 Odjo G. Gouttia
Odjo G. Gouttia Yanqiu Li
Yanqiu Li Gregory G. Oakley
Gregory G. Oakley Aimin Peng
Aimin Peng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 28 September 2022
Sec. Cancer Cell Biology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.904719
This article is part of the Research Topic Protein Kinase and Pseudokinase in Cancer Biology View all 7 articles
Platinum-based chemotherapy is the standard first-line treatment for oral squamous cell carcinoma (OSCC) that is inoperable, recurrent, or metastatic. Platinum sensitivity is a major determinant of patient survival in advanced OSCC. Here, we investigated the involvement of MASTL, a cell cycle kinase that mediates ENSA/ARPP19 phosphorylation and PP2A/B55 inhibition, in OSCC therapy. Interestingly, upregulation of MASTL and ENSA/ARPP19, and downregulation of PP2A/B55, were common in OSCC. MASTL expression was in association with poor patient survival. In established OSCC cell lines, upregulation of MASTL and ENSA, and downregulation of B55 genes, correlated with cisplatin resistance. We further confirmed that stable expression of MASTL in OSCC cells promoted cell survival and proliferation under cisplatin treatment, in an ENSA-dependent manner. Conversely, deletion of MASTL or ENSA, or overexpression of B55α, sensitized cisplatin response, consistent with increased DNA damage accumulation, signaling, and caspase activation. Moreover, GKI-1, the first-in-class small molecule inhibitor of MASTL kinase, phenocopied MASTL depletion in enhancing the outcome of cisplatin treatment in OSCC cells, at a dose substantially lower than that needed to disrupt mitotic entry. Finally, GKI-1 exhibited promising efficacy in a mouse tumor xenograft model, in conjunction with cisplatin therapy.
Oral cancer is the sixth most common cancer worldwide. In the United States, approximately 50,000 new oral cancer cases are diagnosed each year. Over 90% of oral cancer cases are oral squamous cell carcinomas (OSCC) arising from the oral epithelium. Compared to many other major types of cancer, the treatment option and overall survival for oral cancer has not markedly improved over the last 3 decades. While OSCC at early stages can be cured largely by surgery alone, the majority of OSCC cases are diagnosed at later stages (III and IV), and are typically treated with surgery and external radiotherapy, in combination with chemotherapeutic agents. Unfortunately, the prognosis for advanced OSCC, especially those not associated with human papilloma virus (HPV), remains poor. Thus, a major challenge for oral cancer treatment lies in the intrinsic or acquired mechanisms that render tumor cells resistance to radiation and chemotherapy (Casiglia and Woo, 2001; Zygogianni et al., 2011; Rivera, 2015; Ali et al., 2017).
Cisplatin (cis-diaminedichloroplatinum) and other platinum analogs are widely used in the treatment of solid tumors. Cisplatin manifests its cytotoxic effect by inducing inter- and intrastrand crosslinked DNA adducts. These forms of DNA damage disrupt DNA metabolism, especially DNA replication and transcription, thereby suppressing cell proliferation and triggering cell death (Dasari and Tchounwou, 2014; Gau et al., 2019). Importantly, our cells possess a collection of complex and evolutionarily conserved mechanisms to sense, and respond to, the induction of DNA damage (Sancar et al., 2004; Jackson and Bartek, 2009; Lord and Ashworth, 2012). DNA repair is a core element of the DNA damage response (DDR). To date, numerous lesion-specific DNA repair pathways have been characterized, with over 100 DNA repair genes identified. For example, multiple DNA repair mechanisms, including nucleotide excision repair, mismatch repair, double strand break repair and interstrand crosslink repair, have been implicated in cisplatin-induced DNA damage (Sancar et al., 2004). Furthermore, DNA damage activates a signaling cascade, composed of ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related), CHK1 (checkpoint kinase 1), CHK2 (checkpoint kinase 2) kinases and other factors, to engage DNA damage checkpoints and arrest cell cycle progression. Ultimately, if the level of DNA damage overwhelms cellular repair capability, cells will be eliminated through caspase-3-dependent apoptosis and other cell death pathways (Melo and Toczyski, 2002; Shiloh, 2003).
Recent studies in various model systems characterized microtubule-associated serine/threonine kinase like (MASTL, also known as Greatwall) as an important regulator of mitosis. MASTL is activated during mitotic entry via CDK1-mediated phosphorylation, and the kinase activity of MASTL is required for mitotic progression (Yu et al., 2004; Yu et al., 2006; Archambault et al., 2007; Castilho et al., 2009; Peng and Maller, 2010; Voets and Wolthuis, 2010; Vigneron et al., 2011; Blake-Hodek et al., 2012). It has been revealed that, upon activation, MASTL phosphorylates α-endosulfine (ENSA) and cyclic AMP-regulated 19 kDa phosphoprotein (ARPP19). Phosphorylated ENSA and ARPP19 then bind and inhibit PP2A/B55 (protein phosphatase 2A with a B55 targeting subunit) which is the principal phosphatase holoenzyme that dephosphorylates substrates of CDK1 (Castilho et al., 2009; Mochida et al., 2009; Vigneron et al., 2009; Gharbi-Ayachi et al., 2010; Mochida et al., 2010). Furthermore, we reported that MASTL modulates DNA damage signaling and facilitates cell cycle recovery from the G2/M DNA damage checkpoint in Xenopus egg extracts (Peng et al., 2010a; Peng et al., 2011a). Interestingly, we and other groups showed MASTL upregulation in multiple types of cancer, in association with aggressive clinicopathological features (Wang et al., 2014a; Vera et al., 2015; Sun et al., 2017; Tian et al., 2017; Zhuge et al., 2017; Alvarez-Fernandez et al., 2018; Rogers et al., 2018; Uppada et al., 2018; Fatima et al., 2021). In this study, we delineated the MASTL-ENSA/ARPP19-PP2A/B55 pathway as an important determinant of cisplatin resistance and clinical treatment outcome in OSCC, and validated MASTL inhibition as a potentially valuable therapeutic strategy in combinatorial cancer therapy with cisplatin.
