
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 27 May 2022
Sec. Signaling
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.915931
This article is part of the Research TopicMitochondrial signaling in disease: from cancer to neurodegenerationView all 6 articles
 Sandra M. Martín-Guerrero*
Sandra M. Martín-Guerrero* Andrea Markovinovic
Andrea Markovinovic Gábor M. Mórotz
Gábor M. Mórotz Shaakir Salam
Shaakir Salam Wendy Noble
Wendy Noble Christopher C. J. Miller*
Christopher C. J. Miller*Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are two major neurodegenerative diseases. FTD is the second most common cause of dementia and ALS is the most common form of motor neuron disease. These diseases are now known to be linked. There are no cures or effective treatments for FTD or ALS and so new targets for therapeutic intervention are required but this is hampered by the large number of physiological processes that are damaged in FTD/ALS. Many of these damaged functions are now known to be regulated by signaling between the endoplasmic reticulum (ER) and mitochondria. This signaling is mediated by “tethering” proteins that serve to recruit ER to mitochondria. One tether strongly associated with FTD/ALS involves an interaction between the ER protein VAPB and the mitochondrial protein PTPIP51. Recent studies have shown that ER-mitochondria signaling is damaged in FTD/ALS and that this involves breaking of the VAPB-PTPIP51 tethers. Correcting disrupted tethering may therefore correct many other downstream damaged features of FTD/ALS. Here, we review progress on this topic with particular emphasis on targeting of the VAPB-PTPIP51 tethers as a new drug target.
FTD is the second most common form of presenile dementia after Alzheimer’s disease and is clinically, genetically and pathologically linked to the most common form of motor neuron disease, ALS. Thus, significant proportions of FTD and ALS patients display features of both diseases (Ringholz et al., 2005; Wheaton et al., 2007). Likewise, both diseases have a genetic overlap and pathogenic variants in the same genes can cause familial dominantly inherited forms of both FTD and ALS (Ling et al., 2013; Robberecht and Philips, 2013; Abramzon et al., 2020). Finally, both diseases can display similar pathological phenotypes and notably, the accumulation of abnormal aggregates of TAR DNA-binding protein 43 (TDP43) in affected neurons (Arai et al., 2006; Neumann et al., 2006).
Like other major neurodegenerative diseases, there are no cures or even effective treatments for either FTD or ALS and so there is much interest in strategies to identify new therapeutic targets. However, a large number of cellular and physiological processes are damaged in FTD/ALS. These include damage to mitochondria, the endoplasmic reticulum (ER), Ca2+ signaling, lipid metabolism, autophagy, axonal transport and finally both diseases display inflammatory responses within the nervous system (Paillusson et al., 2016; Lau et al., 2018; Markovinovic et al., 2022). This makes it difficult to select which damaged function to prioritise as a drug target. Recently, alterations to signaling between the ER and mitochondria has been a focus of interest and this is because ER-mitochondria signaling regulates many of the damaged functions seen in FTD/ALS (Paillusson et al., 2016; Lau et al., 2018; Markovinovic et al., 2022). This has led to the notion that targeting the ER-mitochondria axis may be a route to correct many damaged FTD/ALS functions and achieve effective disease modification.
It is now widely accepted that organelles communicate with each other; this permits them to respond dynamically to changes in the cellular environment in an orchestrated manner (Cohen et al., 2018; Gordaliza-Alaguero et al., 2019). Communications between the ER and mitochondria represent a particularly important component of organelle signaling since this regulates several key cellular processes. These include bioenergetics, Ca2+ homeostasis, lipid metabolism, mitochondrial biogenesis and trafficking, apoptosis, ER stress responses, autophagy and inflammation (Rowland and Voeltz, 2012; Krols et al., 2016; Paillusson et al., 2016; Csordas et al., 2018; Rieusset, 2018; Perrone et al., 2020; Markovinovic et al., 2022). Additionally, in neurons ER-mitochondria signaling regulates synaptic activity and damage to synaptic function is a defining feature in neurodegenerative diseases including FTD/ALS (Herms and Dorostkar, 2016; Hirabayashi et al., 2017; Spires-Jones et al., 2017; Gomez-Suaga et al., 2019).
