 Tiantian Li1†
Tiantian Li1† Michael Miao
Michael Miao Jianwei Li
Jianwei Li Xiaoheng Liu
Xiaoheng Liu- 1Institute of Biomedical Engineering, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu, China
- 2Division of Oral & Craniofacial Health Sciences, University of North Carolina Adams School of Dentistry, Chapel Hill, NC, United States
- 3Department of Endocrinology and Metabolism, West China Hospital, Sichuan University, Chengdu, China
Vascular calcification (VC) is linked to an increased risk of heart disease, stroke, and atherosclerotic plaque rupture. It is a cell-active process regulated by vascular cells rather than pure passive calcium (Ca) deposition. In recent years, extracellular vesicles (EVs) have attracted extensive attention because of their essential role in the process of VC. Matrix vesicles (MVs), one type of EVs, are especially critical in extracellular matrix mineralization and the early stages of the development of VC. Vascular smooth muscle cells (VSMCs) have the potential to undergo phenotypic transformation and to serve as a nucleation site for hydroxyapatite crystals upon extracellular stimulation. However, it is not clear what underlying mechanism that MVs drive the VSMCs phenotype switching and to result in calcification. This article aims to review the detailed role of MVs in the progression of VC and compare the difference with other major drivers of calcification, including aging, uremia, mechanical stress, oxidative stress, and inflammation. We will also bring attention to the novel findings in the isolation and characterization of MVs, and the therapeutic application of MVs in VC.
Introduction
Vascular calcification (VC) is a prominent clinical pathology of atherosclerosis, diabetes mellitus, hypertension, aging, and chronic kidney disease (CKD), resulting in abnormal calcium phosphate accumulation in the intimal and medial layers of the vessel wall (Xu et al., 2020). After vascular calcification, the stiffness of the vascular wall is increased, and the compliance is decreased, which results in myocardial ischemia, left ventricular hypertrophy, and heart failure (Nitta and Ogawa, 2015). At present, vascular calcification is still lacks effective treatment methods, and the pathogenesis mechanism remains unclear (Herrmann et al., 2020). Therefore, it is necessary to uncover its specific mechanisms and develop therapeutic strategies.
The phenotype switching of the VSMCs has been regarded as the principal driver in the calcification of intimal and medial layers. VSMCs undergo the phenotypic transformation from a differentiated “contractile” into a dedifferentiated “synthetic” proliferative phenotype in the process of vascular calcification. During this phenotypic switching, VSMCs show decreased expression of the contractile markers smooth muscle α-actin (α-SMA), smooth muscle 22α (SM22α), smooth muscle myosin heavy chain 11 (SM-MHC), CNN1, calponin, leiomodin, smoothelin (SMTN), myosin light chain (MYL) and an increase in synthetic marker S100A4, KLF4, vimentin, osteopontin (OPN) (Furmanik et al., 2020). The phenotypic switching VSMCs express higher osteoblast-like markers, such as runt-related transcription factor 2 (Runx2), zinc finger transcription factor (Osterix), muscle segment homeobox 2 (MSX2) (Qi et al., 2019), and is associated with increased proliferation and migration ability (Kapustin and Shanahan, 2016). The osteoblast-like phenotype of VSMCs is regarded as the cellular characteristic factor of vascular calcification. Many factors such as oxidative stress damage, hyperphosphatemic environment, and inflammation increase the indices related to bone formation in VSMCs and promote their transformation into osteoblasts (Voelkl et al., 2018a). On the other hand, a variety of biochemical factors are involved in the phenotype switching of VSMCs, i.e., the growth factors platelet-derived growth factor (PDGF-BB) and transforming growth factor-β1 (TGF-β1) could promote the phenotype switching of VSMCs (Jain et al., 2021).
Matrix vesicles (MVs), one kind of extracellular matrix-derived EVs, are membrane-bound microparticles released by cells, containing various cargo, including proteins, carbohydrates, lipids, DNA and mRNAs, and species of small RNAs, such as microRNAs (miRNAs). The origin and composition of MVs determine their calcification potential (Zazzeroni et al., 2018). Recent evidence showed that extracellular MVs serve as nucleating foci to initiate microcalcification (Demer and Tintut, 2014). MV nanofragments obtained by mechanical rupture could induce rapid mineralization in vitro (Kunitomi et al., 2019). The formation and secretion of MVs and the increase of intracellular alkaline phosphatase (ALP) activity are also involved in the phenotypic transformation and osteoblast-like phenotype transformation of VSMCs (Lin et al., 2016). However, the specific mechanisms and functions of MVs regulating vascular calcification have not been fully elucidated. For example, what is the originating cell that releases MVs in vascular calcification, and how do the pro-calcification MVs get into the recipient cell? On the other hand, increasing evidence shows that EVs have potential therapeutic applications for many diseases. For example, overexpressing a high-affinity variant human PD-1 protein (havPD-1) EVs has been used to reduce cancer cell proliferation and induce apoptosis (Chen et al., 2022). Another therapeutic role of EVs is dependent on their proteins and/or non-coding RNAs, in particular miRNAs delivery ability (de Abreu et al., 2020). It has also been shown that MVs carry miR-199a-3p, which, by regulating GATA-binding 4 acetylation, were able to rescue electric function in engineered and in vivo atria (Aday et al., 2017). Alginate-based microreactors that were loaded with MVs as support for bone-like osteoblast cells were shown to enhance biomineralization in a co-assembled 3D spheroid (Itel et al., 2018). Therefore, engineered MVs have emerged as a potential therapeutic method to treat vascular calcification. In this review, targeted interventions based on MVs to treat vascular calcification are also discussed (New et al., 2013).
Vascular Calcification
Physiological calcification is a normal process that occurs in bones and teeth; however, pathological calcification occurs in soft tissue such as blood vessels, joints, and tumors, in association with aging, advanced atherosclerosis, diabetes, and chronic kidney disease (Jing et al., 2019). The classification of vascular calcification is divided into intimal calcification, media calcification and cardiac valve calcification Figure 1 (Viegas and Simes, 2018). Vascular intimal calcification often occurs in atherosclerotic diseases. Intimal calcification is related to the stability of atherosclerotic plaque and the microcalcifications that contribute to plaque rupture by concentrating mechanical forces within the fibrous cap (Blaser and Aikawa, 2018). After calcification is formed, it is scattered on the inner wall of the blood vessel in the form of a spot or patch (Nakahara et al., 2017). The early-stage shows microcalcification (range: ≥0.5 to <15 μm) and then develops a spot-like calcification (Nakahara et al., 2017). Vascular media calcification often occurs in the internal elastic lamina and is more common in small and medium arteries such as the femoral artery, tibial artery and radial artery, also known as Monckeberg’s medial sclerosis, which is more common in patients with advanced aging, diabetes and CKD (Zazzeroni et al., 2018). Medial calcification decreases vessel compliance, increases pulse wave velocity and systolic hypertension. The occurrence of cardiac valve calcification is related to mechanical stress and inflammation and is commonly seen in metabolic diseases such as diabetes, dyslipidemia, uremia, etc. When cardiac valve calcification occurs, spotted calcium salts are deposited, accompanied by valve fibrosis and inflammatory cell infiltration (Shekar and Budoff, 2018).
