 Mariana A. Amorós
Mariana A. Amorós Esther S. Choi
Esther S. Choi Axel R. Cofré
Axel R. Cofré Nikolay V. Dokholyan
Nikolay V. Dokholyan Marcelo Duzzioni
Marcelo Duzzioni
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 24 August 2022
Sec. Stem Cell Research
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.962881
This article is part of the Research TopicAdvances in iPSC Technology for Disease Modeling and Therapeutic ApplicationsView all 8 articles
The development of cell culture models that recapitulate the etiology and features of nervous system diseases is central to the discovery of new drugs and their translation onto therapies. Neuronal tissues are inaccessible due to skeletal constraints and the invasiveness of the procedure to obtain them. Thus, the emergence of induced pluripotent stem cell (iPSC) technology offers the opportunity to model different neuronal pathologies. Our focus centers on iPSCs derived from amyotrophic lateral sclerosis (ALS) patients, whose pathology remains in urgent need of new drugs and treatment. In this sense, we aim to revise the process to obtain motor neurons derived iPSCs (iPSC-MNs) from patients with ALS as a drug screening model, review current 3D-models and offer a perspective on bioinformatics as a powerful tool that can aid in the progress of finding new pharmacological treatments.
ALS is a complex and currently uncurable neurodegenerative disease that affects both upper and lower motor neurons (MNs). Although considered rare, it is the most common motor neuron disease, with an incidence of 1–2 cases per 100.000 people per year (Logroscino and Piccininni, 2019). This pathology presents a late-onset, with an average of 50–60 years of age, that progresses from muscle weakness towards the patient’s eventual decease due to respiratory muscle failure (Bruijn et al., 2004; Pasinelli and Brown, 2006). It is estimated that 90% of ALS cases have an unspecific etiology, and thus are categorized as apparently sporadic ALS (sALS). Familial ALS (fALS), which represents the remaining 10% of cases, is defined by specific genetic alterations and traced through the patient’s family history. Both categories, although widely used, can be biased and unreliable in their distinction (Brown and Al-Chalabi, 2017; Turner et al., 2017). Factors such as unknown family history, non-paternity and incomplete genetic penetrance, contribute to this misclassification (Robberecht and Philips, 2013). Lastly, the disease’s complexity, due to symptoms’ heterogeneity -rate of progression, onset site, and the presence of various degrees of cognitive dysfunction- confers further difficulties in providing effective treatment.
ALS patients’ life expectancy is about 3–5 years from symptoms onset, with current drug treatments providing limited improvement for select patients (Brown and Al-Chalabi, 2017). In this sense, therapeutic and pharmacological advances have been slow, mainly attributed to the limitations in the disease’s modeling. Neuropathological tissue samples are difficult to obtain, and restricted to rare invasive biopsies and post-mortem tissues (Stan et al., 2006). Furthermore, they model the final stage of the disease and therefore are unreliable for studying its progression. while animal models are useful to elucidate some of the disease’s underlying mechanisms, and most interestingly, to observe their behavioral cues, their translational value for drug development is yet to be proven (Van Norman, 2019). The intrinsic differences between species, including immunological response and anatomical structures, such as the absence of neurological disease-relevant tissues (Eaton and Wishart, 2017) contribute to their limitations. Furthermore, animal models can only replicate hereditable traits, excluding sporadic forms that, as mentioned, represents most diagnosed cases. In this context, the introduction of iPSC technology has allowed an unprecedented opportunity to model nervous system diseases, from both genetic and idiopathic origin. We are certain that advances in 3D cell culture systems coupled with the integration of bioinformatics tools will considerably change our current understanding and therapeutic landscape of ALS.
In broad terms, the pathological hallmark of ALS involves the degeneration of MNs of the motor cortex, spinal anterior horn, and the lateral columns of the spinal cord (Saberi et al., 2015). Patients with ALS exhibit both upper and lower MN disease symptoms such as muscle weakness, hyper reflexivity, spasticity, and/or rigidity (Redler and Dokholyan, 2012). Patients have also reported symptoms involving exercise intolerance and cognitive impairment (Mezzani et al., 2012; Chiò et al., 2017; Cividini et al., 2021). The inexorable progression of muscle weakness leading towards disability, combined with cognitive changes (Phukan et al., 2007), is devastating for both patient and loved ones. Due to the severe impact of the disease, there is an urgent need to understand the genetic and molecular underpinnings of ALS.
From decades of research dedicated to uncovering the genetic basis of ALS, researchers have discovered over 50 genes to be potentially causative or disease-modifying (Mejzini et al., 2019). The major genes described for ALS include C9orf72, SOD1, TARDBP, and FUS. The C9orf72 gene contains a hexanucleotide repeat expansion of the six-letter string of nucleotides GGGGCC. The normal length of the repeat consists of about 2–24 nucleotides, whereas mutation carriers have more than 30 and even up to hundreds or thousands of repeats (Smeyers et al., 2021). C9orf72 accounts for about 33% of fALS and less than 5% of sALS cases in European populations (Zou et al., 2017). SOD1 was the first gene discovered for fALS and codes for superoxide dismutase 1. Although the role of SOD1 is heavily debated, there is evidence for a toxic gain-of-function (Healy et al., 2020). Mutations in SOD1 account for about 30% of fALS, and 2–7% of sALS cases (Bernard et al., 2020). TDP-43 is encoded by the TARDBP gene and is a transcriptional repressor. TARDBP mutations only account for about 3% of fALS (Rutherford et al., 2008). Finally, FUS encodes an RNA binding protein with the mutations accounting for about 1% of fALS and less than 1% of sALS. FUS mutations are associated with juvenile ALS presenting basophilic inclusions (Picher-Martel et al., 2020).
The genes described above share and differ in the disrupted cellular processes. C9orf72, SOD1, TARDBP, and FUS mutations are associated with impaired proteostasis, but other processes, such as cytoskeleton and axon-transport defects, are associated with SOD1, while disturbed RNA metabolism is seen in TARDBP and FUS. There are several other genes with known disease-significance, such as TP73 (Russell et al., 2021), which is implicated in apoptosis (Wood, 2021).
The identification of new genes and mutations is increasing due to technological advances and increased awareness. With the great number of genes implicated in the disease, and even greater number of mutations associated with each gene, it becomes a challenging task to unveil the critical pathological mechanism of ALS. A characteristic molecular feature is the presence of cytoplasmic inclusions. For example, TDP-43 is a protein present in these inclusions, which are shared between both sALS and fALS (Gruzman et al., 2007; Prasad et al., 2019). In this sense, deciphering the mechanisms of protein misfolding and the role of misfolded protein is key to understanding pivotal aspects of ALS pathomechanisms. Interestingly, recent research has attributed soluble misfolded proteins as the toxic player in neurodegeneration rather than the large aggregates (Kirkitadze et al., 2002; Urushitani et al., 2006; Proctor et al., 2016; Choi and Dokholyan, 2021), implying a protective mechanistic role of large aggregates (Zhu et al., 2018). The toxic mechanism of soluble misfolded proteins is unknown, but major cellular processes include dysfunctional RNA metabolism, impaired proteostasis, mitochondrial dysfunction, and excitotoxicity (Ruegsegger and Saxena, 2016; Butti and Patten, 2019; Calió et al., 2020; Gunes et al., 2020). With the complexity of the aberrant cellular processes and unknown disease triggers, it poses a challenge for developing an effective, common therapeutic.
Historically, neurological disease research focused on neurons as a single and isolated functioning unit. However, dismissing the complex interplay of the stroma and other disease-implicated cell types results in a lack of physiological relevance, more so when modeling a systemic disease. ALS possesses great genotypic and phenotypic heterogeneity, where both cell-autonomous and non-cell-autonomous factors drive the diseases’ progression towards common clinical manifestations. MNs and glial cells are in continuous interaction in order to maintain proper tissue homeostasis. Among other functions, glial cells provide trophic factors, myelinate axons, and clear cell debris, that is, they are critical for health maintenance and survival. Transcriptomics data revealed that multiple non-neuronal cell types are vulnerable to dysfunction and are crucial to maintaining neuronal health (Enright et al., 2020; Gleichman and Carmichael, 2020). Glial cells have been implicated in the initiation of ALS, and its progression (Brites and Vaz, 2014). In addition, astrocytes from SOD1 mice are sufficient to kill motor neurons in WT mice supporting non-cell autonomous effects (Nagai et al., 2007) iPSC derived astrocytes from ALS patients showed impaired autophagy through non-cell-autonomous mechanisms (Madill et al., 2017). iPSCs and co-cultures are potentially beneficial to incorporate critical controllers of neuronal health into ALS studies. This involvement of non-neuronal cell types is consistent with the non-cell-autonomous theory that challenges the cell-autonomous theory (Chen et al., 2018; Argueti-Ostrovsky et al., 2021).
The complexity of ALS, evidenced by the number of cellular aberrant processes and its heterogeneity indicates that the disease is most likely caused by multiple factors rather than a single inciting event (Chiò et al., 2018; Mejzini et al., 2019). Improving models of disease by using iPSCs and 3D-cultures can aid in highlighting the most critical cellular processes disturbed in ALS disease pathogenesis and thus, allow for the discovery of new therapeutic compounds.
Briefly, in 2006, Takahashi and Yamanaka reported the development of iPSCs, via retroviral transduction of four transcription factors (OCT3/4, SOX2, KLF4, and c-MYC) delivered into mouse fibroblasts. These cells were characterized as embryonic stem cells (ESCs)-like concerning their morphology, growth pattern, ability to differentiate, express pluripotent cell markers, and form teratomas after being injected into immunodeficient mice (Takahashi and Yamanaka, 2006). Shortly after, iPSCs generated from human somatic cells were introduced (Takahashi et al., 2007; Wernig et al., 2007; Yu et al., 2007; Kim et al., 2008; Park et al., 2008). Most importantly, these cells bypass ethical concerns associated with the destruction of human embryos, and are easily obtained by avoiding invasive procedures (Zarzeczny et al., 2009; Niemiec and Howard, 2020). Furthermore, as bona fide stem cells, their self-renewal capabilities offer a robust supply of unlimited patient-derived cells, which is especially relevant, as the rare-to-obtain neural tissue samples lack the cell quantity needed for drug screening assays. However, there are some disadvantages with their reprogramming efficiency, being as low as 0.1–1% (Mali et al., 2010), and the presence of incomplete reprogrammed cell phenotypes, epigenetic memory, and genomic instability (Mikkelsen et al., 2008; Kim K. et al., 2010; Gore et al., 2011; Liu et al., 2020).
To obtain iPSCs, it is necessary to select an adequate reprogramming cell population, as not all cell types have shown the same reprogramming efficiency, mostly attributed to their characteristic gene expression profile and epigenetic status (Liebau et al., 2013). An attractive cell source is peripheral blood mononuclear cells (PBMCs), on account of both, the non-invasiveness of the extraction procedure and their reprogramming efficiency (Trokovic et al., 2014; Ye and Wang, 2018). The next step is choosing the right combination of reprogramming factors, given that they can be endogenously expressed in some cell types (Kim J. B. et al., 2009). Optionally, reprogramming enhancers can be added (reviewed by Kwon et al., 2017). The nature of the research and technical skills of the laboratory will condition the selection of the reprogramming strategy. Although there are several available methods, not all are deemed suitable for translational medicine. Two broad strategies can be undertaken, an integrative strategy, which as the name suggests, involves the integration of exogenous genetic material, and a non-integrative strategy, also known as footprint free, which uses excisable vectors or molecules that do not integrate into the host genome. The advantage of the first strategy is the persistent expression of the reprogramming factors, however, there are high risks of genomic instability, random integration leading to insertional mutagenesis, reactive transgenes and chromosomal aberrations (Nakagawa et al., 2008; Tong et al., 2011; Takahashi and Yamanaka, 2013). Both viral and non-viral delivery methods are found within this strategy. The viral methods include the classical use of retrovirus (Takahashi and Yamanaka, 2006) and lentivirus (Yu et al., 2007). The non-viral methods include transfecting plasmids (Okita et al., 2008) and transposons (Woltjen et al., 2009). Within the non-integrative strategies, clinical-grade and translational suitable iPSCs can be obtained, mainly through the direct delivery of microRNAs (Anokye-Danso et al., 2011; Kogut et al., 2018), mRNAs (Warren et al., 2010; Yakubov et al., 2010), episomes (Yu et al., 2009), cell-penetrating peptides (Kim D. et al., 2009), small molecules (Hou et al., 2013), and viral vector transduction via sendai virus (SeV) (Fusaki et al., 2009) or adenovirus (Zhou and Freed, 2009). More recently, a CRISPR-Cas9 variant, CRISPR activation (CRISPRa) has been introduced as a reprogramming tool (Weltner et al., 2018) (Figure 1). Some of these non-integrative systems have combined both quality and safety standards by introducing xeno-free culture methods that meet good manufacturing practice standards (GMP) (Chen et al., 2011; Wang et al., 2015). Do to the robustness of the reprogramming process, it is suggested that the chosen method does not significantly affect the differentiation potential of the produced iPSCs (Churko et al., 2017).
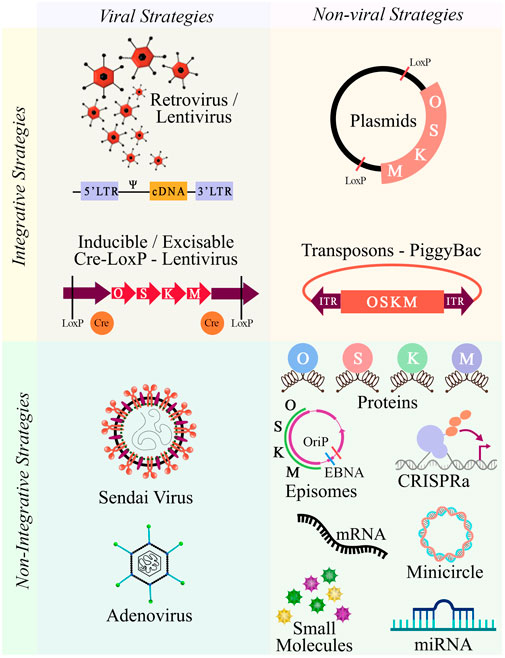
FIGURE 1. Integrative and non-integrative reprogramming strategies. Schematic overview of the available methodologies to dedifferentiate cells towards pluripotency. Abbreviations: O, OCT3/4; S, SOX2; K, KLF4; M, c-MYC.
