 Charlotte EJ. Downes1,2
Charlotte EJ. Downes1,2 Barbara J. McClure1,3
Barbara J. McClure1,3 Daniel P. McDougal2,4
Daniel P. McDougal2,4 Susan L. Heatley1,3,5
Susan L. Heatley1,3,5 John B. Bruning2,4
John B. Bruning2,4 Daniel Thomas1,3
Daniel Thomas1,3 David T. Yeung1,3,6
David T. Yeung1,3,6 Deborah L. White1,2,3,5*
Deborah L. White1,2,3,5*- 1Blood Cancer Program, Precision Cancer Medicine Theme, South Australian Health and Medical Research Institute (SAHMRI), Adelaide, SA, Australia
- 2School of Biological Sciences, Faculty of Sciences, University of Adelaide, Adelaide, SA, Australia
- 3Adelaide Medical School, Faculty of Health and Medical Sciences, University of Adelaide, Adelaide, SA, Australia
- 4Institute for Photonics and Advanced Sensing (IPAS), University of Adelaide, Adelaide, SA, Australia
- 5Australian and New Zealand Children’s Oncology Group (ANZCHOG), Clayton, VIC, Australia
- 6Department of Haematology, Royal Adelaide Hospital and SA Pathology, Adelaide, SA, Australia
Acute lymphoblastic leukemia (ALL) is the most common pediatric cancer, arising from immature lymphocytes that show uncontrolled proliferation and arrested differentiation. Genomic alterations affecting Janus kinase 2 (JAK2) correlate with some of the poorest outcomes within the Philadelphia-like subtype of ALL. Given the success of kinase inhibitors in the treatment of chronic myeloid leukemia, the discovery of activating JAK2 point mutations and JAK2 fusion genes in ALL, was a breakthrough for potential targeted therapies. However, the molecular mechanisms by which these alterations activate JAK2 and promote downstream signaling is poorly understood. Furthermore, as clinical data regarding the limitations of approved JAK inhibitors in myeloproliferative disorders matures, there is a growing awareness of the need for alternative precision medicine approaches for specific JAK2 lesions. This review focuses on the molecular mechanisms behind ALL-associated JAK2 mutations and JAK2 fusion genes, known and potential causes of JAK-inhibitor resistance, and how JAK2 alterations could be targeted using alternative and novel rationally designed therapies to guide precision medicine approaches for these high-risk subtypes of ALL.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy, but despite cure rates now approaching 90% with refined chemotherapy regimens, relapse remains the leading cause of mortality in children (Hunger and Mullighan, 2015; Iacobucci and Mullighan, 2017; Khan et al., 2018). Furthermore, only 30–40% of adult ALL patients achieve long-term remission (Jabbour et al., 2015; Terwilliger and Abdul-Hay, 2017). Over the last decade, technological advances in genomic profiling, such as transcriptome and whole genome sequencing, have transformed risk stratification and treatment approaches for ALL patients by revealing the genomic basis of the disease (Roberts and Mullighan, 2015; Khan et al., 2018). In 2009, large-scale gene expression profiling identified a high-risk B-cell precursor ALL (B-ALL) subtype, termed Philadelphia chromosome-like ALL (Ph-like ALL), which displays a gene expression profile similar to that of Philadelphia chromosome-positive ALL (Ph+ ALL), harbors a high frequency of IKZF1 (IKAROS family zinc finger 1) alterations, but lacks the hallmark BCR::ABL1 (breakpoint cluster region protein/Abelson 1) fusion gene of Ph+ ALL (Mullighan et al., 2009b; Den Boer et al., 2009). Comprehensive genomic profiling studies revealed the diversity of genomic alterations that constitute the heterogeneous genomic landscape of Ph-like ALL (Tasian et al., 2017b; Pui et al., 2017; Khan et al., 2018; Iacobucci and Roberts, 2021). These genomic alterations can include translocations, cryptic rearrangements, mutations, and copy number variations, often in genes that regulate cytokine receptor and kinase signaling pathways. Genes commonly rearranged include ABL1/2, CRLF2 (cytokine receptor like factor 2), EPOR (erythropoietin receptor) and JAK2 (Janus kinase 2). Activating mutations or deletions are usually identified within JAK/STAT (Janus kinase/signal transducer and activator of transcription) or RAS/MAPK (RAS GTP-activating protein/mitogen-activated protein kinase) signaling pathways, although other rare kinase alterations have been reported (Roberts et al., 2012; Roberts et al., 2014a; Roberts and Mullighan, 2015; Tran and Loh, 2016).
JAK2 alterations, including rearrangements and gain-of-function mutations, are associated with poor outcome within the Ph-like ALL subtype (Roberts et al., 2014a). It is unclear why JAK2 alterations are predominantly identified within B-ALL rather than T-cell ALL (T-ALL), although there have been rare reports in T-ALL (Lacronique et al., 1997; Peeters et al., 1997; Cheng et al., 2017; Huang et al., 2020; Kaplan et al., 2021). JAK2 chromosomal rearrangements (JAK2r) which result in JAK2 fusion genes, correlate with some of the lowest survival rates within the Ph-like ALL subtype (Figure 1) (Roberts et al., 2014a; Roberts K. G. et al., 2017). Similar to oncogenic kinase drivers observed in myeloid disorders, JAK2 alterations were identified to be driving ALL lesions, offering renewed hope for precision medicine approaches beyond high intensity combination chemotherapy. Based on the success of tyrosine kinase inhibitors (TKIs) for the treatment of chronic myeloid leukemia (CML) and Ph+ ALL, there is potential for targeted JAK2 inhibitors to improve outcomes for patients with high-risk, JAK2-altered ALL. The semi-selective JAK1/2 inhibitor, ruxolitinib, is currently being assessed in a number of clinical trials (NCT02723994, NCT03117751, NCT03571321, NCT02420717) for the treatment for ALL after promising efficacy was demonstrated in several pre-clinical models (Maude et al., 2012; Roberts et al., 2014a; Roberts KG. et al., 2017).
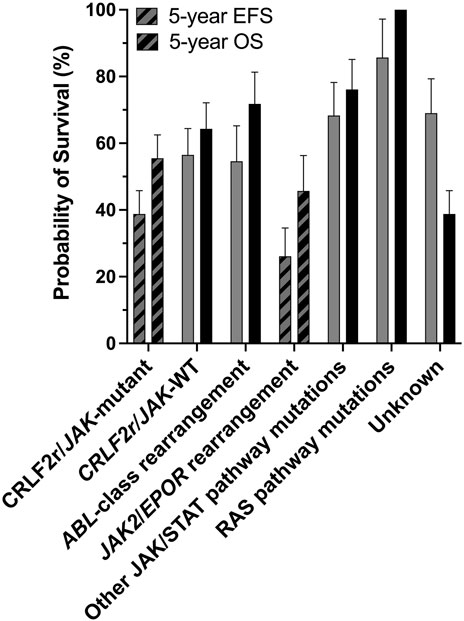
FIGURE 1. The CRLF2r/JAK-mutant and JAK2/EPORr subtypes of Ph-like ALL are associated with poor outcomes. Outcome analyses for different genomic subtypes of Ph-like ALL for all ages combined, probabilities of 5-years event-free survival (EFS) and overall survival (OS) are shown. There are significant differences in the 5-years EFS and OS of CRLF2r/JAK-mutant and JAK2/EPORr cases compared with other Ph-like ALL subtypes. Figure adapted from Roberts et al. (2014a).
In 2011, ruxolitinib was the first JAK inhibitor approved for the treatment of myelofibrosis (MF), a myeloproliferative neoplasm (MPN) that harbors a high frequency of the recurrent activating JAK2 p. V617F point mutation. Although ruxolitinib reduces the symptomatic burden of MF, unfortunately, it does not significantly reduce the mutant allele frequency (Deininger et al., 2015; Greenfield et al., 2018; Bewersdorf et al., 2019). Furthermore, the use of ruxolitinib as a first-line therapy for MF has revealed several clinical limitations (also apparent with a subsequently approved JAK inhibitor fedratinib), which are directly relevant to ALL and are discussed in detail below. There have been few case reports to date documenting successful responses to ruxolitinib in Ph-like ALL, with only one report in JAK2-mutant Ph-like ALL (Mayfield et al., 2017) and four reports in JAK2r Ph-like ALL (Ding et al., 2018; Chen X. et al., 2019; Chen et al., 2022; Rizzuto et al., 2022). It is also difficult to decipher the role of graft-versus-leukemia effect in the context of allogeneic transplant and the “on-target” but “off-cancer” effects of ruxolitinib on the immune system. Here, we comprehensively review the molecular biology and clinical knowledge of JAK2 alterations in ALL. The therapeutic implications of current and future precision medicine approaches for this high-risk subtype of ALL are discussed, emphasizing the need for further lesion-specific molecular insights and a new suite of JAK-targeting approaches.
Normal JAK2 Structure and Function
Wild-type (WT) JAK2 is a non-receptor tyrosine kinase that pre-associates with a variety of type I (containing a WSXWS motif) and type II (lacking a WSXWS motif) cytokine receptors. Upon cytokine binding, both classes of cytokine receptors activate downstream intracellular signaling pathways, predominantly the JAK/STAT signaling pathway (Babon et al., 2014; Morris et al., 2018). The JAK family kinases (comprising JAK1, JAK2, JAK3, and TYK2 (tyrosine kinase 2)) all share a common protein structure comprising seven JAK homology (JH) domains (Gnanasambandan and Sayeski, 2011; Steeghs et al., 2017). The FERM (4.1 protein, ezrin, radixin, moesin) domain and Src homology 2 (SH2)-like (SH2L) domains are N-terminally located (Figure 2A), and mediate protein-protein interactions and cytokine binding respectively (Kesarwani et al., 2015; Leroy and Constantinescu, 2017). The FERM and SH2L domains are required for JAK2 binding to specific juxtamembrane Box1 and Box2 motifs of associated cytokine receptors (Saharinen et al., 2000; Hubbard, 2018; Morris et al., 2018; Raivola et al., 2021). Phosphorylation of conserved tyrosine residues within the FERM domain can also positively or negatively regulate JAK2 activity (Gnanasambandan and Sayeski, 2011; Hammaren et al., 2019b). At the C-terminal end of JAK2 are the catalytic kinase (JH1), and pseudokinase (JH2) domains (Figure 2A) (Kesarwani et al., 2015; Leroy and Constantinescu, 2017). The kinase domain is responsible for catalyzing the phosphorylation of substrate-specific tyrosine residues (Babon et al., 2014; Morris et al., 2018). The pseudokinase domain lies directly upstream of the kinase domain, sharing conserved motifs, but exhibits minimal catalytic activity (Ungureanu et al., 2011; Lupardus et al., 2014). The pseudokinase domain allows a basal level of kinase activity to be maintained in the absence of cytokine through direct inhibition of the kinase domain (Saharinen et al., 2000; Saharinen and Silvennoinen, 2002; Hubbard, 2018). The JH2-SH2 linker region has been hypothesized to stabilize the interaction between the pseudokinase and kinase domains during this JH2-mediated auto-inhibition (Babon et al., 2014; Shan et al., 2014). Release of JH2-mediated auto-inhibition plays an important role in facilitating full JAK2 activation upon cytokine binding, as discussed below, and as such, this mechanism is often exploited by leukemogenic drivers.
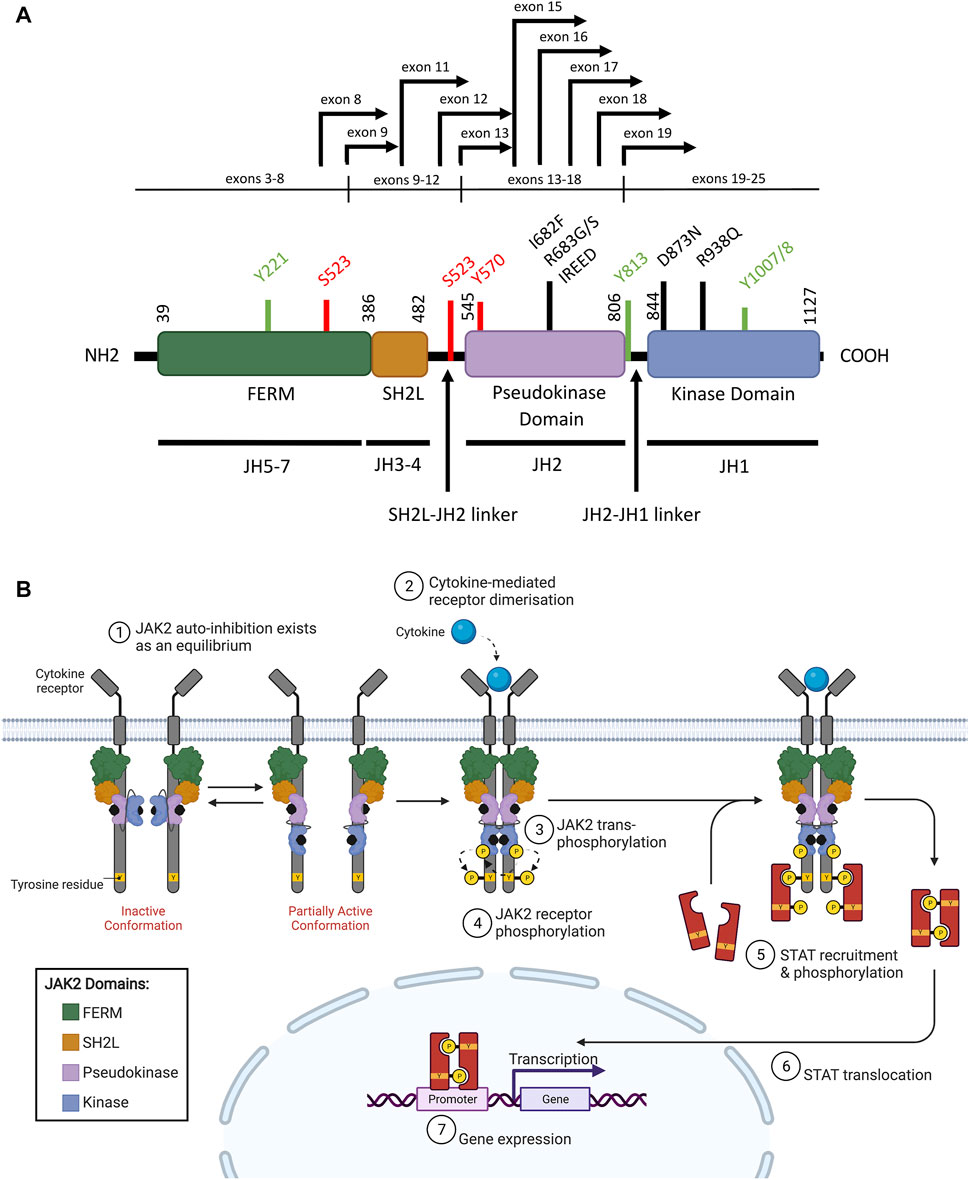
FIGURE 2. WT JAK2 structure and function (A) Schematic representation of the JAK2 domain structure (NCBI reference sequence: NP_004963.1) encoded by the seven JAK homology (JH) domains. The FERM (4.1 protein, ezrin, radixin, moesin), SH2 (Src homology 2)-like (SH2L), pseudokinase (JH2) and kinase (JH1) domains are represented by purple, red, light blue, and dark blue respectively. Key residues for phosphorylation for positive (green) or negative (red) regulation are shown. Mutations commonly associated with ALL (black lines) and JAK2 fusion breakpoints (black arrows) are indicated. Adapted from Silvennoinen and Hubbard (2015b) (Silvennoinen and Hubbard, 2015a). (B) Schematic representation of JAK/STAT signaling pathway activation through JAK2. The JAK2 FERM and SH2L domains associate with the cytoplasmic juxtamembrane motifs of a cytokine receptor (grey) to recruit JAK2 to the cell membrane. The four domains of JAK2 are presented: FERM (green), SH2-like (orange), pseudokinase (JH2, purple), and kinase (JH1, blue). JAK2 is shown bound to ATP (black). The proposed model of JAK2 activation suggests that JAK2 exists in an equilibrium between inactive and partially active conformations. In the inactive conformation (left), the JAK2 kinase domain is inhibited by a JH2-mediated autoinhibitory interaction. In the partially active conformation (right), the JAK2 kinase domain is released from the JH2-mediated auto-inhibition and is available for some limited transphosphorylation. Cytokine (cyan) binding to their cognate receptor promotes receptor dimerisation, which facilitates JAK2 activation by transphosphorylation (arrows). JAK2 then auto-phosphorylates the cytoplasmic region of the receptor creating recruitment sites for cytoplasmic STATs (red). JAK2-mediated STAT phosphorylation facilitates STAT dimerisation. These STAT dimers are then translocated to the nucleus where they regulate gene expression by binding to promoters with STAT-binding sites. Adapted from Hubbard (2018) and “Cytokine Signaling through the JAK-STAT Pathway” (BioRender.com, 2021).
Physiological JAK2 Activation
Precise activation of cytokine receptor signaling via JAK2 is essential for the complex co-ordination of hematopoietic cell proliferation and differentiation (Vainchenker and Constantinescu, 2013; Raivola et al., 2021). In the traditional model of cytokine-induced receptor activation, high affinity cytokine binding to cognate receptor subunits results in conformational changes that facilitate and stabilize receptor dimerization and, in many cases, oligomerization of higher order protein complexes at the cell surface (Livnah et al., 1999; Vainchenker and Constantinescu, 2013). If the cytokine receptor is a homodimer then JAK2 will homodimerize, whereas heterodimeric cytokine receptors enable either JAK2 homodimerization, or heterodimerization with other JAK family members (Morris et al., 2018; Raivola et al., 2021). In contrast, several biochemical studies have reported pre-dimerization of JAK2-associated receptors, including EPOR and human growth hormone receptor (hGHR), in which cytokine binding may facilitate receptor reorientation and subsequent JAK2 activation (Livnah et al., 1999; Constantinescu et al., 2001; Gent et al., 2002; Hammaren et al., 2019b). However, the use of over-expression systems or cysteine cross-linking may have confounded such data by enriching the cell-surface density of expressed receptors (Hubbard, 2018; Wilmes et al., 2020) and thus the mechanism for pre-formed receptor activation remains speculative (Hammaren et al., 2019b). Indeed, recent single-molecule imaging by Wilmes et al. (2020) identified almost no co-trajectories of thrombopoietin receptor (TPOR) (a Type I receptor), EPOR or hGHR acting as a stable “single dimeric molecule” over time in the absence of cytokine (Wilmes et al., 2020).
In a model first proposed by Silvennoinen and Hubbard (2015a), inactive and partially active dimers of JAK2 may exist in an equilibrium at the cell membrane, where the inactive conformation is stabilized by JH2-mediated auto-inhibition (Figure 2B) (Shan et al., 2014; Silvennoinen and Hubbard, 2015a; Hubbard, 2018). In the inactive conformation, the JAK2 pseudokinase domain binds the kinase domain in a front-to-back orientation to inhibit kinase activity, a conformation stabilized by trans-phosphorylation of JAK2 p. S523 and p. Y570 by the JAK2 pseudokinase domain (Saharinen et al., 2000; Saharinen and Silvennoinen, 2002; Bandaranayake et al., 2012; Shan et al., 2014; Hubbard, 2018; Hammaren et al., 2019a). In the partially active conformation, the JAK2 kinase domain is released from the JH2-mediated auto-inhibition potentially through loosening of the linker region between the SH2L and pseudokinase domains (Gnanasambandan and Sayeski, 2011; Shan et al., 2014). The partially active conformation of JAK2 is proposed to support limited trans-phosphorylation in the absence of cytokine to maintain a low, basal level of JAK2 activity (Shan et al., 2014; Silvennoinen and Hubbard, 2015a; Hubbard, 2018). In the traditional model, where JAK2-associated receptors do not exist as pre-formed dimers, auto-inhibition of the JAK2 kinase domain by the pseudokinase domain likely occurs in cis (within the same JAK2 molecule) (Hubbard, 2018). However, the disordered JAK2 JH2-JH1 linker region could be long enough to enable trans phosphorylation of the JAK2 kinase domain in a pre-formed receptor dimer (Hubbard, 2018). Potentially, JAK2 activation requires both cytokine-mediated receptor dimerization and release of the JH2-mediated auto-inhibitory interaction, facilitating trans-phosphorylation of a string of tyrosine residues located on the JAK2 activation loop, including JAK2 p. Y1007 and p. Y1008 (Figure 2A, Figure 2B) (Feng et al., 1997; Chatti et al., 2004; Silvennoinen and Hubbard, 2015a). However, the mechanism by which these individual phosphorylation events activate JAK2 is yet to be fully elucidated (Babon et al., 2014; Hammaren et al., 2019b).
JAK2 dimerization and trans-phosphorylation orientates the overall JAK2 structure to an active, or “DFG-in” conformation, characterized by the positioning of the N-lobe αC helix, and the DFG motif (residues 994–996) at the N-terminus of the activation loop (Figure 3A, Figure 3B) (Shan et al., 2014; Leroy and Constantinescu, 2017). In the active conformation, the DFG motif faces inward to enable hydrophobic interactions with the αC helix and catalytic loop (Shan et al., 2014; Leroy and Constantinescu, 2017). This rotates the αC helix towards the active site for catalysis and extends the activation loop outward to enable substrate binding (Lucet et al., 2006; Babon et al., 2014). This contrasts the inactive, or “DFG-out” conformation of JAK2, where the αC helix is rotated away from the active site and the activation loop is disordered (Figure 3B) (Silvennoinen and Hubbard, 2015b). The active conformation of JAK2 promotes ATP (adenosine triphosphate) to bind within the critical ATP-binding site, which lies between the N- and C-terminal lobes of the kinase domain (Lucet et al., 2006; Bandaranayake et al., 2012). ATP binding is stabilized by hydrogen bonds with residues located in the JAK2 hinge region and positions the terminal phosphates of ATP for phosphoryl transfer (Bandaranayake et al., 2012; Hammaren et al., 2015; Bhullar et al., 2018). A number of residues within the ATP-binding site are conserved between JAK family members, suggesting that they are essential for JAK kinase activity (Lucet et al., 2006; Bhullar et al., 2018). Following activation, JAK2 auto-phosphorylates cytoplasmic receptor tyrosine residues generating docking sites for proteins containing SH2 domains, including STATs (Figure 2B) (Levy and Darnell, 2002; Morris et al., 2018).
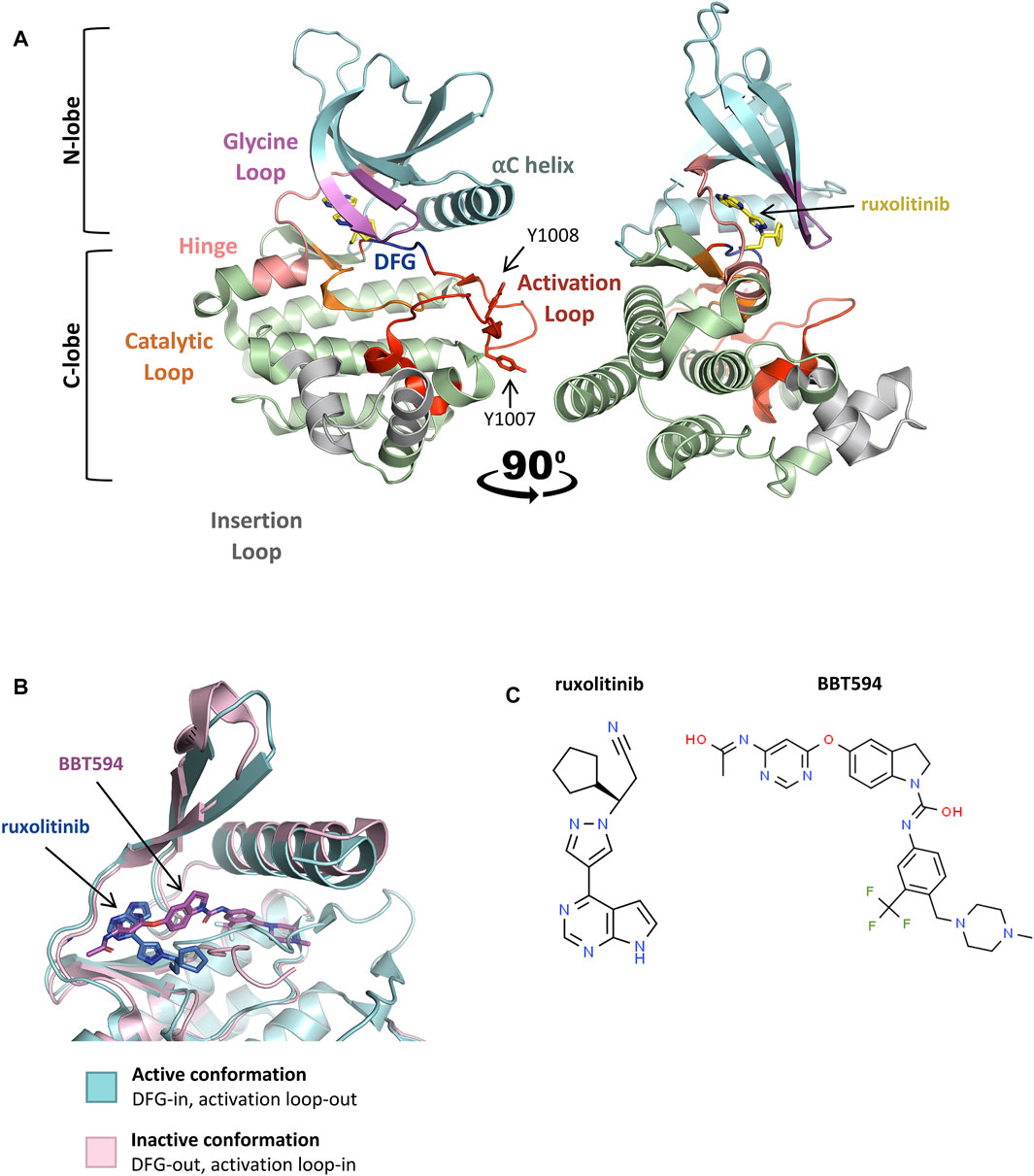
FIGURE 3. Active and inactive conformations of JAK2. (A) Co-crystal structure of the type-I JAK inhibitor, ruxolitinib, bound to the JAK2 kinase domain in the active conformation (PDB: 6VGL). Ruxolitinib (yellow) is presented in ball-and-stick representation with nitrogen atoms in blue. The JAK2 kinase domain is presented in ribbon representation with amino acid side chains shown for essential phosphotyrosine residues, JAK2 p. Y1007 and p. Y1008. The N-terminal lobe (residues 840–931), shown in cyan, comprises a 5-stranded antiparallel β-sheet and one α-helix (αC). The C-terminal lobe (residues 932–1,132), shown in green, comprises 8 α-helices, 3 3/10 helices, and 3 pairs of antiparallel β-strands. The glycine loop is colored in pink, the hinge region between the 2 lobes in peach, the catalytic loop in orange, the activation loop in red, DFG-motif in blue, and the insertion loop in grey. (B) Superimposition of the active (blue) and inactive (pink) conformations of the JAK2 kinase domain ATP-binding site within co-crystal structures bound to JAK inhibitors. Ruxolitinib (dark blue) and the type-II JAK inhibitor, BBT594 (dark pink), are bound to the active (PDB: 6VGL) and inactive (PDB: 3UGC) conformations respectively. JAK inhibitors are presented in ball-and-stick representations with oxygen atoms in red and nitrogen atoms in blue. The JAK2 activation loop is disordered in the inactive conformation. Structures were visualized with PyMOL 2.0 (Schrödinger, LCC). (C) 2D chemical structures of ruxolitinib (ChemSpider, CSID: 25027389) and BBT594 (ChemSpider, CSID: 34980928)
The JAK/STAT Signaling Pathway
The JAK/STAT pathway is the primary signaling pathway activated by JAK2, regulating the transcription of numerous genes involved in critical pleiotropic cell processes, particularly cell proliferation, differentiation and apoptosis (Vainchenker and Constantinescu, 2013; Brachet-Botineau et al., 2020). While many JAK/STAT genes are expressed ubiquitously, mouse knockout and patient data suggest the JAK/STAT pathway is critically involved in stimulatory (rather than inhibitory) responses of immune effector cells in both innate and adaptive immunity, including mucosal cell integrity (Ye et al., 2019). JAK3 is critical for γc receptor signaling in T cells and natural killer cells, resulting in severe immunodeficiency if mutated, whereas JAK1 and TYK2 have more pleiotropic roles (Ye et al., 2019; Musella et al., 2021). JAK2 activation occurs in response to a variety of cytokines and is essential for a plethora of normal cellular functions, particularly those involved in normal hematopoiesis (Levine et al., 2007; Vainchenker and Constantinescu, 2013; Akada et al., 2014). JAK2 knockout is embryonic lethal in mice (Neubauer et al., 1998), and is critical for signaling through homo-dimeric type-I cytokine receptors and some heterodimeric type-I receptors (Morris et al., 2018; Raivola et al., 2021). Juvenile mice with conditional JAK2 homozygous knockout demonstrate a rapid increase in HSC apoptosis and subsequent lethality (Akada et al., 2014; Fasouli and Katsantoni, 2021; Raivola et al., 2021). STAT1 and STAT2 are critical for suppressing intracellular viral and mycobacterial infections through type-I interferon receptors; STAT3 is critical for regulating immunoglobulin E production; STAT4 and STAT6 for CD4+ T-helper 1 and T-helper 2 responses in adaptive immunity, respectively; while STAT5a and STAT5b are more pleiotropic in function with roles in both myeloid cell proliferation and differentiation as well as mammary gland development mediated by prolactin (Benekli et al., 2003; Awasthi et al., 2021).