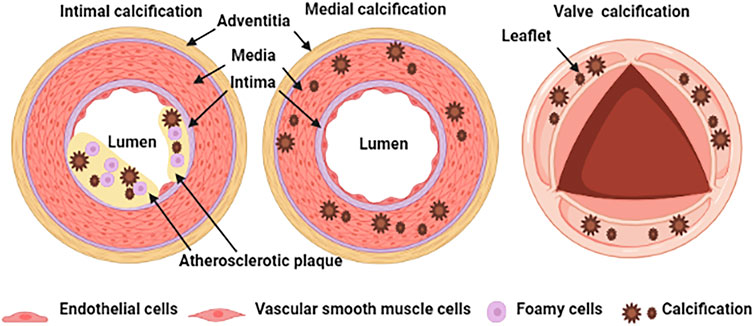
FIGURE 1. Schematic representation of vascular calcification classification.
The main cause of vascular calcification is the ectopic deposition of hydroxyapatite crystals in blood vessel wall cells and extracellular matrix (ECM) (Viegas and Simes, 2018). During the last few decades, vascular calcification had been regarded as a process of accumulating insoluble precipitates of Ca phosphate without a specific cellular biological response. In recent years, increasing evidence shows that vascular cells actively regulate the calcification process (Proudfoot, 2019).
The Contribution of Matrix Vesicles to Vascular Calcification
Many risk factors such as calcium and phosphorus metabolism disorder, inflammation, oxidative stress, apoptosis, autophagy and aging can contribute to vascular calcification. They can also intersect or interact with each other, thereby affecting the occurrence and development of vascular calcification (Pescatore et al., 2019). Vascular calcification is a major complication of CKD patients. This may be due to the decline of renal function, leading to calcium and phosphorus metabolism disorders, which in turn leads to hyperphosphatemia and hypercalcemia. Both hypercalcemia and hyperphosphatemia may promote intimal and media calcification. Hyperphosphatemia may increase the activity of the sodium-dependent cotransporters, PIT-1 and PIT-2, and up-regulate genes associated with matrix mineralization, which eventually leads to vascular calcification (Cozzolino et al., 2019). Interestingly, hypercalcemia and hyperphosphatemia both increase the release of MVs, leading to the deposition of hydroxyapatite in the extracellular matrix (Reynolds et al., 2004).
Inflammatory cells such as macrophages promote vascular calcification by secreting inflammatory cytokines such as TNF-a, IL-1β, and IL-6 (Henaut et al., 2016). Inflammatory cytokines directly promote calcification in VSMCs by down-regulating VSMC-specific genes and up-regulating osteoblastic genes such as BMP2 and ALP. They also indirectly reduce circulating Fetuin-A, MGP, and Klotho levels, further promoting vascular calcification procession in VSMCs (Henaut et al., 2016; Zhang et al., 2018). MVs secreted by pro-inflammatory cells also play pivotal roles in the procession of vascular calcification. These MVs participate in vascular calcification by forming the phosphatidylserine (PS)-annexin A5-S100A9 membrane complex involved in mineralization or by mediating the transport of miRNAs such as miRNA-153 and miRNA-223 (Henaut et al., 2016; Zhang et al., 2018).
Oxidative stress-induced by intracellular calcium and phosphate overloading promotes vascular calcification via inducing signal activation and VSMCs osteogenic phenotype transition (Nguyen et al., 2020). Pro-mineralizing MVs induced calcification of recipient VSMCs by stimulating reactive oxygen species (ROS) production (Chen et al., 2021). Apoptotic cells form a nidus for calcification, releasing apoptotic bodies loaded with calcium that participate in the calcification process by deposition of hydroxyapatite in the extracellular matrix (Cozzolino et al., 2019).
Emerging evidence has demonstrated that autophagy is directly related to vascular calcification. Crucially, autophagy maintains the physiological function of VSMCs by regulating Ca2+ homeostasis (Phadwal et al., 2020). Autophagy-related proteins and protein complexes play important roles in exosome biogenesis (Xu et al., 2018). It has been shown that MVs are rich in annexin-A5, which is also known to have an important role in the formation of mature autophagosomes (Ghislat and Knecht, 2012). Autophagic proteins including LAMP1, LAMP2, and LAMTOR1 were found in MVs released from rat VICs (Cui et al., 2016). In the progression of vascular calcification, MVs may be entwined with the network of autophagic vesicles during the formation or release stage (Cui et al., 2016). Cell autophagy has a special pro-calcification effect than cell apoptosis. It increases the inorganic phosphate (Pi)-induced MVs release with increased ALP activity (Dai et al., 2013). Therefore, inhibiting the autophagy pathway may help to prevent Pi-induced vascular calcification by reducing MVs release (Dai et al., 2013; Cui et al., 2016).
Vascular calcification is associated with aging, which is commonly seen in older adults or in middle-aged subjects affected by premature vascular aging. MVs released from senescent ECs and elderly subjects’ plasma promote calcification of VSMCs, and MV carriage of increased quantities of annexins A2 and A6, BMP2, and Ca (Alique et al., 2017). EVs released from senescent cells can also drive the senescence of neighboring cells in a paracrine manner (Blaser and Aikawa, 2018).
The above-mentioned studies underscore the essential role of MVs, which are totally dependent on their cellular source, in regulating the vascular calcification process. Therefore, the origin of MVs is briefly described below.
The Origin of Matrix Vesicles
EVs encompass a wide variety of vesicles with a lipid bilayer membrane structure released by various kinds of cells in either resting or stress states (Goettsch et al., 2013). The main characteristics of various EVs are described in Table 1. According to the route of biogenesis, size, density, and protein markers, EVs are categorized into exosomes (50–150 nm), microvesicles (100–500 nm), and apoptotic bodies (1,000–5,000 nm) (Mathivanan et al., 2010; Goettsch et al., 2013; Blaser and Aikawa, 2018) (Figure 2). Exosomes are released through the endosomal-sorting complex, where intraluminal vesicles formed by the inward budding of the endosomal membrane are packaged within multivesicular bodies (MVBs). These MVBs then fuse with the plasma membrane to release their enclosed exosomes in a Rab GTPase-dependent manner (Blaser and Aikawa, 2018). Microvesicles are formed by direct budding and fission of the plasma membrane (Mathivanan et al., 2010; Blaser and Aikawa, 2018). Apoptotic bodies are formed by apoptotic cells due to the breakdown of the cytoskeleton, which induces the plasma membrane to bulge, leading to the separation of the plasma membrane (Mathivanan et al., 2010; Blaser and Aikawa, 2018).