Once iPSCs cultures are obtained, their characterization must be performed through a combination of stringent analysis that ensures their translational value, quality, and reproducibility. A quick indicator is their morphology, which should be consistently observed as tightly packed colonies, with smooth and defined borders, composed of small cells with a large nucleus/cytoplasm ratio (Meissner et al., 2007; Takahashi et al., 2007). Regular monitoring of genotoxicity is unavoidable, since genomic alterations are attributed to the effects of the reprogramming method, prolong cell culture, and pluripotency-induction itself (Hong et al., 2013; Liu et al., 2020). Moreover, controls to evaluate the presence of residual exogenous reprogramming factors, footprint, or inefficient plasmid clearance are needed, as it hinders the cells capabilities to differentiate, and predispose them to genomic instability (Yu et al., 2007; Ramos-Mejia et al., 2010; Sommer et al., 2012).
Regarding molecular characterization, robust test-sets often include detection of core-factors and proteins related to pluripotent stem cells (PSCs) and ESCs, such as NANOG, OCT4, SOX2, tumor-related antigen (TRA)-1-60/81, stage-specific embryonic antigen (SSEA)-3/4, and the traditional staining of alkaline phosphatase (ALP) (Boulting et al., 2011).
Furthermore, functional assays must demonstrate their pluripotency by differentiating toward cells belonging to the three-germ layers. Both in vitro and in vivo approaches can be undertaken. The in vitro approach includes molecule-based differentiation or spontaneous embryoid body (EB) generation (Sheridan et al., 2012), while the in vivo approach involves teratoma formation (Müller et al., 2010). Regardless of the method, cell lineage identity is assessed by markers such as GFAP, NESTIN, PAX6 (Ectoderm); AFP, PDX1, GATA4 (Endoderm); and Brachyury (TBXT), FLT1, RUNX1, FOXA2 (Mesoderm) (Figure 2).
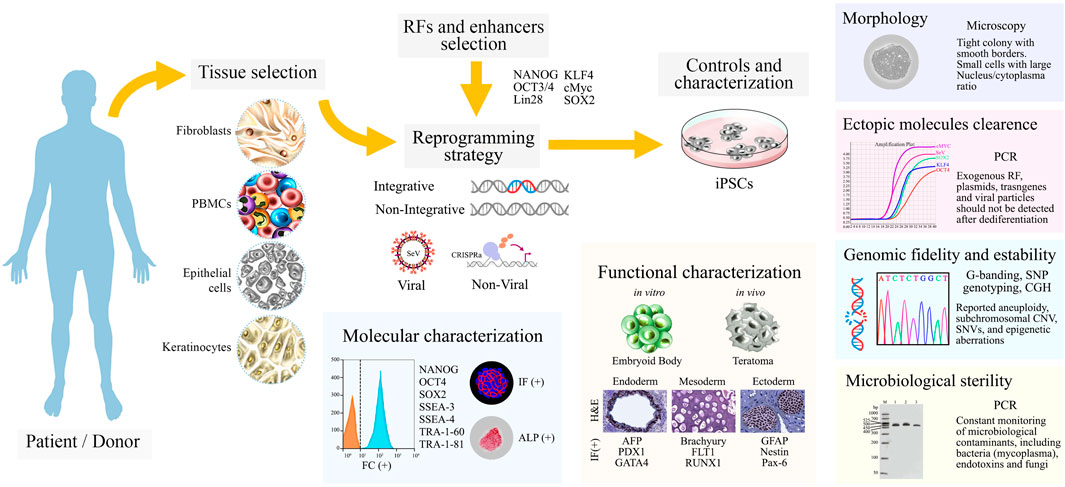
FIGURE 2. A schematic overview from patient’s tissue sample to characterized iPSCs cultures. Patient/donor samples are chosen, the four most frequent tissues are fibroblasts, blood cells, urine track epithelial cells and keratinocytes. Reprogramming factors (RFs), enhancers, and the reprogramming strategy are carefully selected. Once iPSCs cultures are obtained, controls and characterization assays are performed as shown. Abbreviations: SeV, Sendai virus; CRISPRa, CRISPR-Cas9-based gene activation; IF, immunofluorescence; ALP, alkaline phosphatase assay; H&E, hematoxylin and eosin; SNP, single-nucleotide polymorphism; CGH, comparative genomic hybridization; CNV, copy number variation.
Obtaining iPSCs is a laborious and costly process that also requires significant expert training. As experts in the field will agree, high-quality iPSCs will determine the effectiveness and efficiency of the subsequential differentiation process. In this sense, we believe in the importance of incentivizing and joining efforts to establish cell repositories or comprehensive cell libraries. Publicly available and fully characterized iPSCs derived from diverse ALS genetic backgrounds and idiopathic origins can immensely aid the scientific community (Li et al., 2015).
iPSC technology, coupled with a deeper understanding of the ectodermal cell differentiation process has allowed for the production of diverse ALS neural cell types. This includes MNs (Dimos et al., 2008), oligodendrocytes (OLGs) (Ferraiuolo et al., 2016), astrocytes (ACs) (Birger et al., 2019), sensory neurons (SNs) and microglia cells (Pereira et al., 2021). Since multiple pathological mechanisms, both genetic and molecular, converge in the degeneration and death of MNs, most of the published works focus on obtaining and characterizing these MN mono-culture systems.
In broad terms, molecule-based differentiation process recapitulates neurodevelopment. Cells transition towards the ectoderm layer, where both neural stem cells (NSCs) and neural progenitor cells (NPSCs) can be specified by supplementing cultures with appropriate molecules that regulate cell-identity. Current protocols show large variations (reviewed by Faravelli et al., 2014; Sances et al., 2016), however, they all rely on the same stages: induction of neuralization, caudal patterning, ventral patterning, and a final maturation stage.
Neural induction results in the generation of NPCs, parting either from EBs or monolayer cultures, and involves the regulation of several signaling pathways, including the fibroblast growth factor (FGF), bone morphogen protein (BMP), transforming growth factor-β (TGF-β), and notch pathway (Joannides et al., 2007; Du et al., 2015; Maury et al., 2015; Bianchi et al., 2018). The Neuralization process is commonly performed by the synergistic action of dual-SMAD inhibitors (SMADi), comprising SB431542 (SB) -a nodal inhibitor, member of the TGF-β signaling pathway- and LDN193189 (LDN) or noggin -a BMP inhibitor- (Chambers et al., 2009; Fukusumi et al., 2016). Moreover, the efficiency of this process is further increased when combining SMADi with γ-secretase inhibitors and WNT activators (Bianchi et al., 2018). The next stage involves caudalization, which is mainly driven by the effects of retinoic acid (RA) (Li et al., 2005; Qu et al., 2014). The activation of WNT/β-catenin signaling via GSK3β inhibition, either by (2’Z, 3’E)-6-bromoin-dirubin-3’-oxime (BIO) or CHIR99021 (CHIR) optimizes the procedure (Maury et al., 2015; Shimojo et al., 2015). Ventral patterning is orchestrated by a key Smoothened (Smo) agonist, sonic hedgehog (SHH) (Li et al., 2005), or its synthetic alternatives, purmorphamine (PMN) and SAG (Wichterle et al., 2002). Finally, maturation can be guided by neurotrophic factors, including insulin-like growth factor-1 (IGF-1), glial-derived neurotrophic factor (GDNF), and brain-derived neurotrophic factor (BDNF) (Li et al., 2005; Hu and Zhang, 2009; Burkhardt et al., 2013). Lastly, the γ-secretase inhibitor, compound E (Cpd E), displays capabilities to enhance and shorten maturation time (Du et al., 2015) (Figure 3A).
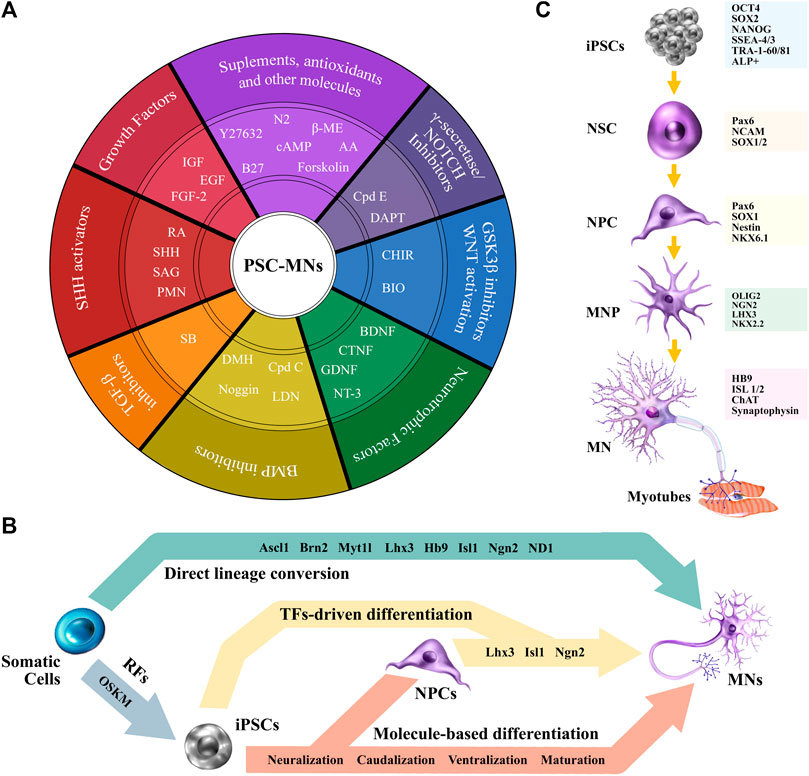
FIGURE 3. From PSCs to MNs. (A) A schematic view of commonly used molecules to differentiate PSCs towards MNs. (B) Differentiation strategies. (C) iPSCs-MN cell markers for characterization. Abbreviations: RFs, reprograming factors; TFs, transcription factors; ß-ME, ß-Mercaptoethanol; AA, ascorbic acid; Cpd E, compound E; cAMP, cyclic adenosine monophosphate; DAPT, N-[(3,5-difluorophenyl)acetyl]-l-alanyl-2- phenyl]glycine-1,1-dimethylethyl ester; GSK3, glycogen synthase kinase 3; WNT, wingless/integrated; CHIR, CHIR99021; BIO, (2′Z, 3′E)-6-bromoin-dirubin-3′-oxime; BDNF, brain-derived neurotrophic factor; CNTF, ciliary neurotrophic factor; GDNF, glial-derived neurotrophic factor; NT-3, neurotrophin-3; BMP, bone morphogen protein; Cpd C, compound C; LDN, LDN193189; RA, retinoic acid; DMH, dorsomorphin; SHH, sonic hedgehog; TGF-ß, transforming growth factor-ß; SB, SB431542; SAG, smoothened agonist; IGF, insulin-like growth factor; FGF-2, fibroblast growth factor-2; EGF, epidermal growth factor; PMN, purmorphamine.
Of great importance, Maury et al. (2015) demonstrated how fine tuning of the combination, concentration, and exposure time of the differentiating factors impact cell specification. Proving that these subtle modifications cue MN progenitors (MNPs) to differentially specify four neuronal subtypes, namely, interneurons, SNs, cranial MNs, and spinal MNs.
Highly relevant to both translational and clinical research is the development of GMP protocols performed under fully humanized conditions. To our knowledge, there is only one report of a fully xeno-free protocol from iPSCs to MN production (Hu et al., 2016).
An alternative strategy to obtain iPSC-MNs involves the combination of molecule-based differentiation with transcriptional coding. After neuralization of iPSCs, NPCs can be transduced with LIM/homeobox protein 3 (Lhx3), Islet-1 (Isl1), and neurogenin 2 (Ngn2) -LIN, either via adenovirus (Hester et al., 2011) or lentivirus (Sepehrimanesh and Ding, 2020). Other protocols bypass neuralization and force the expression of the transcriptional factors directly into iPSCs (Figure 3B). As an example, lower-MNs are obtained by generating LIN-inducible transgenic lines (Fernandopulle et al., 2018). Footprint-free approaches, that avoid genomic integrity concerns, use SeV encoding LIN (Goto et al., 2017), piggyBac transposon vectors (Imamura et al., 2017), and synthetic-mRNA encoding Ngn1, Nng2, Ngn3, NeuroD1 (ND1) and ND2 (Goparaju et al., 2017). Cranial MNs have also been obtained via the ectopic expression of Ngn2, Isl1, and Phox2a (Garone et al., 2019). Recent work shows that overexpression of just Ngn2 coupled with molecular patterning of RA and SAG produces a pure population of lower induced MNs (LiMoNe). LiMoNe cells are obtained with a high-yield, and display both electrophysiological activity and form synaptic contacts with muscle cells (Limone et al., 2022).
Transcriptional factor-driven differentiation shows some advantages over molecule-based differentiation, including an overall reduction in time, cost and technical skills, as well as an increase in culture purity (reviewed by Canals et al., 2021). Moreover, molecule-based differentiation generates cultures that contain mixed-degrees of differentiated cells, which while advantageous as a developmental model, its a concern for drug screening. Therefore, MN-enrichment may be required as an additional step. For this, protocols include gradient centrifugation (Corti et al., 2012), magnetic cell sorting of L1CAM (CD171) (Maciel et al., 2018), and more recently, fluorescent activated cell sorting (Klim et al., 2019). This last strategy allows for efficient enrichment of post-mitotic Hb9::GFP + MNs based on their NCAM+/EpCAM-immunoprofile.
