 Stefano Fumagalli
Stefano Fumagalli Mario Pende
Mario Pende
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 12 August 2022
Sec. Cell Growth and Division
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.949196
This article is part of the Research TopicCell Size Regulation: Molecular Mechanisms and Physiological ImportanceView all 10 articles
Genetic evidence in living organisms from yeast to plants and animals, including humans, unquestionably identifies the Target Of Rapamycin kinase (TOR or mTOR for mammalian/mechanistic) signal transduction pathway as a master regulator of growth through the control of cell size and cell number. Among the mTOR targets, the activation of p70 S6 kinase 1 (S6K1) is exquisitely sensitive to nutrient availability and rapamycin inhibition. Of note, in vivo analysis of mutant flies and mice reveals that S6K1 predominantly regulates cell size versus cell proliferation. Here we review the putative mechanisms of S6K1 action on cell size by considering the main functional categories of S6K1 targets: substrates involved in nucleic acid and protein synthesis, fat mass accumulation, retrograde control of insulin action, senescence program and cytoskeleton organization. We discuss how S6K1 may be involved in the observed interconnection between cell size, regenerative and ageing responses.
At the turn of the second and third millennium, genetic screens for tissue overgrowth in Drosophila flies identify loss-of-function (LOF) mutants falling in two broad categories: hyperplastic with increased cell size, such as Gigas-Tuberous Sclerosis Complex 2 (TSC2), Tuberous Sclerosis Complex 1 (TSC1) and Phosphatase And Tensin Homolog (PTEN) (Huang et al., 1999; Ito and Rubin, 1999; Stocker and Hafen, 2000); and hyperplastic with constant cell size, such as Hippo, Salvador, Merlin, Warts (Pantalacci et al., 2003; Udan et al., 2003; Wu et al., 2003). At the same time, biochemical studies demonstrated that the gene products belong to two signal transduction pathways integrating environmental cues and regulating tissue growth: the mTOR and the Hippo kinase pathways (Ma et al., 2018; Liu and Sabatini, 2020). mTOR mainly senses nutrient availability, whereas the Hippo pathway mainly senses the physical environment. While TSC1, TSC2 and PTEN are negative regulators of the mTOR pathway, LOF mutants in positive regulators, such as Insulin Receptor Substrate (IRS), Phosphoinositide-3-Kinase (PI3K), Akt and mTOR itself, reduce both cell size and number (Leevers et al., 1996; Bohni et al., 1999; Verdu et al., 1999; Zhang et al., 2000; Gao et al., 2002; Zhang et al., 2003). Of note, adult flies lacking the S6K gene have the same cell number as wild type flies but their cell size is reduced, as measured in the eye and wing tissues (Montagne et al., 1999). The differential impact of growth mutants on cell size and cell number points to a loose relationship between these parameters. While the Drosophila fly genome contains a single S6K gene, two orthologous RPS6KB1 and RPS6KB2 genes exist in mammals, encoding the S6K1 and S6K2 proteins, respectively (Banerjee et al., 1990; Kozma et al., 1990; Gout et al., 1998; Shima et al., 1998; Lee-Fruman et al., 1999). Here, we will review the studies detailing the effectors of S6 kinases and the physiological alterations concomitant with the cell size control by this molecular program.
S6K and Akt belong to the AGC family of serine and threonine protein kinases (Pearce et al., 2010). They are activated by phosphorylation on their hydrophobic motifs and T-loop motifs by mTOR and 3-Phosphoinositide Dependent Protein Kinase (PDK1), respectively (Alessi et al., 1997; Alessi et al., 1998; Pullen et al., 1998; Hara et al., 2002; Kim et al., 2002; Sarbassov et al., 2005). However, mTOR resides in different protein complexes when it catalyzes S6K or Akt phosphorylation. While S6K is phosphorylated by mTOR complex 1 (mTORC1), Akt is phosphorylated by mTOR complex 2 (mTORC2). mTORC1 and mTORC2 differ in their sensitivity to the two main anabolic signals from nutrition: small nutrients molecules, such as glucose, amino acids and lipids which enter in circulation after food digestion, and growth factor peptides, such as insulin, that are secreted after food intake and coordinate nutrient usage in peripheral organs. mTORC1 activity requires signals from both nutrients and growth factors, while mTORC2 activity is relatively insensitive to nutrient levels (Liu and Sabatini, 2020). In addition, mTORC2 promotes mTORC1 activity, while being negatively regulated by mTORC1 in a retrocontrol mechanism. This cross-talk and differential sensitivity of mTORC1 and mTORC2 to the anabolic signals explain many physiological adaptations to nutrition. For instance, protein synthesis and cell growth are mainly upregulated by mTORC1 when both insulin and nutrients are present (Pham et al., 2000). Conversely, the metabolic action of insulin on nutrient uptake and gluconeogenesis is mainly regulated by mTORC2, with an excess of nutrients causing insulin resistance through the negative feedback loop of mTORC1 on mTORC2 (Fingar et al., 1993; Tremblay and Marette, 2001).
The convergence of nutrients and insulin signals for mTORC1 activation takes place at the lysosomal membrane, through the regulation of two small GTPase family members Ras Related GTP Binding A/B/C/D (RagA/B/C/D) and Ras Homolog Enriched In Brain (Rheb) that are important for mTORC1 localization and activity, respectively (Liu and Sabatini, 2020). It is becoming increasingly clear that the phosphorylation of protein substrates by mTORC1 is differentially sensitive to upstream regulators. Since the relative activity of mTORC1 towards S6K1 is low, this phosphorylation event is very sensitive to rapamycin treatment and nutritional perturbations that change mTORC1 activity through Rheb and Rag GTP loading (Chung et al., 1992; Kang et al., 2013). Conversely, the phosphorylation of other mTORC1 substrates, such as 4EBPs, is more constitutive and less sensitive to nutritional fluctuations as well as pharmacological inhibition by rapamycin (Choo et al., 2008). Moreover, phosphorylation of certain substrates, such as TFEB, is insensitive to the Rheb-dependent regulation of mTORC1 and exquisitely depends on the regulation at the level of RagC/D (Napolitano et al., 2020). It appears therefore that mTORC1 is capable to integrate a large variety of information from the environment and adapt the output towards distinct classes of substrates depending on the conditions.
S6 kinases are also extensively phosphorylated in the proline rich C-terminus region, a pseudosubstrate domain possibly exerting autoinhibitory effects on kinase activity (Mukhopadhyay et al., 1992; Dennis et al., 1998). The phosphorylation of the C-terminus region has been proposed to relieve the autoinhibitory regulation and expose the kinase for phosphorylation on the hydrophobic motifs and T-loop motifs required for activation and substrate docking. Several proline directed kinases can phosphorylate the C-terminus region of S6K1 in vitro, including Cyclin dependent kinases (Cdk) Cdk5 and Cdk1, Mitogen Activated protein kinases (MAPK) and mTOR itself (Mukhopadhyay et al., 1992; Arif et al., 2019). It is still unclear whether these kinases can compensate each other or whether they directly phosphorylate specific sites in vivo. This model of S6K1 regulation is consistent with the genetic epistasis experiments and pharmacological treatments by rapamycin derivatives, indicating that mTOR-mediated phosphorylation on the hydrophobic motif is absolutely required for S6K1 action on downstream substrates. However, a recent study proposes that Cdk5-mediated S6K1 phosphorylation on the C-terminus region may trigger a conformational switch that directs S6K1 kinase activity towards a different class of substrates (Arif et al., 2019). This appealing and original control still requires further work, namely the in vivo analysis of Cdk5 knockout mice and the generation of phospho-specific antibodies against the Cdk5-directed substrates. Of note, additional Cdk, such as Cdk4 and Cdk6, impinge on mTOR/S6K activity by affecting upstream regulators, such as the lysosomal complexes and TSC, rather than directly on kinase phosphorylation (Martinez-Carreres et al., 2019; Romero-Pozuelo et al., 2020).
Given the peculiar sensitivity of S6K1 to mTORC1 activity, it is not surprising that S6K1 deficient mice mimic a caloric restriction phenotype. They have low insulin levels in their blood, are hypersensitive to action of insulin on glucose tolerance, do not become obese after high fat diet and live longer (Pende et al., 2000; Um et al., 2004; Selman et al., 2009). Of note, all these physiological adaptations are accompanied by a reduction of cell size. The cell volume shrinkage is approximately 15%–35% depending on the cell type. This is less dramatic than the profound cell atrophy due to mTOR deletion, consistent with the idea that S6K1 deletion does not induce autophagy. Similarly, S6K deletion in Drosophila flies rescues the overgrowth phenotype of the Gigas-TSC2 mutation to the level of wild type tissues (Gao et al., 2002). The effect of S6K on cell size can be recapitulated in vitro after genome editing in cultured cells, pointing to a cell autonomous regulation of cell size by S6K1 (Bonucci et al., 2020). The decreased cell size is observed throughout different phases of cell cycle as well as in post-mitotic cells (Ohanna et al., 2005; Bonucci et al., 2020). S6K1 deletion mimics the effect of rapamycin on cell size but not on cell proliferation (Ohanna et al., 2005; Dowling et al., 2010). Conversely, 4EBPs deletion causes resistance to the anti-proliferative action of mTOR inhibitors without affecting cell size (Dowling et al., 2010).