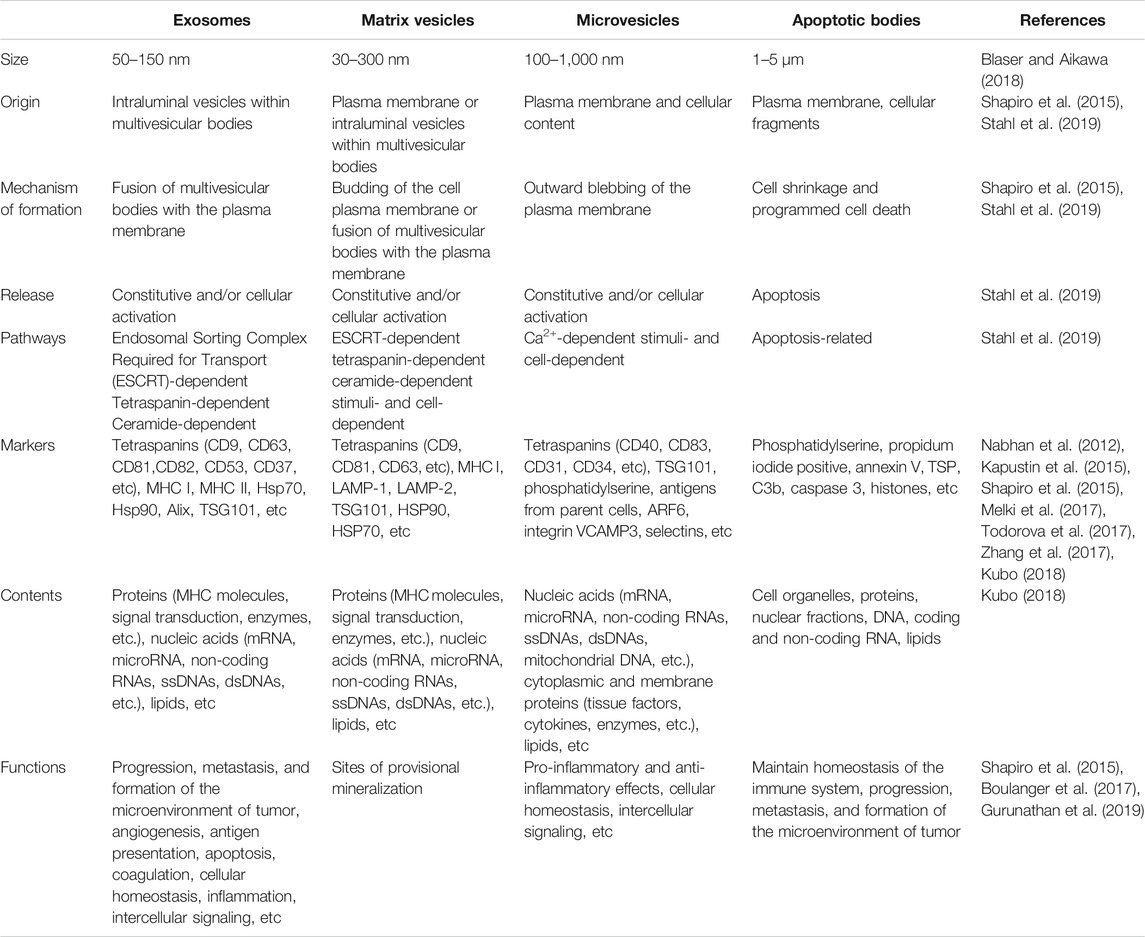
TABLE 1. Main characteristics of exosomes, matrix vesicles, microvesicles, and apoptotic bodies.
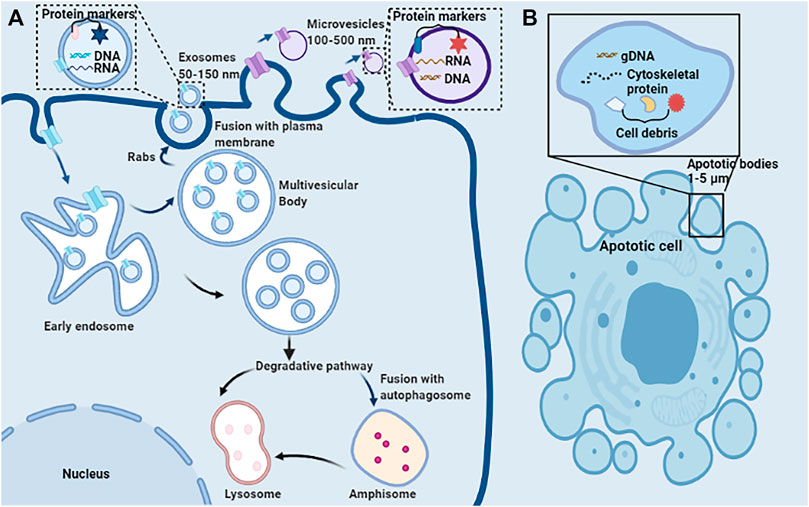
FIGURE 2. Schematic representation of the release, structure, and composition of extracellular vesicles. (A) Direct sprouting and splitting of the plasma membrane deformed into microvesicles (100–500 nm in diameter). In early endosomes, proteins are sequestered in intraluminal vesicles of the larger MVBs. The inward budding of endosomal membranes forms intraluminal vesicles of MVBs. MVBs bud inward and then fuse with the plasma membrane results in the release of their enclosed exosomes (∼50–150 nm) from the cell into the microenvironment. Due to its biophysical properties, MVBs can be degraded by entering the lysosome directly or fusing with autophagosome and then entering the lysosome. (B) Apoptotic or dying cells shrink to produce apoptotic bodies (ABs) (1,000–5,000 nm). ABs are condensed remnants of the apoptotic cell, with nuclear and cytoplasmic components.
In the process of vascular calcification, EVs that are released by macrophages are 30–300 nm in diameter and originate as membranous protrusions in the intima layer (Blaser and Aikawa, 2018). Besides this, VSMCs-derived EVs, with a diameter of 100–150 nm, originating from an exosomal pathway in the media layer, are processed by multivesicular bodies and are enriched in members of the Rab GTPase family (Blaser and Aikawa, 2018). Previous advances have shown that a common mechanism may mediate both intimal and medial vascular calcification, that is, a substantial increase of EVs in the vascular interstitial space, in particular, the small EVs (sEVs, with a size of 40–100 or to 140 nm) which are mainly produced and secreted from arterial VSMCs. These sEVs facilitate the formation of nucleate Ca phosphate (Ca/P) crystals in the form of Hydroxyapatite (Bhat et al., 2020a). Valve homeostasis also depends on appropriate intercellular interactions between valvular endothelial cells (VECs) and valvular interstitial cells (VICs) (Bakhshian Nik et al., 2017). EVs are released by VICs can influence VECs, and transfer of VIC-derived EVs containing perinuclear proteins to VECs cultures reveals intercellular vesicle transfer through the endosomal pathway (Krohn et al., 2016a; Bakhshian Nik et al., 2017).
MVs, another kind of EVs, are approximately 100–300 nm in diameter, membrane-invested particles, and can be located in the ECM (Bottini et al., 2018). Under physiological conditions, MVs can be released from hypertrophic chondrocytes, osteoblasts, odontoblasts, and tenocytes. In addition, some non-skeletal tissue cells such as VSMCs, in dead or dying macrophages, and VICs also release pathological MVs (Bakhshian Nik et al., 2017). Pro-vascular calcification MVs contain reactive oxygen species and pro-inflammatory cytokines. While inhibiting vascular calcification MVs secret anti-inflammatory factors and retards VSMCs to differentiate into osteoblast-like cells (Li et al., 2020). Macrophage-derived MVs express exosomal markers (CD9 and TSG101) and contain PS-annexin A5-S100A9 membrane complex, which promotes hydroxyapatite nucleation and directly contributes to microcalcification in chronic renal disease (New et al., 2013). MVs derived from macrophages enhance ectopic mineralization in the high Ca/Pi environment (Zhang et al., 2018). VSMCs-derived MVs ranging in size from 50 to 500 nm respond by disturbing intracellular calcium homeostasis and enhance mineralization by releasing the key mineralization inhibitor matrix Gla protein loading, enhancing matrix metalloproteinase-2 activity, and forming annexin A6/PS nucleation complexes (Kapustin et al., 2011). On the other hand, circulating EVs could be taken up by recipient VSMCs, which contribute to vascular calcification in CKD, through calcification/osteogenic differentiation and inflammatory status, leading to increased mineral deposition (Viegas et al., 2018).