A direct lineage conversion strategy has been refined, which involves bypassing pluripotent reprogramming, and directly converting somatic cells into other lineages. The process centers on forced expression of transcriptional factors by retroviral transduction. The combination of factors includes Ascl1, Brn2 (also known as Pou3f2), Myt1l, Lhx3, Hb9, Isl1, Ngn2, and ND1. The resulting cells, coined induced MNs (iMNs), showed electrophysiological activity, neuromuscular junction (NMJ) formation, and molecular and functional properties of naïve MNs (Son et al., 2011). Induced Neurons (iNs) can also be produced in this manner by lentiviral infection of Brn2, Ascl1, Myt1l, and ND1 -BAMD. However, these cells did not exhibit a fully mature phenotype (Pang et al., 2011). Notably, there are some drawbacks to this methodology, including their limited yield, as no stem cell nor progenitor cell stages are present, therefore their number is restricted to the parting cell count. Moreover, there is evidence that transdifferentiation efficiency decreases in virtue of the donor’s age (Montana et al., 2013), which becomes an obstacle for modeling late-onset diseases. Furthermore, the use of integrative-viral strategies, as previously mentioned, poses risk of insertional mutagenesis.
As a cautionary tale, both the dedifferentiation and differentiation processes subject cells to metabolic and epigenetic changes that can lead to genomic variations. In addition to this, protocol diversity, un-standardized cell culture technique, and intrinsic variables associated with the cells per-se, contribute to cell culture heterogeneity. Furthermore, PSC lines have shown variating innate degrees of differentiation potential towards a determined cell lineage, attributable to their individual genetic and epigenetic background (Osafune et al., 2008). Remarkably, a strategy to coax iPSCs lines with unsimilar differentiation propensity towards a neural lineage is achieved by SMADi, which is already an established stage of the MN differentiation protocol (Kim D.-S. et al., 2010).
Regardless of the differentiation strategy, cell characterization is central for appropriate downstream research and data analysis. Early neuroectodermal markers such as Pax6, NCAM, and SOX1/2 can be used to identify NSCs (Joannides et al., 2007). For NPCs, there is overlapping of markers, like Pax6 (Chambers et al., 2009), and SOX1 (Bianchi et al., 2018), that can be used along with Nestin and Nkx6.1 (Maury et al., 2015). MNP markers include further overlapping of Pax6, and the increasing expression of Olig2, Ngn2, Lhx3, NKx2.2 (Hu and Zhang, 2009; Maury et al., 2015; Shimojo et al., 2015; Bianchi et al., 2018). Mature MN markers comprise HB9, Isl1/2, ChAT (Hu and Zhang, 2009; Maury et al., 2015), and Synaptophysin, an indicator of possible synaptic connectivity (Bianchi et al., 2018) (Figure 3C). Cytoskeletal pan-neural markers, MAP2 (dendrite), ß-III tubulin [(Tuj1, TUBB2); axon, dendrite and Soma], and neurofilament (axon) (Qu et al., 2014; Bianchi et al., 2018; Sepehrimanesh and Ding, 2020) are also frequently used. The absence of proliferative markers, as Ki67, can further characterize cells in their post-mitotic stages (Maury et al., 2015).
Functional characterization validates the system in study and offers physiological relevance to the generated data. Thus, MNs should be synaptically mature, and exhibit an appropriate electrophysiological activity. Several strategies can be used to assess these characteristics, including patch-clamp, calcium imaging (Prè et al., 2014), optopatch (Kiskinis et al., 2018) and multielectrode array (MEA) (Taga et al., 2021). A central MN ability, indicative of a mature phenotype, is NMJ formation. These are specialized cholinergic synapsis that transmits chemical signals to muscle fibers from the electrical impulse generated by MNs. They can be identified by α-BTX staining of acetylcholine receptors (AChRs) clustering on myotubes juxtaposed to axon terminals (Lin et al., 2019).
Thus far, we have overviewed the process to obtain iPSC-MNs from donor cells samples in the interest of introducing their use in drug screening, and discuss how 3D-modeling and bioinformatics can aid ALS research.
A significant interest in iPSC-ALS modeling is associated with targeting both fALS and sALS (Fujimori et al., 2018; Sun et al., 2018). However, the diversity of the reprogramming methods (Guo et al., 2017a), characterization criteria and standards, and protocols to re-differentiate cells towards neuronal lineages (Hawrot et al., 2020) hinders appropriate comparison of results between the generated data. A crucial tool for increasing confidence in iPSC-ALS modeling is the use of isogenic lineages. These lineages are obtained either from correcting the mutation of the parental-line in study, or by inserting a mutation that correlates with the disease’s phenotype. These cultures should only differ in the genome-edited loci, and can be performed by CRISPR (Deneault et al., 2021), transcription activator-like effector nucleases (TALENs) (Chen et al., 2014), and zinc-finger nucleases (ZFNs) (Kiskinis et al., 2014). Unfortunately, it is not possible to produce isogenic controls when the genetic lesion is not identified, as in some cases of sALS. The lack of isogenic lineages difficult the discerning of the disease’s phenotype from the cells own phenotype. Relevant to ALS drug screening is a novel scalable platform based on CRISPR interference (CRISPRi). This technology centers on robust gene-knockdown that allows for functional characterization of genes that control disease-relevant phenotypes including neuron survival and morphology. Importantly, it enables interrogation of ‘druggable genome’. This technology coupled with isogenic controls could elucidate new disease mechanisms that, in turn, lead to the discovery of novel drug-targets (Tian et al., 2019).
Recent reviews have centered on the use of iPSCs-based drug platforms for neurodegenerative diseases (reviewed by Bonaventura et al., 2021; Ferraiuolo and Maragakis, 2021; Pasteuning-Vuhman et al., 2021) Specifically for ALS, researched drugs include rapamycin (Imamura et al., 2017; Marrone et al., 2018; Osaki et al., 2018), bosutinib (Imamura et al., 2017; Osaki et al., 2018), anacardic acid (Egawa et al., 2012), ropinirole (Fujimori et al., 2018), vardenafil (Osborn et al., 2018), ezogabine (Wainger et al., 2014), GKS2606414 (Szebényi et al., 2021), trolox (Lopez-Gonzalez et al., 2016), 4-aminopyridine (Naujock et al., 2016), kenpaullone (Yang et al., 2013), the cyclin-dependent kinase inhibitors: digoxin, lanatoside C, and proscillaridin (Burkhardt et al., 2013), and the histone deacetylation inhibitors: HDAC6 antisense oligonucleotide-ASO, tubastatin A and ACY-738 (Guo et al., 2017b) (Figure 4). In the limelight, three of these drugs, ropinirole (Morimoto et al., 2019), retigabine (Wainger et al., 2021), and bosutinib (Imamura et al., 2019) are currently in clinical trial.
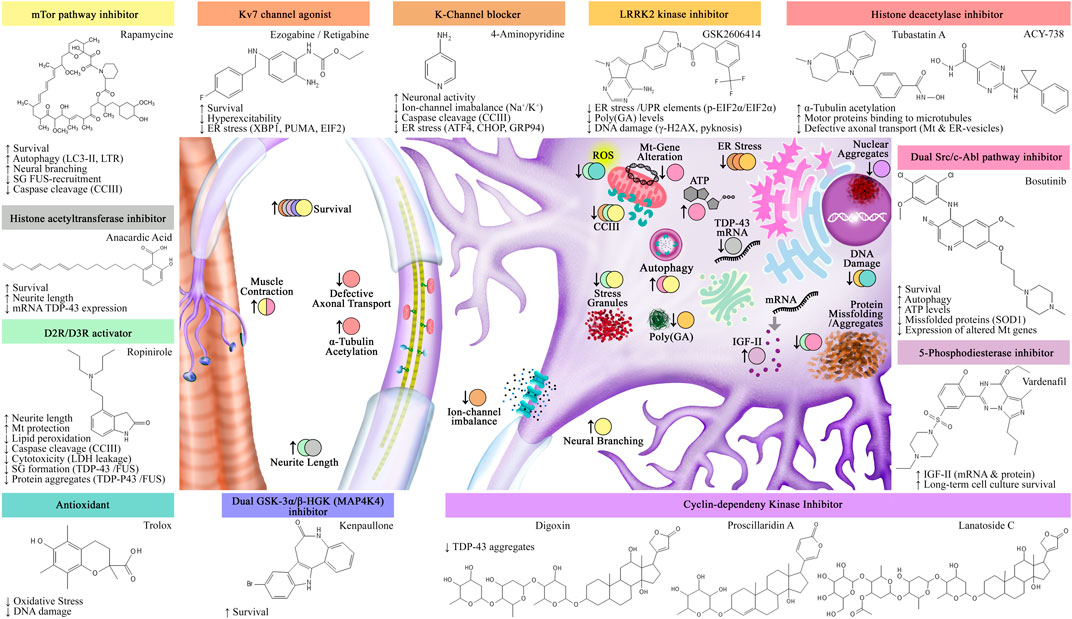
FIGURE 4. Assessed drugs using iPSC-ALS modeling. Identified drugs and their effects in both iPSCs-MNs and muscle fibers. Abbreviations: SG: stress granules; Mt: mitochondria; ER: endoplasmic reticulum; UPR: unfolded protein response.
Two concerns are put forward, the first involves cell-aging or the conservation of the epigenetic drift in iPSCs, as ALS is a late-occurring disease (Pandya and Patani, 2020). Several authors have evidenced iPSCs rejuvenation by their whole-transcriptome RNA sequencing (RNA-seq) (Mertens et al., 2015), nuclear organization, reduced DNA damage, mitochondrial metabolism (Miller et al., 2013), telomere length (Agarwal et al., 2010), and the loss of senescence markers (Lapasset et al., 2011). To overcome this age-setback, artificially induced-aging strategies have been developed. For instance, telomerase inhibition (Vera et al., 2016), and the forced ectopic expression of progerin, a truncated form of the nuclear envelope protein laminin A (Miller et al., 2013). This last molecule was able to phenocopy tissue-specific age-associated features that were later corroborated in vivo (Miller et al., 2013). However, much uncertainty remains as to the degree with which age-related physiology is mimicked.
The second concern involves the possible loss of non-genetic disease-related traits, given that, during the reprogramming process, cells suffer epigenetic reshaping allowing cell-identity pattern erasing. Analogously, the possibility of acquiring unknown and disease-unrelated features can render the model less relevant (Liang and Zhang, 2013). Assessment of DNA-methylation and histone marks from the reprogrammed iPSCs to the patients’ wild-type cells, as well as the use of isogenic controls increases the model’s credibility.
Direct conversion could, to some extent, breach both aging and epigenetic concerns. In this sense, iNs from donors of diverse ages presented age-correlated features including transcriptomic signature, compromised nucleocytoplasmic compartmentalization (Mertens et al., 2015), microRNA profile, and DNA-methylation status (Huh et al., 2016). Aging hallmark preservation has also been observed in iMNs. Nuclear organization (heterochromatin loss and DNA damage), and increased senescence-activated ß-galactosidase activity (Tang et al., 2017) have been reported. However, direct conversion advantages related to cost, time, and epigenetic status should be carefully considered in relation to their limitations. These limitations include culture scale-up and their ability to form organoids.
Variations in cellular behavior, drug response and pathomechanisms can be detected according to the cell culture system used. Three-dimensional (3D) systems are regarded with higher predictive value as they encompass diverse cell type interactions, ECM interplay, and complex microenvironmental cues not found in two-dimension (2D) cultures (Pampaloni et al., 2007; Maltman and Przyborski, 2010).
A wide variety of 3D-culture systems have been implemented in neurosciences. Complex tissue constructs, such as brain region-specific organoids, including cerebral (Lancaster and Knoblich, 2014), midbrain (Nickels et al., 2020), forebrain (Gomes et al., 2020), hypothalamus (Qian et al., 2018), and cerebellum (Muguruma et al., 2015), as well as spinal cord organoids (Hor et al., 2018), spheroids (Paşca et al., 2015; Bowser and Moore, 2019), assembloids (Birey et al., 2017), and brain-on-a-chip technology (Adriani et al., 2017) are on the rise.
A critical component in the generation of these tissues is the presence or absence of a scaffold. The scaffold-free approach involves spontaneously formed and self-organizing multilayer cell aggregates that can eventually produce their own non-cellular elements (Fennema et al., 2013; Raja et al., 2016; Türker et al., 2018). The scaffold-based approach uses a substrate that guides cell behavior and organization through mechanical and chemical cues, allowing the generation of bigger and more complex structures. These substrates are hydrogels derived from a biological source (e.g., Matrigel, Collagen, Gelatin, Alginate, etc.), have a synthetic origin (e.g., Polyglycol Acid, Polyethylene Glycol), or present a combination of both. As each material has its own limitations and advantages, the nature of the study or end-product will help determine the appropriate approach (Reviewed: Murphy et al., 2017; Rey et al., 2020; Roth et al., 2021). Noteworthy, enhanced long-term culture and mature phenotypes of neural cell types have been associated with the use of natural scaffolds, such as Hyaluronan Acid (Zhang et al., 2016; Wu et al., 2017) and decellularized brain-derived ECM (Sood et al., 2019). However, these can compromise xeno-free systems and contribute to variability, as they present batch-to-batch variations in their composition.
Although MNs are predominantly studied, ALS is a systemic non-autonomous and multicellular disease, that cannot be fully represented by a single monolayer-cell lineage. In this sense, 3D-cultures allow the formation of neural networks, with brain-like functions, and multidirectional communication between, for example, neurons and glial cells (Lancaster et al., 2013; Sakaguchi et al., 2019; Yakoub, 2019; Samarasinghe et al., 2021). However, to date, only five 3D-systems have been published for the study of ALS (Table 1).
