 Priyanka1,2*
Priyanka1,2* Pankaj Seth1
Pankaj Seth1- 1Department of Cellular and Molecular Neuroscience, National Brain Research Centre, Gurgaon, India
- 2Current Address—Synaptic and Developmental Plasticity Group, Neurobiology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC, United States
Mortalin is a chaperone protein that regulates physiological functions of cells. Its multifactorial role allows cells to survive pathological conditions. Pharmacological, chemical, and siRNA-mediated downregulation of mortalin increases oxidative stress, mitochondrial dysfunction leading to unregulated inflammation. In addition to its well-characterized function in controlling oxidative stress, mitochondrial health, and maintaining physiological balance, recent evidence from human brain autopsies and cell culture–based studies suggests a critical role of mortalin in attenuating the damage seen in several neurodegenerative diseases. Overexpression of mortalin provides an important line of defense against accumulated proteins, inflammation, and neuronal loss, a key characteristic feature observed in neurodegeneration. Neurodegenerative diseases are a group of progressive disorders, sharing pathological features in Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and HIV-associated neurocognitive disorder. Aggregation of insoluble amyloid beta-proteins and neurofibrillary tangles in Alzheimer’s disease are among the leading cause of neuropathology in the brain. Parkinson’s disease is characterized by the degeneration of dopamine neurons in substantia nigra pars compacta. A substantial synaptic loss leading to cognitive decline is the hallmark of HIV-associated neurocognitive disorder (HAND). Brain autopsies and cell culture studies showed reduced expression of mortalin in Alzheimer’s, Parkinson’s, and HAND cases and deciphered the important role of mortalin in brain cells. Here, we discuss mortalin and its regulation and describe how neurotoxic conditions alter the expression of mortalin and modulate its functions. In addition, we also review the neuroprotective role of mortalin under neuropathological conditions. This knowledge showcases the importance of mortalin in diverse brain functions and offers new opportunities for the development of therapeutic targets that can modulate the expression of mortalin using chemical compounds.
Introduction
Neurodegenerative diseases of the central nervous system (CNS) are highly debilitating illnesses which impose substantial health burden affecting more than one billion people worldwide. These are characterized by the progressive loss of neurons and physiological dysfunction of the brain cells (Gorman, 2008; Chi et al., 2018), especially in diseases such as Alzheimer’s, Parkinson’s, and HIV-induced neurodegeneration. Clinical studies suggest disturbances in cognition (dementia and motor function impairment) and anatomical alterations in the brain, supporting a common disease spectrum, at least with respect to these diseases. The prevalence and incidence of these diseases increase dramatically with age and are also influenced by environmental and genetic factors (Hou et al., 2019). Among various neurodegenerative diseases, Alzheimer’s and Parkinson’s disease share common pathological hallmarks. Currently, more than 45 million people are affected by Alzheimer’s disease (AD) which is expected to grow to over 100 million by 2050. AD typically progresses with age from mild semantic memory problems to higher-order cognitive dysfunctions. Several brain autopsy studies showed severe brain pathology. Accumulation of Lewy bodies, beta-amyloid aggregate formation, and neurofibrillary tangle (NFT) pathology is characteristic of AD, many of these are also observed in the postmortem AD brains. Among all, beta-amyloid cascade theory is one of the major causes of dementia, it is hypothesized that increased production of insoluble beta-amyloid leads to aggregation and collapsed ubiquitin machinery which manifests as plaque formation in the brain. Detailed mechanistic studies indicate that the accumulation of these proteins leads to severe dysfunction of various brain cells and ultimately results in neurodegeneration (Mungenast et al., 2016; Wildburger et al., 2018; Hardy and Selkoe, 2002; Alzheimer’s Disease, 2022).
Parkinson’s disease (PD) is another progressive neurodegenerative disease that develops between the age of 55 and 65 years and is characterized by rigidity, resting tremors, and bradykinesia. It accounts for 10 million cases across the world. Ten percent of patients carry a single gene mutation, and the remaining 90% of patients are sporadic with no family history (Klein and Westenberger, 2012). Substantial reduction in dopaminergic neurons in substantia nigra pars compacta (SNpc) is the most consistent postmortem finding in patient’s brain (Jankovic, 2008; Kouli et al., 2018; Greenland et al., 2018). Dopamine replacement through L-DOPA has proven to reduce the symptoms and effectively manage the disease (Obeso et al., 2017). Together, AD and PD are one of the greatest burdens of age-related disorders. A unifying feature of AD and PD is the abnormal accumulation of proteins, leading to neuronal loss and several pathophysiological changes in the brain. These features include, but are not limited to oxidative stress, mitochondrial dysfunction (increased mitochondrial fragmentation, altered mitochondrial membrane potential, and ATP dysregulation), increased inflammation, and neuronal death.
Neurotropic viruses have neural-invasive and neurovirulent properties that damage the central nervous system (CNS). A large number of viruses have been reported in neurological diseases as they cause severe damage to neuronal cells and activate astrocytes and microglia leading to severe pathological and behavioral alterations. Human immunodeficiency virus-1 (HIV-1) breaches the blood–brain barrier and causes neuronal dysfunction leading to cognitive dysfunctions called HIV-associated neurocognitive disorders (HAND). Approximately 37.7 million people are living with HIV worldwide with more than 42.6% develop HIV-associated neurocognitive dementia (Wang et al., 2020a). HIV-associated neurodegeneration is characterized by impaired neuropsychological test performance and functional impairment and the spectrum ranges from asymptomatic to mild neurocognitive impairment to more severe HIV-associated dementia. The predominate clinical features combine a variety of neurological problems including dementia and difficulty in performing daily tasks (Eggers et al., 2017). HIV and its proteins are known to induce key pathology through altering various physiological functions of brain cells.
Despite of the differential pathological causes, these diseases share common neuro-inflammatory hallmarks, such as increased oxidative stress, accumulation of misfolded proteins into insoluble aggregates, synaptic dysfunctions, apoptosis, mitopathy, and chronic inflammation (Abramov et al., 2020; Chen et al., 2012; Taoufik et al., 2018; Martinez-Vicente, 2017). Dysregulated tripartite connection and accumulation of misfolded proteins are some of the most common underlying causes of neuronal loss and neurodegeneration. In the past few decades, more mechanistic insights into the role of heat shock proteins (HSPs) in the brain cells (in both physiological and pathophysiological conditions) have been observed (Buccellato et al., 2007; Miller and Fort, 2018; Taha et al., 2019).
From a traditional viewpoint, HSPs are evolutionarily conserved proteins known for their pro-survival role (Dukay et al., 2019). These proteins have evolved to cope with the heat stress. Decades of research showed that HSPs are much more important in physiological and pathological functions than their characterized aspects. It is a well-established fact that HSPs are versatile and play an important role besides classical molecular chaperoning (Schopf et al., 2017). They are widely studied and associated with several neurodegenerative diseases. In recent times, HSPs gathered the due recognition for their role in regulating some of the key CNS functions. The way in which HSPs regulate the fundamental functions of brain cells has been intensively studied over the past few decades (San Gil et al., 2017; Miller and Fort, 2018; Walsh et al., 1997). HSPs play important homeostatic functions in CNS notably by regulating oxidative stress, acting as a chaperone in terms of clearing accumulated proteins, maintaining protein conformations, mitochondrial biogenesis, and neuronal survival and functions (Böttinger et al., 2015; Voos and Röttgers, 2002; Lackie et al., 2017; Sweeney et al., 2017). They function virtually at all stages of life and take active participation in regulating stressful conditions and guarding cells from a wide range of deleterious effect of neurotoxicants. There are also growing evidence of HSPs in regulating cell differentiation, neurite outgrowth, and neuronal differentiation through specific pathways.
Unlike other HSPs, interestingly, mortalin is a heat un-inducible protein (Londono et al., 2012). Mortalin is localized in mitochondria, nucleus, endoplasmic reticulum (ER), cytoplasm, and detected in blood as it could be circulated or released in blood (Ran et al., 2000; Jubran et al., 2017). The ramified localization allows mortalin to perform diverse tasks through its interaction with various proteins. This also resulted in different names of mortalin such as HSPA9, Grp75, PBP74, and mtHSP70 (Kaul et al., 2002; Flachbartová and Kovacech, 2013). Mortalin is a highly complex protein that executes a plethora of functions in healthy and disease conditions. This includes maintaining cellular homeostasis, providing metabolic support to the cells, chaperoning of protein which includes folding and refolding of proteins, removing aggregated or misfolded protein, and majorly it is studied in regulating mitochondrial biogenesis (Deocaris et al., 2008; Londono et al., 2012; Dores-Silva et al., 2015; Deocaris et al., 2006). S A Bruschi et al. (1994) originally described mortalin as a mitochondrial stress protein (Bruschi et al., 1993; Bruschi and Lindsay, 1994). During stress, mortalin gets phosphorylated in order to rescue or reduce the cellular stress (Londono et al., 2012). Considering these changes in the cell, mortalin is known as an anti-stress protein, that confers resistance against oxidative stress and apoptosis and participates in reducing neurotoxicity induced by cerebral ischemia, amyloid-β, and synucleinopathy like neurotoxicants (Xu et al., 2009; Qu et al., 2012; Deocaris et al., 2007; Qu et al., 2011). These observations motivated a series of studies examining the role of mortalin under stress in different cell types. During acute to chronic inflammatory period, several savior pathways get dysregulated leading to alteration in the fundamental properties of the cells (Chen et al., 2010; Multhoff et al., 2011; Flusberg and Sorger, 2015). Partial loss or chemical-mediated inhibition of mortalin showed similar inflammatory responses in various types of cells (Honrath et al., 2017; Chiasserini et al., 2011). Such observations led to the foundation of mortalin as a critical regulatory protein associated with cellular homeostasis. Various studies showed a wide range of functions of mortalin, interestingly, mortalin has been observed to play both protective and destructive roles in various cell types. Several studies have reported that the dual role of mortalin could be dependent upon a threshold level of this protein or on its interacting partners. Besides to the explicit role of mortalin in maintaining cellular homeostasis, recently mortalin has been linked with mitochondria which paved the way to the idea of the role of mortalin in mitochondria-related dysfunction and diseases (Jin et al., 2006; Starenki et al., 2019; Mazkereth et al., 2016; Qu et al., 2012). Accordingly, mitochondrial mortalin (mtHSP70/mortalin) have been implicated in several complex mitochondrial functions ranging from maintaining mitochondrial membrane potential to mitochondrial damage or mitophagy.
In this review, we discussed the key mechanistic roles of mortalin in neurodegenerative (AD and PD) and HIV infection, focusing on progress in understanding mortalin as a key aspect of the brain cells as well as in astrocyte–neuron interplay. In this context, we will illuminate recent advances covering the role of mitochondrial mortalin that is not only crucial for mitochondrial biogenesis but is also involved in inflammation induced in several neuropathies. In addition, we have also highlighted the need for in depth research on mortalin in neurodegeneration and viral infection. The review also offers insights into how mortalin can influence the disease progression by activating and binding with various proteins which may open a new avenue for therapeutic development.
Expression of Mortalin and its Role in Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder, accounting for up to 70% of worldwide cases of age-related disorders (Apostolova, 2016). Alzheimer’s disease is a progressive neurodegenerative disorder, characterized by memory loss (dementia), cognitive decline, and behavioral defects such as impaired motor system, speech, and visuospatial orientation (Nelson et al., 2009). AD is a growing health concern. Typically, AD is a complex interplay between genetic and environmental factors, which affects the brain cells and influences their functioning. Clinicopathologic studies found the accumulation of highly insoluble, densely packed filaments—β-amyloid plaques (Aβ), and neurofibrillary tangles (NFTs) in the brain of AD patients. They are considered as a major pathological hallmark of AD and are still relied upon for a pathological diagnosis (Wisniewski et al., 1990). Both these proteins are the major cause of synaptic and neuronal loss in AD patients and strongly correlate with cognitive deficit (Terry et al., 1991; DeKosky et al., 1996).
Various evidence and hypothesis link mitochondrial dysfunction to the pathogenesis of AD. The advent of fluorescence labeling technologies and biochemical assays enabled direct visualization of mitochondrial DNA and morphometry of electron micrographs revealed severe mitochondrial abnormalities in AD with increased oxidative stress in neurons with mitochondrial abnormality (Sayre et al., 1997; Montine et al., 1996; Smith et al., 1997; Xie et al., 2013). A series of reports suggest that the cytotoxic effect of elevated oxidative stress and mitochondrial damage on the neuronal health initiates the vicious cycle of inflammation resulting in neurodegeneration and behavioral changes.
