
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 16 March 2022
Sec. Stem Cell Research
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.851613
This article is part of the Research TopicCell-Based Neurodegenerative Disease ModelingView all 6 articles
 Ting-Jung Lin1,2†
Ting-Jung Lin1,2† Kuang-Chao Cheng1,2†
Kuang-Chao Cheng1,2† Luo-Yun Wu1,2†
Luo-Yun Wu1,2† Wei-Yu Lai2,3
Wei-Yu Lai2,3 Thai-Yen Ling4,5
Thai-Yen Ling4,5 Yung-Che Kuo3*
Yung-Che Kuo3* Yen-Hua Huang2,3,6,7,8,9,10,11*
Yen-Hua Huang2,3,6,7,8,9,10,11*Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive upper and lower motor neuron (MN) degeneration with unclear pathology. The worldwide prevalence of ALS is approximately 4.42 per 100,000 populations, and death occurs within 3–5 years after diagnosis. However, no effective therapeutic modality for ALS is currently available. In recent years, cellular therapy has shown considerable therapeutic potential because it exerts immunomodulatory effects and protects the MN circuit. However, the safety and efficacy of cellular therapy in ALS are still under debate. In this review, we summarize the current progress in cellular therapy for ALS. The underlying mechanism, current clinical trials, and the pros and cons of cellular therapy using different types of cell are discussed. In addition, clinical studies of mesenchymal stem cells (MSCs) in ALS are highlighted. The summarized findings of this review can facilitate the future clinical application of precision medicine using cellular therapy in ALS.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive motor neuron (MN) degeneration in the cortex and spinal cord (Brown and Al-Chalabi 2017). Typically, death occurs in 3–5 years after the diagnosis of ALS due to respiratory system dysfunction (Niedermeyer et al., 2019). The overall crude prevalence and incidence of ALS worldwide were 0.442‱ and 0.159‱, respectively (Xu et al., 2020). Compared with women, men are slightly more frequently affected by ALS.
ALS is believed to result from a combination of genetic and environmental factors (Masrori and Van Damme 2020). ALS exists in two forms: familial ALS (fALS) and sporadic ALS (sALS). fALS exhibits a Mendelian pattern of inheritance and accounts for 5–10% of all cases. The remaining 90–95% of cases that do not have an apparent genetic link are classified as sALS (Kiernan et al., 2011). At the genetic level, more than 20 genes have been identified. Among them, chromosome 9 open reading frame 72 (C9ORF72), fused in sarcoma (FUS), TAR DNA binding protein (TARDBP), and superoxide dismutase 1 (SOD1) genes have been identified as the most common causative genes (Riancho et al., 2019). Beyond genetic factors, the diverse pathological mechanisms of ALS-associated neurodegeneration have been discussed (van Es et al., 2017). The clinical symptoms of ALS are heterogeneous, with main symptoms including limb weakness, muscle atrophy, and fasciculations involving both upper and lower MNs. The onset of ALS can be usually divided into two subtypes: spinal and bulbar (Masrori and Van Damme 2020). In addition to MN symptoms, some patients present with frontotemporal dementia, which may cause cognitive and behavioral changes (Grad et al., 2017).
Currently, edaravone and riluzole are the only two drugs approved by the United States Food and Drug Administration (FDA); however, only riluzole can prolong the lifespan of patients for a short time, and edaravone can improve quality of life (Bensimon et al., 1994; Rothstein 2017). In the past few years, increasing attention has been paid to stem cell therapies for ALS because of the differentiation, neuroprotective, and immunomodulatory properties of stem cells (Oskarsson et al., 2018). Several clinical trials have examined multiple types and resources of stem cells, including pluripotent embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), multipotent mesenchymal stem cells (MSCs), neural precursor cells, and mononuclear cells (MCs). This review discusses the findings of recent studies on cellular therapies for ALS. Both the potential and challenges are discussed to provide insights into future precision cellular therapeutic strategies.
ALS is a complex disease involving multiple genetic risk factors and heterogeneous pathogenic mechanisms. Recent improvements in molecular and genetic technologies have rapidly and substantially advanced the discovery of disease-causing gene mutations and pathways. Understanding disease pathology can maximize the efficacy of treatments and enable the development of innovative therapies for ALS patients.
Familial ALS (fALS) presents with a family history of the disease, while sporadic ALS (sALS) doesn’t have a hereditary pattern. fALS can be inherited as an autosomal dominant, autosomal recessive, or dominant X-linked type. Adult-onset autosomal dominant inheritance is more common in fALS (Siddique and Ajroud-Driss 2011). Currently, more than 150 disease-causing mutations have been reported. According to a meta-analysis (Zou et al., 2017), mutations in C9ORF72 account for 33.7% of European ALS cases, followed by SOD1 at 14.8%, TARDBP at 4.2%, FUS at 2.8%. Different from their Western counterparts, the Asian population witnessed SOD1 mutations at 30% in familial cases, C9ORF72 at 2.3%, TARDBP at 4.2%, and FUS at 2.8%. On the other hand, mutations in C9ORF72, TARDBP, SOD1, and FUS account for less than 8% of sALS cases in both European and Asian populations.
The bulk of the genetic determinants of sALS remains elusive, and it is believed that exogenous environmental factors can activate endogenous retroviruses to initiate neurodegenerative changes (Brown and Al-Chalabi 2015). Studies have reported some key features of ALS pathology, such as the involvement of mutated genes encoding RNA-binding proteins including the TARDBP (Sreedharan et al., 2008), fused in sarcoma (FUS) (Vance et al., 2009), and heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) (Kim et al., 2013). Mutations lead to aberrant RNA metabolism with dysregulated mRNA splicing and transcriptional activity in MNs, thus contributing to protein aggregate formation (Highley et al., 2014; Masuda et al., 2016). A disease hallmark of sALS (up to 97%) cases is TDP-43 proteinopathy (Prasad et al., 2019). Mutations in TDP-43 lead to mislocalization of nuclear TDP-43 to cytoplasmic inclusions and disruption of the autoregulation loop controlling its phase transition (Tziortzouda et al., 2021). The GGGGCC (G4C2) hexanucleotide repeat expansion in C9ORF72 interacts with RNA-binding proteins and impairs their function (Lee K.-H. et al., 2016). In addition, multiple ALS-causing mutations, such as those in ubiquilin 2 (UBQLN2) (Nishitoh et al., 2008) involved in protein clearance pathways, impair proteostasis and exacerbate protein aggregation. To explain how symptoms spread along neuroanatomical pathways in sALS, it is demonstrated that mutant SOD1 propagate between cells in the motor nervous system in a prion-like manner using SOD1G85R mice (Ayers et al., 2016). Protein aggregates in mitochondria lead to the disruption of the electron transport chain (Choi et al., 2019), resulting in increased reactive oxygen species (ROS) production and mitochondrial dysfunction (Le Gall et al., 2020). Oxidative stress occurs when ROS production exceeds a cell’s capacity to alleviate the stress, thus damaging DNA, proteins, and lipids (Yang 2021). Glutamate excitotoxicity is another main cause of oxidative stress. Because excitatory amino acid transporter-2 functions to uptake glutamate (Lauriat and McInnes 2007), its decreased expression on astrocytes might contribute to high glutamate levels (Rothstein et al., 1995; Gibb et al., 2007), thus resulting in an over influx of Ca2+ ions in MNs. The aberrant level of Ca2+ further increases ROS production and causes neuronal degeneration (Wang and Qin 2010).
Impaired DNA repair may also contribute to ALS pathogenesis (Mejzini et al., 2019). Because DNA damage and p53 activation mediate the apoptosis of adult MNs (Martin et al., 2005), diseases associated with the nervous system can arise when DNA repair pathways are compromised. Two of the most widely studied ALS-linked proteins, TDP-43 and FUS, prevent or repair DNA damage. TDP-43 is involved in the nonhomologous end joining (NHEJ)–mediated DNA double-strand break repair pathway, whereas FUS interacts with histone deacetylase 1 to regulate DNA damage responses (Mitra et al., 2019). In addition to impaired DNA repair, the G4C2 repeat expansion in C9ORF72 might alter the intracellular localization of C9ORF72 mRNA, causing nucleocytoplasmic transport defects in ALS (Brown and Al-Chalabi 2017). Although the G4C2 of C9ORF72 is the most common genetic variant in ALS, the underlying mechanisms remain unclear (Cooper-Knock et al., 2014). RanGAP is a key regulator of nucleocytoplasmic transport. It also suppresses G4C2-mediated neurodegeneration in Drosophila (Cooper-Knock et al., 2014; Zhang et al., 2015). Furthermore, the dysregulation of axonal transport and the axonal compartment are involved in the pathophysiology of ALS (Zhang et al., 2016; Chou et al., 2018). Some pathways may be responsible for impaired axonal transport in patients with mutant SOD1 (Zarei et al., 2015). Studies have revealed that mutant SOD1 inhibits the anterograde axonal transport of mitochondria by reducing the level of mitochondrial Rho GTPase 1 (Miro1) (Moller et al., 2017). Moreover, depletions in two motor molecules, dynein and dynactin-1, can disrupt axonal transport and cause MN degeneration (Ikenaka et al., 2012).
Neuronal injury and death in ALS can be attributed to non–cell-autonomous toxicity, indicating that only gene mutations in motor neurons are insufficient to cause the disease. Activation of glial cells, including microglia and astrocytes, mainly characterizes neuroinflammation in ALS (Vahsen et al., 2021). Two phenotypes of microglia have been reported: M1 and M2; M1 is neurotoxic and produces proinflammatory cytokines and ROS, whereas M2 is neuroprotective and secretes neurotrophic molecules and anti-inflammatory cytokines (Brites and Vaz 2014). The neuroprotective phenotype of microglia is observed in the early stage of ALS, and it switches to the neurotoxic phenotype as the disease progresses, causing neuroinflammation. Recently, this dichotomized classification has been challenged, and the microglia phenotype is considered to be a spectrum in reality (Ransohoff 2016). Similar to microglia, two reactive astrocyte types have been documented: A1 neurotoxic type and A2 neuroprotective type. A2 can promote healing after injury, whereas A1 can be deleterious by reducing the expression of the glutamate transporter GLT-1 (Papadeas et al., 2011), impairing the astrocyte lactate efflux transporter, and activating pro-nerve growth factor-p75 receptor signaling (Ferraiuolo et al., 2011). Recently, the inflammatory niche has been reported to activate microglia to secrete TNFα, IL-1α, and C1q, resulting in the conversion of astrocytes from A2 into A1 (Guttenplan et al., 2020). The neuronal microenvironment is determined by cytokines, which are secreted by surrounding glial cells such as microglia and astrocytes, and it plays a pivotal role in neuroinflammation, neurodegeneration, disease onset, and progression. Current cellular therapies mainly aim to replace damaged MNs or influence the diseased microenvironment through neurotrophic factors produced by differentiated astrocytes and microglia.
From a single initiating event to multiple factors, causative pathogenic mechanisms are yet to have integrated interpretations. Because of genetic and phenotypic variations, additional studies should be performed to elucidate and draw conclusions regarding the pathogenic mechanisms of ALS.
Because no effective cure is available for ALS and some patients have slow disease progression, symptomatic support is crucial for minimizing mortality and improving patients’ quality of life in the disease course (Hobson and McDermott 2016). 75% of patients experience the onset of limb weakness and require the initial focus of care, such as physical therapy and assistive aids, to relieve physical symptoms. Moreover, 25% of patients develop bulbar dysfunction, commonly with dysarthria, dysphagia, weakness, or twitching in the muscles of the tongue (Hobson and McDermott 2016; Institute 2021; Masrori and Van Damme 2020). Therefore, early speech therapy and optimization of communication and nutrition should be provided for these patients (Hobson and McDermott 2016). To alleviate specific symptoms, cannabinoids, tizanidine, baclofen, and muscle stretching can treat spasticity; botulinum toxin injects into the salivary gland or anticholinergic medications can treat sialorrhea. Administration with magnesium supplements, quinine sulfate, gabapentin, or carbamazepine may release muscle cramps. Patients with emotional lability can consider medicating selective serotonin reuptake inhibitors, benzodiazepines, amitriptyline, and dextromethorphan hydrobromide/quinidine sulfate (Masrori and Van Damme 2020). In the past half-century, more than 50 randomized controlled trials of proposed disease-modifying drugs have failed to demonstrate positive results, highlighting our poor knowledge regarding the primary mechanisms of ALS (Petrov et al., 2017; Writing-Group and Edaravone-(MCI-186)-ALS-19-Study-Group 2017). Although numerous candidate drugs for ALS have been identified experimentally, they have failed to show efficacy in human clinical trials, except for riluzole and edaravone (Saitoh and Takahashi 2020) (Figure 1). Riluzole, the first drug for ALS treatment, exhibits neuroprotective activity that is possibly related to the presynaptic inhibition of glutamate damage in the central nervous system. A dose-ranging study reported that treatment with riluzole prolonged stage 4 (King’s clinical staging) not by prolonged stage 2 or 3 or by slowed the disease progression in patients with ALS (Fang et al., 2018). Edaravone (also known as MCI-186), a free radical scavenger, could eliminate peroxynitrite and lipid peroxyl radicals as well as protect MNs from damage caused by free radicals and oxidative stress (Okada et al., 2018; Park et al., 2020; Shefner et al., 2020). However, in the first placebo-controlled phase III study of edaravone, no significant difference in revised ALS Functional Rating Scale (ALSFRS-R) scores was observed between patients receiving edaravone and those receiving placebo. Therefore, whether edaravone is safe and effective in a broader population of patients with advanced ALS stages would require further study (Petrov et al., 2017).
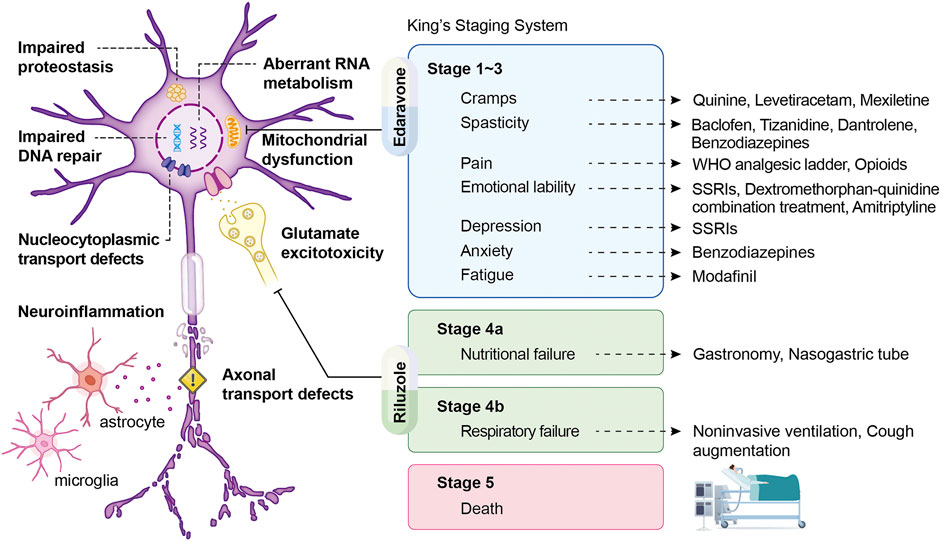
FIGURE 1. Mechanisms of ALS following treatment to their corresponding symptoms according to King’s staging system. Schematic displaying pathology, symptoms, and therapeutic strategies of ALS, with King’s staging system being presented as the illustration of disease progression. The arrow derives from each symptom points to its therapeutic counterparts. In addition, two FDA-approved drugs, edaravone and riluzole, show considerable benefits in stage 1–3 and stage 4 and are aiming at mitochondrial dysfunction and glutamate excitotoxicity, respectively.
Recently, gene therapy arose as a promising treatment option for ALS patients. Gene therapy can be used to replace the mutated gene with the normal copy, knockout targeting RNA to reduce the expression of causative gene, utilize gene addition to introduce a protective or beneficial factor, or edit the mutant genome (Cappella et al., 2019). Gene therapy delivers genetic material to cells for introducing the functional copies of dysfunctional genes, trophic factors, and other disease-modifying genes, using antisense oligonucleotides (ASOs), RNA interference (RNAi), and gene-editing technology to silence the expression of harmful genes (Amado and Davidson 2021). An ALS-specific challenge must reach both the motor cortex and spinal cord. In order to administrate the material across the blood-brain barrier (BBB) to the motor cortex and spinal cord, intravenous, intracerebroventricular (ICV), intrathecal (IT), intraparenchymal, or intranasal are proved to use.