Human oral squamous-cell carcinoma cell line SCC38 (UM-SCC-38), as characterized in previous studies (Brenner et al., 2010; Wang et al., 2012; Khanh et al., 2016), were maintained in Dulbecco’s modified Eagle medium (DMEM, Sigma) supplemented with 10% fetal bovine serum and 1% antibiotics. Human tongue squamous-cell carcinoma Cal27 cells were purchased from ATCC, and maintained in DMEM. CFP-tagged MASTL was constructed to pLZBob, a retroviral vector provided by Dr. James Wahl at the University of Nebraska Medical Center. The cell population stably expressing CFP-MASTL were isolated by G418 selection. Cisplatin and nocodazole were obtained from Sigma, and used for cell treatment as specified in the experiments. GKI-1 was characterized in a previous study (Ocasio et al., 2016). SiRNA targeting MASTL or ENSA was purchased from Integrated DNA Technologies (IDT). These siRNAs were transfected into cells with Lipofectamine RNAiMAX Transfection Reagent, using the protocol recommended by the manufacturer. HA-PPP2R2A (B55α) expression vector, as described in our previous study (Wang et al., 2018), was transfected into cells with Lipofectamine, using the protocol recommended by the manufacturer.
To measure cell growth and sensitivity to cisplatin, cells were treated without or with MASTL or ENSA siRNA for 1 day, and then incubated in cisplatin, with or without GKI-1, at the indicated concentrations for 1–4 days. The numbers of viable cells were counted using a hemocytometer. The concentration of cisplatin that inhibited 50% cell growth (IC50) was calculated as in our previous study (Wang et al., 2012). Briefly, cells were treated with various concentrations of cisplatin, ranging from 1–10 μM. Cell numbers were counted 2 days post treatment. For anchorage-independent cell growth, cells were grown in 0.3% agar on a cushion of 0.6% agar in 35-mm plates, as described previously (Peng et al., 2010b).
The gene expression data for MASTL, ENSA, ARPP19, PPP2R2A and PPP2R2B were obtained from previous studies (Cromer et al., 2004; Ginos et al., 2004; Sengupta et al., 2006; Ye et al., 2008; Estilo et al., 2009; Peng et al., 2011b). The box plot diagrams were generated using Microsoft Excel, with the medium value of the control group set as zero. For the OSCC cell line data analyses, gene expression profiles (GSE36133) were obtained from the Cancer Cell Line Encyclopedia (CCLE) project. The OSCC IC50 data for cisplatin were downloaded from the Genomics of Drug Sensitivity in Cancer (GDSC) database.
Sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting was performed as previously described (Ren et al., 2017). The following primary antibodies were used: anti-MASTL (MABT372, Millipore), anti-active-caspase 3 (ab47131, Abcam), anti-phospho-CHK2 (Thr-68, #2661, Cell Signaling Technology), anti-CHK2 (#6334, Cell Signaling Technology), anti-cleaved PARP1 (#9541, Cell Signaling Technology), anti-ENSA/ARPP19 (ab180513, Abcam), anti-γ-H2AX (Ser-139, SC-517348, Santa Cruz Biotechnology), anti-H2B (#12364, Cell Signaling Technology), anti-H2AX (SC-517336, Santa Cruz Biotechnology), anti-PPP2R2A (B55α, GTX111128, Genetex), anti-SMC1 phospho-Ser-957 (A300-045, Bethyl Laboratories), anti-α-tubulin (from Dr. James Wahl, as in (Wang et al., 2021)). Immunohistochemistry analysis was performed as in a previous study (Wang et al., 2014a). As described in (Rector et al., 2016), OSCC tissues were obtained from the University of Nebraska Medical Center College of Medicine Department of Pathology and Microbiology, with applicable, biographical data and disease-specific medical history obtained from the UNMC College of Medicine Department of Otolaryngology/Head & Neck Surgery. Institutional Review Board (IRB) approval was obtained for the performed experiments. The slides were deparaffinized, and autoclaved for antigen retrieval. Slides were treated with 3% hydrogen peroxide solution, and blocked with 10% normal goat serum in PBS, prior to incubation in primary antibody (anti-MASTL, MABT372, Millipore). Bound antibody was detected with a streptavidin-biotin system suing DAB substrate. Hematoxylin counterstain was performed.
Athymic nude mice were purchased from the Jackson Laboratory, and housed at the UNMC College of Dentistry animal facility. SCC38 cells were implanted into 6-week old mice by a single subcutaneous injection (5 × 105 cells in 100 microliters of sterile PBS). To test the tumor response to chemotherapy, once the tumor size reached 100 mm3, cisplatin (5 mg/kg mouse), with or without GKI-1 (10 mg/kg mouse) were administered intraperitoneally, using a three dose schedule with 3-day intervals. Twenty days after the initial treatment, the mice were euthanized, and tumors were removed and weighed. Tumor lysates were collected for immunoblotting analysis, as described in previous study (Wang et al., 2014a).
Statistical analyses were performed in cell viability assays and in the tumor weight measurements. Briefly, data were analyzed using an unpaired 2-tailed Student’s t test to determine the statistical significance. A p-value less than 0.05 is considered as significant. The association between cisplatin IC50 and gene expression level of MASTL pathway was analyzed by Pearson correlation coefficient. The survival probability in MASTL high or low groups were calculated using the Kaplan-Meier method and compared using the log-rank test.
By surveying previous studies that profiled gene expression in OSCC (Cromer et al., 2004; Ginos et al., 2004; Sengupta et al., 2006; Ye et al., 2008; Estilo et al., 2009; Peng et al., 2011b), we found significantly elevated expression of MASTL in tumor samples, in comparison to normal controls (Figure 1A). Moreover, the combined expression of ENSA and ARPP19 exhibited a similar pattern of upregulation in OSCC (Figure 1B). In contrast, PPP2R2A and PPP2R2B, two B55 subunits, were largely suppressed in OSCC (Figures 1C,D). We then evaluated MASTL expression in HPV- oral cancer patients treated at the UNMC Department of Otolaryngology/Head & Neck Surgery. Compelling to us, the study revealed a highly significant correlation between MASTL upregulation and poor patient survival (Figure 1E).