HSP70 is the central component of the cellular machinery which acts as a quality controller in regulating cellular and molecular mechanisms (translocating many proteins in and out of the cells) and assisting a wide range of protein folding processes (helps in folding and maintaining the native conformation of proteins) (Ruan et al., 2017; Craig, 2018; Cyr and Neupert, 1996). HSP70 are considered as a biomarker for oxidative stress. In the hypoxia model, HSP70 plays a critical role in regulating and reducing the cytotoxic effects of stress. Accumulated and misfolded proteins in AD pathology are also known as chaperonopathy in which chaperones are abnormal in structure and function or their quantitative levels are dysregulated. These observations strongly indicate toward the altered HSP70 machinery. Consistently, many subsequent studies using immunoprecipitation and immunolabeling techniques showed C-terminus HSP70 interacting protein (CHIP) being involved in the metabolism of beta-amyloid precursor protein (βAPP) using the proteasome-dependent pathway (Kumar et al., 2007). In particular, HSP70 is reported in degrading the Tau and Aβ oligomers through the proteasome system (Kundel et al., 2018). Such studies indicate toward the key potential of HSP70 in AD pathology.
In addition to these aggregated proteins pathology, an APOE (apolipoprotein E) gene has been widely studied and associated with the increased risk of AD physiopathology. APOE is also known to disrupt the blood–brain barrier (BBB) integrity and induce severe neuropathological events in AD brain (Chernick et al., 2019). In humans, three alleles of the APOE gene are detected: APOE2, APOE3, and APOE4. They are involved in maintaining synaptic integrity, neurite outgrowth, and clearance of neurotoxicants from the CNS. A hippocampi APOE-knockout mice model showed reduced mortalin expression along with activated glial cells and elevated oxidative stress. The increased oxidative damage and neuronal apoptosis were correlated with the reduced expression of mortalin. A meta-analysis of clinical and autopsy-based studies from the various brain regions showed the increased expression of APOE4 allele (Farrer et al., 1997). Interestingly, a differential (1D and 2D) gel electrophoresis showed differential regulation of mortalin under the APOE-targeted mice model. Various studies showed that out of three APOE variants, APOE3 and APOE4 are highly prevalent among AD patients. Upon further examination, the proteomics analysis found four isoforms of mortalin in the hippocampus of APOE4 transgenic mice. Interestingly, out of four isoforms, particularly “d isoform of mortalin” was increased in AD patients. However, APOE4 transgenic mice showed lower expression of “d isoform of mortalin” in the hippocampus (Osorio et al., 2007). These observations indicate toward the differential regulation of mortalin in the AD hippocampal brain. However, how differential expression of mortalin influences the APOE gene remains to be elucidated.
Oxidative stress plays a major role in AD. There is strong evidence from postmortem and experimental analysis of increased oxidative stress and guided pathways are the major sources of inflammation in AD, which is associated with modulated mortalin levels. Both increased reactive oxygen species (ROS) and nitric oxide are implicated in Aβ-mediated neurotoxicity and disease progression. Increased oxidative stress–mediated inflammation is found to activate apoptotic signal–regulating kinase 1 (ASK1) and JNK which is critical for neuronal stability and its functions (Kadowaki et al., 2005; Song et al., 2014). Interestingly, ASK1 and JNK pathways are also studied in various AD models. The chronic expression of oxidative stress creates an imbalance between oxidant and antioxidant systems which overwhelms the intrinsic antioxidant defense. Growing evidence supports a principle of oxidative stress–mediated mitochondrial dysfunction in AD pathogenesis and it is a well-known fact that mitochondria are a main source of ROS, categorized as mtROS. Aβ treatment or mutant APPs induce mitochondrial fragmentation and increase neuronal dysfunction (Barsoum et al., 2006; Wang et al., 2008). From a cell-based functional screening, a siRNA library found mortalin a major regulator of mitochondrial machinery (Park et al., 2014). Upon chemical (MKT077) and genetic (siRNA) inhibition of mortalin increases oxidative stress and triggers mitochondrial damage, moreover, the induction of beta-amyloid treatment in these mortalin inhibited neurons synergistically increases the neurotoxicity and mitochondrial damage, however, upregulation of mortalin in beta-amyloid–treated neurons remarkably reduces the beta-amyloid–mediated cellular damage (Park et al., 2014). Taken together, such reports support the critical role of mortalin in mitochondrial function and neurodegenerative diseases.
Notably, reduced expression of mortalin has been widely correlated with increased oxidative stress and mitochondrial dysfunction, therefore, an altered level of mortalin could be an indicator of cytotoxicity, considerably, the chronic inhibition of mortalin might be playing a role in inducing neuropathology-like features (Park et al., 2014; Liu et al., 2017; Zhu et al., 2013a). Upon further examination, histopathological analysis showed the reduced expression of mortalin in the human brain autopsy section of AD patients and also in AD mice models (Osorio et al., 2007). Considering mtHSP70/mortalin, recent evidence revealed significant upregulation in Drp-1, one of the major sources of increased mitochondrial fragmentation in AD patients and animal models (Oliver and Reddy, 2019; Park et al., 2014). Mortalin has been consistently correlated with various proteins as its binding partner and they are considered as one of the contributing factors of mortalin versatility, however, immunoprecipitation analysis revealed no direct binding of mortalin with Drp-1 in AD models. These studies suggest that the inhibition of mortalin is not directly regulating the mitochondrial fission machinery, but mortalin may still contribute to mitochondrial dysfunction. The knockdown of mortalin induces failure in regulating oxidative stress and induces alteration in mitochondrial machinery and related genes resulting in mitochondrial dysfunction and fragmentation, which is widely observed in AD pathology (Zhu et al., 2013b; Park et al., 2014).
In AD pathophysiology, beta-amyloid accumulation in mitochondria and its interaction with various mitochondrial proteins facilitates the activation of mitochondrial permeability transition pore (mPTP) which results in the loss of mitochondrial membrane potential (ΔΨm) leading to altered energy production and ROS modulation (Manczak et al., 2006; Pagani and Eckert, 2011; Wang et al., 2020b; Picone et al., 2014). Overexpression of mortalin conferred the inhibition of mPTP activation against beta-amyloid suggesting that mortalin can act as a cyto-protector by suppressing common pathogenic mechanisms (Qu et al., 2011; Qu et al., 2012). According to various studies, we hypothesize that mortalin is not directly regulating the mPTP activation. As per pathway studies, beta-amyloid induces massive calcium influx which further activates calcium release from the endoplasmic reticulum (ER) creating an overload in the cytosolic calcium that leads to mPTP opening. Interestingly, another study reveals that mortalin can regulate the calcium influx from ER through its interaction with a mitochondrial protein-VDAC (Qu et al., 2012; Qu et al., 2011). Hence, overexpression of mortalin under the influence of beta-amyloid might be influencing the calcium influx and regulating the further downstream signaling. Hence, studying the calcium–ER–mortalin pathway would help in understanding the mechanisms responsible for calcium-mediated mitochondrial damage.
Since the discovery of mortalin, it has been established as a protein involved in cellular immortality, various overexpression of mortalin induces immortality in the cells (Wadhwa et al., 2006; Wadhwa et al., 1994; Wadhwa et al., 2004). Mortalin has been widely studied in the cancer cells due to highly enriched expression of mortalin in various cancer cells (Lu et al., 2011; Na et al., 2016). Unlike cancer, reduced expression of mortalin in normal cells under the influence of glucose deprivation activates cell death, however, overexpression of mortalin in these cells inhibits the Bax (pro-apoptotic protein) and reduces the cytochrome C release, one of the key mitochondrial proteins and a master regulator of cell death (Liu et al., 2005), (Qu et al., 2012). In the AD model, beta-amyloid is found to upregulate the Bax expression which further activates neuronal death signaling, whereas the overexpression of mortalin counteracts the Bax expression and increases the Bcl2 (anti-apoptotic) levels (Paradis et al., 1996; Qu et al., 2011). In spite of these exemplary studies, the mechanism of action of mortalin remains unclear, which raises various questions such as—is the basal expression of mortalin sufficient to reduce the neurotoxic effects on brain cells? How and what are the threshold levels of mortalin that can influence the cellular conditions? In addition, there are no reports on the mortalin and beta-amyloid plaques or NFT, whether the overexpression of mortalin can clear the beta-amyloid plaques or it is only attenuating the stress mediated by beta-amyloid. Hence, to understand the plethora of mortalin in AD, such questions need to be explored.
Considering the potential role of mortalin in beta-amyloid–mediated neuronal damage. It is strongly suggestive that modulating mortalin levels either chemically (natural and synthetic) or genetically in AD can prevent beta-amyloid–mediated damage and might reduce the neuronal loss and neurocognitive dysfunction. AD is also defined as proteinopathy, and reduced mortalin expression can also be an additive factor in beta-amyloid plaque formation, failure of ubiquitin machinery, and accumulation of proteins. Understanding the interplay of mortalin and accumulated proteins in AD remains to be elucidated.
Neuropathology of Parkinson’s Disease and Functional Significance of Mortalin
Parkinson’s disease (PD) is the second most common neurodegenerative disease which manifests with aging and presents clinically with parkinsonism. The main features include motor symptoms, abnormal posture, bradykinesia, and resting tremor. In 1817, James Parkinson described the behavioral defects such as shaking palsy in patients which was named after Parkinson, by Jean-Martin Charcot (Parkinson, 2002). Despite the widespread pathology, not all cell types are equally susceptible. PD is typified by major dopaminergic neuronal loss in the substantia nigra pars compacta (SNpc), a major region regulates motor functions. Studies found that reduced dopamine levels in basal ganglia lead to a movement disorder. Other than these, various other brain regions such as locus coeruleus, dorsal raphe nuclei, and post-ganglionic sympathetic neurons were also found to be affected, which contributed to dementia and autonomic dysfunction.
After almost a century, in 1912, Fritz Heinrich Lewy identified aggregated proteins outside the substantia nigra region and named them as Lewy bodies (LBs) (Holdorff et al., 2013). Reduced dopaminergic neurons of SNpc and LB formation are the classical characteristic pathological hallmarks observed in the PD brains. LBs are the abnormal intracellular aggregates containing proteins, such as alpha-synuclein (αSyn) and ubiquitin (Dickson et al., 2009; Holdorff et al., 2013). More than 60% of SNpc neurons are lost before any symptoms occur. The aggregation of alpha-synuclein either in Lewy bodies (LB) or Lewy neuritis (LN) form is considered a diagnostic marker (Halliday and McCann, 2010). In view of having effective treatment and early diagnosis in the field of neurodegeneration, several advances have been made in the past three decades and as a result, Tau protein, α-synuclein, DJ-1, and beta-amyloid in cerebrospinal fluid (CSF) and blood have been identified as potential diagnostic biomarkers for PD (Hong et al., 2010; Parnetti et al., 2013). Alpha-synuclein and LB formation are considered as a central event in PD pathology and a major cause of neuronal loss. Neuronal loss induces chronic neuro-inflammation characterized by reactive oxygen radicals, cytokines, mitochondrial dysfunction, and severely dysregulated kinase signaling. From several clinical studies, these pathological events are considered likely to be a major factor in disease recurrence or progression. Several advances have been made in this direction, and from a histopathological analysis, profound accumulation of α-synuclein in the mitochondria of the postmortem PD brain has been constitutively reported (Devi et al., 2008). Mitochondrial dysfunction is proposed to be an integral player in PD progression. The PD model not only alters the mitochondrial energy production but also plays an important role in inducing mitophagy, a critical event in neurodegeneration.
