 Hamed Alizadeh Pahlavani
Hamed Alizadeh Pahlavani
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 11 August 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.950927
This article is part of the Research Topic Cardiac and Skeletal Muscle Response to Exercise View all 4 articles
Cardiovascular diseases are the most common cause of death in the world. One of the major causes of cardiac death is excessive apoptosis. However, multiple pathways through moderate exercise can reduce myocardial apoptosis. After moderate exercise, the expression of anti-apoptotic proteins such as IGF-1, IGF-1R, p-PI3K, p-Akt, ERK-1/2, SIRT3, PGC-1α, and Bcl-2 increases in the heart. While apoptotic proteins such as PTEN, PHLPP-1, GSK-3, JNK, P38MAPK, and FOXO are reduced in the heart. Exercise-induced mechanical stress activates the β and α5 integrins and subsequently, focal adhesion kinase phosphorylation activates the Akt/mTORC1 and ERK-1/2 pathways, leading to an anti-apoptotic response. One of the reasons for the decrease in exercise-induced apoptosis is the decrease in Fas-ligand protein, Fas-death receptor, TNF-α receptor, Fas-associated death domain (FADD), caspase-8, and caspase-3. In addition, after exercise mitochondrial-dependent apoptotic factors such as Bid, t-Bid, Bad, p-Bad, Bak, cytochrome c, and caspase-9 are reduced. These changes lead to a reduction in oxidative damage, a reduction in infarct size, a reduction in cardiac apoptosis, and an increase in myocardial function. After exercising in the heart, the levels of RhoA, ROCK1, Rac1, and ROCK2 decrease, while the levels of PKCε, PKCδ, and PKCɑ are activated to regulate calcium and prevent mPTP perforation. Exercise has an anti-apoptotic effect on heart failure by increasing the PKA-Akt-eNOS and FSTL1-USP10-Notch1 pathways, reducing the negative effects of CaMKIIδ, and increasing the calcineurin/NFAT pathway. Exercise plays a protective role in the heart by increasing HSP20, HSP27, HSP40, HSP70, HSP72, and HSP90 along with increasing JAK2 and STAT3 phosphorylation. However, research on exercise and factors such as Pim-1, Notch, and FAK in cardiac apoptosis is scarce, so further research is needed. Future research is recommended to discover more anti-apoptotic pathways. It is also recommended to study the synergistic effect of exercise with gene therapy, dietary supplements, and cell therapy for future research.
Cardiovascular diseases (CVDs) are the most common cause of death in many parts of the world. If this condition persists, cardiac mortality will increase in the future (Alizadeh Pahavani et al., 2020). Hypoxia due to atherosclerosis is one of the causes of damage to heart muscle cells and leads to the programmed death of cardiomyocytes called apoptosis (Pahlavani and Veisi, 2018). Apoptosis occurs when cardiomyocytes are exposed to hydrogen peroxide (H2O2) or superoxide anion (
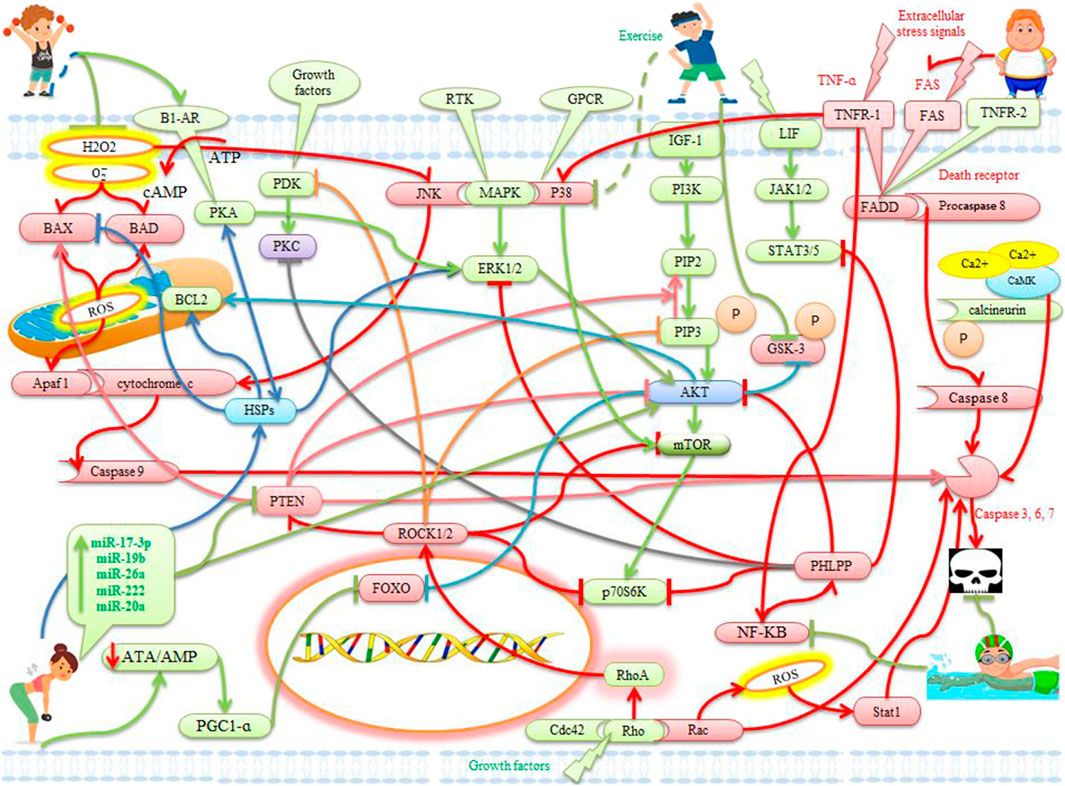
FIGURE 1. The effects of exercise on apoptotic and anti-apoptotic pathways in the heart. Description is available in the text.
Tumor necrosis factor-α (TNF-α) is secreted by mast cells and macrophages, which is dependent on oxidative stress. Global and persistent TNF-α deficiency reduces inflammatory cells and cardiac apoptosis after permanent ischemia or I/R injury. In addition, deletion of TNF-α receptor 1 (TNFR1), but not TNFR2, protects the heart against I/R damage, suggesting that the harmful effect of TNF-α may be mediated by TNFR1. Studies have shown that deletion of TNFR1 reduces apoptosis and suppresses NF-κB, p38, and JNK2 activation, while deletion of TNFR2 increases apoptosis and activation of NF-κB and p38. Activation of TNFR1 and TNFR2 mediates harmful and beneficial signals, respectively (Xia et al., 2016) (Figure 1). In rats short-term (3 days) and long-term exercise (3 weeks) can reduce myocardial apoptosis by reducing TNF-α, caspase-8, and caspase-3. While long-term exercise is more effective than short-term exercise (Meng et al., 2017). There is evidence that in rats apoptotic cells are lower in the exercise group (48–66% of VO2max for 12 weeks) (Qin, Dong, Wang, Xu, Wang, Qu, et al.) with high blood pressure than in the sedentary group. Under exercise conditions, decreased apoptosis is due to decreased Fas-dependent apoptotic pathway factors such as Fas-ligand protein, Fas death receptor, TNFR-1, Fas-associated death domain (FADD), caspase-8, and caspase-3. In addition, under exercise conditions, mitochondrial-dependent apoptotic pathway factors such as Bid, t-Bid, Bad, p-Bad, Bak, cytochrome c, caspase-9, and caspase-3 are reduced. In contrast, increased pathways supporting cardiac survival have been reported with protein levels of IGF-1, IGF-1R, p-PI3K, p-Akt, p-Bad, and Bcl2. These results show the new therapeutic effects of exercise on hearts with high blood pressure to prevent apoptosis (Huang et al., 2012) (Table 1). After I/R in rats, the exercise group (56–66% of VO2max for 10 days) (Qin, Dong, Wang, Xu, Wang, Qu, et al.) compared to the control group decreases caspase-7 (15-fold), TNF-ɑ (2.5-fold), TNF-ɑ receptor (12-fold), and pro-apoptosis factors (Cradd, FADD, NF-KB). In general, depletion of these genes reduces myocardial apoptosis (Dickson et al., 2008) (Figure 1) (Table 1). In obese mice under exercise (48–66% of VO2max for 3 months), compared to obese mice, levels of TNF-ɑ, Fas ligand, Fas receptors, FADD, caspase-8, and caspase-3 and protein levels of Bad, Bax, the Bax to Bcl2 ratio, caspase-9 is less. While extracellular and intracellular apoptosis pathways are more active in obese animals’ hearts. Therefore, exercise (4 weeks) can prevent extracellular and intracellular apoptotic pathways (Lee et al., 2013; Otaka et al., 2018) (Table 1). In rats, the exercise group (56–73 VO2max for 10 weeks) compared with the group without ovaries decreases protein levels of TNF-α, TNFR-1, Fas-ligand protein, Fas receptors, FADD, caspase-8, and caspase-3, as well as t-Bid, Bad, Bak, Bax, cytosolic cytochrome c, and caspase-9. These findings could indicate a new therapeutic effect of exercise training to prevent cardiac apoptosis in postmenopausal women or oophorectomized (Huang et al., 2016) (Table 1). Possible beneficial mechanisms of exercise can be the reduction of oxidative stress and inflammatory factors due to exercise adaptation.
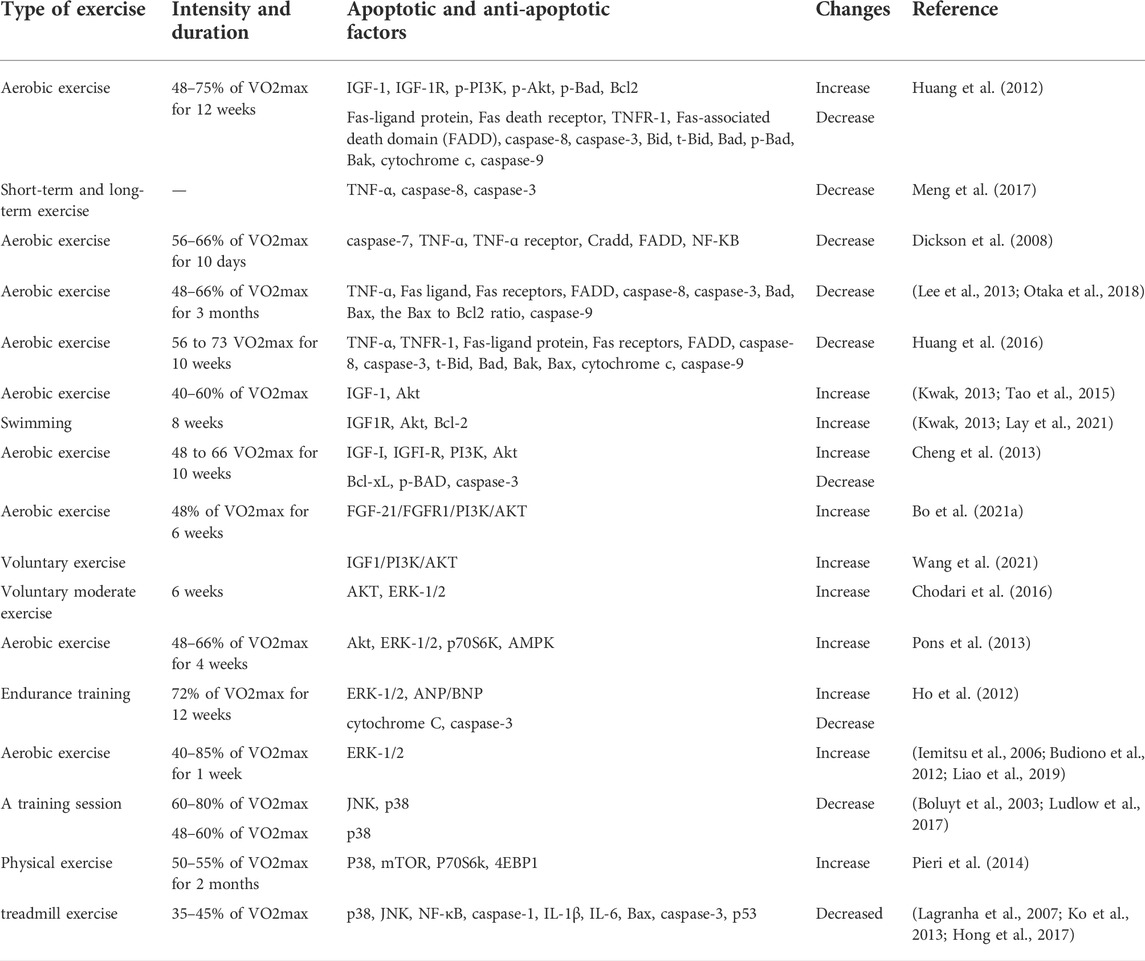
TABLE 1. A summary of exercise studies on the factors affecting cardiac apoptosis.
The Insulin-like growth factor (IGF)-1/phosphoinositide 3-kinases (PI3K)/Akt signaling pathway is activated after stimulation of growth factors, cytokines, hormones, and muscle contractions (Xia et al., 2016). Akt has 3 isoforms called AKT1 and AKT2, AKT3 (Wang et al., 2021) (Table 1). Both Akt family members Akt1 and Akt2 are abundantly expressed in cardiomyocytes (Xia et al., 2016). Among these, AKT1 is known as an isoform-specific to physiological hypertrophy and exercise-induced heart growth (Wang et al., 2021). In transgenic mice, an 80% increase in AKT lead to a 2.2-fold increase in heart weight compared with controls (Wang et al., 2021). Protection of cardiomyocytes via the IGF-1/PI3k/Akt pathway against apoptosis is associated with the inactivation of Bad pro-apoptotic protein. It seems in mitochondria, Akt protects the integrity of the outer membrane (Tao et al., 2015; Xia et al., 2016). Nuclear overexpression of Akt protects cardiomyocyte apoptosis without phosphorylation of cytoplasmic Akt. Nuclear Akt targets in the myocardium include the forkhead box (FOX) transcription factor and serine/threonine kinase (Pim-1) (Xia et al., 2016) (Figures 1,2). In humans, exercise (40–60% of VO2max) increases cardiac expression of IGF-1 and Akt and leads to cardiac hypertrophy and survival (Kwak, 2013; Tao et al., 2015) (Table 1). In older rats, also the reduction in cardiac survival pathway proteins such as IGF1R, Akt, and Bcl-2 improves with exercise (swimming for 8 weeks) (Kwak, 2013; Lay et al., 2021) (Table 1). In addition, levels of IGF-1, IGF-1R, p-PI3K, p-Akt, and Bcl2 are higher in the exercise group (48–66% of VO2max for 12 weeks) than in the sedentary group and the sedentary hypertensive group. As a result, exercise prevents intracellular apoptotic pathways in the hearts of patients with hypertension and increases cardiac survival pathways (Huang et al., 2012) (Table 1). It is reported that in the diabetic rat group the components of hypertrophy and cardiac survival pathways such as IGF-I, IGF-I receptor (IGFI-R), PI3K, Akt, and anti-apoptotic proteins such as Bcl-2, Bcl-xL, and p-BAD decrease significantly, while increases in the diabetic aerobic exercise group (48–66 VO2max for 10 weeks). In addition, caspase-3, the abnormal structure of the myocardium, and an increase in cardiac apoptotic cells are more severe in the diabetic group than in the diabetic group with exercise. These findings emphasize the role of exercise in preventing myocardial apoptosis (Cheng et al., 2013). Aerobic exercise (48% of VO2max for 6 weeks) also significantly reduces oxidative stress (H2O2) and apoptosis by activating the Fibroblast growth factor 21 (FGF-21)/FGFR1/PI3K/AKT pathway, which ultimately improves heart function in mice with myocardial infarction (Bo et al., 2021a) (Figure 1) (Table 1). Hence, other studies have suggested voluntary exercise such as running and swimming as a recommendation to prevent CVD because exercise via the IGF1/PI3K/AKT pathway improves heart function. This pathway can reduce heart damage in a variety of ways, such as cardiomyocyte proliferation and growth, physiological hypertrophy, increased cardiac apoptotic capacity, and inhibition of fibrosis (Wang et al., 2021) (Table 1). The possible mechanism of physiological cardiac hypertrophy in exercise is to increase the demand of cells and tissues of the heart for oxygen and blood supply. Hence the survival, growth and proliferation of heart cells and counteracting apoptosis seems reasonable during exercise.