The mechanisms by which ER-mitochondria communications impact on all these different cellular processes are not properly understood but the two primary functions of ER-mitochondria signaling are delivery of Ca2+ from ER stores to mitochondria and the synthesis of phospholipids (Rowland and Voeltz, 2012; Vance, 2015; Paillusson et al., 2016; Csordas et al., 2018; Markovinovic et al., 2022). It is likely that these primary functions impact on the other downstream roles of ER-mitochondria signaling (Figure 1). Mitochondria require Ca2+ to generate ATP and this is because dehydrogenases in the tricarboxylic acid cycle are Ca2+ dependent. In addition, mitochondrial Ca2+ is involved in the activation of Ca2+-regulated mitochondrial carriers (CaMCs) located in the inner mitochondrial membrane (IMM) (Del Arco et al., 2016). The major route for delivery of this Ca2+ involves its release from ER stores via inositol 1,4,5-trisphosphate (IP3) receptors and uptake into mitochondria by the outer mitochondrial membrane located voltage-dependent anion-selective channel-1 (VDAC1) and the inner membrane located mitochondrial calcium uniporter (MCU) (Rowland and Voeltz, 2012; Paillusson et al., 2016; Csordas et al., 2018). As such, ER-mitochondria signaling regulates bioenergetics and indeed, changes in metabolic demand have been shown to stimulate ER-mitochondria signaling (Gomez-Suaga et al., 2019) (Figure 1). Aside from its release from IP3 receptors, Ca2+ can also be released from Ryanodine receptors in ER for uptake by mitochondria (Csordas et al., 2018). There are also several other subunits to the MCU channel; together these other proteins can all influence ER-mitochondria Ca2+ exchange (Feno et al., 2021).
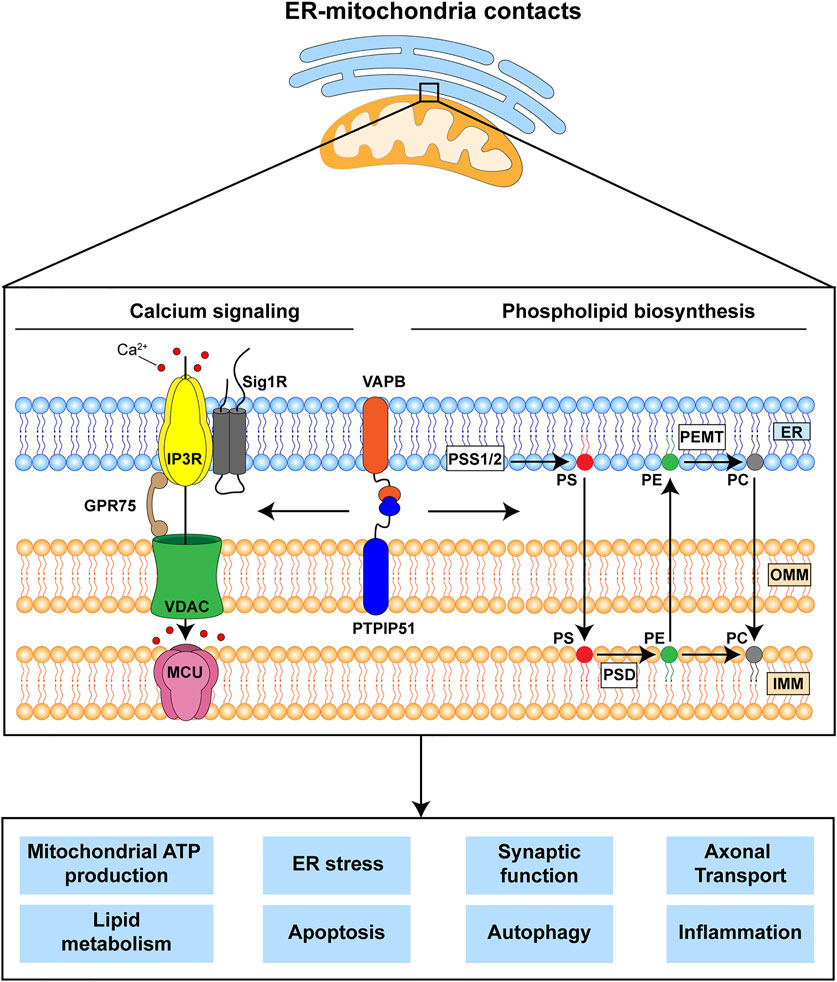
FIGURE 1. The VAPB-PTPIP51 tethers regulate delivery of Ca2+ from ER stores to mitochondria and phospholipid synthesis. These primary functions are believed to impact upon a number of other downstream physiological processes many of which are damaged in FTD/ALS. The VAPB-PTPIP51 interaction facilitates Ca2+ transfer from ER to mitochondria via IP3R-GRP75-VDAC1. Phospholipid synthesis involves initial production of phosphatidylserine (PS) in MAM by PS synthase 1 and 2 (PSS1/2); this is transferred to mitochondria where PS decarboxylase (PSD) converts it to phosphatidylethanolamine (PE). PE can be transferred back to the ER, where phosphatidylethanolamine N-methyltransferase (PEMT) converts it to phosphatidylcholine (PC). Finally, PC can be also transferred back to mitochondria. ER, endoplasmic reticulum; IMM, inner mitochondrial membrane; IP3R, inositol 1,4,5-trisphosphate receptor; GPR75, glucose-regulated protein 75; MCU, mitochondrial Ca2+ uniporter; OMM, outer mitochondrial membrane; Sig1R, Sigma-1 receptor; VDAC1, voltage-dependent anion-selective channel.