Interestingly, mitochondrial complex 1 deficiency has been reported in the substantia nigra of PD patients (Schapira et al., 1989), and is widely associated with increased mitochondrial ROS production, oxidative stress, reduced mitochondrial membrane potential, and mitophagy, along with α-synuclein pathology observed in multiple PD models (Devi et al., 2008; Hsu et al., 2000; Ludtmann et al., 2018). There is indisputable evidence for Parkin, PTEN-induced kinase 1 (PINK1), and DJ-1 mutation in PD patients, and other genes also involved in mitochondrial functions are also closely linked to controlling mitochondrial quality and providing an important connection to neurodegeneration (Cookson, 2012; Bonnet et al., 2012; Savitt et al., 2006). A series of reports on these mutated genes evaluated in various PD models exposed their role in disease progression and set the stage to elucidate the molecular mechanisms and the associated regulatory pathways (Cherian and Divya, 2020; Repici and Giorgini, 2019). Multiple lines of evidence point toward the role of mitochondrial dysfunction and mediated stress in the pathogenesis of PD. In terms of evaluating mitochondrial dysfunction in PD, a biochemical study showed the interaction of mortalin, which is a mitochondrial heat shock protein 70 (HSP70) with key genes involved in Parkinson’s disease such as Parkin, PINK1, and DJ-1 (Ge et al., 2020). Knockdown of DJ-1 is sufficient to disrupt the integrity and functionality of mitochondria and showed reduced protective capacity against cell death (Larsen et al., 2011; Hao et al., 2010). Interestingly, in hematopoietic stem cells (HSCs), mortalin and DJ-1 are found to interact and maintain the HSC properties by regulating mitochondrial health and oxidative stress. In fact, the inhibition of mortalin augments the ROS production in HSCs (Tai-Nagara et al., 2014). Intriguingly, functional impairment and oxidative stress are one of the pathological hallmarks in PD. Supporting the role of mitochondrial mortalin in PD, a shotgun proteomics multidimensional protein identification technology (MudPIT) profiled mitochondrial proteins from PD patients and PD model (DAergic neurons treated with rotenone) and found reduced expression of mortalin. Reduced expression of mortalin in DAergic neurons treated with rotenone induces oxidative stress and mitochondrial dysfunction (Jin et al., 2006). Overexpression and inhibition of mortalin play an important role in PD pathology. Considering these reports, a cohort study in a Spanish population found three variants of the mortalin gene (two missense-R126W and P509S and a 17 kb insertion in intron 8) in PD patients and the German cohort found a mutation in mortalin gene-A476T (De Mena et al., 2009; Burbulla et al., 2010). Cells carrying these variants showed increased proteolytic and oxidative stress, indicating a potential role of mortalin in inducing functional changes in the brain. In-depth consideration of mortalin in mitochondria with respect to PD, reduced expression of mortalin in PD is linked with dysregulated mitochondrial function, which was rescued by the overexpression of wild type mortalin (Jin et al., 2006).
Defects in several cellular systems have been implicated in the early onset of PD which triggers neuronal death. Reduced expression of mortalin-mediated cellular damage can be conceptualized as a relatively early event in neuronal death, through increased oxidative stress which likely participates in the recruitment of cell death activators. For the evidence of mortalin in the early onset of PD (EOPD), a cohort study screened 139 EOPD patients and detected one missense mutation in mortalin (p.L358P) which was not present in 279 control individuals. However, no changes were detected in mitochondrial morphology or related respiratory functions in EOPD patients (Freimann et al., 2013). Further supporting a role of mortalin in PD, a quantitative analysis showed a reduced cytosolic fraction of mortalin in the frontal cortex and in astrocytes of PD patients (Cook et al., 2016). Loss of mortalin in the Drosophila model mimics the PD pathology-like phenotype such as reduced ATP levels, abnormal locomotion, and posture defects (Zhu et al., 2013a). Dopaminergic neurons (DAs) are more sensitive to mortalin loss as compared to non-neuronal cells. α-synuclein treatment in DA neurons showed a reduction in mortalin expression, correlated with increased mitochondrial dysfunction and apoptosis (Basso et al., 2020; De Miranda et al., 2020). Interestingly, elevated levels of mortalin are found to regulate the acute toxicity, but chronic inflammation levels of mortalin are found to be consistently reduced. A serum-based study of PD patients and control individuals negatively correlated the levels of mortalin to α-synuclein levels. Levels of mortalin were lower in PD patients whereas α-synuclein was elevated. Taking reference from these studies, mortalin has been proposed to be used as a potential marker for PD diagnosis (Singh et al., 2018).
In general, the cellular systems have extensively evolved to deal with abnormally folded and aggerated proteins such as α-Syn. In this mechanism, mortalin is one of the key players in the ubiquitin–proteasome system (UPS). PINK1 and Parkin are commonly studied in autosomal recessive PD. PINK1 encodes PTEN-induced serine/threonine kinase 1 and Parkin is encoded by PRKN- E3 ubiquitin ligase Parkin. PINK1/Parkin are one of the well-studied pathways in PD pathogenesis. Several findings firmly establish their role in mitochondrial quality control (MQC). Mitochondria in neurons face a unique challenge in Parkinson’s disease. In in vitro model systems such as Hela cells and DA neurons, knockdown of mortalin triggers mitochondrial dysfunction. Overexpression of Parkin notably rescues the mitochondrial dysfunction that was induced by the knockdown of mortalin (Yang et al., 2011). In in vivo and ex vivo studies, loss of mortalin causes intramitochondrial proteolytic stress which further increases the expression of HSP60, a marker of mitochondrial Unfolded Protein Response (mtUPR). The mtUPR pathway has been widely studied in a C. elegans model of PD. Rotenone, MPP+ (complex I inhibitor), and paraquat (ROS inducer) are known to induce mtUPR and used to chemically induce the parkinsonian model. Importantly, rotenone-induced mtUPR signaling was found to be reduced by the mortalin overexpression (Jin et al., 2006). Chemical inhibition of mortalin in neurons induces proteolytic stress, reminiscent of an α-Syn induced effect. Interestingly, mitochondrial dysfunction induced by the knockdown of mortalin was rescued by the overexpression of Parkin and PINK1 (Burbulla et al., 2014).
Taken together, several conclusions can be drawn: 1) severe mitochondrial dysfunction is reported in the PD model and PD patients; 2) mitochondria-mediated damage plays an important role in inducing neuro-inflammation; 3) mortalin and/or mortalin interacting proteins appear to be affected in PD; 4) mortalin can attenuate the Lewy bodies and α-Syn-induced neuronal toxicity; 5) Parkin and PINK1 can act as mitochondrial protector. These concepts have implications for the development of therapeutics aimed at LBs and α-Syn. Another approach may be to use endogenous activation of mortalin or pharmacological overexpression to reduce oxidative damage and clearance of the aggregated protein.
Differential Role of Mortalin in HIV-1 Neuropathogenesis
Human immunodeficiency virus (HIV) is the causative agent of acquired immunodeficiency syndrome (AIDS). Although it is an immune system diseases as it targets CD4 positive cells, HIV also infects the CNS via a “Trojan horse” model—invading the CNS by breaching the blood–brain barrier (Anderson et al., 2010; Izquierdo-Useros et al., 2010). Once it gains access to brain tissue, HIV-1 target microglial cells, as these cells allow multiplication of HIV-1 and infects other glial cells particularly astrocytes. Several reports found severe neuropathological conditions in HIV/AIDS patients such as memory loss, and dysregulated cognitive and motor functions (Glass et al., 1993; Marino et al., 2020). HIV-1-associated neurocognitive disorders (HAND) represent a range of neurological dysfunction similar to other neurodegenerative diseases such as Alzheimer’s disease (Putatunda et al., 2019). Despite the immense success of combinatorial antiretroviral therapy (cART), various viral proteins are being detected in the cerebrospinal fluid (CSF) and the autopsy brain sections of infected patients (Kaiser et al., 1989; de Almeida et al., 2022). HIV-1 viral proteins (Nef, Vpr, and Tat-transactivator of transcription) have been highlighted in contributing to the ample neuronal loss (Darbinian et al., 2020; Wang et al., 2017; Cheng et al., 2007; Sami Saribas et al., 2017). Among these, HIV-1 Tat and gp120 are highly neurotoxic thereby contributing to the neuronal loss in HIV/AIDS patients.
HSPs have been shown to play an important role in the life cycle of various viruses (Iyer et al., 2021; Gao et al., 2014). Their critical role has been demonstrated in HIV-1 neuropathogenesis either through their interaction with viral proteins and promoting viral replication or providing aid in inducing viral toxicity (Iyer et al., 2021; Pan et al., 2018; Brenner and Wainberg, 2001). Previously, HSP40, co-chaperone of HSP70 was found to interact with the HIV-1 negative regulatory factor (Nef), and this interaction is crucial for Nef-mediated toxicity and found to be involved in viral gene expression and replication (Kumar et al., 2011). The overexpression of HSP70 in several cell lines showed reduced Vpr replication, rescue of the cell cycle arrest, and increased cellular proliferation (Iordanskiy et al., 2004a; Iordanskiy et al., 2004b). HSPs are widely known to interact with several protein partners, which facilitate their multifaceted functions. Co-immunoprecipitation analysis showed direct interaction of Vpr-HSP70 (Iordanskiy et al., 2004a). However, several HSPs and viral protein interactions are known to facilitate viral replication and viral protein formation (Kim and Oglesbee, 2012; Lubkowska et al., 2021; Wan et al., 2020). Perhaps, there might be a further classification of the distinct HSPs which helps in promoting viral replication or others that might play a defensive role against virus production, or it could be possible that same HSPs has different effects under distinct conditions. However, this observation indicates a protective role of HSP70 against HIV. Further efforts are needed to integrate the various proximal pathophysiological mechanisms that have been implicated through HSPs in HIV. For instance, does any particular HSP contribute in reducing HIV-mediated stress or vice versa? Do the stressful events reinforce the expression of HSPs? Due to the wide range of inter-connectedness of HSPs, few HSPs may be an initiating factor of neurodegeneration in HIV. More research is needed to clarify the mechanism of HSPs in viral replication.
Numerous in vitro and in vivo studies have found secreted and circulated Nef in the CSF of infected patients (Valcour et al., 2012; McPhee et al., 1998). Nef is an accessory protein of the human immunodeficiency virus (HIV) (Basmaciogullari and Pizzato, 2014; Olivetta et al., 2016; Jacob et al., 2021). It counteracts host defense through secretory pathways. It is found to be expressed in astrocytes in HIV and has been suggested to play an important role in the pathogenesis of HAND (van Marle et al., 2004; Chompre et al., 2013; Rivera et al., 2019). From an in vivo study, Nef is found to be released in extracellular vesicles and linked with increased HSP70 levels. This suggests a possible role of HSP70 in extracellular vesicles which leads to the release of Nef in astrocytes (Sami Saribas et al., 2017). This could be a potential mechanism for host–virus interactions and viral infection in neighboring cells through the exploitation of the astrocyte functions (Ali et al., 2010; McNamara et al., 2018). A membrane-based study identified various proteins anchored Nef which are required to facilitate Nef binding and secretion, this human brain cDNA library found mortalin as one of the potential interacting proteins with Nef (Kammula et al., 2012). Predominate expression of extracellular Nef has been widely reported in various HIV models and has been linked with deleterious effects. The extracellular release of HSPs in the extracellular milieu is detected in various viral infection and disease conditions (Multhoff, 2007; Kotsiopriftis et al., 2005). Experimental evidence showed “secretion modification region (SMR)” is highly conserved among all HIV-1 clades and is an essential domain for the Nef secretion. Recently, an in silico study found SMR forms a putative binding pocket which assists Nef in exosome release (ex-Nef). Further analysis showed that mortalin is one of the proteins which binds to SMR-pocket and facilitates ex-Nef secretion (Shelton et al., 2012). Previously, SMRwt peptides have been used to antagonize the effect of mortalin on cancer cells and effectively reduce the complement-dependent cell toxicity (Huang et al., 2019; Fishelson and Kirschfink, 2019). The trio of SMR–Nef–mortalin (interaction) is one of the factors known for severe infection in uninfected cells and increased inflammation. This could represent a novel mechanism whereby mortalin can influence the Nef release in cells. Upon verification of mortalin and SMR specification, SMR mutation appears to cause alteration in the SMR-Nef binding domain by reducing the binding affinity and interfere in ex-Nef secretion. Mortalin is likely responsible for the extracellular release of Nef which results in neuronal death. Consistent with this, a fluorescence-based study found reduced ex-Nef secretion in mortalin knockdown cells. Conversely, the overexpression of mortalin in these Jurkat cells positively elevates the ex-Nef levels in the supernatants (Shelton et al., 2012). These findings displayed the role of mortalin in the trafficking of viral and host proteins. However, the in-depth mechanism behind mortalin and ex-Nef secretion remains unclear. Other than mortalin, three proteins were also found to be interacting with Nef in the same study. The effect of these proteins in ex-Nef remains unclear. In order to find the mechanism of mortalin in Nef-induced pathogenesis, a clear understanding of the mechanism of mortalin–Nef interaction is needed.
In this article, we review the evidence of mortalin in ex-Nef secretions. During recent years extracellular vesicles have been widely studied in various diseases and have been implicated in drug delivery and in basic biology to understand vesicular trafficking and signaling. In terms of potential therapeutics, a detailed study of mortalin in various viral conditions may be a more promising approach.