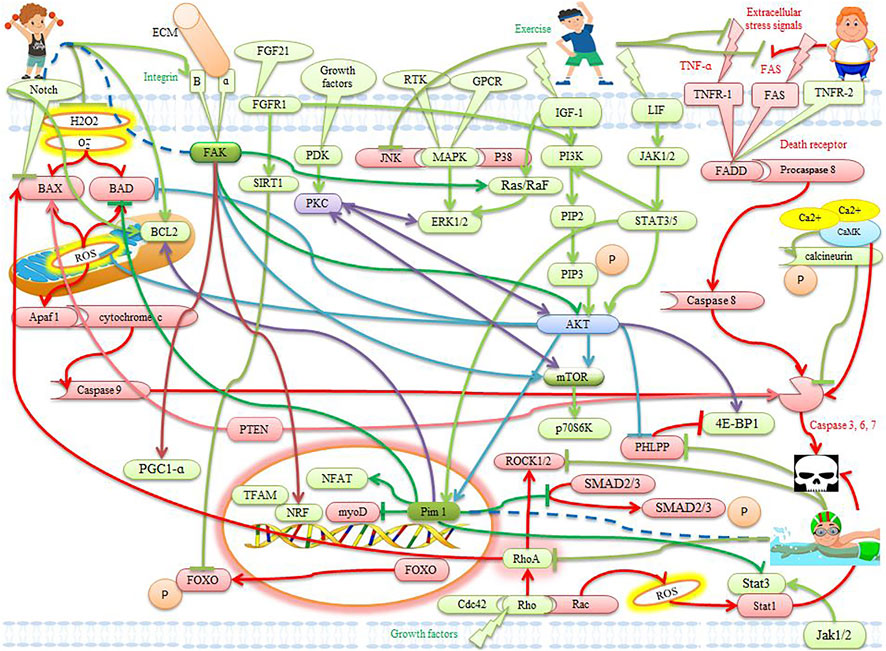
FIGURE 2. The effects of exercise on apoptotic and anti-apoptotic pathways in the heart. Description is available in the text.
Mitogen-activated protein kinases (MAPKs) regulate cell functions such as survival, growth, and differentiation in response to extracellular signals. Extracellular signals lead to phosphorylation and activation of factors such as extracellular signal-regulated kinase 1/2 (ERK-1/2 or p44/42), Jun N-terminal kinases (JNK), and p38 isoforms (Xia et al., 2016). Myocytes are more susceptible to apoptosis in ERK2-deficient mice, suggesting a role for ERK-1/2 in cell survival. However, continuous activation of ERK-1/2 and its nuclear transport contribute to cardiomyocyte apoptosis. In addition, ERK-1/2 is also activated by hydroxyl radicals via Ras/Raf-1 MAPKKK and has a protective role. Mice with cardiac Raf-1 deficiency show myocyte apoptosis at 3 weeks of age and heart failure late in life (Xia et al., 2016). Voluntary moderate exercise (6 weeks) increases the cardiac activity of AKT and ERK-1/2 in diabetic rats, which in turn leads to a reduction in cardiac apoptosis (Chodari et al., 2016). In wild-type mice, exercise (48–66% of VO2max for 4 weeks) reduces the size of infarction by up to 60% by increasing the phosphorylation of kinases such as Akt, ERK-1/2, p70S6K, and AMPK (Pons et al., 2013) (Figure 1) (Table 1). In mice, excessive endurance training (72% of VO2max for 12 weeks) also leads to increased myocyte cross-section, hypertrophy-related pathways (ERK-1/2), hypertrophic markers (ANP/BNP), and decreased pro-apoptotic molecules (cytochrome C and caspase-3) (Ho et al., 2012) (Table 1). It seems also exercise (40–85% of VO2max for 1 week) can significantly improve cardiac ischemic tolerance by increasing ERK-1/2 and inhibiting GSK-3β phosphorylation in mice (Budiono et al., 2012). In mice, increased irisin during exercise (4 weeks) after myocardial infarction with ERK-1/2 phosphorylation has been shown to reduce ventricular dilatation, infarction size, fibrosis, cardiomyocyte apoptosis, and increased angiogenesis. While the effects of irisin are blocked by the ERK-1/2 inhibitor (Liao et al., 2019). An acute training session has been shown to cause time-dependent transient activation of ERK-1/2 in the heart. This activation peaks after 15 and 30 min. Increased levels of ERK-1/2 are reduced to resting levels after 24 h of exercise. This represents a temporary increase in ERK−1/2, which is useful for counteracting cardiac apoptosis. Hence, mice in the 12-weeks exercise program (50–70% of VO2max) showed significant cardiac hypertrophy by activating several MAPK pathways, an effect that gradually diminishes with exercise-induced cardiac hypertrophy (Iemitsu et al., 2006) (Table 1).
Unlike ERK-1/2, JNK and p38 are stress-activated protein kinases (SAPKs) that respond to environmental stresses such as hypoxia, heat, exercise, inflammatory cytokines, and DNA-damaging agents. Mitochondrial JNK is phosphorylated by H2O2 stimulation, leading to mitochondrial outer membrane permeability and cytochrome c release (Xia et al., 2016) (Figure 1). JNK inhibition protects cardiomyocyte apoptosis under I/R conditions and is associated with increased Bcl-2 and Bcl-xl family members. Therefore, inhibition of p38 activity is suggested as a central mechanism for heart protection (Xia et al., 2016). In untrained mice, JNK activity in myocytes increases immediately after a training session, whereas in trained mice, no JNK activation is observed above baseline. Therefore, exercise (60–80% of VO2max) activates the JNK in an intensity-dependent and adaptive manner, and chronic exercise reverses the myocardial JNK (Boluyt et al., 2003) (Table 1). In mice cardiac muscle, higher levels of phosphorylated p38 have been reported immediately after a treadmill run than before and 1 hour after training (48–60% of VO2max). This temporary increase seems to be due to the initial adaptation to exercise (Ludlow et al., 2017). The myocardium of obese rats with a high-fat diet increases phosphorylation levels of P38, mTOR, P70S6k, and 4EBP1 proteins after 2 months of physical exercise (50–55% of VO2max). P38 appears to be involved in mTOR activation and this time has not been sufficient for compatibility (Pieri et al., 2014) (Table 1). In the endoplasmic reticulum, exercise reduces inflammatory factors such as p38/JNK and NF-κB. Exercise then regulates the expression of caspase-1, IL-1β, and IL-6, leading to a decrease in caspase-12 and an increase in Bcl-2 in cardiovascular disease (Hong et al., 2017). In rats, aging induces apoptosis in the myocardium by increasing Bax, increasing p-p38, and suppressing HSP70, while treadmill exercise (35–45% of VO2max) decreases aging-induced apoptosis by increasing HSP70 expression and suppressing p-p38 expression in cardiac myocytes (Ko et al., 2013). In rats, glutamine supplementation with exercise (85% of VO2max) partially prevents p38 and JNK phosphorylation as well as p53 expression and completely reverses the increase in caspase-3 expression (Lagranha et al., 2007) (Table 1). In general, exercise can increase ERK-1/2 in myocytes and decrease JNK and P38 according to severity, compatibility, and location, leading to a decrease in myocyte apoptosis.
Integrins are used to maintain cell adhesion, and tissue integrity, and to transmit cellular external mechanical signals into the cell. Integrins require cytosolic kinases such as focal adhesion kinase (FAK) to transmit signals into the cell. Integrins consist of two subunits α and β. Integrin β1 is the dominant β subunit in the heart. Mice lacking β1 integrin grow into adulthood and are more vulnerable to myocardial dysfunction and myocyte apoptosis, indicating a protective role of β1 integrin against apoptosis (Xia et al., 2016). Stretching-induced FAK activation stimulates hypertrophic growth associated with mitochondrial biogenesis through up-regulation of PGC1-α, NRF, and Tfam (Ferreira et al., 2018) (Figure 2). FAK is involved in activating the Ras/Raf/MEK/ERK pathway to cause concentric hypertrophy (Watson and Baar, 2014). RhoA, on the other hand, is activated by β1 integrin under mechanical stress, leading to Akt activation through FAK activation (Rindom and Vissing, 2016). It also seems cardiac adaptation to exercise is regulated by integrins and FAK activation (Ferreira et al., 2018). In rats exercising (74% of VO2max) on a treadmill, integrin β1, and α5 are increased in cardiac (Burgess et al., 2002; Manso et al., 2009) (Table 2). In the heart, FAK controls the phosphorylation of AKT/GSK-3β and ERK-1/2, and these signaling pathways lead to cell survival and a hypertrophic response after exercise (Brancaccio et al., 2006) (Table 2). In the hearts of trained pigs (5 days a week for 14 weeks), β1 integrin expression is increased (Muthuchamy et al., 2012). FAK absorbs PI3K into membranes in exercise-induced physiological hypertrophy, leading to increased PIP3 and Akt phosphorylation. Akt then activates mTORC1 (Watson and Baar, 2014) (Figure 1). In addition to cardiac muscle in skeletal muscle, FAK phosphorylation and β1 integrin for aerobic and anaerobic exercise (75–98% VO2max three times a week for 8 weeks) increase immediately after exercise (Wilkinson et al., 2008; Flueck et al., 2011; Li et al., 2013; Graham et al., 2015a; Graham et al., 2015b; Franchi et al., 2018) (Table 2). In exercise, cardiac hypertrophy occurs with a proportional increase in collagen and possibly integrin, leading to a highly functional matrix (Burgess et al., 2001). Finally, FAK plays several roles such as myoblast growth, muscle fiber formation, myogenesis, and hypertrophic and anti-apoptotic signals, which reinforces the importance of FAK signaling during muscle growth and homeostasis. However, due to low exercise studies in cardiac tissue, more studies are needed.
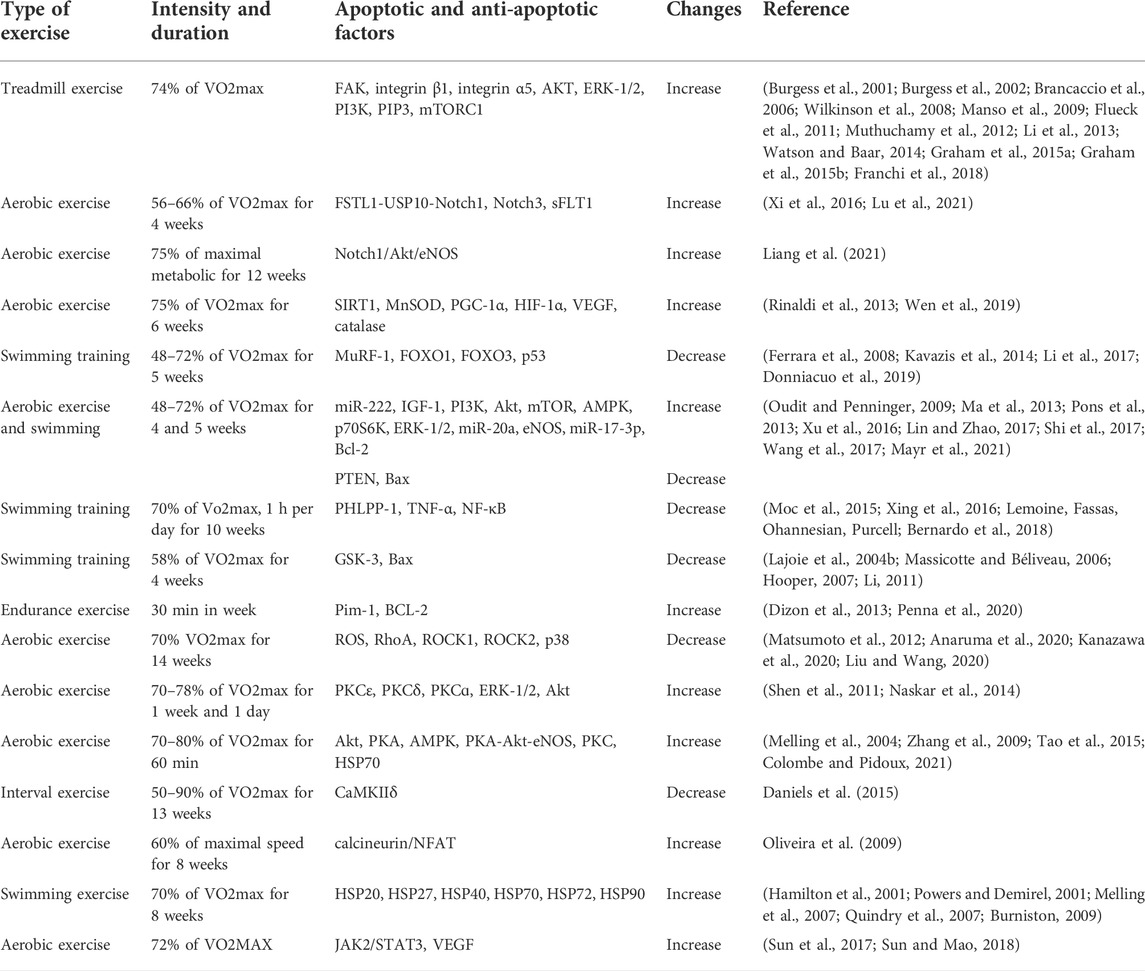
TABLE 2. A summary of exercise studies on the factors affecting cardiac apoptosis.
Notch signaling determines the fate of embryonic cells and adult tissue homeostasis through cell-to-cell interactions. Mammalian cells express five notch ligands (delta-like 1, delta-like 3, delta-like 4, Jagged1, and Jagged2) and four notch receptors (Notch 1–4) (Carey et al., 2007; Xia et al., 2016). Jagged1 and Notch1 are expressed in the adult heart and protect heart tissue against cardiomyopathy, myocardial infarction, pathological hypertrophy, and I-R injury (Cai et al., 2016). In the myocardium, Notch signaling is involved in modulating cardiac survival, cardiac stem cell differentiation, and angiogenesis (Ferrari and Rizzo, 2014). Myocardial Notch1 level is activated after injury and enhances the anti-apoptotic signaling Akt and Bcl-2 and improves heart function after myocardial infarction. In contrast, the removal of Notch1 in cardiomyocytes increases apoptosis after hemodynamic overload and Notch inactivation dramatically exacerbates apoptosis. In addition, preconditioning protects against cardiomyocyte apoptosis by activating PI3K/Akt, Pim-1, and Notch1, and inhibiting p38 (Xia et al., 2016) (Figure 1 and Figure 2) (Table 2). Notch signaling also protects heart tissue through Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling, MnSOD activation, and ROS reduction against myocardial damage (Cai et al., 2016). In addition, Notch3 overexpression in cardiomyocytes plays a protective role against apoptosis (Xia et al., 2016). While Notch3 deficiency indicates low arterial density and increased oxidative stress. Increased expression of anti-angiogenic receptors such as sFLT1 has been reported in mice lacking Notch3 after exercise. While moderate physical activity increases the coronary arteries and capillary density in wild-type mice and capillary density does not change in mice without Notch3 (Ragot et al., 2016). Increased exercise (56–66% of VO2max for 4 weeks)-induced Follistatin-like protein 1 (FSTL1) secretion through the FSTL1-USP10-Notch1 signaling axis has been reported to inhibit myocardial fibrosis in diabetic rats (Xi et al., 2016; Lu et al., 2021) (Table 2). Aerobic exercise (75% of maximal metabolic for 12 weeks) significantly reduces systolic blood pressure, which is independently correlated with skin capillary density and vascular density in humans. While Notch1/Akt/eNOS signaling obstruction almost completely abolishes the protective effect of aerobic exercise on endothelial progenitor cell function (Liang et al., 2021) (Figure 1). In mice, maternal voluntary exercise (3 weeks) reduces congenital heart defects (CHDs), ROS, oxidative stress, and coronary and capillary defects. In this way, the expression of Notch1 and Gata4 cardiac genes is significantly higher in the fetuses of the exercise group compared to the non-exercise group (Saiyin et al., 2019). In addition, Delta-like-1 (DLL-1) is mainly expressed in arterial endothelial cells and plays an important role in stress responses. Serum DLL-1 levels are significantly higher in patients with chronic heart failure than in healthy individuals, and DLL-1 levels have been significantly associated with diastolic dysfunction, decreased exercise capacity, increased levels of C-reactive protein (CRP), and adverse (Norum et al., 2016). In addition, much lower expression of Notch1, Jagged1, and Dll-1 is observed in the muscles of older men (60–75 years) compared with the muscles of young men (18–25 years). However, resistance training increases the expression of Notch1 and decreases the expression of Dll-1. It seems that some of the benefits of resistance training may be achieved through the Notch signaling pathway (Carey et al., 2007). However, due to few sports studies on this subject, more studies are needed.