Cell size fluctuations have been recently proposed to directly affect senescence or stem cell potential (Neurohr et al., 2019; Lengefeld et al., 2021). We will now discuss how the S6K dependent molecular program may contribute to these coordinated responses: protein and lipid mass accumulation, insulin resistance, cytoskeletal dynamics and senescence. The elucidation of the S6K substrates is rapidly progressing thanks to the deep coverage of the phosphoproteomics analysis after rapamycin treatment, mTORC1 or S6K1 genetic deletion (Moritz et al., 2010; Hsu et al., 2011; Yu et al., 2011; Robitaille et al., 2013; Bonucci et al., 2020). A comprehensive and updated list of S6K substrates can be found at the PhosphoSitePlus online tool: https://www.phosphosite.org/substrateSearchViewAction.action?id=1012&type=Protein: the phosphorylation sites and consensus sequences are also adequately indicated there and are therefore omitted in this review. Figure 1 is a graphic representation of the S6K1 signaling network.
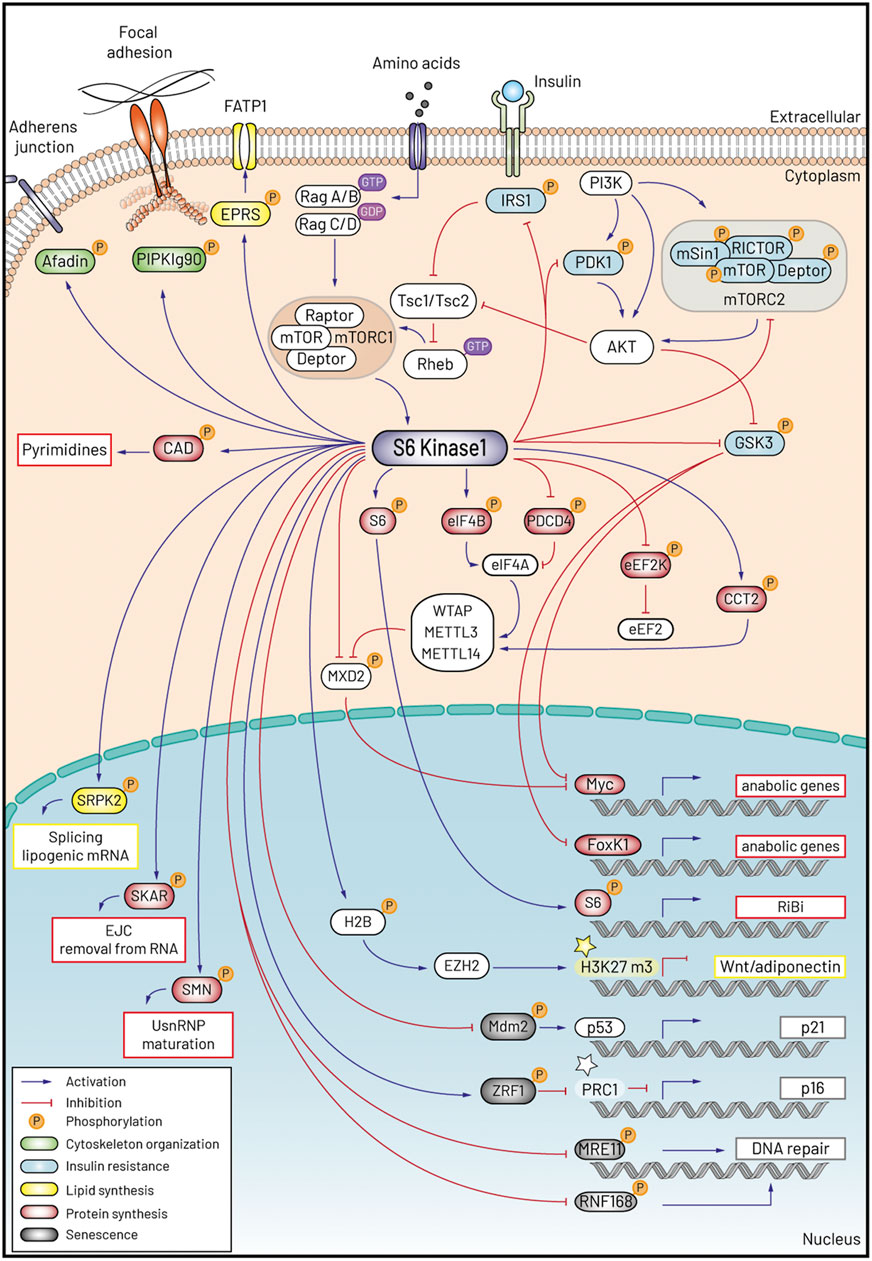
FIGURE 1. Schematic representation of S6K1 signaling and the five programs accounting for the control of cell size and ageing.
Substrates belonging to the protein synthesis machinery represent the most important class of S6K targets for both historic reasons and their high number. S6 kinases were initially isolated due to their enzymatic activity phosphorylating the ribosomes. Phosphorylation of the 40S ribosomal protein S6 (eS6 according to the novel nomenclature (Ban et al., 2014)) occurs in mammals and Xenopus at the C-terminus of the protein (Krieg et al., 1988; Wettenhall et al., 1992). In vivo, S6K1 and S6K2 are the main kinases responsible for eS6 phosphorylation, although residual eS6 phosphorylation by compensating p90 Ribosomal Protein S6 Kinase RPS6KA1-3 (RSK 1-3) downstream of Mitogen Activated protein kinases (MAPK, ERK1 and ERK2) can occur in an S6K null background (Pende et al., 2004). The evidence of eS6 phosphorylation playing a stimulatory role on protein synthesis is correlative and its function in mRNA translation remains elusive. Some insights on a physiological function of eS6 phosphorylation have arisen from the analysis of the S6−/−P knock-in mice where all the five phosphorylatable serines of eS6 are mutated to alanines (Ruvinsky et al., 2005). Although the phenotype of S6−/−P knock-in mice is milder than S6K1−/− mice, they also display small cell size of pancreatic beta cells and muscle cells, suggesting that eS6 phosphorylation participates in cell size control (Pende et al., 2000; Ohanna et al., 2005; Ruvinsky et al., 2005; Ruvinsky et al., 2009). If the phenotype of S6−/−P mice would be consistent with a stimulatory role of eS6 phosphorylation on protein synthesis, it is puzzling that S6−/−P cells from different tissues display higher protein synthesis rates than their wild type (WT) controls, arguing that eS6 phosphorylation has an inhibitory function on mRNA translation (Ruvinsky et al., 2005; Hsieh et al., 2010). Consistently, S6K1 deficient mice do not display a reduction in global protein synthesis (Mieulet et al., 2007). The mechanistic aspects of these effects have not been thoroughly investigated and it cannot be excluded that they arise from the activation of compensatory mechanisms. Some clues come from a recent study that by comparing the translation efficiency of WT and S6−/−P cells provides evidence that eS6 phosphorylation has a mild stimulatory effect on the translation of short mRNAs (Bohlen et al., 2021). Among those, however, an exception is that of 5TOP mRNAs that mainly encode ribosomal proteins and are translated more efficiently in S6−/−P than WT cells. In agreement with this observation, ribosomal protein levels, a proxy of ribosome content, are higher in S6−/−P as compared to WT cell (Bohlen et al., 2021), which is consistent with the difference in the rates of protein synthesis (Ruvinsky et al., 2005). An in vivo study employing S6P−/- mice indicates that in liver of animals fed after fasting eS6 phosphorylation mediates in part the stimulatory effects of S6K on the transcription of genes encoding RiBi (ribosome biogenesis) factors, implicated in the assembly and maturation of ribosomal subunits, contributing therefore to the increase in ribosome synthesis that ensues in response to feeding (Chauvin et al., 2014). It is likely that these effects are due to an extra-ribosomal function of eS6, although it cannot be excluded that phosphorylated ribosomes regulate the synthesis of factors that impact on Ribi mRNA levels.