Inactive STATs exist primarily in the cytoplasm as anti-parallel, inactive homo- and hetero-dimers, formed through interactions between the coiled-coil (CC) domain and the DNA-binding domain of two different STAT monomers (Neculai et al., 2005; Morris et al., 2018). The anti-parallel conformation of these inactive STAT dimers places their SH2 domains on opposing ends, accessible for binding to SH2 phosphotyrosine docking sites on cytokine receptors (Mao et al., 2005; Neculai et al., 2005). The SH2 domains of different STAT proteins determine their affinity for different cytokine receptors (Woldman et al., 2001; Ivashkiv and Hu, 2004). Receptors that activate JAK2 predominantly bind STAT5 and STAT3, which are activated by JAK2-mediated phosphorylation of a single, conserved tyrosine residue at the C-terminal end, Y705 in STAT3 (Schaefer et al., 1997), Y694 in STAT5a (Barber et al., 2001), and Y699 in STAT5b (Azam et al., 1995)). The SH2 domains of each STAT monomer then reorientate to bind this phosphorylation site in the other monomer, facilitating a conformation change to produce parallel, active dimers with exposed DNA-binding domains (Figure 2B) (Babon et al., 2014; Morris et al., 2018). These now active STAT dimers are translocated and retained in the nucleus where they act as transcription factors to regulate gene expression (Schindler and Darnell, 1995; Vainchenker and Constantinescu, 2013). The promoter regions of these genes often harbor conserved STAT-binding motifs with interferon gamma-activated site (GAS)-like core sequences (Kang et al., 2013; Brachet-Botineau et al., 2020). JAK2 also activates other signaling pathways including RAS/MAPK, phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B (PI3K/PKB), and mammalian target of rapamycin (mTOR) pathways (Morris et al., 2018).
Strict regulation of JAK2 activity via a variety of negative feedback loops ensures an appropriate cellular response to cytokines (Babon et al., 2014; Hammaren et al., 2019b). The suppressor of cytokine signaling (SOCS1) and SOCS3 are the key intermolecular JAK2 negative regulators that are upregulated by JAK/STAT signaling (Starr et al., 1997; Babon et al., 2014; Hammaren et al., 2019b). All SOCS family proteins contain a central SH2 domain and a short C-terminal SOCS box domain (Kershaw et al., 2013b; Morris et al., 2018). The SH2 domains of SOCS1/3 bind specific phosphotyrosine motifs to inhibit JAK1/2 and TYK2, but not JAK3 (Babon et al., 2008; Liau et al., 2018). The SOCS box domain recruits the adaptor complex, elonginBC, RING-finger-domain-only protein (RBX2) and E3 ligase scaffolds, Cullins (Babon et al., 2008; Babon et al., 2009; Zhang et al., 2015). These E3 ligase complexes catalyze the polyubiquitination and subsequent proteasomal degradation of proteins bound by the SOCS SH2 domains including JAK2 or more commonly, its associated cytokine receptors (Babon et al., 2009; Babon et al., 2014; Zhang et al., 2015). SOCS1 and SOCS3 also contain a short kinase inhibitory region (KIR) motif upstream of their SH2 domains, which can inhibit JAK2 activity by sterically hindering substrate binding (Krebs and Hilton, 2001; Kershaw et al., 2013b). The KIR is an unstructured domain that by undergoing a conformation change, can bind within the JAK2 hydrophobic substrate binding pocket with non-ATP-competitive kinetics (Krebs and Hilton, 2001; Kershaw et al., 2013a). This enables simultaneous targeting of JAK2-associated receptors for degradation and direct JAK2 inhibition (Kershaw et al., 2013a; Kershaw et al., 2013b; Liau et al., 2018).
Another SH2-domain containing protein, the lymphocyte adaptor protein (LNK or SH2B3), also negatively regulates JAK2 activity (Babon et al., 2014; Morris et al., 2018). LNK inhibits JAK2 activity by directing binding regulatory JAK2 phosphotyrosine residues including JAK2 p. Y813, which lies within the JH1-JH2 linker region (Maslah et al., 2017). LNK may also inhibit signaling activation through JAK2 by competitively binding critical cytoplasmic phosphotyrosine residues on cytokine receptors (Maslah et al., 2017). Furthermore, JAK/STAT signaling can be negatively regulated by protein tyrosine phosphatases (PTPs), which dephosphorylate critical tyrosine residues within JAK2, STATs or JAK2-associated cytokine receptors (Bohmer and Friedrich, 2014). Cytoplasmic phosphatases that regulate JAK2 include protein tyrosine phosphatase non-receptor type 1 (PTPN1 or PTP1B), type 6 (PTPN6 or SHP1) and type 11 (PTPN11 or SHP2) (Babon et al., 2014). PTPN6 is primarily expressed in hematopoietic cells and inhibits JAK2 by binding and dephosphorylating JAK2 p. Y429 within the JAK2 SH2-like domain (Klingmüller et al., 1995; Bohmer and Friedrich, 2014). In contrast, PTPN11 is ubiquitously expressed and can positively or negatively regulate JAK/STAT signaling in different contexts (Hammaren et al., 2019b). JAK/STAT signaling can also be regulated through receptor phosphatases such as protein tyrosine phosphatase receptor type C (PTPRC or CD45) and type T (PTPRT) (Babon et al., 2014; Morris et al., 2018). PTPRT can dephosphorylate STAT3 p. Y705 (Zhang et al., 2007), whereas CD45 is highly expressed in hematopoietic cells and has been demonstrated to dephosphorylate all JAK family proteins in murine cells (Irie-Sasaki et al., 2001), and JAK1 and JAK3 in human cells (Yamada et al., 2002; Bohmer and Friedrich, 2014). The suite of JAK2 regulators highlights the critical role of strict JAK2 control for appropriate responses to cytokine stimulation in normal cells.
JAK2 Mutations in Ph-like ALL
Appropriate regulation of JAK/STAT signaling plays a critical role in the development and functional activation of crucial hematopoietic cells, including hematopoietic stem cells (HSCs) (Wang and Bunting, 2013; Fasouli and Katsantoni, 2021; Raivola et al., 2021). The importance of JAK2 in hematological malignancies became apparent in 2005 after four research groups identified a single missense mutation within the pseudokinase domain of JAK2; JAK2 p. V617F, as the primary driving alteration underlying most MPNs (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005; Silvennoinen and Hubbard, 2015a; Hubbard, 2018). Following identification of JAK2 p. V617F in 2005 (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005), an array of other JAK2 mutations have been identified in MPNs, myeloma, lymphoma, and chronic and acute leukaemias of either myeloid or lymphoid lineage (Lee et al., 2006; Krämer et al., 2007; Furqan et al., 2013; Vainchenker and Constantinescu, 2013; Fasouli and Katsantoni, 2021; Raivola et al., 2021). Gain-of-function mutations in JAK2 have been identified in the high-risk Ph-like ALL subtype, occurring exclusively with rearrangements of CRLF2 (CRLF2r), which lead to CRLF2 overexpression (Roberts et al., 2012; Roberts et al., 2014a; Boer et al., 2017; Tasian et al., 2017b; Pui et al., 2017; Reshmi et al., 2017; Steeghs et al., 2017). Approximately 50% of Ph-like ALL patients harbor CRLF2r, and roughly half of these patients also harbor activating point mutations in JAK1 or JAK2 (Table 1) (Mullighan et al., 2009a; Mullighan et al., 2009c; Russell et al., 2009; Pui et al., 2017; Reshmi et al., 2017). JAK alterations also occur in approximately 20% of Down-Syndrome ALL (DS-ALL) patients, with CRLF2r identified in approximately 60% of DS-ALL patients (Bercovich et al., 2008; Mullighan et al., 2009c; Hertzberg et al., 2010; Schwartzman et al., 2017; Page et al., 2018; Harvey and Tasian, 2020).
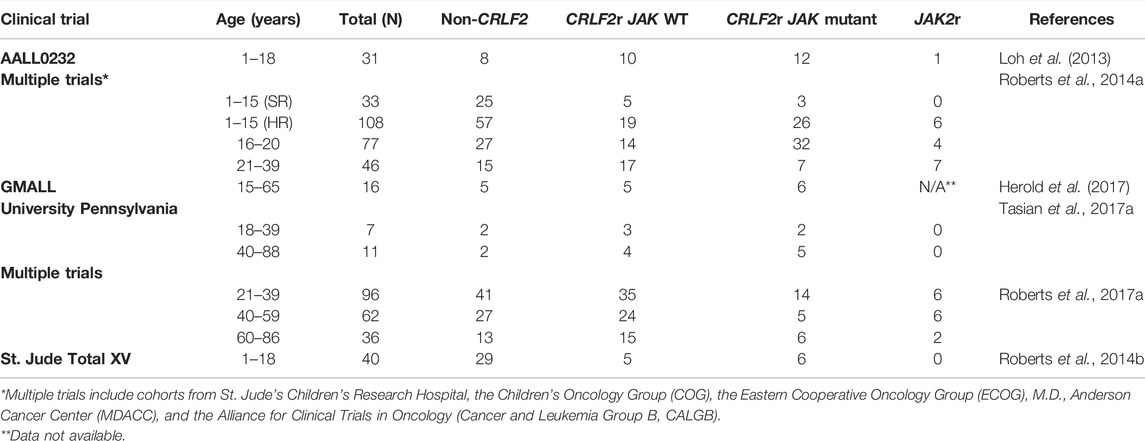
TABLE 1. Frequency of JAK2 mutations and JAK2 rearrangements within Ph-like ALL. Prevalence of CRLF2r/JAK mutant and JAK2r subtypes of Ph-like ALL compared with Ph-like ALL cases without CRLF2 overexpression.
JAK2 Exon 14 Mutations and the Molecular Activation Mechanisms of JAK2 p. V617F
Most JAK2 mutations associated with hematological malignancies encode missense mutations that localize within JAK2 exon 12 of SH2L-JH2 linker region, or within JAK2 exons 14 or 16 of the pseudokinase domain, highlighting these regions as oncogenic hot-spots for mutation (Figure 4A) (Mullighan et al., 2009c; Gnanasambandan and Sayeski, 2011; Silvennoinen and Hubbard, 2015b). Mutations within JAK2 exon 14 associate primarily with MPNs, where JAK2 p. V617F occurs in >95% of patients with polycythemia vera (PV), and ∼60% of patients with essential thrombocythemia (ET) or primary myelofibrosis (PMF) (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005; Vainchenker and Constantinescu, 2013; Silvennoinen and Hubbard, 2015a). Interestingly, the JAK2 p. V617F mutation has not been identified in ALL and only a single JAK2 exon 14 mutation, JAK2 p. L611S, has been reported in an ALL setting (Kratz et al., 2006; Funakoshi-Tago et al., 2009; Gnanasambandan and Sayeski, 2011; Jain et al., 2017; Konoplev et al., 2017). This suggests that JAK2 exon 14 mutations associate primarily with myeloid lineage diseases. Different JAK2 mutants have demonstrated varying affinities to lineage-specific cytokine receptors, which may explain phenotypic differences induced by different JAK2 mutations and their association with either myeloid or lymphoid lineage diseases (Yao et al., 2017).
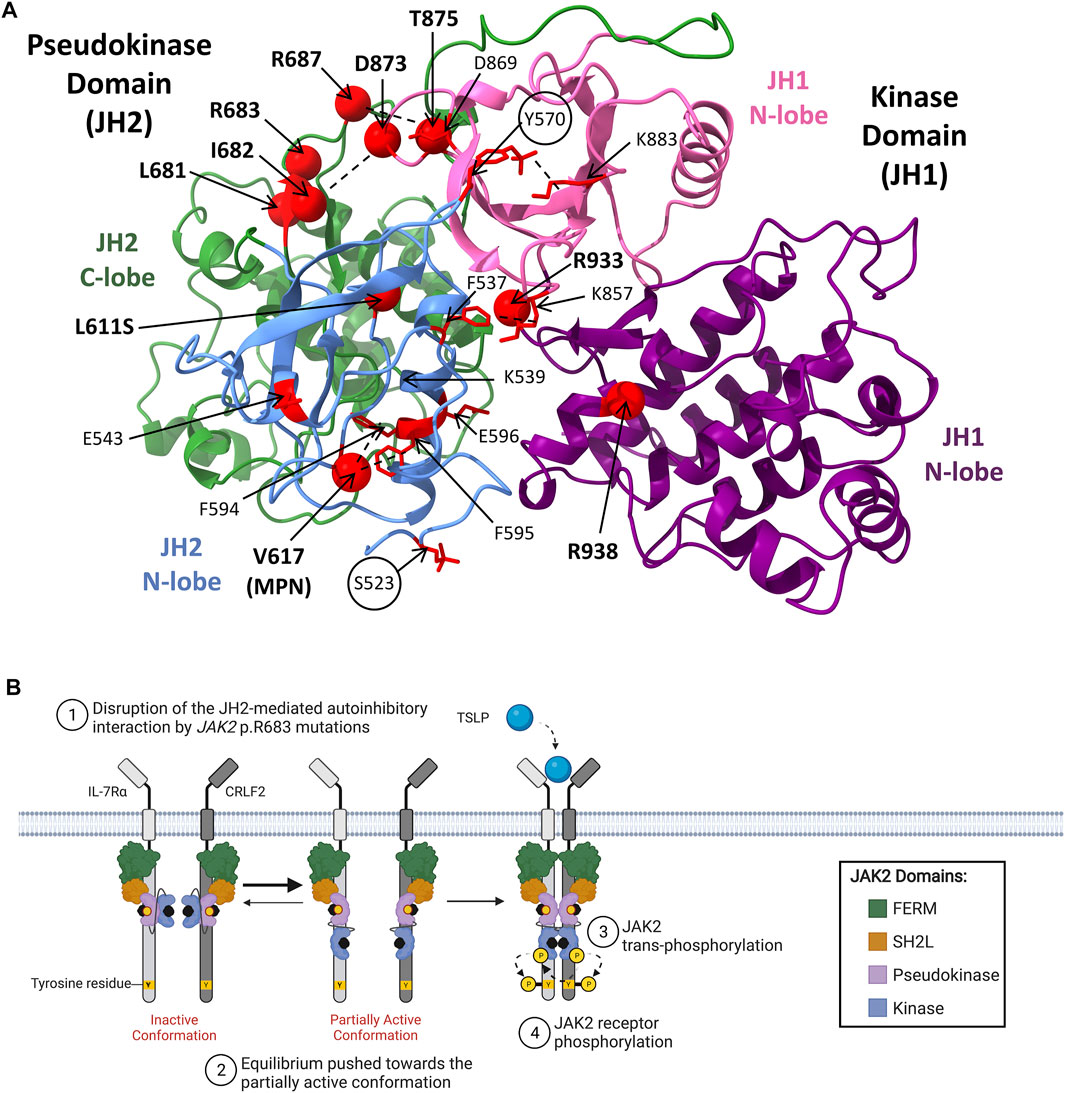
FIGURE 4. JAK2 mutations in hematological malignancies. (A) Model of JAK2 JH2-JH1 interface showing the positions of known activating JAK2 mutations. The JAK2 JH2-JH1 model was generated by Shan et al. (2014) using molecular dynamics simulations and annotated using ChimeraX-1.2.5 (University of California). The JH2 (pseudokinase domain) N-terminal (residues 536–629) and C-terminal (residues 630–839) lobe are colored in light blue and green respectively. The JH1 (kinase domain) N-terminal (residues 840–931) and C-terminal (932–1,132) lobes are colored in pink and purple respectively. Residues that when mutated are known to be activating are shown as red spheres (α carbon). Two critical inhibitory phosphorylation sites, pS523 and pY570, are encircled. Other key residues predicted to be involved in activating JAK2 mechanisms are colored in red and presented with amino acid side chains shown. Proposed interactions are represented by dashed lines. Figure adapted from Shan et al. (2014), Hammarén et al. (2019a), Leroy et al. (2016) and Lupardus et al. (2014). (B) Schematic representation of JAK/STAT signaling pathway activation through mutant-JAK2. CRLF2 (dark grey) heterodimerizes with IL-7Rα (light grey) to form the cytokine receptor for TSLP. The JAK2 FERM and SH2-like domains associate with the cytoplasmic juxtamembrane motifs of the receptor to recruit JAK2 to the cell membrane. The four domains of JAK2 are presented: FERM (green), SH2-like (orange), pseudokinase (JH2, purple), and kinase (JH1, blue). JAK2 is shown bound to ATP (black). The proposed model of mutant-JAK2 activation suggests that mutations such as JAK2 p. R683G (represented by a yellow sphere) disrupt the JH2-mediated autoinhibitory interaction with the kinase domain. This shifts the equilibrium of JAK2 from the inactive, auto-inhibited state towards the partially active state, supporting mutant-JAK2 dimerisation. Although mutant-JAK2 alone remains dependent on cytokine binding, additional mechanisms such as receptor overexpression may promote malignant transformation. Adapted from “Cytokine Signaling through the JAK-STAT Pathway” (BioRender.com, 2021).
Mutagenesis studies have demonstrated that JAK2 p. V617F confers cytokine-independent signaling activation (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Senkevitch and Durum, 2017). Activating JAK2 mutations, including JAK2 p. V617F, were initially predicted to confer cytokine-independent signaling through disruption of the JH2-mediated autoinhibitory interaction, facilitating mutant-JAK2 dimerization (Gnanasambandan and Sayeski, 2011; Ungureanu et al., 2011; Hubbard, 2018; Glassman et al., 2022). In addition, a recent report using single-molecule microscopy demonstrated that the JAK2 p. V617F mutation confers cytokine-independent dimerization of receptor subunits (50% of the maximum level for TPOR, 25% for EPOR and 10% for hGHR), with a stable time-dependent dimer formation similar to cytokine binding (Wilmes et al., 2020). However, introduction of JAK2 p. V617F into JAK2 JH2-JH1 protein fragments revealed that the JAK2 p. V617F mutation only resulted in a 3-fold increase in JAK2 catalytic activity, while deletion of the pseudokinase domain produced a 20-fold increase (Sanz et al., 2011). This suggested that disruption of the JH2-mediated autoinhibitory interaction alone is not sufficient to constitutively activate signaling through JAK2 (Hammaren et al., 2019a; Hammaren et al., 2019b). Leroy et al. (2016) and Glassman et al. (2022) have proposed two molecular mechanisms for JAK2 constitutive activation by JAK2 p. V617F (Leroy et al., 2016; Glassman et al.). The first mechanism destabilizes the JH2-JH1 autoinhibitory interaction through the formation of a π stacking interaction between JAK2 p. V617F and JAK2 p. F595, which stabilizes the JAK2 pseudokinase domain αC helix in a straightened conformation (Bandaranayake et al., 2012; Leroy et al., 2016; Hubbard, 2018). The second mechanism involves the formation of a positive regulatory interaction that favors dimerisation of active JAK2 monomers (Leroy et al., 2016; Glassman et al.). The combination of this positive regulatory interaction, in addition to the disruption of the JH2-mediated autoinhibition, may explain the high driving activity of the JAK2 p. V617F mutation (Hammaren et al., 2019a; Hammaren et al., 2019b).
JAK2Disruption of JH2-Mediated Autoinhibition and the role of CRLF2 Overexpression
In contrast to JAK2 p. V617F, the molecular mechanisms by which JAK2 mutations identified ALL disrupt JH2-mediated autoinhibition and facilitate JAK2 dimerization are yet to be fully elucidated (Hammaren et al., 2019a). JAK2 mutations reported in patients with ALL and DS-ALL are shown in Table 2. The transformative ability of all ALL-associated JAK2 mutations are dependent on cytokine receptor association, suggesting that these mutations are dependent on JAK2 dimerization (Lu et al., 2008; Wernig et al., 2008; Yao et al., 2017; Hammaren et al., 2019b). The majority of ALL-associated JAK2 mutations lie within JAK2 exon 16 (Table 2), where the most frequent mutations are JAK2 p. R683G/S (Mullighan et al., 2009a; Harvey et al., 2010; Pui et al., 2017; Kim et al., 2018). JAK2 exon 16 mutations all localize to the ATP-binding site of the JAK2 pseudokinase domain between the N- and C-terminal lobes (Figure 4A) (Gnanasambandan and Sayeski, 2011; Ungureanu et al., 2011; Bandaranayake et al., 2012). ATP binding to the JAK2 pseudokinase domain is essential for JAK2 activation as mutations within this region are known to suppress JAK2 kinase activity (Hammaren et al., 2015; Hammaren et al., 2019a). JAK2 p. R683 maps to the JAK2 pseudokinase domain β7-β8 loop and forms an ionic interaction with JAK2 p. D873 within the JAK2 kinase domain β2-β3 loop (Figure 4B) (Hammaren et al., 2019a; Hammaren et al., 2019b). Mutations of JAK2 p. R683 (Table 2) are predicted to disrupt this ionic interaction within the JH2-JH1 interface and hinder the JH2-mediated autoinhibitory interaction (Lupardus et al., 2014; Shan et al., 2014; Hammaren et al., 2019a; Hammaren et al., 2019b). Similarly, mutations of JAK2 p. L681 and p. I682 are predicted to alter the positioning of JAK2 p. R683, thereby disrupting JH2-mediated autoinhibition by affecting its interaction with JAK2 p. D873 (Li et al., 2015).
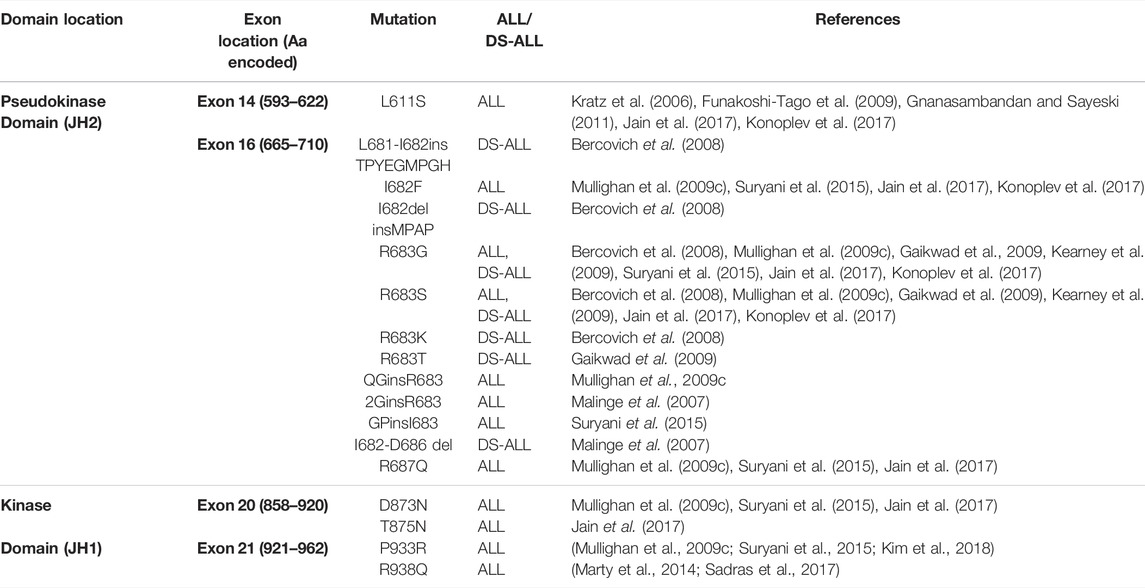
TABLE 2. All reported JAK2 mutations in patients with ALL. The majority of mutations reported in ALL and DS-ALL localize to JAK2 exon 14 or 16, encoding the JAK2 pseudokinase domain (JH2). Some mutations have also been reported to localize to JAK2 exon 20 or 21, encoding the JAK2 kinase domain (JH1). Amino acids (aa.) encoded by each JAK2 exon are shown.
While JAK2 mutations associated with ALL are predicted to disrupt JH2-mediated autoinhibition, these mutations alone are not sufficient to constitutively activate JAK2 (Hammaren et al., 2019a; Hammaren et al., 2019b). Instead, release of this autoinhibitory interaction may support the partially active conformation of JAK2, potentially exposing an interface to facilitate JAK2 dimerization (Figure 4A) (Hubbard, 2018). The high association of JAK1/2 mutations with CRLF2r (Mullighan et al., 2009a; Mullighan et al., 2009c; Russell et al., 2009; Pui et al., 2017; Reshmi et al., 2017) in ALL suggests that these events functionally cooperate to drive lymphoid transformation (Russell et al., 2009; Tasian and Loh, 2011; Kim et al., 2018). CRLF2 overexpression has been demonstrated to increase the proliferation of primary lymphoid progenitors (Russell et al., 2009). However, similar to JAK1/2 mutations, CRLF2 overexpression alone is not sufficient to transform cytokine-dependent cells (Russell et al., 2009; Roll and Reuther, 2010). Several groups discovered that murine pro-B cells expressing human CRLF2 can only drive cytokine-independent proliferation when co-expressed with ALL-associated JAK2 mutations (Mullighan et al., 2009a; Mullighan et al., 2009c; Hertzberg et al., 2010; Roll and Reuther, 2010; Yoda et al., 2010). As further support, a more recent study using transgenic mice demonstrated that while expression of CRLF2 alone in B-lineage hematopoietic cells did not induce B-ALL development, CRLF2 transgenic mice transplanted with fetal liver cells expressing JAK2 p. R683G or JAK2 p. P933R-mutant JAK2 succumbed to ALL disease within 10–20 days post-transplantation (Kim et al., 2018). These studies suggest that CRLF2r and JAK2 mutations cooperate to drive leukaemogenesis (Russell et al., 2009; Tasian and Loh, 2011; Kim et al., 2018), a fact which could be exploited for therapeutic advantage in Ph-like ALL. CRLF2 heterodimerizes with interleukin 7 receptor alpha chain (IL-7Rα) to form the thymic stromal lymphopoietin receptor (TSLPR) (Tasian and Loh, 2011; Bugarin et al., 2015; Page et al., 2018), and CRLF2r highly correlate with increased TSLPR surface expression (Bugarin et al., 2015; Konoplev et al., 2017; Pastorczak et al., 2018). Potentially, the combination of increased TSLPR expression and an increased ratio of JAK2 in the partially active conformation resulting from JAK2 mutations, cooperate to drive a leukaemic transformation.
There have also been some rare activating JAK2 mutations identified in ALL that localize to the JAK2 kinase domain (Mullighan et al., 2009c; Marty et al., 2014; Sadras et al., 2017; Hammaren et al., 2019b). These include JAK2 p. D873N, p. T875N, p. P933R, and p. R938Q (Mullighan et al., 2009c; Marty et al., 2014; Suryani et al., 2015; Jain et al., 2017; Sadras et al., 2017). JAK2 p. D873N and p. T875N that localize to JAK2 exon 20, encoding part of the JAK2 ATP-binding site that lies in the proximity of the glycine loop (Lucet et al., 2006). JAK2 p. D873N is expected to activate JAK2 through loss of its ionic interaction with JAK2 p. R683, weakening the JH2-JH1 autoinhibitory interaction to facilitate JAK2 dimerization (Chen C. et al., 2019; Hammaren et al., 2019a). Likewise, JAK2 p. T875N is proposed to weaken the JH2-JH1 autoinhibitory interaction via an allosteric mechanism involving the disruption of a hydrogen bond with JAK2 p. D873 (Dusa et al., 2010; Gnanasambandan and Sayeski, 2011; Chen C. et al., 2019). JAK2 exon 21 mutations, JAK2 p. P933R and p. R938Q, are also expected to disrupt the JH2-JH1 autoinhibitory interaction but these mutations map to the conserved JAK2 hinge region of the ATP-binding site (Lucet et al., 2006; Marty et al., 2014). The mechanism of JAK2 p. P933R activation is poorly understood, however, JAK2 p.938Q has been proposed to disrupt JH2-mediated autoinhibition through loss of an ionic interaction between JAK2 p. R867 and JAK2 p. D869 (Marty et al., 2014). Overall, all JAK2 mutations reported in ALL are predicted to weaken JH2-mediated autoinhibition, similar to JAK2 p. V617F, likely increasing the probability of receptor dimerization. However, the lack of an additional second molecular mechanism driven by JAK2 exon 16 mutations, unlike JAK2 p. V617F, may explain why ALL-associated JAK2 mutations require CRLF2 overexpression to cooperatively drive malignant transformation and subsequent leukemogenesis.
JAK2 Rearrangements in Ph-like ALL
In addition to JAK2 mutations, JAK2 rearrangements have been associated with various myeloid and lymphoid hematological malignancies (Furqan et al., 2013; Vainchenker and Constantinescu, 2013; Levavi et al., 2019; Raivola et al., 2021). The ETV6::JAK2 (ETS variant transcription factor 6/JAK2) fusion (initially known as TEL::JAK2) was the first JAK2r identified by cytogenics in both ALL and CML patients 1997 and was the first JAK2 alteration demonstrated to induce constitutive activation of JAK2 (Lacronique et al., 1997; Peeters et al., 1997; Raivola et al., 2021). The JAK2 fusion proteins encoded by these JAK2r comprise the N-terminus of a fusion partner and the C-terminus of JAK2 (Figure 4B) (Ho et al., 2010; Babon et al., 2014; Boer and den Boer, 2017). For example, the rearrangement between BCR and JAK2 produces the BCR::JAK2 fusion gene (Figure 5A). All reported JAK2 fusion genes retain JAK2 exons 19-25 encoding the kinase domain (Table 3), however the influence of the 5’ fusion partner gene is not well characterized. A diverse range of JAK2 fusion partner genes have been reported across different lymphoid and myeloid malignancies (Levavi et al., 2019). There have been 94 reported cases of JAK2r in ALL (Table 3), in comparison, only four cases of JAK2r have been reported in MPNs, including BCR::JAK2, PCM1::JAK2 (pericentriolar material 1/JAK2), RPN1::JAK2 (ribophorin 1/JAK2) and PEX14::JAK2 (peroxisomal biogenesis factor 14/JAK2) (Murati et al., 2005; Mark et al., 2006; Elnaggar et al., 2012; Lundberg et al., 2014; He et al., 2016; Levavi et al., 2019). Albeit in very low numbers, JAK2r have also been identified in solid tumors including breast cancer (Quesada et al., 2021) and small lung cancer (Iwakawa et al., 2013; Levavi et al., 2019), but these particular JAK2r have not been reported in any hematological malignancies.