FOXO family consists of four members (FOXO1, FOXO3, FOXO4, and FOXO6) whose activity is inhibited by insulin signaling and growth factor (Webb and Brunet, 2014; Alizadeh Pahlavani, 2022a; Alizadeh Pahlavani, 2022b). FOXO acts as a pro-apoptotic agent through the expression of proteins such as Bim, BAD, Bnip3, FasL, and TRAIL in the apoptotic internal and external pathways. Akt phosphorylates the transcription factors of the FOXO family, leading to their exit from the nucleus and their destruction in the cytosol via the ubiquitin-proteasome pathway (Xia et al., 2016). In drosophila, after exercise (2 weeks), SIRT1 appears to inhibit the ability of FOXO3 to induce cell death, leading to an increase in a variety of antioxidant genes, including MnSOD (Wen et al., 2019). Long-term and moderate exercise (75% of VO2max for 6 weeks) before infarction injury has also been shown to increase FOXO3 and its two downstream targets, MnSOD, and catalase, compared to sedentary rats with infarction (Donniacuo et al., 2019). In rats, it seems that exercise (48–72% of VO2max for 8 weeks) first significantly increases the FOXO3 protein in the heart and then increases antioxidant levels such as Mn-SOD and catalase. Subsequently, an exercise-induced increase in SIRT1 in the heart prevents the role of FOXO3 apoptosis (Ferrara et al., 2008) (Table 2). After myocardial infarction, exercise (75% of VO2max) activates the SIRT1 and SIRT3 pathways, which reduce cardiac apoptosis and oxidative damage by inhibiting p53 and FOXO3 (Donniacuo et al., 2019). Endurance training (swimming for 5 weeks) also can protect the myocardium by activating the SIRT1 pathway and regulating FOXO1 deacetylation (Li et al., 2017). Doxorubicin (DOX) has been reported to increase skeletal and heart myopathy by increasing MuRF-1, FOXO1, and FOXO3 mRNA expression, while short-term exercise training (75% of VO2max) protects against acute Doxorubicin-induced heart toxicity by reducing FOXO. In addition, exercises increase PGC-1α in the heart muscle and are associated with the suppression of FOXO activity. Finally, exercise protects against DOX-induced heart myopathy by reducing FOXO1 and MuRF-1 (Kavazis et al., 2014). In confirmation of this, a study reported that prolonged moderate exercise (75% of VO2max for 6 weeks) increases mRNA levels of PGC-1α, HIF-1α, and VEGF genes, which are involved in mitochondrial biogenesis and angiogenesis, and decreases FOXO3, MuRF-1, and Atrogin-1 mRNA levels. Finally, moderate exercise improves cardiac tissue perfusion and regenerates heart muscle (Rinaldi et al., 2013) (Table 2). Thus exercise through Akt, SIRT3, SIRT1, MnSOD, catalase, PGC-1α, HIF-1α, and VEGF reduces p53, MuRF-1, FOXO1, and FOXO3, then reduces cardiac apoptosis.
Phosphatase and tensin homolog (PTEN) produce phosphatidylinositol-(4,5)-bisphosphate (PIP2) through phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) dephosphorylation and ultimately inhibit Akt activation (Xia et al., 2016). PTEN acts as a negative regulator of PI3K and plays a role in many diseases such as myocardial hypertrophy, heart failure, and preconditioning (Oudit and Penninger, 2009). PTEN expression in cardiomyocytes leads to caspase-3 activation, Bax, and apoptosis. Conversely, specific deletion of PTEN in the heart or PTEN inactivation reduces myocyte apoptosis (Xia et al., 2016). Aerobic exercise works by increasing the activity of IGF-1, PI3Kα, Akt, p70S6K, and decreasing PTEN to maintain heart structure and function in patients with heart failure. Exercise appears to eliminate the inhibitory effect of PTEN on the IGF-1/PI3K/Akt/p70S6K pathway (Oudit and Penninger, 2009) (Table 2). In the myocardium of diabetic mice is increased PTEN expression and PI3K/Akt expression is inhibited. While after training (swimming training for 5 weeks), PTEN expression decreases in the myocardium of diabetic mice, and the PI3K/Akt pathway and miR-222 induction are activated. Therefore, exercise intervention may protect the myocardium under high glucose by activating miR-222 and the PI3K/Akt pathway (Lin and Zhao, 2017) (Figure 1). In mice, exercise (48–72% of VO2max for 4 weeks) also reduces infarction size by up to 60% by increasing phosphorylation of kinases such as Akt, ERK-1/2, p70S6K, AMPK, and GSK3β while reducing levels of PTEN (Pons et al., 2013) (Table 2). These factors protect the heart against myocardial infarction with survival-supporting signaling pathways, reducing phosphatase, and increasing mitochondrial permeability transition pore (mPTP) opening resistance (Pons et al., 2013). In mice with coronary artery disease, VEGF and PTEN are upregulated, while overexpression of miR-20a with exercise (swimming training for 15 weeks) results in decreased PTEN and increased eNOS and VEGF at both transcriptional and translational levels. MiR-20a has been shown to bind specifically to 3′UTR PTEN and induce myocardial cell survival and proliferation by activating PI3K/Akt (Wang et al., 2017) (Table 2). In rats, swimming increases PI3K protein levels, while PTEN and TSC2 decrease by 37 and 22%, respectively, activating the PI3K/AKT/mTOR signaling pathway (Ma et al., 2013). In murine, swimming exercise-induced increase in miR-17-3p leads to inhibition of PTEN and increased hypertrophy, proliferation, and survival in the myocardium. miR-17-3p acts through the tissue inhibitor of metalloproteinase 3 (TIMP3) and the PTEN-AKT pathway to repair myocardium after I/R injury. However, no binding sites for miR-17-3p were detected in the 3′UTR PTEN, indicating that PTEN is not a direct target of miR-17-3p (Shi et al., 2017). In murine, an aerobic exercise-induced increase of miR-17-3p could prevent pathological myocardial dysfunction by inhibiting PTEN because the MEF2C/miR-17-3p/PTEN pathway acts as a new therapeutic strategy for pathological myocardial dysfunction (Xiao et al., 2016; Zhou and Xiao, 2019; Bo et al., 2021b; Rivas et al., 2021) (Figure 1). On the other hand, increased miR-26a after exercise can negatively regulate PTEN and SMAD1 to protect against cardiac damage in diabetic rats, which prevents apoptosis and myocardial atrophy (Icli et al., 2013; Cai et al., 2019). In patients with CAD, there is evidence that after exercise overexpression of miR-19b reduces H2O2-induced apoptosis and negatively regulates PTEN at the protein level. The overexpression of miR-19b and then decreased PTEN may be a new treatment for myocardial injury after (I-R) because the downregulation of PTEN increases Bcl-2, decreased Bax, and decreased caspase-3 (Xu et al., 2016; Mayr et al., 2021). Finally, it seems that exercise by increasing PI3K/Akt, miR-17-3p, miR-19b, miR-26a, miR-222, and miR-20a can lead to a decrease in PTEN and further stimulate myocardial hypertrophy and proliferation.
PH domain leucine-rich repeat protein phosphatase (PHLPP) dephosphorylates and inhibits Akt (Xia et al., 2016). There are two isoforms of PHLPP, called PHLPP-1 and PHLPP-2 (Xing et al., 2016). PHLPP targets include Akt, protein kinase C (PKC), p70S6 kinase 1 (p70S6K), and the RAF/MEK/ERK cascade. Tumor necrosis factor-α (TNF-α) increases PHLPP1 levels in cardiac myocytes through NF-κB transcriptional activity after I/R (Xing et al., 2016; Lemoine, Fassas, Ohannesian, Purcell). PHLPP-1 levels are more in the hearts of older mice than in younger mice and overexpression of PHLPP-1 stops p70S6K and Akt phosphorylation and worsens myocyte apoptosis (Xing et al., 2016). In contrast, the hearts of mice lacking PHLPP-1 increase Akt phosphorylation and decrease infarction size after I/R (Xia et al., 2016) (Figure 1). Mice lacking PHLPP-1 also show excellent response to exercise-induced hypertrophy via the Akt, p70S6Kinase, and ERK pathways. Akt phosphorylation in the hearts of mice lacking PHLPP-1 almost doubles and the physiological hypertrophy resulting from swimming (70% of Vo2max) is exacerbated by an increase in heart size and myocyte cell area (Lemoine, Fassas, Ohannesian, Purcell; Moc et al., 2015; Bernardo et al., 2018) (Figure 1). PHLPP-1 knockout mice underwent forced swimming training for 20 days which resulted in physiological hypertrophy of the heart compared to wild-trained mice (Moc et al., 2015; Bernardo et al., 2018) (Table 2). Decreased PHLPP-1 in cardiomyocytes by increased exercise (1 h per day for 10 weeks)-induced insulin sensitivity was also shown to significantly increase Akt phosphorylation in animals because insulin suppressed PHLPP-1 to increase Akt activation. Therefore, in diabetic people, PHLPP-1 could be a promising therapeutic target for patients with ischemic heart disease because cardiac apoptosis and infarction size decrease and cardiac function increases (Xing et al., 2016). On the other hand, the pathophysiological hypertrophy caused by pressure overload decreases through swimming exercise along with increased heart size, increased cardiomyocyte levels, and increased expression of hypertrophic genes. This reduction in pathophysiological hypertrophy is associated with reduced fibrosis and cell death in mice lacking PHLPP-1. Also, the expression of VEGF and increased capillary density is higher in the myocardium without PHLPP-1 because deletion of PHLPP-1 can increase endothelial tube formation (Moc et al., 2015) (Table 2). Mice lacking PHLPP-1 under exercise conditions show marked physiological hypertrophy compared to wild-type mice, which is observed with increased cardiac muscle cells (Lemoine, Fassas, Ohannesian, Purcell). Mice lacking PHLPP-1 under swimming exercise compared with wild-type mice lead to increased heart size, angiogenesis, increased capillary density and myocyte cell area compared with wild-type mice, and reduced pathological hypertrophy after aortic stenosis (Moc, 2015) (Table 2). In general, the exercise by decreasing PHLPP-1 and increasing mediators such as Akt, p70S6Kinase, and ERK can lead to increased heart size, increased angiogenesis, increased capillary density, and increased hypertrophic genes, decreased apoptosis, decreased fibrosis, and decreased cell death.
Glycogen synthase kinase 3 (GSK-3) is a serine/threonine kinase consisting of two isoforms GSK-3α and GSK-3β and is phosphorylated by insulin. GSK-3 phosphorylation is facilitated by PKB, leading to enzyme inactivation and increased glycogen synthesis. High glycogen content acts as a regulator of GSK-3 and limits further glycogen accumulation (Lajoie et al., 2004a). Akt phosphorylation provides cardiac protection by preconditioning ischemia and inactivating GSK-3, these two factors reduce the size of infarction by a similar amount. GSK-3β-specific deficiency dramatically inhibits apoptosis after myocardial infarction (MI), and conditional deletion of GSK-3α in the heart reduces the Bax/Bcl-2 ratio and apoptosis (Xia et al., 2016). Hyperglycemia and increased glycogen in diabetic mice are associated with dysregulation of PKB and GSK-3. While 13-weeks swimming significantly reduces cardiac glycogen levels and normalizes PKB and GSK-3 in diabetic rats (Lajoie et al., 2004a) (Table 2). Low-dose copper nanoparticles and exercise (swimming, 90 min, 5 days/4 weeks) also significantly prevent infarction through preconditioning and inhibition of GSK-3β (Sharma et al., 2018). In mice with hypertension under training (swimming, 150 min, 5 days/13 weeks) conditions, PKB phosphorylation is significantly improved and is associated with increased GSK-3β phosphorylation. Bax protein expression in the heart of hypertensive mice under training is associated with increased Bcl-2 protein expression, which allows the Bcl-2/Bax ratio to be maintained (Lajoie, 2003). In the left ventricle of mice with hypertension under exercise (70% of VO2max), PKB phosphorylation is significantly increased compared with mice with sedentary hypertension and PKB phosphorylation has significantly correlated with GSK-3β phosphorylation. In addition, the expression of HSP72 and Bcl-2 protein is increased in the left ventricle of mice with hypertension under training, which indicates that the anti-apoptotic mechanism is effective in compensating for the increased expression of Bax pro-apoptotic protein in the myocardium (Lajoie et al., 2004b). In confirmation of these findings was shown that in rats long-term moderate-intensity exercise (58% of VO2max for 4 weeks) can improve myocardial cell anti-apoptotic proteins such as HSP72, Bcl-2, and PKB. While they significantly reduce Bax and GSK-3 apoptotic proteins and inhibit myocardial apoptosis (Lajoie et al., 2004b; Massicotte and Béliveau, 2006; Li, 2011) (Table 2). In other words, exercise-induced heat shock proteins activate the PI3K/AKT pathway, which in turn inhibits GSK-3 (Hooper, 2007) (Figure 1). These data suggest that exercise may protect the heart against the harmful effects of GSK-3 because it temporarily inhibits GSK-3 via the PI3K/AKT pathway. This process protects the myocardium and reduces cardiac apoptosis.