With regard to mortalin playing important role in HIV-1, our histopathological study showed reduced expression of mortalin in HIV-1 human autopsy brain sections. Similar downregulation was observed in the human fetal brain–derived astrocytes (Priyanka et al., 2020). Of note, mortalin was found playing a role in ex-Nef secretion, another HIV-1 viral protein. This creates disparities and raises a few questions; 1) what the expression level of mortalin in Nef condition is, as only the mortalin-Nef interaction was studied earlier. One possible explanation is that mortalin is involved in at least one stage of the viral life cycle such as secretion and/or production and after that, it has been degraded or reduced. In agreement with this, several reports documented the positive role of HSP70 during the early phase of viral replication and binding. During the initial phase of infection, a significant increase in various HSPs have been reported which are found to be reduced at the later stage of viral infection (Pujhari et al., 2019; Csillik et al., 1982). The basic function of mortalin could depend upon the cell type. So far, very few reports documented the role of mortalin in the brain and in neurodegeneration. There are few protective mechanisms that operate in the brain to tackle stress and mortalin is among them. Astrocytes under ischemic stress induce severe mitochondrial damage and oxidative stress which leads to inflammation, and the overexpression of mortalin in these ischemic astrocytes rescued them from oxidative stress and mitochondrial dysfunction (Voloboueva et al., 2008). Astrocytes are an integral part of CNS, a wide range of studies represent the critical role of astrocytes in regulating neuronal functions in both physiological and pathophysiological conditions (Liu et al., 2018; Siracusa et al., 2019). However, astrocytes are found to gain toxic functions and lose a neuroprotective role in various disease conditions, including HIV-1 Tat (Fan and He, 2016; Zhou et al., 2004).
Tat is an early gene product and critical for viral replication and has been associated with inducing apoptosis, oxidative stress, and chronic release of inflammatory toxicants (Agrawal et al., 2012; Tang et al., 2020). Interestingly, Tat protein is known to induce neuronal death via both direct and indirect mechanisms (Buscemi et al., 2007). Moreover, Tat infection alone in an animal model is enough to induce HAND pathology and behavioral defects. Although neurons are not infected by HIV-1, the major cause of neuronal death is via infected astrocytes and microglia. These infected cells release cytokines such as TNF-α and IL-1β, increase ROS and NO production in neurons which results in synaptic dysfunction, neurite retraction, and neuronal death, which is a leading cause of neuropathogenesis and neurodegeneration. In addition, Tat protein is secreted from infected cells and taken up by uninfected bystander cells (Chen et al., 2002). Exogenous Tat expression in astrocytic culture induces dysregulation in their fundamental functions such as dysregulated glutamate transporter causing excitotoxicity and mitochondrial dysfunction (reduced mitochondria size, altered ATP production, and increased mitochondrial fragmentation), which induces neuronal death (Lecoeur et al., 2012; Teodorof-Diedrich and Spector, 2020; Teodorof-Diedrich and Spector, 2018). Given the clear association of mortalin in these functions, it suggests a potential role of mortalin in HIV-1 Tat induced neuronal toxicity. It is, hence, important to emphasize the role of mortalin in astrocyte-mediated neuronal damage in HIV-1 Tat.
While the normal functions of mortalin in brain cells are still unexplored, it is clear that mortalin plays an important role in regulating varied stress, importantly in the mitochondrial context. The kind of mitochondrial dysfunction and morphological changes that were widely found in astrocyte and neuronal culture treated with Tat, was reminiscent of a similar degree of functional and morphological changes as reported in HIV/AIDS patients. A recent study from our laboratory showed that the overexpression of mortalin along with HIV-1 Tat rescued mitochondrial functions (examined by ATP production) and reduced mitochondrial fragmentation, which is a product of Tat inflammation (Priyanka et al., 2020). This study showed a protective effect of mortalin in astrocytes in HIV-1 Tat condition. Overexpression of mortalin was found to degrade the Tat protein in astrocyte culture, leading to the observed protective effects. It is noteworthy that many studies have demonstrated mortalin and various other HSP70 proteins to be involved with the ubiquitin machinery. A mechanistic study showed that co-chaperone carboxyl terminus HSP70/90 (CHIP), an E3 ubiquitin ligase binds with mortalin. In cancerous cells, degradation of proto-Dbl (dbl proto-oncogene product) was found to be promoted by GRP75/mortalin through the CHIP-mediated ubiquitin–proteasome pathway (Niu et al., 2018). The ubiquitin role of mortalin has also been studied in other neurodegenerative diseases (Li et al., 2005; Londono et al., 2012). Treatment of MG132 in transfected astrocytes (with mortalin and Tat) revealed mortalin-mediated Tat degradation via the ubiquitin–proteasome pathway (UPP) (Priyanka et al., 2020). This study also highlighted the ubiquitin role of mortalin. Astrocyte-mediated neuronal damage is a key characteristic feature of Tat, as documented in several studies. Astrocytes-mediated neuronal stress is a major contributing factor in HAND. Neurons exposed to infected astrocytes supernatant induce dendritic spines retraction, axonal disruption, dendritic pruning, mitochondrial dysfunction, and neuronal apoptosis (New et al., 1998; Perry et al., 2005). Interestingly, the indirect examination of neuronal health upon co-overexpression of mortalin and Tat condition showed intact neuronal morphology and reduced neuronal death (Priyanka et al., 2020). These evidence suggests that mortalin is neuroprotective in HAND and can play similar roles in other neurodegenerative diseases.
It is evident that further approaches could improve our understanding in this regard. However, the following questions need further attention—1) how does Tat downregulate mortalin levels in the brain? 2) Which event is occurring first, whether a) mortalin-mediated Tat degradation, or b) mortalin-mediated rescue as both are observed at several levels in astrocytes and in neurons. Considering the evidence, it is a fair assumption that both the events are happening simultaneously or in parallel.
Conclusion and Future Perspectives
This review highlights the multiple roles of mortalin in the physiological and pathophysiological conditions associated with neurodegeneration and viral infection (Table 1). Several reports documented that knockdown of mortalin in normal cells resulted in increased oxidative stress, dysregulated mitochondrial functions and mitochondrial morphology, increased inflammation, and reduced cellular proliferation, whereas overexpression of mortalin increases the antistress property by reducing the ischemic stress and rescuing mitochondrial dysfunction. Such studies suggest the key role of mortalin in regulating physiological functions in neuronal and non-neuronal cells. Mortalin is positioned at the heart of mitochondria metabolism and serves as a key player in maintaining mitochondrial functions via distinct pathways and through its interaction with various proteins, or co-chaperones (Table 2). Here in this review, we highlighted the role of mortalin in the brain in AD, PD, and HAND (Figure 1). In current understanding, reduced expression of mortalin in the brain autopsy sections of AD, PD, and HIV-1 patients suggested its key role in brain cells. However, there is no clear understanding of how mortalin is reduced in these diseases. Various studies in this review highlighted the critical role of mortalin overexpression in attenuating beta-amyloid and alpha-synuclein toxicity in neuronal cells. Interaction of mortalin with various proteins like-Nef and Tat showed its pro-inflammatory and anti-inflammatory role, highlighting its bifunctional role. However, in depth research is warranted to understand these mechanisms better. We also noted that the overexpression of mortalin proved to be critical in reducing the detrimental effects of AD, PD, and HIV-1 on the brain cells. In what may seem like a paradox, mortalin is also essential for viral infection. Expression level of mortalin underpins the health of the cell; for example, excessive mortalin contributes to irregular cellular proliferation observed in cancer cells, whereas inhibition of mortalin promotes oxidative stress, increased mitochondrial functions, and reduced lifespan detected in neurodegenerative diseases such as Parkinson’s, Alzheimer’s, and HIV. Further efforts in elucidating the role of mortalin and its interacting partners is required to fully recognize and exploit mortalin’s protective role against the discussed disorders.
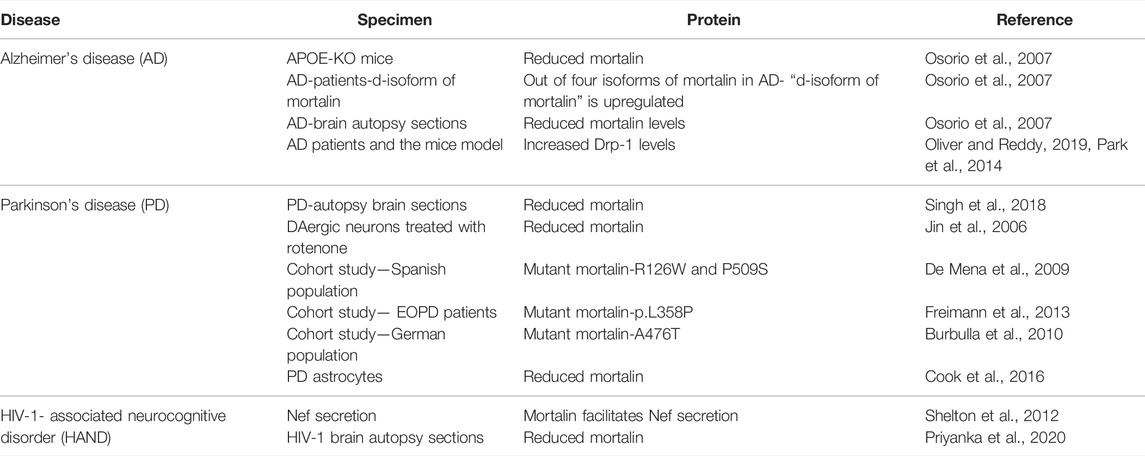
TABLE 1. Tabular overview of proteins modulated in Alzheimer’s disease, Parkinson’s disease, and HIV-1-associated neurocognitive disorder (HAND).

TABLE 2. Schematic representation of protein–protein interaction between mortalin and other proteins under different conditions.
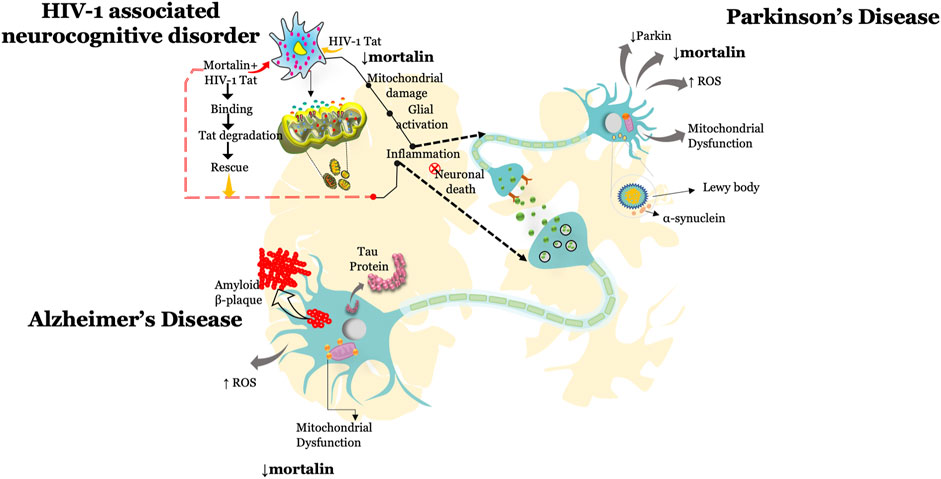
FIGURE 1. Tiff.
Author Contributions
Priyanka designed the structure of the review, performed literature search, wrote the draft, and edited the manuscript. The figures were prepared by Priyanka. PS edited the manuscript, provided valuable suggestions, and approved the submitted version.
Funding
The study was funded by the National Brain Research Centre core funds.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor RW declared a past co-authorship with the author PrS.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge the critical comments and suggestions of Hriday S Pandey and Rituparna Chaudhuri during the preparation of this review. The financial assistance from core grants of the National Brain Research Centre to PS and the student fellowship of Priyanka are acknowledged.