The major hub of protein synthesis control by the mTORC1 pathway is the eIF4F (eukaryotic initiation factor 4F) complex that mediates during the initiation phase the recruitment to the mRNA of the 43S pre-initiation complex, i.e., the 40S ribosomal subunit bound to the ternary complex (eIF2-GTP-Met-tRNA), and the initiation factors eIF1, eIF1A, eIF3, and eIF5 (Merrick and Pavitt, 2018). eIF4F is composed by a scaffold, eIF4G, that interacts with the two other components of the complex, the cap binding protein eIF4E and the RNA helicase eIF4A. During initiation of protein synthesis, eIF4G interacts with the 43S preinitiation complex whereas the eIF4E moiety makes contact with the cap structure found at the 5′ end of mRNAs. This interaction is favoured by eIF4A that unwinds secondary structures proximal to the cap and facilitates therefore binding of the 43S complex to the mRNA. In this process the activity of eIF4A is greatly enhanced by the factor eIF4B (Merrick and Pavitt, 2018). Formation of the eIF4F complex is regulated in an mTORC1 dependent manner by growth factors and nutrients. Thus, under conditions of nutrient scarcity, members of the 4EBP (eIF4E binding protein) family efficiently compete with eIF4G for binding to eIF4E preventing therefore eIF4F complex formation and suppressing protein synthesis. Upon nutrient addition, phosphorylation of 4EBPs at several sites mainly by mTORC1, promotes their dissociation from eIF4E which becomes available for the formation of the eIF4F complex allowing an increase in the rates of protein synthesis (Merrick and Pavitt, 2018). A further layer of regulation on eIF4F formation is provided by S6K. This involves the phosphorylation and subsequent ubiquitin-dependent degradation of programmed cell death 4 (PDCD4), a protein that interferes with eIF4A activity and its binding to eIF4G (Dorrello et al., 2006). Furthermore, phosphorylation of eIF4B by S6K stimulates protein synthesis through a mechanism that may involve recruitment of eIF4B in proximity of eIF4F (Holz et al., 2005; Shahbazian et al., 2006).
Besides translation initiation, elongation is also subjected to regulation by the pathway. An inhibitory effect on protein synthesis occurs through phosphorylation and consequent inactivation of eukaryotic elongation factor 2 (eEF2) that, during elongation, catalyses the translocation of peptidyl-tRNA from the A to the P site of the translating ribosome (Dever and Green, 2012). The kinase responsible for eEF2 phosphorylation is the Ca2+/Calmodulin dependent kinase eEF2 kinase (eEF2K) (Kenney et al., 2014). This response is thought to promote cell survival under conditions of nutrient scarcity by limiting the highly energy-consuming process of protein synthesis (Leprivier et al., 2013; Faller et al., 2015). It has been shown that in response to mitogenic signals S6K phosphorylates and inactivates eEF2K, ensuring therefore high rates of protein synthesis (Wang et al., 2001; Kenney et al., 2014).
S6K might regulate specific mRNA translation through the regulation of their metabolism, including mRNA methylation and splicing. It has been recently shown that in TSC2−/− cells, S6K through the eIF4A-eIF4B axis promotes the translation of the mRNA encoding Wilms tumor 1-associated protein (WTAP), the scaffold component of the m6A methyltransferase complex (MTC). This in turn results in formation of m6A by the MTC components m6A methyltransferase like proteins 3 and 14 (METTL3 and METTL14) on, among others, the mRNA encoding the c-myc suppressor MAX dimerization protein 2 (MXD2), triggering its degradation and leading to activation of c-myc transcriptional activity (Cho et al., 2021). MXD2 can also be directly phosphorylated by S6K1, an event promoting protein ubiquitination and degradation (Huang et al., 2018). A role of eIF4B phosphorylation by S6K in directly promoting translation of c-myc mRNA has also been documented in TSC2−/− cells (Csibi et al., 2014). Taken together, these findings converge on the Myc transcriptional program in mediating some of the growth-promoting actions of S6K.
Moreover, S6K intervenes in the final maturation steps of mRNAs. As introns are removed from pre-RNAs during splicing, a complex forms twenty four nucleotides upstream of exon-exon junctions known as EJC (Exon-Junction complex). mRNAs associate then to the nuclear CAP binding complex (CBP) and are exported from the nucleus into the cytosol. Here mRNAs, still bound to CBP, are inspected by the so-called pioneer round of translation, a quality control step during which EJC are removed unless an upstream premature translation stop codon is present. Successful removal of the EJC commits the mRNA toward productive protein synthesis (Woodward et al., 2017). It has been shown that S6K1 Aly/REF-like target (SKAR), an RNA binding protein phosphorylated by S6K1 (Richardson et al., 2004), is a component of the EJC (Ma et al., 2008). In response to Insulin, S6K1, by interacting with SKAR, is recruited to CBP-bound mRNPs and presumably stimulates the inspection of the associated mRNAs by the pioneer round of translation providing transcripts to support protein synthesis (Ma et al., 2008). It is interesting that knock down of SKAR results in a small cell size phenotype (Richardson et al., 2004). It will be important to analyse the role that SKAR phosphorylation by S6K plays on regulation of cell size as well as on SKAR function as a component of the EJC. Additional splicing factors controlled by S6K will be discussed in the next chapter.
Growth control by mTOR and S6K also relies on protein folding. In Drosophila, the overgrowth phenotype of TOR activation is suppressed by depletion of subunits of chaperonin containing tailless complex polypeptide 1 (CCT), a complex that assists the folding of about 10% of newly synthesized cytosolic proteins (Kim and Choi, 2019). TOR promotes transcription of CCT genes. Of note, CCT subunits are important to maintain the levels of METTL3 and METTL14 in MTC, which contribute to the inhibition of autophagy by catalysing formation of destabilising m6A on mRNAs encoding autophagy effectors (Tang et al., 2021). Some aspects of this regulatory circuit are conserved in mammalian cells. A possible role of S6K in regulating both CCT and MTC functions is supported by the finding that the CCT2 subunit is phosphorylated by S6K (Abe et al., 2009). Interestingly a CCT2 phosphomutant fails to rescue the proliferation defect caused by depletion of CCT2 in human mammary epithelial cells. S6K also phosphorylates ZRF1, the HSP40 component of the ribosome-associated complex (RAC), a universally conserved broad specificity chaperone system that interacts with the 60S ribosomal subunit assist co-translational folding of nascent polypeptides as they emerge from the tunnel of the ribosome (Barilari et al., 2017). Furthermore, studies in yeast suggest that ZRF1 has a role in maintaining protein synthesis fidelity by interacting with the 40S subunit at a region that is involved in decoding (Lee et al., 2016). It will be important to determine the functional relevance of this phosphorylation event on protein synthesis fidelity.
A major cellular function involved in cell growth is the synthesis of precursors employed in macromolecular synthesis. These include the pyrimidine and purine bases that are then employed for the synthesis of nucleosides, the components of DNA and RNA (Zhu and Thompson, 2019). Recent work from the Manning laboratory has shown that de novo synthesis of both pyrimidines and purines is driven by the mTOR pathway (Ben-Sahra et al., 2013; Ben-Sahra et al., 2016). Downstream of mTOR, S6K plays a crucial role on pyrimidines synthesis through the stimulatory phosphorylation of carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase (CAD), the enzyme that catalyses the first three steps of de novo pyrimidine synthesis (Ben-Sahra et al., 2013; Robitaille et al., 2013).
Interestingly, phosphorylation of splicing and translational machineries has been implicated in one of the most striking cellular responses to S6K1 activity, the fat accumulation. In Drosophila, a lipogenic program is sufficient to alter cell size (Porstmann et al., 2008). Although S6K1 has been proposed to directly phosphorylate and regulate the expression of the master regulator of lipid gene expression, the Sterol Regulatory Element-Binding Protein 1 (SREBP1), the experimental evidence is not conclusive (Lewis et al., 2011; Yecies et al., 2011). Two recent studies suggest alternative SREBP1- and transcription-independent mechanisms for the regulation of fat metabolism, involving the Serine-arginine rich protein kinase 2 (SRPK2) and the glutamyl prolyl tRNA synthetase EPRS. SRPK2 regulates serine arginine rich proteins (SR), whose function is to catalyze mRNA splicing by binding exons and recruiting small nuclear ribonucleoproteins (snRNPs) (Lee et al., 2017). S6K1 dependent phosphorylation of SRPK2 in cooperation with an additional Casein Kinase 1 (CK1)-dependent site upregulates nuclear localization of the protein. Nuclear SRPK2 promotes efficient intron splicing of acly, acss2, hmgcs1, mvd, fdft1, scd1, fasn mRNA, thus increasing the stability and the levels of these mRNA coding for proteins involved in lipid synthesis. Conversely, the S6K1-dependent phosphorylation of EPRS acts as a switch for the function of the protein, that becomes a partner of Long-Chain Fatty Acid Transport Protein (FATP1), promoting long chain FA uptake and TG synthesis (Arif et al., 2017). Similar to S6K1 deficient mice, EPRS phosphomutant mice also have reduced fat mass and increased longevity, indicating a participation of the post-translational modification in these phenotypes (Arif et al., 2017). Moreover, when S6K1 receives activating input by Cdk5, additional proteins involved in lipid synthesis can be phosphorylated, including cortactin, lipocalin 2 and coA synthase (Arif et al., 2019). Another original mechanism for the control of lipid synthesis is through epigenetic modifications triggered by S6K1-dependent Histone 2B (H2B) phosphorylation (Yi et al., 2016). In adipocytes, S6K1 can translocate to the nucleus upon treatment with the adipogenic factor Bone Morphogenetic Protein 4 (BMP4). The H2B phosphorylation recruits the Histone-Lysine N-Methyltransferase EZH2 and favours H3K27 trimethylation (H3K27m3), thus suppressing Wnt ligand expression which acts as a brake for adipogenic commitment. The expression of fat-regulating hormones in adipocytes and pancreas, such as adiponectin, insulin and glucagon, may also undergo a similar epigenetic regulation (Yi et al., 2018; Yi et al., 2022).