To target the central nervous system, materials can be delivered naked by using antisense oligonucleotides (ASOs), viral vectors such as an adeno-associated virus, and physical or chemical systems including nanoparticles. Among these, ASOs targeting SOD1 mRNA prolonged survival and improved motor performance in ALS rodent models with SOD1 mutations (Thomsen et al., 2014; McCampbell et al., 2018). Tofersen (BIIB067) is an ASOs targeting SOD1 mRNA by RNase H-dependent degradation (Smith et al., 2006; McCampbell et al., 2018; Rinaldi and Wood 2018) to reduce SOD1 protein concentrations in patients with ALS (Miller et al., 2020). A phase I clinical study revealed that intrathecal delivery of an ASOs targeting SOD1 is safe and no serious adverse events occurred (Miller et al., 2013); another phase I/II clinical study, which is a multiple ascending dose trial, revealed that SOD1 levels are reduced in the cerebrospinal fluid in the patients who received the highest dose of tofersen, and some patients show improvements in clinical function and muscle strength (Miller et al., 2020).
For viral vectors delivery, there is a theoretical advantage that a single treatment can sustain the expression of genetic material in target cells. The adeno-associated viruses have emerged as the leading vectors for CNS delivery due to their ability to transduce terminally differentiated cells and establish nuclear episomes without posing the risk of insertional mutagenesis. Most recently, two patients have intrathecally treated with adeno-associated virus containing an anti-SOD1 microRNA (AAV-miR-SOD1) in an investigator-initiated study. One of the patients shows apparent preservation of strength in his right leg; another patient shows stable scores on a composite measure of ALS function and a stable vital capacity (Mueller et al., 2020). Therefore, the field of gene therapy has the potential for treating MN diseases such as ALS and spinal muscular atrophy. However, the vector should be carefully selected because different viral vectors have pros and cons while delivering the therapeutic gene to the affected region or tissue (O'Connor and Boulis 2012).
Currently, no effective therapeutic options for ALS that can substantially mitigate or even reverse the disease phenotype are available. This may be attributed to our poor knowledge regarding the pathophysiology and variability of ALS (Mitsumoto et al., 2014). Recently, cellular therapy has been the new hope for ALS treatment by targeting multiple pathogenic mechanisms, mainly neuroinflammation (Figure 2). Updated advances, therapeutic potential, and challenges in cellular therapy for ALS are discussed in the following subsections (Tables 1–3).
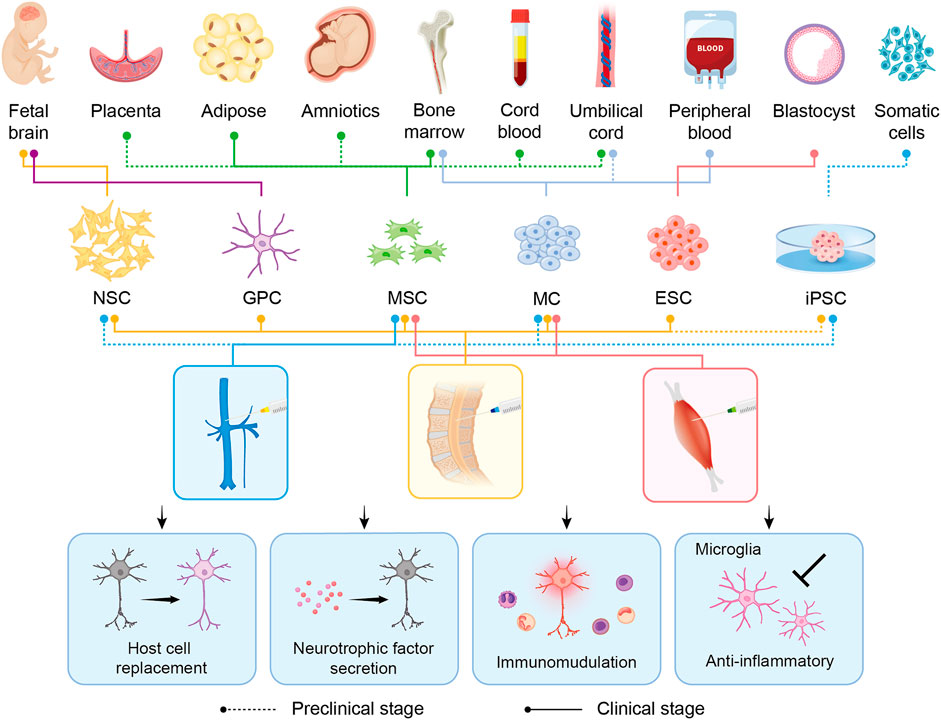
FIGURE 2. Overview of the derivations, intervention methods, and therapeutic benefits of stem cell transplantation in preclinical/clinical studies related to ALS. Six major candidate cell types (NSC, GPC, MSC, MC, ESC, and iPSC) are represented in the schematic. Different derivations and intervention methods of each cell type are indicated by lines of different colors and types. The type of line, dotted or solid, shows the hierarchy, representing the application in preclinical studies alone or in the clinical stage already. In the middle panel, blue, yellow, and red represent intravenous, intrathecal, and intramuscular injection, respectively. The transplanted cells exert their therapeutic benefits through host cell replacement, neurotrophic factor secretion, immunomodulation, and anti-inflammatory. NSC: neural stem cell; GPC: glial progenitor cell; MSC: mesenchymal stem cell; MC: mononuclear cell; ESC: embryonic stem cell; iPSC: induced-pluripotent stem cell.
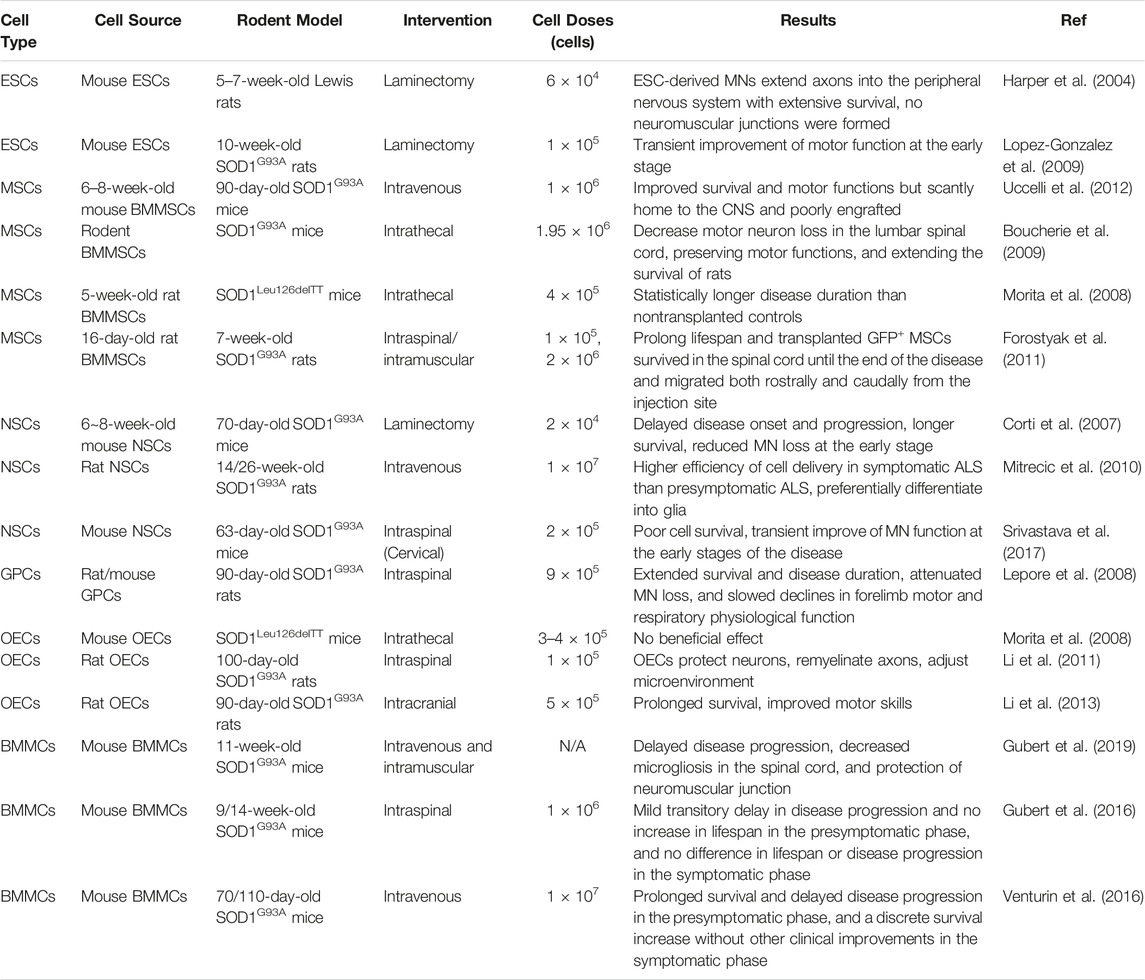
TABLE 1. Preclinical studies of nonhuman cells in rodent models.
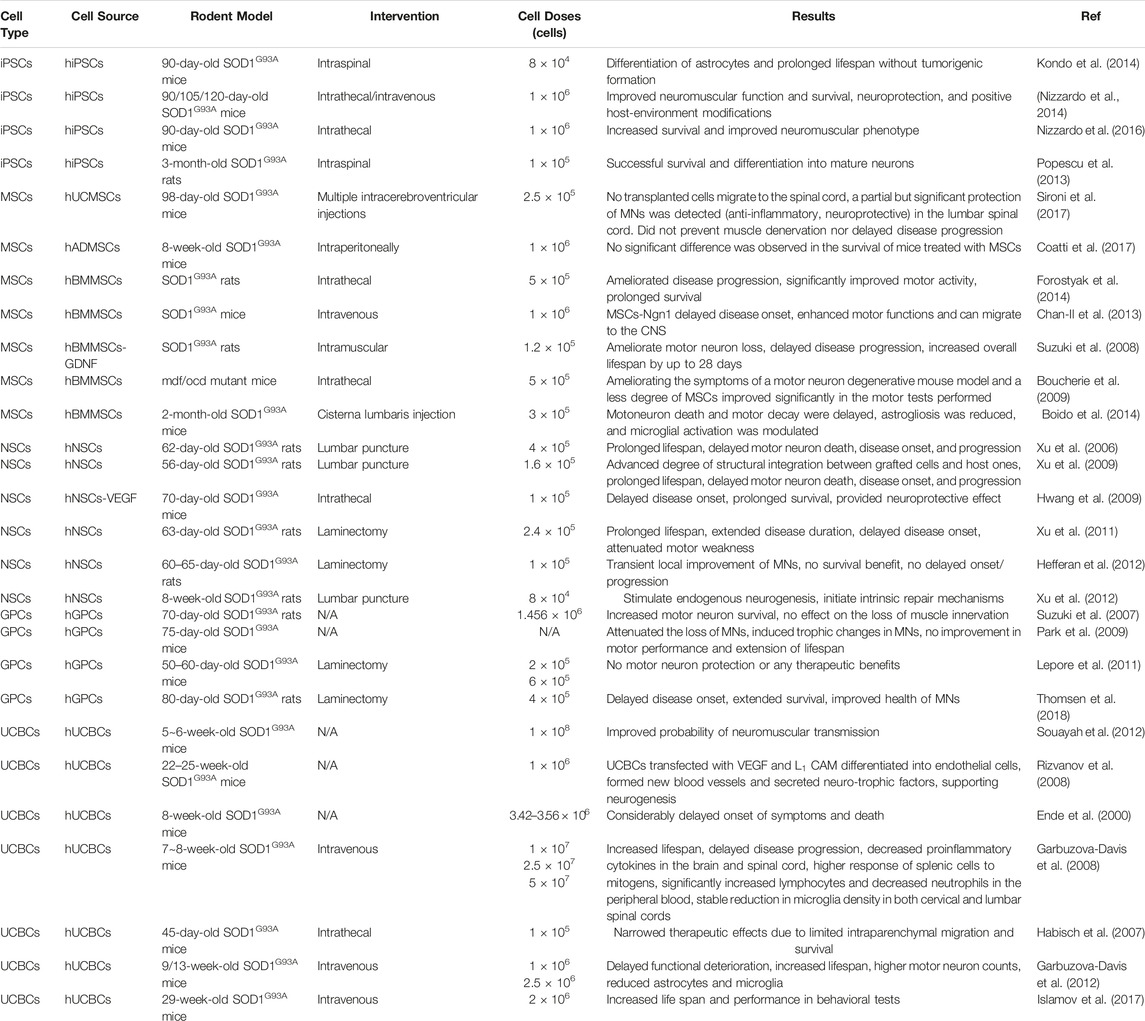
TABLE 2. Preclinical studies of human cells in rodent models.
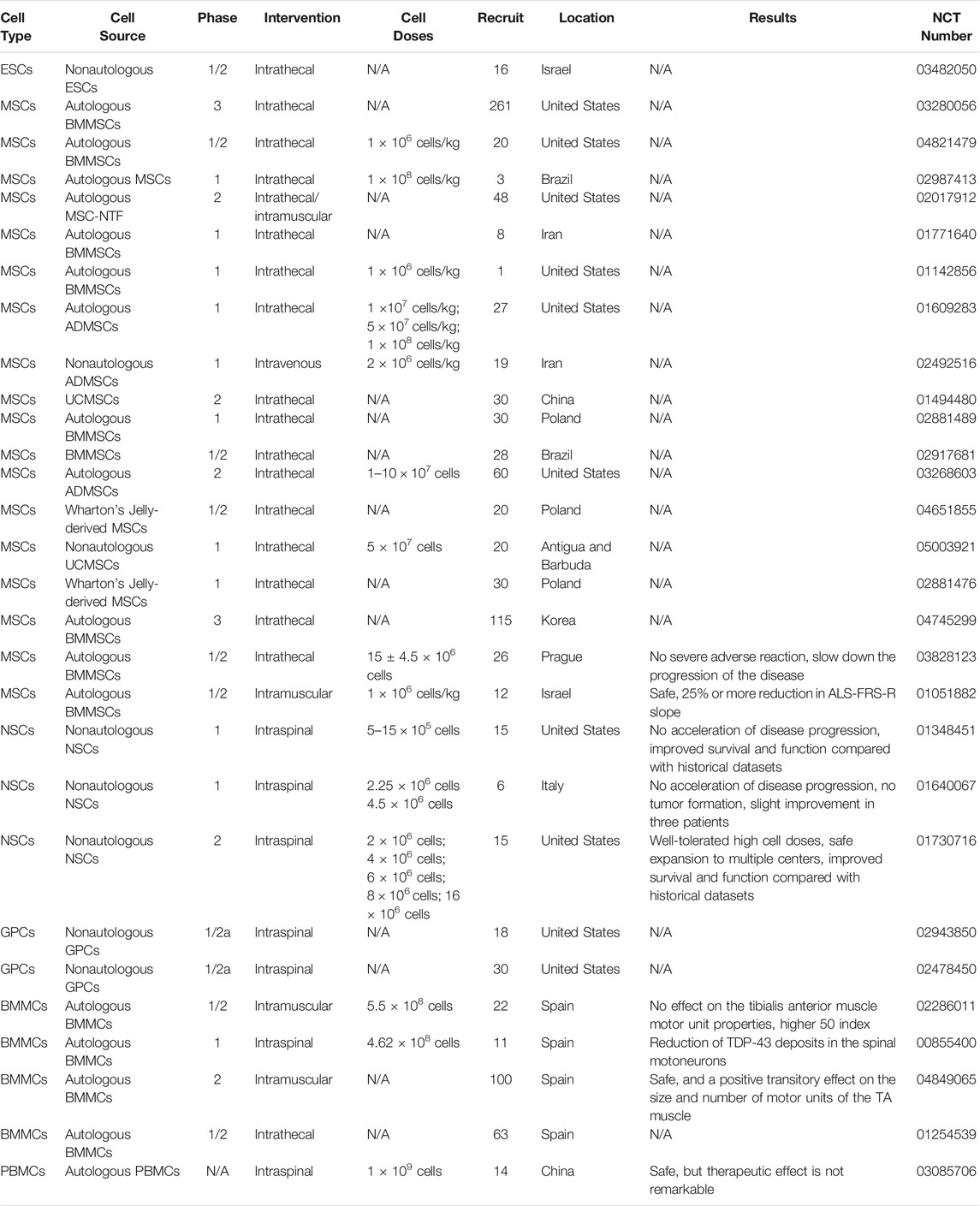
TABLE 3. Clinical trials of cellular therapy in ALS patients.
