kubota, M., Maesako, M., Watanabe, K., Uemura, M., et al. (2013). Copper enhances APP dimerization and promotes Aβ production. Neurosci. Lett. 547, 10–15. doi:10.1016/j.neulet.2013.04.057
Norstrom, E. M., Zhang, C., Tanzi, R., and Sisodia, S. S. (2010). Identification of NEEP21 as a ß-amyloid precursor protein-interacting protein in vivo that modulates amyloidogenic processing in vitro. J. Neurosci. 30, 15677–15685. doi:10.1523/JNEUROSCI.4464-10.2010
O'sullivan, M. L., de Wit, J., Savas, J. N., Comoletti, D., Otto-Hitt, S., Yates, J. R., 3R. D., et al. (2012). FLRT proteins are endogenous latrophilin ligands and regulate excitatory synapse development. Neuron 73, 903–910. doi:10.1016/j.neuron.2012.01.018
Onodera, W., Asahi, T., and Sawamura, N. (2021). Rapid evolution of mammalian APLP1 as a synaptic adhesion molecule. Sci. Rep. 11, 11305. doi:10.1038/s41598-021-90737-y
Ortiz-Sanz, C., Gaminde-Blasco, A., Valero, J., Bakota, L., Brandt, R., Zugaza, J. L., et al. (2020). Early effects of Aβ Oligomers on dendritic spine dynamics and arborization in hippocampal neurons. Front. Synaptic Neurosci. 12, 2. doi:10.3389/fnsyn.2020.00002
Osterfield, M., Egelund, R., Young, L. M., and Flanagan, J. G. (2008). Interaction of amyloid precursor protein with contactins and NgCAM in the retinotectal system. Development 135, 1189–1199. doi:10.1242/dev.007401
Osterhout, J. A., Stafford, B. K., Nguyen, P. L., Yoshihara, Y., and Huberman, A. D. (2015). Contactin-4 mediates axon-target specificity and functional development of the accessory optic system. Neuron 86, 985–999. doi:10.1016/j.neuron.2015.04.005
Parkin, E. T., Watt, N. T., Hussain, I., Eckman, E. A., Eckman, C. B., Manson, J. C., et al. (2007). Cellular prion protein regulates β-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A. 104, 11062–11067. doi:10.1073/pnas.0609621104
Parvathy, S., Hussain, I., Karran, E. H., Turner, A. J., and Hooper, N. M. (1999). Cleavage of Alzheimer's amyloid precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry 38, 9728–9734. doi:10.1021/bi9906827
Peng, X., Williams, J., Smallwood, P. M., and Nathans, J. (2019). Defining the binding interface of amyloid precursor protein (APP) and contactin3 (CNTN3) by site-directed mutagenesis. PLoS one 14, e0219384. doi:10.1371/journal.pone.0219384
Peters, C., Espinoza, M. P., Gallegos, S., Opazo, C., and Aguayo, L. G. (2015). Alzheimer's Aβ interacts with cellular prion protein inducing neuronal membrane damage and synaptotoxicity. Neurobiol. Aging 36, 1369–1377. doi:10.1016/j.neurobiolaging.2014.11.019
Petit, C. S. V., Besnier, L., Morel, E., Rousset, M., and Thenet, S. (2013). Roles of the cellular prion protein in the regulation of cell-cell junctions and barrier function. Tissue Barriers 1, e24377. doi:10.4161/tisb.24377
Prince, M. J., Wimo, A., Guerchet, M. M., Ali, G. C., Wu, Y.-T., and Prina, M. (2015). World alzheimer report 2015 - the global impact of dementia. London.Alzheimer's Dis. Int.