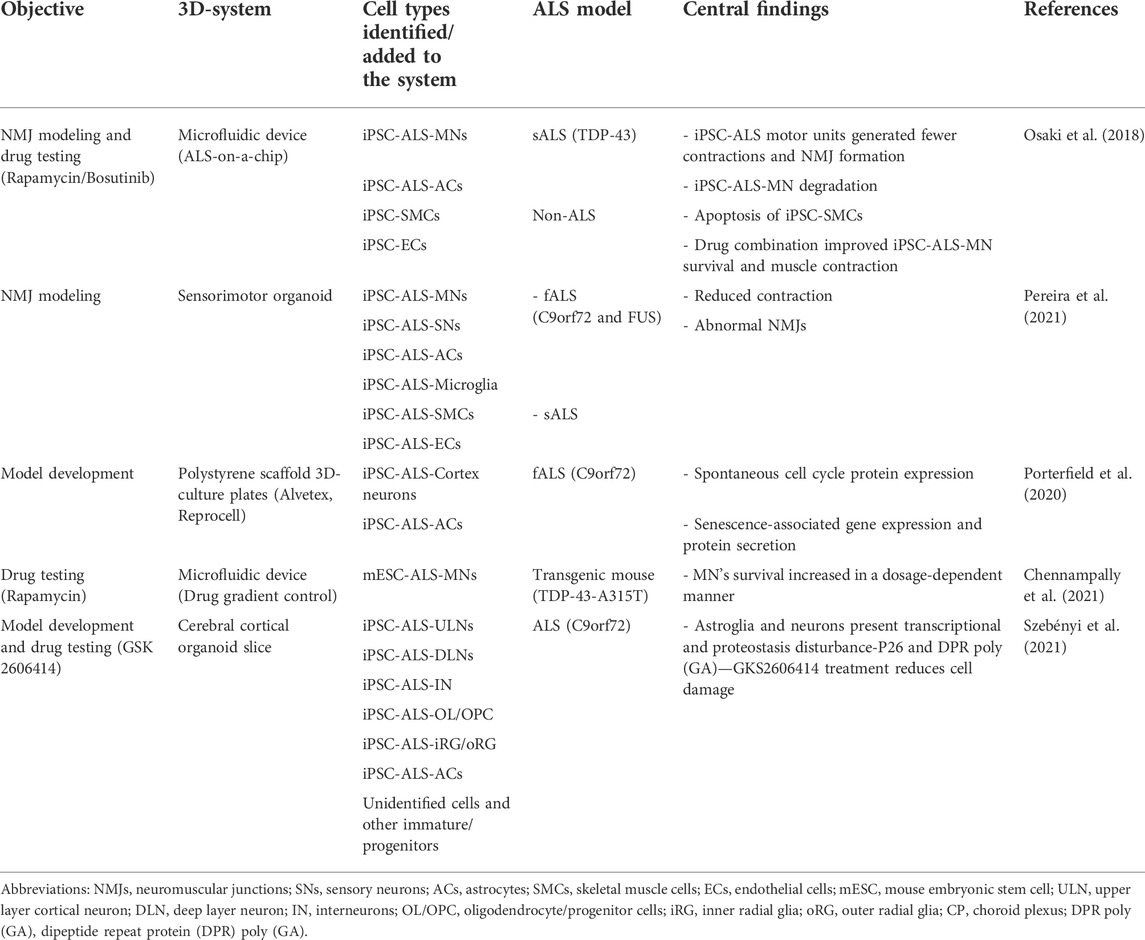
TABLE 1. 3D-culture models implemented in the study of ALS.
Among these systems, organ-on-a-chip, is a microfluidic device that aims to replicate the physiological microenvironment, and allows the study of NMJs between skeletal muscle cells (SMCs) and MNs (Osaki et al., 2018). NMJs are highly relevant for ALS, as its structural disassembly, and muscle denervation are both hallmarks of the disease (Cappello and Francolini, 2017). With this system, Osaki et al. were able to study iPSC-ALS-MNs phenotypical characteristics, neurite elongation speed and length, as well as the positive therapeutic effects of rapamycin and bosutinib. Thus, variables such as axonal outgrowth, synapse formation, muscle contraction, and atrophy can be assessed, highlighting the device’s appealing approach to study drug efficiency for motor unit recovery (Uzel et al., 2016; Osaki et al., 2018).
A more recent model, developed by Pereira et al. (2021), involves the use of iPSCs derived from fALS (C9orf72 and FUS) and sALS. The produced organoids were patterned towards neuromesodermal progenitors. Diverse cell types were identified in these tissue constructs, including MNs, SNs, ACs, microglia, SMCs, vasculature cells and dorsal spinal cord cell derivatives. Unlike the previous model, all cell types present in the system originate from an ALS source. Moreover, these sensorimotor organoids facilitate cell type quantification, enable live-cell imaging, and the study of NMJs, which were found impaired compared to non-pathological controls.
A third model was established using a commercially available 3D-culture plate (Alvetex, Reprocell). Porterfield et al. (2020), studied cortex neurons derived from patients presenting a C9orf72 mutation. They reported that, compared to a 2D-culture-control, phenotype-disease relevant characteristics were observed. These include, spontaneous expression of cyclin D1, dysregulation of cell cycle-associated genes, and the expression of senescence related secretory molecules.
With a different approach, Chennampally et al. (2021), explored the effects of rapamycin in a 3D microfluidic system that allowed for concentration-gradient control while mapping permissive zones of MN survival in the organoid. Here, the chosen disease model was derived from mouse ESCs (mESCs) harboring a TDP-43-A315T mutation. Their findings support that rapamycin dosage (0.4–1.0 µM) can increase MN survival by 40.44% through autophagy regulation of aggregates. As an advantage to the system, drug titration can be performed in a single culture, thus enhancing test throughputs and results.
The newest 3D-model, by Szebényi et al. (2021), involves the development of cortical brain organoids that are subsequently sliced and cultured on fenestrated membranes at the air-liquid interface. These organoid slices contain a consistent microarchitecture, wide cell diversity presenting forebrain signature identity, and functional and mature cortical circuits that display disease-relevant phenotypes. However, mesoderm-derived cells are not detected. The screened drug, GSK2606414, reduced unfolded protein response (UPR) activation and poly (GA) levels. Szebényi et al. (2021) suggested that dormant perinatal or pre-symptomatic cortical vulnerability is present in ALS patients.
A limitation for 3D-culture viability is their lack of vascularization, which hinders the tissue from the adequate circulation of nutrients, bioactive molecules, gaseous exchange, and waste disposal. These factors limit the organoids size, induces extensive cell death in the center or core of the tissue construct, and restricts its culture time (Lancaster et al., 2013; Grebenyuk and Ranga, 2019). However, Prolonged culture time is necessary for stem cells to differentiate and mature and mimic adult tissues. Szebényi et al. (2021) model circumvented vascularization drawback by culturing slices instead of whole organoids. These organoid slices boosted the cells viability because of increased nutrition inflow and suppressed core necrosis. In this manner, cultures were maintained for up to 240 days. However, vasculature components (e.g., ECs and pericytes) are indispensable and relevant to the appropriate differentiation and maturation of NPCs (Koutsakis and Kazanis, 2016; Cakir et al., 2019; Karakatsani et al., 2019). In this regard, two of the aforementioned 3D-models present vascular elements. Pereira et al. (2021) identified the formation of a reticulated pattern of ECs. Albeit, the group did not address its functionality nor its potential to model a blood-brain barrier (BBB), which is highly pertinent for drug screening (Cecchelli et al., 2007). While Osaki et al. (2018) model presents vascular elements, their use of a microfluidic chamber compensated—to some extent—the lack of a circulatory system. Taking a step further, they modeled a BBB-like structure, by incorporating iPSC-ECs and primming them towards a brain-specific EC-phenotype. This barrier exhibited functional characteristics by compromising the therapeutic activity of the tested drugs and allowed the authors to hypothesize its possible role in neuroprotection and muscle contraction.
The lack of standardization in 3D-cultures is a concern, and efforts to increase confidence in organoids are under way. Velasco et al. (2019), have made a major advance in developing a method to standardize individual brain organoids. They established four different protocols to generate both organoids and spheroids that were highly reproducible in cellular composition, even across different lines and batches. Single-cell sequencing confirmed that the organoids remained genetically stable along with their morphology for up to 6 months, regardless of their genetic background. Other groups have also shown reproducibility of organoids as a stable model for studies of late stages of neuronal development (Giandomenico et al., 2021), and use for neurological disorders, including ALS (Pereira et al., 2021), Alzheimer’s (Ghatak et al., 2019), and Leigh syndrome (Romero-Morales et al., 2020). The challenges ahead include consistency of the cellular and non-cellular components, the incorporation of vascular elements, immune cell, and other disease-relevant cell types (e.g., Schwann cells). As well as the development of more sophisticated scaffolds, microfluidic devices, and analysis technology adapted to 3D-systems that allow for high-throughput screening.
The integration of bioinformatic and computational tools close the gap between animal and cell model disadvantages. Furthermore, allows for the understanding of the disease mechanisms by exploring large genetic datasets, identify critical biochemical pathways, and generate hypotheses. Here, we will highlight existing bioinformatic and computational tools and their applications in ALS and iPSC-ALS.
ALS genetics is complex and there is a rapidly growing dataset due to the advancement of technology and increased sampling. Existing bioinformatics tools for understanding ALS genetics are ALSoD (Abel et al., 2012) and ALSGeneScanner (Iacoangeli et al., 2019). ALSoD (Abel et al., 2012) is a freely available database, that serves as a bioinformatics repository and an analysis tool for genotype to phenotype association studies. Since the development of ALSoD, several years of repository enrichment established the evidence for the polygenic and oligogenic basis of Australian sALS (McCann et al., 2021) that would not be possible with bioinformatic analysis. In addition, ALSGeneScanner is a tool that allows non-specialists, and healthcare providers to use an automatic and annotated report for interpreting ALS genetic data. The feasibility of ALSGeneScanner allows for whole-genome and exome sequencing, variant prioritization, and helps to distinguish pathogenic from non-pathogenic variants (Iacoangeli et al., 2019). Bioinformatics has bridged complex data generated from advances in sequencing technology for ALS and continues to advance the discovery of new ALS disease-modifying genes such as EPHA4 and CHGB (Pampalakis et al., 2019). The incorporation of bioinformatics provides increased approachability for non-computational experts through the accessibility of data and automatic data processing. Computational techniques, such as comparative omics, allowed researchers to find novel genetic variants such as SPTLC1 variants and linked the variants to childhood-ALS (Mohassel et al., 2021). Most importantly, the application of bioinformatics and computational tools bolsters discoveries from genetic datasets that would otherwise take many years to connect.
For iPSCs-ALS-MNs, the application of computational techniques such as integrated multi-omic analysis resulted in the identification of novel and known aberrant cellular pathways (Li et al., 2021). Integrated multi-omic analysis distinguished pathogenic versus compensatory disease phenotype pathways, which allows for clearer understanding of disease mechanisms. In disease pathway analyses, cell type heterogeneity from iPSC cultures differentiation can confound investigations. Multi-omics integration from genomics, transcriptomics, epigenomics, and proteomics minimize variations from iPSC differentiation and highlight network-based signatures. Researchers performed comparative multi-omics analysis with selectively vulnerable MN subtypes in ALS, and using iPSCs derived from fALS and sALS patients, and have found dysregulation of lipid metabolism. Lee et al. (2021) further applied targeted metabolomics to confirm lipid metabolism dysregulation and showed pharmacological reduction of arachidonic acid reversed disease-related phenotypes in drosophila and mouse models of ALS (Lee et al., 2021). In this sense, bioinformatics allows for the translation of experimental studies to influence disease treatment and management.
Within mechanistic studies for ALS, errors in protein synthesis, trafficking, and degradation are regularly reported (Van Der Stappen et al., 2005). Protein studies provide insight and value in understanding the mechanism of ALS (McAlary et al., 2020). Bioinformatics aid in the study of protein-protein interactions, which are mapped and analyzed using online databases, such as STRING.db (Szklarczyk et al., 2019). STRING.db predicts not only physical protein-protein interaction but also, functional interactions. Moreover, subsequent pathway analysis is performed with both Gene Ontology (Ashburner et al., 2000) and KEGG (Kanehisa, 2000; Kanehisa et al., 2021). Gene Ontology and KEGG help to elucidate high-level functions of biological systems, identify cellular pathways and the structural locations where these occur. An example of Gene Ontology application comes from Feneberg et al. (2020), who compared human wild-type and mutant TDP-43 interactomes from iPSC-ALS, finding that disrupted protein interactions alter TDP-43 response to oxidative stress.
Beyond protein-protein interactions are cell-cell interactions that are critical for understanding disease phenotypes. The development of complex multicellular models of ALS are pushing the Frontier of in vitro models; however, there is a major lack in cell-cell communication analysis tools. Systemic Elucidation and Assessment of Regulatory Cell-to-cell Interaction Networks (SEARCHIN) is an approach created to identify interactions between multicellular models of disease (Mishra et al., 2020). Mishra et al. identified in ALS models of neuron-astrocyte, a deleterious ligand-receptor pair, amyloid precursor protein (APP), and death receptor-6 (DR6). Therefore, multi-modal integrative bioinformatics approaches like SEARCHIN allow for the study of non-cell-autonomous mechanisms.
Central to this review is the use of iPSC-ALS models for drug discovery. Drugs under investigation, such as the acetyltransferase inhibitor, anacardic acid, was discovered through iPSC-ALS models (Egawa et al., 2012). ropinirole was identified through multi-phenotypic screening with high-content imaging using non-SOD1 fALS models from iPSCs (Fujimori et al., 2018). The combination of iPSC-ALS models and high-throughput drug screening with computational tools accelerates the development of ALS drugs (Lee et al., 2018). Neural networks (Congzhou et al., 2021) can be applied to predict small molecule targets to proteins of interest such as TDP-43, SOD1, and FUS. Drift is another tool from the Dokholyan lab that predicts protein targets of chemical compounds. In drug discovery, high-throughput screening methods are used to select potential candidates, that are computationally assessed though molecular modeling. Moreover, in drug design, the conformational changes induced by ligand binding are often difficult to capture and calculating the configurational entropy may pose a challenge. To circumvent these limitations, flexible docking approaches such as MedusaDock (Yin et al., 2010; Wang and Dokholyan, 2019) allow modeling both ligand and receptor flexibilities. MedusaDock is well-suited for iPSC-ALS model studies as it conformationally samples small molecules. Finally, Erebus (Shirvanyants et al., 2011) validates the binding site of the protein of interest. With the power of computation coupled with better disease-reproducible models such as iPSCs cultured in 3D-systems, the validity of the data is strengthened.