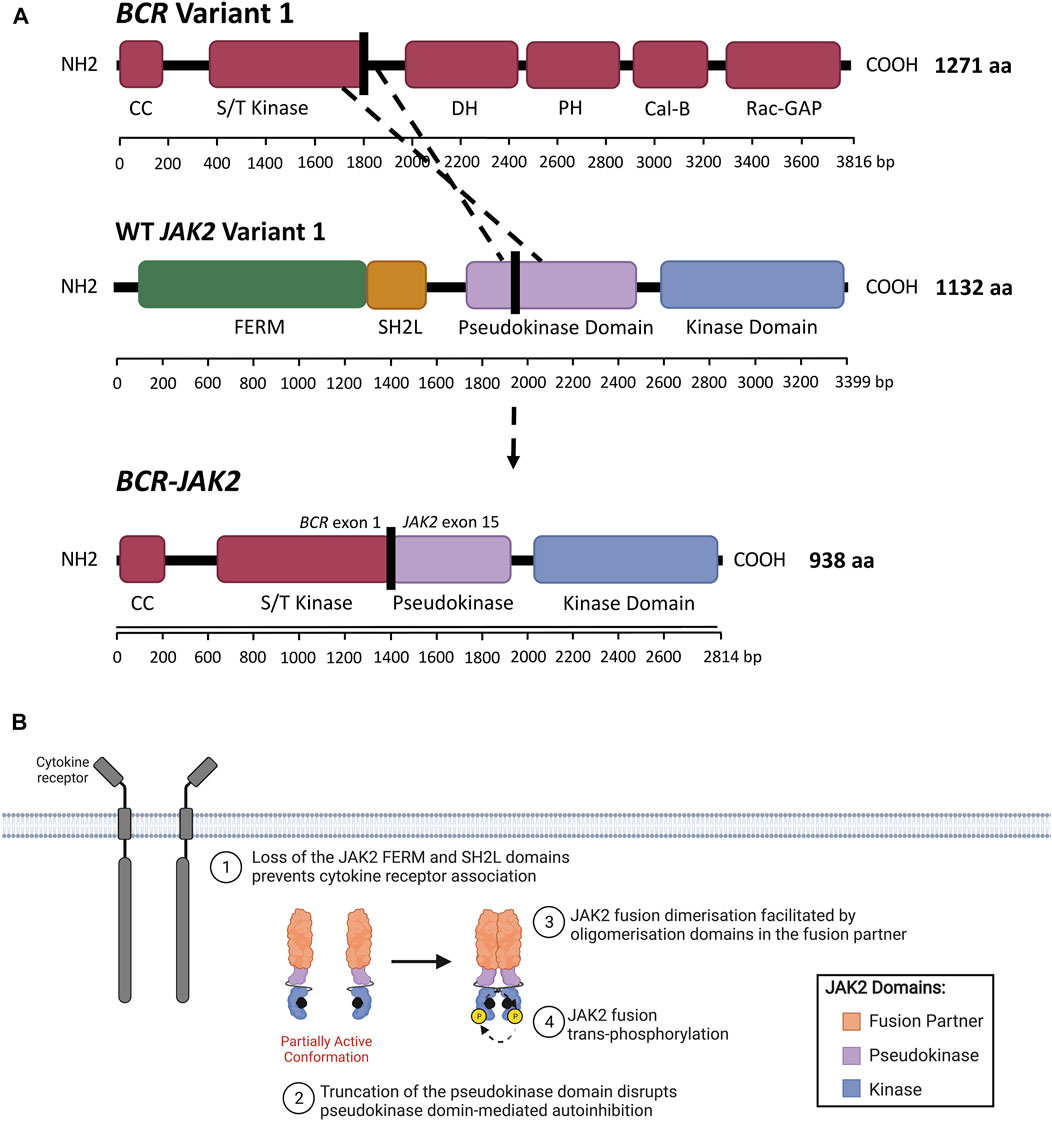
FIGURE 5. JAK2 fusion proteins in ALL. (A) Schematic representation of a genomic rearrangement between JAK2 exon 15 and BCR exon 1 that produces the BCR-JAK2 fusion gene. BCR isoform 1 (encoded by BCR variant 1) contains the following domains: BCR coiled-coil (CC), serine/threonine kinase (S/T kinase), DH (Dbl homology), PH (pleckstrin homology), Cal-B (calcium-dependent lipid-binding) and Rac-GAP (Rac GTPase-activating protein) domains. The BCR DH and PH domains form the Rho-GEF domain (Rho guanine nucleotide exchange factor). JAK2 isoform A (encoded by JAK2 variant 1) contains FERM (4.1 protein, ezrin, radixin, moesin), SH2-like (SH2L, Src homology 2), pseudokinase (JH2) and kinase (JH1) domains. The BCR-JAK2 fusion protein retains the BCR CC and S/T kinase domains, three exons of the JAK2 pseudokinase domain and the full-length JAK2 kinase domain. BCR-JAK2 is predicted to homodimerise via its retained BCR CC motif. Domains encoded by the BCR, JAK2 and BCR-JAK2 transcripts were annotated using InterPro (EMBL-EBI, 2021) (Jones et al., 2014; Blum et al., 2021) and Maru amd Witte (1991). (B) Schematic representation of JAK/STAT signaling pathway activation through JAK2 fusions. All JAK2 fusions comprise of an N-terminal fusion partner (orange) and the full-length JAK2 kinase domain (JH1, blue). The full-length or truncated JAK2 pseudokinase domain (JH2, purple) may also be present or absent in different JAK2 fusions. The absence of the JAK2 FERM and SH2-like domains prevent JAK2 fusions from associating with the cytoplasmic juxtamembrane motifs of cytokine receptors (dark grey). JAK2 fusions are shown bound to ATP (black). The proposed model of JAK2 fusion activation suggests that oligomerization domains within the fusion partner may facilitate JAK2 fusion dimerisation and subsequent trans-phosphorylation, promoting malignant transformation. Adapted from “Cytokine Signaling through the JAK-STAT Pathway” (BioRender.com, 2021).
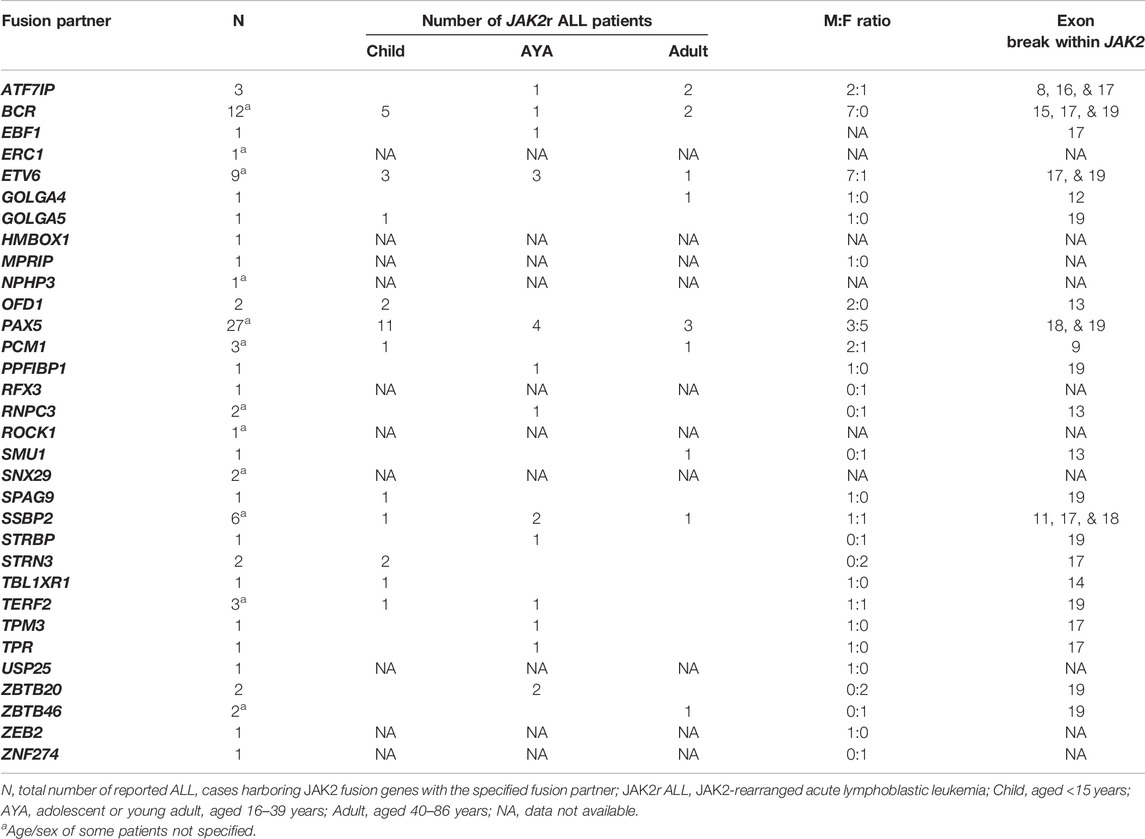
TABLE 3. Reported JAK2 fusion gene partners in patients with JAK2r ALL. For more details and corresponding references see Supplementary Table S1. The JAK2 pseudokinase domain is encoded by JAK2 exons 13-18.
JAK2r in B-ALL are identified exclusively within the Ph-like subtype, occurring in approximately 5% of pediatric Ph-like ALL cases (<15 years) with the highest frequency in young adult patients (16–39 years) (∼14%) (Table 1) (Roberts et al., 2012; Roberts et al., 2014a; Imamura et al., 2016; Roberts K. G. et al., 2017; Boer et al., 2017; Tasian et al., 2017b; Jain et al., 2017; Reshmi et al., 2017). In MPNs, JAK2r are associated with a more aggressive phenotype than fusions involving other kinase genes such as PDGFRA (platelet-derived growth factor receptor A), and long-term remission can often only be achieved after allogenic stem cell transplantation (Allo-SCT) (Schwaab et al., 2015; Schwaab et al., 2020). Similarly, JAK2r in ALL are associated with the poorest outcomes compared with other Ph-like ALL subtypes (Roberts K. G. et al., 2017; Jain et al., 2017; Iacobucci and Roberts, 2021). All reported JAK2 fusion genes retain JAK2 exons 19-15 encoding the kinase domain (Table 3) and the chimeric JAK2 fusion proteins encoded by these JAK2 fusion genes have demonstrated constitutive JAK2 kinase activation (Cuesta-Domínguez et al., 2012; Roberts et al., 2012; Roberts et al., 2014a; Schinnerl et al., 2015; Boer and den Boer, 2017; Steeghs et al., 2017). In contrast to JAK2 mutations, expression of JAK2r in primary murine pre-B cells results in cytokine-independent proliferation, suggesting that JAK2 fusion genes alone are driving genomic lesions in JAK2r ALL (Cuesta-Domínguez et al., 2012; Roberts et al., 2014a; Schinnerl et al., 2015). Over 30 different JAK2 fusion partner genes have been identified in Ph-like ALL to date, the most common of which is PAX5 (Paired box 5) (28.7%) (Table 3, Supplementary Table S1) (Roberts et al., 2012; Roberts et al., 2014a; Yano et al., 2015; Imamura et al., 2016; Roberts K. G. et al., 2017; Boer et al., 2017; Reshmi et al., 2017; Li et al., 2018; Schwab and Harrison, 2018; Gu et al., 2019; Tang et al., 2019). Other commonly identified JAK2 fusion partners in ALL are BCR::JAK2 (12.8%), ETV6::JAK2 (9.6%), SSBP2 (single stranded DNA binding protein 2/JAK2) (6.4%) and ATF7IP (activating transcription factor 7 interacting protein) (3.2%) (Table 3, Supplementary Table S1) (Roberts et al., 2014a; Roberts K. G. et al., 2017).
Similar to other Ph-like ALL subtypes, JAK2r often co-occur with deletions in genes involved in B-cell development including IKZF1 (IKAROS family zinc finger 1) (Mullighan et al., 2008; Mullighan et al., 2009b). The most common IKZF1 alteration associated with Ph-like (and JAK2r) ALL is a deletion of IKZF1 exons 3-6, encoding the dominant negative IK6 isoform of IKAROS, which lacks the N-terminal DNA binding domain (Roberts et al., 2014a; Tran et al., 2018; Shiraz et al., 2020). IKAROS IK6 is unable to bind DNA to regulate the expression of genes required for B-cell differentiation, implying that JAK2r and IKZF1 deletions both drive deregulation of B-cell maturation and promote development of B-ALL (Mullighan et al., 2009b; Harvey et al., 2010; Pui et al., 2017). IKZF1 alterations are also associated with inferior event-free survival in Ph-like ALL patients (Mullighan et al., 2009b; Van der Veer et al., 2013; Roberts et al., 2014a). A number of other genomic alterations also co-occur with JAK2 fusion genes and often involve B-cell pathways, including deletions of PAX5, BTG1 (BTG anti-proliferation factor 1), and CDKN2A/B (cyclin-dependent kinase inhibitor 2A/B) (Roberts et al., 2012; Roberts et al., 2014a; Boer et al., 2015; Kawamura et al., 2015; Roberts K. G. et al., 2017). Deletions of RAG1/2 (recombination-activating gene 1 and 2), VPREB (V-set pre-B cell surrogate light chain 1), EBF1 (EBF transcription factor 1), RUNX1 (RUNX family transcription factor 1), BTLA (B and T lymphocyte associated), CD200 (CD200 molecule) and ETV6 have also been reported to co-occur with JAK2 fusion genes, as well as mutations within IKZF1, KRAS (KRAS proto-oncogene), SETD1 (SET domain-containing 1A), and PTPN11 (Roberts et al., 2012; Roberts et al., 2014a; Boer et al., 2015; Roberts K. G. et al., 2017). Although prognostic significance of some of these alterations have been demonstrated in Ph-like ALL (Tran and Loh, 2016; Roberts K. G. et al., 2017; Pui et al., 2017; Roberts et al., 2018; Tran et al., 2018; Zhang et al., 2019), the influence of these additional alterations on JAK2r patient survival rates is not well elucidated.
Cytokine-Independent Oligomerization
In contrast to JAK2 mutations, the molecular mechanism by which JAK2 fusion genes lead to constitutive JAK2 activation remains largely unknown. The JAK2 regions encoding the full-length JAK2 FERM domain are absent in all reported JAK2 fusion genes (Table 3, Figure 2A). The presence of the JAK2 FERM domain has been demonstrated to be critical for JAK2 localization to the plasma membrane (Zhao et al., 2010) and JAK/STAT signaling activation (Eder-Azanza et al., 2017). The absence of FERM and SH2-like domains in JAK2r likely prevents binding of JAK2 fusions to membrane-associated cytokine receptors, implying these fusion products can promote signaling in the absence of cytokine. Considering the critical role of cytokine-mediated receptor dimerization in WT JAK2 activation (Silvennoinen and Hubbard, 2015a), activation of JAK2 fusions, unlike mutant-JAK2, likely occurs via a mechanism that does not require receptor association. The normal function and tissue specificity of JAK2 fusion partner genes is diverse and their typical expression, or lack of expression, with B-cells is varied (Table 3, Supplementary Table S2). However, the majority of these JAK2 fusion partners have the ability to oligomerize (Supplementary Table S2), suggesting that JAK2 fusion activation occurs through direct homodimerization. The proposed model suggests that JAK2 fusions oligomerize via the presence of oligomerization domains within the N-terminal fusion partner (Medves and Demoulin, 2012). The most common of these oligomerization domains are CC motifs, present in 44% of JAK2 fusion partners reported in ALL, including BCR:JAK2 (Figure 5A) (Cuesta-Domínguez et al., 2012; The UniProt Consortium, 2019). These oligomerization domains may facilitate JAK2 fusion trans-phosphorylation (Figure 5B), however, the quaternary structure of different JAK2 fusions has not yet been elucidated and there are limited published studies investigating the functional impact of JAK2 fusion partner oligomerization domains (Medves and Demoulin, 2012).
Disruption of the BCR CC motif within the BCR:ABL1 fusion has been shown to abrogate the transformative ability of BCR:ABL1 (Beissert et al., 2008; Mian et al., 2009), suggesting that the BCR CC motif is essential for BCR:ABL oligomerization and subsequent constitutive activation. The therapeutic potential of CC mimetics is now being investigated but may be amenable to CC-containing JAK2r (Dixon et al., 2012; Bruno and Lim, 2015; Woessner et al., 2015; Peiris et al., 2020). The helix-loop-helix (HLH) domain, also known as the sterile alpha motif (SAM) or pointed (PNT) domain, is another oligomerization domain that can facilitate self-association (Medves and Demoulin, 2012; Hock and Shimamura, 2017). Deletion of the ETV6 HLH domain has been shown to abrogate the transforming kinase activity of ETV6:LYN (ETV6/tyrosine-protein kinase Lyn) (Takeda et al., 2011) and ETV6:JAK2 (Lacronique et al., 1997) fusion proteins. This suggests that the ETV6 HLH domain may enable constitutive activation of ETV6:JAK2 and EBF1:JAK2 fusions by facilitating JAK2 fusion homodimerization (Medves and Demoulin, 2012; Hock and Shimamura, 2017). Other domains within JAK2 fusion partners that may facilitate JAK2 fusion oligomerization include BR-C, ttk and bab (BTB) domains, scan motifs and LIS1 homology (LisH) domains, but there are likely more oligomerization domains to be identified (Supplementary Table S2) (Poitras et al., 2008; Tijchon et al., 2013). BTB domains are present in ZBTB20 (Zinc finger and BTB domain-containing 20) and ZBTB46, scan motifs in ZNF274 (Zinc finger protein 274), and LisH domains in SSBP2 (single stranded DNA binding protein 2) (Poitras et al., 2008; Tijchon et al., 2013) (Supplementary Table S2).
JAK2 fusions may also be trans-phosphorylated through indirect oligomerization such as via recruitment to larger protein complexes such as centrioles, spliceosomes, nuclear pore complexes (NPCs), or telomere nucleoprotein complexes (Medves and Demoulin, 2012). For example, OFD1:JAK2 ((OFD1 centriole and centriolar satellite protein/JAK2) and PCM1:JAK2 (pericentriolar material 1/JAK2) may be activated by indirect oligomerization at centriolar satellites, as both OFD1 and PCM1 are components of centrioles (Supplementary Table S2) (Medves and Demoulin, 2012; Lee and Stearns, 2013). To support this, a kinase fusion containing the centrosome protein, FGFR1 oncogene partner (FOP), was demonstrated to localize to centriolar satellites where tyrosine phosphorylation was increased (Lee and Stearns, 2013). FOP shares homology with OFD1 and co-localizes with PCM1 (Lee and Stearns, 2013). In addition, some domains and regulatory sites retained within the JAK2 fusion partner could mediate interactions that facilitate JAK2 fusion activation or contribute to leukemogenesis. For example, tyrosine residues within the fusion partner could be phosphorylated and influence intracellular signaling by enabling recruitment of proteins containing SH2 domains (Medves and Demoulin, 2012). However, the significance of these potential interactions to overall cell transformation and disease phenotype is debated (Medves and Demoulin, 2012). Further research is required to understand whether these potential interactions are retained or whether higher order protein complexes can form.
Alternate Mechanisms of JAK2 Fusion Activation
Constitutive activation of the majority of JAK2 fusions likely occurs through a cytokine-independent oligomerization mechanism. However, unlike all other reported JAK2 fusions, PAX5:JAK2 does not harbor an oligomerization domain or self-associate, yet still constitutively activates JAK/STAT signaling similar to other JAK2r (Schinnerl et al., 2015; Sakamoto et al., 2017; Jurado et al., 2022). This suggests that PAX5:JAK2 may be activated via a mechanism distinct from cytokine-independent oligomerization (Schinnerl et al., 2015). PAX5:JAK2 is the only JAK2 fusion protein that has been shown to localize within the nucleus due the presence of a nuclear localization signal (NLS) within the PAX5 fusion partner (Schinnerl et al., 2015). Potentially, PAX5:JAK2 may constitutively activate JAK/STAT signaling by phosphorylation of nuclear STATs (Schinnerl et al., 2015). PAX5:JAK2 also retains the ability of PAX5 to act as a transcription factor, binding and activating PAX5 target loci through its paired domain (Schinnerl et al., 2015; Jurado et al., 2022). Similarly, the majority of JAK2 fusion partners are transcription factors containing DNA-binding domains including CC, HLH, zinc finger C2H2 type, or leucine zippers. Two other JAK2 fusion proteins, ATF7IP:JAK2 and TERF2:JAK2 (Telomeric repeat binding factor 2/JAK2), also contain NLSs but their localization has not been investigated to date, nor has their ability to bind DNA. In addition, many of these DNA-binding JAK2 fusion partners can act as tumor-suppressors, and their DNA-binding domains may also function as oligomerization domains (Medves and Demoulin, 2012). Therefore, oligomerization between JAK2 fusions and their endogenous JAK2 fusion partner may contribute to leukemogenesis by impairing the tumor-suppressive function of the WT JAK2 fusion partner (Medves and Demoulin, 2012). For example, HLH-mediated oligomerization between WT ETV6 and ETV6:JAK2 may reduce the availability of the endogenous ETV6 HLH motif, which normally maintains long-term transcriptional repression of genes by interaction with Polycomb group complexes (De Braekeleer et al., 2012).
Interestingly, a study by Fortschegger et al. (2014) demonstrated that PAX5:JAK2 phosphorylation occurs independently of DNA-binding or trans-phosphorylation by another kinase (Schinnerl et al., 2015). Fortschegger et al. (2014) hypothesized that the absence of the JAK2 pseudokinase domain within PAX5:JAK2 may enable constitutive activation of PAX5:JAK2 by preventing JH2-mediated pseudokinase domain auto-inhibition (Schinnerl et al., 2015). Consistent with this hypothesis, loss of the JH2-JH1 autoinhibitory interaction by either deletion of JAK2 JH2 or the destabilizing JAK2 p. F739R mutation has been shown to increase basal JAK2 kinase activity (Saharinen et al., 2000; Saharinen and Silvennoinen, 2002; Hammaren et al., 2015). Therefore, that truncation or deletion of this domain in JAK2 fusion genes may contribute to constitutive activation of JAK2 fusions such as PAX5:JAK2. The JAK2 pseudokinase domain (encoded by JAK2 exons 13–18) is either absent or truncated in most JAK2 fusion genes (Table 3). Only four JAK2 fusions contain full-length pseudokinase domains, including GOLGA5:JAK2 (Golgin A5/JAK2) (Ding et al., 2018), OFD1:JAK2 (Yano et al., 2015; Imamura et al., 2016), RNPC3:JAK2 (RNA binding region containing 3/JAK2) (Chen X. et al., 2019; Chen et al., 2021), SMU1:JAK2 (SMU DNA Replication Regulator and Spliceosomal Factor/JAK2) (Roberts K. G. et al., 2017) (Table 3) and it is currently unknown whether these fusions display less JAK2 kinase activity in comparison to JAK2 fusions that harbor truncated or deleted pseudokinase domains. The functional effects of different truncations of the JAK2 pseudokinase domain is also unknown.
In addition, one publication reported that JAK2 was highly expressed in pediatric JAK2r B-ALL patients in comparison to non-Ph-like B-ALL patients (Steeghs et al., 2017). JAK2 is expressed at a low level in normal B-cells, in comparison to some JAK2 fusion partner genes that are highly expressed in normal B-cells (Supplementary Table S2). This high-level expression may result from the JAK2r being placed under the control of the JAK2 fusion partner’s promoter. However, no other reports specify whether JAK2r are highly expressed in patients and the importance JAK2r transcript expression levels are currently unknown. Although overexpression may be suggested to contribute the leukaemic potential of JAK2r, overexpression of WT JAK2 alone is not transforming in vitro (Yoda et al., 2010), suggesting that JAK2r transcript expression levels are of marginal importance. Overall, the impact of the fusion partner within JAK2 fusion genes is largely unknown but cytokine-independent oligomerization is predicted to be the driving mechanism behind JAK2 fusion constitutive activity. There are also several other potential mechanisms by which JAK2 rearrangements may contribute to upregulated downstream signaling including loss of JH2-mediated autoinhibition and upregulation of gene expression. Further research assessing the biological phenotypes of different JAK2 fusion partner genes and different breakpoints within JAK2 is required and may potentially reveal novel regulation mechanisms.
JAK2 as a Target for Precision Medicine in ALL
ALL patients harboring JAK2 alterations are currently treated with multi-agent chemotherapy and corticosteroids (Terwilliger and Abdul-Hay, 2017). Allo-SCT following high-dose chemotherapy improves survival in selected patients. (Terwilliger and Abdul-Hay, 2017). However, these intense regimens result in a number of acute and chronic side effects and are accompanied by an increased risk of treatment-related mortality (Senkevitch and Durum, 2017). Intensive chemotherapy regimens are often poorly tolerated in adults and the elderly due to toxicity and an increased occurrence of co-morbidities, a contributor to poor outcomes in these age groups (Terwilliger and Abdul-Hay, 2017). Immunotherapies, such as blinatumomab and chimeric antigen receptor (CAR) T-cell immunotherapy, have proven their effectiveness as salvage therapy in B-ALL (Inaba and Pui, 2019; Zhao et al., 2019). They are now being incorporated into frontline therapy for high risk disease, and may enable the dosage and duration of chemotherapy to be reduced to alleviate toxicity (Inaba and Pui, 2019; Zhao et al., 2019). Underscoring the importance of JAK2 in the pathogenesis of ALL, Roberts et al. (2014a) reported 5-years event-free survival (EFS) rates of 38.8% for CRLF2r/JAK-mutant ALL patients and 26.1% for ALL patients harboring a rearrangement of either JAK2 or EPOR (Figure 1) (Roberts et al., 2014a), subsequently reported as 23.5% in a later study (Roberts K. G. et al., 2017). In both studies, these 5-years EFS were significantly inferior to non-Ph-like ALL subtypes (Roberts et al., 2014a; Roberts K. G. et al., 2017). The poor outcomes associated with JAK2 alterations in ALL highlights the urgent need for more effective and less toxic treatment strategies for these high-risk patients (Roberts and Mullighan, 2015). Targeting of JAK2 with small molecule inhibitors in combination with chemotherapy may be one such therapeutic approach, given the remarkable success of TKIs for the treatment of CML (Ali, 2016).
TKIs as a Paradigm for Targeted Therapy
Direct inhibition of BCR:ABL1 using TKIs has served as a paradigm for the application of targeted therapies (Ali, 2016; Mughal et al., 2016). The first TKI identified to successfully inhibit BCR:ABL1 kinase activity was STI571, now known as imatinib (Druker et al., 1996; Apperley, 2015). This first-generation TKI is classified as a type-II inhibitor, as it inhibits BCR:ABL1 kinase activity by competitively binding the inactive conformation of ABL1 within the ATP-binding site (Druker and Lydon, 2000; Schindler et al., 2000; Rossari et al., 2018). CML patients who achieve a deep molecular response on imatinib therapy for 2 or more years can now expect a normal life expectancy (Gambacorti-Passerini et al., 2011), and 10-years overall survival rates have improved from less than 20% prior to 1982, to now around 83% (Druker et al., 2006; Mughal et al., 2016; Hochhaus et al., 2017). Identification of imatinib resistance, often acquired through the emergence of point mutations within the BCR:ABL1 kinase domain, has since driven the development of second- and third-generation TKIs (Zabriskie et al., 2014; Patel et al., 2017; Pottier et al., 2020; Shoukier et al., 2021). There are now six TKIs that are FDA-approved for the treatment of CML including imatinib, nilotinib, dasatinib, bosutinib, ponatinib and asciminib (Hughes et al., 2019; Shoukier et al., 2021). Incorporation of imatinib into treatment approaches for Ph+ ALL has also drastically improved EFS rates, from 27% to 72% (Senkevitch and Durum, 2017), suggesting that similar approaches may also be successful for the treatment of JAK2-altered ALL. The success of TKIs as a precision medicine approach for targeting BCR:ABL1 in CML and Ph+ ALL launched a new era of discovery into targeted cancer therapies (Sawyers, 2003; Rossari et al., 2018). In particular, the development of small molecule inhibitors of other constitutively active kinases were pursued to potentially treat a variety of other diseases and malignancies (Sawyers, 2003; Zhang et al., 2009; Cohen et al., 2021).
JAK2 as a Therapeutic Target
The identification of the JAK2 p. V617F mutation underlying the majority of MPNs positioned JAK2 as an attractive molecular target for small molecule screening and development (Constantinescu, 2009; Kumar et al., 2009). Targeted JAK2 inhibitors entered clinical development just 6 years following the first report of JAK2 p. V617F (Levine et al., 2007; Pardanani, 2007). In 2011, the semi-selective JAK1/2 inhibitor, ruxolitinib (Figure 3C), was the first JAK2 inhibitor to be FDA-approved for the treatment of MF and hydroxyurea resistant PV (Vannucchi et al., 2015b; Passamonti et al., 2017), followed by approval of the JAK2 specific inhibitor, fedratinib, for the treatment of MF in 2019 (Harrison et al., 2017; Mullally et al., 2020; Venugopal and Mascarenhas, 2020). Both ruxolitinib and fedratinib are classified as type-I JAK inhibitors, competitively binding within the ATP-binding site of JAK2 in the active (DFG-in) conformation (Figure 3A) (Leroy and Constantinescu, 2017). Ruxolitinib therapy can limit further bone marrow fibrosis in JAK2 p. V617F-driven MF and PV (Verstovsek et al., 2017b; Kroger et al., 2021) and multiple studies have shown that ruxolitinib therapy correlates with improved overall survival (Verstovsek et al., 2012a; Vannucchi et al., 2015a; Bose and Verstovsek, 2020; Kroger et al., 2021). However, the significance of this survival benefit is debated due to statistical limitations of the pioneer COMFORT-1 (NCT00952289) and COMFORT-2 (NCT00934544) trials (Passamonti et al., 2015; Cervantes and Pereira, 2017). Despite these limitations, ruxolitinib therapy significantly reduces splenomegaly, which is known to correlate with improved overall survival and can also improve patients’ quality of life (Verstovsek et al., 2012b; Vannucchi et al., 2015a; Harrison et al., 2016; Verstovsek et al., 2017a; Cervantes and Pereira, 2017; Bose and Verstovsek, 2020). Sustained symptomatic reductions have also been reported in MF patients who remain on long-term ruxolitinib therapy (Harrison et al., 2016; Verstovsek et al., 2017b).