S6K1 is involved at multiple levels in the above-mentioned negative feedback mechanism by which an excess of nutrients through the mTORC1/S6K1 axis dowregulates insulin signaling and mTORC2 activity. First, S6K1 acts at the level of IRS proteins which initiate insulin signaling. IRS phosphorylation on Ser and Thr residues by S6K1 and additional kinases contributes to shut down the signal by promoting protein degradation or decreasing the interaction with the receptors (Harrington et al., 2004; Tremblay et al., 2007). Consistently, the increase in insulin sensitivity of S6K1-deficient mice correlates with a decrease in IRS1 Ser/Thr phosphorylation (Um et al., 2004). Second, S6K1 phosphorylates multiple proteins in mTORC1 and mTORC2. S6Ks phosphorylate mTOR itself, however the functional relevance of these phosphorylation sites is unclear (Holz and Blenis, 2005). S6Ks also phosphorylate two components of the mTOR complex 2 (mTORC2), namely rapamycin-insensitive companion of mTOR (RICTOR) and stress-activated MAPK-interacting protein 1 (SIN1) (Dibble et al., 2009; Julien et al., 2010; Treins et al., 2010; Liu et al., 2013), and the Deptor protein present in both mTORC1 and mTORC2 (Gao et al., 2011; Zhao et al., 2011). Although there are some controversies on the function of each phosphorylation event, there is a general consensus that S6Ks act by inhibiting the mTORC2 substrate Akt. Phosphomimetic SIN1 mutants are unable to interact with RICTOR and mTOR, while phospho-ablative mutants increase Akt phosphorylation (Liu et al., 2013). Similarly, two studies have shown a mild inhibitory effect of RICTOR phosphorylation on AKT activity (Dibble et al., 2009; Julien et al., 2010). The reduced half life of Deptor upon its phosphorylation may also concur to decreased Akt activation (Gao et al., 2011; Zhao et al., 2011).
Another level by which S6K1 may suppress Akt activity is through the phosphorylation of PDK1 on the pleckstrin homology domain that impairs the interaction between PDK1 and Akt (Jiang et al., 2022). Interestingly, if S6K1 has clearly a negative role on the metabolic action of insulin and Akt, it can compensate the Akt action on some substrates that are important growth regulators, such as Glycogen Synthase Kinase 3 (GSK3). S6K1 can phosphorylate GSK3 in conditions of mTORC1 overactivation (Zhang et al., 2006), and thus relieve the GSK3-dependent inhibition of two transcription factors promoting growth, c-Myc and Forkhead Box Protein K1 (FoxK1) (He et al., 2018).
Rapamycin treatment has a striking efficacy in delaying the senescence program in cultured cells and preserving their proliferative potential (Leontieva and Blagosklonny, 2013, 2014; Leontieva et al., 2013, 2014; Neurohr et al., 2019; Lengefeld et al., 2021). Primary cells undergoing senescent insults of DNA damage, replication stress, oncogenic mutations and constitutive mTOR activation are protected by rapamycin. Pharmacological inhibition of S6K1 or genetic knock-down/knock-out also rescues the senescent program (Leontieva et al., 2013; Barilari et al., 2017). In TSC1 deficient fibroblasts, S6K1 regulates the expression of the cell cycle inhibitor p16 (Barilari et al., 2017). This is accompanied by the S6K1-dependent phosphorylation of Zrf1, a protein containing a SANT domain for DNA and histone binding and interacting with inhibitor of differentiation 1 (Id1) (Shoji et al., 1995; Richly et al., 2010). Zrf1 has been shown to antagonize the action of polycomb repressive complex 1 (PRC1) that ubiquitinates H2A and silences the Ink4a/ARF locus encoding p16 (Richly et al., 2010). In addition, the Zrf1 partners Id proteins can also suppress the activity of ETS1 and ETS2 transcription factors, which are potent activators of p16 transcription (Shoji et al., 1995). As outlined above, Zrf1 has also cytosolic functions as a co-chaperon forming the ribosome-associated complex (RAC). Although the phosphomutant of Zrf1 delays the senescence program (Barilari et al., 2017), how this post-translational modification affects the molecular function of Zrf1 has not been addressed yet.
S6K1 activity also modulates the DNA damage response, relevant to the induction of senescence. In cells exposed to DNA damaging agents and nutrient-rich conditions, S6K1 contributes to the phosphorylation and cytosolic retention of Mouse double minute homolog 2 (MDM2), the E3 ubiquitin ligase controlling the degradation of p53 (Lai et al., 2010). This potentiates the p53-dependent transcriptional response to DNA damaging agents, and in particular may explain how nutrients and growth factors can favour the pro-apoptotic and pro-senescent function of p53. Moreover, the S6K1-dependent phosphorylation of meiotic recombination 11 protein (MRE11) and Ring Finger Protein 168 (RNF168) triggers the degradation of these proteins crucial for DNA end resection and homologous recombination in the repair of double strand breaks (Piscitello et al., 2018; Xie et al., 2018). This impairs the DNA damage checkpoint and favours the entry of primary fibroblasts into senescence.
The S6K1-dependent hypertrophy of TSC1 mutant kidney epithelial cells correlates with misorientation of cell division leading to cyst deformation of renal tubules (Bonucci et al., 2020). However, micropattern experiments to measure the angle of cell division in single cells demonstrate that aberrant cell size is not sufficient to alter the mitotic angle. The defect of oriented cell division can be observed when cells are adjacent to each other in an epithelial layer. This suggests the importance of adhesion cues in the correct determination of planar polarity. Of note, a comprehensive phosphoproteomics analysis of the inner medullary collecting duct cell line mIMCD3 edited in the TSC1 and S6K1 genes revealed a broad list of putative S6K1 substrates involved in the regulation of the actin cortex and cell adhesion. These include slingshot protein phosphatase 1 (SSH1), phosphatase for the actin depolymerase cofilin; microtubule actin crosslinking factor 1 (Macf1), spectraplakin binding actin and microtubules; phosphatidylinositol 4-Kinase α (PI4K), regulating endocytic trafficking; myosin phosphatase-targeting subunit 1 and 2 (MyPT1 and MyPT2), myosin phosphatases regulating actomyosin contractility; missing in metastasis protein (Mtss1), cortactin-interacting protein. Among the phosphopeptides, Afadin, an actin binding and adapter protein regulating the nectin and cadherin adhesion systems, has been validated as a novel S6K1 substrate. Afadin phosphorylation alters Adherens Junctions in kidney epithelial cells and favours cell migration of breast cancer cells (Elloul et al., 2014; Gao et al., 2017). Of note, it has been suggested that Afadin phosphorylation may also be involved in the negative feed-back regulation of insulin action in adipocytes (Lundh et al., 2019), and Afadin phospho-mutant mice display a mild amelioration of glucose homeostasis at the early stages of high fat diet (Tozzi et al., 2022). Consistent with an important role of the mTOR/S6K1 pathway on cell adhesion, S6K1 phosphorylation down-regulates PI 4 phosphate 5 kinase type I g (PIPKIg90), which mediates together with talin the assembly of focal adhesion (Jafari et al., 2016). As a result of S6K1 action on PIPKIg90, cells become more prone to matrix degradation and invasion.