ESCs are derived from the inner cell mass of a developing blastocyst and possess the ability of self-renewal and pluripotency; in other words, they can develop into any cell type in the human body indefinitely (Young 2011). However, such a powerful capability is a double-edged sword because plasticity not only permits ESCs to be employed in several scientific domains such as human developmental biology, drug discovery, and transplantation therapy but also makes them difficult to control (Thomson et al., 1998; Murry and Keller 2008). Over the last 3 decades, scientists have been decoding the developmental principles of ESCs, in order to convert them into functionally specialized cells. The translation of tuning cell fate from prospect to reality was eventually achieved, thus indicating that ESCs could be artificially induced to differentiate into the ectoderm and further generate specific neurons and glial cells (Wichterle et al., 2002; Jiang et al., 2012). Hence, ESCs have been widely utilized to investigate the mechanisms of ALS by modeling the disease in vitro (Clement et al., 2003; Marchetto et al., 2008). Moreover, ESCs can be used in potential therapeutic molecule screening as an approach for testing its efficacy (Yang et al., 2013). Targeting the replacement of MNs or the neuroprotective functions of glial cells, ESCs are considered to be a potential stem cell resource for ALS treatment.
Studies have reported the possible therapeutic benefits of ESCs. A preclinical study indicated that ESC-derived MNs could be induced to extend axons in the peripheral nervous system but without neuromuscular junctions (Harper et al., 2004). Furthermore, López-González et al. determined that engrafted ESC–derived MNs in SOD1G93A rodent models resulted in the transient recovery of motor function in the early stage of ALS. The potential mechanism involves the trophic support provided by transplanted cells for deteriorating endogenous MNs (Lopez-Gonzalez et al., 2009). In addition to ESC-derived MNs, ESC-derived astrocytes were studied. A phase I/IIa trial recruited 16 patients with ALS to receive human ESC-derived astrocyte treatment (NCT03482050, 2018), aiming for the functional compensation of host astrocytes.
Although ESCs can be useful in cellular therapy, ethical issues and safety concerns have been reported. Ethical problems arise from the destruction of embryos, thus leading to the debate of when human life begins (Lo and Parham 2009). In terms of safety, the tumorigenicity of ESCs is the main issue. Because ESCs share cellular and molecular phenotypes with tumor cell lines, they form teratomas after transplantation (Ben-David and Benvenisty 2011). These uncontrollable uncertainties harbor risks, and ethical controversies must be appropriately addressed. ESCs cannot be used in clinical practice until the aforementioned difficulties are overcome in the future.
Induced Pluripotent Stem Cells have considerably altered the landscape of stem cell research (Malik and Rao 2013). They present ESC-like properties, including unlimited expansion and the capacity to differentiate into three germ layers (Takahashi and Yamanaka 2006). Compared to ESCs, the circumvention of ethical dilemmas and graft rejection strongly drives the potential of iPSC therapy to be a promising approach (Goutman et al., 2019). ALS-related iPSCs were first generated using skin biopsies and were successfully differentiated into MNs (Dimos et al., 2008). In addition, iPSCs from patients with mutated SOD1 (Chen et al., 2014), C9ORF72 (Donnelly et al., 2013), TARDBP (Bilican et al., 2012), FUS (Guo et al., 2017), VABP (Mitne-Neto et al., 2011), and sporadic ALS (Fujimori et al., 2018) have been reported. iPSC-derived MNs or astrocytes present the disease pathology and can thus be used as a disease cell model for studying the pathogenic mechanisms.
The combination of iPSCs and CRISPR/Cas9 facilitates the investigation of disease-causing mutations (Yun and Ha 2020). Because of the high efficiency of disease modeling, iPSC-based drug screening is well developed (Egawa et al., 2012). Among the pharmacological candidates, ropinirole (Fujimori et al., 2018), bosutinib (Imamura et al., 2017), and ezogabine (USAN)/retigabine (INN) (Wainger et al., 2014) are considered potentially beneficial for ALS patients. Compounds such as HDAC6 inhibitors (Guo et al., 2017) and apilimod (Shi et al., 2018) have also been screened by the iPSC platform. In addition, iPSCs have considerable potential for use in the autologous cellular transplantation strategy for ALS. Their ability to differentiate into neural precursor cells provides therapeutic benefits, including the replacement of endogenous cells and the modulation of the host environment in neurodegenerative diseases (Xiao et al., 2015).
Several preclinical studies using skin fibroblast-derived iPSCs have reported the therapeutic potential of iPSCs. A study transplanted human iPSC (hiPSC)-derived neural stem cells (NSCs) into the spinal cord and found that hiPSC-NSCs could survive and differentiate into neurons in the ALS microenvironment (Popescu et al., 2013). In addition, both IT and intravenous injections of hiPSC-NSCs could ameliorate the ALS disease phenotype and increase the lifespan of rodent models, with no side effects (Nizzardo et al., 2014). Novel axonal and collateral sprouting could be induced after engraftment (Nizzardo et al., 2016). Besides, hiPSC-derived glial progenitor cells (GPCs) could induce astrocyte proliferation, prolong the lifespan, and improve lower limb function transiently after being transplanted into the lumbar spinal cord of SOD1G93A mice. Furthermore, AKT-VEGF signaling may be involved in the underlying mechanism of therapeutic effects (Kondo et al., 2014).
Although some preclinical studies have examined the efficacy of iPSCs for ALS treatment, no clinical trial is yet approved. Studies have described some concerns. The first concern is the tumorigenicity of iPSCs. Three tumorigenic scenarios are observed: the formation of teratomas, the active state of reprogramming factors, and genetic abnormalities during in vitro culture. In addition, the heterogeneity of iPSCs contributes to defects in the differentiation of some cell lines, thus resulting in the formation of teratomas (Yamanaka 2020). Second, deriving autologous iPSCs from each individual is expensive and requires a long cell cultivation period. Moreover, allograft approaches may cause immune rejection (Takahashi and Yamanaka 2013; Yamanaka 2020). Despite the existing challenges, iPSC technology holds great potential in many domains related to ALS cellular therapy. For instance, iPSC models can enhance the investigation of disease progression, thus enabling the early diagnosis of ALS; personalized medical strategies for patients with sporadic ALS might be developed through high-throughput screening (Coatti et al., 2015). Although more studies should be performed to determine the feasibility of iPSC engraftment, the application of iPSCs already offers a new path for personalizing ALS cellular therapy.
Nowadays, MSCs are considered a promising tool for ALS cellular therapy due to the following reasons: 1) MSCs can be effectively and safely obtained from somatic tissues and can be easily expanded in vitro culture systems; 2) MSCs can migrate to injury tissue sites, a property called “homing” that is mediated by chemokines; 3) MSCs can release various biological factors to facilitate nervous tissue maintenance and repair; 4) MSCs may develop neuronal phenotypes under appropriate conditions; 5) MSCs have lower ethical concerns than ESCs or fetal-derived stem cells; 6) autologous MSCs can be safely administered without the risk of immune rejection; and 7) MSCs can secrete exosomes that modulate the microenvironment (Karussis et al., 2010; Baglio et al., 2012; Zhou et al., 2021).
Mesenchymal stem cells can be isolated from different somatic tissues including the peripheral blood, cord blood, umbilical cord (Wharton’s jelly), placenta, amniotic fluid, fetal tissues, adipose tissue, dental pulp, skeletal muscle, periosteum, lung, synovial fluid, and cartilage (Wang et al., 2004; Kusuma et al., 2017; Zhuang et al., 2021). Unlike other stem cells, MSC targets could change the cellular environment of vulnerable brain areas when used for treating neurodegenerative diseases in preclinical trials (Chen et al., 2018). The potential therapeutic effects of MSCs on ALS may involve rich trophic factor secretion; enhanced neurogenesis; abnormal protein aggregate clearance; immunomodulation by the increased expression of interleukin (IL)-10 and IL-6, vascular endothelial growth factor (VEGF), and transforming growth factor beta-1 (TGF-β1); and gene delivery or replacement of lost cells (Lewis and Suzuki 2014; Nabavi et al., 2019). MSCs exert neuroprotective effects on glutamate excitotoxicity by inhibiting the expression of the N-methyl-D-aspartate receptor (NMDAR) and by controlling glutamate-related Ca2+ influx (Voulgari-Kokota et al., 2012). Thus, the therapeutic plasticity of MSCs matches the complexity of ALS, making MSCs a strong candidate for ALS treatment.
Mesenchymal stem cells regulate both adaptive and innate immune reactions through paracrine such as TGF-β secretion. An increase in regulatory T cells and Th2 cells play crucial roles in the neuroprotective effect on motor neuronal cell death mechanisms in ALS (Kim S. H. et al., 2018). MSCs can suppress T-cell function, dendritic cell maturation, B-cell proliferation, and natural killer cell cytotoxicity; promote regulatory T cell proliferation; and trigger M1 to M2 macrophage conversion (Shariati et al., 2020). The repeated infusion of MSCs could reduce the deterioration of ALS motor function and prolong life expectancy, whereas a single infusion of MSCs could delay disease progression (Magota et al., 2021a; Magota et al., 2021b). Overexpression of the hepatocyte growth factor, glial cell line-derived neurotrophic factor (GDNF), and insulin-like growth factor in MSCs could substantially reduce the progression of motor dysfunction (Terashima et al., 2020). Intramuscular transplantation of human umbilical cord (hUC)-derived MSCs could ameliorate muscle atrophy and reduce neuromuscular degeneration in the skeletal muscle by reducing intracellular ROS levels (Kook et al., 2020). Repeated ICV injections of hUC-derived MSCs in SOD1G93A mice did not result in the migration of transplanted cells to the spinal cord or delay muscle denervation but provided protection the lumbar spinal cord with anti-inflammatory (IL-4 and IL-10) and neuroprotective (IGF-1) microenvironment instead of the pro-inflammatory (IL-6 and IL-1β) stimuli (Sironi et al., 2017). In patients with ALS, 50% of pericytes, which are responsible for the formation and maintenance of the BBB, from the spinal cord barrier are lost (Coatti et al., 2017). Disruption of the BBB including decreases in the levels of tight junction proteins, immunoglobulin G, and hemosiderin perivascular deposits and the expression of GLUT1 precedes neuroinflammation, MN loss, and motor impairment (Sweeney et al., 2019). In a preclinical study, both adipose-derived pericytes and MSCs were intraperitoneally administered to SOD1G93A mice. The results showed that pericytes significantly prolonged survival in mice, whereas MSCs showed no effect on survival (Coatti et al., 2017). In addition to the loss of the BBB, changes in the ventral horn perineuronal net structure were observed in SOD1 rats; this structure has been recently shown to protect against various toxic insults, and it is affected in neurodegenerative diseases (Cabungcal et al., 2013; Suttkus et al., 2016). Forostyak et al. (2014) demonstrated that the IT delivery of MSCs in SOD1G93A rats preserved the perineuronal net structure and altered cytokine homeostasis accompanied by the prolonged survival of MNs (Forostyak et al., 2014).
Aside from cell transplantation, one of the reasons why MSCs have been highlighted in recent years is their high capacity for secreting exosomes. Exosomes are a kind of extracellular vesicles that are responsible for intercellular communication by carrying numerous molecules, including lipids, DNA, mRNA, miRNA, receptors, growth factors, immunomodulators, and antioxidants, which perform a variety of functions such as neuroprotection and anti-inflammation (Andjus et al., 2020; Sykova et al., 2021). Because of the exosome small size, it can cross the BBB to modulate the disease microenvironment as cell-free therapeutic tools (Bonafede and Mariotti 2017). Previous in vitro studies demonstrated that exosomes produced by adipose-derived MSCs could protect MN-like cells with ALS mutations from oxidative damage (Bonafede et al., 2016), play anti-apoptotic effect (Bonafede et al., 2019), and alleviate SOD1 aggregation and mitochondrial dysfunction (Lee M. et al., 2016). An in vivo study indicated that repeated administration of exosomes from adipose-derived MSCs protects MNs and home to damaged regions in SOD1G93A models (Bonafede et al., 2020). Therefore, exosomes secreted by MSCs might be a novel therapeutic candidate for ALS.
Many clinical studies have examined the safety and efficacy of MSCs. In phase I clinical study, safety evaluations reveal that there are no severe transplant-related adverse events and no structural changes in the brain or the spinal cord such as tumor formation. (Mazzini et al., 2010). Repetitive injections of cells have commonly been used in cellular therapy; for example, repeated injections of autologous bone marrow-derived MSCs (BM-MSCs) are showing no significant adverse events (Nabavi et al., 2019; Siwek et al., 2020). A phase II clinical trial with repeated intrathecal administration of BM-MSCs in patients with ALS is safe and well-tolerated (Petrou et al., 2021). Intriguingly, a case report has shown that intraventricular transplantation of autologous BM-MSCs using the Ommaya reservoir in a patient with ALS is safe, and this device has the potential to make repetitive injections of cells more easily (Baek et al., 2012). Moreover, MSC-neurotrophic factor (NTF) cells (NurOwn®, autologous BM-MSCs) have been induced to secrete high levels of NTFs. NTFs are small, naturally occurring polypeptides that support the development and survival of neurons (Henderson 1996). MSC-NTFs could offer a novel method for simultaneously delivering multiple NTFs to patients with neurodegenerative diseases such as ALS while playing the potential immunomodulatory therapeutic benefits of MSCs (Gothelf et al., 2017). MSC-NTFs cells (NurOwn®) have been successfully used in a phase Ⅱ clinical trial (NCT02017912) and proved its safety and early promising of efficacy in patients with ALS (Berry et al., 2019). Most recently, a randomized phase III controlled clinical trial comparing NurOwn® to placebo has also been completed (NCT03280056) in patients with ALS. The results do not reach statistical significance on the primary endpoint; nevertheless, patients who received NurOwn® have been significantly improved in cerebrospinal biomarkers of neuroinflammation, neurodegeneration, and neurotrophic factor support (Cudkowicz et al., 2021). Although this trial does not support the proposed clinical benefit of cellular therapy for ALS, NurOwn® is warranted for further investigation.
Reducing the expression levels of pro-inflammatory cytokines and increasing anti-inflammatory cytokines are observed after administration of MSCs (Oh et al., 2018). MSCs could stimulate intrinsic neurogenesis, and they released diverse neurotrophic factors and modulated immunoinflammatory processes (Uccelli et al., 2011; Keating 2012). Previously, 12 patients were enrolled to analyze their lymphocyte subsets and cytokine production after MSCs administration and demonstrated that MSCs caused immediate in vivo immunomodulation. This effect of MSCs was observed as early as 4 h after MSC transplantation. For example, the proportions of CD4+/CD25+ regulatory cells and activated dendritic cells are increased and decreased, respectively. Moreover, lymphocytes and lymphocyte proliferation are also reduced at 4 h after MSC transplantation (Karussis et al., 2010). Previous, a clinical trial has combined two cellular therapy protocols: T-cell vaccination (TCV) and BEN (BM-MSCs, effector T cells, and neuroblasts) approach in patients with ALS, and found that the combinational treatment is safe, feasible, and effective. This therapy might restore some dead MNs with clinical significance (Moviglia et al., 2012).
Many limitations still exist in the clinical application of MSCs. Studies examining the effects of MSCs on ALS applied different clinical parameters, and the higher efficacy of MSCs can be ensured by optimizing the treatment timing, MSC types, and treatment route and by examining sex differences in further translational studies. The preparation of BM-MSCs for patients with rapid disease progression is challenging and time-consuming. Thus, other allogeneic sources of MSCs should be identified for this type of therapy (Siwek et al., 2020). Moreover, immunomodulatory effects of ALS patient-derived MSCs are reduced due to their higher levels of pro-inflammatory cytokines. Nevertheless, the therapy effectiveness of autologous MSCs of ALS patients under the pro-inflammatory microenvironment deserves more studies and investigations (Javorkova et al., 2019).
Mononuclear cells (MCs) can be isolated from the peripheral blood, human umbilical cord blood, and bone marrow in the following paragraphs, and they are abbreviated as PBMCs, hUCBMCs, and BMMCs, respectively. Rather than a specific cell type, mononuclear cells are a heterogeneous population composed of hematopoietic stem cells (HSCs), hematopoietic progenitors, a small number of MSCs, and differentiated bone marrow cells including monocytes and lymphocytes (Vasques et al., 2021). With its diverse composition, mononuclear cells are useful considering the multi-etiological and non-cell-autonomous nature of ALS.
Many ALS preclinical trials have tested different mononuclear cell sources, cell doses, and administration routes. First, high doses of irradiation were used in SOD1G93A mouse models to replace the bone marrow with BMMCs (Corti et al., 2004). The results revealed an increase in animal survival. Two studies have suggested that BMMC treatment delayed disease progression, with more favorable outcomes noted in the presymptomatic phase than in the symptomatic phase (Gubert et al., 2016). Administering BMMC therapy through different routes (intravenous and intramuscular) simultaneously not only delayed disease progression but also reduced microgliosis in the spinal cord and protected the neuromuscular junction (Gubert et al., 2019). A phase II clinical study that injected bone marrow-derived HSCs into the anterior part of the spinal cord of 13 patients with ALS. Nine of 13 patients show much better outcomes, indicating that this strategy is safe and effective (Deda et al., 2009). Moreover, in a pilot safety study, Blanquer et al. (NCT00855400) showed a reduction in TDP-43 deposits in the spinal motoneurons of patients (Blanquer et al., 2012). A retrospective controlled study indicated that patient survival was significantly extended by BMMC therapy (Sharma et al., 2015). The safety and feasibility of BMMCs are thereby confirmed, and further research must be conducted to continually investigate the neuroprotective mechanism as well as to determine the best protocol for clinical practice.