Puzzo, D., Bizzoca, A., Loreto, C., Guida, C. A., Gulisano, W., Frasca, G., et al. (2015). Role of F3/contactin expression profile in synaptic plasticity and memory in aged mice. Neurobiol. Aging 36, 1702–1715. doi:10.1016/j.neurobiolaging.2015.01.004
Puzzo, D., Privitera, L., Fa, M., Staniszewski, A., Hashimoto, G., Aziz, F., et al. (2011). Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 69, 819–830. doi:10.1002/ana.22313
Quintela-López, T., Ortiz-Sanz, C., Serrano-Regal, M. P., Gaminde-Blasco, A., Valero, J., Baleriola, J., et al. (2019). Aβ oligomers promote oligodendrocyte differentiation and maturation via integrin β1 and Fyn kinase signaling. Cell. Death Dis. 10, 445. doi:10.1038/s41419-019-1636-8
Rahman, M. M., Westermark, G. T., Zetterberg, H., Härd, T., and Sandgren, M. (2018). Protofibrillar and fibrillar amyloid-β binding proteins in cerebrospinal fluid. J. Alzheimers Dis. 66, 1053–1064. doi:10.3233/JAD-180596
Rasmussen, K. K., Falkesgaard, M. H., Winther, M., Roed, N. K., Quistgaard, C. L., Teisen, M. N., et al. (2018). NCAM2 fibronectin type-III domains form a rigid structure that binds and activates the fibroblast growth factor receptor. Sci. Rep. 8, 8957. doi:10.1038/s41598-018-27089-7
Reissner, C., Runkel, F., and Missler, M. (2013). Neurexins. Genome Biol. 14, 213. doi:10.1186/gb-2013-14-9-213
Reitz, C., Conrad, C., Roszkowski, K., Rogers, R. S., and Mayeux, R. (2012). Effect of genetic variation in LRRTM3 on risk of Alzheimer disease. Arch. Neurol. 69, 894–900. doi:10.1001/archneurol.2011.2463
Rice, H. C., Young-Pearse, T. L., and Selkoe, D. J. (2013). Systematic evaluation of candidate ligands regulating ectodomain shedding of amyloid precursor protein. Biochemistry 52, 3264–3277. doi:10.1021/bi400165f
Riemenschneider, M., Klopp, N., Xiang, W., Wagenpfeil, S., Vollmert, C., Müller, U., et al. (2004). Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology 63, 364–366. doi:10.1212/01.wnl.0000130198.72589.69
Ristori, E., Cicaloni, V., Salvini, L., Tinti, L., Tinti, C., Simons, M., et al. (2020). Amyloid-β precursor protein APP down-regulation alters actin cytoskeleton-interacting proteins in endothelial cells. Cells 9, E2506. doi:10.3390/cells9112506
Rohan De Silva, H. A., Jen, A., Wickenden, C., Jen, L. S., Wilkinson, S. L., and Patel, A. J. (1997). Cell-specific expression of beta-amyloid precursor protein isoform mRNAs and proteins in neurons and astrocytes. Brain Res. Mol. Brain Res. 47, 147–156. doi:10.1016/s0169-328x(97)00045-4
Roucou, X., and Leblanc, A. C. (2005). Cellular prion protein neuroprotective function: Implications in prion diseases. J. Mol. Med. 83, 3–11. doi:10.1007/s00109-004-0605-5
Rushworth, J. V., Griffiths, H. H., Watt, N. T., and Hooper, N. M. (2013). Prion protein-mediated toxicity of amyloid-β oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 288, 8935–8951. doi:10.1074/jbc.M112.400358
Sabo, S., Lambert, M. P., Kessey, K., Wade, W., Krafft, G., Klein, W. L., et al. (1995). Interaction of beta-amyloid peptides with integrins in a human nerve cell line. Neurosci. Lett. 184, 25–28. doi:10.1016/0304-3940(94)11159-g
Sakono, M., and Zako, T. (2010). Amyloid oligomers: formation and toxicity of abeta oligomers. FEBS J. 277, 1348–1358. doi:10.1111/j.1742-4658.2010.07568.x
Sandbrink, R., Masters, C. L., and Beyreuther, K. (1996). APP gene family alternative splicing generates functionally related isoforms. Ann. N. Y. Acad. Sci. 777, 281–287. doi:10.1111/j.1749-6632.1996.tb34433.x
Santuccione, A., Sytnyk, V., Leshchyns'Ka, I., and Schachner, M. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell. Biol. 169, 341–354. doi:10.1083/jcb.200409127
Sathe, G., Mangalaparthi, K. K., Jain, A., Darrow, J., Troncoso, J., Albert, M., et al. (2020). Multiplexed phosphoproteomic study of brain in patients with Alzheimer's disease and age-matched cognitively healthy controls. Omics 24, 216–227. doi:10.1089/omi.2019.0191