It has been recently proposed that cell size alterations may influence important cell fate decisions, such as senescence or regenerative responses (Neurohr et al., 2019; Lengefeld et al., 2021). It is likely that mTOR and S6K affects physical properties of the cells and define spatially organized microdomains, therefore affecting molecular machineries involved in growth and ageing. Several evidences are consistent with this possibility, though the causal relationships will require further studies. It has been shown that inhibition of mTOR by rapamycin treatment is associated with a decrease of macromolecular crowding and viscosity of the cytosol due to loss of ribosomes, a result of suppression of ribosome biogenesis and increased ribophagy (Delarue et al., 2018). Although it is not defined whether the decrease in ribosome contents is responsible of the decrease in cell size caused by rapamycin, it is clear that the ensuing dilution of the cytosol increases the diffusion of macromolecules. This may favour signaling processes but also impair the assembly of biomolecular condensates. The disproportionate growth of senescent cells, which is associated with a gradual loss of biosynthetic capacity, may also lead eventually to the dilution of the cytosol (Neurohr et al., 2019). The implication of S6K in this phenomenon is suggested by the observation that S6K inactivation prevents senescence of TSC1−/− cells (Barilari et al., 2017). S6K might contribute to a level of cytoplasmic crowding appropriate to regulate the on-demand formation of biomolecular condensates that perform crucial cellular functions. This would require a correct balance between the synthesis of crowding agents and other S6K dependent and independent functions that contribute to cell growth. Loss of this coupling would be expected to lead then to variations in the concentration of crowder and consequent alterations in cellular homeostasis.
In recent years, S6K activity has been implicated in the formation of biomolecular condensates, defined as dynamic membraneless subcellular compartments with liquid like properties which perform specific functions and assemble through phase separation. These include among others, nucleoli, Cajal bodies, promyelocytic leukaemia bodies (PML) in the nucleus, and P bodies, stress granules, or synaptic densities in the cytosol (Banani et al., 2017). For instance, stress granules (SGs) formation in response to oxidative stress and heath shock is under the control of the mTOR pathway and sensitive to S6K inhibition (Sfakianos et al., 2018). Although the mechanism has not been elucidated, it probably implicates phosphorylation of SG components that modulate protein-protein interactions and favour the formation of the condensates. Another example is the phosphorylation of the Survival motor Neuron protein (SMN) by S6K. This promotes efficient condensation by liquid liquid phase separation of the SMN complex in Cajal bodies (CBs) (Schilling et al., 2021), thus favouring the final maturation of ribonucleoproteins containing small nuclear U RNAs (UsnRNPs) and assisting the different steps of pre-mRNA splicing (Raimer et al., 2017). Interestingly SMN has SMN-complex independent functions. It has been shown that in fibroblasts a population of SMN is found associated with caveolin at the plasma membrane (Gabanella et al., 2016). Under conditions of recovery from energy stress, SMN at newly-formed membrane protrusion mobilizes inactive membrane-bound ribosomes that are then engaged in local protein synthesis in order to provide proteins required for the stabilization and maintenance of these structures. It will be interesting to determine the dependency of this process on the mTOR/S6K pathway, as well as the elucidation of additional microdomains influenced by these kinases.
As outlined in the previous chapter, the control of cytoskeleton dynamics may present an underappreciated mechanism pervading the whole cell and coordinating biological responses in relationship with cell volume regulation by S6K. In addition, specific metabolite levels and pathway activities may underlie cell volume alterations. In skeletal muscle, it is well established that muscle fiber types relying on different energy sources have different sizes (van Wessel et al., 2010; Bourdeau Julien et al., 2018). Mitochondrial functionality has been associated with the establishment of an optimal cell size (Miettinen et al., 2014; Miettinen and Bjorklund, 2017). Of note, the small size of the S6K1 mutant muscles correlate with alterations in the AMP/ATP ratios (Aguilar et al., 2007), and AMPK activity is a direct target of S6K (Dagon et al., 2012). In addition, the phosphorylation of Bcl-2-associated agonist of cell death (BAD) by Akt and S6K might be involved in reprogramming energy metabolism (Harada et al., 2001; Gimenez-Cassina et al., 2014). Future studies will address the following outstanding questions: What physical properties of the cell, metabolic status and cystoskeleton modifications may contribute to the whole cell sensing of its size in relation with S6K activity? What S6K1 specific substrates, that cannot be compensated by S6K2 or Rsk activities, dictate the cell size adaptations? Can these mechanisms be targeted in age-related pathological conditions?
MP and SF conceptualized, researched, and wrote this review.
Research related to the subject of this review was supported by grants from European Research Council, Inserm Agemed, ANR and RHU-Cosy to MP.
We apologize to colleagues whose work we were unable to discuss due to space constraints. We thank Bertsy Goic for her expert iconography support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abe, Y., Yoon, S. O., Kubota, K., Mendoza, M. C., Gygi, S. P., Blenis, J., et al. (2009). p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J. Biol. Chem. 284, 14939–14948. doi:10.1074/jbc.M900097200
Aguilar, V., Alliouachene, S., Sotiropoulos, A., Sobering, A., Athea, Y., Djouadi, F., et al. (2007). S6 kinase deletion suppresses muscle growth adaptations to nutrient availability by activating AMP kinase. Cell Metab. 5, 476–487. doi:10.1016/j.cmet.2007.05.006
Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney, P. R., Reese, C. B., et al. (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 7, 261–269. doi:10.1016/s0960-9822(06)00122-9
Alessi, D. R., Kozlowski, M. T., Weng, Q. P., Morrice, N., and Avruch, J. (1998). 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 8, 69–81. doi:10.1016/s0960-9822(98)70037-5
Arif, A., Terenzi, F., Potdar, A. A., Jia, J., Sacks, J., China, A., et al. (2017). EPRS is a critical mTORC1-S6K1 effector that influences adiposity in mice. Nature 542, 357–361. doi:10.1038/nature21380
Arif, A., Jia, J., Willard, B., Li, X., and Fox, P. L. (2019). Multisite phosphorylation of S6K1 directs a kinase phospho-code that determines substrate selection. Mol. Cell 73, 446–457. doi:10.1016/j.molcel.2018.11.017
Ban, N., Beckmann, R., Cate, J. H., Dinman, J. D., Dragon, F., Ellis, S. R., et al. (2014). A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 24, 165–169. doi:10.1016/j.sbi.2014.01.002
Banani, S. F., Lee, H. O., Hyman, A. A., and Rosen, M. K. (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298. doi:10.1038/nrm.2017.7
Banerjee, P., Ahmad, M. F., Grove, J. R., Kozlosky, C., Price, D. J., Avruch, J., et al. (1990). Molecular structure of a major insulin/mitogen-activated 70-kDa S6 protein kinase. Proc. Natl. Acad. Sci. U. S. A. 87, 8550–8554. doi:10.1073/pnas.87.21.8550
Barilari, M., Bonfils, G., Treins, C., Koka, V., De Villeneuve, D., Fabrega, S., et al. (2017). ZRF1 is a novel S6 kinase substrate that drives the senescence programme. EMBO J. 36, 736–750. doi:10.15252/embj.201694966
Ben-Sahra, I., Howell, J. J., Asara, J. M., and Manning, B. D. (2013). Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328. doi:10.1126/science.1228792
Ben-Sahra, I., Hoxhaj, G., Ricoult, S. J. H., Asara, J. M., and Manning, B. D. (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. doi:10.1126/science.aad0489