The second primary function is to synthesise phospholipids. Although most phospholipids are produced in the ER, some are synthesised by enzymes that are located in both ER and mitochondria; for these, precursor exchange between the two organelles is required. Indeed, two of the most abundant phospholipids in mammalian cells, phosphatidylcholine and phosphatidylethanolamine are produced at the ER-mitochondria axis (Rowland and Voeltz, 2012; Vance, 2015; Paillusson et al., 2016; Csordas et al., 2018) (Figure 1).
ER-mitochondria signaling involves close physical contacts between the two organelles such that up to approximately 20% of the mitochondrial surface is closely apposed (distances of about 10–30 nm) to ER membranes (Csordas et al., 2006; Paillusson et al., 2016; Csordas et al., 2018; Markovinovic et al., 2022). These regions of ER are termed mitochondria-associated ER membranes (MAM). The mechanisms by which these contacts form are not fully understood but it is generally agreed that it involves “tethering” proteins that serve to recruit regions of ER to the mitochondrial surface. A number of different tethers have now been described and it is possible that different tethers serve to recruit different domains of ER to mitochondria e.g., rough and smooth, and sheets and tubules of ER; also there are proteins that act to regulate the interactions of tethers and their functions. Such tethers and regulators have recently been reviewed (Csordas et al., 2018; Markovinovic et al., 2022). The tethering proteins most strongly linked to FTD/ALS involve an interaction between the integral ER protein vesicle-associated membrane protein-associated protein B (VAPB) and the outer mitochondrial membrane protein, protein tyrosine phosphatase interacting protein-51 (PTPIP51) (De Vos et al., 2012; Stoica et al., 2014). The VAPB-PTPIP51 tethers regulate IP3 receptor mediated delivery of Ca2+ to mitochondria, phospholipid synthesis and synaptic activity (De Vos et al., 2012; Gomez-Suaga et al., 2019; Yeo et al., 2021).
A number of genes have now been identified as causal for familial inherited forms of FTD/ALS (Abramzon et al., 2020). Several of these have been shown to disrupt ER-mitochondria contacts and/or mitochondrial Ca2+ delivery. These include mutant SIGMAR1 encoding the Sigma-1 receptor, mutant SOD1 encoding Cu/Zn superoxide dismutase-1 (SOD1), mutant TARDBP encoding TDP43, mutant FUS encoding fused in sarcoma and mutant C9orf72 (Figure 2) (Stoica et al., 2014; Bernard-Marissal et al., 2015; Dafinca et al., 2016; Gregianin et al., 2016; Stoica et al., 2016; Watanabe et al., 2016; Dafinca et al., 2020; Gomez-Suaga et al., 2022). The Sigma-1 receptor is an ER protein that functions as a chaperone for IP3 receptors to facilitate delivery of Ca2+ to mitochondria; the disease-causing alterations are loss of function mutations (Bernard-Marissal et al., 2015; Gregianin et al., 2016; Watanabe et al., 2016). Mutant SOD1 damages ER-mitochondria signaling via disruption of Sigma-1 receptor function (Watanabe et al., 2016). TDP43 accumulations form the hallmark pathology of FTD/ALS but FUS is now also known to be a widespread pathology of FTD/ALS (Spires-Jones et al., 2017; Tyzack et al., 2019). Mutations in the C9orf72 gene cause most familial FTD/ALS cases (Dejesus-Hernandez et al., 2011; Renton et al., 2011). The mutations involve expansion of an intronic hexanucleotide repeat which is translated into dipeptide repeat (DPR) proteins, some of which have been shown to be neurotoxic (Kwon et al., 2014; Mizielinska et al., 2014; Wen et al., 2014).