References
Alzheimer's Disease (2022). 2022 Alzheimer's Disease Facts and Figures. Alzheimers Dement. 18(4), 700–789. doi:10.1002/alz.12638
Abramov, A. Y., Potapova, E. V., Dremin, V. V., and Dunaev, A. V. (2020). Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life (Basel) 10 (7), 101. doi:10.3390/life10070101
Agrawal, L., Louboutin, J.-P., Reyes, B. A. S., Van Bockstaele, E. J., and Strayer, D. S. (2012). HIV-1 Tat Neurotoxicity: a Model of Acute and Chronic Exposure, and Neuroprotection by Gene Delivery of Antioxidant Enzymes. Neurobiol. Dis. 45 (2), 657–670. doi:10.1016/j.nbd.2011.10.005
Ali, S. A., Huang, M.-B., Campbell, P. E., Roth, W. W., Campbell, T., Khan, M., et al. (2010). Genetic Characterization of HIV Type 1 Nef-Induced Vesicle Secretion. AIDS Res. Hum. Retroviruses 26 (2), 173–192. doi:10.1089/aid.2009.0068
Anderson, D. J., Politch, J. A., Nadolski, A. M., Blaskewicz, C. D., Pudney, J., and Mayer, K. H. (2010). Targeting Trojan Horse Leukocytes for HIV Prevention. AIDS 24 (2), 163–187. doi:10.1097/qad.0b013e32833424c8
Apostolova, L. G. (2016). Alzheimer Disease. Contin. (Minneap Minn) 22 (2 Dementia), 419–434. doi:10.1212/con.0000000000000307
Barsoum, M. J., Yuan, H., Gerencser, A. A., Liot, G., Kushnareva, Y., Gräber, S., et al. (2006). Nitric Oxide-Induced Mitochondrial Fission Is Regulated by Dynamin-Related GTPases in Neurons. EMBO J. 25 (16), 3900–3911. doi:10.1038/sj.emboj.7601253
Basmaciogullari, S. p., and Pizzato, M. (2014). The Activity of Nef on HIV-1 Infectivity. Front. Microbiol. 5, 232. doi:10.3389/fmicb.2014.00232
Basso, V., Marchesan, E., and Ziviani, E. (2020). A Trio Has Turned into a Quartet: DJ-1 Interacts with the IP3R-Grp75-VDAC Complex to Control ER-Mitochondria Interaction. Cell Calcium 87, 102186. doi:10.1016/j.ceca.2020.102186
Bonnet, A. M., Jutras, M. F., Czernecki, V., Corvol, J. C., and Vidailhet, M. (2012). Nonmotor Symptoms in Parkinson's Disease in 2012: Relevant Clinical Aspects. Park. Dis. 2012, 198316. doi:10.1155/2012/198316
Böttinger, L., Oeljeklaus, S., Guiard, B., Rospert, S., Warscheid, B., and Becker, T. (2015). Mitochondrial Heat Shock Protein (Hsp) 70 and Hsp10 Cooperate in the Formation of Hsp60 Complexes. J. Biol. Chem. 290 (18), 11611–11622. doi:10.1074/jbc.m115.642017
Brenner, B. G., and Wainberg, Z. (2001). Heat Shock Proteins: Novel Therapeutic Tools for HIV-Infection? Expert Opin. Biol. Ther. 1 (1), 67–77. doi:10.1517/14712598.1.1.67
Bruschi, S. A., and Lindsay, J. G. (1994). Mitochondrial Stress Protein Actions during Chemically Induced Renal Proximal Tubule Cell Death. Biochem. Cell Biol. 72 (11-12), 663–667. doi:10.1139/o94-087
Bruschi, S. A., West, K. A., Crabb, J. W., Gupta, R. S., and Stevens, J. L. (1993). Mitochondrial HSP60 (P1 Protein) and a HSP70-like Protein (Mortalin) are Major Targets for Modification during S-(1,1,2,2-tetrafluoroethyl)-L-cysteine-induced Nephrotoxicity. J. Biol. Chem. 268 (31), 23157–23161. doi:10.1016/s0021-9258(19)49440-4
Buccellato, M. A., Carsillo, T., Traylor, Z., and Oglesbee, M. (2007). Heat Shock Protein Expression in Brain: A Protective Role Spanning Intrinsic Thermal Resistance and Defense against Neurotropic Viruses. Prog. Brain Res. 162, 395–415. doi:10.1016/s0079-6123(06)62019-0
Burbulla, L. F., Fitzgerald, J. C., Stegen, K., Westermeier, J., Thost, A.-K., Kato, H., et al. (2014). Mitochondrial Proteolytic Stress Induced by Loss of Mortalin Function is Rescued by Parkin and PINK1. Cell Death Dis. 5, e1180. doi:10.1038/cddis.2014.103
Burbulla, L. F., Schelling, C., Kato, H., Rapaport, D., Woitalla, D., Schiesling, C., et al. (2010). Dissecting the Role of the Mitochondrial Chaperone Mortalin in Parkinson's Disease: Functional Impact of Disease-Related Variants on Mitochondrial Homeostasis. Hum. Mol. Genet. 19 (22), 4437–4452. doi:10.1093/hmg/ddq370
Buscemi, L., Ramonet, D., and Geiger, J. D. (2007). Human Immunodeficiency Virus Type-1 Protein Tat Induces Tumor Necrosis Factor-α-Mediated Neurotoxicity. Neurobiol. Dis. 26 (3), 661–670. doi:10.1016/j.nbd.2007.03.004
Chen, D., Wang, M., Zhou, S., and Zhou, Q. (2002). HIV-1 Tat Targets Microtubules to Induce Apoptosis, a Process Promoted by the Pro-apoptotic Bcl-2 Relative Bim. EMBO J. 21 (24), 6801–6810. doi:10.1093/emboj/cdf683
Chen, L. S., Balakrishnan, K., and Gandhi, V. (2010). Inflammation and Survival Pathways: Chronic Lymphocytic Leukemia as a Model System. Biochem. Pharmacol. 80 (12), 1936–1945. doi:10.1016/j.bcp.2010.07.039
Chen, X., Guo, C., and Kong, J. (2012). Oxidative Stress in Neurodegenerative Diseases. Neural Regen. Res. 7 (5), 376–385. doi:10.3969/j.issn.1673-5374.2012.05.009
Cheng, X., Mukhtar, M., Acheampong, E. A., Srinivasan, A., Rafi, M., Pomerantz, R. J., et al. (2007). HIV-1 Vpr Potently Induces Programmed Cell Death in the CNSin Vivo. DNA Cell Biol. 26 (2), 116–131. doi:10.1089/dna.2006.0541
Cherian, A., and Divya, K. P. (2020). Genetics of Parkinson's Disease. Acta Neurol. Belg 120 (6), 1297–1305. doi:10.1007/s13760-020-01473-5
Chernick, D., Ortiz-Valle, S., Jeong, A., Qu, W., and Li, L. (2019). Peripheral Versus Central Nervous System APOE in Alzheimer's Disease: Interplay across the Blood-Brain Barrier. Neurosci. Lett. 708, 134306. doi:10.1016/j.neulet.2019.134306
Chi, H., Chang, H. Y., and Sang, T. K. (2018). Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 19 (10), 3082. doi:10.3390/ijms19103082
Chiasserini, D., Tozzi, A., de Iure, A., Tantucci, M., Susta, F., Orvietani, P. L., et al. (2011). Mortalin Inhibition in Experimental Parkinson's Disease. Mov. Disord. 26 (9), 1639–1647. doi:10.1002/mds.23647
Chompre, G., Cruz, E., Maldonado, L., Rivera-Amill, V., Porter, J. T., and Noel, R. J. (2013). Astrocytic Expression of HIV-1 Nef Impairs Spatial and Recognition Memory. Neurobiol. Dis. 49, 128–136. doi:10.1016/j.nbd.2012.08.007
Cook, T. J., Hoekstra, J. G., Eaton, D. L., and Zhang, J. (2016). Mortalin Is Expressed by Astrocytes and Decreased in the Midbrain of Parkinson's Disease Patients. Brain Pathol. 26 (1), 75–81. doi:10.1111/bpa.12274
Cookson, M. R. (2012). Parkinsonism Due to Mutations in PINK1, Parkin, and DJ-1 and Oxidative Stress and Mitochondrial Pathways. Cold Spring Harb. Perspect. Med. 2 (9), a009415. doi:10.1101/cshperspect.a009415
Craig, E. A. (2018). Hsp70 at the Membrane: Driving Protein Translocation. BMC Biol. 16 (1), 11. doi:10.1186/s12915-017-0474-3
Csillik, B., Knyihár-Csillik, E., and Szücs, A. (1982). Treatment of Chronic Pain Syndromes with Iontophoresis of vinca Alkaloids to the Skin of Patients. Neurosci. Lett. 31 (1), 87–90. doi:10.1016/0304-3940(82)90059-3
Cyr, D. M., and Neupert, W. (1996). Roles for Hsp70 in Protein Translocation across Membranes of Organelles. EXS 77, 25–40. doi:10.1007/978-3-0348-9088-5_3
Darbinian, N., Darbinyan, A., Merabova, N., Selzer, M. E., and Amini, S. (2020). HIV-1 and HIV-1-Tat Induce Mitochondrial DNA Damage in Human Neurons. J. HIV AIDS 6 (1). doi:10.16966/2380-5536.176
de Almeida, S. M., Beltrame, M. P., Tang, B., Rotta, I., Schluga, Y., Justus, J. L. P., et al. (2022). Main Lymphocyte Subpopulations in Cerebrospinal Fluid and Peripheral Blood in HIV-1 Subtypes C and B. J. Neurovirol. doi:10.1007/s13365-022-01054-7
De Mena, L., Coto, E., Sánchez-Ferrero, E., Ribacoba, R., Guisasola, L. M., Salvador, C., et al. (2009). Mutational Screening of the Mortalin Gene (HSPA9) in Parkinson's Disease. J. Neural Transm. 116 (10), 1289–1293. doi:10.1007/s00702-009-0273-2
De Miranda, B. R., Rocha, E. M., Castro, S. L., and Greenamyre, J. T. (2020). Protection from α-Synuclein Induced Dopaminergic Neurodegeneration by Overexpression of the Mitochondrial Import Receptor TOM20. npj Park. Dis. 6 (1), 38. doi:10.1038/s41531-020-00139-6
DeKosky, S. T., Scheff, S. W., and Styren, S. D. (1996). Structural Correlates of Cognition in Dementia: Quantification and Assessment of Synapse Change. Neurodegeneration 5 (4), 417–421. doi:10.1006/neur.1996.0056
Deocaris, C. C., Kaul, S. C., and Wadhwa, R. (2008). From Proliferative to Neurological Role of an Hsp70 Stress Chaperone, Mortalin. Biogerontology 9 (6), 391–403. doi:10.1007/s10522-008-9174-2
Deocaris, C. C., Kaul, S. C., and Wadhwa, R. (2006). On the Brotherhood of the Mitochondrial Chaperones Mortalin and Heat Shock Protein 60. Cell Stress Chaper 11 (2), 116–128. doi:10.1379/csc-144r.1
Deocaris, C. C., Widodo, N., Ishii, T., Kaul, S. C., and Wadhwa, R. (2007). Functional Significance of Minor Structural and Expression Changes in Stress Chaperone Mortalin. Ann. N. Y. Acad. Sci. 1119, 165–175. doi:10.1196/annals.1404.007
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 283 (14), 9089–9100. doi:10.1074/jbc.m710012200
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., et al. (2009). Neuropathological Assessment of Parkinson's Disease: Refining the Diagnostic Criteria. Lancet Neurology 8 (12), 1150–1157. doi:10.1016/s1474-4422(09)70238-8
Dores-Silva, P. R., Barbosa, L. R. S., Ramos, C. H. I., and Borges, J. C. (2015). Human Mitochondrial Hsp70 (Mortalin): Shedding Light on ATPase Activity, Interaction with Adenosine Nucleotides, Solution Structure and Domain Organization. PLoS One 10 (1), e0117170. doi:10.1371/journal.pone.0117170
Dukay, B., Csoboz, B., and Tóth, M. E. (2019). Heat-Shock Proteins in Neuroinflammation. Front. Pharmacol. 10, 920. doi:10.3389/fphar.2019.00920
Eggers, C., Csoboz, B., Arendt, G., Hahn, K., Husstedt, I. W., Maschke, M., et al. (2017). HIV-1-associated Neurocognitive Disorder: Epidemiology, Pathogenesis, Diagnosis, and Treatment. J. Neurol. 264 (8), 1715–1727. doi:10.1007/s00415-017-8503-2
Fan, Y., and He, J. J. (2016). HIV-1 Tat Promotes Lysosomal Exocytosis in Astrocytes and Contributes to Astrocyte-Mediated Tat Neurotoxicity. J. Biol. Chem. 291 (43), 22830–22840. doi:10.1074/jbc.m116.731836