Bohlen, J., Roiuk, M., and Teleman, A. A. (2021). Phosphorylation of ribosomal protein S6 differentially affects mRNA translation based on ORF length. Nucleic Acids Res. 49, 13062–13074. doi:10.1093/nar/gkab1157
Bohni, R., Riesgo-Escovar, J., Oldham, S., Brogiolo, W., Stocker, H., Andruss, B. F., et al. (1999). Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 97, 865–875. doi:10.1016/s0092-8674(00)80799-0
Bonucci, M., Kuperwasser, N., Barbe, S., Koka, V., de Villeneuve, D., Zhang, C., et al. (2020). mTOR and S6K1 drive polycystic kidney by the control of Afadin-dependent oriented cell division. Nat. Commun. 11, 3200. doi:10.1038/s41467-020-16978-z
Bourdeau Julien, I., Sephton, C. F., and Dutchak, P. A. (2018). Metabolic networks influencing skeletal muscle fiber composition. Front. Cell Dev. Biol. 6, 125. doi:10.3389/fcell.2018.00125
Chauvin, C., Koka, V., Nouschi, A., Mieulet, V., Hoareau-Aveilla, C., Dreazen, A., et al. (2014). Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 33, 474–483. doi:10.1038/onc.2012.606
Cho, S., Lee, G., Pickering, B. F., Jang, C., Park, J. H., He, L., et al. (2021). mTORC1 promotes cell growth via m(6)A-dependent mRNA degradation. Mol. Cell 81, 2064–2075.e8. doi:10.1016/j.molcel.2021.03.010
Choo, A. Y., Yoon, S. O., Kim, S. G., Roux, P. P., and Blenis, J. (2008). Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. U. S. A. 105, 17414–17419. doi:10.1073/pnas.0809136105
Chung, J., Kuo, C. J., Crabtree, G. R., and Blenis, J. (1992). Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69, 1227–1236. doi:10.1016/0092-8674(92)90643-q
Csibi, A., Lee, G., Yoon, S. O., Tong, H., Ilter, D., Elia, I., et al. (2014). The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 24, 2274–2280. doi:10.1016/j.cub.2014.08.007
Dagon, Y., Hur, E., Zheng, B., Wellenstein, K., Cantley, L. C., Kahn, B. B., et al. (2012). p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin's effect on food intake. Cell Metab. 16, 104–112. doi:10.1016/j.cmet.2012.05.010
Delarue, M., Brittingham, G. P., Pfeffer, S., Surovtsev, I. V., Pinglay, S., Kennedy, K. J., et al. (2018). mTORC1 controls phase separation and the biophysical properties of the cytoplasm by tuning crowding. Cell 174, 338–349. doi:10.1016/j.cell.2018.05.042
Dennis, P. B., Pullen, N., Pearson, R. B., Kozma, S. C., and Thomas, G. (1998). Phosphorylation sites in the autoinhibitory domain participate in p70(s6k) activation loop phosphorylation. J. Biol. Chem. 273, 14845–14852. doi:10.1074/jbc.273.24.14845
Dever, T. E., and Green, R. (2012). The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb. Perspect. Biol. 4, a013706. doi:10.1101/cshperspect.a013706
Dibble, C. C., Asara, J. M., and Manning, B. D. (2009). Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 29, 5657–5670. doi:10.1128/MCB.00735-09
Dorrello, N. V., Peschiaroli, A., Guardavaccaro, D., Colburn, N. H., Sherman, N. E., Pagano, M., et al. (2006). S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471. doi:10.1126/science.1130276
Dowling, R. J., Topisirovic, I., Alain, T., Bidinosti, M., Fonseca, B. D., Petroulakis, E., et al. (2010). mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176. doi:10.1126/science.1187532
Elloul, S., Kedrin, D., Knoblauch, N. W., Beck, A. H., and Toker, A. (2014). The adherens junction protein afadin is an AKT substrate that regulates breast cancer cell migration. Mol. Cancer Res. 12, 464–476. doi:10.1158/1541-7786.MCR-13-0398
Faller, W. J., Jackson, T. J., Knight, J. R., Ridgway, R. A., Jamieson, T., Karim, S. A., et al. (2015). mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517, 497–500. doi:10.1038/nature13896
Fingar, D. C., Hausdorff, S. F., Blenis, J., and Birnbaum, M. J. (1993). Dissociation of pp70 ribosomal protein S6 kinase from insulin-stimulated glucose transport in 3T3-L1 adipocytes. J. Biol. Chem. 268, 3005–3008. doi:10.1016/s0021-9258(18)53873-4
Gabanella, F., Pisani, C., Borreca, A., Farioli-Vecchioli, S., Ciotti, M. T., Ingegnere, T., et al. (2016). SMN affects membrane remodelling and anchoring of the protein synthesis machinery. J. Cell Sci. 129, 804–816. doi:10.1242/jcs.176750
Gao, X., Zhang, Y., Arrazola, P., Hino, O., Kobayashi, T., Yeung, R. S., et al. (2002). Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 4, 699–704. doi:10.1038/ncb847
Gao, D., Inuzuka, H., Tan, M. K., Fukushima, H., Locasale, J. W., Liu, P., et al. (2011). mTOR drives its own activation via SCF(βTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 44, 290–303. doi:10.1016/j.molcel.2011.08.030
Gao, L., Yang, Z., Hiremath, C., Zimmerman, S. E., Long, B., Brakeman, P. R., et al. (2017). Afadin orients cell division to position the tubule lumen in developing renal tubules. Development 144, 3511–3520. doi:10.1242/dev.148908
Gimenez-Cassina, A., Garcia-Haro, L., Choi, C. S., Osundiji, M. A., Lane, E. A., Huang, H., et al. (2014). Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell Metab. 19, 272–284. doi:10.1016/j.cmet.2013.12.001
Gout, I., Minami, T., Hara, K., Tsujishita, Y., Filonenko, V., Waterfield, M. D., et al. (1998). Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase beta containing a proline-rich region. J. Biol. Chem. 273, 30061–30064. doi:10.1074/jbc.273.46.30061
Hara, K., Maruki, Y., Long, X., Yoshino, K., Oshiro, N., Hidayat, S., et al. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189. doi:10.1016/s0092-8674(02)00833-4
Harada, H., Andersen, J. S., Mann, M., Terada, N., and Korsmeyer, S. J. (2001). p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. U. S. A. 98, 9666–9670. doi:10.1073/pnas.171301998
Harrington, L. S., Findlay, G. M., Gray, A., Tolkacheva, T., Wigfield, S., Rebholz, H., et al. (2004). The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223. doi:10.1083/jcb.200403069
He, L., Gomes, A. P., Wang, X., Yoon, S. O., Lee, G., Nagiec, M. J., et al. (2018). mTORC1 promotes metabolic reprogramming by the suppression of GSK3-dependent Foxk1 phosphorylation. Mol. Cell 70, 949–960. doi:10.1016/j.molcel.2018.04.024
Holz, M. K., and Blenis, J. (2005). Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 280, 26089–26093. doi:10.1074/jbc.M504045200
Holz, M. K., Ballif, B. A., Gygi, S. P., and Blenis, J. (2005). mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580. doi:10.1016/j.cell.2005.10.024
Hsieh, A. C., Costa, M., Zollo, O., Davis, C., Feldman, M. E., Testa, J. R., et al. (2010). Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17, 249–261. doi:10.1016/j.ccr.2010.01.021
Hsu, P. P., Kang, S. A., Rameseder, J., Zhang, Y., Ottina, K. A., Lim, D., et al. (2011). The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322. doi:10.1126/science.1199498
Huang, H., Potter, C. J., Tao, W., Li, D. M., Brogiolo, W., Hafen, E., et al. (1999). PTEN affects cell size, cell proliferation and apoptosis during Drosophila eye development. Development 126, 5365–5372. doi:10.1242/dev.126.23.5365
Huang, Y., Hu, K., Zhang, S., Dong, X., Yin, Z., Meng, R., et al. (2018). S6K1 phosphorylation-dependent degradation of Mxi1 by beta-Trcp ubiquitin ligase promotes Myc activation and radioresistance in lung cancer. Theranostics 8, 1286–1300. doi:10.7150/thno.22552
Ito, N., and Rubin, G. M. (1999). gigas, a Drosophila homolog of tuberous sclerosis gene product-2, regulates the cell cycle. Cell 96, 529–539. doi:10.1016/s0092-8674(00)80657-1
Jafari, N., Zheng, Q., Li, L., Li, W., Qi, L., Xiao, J., et al. (2016). p70S6K1 (S6K1)-mediated phosphorylation regulates phosphatidylinositol 4-phosphate 5-kinase type I gamma degradation and cell invasion. J. Biol. Chem. 291, 25729–25741. doi:10.1074/jbc.M116.742742
Jiang, Q., Zhang, X., Dai, X., Han, S., Wu, X., Wang, L., et al. (2022). S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions. Nat. Commun. 13, 1548. doi:10.1038/s41467-022-28910-8
Julien, L. A., Carriere, A., Moreau, J., and Roux, P. P. (2010). mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell. Biol. 30, 908–921. doi:10.1128/MCB.00601-09