Regarding the clinical translation of bioinformatics, analysis of datasets investigating immune cells and gene alterations provided potential biomarkers for the much needed monitoring of ALS disease and treatment response (Xie et al., 2021). Investigations for unique ALS gene signatures compared to non-ALS controls led researchers to predict disease occurrence based on the combination of neurofilament-light (Nf-L) and neuroinflammatory biomarkers in the serum and cerebral spinal fluid these patients (Brodovitch et al., 2021). To date, the most accurate biomarker for ALS is the unique combination of Nf-L and neuroinflammatory markers in the serum and cerebral spinal fluid. Nf-L and cytokines possess long-term stability providing prognostic value as biomarkers for patients with neurogenerative symptoms (Huang et al., 2020). Also, the development of genetic bioinformatic tools affect clinical decisions such as genetic counseling and disease-risk assessment (Al-Chalabi et al., 2017). As stated, ALS presents a late-onset, however the disease may manifest much earlier, thus the discovery of specific and stage-sensitive biomarkers can aid the timely identification of the disease to efficiently intervene early symptoms and monitor the pathologies’ progression.
ALS is a complex disease without a clear understanding of targetable disease-curable pathways. Therefore, it is imperative to highlight the critical axis for ALS pathogenesis. The application of bioinformatics and computational approaches such as ALSoD, ALSGeneScanner, multi-omics analysis, STRING.db, SEARCHIN, neural networks, DRIFT, and MedusaDock pushes the Frontier of our understanding of ALS. SEARCHIN applied to iPSC co-cultures of ALS patients elucidated our understanding of cell-cell interactions, similarly STRING.db permits the use of proteins extracted from iPSC-ALS to understand specific protein-protein interactions. Neural networks, DRIFT, Erebus, and MedusaDock allow for identification of new drugs, prediction of drug targets to proteins of interest, binding site validation, and docking and modeling of proteins or small molecules with iPSC-ALS cultures for in vivo validation experiments (Figure 5). Bioinformatic and computational techniques paired with iPSC-ALS experiments empowers streamlining drug discovery of iPSC-ALS cultures.
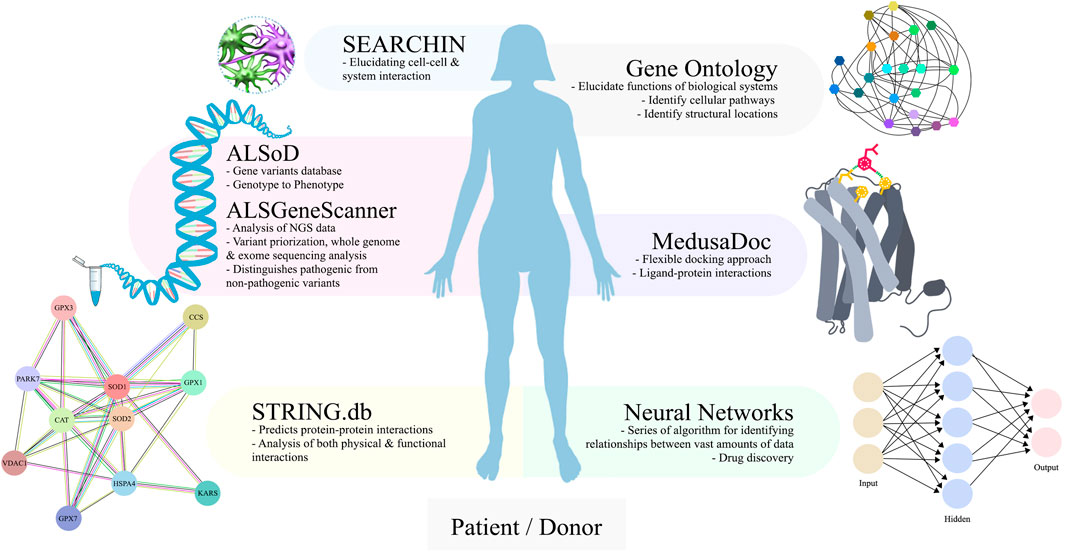
FIGURE 5. An overview of bioinformatics and computational tools that can be paired to iPSC-ALS research. For ALS genetics, bioinformatics repositories provide automated analysis for genotype to phenotype association studies (ALSoD) and interpretation of complex genetics data (ALSGeneScanner). For iPSC-ALS, protein-protein interactions can be mapped with subsequent pathway analysis (STRING.db). Scaling up to multicellular systems requires increased analysis techniques that are available (SEARCHIN). For iPSC-ALS drug discovery, small molecule targets to proteins of interest can be identified (Neural Networks) and docked with the ligand (MedusaDock).
Since their discovery, iPSCs’ public interest has centered on their use as therapeutic agents. However, unlike their multipotent counterparts, the mesenchymal stem cells, iPSCs’ clinical translation is still in preliminary stages. The lagging in the implementation of iPSCs in a clinical setting is due to the discussed limitations that include genetic instability, lack of standardized production, quality control, and robust xeno-free clinical-grade protocols. We believe that iPSCs’ true impact resides in their use as disease models and drug screening platform for pharmaceutical development. In addition to their clear advantage to model the donors’ genetic and epigenetic identity, iPSCs allow for scaling-up and high-throughput drug screening processes. Perhaps one of their most exciting features, is their capability to bypass animal experimentation in drug trials, which translates to a substantial reduction of the time, cost and ethical concerns associated with animal use. However, for this system to be widely implemented in the drug discovery field, iPSCs culture standardization demands must be rigorously met. Defined differentiation protocols, along with stringent characterization and use of isogenic controls can significantly assist in the systems reliability.
While continuous improvement of iPSC production and a deeper understanding of the cell differentiation process allows us to search for shared protocol standards and define criteria, a new set of tools involving 3D-culture and bioinformatics are emerging. Using 3D-cultures we can increase the resemblance of the in vivo physiological microenvironments, and thus, greatly enhance our understanding of the diseases’ biology and drug response. While bioinformatics can aid us in unveiling the diseases’ pathological mechanisms and in predicting new therapeutic compounds and their combinations for effective treatments. The incorporation of both these tools have the potential to shift the current limitations of ALS research and spark the discovery of new drugs and treatments. However, despite these clear advantages, 3D-modeling and bioinformatics integration to iPSCs-ALS research is rarely found. From scarce postmortem samples to in bulk-patient specific iPSCs that differentiate into any cell type of the brain, in vitro technology and their analysis tools have moved forward with giant leaps. We are now in need of the next step, towards the third dimension and integrated in silico analysis.
Illustrations were created using Adobe Photoshop (2020), and chemical molecules were modeled with PubChem Sketcher V2.4 (2021) by MA.
Conceptualization: MA, AC, ND, and MD. Researching data: MA, EC, and AC. Writing—first draft: MA and EC. Review and Editing: MA, EC, ND, and MD. Visualization and illustrations: MA. Supervision: MA, ND, and MD.
We acknowledge support from the National Institutes for Health (1R35 GM134864), the Passan Foundation, the Ministry of Health of Brazil (325000.475256/2017-54), and INOVA FIOCRUZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abel, O., Powell, J. F., Andersen, P. M., and Al-Chalabi, A. (2012). ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 33, 1345–1351. doi:10.1002/humu.22157
Adriani, G., Ma, D., Pavesi, A., Kamm, R. D., and Goh, E. L. K. (2017). A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood–brain barrier. Lab. Chip 17, 448–459. doi:10.1039/C6LC00638H
Agarwal, S., Loh, Y., McLoughlin, E. M., Huang, J., Park, I., Miller, J. D., et al. (2010). Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 464, 292–296. doi:10.1038/nature08792
Al-Chalabi, A., van den Berg, L. H., and Veldink, J. (2017). Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 13, 96–104. doi:10.1038/nrneurol.2016.182
Anokye-Danso, F., Trivedi, C. M., Juhr, D., Gupta, M., Cui, Z., Tian, Y., et al. (2011). Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell. Stem Cell. 8, 376–388. doi:10.1016/j.stem.2011.03.001
Argueti-Ostrovsky, S., Alfahel, L., Kahn, J., and Israelson, A. (2021). All roads lead to rome: Different molecular players converge to common toxic pathways in neurodegeneration. Cells 10, 2438. doi:10.3390/cells10092438
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: Tool for the unification of biology. The gene Ontology consortium. Nat. Genet. 25, 25–29. doi:10.1038/75556
Bernard, E., Pegat, A., Svahn, J., Bouhour, F., Leblanc, P., Millecamps, S., et al. (2020). Clinical and molecular landscape of ALS patients with SOD1 mutations: Novel pathogenic variants and novel phenotypes. A single ALS center study. Int. J. Mol. Sci. 21, 6807. doi:10.3390/ijms21186807
Bianchi, F., Malboubi, M., Li, Y., George, J. H., Jerusalem, A., Szele, F., et al. (2018). Rapid and efficient differentiation of functional motor neurons from human iPSC for neural injury modelling. Stem Cell. Res. 32, 126–134. doi:10.1016/j.scr.2018.09.006
Birey, F., Andersen, J., Makinson, C. D., Islam, S., Wei, W., Huber, N., et al. (2017). Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59. doi:10.1038/nature22330
Birger, A., Ben-Dor, I., Ottolenghi, M., Turetsky, T., Gil, Y., Sweetat, S., et al. (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 50, 274–289. doi:10.1016/j.ebiom.2019.11.026
Bonaventura, G., Iemmolo, R., Attaguile, G. A., La Cognata, V., Pistone, B. S., Raudino, G., et al. (2021). iPSCs: A preclinical drug research tool for neurological disorders. Int. J. Mol. Sci. 22, 4596. doi:10.3390/ijms22094596
Boulting, G. L., Kiskinis, E., Croft, G. F., Amoroso, M. W., Oakley, D. H., Wainger, B. J., et al. (2011). A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 29, 279–286. doi:10.1038/nbt.1783
Bowser, D. A., and Moore, M. J. (2019). Biofabrication of neural microphysiological systems using magnetic spheroid bioprinting. Biofabrication 12, 015002. doi:10.1088/1758-5090/ab41b4
Brites, D., and Vaz, A. R. (2014). Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 8, 117–124. doi:10.3389/fncel.2014.00117
Brodovitch, A., Boucraut, J., Delmont, E., Parlanti, A., Grapperon, A.-M., Attarian, S., et al. (2021). Combination of serum and CSF neurofilament-light and neuroinflammatory biomarkers to evaluate ALS. Sci. Rep. 11, 703. doi:10.1038/s41598-020-80370-6
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172. doi:10.1056/NEJMra1603471
Bruijn, L. I., Miller, T. M., and Cleveland, D. W. (2004). Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 27, 723–749. doi:10.1146/annurev.neuro.27.070203.144244
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 355–364. doi:10.1016/j.mcn.2013.07.007
Butti, Z., and Patten, S. A. (2019). RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 9, 712. doi:10.3389/fgene.2018.00712
Cakir, B., Xiang, Y., Tanaka, Y., Kural, M. H., Parent, M., Kang, Y.-J., et al. (2019). Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175. doi:10.1038/s41592-019-0586-5
Calió, M. L., Henriques, E., Siena, A., Bertoncini, C. R. A., Gil-Mohapel, J., and Rosenstock, T. R. (2020). Mitochondrial dysfunction, neurogenesis, and epigenetics: Putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front. Neurosci. 14, 679. doi:10.3389/fnins.2020.00679
Canals, I., Quist, E., and Ahlenius, H. (2021). Transcription factor-based strategies to generate neural cell types from human pluripotent stem cells. Cell. Reprogr. 23, 206–220. doi:10.1089/cell.2021.0045
Cappello, V., and Francolini, M. (2017). Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 18, 2092. doi:10.3390/ijms18102092
Cecchelli, R., Berezowski, V., Lundquist, S., Culot, M., Renftel, M., Dehouck, M.-P., et al. (2007). Modelling of the blood–brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 6, 650–661. doi:10.1038/nrd2368
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi:10.1038/nbt.1529
Chen, G., Gulbranson, D. R., Hou, Z., Bolin, J. M., Ruotti, V., Probasco, M. D., et al. (2011). Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 8, 424–429. doi:10.1038/nmeth.1593
Chen, H., Kankel, M. W., Su, S. C., Han, S. W. S., and Ofengeim, D. (2018). Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell. Death Differ. 25, 648–662. doi:10.1038/s41418-018-0060-4
Chen, H., Qian, K., Du, Z., Cao, J., Petersen, A., Liu, H., et al. (2014). Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell. Stem Cell. 14, 796–809. doi:10.1016/j.stem.2014.02.004
Chennampally, P., Sayed-Zahid, A., Soundararajan, P., Sharp, J., Cox, G. A., Collins, S. D., et al. (2021). A microfluidic approach to rescue ALS motor neuron degeneration using rapamycin. Sci. Rep. 11, 18168. doi:10.1038/s41598-021-97405-1
Chiò, A., Mazzini, L., D’Alfonso, S., Corrado, L., Canosa, A., Moglia, C., et al. (2018). The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 91, e635–e642. doi:10.1212/WNL.0000000000005996
Chiò, A., Mora, G., and Lauria, G. (2017). Pain in amyotrophic lateral sclerosis. Lancet. Neurol. 16, 144–157. doi:10.1016/S1474-4422(16)30358-1
Choi, E. S., and Dokholyan, N. V. (2021). SOD1 oligomers in amyotrophic lateral sclerosis. Curr. Opin. Struct. Biol. 66, 225–230. doi:10.1016/j.sbi.2020.12.002
Churko, J. M., Lee, J., Ameen, M., Gu, M., Venkatasubramanian, M., Diecke, S., et al. (2017). Transcriptomic and epigenomic differences in human induced pluripotent stem cells generated from six reprogramming methods. Nat. Biomed. Eng. 1, 826–837. doi:10.1038/s41551-017-0141-6
Cividini, C., Basaia, S., Spinelli, E. G., Canu, E., Castelnovo, V., Riva, N., et al. (2021). Amyotrophic lateral sclerosis–frontotemporal dementia: Shared and divergent neural correlates across the clinical spectrum. Neurology 98, e402–e415. doi:10.1212/WNL.0000000000013123
Congzhou, M. S., Wang, J., and Dokholyan, N. V. (2021). NeuralDock: Rapid and conformation-agnostic docking of small molecules. Front. Mol. Biosci. 22 (9), 867241. bioRxiv. doi:10.3389/fmolb.2022.867241
Corti, S., Nizzardo, M., Simone, C., Falcone, M., Nardini, M., Ronchi, D., et al. (2012). Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 4, 165ra162–32. doi:10.1126/scitranslmed.3004108
Deneault, E., Chaineau, M., Nicouleau, M., Castellanos Montiel, M. J., Franco Flores, A. K., Haghi, G., et al. (2021). A streamlined CRISPR workflow to introduce mutations and generate isogenic iPSCs for modeling amyotrophic lateral sclerosis. Methods 203, 297–310. doi:10.1016/j.ymeth.2021.09.002
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can Be differentiated into motor neurons. Science 321, 1218–1221. doi:10.1126/science.1158799