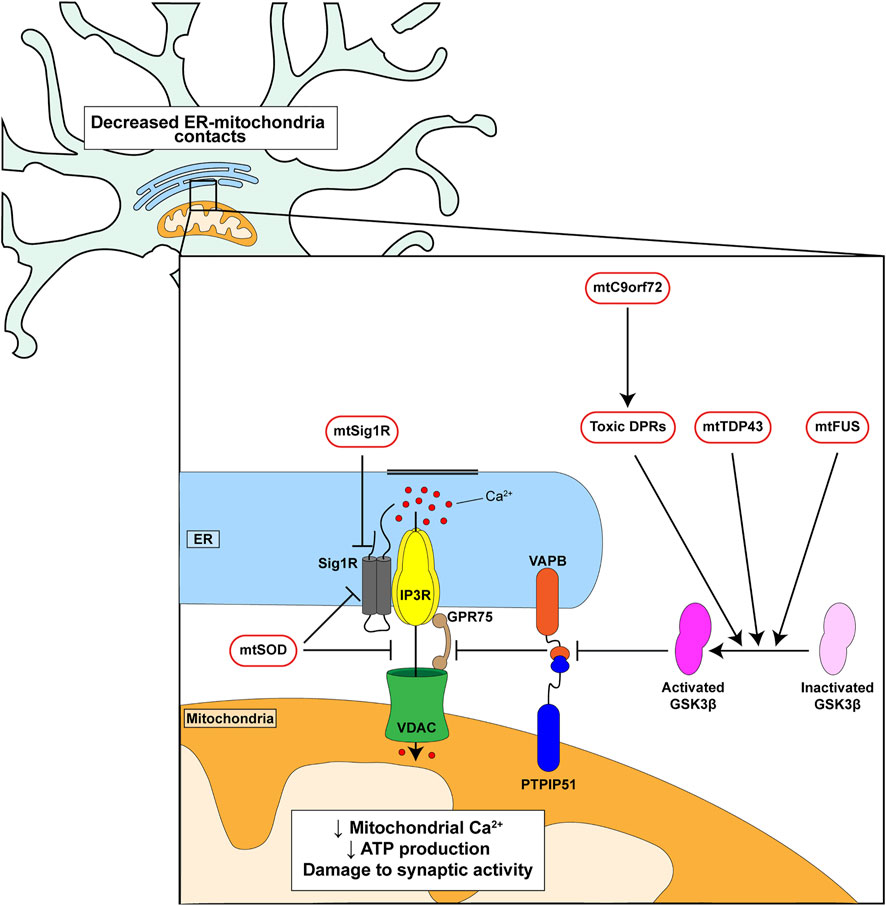
FIGURE 2. Genetic insults linked to familial FTD/ALS disrupt ER-mitochondria signaling and the VAPB-PTPIP51 interaction. C9orf72-derived toxic DPRs, mutant TDP43 and mutant FUS all activate GSK3β which in turn disrupt binding of VAPB to PTPIP51. Mutant Sigma1 receptor perturbs IP3 receptor mediated delivery of Ca2+ from ER to mitochondria. Mutant SOD1 may act directly on the Sigma-1 receptor and/or target the IP3 receptor-VDAC1 interaction.
Mutant C9orf72, TDP43 and FUS all disrupt ER-mitochondria contacts and Ca2+ exchange via an effect on the VAPB-PTPIP51 tethers. For C9orf72 this involves the toxic DPRs (Figure 2) (Stoica et al., 2014; Stoica et al., 2016; Gomez-Suaga et al., 2022). Moreover, this breaking of the VAPB-PTPIP51 tethers is an early feature that appears before disease onset in mutant C9orf72 transgenic mice; early pathogenic changes are believed to be the most important so this finding supports the notion that disruption of the VAPB-PTPIP51 tethers contributes in a major way to disease (Gomez-Suaga et al., 2022). In addition, damage to the VAPB-PTPIP51 tethers by mutant TDP43, FUS and C9orf72-derived DPRs involves activation of glycogen synthase kinase-3β (GSK3β) (Stoica et al., 2014; Stoica et al., 2016; Gomez-Suaga et al., 2022) (Figure 2). GSK3β is a modulator of the VAPB-PTPIP51 interaction; activation disrupts whereas inhibition stimulates binding (Stoica et al., 2014; Stoica et al., 2016). GSK3β is strongly implicated in dementia and ALS and so GSK3β inhibitors may prove to be therapeutic for FTD/ALS (Llorens-Martin et al., 2014; Lauretti et al., 2020). Thus, disruption of ER-mitochondria contacts and signaling is a feature of several familial FTD/ALS linked genes and where studied, this involves breaking of the VAPB-PTPIP51 tethers.
The findings that several FTD/ALS linked genes disrupt ER-mitochondria signaling and that this signaling regulates many damaged functions, suggests that correcting this disruption may remedy other downstream disease features (Paillusson et al., 2016) (Figure 1). Enhancing ER-mitochondria contacts and signaling may therefore be broadly therapeutic. GSK3β inhibitors provide an obvious solution but whilst a number of these have shown beneficial effects in disease models, none have so far made it to the clinic as treatments for neurodegenerative disorders. This may be because GSK3β has functions outside of the nervous system and inhibition of these other functions may be detrimental. Another route involves use of Sigma-1 receptor agonists since loss of SIGMA1R causes familial FTD/ALS (Bernard-Marissal et al., 2015; Gregianin et al., 2016; Watanabe et al., 2016). Several Sigma1 receptor agonists have proved to be beneficial in cell and animal models of neurodegenerative diseases including FTD/ALS and one, Anavex2-73 is being tested in clinical trials (Watanabe et al., 2016; Ryskamp et al., 2019). A further route involves identifying novel agents that stimulate ER-mitochondria contacts and screens for the identification of such molecules are discussed below. However, it must be stressed that a strong reinforcement of ER-mitochondria tethering is likely to be detrimental to neurons as it could induce Ca2+ overload in mitochondria which can be a signal for apoptosis (Paillusson et al., 2016; Csordas et al., 2018; Markovinovic et al., 2022).