There is evidence that Akt1 plays a role in the proliferation of heart stem cells and cardiomyocytes, primarily by activating of serine/threonine-protein kinase-1 (Pim-1)-dependent pathway (Mann and Rosenzweig, 2012). Pim-1 regulates many factors of proliferation, survival, and cell cycle (Bishopric, 2012; Liu et al., 2019). Myocyte survival is modulated by Pim-1, heme oxygenase-1, and hypoxia-inducible factor-1α (HIF-α), which act in concert with the Akt signaling network (Oshima et al., 2008). Pim-1 is an oncoprotein that is overexpressed in the lungs of patients with pulmonary arterial hypertension (PAH) and is involved in cell proliferation by activating the nuclear factor of the activated T cells (NFAT)/STAT3 signaling pathway. Plasma levels of Pim-1 in PAH are higher than in the control group and correlate with traditional PAH markers. Pim-1 levels are an independent predictor of mortality and a promising new biomarker in PAH (Renard et al., 2013; Magne et al., 2015; Vaillancourt et al., 2015). The inhibition of Pim-1 activity by genetic manipulation exacerbates cardiomyocyte apoptosis, while Pim-1 expression significantly reduces infarction size in mice. The specific expression of Pim-1 in the heart increases the levels of Bcl-2 and Bcl-xl proteins, which protect against mitochondrial damage due to oxidative stress and Bid pre-apoptotic protein. Inhibition of Pim-1-dependent apoptosis by post-conditioning supports the important role of Pim-1 in regulating cardiac survival after pathological injury (Penna et al., 2009; Xia et al., 2016; Weeks et al., 2017). In addition, Pim-1 significantly leads to the proliferation of myoblasts and stops the acceleration of apoptosis, indicating that Pim-1 is vital for the survival and enhancement of myoblasts (Liu et al., 2019). Physical activity also activates factors to protect the heart. These factors such as Bad/Bax and Pim-1 also participate in the closure of mitochondrial permeability transition pore (mPTP), which is associated with cardiac protection by preventing the opening of mPTP and reducing mitochondrial swelling (Dizon et al., 2013; Penna et al., 2020) (Table 2). Therefore, exercise (70–86% of VO2max) reduces and increases the pre-apoptotic and anti-apoptotic proteins of Bax and BCL-2 in rat hearts (Alizadeh et al., 2017; Pahlavani and Veisi, 2018; Alizadeh Pahavani et al., 2020) (Figure 2). Pim-1 also inhibits the phosphorylation of Smad2 and Smad3, prevents their transfer to the nucleus, and suppresses the expression of TGFβ pathway genes. In addition, Pim-1 maintains telomere length in the heart by inhibiting the phosphorylation of Smad2 and Smad3, as it prevents the suppression of telomerase reverse transcriptase (Ebeid et al., 2021). Since exercise studies on Pim-1 have been reported rarely, and on the other hand, upstream and downstream factors of Pim-1, such as Akt and BCL-2, change with exercise, further studies are needed.
Small GTPases are subdivided into subtypes of Rho, Rac, and cell division control protein 42 (Cdc42). Overexpression of Ras homolog family member A (RhoA), a GTPase of the Rho subfamily, stimulates heart failure and mitochondrial apoptosis due to increased Bax. In contrast, the relative increase in RhoA and subsequent activation of pro-survival kinases such as Rho, FAK, PI3K/Akt, and PKD play a protective role against myocyte apoptosis. The beneficial role of RhoA through the removal of endogenous RhoA in the heart is characterized by an exacerbation of I/R damage and activation of caspase-3 and apoptosis (Xia et al., 2016). There is evidence that Rac plays a destructive role in cardiomyocyte apoptosis because the removal of Rac1 leads to a decrease in mitochondrial reactive oxygen species (ROS) as well as myocardial dysfunction. In addition, treatment with a Rac1 inhibitor improves cardiomyocyte apoptosis and heart function in type 2 diabetic rats. In contrast, Rac1 expression in the heart leads to the production of superoxide and activation of caspase, pathological hypertrophy, or myocardial dilatation (Xia et al., 2016). In addition, Rho-associated kinase (ROCK) leads to PTEN activation, which in turn reduces PIP3, PDK, and AKT phosphorylation. ROCK also is capable of negatively modulating mTOR and p70S6K, and both proteins are involved in cell growth and survival (Anaruma et al., 2020). When ROCK1 is expressed in the heart, it automatically activates caspase-3, apoptosis, and myocardial fibrosis in vivo. In addition, ROCK1 deficiency inhibits myocyte apoptosis, and increased ROCK1 expression causes apoptosis after pressure overload or pathological hypertrophy. ROCK2 appears to play a similar detrimental role to ROCK1 in apoptosis because ROCK2-specific cardiac knockout reduces oxidative stress, myocyte hypertrophy, and apoptosis after transverse aortic stenosis (Xia et al., 2016). ROCK has been reported to be essential for pathological cardiac hypertrophy and is responsible for transmitting p38 to the nucleus. Thus, ROCK and p38 are active in pathological hypertrophy (Anaruma et al., 2020).
Pathological hypertrophic changes are modulated by stimulation of physiological hypertrophic activation by mechanical overloads such as exercise. Swimming exercise induces pathways such as ERK-1/2 activation and AKT/mTOR while no ROCK protein activation is observed. Thus, the factors activated during swimming are the PI3-K/AKT pathway without the participation of ROCK and p38 (Anaruma et al., 2020) (Table 2). In rats, RhoA, ROCK1, and ROCK2 levels are significantly higher in the exhausting exercise than in the without exercise group. While these factors in the preconditioning exercise are significantly less than the exhausting exercise. In the preconditioning exercise, Bcl-2/Bax ratio is significantly higher than in the exhausting exercise. These findings suggest that preconditioning exercise has a protective effect against myocardial injury and improves heart function in mice. The mechanism of this improvement may be related to the Rho/ROCK pathway (Liu and Wang, 2020). High levels of ROCK2 have been seen in mice with myocardial infarction, whereas ROCK2 is significantly reduced with the post-infarction exercise (8 weeks) (Kanazawa et al., 2020) (Table 2). It seems there is an association between ROS and the RhoA/ROCK pathway because inflammatory atherosclerotic lesions show an increase in ROCK, while administration of a RhoA/ROCK inhibitor reduces atherosclerotic plaque size and apoptosis in rats. RhoA mRNA expression is also reduced in endothelial cells of trained hearts (70% VO2max for 14 weeks) (Matsumoto et al., 2012) (Figures 1,2) (Table 2). In addition, RhoA gene expression levels increase in patients with cardiovascular disease while decreasing with exercise (70% of VO2max). Therefore, this pathway may be one of the positive mechanisms of cardiac protection due to exercise (Matsumoto et al., 2007). RhoA and ROCK have been reported to be involved in the pathogenesis of the cardiovascular disease, while 8 weeks of exercise reduces RhoA and ROCK gene expression. Therefore, it indicates the inhibitory effect of exercise on the RhoA/ROCK pathway in mice (Matsumoto et al., 2013). During physical activity (70% of VO2max), physical stresses such as shear stress activate mechanical transfer mechanisms in endothelial cells and smooth muscle that bind to small GTPases such as RhoA. They stimulate various pathways such as FAK/integrin, c-Src, PI3K, and AKT. These mechanisms regulate anti-atherogenic genes by modifying calcium management and vascular myogenic response to pressure by promoting anti-apoptotic signals (Kojda and Hambrecht, 2005). In addition, Cdc42, a subfamily of small GTPases, inhibits cardiomyocyte apoptosis. While removal of Cdc42 in the heart exacerbates apoptosis (Xia et al., 2016). Cdc42 also inhibits the growth response of the heart to physiological and pathological stimuli (Maillet et al., 2009). For example, 8 weeks of exercise reduces protein levels and RhoA, Rac1, and Cdc42 mRNA levels in the heart (Yan and Li, 2012; Tao et al., 2015) (Figure 1) (Table 2). Mice lacking Cdc42 experience more exercise-induced cardiac hypertrophy and develop heart failure and sudden death more rapidly than controls. Thus Cdc42 is an anti-hypertrophic mediator that acts on exercise-induced physiological hypertrophy to limit heart growth. In addition, Cdc42 is potentially one of the heart’s protective pathways, while removal of Cdc42 in strenuous exercise leads to arrhythmias and sudden death or increased hypertrophy (Maillet et al., 2009). However, more studies are needed in this area.
The family of protein kinase C (PKC) consists of at least 12 isoforms. In cardiomyocytes, PKC consists of three main subgroups: the calcium-dependent group (α, βI, βII, and γ), the calcium-independent group (δ, ε, η, θ, and possibly μ), and the atypical PKC (ζ And τ/λ) (Shen et al., 2011). PKC activation is performed by phosphorylation of PDK1 and mTORC2 (Xia et al., 2016). PKCδ or PKCε cause cardiomyocyte hypertrophy by activating ERK-1/2. In addition, concomitant deficiency of PKCδ and PKCε increases pathological cardiac hypertrophy due to pressure overload by inhibiting ERK-1/2. Lack of overexpression of PKCε has also been shown to induce apoptosis in cardiomyocytes, indicating the importance of PKCε for maintaining cell survival (Xia et al., 2016). In mice, PKC-ε signaling pathways are critical for cardioprotection and Akt may be a downstream factor for resistance to myocardial injury because selective inhibition of PKC-ε inhibits Akt activity and blocks sustained cardiac protection (Zhou et al., 2002). In rats, the expression of several isoforms of PKC changes after repeated exercises (70–78% of VO2max for 1 week and 1 day) in the heart, and inhibition of PKC before exercise reverses the improvement of myocardial infarction (Frasier et al., 2011). In the hearts of mice after one exercise (65–70% of VO2max) session is increased activation of PKCε, PKCδ, and PKCɑ. While PKC inhibitors suppress exercise-induced myocardial infarction size reduction. Evidence suggests that PKCɑ acts as a regulator of Ca2+ and myocardial contraction in cardiomyocytes (Shen et al., 2011) (Table 2). Mice lacking PKCα show an increase in heart contraction (Xia et al., 2016). In rats, during exercise (70% of VO2max for 5 days) PKC is activated by a slight increase in cytosolic Ca2+. Indeed, transient Ca2+ exposure limits myocardial infarction through PKC activation (French et al., 2008). In mice, during exercise (swimming exercise for 3 weeks) PKCα phosphorylation is involved in cardiac hypertrophy via ERK-1/2, while PKCε activation has a positive effect on ventricular hypertrophy and acts as protection against apoptosis after pre-conditioning. By quitting exercise, inhibition of PKCα leads to inactivation of ERK-1/2, Akt, and activation of caspase-9, indicating the role of PKCα survival in the heart after exercise. In addition, the exercise leads to the reactivation of PKCα and the downstream Akt and ERK-1/2 pathways for improving cardiac function during pathological hypertrophy (Naskar et al., 2014) (Table 2). In rabbits, activation of adenosine receptors and/or opioids during exercise appears to protect the heart through PKC-dependent mechanisms (Miura et al., 2000; Frasier et al., 2011). Under exercise conditions, ROS induced by preconditioning is likely to regulate PKC activity and inhibit mPTP perforating (Zhou et al., 2015). PKC also is activated directly by exercise by producing ROS without the need to interact with a specific receptor, while concomitant administration of a PKC inhibitor destroys cardiac protection (Penna et al., 2020). Overall, studies show that exercise-induced cardiac protection includes Akt, PKCα, PKCδ, PKCε, and ERK-1/2 (Penna et al., 2020) (Figures 1,2). Therefore, exercise can activate calcium-dependent and calcium-independent PKC, leading to cardiac survival factors.
Cyclic AMP-dependent protein kinase (PKA) is inactively composed of 2 regulatory subunits and 2 catalytic subunits. With hormone stimulation, the G protein of the α subunit binds to adenylyl cyclase (AC) to convert ATP to cAMP, which then releases and activates PKA catalytic subunits by altering the composition of the regulatory subunits (Xia et al., 2016). In addition, PKA exists in the form of multiple isoforms (RIα, RIβ, RIIα, RIIβ, Ca, Cβ, Cγ, and PRKX). In the heart, isoforms of RIα, RIIα, Ca, and also to a lesser extent Cβ are expressed (Colombe and Pidoux, 2021). It seems decreased PKA activity in the cytoplasm and myofilaments leads to impaired myocardial contraction. In contrast, PKA protective roles have been reported in the heart because PKA activation leads to mitochondrial fusion and cellular protection. Inhibition of apoptosis has been reported to be stopped in the model of diabetic cardiomyopathies with the PKA inhibitor. These findings indicate the role of PKA in the regulation of apoptosis (Xia et al., 2016). Immediately after MI, PKA activity increases heart pump balance and regulates MI-induced cardiac dysfunction. PKA signaling demonstrates cardiac protective effects to limit infarction size by inhibiting Rho-kinase (Colombe and Pidoux, 2021). In rats, high levels of catecholamines in the trained heart (75% of VO2max for 60 min) increase PKA and PKC activity (Melling et al., 2004) (Table 2). In mice, activation of vascular eNOS during exercise (80% of VO2max for 50 min) can be regulated by several signaling pathways including Akt, PKA, and/or AMPK (Zhang et al., 2009; Tao et al., 2015). The PKA-Akt-eNOS pathway reduces harmful heart remodeling and protects heart function (Colombe and Pidoux, 2021). The overall effects of exercise are accompanied by the normalization of myofilament sensitivity to calcium via PKA, as characterized by increased β1-adrenoceptor protein (48%) and cAMP levels (36%) (De Waard et al., 2007). After exercise, PKA phosphorylates stimulation-contraction substrate substrates and modulates chronotropic, inotropic, and lusitropic positive responses (Melling et al., 2006; Colombe and Pidoux, 2021) (Table 2). This process involves phosphorylation of PKA, L-type calcium channels (LTCCs), ryanodine receptor (RyR), and phospholamban (PLB), which increases calcium release and heart rate. In myocardial cells, PKA is the primary regulator of calcium through LTCCs phosphorylation. However, acute PKA activation causes cardiac adaptation (e.g., war or escape response) while chronic activation causes adverse heart regeneration (Colombe and Pidoux, 2021). On the other hand, increased cAMP levels after exercise (70% of VO2max) as an upstream PKA activator lead to improved myocardial function during I-R injury. This cardiac protection may be due to cAMP-induced PKA activity that induces HSP70 expression (Melling et al., 2004). PKA plays an essential role in the early regulation of exercise-induced HSP70 gene expression in DNA binding (Melling et al., 2004) (Table 2). Exercise (70% of VO2max) induces the expression of the HSP70 gene as the expression of a protective heart protein by activating the heat shock transcription factor (HSF1). While administration of a PKA inhibitor suppresses exercise-induced HSP70 gene expression and demonstrates the role of PKA in regulating HSF1 activation in vivo (Melling et al., 2006). HSF1 is also phosphorylated on the two serine residues ERK-1/2 and JNK/SAPK and then prevents myocardial apoptosis (Melling et al., 2006). Exercise increases the frequency of dissolved HSP20 in the heart 2.5-fold, and HSP20 phosphorylation plays a compensatory role in heart disease through PKA/PKG-dependent protein kinases (Fan and Kranias, 2011). Therefore, HSP20 phosphorylation by PKA regulation shows cardiac protection after I/R injury (Colombe and Pidoux, 2021). In addition, PKA regulates many signaling pathways such as ERK-1/2 during exercise (75% of VO2max) (Melling et al., 2004) (Figures 1,2, and Figure 3) (Table 2). In general, appears exercise prevents cardiac apoptosis by activating PKA.
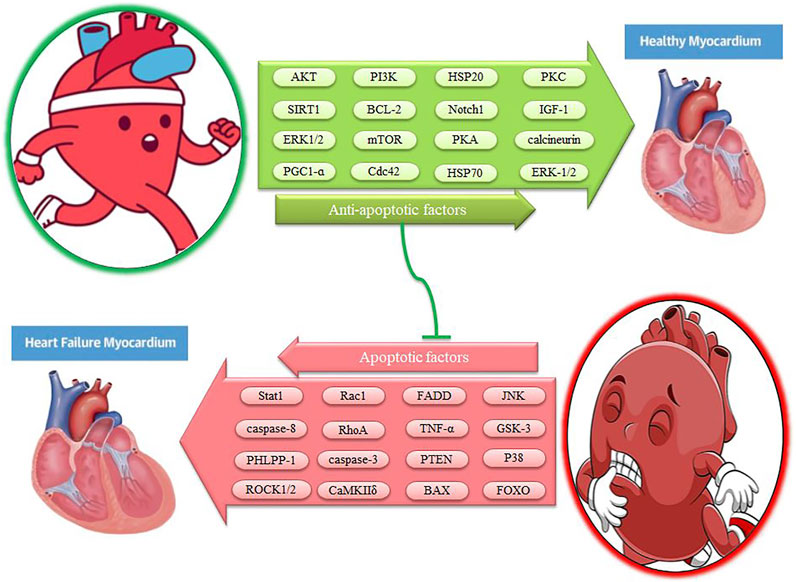
FIGURE 3. The effect of exercise on myocardial apoptotic factors. Description is available in the text.