Farrer, L., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease. a Meta-Analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278 (16), 1349–1356. doi:10.1001/jama.278.16.1349
Fishelson, Z., and Kirschfink, M. (2019). Complement C5b-9 and Cancer: Mechanisms of Cell Damage, Cancer Counteractions, and Approaches for Intervention. Front. Immunol. 10, 752. doi:10.3389/fimmu.2019.00752
Flachbartová, Z., and Kovacech, B. (2013). Mortalin - a Multipotent Chaperone Regulating Cellular Processes Ranging from Viral Infection to Neurodegeneration. Acta Virol. 57 (1), 3–15. doi:10.4149/av_2013_01_3
Flusberg, D. A., and Sorger, P. K. (2015). Surviving Apoptosis: Life-Death Signaling in Single Cells. Trends Cell Biol. 25 (8), 446–458. doi:10.1016/j.tcb.2015.03.003
Freimann, K., Zschiedrich, K., Brüggemann, N., Grünewald, A., Pawlack, H., Hagenah, J., et al. (2013). Mortalin Mutations are Not a Frequent Cause of Early-Onset Parkinson Disease. Neurobiol. Aging 34 (11), 2694–2720. doi:10.1016/j.neurobiolaging.2013.05.021
Gao, J., Xiao, S., Liu, X., Wang, L., Ji, Q., Mo, D., et al. (2014). Inhibition of HSP70 Reduces Porcine Reproductive and Respiratory Syndrome Virus Replication in Vitro. BMC Microbiol. 14, 64. doi:10.1186/1471-2180-14-64
Ge, P., Dawson, V. L., and Dawson, T. M. (2020). PINK1 and Parkin Mitochondrial Quality Control: A Source of Regional Vulnerability in Parkinson's Disease. Mol. Neurodegener. 15 (1), 20. doi:10.1186/s13024-020-00367-7
Glass, J. D., Wesselingh, S. L., Selnes, O. A., and McArthur, J. C. (1993). Clinical‐neuropathologic Correlation in HIV‐associated Dementia. Neurology 43 (11), 2230. doi:10.1212/wnl.43.11.2230
Gorman, A. M. (2008). Neuronal Cell Death in Neurodegenerative Diseases: Recurring Themes Around Protein Handling. J. Cell Mol. Med. 12 (6A), 2263–2280. doi:10.1111/j.1582-4934.2008.00402.x
Greenland, J. C., and Barker, R. A. (2018). “The Differential Diagnosis of Parkinson's Disease,” in Parkinson's Disease: Pathogenesis and Clinical Aspects. Editors T. B. Stoker, and J. C. Greenland (Brisbane (AU): Codonpublications). doi:10.15586/codonpublications.parkinsonsdisease.2018.ch6
Halliday, G. M., and McCann, H. (2010). The Progression of Pathology in Parkinson's Disease. Ann. N. Y. Acad. Sci. 1184, 188–195. doi:10.1111/j.1749-6632.2009.05118.x
Hao, L.-Y., Giasson, B. I., and Bonini, N. M. (2010). DJ-1 is Critical for Mitochondrial Function and Rescues PINK1 Loss of Function. Proc. Natl. Acad. Sci. U.S.A. 107 (21), 9747–9752. doi:10.1073/pnas.0911175107
Hardy, J., and Selkoe, D. J. (2002). The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science 297 (5580), 353–356. doi:10.1126/science.1072994
Holdorff, B., Rodrigues e Silva, A. M., and Dodel, R. (2013). Centenary of Lewy Bodies (1912-2012). J. Neural Transm. 120 (4), 509–516. doi:10.1007/s00702-013-0984-2
Hong, Z., Shi, M., Chung, K. A., Quinn, J. F., Peskind, E. R., Galasko, D., et al. (2010). DJ-1 and Alpha-Synuclein in Human Cerebrospinal Fluid as Biomarkers of Parkinson's Disease. Brain 133 (Pt 3), 713–726. doi:10.1093/brain/awq008
Honrath, B., Metz, I., Bendridi, N., Rieusset, J., Culmsee, C., and Dolga, A. M. (2017). Glucose-regulated Protein 75 Determines ER-Mitochondrial Coupling and Sensitivity to Oxidative Stress in Neuronal Cells. Cell Death Discov. 3, 17076. doi:10.1038/cddiscovery.2017.76
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 15 (10), 565–581. doi:10.1038/s41582-019-0244-7
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., et al. (2000). α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. Am. J. Pathology 157 (2), 401–410. doi:10.1016/s0002-9440(10)64553-1
Huang, M.-B., Wu, J. Y., Lillard, J., and Bond, V. C. (2019). SMR Peptide Antagonizes Mortalin Promoted Release of Extracellular Vesicles and Affects Mortalin Protection from Complement-dependent Cytotoxicity in Breast Cancer Cells and Leukemia Cells. Oncotarget 10 (52), 5419–5438. doi:10.18632/oncotarget.27138
Iordanskiy, S., Zhao, Y., DiMarzio, P., Agostini, I., Dubrovsky, L., and Bukrinsky, M. (2004). Heat-shock Protein 70 Exerts Opposing Effects on Vpr-dependent and Vpr-independent HIV-1 Replication in Macrophages. Blood 104 (6), 1867–1872. doi:10.1182/blood-2004-01-0081
Iordanskiy, S., Zhao, Y., Dubrovsky, L., Iordanskaya, T., Chen, M., Liang, D., et al. (2004). Heat Shock Protein 70 Protects Cells from Cell Cycle Arrest and Apoptosis Induced by Human Immunodeficiency Virus Type 1 Viral Protein R. J. Virol. 78 (18), 9697–9704. doi:10.1128/jvi.78.18.9697-9704.2004
Iyer, K., Chand, K., Mitra, A., Trivedi, J., and Mitra, D. (2021). Diversity in Heat Shock Protein Families: Functional Implications in Virus Infection with a Comprehensive Insight of Their Role in the HIV-1 Life Cycle. Cell Stress Chaperones 26 (5), 743–768. doi:10.1007/s12192-021-01223-3
Izquierdo-Useros, N., Naranjo-Gómez, M., Erkizia, I., Puertas, M. C., Borràs, F. E., Blanco, J., et al. (2010). HIV and Mature Dendritic Cells: Trojan Exosomes Riding the Trojan Horse? PLoS Pathog. 6 (3), e1000740. doi:10.1371/journal.ppat.1000740
Jacob, R. A., Edgar, C. R., Prévost, J., Trothen, S. M., Lurie, A., Mumby, M. J., et al. (2021). The HIV-1 Accessory Protein Nef Increases Surface Expression of the Checkpoint Receptor Tim-3 in Infected CD4+ T Cells. J. Biol. Chem. 297 (3), 101042. doi:10.1016/j.jbc.2021.101042
Jankovic, J. (2008). Parkinson's Disease: Clinical Features and Diagnosis. J. Neurology, Neurosurg. Psychiatry 79 (4), 368–376. doi:10.1136/jnnp.2007.131045
Jin, J., Hulette, C., Wang, Y., Zhang, T., Pan, C., Wadhwa, R., et al. (2006). Proteomic Identification of a Stress Protein, Mortalin/mthsp70/GRP75. Mol. Cell. Proteomics 5 (7), 1193–1204. doi:10.1074/mcp.m500382-mcp200
Jubran, R., Kocsis, J., Garam, N., Maláti, É., Gombos, T., Barabás, L., et al. (2017). Circulating Mitochondrial Stress 70 Protein/mortalin and Cytosolic Hsp70 in Blood: Risk Indicators in Colorectal Cancer. Int. J. Cancer 141 (11), 2329–2335. doi:10.1002/ijc.30918
Kadowaki, H., Nishitoh, H., Urano, F., Sadamitsu, C., Matsuzawa, A., Takeda, K., et al. (2005). Amyloid β Induces Neuronal Cell Death through ROS-Mediated ASK1 Activation. Cell Death Differ. 12 (1), 19–24. doi:10.1038/sj.cdd.4401528
Kaiser, R., Dörries, R., Lüer, W., Poser, S., Pohle, H. D., Felgenhauer, K., et al. (1989). Analysis of Oligoclonal Antibody Bands against Individual HIV Structural Proteins in the CSF of Patients Infected with HIV. J. Neurol. 236 (3), 157–160. doi:10.1007/bf00314332
Kammula, E. C., Mötter, J., Gorgels, A., Jonas, E., Hoffmann, S., and Willbold, D. (2012). Brain Transcriptome-wide Screen for HIV-1 Nef Protein Interaction Partners Reveals Various Membrane-Associated Proteins. PLoS One 7 (12), e51578. doi:10.1371/journal.pone.0051578
Kaul, S. C., Taira, K., Pereira-Smith, O. M., and Wadhwa, R. (2002). Mortalin: Present and Prospective. Exp. Gerontol. 37 (10-11), 1157–1164. doi:10.1016/s0531-5565(02)00135-3
Kim, M. Y., and Oglesbee, M. (2012). Virus-heat Shock Protein Interaction and a Novel axis for Innate Antiviral Immunity. Cells 1 (3), 646–666. doi:10.3390/cells1030646
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson's Disease. Cold Spring Harb. Perspect. Med. 2 (1), a008888. doi:10.1101/cshperspect.a008888
Kotsiopriftis, M., Tanner, J. E., and Alfieri, C. (2005). Heat Shock Protein 90 Expression in Epstein-Barr Virus-Infected B Cells Promotes γδ T-Cell Proliferation in Vitro. J. Virol. 79 (11), 7255–7261. doi:10.1128/jvi.79.11.7255-7261.2005
Kouli, A., Torsney, K. M., and Kuan, W. L. (2018). “Parkinson's Disease: Etiology, Neuropathology, and Pathogenesis,” in Parkinson's Disease: Pathogenesis and Clinical Aspects. Editors T.B. Stoker, and J.C. Greenland (Brisbane (AU): Codonpublications). doi:10.15586/codonpublications.parkinsonsdisease.2018.ch1
Kumar, M., Rawat, P., Khan, S. Z., Dhamija, N., Chaudhary, P., Ravi, D. S., et al. (2011). Reciprocal Regulation of Human Immunodeficiency Virus-1 Gene Expression and Replication by Heat Shock Proteins 40 and 70. J. Mol. Biol. 410 (5), 944–958. doi:10.1016/j.jmb.2011.04.005
Kumar, P., Ambasta, R. K., Veereshwarayya, V., Rosen, K. M., Kosik, K. S., Band, H., et al. (2007). CHIP and HSPs Interact with β-APP in a Proteasome-dependent Manner and Influence Aβ Metabolism. Hum. Mol. Genet. 16 (7), 848–864. doi:10.1093/hmg/ddm030
Kundel, F., De, S., Flagmeier, P., Horrocks, M. H., Kjaergaard, M., Shammas, S. L., et al. (2018). Hsp70 Inhibits the Nucleation and Elongation of Tau and Sequesters Tau Aggregates with High Affinity. ACS Chem. Biol. 13 (3), 636–646. doi:10.1021/acschembio.7b01039
Lackie, R. E., Maciejewski, A., Ostapchenko, V. G., Marques-Lopes, J., Choy, W.-Y., Duennwald, M. L., et al. (2017). The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 11, 254. doi:10.3389/fnins.2017.00254
Larsen, N. J., Ambrosi, G., Mullett, S. J., Berman, S. B., and Hinkle, D. A. (2011). DJ-1 Knock-Down Impairs Astrocyte Mitochondrial Function. Neuroscience 196, 251–264. doi:10.1016/j.neuroscience.2011.08.016
Lecoeur, H., Borgne-Sanchez, A., Chaloin, O., El-Khoury, R., Brabant, M., Langonné, A., et al. (2012). HIV-1 Tat Protein Directly Induces Mitochondrial Membrane Permeabilization and Inactivates Cytochrome C Oxidase. Cell Death Dis. 3, e282. doi:10.1038/cddis.2012.21
Li, H. M., Niki, T., Taira, T., Iguchi-Ariga, S. M. M., and Ariga, H. (2005). Association of DJ-1 with Chaperones and Enhanced Association and Colocalization with Mitochondrial Hsp70 by Oxidative Stress. Free Radic. Res. 39 (10): 1091–1099. doi:10.1080/10715760500260348
Liu, C.-Y., Yang, Y., Ju, W.-N., Wang, X., and Zhang, H.-L. (2018). Emerging Roles of Astrocytes in Neuro-Vascular Unit and the Tripartite Synapse With Emphasis on Reactive Gliosis in the Context of Alzheimer's Disease. Front. Cell. Neurosci. 12, 193. doi:10.3389/fncel.2018.00193