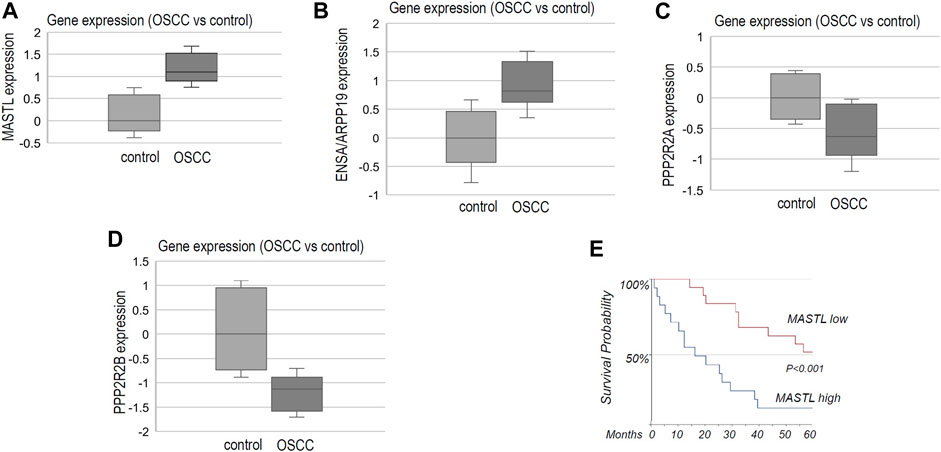
FIGURE 1. The MASTL-ENSA/ARPP19-PP2A/B55 pathway is dysregulated in OSCC, in association with patient survival. (A) The gene expression data for MASTL, ENSA, ARPP19, PPP2R2A and PPP2R2B were obtained, as described in Materials and Methods. The expression level of MASTL in control (normal, n = 44) or OSCC tumor tissues (n = 114) was shown in the box plot diagram (in log2 scale). (B) The expression level of ENSA + ARPP19 in control (normal, n = 53) or OSCC tumor tissues (n = 137) was shown in the box plot diagram (in log2 scale). (C) The expression level of PPP2R2A in control (normal, n = 39) or OSCC tumor tissues (n = 132) was shown in the box plot diagram (in log2 scale). (D) The expression level of PPP2R2B in control (normal, n = 48) or OSCC tumor tissues (n = 89) was shown in the box plot diagram (in log2 scale). (E) As described in Materials and Methods, OSCC tumor samples were obtained for 35 HPV- oral patients who were treated at the University of Nebraska Medical Center. Immunohistochemistry was performed to detect MASTL expression. The survival probability was shown for patients with high or low levels of MASTL expression.
We analyzed the Cancer Cell Line Encyclopedia (CCLE) database for a potential connection between cisplatin resistance and the expression of MASTL and its downstream factors. In an array of OSCC cell lines with different levels of cisplatin resistance, a higher IC50 of cisplatin was in general correlation with higher expression of MASTL, ENSA and ARPP19, but with lower PPP2R2A (B55α) and PPP2R2B (B55β) expression (Figure 2A). We then showed that the overall expression scores of the MASTL pathway, as calculated by log2 (MASTL) + log2 (ENSA) + log2 (ARPP19)—log2 (PPP2R2A)—log2 (PPP2R2B), are in strong positive association with cisplatin resistance in the collection of OSCC cell lines (Figure 2B).
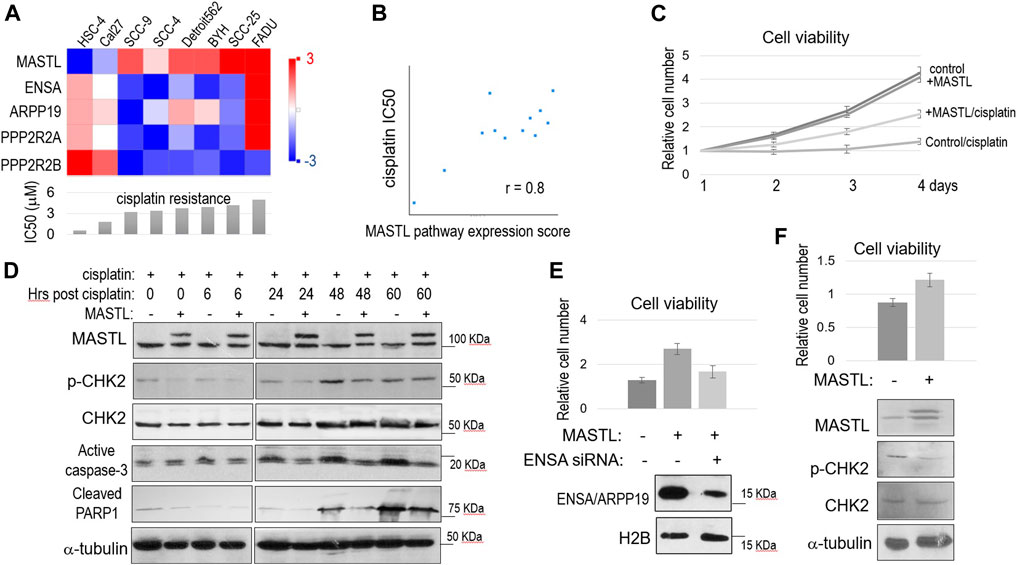
FIGURE 2. The MASTL-ENSA/ARPP19-PP2A/B55 pathway mediates cisplatin resistance in OSCC. (A) The expression levels of MASTL, ENSA, ARPP19, PPP2R2A, and PPP2R2B in various OSCC cell lines were obtained, and shown in the heatmap graph, as described in Materials and Methods. The IC50 of cisplatin was shown in the below panel. (B) The expression levels of MASTL, ENSA, ARPP19, PPP2R2A, and PPP2R2B in OSCC cell lines were obtained as in panel (A). The MASTL pathway expression score was calculated by MASTL + ENSA + ARPP19-PPP2R2A-PPP2R2B (log2 values), and correlated with cisplatin IC50 values, as described in Materials and Methods. (C) Cell viability assay was performed as in Materials and Methods. SCC38 cells with or without stable expression of CFP-MASTL were incubated with cisplatin (3.3 μM) from day 1. The cell numbers at days 2–4 were normalized to that at day 1 (untreated). The mean values and standard derivations, from three independent experiments, were shown. (D) SCC38 cells without or with stably expression of CFP-MASTL were treated with cisplatin (5 μM) for 2 h, and allowed for recovery for the indicated numbers of hours. Cells were harvested and analyzed by immunoblotting, using the indicated antibodies. (E) SCC38 cells with or without CFP-MASTL expression or ENSA siRNA-treatment were cultured in the presence of cisplatin (3.3 μM). After 3 days of cisplatin treatment, cell viability was measured, as in panel (C). Mean values and standard deviations were calculated from three independent experiments, and shown in the upper panel. The effect of ENSA knockdown was shown by immunoblotting, in the lower panel, using antibodies that recognize H2B and both ENSA/ARPP19. (F) Cal27 cells transfected with control GFP or GFP-MASTL expression vectors were treated with cisplatin (3.3 μM). The cell viability after 2 days incubation was determined, and normalized to that of pre-treatment. The mean values and standard deviations were calculated from three independent experiments, and shown in the upper panel. Cells were analyzed by immunoblotting using the indicated antibodies, and shown in the lower panel.