Sato, T., Tang, T.-C., Reubins, G., Fei, J. Z., Fujimoto, T., Kienlen-Campard, P., et al. (2009). A helix-to-coil transition at the ε-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc. Natl. Acad. Sci. U. S. A. 106, 1421–1426. doi:10.1073/pnas.0812261106
Schettini, G., Govoni, S., Racchi, M., and Rodriguez, G. (2010). Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: Signal transduction and/or transcriptional role – relevance for alzheimer pathology. J. Neurochem. 115, 1299–1308. doi:10.1111/j.1471-4159.2010.07044.x
Scheuermann, S., Hambsch, B., Hesse, L., Stumm, J., Schmidt, C., Beher, D., et al. (2001). Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J. Biol. Chem. 276, 33923–33929. doi:10.1074/jbc.M105410200
Schilling, S., Mehr, A., Ludewig, S., Stephan, J., Zimmermann, M., August, A., et al. (2017). APLP1 is a synaptic cell adhesion molecule, supporting maintenance of dendritic spines and basal synaptic transmission. J. Neurosci. 37, 5345–5365. doi:10.1523/JNEUROSCI.1875-16.2017
Schmidt, V., Baum, K., Lao, A., Rateitschak, K., Schmitz, Y., Teichmann, A., et al. (2012). Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer's disease. EMBO J. 31, 187–200. doi:10.1038/emboj.2011.352
Schmitt-Ulms, G., Hansen, K., Liu, J., Cowdrey, C., Yang, J., Dearmond, S. J., et al. (2004). Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat. Biotechnol. 22, 724–731. doi:10.1038/nbt969
Schroeder, A., and De Wit, J. (2018). Leucine-rich repeat-containing synaptic adhesion molecules as organizers of synaptic specificity and diversity. Exp. Mol. Med. 50, 10. doi:10.1038/s12276-017-0023-8
Scott-Mckean, J. J., Surewicz, K., Choi, J.-K., Ruffin, V. A., Salameh, A. I., Nieznanski, K., et al. (2016). Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Aβ oligomers: Implications for novel therapeutic strategy in Alzheimer's disease. Neurobiol. Dis. 91, 124–131. doi:10.1016/j.nbd.2016.03.001
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608. doi:10.15252/emmm.201606210
Selkoe, D. J. (1991). The molecular pathology of Alzheimer's disease. Neuron 6, 487–498. doi:10.1016/0896-6273(91)90052-2
Shaked, G. M., Kummer, M. P., Lu, D. C., Galvan, V., Bredesen, D. E., Koo, E. H., et al. (2006). Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597-624). FASEB J. 20, 1254–1256. doi:10.1096/fj.05-5032fje
Shih, W., and Yamada, S. (2012). N-cadherin-mediated cell–cell adhesion promotes cell migration in a three-dimensional matrix. J. Cell. Sci. 125, 3661–3670. doi:10.1242/jcs.103861
Shimazaki, K., Hosoya, H., Takeda, Y., Kobayashi, S., and Watanabe, K. (1998). Age-related decline of F3/Contactin in rat hippocampus. Neurosci. Lett. 245, 117–120. doi:10.1016/s0304-3940(98)00179-7
Shimoda, Y., and Watanabe, K. (2009). Contactins: Emerging key roles in the development and function of the nervous system. Contactins. Cell. Adhesion Migr. 3, 64–70. doi:10.4161/cam.3.1.7764
Shu, J., Li, N., Wei, W., and Zhang, L. (2022). Detection of molecular signatures and pathways shared by Alzheimer’s disease and type 2 diabetes. Gene 810, 146070. doi:10.1016/j.gene.2021.146070
Siano, G., Caiazza, M. C., Ollà, I., Varisco, M., Madaro, G., Quercioli, V., et al. (2019). Identification of an ERK inhibitor as a therapeutic drug against tau aggregation in a new cell-based assay. Front. Cell. Neurosci. 13, 386. doi:10.3389/fncel.2019.00386
Small, D., Nurcombe, V., Reed, G., Clarris, H., Moir, R., Beyreuther, K., et al. (1994). A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J. Neurosci. 14, 2117–2127. doi:10.1523/jneurosci.14-04-02117.1994
So, P. P., Zeldich, E., Seyb, K. I., Huang, M. M., Concannon, J. B., King, G. D., et al. (2012). Lowering of amyloid beta peptide production with a small molecule inhibitor of amyloid-β precursor protein dimerization. Am. J. Neurodegener. Dis. 1, 75–87.