In calcified aortic valve disease (CAVD), the phenotypic changes of VICs play a vital role in ECM remodeling and mineral deposition (Bakhshian Nik et al., 2017). Under pathological conditions, such as hyperphosphatemia, VICs can differentiate into two distinct phenotypes: an activated myofibroblast-like VIC (aVIC) or an osteoblast-like VIC (oVIC), which are responsible for the active deposition of Ca in CAVD (Hjortnaes et al., 2015). In this calcifying milieu, pro-calcific VIC-derived EVs are similar to MVs from chondrocytes and VSMCs, showing elevated annexins A2, A5 and A6 (Bakhshian Nik et al., 2017). In the standard Ca/Pi calcification environment, the expression of vascular calcification-associated protein annexin A6 and Ca content were significantly up-regulated in rat VICs derived MVs (Cui et al., 2017). Through bioinformatics analysis techniques, MVs from calcified VICs were analyzed to reveal calcification regulators and exosome markers, including CD9, CD63, LAMP-1, and LAMP-2 (Cui et al., 2017).
Based on the above content, MVs should not be confused with microvesicles according to their different biogenesis type, particle size, and functional location.
Strategies and Opportunities to Modify Matrix Vesicles to Treat Vascular Calcification
Nucleation and Crystallization of Minerals
Under pathological conditions, such as inflammation or hyperlipidemia, the increased expression and activity of ALP will promote calcification, stimulating the release of phosphate from biological sources (for example, ATP) for mineral nucleation (Blaser and Aikawa, 2018). In this process, the multiligand sorting receptor sortilin in EVs regulates the release of EVs and drives the activated ALP to load into calcifying EVs in a Rab11-dependent manner (Blaser and Aikawa, 2018). Importantly, MVs are a cell product that can be used as sites for hydroxyapatite crystal precipitation. The origin and content of MVs are essential factors in determining their mineralization potential (Kapustin et al., 2011). MVs can obtain mineral compounds to maintain intracellular mineral metabolic homeostasis and participate in the process of vascular calcification by promoting mineral deposition sites formation (Zhang et al., 2018). Elevated extracellular Ca and cytosolic Ca alterations are necessary to produce the mineralization-competent MVs (Kapustin et al., 2011). In the presence of high Ca, VSMCs can release more mineralization-competent MVs, which are filled with microspicules of crystalline mineral, predominantly composed of Ca, which would lead to matrix mineralization (Reynolds et al., 2004). Unlike the cell membrane composition, MVs are rich in annexins, sphingomyelins, and PS, which are vital mineral nucleation sites on the MVs membrane (Genge et al., 2007). Upon the stimulation of Ca and phosphate, calcification inhibitor is suppressed, and annexins/PS nucleation complexes is formed, providing hydroxyapatite nucleation sites (Kapustin et al., 2011; New et al., 2013). Meanwhile, the MVs membrane also contains calcification inhibitors such as fetuin-A and MGP, which prevent mineral nucleation (Reynolds et al., 2004). In calcifying EVs, the homeostasis between calcification inhibitors and promoters is broken, resulting in a loss of mineralization inhibition.
MiRNAs in Matrix Vesicles Regulate the Process of Vascular Calcification
For the reason that the lipid bilayer of EVs can protect the small RNAs from degradation by extracellular ribonuclease and proteases (Blaser and Aikawa, 2018), small RNAs such as ribosomal RNA (rRNA), miRNAs, and transfer RNA (tRNA) are enriched in EVs. Interestingly, the total miRNA contained in EVs is higher, while the total RNA is less than the donor cells (Chaturvedi et al., 2015). MiRNAs in EVs could be transported and regulate the proliferation and differentiation of recipient cells. Furthermore, it also mediates intercellular communication during the vascular calcification process (Goettsch et al., 2013). In uraemic rats EVs, the expression of miR-221 and miR-222 were significantly enhanced, and the levels of miR-143 and miR-145 were significantly reduced. These EVs can participate in the phenotypic switch and the calcification of VSMCs via enhanced AKT signaling and PiT-1 expression (Freise et al., 2021). Endothelial cell-derived EVs are selectively loaded with miR-126, miR-143, and miR-145, which participate in VSMCs phenotype regulation by mediating paracrine signaling across cells within the vessel wall (Krohn et al., 2016a). Bone marrow mesenchymal stem cells derived exosomes inhibited high P-induced calcification in VSMCs through modifying miRNA profiles. The mTOR, MAPK, and Wnt signaling pathways were involved in this process (Guo et al., 2019). On the other hand, many miRNAs in EVs are also involved in inhibiting the function of vascular calcification. MiR-30, miR-125-b, miR-143, miR-145 and miR-155 loaded into calcifying VSMCs-derived EVs can influence the expression of a specific set of osteogenic markers such as Smad1, RUNX-2, ALP, and osterix, and change in the concentrations of these miRNAs in EVs lead to shifts in Ca and MAPK signaling pathways implicated in SMC-mediated calcification (Krohn et al., 2016a). Melatonin-induced exosomes miR-204 and miR-211 inhibit the osteogenic differentiation of VSMCs by reducing RUNX2 and BMP2 expression and ALP activity (Xu et al., 2020). Exosome-derived miR-324-3p regulate the expression levels of the IGF1R, MAP2K1 and PIK3CA proteins in the mouse smooth muscle cell line (MOVAS-1) and inhibit coronary artery calcification (Pan et al., 2020).
In the mineralization environment, especially the vascular calcification process, some miRNAs in MVs facilitate deposition of Ca orthophosphate, which can change Ca phosphate structure from an amorphous form to more crystalline structures, such as hydroxyapatite; some miRNAs have the opposite effect, inhibiting mineral deposition (Krohn et al., 2016a). In VMSCs from CKD rats, miR-667, miR-702, miR-3562, miR-3568, and miR-3584 are highly concentrated in MVs (Chaturvedi et al., 2015). Some miRNAs contained in MVs can regulate the phenotype of recipient cells. These studies indicate that the dysregulation of miRNAs in MVs may promote vascular calcification by causing abnormal mineral deposits, promoting hydroxyapatite’s formation, or by driving the transformation of VSMCs phenotype from a contractile to a synthetic and osteochondrogenic phenotype. MVs also act as carriers to transfer dysregulated miRNAs from cell to cell to induce changes in the function of the recipient cells, which leads to vascular calcification.
Proteins Associated With Vascular Calcification in Matrix Vesicles
MVs contain a large number of proteins, each of which has specific characteristics and performs its specific functions in mineral deposits. The endogenous protein in MVs could be divided into three groups, inhibitors, promoters, and other proteins. The inhibitors in MVs, such as MGP, Gla-rich protein (GRP), and fetoglobulin A (fetuin-A) (Kapustin et al., 2015), suppress the calcification, while the promoters in MVs such as annexin family, sortilin, and S100 protein family aggregate the calcification. Finally, other proteins functionally regulate MVs secretion.