We sought to investigate the functional impact of MASTL upregulation in OSCC cells. SCC38 was selected for the study, as this HPV- OSCC cell line was previously characterized to be highly resistant to cisplatin, consistent with the poor clinical treatment outcome of the patient from whom this cell line was derived (Wang et al., 2012). We generated SCC38 cells that stably expressed recombinant MASTL to a level similar to endogenous MASTL. Compared to the control SCC38 cells, SCC38 harboring 2-fold MASTL upregulation exhibited a very strong proliferative advantage in the presence of cisplatin (Figure 2C). In comparison, both cell lines showed near identical rates of cell proliferation without cisplatin, suggesting specific function of MASTL in the cellular response to cisplatin (Figure 2C). Moreover, MASTL overexpression reduced DNA damage signaling, as shown by CHK2 phosphorylation, and the induction of cell death, as indicated by active caspase-3 and PARP1 cleavage, in response to cisplatin (Figure 2D). The function of MASTL in conferring cisplatin resistance was mediated by ENSA, as ENSA knockdown reversed the effect of MASTL expression (Figure 2E). We determined that ENSA was more dominantly expressed in these cells, compared to ARPP19, because knockdown of ENSA significantly reduced the total expression of ENSA and ARPP19 (Figure 2E). Finally, we sought to confirm the effect of MASTL expression in Cal27 cells that featured relatively low endogenous MASTL expression and high cisplatin sensitivity (Figure 2A). Expression of exogenous MASTL in Cal27 increased cell viability and reduced CHK2 phosphorylation, in the presence of cisplatin (Figure 2F), consistent with the observations in SCC38.
To the contrary of MASTL overexpression, partial depletion of MASTL expression using siRNA reduced SCC38 cell viability after cisplatin treatment (Figure 3A). Depletion of ENSA exhibited a similar outcome, whereas simultaneous depletion of MASTL and ENSA showed no additive effect, in comparison with single depletion (Figure 3A). Thus, MASTL and ENSA acted in the same pathway to mediate cisplatin resistance. At the molecular level, MASTL depletion led to enhanced DNA damage (γ-H2AX), signaling (CHK2 phosphorylation), and cell death (caspase-3 activation and PARP1 cleavage, Figure 3B). Similar molecular events were observed in SCC38 cells with upregulation of B55α (Figure 3C), indicating that the function of MASTL was indeed mediated by PP2A/B55 suppression through ENSA/ARPP19. To this end, we confirmed also that ENSA depletion enhanced H2AX phosphorylation, caspase-3 activation, and PARP1 cleavage post-cisplatin (Figure 3D).
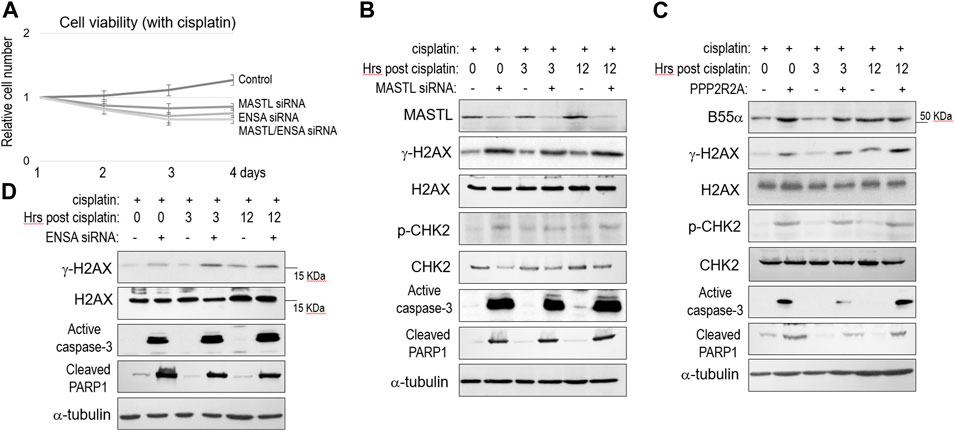
FIGURE 3. The MASTL-ENSA/ARPP19-PP2A/B55 pathway modulates the cisplatin response in OSCC. (A) Cell viability assay was performed as in Materials and Methods. SCC38 cells with or without MASTL or ENSA siRNA were incubated with cisplatin (3.3 μM) from day 1. The cell numbers at days 2–4 were normalized to that at day 1 (untreated). The mean values and standard derivations, from three independent experiments, were shown. (B) SCC38 cells with or without MASTL siRNA were treated with cisplatin (5 μM) for 2 h, and allowed for recovery for the indicated numbers of hours. Cells were harvested and analyzed by immunoblotting, using the indicated antibodies. (C) SCC38 cells with or without HA-PPP2R2A expression were treated with cisplatin (5 μM) for 2 h, and allowed for recovery for the indicated numbers of hours. Cells were harvested and analyzed by immunoblotting, using the indicated antibodies. (D) SCC38 cells with or without ENSA siRNA were treated with cisplatin (5 μM) for 2 h, and allowed for recovery for the indicated numbers of hours. Cells were harvested and analyzed by immunoblotting, using the indicated antibodies.
Ocasio et al. discovered the first small molecule inhibitor of MASTL kinase, named GKI-1 (Greatwall kinase inhibitor-1) (Ocasio et al., 2016). That previous study confirmed the cellular efficacy of GKI-1, including the reduction of ENSA/ARPP19 phosphorylation (Ocasio et al., 2016). Interestingly, we showed that GKI-1 sensitized SCC38 cells to cisplatin (Figure 4A). GKI-1 alone moderately impacted cell proliferation, but a combination of GKI-1 and cisplatin synergistically suppressed SCC38 viability (Figure 4A). IC50 analysis showed comparable levels of cisplatin sensitization by GKI-1 and the depletion of MASTL or ENSA (Figure 4B). Moreover, the effect of GKI-1/cisplatin combination was confirmed by spheroid formation, which was much more efficiently hindered by the combination than by cisplatin alone (Figures 4C,D). Given the role of MASTL in mitotic progression, we also sought to determine the effect of GKI-1 in preventing mitosis. Only at a high concentration (50 μM) was GKI-1 capable of blocking mitotic entry (Figure 4E). By comparison, cisplatin sensitization was readily achieved by GKI-1 at 10 μM.