Unfortunately, the use of ruxolitinib and fedratinib in MF has revealed a number of issues related to JAK2 as a therapeutic target and the consequences of type-I JAK2 inhibition. Ruxolitinib does not significantly reduce the mutant allele frequency. In a study by Deininger et al. (2015), ruxolitinib treatment reduced the JAK2 p. V617F allele burden by >50% in only 12% of 236 MF patients (Deininger et al., 2015). Several studies have also reported a lack of significant spleen responses in a proportion of patients, where there was a less than 35% reduction in spleen volume (Harrison et al., 2012; Harrison et al., 2016; Verstovsek et al., 2017b; Gupta et al., 2020; Palandri et al., 2020). Furthermore, the majority of ruxolitinib-treated MF patients discontinue therapy due to dose-dependent adverse events, including thrombocytopenia and anemia (Harrison et al., 2016; Kuykendall et al., 2018; Bewersdorf et al., 2019; Palandri et al., 2020). The toxicity associated with ruxolitinib may be due to suppression of other JAK family kinases, with 6-fold selectivity over TYK2 and 130-fold selectivity over JAK3 (Quintas-Cardama et al., 2010). In addition, treatment discontinuation has been associated with severe ruxolitinib discontinuation syndrome, which is most likely caused by a rebound cytokine storm driven by the sudden release of accumulated phosphorylated JAK2 (pJAK2) (Coltro et al., 2017; Tvorogov et al., 2018; Palandri et al., 2021; Ross et al., 2021). Tvorogov et al. (2018) suggested that ruxolitinib binding promotes pJAK2 accumulation by preventing JAK2 dephosphorylation and degradation (Tvorogov et al., 2018; Ross et al., 2021). Despite the dose-dependent toxicity, low efficacy and the withdrawal syndrome associated with ruxolitinib therapy in MPNs, ruxolitinib remains the best available therapy (BAT) for MF and therefore, may be beneficial for ALL patients harboring JAK2 alterations.
Resistance to JAK2 Inhibitors
Introduction of TKIs into front-line combination therapies for Ph+ ALL has improved long-term outcomes primarily by improving complete remission rates, enabling more patients to become eligible for Allo-SCT (Bassan et al., 2010; Brissot et al., 2015; Chalandon et al., 2015). Therefore, despite the ongoing clinical challenges associated with JAK2 inhibition in the setting of MPNs, JAK2 inhibition may still reduce symptomatic burden of JAK2-altered ALL and improve outcomes by bridging more patients to Allo-SCT. However, the development of treatment resistance to kinase inhibitors is, unfortunately, a well-established occurrence following long-term targeted therapy in both hematologic malignancies and solid tumors (Gross et al., 2015; Bhullar et al., 2018; Pottier et al., 2020). The majority of Ph+ ALL patients treated with TKI who do not undergo Allo-SCT will ultimately relapse (Bassan et al., 2010; Fielding et al., 2014; Chalandon et al., 2015). Approximately 70–80% of Ph+ ALL patients who relapse following imatinib therapy harbor emergent mutations within the region encoding the ABL1 kinase domain of BCR:ABL1 (Pfeifer et al., 2007; Pfeifer et al., 2012; Soverini et al., 2014). Most imatinib-resistant mutations retain sensitivity to second-generation TKIs, including dasatinib, nilotinib and bosutinib, however resistance to these inhibitors can also occur via mutations such as ABL1 p. T315I (Hochhaus et al., 2020). Similar to TKIs, all clinically available JAK2 inhibitors are ATP mimetics and there are concerns that incorporation of JAK2 inhibitors into treatment approaches for JAK2r ALL will lead to the development of resistance (Miller et al., 2014; Meyer, 2017).
The majority of ruxolitinib-treated MF patients lose their response over time, with a 3-years median duration of response (Harrison et al., 2016; Verstovsek et al., 2017b). The emergence of ruxolitinib-resistant mutations was initially suspected to underlie relapse of MF these patients, similar to the emergence of resistant mutations in TKI-treated CML and Ph+ ALL. JAK2 inhibitor-resistant mutations within JAK2 have been identified primarily through in vitro random mutagenesis screens of JAK2 (Hornakova et al., 2011; Deshpande et al., 2012; Weigert et al., 2012; Kesarwani et al., 2015; Wu et al., 2015). Screens performed in vitro by Kesarwani et al. (2015) identified 39 different JAK2 mutations spanning across all domains of JAK2 (FERM, SH2, pseudokinase, and kinase) that conferred resistance to ruxolitinib (Kesarwani et al., 2015). The JAK2 p. Y931C mutation, homologous to the activating JAK1 p. F958C mutation, was the first JAK2 mutation identified to confer resistance to ruxolitinib and has been detected by in vitro screens from multiple groups (Hornakova et al., 2011). Several other JAK2 mutations that confer resistance to ruxolitinib have been identified by saturation mutagenesis screens using cell lines expressing JAK2 p. V617F or CRLF2/JAK2 p. R683F (Deshpande et al., 2012; Weigert et al., 2012). All ruxolitinib-resistant JAK2 mutations localize to the ATP/ruxolitinib binding site of the JAK2 kinase domain and confer cross-resistance to multiple type-I JAK inhibitors, suggesting that the ATP/ruxolitinib binding site is susceptible to JAK inhibitor-resistant mutations (Deshpande et al., 2012; Weigert et al., 2012; Downes et al., 2021).
However, despite in vitro predictions, clinical resistance to ruxolitinib in MF has not been reported to associate with any JAK2 point mutations. This may be due to an insufficient selective pressure related to the low specificity and high toxicity of ruxolitinib (Downes et al., 2021; Ross et al., 2021). The absence of any JAK2 point mutations in MF patients who acquired resistance to ruxolitinib suggests a role for a mutation-independent mechanism that enables persistent JAK/STAT signaling in the setting of long-term JAK2 inhibition (Koppikar et al., 2012; Harrison et al., 2020a; Ross et al., 2021). Ruxolitinib resistance in MF has been modelled in vitro by culturing cell lines expressing JAK2 p. V617F long-term with ruxolitinib and demonstrated that ruxolitinib resistance occurs due to heterodimeric activation of JAK2 p. V617F pJAK2 by other JAK family members, a mechanism now known as ruxolitinib persistence (Andraos et al., 2012; Koppikar et al., 2012; Tvorogov et al., 2018). Interestingly, ruxolitinib persistent cells could be re-sensitized following ruxolitinib withdrawal (Koppikar et al., 2012), consistent with a number of clinical reports following ruxolitinib rechallenging (Gisslinger et al., 2014; Gerds et al., 2018). However, this ruxolitinib persistence mechanism is not predicted to occur in JAK2r ALL as a recent study modelling acquired ruxolitinib resistance in JAK2r ALL in vitro identified emergent JAK inhibitor-resistant JAK2 point mutations (Downes et al., 2021). Interestingly, one of these acquired mutations, JAK2 p. G993A, also conferred resistance to the type-II JAK inhibitor, CHZ-868 (Downes et al., 2021). However, ruxolitinib resistance has not yet been reported in any ongoing clinical trials for ALL. There has only been one report of primary B-ALL leukemia cells harboring a JAK2 kinase domain mutation and these cells demonstrated a reduced sensitivity to ruxolitinib (Sadras et al., 2017).
Progress of Targeted Therapies for JAK2-Altered ALL
There were high expectations for ruxolitinib following its FDA-approval for MPNs in 2011 but unfortunately, ruxolitinib therapy has not matched the success of TKIs for CML. Consistent with reports of adverse events, JAK2 is a difficult protein to potently inhibit without toxic side effects as it plays an essential role in several normal cellular functions, including hematopoiesis (Levine et al., 2007; Vainchenker and Constantinescu, 2013; Akada et al., 2014). JAK2 conditional knockout mice display severely impaired erythropoiesis (Akada et al., 2014; Grisouard et al., 2014; Fasouli and Katsantoni, 2021), whereas the myeloid-erythroid system of ABL1 knockout mice appears normal (Hardin et al., 1995; Walz et al., 2008). All JAK2 inhibitors currently in development also target the JAK2 ATP-binding site, which is highly conserved across the JAK family and other kinases (Lucet et al., 2006; Singer et al., 2019). Imatinib also binds within the highly conserved ATP-binding site of ABL1, however, ruxolitinib inhibits a significantly higher number of kinases compared to imatinib (Davis et al., 2011), which may contribute to ruxolitinib’s increased treatment-related toxicity. Furthermore, clinical resistance to ruxolitinib occurs primarily through heterodimeric activation (Andraos et al., 2012; Koppikar et al., 2012; Tvorogov et al., 2018), rather than the emergence of point mutations, enabling therapeutic resistance despite ruxolitinib binding to WT and/or JAK2 p. V617F-mutant JAK2. The adverse events associated with ruxolitinib therapy in MPNs suggests that similar clinical challenges will be observed when incorporating ruxolitinib into treatment approaches for JAK2-altered ALL.
Despite these limitations, the efficacy of JAK2 inhibitors has been demonstrated in several pre-clinical models of JAK2-mutant (Bercovich et al., 2008; Mullighan et al., 2009c; Yoda et al., 2010; Tasian et al., 2012; Van Bodegom et al., 2012; Wu et al., 2015; Steeghs et al., 2017) and JAK2r (Maude et al., 2012; Chase et al., 2013; Boer and den Boer, 2017; Downes et al., 2021) ALL. Type-I JAK2 inhibitors have been demonstrated to reduce cell proliferation and STAT5 phosphorylation in cell lines co-expressing JAK2 p. R683 mutations and either EPOR or CRLF2 (Bercovich et al., 2008; Mullighan et al., 2009c; Yoda et al., 2010; Tasian et al., 2012). However, there have been limited ex vivo studies assessing the efficacy of JAK2 inhibition in primary CRLF2r/JAK2-mutant ALL cells. Importantly, Steeghs et al. (2017) demonstrated that the ex vivo efficacy of ruxolitinib in CRLF2r/JAK2-mutant primary ALL cells was highly dependent on the addition of human TSLP (Steeghs et al., 2017). Human TSLPR cannot be activated by mouse TSLP (Van Bodegom et al., 2012; Francis et al., 2016) yet despite this dependence, patient-derived xenograft (PDX) models of CRLF2r/JAK2-mutant ALL cells have been generated in NSG mice (Maude et al., 2012; Suryani et al., 2015; Tasian SK. et al., 2017). This suggests the activation of alternative signaling pathways, such as RAS/MAPK, PI3K/PKB and mTOR, arguing against JAK2 inhibition as a precision medicine strategy for in CRLF2r/JAK2-mutant ALL (Winter et al., 2014; Tasian SK. et al., 2017; Steeghs et al., 2017). Furthermore, the dependence of JAK2 mutations on human TSLPR activation suggests that conventional patient-derived xenograft (PDX) models of CRLF2r/JAK2-mutant ALL are not suitable to assess the efficacy of JAK2 inhibition (Francis et al., 2016; Steeghs et al., 2017; Kim et al., 2018). Consistent with this principle, ruxolitinib has only exhibited a low efficacy in PDX models of CRLF2r/JAK2-mutant ALL, despite reductions in peripheral blood and splenic blast counts (Maude et al., 2012). CRLF2r/JAK2-mutant PDX models engineered by Francis et al. (2016) to produce human TSLP may prove to be more clinically relevant models, enabling the in vivo efficacy of JAK2 inhibition for CRLF2r/JAK2-mutant ALL to be determined (Francis et al., 2016).
In contrast to CRLF2r/JAK2 mutant primary cells, Roberts et al. (2014b) and Steeghs et al. (2017) have demonstrated that ruxolitinib treatment of JAK2r primary leukemic cells can significantly reduce cell viability and STAT5 phosphorylation (Roberts et al., 2014a; Steeghs et al., 2017). Similar results were also shown using murine B-cells transduced to express JAK2 fusions (BCR::JAK2, ETV6::JAK2, PAX5::JAK2, GOLGA4::JAK2, or ATF7IP::JAK2) (Cuesta-Domínguez et al., 2012; Marit et al., 2012; Chase et al., 2013; Schinnerl et al., 2015; Downes et al., 2021; Downes et al., 2022), and PAX5::JAK2 expressing Arf−/− murine pre-B cell models (Roberts et al., 2014a). Unlike PDX models of CRLF2r/JAK2-mutant ALL, the efficacy of ruxolitinib has also been demonstrated in vivo using PDX models of JAK2r ALL, where ruxolitinib treatment reduced peripheral blood blast counts and tumor burden (Roberts et al., 2012; Roberts et al., 2014a; Roberts KG. et al., 2017). An additive effect was also observed when used in combination with dexamethasone (Roberts KG. et al., 2017). However, ruxolitinib treatment did not induce complete remission in PDX models of JAK2r ALL (Maude et al., 2012; Roberts et al., 2012; Roberts et al., 2014a; Roberts KG. et al., 2017). This suggests that JAK2 inhibition in combination with chemotherapy may improve outcomes for ALL patients harboring JAK2r (Downes et al., 2021). This also suggests JAK2 inhibitors may also be an effective precision medicine strategy for CRLF2r/JAK2-mutant ALL, but more clinically relevant in vivo models of CRLF2r/JAK2-mutant ALL, that include human TSLP, are required to assess their efficacy.
Ruxolitinib is the only JAK inhibitor known to be undergoing clinical assessment in an ALL setting. High clinical effectiveness of ruxolitinib in combination with multi-agent chemotherapy has been reported in only small number of patients with either CRLF2r/JAK-mutant or JAK2r ALL (Schrappe et al., 2012; Roberts et al., 2014a; Schwab et al., 2016; Mayfield et al., 2017; Ding et al., 2018; Chen X. et al., 2019; Chen et al., 2022; Rizzuto et al., 2022). A phase 2 clinical trial (NCT02723994) led by the Children’s Oncology Group is currently assessing ruxolitinib in combination with chemotherapy for the treatment of ALL patients harboring CRLF2 and/or JAK pathway alterations (Senkevitch and Durum, 2017). Results from the phase I of this trial recently reported no dose-limiting toxicity up to 50 mg/m2 dosed day 1–14 of a 28 days cycle, as well as continuous dosing at 40 mg/m2 post-induction chemotherapy (Tasian et al., 2018). Ruxolitinib therapy was also well tolerated and induced morphologic remission in a case report of a child with chemo-resistant JAK2r ALL and induction failure (Ding et al., 2018; Tasian et al., 2018). A phase 3 clinical trial (NCT03117751) is now investigating ruxolitinib/chemotherapy combination in patients with JAK-STAT signaling activation (Harvey and Tasian, 2020). There are also a number of other phase 1/2 clinical trials (NCT02420717, NCT03571321) assessing ruxolitinib for the treatment of Ph-like ALL harboring JAK/STAT pathway alterations. Early findings suggest that JAK inhibitors in combination with chemotherapy may improve outcomes for patients with these high-risk ALL subtypes, but we await the results of ongoing trials.
Additional/Alternate Type-I JAK2 Inhibitors in Clinical Development
The myelosuppression resulting from ruxolitinib treatment in MF suggests that this should also be expected when including ruxolitinib in ALL treatment regimens. To attempt to overcome these limitations, several other JAK2 inhibitors have been assessed in clinical trials, but most have been discontinued primarily due to toxicity (Sonbol et al., 2013; Bose and Verstovsek, 2017). Current clinical studies of JAK2 inhibitors and their specificities are shown in Table 4. All JAK2 inhibitors being assessed clinically are type-I inhibitors, targeting the ATP-binding site of JAK2 in the active conformation. As the ATP-binding site is highly conserved among kinases, off-target suppression of JAK1 has been proposed to contribute to the myelosuppression and opportunistic infections associated with ruxolitinib therapy (Singer et al., 2016). Fedratinib is the most selective JAK2 inhibitor currently available and is likely less immunosuppressive than ruxolitinib due to weaker inhibition of JAK1 (Mullally et al., 2020; Talpaz and Kiladjian, 2021). Fedratinib has been demonstrated to significantly reduce splenomegaly and symptom burden in patients with either intermediate- or high-risk MF (Pardanani et al., 2011; Pardanani et al., 2015; Harrison et al., 2017; Mullally et al., 2020; Talpaz and Kiladjian, 2021). It is recent FDA-approval may reveal whether more selective JAK2 inhibition improves therapy-associated thrombocytopenia and anemia (Talpaz and Kiladjian, 2021).

TABLE 4. Clinical studies of type-I JAK2 inhibitors in hematological malignancies and their specificities. Myelofibrosis (MF), polycythemia vera (PV), essential thrombocythemia (ET), chronic myeloid leukemia (CML), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), small lymphocytic leukemia (SLL), acute lymphoblastic leukemia (ALL). Adapted from Raivola et al. (2021)(Raivola et al., 2021) and Vainchenker et al. (2018)(Vainchenker et al., 2018).
Unfortunately, fedratinib has also been associated with dose-dependent thrombocytopenia and anemia, in addition to gastrointestinal adverse events (Pardanani et al., 2011; Mullally et al., 2020). Approval of fedratinib also includes a “black box warning” on the risk of serious and fatal Wernicke encephalopathy (WE), a neurodegenerative condition traditionally caused by thiamine deficiency (Bewersdorf et al., 2019; Mullally et al., 2020). Suspected treatment-associated cases of WE resulted in the FDA issuing a clinical hold on fedratinib between 2013-2017, however these cases were ultimately determined to not be caused by fedratinib therapy (Bewersdorf et al., 2019; Mullally et al., 2020). The thrombocytopenia and anemia associated with fedratinib may be related to its on-target inhibition of WT JAK2 and off-target inhibition of FMS-like tyrosine kinase 3 (FLT3) and bromodomain-containing protein 4 (BRD4) (Talpaz and Kiladjian, 2021). It is currently unknown whether fedratinib is more effective than ruxolitinib as a first-line treatment for MF and no clinical trials are currently planned to assess these inhibitors head-to-head. However, a phase 2 study evaluating the efficacy of fedratinib for the treatment of ruxolitinib relapsed, refractory, or intolerant MF demonstrated significant reductions in splenomegaly and symptomatic burden (Harrison et al., 2020b; Talpaz and Kiladjian, 2021). In addition, fedratinib has been shown to bind both the ATP-binding site and the less conserved substrate-binding site of JAK2, a site which may be less prone to acquiring inhibitor-resistant mutations (Kesarwani et al., 2015). The high specificity of fedratinib for JAK2 suggests that it may associate with less dose-limiting toxicity in comparison to ruxolitinib and therefore, may be a more efficacious JAK inhibitor for the treatment of JAK2-altered ALL when used in combination with chemotherapy.
Other type-I JAK2 inhibitors currently being assessed in phase 3 clinical trials include momelotinib and pacritinib. Momelotinib is also type-I JAK1/2 specific inhibitor, similar to ruxolitinib, that was expected to improve symptoms of therapy-induced anemia by also inhibiting activin A receptor type 1 (ACVR1) (Asshoff et al., 2017). Unfortunately, momelotinib was not found to be superior at reducing symptom burden compared with ruxolitinib and consistent with this lower efficacy, momelotinib therapy was associated with fewer reports of anemia (Mesa et al., 2017a; Harrison et al., 2018; Bassiony et al., 2020). Another type-I JAK inhibitor assessed in phase 3 clinical trials is pacritinib, a type-I JAK2/FLT3 inhibitor that, similar to fedratinib, does not inhibit JAK1 (Talpaz and Kiladjian, 2021). Pacritinib therapy was demonstrated to be superior to the BAT at reducing splenomegaly (Mesa et al., 2017b; Mascarenhas et al., 2018) and was mostly non-myelosuppressive, likely due to weaker inhibition of JAK1 (Singer et al., 2016; Mesa et al., 2017b; Talpaz and Kiladjian, 2021). Pacritinib was also well-tolerated in patients with severe thrombocytopenia, suggesting that pacritinib may be beneficial for patients with anemia (Gerds et al., 2019; Harrison et al., 2020a; Bassiony et al., 2020; Verstovsek et al., 2021). To note, the efficacy of momelotinib and pacritinib for patients previously treated with ruxolitinib has not been reported (Harrison et al., 2020a). Fedratinib and pacritinib may reduce the leukaemic burden of JAK2-alterated ALL with superior or equivalent efficacy to ruxolitinib, potential improving complete remission rates and associating with less treatment-associated adverse events.
Type-II JAK Inhibitors
In contrast to type-I JAK inhibitors, type-II JAK inhibitors bind the ATP-binding site of JAK2 in the inactive (DFG-out) conformation (Figure 3B) (Leroy and Constantinescu, 2017). Although type-II JAK inhibitors are still ATP-competitive, they are more specific for JAK2 by also binding a less conserved allosteric pocket, potentially minimizing toxicity by reduced inhibition of other kinases (Li et al., 2019). This type-II binding mode is similar to the inhibition of BCR:ABL1 with imatinib (Druker and Lydon, 2000; Schindler et al., 2000). The first type-II JAK inhibitor identified was BBT594 (Figure 3C), which was originally designed to inhibit BCR:ABL1 harboring the TKI-resistant ABL1 p. T315I-mutation (Andraos et al., 2012). BBT594 inhibited STAT5 phosphorylation in cell models expressing either TEL::JAK2 or JAK2 p. V617F-mutant JAK2, albeit with low specificity and limited potency (Andraos et al., 2012). These findings prompted the development of the only other type-II JAK inhibitor, CHZ868 (Meyer et al., 2015; Li et al., 2019). CHZ868 potently inhibited JAK2 with a high selectivity over other JAK family members (Meyer et al., 2015; Wu et al., 2015). Promising pre-clinical studies showed that CHZ868 not only improved survival and leukaemic burden in vivo models of MPN and B-ALL, but preferentially inhibited JAK2 p. V617F-mutant JAK2 hematopoietic cells over WT JAK2 cells (Meyer et al., 2015; Wu et al., 2015). CHZ868 also reduced the JAK2 p. V617F allele burden in these MPN models, which is not observed with type-I inhibitor treatment (Meyer et al., 2015). The potent activity of type-II inhibitors against JAK2 p. V617F-mutant hematopoietic cells suggests that they may be a more effective than type-I JAK inhibitors for the treatment of JAK2-altered ALL.
By binding the inactive conformation of JAK2, type-II JAK inhibitors may also prevent the development of resistance through the persistent JAK/STAT signaling that is associated with ruxolitinib therapy. Type-II JAK inhibitors have been demonstrated to reduce the proliferation of ruxolitinib-persistent cell models and inhibit JAK2 activation loop phosphorylation (Andraos et al., 2012; Koppikar et al., 2012; Meyer et al., 2015; Tvorogov et al., 2018). The inhibition of JAK2 activation loop phosphorylation by CHZ868 has been suggested to prevent accumulation of pJAK2, preventing heterodimeric JAK2 activation and subsequent inhibitor persistence (Meyer et al., 2015; Tvorogov et al., 2018). CHZ868 did not facilitate the accumulation of phosphorylated JAK2 in hematopoietic cell lines or primary JAK2 p. V617F cells (Tvorogov et al., 2018). Furthermore, CHZ868 withdrawal was not associated with a rebound in STAT5 signaling (Tvorogov et al., 2018), suggesting that type-II JAK2 inhibition may not be associated with withdrawal syndrome (Meyer et al., 2015; Wu et al., 2015; Tvorogov et al., 2018). However, one JAK2 mutation (JAK2 p. L884P) confers resistance to both BBT594 and CHZ868, suggesting that resistance to type-II JAK inhibitors may still occur through mutations within the JAK2 ATP-binding site (Wu et al., 2015; Leroy and Constantinescu, 2017). Unfortunately, the potent activity of type-II JAK inhibitors also risks stronger suppression of normal hematopoiesis through greater inhibition of WT JAK2, but without clinical assessment it is unknown whether type-II JAK inhibitors would result in more or less pronounced cytopenia (Vainchenker et al., 2018; Ross et al., 2021). The risk of severe cytopenia may underlie why neither CHZ868 nor BBT594 were pursued for further drug development (Andraos et al., 2012; Wu et al., 2015). Further research is needed to determine whether type-II JAK inhibitors are clinically viable and their susceptibility to persistence or resistance. The development of type-II JAK inhibitors may be an effective therapeutic approach for JAK2-altered ALL, enabling JAK2 inhibition without risking the withdrawal syndrome and disease persistence that is associated with type-I JAK inhibitors.
Allosteric JAK Inhibitors (Type-III JAK Inhibitors)
Type-II JAK inhibitors still target the highly conserved ATP-binding site of JAK2, which may lead to toxicity due to off-target effects on other kinases. In contrast, allosteric JAK inhibitors, also referred to as type-III JAK inhibitors, bind less conserved allosteric pockets outside of the JAK2 ATP-binding site (Leroy and Constantinescu, 2017). In addition to the substrate binding site, three potentially targetable allosteric sites of the JAK2 kinase domain have been computationally identified, but inhibitors of these sites have not yet been verified (Kesarwani et al., 2015; Leroy and Constantinescu, 2017). These allosteric sites are less conserved than the ATP-binding site and therefore, may offer greater selectivity and potency compared with type-I or type-II JAK inhibitors (Kesarwani et al., 2015). Two non-ATP-competitive JAK inhibitors, ON044580 and LS104, have been described as allosteric JAK inhibitors and demonstrated efficacy in vitro against JAK2 p. V617F transformed cell lines and primary patient cells (Lipka et al., 2008; Jatiani et al., 2010). The allosteric binding mechanisms of these compounds and their in vivo efficacy were never determined, although they did demonstrate substrate-competitive binding modes (Lipka et al., 2008; Jatiani et al., 2010; Raivola et al., 2021). The JAK2 substrate-binding site may be less susceptible to inhibitor-resistant mutations than other allosteric sites as mutations may prevent substrate binding, which is essential for the downstream signaling activation (Kesarwani et al., 2015). To support this hypothesis, no JAK inhibitor-resistant mutations have been identified within the substrate-binding site by random mutagenesis of JAK2 (Kesarwani et al., 2015). These findings suggest that the JAK2 substrate-binding site may be a novel and effective targetable site for JAK2-alterated malignancies, and long-term therapy may not result in the development of resistance (Kesarwani et al., 2015).
Targeted JAK2 Degradation
Direct targeting of JAK2 by proteolysis-targeting chimeras (PROTACS) have recently emerged as an approach to limit withdrawal syndrome and ruxolitinib persistence, whilst still inhibiting signaling activation through JAK2 (Shah et al., 2020; Chang et al., 2021). PROTACS comprise three distinct components: a ligand for E3 ligase, a ligand for the protein of interest, and a linker to couple the two functional ligands (Kargbo, 2021). JAK2 PROTACs facilitate the formation of E3-PROTAC-JAK2 complexes, inducing E3 ligase-mediated ubiquitination and subsequent proteasomal degradation of JAK2 (Kargbo, 2021). Current JAK2 PROTACs have been designed using the full-length, or a portion, of known type-I JAK inhibitors as the JAK2-targeting ligand and have been shown to induce JAK2 ubiquitination and degradation in leukaemic cell lines (Shah et al., 2020; Chang et al., 2021; Kargbo, 2021). Strikingly, Chang et al. (2021) demonstrated significant reductions in leukaemic burden in vivo CRLF2r ALL models treated with JAK2 PROTACS, but not ruxolitinib monotherapy (Chang et al., 2021). The superior activity of these PROTACs was attributed to both JAK2 inhibition and targeted degradation of proteins including JAK1/2/3, TKY2, IKZF1/3, and G1 to S Phase Transition 1 (GSPT1) (Chang et al., 2021). Degradation of multiple targets with a single PROTAC establishes the basis for PROTACs with modifiable specificity and high efficacy in malignancies driven by JAK-STAT. However, this degradation of multiple targets may result in toxic side effects if PROTAC target proteins are required for hematopoiesis or B-cell maintenance. Furthermore, the clinical viability of JAK2 PROTACs is yet to be determined and, in the absence of PROTAC ligands specific for mutant-JAK2 or JAK2 fusions, JAK2 PROTACS may induce anemia and thrombocytopenia due to degradation of WT JAK2, therefore, further investigations are required.