Calcium signaling regulates several biological processes such as cell survival and death. Intracellular calcium ions (Ca2+) bind to calcium-calmodulin (CaM) and activate calcineurin phosphatase. Calcineurin binds to Ca2+/CaM with high affinity and is activated with moderate levels of calcium ions, while CaMKII is not very sensitive to calcium ion signals and is activated with high levels of calcium ions (Xia et al., 2016). Calcium/calmodulin-dependent kinases consist of three classes: CaMKI, CaMKII, and CaMKIV. In patients’ myocardium, CaMKII plays a major role in processes such as unfavorable remodeling, arrhythmogenesis, interstitial fibrosis, and apoptosis (Beckendorf et al., 2018). CaMKII is characterized by four isoforms α, β, γ, δ. CaMKIIδ and CaMKIIγ isoforms are present in heart tissue, while the δ isoform is about 2.5 times more important than the isoform CaMKIIγ (Beckendorf et al., 2018). CaMKII activity in human heart failure is up-regulated up to 3-fold and CaMKIIδ expression is increased up to 2-fold (Beckendorf et al., 2018). In the heart, activation of the CaMKIIδC- KB kinase inhibitor (IKK)-NF-κB axis leads to increased expression of TNF-α, and inhibition of IKK or TNF-α is sufficient to reduce the size of myocardial infarction (Beckendorf et al., 2018). CaMKIIδ activity is positively regulated in the myocardium of patients and enhances pathological signals such as hypertrophy, fibrosis, and apoptosis (Daniels et al., 2015). Therefore, excessive CaMKII leads to cardiac pathological hypertrophy, heart failure, and arrhythmia (Bernardo et al., 2018). In contrast, exercise (50–90% of VO2max for 13 weeks) eliminates the negative effects of CaMKIIδ on heart failure because intermittent aerobic exercise reduces CaMKIIδ levels and improves heart function in mice (Daniels et al., 2015) (Table 2). Swimming (6 weeks) may also reverse O-GlcNAcylation-mediated CaMKII activation in type I diabetic mice and ultimately improve heart condition (Bennett et al., 2013; Zhang, 2017). In rats, the expression of CaMKII in the moderate-intensity endurance training (48–60% of VO2max for 6 weeks) group is significantly lower than in the control group and can be effective in some factors in the prevention of cardiovascular disease (Khajehlandi et al., 2020). These results indicate the decreasing of CaMKII as a promising pathway to reduce the progression of heart failure (Kreusser et al., 2014). In addition, CaMKII is also involved in adaptive responses after aerobic exercise and is essential for an adequate cardiac response to a physiological stimulus (Beckendorf et al., 2018). Aerobic exercise (52–78% of VO2max) is associated with increased cardiac myocyte contraction, greater calcium amplitude, faster relaxation, PLB phosphorylation, increased SERCA2, and increased activation of CaMKIIδ (Oliveira et al., 2009; Bernardo et al., 2018) (Table 2). Many of these effects are decreased by a CaMKII inhibitor or are decreased in heart failure (Oliveira et al., 2009; Bernardo et al., 2018). Under normal conditions when CaMKIIδ phosphorylation is inhibited, exercise-induced improvements in heart contraction and calcium displacement are reversed and reduced. In this way, CaMKIIδ significantly contributes to cardiomyocyte adaptation to regular exercise (50–90% of VO2max) (Kemi et al., 2007) (Figures 1–3). Thus, some of the consequences of CaMKII may be adaptive, while other sustained functions of CaMKII may be detrimental to cardiac integrity and function (Beckendorf et al., 2018). After exercise (endurance swimming training), calmodulin-deficient mice show hypertrophy with increased calcineurin expression, but there are no signs of adverse heart regeneration (Kreusser et al., 2014). Cardiac calcineurin leads to transcription of hypertrophic genes through dephosphorylation of NFAT. Continuous activation of calcineurin protects against cardiac apoptosis, and decreased calcineurin increases apoptosis through an NFAT-dependent mechanism in a model of mice with dilated cardiomyopathy. Thus calcineurin/NFAT is involved in anti-apoptotic settings (Xia et al., 2016). In mice, inactivation of pathological hypertrophy via the calcineurin/NFAT pathway by exercise (60% of maximal speed for 8 weeks) leads to an anti-apoptotic effect on heart failure (Oliveira et al., 2009) (Table 2). In trained mice hearts (59–70% of VO2max for 10 weeks), p-ERK-1/2 and calcineurin increase significantly, leading to physiological left ventricular hypertrophy (Eto et al., 2000; Nakamura et al., 2002). However, there is evidence that physiological hypertrophy of the heart is not affected in response to exercise in NFAT2-deficient mice because, after 4 weeks of voluntary exercise, a cardiac growth response is observed in NFAT2-deficient mice (Bourajjaj et al., 2008). Therefore, exercise can prevent myocardial apoptosis by modulating calcineurin and CaMK and cause physiological cardiac hypertrophy.
Heat shock proteins (HSPs) in cells make up about 5–10% of total proteins, while under stress they increase by up to 15%. HSPs are often found in the cytosol, nucleus, mitochondria, and endoplasmic reticulum (ER), while they are released into the extracellular matrix during cell death or attach freely to the membrane (Wu, Chen, Liu, Liu, Wang, Cheng, et al.). HSPs are involved in maintaining cellular homeostasis and protecting heart cells during periods of acute stress, hypoxia, heat shock, and energy depletion (Powers and Demirel, 2001; Wu, Chen, Liu, Liu, Wang, Cheng, et al.). In rats, HSP20 expression is altered by different models of heart damage, and both HSP20 mRNA and protein levels have been reported to decrease in response to ischemia. While overexpression of HSP20 shows cardiac protective effects (Yin et al., 2019). In the myocardial infarction model, HSP20-regulated exosomes increase cell survival by activating Akt and decreasing mRNA levels of inflammatory factors such as TNF-α and IL-1β. Increased HSP20 expression leads to increased Bcl2 to Bax ratio and inhibition of caspase-3, and myocardial apoptosis (Wu, Chen, Liu, Liu, Wang, Cheng, et al.). In Mice, HSP27 expression is high in mammals after 7 days of MI, whereas it decreases at 14 days after MI. Overexpression of HSP27 increases myocardial infarction tolerance, while HSP27 deficiency exacerbates injury and worsens heart function. Overexpression of HSP27 stops the activation of caspase-3, ROS, the NF-κB-induced inflammatory response, and the degradation of troponin I (cTnI) and cTnT in the myocardium (Wu, Chen, Liu, Liu, Wang, Cheng, et al.; Wang et al., 2019). In rats, cardiac HSP20 is increased with endurance exercise (70–75% of VO2max for 6 weeks), and HSP20 phosphorylation has been identified as a therapeutic target for heart disease (Burniston, 2009) (Table 2). In mice, swimming for 3 weeks also reduces cardiomyocyte apoptosis by inducing extracellular vesicles by activating ERK/HSP27 signaling (Bei et al., 2017). During exercise (65–75% VO2max for 10–12 weeks), HSP72 is important in preventing apoptosis, necrosis, and oxidative damage due to I/R in rat myocytes. HSP72 may help protect cells against a variety of stresses by preventing the accumulation of defective proteins, helping to re-fold damaged proteins (Powers and Demirel, 2001) (Table 2). In rats, after acute exercise (70% of VO2max for 8 weeks), myocardial HSP70 mRNA expression is stimulated in the presence of ERK-1/2 activation. Thus, exercise leads to myocardial adaptation, the onset of proliferative and protective responses (Melling et al., 2007) (Figures 1, 3). Endurance exercise has been shown to increase myocardial HSP72 by 400–500% in young adult rats (Powers and Demirel, 2001). In addition, exercise (70% of VO2max) in a warm environment significantly increases myocardial HSP72 content compared to inactive rats. While exercising in a cold environment prevents the increase in HSP72 caused by exercise in the myocardium. However, after myocardial I/R, infarction size and caspase-3 activity decrease in both exercise groups compared with sedentary animals (Quindry et al., 2007) (Table 2). There is evidence that relative levels of HSP90, HSP72, and HSP40 are higher in rats exercising (70% of VO2max) in warm water compared with controls and cold water (Hamilton et al., 2001) (Table 2). Therefore, exercise (66% of VO2max) in addition to increasing other anti-apoptotic agents, leads to dynamic changes in heat shock proteins to protect the heart that is affected by body temperature (Harris and Starnes, 2001). In general, it seems exercise improves heart function through HSP20, HSP27, HSP40, HSP70, HSP72, HSP90, and other anti-apoptotic agents.
Expression of all seven STAT1-7 family members has been reported in cardiomyocytes. Among them, STAT1 and STAT3 have opposite effects on post-ischemic heart cell survival. In the heart of animals under ischemic conditions, STAT1 activation is induced by the production of ROS, which is detrimental to cardiomyocytes through induction of apoptosis and inhibition of autophagy. STAT1 transcription activity is encoded on caspase-1 promoters and also enhances Fas and FasL, which increase cell-level death. In addition, STAT1 induces p53 and Bax transcriptional activity and inhibits Bcl2 (Carroll et al., 2013; Eid et al., 2018). STAT3 is present in most parts of the heart such as the plasma membrane, cytosol, nucleus, and mitochondria. STAT3 is an important cardioprotective agent after ischemia or hypoxia (Eid et al., 2018). STAT3 is activated by a range of cytokines including IL-6 and IL-10, as well as growth factors such as IGF-I, liver growth factor (LGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and Basic fibroblast growth factor (bFGF), ANG II, and mechanical traction (RhoA) (Trenerry et al., 2007; Haghikia et al., 2011). In rats, the cardioprotective effect of STAT3 is mediated by the positive regulation of anti-apoptotic proteins such as BCL-XL, BCL-2, and HSP70. STAT3 also contains the effects of antioxidant proteins such as MnSOD and metallothioneins and enhances angiogenic factors such as VEGF and VE-cadherin. STAT3 prevents the opening of mPTP by activating PI3K, AKT, and ERK-1/2 components and direct regulation of BCL-2. In addition, STAT3 maintains mitochondrial energy and function by improving complex 1 and reducing ROS formation. However, phosphorylation of JAK2 and STAT3 is significantly reduced in human heart failure, which shows an important role in heart regeneration (Eid et al., 2018) (Figures 2, 3). Ischemic preconditioning with exercise in cardiomyocytes activates the JAK/STAT pathway after mechanical traction and pressure overload. This pathway then protects the heart against I/R injury (Boengler et al., 2008; Eid et al., 2018; Zhang et al., 2021). In rats, moderate exercise increases p-JAK2 and p-STAT3 expression and decreases caspase-3 and myocardial apoptosis. While strenuous exercise increases the expression of caspase-3, p-JAK2, and p-STAT3 in the heart (Sun et al., 2017) (Table 2). In addition, after STAT3 phosphorylation, transcription of various hypertrophic factors such as natriuretic peptide type A and natriuretic peptide type B is enhanced (Geng et al., 2019). Rapid and transient activation of STAT3 occurs 2 h after exercise with transient transfer to the nucleus. This process is followed by downstream transcription events such as c-FOS (800-fold), JUNB (38-fold), c-MYC (140-fold), and VEGF (4-fold). Therefore, STAT3 signaling helps to regenerate and adapt after exercise training (Trenerry et al., 2007; Trenerry et al., 2008). In rats, exercise preconditioning (72% of VO2MAX) also significantly reduces myocardial ischemic injury through decreased serum cTnI levels, decreased infarction area, decreased myocardial apoptosis, increased Bcl-2 levels, decreased caspase-3 levels, and increased JAK2 and STAT3 phosphorylation. In this way, exercise preconditioning plays a protective role in the heart by activating the JAK2/STAT3 signaling pathway and reducing myocardial cell apoptosis (Sun and Mao, 2018) (Table 2).
Cardiovascular disease is one of the leading causes of death in the world. Excessive intracellular and extracellular apoptosis is one of the triggers for these deaths. However, several factors, such as aerobic exercise (30–75% of VO2max), swimming, short-time (3 days) exercise, and long-time (3 weeks) exercise, can reduce or reverse this process. After exercise, cardiac expression of IGF-1, IGF-1R, p-PI3K, p-Akt, and Bcl2 increases, and these factors also leads to hypertrophy, survival, and cardioprotection (Figures 1–3). After exercise, miR-222, miR-17-3p, MiR-20a, and miR-26a can negatively regulate PTEN to protect the heart against myocardial damage because PTEN inhibits Akt activation and leads to a decrease in Bcl- 2, increases Bax, caspase-3, and apoptosis. Exercise-induced decrease in PHLPP-1 significantly stimulates cardiac hypertrophy through Akt, p70S6Kinase, and ERK, leading to a reduction in pathological hypertrophy, a reduction in cardiac apoptosis, and a reduction in infarct size. The PI3K/AKT pathway then inhibits the GSK-3 apoptotic agent. After exercise, increased anti-apoptotic agents such as Akt, SIRT3, and PGC-1α suppress FOXO activity, which in turn reduces cardiac apoptosis. Exercise can also increase ERK1/2 in myocytes, decrease JNK and P38MAPK, and reduce myocyte apoptosis. During exercise, mechanical stress stimulates β1 and α5 integrins and then activates PI3K/PIP3/Akt/mTORC1 and ERK-1/2 through FAK phosphorylation, leading to cell survival and a hypertrophic response (Figures 1–3). This study shows that the PI3K/Akt pathway is one of the most important pathways to combat apoptosis and protect the heart.
Reduction of exercise-induced apoptosis also occurs through decreased levels of Fas ligand-protein, Fas death receptor, TNF-α, TNFR-1, FADD, caspase-8, and caspase-3. During exercise, also levels of RhoA, ROCK1, Rac1, Cdc42, and ROCK2 are significantly reduced while the Bcl-2/Bax ratio is significantly increased. Under training conditions, PKCε, PKCδ, and PKCɑ are activated, and PKCɑ acts as a Ca2+ regulator in the heart and prevents mPTP perforation (Figures 1, 3). During exercise, the PKA-Akt-eNOS pathway protects heart function by inhibiting Rho-kinase and phosphorylation of HSP20 and HSP70 and regulating ERK-1/2 via PKA. Moderate physical activity inhibits myocardial fibrosis by expressing Notch1 cardiac genes, the FSTL1-USP10-Notch1 signaling axis. Exercise also eliminates the negative effects of CaMKIIδ through O-GlcNAcylation on heart failure. Inactivation of exercise-induced pathological hypertrophy via the calcineurin/NFAT pathway leads to an anti-apoptotic effect on heart failure. After acute exercise, expression of Hsp20, HSP27, HSP40, HSP70, HSP72, and HSP90 in the myocardium via JAK2/STAT3 phosphorylation, ERK-1/2 activation leads to myocardial adaptation and proliferative and protective responses (Figure 2, 3). Finally, a variety of exercises block the signaling pathways of intracellular and extracellular apoptosis through various pathways. Since the little study has been done on Pim-1 and FAK, further studies on the effect of exercise on Pim-1 and FAK are needed. Future studies should also try to find other pathways related to cardiac apoptosis. It is also recommended to study the synergistic effect of exercise along with other methods such as gene therapy, dietary supplements, and cell therapy for future research.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alizadeh Pahavani, H., Rajabi, H., Nabiuni, M., Motamedi, P., Khaledi, N., and Tayanloo, A. (2020). The effect of aerobic exercise with medium and high intensity on the gene expression of Bax (BCL2 associated X) and Bcl-2 (B-cell lymphoma 2) markers in rat myocard after ischemic-reperfusion. Tehran: Sport Physiology, 31–44. 12(45).