Kang, S. A., Pacold, M. E., Cervantes, C. L., Lim, D., Lou, H. J., Ottina, K., et al. (2013). mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 1236566. doi:10.1126/science.1236566
Kenney, J. W., Moore, C. E., Wang, X., and Proud, C. G. (2014). Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 55, 15–27. doi:10.1016/j.jbior.2014.04.003
Kim, A. R., and Choi, K. W. (2019). TRiC/CCT chaperonins are essential for organ growth by interacting with insulin/TOR signaling in Drosophila. Oncogene 38, 4739–4754. doi:10.1038/s41388-019-0754-1
Kim, D. H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., et al. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. doi:10.1016/s0092-8674(02)00808-5
Kozma, S. C., Ferrari, S., Bassand, P., Siegmann, M., Totty, N., Thomas, G., et al. (1990). Cloning of the mitogen-activated S6 kinase from rat liver reveals an enzyme of the second messenger subfamily. Proc. Natl. Acad. Sci. U. S. A. 87, 7365–7369. doi:10.1073/pnas.87.19.7365
Krieg, J., Hofsteenge, J., and Thomas, G. (1988). Identification of the 40 S ribosomal protein S6 phosphorylation sites induced by cycloheximide. J. Biol. Chem. 263, 11473–11477. doi:10.1016/s0021-9258(18)37981-x
Lai, K. P., Leong, W. F., Chau, J. F., Jia, D., Zeng, L., Liu, H., et al. (2010). S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 29, 2994–3006. doi:10.1038/emboj.2010.166
Lee, K., Sharma, R., Shrestha, O. K., Bingman, C. A., and Craig, E. A. (2016). Dual interaction of the Hsp70 J-protein cochaperone Zuotin with the 40S and 60S ribosomal subunits. Nat. Struct. Mol. Biol. 23, 1003–1010. doi:10.1038/nsmb.3299
Lee, G., Zheng, Y., Cho, S., Jang, C., England, C., Dempsey, J. M., et al. (2017). Post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell 171, 1545–1558. doi:10.1016/j.cell.2017.10.037
Lee-Fruman, K. K., Kuo, C. J., Lippincott, J., Terada, N., and Blenis, J. (1999). Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 18, 5108–5114. doi:10.1038/sj.onc.1202894
Leevers, S. J., Weinkove, D., MacDougall, L. K., Hafen, E., and Waterfield, M. D. (1996). The drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 15, 6584–6594. doi:10.1002/j.1460-2075.1996.tb01049.x
Lengefeld, J., Cheng, C. W., Maretich, P., Blair, M., Hagen, H., McReynolds, M. R., et al. (2021). Cell size is a determinant of stem cell potential during aging. Sci. Adv. 7, eabk0271. doi:10.1126/sciadv.abk0271
Leontieva, O. V., and Blagosklonny, M. V. (2013). CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell cycle 12, 3063–3069. doi:10.4161/cc.26130
Leontieva, O. V., and Blagosklonny, M. V. (2014). Gerosuppression in confluent cells. Aging 6, 1010–1018. doi:10.18632/aging.100714
Leontieva, O. V., Demidenko, Z. N., and Blagosklonny, M. V. (2013). S6K in geroconversion. Cell cycle 12, 3249–3252. doi:10.4161/cc.26248
Leontieva, O. V., Demidenko, Z. N., and Blagosklonny, M. V. (2014). Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc. Natl. Acad. Sci. U. S. A. 111, 8832–8837. doi:10.1073/pnas.1405723111
Leprivier, G., Remke, M., Rotblat, B., Dubuc, A., Mateo, A. R., Kool, M., et al. (2013). The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153, 1064–1079. doi:10.1016/j.cell.2013.04.055
Lewis, C. A., Griffiths, B., Santos, C. R., Pende, M., and Schulze, A. (2011). Genetic ablation of S6-kinase does not prevent processing of SREBP1. Adv. Enzyme Regul. 51, 280–290. doi:10.1016/j.advenzreg.2010.09.001
Liu, G. Y., and Sabatini, D. M. (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203. doi:10.1038/s41580-019-0199-y
Liu, P., Gan, W., Inuzuka, H., Lazorchak, A. S., Gao, D., Arojo, O., et al. (2013). Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 15, 1340–1350. doi:10.1038/ncb2860
Lundh, M., Petersen, P. S., Isidor, M. S., Kazoka-Sorensen, D. N., Plucinska, K., Shamsi, F., et al. (2019). Afadin is a scaffold protein repressing insulin action via HDAC6 in adipose tissue. EMBO Rep. 20, e48216. doi:10.15252/embr.201948216
Ma, X. M., Yoon, S. O., Richardson, C. J., Julich, K., and Blenis, J. (2008). SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 133, 303–313. doi:10.1016/j.cell.2008.02.031
Ma, S., Meng, Z., Chen, R., and Guan, K. L. (2018). The hippo pathway: Biology and pathophysiology. Annu. Rev. Biochem. 88, 577–604. doi:10.1146/annurev-biochem-013118-111829
Martinez-Carreres, L., Puyal, J., Leal-Esteban, L. C., Orpinell, M., Castillo-Armengol, J., Giralt, A., et al. (2019). CDK4 regulates lysosomal function and mTORC1 activation to promote cancer cell survival. Cancer Res. 79, 5245–5259. doi:10.1158/0008-5472.CAN-19-0708
Merrick, W. C., and Pavitt, G. D. (2018). Protein synthesis initiation in eukaryotic cells. Cold Spring Harb. Perspect. Biol. 10, a033092. doi:10.1101/cshperspect.a033092
Miettinen, T. P., and Bjorklund, M. (2017). Mitochondrial function and cell size: An allometric relationship. Trends Cell Biol. 27, 393–402. doi:10.1016/j.tcb.2017.02.006
Miettinen, T. P., Pessa, H. K., Caldez, M. J., Fuhrer, T., Diril, M. K., Sauer, U., et al. (2014). Identification of transcriptional and metabolic programs related to mammalian cell size. Curr. Biol. 24, 598–608. doi:10.1016/j.cub.2014.01.071
Mieulet, V., Roceri, M., Espeillac, C., Sotiropoulos, A., Ohanna, M., Oorschot, V., et al. (2007). S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am. J. Physiol. Cell Physiol. 293, C712–C722. doi:10.1152/ajpcell.00499.2006
Montagne, J., Stewart, M. J., Stocker, H., Hafen, E., Kozma, S. C., Thomas, G., et al. (1999). Drosophila S6 kinase: a regulator of cell size. Science 285, 2126–2129. doi:10.1126/science.285.5436.2126
Moritz, A., Li, Y., Guo, A., Villen, J., Wang, Y., MacNeill, J., et al. (2010). Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 3, ra64. doi:10.1126/scisignal.2000998
Mukhopadhyay, N. K., Price, D. J., Kyriakis, J. M., Pelech, S., Sanghera, J., Avruch, J., et al. (1992). An array of insulin-activated, proline-directed serine/threonine protein kinases phosphorylate the p70 S6 kinase. J. Biol. Chem. 267, 3325–3335. doi:10.1016/s0021-9258(19)50735-9
Napolitano, G., Di Malta, C., Esposito, A., de Araujo, M. E. G., Pece, S., Bertalot, G., et al. (2020). A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature 585, 597–602. doi:10.1038/s41586-020-2444-0
Neurohr, G. E., Terry, R. L., Lengefeld, J., Bonney, M., Brittingham, G. P., Moretto, F., et al. (2019). Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell 176, 1083–1097. doi:10.1016/j.cell.2019.01.018
Ohanna, M., Sobering, A. K., Lapointe, T., Lorenzo, L., Praud, C., Petroulakis, E., et al. (2005). Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat. Cell Biol. 7, 286–294. doi:10.1038/ncb1231
Pantalacci, S., Tapon, N., and Leopold, P. (2003). The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 5, 921–927. doi:10.1038/ncb1051
Pearce, L. R., Komander, D., and Alessi, D. R. (2010). The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22. doi:10.1038/nrm2822
Pende, M., Kozma, S. C., Jaquet, M., Oorschot, V., Burcelin, R., Le Marchand-Brustel, Y., et al. (2000). Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 408, 994–997. doi:10.1038/35050135
Pende, M., Um, S. H., Mieulet, V., Sticker, M., Goss, V. L., Mestan, J., et al. (2004). S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5'-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol. 24, 3112–3124. doi:10.1128/mcb.24.8.3112-3124.2004
Pham, P. T., Heydrick, S. J., Fox, H. L., Kimball, S. R., Jefferson, L. S., Lynch, C. J., et al. (2000). Assessment of cell-signaling pathways in the regulation of mammalian target of rapamycin (mTOR) by amino acids in rat adipocytes. J. Cell. Biochem. 79, 427–441. doi:10.1002/1097-4644(20001201)79:3<427::aid-jcb80>3.0.co;2-0
Piscitello, D., Varshney, D., Lilla, S., Vizioli, M. G., Reid, C., Gorbunova, V., et al. (2018). AKT overactivation can suppress DNA repair via p70S6 kinase-dependent downregulation of MRE11. Oncogene 37, 427–438. doi:10.1038/onc.2017.340
Porstmann, T., Santos, C. R., Griffiths, B., Cully, M., Wu, M., Leevers, S., et al. (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236. doi:10.1016/j.cmet.2008.07.007