Inhibition of heat shock protein 90 (HSP90) has also been explored as a less targeted approach to degrade JAK2 (Brkic and Meyer, 2021; Raivola et al., 2021). Degradation of JAK2 is reduced through stabilization by chaperones including HSP90 (Bose and Verstovsek, 2017; Ross et al., 2021). HSP90 inhibitors, such as PU-H71 and AUY922, degrade JAK2 and inhibit downstream signaling in cell lines expressing JAK2 p. V617F (Marubayashi et al., 2010; Fiskus et al., 2011) and cells harboring JAK inhibitor-resistant mutations (Weigert et al., 2012). HSP90 inhibitor monotherapy (Marubayashi et al., 2010), or in combination with a JAK2 inhibitor (Fiskus et al., 2011; Bhagwat et al., 2014), also significantly reduced leukaemic burden in JAK2 p. V617F murine models. Early phase clinical trials assessing PU-H71 (Speranza et al., 2018) and AUY922(Hobbs et al., 2018) demonstrated reductions in splenomegaly, however, both trials were terminated due to toxicity. Histone deacetylase (HDAC) inhibitors, such as panobinostat and vorinostat, have also been investigated as a strategy to inhibit HSP90 activity by promoting HSP90 hyperacetylation (Brkic and Meyer, 2021). The efficacy of HDAC inhibitors has been demonstrated in vitro and in vivo models of JAK2 p. V617F-mutant MPN, particularly in combination with a JAK inhibitor (Wang et al., 2009; Akada et al., 2012; Evrot et al., 2013). However, HDAC inhibitors in combination with ruxolitinib were not superior to ruxolitinib alone in recent phase 1/2 MPN clinicals trials (Bose et al., 2019; Mascarenhas et al., 2020). Similar to AUY922, vorinostat was also associated with adverse effects (Andersen et al., 2013). This suggests that HSP90 inhibition may be too toxic to be clinically viable. However, the more tolerable HDAC inhibitor, givinostat, is expected to be assessed in a phase 3 clinical trial for the treatment of PV, suggesting that givinostat may be a potential therapeutic approach for the treatment of JAK2-altered ALL when in combination with chemotherapy or type-I JAK2 inhibition (Chifotides et al., 2020).
Discussion - Future Outlooks
The CRLF2/JAK2-mutant and JAK2r subtypes of ALL correlate with poor prognosis and targeted JAK2 inhibition remains a feasible precision medicine approach. Inhibition of mutant-JAK2 or JAK2 fusions using targeted therapeutic strategies would abolish the resulting constitutively active JAK/STAT signaling. Such approaches may improve patient outcomes by increasing complete remission rates, enabling more patients to be eligible for allogeneic transplantation therapy. Current precision medicine approaches that are being investigated for the treatment of JAK2-altered ALL are shown in Table 5. To date, only type-I JAK inhibitors have been tested in vivo for the treatment of JAK2-altered ALL with most clinical data involving ruxolitinib thus far. Since the approval of ruxolitinib for the treatment of MF, development of other type-I JAK inhibitors has focused on reducing the treatment-related myelosuppression associated with ruxolitinib therapy. However, momelotinib has not demonstrated superior efficacy compared with ruxolitinib, and no clinical trials are planned to assess fedratinib or pacritinib against ruxolitinib head-to-head. Nonetheless, the recent FDA approval of fedratinib for the treatment of MPNs may, in future, give some indication as to whether highly specific JAK2 inhibition can improve treatment-related thrombocytopenia and anemia. Several studies have also assessed ruxolitinib combination therapies with other disease-modifying agents to improve side effects and therapeutic responses through synergistic activities (Böhm et al., 2021). The vast array of ruxolitinib combination therapies have been extensively reviewed by Kuykendall et al. (2020) and includes HDAC inhibitors, DNA methyltransferases inhibitors, erythropoiesis-stimulating agents, BCL-2 (B-cell lymphoma 2) inhibitors, BET (bromodomain and extra-terminal protein) inhibitors, and many others (Kuykendall et al., 2020).
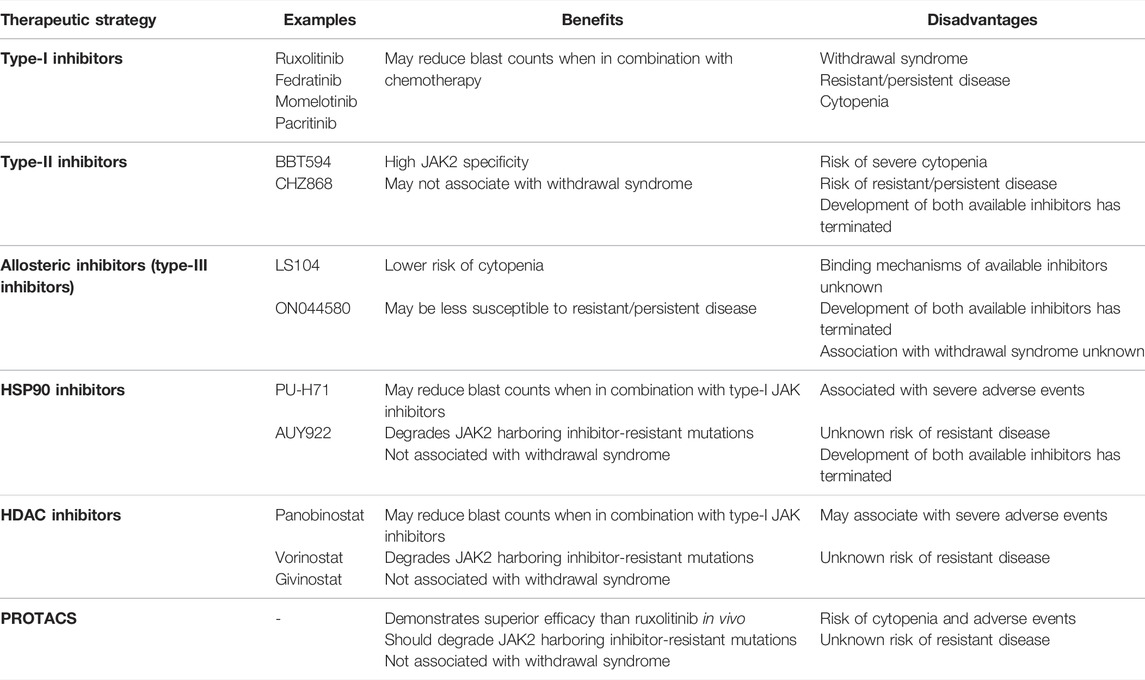
TABLE 5. Current precision medicine approaches to target JAK2 in JAK2-altered ALL and their associated benefits and disadvantages.
Type-II JAK inhibitors offer several potential advantages over type-I inhibition. Type-II JAK inhibitors offer the opportunity to specifically target JAK2 by binding an additional allosteric site, and the potential to prevent withdrawal syndrome by binding the inactive conformation of JAK2. Although clinical development of CHZ868 was not pursued, pre-clinical studies demonstrated promising results. Research and development should continue to determine the clinical viability of type-II JAK inhibitors. JAK2 PROTACs, which degrade ligand bound-JAK2, also offer another potentially efficacious approach and may prevent withdrawal syndrome. Recent research demonstrating the superior efficacy of JAK2 PROTACS over ruxolitinib in murine models suggests that JAK2 PROTACs may be an effective therapeutic strategy for the treatment of JAK2-altered malignancies, and future clinical evaluation is warranted. Allosteric inhibitors are also another possible approach to more specifically target JAK2 by binding regions that are less conserved among other kinases. The field of allosteric JAK inhibitors are still in their relative infancy but the discovery that fedratinib binds to both the ATP- and substrate-binding sites of JAK2 suggests that this is promising area for future development. Allosteric JAK inhibitors used in combination with type-I JAK inhibitors, or inhibitors targeting the substrate-binding site, may also impede the development of acquired resistance.
The ATP-binding site of the JAK2 pseudokinase domain could potentially be targetable with small molecular inhibitors to stabilize the JH2-JH1 autoinhibitory interaction. To support this hypothesis, targeting of the TYK2 pseudokinase domain has been demonstrated to inhibit TYK2 activation (Tokarski et al., 2015). However, it is unknown whether this approach is translatable to JAK2 as targeting this site may actually activate JAK2 by preventing JAK2 p. S523 and p. Y570 phosphorylation, destabilizing the JH2-JH1 autoinhibitory interaction. No compounds have been identified to bind to the JAK2 pseudokinase domain ATP-binding site to date. In addition, point mutations within the JAK2 FERM domain have been demonstrated to abolish the JAK2 association with TPOR (Royer et al., 2005), indicating that the interface between the JAK2 FERM-SH2 domains and the cytoplasmic region of the associated cytokine receptors may be a novel targetable site. Inhibiting the association of JAK2 with cytokine receptors may prevent JAK2 dimerization and subsequent activation. This approach may also reduce toxic side effects by not only specifically targeting JAK2, but also specific receptors and their complexes with JAK2. Another precision medicine approach for CRLF2/JAK2-mutant ALL could also include strategies that inhibit TSLPR dimerization or activation (Markovic and Savvides, 2020) potentially by using antagonistic monoclonal antibodies (Zhang et al., 2011; Mohamed et al., 2021; Numazaki et al., 2021), inhibitors (Van Rompaey et al., 2017), or CAR T-cells (Qin et al., 2015). Therapeutic approaches that offer more selectivity for JAK2 should improve the toxic side effects associated with JAK2 inhibition. However, due to the inherent essential role of WT JAK2 in normal hematopoiesis, therapeutic approaches that do not offer selectivity for JAK2 alterations over WT JAK2 will likely associate with a risk of anemia and thrombocytopenia.
Future drug design approaches that are specific for mutant-JAK2 or JAK2 fusions would ultimately provide less toxic and more effective therapies for MPN and ALL patients harboring JAK2 alterations. If JAK2 mutations or JAK2r are overexpressed in ALL patients, there are potentially therapeutic strategies that can influence gene expression. One such approach may be through BET inhibitors, which have been shown to disrupt super-enhancers, promoter enhancers often associated with oncogenes (Crump et al., 2021). Additionally, the majority of JAK2 mutations associated with ALL lie within JAK2 exon 16, localizing to the ATP-binding site of the JAK2 pseudokinase domain. Several lines of evidence suggest that allosteric inhibitors could target the JAK2 pseudokinase domain ATP-binding site, and there is potential for these inhibitors to be designed to target specific JAK2 mutants. In particular, it has been postulated that JAK2 p. R683 mutations disrupt the pseudokinase domain-mediated autoinhibitory interaction, therefore inhibitors that can stabilize this interaction may be able to overcome the effects of these mutations. The recent discovery that apposing JAK1 monomers dimerize via their pseudokinase domains also positions this dimerisation interface as a potentially novel targetable site (Glassman et al., 2022). JAK2 dimerisation may also be mediated via the pseudokinase domains however the full-length structure of JAK2 is yet to be determined. Future development of JAK2 inhibitors that aim to either stabilize the pseudokinase domain-mediated autoinhibitory interaction, or hinder pseudokinase-domain mediated dimerisation, may prove to be effective new therapeutic approaches to target JAK2. Importantly, targeting JAK2 dimerisation, rather than its activation, through such precision medicine strategies may prevent JAK2 heterodimerization as a mechanism of drug resistance.
Therapeutic targeting of the JAK2 pseudokinase domain also offers opportunities to target JAK2 proteins that harbor specific mutations within this region. The molecular activation mechanisms of ALL-associated JAK2 p. R683 mutations are yet to be fully elucidated but further structural research may inform future drug design strategies to target JAK2 p. R683-mutant JAK2. However, in contrast to JAK2 mutations, the majority of JAK2 fusions do not retain the ATP-binding site of the JAK2 pseudokinase domain (encoded by JAK2 exons 13–15). This restricts targetable regions within the JAK2 portion of the fusion to the kinase domain. Therapeutic approaches that target the JAK2 kinase domain will not have the ability to distinguish between JAK2 fusions and WT JAK2, preventing potent inhibition of leukaemic cell growth without toxic side effects. Future work to elucidate the structure of different JAK2 fusions may potentially reveal novel targetable sites that are specific for JAK2 fusions over WT JAK2. A deeper understanding of the cytokine-independent oligomerization mechanism that is hypothesized to underlie the constitutive activation of JAK2 fusions may also reveal novel targetable sites. In particular, there may be similar oligomerization domains within the JAK2 fusion partners, such as the CC and HLH motifs. Inhibitors that can bind these motifs and prevent JAK2 fusion dimerization may be a potential therapeutic strategy to achieve specificity for JAK2 fusions and reduce the toxic side effects observed with traditional JAK2 inhibition.
Ultimately, future approaches targeting JAK2 need to 1) target less conserved regions to achieve a higher specificity for JAK2 over other JAK family members, 2) avoid other kinases with potential for off-target toxicity, 3) inhibit inactive rather that active conformation and 4) target mutant forms of JAK vs. WT using allosteric or novel domain-domain inhibition. Combination strategies using type-I JAK inhibitors or PROTACs may also offer improved efficacy over ruxolitinib. Future research into these therapeutic approaches, and the design of inhibitors targeting mutant-JAK2 and JAK2-fusions, is urgently needed to improve outcomes for the high-risk JAK2-altered subtypes of ALL.
Author Contributions
CD conceived the review, performed the literature analysis and wrote the manuscript. BM, DM, SH, JB, DT, DY and DW critically reviewed and edited the manuscript. BM and DW supervised CD, and all authors approved the manuscript.
Funding
This study was supported in part by grants from the National Health and Medical Research Council, Australia (NHRMC) (APP1057746); the Australian Genomics Health Alliance (AGHA), the Leukaemia Foundation, Cancer Council SA Beat Cancer Project and Bristol-Meyers Squibb Company to DLW; and Contributing Haematologist Committee (CHC) to DTY. Both DLW and DTY are NHMRC Fellows. SLH is a Kids’ Cancer Project Postdoctoral Fellow. DT receives funding from Leukemia and Lymphoma Society, Leukaemia Foundation, Snowdome, NHRMC and Medical Research Futures Fund. CEJD and DPM are supported by a research training program (RTP) stipends from the University of Adelaide and Australian Government.
Conflict of Interest
DW receives research support from BMS, Honoraria from Amgen. DY receives research support from Novartis, Ariad and BMS, Honoraria from Novartis, BMS, Amgen and Pfizer. None of these agencies have had a role in the preparation of this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.942053/full#supplementary-material
References
Akada, H., Akada, S., Gajra, A., Bair, A., Graziano, S., Hutchison, R. E., et al. (2012). Efficacy of Vorinostat in a Murine Model of Polycythemia Vera. Blood 119, 3779–3789. doi:10.1182/blood-2011-02-336743
Akada, H., Akada, S., Hutchison, R. E., Sakamoto, K., Wagner, K.-U., and Mohi, G. (2014). Critical Role of Jak2 in the Maintenance and Function of Adult Hematopoietic Stem Cells. Stem Cells 32, 1878–1889. doi:10.1002/stem.1711
Ali, M. A. M. (2016). Chronic Myeloid Leukemia in the Era of Tyrosine Kinase Inhibitors: An Evolving Paradigm of Molecularly Targeted Therapy. Mol. Diagn Ther. 20, 315–333. doi:10.1007/s40291-016-0208-1
Andersen, C. L., Mcmullin, M. F., Ejerblad, E., Zweegman, S., Harrison, C., Fernandes, S., et al. (2013). A Phase II Study of Vorinostat (MK-0683) in Patients with Polycythaemia Vera and Essential Thrombocythaemia. Br. J. Haematol. 162, 498–508. doi:10.1111/bjh.12416
Andraos, R., Qian, Z., Bonenfant, D., Rubert, J., Vangrevelinghe, E., Scheufler, C., et al. (2012). Modulation of Activation-Loop Phosphorylation by JAK Inhibitors Is Binding Mode Dependent. Cancer Discov. 2, 512–523. doi:10.1158/2159-8290.cd-11-0324
Apperley, J. F. (2015). Chronic Myeloid Leukaemia. Lancet 385, 1447–1459. doi:10.1016/s0140-6736(13)62120-0
Asshoff, M., Petzer, V., Warr, M. R., Haschka, D., Tymoszuk, P., Demetz, E., et al. (2017). Momelotinib Inhibits ACVR1/ALK2, Decreases Hepcidin Production, and Ameliorates Anemia of Chronic Disease in Rodents. Blood 129, 1823–1830. doi:10.1182/blood-2016-09-740092
Awasthi, N., Liongue, C., and Ward, A. C. (2021). STAT Proteins: a Kaleidoscope of Canonical and Non-canonical Functions in Immunity and Cancer. J. Hematol. Oncol. 14, 198. doi:10.1186/s13045-021-01214-y
Azam, M., Erdjument-Bromage, H., Kreider, B. L., Xia, M., Quelle, F., Basu, R., et al. (1995). Interleukin-3 Signals through Multiple Isoforms of Stat5. EMBO J. 14, 1402–1411. doi:10.1002/j.1460-2075.1995.tb07126.x
Babon, J. J., Lucet, I. S., Murphy, J. M., Nicola, N. A., and Varghese, L. N. (2014). The Molecular Regulation of Janus Kinase (JAK) Activation. Biochem. J. 462, 1–13. doi:10.1042/bj20140712
Babon, J. J., Sabo, J. K., Soetopo, A., Yao, S., Bailey, M. F., Zhang, J.-G., et al. (2008). The SOCS Box Domain of SOCS3: Structure and Interaction with the elonginBC-Cullin5 Ubiquitin Ligase. J. Mol. Biol. 381, 928–940. doi:10.1016/j.jmb.2008.06.038
Babon, J. J., Sabo, J. K., Zhang, J.-G., Nicola, N. A., and Norton, R. S. (2009). The SOCS Box Encodes a Hierarchy of Affinities for Cullin5: Implications for Ubiquitin Ligase Formation and Cytokine Signalling Suppression. J. Mol. Biol. 387, 162–174. doi:10.1016/j.jmb.2009.01.024
Bandaranayake, R. M., Ungureanu, D., Shan, Y., Shaw, D. E., Silvennoinen, O., and Hubbard, S. R. (2012). Crystal Structures of the JAK2 Pseudokinase Domain and the Pathogenic Mutant V617F. Nat. Struct. Mol. Biol. 19, 754–759. doi:10.1038/nsmb.2348
Barber, D. L., Beattie, B. K., Mason, J. M., Nguyen, M. H.-H., Yoakim, M., Neel, B. G., et al. (2001). A Common Epitope Is Shared by Activated Signal Transducer and Activator of Transcription-5 (STAT5) and the Phosphorylated Erythropoietin Receptor: Implications for the Docking Model of STAT Activation. Blood 97, 2230–2237. doi:10.1182/blood.v97.8.2230
Bassan, R., Rossi, G., Pogliani, E. M., Di Bona, E., Angelucci, E., Cavattoni, I., et al. (2010). Chemotherapy-phased Imatinib Pulses Improve Long-Term Outcome of Adult Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Northern Italy Leukemia Group Protocol 09/00. Jco 28, 3644–3652. doi:10.1200/jco.2010.28.1287
Bassiony, S., Harrison, C. N., and Mclornan, D. P. (2020). Evaluating the Safety, Efficacy, and Therapeutic Potential of Momelotinib in the Treatment of Intermediate/High-Risk Myelofibrosis: Evidence to Date. Tcrm 16, 889–901. doi:10.2147/tcrm.s258704
Baxter, E. J., Scott, L. M., Campbell, P. J., East, C., Fourouclas, N., Swanton, S., et al. (2005). Acquired Mutation of the Tyrosine Kinase JAK2 in Human Myeloproliferative Disorders. Lancet 365, 1054–1061. doi:10.1016/s0140-6736(05)71142-9
Beissert, T., Hundertmark, A., Kaburova, V., Travaglini, L., Mian, A. A., Nervi, C., et al. (2008). Targeting of the N-Terminal Coiled Coil Oligomerization Interface by a Helix-2 Peptide Inhibits Unmutated and Imatinib-Resistant BCR/ABL. Int. J. Cancer 122, 2744–2752. doi:10.1002/ijc.23467
Benekli, M., Baer, M. R., Baumann, H., and Wetzler, M. (2003). Signal Transducer and Activator of Transcription Proteins in Leukemias. Blood 101, 2940–2954. doi:10.1182/blood-2002-04-1204
Bercovich, D., Ganmore, I., Scott, L. M., Wainreb, G., Birger, Y., Elimelech, A., et al. (2008). Mutations of JAK2 in Acute Lymphoblastic Leukaemias Associated with Down's Syndrome. Lancet 372, 1484–1492. doi:10.1016/s0140-6736(08)61341-0
Bewersdorf, J. P., Jaszczur, S. M., Afifi, S., Zhao, J. C., and Zeidan, A. M. (2019). Beyond Ruxolitinib: Fedratinib and Other Emergent Treatment Options for Myelofibrosis. Cmar 11, 10777–10790. doi:10.2147/cmar.s212559
Bhagwat, N., Koppikar, P., Keller, M., Marubayashi, S., Shank, K., Rampal, R., et al. (2014). Improved Targeting of JAK2 Leads to Increased Therapeutic Efficacy in Myeloproliferative Neoplasms. Blood 123, 2075–2083. doi:10.1182/blood-2014-01-547760
Bhullar, K. S., Lagarón, N. O., Mcgowan, E. M., Parmar, I., Jha, A., Hubbard, B. P., et al. (2018). Kinase-targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 17, 48. doi:10.1186/s12943-018-0804-2
Blum, M., Chang, H.-Y., Chuguransky, S., Grego, T., Kandasaamy, S., Mitchell, A., et al. (2021). The InterPro Protein Families and Domains Database: 20 Years on. Nucleic Acids Res. 49, D344–D354. doi:10.1093/nar/gkaa977
Boer, J. M., Marchante, J. R., Evans, W. E., Horstmann, M. A., Escherich, G., Pieters, R., et al. (2015). BCR-ABL1-like Cases in Pediatric Acute Lymphoblastic Leukemia: A Comparison between DCOG/Erasmus MC and COG/St. Jude Signatures. Haematologica 100, e354–7. doi:10.3324/haematol.2015.124941
Boer, J. M., and Den Boer, M. L. (2017). BCR-ABL1 -like Acute Lymphoblastic Leukaemia: From Bench to Bedside. Eur. J. Cancer 82, 203–218. doi:10.1016/j.ejca.2017.06.012
Boer, J. M., Steeghs, E. M. P., Marchante, J. R. M., Boeree, A., Beaudoin, J. J., Beverloo, H. B., et al. (2017). Tyrosine Kinase Fusion Genes in Pediatric BCR-ABL1-like Acute Lymphoblastic Leukemia. Oncotarget 8, 4618–4628. doi:10.18632/oncotarget.13492
Bӧhm, J. W., Sia, K. C. S., Jones, C., Evans, K., Mariana, A., Pang, I., et al. (2021). Combination Efficacy of Ruxolitinib with Standard-Of-Care Drugs in CRLF2-Rearranged Ph-like Acute Lymphoblastic Leukemia. Leukemia 35, 3101–3112. doi:10.1038/s41375-021-01248-8
Böhmer, F.-D., and Friedrich, K. (2014). Protein Tyrosine Phosphatases as Wardens of STAT Signaling. JAK-STAT 3, e28087. doi:10.4161/jkst.28087
Bose, P., Swaminathan, M., Pemmaraju, N., Ferrajoli, A., Jabbour, E. J., Daver, N. G., et al. (2019). A Phase 2 Study of Pracinostat Combined with Ruxolitinib in Patients with Myelofibrosis. Leukemia Lymphoma 60, 1767–1774. doi:10.1080/10428194.2018.1543876
Bose, P., and Verstovsek, S. (2020). JAK Inhibition for the Treatment of Myelofibrosis: Limitations and Future Perspectives. Hemasphere 4, e424. doi:10.1097/hs9.0000000000000424
Bose, P., and Verstovsek, S. (2017). JAK2 Inhibitors for Myeloproliferative Neoplasms: what Is Next? Blood 130, 115–125. doi:10.1182/blood-2017-04-742288
Brachet-Botineau, M., Polomski, M., Neubauer, H. A., Juen, L., Hédou, D., Viaud-Massuard, M. C., et al. (2020). Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers (Basel) 12. doi:10.3390/cancers12010240
Brissot, E., Labopin, M., Beckers, M. M., Socie, G., Rambaldi, A., Volin, L., et al. (2015). Tyrosine Kinase Inhibitors Improve Long-Term Outcome of Allogeneic Hematopoietic Stem Cell Transplantation for Adult Patients with Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia. Haematologica 100, 392–399. doi:10.3324/haematol.2014.116954
Brkic, S., and Meyer, S. C. (2021). Challenges and Perspectives for Therapeutic Targeting of Myeloproliferative Neoplasms. Hemasphere 5, e516. doi:10.1097/hs9.0000000000000516
Bruno, B. J., and Lim, C. S. (2015). Inhibition of Bcr-Abl in Human Leukemic Cells with a Coiled-Coil Protein Delivered by a Leukemia-specific Cell-Penetrating Peptide. Mol. Pharm. 12, 1412–1421. doi:10.1021/mp500701u
Bugarin, C., Sarno, J., Palmi, C., Savino, A. M., Te Kronnie, G., Dworzak, M., et al. (2015). Fine Tuning of Surface CRLF2 Expression and its Associated Signaling Profile in Childhood B-Cell Precursor Acute Lymphoblastic Leukemia. Haematologica 100, e229–e232. doi:10.3324/haematol.2014.114447
Cervantes, F., and Pereira, A. (2017). Does Ruxolitinib Prolong the Survival of Patients with Myelofibrosis? Blood 129, 832–837. doi:10.1182/blood-2016-11-731604
Chalandon, Y., Thomas, X., Hayette, S., Cayuela, J.-M., Abbal, C., Huguet, F., et al. (2015). Randomized Study of Reduced-Intensity Chemotherapy Combined with Imatinib in Adults with Ph-Positive Acute Lymphoblastic Leukemia. Blood 125, 3711–3719. doi:10.1182/blood-2015-02-627935
Chang, Y., Min, J., Jarusiewicz, J. A., Actis, M., Yu-Chen Bradford, S., Mayasundari, A., et al. (2021). Degradation of Janus Kinases in CRLF2-Rearranged Acute Lymphoblastic Leukemia. Blood 138, 2313–2326. doi:10.1182/blood.2020006846
Chase, A., Bryant, C., Score, J., Haferlach, C., Grossmann, V., Schwaab, J., et al. (2013). Ruxolitinib as Potential Targeted Therapy for Patients with JAK2 Rearrangements. Haematologica 98, 404–408. doi:10.3324/haematol.2012.067959
Chatti, K., Farrar, W. L., and Duhé, R. J. (2004). Tyrosine Phosphorylation of the Janus Kinase 2 Activation Loop Is Essential for a High-Activity Catalytic State but Dispensable for a Basal Catalytic State. Biochemistry 43, 4272–4283. doi:10.1021/bi036109b
Chen, C., Li, F., Ma, M.-M., Zhang, S., Liu, Y., Yan, Z.-L., et al. (2019a). Roles of T875N Somatic Mutation in the Activity, Structural Stability of JAK2 and the Transformation of OCI-AML3 Cells. Int. J. Biol. Macromol. 137, 1030–1040. doi:10.1016/j.ijbiomac.2019.07.065
Chen, X., Wang, F., Zhang, Y., Ma, X., Liu, M., Cao, P., et al. (2019b). Identification of RNPC3 as a Novel JAK2 Fusion Partner Gene in B-Acute Lymphoblastic Leukemia Refractory to Combination Therapy Including Ruxolitinib. Mol. Genet. Genomic Med. 8, e1110. doi:10.1002/mgg3.1110
Chen, X., Wang, F., Zhang, Y., Ma, X., Cao, P., Yuan, L., et al. (2021). Fusion Gene Map of Acute Leukemia Revealed by Transcriptome Sequencing of a Consecutive Cohort of 1000 Cases in a Single Center. Blood Cancer J. 11, 112. doi:10.1038/s41408-021-00504-5
Chen, X., Yuan, L., Zhou, J., Wang, F., Zhang, Y., Ma, X., et al. (2022). Sustained Remission after Ruxolitinib and Chimeric Antigen Receptor T‐cell Therapy Bridged to a Second Allogeneic Hematopoietic Stem Cell Transplantation for Relapsed Philadelphia Chromosome‐like B‐cell Precursor Acute Lymphoblastic Leukemia with Novel NPHP3‐JAK2 Fusion. Genes Chromosomes Cancer 61, 55–58. doi:10.1002/gcc.22995
Cheng, Z., Yi, Y., Xie, S., Yu, H., Peng, H., and Zhang, G. (2017). The Effect of the JAK2 Inhibitor TG101209 against T Cell Acute Lymphoblastic Leukemia (T-ALL) Is Mediated by Inhibition of JAK-STAT Signaling and Activation of the Crosstalk between Apoptosis and Autophagy Signaling. Oncotarget 8, 106753–106763. doi:10.18632/oncotarget.22053
Chifotides, H. T., Bose, P., and Verstovsek, S. (2020). Givinostat: an Emerging Treatment for Polycythemia Vera. Expert Opin. Investigational Drugs 29, 525–536. doi:10.1080/13543784.2020.1761323
Cohen, P., Cross, D., and Jänne, P. A. (2021). Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 20, 551–569. doi:10.1038/s41573-021-00195-4
Coltro, G., Mannelli, F., Guglielmelli, P., Pacilli, A., Bosi, A., and Vannucchi, A. M. (2017). A Life-Threatening Ruxolitinib Discontinuation Syndrome. Am. J. Hematol. 92, 833–838. doi:10.1002/ajh.24775
Constantinescu, S. N. (2009). A New Era for Small Molecule Screening: from New Targets, Such as JAK2 V617F, to Complex Cellular Screens. J. Cell. Mol. Med. 13, 212–214. doi:10.1111/j.1582-4934.2008.00666.x
Constantinescu, S. N., Keren, T., Socolovsky, M., Nam, H.-s., Henis, Y. I., and Lodish, H. F. (2001). Ligand-independent Oligomerization of Cell-Surface Erythropoietin Receptor Is Mediated by the Transmembrane Domain. Proc. Natl. Acad. Sci. U.S.A. 98, 4379–4384. doi:10.1073/pnas.081069198
Crump, N. T., Ballabio, E., Godfrey, L., Thorne, R., Repapi, E., Kerry, J., et al. (2021). BET Inhibition Disrupts Transcription but Retains Enhancer-Promoter Contact. Nat. Commun. 12, 223. doi:10.1038/s41467-020-20400-z
Cuesta-Domínguez, Á., Ortega, M., Ormazábal, C., Santos-Roncero, M., Galán-Díez, M., Steegmann, J. L., et al. (2012). Transforming and Tumorigenic Activity of JAK2 by Fusion to BCR: Molecular Mechanisms of Action of a Novel BCR-JAK2 Tyrosine-Kinase. PLoS One 7, e32451. doi:10.1371/journal.pone.0032451
Davis, M. I., Hunt, J. P., Herrgard, S., Ciceri, P., Wodicka, L. M., Pallares, G., et al. (2011). Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 29, 1046–1051. doi:10.1038/nbt.1990
De Braekeleer, E., Douet-Guilbert, N., Morel, F., Le Bris, M.-J., Basinko, A., and De Braekeleer, M. (2012). ETV6 Fusion Genes in Hematological Malignancies: A Review. Leukemia Res. 36, 945–961. doi:10.1016/j.leukres.2012.04.010
Deininger, M., Radich, J., Burn, T. C., Huber, R., Paranagama, D., and Verstovsek, S. (2015). The Effect of Long-Term Ruxolitinib Treatment on JAK2p.V617F Allele Burden in Patients with Myelofibrosis. Blood 126, 1551–1554. doi:10.1182/blood-2015-03-635235
Den Boer, M. L., van Slegtenhorst, M., De Menezes, R. X., Cheok, M. H., Buijs-Gladdines, J. G., Peters, S. T., et al. (2009). A Subtype of Childhood Acute Lymphoblastic Leukaemia with Poor Treatment Outcome: a Genome-wide Classification Study. Lancet Oncol. 10, 125–134. doi:10.1016/s1470-2045(08)70339-5
Deshpande, A., Reddy, M. M., Schade, G. O. M., Ray, A., Chowdary, T. K., Griffin, J. D., et al. (2012). Kinase Domain Mutations Confer Resistance to Novel Inhibitors Targeting JAK2V617F in Myeloproliferative Neoplasms. Leukemia 26, 708–715. doi:10.1038/leu.2011.255