Alizadeh Pahlavani, H. (2022). Exercise therapy for people with sarcopenic obesity: Myokines and adipokines as effective actors. Front. Endocrinol. 148, 811751. doi:10.3389/fendo.2022.811751
Alizadeh Pahlavani, H. (2022). Exercise therapy for people with sarcopenic obesity: Myokines and adipokines as effective actors. Front. Endocrinol. 13, 811751. doi:10.3389/fendo.2022.811751
Alizadeh, P. H., Rajabi, H., Nabiuni, M., Motamedi, P., and Khaledi, N. (2017). The effect of aerobic exercise and melatonin consumption on the expression of bax and BCL-2 markers in rat myocard after ischemia-reperfusion. J. Isfahan Med. Sch. 35 (423), 318–325. doi:10.34172/mj.2019.035
Anaruma, C. P., Pereira, R. M., da Cruz Rodrigues, K. C., da Silva, A. S. R., Cintra, D. E., Ropelle, E. R., et al. (2020). Rock protein as cardiac hypertrophy modulator in obesity and physical exercise. Life Sci. 254, 116955. doi:10.1016/j.lfs.2019.116955
Beckendorf, J., van den Hoogenhof, M. M., and Backs, J. (2018). Physiological and unappreciated roles of CaMKII in the heart. Basic Res. Cardiol. 113 (4), 29. doi:10.1007/s00395-018-0688-8
Bei, Y., Xu, T., Lv, D., Yu, P., Xu, J., Che, L., et al. (2017). Exercise-induced circulating extracellular vesicles protect against cardiac ischemia–reperfusion injury. Basic Res. Cardiol. 112 (4), 38. doi:10.1007/s00395-017-0628-z
Bennett, C. E., Johnsen, V. L., Shearer, J., and Belke, D. D. (2013). Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci. 92 (11), 657–663. doi:10.1016/j.lfs.2012.09.007
Bernardo, B. C., Ooi, J. Y., Weeks, K. L., Patterson, N. L., and McMullen, J. R. (2018). Understanding key mechanisms of exercise-induced cardiac protection to mitigate disease: Current knowledge and emerging concepts. Physiol. Rev. 98 (1), 419–475. doi:10.1152/physrev.00043.2016
Bishopric, N. H. (2012). A longer, better ride with engineered stem cells. Washington, DC: American College of Cardiology Foundation, 1288–1290.
Bo, B., Zhou, Y., Zheng, Q., Wang, G., Zhou, K., Wei, J., et al. (2021). The molecular mechanisms associated with aerobic exercise-induced cardiac regeneration. Biomolecules 11 (1), 19. doi:10.3390/biom11010019
Bo, W., Ma, Y., Xi, Y., Liang, Q., Cai, M., and Tian, Z. (2021). The roles of FGF21 and ALCAT1 in aerobic exercise-induced cardioprotection of postmyocardial infarction mice. Oxidative Med. Cell. Longev. 2021, 1–17. doi:10.1155/2021/8996482
Boengler, K., Hilfiker-Kleiner, D., Drexler, H., Heusch, G., and Schulz, R. (2008). The myocardial JAK/STAT pathway: From protection to failure. Pharmacol. Ther. 120 (2), 172–185. doi:10.1016/j.pharmthera.2008.08.002
Boluyt, M., Loyd, A., Roth, M., Randall, M., and Song, E. (2003). Activation of JNK in rat heart by exercise: Effect of training. Am. J. Physiol. Heart Circ. Physiol. 285 (6), H2639–H2647. doi:10.1152/ajpheart.00596.2003
Bourajjaj, M., Armand, A-S., da Costa Martins, P. A., Weijts, B., van der Nagel, R., Heeneman, S., et al. (2008). NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J. Biol. Chem. 283 (32), 22295–22303. doi:10.1074/jbc.M801296200
Brancaccio, M., Hirsch, E., Notte, A., Selvetella, G., Lembo, G., Tarone, G., et al. (2006). Integrin signalling: The tug-of-war in heart hypertrophy. Cardiovasc. Res. 70 (3), 422–433. doi:10.1016/j.cardiores.2005.12.015
Budiono, B. P., See Hoe, L. E., Peart, J. N., Sabapathy, S., Ashton, K. J., Haseler, L. J., et al. (2012). Voluntary running in mice beneficially modulates myocardial ischemic tolerance, signaling kinases, and gene expression patterns. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302 (9), R1091–R1100. doi:10.1152/ajpregu.00406.2011
Burgess, M. L., McCrea, J. C., and Hedrick, H. L. (2001). Age-associated changes in cardiac matrix and integrins. Mech. Ageing Dev. 122 (15), 1739–1756. doi:10.1016/s0047-6374(01)00296-2
Burgess, M. L., Terracio, L., Hirozane, T., and Borg, T. K. (2002). Differential integrin expression by cardiac fibroblasts from hypertensive and exercise-trained rat hearts. Cardiovasc. Pathol. 11 (2), 78–87. doi:10.1016/s1054-8807(01)00104-1
Burniston, J. G. (2009). Adaptation of the rat cardiac proteome in response to intensity-controlled endurance exercise. Proteomics 9 (1), 106–115. doi:10.1002/pmic.200800268
Cai, S., Tao, X., Long, Y., Xia, K., and Zhang, Y. (2019). Effect of miR-26a on diabetic rats with myocardial injury by targeting PTEN. Eur. Rev. Med. Pharmacol. Sci. 23 (3), 304–311. doi:10.26355/eurrev_201908_18661
Cai, W., Yang, X., Han, S., Guo, H., Zheng, Z., Wang, H., et al. (2016). Notch1 pathway protects against burn-induced myocardial injury by repressing reactive oxygen species production through JAK2/STAT3 signaling. Oxidative Med. Cell. Longev. 2016, 5638943. doi:10.1155/2016/5638943
Carey, K. A., Farnfield, M. M., Tarquinio, S. D., and Cameron-Smith, D. (2007). Impaired expression of Notch signaling genes in aged human skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 62 (1), 9–17. doi:10.1093/gerona/62.1.9
Carroll, C. J., Sayan, B. S., Bailey, S. G., McCormick, J., Stephanou, A., Latchman, D. S., et al. (2013). Regulation of myocardial interleukin-6 expression by p53 and STAT1. J. Interferon Cytokine Res. 33 (9), 542–548. doi:10.1089/jir.2012.0165
Cheng, S-M., Ho, T-J., Yang, A-L., Chen, I-J., Kao, C-L., Wu, F-N., et al. (2013). Exercise training enhances cardiac IGFI-R/PI3K/Akt and Bcl-2 family associated pro-survival pathways in streptozotocin-induced diabetic rats. Int. J. Cardiol. 167 (2), 478–485. doi:10.1016/j.ijcard.2012.01.031
Chodari, L., Mohammadi, M., Mohaddes, G., Alipour, M. R., Ghorbanzade, V., Dariushnejad, H., et al. (2016). Testosterone and voluntary exercise, alone or together increase cardiac activation of AKT and ERK1/2 in diabetic rats. Arq. Bras. Cardiol. 107, 532–541. doi:10.5935/abc.20160174
Colombe, A-S., and Pidoux, G. (2021). Cardiac cAMP-PKA signaling compartmentalization in myocardial infarction. Cells 10 (4), 922. doi:10.3390/cells10040922
Daniels, L., Bell, J. R., Delbridge, L., McDonald, F. J., Lamberts, R. R., Erickson, J. R., et al. (2015). The role of CaMKII in diabetic heart dysfunction. Heart fail. Rev. 20 (5), 589–600. doi:10.1007/s10741-015-9498-3
De Waard, M. C., Van Der Velden, J., Bito, V., Ozdemir, S., Biesmans, L., Boontje, N. M., et al. (2007). Early exercise training normalizes myofilament function and attenuates left ventricular pump dysfunction in mice with a large myocardial infarction. Circ. Res. 100 (7), 1079–1088. doi:10.1161/01.RES.0000262655.16373.37
Dickson, E. W., Hogrefe, C. P., Ludwig, P. S., Ackermann, L. W., Stoll, L. L., Denning, G. M., et al. (2008). Exercise enhances myocardial ischemic tolerance via an opioid receptor-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 294 (1), H402–H408. doi:10.1152/ajpheart.00280.2007
Dizon, L. A., Seo, D. Y., Kim, H. K., Kim, N., Ko, K. S., Rhee, B. D., et al. (2013). Exercise perspective on common cardiac medications. Integr. Med. Res. 2 (2), 49–55. doi:10.1016/j.imr.2013.04.006
Dong, Y., Chen, H., Gao, J., Liu, Y., Li, J., Wang, J., et al. (2019). Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 136, 27–41. doi:10.1016/j.yjmcc.2019.09.001
Donniacuo, M., Urbanek, K., Nebbioso, A., Sodano, L., Gallo, L., Altucci, L., et al. (2019). Cardioprotective effect of a moderate and prolonged exercise training involves sirtuin pathway. Life Sci. 222, 140–147. doi:10.1016/j.lfs.2019.03.001
Ebeid, D. E., Khalafalla, F. G., Broughton, K. M., Monsanto, M. M., Esquer, C. Y., Sacchi, V., et al. (2021). Pim1 maintains telomere length in mouse cardiomyocytes by inhibiting TGFβ signalling. Cardiovasc. Res. 117 (1), 201–211. doi:10.1093/cvr/cvaa066
Eid, R. A., Alkhateeb, M. A., Eleawa, S., Al-Hashem, F. H., Al-Shraim, M., El-Kott, A. F., et al. (2018). Cardioprotective effect of ghrelin against myocardial infarction-induced left ventricular injury via inhibition of SOCS3 and activation of JAK2/STAT3 signaling. Basic Res. Cardiol. 113 (2), 13. doi:10.1007/s00395-018-0671-4
Eto, Y., Yonekura, K., Sonoda, M., Arai, N., Sata, M., Sugiura, S., et al. (2000). Calcineurin is activated in rat hearts with physiological left ventricular hypertrophy induced by voluntary exercise training. Circulation 101 (18), 2134–2137. doi:10.1161/01.cir.101.18.2134
Fan, G-C., and Kranias, E. G. (2011). Small heat shock protein 20 (HspB6) in cardiac hypertrophy and failure. J. Mol. Cell. Cardiol. 51 (4), 574–577. doi:10.1016/j.yjmcc.2010.09.013
Ferrara, N., Rinaldi, B., Corbi, G., Conti, V., Stiuso, P., Boccuti, S., et al. (2008). Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res. 11 (1), 139–150. doi:10.1089/rej.2007.0576
Ferrari, R., and Rizzo, P. (2014). The notch pathway: A novel target for myocardial remodelling therapy? Eur. Heart J. 35 (32), 2140–2145. doi:10.1093/eurheartj/ehu244
Ferreira, R., Nogueira-Ferreira, R., Trindade, F., Vitorino, R., Powers, S. K., Moreira-Gonçalves, D., et al. (2018). Sugar or fat: The metabolic choice of the trained heart. Metabolism. 87, 98–104. doi:10.1016/j.metabol.2018.07.004
Flueck, M., Eyeang-Békalé, N., Héraud, A., Girard, A., Gimpl, M., Seynnes, O., et al. (2011). Load-sensitive adhesion factor expression in the elderly with skiing: Relation to fiber type and muscle strength. Scand. J. Med. Sci. Sports 21, 29–38. doi:10.1111/j.1600-0838.2011.01339.x
Franchi, M. V., Ruoss, S., Valdivieso, P., Mitchell, K. W., Smith, K., Atherton, P. J., et al. (2018). Regional regulation of focal adhesion kinase after concentric and eccentric loading is related to remodelling of human skeletal muscle. Acta Physiol. 223 (3), e13056. doi:10.1111/apha.13056
Frasier, C. R., Moore, R. L., and Brown, D. A. (2011). Exercise-induced cardiac preconditioning: How exercise protects your achy-breaky heart. J. Appl. Physiol. 111 (3), 905–915. doi:10.1152/japplphysiol.00004.2011
French, J. P., Hamilton, K. L., Quindry, J. C., Lee, Y., Upchurch, P. A., Powers, S. K., et al. (2008). Exercise-induced protection against myocardial apoptosis and necrosis: MnSOD, calcium-handling proteins, and calpain. FASEB J. 22 (8), 2862–2871. doi:10.1096/fj.07-102541
Geng, Z., Fan, W-Y., Zhou, B., Ye, C., Tong, Y., Zhou, Y-B., et al. (2019). FNDC5 attenuates obesity-induced cardiac hypertrophy by inactivating JAK2/STAT3-associated inflammation and oxidative stress. J. Transl. Med. 17 (1), 107. doi:10.1186/s12967-019-1857-8
Gill, C., Mestril, R., and Samali, A. (2002). Losing heart: The role of apoptosis in heart disease—a novel therapeutic target? FASEB J. 16 (2), 135–146. doi:10.1096/fj.01-0629com
Graham, Z. A., Gallagher, P. M., and Cardozo, C. P. (2015). Focal adhesion kinase and its role in skeletal muscle. J. Muscle Res. Cell Motil. 36 (4), 305–315. doi:10.1007/s10974-015-9415-3
Graham, Z. A., Touchberry, C. D., Gupte, A. A., Bomhoff, G. L., Geiger, P. C., Gallagher, P. M., et al. (2015). Changes in α7β1 integrin signaling after eccentric exercise in heat-shocked rat soleus. Muscle Nerve 51 (4), 562–568. doi:10.1002/mus.24324
Habibi, P., Alihemmati, A., NourAzar, A., Yousefi, H., Mortazavi, S., Ahmadiasl, N., et al. (2016). Expression of the Mir-133 and Bcl-2 could be affected by swimming training in the heart of ovariectomized rats. Iran. J. Basic Med. Sci. 19 (4), 381–387.
Haghikia, A., Stapel, B., Hoch, M., and Hilfiker-Kleiner, D. (2011). STAT3 and cardiac remodeling. Heart fail. Rev. 16 (1), 35–47. doi:10.1007/s10741-010-9170-x
Hamilton, K. L., Powers, S. K., Sugiura, T., Kim, S., Lennon, S., Tumer, N., et al. (2001). Short-term exercise training can improve myocardial tolerance to I/R without elevation in heat shock proteins. Am. J. Physiol. Heart Circ. Physiol. 281 (3), H1346–H1352. doi:10.1152/ajpheart.2001.281.3.H1346
Harris, M. B., and Starnes, J. W. (2001). Effects of body temperature during exercise training on myocardial adaptations. Am. J. Physiol. Heart Circ. Physiol. 280 (5), H2271–H2280. doi:10.1152/ajpheart.2001.280.5.H2271
Ho, T-J., Huang, C-C., Huang, C-Y., and Lin, W-T. (2012). Fasudil, a Rho-kinase inhibitor, protects against excessive endurance exercise training-induced cardiac hypertrophy, apoptosis and fibrosis in rats. Eur. J. Appl. Physiol. 112 (8), 2943–2955. doi:10.1007/s00421-011-2270-z
Hong, J., Kim, K., Kim, J-H., and Park, Y. (2017). The role of endoplasmic reticulum stress in cardiovascular disease and exercise. Int. J. Vasc. Med. 2017, 2049217. doi:10.1155/2017/2049217
Hooper, P. L. (2007). Insulin signaling, GSK-3, heat shock proteins and the natural history of type 2 diabetes mellitus: A hypothesis. Metab. Syndr. Relat. Disord. 5 (3), 220–230. doi:10.1089/met.2007.0005
Huang, C-Y., Lin, Y-Y., Hsu, C-C., Cheng, S-M., Shyu, W-C., Ting, H., et al. (2016). Antiapoptotic effect of exercise training on ovariectomized rat hearts. J. Appl. Physiol. 121 (2), 457–465. doi:10.1152/japplphysiol.01042.2015
Huang, C-Y., Yang, A-L., Lin, Y-M., Wu, F-N., Lin, J. A., Chan, Y-S., et al. (2012). Anti-apoptotic and pro-survival effects of exercise training on hypertensive hearts. J. Appl. Physiol. 112 (5), 883–891. doi:10.1152/japplphysiol.00605.2011
Icli, B., Wara, A., Moslehi, J., Sun, X., Plovie, E., Cahill, M., et al. (2013). MicroRNA-26a regulates pathological and physiological angiogenesis by targeting BMP/SMAD1 signaling. Circ. Res. 113 (11), 1231–1241. doi:10.1161/CIRCRESAHA.113.301780
Iemitsu, M., Maeda, S., Jesmin, S., Otsuki, T., Kasuya, Y., Miyauchi, T., et al. (2006). Activation pattern of MAPK signaling in the hearts of trained and untrained rats following a single bout of exercise. J. Appl. Physiol. 101 (1), 151–163. doi:10.1152/japplphysiol.00392.2005
Kanazawa, M., Matsumoto, Y., Takahashi, K., Suzuki, H., Uzuka, H., Nishimiya, K., et al. (2020). Treadmill exercise prevents reduction of bone mineral density after myocardial infarction in apolipoprotein E-deficient mice. Eur. J. Prev. Cardiol. 27 (1), 28–35. doi:10.1177/2047487319834399
Kavazis, A. N., Smuder, A. J., and Powers, S. K. (2014). Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 117 (3), 223–230. doi:10.1152/japplphysiol.00210.2014
Kemi, O. J., Ellingsen, Ø., Ceci, M., Grimaldi, S., Smith, G. L., Condorelli, G., et al. (2007). Aerobic interval training enhances cardiomyocyte contractility and Ca2+ cycling by phosphorylation of CaMKII and Thr-17 of phospholamban. J. Mol. Cell. Cardiol. 43 (3), 354–361. doi:10.1016/j.yjmcc.2007.06.013
Khajehlandi, M., Bolboli, L., Siahkuhian, M., Rami, M., and Tabandeh, M. (2020). Effect of moderate intensity exercise on HDAC4 and CaMKII genes expression in myocardium of male rats. KAUMS J. (FEYZ). 24 (4), 357–365.