A number of cellular assays for monitoring the strength of ER-mitochondria contacts and signaling have been devised and reported. The first involves use of split or dimer-dependent fluorescent proteins such as enhanced green fluorescent protein (EGFP). Here, split or dimer dependent EGFP moieties are directed to ER and mitochondria respectively via ER and mitochondria targeting sequences. Close associations between the EGFP moieties at MAM analogous to those seen in fluorescence resonance energy transfer (FRET) assays generate signals which can be quantified after application of potential therapeutics to the media in drug screens (Alford et al., 2012; Cieri et al., 2018; Kakimoto et al., 2018; Yang et al., 2018; Calì and Brini, 2021). Such assays have already facilitated the identification of the flavonoid luteolin as a stimulator of ER-mitochondria contacts; luteolin was identified via its ability to stimulate mitochondrial ATP production but a secondary split-EGFP assay was used to show it influences ER-mitochondria contacts (Naia et al., 2021).
An extension of such cellular assays involves monitoring how potential therapeutics might influence the interaction of known tethering proteins involved in FTD/ALS such as VAPB and PTPIP51. Here, split or dimer dependent EGFP moieties are fused to VAPB and PTPIP51 and signals again quantified after application of drugs to the media. Similar approaches could involve bioluminescence resonance energy transfer (BRET) assays such as Nanoluc Binary Technology (NanoBiT) luciferase complementation assays (Dale et al., 2019). Nanoluc is derived from Oplophorus gracilirostris (deep sea shrimp) luciferase and is genetically engineered for minimal size and optimal performance in luciferase assays. Complementation assays to monitor the strength of protein-protein interactions involve fusion of fragments of Nanoluc (LargeBiT and SmallBiT) to the proteins of interest (VAPB and PTPIP51). Readouts for NanoBiT assays are performed without cell lysis so the signals obtained represent the strength of protein-protein interaction in living cells.
Finally, proximity ligation assays (PLAs) can be used quantify the strength of ER-mitochondria contacts and the VAPB-PTPIP51 interaction in drug screens. The distances detected by proximity ligation assays are similar to those detected by FRET (i.e., approximately 30 nm) (Soderberg et al., 2006). Such proximity ligation assays have already been used to quantify ER-mitochondria contacts and binding of VAPB to PTPIP51 (De Vos et al., 2012; Hedskog et al., 2013; Stoica et al., 2014; Bernard-Marissal et al., 2015; Stoica et al., 2016; Gomez-Suaga et al., 2019; Gomez-Suaga et al., 2022).
One disadvantage of the above cellular assays is that the primary target of any novel drug is not clear. For example, it could be the VAPB-PTPIP51 interaction itself or some upstream regulator such as GSK3β. Deconvolution of the mechanism of action of any identified drug is thus required and this can be time consuming.
As an alternative to the above cellular assays, in vitro binding assays with purified recombinant tethering proteins such as VAPB and PTPIP51 can be employed to screen for small molecules that enhance the interaction. Clearly, such assays only identify molecules that act directly on the tethers so further deconvolution work is less labour intensive. One route would be to use FRET based methods to monitor the strength of the VAPB-PTPIP51 interaction in vitro after application of drug. As an extension, fragment-based drug discovery methods could also be applied. Traditional small molecule drug screens involve use of millions of compounds but fragment-based screens utilise much smaller libraries containing very low molecular mass molecules termed “fragments”. These have low complexity which can enable them to bind to key areas of the protein(s) of interest. Once lead “fragments” have been identified, they can then undergo medicinal chemistry to increase affinity and biological potency (Erlanson et al., 2016). Modulating protein-protein interactions is considered a relatively difficult drug target but the use of fragment based methods is enabling rapid progress in this area (Modell et al., 2016; Valenti et al., 2019).
New drug targets for FTD/ALS are required and ER-mitochondria signaling represents a particularly attractive one. This is because: 1) ER-mitochondria signaling is damaged in FTD/ALS and where studied is an early disease feature; early pathogenic changes are believed to be the most important. 2) ER-mitochondria signaling regulates many of the other damaged features of FTD/ALS so correcting this damage may be broadly beneficial. 3) VAPB and PTPIP51 have been identified as ER-mitochondria tethers that are disrupted in FTD/ALS (Stoica et al., 2014; Bernard-Marissal et al., 2015; Dafinca et al., 2016; Gregianin et al., 2016; Stoica et al., 2016; Watanabe et al., 2016; Dafinca et al., 2020; Gomez-Suaga et al., 2022). The VAPB-PTPIP51 interaction thus represents a defined molecular target for drug intervention.
Interestingly, damage to ER-mitochondria tethering and signaling has also been described for other neurodegenerative diseases including Alzheimer’s disease and Parkinson’s disease (Paillusson et al., 2016; Markovinovic et al., 2022). However, for these diseases there is evidence that damage may involve increased or reduced ER-mitochondria contacts and signaling (Paillusson et al., 2016; Markovinovic et al., 2022). Thus, it is possible that inhibitors of ER-mitochondria tethering may also have therapeutic potential. Whatever the precise mechanism, the screens described above may be beneficial in identifying such molecules.
All authors contributed to the design and drafting of this mini review. All authors edited the manuscript.