Ding, Y. Y., Stern, J. W., Jubelirer, T. F., Wertheim, G. B., Lin, F., Chang, F., et al. (2018). Clinical Efficacy of Ruxolitinib and Chemotherapy in a Child with Philadelphia Chromosome-like Acute Lymphoblastic Leukemia with GOLGA5-JAK2 Fusion and Induction Failure. Haematologica 103, e427–e431. doi:10.3324/haematol.2018.192088
Dixon, A. S., Miller, G. D., Bruno, B. J., Constance, J. E., Woessner, D. W., Fidler, T. P., et al. (2012). Improved Coiled-Coil Design Enhances Interaction with Bcr-Abl and Induces Apoptosis. Mol. Pharm. 9, 187–195. doi:10.1021/mp200461s
Downes, C. E. J., Mcclure, B. J., Bruning, J. B., Page, E., Breen, J., Rehn, J., et al. (2021). Acquired JAK2 Mutations Confer Resistance to JAK Inhibitors in Cell Models of Acute Lymphoblastic Leukemia. NPJ Precis. Oncol. 5, 75. doi:10.1038/s41698-021-00215-x
Downes, C. E. J., Rehn, J., Heatley, S. L., Yeung, D., Mcclure, B. J., and White, D. L. (2022). Identification of a Novel GOLGA4-JAK2 Fusion Gene in B‐cell Acute Lymphoblastic Leukaemia. Br. J. Haematol. 196, 700–705. doi:10.1111/bjh.17910
Druker, B. J., Guilhot, F., O'brien, S. G., Gathmann, I., Kantarjian, H., Gattermann, N., et al. (2006). Five-year Follow-Up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N. Engl. J. Med. 355, 2408–2417. doi:10.1056/nejmoa062867
Druker, B. J., and Lydon, N. B. (2000). Lessons Learned from the Development of an Abl Tyrosine Kinase Inhibitor for Chronic Myelogenous Leukemia. J. Clin. Invest. 105, 3–7. doi:10.1172/jci9083
Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., et al. (1996). Effects of a Selective Inhibitor of the Abl Tyrosine Kinase on the Growth of Bcr-Abl Positive Cells. Nat. Med. 2, 561–566. doi:10.1038/nm0596-561
Dusa, A., Mouton, C., Pecquet, C., Herman, M., and Constantinescu, S. N. (2010). JAK2 V617F Constitutive Activation Requires JH2 Residue F595: a Pseudokinase Domain Target for Specific Inhibitors. PLoS One 5, e11157. doi:10.1371/journal.pone.0011157
Eder-Azanza, L., Hurtado, C., Navarro-Herrera, D., Aranaz, P., Novo, F. J., and Vizmanos, J. L. (2017). p.Y317H Is a New JAK2 Gain-Of-Function Mutation Affecting the FERM Domain in a Myelofibrosis Patient with CALR Mutation. Haematologica 102, e328–e331. doi:10.3324/haematol.2017.166439
Elnaggar, M. M., Agersborg, S., Sahoo, T., Girgin, A., Ma, W., Rakkhit, R., et al. (2012). BCR-JAK2 Fusion as a Result of a Translocation (9;22)(p24;q11.2) in a Patient with CML-like Myeloproliferative Disease. Mol. Cytogenet 5, 23. doi:10.1186/1755-8166-5-23
Evrot, E., Ebel, N., Romanet, V., Roelli, C., Andraos, R., Qian, Z., et al. (2013). JAK1/2 and Pan-Deacetylase Inhibitor Combination Therapy Yields Improved Efficacy in Preclinical Mouse Models of JAK2V617F-Driven Disease. Clin. Cancer Res. 19, 6230–6241. doi:10.1158/1078-0432.ccr-13-0905
Fasouli, E. S., and Katsantoni, E. (2021). JAK-STAT in Early Hematopoiesis and Leukemia. Front. Cell Dev. Biol. 9, 669363. doi:10.3389/fcell.2021.669363
Feng, J., Witthuhn, B. A., Matsuda, T., Kohlhuber, F., Kerr, I. M., and Ihle, J. N. (1997). Activation of Jak2 Catalytic Activity Requires Phosphorylation of Y1007 in the Kinase Activation Loop. Mol. Cell Biol. 17, 2497–2501. doi:10.1128/mcb.17.5.2497
Fielding, A. K., Rowe, J. M., Buck, G., Foroni, L., Gerrard, G., Litzow, M. R., et al. (2014). UKALLXII/ECOG2993: Addition of Imatinib to a Standard Treatment Regimen Enhances Long-Term Outcomes in Philadelphia Positive Acute Lymphoblastic Leukemia. Blood 123, 843–850. doi:10.1182/blood-2013-09-529008
Fiskus, W., Verstovsek, S., Manshouri, T., Rao, R., Balusu, R., Venkannagari, S., et al. (2011). Heat Shock Protein 90 Inhibitor Is Synergistic with JAK2 Inhibitor and Overcomes Resistance to JAK2-TKI in Human Myeloproliferative Neoplasm Cells. Clin. Cancer Res. 17, 7347–7358. doi:10.1158/1078-0432.ccr-11-1541
Francis, O. L., Milford, T.-A. M., Martinez, S. R., Baez, I., Coats, J. S., Mayagoitia, K., et al. (2016). A Novel Xenograft Model to Study the Role of TSLP-Induced CRLF2 Signals in Normal and Malignant Human B Lymphopoiesis. Haematologica 101, 417–426. doi:10.3324/haematol.2015.125336
Funakoshi-Tago, M., Tago, K., Sumi, K., Abe, M., Aizu-Yokota, E., Oshio, T., et al. (2009). The Acute Lymphoblastic Leukemia-Associated JAK2 L611S Mutant Induces Tumorigenesis in Nude Mice. J. Biol. Chem. 284, 12680–12690. doi:10.1074/jbc.m808879200
Furqan, M., Mukhi, N., Lee, B., and Liu, D. (2013). Dysregulation of JAK-STAT Pathway in Hematological Malignancies and JAK Inhibitors for Clinical Application. Biomark. Res. 1, 5. doi:10.1186/2050-7771-1-5
Gaikwad, A., Rye, C. L., Devidas, M., Heerema, N. A., Carroll, A. J., Izraeli, S., et al. (2009). Prevalence and Clinical Correlates ofJAK2mutations in Down Syndrome Acute Lymphoblastic Leukaemia. Br. J. Haematol. 144, 930–932. doi:10.1111/j.1365-2141.2008.07552.x
Gambacorti-Passerini, C., Antolini, L., Mahon, F.-X., Guilhot, F., Deininger, M., Fava, C., et al. (2011). Multicenter Independent Assessment of Outcomes in Chronic Myeloid Leukemia Patients Treated with Imatinib. J. Natl. Cancer Inst. 103, 553–561. doi:10.1093/jnci/djr060
Gent, J., van Kerkhof, P., Roza, M., Bu, G., Strous, G. J., and Strous, G. J. (2002). Ligand-independent Growth Hormone Receptor Dimerization Occurs in the Endoplasmic Reticulum and Is Required for Ubiquitin System-dependent Endocytosis. Proc. Natl. Acad. Sci. U.S.A. 99, 9858–9863. doi:10.1073/pnas.152294299
Gerds, A., Su, D., Martynova, A., Pannell, B., Mukherjee, S., O'neill, C., et al. (2018). Ruxolitinib Rechallenge Can Improve Constitutional Symptoms and Splenomegaly in Patients with Myelofibrosis: A Case Series. Clin. Lymphoma Myeloma Leukemia 18, e463–e468. doi:10.1016/j.clml.2018.06.025
Gerds, A. T., Savona, M. R., Scott, B. L., Talpaz, M., Egyed, M., Harrison, C. N., et al. (2019). Results of PAC203: A Randomized Phase 2 Dose-Finding Study and Determination of the Recommended Dose of Pacritinib. Blood 134, 667. doi:10.1182/blood-2019-129293
Gisslinger, H., Schalling, M., Gisslinger, B., Skrabs, C., Müllauer, L., and Kralovics, R. (2014). Restoration of Response to Ruxolitinib upon Brief Withdrawal in Two Patients with Myelofibrosis. Am. J. Hematol. 89, 344–346. doi:10.1002/ajh.23637
Glassman, C. R., Tsutsumi, N., Saxton, R. A., Lupardus, P. J., Jude, K. M., and Garcia, K. C. (2022). Structure of a Janus Kinase Cytokine Receptor Complex Reveals the Basis for Dimeric Activation. Science 10, 1126. doi:10.1126/science.abn8933
Gnanasambandan, K., and Sayeski, P. P. (2011). A Structure-Function Perspective of Jak2 Mutations and Implications for Alternate Drug Design Strategies: The Road Not Taken. Cmc 18, 4659–4673. doi:10.2174/092986711797379267
Greenfield, G., Mcpherson, S., Mills, K., and Mcmullin, M. F. (2018). The Ruxolitinib Effect: Understanding How Molecular Pathogenesis and Epigenetic Dysregulation Impact Therapeutic Efficacy in Myeloproliferative Neoplasms. J. Transl. Med. 16, 360. doi:10.1186/s12967-018-1729-7
Grisouard, J., Hao-Shen, H., Dirnhofer, S., Wagner, K.-U., and Skoda, R. C. (2014). Selective Deletion of Jak2 in Adult Mouse Hematopoietic Cells Leads to Lethal Anemia and Thrombocytopenia. Haematologica 99, e52–e54. doi:10.3324/haematol.2013.100016
Gross, S., Rahal, R., Stransky, N., Lengauer, C., and Hoeflich, K. P. (2015). Targeting Cancer with Kinase Inhibitors. J. Clin. Invest. 125, 1780–1789. doi:10.1172/jci76094
Gu, Z., Churchman, M. L., Roberts, K. G., Moore, I., Zhou, X., Nakitandwe, J., et al. (2019). PAX5-driven Subtypes of B-Progenitor Acute Lymphoblastic Leukemia. Nat. Genet. 51, 296–307. doi:10.1038/s41588-018-0315-5
Gupta, V., Cerquozzi, S., Foltz, L., Hillis, C., Devlin, R., Elsawy, M., et al. (2020). Patterns of Ruxolitinib Therapy Failure and its Management in Myelofibrosis: Perspectives of the Canadian Myeloproliferative Neoplasm Group. JCO Oncol. Pract. 16, 351–359. doi:10.1200/jop.19.00506
Hammarén, H. M., Ungureanu, D., Grisouard, J., Skoda, R. C., Hubbard, S. R., and Silvennoinen, O. (2015). ATP Binding to the Pseudokinase Domain of JAK2 Is Critical for Pathogenic Activation. Proc. Natl. Acad. Sci. U.S.A. 112, 4642–4647. doi:10.1073/pnas.1423201112
Hammarén, H. M., Virtanen, A. T., Abraham, B. G., Peussa, H., Hubbard, S. R., and Silvennoinen, O. (2019a). Janus Kinase 2 Activation Mechanisms Revealed by Analysis of Suppressing Mutations. J. Allergy Clin. Immunol. 143, 1549–1559. e1546. doi:10.1016/j.jaci.2018.07.022
Hammarén, H. M., Virtanen, A. T., Raivola, J., and Silvennoinen, O. (2019b). The Regulation of JAKs in Cytokine Signaling and its Breakdown in Disease. Cytokine 118, 48–63. doi:10.1016/j.cyto.2018.03.041
Hardin, J. D., Boast, S., Schwartzberg, P. L., Lee, G., Alt, F. W., Stall, A. M., et al. (1995). Bone Marrow B Lymphocyte Development in C-Abl-Deficient Mice. Cell. Immunol. 165, 44–54. doi:10.1006/cimm.1995.1185
Harrison, C., Kiladjian, J.-J., Al-Ali, H. K., Gisslinger, H., Waltzman, R., Stalbovskaya, V., et al. (2012). JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 366, 787–798. doi:10.1056/nejmoa1110556
Harrison, C. N., Schaap, N., and Mesa, R. A. (2020a). Management of Myelofibrosis after Ruxolitinib Failure. Ann. Hematol. 99, 1177–1191. doi:10.1007/s00277-020-04002-9
Harrison, C. N., Schaap, N., Vannucchi, A. M., Kiladjian, J.-J., Tiu, R. V., Zachee, P., et al. (2017). Janus Kinase-2 Inhibitor Fedratinib in Patients with Myelofibrosis Previously Treated with Ruxolitinib (JAKARTA-2): a Single-Arm, Open-Label, Non-randomised, Phase 2, Multicentre Study. Lancet Haematol. 4, e317–e324. doi:10.1016/s2352-3026(17)30088-1
Harrison, C. N., Schaap, N., Vannucchi, A. M., Kiladjian, J. J., Jourdan, E., Silver, R. T., et al. (2020b). Fedratinib in Patients with Myelofibrosis Previously Treated with Ruxolitinib: An Updated Analysis of the JAKARTA2 Study Using Stringent Criteria for Ruxolitinib Failure. Am. J. Hematol. 95, 594–603. doi:10.1002/ajh.25777
Harrison, C. N., Vannucchi, A. M., Platzbecker, U., Cervantes, F., Gupta, V., Lavie, D., et al. (2018). Momelotinib versus Best Available Therapy in Patients with Myelofibrosis Previously Treated with Ruxolitinib (SIMPLIFY 2): a Randomised, Open-Label, Phase 3 Trial. Lancet Haematol. 5, e73–e81. doi:10.1016/s2352-3026(17)30237-5
Harrison, C. N., Vannucchi, A. M., Vannucchi, A. M., Kiladjian, J.-J., Al-Ali, H. K., Gisslinger, H., et al. (2016). Long-term Findings from COMFORT-II, a Phase 3 Study of Ruxolitinib vs Best Available Therapy for Myelofibrosis. Leukemia 30, 1701–1707. doi:10.1038/leu.2016.148
Harvey, R. C., Mullighan, C. G., Chen, I.-M., Wharton, W., Mikhail, F. M., Carroll, A. J., et al. (2010). Rearrangement of CRLF2 Is Associated with Mutation of JAK Kinases, Alteration of IKZF1, Hispanic/Latino Ethnicity, and a Poor Outcome in Pediatric B-Progenitor Acute Lymphoblastic Leukemia. Blood 115, 5312–5321. doi:10.1182/blood-2009-09-245944
Harvey, R. C., and Tasian, S. K. (2020). Clinical Diagnostics and Treatment Strategies for Philadelphia Chromosome-like Acute Lymphoblastic Leukemia. Blood Adv. 4, 218–228. doi:10.1182/bloodadvances.2019000163
He, R., Greipp, P. T., Rangan, A., Mai, M., Chen, D., Reichard, K. K., et al. (2016). BCR-JAK2 Fusion in a Myeloproliferative Neoplasm with Associated Eosinophilia. Cancer Genet. 209, 223–228. doi:10.1016/j.cancergen.2016.03.002
Herold, T., Schneider, S., Metzeler, K. H., Neumann, M., Hartmann, L., Roberts, K. G., et al. (2017). Adults with Philadelphia Chromosome-like Acute Lymphoblastic Leukemia Frequently Have IGH-CRLF2 and JAK2 Mutations, Persistence of Minimal Residual Disease and Poor Prognosis. Haematologica 102, 130–138. doi:10.3324/haematol.2015.136366
Hertzberg, L., Vendramini, E., Ganmore, I., Cazzaniga, G., Schmitz, M., Chalker, J., et al. (2010). Down Syndrome Acute Lymphoblastic Leukemia, a Highly Heterogeneous Disease in Which Aberrant Expression of CRLF2 Is Associated with Mutated JAK2: a Report from the International BFM Study Group. Blood 115, 1006–1017. doi:10.1182/blood-2009-08-235408
Ho, K., Valdez, F., Garcia, R., and Tirado, C. A. (2010). JAK2 Translocations in Hematological Malignancies: Review of the Literature. J. Assoc. Genet. Technol. 36, 107–109.
Hobbs, G. S., Hanasoge Somasundara, A. V., Kleppe, M., Litvin, R., Arcila, M., Ahn, J., et al. (2018). Hsp90 Inhibition Disrupts JAK-STAT Signaling and Leads to Reductions in Splenomegaly in Patients with Myeloproliferative Neoplasms. Haematologica 103, e5–e9. doi:10.3324/haematol.2017.177600
Hochhaus, A., Baccarani, M., Silver, R. T., Schiffer, C., Apperley, J. F., Cervantes, F., et al. (2020). European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia 34, 966–984. doi:10.1038/s41375-020-0776-2
Hochhaus, A., Larson, R. A., Guilhot, F., Radich, J. P., Branford, S., Hughes, T. P., et al. (2017). Long-term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 376, 917–927. doi:10.1056/nejmoa1609324
Hock, H., and Shimamura, A. (2017). ETV6 in Hematopoiesis and Leukemia Predisposition. Seminars Hematol. 54, 98–104. doi:10.1053/j.seminhematol.2017.04.005
Hornakova, T., Springuel, L., Devreux, J., Dusa, A., Constantinescu, S. N., Knoops, L., et al. (2011). Oncogenic JAK1 and JAK2-Activating Mutations Resistant to ATP-Competitive Inhibitors. Haematologica 96, 845–853. doi:10.3324/haematol.2010.036350
Huang, X., Celiker, M., Guarini, L., Patel, S., and Chen, N. N. (2020). TBL1XR1-JAK2: a Novel Fusion in a Pediatric T Cell Acute Lymphoblastic Leukemia Patient with Increased Absolute Eosinophil Count. J. Hematop. 13, 259–263. doi:10.1007/s12308-020-00413-9
Hubbard, S. R. (2018). Mechanistic Insights into Regulation of JAK2 Tyrosine Kinase. Front. Endocrinol. 8, 1–7. doi:10.3389/fendo.2017.00361
Hughes, T. P., Mauro, M. J., Cortes, J. E., Minami, H., Rea, D., Deangelo, D. J., et al. (2019). Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 381, 2315–2326. doi:10.1056/nejmoa1902328
Hunger, S. P., and Mullighan, C. G. (2015). Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 373, 1541–1552. doi:10.1056/nejmra1400972
Iacobucci, I., and Roberts, K. G. (2021). Genetic Alterations and Therapeutic Targeting of Philadelphia-like Acute Lymphoblastic Leukemia. Genes (Basel) 12. doi:10.3390/genes12050687
Iacobucci, I., and Mullighan, C. G. (2017). Genetic Basis of Acute Lymphoblastic Leukemia. Jco 35, 975–983. doi:10.1200/jco.2016.70.7836
Imamura, T., Kiyokawa, N., Kato, M., Imai, C., Okamoto, Y., Yano, M., et al. (2016). Characterization of Pediatric Philadelphia-negative B-Cell Precursor Acute Lymphoblastic Leukemia with Kinase Fusions in Japan. Blood Cancer J. 6, e419. doi:10.1038/bcj.2016.28
Inaba, H., and Pui, C.-H. (2019). Immunotherapy in Pediatric Acute Lymphoblastic Leukemia. Cancer Metastasis Rev. 38, 595–610. doi:10.1007/s10555-019-09834-0
Irie-Sasaki, J., Sasaki, T., Matsumoto, W., Opavsky, A., Cheng, M., Welstead, G., et al. (2001). CD45 Is a JAK Phosphatase and Negatively Regulates Cytokine Receptor Signalling. Nature 409, 349–354. doi:10.1038/35053086
Ivashkiv, L. B., and Hu, X. (2004). Signaling by STATs. Arthritis Res. Ther. 6, 159–168. doi:10.1186/ar1197
Iwakawa, R., Takenaka, M., Kohno, T., Shimada, Y., Totoki, Y., Shibata, T., et al. (2013). Genome‐wide Identification of Genes with Amplification And/or Fusion in Small Cell Lung Cancer. Genes Chromosom. Cancer 52, 802–816. doi:10.1002/gcc.22076
Jabbour, E., O'brien, S., Konopleva, M., and Kantarjian, H. (2015). New Insights into the Pathophysiology and Therapy of Adult Acute Lymphoblastic Leukemia. Cancer 121, 2517–2528. doi:10.1002/cncr.29383
Jain, N., Roberts, K. G., Jabbour, E., Patel, K., Eterovic, A. K., Chen, K., et al. (2017). Ph-like Acute Lymphoblastic Leukemia: a High-Risk Subtype in Adults. Blood 129, 572–581. doi:10.1182/blood-2016-07-726588
James, C., Ugo, V., Le Couédic, J.-P., Staerk, J., Delhommeau, F., Lacout, C., et al. (2005). A Unique Clonal JAK2 Mutation Leading to Constitutive Signalling Causes Polycythaemia Vera. Nature 434, 1144–1148. doi:10.1038/nature03546
Jatiani, S. S., Cosenza, S. C., Reddy, M. V. R., Ha, J. H., Baker, S. J., Samanta, A. K., et al. (2010). A Non-ATP-competitive Dual Inhibitor of JAK2V617F and BCR-Ablt315i Kinases: Elucidation of a Novel Therapeutic Spectrum Based on Substrate Competitive Inhibition. Genes & Cancer 1, 331–345. doi:10.1177/1947601910371337
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., Mcanulla, C., et al. (2014). InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 30, 1236–1240. doi:10.1093/bioinformatics/btu031
Jurado, S., Fedl, A. S., Jaritz, M., Kostanova-Poliakova, D., Malin, S. G., Mullighan, C. G., et al. (2022). The PAX5-JAK2 Translocation Acts as Dual-Hit Mutation that Promotes Aggressive B-Cell Leukemia via Nuclear STAT5 Activation. EMBO J. 10, 15252e108397. doi:10.15252/embj.2021108397
Kang, K., Robinson, G. W., and Hennighausen, L. (2013). Comprehensive Meta-Analysis of Signal Transducers and Activators of Transcription (STAT) Genomic Binding Patterns Discerns Cell-specific Cis-Regulatory Modules. BMC Genomics 14, 4. doi:10.1186/1471-2164-14-4
Kaplan, H. G., Bifulco, C. B., Jin, R., Scanlan, J. M., and Corwin, D. (2021). Treatment of PCM1-JAK2 Fusion Tyrosine Kinase Gene-Related Acute Lymphoblastic Leukemia with Stem Cell Transplantation. Future Rare Dis. 1, FRD10. doi:10.2217/frd-2021-0006
Kargbo, R. B. (2021). Degradation of Janus Kinase for Potential Application in Immune Response Therapeutics. ACS Med. Chem. Lett. 12, 316–317. doi:10.1021/acsmedchemlett.1c00058
Kawamura, M., Taki, T., Kaku, H., Ohki, K., and Hayashi, Y. (2015). Identification ofSPAG9as a novelJAK2fusion Partner Gene in Pediatric Acute Lymphoblastic Leukemia with T(9;17)(p24;q21). Genes Chromosom. Cancer 54, 401–408. doi:10.1002/gcc.22251
Kearney, L., Gonzalez De Castro, D., Yeung, J., Procter, J., Horsley, S. W., Eguchi-Ishimae, M., et al. (2009). Specific JAK2 Mutation (JAK2R683) and Multiple Gene Deletions in Down Syndrome Acute Lymphoblastic Leukemia. Blood 113, 646–648. doi:10.1182/blood-2008-08-170928
Kershaw, N. J., Murphy, J. M., Liau, N. P. D., Varghese, L. N., Laktyushin, A., Whitlock, E. L., et al. (2013a). SOCS3 Binds Specific Receptor-JAK Complexes to Control Cytokine Signaling by Direct Kinase Inhibition. Nat. Struct. Mol. Biol. 20, 469–476. doi:10.1038/nsmb.2519
Kershaw, N. J., Murphy, J. M., Lucet, I. S., Nicola, N. A., and Babon, J. J. (2013b). Regulation of Janus Kinases by SOCS Proteins. Biochem. Soc. Trans. 41, 1042–1047. doi:10.1042/bst20130077
Kesarwani, M., Huber, E., Kincaid, Z., Evelyn, C. R., Biesiada, J., Rance, M., et al. (2015). Targeting Substrate-Site in Jak2 Kinase Prevents Emergence of Genetic Resistance. Sci. Rep. 5. doi:10.1038/srep14538
Khan, M., Siddiqi, R., and Tran, T. H. (2018). Philadelphia Chromosome-like Acute Lymphoblastic Leukemia: A Review of the Genetic Basis, Clinical Features, and Therapeutic Options. Seminars Hematol. 55, 235–241. doi:10.1053/j.seminhematol.2018.05.001
Kim, S.-K., Knight, D. A., Jones, L. R., Vervoort, S., Ng, A. P., Seymour, J. F., et al. (2018). JAK2 Is Dispensable for Maintenance of JAK2 Mutant B-Cell Acute Lymphoblastic Leukemias. Genes Dev. 32, 849–864. doi:10.1101/gad.307504.117
Klingmüller, U., Lorenz, U., Cantley, L. C., Neel, B. G., and Lodish, H. F. (1995). Specific Recruitment of SH-PTP1 to the Erythropoietin Receptor Causes Inactivation of JAK2 and Termination of Proliferative Signals. Cell 80, 729–738. doi:10.1016/0092-8674(95)90351-8
Konoplev, S., Lu, X., Konopleva, M., Jain, N., Ouyang, J., Goswami, M., et al. (2017). CRLF2-Positive B-Cell Acute Lymphoblastic Leukemia in Adult Patients. Am. J. Clin. Pathol. 147, 357–363. doi:10.1093/ajcp/aqx005
Koppikar, P., Bhagwat, N., Kilpivaara, O., Manshouri, T., Adli, M., Hricik, T., et al. (2012). Heterodimeric JAK-STAT Activation as a Mechanism of Persistence to JAK2 Inhibitor Therapy. Nature 489, 155–159. doi:10.1038/nature11303
Kralovics, R., Passamonti, F., Buser, A. S., Teo, S.-S., Tiedt, R., Passweg, J. R., et al. (2005). A Gain-Of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 352, 1779–1790. doi:10.1056/nejmoa051113
Krämer, A., Reiter, A., Kruth, J., Erben, P., Hochhaus, A., Müller, M., et al. (2007). JAK2-V617F Mutation in a Patient with Philadelphia-chromosome-positive Chronic Myeloid Leukaemia. Lancet Oncol. 8, 658–660. doi:10.1016/s1470-2045(07)70206-1
Kratz, C. P., Böll, S., Kontny, U., Schrappe, M., Niemeyer, C. M., and Stanulla, M. (2006). Mutational Screen Reveals a Novel JAK2 Mutation, L611S, in a Child with Acute Lymphoblastic Leukemia. Leukemia 20, 381–383. doi:10.1038/sj.leu.2404060
Krebs, D. L., and Hilton, D. J. (2001). SOCS Proteins: Negative Regulators of Cytokine Signaling. Stem Cells 19, 378–387. doi:10.1634/stemcells.19-5-378
Kröger, N., Sbianchi, G., Sirait, T., Wolschke, C., Beelen, D., Passweg, J., et al. (2021). Impact of Prior JAK-Inhibitor Therapy with Ruxolitinib on Outcome after Allogeneic Hematopoietic Stem Cell Transplantation for Myelofibrosis: a Study of the CMWP of EBMT. Leukemia 35, 3551–3560. doi:10.1038/s41375-021-01276-4
Kumar, C., Purandare, A. V., Lee, F. Y., and Lorenzi, M. V. (2009). Kinase Drug Discovery Approaches in Chronic Myeloproliferative Disorders. Oncogene 28, 2305–2313. doi:10.1038/onc.2009.107
Kuykendall, A. T., Horvat, N. P., Pandey, G., Komrokji, R., and Reuther, G. W. (2020). Finding a Jill for JAK: Assessing Past, Present, and Future JAK Inhibitor Combination Approaches in Myelofibrosis, Cancers (Basel) 12, 2278. doi:10.3390/cancers12082278
Kuykendall, A. T., Shah, S., Talati, C., Al Ali, N., Sweet, K., Padron, E., et al. (2018). Between a Rux and a Hard Place: Evaluating Salvage Treatment and Outcomes in Myelofibrosis after Ruxolitinib Discontinuation. Ann. Hematol. 97, 435–441. doi:10.1007/s00277-017-3194-4
Lacronique, V., Boureux, A., Della Valle, V., Poirel, H., Quang, C. T., Mauchauffé, M., et al. (1997). A TEL-JAK2 Fusion Protein with Constitutive Kinase Activity in Human Leukemia. Science 278, 1309–1312. doi:10.1126/science.278.5341.1309
Lee, J. W., Kim, Y. G., Soung, Y. H., Han, K. J., Kim, S. Y., Rhim, H. S., et al. (2006). The JAK2 V617F Mutation in De Novo Acute Myelogenous Leukemias. Oncogene 25, 1434–1436. doi:10.1038/sj.onc.1209163
Lee, J. Y., and Stearns, T. (2013). FOP Is a Centriolar Satellite Protein Involved in Ciliogenesis. PLoS One 8, e58589. doi:10.1371/journal.pone.0058589
Leroy, E., and Constantinescu, S. N. (2017). Erratum: Rethinking JAK2 Inhibition: towards Novel Strategies of More Specific and Versatile Janus Kinase Inhibition. Leukemia 31, 2853. doi:10.1038/leu.2017.158
Leroy, E., Dusa, A., Colau, D., Motamedi, A., Cahu, X., Mouton, C., et al. (2016). Uncoupling JAK2 V617F Activation from Cytokine-Induced Signalling by Modulation of JH2 αC Helix. Biochem. J. 473, 1579–1591. doi:10.1042/bcj20160085
Levavi, H., Tripodi, J., Marcellino, B., Mascarenhas, J., Jones, A. V., Cross, N. C. P., et al. (2019). A Novel t(1;9)(p36;p24.1) JAK2 Translocation and Review of the Literature. Acta Haematol. 142, 105–112. doi:10.1159/000498945
Levine, R. L., Pardanani, A., Tefferi, A., and Gilliland, D. G. (2007). Role of JAK2 in the Pathogenesis and Therapy of Myeloproliferative Disorders. Nat. Rev. Cancer 7, 673–683. doi:10.1038/nrc2210
Levine, R. L., Wadleigh, M., Cools, J., Ebert, B. L., Wernig, G., Huntly, B. J. P., et al. (2005). Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell 7, 387–397. doi:10.1016/j.ccr.2005.03.023
Levy, D. E., and Darnell, J. E. (2002). Stats: Transcriptional Control and Biological Impact. Nat. Rev. Mol. Cell Biol. 3, 651–662. doi:10.1038/nrm909
Li, B., Rampal, R. K., and Xiao, Z. (2019). Targeted Therapies for Myeloproliferative Neoplasms. Biomark. Res. 7, 15. doi:10.1186/s40364-019-0166-y
Li, F., Li, W., Liu, Y., Tong, Y.-X., Zhou, P., Wang, L., et al. (2015). Effects of the I682F Mutation on JAK2's Activity, Structure and Stability. Int. J. Biol. Macromol. 79, 118–125. doi:10.1016/j.ijbiomac.2015.04.063
Li, J. F., Dai, Y. T., Lilljebjörn, H., Shen, S. H., Cui, B. W., Bai, L., et al. (2018). Transcriptional Landscape of B Cell Precursor Acute Lymphoblastic Leukemia Based on an International Study of 1,223 Cases. Proc. Natl. Acad. Sci. U. S. A. 115, e11711–e11720. doi:10.1073/pnas.1814397115
Liau, N. P. D., Laktyushin, A., Lucet, I. S., Murphy, J. M., Yao, S., Whitlock, E., et al. (2018). The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat. Commun. 9, 1558. doi:10.1038/s41467-018-04013-1
Lipka, D. B., Hoffmann, L. S., Heidel, F., Markova, B., Blum, M.-C., Breitenbuecher, F., et al. (2008). LS104, a Non-ATP-competitive Small-Molecule Inhibitor of JAK2, Is Potently Inducing Apoptosis in JAK2V617F-Positive Cells. Mol. Cancer Ther. 7, 1176–1184. doi:10.1158/1535-7163.mct-07-2215
Livnah, O., Stura, E. A., Middleton, S. A., Johnson, D. L., Jolliffe, L. K., and Wilson, I. A. (1999). Crystallographic Evidence for Preformed Dimers of Erythropoietin Receptor before Ligand Activation. Science 283, 987–990. doi:10.1126/science.283.5404.987
Loh, M. L., Zhang, J., Harvey, R. C., Roberts, K., Payne-Turner, D., Kang, H., et al. (2013). Tyrosine Kinome Sequencing of Pediatric Acute Lymphoblastic Leukemia: a Report from the Children's Oncology Group TARGET Project. Blood 121, 485–488. doi:10.1182/blood-2012-04-422691
Lu, X., Huang, L. J.-S., and Lodish, H. F. (2008). Dimerization by a Cytokine Receptor Is Necessary for Constitutive Activation of JAK2V617F. J. Biol. Chem. 283, 5258–5266. doi:10.1074/jbc.m707125200
Lucet, I. S., Fantino, E., Styles, M., Bamert, R., Patel, O., Broughton, S. E., et al. (2006). The Structural Basis of Janus Kinase 2 Inhibition by a Potent and Specific Pan-Janus Kinase Inhibitor. Blood 107, 176–183. doi:10.1182/blood-2005-06-2413
Lundberg, P., Takizawa, H., Kubovcakova, L., Guo, G., Hao-Shen, H., Dirnhofer, S., et al. (2014). Myeloproliferative Neoplasms Can Be Initiated from a Single Hematopoietic Stem Cell Expressing JAK2-V617f. J. Exp. Med. 211, 2213–2230. doi:10.1084/jem.20131371
Lupardus, P. J., Ultsch, M., Wallweber, H., Bir Kohli, P., Johnson, A. R., and Eigenbrot, C. (2014). Structure of the Pseudokinase-Kinase Domains from Protein Kinase TYK2 Reveals a Mechanism for Janus Kinase (JAK) Autoinhibition. Proc. Natl. Acad. Sci. U.S.A. 111, 8025–8030. doi:10.1073/pnas.1401180111
Malinge, S., Ben-Abdelali, R., Settegrana, C., Radford-Weiss, I., Debre, M., Beldjord, K., et al. (2007). Novel Activating JAK2 Mutation in a Patient with Down Syndrome and B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 109, 2202–2204. doi:10.1182/blood-2006-09-045963
Mao, X., Ren, Z., Parker, G. N., Sondermann, H., Pastorello, M. A., Wang, W., et al. (2005). Structural Bases of Unphosphorylated STAT1 Association and Receptor Binding. Mol. Cell 17, 761–771. doi:10.1016/j.molcel.2005.02.021
Marit, M. R., Chohan, M., Matthew, N., Huang, K., Kuntz, D. A., Rose, D. R., et al. (2012). Random Mutagenesis Reveals Residues of JAK2 Critical in Evading Inhibition by a Tyrosine Kinase Inhibitor. PLoS One 7, e43437. doi:10.1371/journal.pone.0043437
Mark, H. F. L., Sotomayor, E. A., Nelson, M., Chaves, F., Sanger, W. G., Kaleem, Z., et al. (2006). Chronic Idiopathic Myelofibrosis (CIMF) Resulting from a Unique 3;9 Translocation Disrupting the Janus Kinase 2 (JAK2) Gene. Exp. Mol. Pathology 81, 217–223. doi:10.1016/j.yexmp.2006.07.004
Markovic, I., and Savvides, S. N. (2020). Modulation of Signaling Mediated by TSLP and IL-7 in Inflammation, Autoimmune Diseases, and Cancer. Front. Immunol. 11, 1557.