Pullen, N., Dennis, P. B., Andjelkovic, M., Dufner, A., Kozma, S. C., Hemmings, B. A., et al. (1998). Phosphorylation and activation of p70s6k by PDK1. Science 279, 707–710. doi:10.1126/science.279.5351.707
Raimer, A. C., Gray, K. M., and Matera, A. G. (2017). SMN - a chaperone for nuclear RNP social occasions? RNA Biol. 14, 701–711. doi:10.1080/15476286.2016.1236168
Richardson, C. J., Broenstrup, M., Fingar, D. C., Julich, K., Ballif, B. A., Gygi, S., et al. (2004). SKAR is a specific target of S6 kinase 1 in cell growth control. Curr. Biol. 14, 1540–1549. doi:10.1016/j.cub.2004.08.061
Richly, H., Rocha-Viegas, L., Ribeiro, J. D., Demajo, S., Gundem, G., Lopez-Bigas, N., et al. (2010). Transcriptional activation of polycomb-repressed genes by ZRF1. Nature 468, 1124–1128. doi:10.1038/nature09574
Robitaille, A. M., Christen, S., Shimobayashi, M., Cornu, M., Fava, L. L., Moes, S., et al. (2013). Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323. doi:10.1126/science.1228771
Romero-Pozuelo, J., Figlia, G., Kaya, O., Martin-Villalba, A., and Teleman, A. A. (2020). Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 31, 107504. doi:10.1016/j.celrep.2020.03.068
Ruvinsky, I., Sharon, N., Lerer, T., Cohen, H., Stolovich-Rain, M., Nir, T., et al. (2005). Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 19, 2199–2211. doi:10.1101/gad.351605
Ruvinsky, I., Katz, M., Dreazen, A., Gielchinsky, Y., Saada, A., Freedman, N., et al. (2009). Mice deficient in ribosomal protein S6 phosphorylation suffer from muscle weakness that reflects a growth defect and energy deficit. PloS one 4, e5618. doi:10.1371/journal.pone.0005618
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi:10.1126/science.1106148
Schilling, M., Prusty, A. B., Boysen, B., Oppermann, F. S., Riedel, Y. L., Husedzinovic, A., et al. (2021). TOR signaling regulates liquid phase separation of the SMN complex governing snRNP biogenesis. Cell Rep. 35, 109277. doi:10.1016/j.celrep.2021.109277
Selman, C., Tullet, J. M., Wieser, D., Irvine, E., Lingard, S. J., Choudhury, A. I., et al. (2009). Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144. doi:10.1126/science.1177221
Sfakianos, A. P., Mellor, L. E., Pang, Y. F., Kritsiligkou, P., Needs, H., Abou-Hamdan, H., et al. (2018). The mTOR-S6 kinase pathway promotes stress granule assembly. Cell Death Differ. 25, 1766–1780. doi:10.1038/s41418-018-0076-9
Shahbazian, D., Roux, P. P., Mieulet, V., Cohen, M. S., Raught, B., Taunton, J., et al. (2006). The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25, 2781–2791. doi:10.1038/sj.emboj.7601166
Shima, H., Pende, M., Chen, Y., Fumagalli, S., Thomas, G., Kozma, S. C., et al. (1998). Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 17, 6649–6659. doi:10.1093/emboj/17.22.6649
Shoji, W., Inoue, T., Yamamoto, T., and Obinata, M. (1995). MIDA1, a protein associated with Id, regulates cell growth. J. Biol. Chem. 270, 24818–24825. doi:10.1074/jbc.270.42.24818
Stocker, H., and Hafen, E. (2000). Genetic control of cell size. Curr. Opin. Genet. Dev. 10, 529–535. doi:10.1016/s0959-437x(00)00123-4
Tang, H. W., Weng, J. H., Lee, W. X., Hu, Y., Gu, L., Cho, S., et al. (2021). mTORC1-chaperonin CCT signaling regulates m(6)A RNA methylation to suppress autophagy. Proc. Natl. Acad. Sci. U. S. A. 118, e2021945118. doi:10.1073/pnas.2021945118
Tozzi, M., Brown, E. L., Petersen, P. S. S., Lundh, M., Isidor, M. S., Plucinska, K., et al. (2022). Dynamic interplay between Afadin(S1795) phosphorylation and diet regulates glucose homeostasis in obese mice. J. Physiol. 600, 885–902. doi:10.1113/JP281657
Treins, C., Warne, P. H., Magnuson, M. A., Pende, M., and Downward, J. (2010). Rictor is a novel target of p70 S6 kinase-1. Oncogene 29, 1003–1016. doi:10.1038/onc.2009.401
Tremblay, F., and Marette, A. (2001). Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem. 276, 38052–38060. doi:10.1074/jbc.M106703200
Tremblay, F., Brule, S., Hee Um, S., Li, Y., Masuda, K., Roden, M., et al. (2007). Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 104, 14056–14061. doi:10.1073/pnas.0706517104
Udan, R. S., Kango-Singh, M., Nolo, R., Tao, C., and Halder, G. (2003). Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 5, 914–920. doi:10.1038/ncb1050
Um, S. H., Frigerio, F., Watanabe, M., Picard, F., Joaquin, M., Sticker, M., et al. (2004). Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205. doi:10.1038/nature02866
van Wessel, T., de Haan, A., van der Laarse, W. J., Jaspers, R. T., and de HAAn, A. (2010). The muscle fiber type-fiber size paradox: hypertrophy or oxidative metabolism? Eur. J. Appl. Physiol. 110, 665–694. doi:10.1007/s00421-010-1545-0
Verdu, J., Buratovich, M. A., Wilder, E. L., and Birnbaum, M. J. (1999). Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat. Cell Biol. 1, 500–506. doi:10.1038/70293
Wang, X., Li, W., Williams, M., Terada, N., Alessi, D. R., Proud, C. G., et al. (2001). Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20, 4370–4379. doi:10.1093/emboj/20.16.4370
Wettenhall, R. E., Erikson, E., and Maller, J. L. (1992). Ordered multisite phosphorylation of Xenopus ribosomal protein S6 by S6 kinase II. J. Biol. Chem. 267, 9021–9027. doi:10.1016/s0021-9258(19)50382-9
Woodward, L. A., Mabin, J. W., Gangras, P., and Singh, G. (2017). The exon junction complex: a lifelong guardian of mRNA fate. Wiley Interdiscip. Rev. RNA 8. doi:10.1002/wrna.1411
Wu, S., Huang, J., Dong, J., and Pan, D. (2003). hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456. doi:10.1016/s0092-8674(03)00549-x
Xie, X., Hu, H., Tong, X., Li, L., Liu, X., Chen, M., et al. (2018). The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nat. Cell Biol. 20, 320–331. doi:10.1038/s41556-017-0033-8
Yecies, J. L., Zhang, H. H., Menon, S., Liu, S., Yecies, D., Lipovsky, A. I., et al. (2011). Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 14, 21–32. doi:10.1016/j.cmet.2011.06.002
Yi, S. A., Um, S. H., Lee, J., Yoo, J. H., Bang, S. Y., Park, E. K., et al. (2016). S6K1 phosphorylation of H2B mediates EZH2 trimethylation of H3: A determinant of early adipogenesis. Mol. Cell 62, 443–452. doi:10.1016/j.molcel.2016.03.011
Yi, S. A., Lee, J., Park, J. W., Han, J., Lee, M. G., Nam, K. H., et al. (2018). S6K1 controls epigenetic plasticity for the expression of pancreatic α/β cell marker genes. J. Cell. Biochem. 119, 6674–6683. doi:10.1002/jcb.26853
Yi, S. A., Jeon, Y. J., Lee, M. G., Nam, K. H., Ann, S., Lee, J., et al. (2022). S6K1 controls adiponectin expression by inducing a transcriptional switch: BMAL1-to-EZH2. Exp. Mol. Med. 54, 324–333. doi:10.1038/s12276-022-00747-7
Yu, Y., Yoon, S. O., Poulogiannis, G., Yang, Q., Ma, X. M., Villen, J., et al. (2011). Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326. doi:10.1126/science.1199484
Zhang, H., Stallock, J. P., Ng, J. C., Reinhard, C., and Neufeld, T. P. (2000). Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 14, 2712–2724. doi:10.1101/gad.835000
Zhang, Y., Gao, X., Saucedo, L. J., Ru, B., Edgar, B. A., Pan, D., et al. (2003). Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 5, 578–581. doi:10.1038/ncb999
Zhang, H. H., Lipovsky, A. I., Dibble, C. C., Sahin, M., and Manning, B. D. (2006). S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell 24, 185–197. doi:10.1016/j.molcel.2006.09.019
Zhao, Y., Xiong, X., and Sun, Y. (2011). DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 44, 304–316. doi:10.1016/j.molcel.2011.08.029
Keywords: mTOR, signal transduction, nutrition, growth, ageing, senescence
Citation: Fumagalli S and Pende M (2022) S6 kinase 1 at the central node of cell size and ageing. Front. Cell Dev. Biol. 10:949196. doi: 10.3389/fcell.2022.949196
Received: 20 May 2022; Accepted: 07 July 2022;
Published: 12 August 2022.
Edited by:
Evgeny Zatulovskiy, Stanford University, United StatesReviewed by:
Shuyuan Zhang, Stanford University, United StatesCopyright © 2022 Fumagalli and Pende. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Fumagalli, c3RlZmFuby5mdW1hZ2FsbGlAaW5zZXJtLmZy; Mario Pende, TWFyaW8ucGVuZGVAaW5zZXJtLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.