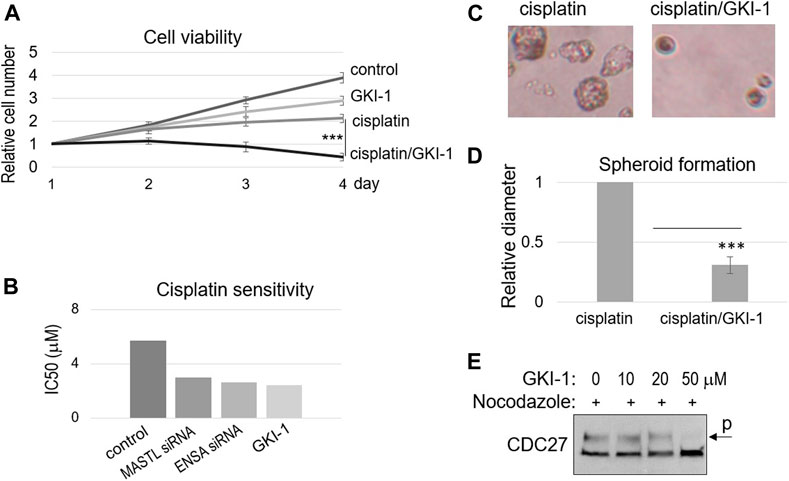
FIGURE 4. GKI-1 sensitizes OSCC cells to cisplatin. (A) SCC38 cells were treated with or without GKI-1 (10 μM) and cisplatin (3.3 μM) for 3 days. The cell viability was studied as described in Materials and Methods. The cell numbers at days 2–4 were normalized to that at day 1 (untreated). The mean values and standard derivations, from three independent experiments, were shown. (B) SCC38 cells were treated with MASTL siRNA, ENSA siRNA, or GKI-1, as indicated. IC50 of cisplatin was determined, as described in Materials and Methods, and shown. (C,D) SCC38 cells treated with GKI-1 and cisplatin were cultured in soft agar for 14 days. Representative spheroid formation was shown in panel C, and average diameters were shown in panel D (N > 20, p < 0.001). (E) SCC38 cells were treated with nocodazole (1 μg/ml) for 12 h, so that cells entered mitosis will be arrested/trapped in mitosis. The cells were also treated with various concentrations of GKI-1, as indicated, to prevent mitotic entry. Cells were harvested and analyzed by immunoblotting for CDC27. The phosphorylation of CDC27 retards its gel migration, and is commonly used as a marker of mitosis.
To further evaluate the therapeutic efficacy of GKI-1, we established a xenograft tumor model in mice, using SCC38 cells. These tumors were treated with either cisplatin alone or cisplatin/GKI-1. The combination treatment consistently resulted in more reduction of tumor volume (Figures 5A,B). We analyzed the tumor lysates and detected elevated levels of DNA damage signaling (CHK2 and SMC1 phosphorylation) and cell death (active caspase-3, Figure 5C).
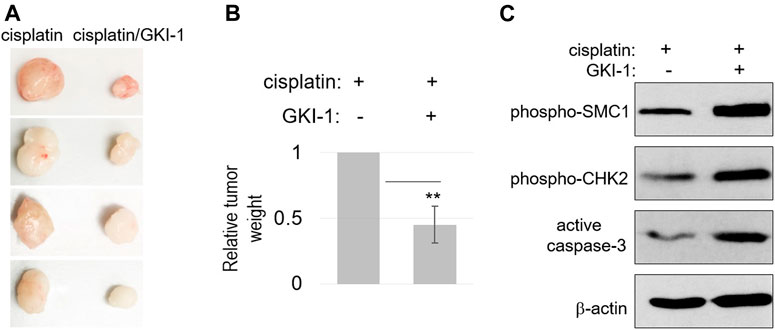
FIGURE 5. GKI-1 enhances the tumor response to cisplatin in vivo. (A–C) SCC38 xenograft tumor model was established, as described in Materials and Methods. Once tumor volume reached 100mm3, three doses of cisplatin and GKI-1 were administered Intraperitoneally with 3-day intervals. At the end of the study, tumors were excited and weighted. Tumor photos were shown in panel (A). The mean values of tumor weight (n = 4), standard deviation, and power analysis (2-tailed t-test) were shown in panel (B). Tumor analysis by immunoblotting was shown in panel (C).
We showed in the current study that MASTL and its downstream substrates ENSA and ARPP19 are frequently upregulated in OSCC, whereas B55 subunits of PP2A are downregulated. Thus, the MASTL-ENSA/ARPP19-PP2A/B55 pathway is of strong interest to the pathophysiology of OSCC. OSCC patients with high levels of MASTL expression suffered adverse treatment outcome, as indicated by shorter overall survival. Notably, MASTL upregulation has been observed also in breast, colon, and other types of cancer (Castro and Lorca, 2018; Marzec and Burgess, 2018; Fatima et al., 2020). In breast cancer, MASTL overexpression promoted chromosomal instability, and was correlated with disease progression and poor prognosis. Conversely, ablation of MASTL expression impaired the proliferation and metastasis of MDA-MB-231 breast cancer cells in vitro and in vivo (Vera et al., 2015; Zhuge et al., 2017; Alvarez-Fernandez et al., 2018; Rogers et al., 2018; Yoon et al., 2018). Thus, MASTL is likely to play a broad role in promoting tumor progression in various types of cancer, and the translational potential of MASTL targeting can be extended beyond OSCC.
During mitotic progression, MASTL functions by phosphorylating ENSA/ARPP19, which then inhibits PP2A/B55, preventing it from dephosphorylating CDK substrates. This mode of MASTL function is evolutionarily conserved, as a similar pathway has been reported in other vertebrate and invertebrate organisms, such as frog, fly, and yeast (Glover, 2012; Wang et al., 2014b; Vigneron et al., 2016; Castro and Lorca, 2018). It shall be noted, however, that additional, and distinct, mechanisms have been implicated for MASTL. For example, MASTL promoted AKT phosphorylation in MDA-MB-231 cells, in a manner that was not related to the decreased PP2A/B55 activity through ENSA/ARPP19 phosphorylation (Vera et al., 2015). Furthermore, MASTL was shown to promote cell contractility and motility independent of its kinase activity (Taskinen et al., 2020).
In this study, we highlighted the therapeutic potential of MASTL targeting in OSCC, in conjunction with cisplatin. Cisplatin and other platinum-based drugs are fundamental chemotherapeutics in the treatment of OSCC and other solid tumors. In advanced OSCC, the therapeutic response to cisplatin is a major determinant of treatment outcome and patient survival. Of a strong interest to us, analyses of data obtained in a panel of OSCC cell lines established a robust correlation between cisplatin resistance and the expression profiles of MASTL and its downstream factors. High levels of MASTL, ENSA, and ARPP19 expression, and low levels of PPP2R2A and PPP2R2B, were indicatives of cisplatin resistance. Among them, PPP2R2B and MASTL exhibited the highest predictive values. We believe that the role of MASTL in cisplatin response is consistent with previous studies that connected MASTL to the cell cycle recovery from DNA damage (Peng et al., 2010a; Peng et al., 2011a; Wang et al., 2014a; Wong et al., 2016). Indeed, MASTL overexpression in OSCC reduced cisplatin-induced DNA damage signaling and caspase activation, whereas MASTL depletion elevated the levels of DNA damage signaling and cell death. We presented further evidence to show that MASTL mediated cisplatin resistance via its downstream ENSA/ARPP19 and PP2A/B55. MASTL/ENSA double depletion did not further enhance cisplatin sensitivity over the single depletion; ENSA knockdown, or B55 expression, elicited similar effects in DNA damage signaling and caspase activation, as MASTL depletion. Taken together, our studies defined an important role of MASTL in conferring tumor cell resistance to cisplatin. We believe that this function of MASTL is mediated largely by its canonical downstream ENSA/ARPP19 phosphorylation and PP2A/B55 inhibition. Future studies are needed to reveal specific phospho-substrates of B55 that potentially mediate DNA repair, cell cycle progression, and cell death post cisplatin treatment.