Marty, C., Saint-Martin, C., Pecquet, C., Grosjean, S., Saliba, J., Mouton, C., et al. (2014). Germ-line JAK2 Mutations in the Kinase Domain Are Responsible for Hereditary Thrombocytosis and Are Resistant to JAK2 and HSP90 Inhibitors. Blood 123, 1372–1383. doi:10.1182/blood-2013-05-504555
Maru, Y., and Witte, O. N. (1991). The BCR Gene Encodes a Novel Serine/threonine Kinase Activity within a Single Exon. Cell 67, 459–468. doi:10.1016/0092-8674(91)90521-y
Marubayashi, S., Koppikar, P., Taldone, T., Abdel-Wahab, O., West, N., Bhagwat, N., et al. (2010). HSP90 Is a Therapeutic Target in JAK2-dependent Myeloproliferative Neoplasms in Mice and Humans. J. Clin. Invest. 120, 3578–3593. doi:10.1172/jci42442
Mascarenhas, J., Hoffman, R., Talpaz, M., Gerds, A. T., Stein, B., Gupta, V., et al. (2018). Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients with Myelofibrosis. JAMA Oncol. 4, 652–659. doi:10.1001/jamaoncol.2017.5818
Mascarenhas, J., Marcellino, B. K., Lu, M., Kremyanskaya, M., Fabris, F., Sandy, L., et al. (2020). A Phase I Study of Panobinostat and Ruxolitinib in Patients with Primary Myelofibrosis (PMF) and Post-polycythemia Vera/essential Thrombocythemia Myelofibrosis (Post-PV/ET MF). Leukemia Res. 88, 106272. doi:10.1016/j.leukres.2019.106272
Maslah, N., Cassinat, B., Verger, E., Kiladjian, J.-J., and Velazquez, L. (2017). The Role of LNK/SH2B3 Genetic Alterations in Myeloproliferative Neoplasms and Other Hematological Disorders. Leukemia 31, 1661–1670. doi:10.1038/leu.2017.139
Maude, S. L., Tasian, S. K., Vincent, T., Hall, J. W., Sheen, C., Roberts, K. G., et al. (2012). Targeting JAK1/2 and mTOR in Murine Xenograft Models of Ph-like Acute Lymphoblastic Leukemia. Blood 120, 3510–3518. doi:10.1182/blood-2012-03-415448
Mayfield, J. R., Czuchlewski, D. R., Gale, J. M., Matlawska-Wasowska, K., Vasef, M. A., and Nickl, C. (2017). Integration of Ruxolitinib into Dose-Intensified Therapy Targeted against a Novel JAK2 F694L Mutation in B-Precursor Acute Lymphoblastic Leukemia. Pediatr. Blood Cancer 64. doi:10.1002/pbc.26328
Medves, S., and Demoulin, J.-B. (2012). Tyrosine Kinase Gene Fusions in Cancer: Translating Mechanisms into Targeted Therapies. J. Cell. Mol. Med. 16, 237–248. doi:10.1111/j.1582-4934.2011.01415.x
Mesa, R. A., Kiladjian, J.-J., Catalano, J. V., Devos, T., Egyed, M., Hellmann, A., et al. (2017a). SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib versus Ruxolitinib in Janus Kinase Inhibitor-Naïve Patients with Myelofibrosis. Jco 35, 3844–3850. doi:10.1200/jco.2017.73.4418
Mesa, R. A., Vannucchi, A. M., Mead, A., Egyed, M., Szoke, A., Suvorov, A., et al. (2017b). Pacritinib versus Best Available Therapy for the Treatment of Myelofibrosis Irrespective of Baseline Cytopenias (PERSIST-1): an International, Randomised, Phase 3 Trial. Lancet Haematol. 4, e225–e236. doi:10.1016/s2352-3026(17)30027-3
Meyer, S. C., Keller, M. D., Chiu, S., Koppikar, P., Guryanova, O. A., Rapaport, F., et al. (2015). CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 28, 15–28. doi:10.1016/j.ccell.2015.06.006
Meyer, S. C. (2017). Mechanisms of Resistance to JAK2 Inhibitors in Myeloproliferative Neoplasms. Hematology/Oncology Clin. N. Am. 31, 627–642. doi:10.1016/j.hoc.2017.04.003
Mian, A. A., Oancea, C., Zhao, Z., Ottmann, O. G., and Ruthardt, M. (2009). Oligomerization Inhibition, Combined with Allosteric Inhibition, Abrogates the Transformation Potential of T315I-Positive BCR/ABL. Leukemia 23, 2242–2247. doi:10.1038/leu.2009.194
Miller, G. D., Bruno, B. J., and Lim, C. S. (2014). Resistant Mutations in CML and Ph(+)ALL - Role of Ponatinib. Biologics 8, 243–254. doi:10.2147/BTT.S50734
Mohamed, S. M. A., Wohlmann, A., Schofield, P., Sia, K. C. S., Mccalmont, H., Savvides, S. N., et al. (2021). A Recombinant Antibody Fragment Directed to the Thymic Stromal Lymphopoietin Receptor (CRLF2) Efficiently Targets Pediatric Philadelphia Chromosome-like Acute Lymphoblastic Leukemia. Int. J. Biol. Macromol. 190, 214–223. doi:10.1016/j.ijbiomac.2021.08.194
Morris, R., Kershaw, N. J., and Babon, J. J. (2018). The Molecular Details of Cytokine Signaling via the JAK/STAT Pathway. Protein Sci. 27, 1984–2009. doi:10.1002/pro.3519
Mughal, T. I., Radich, J. P., Deininger, M. W., Apperley, J. F., Hughes, T. P., Harrison, C. J., et al. (2016). Chronic Myeloid Leukemia: Reminiscences and Dreams. Haematologica 101, 541–558. doi:10.3324/haematol.2015.139337
Mullally, A., Hood, J., Harrison, C., and Mesa, R. (2020). Fedratinib in Myelofibrosis. Blood Adv. 4, 1792–1800. doi:10.1182/bloodadvances.2019000954
Mullighan, C. G., Collins-Underwood, J. R., Phillips, L. A. A., Loudin, M. G., Liu, W., Zhang, J., et al. (2009a). Rearrangement of CRLF2 in B-Progenitor- and Down Syndrome-Associated Acute Lymphoblastic Leukemia. Nat. Genet. 41, 1243–1246. doi:10.1038/ng.469
Mullighan, C. G., Miller, C. B., Radtke, I., Phillips, L. A., Dalton, J., Ma, J., et al. (2008). BCR-ABL1 Lymphoblastic Leukaemia Is Characterized by the Deletion of Ikaros. Nature 453, 110–114. doi:10.1038/nature06866
Mullighan, C. G., Su, X., Zhang, J., Radtke, I., Phillips, L. A. A., Miller, C. B., et al. (2009b). Deletion ofIKZF1and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 360, 470–480. doi:10.1056/nejmoa0808253
Mullighan, C. G., Zhang, J., Harvey, R. C., Collins-Underwood, J. R., Schulman, B. A., Phillips, L. A., et al. (2009c). JAK Mutations in High-Risk Childhood Acute Lymphoblastic Leukemia. Proc. Natl. Acad. Sci. U.S.A. 106, 9414–9418. doi:10.1073/pnas.0811761106
Murati, A., Gelsi-Boyer, V., Adélaïde, J., Perot, C., Talmant, P., Giraudier, S., et al. (2005). PCM1-JAK2 Fusion in Myeloproliferative Disorders and Acute Erythroid Leukemia with T(8;9) Translocation. Leukemia 19, 1692–1696. doi:10.1038/sj.leu.2403879
Musella, M., Galassi, C., Manduca, N., and Sistigu, A. (2021). The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation. Biol. (Basel) 10. doi:10.3390/biology10090856
Neculai, D., Neculai, A. M., Verrier, S., Straub, K., Klumpp, K., Pfitzner, E., et al. (2005). Structure of the Unphosphorylated STAT5a Dimer*. J. Biol. Chem. 280, 40782–40787. doi:10.1074/jbc.m507682200
Neubauer, H., Cumano, A., Müller, M., Wu, H., Huffstadt, U., and Pfeffer, K. (1998). Jak2 Deficiency Defines an EssentialDevelopmental Checkpoint in DefinitiveHematopoiesis. Cell 93, 397–409. doi:10.1016/s0092-8674(00)81168-x
Numazaki, M., Abe, M., Hanaoka, K., Imamura, E., Maeda, M., Kimura, A., et al. (2021). ASP7266, a Novel Antibody against Human Thymic Stromal Lymphopoietin Receptor for the Treatment of Allergic Diseases. J. Pharmacol. Exp. Ther. 10, 1124. doi:10.1124/jpet.121.000686
Page, E. C., Heatley, S. L., Yeung, D. T., Thomas, P. Q., and White, D. L. (2018). Precision Medicine Approaches May Be the Future for CRLF2 Rearranged Down Syndrome Acute Lymphoblastic Leukaemia Patients. Cancer Lett. 432, 69–74. doi:10.1016/j.canlet.2018.05.045
Palandri, F., Breccia, M., Bonifacio, M., Polverelli, N., Elli, E. M., Benevolo, G., et al. (2020). Life after Ruxolitinib: Reasons for Discontinuation, Impact of Disease Phase, and Outcomes in 218 Patients with Myelofibrosis. Cancer 126, 1243–1252. doi:10.1002/cncr.32664
Palandri, F., Palumbo, G. A., Elli, E. M., Polverelli, N., Benevolo, G., Martino, B., et al. (2021). Ruxolitinib Discontinuation Syndrome: Incidence, Risk Factors, and Management in 251 Patients with Myelofibrosis. Blood Cancer J. 11, 4. doi:10.1038/s41408-020-00392-1
Pardanani, A., Gotlib, J. R., Jamieson, C., Cortes, J. E., Talpaz, M., Stone, R. M., et al. (2011). Safety and Efficacy of TG101348, a Selective JAK2 Inhibitor, in Myelofibrosis. Jco 29, 789–796. doi:10.1200/jco.2010.32.8021
Pardanani, A., Harrison, C., Cortes, J. E., Cervantes, F., Mesa, R. A., Milligan, D., et al. (2015). Safety and Efficacy of Fedratinib in Patients with Primary or Secondary Myelofibrosis. JAMA Oncol. 1, 643–651. doi:10.1001/jamaoncol.2015.1590
Pardanani, A. (2007). JAK2 Inhibitor Therapy in Myeloproliferative Disorders: Rationale, Preclinical Studies and Ongoing Clinical Trials. Leukemia 22, 23–30. doi:10.1038/sj.leu.2404948
Passamonti, F., Griesshammer, M., Palandri, F., Egyed, M., Benevolo, G., Devos, T., et al. (2017). Ruxolitinib for the Treatment of Inadequately Controlled Polycythaemia Vera without Splenomegaly (RESPONSE-2): a Randomised, Open-Label, Phase 3b Study. Lancet Oncol. 18, 88–99. doi:10.1016/s1470-2045(16)30558-7
Passamonti, F., Vannucchi, A. M., Cervantes, F., Harrison, C., Morra, E., Kantarjian, H., et al. (2015). Ruxolitinib and Survival Improvement in Patients with Myelofibrosis. Leukemia 29, 739–740. doi:10.1038/leu.2014.282
Pastorczak, A., Sedek, L., Braun, M., Madzio, J., Sonsala, A., Twardoch, M., et al. (2018). Surface Expression of Cytokine Receptor-like Factor 2 Increases Risk of Relapse in Pediatric Acute Lymphoblastic Leukemia Patients Harboring IKZF1 Deletions. Oncotarget 9, 25971–25982. doi:10.18632/oncotarget.25411
Patel, A. B., O’Hare, T., and Deininger, M. W. (2017). Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematology/Oncology Clin. N. Am. 31, 589–612. doi:10.1016/j.hoc.2017.04.007
Peeters, P., Raynaud, S. D., Cools, J., Wlodarska, I., Grosgeorge, J., Philip, P., et al. (1997). Fusion of TEL, the ETS-Variant Gene 6 (ETV6), to the Receptor-Associated Kinase JAK2 as a Result of T(9; 12) in a Lymphoid and T(9; 15; 12) in a Myeloid Leukemia. Blood 90, 2535–2540. doi:10.1182/blood.v90.7.2535
Peiris, M. N., Meyer, A. N., Nelson, K. N., Bisom-Rapp, E. W., and Donoghue, D. J. (2020). Oncogenic Fusion Protein BCR-FGFR1 Requires the Breakpoint Cluster Region-Mediated Oligomerization and Chaperonin Hsp90 for Activation. Haematologica 105, 1262–1273. doi:10.3324/haematol.2019.220871
Pfeifer, H., Lange, T., Wystub, S., Wassmann, B., Maier, J., Binckebanck, A., et al. (2012). Prevalence and Dynamics of Bcr-Abl Kinase Domain Mutations during Imatinib Treatment Differ in Patients with Newly Diagnosed and Recurrent Bcr-Abl Positive Acute Lymphoblastic Leukemia. Leukemia 26, 1475–1481. doi:10.1038/leu.2012.5
Pfeifer, H., Wassmann, B., Pavlova, A., Wunderle, L., Oldenburg, J., Binckebanck, A., et al. (2007). Kinase Domain Mutations of BCR-ABL Frequently Precede Imatinib-Based Therapy and Give Rise to Relapse in Patients with De Novo Philadelphia-positive Acute Lymphoblastic Leukemia (Ph+ ALL). Blood 110, 727–734. doi:10.1182/blood-2006-11-052373
Poitras, J. L., Cin, P. D., Aster, J. C., Deangelo, D. J., and Morton, C. C. (2008). NovelSSBP2-JAK2fusion Gene Resulting from a t(5;9)(q14.1;p24.1) in Pre-B Acute Lymphocytic Leukemia. Genes Chromosom. Cancer 47, 884–889. doi:10.1002/gcc.20585
Pottier, C., Fresnais, M., Gilon, M., Jérusalem, G., Longuespée, R., and Sounni, N. E. (2020). Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers (Basel) 12. doi:10.3390/cancers12030731
Pui, C.-H., Roberts, K. G., Yang, J. J., and Mullighan, C. G. (2017). Philadelphia Chromosome-like Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leukemia 17, 464–470. doi:10.1016/j.clml.2017.03.299
Qin, H., Cho, M., Haso, W., Zhang, L., Tasian, S. K., Oo, H. Z., et al. (2015). Eradication of B-ALL Using Chimeric Antigen Receptor-Expressing T Cells Targeting the TSLPR Oncoprotein. Blood 126, 629–639. doi:10.1182/blood-2014-11-612903
Quesada, A. E., Zhang, Y., Ptashkin, R., Ho, C., Horwitz, S., Benayed, R., et al. (2021). Next Generation Sequencing of Breast Implant‐associated Anaplastic Large Cell Lymphomas Reveals a Novel STAT3‐JAK2 Fusion Among Other Activating Genetic Alterations within the JAK‐STAT Pathway. Breast J. 27, 314–321. doi:10.1111/tbj.14205
Quintás-Cardama, A., Vaddi, K., Liu, P., Manshouri, T., Li, J., Scherle, P. A., et al. (2010). Preclinical Characterization of the Selective JAK1/2 Inhibitor INCB018424: Therapeutic Implications for the Treatment of Myeloproliferative Neoplasms. Blood 115, 3109–3117. doi:10.1182/blood-2009-04-214957
Raivola, J., Haikarainen, T., Abraham, B. G., and Silvennoinen, O. (2021). Janus Kinases in Leukemia. Cancers (Basel) 13. doi:10.3390/cancers13040800
Reshmi, S. C., Harvey, R. C., Roberts, K. G., Stonerock, E., Smith, A., Jenkins, H., et al. (2017). Targetable Kinase Gene Fusions in High-Risk B-ALL: a Study from the Children's Oncology Group. Blood 129, 3352–3361. doi:10.1182/blood-2016-12-758979
Rizzuto, G., Leoncin, M., Imbergamo, S., Taurino, D., Mico, M. C., Tosi, M., et al. (2022). Sequential Allogeneic Transplantation and Ruxolitinib Maintenance for a Synchronous PCM1-JAK2 Positive Myeloid Sarcoma and Acute B-Lymphoblastic Leukemia. Clin. Case Rep. 10, e05212. doi:10.1002/ccr3.5212
Roberts, K. G., Yang, Y. L., Payne-Turner, D., Lin, W., Files, J. K., Dickerson, K., et al. (2017b). Oncogenic Role and Therapeutic Targeting of ABL-Class and JAK-STAT Activating Kinase Alterations in Ph-like ALL. Blood Adv. 1, 1657–1671. doi:10.1182/bloodadvances.2017011296
Roberts, K. G., Gu, Z., Payne-Turner, D., Mccastlain, K., Harvey, R. C., Chen, I.-M., et al. (2017a). High Frequency and Poor Outcome of Philadelphia Chromosome-like Acute Lymphoblastic Leukemia in Adults. Jco 35, 394–401. doi:10.1200/jco.2016.69.0073
Roberts, K. G., Li, Y., Payne-Turner, D., Harvey, R. C., Yang, Y.-L., Pei, D., et al. (2014a). Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 371, 1005–1015. doi:10.1056/nejmoa1403088
Roberts, K. G., Morin, R. D., Zhang, J., Hirst, M., Zhao, Y., Su, X., et al. (2012). Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell 22, 153–166. doi:10.1016/j.ccr.2012.06.005
Roberts, K. G., and Mullighan, C. G. (2015). Genomics in Acute Lymphoblastic Leukaemia: Insights and Treatment Implications. Nat. Rev. Clin. Oncol. 12, 344–357. doi:10.1038/nrclinonc.2015.38
Roberts, K. G., Pei, D., Campana, D., Payne-Turner, D., Li, Y., Cheng, C., et al. (2014b). Outcomes of Children with BCR-ABL1-like Acute Lymphoblastic Leukemia Treated with Risk-Directed Therapy Based on the Levels of Minimal Residual Disease. Jco 32, 3012–3020. doi:10.1200/jco.2014.55.4105
Roberts, K. G., Reshmi, S. C., Harvey, R. C., Chen, I.-M., Patel, K., Stonerock, E., et al. (2018). Genomic and Outcome Analyses of Ph-like ALL in NCI Standard-Risk Patients: a Report from the Children's Oncology Group. Blood 132, 815–824. doi:10.1182/blood-2018-04-841676
Roll, J. D., and Reuther, G. W. (2010). CRLF2 and JAK2 in B-Progenitor Acute Lymphoblastic Leukemia: A Novel Association in Oncogenesis: Figure 1. Cancer Res. 70, 7347–7352. doi:10.1158/0008-5472.can-10-1528
Ross, D. M., Babon, J. J., Tvorogov, D., and Thomas, D. (2021). Persistence of Myelofibrosis Treated with Ruxolitinib: Biology and Clinical Implications. haematol 106, 1244–1253. doi:10.3324/haematol.2020.262691
Rossari, F., Minutolo, F., and Orciuolo, E. (2018). Past, Present, and Future of Bcr-Abl Inhibitors: from Chemical Development to Clinical Efficacy. J. Hematol. Oncol. 11, 84. doi:10.1186/s13045-018-0624-2
Royer, Y., Staerk, J., Costuleanu, M., Courtoy, P. J., and Constantinescu, S. N. (2005). Janus Kinases Affect Thrombopoietin Receptor Cell Surface Localization and Stability. J. Biol. Chem. 280, 27251–27261. doi:10.1074/jbc.m501376200
Russell, L. J., Capasso, M., Vater, I., Akasaka, T., Bernard, O. A., Calasanz, M. J., et al. (2009). Deregulated Expression of Cytokine Receptor Gene, CRLF2, Is Involved in Lymphoid Transformation in B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 114, 2688–2698. doi:10.1182/blood-2009-03-208397
Sadras, T., Heatley, S. L., Kok, C. H., Mcclure, B. J., Yeung, D., Hughes, T. P., et al. (2017). A Novel Somatic JAK2 Kinase-Domain Mutation in Pediatric Acute Lymphoblastic Leukemia with Rapid On-Treatment Development of LOH. Cancer Genet. 216-217, 86–90. doi:10.1016/j.cancergen.2017.07.008
Saharinen, P., and Silvennoinen, O. (2002). The Pseudokinase Domain Is Required for Suppression of Basal Activity of Jak2 and Jak3 Tyrosine Kinases and for Cytokine-Inducible Activation of Signal Transduction. J. Biol. Chem. 277, 47954–47963. doi:10.1074/jbc.m205156200
Saharinen, P., Takaluoma, K., and Silvennoinen, O. (2000). Regulation of the Jak2 Tyrosine Kinase by its Pseudokinase Domain. Mol. Cell Biol. 20, 3387–3395. doi:10.1128/mcb.20.10.3387-3395.2000
Sakamoto, K., Imamura, T., Kanayama, T., Yano, M., Asai, D., Deguchi, T., et al. (2017). Ph-like Acute Lymphoblastic Leukemia with a novelPAX5-KIDINS220fusion Transcript. Genes Chromosom. Cancer 56, 278–284. doi:10.1002/gcc.22433
Sanz, A., Ungureanu, D., Pekkala, T., Ruijtenbeek, R., Touw, I. P., Hilhorst, R., et al. (2011). Analysis of Jak2 Catalytic Function by Peptide Microarrays: the Role of the JH2 Domain and V617F Mutation. PLoS One 6, e18522. doi:10.1371/journal.pone.0018522
Sawyers, C. L. (2003). Opportunities and Challenges in the Development of Kinase Inhibitor Therapy for Cancer. Genes Dev. 17, 2998–3010. doi:10.1101/gad.1152403
Schaefer, T. S., Sanders, L. K., Park, O. K., and Nathans, D. (1997). Functional Differences between Stat3alpha and Stat3beta. Mol. Cell Biol. 17, 5307–5316. doi:10.1128/mcb.17.9.5307
Schindler, C., and Darnell, J. E. (1995). Transcriptional Responses to Polypeptide Ligands: The JAK-STAT Pathway. Annu. Rev. Biochem. 64, 621–652. doi:10.1146/annurev.bi.64.070195.003201
Schindler, T., Bornmann, W., Pellicena, P., Miller, W. T., Clarkson, B., and Kuriyan, J. (2000). Structural Mechanism for STI-571 Inhibition of Abelson Tyrosine Kinase. Science 289, 1938–1942. doi:10.1126/science.289.5486.1938
Schinnerl, D., Fortschegger, K., Kauer, M., Marchante, J. R. M., Kofler, R., Den Boer, M. L., et al. (2015). The Role of the Janus-Faced Transcription Factor PAX5-JAK2 in Acute Lymphoblastic Leukemia. Blood 125, 1282–1291. doi:10.1182/blood-2014-04-570960
Schrappe, M., Hunger, S. P., Pui, C.-H., Saha, V., Gaynon, P. S., Baruchel, A., et al. (2012). Outcomes after Induction Failure in Childhood Acute Lymphoblastic Leukemia. N. Engl. J. Med. 366, 1371–1381. doi:10.1056/nejmoa1110169
Schwaab, J., Knut, M., Haferlach, C., Metzgeroth, G., Horny, H.-P., Chase, A., et al. (2015). Limited Duration of Complete Remission on Ruxolitinib in Myeloid Neoplasms with PCM1-JAK2 and BCR-JAK2 Fusion Genes. Ann. Hematol. 94, 233–238. doi:10.1007/s00277-014-2221-y
Schwaab, J., Naumann, N., Luebke, J., Jawhar, M., Somervaille, T. C. P., Williams, M. S., et al. (2020). Response to Tyrosine Kinase Inhibitors in Myeloid Neoplasms Associated with PCM1 ‐ JAK2 , BCR‐JAK2 and ETV6‐ABL1 Fusion Genes. Am. J. Hematol. 95, 824–833. doi:10.1002/ajh.25825
Schwab, C., and Harrison, C. J. (2018). Advances in B-Cell Precursor Acute Lymphoblastic Leukemia Genomics. Hemasphere 2, e53. doi:10.1097/hs9.0000000000000053
Schwab, C., Ryan, S. L., Chilton, L., Elliott, A., Murray, J., Richardson, S., et al. (2016). EBF1-PDGFRB Fusion in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL): Genetic Profile and Clinical Implications. Blood 127, 2214–2218. doi:10.1182/blood-2015-09-670166
Schwartzman, O., Savino, A. M., Gombert, M., Palmi, C., Cario, G., Schrappe, M., et al. (2017). Suppressors and Activators of JAK-STAT Signaling at Diagnosis and Relapse of Acute Lymphoblastic Leukemia in Down Syndrome. Proc. Natl. Acad. Sci. U. S. A. 114, E4030–E4039. doi:10.1073/pnas.1702489114
Senkevitch, E., and Durum, S. (2017). The Promise of Janus Kinase Inhibitors in the Treatment of Hematological Malignancies. Cytokine 98, 33–41. doi:10.1016/j.cyto.2016.10.012
Shah, R. R., Redmond, J. M., Mihut, A., Menon, M., Evans, J. P., Murphy, J. A., et al. (2020). Hi-JAK-ing the Ubiquitin System: The Design and Physicochemical Optimisation of JAK PROTACs. Bioorg. Med. Chem. 28, 115326. doi:10.1016/j.bmc.2020.115326
Shan, Y., Gnanasambandan, K., Ungureanu, D., Kim, E. T., Hammarén, H., Yamashita, K., et al. (2014). Molecular Basis for Pseudokinase-dependent Autoinhibition of JAK2 Tyrosine Kinase. Nat. Struct. Mol. Biol. 21, 579–584. doi:10.1038/nsmb.2849
Shiraz, P., Payne, K. J., and Muffly, L. (2020). The Current Genomic and Molecular Landscape of Philadelphia-like Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 21. doi:10.3390/ijms21062193
Shoukier, M., Kubiak, M., and Cortes, J. (2021). Review of New-Generation Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia. Curr. Oncol. Rep. 23, 91. doi:10.1007/s11912-021-01087-x