Inhibitors in Matrix Vesicles
MGP is a 15-kDa vitamin K-dependent mineral binding protein, which is widely expressed in VSMCs and chondrocytes (Bjorklund et al., 2020). MGP is known as an effective inhibitor of Ca phosphate precipitation, which can inhibit aortic and cartilage calcification (Xiao et al., 2021). The reason why MGP exerts an anti-calcfication role is that carboxylated and phosphorylated MGP could eliminate the calcification effect of various bone morphogenetic proteins (BMPs) such as BMP-2 and BMP-4 (Houben et al., 2016; Bjorklund et al., 2020; Khan et al., 2021). In addition, MGP-fetuin-A complex inhibits ectopic mineralization by combining with alkaline Ca phosphate crystals (Xiao et al., 2021). MGP is a potent inhibitor of ECM mineralization; deficiency of MGP initiates arterial calcification (Khavandgar et al., 2014). Deposits of hydroxyapatite minerals were shown in the arterial wall of MGP-deficient (Mgp−/−) mice, which died within 2 months (Khavandgar et al., 2014). The anti-calcification function of MGP is most likely related to the formation of MVs (Bjorklund et al., 2020). The elevated Ca concentration decreases the MGP expression in MVs, while a similar result is not observed upon high concentration of phosphate (Houben et al., 2016).
GRP (Gla-rich protein) is an inhibitor of vascular calcification involved in Ca homeostasis, and γ-carboxylated GRP could inhibit calcification and osteochondrogenic differentiation by α-SMA upregulation and osteopontin downregulation (Viegas et al., 2015). GRP was expressed at protein and mRNA levels in EVs released by macrophages, which may act as vehicles for ECM transport and information transmission (Viegas et al., 2017). It prevents Ca-induced signaling pathways and directly binds to minerals to inhibit crystal formation (Viegas et al., 2015). Ucma/GRP (Upper zone of growth plate and cartilage matrix-associated protein/Gla-rich protein) can directly interact with BMP2 and regulate phosphate-induced mineralization of VSMCs by participating in BMP-2-SMAD1/5/8 osteo/chondrogenic signaling pathway in vitro (Willems et al., 2018). GRP can form a large complex with MGP and fetuin-A, which is loaded in noncalcified EVs; the ratio of the complex is decreased in the high Ca-loaded vesicles (Xiao et al., 2021). Additionally, they can also combine with minerals to form a fetuin-mineral complex called colloids calciprotein particles (CPP), which greatly affects the stability of minerals (Xiao et al., 2021). Circulating CPPs and EVs, which contain lower levels of fetuin-A and GRP, are determinants of vascular calcification in CKD, with the capacity to modulate VSMCs responses through increased osteochondrogenic differentiation and inflammation, leading to increased mineral deposition (Viegas et al., 2018).
Fetuin-A is a plasma glycoprotein from the family of cystatin protease inhibitors with a TGF-β cytokine-binding motif and a Ca phosphate-binding site that can inhibit the precipitation of basic calcium phosphate in vitro (Khan et al., 2021). Fetuin-A is one of the biomarkers acting as calcification inhibitors, and it plays an important role in ectopic calcification. The possible mechanisms of fetuin-A involved in ectopic vascular calcification include inhibiting spontaneous mineral nucleation and growth and inhibiting the formation of hydroxyapatite crystals; combining clusters of Ca and phosphate to stabilize these ions and preventing cellular uptake; mediating the formation of protein mineral CPP (Icer and Yildiran, 2021). Fetuin-A can be loaded into MVs, which maintains the phenotype of healthy VSMCs and also binds minerals and stabilizes them against further growth, preventing vesicles from mineralizing (Chen and Moe, 2015; Kapustin et al., 2015).
Osteoprotegerin (OPG) is known to be a soluble tumor necrosis factor (TNF) superfamily receptor that inhibits the actions of the cytokine receptor activator of nuclear factor kappa-B ligand (RANKL) (Montanez-Barragan et al., 2014). OPG was also detected in VSMCs- derived MVs and co-located with annexin A6 (Cui et al., 2016). Under physiological conditions, OPG can be secreted by vesicles released from viable or apoptotic VSMCs and directly inhibit VSMCs mineralization by limiting MVs driven mineral nucleation and hydroxyapatite deposition in the vascular wall (Cui et al., 2016).
Promoters in Matrix Vesicles
As the membrane factors of MVs, the annexin family members A2, A5, and A6 mediate Ca influx into MVs and are well-known calcification promoters (Kapustin et al., 2011). The massive enrichment of various annexins in calcifying MVs may be a key factor in regulating MVs release and calcification potential in the cardiovascular system. For example, annexin A2 could bind to fetuin-A at the cell membrane of VSMCs in the presence of high calcium and contribute to MVs-mediated calcification. Additionally, the high level of annexin A2 might also be one of the reasons that end-stage renal disease-exosomes promote VSMCs calcification (Lin et al., 2020). Annexin A6 was enriched in calcifying VIC-derived MVs, and these MVs may promote aortic valve calcification in end-stage renal disease (Cui et al., 2017). Most importantly, annexin A6, the largest member of the annexin family, is present highly in MVs. Biochemical analyses revealed annexin A6 could have three different localizations in MVs during physiological mineralization. They are Ca2+-bound annexin A6 interacting with the inner leaflet of the MVs membrane; annexin A6 localized on the surface of the outer leaflet; and annexin A6 inserted in the membrane’s hydrophobic bilayer and co-localized in lipid domains enriched in cholesterol (Veschi et al., 2020). Moreover, annexin A6 and Ca induced PS exposure on the MVs surface and formed of annexin A6/PS nucleation complexes, thus providing hydroxyapatite nucleation sites (Hjortnaes et al., 2015).
Sortilin is a sorting receptor that directs target proteins, including growth factors, signaling receptors, and enzymes, to the secretary or endocytic compartments of cells. Sortilin expression levels are atypically high in calcified arteries in CKD and atherosclerosis disease. Moreover, sortilin deficiency reduces vascular but not skeletal calcification in vivo, indicating that sortilin might be a key regulator of vascular calcification (Goettsch et al., 2016). Sortilin plays a direct role in ectopic calcification and has become a potential cardiovascular risk biomarker and a drug target for cardiovascular disease (Gebraad et al., 2018). In older men aged ≥50 years, higher serum sortilin levels are associated with a higher risk of major adverse cerebrovascular and cardiovascular events (MACCE) and severe abdominal aortic calcification (AAC) (Goettsch et al., 2017). The receptors for AGEs (RAGE) down-regulated sortilin and mediated the formation of microcalcification, whereas galectin-3 up-regulated sortilin and induced macrocalcification (Sun et al., 2019).
Sortilin could traffic and load the calcification protein TNAP into VSMC-derived EVs, conferring its calcification potential (Itoh et al., 2018). Sortilin forms homodimers in the extracellular and intracellular domains with intermolecular disulfide bonds, which likely facilitate the trafficking of sortilin to the EVs (Itoh et al., 2018). Sortilin could accelerate the formation of MVs aggregates in the early stage of calcification and affect the microcalcification signals (Sun et al., 2019). In a diabetic apolipoprotein E-deficient background (ApoE−/−) mouse model, mouse tail vein injection of Nε-Carboxymethyl-lysine (CML)-induced MVs originated from VSMCs-derived obviously aggravated diabetic atherosclerotic calcification (Jing et al., 2020). High concentrations of CML significantly promoted the release of MVs from VSMCs and the recruitment of sortilin to MVs (Jing et al., 2020).