Du, Z.-W., Chen, H., Liu, H., Lu, J., Qian, K., Huang, C.-L., et al. (2015). Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 6, 6626. doi:10.1038/ncomms7626
Eaton, S. L., and Wishart, T. M. (2017). Bridging the gap: Large animal models in neurodegenerative research. Mamm. Genome 28, 324–337. doi:10.1007/s00335-017-9687-6
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4, 145ra104. doi:10.1126/scitranslmed.3004052
Enright, H. A., Lam, D., Sebastian, A., Sales, A. P., Cadena, J., Hum, N. R., et al. (2020). Functional and transcriptional characterization of complex neuronal co-cultures. Sci. Rep. 10, 11007. doi:10.1038/s41598-020-67691-2
Faravelli, I., Bucchia, M., Rinchetti, P., Nizzardo, M., Simone, C., Frattini, E., et al. (2014). Motor neuron derivation from human embryonic and induced pluripotent stem cells: Experimental approaches and clinical perspectives. Stem Cell. Res. Ther. 5, 87. doi:10.1186/scrt476
Feneberg, E., Gordon, D., Thompson, A. G., Finelli, M. J., Dafinca, R., Candalija, A., et al. (2020). An ALS-linked mutation in TDP-43 disrupts normal protein interactions in the motor neuron response to oxidative stress. Neurobiol. Dis. 144, 105050. doi:10.1016/j.nbd.2020.105050
Fennema, E., Rivron, N., Rouwkema, J., van Blitterswijk, C., and de Boer, J. (2013). Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 31, 108–115. doi:10.1016/j.tibtech.2012.12.003
Fernandopulle, M. S., Prestil, R., Grunseich, C., Wang, C., Gan, L., and Ward, M. E. (2018). Transcription factor–mediated differentiation of human iPSCs into neurons. Curr. Protoc. Cell. Biol. 79, e51–48. doi:10.1002/cpcb.51
Ferraiuolo, L., and Maragakis, N. J. (2021). Mini-Review: Induced pluripotent stem cells and the search for new cell-specific ALS therapeutic targets. Neurosci. Lett. 755, 135911. doi:10.1016/j.neulet.2021.135911
Ferraiuolo, L., Meyer, K., Sherwood, T. W., Vick, J., Likhite, S., Frakes, A., et al. (2016). Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 113, E6496–E6505. doi:10.1073/pnas.1607496113
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi:10.1038/s41591-018-0140-5
Fukusumi, H., Shofuda, T., Bamba, Y., Yamamoto, A., Kanematsu, D., Handa, Y., et al. (2016). Establishment of human neural progenitor cells from human induced pluripotent stem cells with diverse tissue origins. Stem Cells Int. 2016, 1–10. doi:10.1155/2016/7235757
Fusaki, N., Ban, H., Nishiyama, A., Saeki, K., and Hasegawa, M. (2009). Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 85, 348–362. doi:10.2183/pjab.85.348
Garone, M. G., de Turris, V., Soloperto, A., Brighi, C., De Santis, R., Pagani, F., et al. (2019). Conversion of human induced pluripotent stem cells (iPSCs) into functional spinal and cranial motor neurons using piggyBac vectors. J. Vis. Exp. 147, 1–13. doi:10.3791/59321
Ghatak, S., Dolatabadi, N., Trudler, D., Zhang, X., Wu, Y., Mohata, M., et al. (2019). Mechanisms of hyperexcitability in Alzheimer's disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. Elife 8, e50333–22. doi:10.7554/eLife.50333
Giandomenico, S. L., Sutcliffe, M., and Lancaster, M. A. (2021). Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 16, 579–602. doi:10.1038/s41596-020-00433-w
Gleichman, A. J., and Carmichael, S. T. (2020). Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiol. Dis. 142, 104957. doi:10.1016/j.nbd.2020.104957
Gomes, A. R., Fernandes, T. G., Vaz, S. H., Silva, T. P., Bekman, E. P., Xapelli, S., et al. (2020). Modeling rett syndrome with human patient-specific forebrain organoids. Front. Cell. Dev. Biol. 8, 610427. doi:10.3389/fcell.2020.610427
Goparaju, S. K., Kohda, K., Ibata, K., Soma, A., Nakatake, Y., Akiyama, T., et al. (2017). Rapid differentiation of human pluripotent stem cells into functional neurons by mRNAs encoding transcription factors. Sci. Rep. 7, 42367. doi:10.1038/srep42367
Gore, A., Li, Z., Fung, H., Young, J. E., Agarwal, S., Antosiewicz-Bourget, J., et al. (2011). Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63–67. doi:10.1038/nature09805
Goto, K., Imamura, K., Komatsu, K., Mitani, K., Aiba, K., Nakatsuji, N., et al. (2017). Simple derivation of spinal motor neurons from ESCs/iPSCs using sendai virus vectors. Mol. Ther. Methods Clin. Dev. 4, 115–125. doi:10.1016/j.omtm.2016.12.007
Grebenyuk, S., and Ranga, A. (2019). Engineering organoid vascularization. Front. Bioeng. Biotechnol. 7, 39–12. doi:10.3389/fbioe.2019.00039
Gruzman, A., Wood, W. L., Alpert, E., Prasad, M. D., Miller, R. G., Rothstein, J. D., et al. (2007). Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 104, 12524–12529. doi:10.1073/pnas.0705044104
Gunes, Z. I., Kan, V. W. Y., Ye, X., and Liebscher, S. (2020). Exciting complexity: The role of motor circuit elements in ALS pathophysiology. Front. Neurosci. 14, 573. doi:10.3389/fnins.2020.00573
Guo, W., Fumagalli, L., Prior, R., and Van Den Bosch, L. (2017a). Current advances and limitations in modeling ALS/FTD in a dish using induced pluripotent stem cells. Front. Neurosci. 11, 671. doi:10.3389/fnins.2017.00671
Guo, W., Naujock, M., Fumagalli, L., Vandoorne, T., Baatsen, P., Boon, R., et al. (2017b). HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8, 861. doi:10.1038/s41467-017-00911-y
Hawrot, J., Imhof, S., and Wainger, B. J. (2020). Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol. Dis. 134, 104680. doi:10.1016/j.nbd.2019.104680
Healy, E. F., Roth-Rodriguez, A., and Toledo, S. (2020). A model for gain of function in superoxide dismutase. Biochem. Biophys. Rep. 21, 100728. doi:10.1016/j.bbrep.2020.100728
Hester, M. E., Murtha, M. J., Song, S., Rao, M., Miranda, C. J., Meyer, K., et al. (2011). Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. 19, 1905–1912. doi:10.1038/mt.2011.135
Hong, S. G., Dunbar, C. E., and Winkler, T. (2013). Assessing the risks of genotoxicity in the therapeutic development of induced pluripotent stem cells. Mol. Ther. 21, 272–281. doi:10.1038/mt.2012.255
Hor, J. H., Soh, E. S.-Y., Tan, L. Y., Lim, V. J. W., Santosa, M. M., Winanto, W., et al. (2018). Cell cycle inhibitors protect motor neurons in an organoid model of Spinal Muscular Atrophy. Cell. Death Dis. 9, 1100. doi:10.1038/s41419-018-1081-0
Hou, P., Li, Y., Zhang, X., Liu, C., Guan, J., Li, H., et al. (2013). Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341, 651–654. doi:10.1126/science.1239278
Hu, B.-Y., and Zhang, S.-C. (2009). Differentiation of spinal motor neurons from pluripotent human stem cells. Nat. Protoc. 4, 1295–1304. doi:10.1038/nprot.2009.127
Hu, W., He, Y., Xiong, Y., Lu, H., Chen, H., Hou, L., et al. (2016). Derivation, expansion, and motor neuron differentiation of human-induced pluripotent stem cells with non-integrating episomal vectors and a defined xenogeneic-free culture system. Mol. Neurobiol. 53, 1589–1600. doi:10.1007/s12035-014-9084-z
Huang, F., Zhu, Y., Hsiao‐Nakamoto, J., Tang, X., Dugas, J. C., Moscovitch‐Lopatin, M., et al. (2020). Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 7, 1103–1116. doi:10.1002/acn3.51078
Huh, C. J., Zhang, B., Victor, M. B., Dahiya, S., Batista, L. F. Z., Horvath, S., et al. (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. Elife 5, e18648–14. doi:10.7554/eLife.18648
Iacoangeli, A., Al Khleifat, A., Sproviero, W., Shatunov, A., Jones, A. R., Opie-Martin, S., et al. (2019). ALSgeneScanner: A pipeline for the analysis and interpretation of DNA sequencing data of ALS patients. Amyotroph. Lateral Scler. Front. Degener. 20, 207–215. doi:10.1080/21678421.2018.1562553
Imamura, K., Izumi, Y., Banno, H., Uozumi, R., Morita, S., Egawa, N., et al. (2019). Induced pluripotent stem cell–based drug repurposing for amyotrophic lateral sclerosis medicine (iDReAM) study: Protocol for a phase I dose escalation study of bosutinib for amyotrophic lateral sclerosis patients. BMJ Open 9, e033131. doi:10.1136/bmjopen-2019-033131
Imamura, K., Izumi, Y., Watanabe, A., Tsukita, K., Woltjen, K., Yamamoto, T., et al. (2017). The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 9, eaaf3962–11. doi:10.1126/scitranslmed.aaf3962
Joannides, A. J., Fiore‐Hériché, C., Battersby, A. A., Athauda‐Arachchi, P., Bouhon, I. A., Williams, L., et al. (2007). A scaleable and defined system for generating neural stem cells from human embryonic stem cells. Stem Cells 25, 731–737. doi:10.1634/stemcells.2006-0562
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M., and Tanabe, M. (2021). KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551. doi:10.1093/nar/gkaa970
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi:10.1093/nar/28.1.27
Karakatsani, A., Shah, B., and Ruiz de Almodovar, C. (2019). Blood vessels as regulators of neural stem cell properties. Front. Mol. Neurosci. 12, 85–11. doi:10.3389/fnmol.2019.00085
Kim, D.-S., Lee, J. S., Leem, J. W., Huh, Y. J., Kim, J. Y., Kim, H.-S., et al. (2010a). Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell. Rev. Rep. 6, 270–281. doi:10.1007/s12015-010-9138-1
Kim, D., Kim, C.-H., Moon, J., Chung, Y., Chang, M., Han, B., et al. (2009a). Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell. Stem Cell. 4, 472–476. doi:10.1016/j.stem.2009.05.005
Kim, J. B., Greber, B., Araúzo-Bravo, M. J., Meyer, J., Park, K. I., Zaehres, H., et al. (2009b). Direct reprogramming of human neural stem cells by OCT4. Nature 461, 649–643. doi:10.1038/nature08436
Kim, J. B., Zaehres, H., Wu, G., Gentile, L., Ko, K., Sebastiano, V., et al. (2008). Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 454, 646–650. doi:10.1038/nature07061
Kim, K., Doi, A., Wen, B., Ng, K., Zhao, R., Cahan, P., et al. (2010b). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. doi:10.1038/nature09342
Kirkitadze, M. D., Bitan, G., and Teplow, D. B. (2002). Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res. 69, 567–577. doi:10.1002/jnr.10328
Kiskinis, E., Kralj, J. M., Zou, P., Weinstein, E. N., Zhang, H., Tsioras, K., et al. (2018). All-optical electrophysiology for high-throughput functional characterization of a human iPSC-derived motor neuron model of ALS. Stem Cell. Rep. 10, 1991–2004. doi:10.1016/j.stemcr.2018.04.020
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell. Stem Cell. 14, 781–795. doi:10.1016/j.stem.2014.03.004
Klim, J. R., Williams, L. A., Limone, F., Guerra San Juan, I., Davis-Dusenbery, B. N., Mordes, D. A., et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179. doi:10.1038/s41593-018-0300-4
Kogut, I., McCarthy, S. M., Pavlova, M., Astling, D. P., Chen, X., Jakimenko, A., et al. (2018). High-efficiency RNA-based reprogramming of human primary fibroblasts. Nat. Commun. 9, 745. doi:10.1038/s41467-018-03190-3
Koutsakis, C., and Kazanis, I. (2016). How necessary is the vasculature in the life of neural stem and progenitor cells? Evidence from evolution, development and the adult nervous system. Front. Cell. Neurosci. 10, 35–37. doi:10.3389/fncel.2016.00035
Kwon, D., Ji, M., Lee, S., Seo, K. W., and Kang, K.-S. (2017). Reprogramming enhancers in somatic cell nuclear transfer, iPSC technology, and direct conversion. Stem Cell. Rev. Rep. 13, 24–34. doi:10.1007/s12015-016-9697-x
Lancaster, M. A., and Knoblich, J. A. (2014). Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340. doi:10.1038/nprot.2014.158
Lancaster, M. A., Renner, M., Martin, C.-A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. doi:10.1038/nature12517
Lapasset, L., Milhavet, O., Prieur, A., Besnard, E., Babled, A., Aït-Hamou, N., et al. (2011). Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes. Dev. 25, 2248–2253. doi:10.1101/gad.173922.111
Lee, H., Lee, J. J., Park, N. Y., Dubey, S. K., Kim, T., Ruan, K., et al. (2021). Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS. Nat. Neurosci. 24, 1673–1685. doi:10.1038/s41593-021-00944-z
Lee, J.-H., Liu, J.-W., Lin, S.-Z., Harn, H.-J., and Chiou, T.-W. (2018). Advances in patient-specific induced pluripotent stem cells shed light on drug discovery for amyotrophic lateral sclerosis. Cell. Transpl. 27, 1301–1312. doi:10.1177/0963689718785154
Li, J., Lim, R. G., Kaye, J. A., Dardov, V., Coyne, A. N., Wu, J., et al. (2021). An integrated multi-omic analysis of iPSC-derived motor neurons from C9ORF72 ALS patients. iScience 24, 103221. doi:10.1016/j.isci.2021.103221
Li, X.-J., Du, Z.-W., Zarnowska, E. D., Pankratz, M., Hansen, L. O., Pearce, R. A., et al. (2005). Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 23, 215–221. doi:10.1038/nbt1063
Li, Y., Balasubramanian, U., Cohen, D., Zhang, P.-W., Mosmiller, E., Sattler, R., et al. (2015). A comprehensive library of familial human amyotrophic lateral sclerosis induced pluripotent stem cells. PLoS One 10, e0118266. doi:10.1371/journal.pone.0118266
Liang, G., and Zhang, Y. (2013). Genetic and epigenetic variations in iPSCs: Potential causes and implications for application. Cell. Stem Cell. 13, 149–159. doi:10.1016/j.stem.2013.07.001
Liebau, S., Mahaddalkar, P. U., Kestler, H. A., Illing, A., Seufferlein, T., and Kleger, A. (2013). A hierarchy in reprogramming capacity in different tissue microenvironments: What we know and what we need to know. Stem Cells Dev. 22, 695–706. doi:10.1089/scd.2012.0461
Limone, F., Mitchell, J. M., San Juan, I. G., Smith, J. L. M., Raghunathan, K., Couto, A., et al. (2022). Efficient generation of lower induced Motor Neurons by coupling Ngn2 expression with developmental cues. bioRxiv, 2022.01.12.476020. Available at: http://biorxiv.org/content/early/2022/01/12/2022.01.12.476020.abstract (Accessed June 30, 2022).