Soba, P., Eggert, S., Wagner, K., Zentgraf, H., Siehl, K., Kreger, S., et al. (2005). Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 24, 3624–3634. doi:10.1038/sj.emboj.7600824
Sondag, C. M., and Combs, C. K. (2004). Amyloid precursor protein mediates proinflammatory activation of monocytic lineage cells. J. Biol. Chem. 279, 14456–14463. doi:10.1074/jbc.M313747200
Stahl, R., Schilling, S., Soba, P., Rupp, C., Hartmann, T., Wagner, K., et al. (2014). Shedding of APP limits its synaptogenic activity and cell adhesion properties. Front. Cell. Neurosci. 8, 410. doi:10.3389/fncel.2014.00410
Strekalova, H., Buhmann, C., Kleene, R., Eggers, C., Saffell, J., Hemperly, J., et al. (2006). Elevated levels of neural recognition molecule L1 in the cerebrospinal fluid of patients with Alzheimer disease and other dementia syndromes. Neurobiol. Aging 27, 1–9. doi:10.1016/j.neurobiolaging.2004.11.013
Sushma, , and Mondal, A. C. (2019). Role of GPCR signaling and calcium dysregulation in Alzheimer's disease. Mol. Cell. Neurosci. 101, 103414. doi:10.1016/j.mcn.2019.103414
Sytnyk, V., Leshchyns’Ka, I., and Schachner, M. (2017). Neural cell adhesion molecules of the immunoglobulin superfamily regulate synapse formation, maintenance, and function. Trends Neurosci. 40, 295–308. doi:10.1016/j.tins.2017.03.003
Tachi, N., Hashimoto, Y., Nawa, M., and Matsuoka, M. (2010). TAG-1 is an inhibitor of TGFbeta2-induced neuronal death via amyloid beta precursor protein. Biochem. Biophys. Res. Commun. 394, 119–125. doi:10.1016/j.bbrc.2010.02.127
Takada, Y., Ye, X., and Simon, S. (2007). The integrins. Genome Biol. 8, 215. doi:10.1186/gb-2007-8-5-215
Takei, N., Sobu, Y., Kimura, A., Urano, S., Piao, Y., Araki, Y., et al. (2015). Cytoplasmic fragment of alcadein α generated by regulated intramembrane proteolysis enhances amyloid β-protein precursor (APP) transport into the late secretory pathway and facilitates APP cleavage. J. Biol. Chem. 290, 987–995. doi:10.1074/jbc.M114.599852
Tan, J. Z. A., and Gleeson, P. A. (2019). The trans-Golgi network is a major site for α-secretase processing of amyloid precursor protein in primary neurons. J. Biol. Chem. 294, 1618–1631. doi:10.1074/jbc.RA118.005222
Tang, S.-Y., Liu, D.-X., Li, Y., Wang, K.-J., Wang, X.-F., Su, Z.-K., et al. (2020). Caspr1 facilitates sAPPα production by regulating α-secretase ADAM9 in brain endothelial cells. Front. Mol. Neurosci. 13, 23. doi:10.3389/fnmol.2020.00023
Tanzi, R. E., Gusella, J. F., Watkins, P. C., Bruns, G. A., St George-Hyslop, P., van Keuren, M. L., et al. (1987). Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the alzheimer locus. Science 235, 880–884. doi:10.1126/science.2949367
Tian, X., Liu, Z., Niu, B., Zhang, J., Tan, T. K., Lee, S. R., et al. (20112011). E-cadherin/β-catenin complex and the epithelial barrier. J. Biomed. Biotechnol., 567305. doi:10.1155/2011/567305
Todaro, L., Puricelli, L., Gioseffi, H., Guadalupe Pallotta, M. A., Lastiri, J., Balde de Kier Joffé, E., et al. (2004). Neural cell adhesion molecule in human serum Increased levels in dementia of the Alzheimer type. Neurobiol. Dis. 15, 387–393. doi:10.1016/j.nbd.2003.11.014