Next, in a preclinical trial, Ende et al. reported that the use of hUCBMCs in SOD1G93A mouse models considerably delayed the onset of ALS symptoms and death (Ende et al., 2000). The optimal dose of hUCBMCs was found to be 2.5 ×
As for PBMCs, only a few trials have examined their safety and feasibility for the treatment of ALS. Because PBMCs can release growth factors and regulate the host immune system, they are suitable candidates for cellular therapy in ALS. A small pilot study demonstrated no adverse effects in patients with ALS who are infused with granulocyte-colony stimulating factor–mobilized peripheral blood stem cells (Cashman et al., 2008). Another study that transplanted bone marrow cells and PBMCs separately also confirmed their safety (Martinez et al., 2012). Notably, a recent clinical trial including 14 patients intraspinal transplanted PBMCs into the subarachnoid space and observed no severe transplant-related adverse events (Li et al., 2017). In addition, a case study has shown that no significant clinical changes in ALS patients who transplanted with PBMCs (Janson et al., 2001); allogeneic transplantation of peripheral blood-HSCs in patients with ALS has revealed that cells can enter injured sites of motoneuron in CNS and engraft as immunomodulatory cells, but no clinical benefits (Appel et al., 2008). Based on these safety results, one of the studies provides an aspect that peripheral blood-HSCs could serve as a cellular vehicle for future CNS gene therapy (Appel et al., 2008) and another study suggests that genetically engineered PBMCs could optimize the PBMCs secreting various growth factors to support the injured sites (Janson et al., 2001). Finally, although these studies have been reported the safety of PBMCs, further preclinical studies and larger trials are required to assess their therapeutic efficacy.
The term “neural precursor cell” refers to both neural stem and progenitor cells whose progeny are mature neurons and glial cells (Dibajnia and Morshead 2013). During embryonic development, neuroepithelial cells, which function as NSCs, can be divided into early neurons and radial glial (RG) cells (Gotz and Huttner 2005; Kriegstein and Alvarez-Buylla 2009). RG cells may develop into neurons directly or develop into neurons and glia indirectly through intermediate progenitor cells including GPCs and white matter progenitor cells (Kriegstein and Alvarez-Buylla 2009). Although most of the progenitors can only differentiate into limited phenotypes of glial cells, GPCs can generate both type 1- and type 2-astrocytes and oligodendrocytes (Han et al., 2004; Martins-Macedo et al., 2021). With their multipotent feature, NSCs and GPCs have been frequently discussed as a possible cellular therapy for ALS.
Many studies have discussed the therapeutic benefits of NSCs in recent years. Because NSCs can differentiate into neurons, they can replace lost or injured neurons, contributing to the reconstruction of motor circuits (Lepore and Maragakis 2007). NSCs can restore the diseased microenvironment of the CNS through the secretion of neurotrophic factors. Modulation of neuronal plasticity increases the survival and regeneration of endogenous neurons (Kim M. J. et al., 2018), promoting dendritic regrowth (Han et al., 2019), inhibiting apoptotic cell death, and enhancing injured axon regeneration (Keefe et al., 2017). Finally, NSCs release immune-modulatory molecules in the CNS, with a consequential reduction in CD3+ T cells and an increase in CD25+ and CD25+/CD62L+ Treg cells (Pluchino and Cossetti 2013). NSCs reduce the proportion of activated macrophages (Pluchino and Cossetti 2013) and interact with mononuclear phagocytes by scavenging extracellular succinate, thus affecting immune responses and suppressing inflammation (Peruzzotti-Jametti et al., 2018).
Preclinical studies have examined the safety and efficacy of NSC implantation. Transplanted rodent NSCs (rNSCs) in SOD1G93A mice showed therapeutic potential through neurogenesis and neurotrophin secretion (Corti et al., 2007). In addition, the efficiency of rNSCs administered through the blood delivery route was higher in symptomatic models (13%) than in presymptomatic models (6%) (Mitrecic et al., 2010). Another study using human NSCs (hNSCs) derived from aborted fetuses reported that hNSCs extensively differentiated into neurons and released GDNF and BDNF, leading to the delay of disease onset and the extension of lifespan (Xu et al., 2006). The combination of FK506 with rapamycin can serve as an effective immunosuppressive regimen (Yan et al., 2006). Grafted hNSCs can not only modulate the motor circuits but also engage in crosstalk with the host ependyma, thus enabling intrinsic repair (Xu et al., 2009; Xu et al., 2012). Moreover, dual transplantations of hNSCs in the cervical and lumbar spinal cord were more effective than single transplantation (Xu et al., 2011), and overexpression of VEGF may significantly improve survival (Hwang et al., 2009). However, the poor therapeutic benefits of transplantation were also reported with neither survival benefit nor delayed disease progression (Hefferan et al., 2012). The reduced survival of engrafted hNSCs may be due to the hostile ALS microenvironment; thus, additional approaches to protect these cells are needed (Srivastava et al., 2017).
To support the translation of NSC therapy from bench to bedside, several clinical trials have been conducted. The first FDA-approved phase I trial was conducted in 2009 (NCT01348451). No major complications were noted after lumbar injections, and most of the adverse events could be attributed to the toxicities of immunosuppressant drugs or the injection procedure itself (Glass et al., 2012); this finding is consistent with that of another phase I study (NCT01640067) (Mazzini et al., 2015). Cervical injections were also well-tolerated, whereas the dual targeting group (both lumbar and cervical) showed slowed disease progression. The transplantation of hNSCs might be beneficial to patients with spinal symptom onset in the early stage of ALS (Tadesse et al., 2014). Autopsies on seven patients showed that transplanted hNSCs could survive up to 2.5 years posttransplant with “nests” consisting of donor cells near the injection site (Glass et al., 2016). Another open-label trial (NCT01730716) indicated that hNSCs could be safely injected at high doses, and the procedure could be expanded to multiple surgical centers (Goutman et al., 2018). Compared with historical controls, transplantation significantly increased the revised ALS Functional Rating Scale scores, which indicated possible efficacy (Mazzini et al., 2015).
Although most studies have reported positive results, several difficulties should be overcome before NSCs become a potent therapeutic tool for ALS. For instance, the poor migration of NSCs is an obstacle. NSCs have a tendency to form cell clusters close to the injection site, which is further enhanced through CXCR4 and adhesion molecules including integrins, selectins, and immunoglobulins (Gonzalez et al., 2016). In addition to the endogenous mechanism, transplantation itself may have latent risks; for example, clots formed by NSCs in vivo can cause secondary damage to injected tissues under high-density transplantation (Tang et al., 2017). To summarize, preclinical and clinical studies have provided the framework of engraftment; with more solid knowledge acquired, NSCs may become a preponderant candidate of cellular therapy for ALS.
GPCs are multipotent cells that can differentiate into oligodendrocytes, as well as type 1 and type 2 astrocytes. Astrocytes and oligodendrocytes have different characteristics, but both play vital roles in ALS pathology. Astrocytes drive MN development, including synaptogenesis and maturation (Allen and Lyons 2018), and they maintain the neuronal microenvironment by mediating ion concentrations, secreting neurotrophic factors, and uptaking neurotransmitters (Liddelow and Barres 2015; Halpern et al., 2019; Izrael et al., 2020). They also provide neuronal protection in ALS models, with preference for the spreading of TDP-43 aggregation from MNs to astrocytes along with better tolerability to aggregates (Smethurst et al., 2020). In addition to protecting MNs from neurotoxicity, astrocytes release exosomes that express neuroprotective proteins with anti-oxidative, anti-apoptotic, and anti-inflammatory effects. With the ability to selectively target neurons, astrocyte-derived exosomes have therapeutic potential by transferring neuroglobin to neurons (Venturini et al., 2019).
Oligodendrocytes play roles in the pathophysiology of ALS (Ferraiuolo et al., 2016). Functioning as myelinating cells, they generate myelin sheaths around axons, thus establishing saltatory conduction and providing both physical protection and metabolic support for neurons (Philips et al., 2013). After myelination, the rate of lactate fermentation increases as a predominant energy source (Funfschilling et al., 2012). Lactate is transported abundantly through monocarboxylate transporter-1 (MCT1), which is highly expressed in oligodendrocytes (Lee et al., 2012), In the disease model of sporadic ALS, MCT1 expression is reduced, causing the death of MNs consequently through two pathways: the deleterious effect on neuronal survival due to MCT1 dysfunction or the homeostatic failure of iron, leading to macrophage cytotoxicity and neuronal ferroptosis (Tognatta and Miller 2016). Because the dysfunction of oligodendrocytes is one of the contributing factors of MN degeneration, the potential of transplanted GPCs to generate oligodendrocytes is now emerging (Walczak et al., 2011).
Targeting therapeutic potential through the differentiation of astrocytes and oligodendrocytes, transplantations of GPCs were conducted in several studies. Engrafted GPCs in the cervical spinal cord of SOD1G93A rats could efficiently differentiate into astrocytes and exert neuroprotection by preventing the loss of GLT1, thus prolonging survival and attenuating the degeneration of forelimb and respiratory MNs (Lepore et al., 2008). However, transplanted human GPCs (hGPCs, also known as Q-Cells) in ALS rodent models did not exert any therapeutic benefits (Lepore et al., 2011). By contrast, several preclinical studies have genetically modified hGPCs to overexpress neurotrophic factors. GDNF-overexpressing hGPCs could survive, integrate, and release GDNF in the spinal cord after transplantation (Klein et al., 2005). The efficient delivery of GDNF significantly enhanced MN survival but had no beneficial effect on functional outcomes (Suzuki et al., 2007), which is consistent with the result of another study (Park et al., 2009). Transplantation of GDNF-overexpressing hGPCs in the SOD1G93A rat motor cortex could delay disease onset and extend lifespan by supporting lower MNs through the releasing GDNF. These cells could also survive and release GDNF in the cortex of cynomolgus macaques (Thomsen et al., 2018). In 2019, a phase I/IIa study (NCT02943850) unilaterally injected hGPCs into the lumbar region of 18 participants. Moreover, a phase I/IIa study (NCT02478450) is currently examining both the safety and efficacy of Q-Cell transplantation in ALS patients and is estimated to be completed in 2024.
Olfactory ensheathing cells (OECs) are a unique class of glial cells because they locate in both the central and peripheral nervous system (Su and He 2010), and could promote axon regeneration in the spinal cord in vivo model (Ramón-Cueto and Nieto-Sampedro 1994). Although a preclinical study reveals that OECs intrathecally transplanted into ALS rodent models show no significant benefits than that of in the control rodent (Morita et al., 2008), Li et al. demonstrated that OECs play a neuroprotective function (Li et al., 2011) and prolong survival after the injection of OECs into SOD1G93A rats (Li et al., 2013). As for clinical application, transplantation of OECs has demonstrated that they can slow down the ALS disease progression (Huang et al., 2008). Additionally, a study has shown that 507 ALS patients exhibit partial neurological functional recovery after receiving multiple OECs administration (Chen et al., 2012). However, several authors query the efficacy of OECs treatment because no clear evidences of axonal regeneration, neuronal differentiation, and myelination are seen after post-mortem neuropathologic analyses (Giordana et al., 2010). A prospective study has found no clinical benefits for patients as well (Piepers and van den Berg 2010), which is in line with another observational study showing negative or conflicting outcomes (Gamez et al., 2010). Besides, the safety is still in doubt due to the reported disabling side-effects (Chew et al., 2007). The conflicting results suggested that OECs transplantation should not yet be considered as an alternative treatment for ALS.
In this review, we summarize potential cellular therapies for ALS, including the pluripotent stem cells ESCs and iPSCs; multipotent stem cells such as MSCs and neuron-related progenitors, as well as MCs. Although ESCs/iPSCs could differentiate into three germ layers, tumor formation risk is the major concern in clinical practice. By contrast, MSCs, neuron-related progenitors, and MCs are promising because of their capabilities of microenvironment modulation and neuroprotection through rich trophic factor secretion, enhanced neurogenesis, abnormal protein aggregate clearance, and immune modulation (Figure 2). The current preclinical studies and clinical trials of cellular therapy on ALS are summarized (Tables 1–3). Among them, MSCs account for the majority of all cell types (Figure 3A). MSCs hold great potential since two phase III studies were conducted in 2017 and 2021 (Figure 3B). Similar to MSCs, mononuclear cells are emerging cellular therapy with one phase II study preparing, whereas neural precursor cells require further clinical research (Figure 3C). Generally, the abundant emergence of clinical trials in recent years reveals the growing interest in the actual application of cellular therapy for ALS. Although most trials remain in the early stages, more advanced studies will be performed, and the cellular therapy platform will optimize the dose and types of different stem cells for future clinical treatment for ALS.
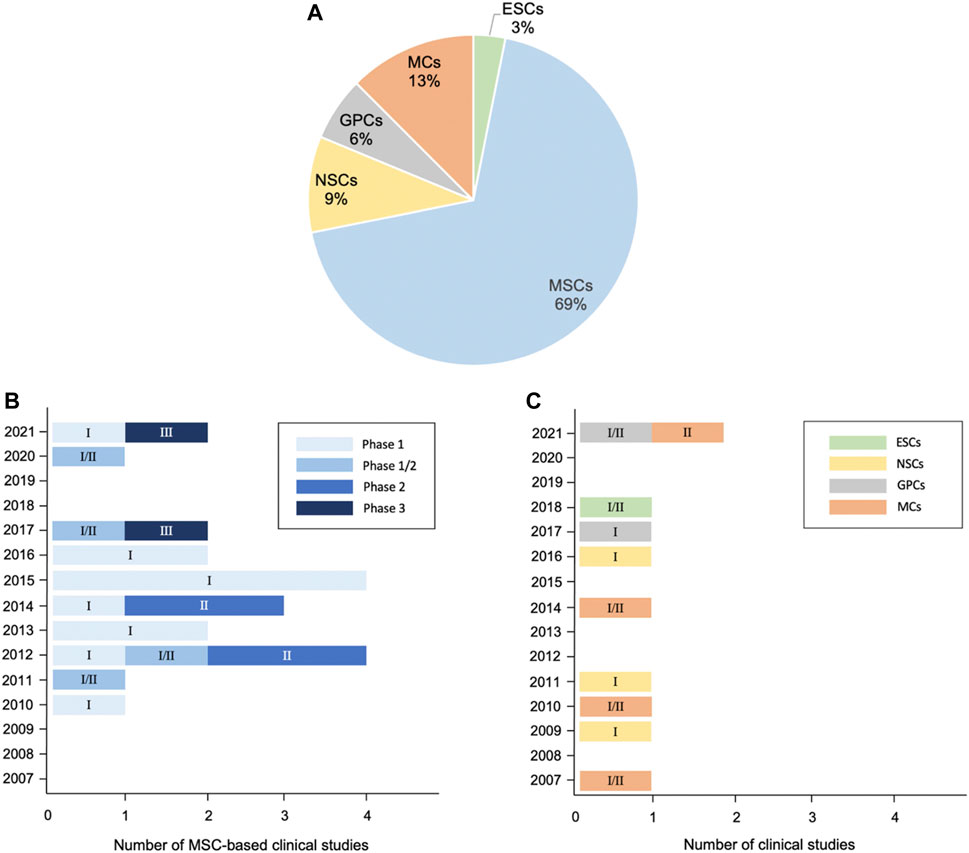
FIGURE 3. Distribution of cellular therapy for ALS in clinical trials from 2007 to 2021. (A) Pie chart: analysis of cell types. (B) Trends of MSC-based applications regarding clinical phase. (C) Trends of ESC-, neural precursor cell-, and MC-based applications regarding clinical phase. 32 clinical trials have been registered as of October 2021, excluding studies with suspended, temporarily not available, terminated, withdrawn, available, and no longer available status according to clinicaltrials.gov. Numbers in the bars of Figures 3B,C indicate the corresponding phase of clinical trials.
Conception and design: T-JL, G-CC, L-YW, T-YL, Y-CK, and Y-HH. Writing, review, and/or revision of the manuscript: T-JL, G-CC, L-YW, W-YL, T-YL, Y-CK, and Y-HH. Study supervision: T-YL, Y-CK, and Y-HH.
This work was financially supported by research grants from the Ministry of Science and Technology, Taiwan (Grant numbers: MOST110-2314-B-038-132, and MOST110-2314-B-038-034) to Y-HH, and the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan (Grant numbers: DP2-109-21121-01-T-03-02 and DP2-110-21121-01-T-03-02); Taipei Medical University (Grant number: TMU109-AE1-B02) to Y-CK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors acknowledge the academic and science graphic illustration service provided by TMU Office of Research and Development at Taipei Medical University. This manuscript was edited by Wallace Academic Editing.