Work in our research group is funded by grants from the UK Medical Research Council (MR/R022666/1), Alzheimer’s Research UK and the Alzheimer’s Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abramzon, Y. A., Fratta, P., Traynor, B. J., and Chia, R. (2020). The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 14, 42. doi:10.3389/fnins.2020.00042
Alford, S. C., Ding, Y., Simmen, T., and Campbell, R. E. (2012). Dimerization-dependent Green and Yellow Fluorescent Proteins. ACS Synth. Biol. 1, 569–575. doi:10.1021/sb300050j
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophysical Res. Commun. 351, 602–611. doi:10.1016/j.bbrc.2006.10.093
Bernard-Marissal, N., Médard, J.-J., Azzedine, H., and Chrast, R. (2015). Dysfunction in Endoplasmic Reticulum-Mitochondria Crosstalk Underlies SIGMAR1 Loss of Function Mediated Motor Neuron Degeneration. Brain 138, 875–890. doi:10.1093/brain/awv008
Calì, T., and Brini, M. (2021). Quantification of Organelle Contact Sites by Split-GFP-Based Contact Site Sensors (SPLICS) in Living Cells. Nat. Protoc. 16, 5287–5308. doi:10.1038/s41596-021-00614-1
Cieri, D., Vicario, M., Giacomello, M., Vallese, F., Filadi, R., Wagner, T., et al. (2018). SPLICS: a Split Green Fluorescent Protein-Based Contact Site Sensor for Narrow and Wide Heterotypic Organelle Juxtaposition. Cell Death Differ. 25, 1131–1145. doi:10.1038/s41418-017-0033-z
Cohen, S., Valm, A. M., and Lippincott-Schwartz, J. (2018). Interacting Organelles. Curr. Opin. Cell Biol. 53, 84–91. doi:10.1016/j.ceb.2018.06.003
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and Functional Features and Significance of the Physical Linkage between ER and Mitochondria. J. Cell Biol. 174, 915–921. doi:10.1083/jcb.200604016
Csordás, G., Weaver, D., and Hajnóczky, G. (2018). Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 28, 523–540. doi:10.1016/j.tcb.2018.02.009
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A. A., et al. (2020). Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 14 (5), 892–908. doi:10.1016/j.stemcr.2020.03.023
Dafinca, R., Scaber, J., Ababneh, N. A., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells. 34, 2063–2078. doi:10.1002/stem.2388
Dale, N. C., Johnstone, E. K. M., White, C. W., and Pfleger, K. D. G. (2019). NanoBRET: The Bright Future of Proximity-Based Assays. Front. Bioeng. Biotechnol. 7, 56. doi:10.3389/fbioe.2019.00056
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K.-F., Ackerley, S., et al. (2012). VAPB Interacts with the Mitochondrial Protein PTPIP51 to Regulate Calcium Homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi:10.1093/hmg/ddr559
Dejesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. 72, 245–256. doi:10.1016/j.neuron.2011.09.011
Del Arco, A., Contreras, L., Pardo, B., and Satrustegui, J. (2016). Calcium Regulation of Mitochondrial Carriers. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1863 (10), 2413–2421. doi:10.1016/j.bbamcr.2016.03.024
Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W., and Jhoti, H. (2016). Twenty Years on: the Impact of Fragments on Drug Discovery. Nat. Rev. Drug Discov. 15 (9), 605–619. doi:10.1038/nrd.2016.109
Feno, S., Rizzuto, R., Raffaello, A., and Vecellio Reane, D. (2021). The Molecular Complexity of the Mitochondrial Calcium Uniporter. Cell Calcium. 93, 102322. doi:10.1016/j.ceca.2020.102322
Gomez‐Suaga, P., Mórotz, G. M., Markovinovic, A., Martín‐Guerrero, S. M., Preza, E., Arias, N., et al. (2022). Disruption of ER‐mitochondria Tethering and Signalling in C9orf72 ‐associated Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Aging Cell. 21 (2), e13549. doi:10.1111/acel.13549
Gómez-Suaga, P., Pérez-Nievas, B. G., Glennon, E. B., Lau, D. H. W., Paillusson, S., Mórotz, G. M., et al. (2019). The VAPB-PTPIP51 Endoplasmic Reticulum-Mitochondria Tethering Proteins Are Present in Neuronal Synapses and Regulate Synaptic Activity. Acta Neuropathol. Commun. 7, 35. doi:10.1186/s40478-019-0688-4
Gordaliza‐Alaguero, I., Cantó, C., and Zorzano, A. (2019). Metabolic Implications of Organelle-Mitochondria Communication. EMBO Rep. 20, e47928. doi:10.15252/embr.201947928