Ulbrich, S., Janning, P., Seidel, R., Matschke, J., Gonsberg, A., Jung, S., et al. (2018). Alterations in the brain interactome of the intrinsically disordered N-terminal domain of the cellular prion protein (PrPC) in Alzheimer’s disease. PLoS one 13, e0197659. doi:10.1371/journal.pone.0197659
Um, J. I. W., Kaufman, A. D. A. M. C., Kostylev, M., Heiss, J. A. C. Q. U. E. L. I. N. E. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron 79, 887–902. doi:10.1016/j.neuron.2013.06.036
Varoqueaux, F., Jamain, S., and Brose, N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell. Biol. 83, 449–456. doi:10.1078/0171-9335-00410
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi:10.1126/science.286.5440.735
Venkatasubramaniam, A., Drude, A., and Good, T. (2014). Role of N-terminal residues in Aβ interactions with integrin receptor and cell surface. Biochim. Biophys. Acta 1838, 2568–2577. doi:10.1016/j.bbamem.2014.06.011
Verdier, Y., Huszár, E., Penke, B., Penke, Z., Woffendin, G., Scigelova, M., et al. (2005). Identification of synaptic plasma membrane proteins co-precipitated with fibrillar β-amyloid peptide. J. Neurochem. 94, 617–628. doi:10.1111/j.1471-4159.2005.03158.x
Walsh, D. M., Minogue, A. M., Sala Frigerio, C., Fadeeva, J. V., Wasco, W., Selkoe, D. J., et al. (2007). The APP family of proteins: Similarities and differences. Biochem. Soc. Trans. 35, 416–420. doi:10.1042/BST0350416
Wang, H., Ren, C. H., Gunawardana, C. G., and Schmitt-Ulms, G. (2013). Overcoming barriers and thresholds – signaling of oligomeric Aβ through the prion protein to Fyn. Mol. Neurodegener. 8, 24. doi:10.1186/1750-1326-8-24
Wang, X.-L., and Li, L. (2021). Cell type-specific potential pathogenic genes and functional pathways in Alzheimer’s Disease. BMC Neurol. 21, 381. doi:10.1186/s12883-021-02407-1
Wang, Y., and Ha, Y. (2004). The x-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol. Cell. 15, 343–353. doi:10.1016/j.molcel.2004.06.037
Wang, Z., Jackson, R. J., Hong, W., Taylor, W. M., Corbett, G. T., Moreno, A., et al. (2017). Human brain-derived Aβ oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J. Neurosci. 37, 11947–11966. doi:10.1523/JNEUROSCI.2009-17.2017
Watts, J. C., Bourkas, M. E. C., and Arshad, H. (2018). The function of the cellular prion protein in health and disease. Acta Neuropathol. 135, 159–178. doi:10.1007/s00401-017-1790-y
Weledji, E. P., and Assob, J. C. (2014). The ubiquitous neural cell adhesion molecule (N-CAM). Ann. Med. Surg. 3, 77–81. doi:10.1016/j.amsu.2014.06.014
Whitehouse, I. J., Miners, J. S., Glennon, E. B. C., Kehoe, P. G., Love, S., Kellett, K. A. B., et al. (2013). Prion protein is decreased in Alzheimer's brain and inversely correlates with BACE1 activity, amyloid-β levels and Braak stage. PLoS one 8, e59554. doi:10.1371/journal.pone.0059554
Winther, M., Berezin, V., and Walmod, P. S. (2012). NCAM2/OCAM/RNCAM: Cell adhesion molecule with a role in neuronal compartmentalization. Int. J. Biochem. Cell. Biol. 44, 441–446. doi:10.1016/j.biocel.2011.11.020