Allen, N. J., and Lyons, D. A. (2018). Glia as Architects of central Nervous System Formation and Function. Science 362, 181–185. doi:10.1126/science.aat0473
Amado, D. A., and Davidson, B. L. (2021). Gene Therapy for ALS: A Review. Mol. Ther. 29, 3345–3358. doi:10.1016/j.ymthe.2021.04.008
Andjus, P., Kosanović, M., Milićević, K., Gautam, M., Vainio, S. J., Jagečić, D., et al. (2020). Extracellular Vesicles as Innovative Tool for Diagnosis, Regeneration and Protection against Neurological Damage. Int. J. Mol. Sci. 216859. doi:10.3390/ijms21186859
Appel, S. H., Engelhardt, J. I., Henkel, J. S., Siklos, L., Beers, D. R., Yen, A. A., et al. (2008). Hematopoietic Stem Cell Transplantation in Patients with Sporadic Amyotrophic Lateral Sclerosis. Neurology 71, 1326–1334. doi:10.1212/01.wnl.0000327668.43541.22
Ayers, J. I., Fromholt, S. E., O’Neal, V. M., Diamond, J. H., and Borchelt, D. R. (2016). Prion-like Propagation of Mutant SOD1 Misfolding and Motor Neuron Disease Spread along Neuroanatomical Pathways. Acta Neuropathol. 131, 103–114. doi:10.1007/s00401-015-1514-0
Baek, W., Kim, Y. S., Koh, S. H., Lim, S. W., Kim, H. Y., Yi, H. J., et al. (2012). Stem Cell Transplantation into the Intraventricular Space via an Ommaya Reservoir in a Patient with Amyotrophic Lateral Sclerosis. J. Neurosurg. Sci. 56, 261–263.
Baglio, S. R., Pegtel, D. M., and Baldini, N. (2012). Mesenchymal Stem Cell Secreted Vesicles Provide Novel Opportunities in (Stem) Cell-free Therapy. Front. Physio. 3, 359. doi:10.3389/fphys.2012.00359
Ben-David, U., and Benvenisty, N. (2011). The Tumorigenicity of Human Embryonic and Induced Pluripotent Stem Cells. Nat. Rev. Cancer 11, 268–277. doi:10.1038/nrc3034
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 330, 585–591. doi:10.1056/NEJM199403033300901
Berry, J. D., Cudkowicz, M. E., Windebank, A. J., Staff, N. P., Owegi, M., Nicholson, K., et al. (2019). NurOwn, Phase 2, Randomized, Clinical Trial in Patients with ALS. Neurology 93, e2294–e2305. doi:10.1212/wnl.0000000000008620
Bilican, B., Serio, A., Barmada, S. J., Nishimura, A. L., Sullivan, G. J., Carrasco, M., et al. (2012). Mutant Induced Pluripotent Stem Cell Lines Recapitulate Aspects of TDP-43 Proteinopathies and Reveal Cell-specific Vulnerability. Proc. Natl. Acad. Sci. 109, 5803–5808. doi:10.1073/pnas.1202922109
Blanquer, M., Moraleda, J. M., Iniesta, F., Gómez-Espuch, J., Meca-Lallana, J., Villaverde, R., et al. (2012). Neurotrophic Bone Marrow Cellular Nests Prevent Spinal Motoneuron Degeneration in Amyotrophic Lateral Sclerosis Patients: a Pilot Safety Study. Stem Cells. 30, 1277–1285. doi:10.1002/stem.1080
Boido, M., Piras, A., Valsecchi, V., Spigolon, G., Mareschi, K., Ferrero, I., et al. (2014). Human Mesenchymal Stromal Cell Transplantation Modulates Neuroinflammatory Milieu in a Mouse Model of Amyotrophic Lateral Sclerosis. Cytotherapy. 16, 1059–1072. doi:10.1016/j.jcyt.2014.02.003
Bonafede, R., Brandi, J., Manfredi, M., Scambi, I., Schiaffino, L., Merigo, F., et al. (2019). The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis. Cells 8, 1087. doi:10.3390/cells8091087
Bonafede, R., and Mariotti, R. (2017). ALS Pathogenesis and Therapeutic Approaches: The Role of Mesenchymal Stem Cells and Extracellular Vesicles. Front. Cell Neurosci. 11, 80. doi:10.3389/fncel.2017.00080
Bonafede, R., Scambi, I., Peroni, D., Potrich, V., Boschi, F., Benati, D., et al. (2016). Exosome Derived from Murine Adipose-Derived Stromal Cells: Neuroprotective Effect on In Vitro Model of Amyotrophic Lateral Sclerosis. Exp. Cell Res. 340, 150–158. doi:10.1016/j.yexcr.2015.12.009
Bonafede, R., Turano, E., Scambi, I., Busato, A., Bontempi, P., Virla, F., et al. (2020). ASC-exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS. Int. J. Mol. Sci. 21, 3651. doi:10.3390/ijms21103651
Boucherie, C., Schäfer, S., Lavand'homme, P., Maloteaux, J.-M., and Hermans, E. (2009). Chimerization of Astroglial Population in the Lumbar Spinal Cord after Mesenchymal Stem Cell Transplantation Prolongs Survival in a Rat Model of Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 87, 2034–2046. doi:10.1002/jnr.22038
Brites, D., and Vaz, A. R. (2014). Microglia Centered Pathogenesis in ALS: Insights in Cell Interconnectivity. Front. Cell Neurosci. 8, 117. doi:10.3389/fncel.2014.00117
Brown, R. H., and Al-Chalabi, A. (2015). Endogenous Retroviruses in ALS: A Reawakening? Sci. Transl Med. 7, 307fs40. doi:10.1126/scitranslmed.aad3533
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 377, 162–172. doi:10.1056/NEJMra1603471
Cabungcal, J.-H., Steullet, P., Morishita, H., Kraftsik, R., Cuenod, M., Hensch, T. K., et al. (2013). Perineuronal Nets Protect Fast-Spiking Interneurons against Oxidative Stress. Proc. Natl. Acad. Sci. 110, 9130–9135. doi:10.1073/pnas.1300454110
Cappella, M., Ciotti, C., Cohen-Tannoudji, M., and Biferi, M. G. (2019). Gene Therapy for ALS-A Perspective. Int. J. Mol. Sci., 20, 4388. doi:10.3390/ijms20184388
Cashman, N., Tan, L.-Y., Krieger, C., Mädler, B., Mackay, A., Mackenzie, I., et al. (2008). Pilot Study of Granulocyte colony Stimulating Factor (G-CSF)-mobilized Peripheral Blood Stem Cells in Amyotrophic Lateral Sclerosis (ALS). Muscle Nerve. 37, 620–625. doi:10.1002/mus.20951
Chan-Il, C., Young-Don, L., Heejaung, K., Kim, S. H., Suh-Kim, H., and Kim, S. S. (2013). Neural Induction with Neurogenin 1 Enhances the Therapeutic Potential of Mesenchymal Stem Cells in an Amyotrophic Lateral Sclerosis Mouse Model. Cell Transplant. 22, 855–870. doi:10.3727/096368912X637019
Chen, H., Qian, K., Du, Z., Cao, J., Petersen, A., Liu, H., et al. (2014). Modeling ALS with iPSCs Reveals that Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell. 14, 796–809. doi:10.1016/j.stem.2014.02.004
Chen, L., Chen, D., Xi, H., Wang, Q., Liu, Y., Zhang, F., et al. (2012). Olfactory Ensheathing Cell Neurorestorotherapy for Amyotrophic Lateral Sclerosis Patients: Benefits from Multiple Transplantations. Cell Transplant. 21, S65–S77. doi:10.3727/096368912x633789
Chen, X., Wang, S., and Cao, W. (2018). Mesenchymal Stem Cell-Mediated Immunomodulation in Cell Therapy of Neurodegenerative Diseases. Cell Immunol. 326, 8–14. doi:10.1016/j.cellimm.2017.06.006
Chew, S., Khandji, A. G., Montes, J., Mitsumoto, H., and Gordon, P. H. (2007). Olfactory Ensheathing Glia Injections in Beijing: Misleading Patients with ALS. Amyotroph. Lateral Scler. 8, 314–316. doi:10.1080/17482960701419315
Choi, S. Y., Lopez-Gonzalez, R., Krishnan, G., Phillips, H. L., Li, A. N., Seeley, W. W., et al. (2019). C9ORF72-ALS/FTD-associated Poly(GR) Binds Atp5a1 and Compromises Mitochondrial Function In Vivo. Nat. Neurosci. 22, 851–862. doi:10.1038/s41593-019-0397-0
Chou, C.-C., Zhang, Y., Umoh, M. E., Vaughan, S. W., Lorenzini, I., Liu, F., et al. (2018). TDP-43 Pathology Disrupts Nuclear Pore Complexes and Nucleocytoplasmic Transport in ALS/FTD. Nat. Neurosci. 21, 228–239. doi:10.1038/s41593-017-0047-3
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillée, S., Rule, M., et al. (2003). Wild-type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science 302, 113–117. doi:10.1126/science.1086071
Coatti, G. C., Beccari, M. S., Olávio, T. R., Mitne-Neto, M., Okamoto, O. K., and Zatz, M. (2015). Stem Cells for Amyotrophic Lateral Sclerosis Modeling and Therapy: Myth or Fact? Cytometry 87, 197–211. doi:10.1002/cyto.a.22630
Coatti, G. C., Frangini, M., Valadares, M. C., Gomes, J. P., Lima, N. O., Cavaçana, N., et al. (2017). Pericytes Extend Survival of ALS SOD1 Mice and Induce the Expression of Antioxidant Enzymes in the Murine Model and in IPSCs Derived Neuronal Cells from an ALS Patient. Stem Cell Rev. Rep. 13, 686–698. doi:10.1007/s12015-017-9752-2
Cooper-Knock, J., Walsh, M. J., Higginbottom, A., Robin Highley, J., Dickman, M. J., Edbauer, D., et al. (2014). Sequestration of Multiple RNA Recognition Motif-Containing Proteins by C9orf72 Repeat Expansions. Brain. 137, 2040–2051. doi:10.1093/brain/awu120
Cordes, A.-L., Jahn, K., Hass, R., Schwabe, K., Weissinger, E. M., Ganser, A., et al. (2011). Intramedullary Spinal Cord Implantation of Human CD34+umbilical Cord-Derived Cells in ALS. Amyotroph. Lateral Scler. 12, 325–330. doi:10.3109/17482968.2011.580850
Corti, S., Locatelli, F., Donadoni, C., Guglieri, M., Papadimitriou, D., Strazzer, S., et al. (2004). Wild-type Bone Marrow Cells Ameliorate the Phenotype of SOD1-G93a ALS Mice and Contribute to CNS, Heart and Skeletal Muscle Tissues. Brain. 127, 2518–2532. doi:10.1093/brain/awh273
Corti, S., Locatelli, F., Papadimitriou, D., Del Bo, R., Nizzardo, M., Nardini, M., et al. (2007). Neural Stem Cells LewisX + CXCR4 + Modify Disease Progression in an Amyotrophic Lateral Sclerosis Model. Brain. 130, 1289–1305. doi:10.1093/brain/awm043
Cudkowicz, M. E., Lindborg, S. R., Goyal, N. A., Miller, R. G., Burford, M. J., Berry, J. D., et al. (2021). A Randomized Placebo-Controlled Phase 3 Study of Mesenchymal Stem Cells Induced to Secrete High Levels of Neurotrophic Factors in Amyotrophic Lateral Sclerosis. Muscle Nerve 10, 27472. doi:10.1002/mus.27472
Deda, H., Inci, M., Kürekçi, A., Sav, A., Kayıhan, K., Özgün, E., et al. (2009). Treatment of Amyotrophic Lateral Sclerosis Patients by Autologous Bone Marrow-Derived Hematopoietic Stem Cell Transplantation: a 1-year Follow-Up. Cytotherapy. 11, 18–25. doi:10.1080/14653240802549470
Dibajnia, P., and Morshead, C. M. (2013). Role of Neural Precursor Cells in Promoting Repair Following Stroke. Acta Pharmacol. Sin. 34, 78–90. doi:10.1038/aps.2012.107
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons. Science 321, 1218–1221. doi:10.1126/science.1158799
Donnelly, C. J., Zhang, P.-W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron. 80, 415–428. doi:10.1016/j.neuron.2013.10.015
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug Screening for ALS Using Patient-specific Induced Pluripotent Stem Cells. Sci. Transl Med. 4, 145ra104. doi:10.1126/scitranslmed.3004052
Ende, N., Weinstein, F., Chen, R., and Ende, M. (2000). Human Umbilical Cord Blood Effect on Sod Mice (Amyotrophic Lateral Sclerosis). Life Sci. 67, 53–59. doi:10.1016/s0024-3205(00)00602-0
Fang, T., Al Khleifat, A., Meurgey, J.-H., Jones, A., Leigh, P. N., Bensimon, G., et al. (2018). Stage at Which Riluzole Treatment Prolongs Survival in Patients with Amyotrophic Lateral Sclerosis: a Retrospective Analysis of Data from a Dose-Ranging Study. Lancet Neurol. 17, 416–422. doi:10.1016/S1474-4422(18)30054-1
Ferraiuolo, L., Higginbottom, A., Heath, P. R., Barber, S., Greenald, D., Kirby, J., et al. (2011). Dysregulation of Astrocyte-Motoneuron Cross-Talk in Mutant Superoxide Dismutase 1-related Amyotrophic Lateral Sclerosis. Brain. 134, 2627–2641. doi:10.1093/brain/awr193
Ferraiuolo, L., Meyer, K., Sherwood, T. W., Vick, J., Likhite, S., Frakes, A., et al. (2016). Oligodendrocytes Contribute to Motor Neuron Death in ALS via SOD1-dependent Mechanism. Proc. Natl. Acad. Sci. USA 113, E6496–E6505. doi:10.1073/pnas.1607496113
Forostyak, S., Homola, A., Turnovcova, K., Svitil, P., Jendelova, P., and Sykova, E. (2014). Intrathecal Delivery of Mesenchymal Stromal Cells Protects the Structure of Altered Perineuronal Nets in SOD1 Rats and Amends the Course of ALS. Stem Cells. 32, 3163–3172. doi:10.1002/stem.1812
Forostyak, S., Jendelova, P., Kapcalova, M., Arboleda, D., and Sykova, E. (2011). Mesenchymal Stromal Cells Prolong the Lifespan in a Rat Model of Amyotrophic Lateral Sclerosis. Cytotherapy. 13, 1036–1046. doi:10.3109/14653249.2011.592521
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling Sporadic ALS in iPSC-Derived Motor Neurons Identifies a Potential Therapeutic Agent. Nat. Med. 24, 1579–1589. doi:10.1038/s41591-018-0140-5
Fünfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J., et al. (2012). Glycolytic Oligodendrocytes Maintain Myelin and Long-Term Axonal Integrity. Nature 485, 517–521. doi:10.1038/nature11007
Gamez, J., Carmona, F., Raguer, N., Ferrer-Sancho, J., Martín-Henao, G. A., Martí-Beltrán, S., et al. (2010). Cellular Transplants in Amyotrophic Lateral Sclerosis Patients: an Observational Study. Cytotherapy. 12, 669–677. doi:10.3109/14653241003774037
Garbuzova-Davis, S., Rodrigues, M. C. O., Mirtyl, S., Turner, S., Mitha, S., Sodhi, J., et al. (2012). Multiple Intravenous Administrations of Human Umbilical Cord Blood Cells Benefit in a Mouse Model of ALS. PLoS One. 7, e31254. doi:10.1371/journal.pone.0031254
Garbuzova-Davis, S., Sanberg, C. D., Kuzmin-Nichols, N., Willing, A. E., Gemma, C., Bickford, P. C., et al. (2008). Human Umbilical Cord Blood Treatment in a Mouse Model of ALS: Optimization of Cell Dose. PLoS One. 3, e2494. doi:10.1371/journal.pone.0002494
Gibb, S. L., Boston-Howes, W., Lavina, Z. S., Gustincich, S., Brown, R. H., Pasinelli, P., et al. (2007). A Caspase-3-Cleaved Fragment of the Glial Glutamate Transporter EAAT2 Is Sumoylated and Targeted to Promyelocytic Leukemia Nuclear Bodies in Mutant SOD1-Linked Amyotrophic Lateral Sclerosis. J. Biol. Chem. 282, 32480–32490. doi:10.1074/jbc.M704314200
Giordana, M. T., Grifoni, S., Votta, B., Magistrello, M., Vercellino, M., Pellerino, A., et al. (2010). Neuropathology of Olfactory Ensheathing Cell Transplantation into the Brain of Two Amyotrophic Lateral Sclerosis (ALS) Patients. Brain Pathol. 20, 730–737. doi:10.1111/j.1750-3639.2009.00353.x