Ko, I-G., Kim, S-E., Kim, C-J., and Jee, Y-S. (2013). Treadmill exercise alleviates aging-induced apoptosis in rat cardiac myocytes. Int. J. gerontology 7 (3), 152–157. doi:10.1016/j.ijge.2013.01.001
Kojda, G., and Hambrecht, R. (2005). Molecular mechanisms of vascular adaptations to exercise. Physical activity as an effective antioxidant therapy? Cardiovasc. Res. 67 (2), 187–197. doi:10.1016/j.cardiores.2005.04.032
Kreusser, M. M., Lehmann, L. H., Keranov, S., Hoting, M-O., Oehl, U., Kohlhaas, M., et al. (2014). Cardiac CaM Kinase II genes δ and γ contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation 130 (15), 1262–1273. doi:10.1161/CIRCULATIONAHA.114.006185
Kwak, H-B. (2013). Effects of aging and exercise training on apoptosis in the heart. J. Exerc. Rehabil. 9 (2), 212–219. doi:10.12965/jer.130002
Lagranha, C. J., Hirabara, S. M., Curi, R., and Pithon-Curi, T. C. (2007). Glutamine supplementation prevents exercise-induced neutrophil apoptosis and reduces p38 MAPK and JNK phosphorylation and p53 and caspase 3 expression. Cell biochem. Funct. 25 (5), 563–569. doi:10.1002/cbf.1421
Lajoie, C., Calderone, A., and Béliveau, L. (2004). Exercise training enhanced the expression of myocardial proteins related to cell protection in spontaneously hypertensive rats. Pflugers Arch. 449 (1), 26–32. doi:10.1007/s00424-004-1307-0
Lajoie, C., Calderone, A., Trudeau, F., Lavoie, N., Massicotte, G., Gagnon, S., et al. (2004). Exercise training attenuated the PKB and GSK-3 dephosphorylation in the myocardium of ZDF rats. J. Appl. Physiol. 96 (5), 1606–1612. doi:10.1152/japplphysiol.00853.2003
Lajoie, C. (2003). Exercice et protection myocardique chez des rats genetiquement diabetiques ou hypertendus (French text).
Lay, S., Kuo, W-W., Shibu, M. A., Ho, T-J., Cheng, S-M., Day, C. H., et al. (2021). Exercise training restores IGFIR survival signaling in d-galactose induced-aging rats to suppress cardiac apoptosis. J. Adv. Res. 28, 35–41. doi:10.1016/j.jare.2020.06.015
Lee, S-D., Shyu, W-C., Cheng, I-S., Kuo, C-H., Chan, Y-S., Lin, Y-M., et al. (2013). Effects of exercise training on cardiac apoptosis in obese rats. Nutr. Metab. Cardiovasc. Dis. 23 (6), 566–573. doi:10.1016/j.numecd.2011.11.002
Lemoine, K. A., Fassas, J. M., Ohannesian, S. H., and Purcell, N. H. (2021). On the PHLPPside: Emerging roles of PHLPP phosphatases in the heart. Cell. Signal. 86, 110097. doi:10.1016/j.cellsig.2021.110097
Li, F. (2011). Exercise training influence on myocardial apoptosis of spontaneously hypertensive rat's protein expression. J. Shenyang Sport Univ. 03, 1.
Li, R., Narici, M. V., Erskine, R. M., Seynnes, O. R., Rittweger, J., Pišot, R., et al. (2013). Costamere remodeling with muscle loading and unloading in healthy young men. J. Anat. 223 (5), 525–536. doi:10.1111/joa.12101
Li, X., Han, X., Zhang, H., Tan, H., and Han, S. (2017). SIRT1 signaling pathway mediated the protective effects on myocardium of rats after endurance training and acute exhaustive exercise. Zhonghua xin xue guan bing za zhi 45 (6), 501–506. doi:10.3760/cma.j.issn.0253-3758.2017.06.012
Liang, J., Zhang, X., Xia, W., Tong, X., Qiu, Y., Qiu, Y., et al. (2021). Promotion of aerobic exercise induced angiogenesis is associated with decline in blood pressure in hypertension: Result of EXCAVATION-CHN1. Hypertension 77 (4), 1141–1153. doi:10.1161/HYPERTENSIONAHA.120.16107
Liao, Q., Qu, S., Tang, L-X., Li, L-P., He, D-F., Zeng, C-Y., et al. (2019). Irisin exerts a therapeutic effect against myocardial infarction via promoting angiogenesis. Acta Pharmacol. Sin. 40 (10), 1314–1321. doi:10.1038/s41401-019-0230-z
Lin, Y., and Zhao, L. (2017). Effect and mechanism of exercise-induced miR-222 in the protection of myocardial damage in diabetic mice. Chin. J. Endocrinol. Metabolism 33 (1), 56–61.
Liu, X., and Wang, G. (2020). Exercise preconditioning for myocardial injury in rats after exhaustive exercise based on Rho/ROCK pathway. Chin. J. Tissue Eng. Res. 24 (11), 1714.
Liu, Y., Shang, Y., Yan, Z., Li, H., Wang, Z., Liu, Z., et al. (2019). Pim1 kinase positively regulates myoblast behaviors and skeletal muscle regeneration. Cell Death Dis. 10 (10), 773. doi:10.1038/s41419-019-1993-3
Lu, L., Ma, J., Liu, Y., Shao, Y., Xiong, X., Duan, W., et al. (2021). FSTL1-USP10-Notch1 signaling Axis protects against myocardial infarction through inhibition of myocardial fibrosis in diabetic mice. Front. cell Dev. Biol. 9, 757068. doi:10.3389/fcell.2021.757068
Ludlow, A. T., Gratidao, L., Ludlow, L. W., Spangenburg, E. E., and Roth, S. M. (2017). Acute exercise activates p38 MAPK and increases the expression of telomere-protective genes in cardiac muscle. Exp. Physiol. 102 (4), 397–410. doi:10.1113/EP086189
Ma, Z., Qi, J., Meng, S., Wen, B., and Zhang, J. (2013). Swimming exercise training-induced left ventricular hypertrophy involves microRNAs and synergistic regulation of the PI3K/AKT/mTOR signaling pathway. Eur. J. Appl. Physiol. 113 (10), 2473–2486. doi:10.1007/s00421-013-2685-9
Magne, J., Pibarot, P., Sengupta, P. P., Donal, E., Rosenhek, R., Lancellotti, P., et al. (2015). Pulmonary hypertension in valvular disease: A comprehensive review on pathophysiology to therapy from the HAVEC group. JACC. Cardiovasc. Imaging 8 (1), 83–99. doi:10.1016/j.jcmg.2014.12.003
Maillet, M., Lynch, J. M., Sanna, B., York, A. J., Zheng, Y., Molkentin, J. D., et al. (2009). Cdc42 is an antihypertrophic molecular switch in the mouse heart. J. Clin. Invest. 119 (10), 3079–3088. doi:10.1172/JCI37694
Mann, N., and Rosenzweig, A. (2012). Can exercise teach us how to treat heart disease? Circulation 126 (22), 2625–2635. doi:10.1161/CIRCULATIONAHA.111.060376
Manso, A. M., Kang, S-M., and Ross, R. S. (2009). Integrins, focal adhesions, and cardiac fibroblasts. J. Investig. Med. 57 (8), 856–860. doi:10.2310/JIM.0b013e3181c5e61f
Massicotte, S. G., and Béliveau, L. (2006). Exercise training attenuated the PKB and GSK-3. Circ. Res. 98 (4), 443–445.
Matsumoto, A., Manthey, H. D., Marsh, S. A., Fassett, R. G., De Haan, J. B., Rolfe, B. E., et al. (2013). Effects of exercise training and RhoA/ROCK inhibition on plaque in ApoE−/− mice. Int. J. Cardiol. 167 (4), 1282–1288. doi:10.1016/j.ijcard.2012.03.172
Matsumoto, A., Mason, S. R., Flatscher-Bader, T., Ward, L. C., Marsh, S. A., Wilce, P. A., et al. (2012). Effects of exercise and antioxidant supplementation on endothelial gene expression. Int. J. Cardiol. 158 (1), 59–65. doi:10.1016/j.ijcard.2010.12.104
Matsumoto, A., Ward, L., Wilce, P., Mason, S., Marsh, S., and Coombes, J. (2007). Effects of exercise on cardioprotection–down-regulation of Ras homolog gene family member A. Asia. Pac J. Clin. Nutr. 16 (3), S100.
Mayr, B., Müller, E. E., Schäfer, C., Droese, S., Schönfelder, M., Niebauer, J., et al. (2021). Exercise-induced changes in miRNA expression in coronary artery disease. Clin. Chem. Lab. Med. 59 (10), 1719–1727. doi:10.1515/cclm-2021-0164
Mediano, M. F., Leifer, E. S., Cooper, L. S., Keteyian, S. J., Kraus, W. E., Mentz, R. J., et al. (2018). Influence of baseline physical activity level on exercise training response and clinical outcomes in heart failure: The HF-action trial. JACC. Heart Fail. 6 (12), 1011–1019. doi:10.1016/j.jchf.2018.09.012
Melling, C. J., Krause, M. P., and Noble, E. G. (2006). PKA-mediated ERK1/2 inactivation and hsp70 gene expression following exercise. J. Mol. Cell. Cardiol. 41 (5), 816–822. doi:10.1016/j.yjmcc.2006.05.010
Melling, C. J., Thorp, D. B., Milne, K. J., Krause, M. P., and Noble, E. G. (2007). Exercise-mediated regulation of Hsp70 expression following aerobic exercise training. Am. J. Physiol. Heart Circ. Physiol. 293 (6), H3692–H3698. doi:10.1152/ajpheart.00827.2007
Melling, C. J., Thorp, D. B., and Noble, E. G. (2004). Regulation of myocardial heat shock protein 70 gene expression following exercise. J. Mol. Cell. Cardiol. 37 (4), 847–855. doi:10.1016/j.yjmcc.2004.05.021
Meng, D., Li, P., Huang, X., Jiang, M-H., and Cao, X-B. (2017). Protective effects of short-term and long-term exercise preconditioning on myocardial injury in rats. Zhongguo ying yong sheng li xue za zhi= Zhongguo yingyong shenglixue zazhi= Chin. J. Appl. physiology. 33 (6), 531–534. doi:10.12047/j.cjap.5601.2017.126
Miura, T., Liu, Y., Kita, H., Ogawa, T., and Shimamoto, K. (2000). Roles of mitochondrial ATP-sensitive K channels and PKC in anti-infarct tolerance afforded by adenosine A1receptor activation. J. Am. Coll. Cardiol. 35 (1), 238–245. doi:10.1016/s0735-1097(99)00493-3
Moc, C., Taylor, A. E., Chesini, G. P., Zambrano, C. M., Barlow, M. S., Zhang, X., et al. (2015). Physiological activation of Akt by PHLPP1 deletion protects against pathological hypertrophy. Cardiovasc. Res. 105 (2), 160–170. doi:10.1093/cvr/cvu243
Muthuchamy, M., Chakraborty, S., Wu, X., Sarin, V., and Heaps, C. L. (2012). Exercise-induced changes in mechanotransduction improve myocardial function in a porcine model of chronic occlusion. Greenville: Am Heart Assoc.
Nakamura, A., Yoshida, K., Takeda, S. I., Dohi, N., and Ikeda, S-I. (2002). Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 520 (1-3), 18–24. doi:10.1016/s0014-5793(02)02739-4
Naskar, S., Datta, K., Mitra, A., Pathak, K., Datta, R., Bansal, T., et al. (2014). Differential and conditional activation of PKC-isoforms dictates cardiac adaptation during physiological to pathological hypertrophy. PloS One 9 (8), e104711. doi:10.1371/journal.pone.0104711
Norum, H. M., Gullestad, L., Abraityte, A., Broch, K., Aakhus, S., Aukrust, P., et al. (2016). Increased serum levels of the notch ligand DLL1 are associated with diastolic dysfunction, reduced exercise capacity, and adverse outcome in chronic heart failure. J. Card. Fail. 22 (3), 218–223. doi:10.1016/j.cardfail.2015.07.012
Oliveira, R., Ferreira, J., Gomes, E., Paixao, N., Rolim, N., Medeiros, A., et al. (2009). Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J. Physiol. 587 (15), 3899–3910. doi:10.1113/jphysiol.2009.173948
Oshima, Y., Ouchi, N., Sato, K., Izumiya, Y., Pimentel, D. R., Walsh, K., et al. (2008). Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation 117 (24), 3099–3108. doi:10.1161/CIRCULATIONAHA.108.767673
Otaka, N., Shibata, R., Ohashi, K., Uemura, Y., Kambara, T., Enomoto, T., et al. (2018). Myonectin is an exercise-induced myokine that protects the heart from ischemia-reperfusion injury. Circ. Res. 123 (12), 1326–1338. doi:10.1161/CIRCRESAHA.118.313777
Oudit, G. Y., and Penninger, J. M. (2009). Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 82 (2), 250–260. doi:10.1093/cvr/cvp014
Pahlavani, H. A., and Veisi, A. (2018). The effect of aerobic and anaerobic training with melatonin consumption on the expression of apoptotic genes BAX and BCL2 myocardial in rats after ischemic reperfusion.