Woo, J. A., Zhao, X., Khan, H., Penn, C., Wang, X., Joly-Amado, A., et al. (2015). Slingshot-Cofilin activation mediates mitochondrial and synaptic dysfunction via Aβ ligation to β1-integrin conformers. Cell. Death Differ. 22, 921–934. doi:10.1038/cdd.2015.5
Wright, S., Malinin, N. L., Powell, K. A., Yednock, T., Rydel, R. E., Griswold-Prenner, I., et al. (2007). Alpha2beta1 and alphaVbeta1 integrin signaling pathways mediate amyloid-beta-induced neurotoxicity. Neurobiol. Aging 28, 226–237. doi:10.1016/j.neurobiolaging.2005.12.002
Wulf, M. A., Senatore, A., and Aguzzi, A. (2017). The biological function of the cellular prion protein: An update. BMC Biol. 15, 34. doi:10.1186/s12915-017-0375-5
Xia, M., Cheng, X., Yi, R., Gao, D., and Xiong, J. (2016). The binding receptors of Aβ: An alternative therapeutic target for Alzheimer’s disease. Mol. Neurobiol. 53, 455–471. doi:10.1007/s12035-014-8994-0
Xu, X.-F., Wang, Y.-C., Zong, L., Chen, Z.-Y., and Li, Y. (2018). Elevating integrin-linked kinase expression has rescued hippocampal neurogenesis and memory deficits in an AD animal model. Brain Res. 1695, 65–77. doi:10.1016/j.brainres.2018.05.024
Yamazaki, T., Koo, E. H., and Selkoe, D. J. (1997). Cell surface amyloid β-protein precursor colocalizes with β1 integrins at substrate contact sites in neural cells. J. Neurosci. 17, 1004–1010. doi:10.1523/jneurosci.17-03-01004.1997
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., et al. (1999). Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 402, 533–537. doi:10.1038/990107
Yin, Z., Raj, D., Saiepour, N., van Dam, D., Brouwer, N., Holtman, I. R., et al. (2017). Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer's disease. Neurobiol. Aging 55, 115–122. doi:10.1016/j.neurobiolaging.2017.03.021
Young-Pearse, T. L., Chen, A. C., Chang, R., Marquez, C., and Selkoe, D. J. (2008). Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 3, 15. doi:10.1186/1749-8104-3-15
Yu, Y., Li, Y., and Zhang, Y. (2016). Yeast two-hybrid screening for proteins that interact with the extracellular domain of amyloid precursor protein. Neurosci. Bull. 32, 171–176. doi:10.1007/s12264-016-0021-1
Zhang, Y.-W., Thompson, R., Zhang, H., and Xu, H. (2011). APP processing in Alzheimer's disease. Mol. Brain 4, 3. doi:10.1186/1756-6606-4-3
Keywords: cell adhesion molecule (CAM), Alzheimer’s disease, amyloid precursor protein (APP), amyloid-beta, immunoglobulin superfamily, integrin, prion protein (PrP), neurexin
Citation: Pfundstein G, Nikonenko AG and Sytnyk V (2022) Amyloid precursor protein (APP) and amyloid β (Aβ) interact with cell adhesion molecules: Implications in Alzheimer’s disease and normal physiology. Front. Cell Dev. Biol. 10:969547. doi: 10.3389/fcell.2022.969547
Received: 15 June 2022; Accepted: 07 July 2022;
Published: 26 July 2022.
Edited by:
Claudia Tanja Mierke, Leipzig University, GermanyReviewed by:
Ilaria Canobbio, University of Pavia, ItalyCopyright © 2022 Pfundstein, Nikonenko and Sytnyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir Sytnyk, di5zeXRueWtAdW5zdy5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.