Gregianin, E., Pallafacchina, G., Zanin, S., Crippa, V., Rusmini, P., Poletti, A., et al. (2016). Loss-of-function Mutations in theSIGMAR1gene Cause Distal Hereditary Motor Neuropathy by Impairing ER-Mitochondria Tethering and Ca2+signalling. Hum. Mol. Genet. 25, 3741–3753. doi:10.1093/hmg/ddw220
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the Endoplasmic Reticulum-Mitochondria Interface in Alzheimer's Disease and Related Models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi:10.1073/pnas.1300677110
Herms, J., and Dorostkar, M. M. (2016). Dendritic Spine Pathology in Neurodegenerative Diseases. Annu. Rev. Pathol. Mech. Dis. 11, 221–250. doi:10.1146/annurev-pathol-012615-044216
Hirabayashi, Y., Kwon, S.-K., Paek, H., Pernice, W. M., Paul, M. A., Lee, J., et al. (2017). ER-mitochondria Tethering by PDZD8 Regulates Ca2+dynamics in Mammalian Neurons. Science. 358, 623–630. doi:10.1126/science.aan6009
Kakimoto, Y., Tashiro, S., Kojima, R., Morozumi, Y., Endo, T., and Tamura, Y. (2018). Visualizing Multiple Inter-organelle Contact Sites Using the Organelle-Targeted Split-GFP System. Sci. Rep. 8 (1), 6175. doi:10.1038/s41598-018-24466-0
Krols, M., van Isterdael, G., Asselbergh, B., Kremer, A., Lippens, S., Timmerman, V., et al. (2016). Mitochondria-associated Membranes as Hubs for Neurodegeneration. Acta Neuropathol. 131, 505–523. doi:10.1007/s00401-015-1528-7
Kwon, I., Xiang, S., Kato, M., Wu, L., Theodoropoulos, P., Wang, T., et al. (2014). Poly-dipeptides Encoded by the C9orf72 Repeats Bind Nucleoli, Impede RNA Biogenesis, and Kill Cells. Science. 345 (6201), 1139–1145. doi:10.1126/science.1254917
Lau, D. H. W., Hartopp, N., Welsh, N. J., Mueller, S., Glennon, E. B., Mórotz, G. M., et al. (2018). Disruption of ER−mitochondria Signalling in Fronto-Temporal Dementia and Related Amyotrophic Lateral Sclerosis. Cell Death Dis. 9, 327. doi:10.1038/s41419-017-0022-7
Lauretti, E., Dincer, O., and Praticò, D. (2020). Glycogen Synthase Kinase-3 Signaling in Alzheimer's Disease. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1867 (5), 118664. doi:10.1016/j.bbamcr.2020.118664
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron. 79, 416–438. doi:10.1016/j.neuron.2013.07.033
Llorens-Martín, M., Jurado, J., Hernández, F., and Ávila, J. (2014). GSK-3β, a Pivotal Kinase in Alzheimer Disease. Front. Mol. Neurosci. 7, 46. doi:10.3389/fnmol.2014.00046
Markovinovic, A., Greig, J., Martín-Guerrero, S. M., Salam, S., and Paillusson, S. (2022). Endoplasmic Reticulum-Mitochondria Signaling in Neurons and Neurodegenerative Diseases. J. Cell Sci. 135 (3). doi:10.1242/jcs.248534
Mizielinska, S., Grönke, S., Niccoli, T., Ridler, C. E., Clayton, E. L., Devoy, A., et al. (2014). C9orf72 Repeat Expansions Cause Neurodegeneration in Drosophila through Arginine-Rich Proteins. Science. 345, 1192–1194. doi:10.1126/science.1256800
Modell, A. E., Blosser, S. L., and Arora, P. S. (2016). Systematic Targeting of Protein-Protein Interactions. Trends Pharmacol. Sci. 37, 702–713. doi:10.1016/j.tips.2016.05.008
Naia, L., Pinho, C. M., Dentoni, G., Liu, J., Leal, N. S., Ferreira, D. M. S., et al. (2021). Neuronal Cell-Based High-Throughput Screen for Enhancers of Mitochondrial Function Reveals Luteolin as a Modulator of Mitochondria-Endoplasmic Reticulum Coupling. BMC Biol. 19 (1), 57. doi:10.1186/s12915-021-00979-5
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science. 314, 130–133. doi:10.1126/science.1134108
Paillusson, S., Stoica, R., Gomez-Suaga, P., Lau, D. H. W., Mueller, S., Miller, T., et al. (2016). There's Something Wrong with My MAM; the ER-Mitochondria axis and Neurodegenerative Diseases. Trends Neurosci. 39, 146–157. doi:10.1016/j.tins.2016.01.008
Perrone, M., Caroccia, N., Genovese, I., Missiroli, S., Modesti, L., Pedriali, G., et al. (2020). The Role of Mitochondria-Associated Membranes in Cellular Homeostasis and Diseases. Int. Rev. Cell Mol. Biol. 350, 119–196. doi:10.1016/bs.ircmb.2019.11.002
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 72, 257–268. doi:10.1016/j.neuron.2011.09.010