Glass, J. D., Boulis, N. M., Johe, K., Rutkove, S. B., Federici, T., Polak, M., et al. (2012). Lumbar Intraspinal Injection of Neural Stem Cells in Patients with Amyotrophic Lateral Sclerosis: Results of a Phase I Trial in 12 Patients. Stem Cells. 30, 1144–1151. doi:10.1002/stem.1079
Glass, J. D., Hertzberg, V. S., Boulis, N. M., Riley, J., Federici, T., Polak, M., et al. (2016). Transplantation of Spinal Cord-Derived Neural Stem Cells for ALS. Neurology 87, 392–400. doi:10.1212/WNL.0000000000002889
Gonzalez, R., H. Hamblin, M., and Lee, J.-P. (2016). Neural Stem Cell Transplantation and CNS Diseases. CNS Neurol. Disord.-DR. 15, 881–886. doi:10.2174/1871527315666160815164247
Gothelf, Y., Kaspi, H., Abramov, N., and Aricha, R. (2017). miRNA Profiling of NurOwn: Mesenchymal Stem Cells Secreting Neurotrophic Factors. Stem Cell Res. Ther. 8, 249. doi:10.1186/s13287-017-0692-1
Götz, M., and Huttner, W. B. (2005). The Cell Biology of Neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788. doi:10.1038/nrm1739
Goutman, S. A., Brown, M. B., Glass, J. D., Boulis, N. M., Johe, K., Hazel, T., et al. (2018). Long-term Phase 1/2 Intraspinal Stem Cell Transplantation Outcomes in ALS. Ann. Clin. Transl Neurol. 5, 730–740. doi:10.1002/acn3.567
Goutman, S. A., Savelieff, M. G., Sakowski, S. A., and Feldman, E. L. (2019). Stem Cell Treatments for Amyotrophic Lateral Sclerosis: a Critical Overview of Early Phase Trials. Expert Opin. Investig. Drugs 28, 525–543. doi:10.1080/13543784.2019.1627324
Grad, L. I., Rouleau, G. A., Ravits, J., and Cashman, N. R. (2017) Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb Perspect. Med. 7, a024117. doi:10.1101/cshperspect.a024117
Gubert, F., Bonacossa-Pereira, I., Decotelli, A. B., Furtado, M., Vasconcelos-Dos-Santos, A., Mendez-Otero, R., et al. (2019). Bone-marrow Mononuclear Cell Therapy in a Mouse Model of Amyotrophic Lateral Sclerosis: Functional Outcomes from Different Administration Routes. Brain Res. 1712, 73–81. doi:10.1016/j.brainres.2019.02.003
Gubert, F., Decotelli, A. B., Bonacossa-Pereira, I., Figueiredo, F. R., Zaverucha-do-Valle, C., Tovar-Moll, F., et al. (2016). Intraspinal Bone-Marrow Cell Therapy at Pre- and Symptomatic Phases in a Mouse Model of Amyotrophic Lateral Sclerosis. Stem Cell Res. Ther. 7, 41. doi:10.1186/s13287-016-0293-4
Guo, W., Naujock, M., Fumagalli, L., Vandoorne, T., Baatsen, P., Boon, R., et al. (2017). HDAC6 Inhibition Reverses Axonal Transport Defects in Motor Neurons Derived from FUS-ALS Patients. Nat. Commun. 8, 861. doi:10.1038/s41467-017-00911-y
Guttenplan, K. A., Weigel, M. K., Adler, D. I., Couthouis, J., Liddelow, S. A., Gitler, A. D., et al. (2020). Knockout of Reactive Astrocyte Activating Factors Slows Disease Progression in an ALS Mouse Model. Nat. Commun. 11, 3753. doi:10.1038/s41467-020-17514-9
Habisch, H.-J., Janowski, M., Binder, D., Kuzma-Kozakiewicz, M., Widmann, A., Habich, A., et al. (2007). Intrathecal Application of Neuroectodermally Converted Stem Cells into a Mouse Model of ALS: Limited Intraparenchymal Migration and Survival narrows Therapeutic Effects. J. Neural Transm. 114, 1395–1406. doi:10.1007/s00702-007-0748-y
Halpern, M., Brennand, K. J., and Gregory, J. (2019). Examining the Relationship between Astrocyte Dysfunction and Neurodegeneration in ALS Using hiPSCs. Neurobiol. Dis. 132, 104562. doi:10.1016/j.nbd.2019.104562
Han, Q., Ordaz, J. D., Liu, N.-K., Richardson, Z., Wu, W., Xia, Y., et al. (2019). Descending Motor Circuitry Required for NT-3 Mediated Locomotor Recovery after Spinal Cord Injury in Mice. Nat. Commun. 10, 5815. doi:10.1038/s41467-019-13854-3
Han, S. S. W., Liu, Y., Tyler-Polsz, C., Rao, M. S., and Fischer, I. (2004). Transplantation of Glial-Restricted Precursor Cells into the Adult Spinal Cord: Survival, Glial-specific Differentiation, and Preferential Migration in white Matter. Glia. 45, 1–16. doi:10.1002/glia.10282
Harper, J. M., Krishnan, C., Darman, J. S., Deshpande, D. M., Peck, S., Shats, I., et al. (2004). Axonal Growth of Embryonic Stem Cell-Derived Motoneurons In Vitro and in Motoneuron-Injured Adult Rats. Proc. Natl. Acad. Sci. 101, 7123–7128. doi:10.1073/pnas.0401103101
Hefferan, M. P., Galik, J., Kakinohana, O., Sekerkova, G., Santucci, C., Marsala, S., et al. (2012). Human Neural Stem Cell Replacement Therapy for Amyotrophic Lateral Sclerosis by Spinal Transplantation. PLoS One. 7, e42614. doi:10.1371/journal.pone.0042614
Henderson, C. E. (1996). Role of Neurotrophic Factors in Neuronal Development. Curr. Opin. Neurobiol. 6, 64–70. doi:10.1016/s0959-4388(96)80010-9
Highley, J. R., Kirby, J., Jansweijer, J. A., Webb, P. S., Hewamadduma, C. A., Heath, P. R., et al. (2014). Loss of Nuclear TDP-43 in Amyotrophic Lateral Sclerosis (ALS) Causes Altered Expression of Splicing Machinery and Widespread Dysregulation of RNA Splicing in Motor Neurones. Neuropathol. Appl. Neurobiol. 40, 670–685. doi:10.1111/nan.12148
Hobson, E. V., and McDermott, C. J. (2016). Supportive and Symptomatic Management of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 12, 526–538. doi:10.1038/nrneurol.2016.111
Huang, H., Chen, L., Xi, H., Wang, H., Zhang, J., Zhang, F., et al. (2008). Fetal Olfactory Ensheathing Cells Transplantation in Amyotrophic Lateral Sclerosis Patients: a Controlled Pilot Study. Clin. Transpl. 22, 710–718. doi:10.1111/j.1399-0012.2008.00865.x
Hwang, D. H., Lee, H. J., Park, I. H., Seok, J. I., Kim, B. G., Joo, I. S., et al. (2009). Intrathecal Transplantation of Human Neural Stem Cells Overexpressing VEGF Provide Behavioral Improvement, Disease Onset Delay and Survival Extension in Transgenic ALS Mice. Gene Ther. 16, 1234–1244. doi:10.1038/gt.2009.80
Ikenaka, K., Katsuno, M., Kawai, K., Ishigaki, S., Tanaka, F., and Sobue, G. (2012). Disruption of Axonal Transport in Motor Neuron Diseases. Int. J. Mol. Sci. 13, 1225–1238. doi:10.3390/ijms13011225
Imamura, K., Izumi, Y., Watanabe, A., Tsukita, K., Woltjen, K., Yamamoto, T., et al. (2017). The Src/c-Abl Pathway Is a Potential Therapeutic Target in Amyotrophic Lateral Sclerosis. Sci. Transl. Med., 9, eaaf3962. doi:10.1126/scitranslmed.aaf3962
Institute, T. A. T. D. (2021). Limb and Bulbar Onset ALS the ALS Therapy Development Institute: The ALS Therapy Development Institute. . https://www.als.net/news/science-sunday-limb-and-bulbar-onset-als/.Available from
Islamov, R. R., Rizvanov, A. A., Fedotova, V. Y., Izmailov, A. A., Safiullov, Z. Z., Garanina, E. E., et al. (2017). Tandem Delivery of Multiple Therapeutic Genes Using Umbilical Cord Blood Cells Improves Symptomatic Outcomes in ALS. Mol. Neurobiol. 54, 4756–4763. doi:10.1007/s12035-016-0017-x
Izrael, M., Slutsky, S. G., and Revel, M. (2020). Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Front. Neurosci. 14, 824. doi:10.3389/fnins.2020.00824
Janson, C. G., Ramesh, T. M., During, M. J., Leone, P., and Heywood, J. (2001). Human Intrathecal Transplantation of Peripheral Blood Stem Cells in Amyotrophic Lateral Sclerosis. J. Hematotherapy Stem Cell Res. 10, 913–915. doi:10.1089/152581601317211015
Javorkova, E., Matejckova, N., Zajicova, A., Hermankova, B., Hajkova, M., Bohacova, P., et al. (2019). Immunomodulatory Properties of Bone Marrow Mesenchymal Stem Cells from Patients with Amyotrophic Lateral Sclerosis and Healthy Donors. J. Neuroimmune Pharmacol. 14, 215–225. doi:10.1007/s11481-018-9812-7
Jiang, Y., Zhang, M.-J., and Hu, B.-Y. (2012). Specification of Functional Neurons and Glia from Human Pluripotent Stem Cells. Protein Cell. 3, 818–825. doi:10.1007/s13238-012-2086-6
Karussis, D., Karageorgiou, C., Vaknin-Dembinsky, A., Gowda-Kurkalli, B., Gomori, J. M., Kassis, I., et al. (2010). Safety and Immunological Effects of Mesenchymal Stem Cell Transplantation in Patients with Multiple Sclerosis and Amyotrophic Lateral Sclerosis. Arch. Neurol. 67, 1187–1194. doi:10.1001/archneurol.2010.248
Keating, A. (2012). Mesenchymal Stromal Cells: New Directions. Cell Stem Cell. 10, 709–716. doi:10.1016/j.stem.2012.05.015
Keefe, K. M., Sheikh, I. S., and Smith, G. M. (2017). Targeting Neurotrophins to Specific Populations of Neurons: NGF, BDNF, and NT-3 and Their Relevance for Treatment of Spinal Cord Injury. Int. J. Mol. Sci., 18, 548. doi:10.3390/ijms18030548
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic Lateral Sclerosis. The Lancet 377, 942–955. doi:10.1016/S0140-6736(10)61156-7
Kim, H. J., Kim, N. C., Wang, Y.-D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in Prion-like Domains in hnRNPA2B1 and hnRNPA1 Cause Multisystem Proteinopathy and ALS. Nature 495, 467–473. doi:10.1038/nature11922
Kim, M. J., Vargas, M. R., Harlan, B. A., Killoy, K. M., Ball, L. E., Comte-Walters, S., et al. (2018a). Nitration and Glycation Turn Mature NGF into a Toxic Factor for Motor Neurons: A Role for p75NTRand RAGE Signaling in ALS. Antioxid. Redox Signaling 28, 1587–1602. doi:10.1089/ars.2016.6966
Kim, S. H., Oh, K.-W., Jin, H. K., and Bae, J.-S. (2018b). Immune Inflammatory Modulation as a Potential Therapeutic Strategy of Stem Cell Therapy for ALS and Neurodegenerative Diseases. BMB Rep. 51, 545–546. doi:10.5483/bmbrep.2018.51.11.255
Klein, S. M., Behrstock, S., McHugh, J., Hoffmann, K., Wallace, K., Suzuki, M., et al. (2005). GDNF Delivery Using Human Neural Progenitor Cells in a Rat Model of ALS. Hum. Gene Ther. 16, 509–521. doi:10.1089/hum.2005.16.509
Kondo, T., Funayama, M., Tsukita, K., Hotta, A., Yasuda, A., Nori, S., et al. (2014). Focal Transplantation of Human iPSC-Derived Glial-Rich Neural Progenitors Improves Lifespan of ALS Mice. Stem Cell Rep. 3, 242–249. doi:10.1016/j.stemcr.2014.05.017
Kook, M. G., Lee, S., Shin, N., Kong, D., Kim, D.-H., Kim, M.-S., et al. (2020). Repeated Intramuscular Transplantations of hUCB-MSCs Improves Motor Function and Survival in the SOD1 G93A Mice through Activation of AMPK. Sci. Rep. 10, 1572. doi:10.1038/s41598-020-58221-1
Kriegstein, A., and Alvarez-Buylla, A. (2009). The Glial Nature of Embryonic and Adult Neural Stem Cells. Annu. Rev. Neurosci. 32, 149–184. doi:10.1146/annurev.neuro.051508.135600
Kusuma, G. D., Carthew, J., Lim, R., and Frith, J. E. (2017). Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem Cell Dev. 26, 617–631. doi:10.1089/scd.2016.0349
Lauriat, T. L., and McInnes, L. A. (2007). EAAT2 Regulation and Splicing: Relevance to Psychiatric and Neurological Disorders. Mol. Psychiatry 12, 1065–1078. doi:10.1038/sj.mp.4002065
Le Gall, L., Anakor, E., Connolly, O., Vijayakumar, U. G., Duddy, W. J., and Duguez, S. (2020). Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 10, 101. doi:10.3390/jpm10030101
Lee, K.-H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016a). C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell. 167, 774. doi:10.1016/j.cell.2016.10.002
Lee, M., Ban, J.-J., Kim, K. Y., Jeon, G. S., Im, W., Sung, J.-J., et al. (2016b). Adipose-derived Stem Cell Exosomes Alleviate Pathology of Amyotrophic Lateral Sclerosis In Vitro. Biochem. Biophysical Res. Commun. 479, 434–439. doi:10.1016/j.bbrc.2016.09.069
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 487, 443–448. doi:10.1038/nature11314
Lepore, A. C., O'Donnell, J., Kim, A. S., Williams, T., Tuteja, A., Rao, M. S., et al. (2011). Human Glial-Restricted Progenitor Transplantation into Cervical Spinal Cord of the SOD1G93A Mouse Model of ALS. PLoS One. 6, e25968. doi:10.1371/journal.pone.0025968
Lepore, A. C., Rauck, B., Dejea, C., Pardo, A. C., Rao, M. S., Rothstein, J. D., et al. (2008). Focal Transplantation-Based Astrocyte Replacement Is Neuroprotective in a Model of Motor Neuron Disease. Nat. Neurosci. 11, 1294–1301. doi:10.1038/nn.2210
Lepore, A., and Maragakis, N. (2007). Targeted Stem Cell Transplantation Strategies in ALS. Neurochem. Int. 50, 966–975. doi:10.1016/j.neuint.2006.09.005
Lewis, C. M., and Suzuki, M. (2014). Therapeutic Applications of Mesenchymal Stem Cells for Amyotrophic Lateral Sclerosis. Stem Cell Res. Ther. 5, 32. doi:10.1186/scrt421
Li, Y., Chen, L., Zhao, Y., Bao, J., Xiao, J., Liu, J., et al. (2013). Intracranial Transplant of Olfactory Ensheathing Cells Can Protect Both Upper and Lower Motor Neurons in Amyotrophic Lateral Sclerosis. Cell Transplant. 22 Suppl 1 (22 Suppl. l), S51–S65. doi:10.3727/096368913x672208
Li, Y., Bao, J., Khatibi, N. H., Chen, L., Wang, H., Duan, Y., et al. (2011). Olfactory Ensheathing Cell Transplantation into Spinal Cord Prolongs the Survival of Mutant SOD1G93A ALS Rats through Neuroprotection and Remyelination. Anat. Rec. 294, 847–857. doi:10.1002/ar.21362
Liddelow, S., and Barres, B. (2015). SnapShot: Astrocytes in Health and Disease. Cell. 162, 1170. doi:10.1016/j.cell.2015.08.029
Liu, J., Li, X.-y., Liang, Z.-h., Han, C., Wei, W.-j., Song, C.-l., et al. (2017). Transplantation of Autologous Peripheral Blood Mononuclear Cells in the Subarachnoid Space for Amyotrophic Lateral Sclerosis: a Safety Analysis of 14 Patients. Neural Regen. Res. 12, 493–498. doi:10.4103/1673-5374.202918
Lo, B., and Parham, L. (2009). Ethical Issues in Stem Cell Research. Endocr. Rev. 30, 204–213. doi:10.1210/er.2008-0031
López-González, R., Kunckles, P., and Velasco, I. (2009). Transient Recovery in a Rat Model of Familial Amyotrophic Lateral Sclerosis after Transplantation of Motor Neurons Derived from Mouse Embryonic Stem Cells. Cell Transplant. 18, 1171–1181. doi:10.3727/096368909X12483162197123