Lin, C.-Y., Yoshida, M., Li, L.-T., Ikenaka, A., Oshima, S., Nakagawa, K., et al. (2019). iPSC-derived functional human neuromuscular junctions model the pathophysiology of neuromuscular diseases. JCI Insight 4, 124299. doi:10.1172/jci.insight.124299
Liu, X., Li, C., Zheng, K., Zhao, X., Xu, X., Yang, A., et al. (2020). Chromosomal aberration arises during somatic reprogramming to pluripotent stem cells. Cell. Div. 15, 12. doi:10.1186/s13008-020-00068-z
Logroscino, G., and Piccininni, M. (2019). Amyotrophic lateral sclerosis descriptive epidemiology: The origin of geographic difference. Neuroepidemiology 52, 93–103. doi:10.1159/000493386
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72 -related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. doi:10.1016/j.neuron.2016.09.015
Maciel, R., Bis, D. M., Rebelo, A. P., Saghira, C., Züchner, S., and Saporta, M. A. (2018). The human motor neuron axonal transcriptome is enriched for transcripts related to mitochondrial function and microtubule-based axonal transport. Exp. Neurol. 307, 155–163. doi:10.1016/j.expneurol.2018.06.008
Madill, M., McDonagh, K., Ma, J., Vajda, A., McLoughlin, P., O’Brien, T., et al. (2017). Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 10, 22. doi:10.1186/s13041-017-0300-4
Mali, P., Chou, B., Yen, J., Ye, Z., Zou, J., Dowey, S., et al. (2010). Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency‐associated genes. Stem Cells 28, 713–720. doi:10.1002/stem.402
Maltman, D. J., and Przyborski, S. A. (2010). Developments in three-dimensional cell culture technology aimed at improving the accuracy of in vitro analyses. Biochem. Soc. Trans. 38, 1072–1075. doi:10.1042/BST0381072
Marrone, L., Poser, I., Casci, I., Japtok, J., Reinhardt, P., Janosch, A., et al. (2018). Isogenic FUS-eGFP iPSC reporter lines enable quantification of FUS stress granule pathology that is rescued by drugs inducing autophagy. Stem Cell. Rep. 10, 375–389. doi:10.1016/j.stemcr.2017.12.018
Maury, Y., Côme, J., Piskorowski, R. A., Salah-Mohellibi, N., Chevaleyre, V., Peschanski, M., et al. (2015). Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 33, 89–96. doi:10.1038/nbt.3049
McAlary, L., Chew, Y. L., Lum, J. S., Geraghty, N. J., Yerbury, J. J., and Cashman, N. R. (2020). Amyotrophic lateral sclerosis: Proteins, proteostasis, prions, and promises. Front. Cell. Neurosci. 14, 581907. doi:10.3389/fncel.2020.581907
McCann, E. P., Henden, L., Fifita, J. A., Zhang, K. Y., Grima, N., Bauer, D. C., et al. (2021). Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J. Med. Genet. 58, 87–95. doi:10.1136/jmedgenet-2020-106866
Meissner, A., Wernig, M., and Jaenisch, R. (2007). Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat. Biotechnol. 25, 1177–1181. doi:10.1038/nbt1335
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 13, 1310. doi:10.3389/fnins.2019.01310
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Böhnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell. Stem Cell. 17, 705–718. doi:10.1016/j.stem.2015.09.001
Mezzani, A., Pisano, F., Cavalli, A., Tommasi, M. A., Corrà, U., Colombo, S., et al. (2012). Reduced exercise capacity in early-stage amyotrophic lateral sclerosis: Role of skeletal muscle. Amyotroph. Lateral Scler. 13, 87–94. doi:10.3109/17482968.2011.601463
Mikkelsen, T. S., Hanna, J., Zhang, X., Ku, M., Wernig, M., Schorderet, P., et al. (2008). Dissecting direct reprogramming through integrative genomic analysis. Nature 454, 49–55. doi:10.1038/nature07056
Miller, J. D., Ganat, Y. M., Kishinevsky, S., Bowman, R. L., Liu, B., Tu, E. Y., et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell. Stem Cell. 13, 691–705. doi:10.1016/j.stem.2013.11.006
Mishra, V., Re, D. B., Le Verche, V., Alvarez, M. J., Vasciaveo, A., Jacquier, A., et al. (2020). Systematic elucidation of neuron-astrocyte interaction in models of amyotrophic lateral sclerosis using multi-modal integrated bioinformatics workflow. Nat. Commun. 11, 5579. doi:10.1038/s41467-020-19177-y
Mohassel, P., Donkervoort, S., Lone, M. A., Nalls, M., Gable, K., Gupta, S. D., et al. (2021). Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat. Med. 27, 1197–1204. doi:10.1038/s41591-021-01346-1
Montana, C. L., Kolesnikov, A. V., Shen, S. Q., Myers, C. A., Kefalov, V. J., and Corbo, J. C. (2013). Reprogramming of adult rod photoreceptors prevents retinal degeneration. Proc. Natl. Acad. Sci. U. S. A. 110, 1732–1737. doi:10.1073/pnas.1214387110
Morimoto, S., Takahashi, S., Fukushima, K., Saya, H., Suzuki, N., Aoki, M., et al. (2019). Ropinirole hydrochloride remedy for amyotrophic lateral sclerosis – protocol for a randomized, double-blind, placebo-controlled, single-center, and open-label continuation phase I/IIa clinical trial (ROPALS trial). Regen. Ther. 11, 143–166. doi:10.1016/j.reth.2019.07.002
Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., and Sasai, Y. (2015). Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell. Rep. 10, 537–550. doi:10.1016/j.celrep.2014.12.051
Müller, F.-J., Goldmann, J., Löser, P., and Loring, J. F. (2010). A call to standardize teratoma assays used to define human pluripotent cell lines. Cell. Stem Cell. 6, 412–414. doi:10.1016/j.stem.2010.04.009
Murphy, A. R., Laslett, A., O’Brien, C. M., and Cameron, N. R. (2017). Scaffolds for 3D in vitro culture of neural lineage cells. Acta Biomater. 54, 1–20. doi:10.1016/j.actbio.2017.02.046
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi:10.1038/nn1876
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., et al. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106. doi:10.1038/nbt1374
Naujock, M., Stanslowsky, N., Bufler, S., Naumann, M., Reinhardt, P., Sterneckert, J., et al. (2016). 4‐Aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient‐derived induced pluripotent stem cells. Stem Cells 34, 1563–1575. doi:10.1002/stem.2354
Nickels, S. L., Modamio, J., Mendes-Pinheiro, B., Monzel, A. S., Betsou, F., and Schwamborn, J. C. (2020). Reproducible generation of human midbrain organoids for in vitro modeling of Parkinson’s disease. Stem Cell. Res. 46, 101870. doi:10.1016/j.scr.2020.101870
Niemiec, E., and Howard, H. C. (2020). Ethical issues related to research on genome editing in human embryos. Comput. Struct. Biotechnol. J. 18, 887–896. doi:10.1016/j.csbj.2020.03.014
Okita, K., Nakagawa, M., Hyenjong, H., Ichisaka, T., and Yamanaka, S. (2008). Generation of mouse induced pluripotent stem cells without viral vectors. Science 322, 949–953. doi:10.1126/science.1164270
Osafune, K., Caron, L., Borowiak, M., Martinez, R. J., Fitz-Gerald, C. S., Sato, Y., et al. (2008). Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 26, 313–315. doi:10.1038/nbt1383
Osaki, T., Uzel, S. G. M., and Kamm, R. D. (2018). Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4, eaat5847–16. doi:10.1126/sciadv.aat5847
Osborn, T. M., Beagan, J., and Isacson, O. (2018). Increased motor neuron resilience by small molecule compounds that regulate IGF-II expression. Neurobiol. Dis. 110, 218–230. doi:10.1016/j.nbd.2017.11.002
Pampalakis, G., Mitropoulos, K., Xiromerisiou, G., Dardiotis, E., Deretzi, G., Anagnostouli, M., et al. (2019). New molecular diagnostic trends and biomarkers for amyotrophic lateral sclerosis. Hum. Mutat. 40, 361–373. doi:10.1002/humu.23697
Pampaloni, F., Reynaud, E. G., and Stelzer, E. H. K. (2007). The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell. Biol. 8, 839–845. doi:10.1038/nrm2236
Pandya, V. A., and Patani, R. (2020). Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 143, 1057–1072. doi:10.1093/brain/awz360
Pang, Z. P., Yang, N., Vierbuchen, T., Ostermeier, A., Fuentes, D. R., Yang, T. Q., et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. doi:10.1038/nature10202
Park, I.-H., Zhao, R., West, J. A., Yabuuchi, A., Huo, H., Ince, T. A., et al. (2008). Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146. doi:10.1038/nature06534
Paşca, A. M., Sloan, S. A., Clarke, L. E., Tian, Y., Makinson, C. D., Huber, N., et al. (2015). Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678. doi:10.1038/nmeth.3415
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 7, 710–723. doi:10.1038/nrn1971
Pasteuning-Vuhman, S., de Jongh, R., Timmers, A., and Pasterkamp, R. J. (2021). Towards advanced iPSC-based drug development for neurodegenerative disease. Trends Mol. Med. 27, 263–279. doi:10.1016/j.molmed.2020.09.013
Pereira, J. D., DuBreuil, D. M., Devlin, A.-C., Held, A., Sapir, Y., Berezovski, E., et al. (2021). Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat. Commun. 12, 4744. doi:10.1038/s41467-021-24776-4
Phukan, J., Pender, N. P., and Hardiman, O. (2007). Cognitive impairment in amyotrophic lateral sclerosis. Lancet. Neurol. 6, 994–1003. doi:10.1016/S1474-4422(07)70265-X
Picher-Martel, V., Brunet, F., Dupré, N., and Chrestian, N. (2020). The occurrence of FUS mutations in pediatric amyotrophic lateral sclerosis: A case report and review of the literature. J. Child. Neurol. 35, 556–562. doi:10.1177/0883073820915099
Porterfield, V., Khan, S. S., Foff, E. P., Koseoglu, M. M., Blanco, I. K., Jayaraman, S., et al. (2020). A three-dimensional dementia model reveals spontaneous cell cycle re-entry and a senescence-associated secretory phenotype. Neurobiol. Aging 90, 125–134. doi:10.1016/j.neurobiolaging.2020.02.011
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12, 25. doi:10.3389/fnmol.2019.00025
Prè, D., Nestor, M. W., Sproul, A. A., Jacob, S., Koppensteiner, P., Chinchalongporn, V., et al. (2014). A time course analysis of the electrophysiological properties of neurons differentiated from human induced pluripotent stem cells (iPSCs). PLoS One 9, e103418. doi:10.1371/journal.pone.0103418
Proctor, E. A., Fee, L., Tao, Y., Redler, R. L., Fay, J. M., Zhang, Y., et al. (2016). Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 113, 614–619. doi:10.1073/pnas.1516725113
Qian, X., Jacob, F., Song, M. M., Nguyen, H. N., Song, H., and Ming, G. (2018). Generation of human brain region–specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 13, 565–580. doi:10.1038/nprot.2017.152
Qu, Q., Li, D., Louis, K. R., Li, X., Yang, H., Sun, Q., et al. (2014). High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat. Commun. 5, 3449. doi:10.1038/ncomms4449
Raja, W. K., Mungenast, A. E., Lin, Y.-T., Ko, T., Abdurrob, F., Seo, J., et al. (2016). Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate alzheimer’s disease phenotypes. PLoS One 11, e0161969. doi:10.1371/journal.pone.0161969
Ramos-Mejia, V., Muñoz-Lopez, M., Garcia-Perez, J. L., and Menendez, P. (2010). iPSC lines that do not silence the expression of the ectopic reprogramming factors may display enhanced propensity to genomic instability. Cell. Res. 20, 1092–1095. doi:10.1038/cr.2010.125
Redler, R. L., and Dokholyan, N. V. (2012). “The complex molecular biology of amyotrophic lateral sclerosis (ALS),” in Progress in molecular Biology and translational science (Amsterdam: Elsevier), 215–262. doi:10.1016/B978-0-12-385883-2.00002-3
Rey, F., Barzaghini, B., Nardini, A., Bordoni, M., Zuccotti, G. V., Cereda, C., et al. (2020). Advances in tissue engineering and innovative fabrication techniques for 3-D-structures: Translational applications in neurodegenerative diseases. Cells 9, 1636. doi:10.3390/cells9071636
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi:10.1038/nrn3430
Romero-Morales, A., Rastogi, A., Temuri, H., Rasmussen, M., McElroy, G. S., Hsu, L., et al. (2020). Human iPSC-derived cerebral organoids model features of Leigh syndrome and reveal abnormal corticogenesis. Development. 149 (20), dev199914. SSRN Journal. doi:10.1242/dev.199914
Roth, J. G., Huang, M. S., Li, T. L., Feig, V. R., Jiang, Y., Cui, B., et al. (2021). Advancing models of neural development with biomaterials. Nat. Rev. Neurosci. 22, 593–615. doi:10.1242/dev.199914
Ruegsegger, C., and Saxena, S. (2016). Proteostasis impairment in ALS. Brain Res. 1648, 571–579. doi:10.1016/j.brainres.2016.03.032