Rieusset, J. (2018). Mitochondria-associated Membranes (MAMs): An Emerging Platform Connecting Energy and Immune Sensing to Metabolic Flexibility. Biochem. Biophysical Res. Commun. 500, 35–44. doi:10.1016/j.bbrc.2017.06.097
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and Patterns of Cognitive Impairment in Sporadic ALS. Neurology. 65, 586–590. doi:10.1212/01.wnl.0000172911.39167.b6
Robberecht, W., and Philips, T. (2013). The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi:10.1038/nrn3430
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic Reticulum-Mitochondria Contacts: Function of the Junction. Nat. Rev. Mol. Cell. Biol. 13, 607–615. doi:10.1038/nrm3440
Ryskamp, D. A., Korban, S., Zhemkov, V., Kraskovskaya, N., and Bezprozvanny, I. (2019). Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 13, 862. doi:10.3389/fnins.2019.00862
Söderberg, O., Gullberg, M., Jarvius, M., Ridderstråle, K., Leuchowius, K.-J., Jarvius, J., et al. (2006). Direct Observation of Individual Endogenous Protein Complexes In Situ by Proximity Ligation. Nat. Methods. 3, 995–1000. doi:10.1038/nmeth947
Spires-Jones, T. L., Attems, J., and Thal, D. R. (2017). Interactions of Pathological Proteins in Neurodegenerative Diseases. Acta Neuropathol. 134, 187–205. doi:10.1007/s00401-017-1709-7
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K.-F., et al. (2014). ER-mitochondria Associations Are Regulated by the VAPB-PTPIP51 Interaction and Are Disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996. doi:10.1038/ncomms4996
Stoica, R., Paillusson, S., Gomez‐Suaga, P., Mitchell, J. C., Lau, D. H., Gray, E. H., et al. (2016). ALS/FTD ‐associated FUS Activates GSK ‐3β to Disrupt the VAPB - PTPIP 51 Interaction and ER -mitochondria Associations. EMBO Rep. 17, 1326–1342. doi:10.15252/embr.201541726
Tyzack, G. E., Luisier, R., Taha, D. M., Neeves, J., Modic, M., Mitchell, J. S., et al. (2019). Widespread FUS Mislocalization Is a Molecular Hallmark of Amyotrophic Lateral Sclerosis. Brain. 142, 2572–2580. doi:10.1093/brain/awz217
Valenti, D., Hristeva, S., Tzalis, D., and Ottmann, C. (2019). Clinical Candidates Modulating Protein-Protein Interactions: The Fragment-Based Experience. Eur. J. Med. Chem. 167, 76–95. doi:10.1016/j.ejmech.2019.01.084
Vance, J. E. (2015). Phospholipid Synthesis and Transport in Mammalian Cells. Traffic. 16, 1–18. doi:10.1111/tra.12230
Watanabe, S., Ilieva, H., Tamada, H., Nomura, H., Komine, O., Endo, F., et al. (2016). Mitochondria‐associated Membrane Collapse Is a Common Pathomechanism in SIGMAR 1 ‐ and SOD 1 ‐linked ALS. EMBO Mol. Med. 8, 1421–1437. doi:10.15252/emmm.201606403
Wen, X., Tan, W., Westergard, T., Krishnamurthy, K., Markandaiah, S. S., Shi, Y., et al. (2014). Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates that Initiate In Vitro and In Vivo Neuronal Death. Neuron. 84, 1213–1225. doi:10.1016/j.neuron.2014.12.010
Wheaton, M. W., Salamone, A. R., Mosnik, D. M., McDonald, R. O., Appel, S. H., Schmolck, H. I., et al. (2007). Cognitive Impairment in Familial ALS. Neurology. 69, 1411–1417. doi:10.1212/01.wnl.0000277422.11236.2c
Yang, Z., Zhao, X., Xu, J., Shang, W., and Tong, C. (2018). A Novel Fluorescent Reporter Detects Plastic Remodeling of Mitochondria-ER Contact Sites. J. Cell Sci. 131. doi:10.1242/jcs.208686
Keywords: mitochondria, endoplasmic reticulum, amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), therapeutic targets, ER-mitochondria contact
Citation: Martín-Guerrero SM, Markovinovic A, Mórotz GM, Salam S, Noble W and Miller CCJ (2022) Targeting ER-Mitochondria Signaling as a Therapeutic Target for Frontotemporal Dementia and Related Amyotrophic Lateral Sclerosis. Front. Cell Dev. Biol. 10:915931. doi: 10.3389/fcell.2022.915931
Received: 08 April 2022; Accepted: 05 May 2022;
Published: 27 May 2022.
Edited by:
Riccardo Filadi, National Research Council (CNR), ItalyReviewed by:
Paola Pizzo, University of Padua, ItalyCopyright © 2022 Martín-Guerrero, Markovinovic, Mórotz, Salam, Noble and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra M. Martín-Guerrero, c2FuZHJhLm1hcnRpbl9ndWVycmVyb0BrY2wuYWMudWs=; Christopher C. J. Miller, Y2hyaXMubWlsbGVyQGtjbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.