Silvennoinen, O., and Hubbard, S. R. (2015a). Molecular Insights into Regulation of JAK2 in Myeloproliferative Neoplasms. Blood 125, 3388–3392. doi:10.1182/blood-2015-01-621110
Silvennoinen, O., and Hubbard, S. R. (2015b). Targeting the Inactive Conformation of JAK2 in Hematological Malignancies. Cancer Cell 28, 1–2. doi:10.1016/j.ccell.2015.06.010
Singer, J., Al-Fayoumi, S., Ma, H., Komrokji, R., Mesa, R., and Verstovsek, S. (2016). Comprehensive Kinase Profile of Pacritinib, a Nonmyelosuppressive Janus Kinase 2 Inhibitor. Jep Vol. 8, 11–19. doi:10.2147/jep.s110702
Singer, J. W., Al-Fayoumi, S., Taylor, J., Velichko, S., and O’Mahony, A. (2019). Comparative Phenotypic Profiling of the JAK2 Inhibitors Ruxolitinib, Fedratinib, Momelotinib, and Pacritinib Reveals Distinct Mechanistic Signatures. PLoS One 14, e0222944. doi:10.1371/journal.pone.0222944
Sonbol, M. B., Firwana, B., Zarzour, A., Morad, M., Rana, V., and Tiu, R. V. (2013). Comprehensive Review of JAK Inhibitors in Myeloproliferative Neoplasms. Ther. Adv. Hematol. 4, 15–35. doi:10.1177/2040620712461047
Soverini, S., De Benedittis, C., Papayannidis, C., Paolini, S., Venturi, C., Iacobucci, I., et al. (2014). Drug Resistance and BCR-ABL Kinase Domain Mutations in Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia from the Imatinib to the Second-Generation Tyrosine Kinase Inhibitor Era: The Main Changes Are in the Type of Mutations, but Not in the Fr. Cancer 120, 1002–1009. doi:10.1002/cncr.28522
Speranza, G., Anderson, L., Chen, A. P., Do, K., Eugeni, M., Weil, M., et al. (2018). First-in-human Study of the Epichaperome Inhibitor PU-H71: Clinical Results and Metabolic Profile. Invest New Drugs 36, 230–239. doi:10.1007/s10637-017-0495-3
Starr, R., Willson, T. A., Viney, E. M., Murray, L. J. L., Rayner, J. R., Jenkins, B. J., et al. (1997). A Family of Cytokine-Inducible Inhibitors of Signalling. Nature 387, 917–921. doi:10.1038/43206
Steeghs, E. M. P., Jerchel, I. S., de Goffau-Nobel, W., Hoogkamer, A. Q., Boer, J. M., Boeree, A., et al. (2017). JAK2 Aberrations in Childhood B-Cell Precursor Acute Lymphoblastic Leukemia. Oncotarget 8, 89923–89938. doi:10.18632/oncotarget.21027
Suryani, S., Bracken, L. S., Harvey, R. C., Sia, K. C. S., Carol, H., Chen, I.-M., et al. (2015). Evaluation of the In Vitro and In Vivo Efficacy of the JAK Inhibitor AZD1480 against JAK-Mutated Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 14, 364–374. doi:10.1158/1535-7163.mct-14-0647
Takeda, Y., Nakaseko, C., Tanaka, H., Takeuchi, M., Yui, M., Saraya, A., et al. (2011). Direct Activation of STAT5 by ETV6-LYN Fusion Protein Promotes Induction of Myeloproliferative Neoplasm with Myelofibrosis. Br. J. Haematol. 153, 589–598. doi:10.1111/j.1365-2141.2011.08663.x
Talpaz, M., and Kiladjian, J.-J. (2021). Fedratinib, a Newly Approved Treatment for Patients with Myeloproliferative Neoplasm-Associated Myelofibrosis. Leukemia 35, 1–17. doi:10.1038/s41375-020-0954-2
Tang, G., Sydney Sir Philip, J. K., Weinberg, O., Tam, W., Sadigh, S., Lake, J. I., et al. (2019). Hematopoietic Neoplasms with 9p24/JAK2 Rearrangement: a Multicenter Study. Mod. Pathol. 32, 490–498. doi:10.1038/s41379-018-0165-9
Tasian, S. K., Teachey, D. T., Li, Y., Shen, F., Harvey, R. C., Chen, I. M., et al. (2017c). Potent Efficacy of Combined PI3K/mTOR and JAK or ABL Inhibition in Murine Xenograft Models of Ph-like Acute Lymphoblastic Leukemia. Blood 129, 177–187. doi:10.1182/blood-2016-05-707653
Tasian, S. K., Assad, A., Hunter, D. S., Du, Y., and Loh, M. L. (2018). A Phase 2 Study of Ruxolitinib with Chemotherapy in Children with Philadelphia Chromosome-like Acute Lymphoblastic Leukemia (INCB18424-269/AALL1521): Dose-Finding Results from the Part 1 Safety Phase. Blood 132, 555. doi:10.1182/blood-2018-99-110221
Tasian, S. K., Doral, M. Y., Borowitz, M. J., Wood, B. L., Chen, I.-M., Harvey, R. C., et al. (2012). Aberrant STAT5 and PI3K/mTOR Pathway Signaling Occurs in Human CRLF2-Rearranged B-Precursor Acute Lymphoblastic Leukemia. Blood 120, 833–842. doi:10.1182/blood-2011-12-389932
Tasian, S. K., Hurtz, C., Wertheim, G. B., Bailey, N. G., Lim, M. S., Harvey, R. C., et al. (2017a). High Incidence of Philadelphia Chromosome-like Acute Lymphoblastic Leukemia in Older Adults with B-ALL. Leukemia 31, 981–984. doi:10.1038/leu.2016.375
Tasian, S. K., Loh, M. L., and Hunger, S. P. (2017b). Philadelphia Chromosome-like Acute Lymphoblastic Leukemia. Blood 130, 2064–2072. doi:10.1182/blood-2017-06-743252
Tasian, S., and Loh, M. L. (2011). Understanding the Biology of CRLF2-Overexpressing Acute Lymphoblastic Leukemia. Crit. Rev. Oncog. 16, 13–24. doi:10.1615/critrevoncog.v16.i1-2.30
Tefferi, A., and Pardanani, A. (2011). Serious Adverse Events during Ruxolitinib Treatment Discontinuation in Patients with Myelofibrosis. Mayo Clin. Proc. 86, 1188–1191. doi:10.4065/mcp.2011.0518
Terwilliger, T., and Abdul-Hay, M. (2017). Acute Lymphoblastic Leukemia: a Comprehensive Review and 2017 Update. Blood Cancer J. 7, e577. doi:10.1038/bcj.2017.53
The Uniprot Consortium (2019). UniProt: a Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 47, D506–D515. doi:10.1093/nar/gky1049
Tijchon, E., Havinga, J., Van Leeuwen, F. N., and Scheijen, B. (2013). B-Lineage Transcription Factors and Cooperating Gene Lesions Required for Leukemia Development. Leukemia 27, 541–552. doi:10.1038/leu.2012.293
Tokarski, J. S., Zupa-Fernandez, A., Tredup, J. A., Pike, K., Chang, C., Xie, D., et al. (2015). Tyrosine Kinase 2-mediated Signal Transduction in T Lymphocytes Is Blocked by Pharmacological Stabilization of its Pseudokinase Domain. J. Biol. Chem. 290, 11061–11074. doi:10.1074/jbc.m114.619502
Tran, T. H., Harris, M. H., Nguyen, J. V., Blonquist, T. M., Stevenson, K. E., Stonerock, E., et al. (2018). Prognostic Impact of Kinase-Activating Fusions and IKZF1 Deletions in Pediatric High-Risk B-Lineage Acute Lymphoblastic Leukemia. Blood Adv. 2, 529–533. doi:10.1182/bloodadvances.2017014704
Tran, T. H., and Loh, M. L. (20162016). Ph-like Acute Lymphoblastic Leukemia. ASH Educ. Book 2016, 561–566. doi:10.1182/asheducation-2016.1.561
Tvorogov, D., Thomas, D., Liau, N. P. D., Dottore, M., Barry, E. F., Lathi, M., et al. (2018). Accumulation of JAK Activation Loop Phosphorylation Is Linked to Type I JAK Inhibitor Withdrawal Syndrome in Myelofibrosis. Sci. Adv. 4, eaat3834–12. doi:10.1126/sciadv.aat3834
Ungureanu, D., Wu, J., Pekkala, T., Niranjan, Y., Young, C., Jensen, O. N., et al. (2011). The Pseudokinase Domain of JAK2 Is a Dual-Specificity Protein Kinase that Negatively Regulates Cytokine Signaling. Nat. Struct. Mol. Biol. 18, 971–976. doi:10.1038/nsmb.2099
Vainchenker, W., and Constantinescu, S. N. (2013). JAK/STAT Signaling in Hematological Malignancies. Oncogene 32, 2601–2613. doi:10.1038/onc.2012.347
Vainchenker, W., Leroy, E., Gilles, L., Marty, C., Plo, I., and Constantinescu, S. N. (2018). JAK Inhibitors for the Treatment of Myeloproliferative Neoplasms and Other Disorders. F1000Res 7, 82. doi:10.12688/f1000research.13167.1
Van Bodegom, D., Zhong, J., Kopp, N., Dutta, C., Kim, M.-S., Bird, L., et al. (2012). Differences in Signaling through the B-Cell Leukemia Oncoprotein CRLF2 in Response to TSLP and through Mutant JAK2. Blood 120, 2853–2863. doi:10.1182/blood-2012-02-413252
Van Der Veer, A., Waanders, E., Pieters, R., Willemse, M. E., Van Reijmersdal, S. V., Russell, L. J., et al. (2013). Independent Prognostic Value of BCR-ABL1-like Signature and IKZF1 Deletion, but Not High CRLF2 Expression, in Children with B-Cell Precursor ALL. Blood 122, 2622–2629. doi:10.1182/blood-2012-10-462358
Van Rompaey, D., Verstraete, K., Peelman, F., Savvides, S. N., Augustyns, K., Van Der Veken, P., et al. (2017). Virtual Screening for Inhibitors of the Human TSLP:TSLPR Interaction. Sci. Rep. 7, 17211. doi:10.1038/s41598-017-17620-7
Vannucchi, A. M., Kantarjian, H. M., Kiladjian, J.-J., Gotlib, J., Cervantes, F., Mesa, R. A., et al. (2015a). A Pooled Analysis of Overall Survival in COMFORT-I and COMFORT-II, 2 Randomized Phase III Trials of Ruxolitinib for the Treatment of Myelofibrosis. Haematologica 100, 1139–1145. doi:10.3324/haematol.2014.119545
Vannucchi, A. M., Kiladjian, J. J., Griesshammer, M., Masszi, T., Durrant, S., Passamonti, F., et al. (2015b). Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. N. Engl. J. Med. 372, 426–435. doi:10.1056/nejmoa1409002
Venugopal, S., and Mascarenhas, J. (2020). Novel Therapeutics in Myeloproliferative Neoplasms. J. Hematol. Oncol. 13, 162. doi:10.1186/s13045-020-00995-y
Verstovsek, S., Gotlib, J., Mesa, R. A., Vannucchi, A. M., Kiladjian, J.-J., Cervantes, F., et al. (2017a). Long-term Survival in Patients Treated with Ruxolitinib for Myelofibrosis: COMFORT-I and -II Pooled Analyses. J. Hematol. Oncol. 10, 156. doi:10.1186/s13045-017-0527-7
Verstovsek, S., Kantarjian, H. M., Estrov, Z., Cortes, J. E., Thomas, D. A., Kadia, T., et al. (2012a). Long-term Outcomes of 107 Patients with Myelofibrosis Receiving JAK1/JAK2 Inhibitor Ruxolitinib: Survival Advantage in Comparison to Matched Historical Controls. Blood 120, 1202–1209. doi:10.1182/blood-2012-02-414631
Verstovsek, S., Mesa, R. A., Gotlib, J., Levy, R. S., Gupta, V., Dipersio, J. F., et al. (2012b). A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 366, 799–807. doi:10.1056/nejmoa1110557
Verstovsek, S., Mesa, R. A., Mesa, R. A., Gotlib, J., Gupta, V., DiPersio, J. F., et al. (2017b). Long-term Treatment with Ruxolitinib for Patients with Myelofibrosis: 5-year Update from the Randomized, Double-Blind, Placebo-Controlled, Phase 3 COMFORT-I Trial. J. Hematol. Oncol. 10, 55. doi:10.1186/s13045-017-0417-z
Verstovsek, S., Mesa, R., Talpaz, M., Kiladjian, J.-J., Harrison, C. N., Oh, S. T., et al. (2021). Retrospective Analysis of Pacritinib in Patients with Myelofibrosis and Severe Thrombocytopenia. haematol 2021. doi:10.3324/haematol.2021.279415
Walz, C., Cross, N. C. P., Van Etten, R. A., and Reiter, A. (2008). Comparison of Mutated ABL1 and JAK2 as Oncogenes and Drug Targets in Myeloproliferative Disorders. Leukemia 22, 1320–1334. doi:10.1038/leu.2008.133
Wang, Y., Fiskus, W., Chong, D. G., Buckley, K. M., Natarajan, K., Rao, R., et al. (2009). Cotreatment with Panobinostat and JAK2 Inhibitor TG101209 Attenuates JAK2V617F Levels and Signaling and Exerts Synergistic Cytotoxic Effects against Human Myeloproliferative Neoplastic Cells. Blood 114, 5024–5033. doi:10.1182/blood-2009-05-222133
Wang, Z., and Bunting, K. D. (2013). STAT5 in Hematopoietic Stem Cell Biology and Transplantation. JAK-STAT 2, e27159. doi:10.4161/jkst.27159
Weigert, O., Lane, A. A., Bird, L., Kopp, N., Chapuy, B., Van Bodegom, D., et al. (2012). Genetic Resistance to JAK2 Enzymatic Inhibitors Is Overcome by HSP90 Inhibition. J. Exp. Med. 209, 259–273. doi:10.1084/jem.20111694
Wernig, G., Gonneville, J. R., Crowley, B. J., Rodrigues, M. S., Reddy, M. M., Hudon, H. E., et al. (2008). The Jak2V617F Oncogene Associated with Myeloproliferative Diseases Requires a Functional FERM Domain for Transformation and for Expression of the Myc and Pim Proto-Oncogenes. Blood 111, 3751–3759. doi:10.1182/blood-2007-07-102186
Wilmes, S., Hafer, M., Vuorio, J., Tucker, J. A., Winkelmann, H., Löchte, S., et al. (2020). Mechanism of Homodimeric Cytokine Receptor Activation and Dysregulation by Oncogenic Mutations. Science 367, 643–652. doi:10.1126/science.aaw3242
Winter, P. S., Sarosiek, K. A., Lin, K. H., Meggendorfer, M., Schnittger, S., Letai, A., et al. (2014). RAS Signaling Promotes Resistance to JAK Inhibitors by Suppressing BAD-Mediated Apoptosis. Sci. Signal 7, ra122. doi:10.1126/scisignal.2005301
Woessner, D. W., Eiring, A. M., Bruno, B. J., Zabriskie, M. S., Reynolds, K. R., Miller, G. D., et al. (2015). A Coiled-Coil Mimetic Intercepts BCR-ABL1 Dimerization in Native and Kinase-Mutant Chronic Myeloid Leukemia. Leukemia 29, 1668–1675. doi:10.1038/leu.2015.53
Woldman, I., Varinou, L., Ramsauer, K., Rapp, B., and Decker, T. (2001). The Stat1 Binding Motif of the Interferon-γ Receptor Is Sufficient to Mediate Stat5 Activation and its Repression by SOCS3. J. Biol. Chem. 276, 45722–45728. doi:10.1074/jbc.m105320200
Wu, S.-C., Li, L. S., Kopp, N., Montero, J., Chapuy, B., Yoda, A., et al. (2015). Activity of the Type II JAK2 Inhibitor CHZ868 in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 28, 29–41. doi:10.1016/j.ccell.2015.06.005
Yamada, T., Zhu, D., Saxon, A., and Zhang, K. (2002). CD45 Controls Interleukin-4-Mediated IgE Class Switch Recombination in Human B Cells through its Function as a Janus Kinase Phosphatase. J. Biol. Chem. 277, 28830–28835. doi:10.1074/jbc.m201781200
Yano, M., Imamura, T., Asai, D., Kiyokawa, N., Nakabayashi, K., Matsumoto, K., et al. (2015). Identification of Novel Kinase Fusion Transcripts in Paediatric B Cell Precursor Acute Lymphoblastic Leukaemia withIKZF1deletion. Br. J. Haematol. 171, 813–817. doi:10.1111/bjh.13757
Yao, H., Ma, Y., Hong, Z., Zhao, L., Monaghan, S. A., Hu, M.-C., et al. (2017). Activating JAK2 Mutants Reveal Cytokine Receptor Coupling Differences that Impact Outcomes in Myeloproliferative Neoplasm. Leukemia 31, 2122–2131. doi:10.1038/leu.2017.1
Ye, L., Schnepf, D., and Staeheli, P. (2019). Interferon-λ Orchestrates Innate and Adaptive Mucosal Immune Responses. Nat. Rev. Immunol. 19, 614–625. doi:10.1038/s41577-019-0182-z
Yoda, A., Yoda, Y., Chiaretti, S., Bar-Natan, M., Mani, K., Rodig, S. J., et al. (2010). Functional Screening Identifies CRLF2 in Precursor B-Cell Acute Lymphoblastic Leukemia. Proc. Natl. Acad. Sci. U.S.A. 107, 252–257. doi:10.1073/pnas.0911726107
Zabriskie, M. S., Eide, C. A., Tantravahi, S. K., Vellore, N. A., Estrada, J., Nicolini, F. E., et al. (2014). BCR-ABL1 Compound Mutations Combining Key Kinase Domain Positions Confer Clinical Resistance to Ponatinib in Ph Chromosome-Positive Leukemia. Cancer Cell 26, 428–442. doi:10.1016/j.ccr.2014.07.006
Zhang, F., Huang, G., Hu, B., Song, Y., and Shi, Y. (2011). A Soluble Thymic Stromal Lymphopoietin (TSLP) Antagonist, TSLPR-Immunoglobulin, Reduces the Severity of Allergic Disease by Regulating Pulmonary Dendritic Cells. Clin. Exp. Immunol. 164, 256–264. doi:10.1111/j.1365-2249.2011.04328.x
Zhang, J. G., Farley, A., Nicholson, S. E., Willson, T. A., Zugaro, L. M., Simpson, R. J., et al. (2015). Correction for Zhang et al., The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. U. S. A. 112, E2979. doi:10.1073/pnas.1507812112
Zhang, J., Yang, P. L., and Gray, N. S. (2009). Targeting Cancer with Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 9, 28–39. doi:10.1038/nrc2559
Zhang, W., Kuang, P., and Liu, T. (2019). Prognostic Significance of CDKN2A/B Deletions in Acute Lymphoblastic Leukaemia: a Meta-Analysis. Ann. Med. 51, 28–40. doi:10.1080/07853890.2018.1564359
Zhang, X., Guo, A., Yu, J., Possemato, A., Chen, Y., Zheng, W., et al. (2007). Identification of STAT3 as a Substrate of Receptor Protein Tyrosine Phosphatase T. Proc. Natl. Acad. Sci. U.S.A. 104, 4060–4064. doi:10.1073/pnas.0611665104
Zhao, J., Song, Y., and Liu, D. (2019). Recent Advances on Blinatumomab for Acute Lymphoblastic Leukemia. Exp. Hematol. Oncol. 8, 28. doi:10.1186/s40164-019-0152-y
Zhao, L., Ma, Y., Seemann, J., and Huang, L. J.-s. (2010). A Regulating Role of the JAK2 FERM Domain in Hyperactivation of JAK2(V617F). Biochem. J. 426, 91–98. doi:10.1042/bj20090615
Glossary
ABL1 Abelson 1
ACVR1 Activin A Receptor Type 1
ALK ALK Receptor Tyrosine Kinase
Allo-SCT Allogenic Stem Cell Transplantation
ATF7IP Activating Transcription Factor 7 Interacting Protein
ATP Adenosine Triphosphate
ALL Acute Lymphoblastic Leukemia
B-ALL B-cell Acute Lymphoblastic Leukemia
BAT Best Available Therapy
BCL-2 B-cell Lymphoma 2
BCR Breakpoint Cluster Region Protein
BET Bromodomain and Extra-terminal Protein
BRD4 Bromodomain-Containing Protein 4
BTB BR-C, ttk and bab
BTG1 BTG Anti-Proliferation Factor 1
BTLA B and T Lymphocyte Associated
CAR Chimeric Antigen Receptor
CDKN2 Cyclin-Dependent Kinase Inhibitor 2
CD200 CD200 Molecule
CML Chronic Myeloid Leukemia
C-Terminus Carboxyl Terminus
CC Coiled Coil
CRLF2 Cytokine Receptor-Like Factor 2
CRLF2r CRLF2-Rearrangment/Rearranged
DS-ALL Down-Syndrome Acute Lymphoblastic Leukemia
EBF1 EBF Transcription Factor 1
ET Essential Thrombocythemia
EPOR Erythropoietin Receptor
ETV6 ETS Variant Transcription Factor 6
FERM 4.1 Protein, Ezrin, Radixin, Moesin
FGFR1 Fibroblast Growth Factor Receptor 1
FLT3 FMS-like Tyrosine Kinase 3
FOP FGFR1 Oncogene Partner
GAP GTPase Activating Protein
GAS Gamma-Activated Site
GEF Guanidine Nucleotide Exchange factor
GHR GH Receptor
GOLGA Golgin Subfamily A
GSPT1 G1 to S Phase Transition 1
HDAC Histone Deacetylase
hGHR Human Growth Hormone Receptor
HLH Helix-Loop-Helix
HMBOX1 Homeobox Containing 1
HR High-Risk
HSC Hematopoietic Stem Cell
HSP Heat Shock Protein
IL-7Rα Interleukin 7 Receptor Alpha Chain
IKZF1 IKAROS Family Zinc Finger 1
JAK Janus Kinase
JAK2 Janus Kinase 2
JAK2r JAK2-Rearrangment/Rearranged
JH JAK Homology
KIR Kinase Inhibitory Region
KRAS KRAS Proto-Oncogene
LisH LIS1 Homology
LNK Lymphocyte Adaptor Protein
MAPK Mitogen-Activated Protein Kinase
MF Myelofibrosis
MPN Myeloproliferative Neoplasms
MPRIP Myosin Phosphatase Rho Interacting Protein
mTOR Mammalian Target Of Rapamycin
NTRK3 Neurotrophic Receptor Tyrosine Kinase 3
NPC Nuclear Pore Complexes
N-terminus Amino Terminus
NLS Nuclear Localization Signal
OFD1 OFD1 Centriole and Centriolar Satellite Protein
PAX5 Paired Box 5
PCM1 Pericentriolar Material 1
PDGFRA Platelet-Derived Growth Factor Receptor A
PDX Patient-Derived Xenograft
PI3K Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Phosphatidylinositol-4,5-Bisphosphate 3-Kinase
Ph+ ALL Ph-positive Acute Lymphoblastic Leukemia
Ph-like ALL Ph-like Acute Lymphoblastic Leukemia
PI3K Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Phosphatidylinositol-4,5-Bisphosphate 3-Kinase
PKB Protein Kinase B
PMF Primary Myelofibrosis
PNT Pointed Domain
PPFIBP PTPRF Interacting Protein
PROTACS Proteolysis-Targeting Chimeras
PTP Protein Tyrosine Phosphatase
PTPN Protein Tyrosine Phosphatase Non-Receptor
PTPR Protein Tyrosine Phosphatase Receptor
PV Polycythemia Vera
RAG Recombination-Activating Gene
RAS RAS GFP-Activating Protein
RBX2 RING-Finger-Domain-Only Protein
RFX3 Regulatory Factor X3
RNPC3 RNA Binding Region Containing 3
ROCK Rho Associated Coiled-Coil Containing Protein Kinase
RUNX1 RUNX Family Transcription Factor 1
SAM Sterile Alpha Motif
SETD1 SET Domain-Containing 1A
SH2 Src Homology 2
SH2L SH2-like
SLL Small Lymphocytic Leukemia
SMU1 SMU1 DNA Replication Regulator and Spliceosomal Factor
SNX29 Sorting Nexin 29
SOCS Suppressor Of Cytokine Signaling
SPAG9 Sperm Associated Antigen 9
SR Standard Risk
SSBP2 Single Stranded DNA Binding Protein 2
STAT Signal Transducer and Activator of Transcription
STRBP Spermatid Perinuclear RNA Binding Protein
STRN3 Striatin 3
T-ALL T-cell Acute Lymphoblastic Leukemia
TBL1XR1 TBL1Z Receptor
TERF2 Telomeric Repeat Binding Factor 2
TKI Tyrosine Kinase Inhibitor
TPM3 Tropomyosin 3
TPOR Thrombopoietin Receptor
TPR Translocated Promoter Region
TSLPR Thymic Stromal Lymphopoietin Receptor
TYK2 Tyrosine Kinase 2
USP25 Ubiquitin Specific Peptidase 25
VPREB V-set Pre-B Cell Surrogate Light Chain 1
WE Wernicke Encephalopathy
WT Wild-Type
ZBTB Zinc Finger and BTB Domain-Containing
ZEB2 Zinc Finger E-box Binding Homeobox 2
ZMYM2 Zinc Finger MYM-type Protein 2
ZNF Zinc Finger Protein
Keywords: leukemia, Janus kinases, kinase inhibitor, JAK2, targeted therapy, acute lymphoblastic leukemia
Citation: Downes CE, McClure BJ, McDougal DP, Heatley SL, Bruning JB, Thomas D, Yeung DT and White DL (2022) JAK2 Alterations in Acute Lymphoblastic Leukemia: Molecular Insights for Superior Precision Medicine Strategies. Front. Cell Dev. Biol. 10:942053. doi: 10.3389/fcell.2022.942053
Received: 12 May 2022; Accepted: 16 June 2022;
Published: 12 July 2022.
Edited by:
Rosa Ma Montes Lorenzo, University of Granada, SpainReviewed by:
Alexandra Bacquelaine Veloso, KU Leuven, BelgiumKrista Verhoeft, VIB KU Leuven Center for Cancer Biology, Belgium
Copyright © 2022 Downes, McClure, McDougal, Heatley, Bruning, Thomas, Yeung and White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah L. White, ZGVib3JhaC53aGl0ZUBzYWhtcmkuY29t