Liu, Y., Liu, W., Song, X. D., and Zuo, J. (2005). Effect of GRP75/mthsp70/PBP74/mortalin Overexpression on Intracellular ATP Level, Mitochondrial Membrane Potential and ROS Accumulation Following Glucose Deprivation in PC12 Cells. Mol. Cell Biochem. 268 (1-2), 45–51. doi:10.1007/s11010-005-2996-1
Liu, Y., Hu, Y., E, Q., Zuo, J., Yang, L., and Liu, W. (2017). Salvianolic Acid B Inhibits Mitochondrial Dysfunction by Up-Regulating Mortalin. Sci. Rep. 7, 43097. doi:10.1038/srep43097
Londono, C., Osorio, C., Gama, V., and Alzate, O. (2012). Mortalin, Apoptosis, and Neurodegeneration. Biomolecules 2 (1), 143–164. doi:10.3390/biom2010143
Lu, W.-J., Lee, N. P., Kaul, S. C., Lan, F., Poon, R. T. P., Wadhwa, R., et al. (2011). Mortalin-p53 Interaction in Cancer Cells is Stress Dependent and Constitutes a Selective Target for Cancer Therapy. Cell Death Differ. 18 (6), 1046–1056. doi:10.1038/cdd.2010.177
Lubkowska, A., Pluta, W., Strońska, A., and Lalko, A. (2021). Role of Heat Shock Proteins (HSP70 and HSP90) in Viral Infection. Int. J. Mol. Sci. 22 (17). doi:10.3390/ijms22179366
Ludtmann, M. H. R., Angelova, P. R., Horrocks, M. H., Choi, M. L., Rodrigues, M., Baev, A. Y., et al. (2018). α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson's Disease. Nat. Commun. 9 (1), 2293. doi:10.1038/s41467-018-04422-2
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a Direct Site of Aβ Accumulation in Alzheimer's Disease Neurons: Implications for Free Radical Generation and Oxidative Damage in Disease Progression. Hum. Mol. Genet. 15 (9), 1437–1449. doi:10.1093/hmg/ddl066
Marino, J., Wigdahl, B., and Nonnemacher, M. R. (2020). Extracellular HIV-1 Tat Mediates Increased Glutamate in the CNS Leading to Onset of Senescence and Progression of HAND. Front. Aging Neurosci. 12, 168. doi:10.3389/fnagi.2020.00168
Martinez-Vicente, M. (2017). Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 10, 64. doi:10.3389/fnmol.2017.00064
Mazkereth, N., Rocca, F., Schubert, J.-R., Geisler, C., Hillman, Y., Egner, A., et al. (2016). Complement Triggers Relocation of Mortalin/GRP75 from Mitochondria to the Plasma Membrane. Immunobiology 221 (12), 1395–1406. doi:10.1016/j.imbio.2016.07.005
McNamara, R. P., Costantini, L. M., Myers, T. A., Schouest, B., Maness, N. J., Griffith, J. D., et al. (2018). Nef Secretion into Extracellular Vesicles or Exosomes is Conserved Across Human and Simian Immunodeficiency Viruses. mBio 9 (1). doi:10.1128/mBio.02344-17
McPhee, D. A., Greenway, A. L., Holloway, G., Smith, K., Deacon, N., Pemberton, L., et al. (1998). Anomalies in Nef Expression within the Central Nervous System of HIV-1 Positive Individuals/AIDS Patients with or Without AIDS Dementia Complex. J. Neurovirol 4 (3), 291–300. doi:10.3109/13550289809114530
Miller, D. J., and Fort, P. E. (2018). Heat Shock Proteins Regulatory Role in Neurodevelopment. Front. Neurosci. 12, 821. doi:10.3389/fnins.2018.00821
Montine, T. J., Amarnath, V., Martin, M. E., Strittmatter, W. J., and Graham, D. G. (1996). E-4-hydroxy-2-nonenal Is Cytotoxic and Cross-Links Cytoskeletal Proteins in P19 Neuroglial Cultures. Am. J. Pathol. 148 (1), 89–93.
Multhoff, G., Molls, M., and Radons, J. (2011). Chronic Inflammation in Cancer Development. Front. Immunol. 2, 98. doi:10.3389/fimmu.2011.00098
Multhoff, G. (2007). Heat Shock Protein 70 (Hsp70): Membrane Location, Export and Immunological Relevance. Methods 43 (3), 229–237. doi:10.1016/j.ymeth.2007.06.006
Mungenast, A. E., Siegert, S., and Tsai, L.-H. (2016). Modeling Alzheimer's Disease with Human Induced Pluripotent Stem (iPS) Cells. Mol. Cell. Neurosci. 73, 13–31. doi:10.1016/j.mcn.2015.11.010
Na, Y., Kaul, S. C., Ryu, J., Lee, J.-S., Ahn, H. M., Kaul, Z., et al. (2016). Stress Chaperone Mortalin Contributes to Epithelial-To-Mesenchymal Transition and Cancer Metastasis. Cancer Res. 76 (9), 2754–2765. doi:10.1158/0008-5472.can-15-2704
Nelson, P. T., Braak, H., and Markesbery, W. R. (2009). Neuropathology and Cognitive Impairment in Alzheimer Disease: A Complex but Coherent Relationship. J. Neuropathol. Exp. Neurol. 68 (1), 1–14. doi:10.1097/nen.0b013e3181919a48
New, D. R., Maggirwar, S. B., Epstein, L. G., Dewhurst, S., and Gelbard, H. A. (1998). HIV-1 Tat Induces Neuronal Death via Tumor Necrosis Factor-α and Activation of Non-N-methyl-d-aspartate Receptors by a NFκB-Independent Mechanism. J. Biol. Chem. 273 (28), 17852–17858. doi:10.1074/jbc.273.28.17852
Niu, X., Su, L., Qi, S., Gao, Z., Zhang, Q., and Zhang, S. (2018). GRP75 Modulates Oncogenic Dbl-Driven Endocytosis Derailed via the CHIP-Mediated Ubiquitin Degradation Pathway. Cell Death Dis. 9 (10), 971. doi:10.1038/s41419-018-1039-2
Obeso, J. A., Stamelou, M., Goetz, C. G., Poewe, W., Lang, A. E., Weintraub, D., et al. (2017). Past, Present, and Future of Parkinson's Disease: A Special Essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 32 (9), 1264–1310. doi:10.1002/mds.27115
Oliver, D., and Reddy, P. H. (2019). Dynamics of Dynamin-Related Protein 1 in Alzheimer's Disease and Other Neurodegenerative Diseases. Cells 8 (9). doi:10.3390/cells8090961
Olivetta, E., Arenaccio, C., Manfredi, F., Anticoli, S., and Federico, M. (2016). The Contribution of Extracellular Nef to HIV-Induced Pathogenesis. Curr. Drug Targets 17 (1), 46–53. doi:10.2174/1389450116666151001110126
Osorio, C., Sullivan, P. M., He, D. N., Mace, B. E., Ervin, J. F., Strittmatter, W. J., et al. (2007). Mortalin is Regulated by APOE in hippocampus of AD Patients and by Human APOE in TR Mice. Neurobiol. Aging 28 (12), 1853–1862. doi:10.1016/j.neurobiolaging.2006.08.011
Pagani, L., and Eckert, A. (2011). Amyloid-Beta Interaction with Mitochondria. Int. J. Alzheimers Dis. 2011, 925050. doi:10.4061/2011/925050
Pan, X., Lin, J., Zeng, X., Li, W., Wu, W., Lu, W. Z., et al. (2018). Heat Shock Factor 1 Suppresses the HIV-Induced Inflammatory Response by Inhibiting Nuclear Factor-κB. Cell. Immunol. 327, 26–35. doi:10.1016/j.cellimm.2018.01.015
Paradis, E., Douillard, H., Koutroumanis, M., Goodyer, C., and LeBlanc, A. (1996). Amyloid β Peptide of Alzheimer's Disease Downregulates Bcl-2 and Upregulates Bax Expression in Human Neurons. J. Neurosci. 16 (23), 7533–7539. doi:10.1523/jneurosci.16-23-07533.1996
Park, S. J., Shin, J. H., Jeong, J. I., Song, J. H., Jo, Y. K., Kim, E. S., et al. (2014). Down-regulation of Mortalin Exacerbates Aβ-Mediated Mitochondrial Fragmentation and Dysfunction. J. Biol. Chem. 289 (4), 2195–2204. doi:10.1074/jbc.m113.492587
Parkinson, J. (2002). An Essay on the Shaking Palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 14 (2), 223–222. doi:10.1176/jnp.14.2.223
Parnetti, L., Castrioto, A., Chiasserini, D., Persichetti, E., Tambasco, N., El-Agnaf, O., et al. (2013). Cerebrospinal Fluid Biomarkers in Parkinson Disease. Nat. Rev. Neurol. 9 (3), 131–140. doi:10.1038/nrneurol.2013.10
Perry, S. W., Norman, J. P., Litzburg, A., Zhang, D., Dewhurst, S., and Gelbard, H. A. (2005). HIV-1 Transactivator of Transcription Protein Induces Mitochondrial Hyperpolarization and Synaptic Stress Leading to Apoptosis. J. Immunol. 174 (7), 4333–4344. doi:10.4049/jimmunol.174.7.4333
Picone, P., Nuzzo, D., Caruana, L., Scafidi, V., and Di Carlo, M. (2014). Mitochondrial Dysfunction: Different Routes to Alzheimer's Disease Therapy. Oxid. Med. Cell Longev. 2014, 780179. doi:10.1155/2014/780179
Priyanka, S, Wadhwa, R., Chaudhuri, R., Nag, T. C., and Seth, P. (2020). Novel Role of Mortalin in Attenuating HIV-1 Tat-Mediated Astrogliosis. J. Neuroinflammation 17 (1), 276. doi:10.1186/s12974-020-01912-3
Pujhari, S., Brustolin, M., Macias, V. M., Nissly, R. H., Nomura, M., Kuchipudi, S. V., et al. (2019). Heat Shock Protein 70 (Hsp70) Mediates Zika Virus Entry, Replication, and Egress from Host Cells. Emerg. Microbes Infect. 8 (1), 8–16. doi:10.1080/22221751.2018.1557988
Putatunda, R., Ho, W. Z., and Hu, W. (2019). HIV-1 and Compromised Adult Neurogenesis: Emerging Evidence for a New Paradigm of HAND Persistence. AIDS Rev. 21 (1), 11–22. doi:10.24875/AIDSRev.19000003
Qu, M., Zhou, Z., Chen, C., Li, M., Pei, L., Yang, J., et al. (2012). Inhibition of Mitochondrial Permeability Transition Pore Opening is Involved in the Protective Effects of Mortalin Overexpression against Beta-Amyloid-Induced Apoptosis in SH-SY5Y Cells. Neurosci. Res. 72 (1), 94–102. doi:10.1016/j.neures.2011.09.009
Qu, M., Zhou, Z., Xu, S., Chen, C., Yu, Z., and Wang, D. (2011). Mortalin Overexpression Attenuates Beta-Amyloid-Induced Neurotoxicity in SH-SY5Y Cells. Brain Res. 1368, 336–345. doi:10.1016/j.brainres.2010.10.068
Ran, Q., Wadhwa, R., Kawai, R., Kaul, S. C., Sifers, R. N., Bick, R. J., et al. (2000). Extramitochondrial Localization of mortalin/mthsp70/PBP74/GRP75. Biochem. Biophysical Res. Commun. 275 (1), 174–179. doi:10.1006/bbrc.2000.3237
Repici, M., and Giorgini, F. (2019). DJ-1 in Parkinson's Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 8 (9). doi:10.3390/jcm8091377
Rivera, J., Isidro, R. A., Loucil-Alicea, R. Y., Cruz, M. L., Appleyard, C. B., Isidro, A. A., et al. (2019). Infusion of HIV-1 Nef-Expressing Astrocytes into the Rat hippocampus Induces Enteropathy and Interstitial Pneumonitis and Increases Blood-Brain-Barrier Permeability. PLoS One 14 (11), e0225760. doi:10.1371/journal.pone.0225760
Ruan, L., Zhou, C., Jin, E., Kucharavy, A., Zhang, Y., Wen, Z., et al. (2017). Cytosolic Proteostasis through Importing of Misfolded Proteins into Mitochondria. Nature 543 (7645), 443–446. doi:10.1038/nature21695
Sami Saribas, A., Cicalese, S., Ahooyi, T. M., Khalili, K., Amini, S., and Sariyer, I. K. (2017). HIV-1 Nef Is Released in Extracellular Vesicles Derived from Astrocytes: Evidence for Nef-Mediated Neurotoxicity. Cell Death Dis. 8 (1), e2542. doi:10.1038/cddis.2016.467