The S100 protein family has 21 members, which can be released from monocytes, VSMCs, and ECs in response to cellular stress stimuli. Some members of the S100 proteins family, such as S100A8, S100A9, and S100A12 have homologous structures and functions, commonly known as S100/calgranulins, and are closely related to cardiovascular disease (Xiao et al., 2020). These proteins bind with their receptors, such as advanced glycation end products (RAGE), scavenger receptors (CD36), and toll-like receptor 4 (TLR-4), contributing to the cellular response in vascular inflammation, vascular oxidative stress, and vascular calcification progression (Xiao et al., 2020). Among the S100 protein family, S100A9 is closely related to cardiovascular calcifying MVs. S100A9 is the Ca2+-binding neutrophil cytosolic protein, which is strongly expressed in calcifying areas, the surrounding extracellular matrix, and calcifying MVs (McCormick et al., 2005). S100A9 in atherosclerotic plaque and calcifying MVs may be involved in the pathogenesis of atherosclerosis and dystrophic calcification by influencing redox- and Ca2+-dependent processes (McCormick et al., 2005). Macrophage S100A9 plays an important role in vascular calcification in diabetes mellitus. When exposed to high glucose, pro-inflammatory macrophages-derived calcific EVs through the S100A9-RAGE axis contribute to the excessive microcalcification formation within plaques (Kawakami et al., 2020).
MMP-2, also known as gelatinase A, was involved in the process of vascular calcification. MMP-2 facilitates vascular calcification by catalyzing matrix degradation (Kapustin and Shanahan, 2016). The active form of MMP-2 is also found in VSMCs derived MVs, and elevated levels of extracellular Ca can induce enhanced MMP-2 activity. Inhibiting the activity of MMP-2 loaded in MVs can prevent vascular calcification (Kapustin and Shanahan, 2012).
Transglutaminase 2 (TG2) is a Ca-dependent enzyme that can facilitate cell-ECM interaction through integrins (Chen et al., 2013). TG2 becomes the core of inducing the arterial calcification program by promoting VSMCs transdifferentiation to osteoblasts and chondrocytes (Johnson et al., 2008). The expression of TG2 is increased in both VSMCs and MVs isolated from CKD rats. The cross-linking of the ECM with TG2 leads to enhanced cell-ECM and MVs-ECM attachment, induced phenotypic switch of VSMCs to contractile “osteoblast-like” cells, and reduced function of calcification inhibitors (Chen et al., 2013).
The phosphatase orphan 1 (PHOSPHO1), contained in MVs, as an additional inorganic phosphate (Pi) supplier was involved in the first step of MVs-mediated initiation of mineralization (Kiffer-Moreira et al., 2013; Bobryshev et al., 2014).
The Regulator Controlling Matrix Vesicles Secretion
Sphingomyelin phosphodiesterase-3 (SMPD3) is not only a key signal molecule in the process of biomineralization but also a regulator of exosomal biogenesis. In VSMCs, elevated extracellular Ca increased SMPD3 expression. In turn, the up-regulation of SMPD3 could regulate exosome secretion and fetuin-A recycling (Kapustin et al., 2015). Increased ceramide in cell membrane or cytoplasm through the SMPD3 pathway may induce sEVs biogenesis, initiating arterial calcification (Bhat et al., 2020a). Lysosomal overexpression of Smpd1 gene specifically in VSMCs may reduce lysosome-MVB interactions, which may reduce lysosome degradation of MVBs and increase the fusion of MVBs with the plasma membrane to release sEVs, and consequently leading to medial arterial calcification (Bhat et al., 2020a).
High mobility group box 1 (HMGB1), a nuclear protein that binds to chromatin, is usually released into the extracellular space as a damage-associated molecular pattern (DAMP) when cells are activated, damaged, or die (Chen et al., 2017). HMGB1 is also a cytokine associated with biological mineralization and could induce macrophages to secrete MVs through the RAGE/p38MAPK/nSMase2 signaling pathway to enhance ectopic mineralization (Chen et al., 2016). BMSC-derived exosomes alleviate high phosphate-induced aortic calcification and ameliorate vascular function via the SIRT6–HMGB1 deacetylation (Wei et al., 2021).
Microtubule stabilization inhibited hyperphosphatemia-induced vascular calcification by down-regulation of osteogenic signal and attenuation of MVs release (Lee et al., 2014). Mucolipin 1 Gene deletion in mice increased sEVs secretion by inhibiting lysosomes and MVB interactions in arterial stiffening and medial calcification. The expression of exosome/sEVs markers, CD63, and annexin A2 were significantly increased in the coronary arterial wall of Mcoln1−/− mice (Bhat et al., 2020b). Endoplasmic reticulum (ER) stress induces calcification of VSMCs in vitro and modifies VSMCs phenotype by increasing the release of Grp78-loaded calcifying EVs (Furmanik et al., 2021).
The interaction between EVs and ECM accelerates the extracellular accumulation and aggregation of calcifying EVs. In human atherosclerotic plaque formation, a large amount of disorganized collagen will accumulate before calcification begins. However, the content of collagen is inversely proportional to the size of microcalcifications: when collagen is degraded, EVs can aggregate, nucleate hydroxyapatite and form microcalcifications (Blaser and Aikawa, 2018). Many high levels of collagen-binding proteins such as ALP, proteoglycan link proteins, hyaluronic-acid binding regions, and annexins are also found in calcifying EVs (Blaser and Aikawa, 2018). It indicated that the collagen protein inhibits the accumulation of EVs. The MVs secreted by VSMCs can locate close to collagen fibrils and interact with collagen type I (COL1) in VSMCs under calcifying conditions (Tyson et al., 2003). Fetuin-A, MGP, S100A9, and annexins have emerged as active regulators of this interaction (Reynolds et al., 2004). 8-degree-polymerized oligogalacturonic acid prevents vascular calcification development by inhibiting the osteogenic marker expression and dissociating the direct interaction between MVs and COL1 (Hodroge et al., 2017). In particular, this interaction can be inhibited by masking the GFOGER sequence, which reveals the specific areas on the collagen fibers that bind to EVs (Krohn et al., 2016b; Hodroge et al., 2017). The collagen receptor discoidin domain receptor-1 (DDR-1) can regulate collagen deposition and release of calcifying EVs by VSMCs through the TGF-β pathway. In calcification media, DDR-1−/− VSMCs increased the release of TGF-β1, which can stimulate P38 phosphorylation and inhibit the activation of SMPD3, thereby promoting EVs-mediated calcification and collagen matrix production (Krohn et al., 2016b).