Protein kinases are potentially druggable, and many of them have been extensively investigated as therapeutic targets in cancer (Bhullar et al., 2018; Cohen et al., 2021). For example, cell cycle kinases, such as cyclin-dependent kinases (CDK), polo-like kinases (PLK) and Aurora Kinases, are essential for cell proliferation. As a result, numerous pharmacological agents that inhibit these cell cycle kinases are either clinically approved for cancer treatment, or under clinical development toward FDA-approval (Taylor and Peters, 2008; Lapenna and Giordano, 2009; Otto and Sicinski, 2017). Along this line, MASTL is likely to yield translational potentials, given its involvement in cell cycle progression. Importantly, our study strongly indicated the therapeutic benefit of the combinatorial treatment composed of MASTL targeting and cisplatin. Using GKI-1, the first-in-class small molecule inhibitor of MASTL, we validated the synergistic effect between MASTL inhibition and cisplatin treatment. The results obtained in OSCC cells using GKI-1 were consistent with those observed with MASTL depletion. Intriguingly, the concentration of GKI-1 that conferred cisplatin sensitization was substantially lower than that required to block mitotic progression. This finding suggested a more promising application of MASTL targeting in cisplatin sensitization than in suppressing cell proliferation. A possible explanation for this differential dose requirement is that the kinase activity of MASTL is much lower in interphase than in mitosis, hence suppressing MASTL in interphase for cisplatin sensitization represents a more effective way of intervention. Finally, we confirmed the efficacy of GKI-1/cisplatin treatment using a xenograft tumor model, presenting the first in vivo evidence for the anti-cancer application of this compound. To propel future development of MASTL targeting in cancer, characterization of the next generation of MASTL inhibitors with better potency and specificity is a crucial task (Kim et al., 2020; Kang et al., 2021). On the other hand, better understanding of how MASTL functions in cell proliferation, in DNA damage responses, and in oncogenic signaling, will provide necessary guidance for the clinical applications of MASTL targeting.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the UNMC IACUC.
OG, GO, and AP conceived the study. OG, JZ, YL, MZ, LW, and AP performed the experiments, and analyzed the data. OG and AP contributed to the writing and data presentation of the manuscript. All authors contributed to the article and approved the submitted version.
AP is supported by the funding from the National Institutes of Health (CA233037; DE030427).
We thank Drs. James Wahl (University of Nebraska Medical Center, United States) and Cory Ocasio (University of Sussex, UK) for reagents.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ali, J., Sabiha, B., Jan, H. U., Haider, S. A., Khan, A. A., and Ali, S. S. (2017). Genetic etiology of oral cancer. Oral Oncol. 70, 23–28. doi:10.1016/j.oraloncology.2017.05.004
Alvarez-Fernandez, M., Sanz-Flores, M., Sanz-Castillo, B., Salazar-Roa, M., Partida, D., Zapatero-Solana, E., et al. (2018). Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 25 (5), 828–840. doi:10.1038/s41418-017-0024-0
Archambault, V., Zhao, X., White-Cooper, H., Carpenter, A. T. C., and Glover, D. M. (2007). Mutations in drosophila Greatwall/Scant reveal its roles in mitosis and meiosis and interdependence with polo kinase. PLoS Genet. 3 (11), e200–2179. doi:10.1371/journal.pgen.0030200
Bhullar, K. S., Lagaron, N. O., McGowan, E. M., Parmar, I., Jha, A., Hubbard, B. P., et al. (2018). Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 17 (1), 48. doi:10.1186/s12943-018-0804-2
Blake-Hodek, K. A., Williams, B. C., Zhao, Y., Castilho, P. V., Chen, W., Mao, Y., et al. (2012). Determinants for activation of the atypical AGC kinase Greatwall during M phase entry. Mol. Cell. Biol. 32 (8), 1337–1353. doi:10.1128/MCB.06525-11
Brenner, J. C., Graham, M. P., Kumar, B., Saunders, L. M., Kupfer, R., Lyons, R. H., et al. (2010). Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head. Neck 32 (4), 417–426. doi:10.1002/hed.21198
Casiglia, J., and Woo, S. B. (2001). A comprehensive review of oral cancer. Gen. Dent. 49 (1), 72–82. https://pubmed.ncbi.nlm.nih.gov/12004680/.