San Gil, R., Ooi, L., Yerbury, J. J., and Ecroyd, H. (2017). The Heat Shock Response in Neurons and Astroglia and its Role in Neurodegenerative Diseases. Mol. Neurodegener. 12 (1), 65. doi:10.1186/s13024-017-0208-6
Savitt, J. M., Dawson, V. L., and Dawson, T. M. (2006). Diagnosis and Treatment of Parkinson Disease: Molecules to Medicine. J. Clin. Investigation 116 (7), 1744–1754. doi:10.1172/jci29178
Sayre, L. M., Zelasko, D. A., Harris, P. L., Perry, G., Salomon, R. G., and Smith, M. A. (1997). 4-Hydroxynonenal-derived Advanced Lipid Peroxidation End Products are Increased in Alzheimer's Disease. J. Neurochem. 68 (5), 2092–2097. doi:10.1046/j.1471-4159.1997.68052092.x
Schapira, A. H. V., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (1989). Mitochondrial Complex I Deficiency in Parkinson's Disease. Lancet 333 (8649), 1269. doi:10.1016/s0140-6736(89)92366-0
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol. 18 (6), 345–360. doi:10.1038/nrm.2017.20
Shelton, M. N., Huang, M.-B., Ali, S. A., Powell, M. D., and Bond, V. C. (2012). Secretion Modification Region-Derived Peptide Disrupts HIV-1 Nef's Interaction with Mortalin and Blocks Virus and Nef Exosome Release. J. Virol. 86 (1), 406–419. doi:10.1128/jvi.05720-11
Singh, A. P., Bajaj, T., Gupta, D., Singh, S. B., Chakrawarty, A., Goyal, V., et al. (2018). Serum Mortalin Correlated with α-Synuclein as Serum Markers in Parkinson's Disease: A Pilot Study. Neuromol Med. 20 (1), 83–89. doi:10.1007/s12017-017-8475-5
Siracusa, R., Fusco, R., and Cuzzocrea, S. (2019). Astrocytes: Role and Functions in Brain Pathologies. Front. Pharmacol. 10, 1114. doi:10.3389/fphar.2019.01114
Smith, M. A., Richey Harris, P. L., Sayre, L. M., Beckman, J. S., and Perry, G. (1997). Widespread Peroxynitrite-Mediated Damage in Alzheimer's Disease. J. Neurosci. 17 (8), 2653–2657. doi:10.1523/jneurosci.17-08-02653.1997
Song, J., Park, K., Lee, W., and Lee, J. (2014). Apoptosis Signal Regulating Kinase 1 (ASK1): Potential as a Therapeutic Target for Alzheimer's Disease. Ijms 15 (2), 2119–2129. doi:10.3390/ijms15022119
Starenki, D., Sosonkina, N., Hong, S. K., Lloyd, R. V., and Park, J. I. (2019). Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int. J. Mol. Sci. 20 (9), 2069. doi:10.3390/ijms20092069
Sweeney, P., Park, H., Baumann, M., Dunlop, J., Frydman, J., Kopito, R., et al. (2017). Protein Misfolding in Neurodegenerative Diseases: Implications and Strategies. Transl. Neurodegener. 6, 6. doi:10.1186/s40035-017-0077-5
Taha, E. A., Ono, K., and Eguchi, T. (2019). Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 20 (18), 4588. doi:10.3390/ijms20184588
Tai-Nagara, I., Matsuoka, S., Ariga, H., and Suda, T. (2014). Mortalin and DJ-1 Coordinately Regulate Hematopoietic Stem Cell Function through the Control of Oxidative Stress. Blood 123 (1), 41–50. doi:10.1182/blood-2013-06-508333
Tang, X., Lu, H., and Ramratnam, B. (2020). Neurotoxicity of HIV-1 Tat is Attributed to its Penetrating Property. Sci. Rep. 10 (1), 14002. doi:10.1038/s41598-020-70950-x
Taoufik, E., Kouroupi, G., Zygogianni, O., and Matsas, R. (2018). Synaptic Dysfunction in Neurodegenerative and Neurodevelopmental Diseases: An Overview of Induced Pluripotent Stem-Cell-Based Disease Models. Open Biol. 8 (9), 180138. doi:10.1098/rsob.180138
Teodorof-Diedrich, C., and Spector, S. A. (2020). Human Immunodeficiency Virus Type 1 and Methamphetamine-Mediated Mitochondrial Damage and Neuronal Degeneration in Human Neurons. J. Virol. 94 (20), e00924-20. doi:10.1128/JVI.00924-20
Teodorof-Diedrich, C., and Spector, S. A. (2018). Human Immunodeficiency Virus Type 1 Gp120 and Tat Induce Mitochondrial Fragmentation and Incomplete Mitophagy in Human Neurons. J. Virol. 92 (22), e00993. doi:10.1128/JVI.00993-18
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical Basis of Cognitive Alterations in Alzheimer's Disease: Synapse Loss is the Major Correlate of Cognitive Impairment. Ann. Neurol. 30 (4), 572–580. doi:10.1002/ana.410300410
Valcour, V., Chalermchai, T., Sailasuta, N., Marovich, M., Lerdlum, S., Suttichom, D., et al. (2012). Central Nervous System Viral Invasion and Inflammation during Acute HIV Infection. J. Infect. Dis. 206 (2), 275–282. doi:10.1093/infdis/jis326
van Marle, G., Henry, S., Todoruk, T., Sullivan, A., Silva, C., Rourke, S. B., et al. (2004). Human Immunodeficiency Virus Type 1 Nef Protein Mediates Neural Cell Death: A Neurotoxic Role for IP-10. Virology 329 (2), 302–318. doi:10.1016/j.virol.2004.08.024
Voloboueva, L. A., Duan, M., Ouyang, Y., Emery, J. F., Stoy, C., and Giffard, R. G. (2008). Overexpression of Mitochondrial Hsp70/Hsp75 Protects Astrocytes against Ischemic Injury in Vitro. J. Cereb. Blood Flow. Metab. 28 (5), 1009–1016. doi:10.1038/sj.jcbfm.9600600
Voos, W., and Röttgers, K. (2002). Molecular Chaperones as Essential Mediators of Mitochondrial Biogenesis. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1592 (1), 51–62. doi:10.1016/s0167-4889(02)00264-1
Wadhwa, R., Kaul, S. C., and Mitsui, Y. (1994). Cellular Mortality to Immortalization: Mortalin. Cell Struct. Funct. 19 (1), 1–10. doi:10.1247/csf.19.1
Wadhwa, R., Takano, S., Kaur, K., Deocaris, C. C., Pereira-Smith, O. M., Reddel, R. R., et al. (2006). Upregulation of mortalin/mthsp70/Grp75 Contributes to Human Carcinogenesis. Int. J. Cancer 118 (12), 2973–2980. doi:10.1002/ijc.21773
Wadhwa, R., Takano, S., Taira, K., and Kaul, S. C. (2004). Reduction in Mortalin Level by its Antisense Expression Causes Senescence-like Growth Arrest in Human Immortalized Cells. J. Gene Med. 6 (4), 439–444. doi:10.1002/jgm.530
Walsh, D., Li, Z., Wu, Y., and Nagata, K. (1997). Heat Shock and the Role of the HSPs during Neural Plate Induction in Early Mammalian CNS and Brain Development. CMLS, Cell. Mol. life. Sci. 53 (2), 198–211. doi:10.1007/pl00000592
Wan, Q., Song, D., Li, H., and He, M.-l. (2020). Stress Proteins: the Biological Functions in Virus Infection, Present and Challenges for Target-Based Antiviral Drug Development. Sig Transduct. Target Ther. 5 (1), 125. doi:10.1038/s41392-020-00233-4
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria Dysfunction in the Pathogenesis of Alzheimer's Disease: Recent Advances. Mol. Neurodegener. 15 (1), 30. doi:10.1186/s13024-020-00376-6
Wang, X., Zuo, X., Kucejova, B., and Chen, X. J. (2008). Reduced Cytosolic Protein Synthesis Suppresses Mitochondrial Degeneration. Nat. Cell Biol. 10 (9), 1090–1097. doi:10.1038/ncb1769
Wang, Y., Liu, M., Lu, Q., Farrell, M., Lappin, J. M., Shi, J., et al. (2020). Global Prevalence and Burden of HIV-Associated Neurocognitive Disorder. Neurology 95 (19), e2610–e2621. doi:10.1212/wnl.0000000000010752
Wang, Y., Santerre, M., Tempera, I., Martin, K., Mukerjee, R., and Sawaya, B. E. (2017). HIV-1 Vpr Disrupts Mitochondria Axonal Transport and Accelerates Neuronal Aging. Neuropharmacology 117, 364–375. doi:10.1016/j.neuropharm.2017.02.008
Wildburger, N. C., Gyngard, F., Guillermier, C., Patterson, B. W., Elbert, D., Mawuenyega, K. G., et al. (2018). Amyloid-β Plaques in Clinical Alzheimer's Disease Brain Incorporate Stable Isotope Tracer in Vivo and Exhibit Nanoscale Heterogeneity. Front. Neurol. 9, 169. doi:10.3389/fneur.2018.00169
Wisniewski, K. E., Maslinska, D., Kitaguchi, T., Kim, K. S., Goebel, H. H., and Haltia, M. (1990). Topographic Heterogeneity of Amyloid B-Protein Epitopes in Brains with Various Forms of Neuronal Ceroid Lipofuscinoses Suggesting Defective Processing of Amyloid Precursor Protein. Acta Neuropathol. 80 (1), 26–34. doi:10.1007/bf00294218
Xie, H., Guan, J., Borrelli, L. A., Xu, J., Serrano-Pozo, A., and Bacskai, B. J. (2013). Mitochondrial Alterations Near Amyloid Plaques in an Alzheimer's Disease Mouse Model. J. Neurosci. 33 (43), 17042–17051. doi:10.1523/jneurosci.1836-13.2013
Xu, L., Voloboueva, L. A., Ouyang, Y., and Giffard, R. G. (2009). Overexpression of Mitochondrial Hsp70/Hsp75 in Rat Brain Protects Mitochondria, Reduces Oxidative Stress, and Protects from Focal Ischemia. J. Cereb. Blood Flow. Metab. 29 (2), 365–374. doi:10.1038/jcbfm.2008.125
Yang, H., Zhou, X., Liu, X., Yang, L., Chen, Q., Zhao, D., et al. (2011). Mitochondrial Dysfunction Induced by Knockdown of Mortalin is Rescued by Parkin. Biochem. Biophysical Res. Commun. 410 (1), 114–120. doi:10.1016/j.bbrc.2011.05.116
Zhou, B. Y., Liu, Y., oh Kim, B., Xiao, Y., and He, J. (2004). Astrocyte Activation and Dysfunction and Neuron Death by HIV-1 Tat Expression in Astrocytes. Mol. Cell. Neurosci. 27 (3), 296–305. doi:10.1016/j.mcn.2004.07.003
Zhu, J.-Y., Vereshchagina, N., Sreekumar, V., Burbulla, L. F., Costa, A. C., Daub, K. J., et al. (2013). Knockdown of Hsc70-5/mortalin Induces Loss of Synaptic Mitochondria in a Drosophila Parkinson's Disease Model. PLoS One 8 (12), e83714. doi:10.1371/journal.pone.0083714
Keywords: mortalin, HIV-human immunodeficiency virus, Alzheimer’s disease, viral infection, Parkinson’s disease
Citation: Priyanka and Seth P (2022) Insights Into the Role of Mortalin in Alzheimer’s Disease, Parkinson’s Disease, and HIV-1-Associated Neurocognitive Disorders. Front. Cell Dev. Biol. 10:903031. doi: 10.3389/fcell.2022.903031
Received: 23 March 2022; Accepted: 06 May 2022;
Published: 04 July 2022.
Edited by:
Renu Wadhwa, National Institute of Advanced Industrial Science and Technology (AIST), JapanReviewed by:
Sidharth Mehan, Indo-Soviet Friendship College of Pharmacy, IndiaChae-Ok Yun, Hanyang University, South Korea
Sunil Kaul, National Institute of Advanced Industrial Science and Technology (AIST), Japan
Copyright © 2022 Priyanka and Seth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Priyanka, a2h1c2hpLm11a3RlZUBnbWFpbC5jb20=