The Signal Pathway Involved in Matrix Vesicles Regulating Vascular Calcification
During the vascular calcification process, the production and release of cellular MVs is the first step, while the uptake of MVs by recipient cells is also essential. The detailed molecular mechanisms regulating MVs secretion and inclusions in vascular calcification are quite complicated. For instance, uraemic EVs augment Ca- and phosphate-induced osteogenic transdifferentiation of VSMCs via activated AKT and ERK signaling and PiT-1 expression (Freise et al., 2021). The lysosomal sphingolipid/ceramide pathway may be crucial participants in the secretion of sEVs and phenotypic switch in arterial VSMCs (Bhat et al., 2020a). Warfarin is known to be an oral anticoagulant and an effective inducer of calcification, which could increase EVs release and VSMCs calcification via the PERK-ATF4 ER stress pathway (Furmanik et al., 2021). The autophagy pathway may be a key participant in the regulation of MVs-mediated vascular calcification progression (Dai et al., 2013). Through bioinformatic analysis using Ingenuity Pathway Analysis (IPA) revealed the up-regulation of key signaling pathways crucial to cardiovascular function in calcifying VIC-derived MVs in end-stage renal disease, including aldosterone, P2Y purinergic receptor signaling, and thrombin signaling pathways, Rho signaling, and metal binding (Cui et al., 2017). Cellular-derived MVs with characteristics of exosomes and low fetuin-A content from calcifying VSMCs of rats with CKD enhanced the calcification of recipient VSMCs by inducing cell signaling changes and phenotypic switch of recipient VSMCs (Chen et al., 2018). Several major cell signaling pathways were involved in this process: 1) mitogen-activated protein kinase (MAPK) signaling specifically, increased MEK1 and ERK signaling; 2) increased intracellular Ca from sarcoplasmic reticulum stress; 3) increased NADPH oxidase (NOX) and superoxide dismutase (SOD) activity; 4) activation and increased intracellular Ca, leading to reducing VSMCs calcification (Chen et al., 2018). EVs secreted from both patients with acute coronary syndrome and senescent ECs induce early senescence of recipient endothelial cells and cellular oxidative stress. MAPK, Akt, and p53 signaling pathways may be involved in this process (Blaser and Aikawa, 2018).
Toolbox: Isolation and Validation of Matrix Vesicles
Ultracentrifugation
UC is the most commonly used method for MVs purification at present. Differential centrifugation can be used to separate vesicles of similar sizes. UC is usually used in conjunction with sucrose density gradient centrifugation to separate low-abundance small vesicles (Hutcheson et al., 2014). Although this combined method can obtain highly purified MVs, it also has some disadvantages: the steps are complicated with an inconsistent recovery rate. Most importantly, repeated centrifugation is likely to disrupt vesicles and reduce their quality. Some soluble proteins may contaminate the sample by forming clumps with the vesicles, potentially impacting proteomic and RNA content analysis (Lobb et al., 2015).
Ultrafiltration
MVs can also be obtained by selective separation of samples with ultrafiltration membranes of different pore sizes. Compared to UC, UF is less time-consuming and does not require the use of special equipment (Cheruvanky et al., 2007). The combination of UF and size exclusion chromatography (UF/SEC) is superior to the traditional UC method in terms of production time, standardization, scalability, and vesicle yield. Compared with traditional UC, using UF/SEC, approximately 400 times more small extracellular vesicles (sEVs) per ml of media were recovered, and upscaling this process further increases sEVs yield by about 3-fold (Cardoso et al., 2021).
Magnetic Bead Sorting
CD63, CD31, CD9, 70 kDa heat shock proteins (HSP70), and tumor susceptibility gene 101 (TSG101) are the well-known marker genes of MVs. After the MVs are incubated with magnetic beads coated with anti-label antibodies, they can be adsorbed and separated. Using a process of immunoprecipitation based on polymeric beads that were modified with an antibody for tissue nonspecific alkaline phosphatase (TNALP), MVs were isolated from an organotypic culture model (Iordachescu et al., 2018). The magnetic bead sorting method has the advantages of high specificity, simple operation and can maintain the complete shape of MVs (Kluszczynska et al., 2019). However, antigen-antibody binding requires sufficient time for action, so this method generally has the problem of time-consuming operation and low extraction efficiency (Lane et al., 2017), and it may also be affected by the pH value and salt concentration of the separation solution.
Polymer-Based Precipitation
Because of its simplicity and feasibility, polymer-based precipitation such as polyethylene glycol (PEG) precipitation has gained popularity. After incubating with PEG at 4°C, the MVs are precipitated and recovered by filtration or centrifugation. Although this method is effective in obtaining small EVs (sEVs), some of the non-sEV contaminants are also precipitated at the same time (Nakai et al., 2016). Therefore, this method is not commonly used due to the shortcomings of low vesicle recovery rate, low purity, and uneven particle size of isolated MVs (Lobb et al., 2015).
In recent years, various extraction kits have been used for the isolation of MVs. These kits are increasingly popular because of their simple operation, high extraction efficiency, and the ability to extract urine, blood, cell supernatant, and other samples separately. Researchers have developed other methods, including HPLC (high-performance liquid chromatography)-based protocols (Zeringer et al., 2015), affinity-capture methods (Iordachescu et al., 2018), microfluidic-based isolation of vesicles methods (Wang et al., 2013), acoustic-based purification methods (Lee et al., 2015), etc. Each of these separation methods has its unique advantages, but their disadvantages also limit their wide use.
The Identification Methods of Matrix Vesicles
Nanoparticle Tracking Analysis
NTA is one of the most commonly used methods to count the number of MV particles. NTA can analyze the size of small vesicles particles in batches and obtain a particle size distribution curve (Hoshino et al., 2020). It is easy to operate and can protect the structure and function of MVs from damage. However, the concentration of the sample particles greatly influences the results. Therefore, to obtain stable and reliable results, the sample needs to be diluted to an appropriate concentration.
Protein Marker Detection
Enzyme-linked immunosorbent assay (ELISA), flow cytometry and western blot can be used to detect MVs protein markers. Commonly used markers include CD31, CD9, CD63, annexins, integrin receptors, TSG101 and HSP70 (Shapiro et al., 2015).
Quantitative Real Time-Polymerase Chain Reaction Analysis
qRT-PCR is the most sensitive and reliable method to detect gene expression. MVs contain a large number of microRNAs and these microRNAs play an important role in the formation of mineral nucleation sites and the osteogenic differentiation of VSMCs. qRT-PCR is an indispensable method for studying these microRNAs (Sohn et al., 2015). At the same time, a bioanalyzer and droplet digital PCR(ddPCR) can be used to detect the RNA quality and quantity of MVs load.
Electron Microscopy
Transmission electron microscopy (TEM) is a standard morphological detection method for MVs. The particle size of MVs can be observed by TEM, and the MVs are spherical and cup holder-like structures (Greening et al., 2015). Now some laboratories are also beginning to use atomic force microscopes to look at the shape of vesicles. Density-dependent scanning electron microscopy (SEM) can image calcifying EVs in-situ, and assess their size, density, and biophysical accumulation in calcified tissues, hydrogels, or matrices (Blaser and Aikawa, 2018). With the help of super-resolution microscopy, it is possible to obtain in situ imaging of vesicles using confocal light-based approaches (Blaser and Aikawa, 2018).
Immunogold Labeling
Immunogold labeling is a kind of immunolabeling technology that uses colloidal gold as a tracer to participate in the antigen-antibody response. Exosomes were incubated with primary antibody and the secondary antibody conjugated to gold particles and then observed under an electron microscope after washing and counter staining (Hannafon et al., 2016). It has the advantages of simple and quick operation, high sensitivity, and stable markers.
Discussion
Vascular calcification is a complex, active and highly regulated biological process. As important transporters for material transport and intercellular communication, MVs are closely related to vascular calcification. MVs can participate in vascular calcification by promoting the phenotypic transformation of VSMCs, regulating mineral deposits, mediating microRNAs transport, regulating cell signaling pathways (Figure 3).