Russell, K. L., Downie, J. M., Gibson, S. B., Tsetsou, S., Keefe, M. D., Duran, J. A., et al. (2021). Pathogenic effect of TP73 gene variants in people with amyotrophic lateral sclerosis. Neurology 97, e225–e235. doi:10.1212/WNL.0000000000012285
Rutherford, N. J., Zhang, Y.-J., Baker, M., Gass, J. M., Finch, N. A., Xu, Y.-F., et al. (2008). Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4, e1000193. doi:10.1371/journal.pgen.1000193
Saberi, S., Stauffer, J. E., Schulte, D. J., and Ravits, J. (2015). Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 33, 855–876. doi:10.1016/j.ncl.2015.07.012
Sakaguchi, H., Ozaki, Y., Ashida, T., Matsubara, T., Oishi, N., Kihara, S., et al. (2019). Self-organized synchronous calcium transients in a cultured human neural network derived from cerebral organoids. Stem Cell. Rep. 13, 458–473. doi:10.1016/j.stemcr.2019.05.029
Samarasinghe, R. A., Miranda, O. A., Buth, J. E., Mitchell, S., Ferando, I., Watanabe, M., et al. (2021). Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 24, 1488–1500. doi:10.1038/s41593-021-00906-5
Sances, S., Bruijn, L. I., Chandran, S., Eggan, K., Ho, R., Klim, J. R., et al. (2016). Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 19, 542–553. doi:10.1038/nn.4273
Sepehrimanesh, M., and Ding, B. (2020). Generation and optimization of highly pure motor neurons from human induced pluripotent stem cells via lentiviral delivery of transcription factors. Am. J. Physiol. Cell. Physiol. 319, C771–C780. doi:10.1152/ajpcell.00279.2020
Sheridan, S. D., Surampudi, V., and Rao, R. R. (2012). Analysis of embryoid bodies derived from human induced pluripotent stem cells as a means to assess pluripotency. Stem Cells Int. 2012, 738910. doi:10.1155/2012/738910
Shimojo, D., Onodera, K., Doi-Torii, Y., Ishihara, Y., Hattori, C., Miwa, Y., et al. (2015). Rapid, efficient, and simple motor neuron differentiation from human pluripotent stem cells. Mol. Brain 8, 79. doi:10.1186/s13041-015-0172-4
Shirvanyants, D., Alexandrova, A. N., and Dokholyan, N. V. (2011). Rigid substructure search. Bioinformatics 27, 1327–1329. doi:10.1093/bioinformatics/btr129
Smeyers, J., Banchi, E.-G., and Latouche, M. (2021). C9ORF72: What it is, what it does, and why it matters. Front. Cell. Neurosci. 15, 661447. doi:10.3389/fncel.2021.661447
Sommer, C. A., Christodoulou, C., Gianotti-Sommer, A., Shen, S. S., Sailaja, B. S., Hezroni, H., et al. (2012). Residual expression of reprogramming factors affects the transcriptional program and epigenetic signatures of induced pluripotent stem cells. PLoS One 7, e51711. doi:10.1371/journal.pone.0051711
Son, E. Y., Ichida, J. K., Wainger, B. J., Toma, J. S., Rafuse, V. F., Woolf, C. J., et al. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell. Stem Cell. 9, 205–218. doi:10.1016/j.stem.2011.07.014
Sood, D., Cairns, D. M., Dabbi, J. M., Ramakrishnan, C., Deisseroth, K., Black, L. D., et al. (2019). Functional maturation of human neural stem cells in a 3D bioengineered brain model enriched with fetal brain-derived matrix. Sci. Rep. 9, 17874. doi:10.1038/s41598-019-54248-1
Stan, A. D., Ghose, S., Gao, X., Roberts, R. C., Lewis-Amezcua, K., Hatanpaa, K. J., et al. (2006). Human postmortem tissue: What quality markers matter? Brain Res. 1123, 1–11. doi:10.1016/j.brainres.2006.09.025
Sun, X., Song, J., Huang, H., Chen, H., and Qian, K. (2018). Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells. Stem Cell. Res. Ther. 9, 315. doi:10.1186/s13287-018-1048-1
Szebényi, K., Wenger, L. M. D., Sun, Y., Dunn, A. W. E., Limegrover, C. A., Gibbons, G. M., et al. (2021). Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat. Neurosci. 24, 1542–1554. doi:10.1038/s41593-021-00923-4
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi:10.1093/nar/gky1131
Taga, A., Habela, C. W., Johns, A., Liu, S., O’Brien, M., and Maragakis, N. J. (2021). Establishment of an electrophysiological platform for modeling ALS with regionally-specific human pluripotent stem cell-derived astrocytes and neurons. JoVE. 174, e62726. doi:10.3791/62726
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131, 861–872. doi:10.1016/j.cell.2007.11.019
Takahashi, K., and Yamanaka, S. (2013). Induced pluripotent stem cells in medicine and biology. Development 140, 2457–2461. doi:10.1242/dev.092551
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663–676. doi:10.1016/j.cell.2006.07.024
Tang, Y., Liu, M.-L., Zang, T., and Zhang, C.-L. (2017). Direct reprogramming rather than iPSC-based reprogramming maintains aging hallmarks in human motor neurons. Front. Mol. Neurosci. 10, 359. doi:10.3389/fnmol.2017.00359
Tian, R., Gachechiladze, M. A., Ludwig, C. H., Laurie, M. T., Hong, J. Y., Nathaniel, D., et al. (2019). CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron 104, 239–255. e12. doi:10.1016/j.neuron.2019.07.014
Tong, M., Lv, Z., Liu, L., Zhu, H., Zheng, Q.-Y., Zhao, X.-Y., et al. (2011). Mice generated from tetraploid complementation competent iPS cells show similar developmental features as those from ES cells but are prone to tumorigenesis. Cell. Res. 21, 1634–1637. doi:10.1038/cr.2011.143
Trokovic, R., Weltner, J., Nishimura, K., Ohtaka, M., Nakanishi, M., Salomaa, V., et al. (2014). Advanced feeder-free generation of induced pluripotent stem cells directly from blood cells. Stem Cells Transl. Med. 3, 1402–1409. doi:10.5966/sctm.2014-0113
Türker, E., Demirçak, N., and Arslan-Yildiz, A. (2018). Scaffold-free three-dimensional cell culturing using magnetic levitation. Biomater. Sci. 6, 1745–1753. doi:10.1039/C8BM00122G
Turner, M. R., Al-Chalabi, A., Chio, A., Hardiman, O., Kiernan, M. C., Rohrer, J. D., et al. (2017). Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 88, 1042–1044. doi:10.1136/jnnp-2017-315995
Urushitani, M., Sik, A., Sakurai, T., Nukina, N., Takahashi, R., and Julien, J.-P. (2006). Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 9, 108–118. doi:10.1038/nn1603
Uzel, S. G. M., Platt, R. J., Subramanian, V., Pearl, T. M., Rowlands, C. J., Chan, V., et al. (2016). Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci. Adv. 2, e1501429. doi:10.1126/sciadv.1501429
Van Der Stappen, A., Van Den Hauwe, L., Merlevede, K., Degryse, H., Van Goethem, J., and Parizel, P. M. (2005). Amyotrophic lateral sclerosis. Tijdschr. Geneeskd. 61, 352–353. doi:10.2143/tvg.61.5.1002112
Van Norman, G. A. (2019). Limitations of animal studies for predicting toxicity in clinical trials. JACC. Basic Transl. Sci. 4, 845–854. doi:10.1016/j.jacbts.2019.10.008
Velasco, S., Kedaigle, A. J., Simmons, S. K., Nash, A., Rocha, M., Quadrato, G., et al. (2019). Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527. doi:10.1038/s41586-019-1289-x
Vera, E., Bosco, N., and Studer, L. (2016). Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell. Rep. 17, 1184–1192. doi:10.1016/j.celrep.2016.09.062
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S. W., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell. Rep. 7, 1–11. doi:10.1016/j.celrep.2014.03.019
Wainger, B. J., Macklin, E. A., Vucic, S., McIlduff, C. E., Paganoni, S., Maragakis, N. J., et al. (2021). Effect of ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis: A randomized clinical trial. JAMA Neurol. 78, 186–196. doi:10.1001/jamaneurol.2020.4300
Wang, J., and Dokholyan, N. V. (2019). MedusaDock 2.0: Efficient and accurate protein-ligand docking with constraints. J. Chem. Inf. Model. 59, 2509–2515. doi:10.1021/acs.jcim.8b00905
Wang, J., Hao, J., Bai, D., Gu, Q., Han, W., Wang, L., et al. (2015). Generation of clinical-grade human induced pluripotent stem cells in Xeno-free conditions. Stem Cell. Res. Ther. 6, 223. doi:10.1186/s13287-015-0206-y
Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y., Li, H., Lau, F., et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell. Stem Cell. 7, 618–630. doi:10.1016/j.stem.2010.08.012
Weltner, J., Balboa, D., Katayama, S., Bespalov, M., Krjutškov, K., Jouhilahti, E.-M., et al. (2018). Human pluripotent reprogramming with CRISPR activators. Nat. Commun. 9, 2643. doi:10.1038/s41467-018-05067-x
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., et al. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324. doi:10.1038/nature05944
Wichterle, H., Lieberam, I., Porter, J. A., and Jessell, T. M. (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell. 110, 385–397. doi:10.1016/S0092-8674(02)00835-8
Woltjen, K., Michael, I. P., Mohseni, P., Desai, R., Mileikovsky, M., Hämäläinen, R., et al. (2009). piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770. doi:10.1038/nature07863
Wood, H. (2021). TP73 variants implicate apoptosis in amyotrophic lateral sclerosis pathogenesis. Nat. Rev. Neurol. 17, 462. doi:10.1038/s41582-021-00532-6
Wu, S., Xu, R., Duan, B., and Jiang, P. (2017). Three-dimensional hyaluronic acid hydrogel-based models for in vitro human iPSC-derived NPC culture and differentiation. J. Mat. Chem. B 5, 3870–3878. doi:10.1039/C7TB00721C
Xie, Y., Luo, X., He, H., and Tang, M. (2021). Novel insight into the role of immune dysregulation in amyotrophic lateral sclerosis based on bioinformatic analysis. Front. Neurosci. 15, 657465. doi:10.3389/fnins.2021.657465
Yakoub, A. (2019). Cerebral organoids exhibit mature neurons and astrocytes and recapitulate electrophysiological activity of the human brain. Neural Regen. Res. 14, 757–761. doi:10.4103/1673-5374.249283
Yakubov, E., Rechavi, G., Rozenblatt, S., and Givol, D. (2010). Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Commun. 394, 189–193. doi:10.1016/j.bbrc.2010.02.150
Yang, Y. M., Gupta, S. K., Kim, K. J., Powers, B. E., Cerqueira, A., Wainger, B. J., et al. (2013). A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell. Stem Cell. 12, 713–726. doi:10.1016/j.stem.2013.04.003
Ye, H., and Wang, Q. (2018). Efficient generation of non-integration and feeder-free induced pluripotent stem cells from human peripheral blood cells by sendai virus. Cell. Physiol. biochem. 50, 1318–1331. doi:10.1159/000494589
Yin, S., Ding, F., and Dokholyan, N. V. (2010). “Modeling mutations in proteins using medusa and discrete molecule dynamics,” in Introduction to protein structure prediction (Hoboken, NJ, USA: John Wiley & Sons), 453–476. doi:10.1002/9780470882207.ch20
Yu, J., Hu, K., Smuga-Otto, K., Tian, S., Stewart, R., Slukvin, I. I., et al. (2009). Human induced pluripotent stem cells free of vector and transgene sequences. Science 324, 797–801. doi:10.1126/science.1172482
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. doi:10.1126/science.1151526
Zarzeczny, A., Scott, C., Hyun, I., Bennett, J., Chandler, J., Chargé, S., et al. (2009). iPS cells: Mapping the policy issues. Cell. 139, 1032–1037. doi:10.1016/j.cell.2009.11.039
Zhang, Z.-N., Freitas, B. C., Qian, H., Lux, J., Acab, A., Trujillo, C. A., et al. (2016). Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc. Natl. Acad. Sci. U. S. A. 113, 3185–3190. doi:10.1073/pnas.1521255113
Zhou, W., and Freed, C. R. (2009). Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 27, 2667–2674. doi:10.1002/stem.201
Zhu, C., Beck, M. V., Griffith, J. D., Deshmukh, M., and Dokholyan, N. V. (2018). Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 115, 4661–4665. doi:10.1073/pnas.1800187115
Keywords: amyotrophic lateral sclerosis (ALS), induced pluripotent stem cells (iPSCs), drug screening, in vitro disease model, 3D cell culture, bioinformactics, motor neuron differentiation
Citation: Amorós MA, Choi ES, Cofré AR, Dokholyan NV and Duzzioni M (2022) Motor neuron-derived induced pluripotent stem cells as a drug screening platform for amyotrophic lateral sclerosis. Front. Cell Dev. Biol. 10:962881. doi: 10.3389/fcell.2022.962881
Received: 06 June 2022; Accepted: 20 July 2022;
Published: 24 August 2022.
Edited by:
Helen Cristina Miranda, Case Western Reserve University, United StatesReviewed by:
Christopher Grunseich, National Institutes of Health (NIH), United StatesCopyright © 2022 Amorós, Choi, Cofré, Dokholyan and Duzzioni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana A. Amorós, bWFyaWFuYS5hbW9yb3NAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.