Penna, C., Alloatti, G., and Crisafulli, A. (2020). Mechanisms involved in cardioprotection induced by physical exercise. Antioxid. Redox Signal. 32 (15), 1115–1134. doi:10.1089/ars.2019.8009
Penna, C., Perrelli, M-G., Raimondo, S., Tullio, F., Merlino, A., Moro, F., et al. (2009). Postconditioning induces an anti-apoptotic effect and preserves mitochondrial integrity in isolated rat hearts. Biochim. Biophys. Acta 1787 (7), 794–801. doi:10.1016/j.bbabio.2009.03.013
Pieri, B., Souza, D., Luciano, T., Marques, S., Pauli, J., Silva, A., et al. (2014). Effects of physical exercise on the P38MAPK/REDD1/14-3-3 pathways in the myocardium of diet-induced obesity rats. Hormone Metabolic Res. 46 (09), 621–627. doi:10.1055/s-0034-1371824
Pons, S., Martin, V., Portal, L., Zini, R., Morin, D., Berdeaux, A., et al. (2013). Regular treadmill exercise restores cardioprotective signaling pathways in obese mice independently from improvement in associated co-morbidities. J. Mol. Cell. Cardiol. 54, 82–89. doi:10.1016/j.yjmcc.2012.11.010
Powers, S. K., and Demirel, H. (2001). Exercise, heat shock proteins, and myocardial protection from IR injury. Med. Sci. Sports Exerc. 33 (3), 386–392. doi:10.1097/00005768-200103000-00009
Qin, F., Dong, Y., Wang, S., Xu, M., Wang, Z., Qu, C., et al. (2020). Maximum oxygen consumption and quantification of exercise intensity in untrained male Wistar rats. Sci. Rep. 10 (1), 11520. doi:10.1038/s41598-020-68455-8
Quindry, J. C., Hamilton, K. L., French, J. P., Lee, Y., Murlasits, Z., Tumer, N., et al. (2007). Exercise-induced HSP-72 elevation and cardioprotection against infarct and apoptosis. J. Appl. Physiol. 103 (3), 1056–1062. doi:10.1152/japplphysiol.00263.2007
Ragot, H., Guinemer, S., Delacroix, C., Da Costa, I., Polidano, E., Merval, R., et al. (2016). Notch3 modulates the cardiovascular adaptation to output-overload. Circulation 134 (1), A19794–A.
Renard, S., Paulin, R., Breuils-Bonnet, S., Simard, S., Pibarot, P., Bonnet, S., et al. (2013). Pim-1: A new biomarker in pulmonary arterial hypertension. Pulm. Circ. 3 (1), 74–81. doi:10.4103/2045-8932.109917
Rinaldi, B., Donniacuo, M., Sodano, L., Gritti, G., Signoriello, S., Parretta, E., et al. (2013). Effects of sildenafil on the gastrocnemius and cardiac muscles of rats in a model of prolonged moderate exercise training. PLoS One 8 (7), e69954. doi:10.1371/journal.pone.0069954
Rindom, E., and Vissing, K. (2016). Mechanosensitive molecular networks involved in transducing resistance exercise-signals into muscle protein accretion. Front. Physiol. 7, 547. doi:10.3389/fphys.2016.00547
Rivas, D. A., Peng, F., Benard, T., Ramos da Silva, A. S., Fielding, R. A., Margolis, L. M., et al. (2021). miR-19b-3p is associated with a diametric response to resistance exercise in older adults and regulates skeletal muscle anabolism via PTEN inhibition. Am. J. Physiol. Cell Physiol. 321, C977–C991. doi:10.1152/ajpcell.00190.2021
Saiyin, T., Engineer, A., Greco, E. R., Kim, M. Y., Lu, X., Jones, D. L., et al. (2019). Maternal voluntary exercise mitigates oxidative stress and incidence of congenital heart defects in pre-gestational diabetes. J. Cell. Mol. Med. 23 (8), 5553–5565. doi:10.1111/jcmm.14439
Sharma, A. K., Kumar, A., Taneja, G., Nagaich, U., Deep, A., Datusalia, A. K., et al. (2018). Combined and individual strategy of exercise generated preconditioning and low dose copper nanoparticles serve as superlative approach to ameliorate ISO-induced myocardial infarction in rats. Pharmacol. Rep. 70 (4), 789–795. doi:10.1016/j.pharep.2018.02.023
Shen, Y-J., Pan, S-S., Zhuang, T., and Wang, F-J. (2011). Exercise preconditioning initiates late cardioprotection against isoproterenol-induced myocardial injury in rats independent of protein kinase C. J. Physiol. Sci. 61 (1), 13–21. doi:10.1007/s12576-010-0116-9
Shi, J., Bei, Y., Kong, X., Liu, X., Lei, Z., Xu, T., et al. (2017). miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics 7 (3), 664–676. doi:10.7150/thno.15162
Sun, X., and Mao, J. (2018). Role of Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway in cardioprotection of exercise preconditioning. Eur. Rev. Med. Pharmacol. Sci. 22 (15), 4975–4986. doi:10.26355/eurrev_201808_15638
Sun, X-J., Feng, W-L., and Hou, N. (2017). The role of JAK2/STAT3 signaling pathway in exercise preconditioning against myocardial apoptosis. Zhongguo Ying Yong Sheng li xue za zhi= Zhongguo Yingyong Shenglixue Zazhi= Chin. J. Appl. Physiology. 33 (5), 393–397. doi:10.12047/j.cjap.5473.2017.095
Tao, L., Bei, Y., Zhang, H., Xiao, J., and Li, X. (2015). Exercise for the heart: Signaling pathways. Oncotarget 6 (25), 20773–20784. doi:10.18632/oncotarget.4770
Teringova, E., and Tousek, P. (2017). Apoptosis in ischemic heart disease. J. Transl. Med. 15 (1), 87. doi:10.1186/s12967-017-1191-y
Trenerry, M. K., Carey, K. A., Ward, A. C., and Cameron-Smith, D. (2007). STAT3 signaling is activated in human skeletal muscle following acute resistance exercise. J. Appl. Physiol. 102 (4), 1483–1489. doi:10.1152/japplphysiol.01147.2006
Trenerry, M. K., Carey, K. A., Ward, A. C., Farnfield, M. M., and Cameron-Smith, D. (2008). Exercise-induced activation of STAT3 signaling is increased with age. Rejuvenation Res. 11 (4), 717–724. doi:10.1089/rej.2007.0643
Vaillancourt, M., Ruffenach, G., Meloche, J., and Bonnet, S. (2015). Adaptation and remodelling of the pulmonary circulation in pulmonary hypertension. Can. J. Cardiol. 31 (4), 407–415. doi:10.1016/j.cjca.2014.10.023
Von Harsdorf, R. D., Li, P-F., and Dietz, R. (1999). Signaling pathways in reactive oxygen species–induced cardiomyocyte apoptosis. Circulation 99 (22), 2934–2941. doi:10.1161/01.cir.99.22.2934
Wang, D., Wang, Y., Ma, J., Wang, W., Sun, B., Zheng, T., et al. (2017). MicroRNA-20a participates in the aerobic exercise-based prevention of coronary artery disease by targeting PTEN. Biomed. Pharmacother. 95, 756–763. doi:10.1016/j.biopha.2017.08.086
Wang, H., Xie, Y., Guan, L., Elkin, K., and Xiao, J. (2021). Targets identified from exercised heart: Killing multiple birds with one stone. npj Regen. Med. 6 (1), 23. doi:10.1038/s41536-021-00128-0
Wang, Y., Liu, J., Kong, Q., Cheng, H., Tu, F., Yu, P., et al. (2019). Cardiomyocyte-specific deficiency of HSPB1 worsens cardiac dysfunction by activating NFκB-mediated leucocyte recruitment after myocardial infarction. Cardiovasc. Res. 115 (1), 154–167. doi:10.1093/cvr/cvy163
K. Watson, and K. Baar (Editors) (2014). “mTOR and the health benefits of exercise,” Seminars in cell & developmental biology (Elsevier).
Webb, A. E., and Brunet, A. (2014). FOXO transcription factors: Key regulators of cellular quality control. Trends biochem. Sci. 39 (4), 159–169. doi:10.1016/j.tibs.2014.02.003
Weeks, K. L., Bernardo, B. C., Ooi, J. Y., Patterson, N. L., and McMullen, J. R. (2017). The IGF1-PI3K-Akt signaling pathway in mediating exercise-induced cardiac hypertrophy and protection. Exerc. Cardiovasc. Dis. Prev. Treat. 1, 187–210.
Wen, D-T., Zheng, L., Li, J-X., Lu, K., and Hou, W-Q. (2019). The activation of cardiac dSir2-related pathways mediates physical exercise resistance to heart aging in old Drosophila. Aging (Albany NY) 11 (17), 7274–7293. doi:10.18632/aging.102261
Wilkinson, S. B., Phillips, S. M., Atherton, P. J., Patel, R., Yarasheski, K. E., Tarnopolsky, M. A., et al. (2008). Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J. Physiol. 586 (15), 3701–3717. doi:10.1113/jphysiol.2008.153916
Wu, J., Chen, S., Liu, Y., Liu, Z., Wang, D., Cheng, Y., et al. (2020). Therapeutic perspectives of heat shock proteins and their protein-protein interactions in myocardial infarction. Pharmacol. Res. 160, 105162. doi:10.1016/j.phrs.2020.105162
Xi, Y., Gong, D-W., and Tian, Z. (2016). FSTL1 as a potential mediator of exercise-induced cardioprotection in post-myocardial infarction rats. Sci. Rep. 6 (1), 32424. doi:10.1038/srep32424
Xia, P., Liu, Y., and Cheng, Z. (2016). Signaling pathways in cardiac myocyte apoptosis. Biomed. Res. Int. 2016, 9583268. doi:10.1155/2016/9583268
Xiao, J., Shi, J., Bei, Y., Xu, T., Wang, H., Chen, P., et al. (2016). TCTAP A-038 miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. J. Am. Coll. Cardiol. 67 (16), S18. doi:10.1016/j.jacc.2016.03.052
Xing, Y., Sun, W., Wang, Y., Gao, F., and Ma, H. (2016). Mutual inhibition of insulin signaling and PHLPP-1 determines cardioprotective efficiency of Akt in aged heart. Aging (Albany NY) 8 (5), 873–888. doi:10.18632/aging.100933
Xu, J., Tang, Y., Bei, Y., Ding, S., Che, L., Yao, J., et al. (2016). miR-19b attenuates H2O2-induced apoptosis in rat H9C2 cardiomyocytes via targeting PTEN. Oncotarget 7 (10), 10870–10878. doi:10.18632/oncotarget.7678
Yan, L., and Li, Z. (2012). The effect of exercise on the Rho GTPases in rat myocardium. Beijing: Journal of Beijing Sport University. 03.
Yin, R., You, H., Wu, Y., Ye, F., Gu, W., Shen, J., et al. (2019). Knocking down PFL can improve myocardial ischemia/reperfusion injury in rats by up-regulating heat shock protein-20. Eur. Rev. Med. Pharmacol. Sci. 23 (17), 7619–7627. doi:10.26355/eurrev_201909_18885
Zhang, L., Wang, X., Zhang, H., Feng, M., Ding, J., Zhang, B., et al. (2021). Exercise-induced peptide EIP-22 protect myocardial from ischaemia/reperfusion injury via activating JAK2/STAT3 signalling pathway. J. Cell. Mol. Med. 25 (7), 3560–3572. doi:10.1111/jcmm.16441
Zhang, P. (2017). CaMKII: The molecular villain that aggravates cardiovascular disease. Exp. Ther. Med. 13 (3), 815–820. doi:10.3892/etm.2017.4034
Zhang, Q. J., McMillin, S. L., Tanner, J. M., Palionyte, M., Abel, E. D., Symons, J. D., et al. (2009). Endothelial nitric oxide synthase phosphorylation in treadmill-running mice: Role of vascular signalling kinases. J. Physiol. 587 (15), 3911–3920. doi:10.1113/jphysiol.2009.172916
Zhao, D., Sun, Y., Tan, Y., Zhang, Z., Hou, Z., Gao, C., et al. (2018). Short-duration swimming exercise after myocardial infarction attenuates cardiac dysfunction and regulates mitochondrial quality control in aged mice. Oxidative Med. Cell. Longev. 2018, 4079041. doi:10.1155/2018/4079041
Zhou, H-Z., Karliner, J. S., and Gray, M. O. (2002). Moderate alcohol consumption induces sustained cardiac protection by activating PKC-ε and Akt. Am. J. Physiol. Heart Circ. Physiol. 283 (1), H165–H174. doi:10.1152/ajpheart.00408.2001
Zhou, Q., and Xiao, J. (2019). P5395 exercise-induced miR-17-3p prevents pathological myocardial dysfunction. Eur. Heart J. 40 (1), ehz746–0355. doi:10.1093/eurheartj/ehz746.0355
Zhou, T., Chuang, C-C., and Zuo, L. (2015). Molecular characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. Biomed. Res. Int. 2015, 864946. doi:10.1155/2015/864946
AC adenylyl cyclase
Apaf-1 apoptosis protease activating factor 1
bFGF Basic fibroblast growth factor
C/EBPβ CCAAT/enhancer-binding protein β
CaM calcium-calmodulin
cAMP cyclic AMP
Cdc42 cell division control protein 42
CHDs congenital heart defects
CRP C-reactive protein
CVDs Cardiovascular diseases
DLL-1 Delta-like-1
ECM extracellular matrix
EGF epidermal growth factor
ERK-1/2 extracellular signal-regulated kinase 1/2
FADD Fas-associated death domain
FAK focal adhesion kinase
FasL Fas ligand
FSTL1 Follistatin-like protein 1
GAPs GTPase activators
GEFs guanine exchange factors
GPCRs G protein-coupled receptors
GSK-3 glycogen synthase kinase 3
H2O2 hydrogen peroxide
HIF-α hypoxia-inducible factor-1α
HSF1 heat shock transcription factor
HSPs Heat shock proteins
IGF-1 Insulin-like growth factor-1
IGFI-R IGF-I receptor
IKK KB kinase inhibitor
IKKB inhibitor of nuclear factor-kappa B kinase subunit beta
JAK2 Janus kinase 2
JNK Jun N-terminal kinases
LGF liver growth factor
LTCC L-type calcium channel
MAPKs Mitogen-activated protein kinases
MI myocardial infarction
MnSOD superoxide dismutase
mPTP mitochondrial permeability transition pore
MST1 macrophage stimulating protein 1
NFAT nuclear factor of activated T cells
Nrf2/Mts nuclear factor erythroid 2-related factor/metallothioneins
p70S6K p70S6 kinase 1
PAH pulmonary arterial hypertension
PDGF platelet-derived growth factor
PHLPP PH domain leucine-rich repeat protein phosphatase
Pim-1 serine/threonine-protein kinase-1
PKA Cyclic AMP-dependent protein kinase
PKB/Akt protein kinase B
PKC protein kinase C
PLB phospholamban
PP2C protein phosphatase 2C
PTEN phosphatase and tensin homolog
RhoA Ras homolog gene family-A
ROCK Rho-associated protein kinase
ROS reactive oxygen species
RyR ryanodine receptor
SAPKs stress-activated protein kinases
SERCA2 sarcoplasmic reticulum Ca2+-ATPase
SIRT1 Sirtuin 1
STAT1 signal transducer and activator of transcription 1
STAT3 signal transducer and activator of transcription 3
TIMP3 tissue inhibitor of metalloproteinase 3
TNFR1 TNF-α receptor 1
TNF-α tumor necrosis factor-α
TnI troponin I
TRAIL TNF-related apoptosis-inducing ligand
TRAILR1/2 TRAIL receptor 1/2
Keywords: exercise, apoptotic, cardiovascular diseases, cardiomyocytes, signaling, signaling pathways
Citation: Pahlavani HA (2022) Exercise-induced signaling pathways to counteracting cardiac apoptotic processes. Front. Cell Dev. Biol. 10:950927. doi: 10.3389/fcell.2022.950927
Received: 23 May 2022; Accepted: 15 July 2022;
Published: 11 August 2022.
Edited by:
Ashley Smuder, University of Florida, United StatesReviewed by:
Daniel Peña-Oyarzun, Pontifical Catholic University of Chile, ChileCopyright © 2022 Pahlavani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hamed Alizadeh Pahlavani, aGEuYWxpemFkZWhAY2Z1LmFjLmly
†ORCID: Hamed Alizadeh Pahlavani, orcid.org/0000-0002-2313-9684
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.