Magota, H., Sasaki, M., Kataoka-Sasaki, Y., Oka, S., Ukai, R., Kiyose, R., et al. (2021a). Intravenous Infusion of Mesenchymal Stem Cells Delays Disease Progression in the SOD1G93A Transgenic Amyotrophic Lateral Sclerosis Rat Model. Brain Res. 1757, 147296. doi:10.1016/j.brainres.2021.147296
Magota, H., Sasaki, M., Kataoka-Sasaki, Y., Oka, S., Ukai, R., Kiyose, R., et al. (2021b). Repeated Infusion of Mesenchymal Stem Cells Maintain the Condition to Inhibit Deteriorated Motor Function, Leading to an Extended Lifespan in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis. Mol. Brain 14, 76. doi:10.1186/s13041-021-00787-6
Malik, N., and Rao, M. S. (2013). A Review of the Methods for Human iPSC Derivation. Methods Mol. Biol. 997, 23–33. doi:10.1007/978-1-62703-348-0
Marchetto, M. C. N., Muotri, A. R., Mu, Y., Smith, A. M., Cezar, G. G., and Gage, F. H. (2008). Non-Cell-Autonomous Effect of Human SOD1G37R Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells. Cell Stem Cell. 3, 649–657. doi:10.1016/j.stem.2008.10.001
Martin, L. J., Chen, K., and Liu, Z. (2005). Adult Motor Neuron Apoptosis Is Mediated by Nitric Oxide and Fas Death Receptor Linked by DNA Damage and P53 Activation. J. Neurosci. 25, 6449–6459. doi:10.1523/JNEUROSCI.0911-05.2005
Martínez, H. R., Molina-Lopez, J. F., González-Garza, M. T., Moreno-Cuevas, J. E., Caro-Osorio, E., Gil-Valadez, A., et al. (2012). Stem Cell Transplantation in Amyotrophic Lateral Sclerosis Patients: Methodological Approach, Safety, and Feasibility. Cell Transplant. 21, 1899–1907. doi:10.3727/096368911X582769
Martins-Macedo, J., Lepore, A. C., Domingues, H. S., Salgado, A. J., Gomes, E. D., and Pinto, L. (2021). Glial Restricted Precursor Cells in central Nervous System Disorders: Current Applications and Future Perspectives. Glia. 69, 513–531. doi:10.1002/glia.23922
Masrori, P., and Van Damme, P. (2020). Amyotrophic Lateral Sclerosis: a Clinical Review. Eur. J. Neurol. 27, 1918–1929. doi:10.1111/ene.14393
Masuda, A., Takeda, J. i., and Ohno, K. (2016). FUS -mediated Regulation of Alternative RNA Processing in Neurons: Insights from Global Transcriptome Analysis. WIREs RNA 7, 330–340. doi:10.1002/wrna.1338
Mazzini, L., Ferrero, I., Luparello, V., Rustichelli, D., Gunetti, M., Mareschi, K., et al. (2010). Mesenchymal Stem Cell Transplantation in Amyotrophic Lateral Sclerosis: A Phase I Clinical Trial. Exp. Neurol. 223, 229–237. doi:10.1016/j.expneurol.2009.08.007
Mazzini, L., Gelati, M., Profico, D., Sgaravizzi, G., Projetti Pensi, M., Muzi, G., et al. (2015). Human Neural Stem Cell Transplantation in ALS: Initial Results from a Phase I Trial. J. Translational Med. 13, 17. doi:10.1186/s12967-014-0371-2
McCampbell, A., Cole, T., Wegener, A. J., Tomassy, G. S., Setnicka, A., Farley, B. J., et al. (2018). Antisense Oligonucleotides Extend Survival and Reverse Decrement in Muscle Response in ALS Models. J. Clin. Invest. 128, 3558–3567. doi:10.1172/jci99081
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 13, 1310. doi:10.3389/fnins.2019.01310
Miller, T., Cudkowicz, M., Shaw, P. J., Andersen, P. M., Atassi, N., Bucelli, R. C., et al. (2020). Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 383, 109–119. doi:10.1056/NEJMoa2003715
Miller, T. M., Pestronk, A., David, W., Rothstein, J., Simpson, E., Appel, S. H., et al. (2013). An Antisense Oligonucleotide against SOD1 Delivered Intrathecally for Patients with SOD1 Familial Amyotrophic Lateral Sclerosis: a Phase 1, Randomised, First-In-Man Study. Lancet Neurol. 12, 435–442. doi:10.1016/S1474-4422(13)70061-9
Mitne-Neto, M., Machado-Costa, M., Marchetto, M. C. N., Bengtson, M. H., Joazeiro, C. A., Tsuda, H., et al. (2011). Downregulation of VAPB Expression in Motor Neurons Derived from Induced Pluripotent Stem Cells of ALS8 Patients. Hum. Mol. Genet. 20, 3642–3652. doi:10.1093/hmg/ddr284
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor Neuron Disease-Associated Loss of Nuclear TDP-43 Is Linked to DNA Double-Strand Break Repair Defects. Proc. Natl. Acad. Sci. USA 116, 4696–4705. doi:10.1073/pnas.1818415116
Mitrecic, D., Nicaise, C., Gajovic, S., and Pochet, R. (2010). Distribution, Differentiation, and Survival of Intravenously Administered Neural Stem Cells in a Rat Model of Amyotrophic Lateral Sclerosis. Cell Transplant. 19, 537–548. doi:10.3727/096368910X498269
Mitsumoto, H., Brooks, B. R., and Silani, V. (2014). Clinical Trials in Amyotrophic Lateral Sclerosis: Why So many Negative Trials and How Can Trials Be Improved? Lancet Neurol. 13, 1127–1138. doi:10.1016/S1474-4422(14)70129-2
Moller, A., Bauer, C. S., Cohen, R. N., Webster, C. P., and De Vos, K. J. (2017). Amyotrophic Lateral Sclerosis-Associated Mutant SOD1 Inhibits Anterograde Axonal Transport of Mitochondria by Reducing Miro1 Levels. Hum. Mol. Genet. 26, 4668–4679. doi:10.1093/hmg/ddx348
Morita, E., Watanabe, Y., Ishimoto, M., Nakano, T., Kitayama, M., Yasui, K., et al. (2008). A Novel Cell Transplantation Protocol and its Application to an ALS Mouse Model. Exp. Neurol. 213, 431–438. doi:10.1016/j.expneurol.2008.07.011
Moviglia, G. A., Moviglia-Brandolino, M. T., Varela, G. S., Albanese, G., Piccone, S., Echegaray, G., et al. (2012). Feasibility, Safety, and Preliminary Proof of Principles of Autologous Neural Stem Cell Treatment Combined with T-Cell Vaccination for ALS Patients. Cell Transplant. 21 Suppl 1, S57–S63. doi:10.3727/096368912X633770
Mueller, C., Berry, J. D., McKenna-Yasek, D. M., Gernoux, G., Owegi, M. A., Pothier, L. M., et al. (2020). SOD1Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 383, 151–158. doi:10.1056/NEJMoa2005056
Murry, C. E., and Keller, G. (2008). Differentiation of Embryonic Stem Cells to Clinically Relevant Populations: Lessons from Embryonic Development. Cell. 132, 661–680. doi:10.1016/j.cell.2008.02.008
Nabavi, S. M., Arab, L., Jarooghi, N., Bolurieh, T., Abbasi, F., Mardpour, S., et al. (2019). Safety, Feasibility of Intravenous and Intrathecal Injection of Autologous Bone Marrow Derived Mesenchymal Stromal Cells in Patients with Amyotrophic Lateral Sclerosis: An Open Label Phase I Clinical Trial. Cell J. 20, 592–598. doi:10.22074/cellj.2019.5370
Niedermeyer, S., Murn, M., and Choi, P. J. (2019). Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest. 155, 401–408. doi:10.1016/j.chest.2018.06.035
Nishitoh, H., Kadowaki, H., Nagai, A., Maruyama, T., Yokota, T., Fukutomi, H., et al. (2008). ALS-linked Mutant SOD1 Induces ER Stress- and ASK1-dependent Motor Neuron Death by Targeting Derlin-1. Genes Dev. 22, 1451–1464. doi:10.1101/gad.1640108
Nizzardo, M., Bucchia, M., Ramirez, A., Trombetta, E., Bresolin, N., Comi, G. P., et al. (2016). iPSC-derived LewisX+CXCR4+β1-Integrin+ Neural Stem Cells Improve the Amyotrophic Lateral Sclerosis Phenotype by Preserving Motor Neurons and Muscle Innervation in Human and Rodent Models. Hum. Mol. Genet. 25, 3152–3163. doi:10.1093/hmg/ddw163
Nizzardo, M., Simone, C., Rizzo, F., Ruggieri, M., Salani, S., Riboldi, G., et al. (2014). Minimally Invasive Transplantation of iPSC-Derived ALDHhiSSCloVLA4+ Neural Stem Cells Effectively Improves the Phenotype of an Amyotrophic Lateral Sclerosis Model. Hum. Mol. Genet. 23, 342–354. doi:10.1093/hmg/ddt425
O'Connor, D. M., and Boulis, N. M. (2012). Cellular and Molecular Approaches to Motor Neuron Therapy in Amyotrophic Lateral Sclerosis and Spinal Muscular Atrophy. Neurosci. Lett. 527, 78–84. doi:10.1016/j.neulet.2012.04.079
Oh, K.-W., Noh, M.-Y., Kwon, M.-S., Kim, H. Y., Oh, S.-i., Park, J., et al. (2018). Repeated Intrathecal Mesenchymal Stem Cells for Amyotrophic Lateral Sclerosis. Ann. Neurol. 84, 361–373. doi:10.1002/ana.25302
Okada, M., Yamashita, S., Ueyama, H., Ishizaki, M., Maeda, Y., and Ando, Y. (2018). Long-term Effects of Edaravone on Survival of Patients with Amyotrophic Lateral Sclerosis. eNeurologicalSci. 11, 11–14. doi:10.1016/j.ensci.2018.05.001
Oskarsson, B., Gendron, T. F., and Staff, N. P. (2018). Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clinic Proc. 93, 1617–1628. doi:10.1016/j.mayocp.2018.04.007
Papadeas, S. T., Kraig, S. E., O'Banion, C., Lepore, A. C., and Maragakis, N. J. (2011). Astrocytes Carrying the Superoxide Dismutase 1 (SOD1G93A) Mutation Induce Wild-type Motor Neuron Degeneration In Vivo. Proc. Natl. Acad. Sci. 108, 17803–17808. doi:10.1073/pnas.1103141108
Park, J.-M., Kim, S.-Y., Park, D., and Park, J.-S. (2020). Effect of Edaravone Therapy in Korean Amyotrophic Lateral Sclerosis (ALS) Patients. Neurol. Sci. 41, 119–123. doi:10.1007/s10072-019-04055-3
Park, S., Kim, H.-T., Yun, S., Kim, I.-S., Lee, J., Lee, I.-S., et al. (2009). Growth Factor-Expressing Human Neural Progenitor Cell Grafts Protect Motor Neurons but Do Not Ameliorate Motor Performance and Survival in ALS Mice. Exp. Mol. Med. 41, 487–500. doi:10.3858/emm.2009.41.7.054
Peruzzotti-Jametti, L., Bernstock, J. D., Vicario, N., Costa, A. S. H., Kwok, C. K., Leonardi, T., et al. (2018). Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell. 22, 355–368. doi:10.1016/j.stem.2018.01.020
Petrou, P., Kassis, I., Yaghmour, N. E., Ginzberg, A., and Karussis, D. (2021). A Phase II Clinical Trial with Repeated Intrathecal Injections of Autologous Mesenchymal Stem Cells in Patients with Amyotrophic Lateral Sclerosis. Front. Biosci. (Landmark Ed. 26, 693–706. doi:10.52586/4980
Petrov, D., Mansfield, C., Moussy, A., and Hermine, O. (2017). ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 9, 68. doi:10.3389/fnagi.2017.00068
Philips, T., Bento-Abreu, A., Nonneman, A., Haeck, W., Staats, K., Geelen, V., et al. (2013). Oligodendrocyte Dysfunction in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain. 136, 471–482. doi:10.1093/brain/aws339
Piepers, S., and van den Berg, L. H. (2010). No Benefits from Experimental Treatment with Olfactory Ensheathing Cells in Patients with ALS. Amyotroph. Lateral Scler. 11, 328–330. doi:10.3109/17482961003663555
Pluchino, S., and Cossetti, C. (2013). How Stem Cells Speak with Host Immune Cells in Inflammatory Brain Diseases. Glia. 61, 1379–1401. doi:10.1002/glia.22500
Popescu, I. R., Nicaise, C., Liu, S., Bisch, G., Knippenberg, S., Daubie, V., et al. (2013). Neural Progenitors Derived from Human Induced Pluripotent Stem Cells Survive and Differentiate upon Transplantation into a Rat Model of Amyotrophic Lateral Sclerosis. Stem Cell Transl Med. 2, 167–174. doi:10.5966/sctm.2012-0042
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 12, 25. doi:10.3389/fnmol.2019.00025
Ramón-Cueto, A., and Nieto-Sampedro, M. (1994). Regeneration into the Spinal Cord of Transected Dorsal Root Axons Is Promoted by Ensheathing Glia Transplants. Exp. Neurol. 127, 232–244. doi:10.1006/exnr.1994.1099
Ransohoff, R. M. (2016). A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 19, 987–991. doi:10.1038/nn.4338
Riancho, J., Gonzalo, I., Ruiz-Soto, M., and Berciano, J. (2019). Why Do Motor Neurons Degenerate? Actualisation in the Pathogenesis of Amyotrophic Lateral Sclerosis. Neurología (English Edition) 34, 27–37. doi:10.1016/j.nrl.2015.12.00110.1016/j.nrleng.2015.12.019
Rinaldi, C., and Wood, M. J. A. (2018). Antisense Oligonucleotides: the Next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol. 14, 9–21. doi:10.1038/nrneurol.2017.148
Rizvanov, A. A., Kiyasov, A. P., Gaziziov, I. M., Yilmaz, T. S., Kaligin, M. S., Andreeva, D. I., et al. (2008). Human Umbilical Cord Blood Cells Transfected with VEGF and L1CAM Do Not Differentiate into Neurons but Transform into Vascular Endothelial Cells and Secrete Neuro-Trophic Factors to Support Neuro-Genesis-A Novel Approach in Stem Cell Therapy. Neurochem. Int. 53, 389–394. doi:10.1016/j.neuint.2008.09.011
Rothstein, J. D. (2017). Edaravone: A New Drug Approved for ALS. Cell. 171, 725. doi:10.1016/j.cell.2017.10.011
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J., and Kuncl, R. W. (1995). Selective Loss of Glial Glutamate Transporter GLT-1 in Amyotrophic Lateral Sclerosis. Ann. Neurol. 38, 73–84. doi:10.1002/ana.410380114
Saitoh, Y., and Takahashi, Y. (2020). Riluzole for the Treatment of Amyotrophic Lateral Sclerosis. Neurodegenerative Dis. Manag. 10, 343–355. doi:10.2217/nmt-2020-0033
Shariati, A., Nemati, R., Sadeghipour, Y., Yaghoubi, Y., Baghbani, R., Javidi, K., et al. (2020). Mesenchymal Stromal Cells (MSCs) for Neurodegenerative Disease: A Promising Frontier. Eur. J. Cell Biol. 99, 151097. doi:10.1016/j.ejcb.2020.151097
Sharma, A. K., Sane, H. M., Paranjape, A. A., Gokulchandran, N., Nagrajan, A., D'sa, M., et al. (2015). The Effect of Autologous Bone Marrow Mononuclear Cell Transplantation on the Survival Duration in Amyotrophic Lateral Sclerosis - a Retrospective Controlled Study. Am. J. Stem Cell 4, 50–65.
Shefner, J., Heiman-Patterson, T., Pioro, E. P., Wiedau-Pazos, M., Liu, S., Zhang, J., et al. (2020). Long-term Edaravone Efficacy in Amyotrophic Lateral Sclerosis: Post-hoc Analyses of Study 19 (MCI186-19). Muscle Nerve. 61, 218–221. doi:10.1002/mus.26740
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W.-H., Hung, S.-T., et al. (2018). Haploinsufficiency Leads to Neurodegeneration in C9ORF72 ALS/FTD Human Induced Motor Neurons. Nat. Med. 24, 313–325. doi:10.1038/nm.4490
Siddique, T., and Ajroud-Driss, S. (2011). Familial Amyotrophic Lateral Sclerosis, a Historical Perspective. Acta Myol. 30, 117–120.