Castilho, P. V., Williams, B. C., Mochida, S., Zhao, Y., and Goldberg, M. L. (2009). The M phase kinase Greatwall (Gwl) promotes inactivation of PP2A/B55delta, a phosphatase directed against CDK phosphosites. Mol. Biol. Cell 20 (22), 4777–4789. doi:10.1091/mbc.e09-07-0643
Castro, A., and Lorca, T. (2018). Greatwall kinase at a glance. J. Cell Sci. 131 (20), jcs222364. doi:10.1242/jcs.222364
Cohen, P., Cross, D., and Janne, P. A. (2021). Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 20 (7), 551–569. doi:10.1038/s41573-021-00195-4
Cromer, A., Carles, A., Millon, R., Ganguli, G., Chalmel, F., Lemaire, F., et al. (2004). Identification of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis. Oncogene 23 (14), 2484–2498. doi:10.1038/sj.onc.1207345
Dasari, S., and Tchounwou, P. B. (2014). Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378. doi:10.1016/j.ejphar.2014.07.025
Estilo, C. L., O-charoenrat, P., Talbot, S., Socci, N. D., Carlson, D. L., Ghossein, R., et al. (2009). Oral tongue cancer gene expression profiling: Identification of novel potential prognosticators by oligonucleotide microarray analysis. BMC Cancer 9, 11. doi:10.1186/1471-2407-9-11
Fatima, I., Barman, S., Uppada, J., Chauhan, S., Rauth, S., Rachagani, S., et al. (2021). MASTL regulates EGFR signaling to impact pancreatic cancer progression. Oncogene 40 (38), 5691–5704. doi:10.1038/s41388-021-01951-x
Fatima, I., Singh, A. B., and Dhawan, P. (2020). Mastl: A novel therapeutic target for cancer malignancy. Cancer Med. 9 (17), 6322–6329. doi:10.1002/cam4.3141
Gau, M., Karabajakian, A., Reverdy, T., Neidhardt, E. M., and Fayette, J. (2019). Induction chemotherapy in head and neck cancers: Results and controversies. Oral Oncol. 95, 164–169. doi:10.1016/j.oraloncology.2019.06.015
Gharbi-Ayachi, A., Labbe, J. C., Burgess, A., Vigneron, S., Strub, J. M., Brioudes, E., et al. (2010). The substrate of greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330 (6011), 1673–1677. doi:10.1126/science.1197048
Ginos, M. A., Page, G. P., Michalowicz, B. S., Patel, K. J., Volker, S. E., Pambuccian, S. E., et al. (2004). Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 64 (1), 55–63. doi:10.1158/0008-5472.can-03-2144
Glover, D. M. (2012). The overlooked greatwall: A new perspective on mitotic control. Open Biol. 2 (3), 120023. doi:10.1098/rsob.120023
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461 (7267), 1071–1078. doi:10.1038/nature08467
Kang, M., Kim, C., Leem, J., Kim, Y. H., Kwon, Y. J., Yoon, Y. N., et al. (2021). Discovery and characterization of a novel MASTL inhibitor MKI-2 targeting MASTL-pp2a in breast cancer cells and oocytes. Pharm. (Basel) 14 (7), 647. doi:10.3390/ph14070647
Khanh, V., Luong, L. W., Roberts, B. J., Wahl, J. K., and Peng, A. (2016). Cell fate determination in cisplatin resistance and chemosensitization. Oncotarget 7 (17), 23383–23394. doi:10.18632/oncotarget.8110
Kim, A. Y., Yoon, Y. N., Leem, J., Lee, J. Y., Jung, K. Y., Kang, M., et al. (2020). MKI-1, a novel small-molecule inhibitor of MASTL, exerts antitumor and radiosensitizer activities through PP2A activation in breast cancer. Front. Oncol. 10, 571601. doi:10.3389/fonc.2020.571601
Lapenna, S., and Giordano, A. (2009). Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 8 (7), 547–566. doi:10.1038/nrd2907
Lord, C. J., and Ashworth, A. (2012). The DNA damage response and cancer therapy. Nature 481 (7381), 287–294. doi:10.1038/nature10760
Marzec, K., and Burgess, A. (2018). The oncogenic functions of MASTL kinase. Front. Cell Dev. Biol. 6, 162. doi:10.3389/fcell.2018.00162
Melo, J., and Toczyski, D. (2002). A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 14 (2), 237–245. doi:10.1016/s0955-0674(02)00312-5
Mochida, S., Ikeo, S., Gannon, J., and Hunt, T. (2009). Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. Embo J. 28 (18), 2777–2785. doi:10.1038/emboj.2009.238
Mochida, S., Maslen, S. L., Skehel, M., and Hunt, T. (2010). Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330 (6011), 1670–1673. doi:10.1126/science.1195689
Ocasio, C. A., Rajasekaran, M. B., Walker, S., Le Grand, D., Spencer, J., Pearl, F. M. G., et al. (2016). A first generation inhibitor of human Greatwall kinase, enabled by structural and functional characterisation of a minimal kinase domain construct. Oncotarget 7 (44), 71182–71197. doi:10.18632/oncotarget.11511
Otto, T., and Sicinski, P. (2017). Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17 (2), 93–115. doi:10.1038/nrc.2016.138
Peng, A., and Maller, J. L. (2010). Serine/threonine phosphatases in the DNA damage response and cancer. Oncogene 29 (45), 5977–5988. doi:10.1038/onc.2010.371
Peng, A. M., Lewellyn, A. L., Schiemann, W. P., and Maller, J. L. (2010). Repo-man controls a protein phosphatase 1-dependent threshold for DNA damage checkpoint Activation. Curr. Biol. 20 (5), 387–396. doi:10.1016/j.cub.2010.01.020
Peng, A. M., Yamamoto, T. M., Goldberg, M. L., and Maller, J. L. (2010). A novel role for greatwall kinase in recovery from DNA damage. Cell Cycle 9 (21), 4364–4369. doi:10.4161/cc.9.21.13632
Peng, A., Wang, L., and Fisher, L. A. (2011). Greatwall and Polo-like kinase 1 coordinate to promote checkpoint recovery. J. Biol. Chem. 286 (33), 28996–29004. doi:10.1074/jbc.M111.257121
Peng, C. H., Liao, C. T., Peng, S. C., Chen, Y. J., Cheng, A. J., Juang, J. L., et al. (2011). A novel molecular signature identified by systems genetics approach predicts prognosis in oral squamous cell carcinoma. PLoS One 6 (8), e23452. doi:10.1371/journal.pone.0023452
Rector, J., Kapil, S., Treude, K. J., Kumm, P., Glanzer, J. G., Byrne, B. M., et al. (2016). S4S8-RPA phosphorylation as an indicator of cancer progression in oral squamous cell carcinomas. Oncotarget 8, 9243–9250. doi:10.18632/oncotarget.14001
Ren, D., Fisher, L. A., Zhao, J., Wang, L., Williams, B. C., Goldberg, M. L., et al. (2017). Cell cycle-dependent regulation of greatwall kinase by protein phosphatase 1 and regulatory subunit 3B. J. Biol. Chem. 292, 10026–10034. doi:10.1074/jbc.M117.778233
Rivera, C. (2015). Essentials of oral cancer. Int. J. Clin. Exp. Pathol. 8 (9), 11884–11894. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4637760/.
Rogers, S., McCloy, R. A., Parker, B. L., Gallego-Ortega, D., Law, A. M. K., Chin, V. T., et al. (2018). MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 37, 4518–4533. doi:10.1038/s41388-018-0295-z
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K., and Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85. doi:10.1146/annurev.biochem.73.011303.073723
Sengupta, S., den Boon, J. A., Chen, I. H., Newton, M. A., Dahl, D. B., Chen, M., et al. (2006). Genome-wide expression profiling reveals EBV-associated inhibition of MHC class I expression in nasopharyngeal carcinoma. Cancer Res. 66 (16), 7999–8006. doi:10.1158/0008-5472.CAN-05-4399
Shiloh, Y. (2003). ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 3 (3), 155–168. doi:10.1038/nrc1011
Sun, X. J., Li, Y. L., Wang, L. G., Liu, L. Q., Ma, H., Hou, W. H., et al. (2017). Mastl overexpression is associated with epithelial to mesenchymal transition and predicts a poor clinical outcome in gastric cancer. Oncol. Lett. 14 (6), 7283–7287. doi:10.3892/ol.2017.7155
Taskinen, M. E., Narva, E., Conway, J. R. W., Hinojosa, L. S., Lilla, S., Mai, A., et al. (2020). MASTL promotes cell contractility and motility through kinase-independent signaling. J. Cell Biol. 219 (6), e201906204. doi:10.1083/jcb.201906204
Taylor, S., and Peters, J. M. (2008). Polo and Aurora kinases: Lessons derived from chemical biology. Curr. Opin. Cell Biol. 20 (1), 77–84. doi:10.1016/j.ceb.2007.11.008
Tian, J., Lin, Y., and Yu, J. (2017). E2F8 confers cisplatin resistance to ER+ breast cancer cells via transcriptionally activating MASTL. Biomed. Pharmacother. 92, 919–926. doi:10.1016/j.biopha.2017.05.118
Uppada, S. B., Gowrikumar, S., Ahmad, R., Kumar, B., Szeglin, B., Chen, X., et al. (2018). MASTL induces Colon Cancer progression and Chemoresistance by promoting Wnt/β-catenin signaling. Mol. Cancer 17 (1), 111. doi:10.1186/s12943-018-0848-3
Vera, J., Lartigue, L., Vigneron, S., Gadea, G., Gire, V., Del Rio, M., et al. (2015). Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. Elife 4, e10115. doi:10.7554/eLife.10115
Vigneron, S., Brioudes, E., Burgess, A., Labbe, J. C., Lorca, T., and Castro, A. (2009). Greatwall maintains mitosis through regulation of PP2A. EMBO J. 28 (18), 2786–2793. doi:10.1038/emboj.2009.228
Vigneron, S., Gharbi-Ayachi, A., Raymond, A. A., Burgess, A., Labbe, J. C., Labesse, G., et al. (2011). Characterization of the mechanisms controlling greatwall activity. Mol. Cell. Biol. 31, 2262–2275. doi:10.1128/MCB.00753-10
Vigneron, S., Robert, P., Hached, K., Sundermann, L., Charrasse, S., Labbe, J. C., et al. (2016). The master Greatwall kinase, a critical regulator of mitosis and meiosis. Int. J. Dev. Biol. 60 (7-8-9), 245–254. doi:10.1387/ijdb.160155tl
Voets, E., and Wolthuis, R. M. (2010). MASTL is the human orthologue of Greatwall kinase that facilitates mitotic entry, anaphase and cytokinesis. Cell Cycle 9, 3591–3601. doi:10.4161/cc.9.17.12832
Wang, F., Gouttia, O. G., Wang, L., and Peng, A. (2021). PARP1 upregulation in recurrent oral cancer and treatment resistance. Front. Cell Dev. Biol. 9, 804962. doi:10.3389/fcell.2021.804962
Wang, F., Zhu, S., Fisher, L. A., Wang, W., Oakley, G. G., Li, C., et al. (2018). Protein interactomes of protein phosphatase 2A B55 regulatory subunits reveal B55-mediated regulation of replication protein A under replication stress. Sci. Rep. 8 (1), 2683. doi:10.1038/s41598-018-21040-6
Wang, L., Luong, V. Q., Giannini, P. J., and Peng, A. (2014). Mastl kinase, a promising therapeutic target, promotes cancer recurrence. Oncotarget 5 (22), 11479–11489. doi:10.18632/oncotarget.2565
Wang, L., Mosel, A. J., Oakley, G. G., and Peng, A. (2012). Deficient DNA damage signaling leads to chemoresistance to Cisplatin in oral cancer. Mol. Cancer Ther. 11 (11), 2401–2409. doi:10.1158/1535-7163.MCT-12-0448
Wang, P., Malumbres, M., and Archambault, V. (2014). The Greatwall-PP2A axis in cell cycle control. Methods Mol. Biol. 1170, 99–111. doi:10.1007/978-1-4939-0888-2_6
Wong, P. Y., Ma, H. T., Lee, H. j., and Poon, R. Y. C. (2016). MASTL(Greatwall) regulates DNA damage responses by coordinating mitotic entry after checkpoint recovery and APC/C activation. Sci. Rep. 6, 22230. doi:10.1038/srep22230
Ye, H., Yu, T., Temam, S., Ziober, B. L., Wang, J., Schwartz, J. L., et al. (2008). Transcriptomic dissection of tongue squamous cell carcinoma. BMC Genomics 9, 69. doi:10.1186/1471-2164-9-69
Yoon, Y. N., Choe, M. H., Jung, K. Y., Hwang, S. G., Oh, J. S., and Kim, J. S. (2018). MASTL inhibition promotes mitotic catastrophe through PP2A activation to inhibit cancer growth and radioresistance in breast cancer cells. BMC Cancer 18 (1), 716. doi:10.1186/s12885-018-4600-6
Yu, J. T., Fleming, S. L., Williams, B., Williams, E. V., Li, Z., Somma, P., et al. (2004). Greatwall kinase: A nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. J. Cell Biol. 164 (4), 487–492. doi:10.1083/jcb.200310059
Yu, J. T., Zhao, Y., Li, Z., Galas, S., and Goldberg, M. L. (2006). Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol. Cell 22 (1), 83–91. doi:10.1016/j.molcel.2006.02.022
Zhuge, B. Z., Du, B. R., Meng, X. L., and Zhang, Y. Q. (2017). MASTL is a potential poor prognostic indicator in ER+ breast cancer. Eur. Rev. Med. Pharmacol. Sci. 21 (10), 2413–2420. https://www.europeanreview.org/article/12813.
Keywords: MASTL, greatwall kinase, DNA damage, oral cancer, cisplatin, OSCC
Citation: Gouttia OG, Zhao J, Li Y, Zwiener MJ, Wang L, Oakley GG and Peng A (2022) The MASTL-ENSA-PP2A/B55 axis modulates cisplatin resistance in oral squamous cell carcinoma. Front. Cell Dev. Biol. 10:904719. doi: 10.3389/fcell.2022.904719
Received: 25 March 2022; Accepted: 31 August 2022;
Published: 28 September 2022.
Edited by:
Jiancheng Hu, National Cancer Centre Singapore, SingaporeCopyright © 2022 Gouttia, Zhao, Li, Zwiener, Wang, Oakley and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aimin Peng, YWltaW4ucGVuZ0B1bm1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.