Sironi, F., Vallarola, A., Violatto, M. B., Talamini, L., Freschi, M., De Gioia, R., et al. (2017). Multiple Intracerebroventricular Injections of Human Umbilical Cord Mesenchymal Stem Cells Delay Motor Neurons Loss but Not Disease Progression of SOD1G93A Mice. Stem Cell Res. 25, 166–178. doi:10.1016/j.scr.2017.11.005
Siwek, T., Jezierska-Woźniak, K., Maksymowicz, S., Barczewska, M., Sowa, M., Badowska, W., et al. (2020). Repeat Administration of Bone Marrow-Derived Mesenchymal Stem Cells for Treatment of Amyotrophic Lateral Sclerosis. Med. Sci. Monit. 26, e927484. doi:10.12659/MSM.927484
Smethurst, P., Risse, E., Tyzack, G. E., Mitchell, J. S., Taha, D. M., Chen, Y.-R., et al. (2020). Distinct Responses of Neurons and Astrocytes to TDP-43 Proteinopathy in Amyotrophic Lateral Sclerosis. Brain. 143, 430–440. doi:10.1093/brain/awz419
Smith, R. A., Miller, T. M., Yamanaka, K., Monia, B. P., Condon, T. P., Hung, G., et al. (2006). Antisense Oligonucleotide Therapy for Neurodegenerative Disease. J. Clin. Invest. 116, 2290–2296. doi:10.1172/jci25424
Souayah, N., Coakley, K. M., Chen, R., Ende, N., and McArdle, J. J. (2012). Defective Neuromuscular Transmission in the SOD1G93A Transgenic Mouse Improves after Administration of Human Umbilical Cord Blood Cells. Stem Cell Rev. Rep. 8, 224–228. doi:10.1007/s12015-011-9281-3
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 319, 1668–1672. doi:10.1126/science.1154584
Srivastava, A. K., Gross, S. K., Almad, A. A., Bulte, C. A., Maragakis, N. J., and Bulte, J. W. M. (2017). Serial In Vivo Imaging of Transplanted Allogeneic Neural Stem Cell Survival in a Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 289, 96–102. doi:10.1016/j.expneurol.2016.12.011
Su, Z., and He, C. (2010). Olfactory Ensheathing Cells: Biology in Neural Development and Regeneration. Prog. Neurobiol. 92, 517–532. doi:10.1016/j.pneurobio.2010.08.008
Suttkus, A., Morawski, M., and Arendt, T. (2016). Protective Properties of Neural Extracellular Matrix. Mol. Neurobiol. 53, 73–82. doi:10.1007/s12035-014-8990-4
Suzuki, M., McHugh, J., Tork, C., Shelley, B., Hayes, A., Bellantuono, I., et al. (2008). Direct Muscle Delivery of GDNF with Human Mesenchymal Stem Cells Improves Motor Neuron Survival and Function in a Rat Model of Familial ALS. Mol. Ther. 16, 2002–2010. doi:10.1038/mt.2008.197
Suzuki, M., McHugh, J., Tork, C., Shelley, B., Klein, S. M., Aebischer, P., et al. (2007). GDNF Secreting Human Neural Progenitor Cells Protect Dying Motor Neurons, but Not Their Projection to Muscle, in a Rat Model of Familial ALS. PLoS One. 2, e689. doi:10.1371/journal.pone.0000689
Sweeney, M. D., Zhao, Z., Montagne, A., Nelson, A. R., and Zlokovic, B. V. (2019). Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 99, 21–78. doi:10.1152/physrev.00050.2017
Sykova, E., Cizkova, D., and Kubinova, S. (2021). Mesenchymal Stem Cells in Treatment of Spinal Cord Injury and Amyotrophic Lateral Sclerosis. Front. Cell Dev. Biol. 9, 695900. doi:10.3389/fcell.2021.695900
Tadesse, T., Gearing, M., Senitzer, D., Saxe, D., Brat, D. J., Bray, R., et al. (2014). Analysis of Graft Survival in a Trial of Stem Cell Transplant in ALS. Ann. Clin. Transl Neurol. 1, 900–908. doi:10.1002/acn3.134
Takahashi, K., and Yamanaka, S. (2013). Induced Pluripotent Stem Cells in Medicine and Biology. Development 140, 2457. doi:10.1242/dev.092551
Takahashi, K., and Yamanaka, S. (2006). Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 126, 663–676. doi:10.1016/j.cell.2006.07.024
Tang, Y., Yu, P., and Cheng, L. (2017). Current Progress in the Derivation and Therapeutic Application of Neural Stem Cells. Cell Death Dis. 8, e3108. doi:10.1038/cddis.2017.504
Terashima, T., Kobashi, S., Watanabe, Y., Nakanishi, M., Honda, N., Katagi, M., et al. (2020). Enhancing the Therapeutic Efficacy of Bone Marrow-Derived Mononuclear Cells with Growth Factor-Expressing Mesenchymal Stem Cells for ALS in Mice. iScience. 23, 101764. doi:10.1016/j.isci.2020.101764
Thomsen, G. M., Avalos, P., Ma, A. A., Alkaslasi, M., Cho, N., Wyss, L., et al. (2018). Transplantation of Neural Progenitor Cells Expressing Glial Cell Line-Derived Neurotrophic Factor into the Motor Cortex as a Strategy to Treat Amyotrophic Lateral Sclerosis. Stem Cells. 36, 1122–1131. doi:10.1002/stem.2825
Thomsen, G. M., Gowing, G., Latter, J., Chen, M., Vit, J.-P., Staggenborg, K., et al. (2014). Delayed Disease Onset and Extended Survival in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis after Suppression of Mutant SOD1 in the Motor Cortex. J. Neurosci. 34, 15587–15600. doi:10.1523/jneurosci.2037-14.2014
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 282, 1145–1147. doi:10.1126/science.282.5391.1145
Tognatta, R., and Miller, R. H. (2016). Contribution of the Oligodendrocyte Lineage to CNS Repair and Neurodegenerative Pathologies. Neuropharmacology. 110, 539–547. doi:10.1016/j.neuropharm.2016.04.026
Tziortzouda, P., Van Den Bosch, L., and Hirth, F. (2021). Triad of TDP43 Control in Neurodegeneration: Autoregulation, Localization and Aggregation. Nat. Rev. Neurosci. 22, 197–208. doi:10.1038/s41583-021-00431-1
Uccelli, A., Laroni, A., and Freedman, M. S. (2011). Mesenchymal Stem Cells for the Treatment of Multiple Sclerosis and Other Neurological Diseases. Lancet Neurol. 10, 649–656. doi:10.1016/S1474-4422(11)70121-1
Uccelli, A., Milanese, M., Principato, M. C., Morando, S., Bonifacino, T., Vergani, L., et al. (2012). Intravenous Mesenchymal Stem Cells Improve Survival and Motor Function in Experimental Amyotrophic Lateral Sclerosis. Mol. Med. 18, 794–804. doi:10.2119/molmed.2011.00498
Vahsen, B. F., Gray, E., Thompson, A. G., Ansorge, O., Anthony, D. C., Cowley, S. A., et al. (2021). Non-neuronal Cells in Amyotrophic Lateral Sclerosis - from Pathogenesis to Biomarkers. Nat. Rev. Neurol. 17, 333–348. doi:10.1038/s41582-021-00487-8
van Es, M. A., Hardiman, O., Chio, A., Al-Chalabi, A., Pasterkamp, R. J., Veldink, J. H., et al. (2017). Amyotrophic Lateral Sclerosis. The Lancet 390, 2084–2098. doi:10.1016/S0140-6736(17)31287-4
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 323, 1208–1211. doi:10.1126/science.1165942
Vasques, J. F., Teixeira Pinheiro, L. C., de Jesus Goncalves, R. G., Mendez-Otero, R., and Gubert, F. (2021). “Cell-Based Research and Therapy for Amyotrophic Lateral Sclerosis: Promises and Challenges,”. Amyotrophic Lateral Sclerosis. Editor T. Araki Brisbane, AU: Exon Publications,121-139. doi:10.36255/exonpublications.amyotrophiclateralsclerosis.celltherapy.2021
Venturin, G. T., Greggio, S., Zanirati, G., Marinowic, D. R., de Oliveira, I. M., Pêgas Henriques, J. A., et al. (2016). Transplantation of Bone Marrow Mononuclear Cells Prolongs Survival, Delays Disease Onset and Progression and Mitigates Neuronal Loss in Pre-symptomatic, but Not Symptomatic ALS Mice. Neurosci. Lett. 633, 182–188. doi:10.1016/j.neulet.2016.09.030
Venturini, A., Passalacqua, M., Pelassa, S., Pastorino, F., Tedesco, M., Cortese, K., et al. (2019). Exosomes from Astrocyte Processes: Signaling to Neurons. Front. Pharmacol. 10, 1452. doi:10.3389/fphar.2019.01452
Voulgari-Kokota, A., Fairless, R., Karamita, M., Kyrargyri, V., Tseveleki, V., Evangelidou, M., et al. (2012). Mesenchymal Stem Cells Protect CNS Neurons against Glutamate Excitotoxicity by Inhibiting Glutamate Receptor Expression and Function. Exp. Neurol. 236, 161–170. doi:10.1016/j.expneurol.2012.04.011
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S. W., Sandoe, J., et al. (2014). Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons. Cell Rep. 7, 1–11. doi:10.1016/j.celrep.2014.03.019
Walczak, P., All, A. H., Rumpal, N., Gorelik, M., Kim, H., Maybhate, A., et al. (2011). Human Glial-Restricted Progenitors Survive, Proliferate, and Preserve Electrophysiological Function in Rats with Focal Inflammatory Spinal Cord Demyelination. Glia. 59, 499–510. doi:10.1002/glia.21119
Wang, H. S., Hung, S. C., Peng, S. T., Huang, C. C., Wei, H. M., Guo, Y. J., et al. (2004). Mesenchymal Stem Cells in the Wharton's Jelly of the Human Umbilical Cord. Stem Cells. 22, 1330–1337. doi:10.1634/stemcells.2004-0013
Wang, Y., and Qin, Z.-h. (2010). Molecular and Cellular Mechanisms of Excitotoxic Neuronal Death. Apoptosis. 15, 1382–1402. doi:10.1007/s10495-010-0481-0
Wichterle, H., Lieberam, I., Porter, J. A., and Jessell, T. M. (2002). Directed Differentiation of Embryonic Stem Cells into Motor Neurons. Cell. 110, 385–397. doi:10.1016/s0092-8674(02)00835-8
Writing-GroupEdaravone-(MCI-186)-ALS-19-Study-Group (2017). Safety and Efficacy of Edaravone in Well Defined Patients with Amyotrophic Lateral Sclerosis: a Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 16, 505–512. doi:10.1016/S1474-4422(17)30115-1
Xiao, J., Yang, R., Biswas, S., Qin, X., Zhang, M., and Deng, W. (2015). Mesenchymal Stem Cells and Induced Pluripotent Stem Cells as Therapies for Multiple Sclerosis. Int. J. Mol. Sci. 16, 9283–9302. doi:10.3390/ijms16059283
Xu, L., Liu, T., Liu, L., Yao, X., Chen, L., Fan, D., et al. (2020). Global Variation in Prevalence and Incidence of Amyotrophic Lateral Sclerosis: a Systematic Review and Meta-Analysis. J. Neurol. 267, 944–953. doi:10.1007/s00415-019-09652-y
Xu, L., Mahairaki, V., and Koliatsos, V. E. (2012). Host Induction by Transplanted Neural Stem Cells in the Spinal Cord: Further Evidence for an Adult Spinal Cord Neurogenic Niche. Regenerative Med. 7, 785–797. doi:10.2217/rme.12.76
Xu, L., Ryugo, D. K., Pongstaporn, T., Johe, K., and Koliatsos, V. E. (2009). Human Neural Stem Cell Grafts in the Spinal Cord of SOD1 Transgenic Rats: Differentiation and Structural Integration into the Segmental Motor Circuitry. J. Comp. Neurol. 514, 297–309. doi:10.1002/cne.22022
Xu, L., Shen, P., Hazel, T., Johe, K., and Koliatsos, V. E. (2011). Dual Transplantation of Human Neural Stem Cells into Cervical and Lumbar Cord Ameliorates Motor Neuron Disease in SOD1 Transgenic Rats. Neurosci. Lett. 494, 222–226. doi:10.1016/j.neulet.2011.03.017
Xu, L., Yan, J., Chen, D., Welsh, A. M., Hazel, T., Johe, K., et al. (2006). Human Neural Stem Cell Grafts Ameliorate Motor Neuron Disease in SOD-1 Transgenic Rats. Transplantation 82, 865–875. doi:10.1097/01.tp.0000235532.00920.7a
Yamanaka, S. (2020). Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell. 27, 523–531. doi:10.1016/j.stem.2020.09.014
Yan, J., Xu, L., Welsh, A. M., Chen, D., Hazel, T., Johe, K., et al. (2006). Combined Immunosuppressive Agents or CD4 Antibodies Prolong Survival of Human Neural Stem Cell Grafts and Improve Disease Outcomes in Amyotrophic Lateral Sclerosis Transgenic Mice. Stem Cells. 24, 1976–1985. doi:10.1634/stemcells.2005-0518
Yang, E. J. (2021). A Novel Supplement Attenuates Oxidative Stress-Induced TDP-43-Related Pathogenesis in TDP-43-Expressed Cells. Evid. Based Complement. Alternat Med. 2021, 6773260. doi:10.1155/2021/6773260
Yang, Y. M., Gupta, S. K., Kim, K. J., Powers, B. E., Cerqueira, A., Wainger, B. J., et al. (2013). A Small Molecule Screen in Stem-Cell-Derived Motor Neurons Identifies a Kinase Inhibitor as a Candidate Therapeutic for ALS. Cell Stem Cell. 12, 713–726. doi:10.1016/j.stem.2013.04.003
Young, R. A. (2011). Control of the Embryonic Stem Cell State. Cell. 144, 940–954. doi:10.1016/j.cell.2011.01.032
Yun, Y., and Ha, Y. (2020). CRISPR/Cas9-Mediated Gene Correction to Understand ALS. Int. J. Mol. Sci. 21, 3801. doi:10.3390/ijms21113801
Zarei, S., Carr, K., Reiley, L., Diaz, K., Guerra, O., Altamirano, P., et al. (2015). A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int. 6, 171. doi:10.4103/2152-7806.169561
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 Repeat Expansion Disrupts Nucleocytoplasmic Transport. Nature 525, 56–61. doi:10.1038/nature14973
Zhang, K., Grima, J. C., Rothstein, J. D., and Lloyd, T. E. (2016). Nucleocytoplasmic Transport inC9orf72-Mediated ALS/FTD. Nucleus. 7, 132–137. doi:10.1080/19491034.2016.1172152
Zhou, Q., Yuan, M., Qiu, W., Cao, W., and Xu, R. (2021). Preclinical Studies of Mesenchymal Stem Cells Transplantation in Amyotrophic Lateral Sclerosis: a Systemic Review and Metaanalysis. Neurol. Sci. 42, 3637–3646. doi:10.1007/s10072-020-05036-7
Zhuang, W.-Z., Lin, Y.-H., Su, L.-J., Wu, M.-S., Jeng, H.-Y., Chang, H.-C., et al. (2021). Mesenchymal Stem/stromal Cell-Based Therapy: Mechanism, Systemic Safety and Biodistribution for Precision Clinical Applications. J. Biomed. Sci. 28, 28. doi:10.1186/s12929-021-00725-7
Zola, H., Fusco, M., Macardle, P. J., Flego, L., and Roberton, D. (1995). Expression of Cytokine Receptors by Human Cord Blood Lymphocytes: Comparison with Adult Blood Lymphocytes. Pediatr. Res. 38, 397–403. doi:10.1203/00006450-199509000-00021
Keywords: ALS, mesenchymal stem cell, motor neuron degeneration, precision medicine, cellular therapy
Citation: Lin T-J, Cheng K-C, Wu L-Y, Lai W-Y, Ling T-Y, Kuo Y-C and Huang Y-H (2022) Potential of Cellular Therapy for ALS: Current Strategies and Future Prospects. Front. Cell Dev. Biol. 10:851613. doi: 10.3389/fcell.2022.851613
Received: 10 January 2022; Accepted: 15 February 2022;
Published: 16 March 2022.
Edited by:
Yohan Oh, Hanyang University, South KoreaReviewed by:
Susanne Petri, Hannover Medical School, GermanyCopyright © 2022 Lin, Cheng, Wu, Lai, Ling, Kuo and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yen-Hua Huang, cml0YTEyMDRAdG11LmVkdS50dw==; Yung-Che Kuo, czAzMjcxQHRtdS5lZHUudHc=
†These authors contributed equally to this work and shared first authorship.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.