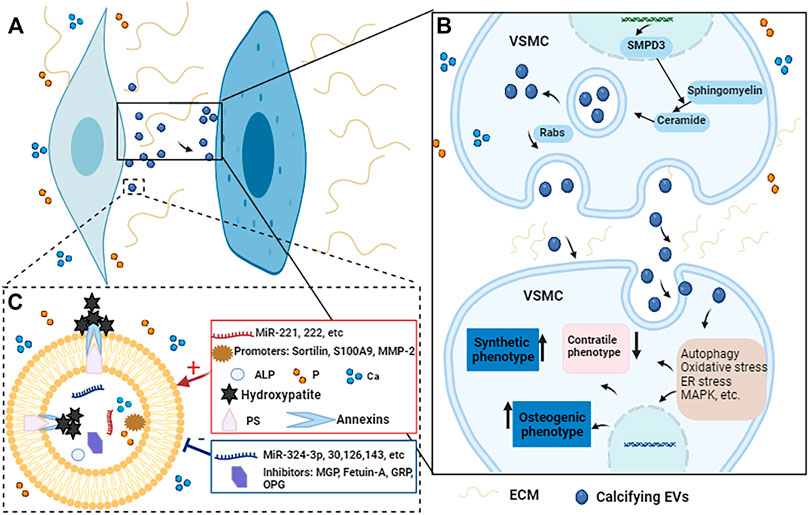
FIGURE 3. The potential regulatory mechanisms of calcifying EVs regulate the phenotypic switch and osteogenic differentiation of VSMCs during vascular calcification. (A) When VSMCs are exposed to elevated Ca and P conditions, the calcifying EVs it releases act on the recipient VSMCs to change their phenotype and biological function. (B) Under pro-calcific milieu, SMPD3 can enhance the secretion of exosomes, which are secreted in a Rabs-dependent manner. Calcifying EVs can regulate the phenotypic transformation and osteogenic differentiation of recipient VSMCs through multiple signaling pathways such as autophagy, oxidative stress, ER stress, and MAPK, etc. (C) Moreover, the composition of calcifying EVs determines its potential to load hydroxyapatite. When EVs are exposed to high Ca and P conditions, the balance between calcification promoters and inhibitors in EVs is broken, increasing the content of promoters such as Ca, P, microRNAs, and annexins. At the same time, the concentration of some inhibitors such as MGP, fetuin-A, and some microRNAs is reduced. Therefore, the mineral concentration gradient between the intra- and extra-vesicular spaces drives the influx of P and Ca into EVs via suitable transporters to form mineral nucleation sites. The annexins and PS complexes provide hydroxyapatite nucleation sites, leading to calcifying EVs and microcalcification.
MVs are endogenous nanovesicles secreted by living cells. Therefore, they are more suitable as therapeutic drug carriers due to their advantages of small particle size, low toxicity, non-immunogenicity, good permeability, and high targeting. Recent studies have shown that VSMCs-derived MVs have shown positive results in many disease models, such as reducing the size of myocardial infarction and improving the inflammation caused after myocardial infarction, resolving pulmonary hypertension, ameliorating renal fibrosis, and restoring neurovascular functions and plasticity (Blaser and Aikawa, 2018). In addition to using MVs as delivery vehicles, the contents of calcifying EVs themselves have also been proven to be an essential source of drug targets (Blaser and Aikawa, 2018). Therefore, MVs may be a new treatment for vascular calcification.
Additionally, researchers have successfully used exosomes to carry small molecules, nucleic acids, and proteins to treat various diseases in recent years. It may be possible to use exogenous engineered EVs as a viable therapeutic strategy for vascular calcification, whereby inhibitors of vascular calcification could be efficiently conveyed into atherosclerotic plaques or vascular microcalcification areas. However, the use of EVs as a nano-drug delivery system is still in its infancy in treating diseases, and researchers are still facing many problems and challenges, such as the low rate of drug encapsulation in EVs (Gaurav et al., 2021). So far, there is not a feasible method to treat vascular calcification. Some potential medications that address vascular calcification have been studied only in preclinical setups or lack extensive clinical research. Potential medications and treatment methods for addressing vascular calcification are listed in Table 2. The role of MVs in the occurrence and development of vascular calcification has not been clearly studied. We have not fully understood, under pathophysiological conditions, how the effective or key molecules in MVs change. This may be due to the disrupted balance between the promoters and inhibitors, the content of mirRNA-30, 125b, 126, 143, 145, fetuin-A, MGP, and GRP that inhibit calcification in MVs is reduced; in contrast, mirRNA-221, 222, annexins, MMP-2 are increased. How do the effective molecules interact with each other? For example, PS exposed on the outside of MVs membrane can bind to GRP and interact with annexins to form complexes that determine the formation of nucleation sites. How are the effective molecules packaged into the MVs? MVs are small in size, easy to penetrate biofilm, and lipid bilayer membrane structure can protect the small RNAs and proteins in EVs from degradation by extracellular ribonuclease and proteases. MVs can enrich signal molecules and make them have a higher local concentration. How are MVs generated and released? SMPD3 is a key protein regulating MVs production. The small Rab GTPases such as Rab11, Rab27, Rab35 regulate MVs secretion by different ways mediating the transport of MVBs toward the plasma membrane. How are the targeted transport of MVs regulated, and how do target cells recruit and activate MVs? This may be because in pathological environments, MVs can induce receptor-mediated signal transduction via surface-binding ligands; deliver specific functional proteins and miRNAs to target cells through cells surface receptors. In order to find out the specific treatment methods of vascular calcification based on MVs for vascular calcification in future studies, we can focus on mineralization-related proteins and miRNAs in MVs, and we could use MVs as carriers to develop safer and more effective nanomedicines.

TABLE 2. Potential medications and treatment methods to treat vascular calcification.
Author Contributions
TL, HY, and XL designed the scope of the review. TL and HY wrote the manuscript. DZ, TF, MM, and JL performed the document searching and prepared the figures. XL and JL guided the planning and critically edited the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (11932014 and 31971239).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aday, S., Zoldan, J., Besnier, M., Carreto, L., Saif, J., Fernandes, R., et al. (2017). Synthetic Microparticles Conjugated with VEGF165 Improve the Survival of Endothelial Progenitor Cells via microRNA-17 Inhibition. Nat. Commun. 8 (1), 747. doi:10.1038/s41467-017-00746-7
Alique, M., Ruíz-Torres, M. P., Bodega, G., Noci, M. V., Troyano, N., Bohórquez, L., et al. (2017). Microvesicles from the Plasma of Elderly Subjects and from Senescent Endothelial Cells Promote Vascular Calcification. Aging 9 (3), 778–789. doi:10.18632/aging.101191
Bakhshian Nik, A., Hutcheson, J. D., and Aikawa, E. (2017). Extracellular Vesicles as Mediators of Cardiovascular Calcification. Front. Cardiovasc. Med. 4, 78. doi:10.3389/fcvm.2017.00078
Bhat, O. M., Yuan, X., Cain, C., Salloum, F. N., and Li, P. L. (2020a). Medial Calcification in the Arterial wall of Smooth Muscle Cell‐specific Smpd1 Transgenic Mice: A Ceramide‐mediated Vasculopathy. J. Cel Mol Med 24 (1), 539–553. doi:10.1111/jcmm.14761
Bhat, O. M., Yuan, X., Camus, S., Salloum, F. N., and Li, P.-L. (2020b). Abnormal Lysosomal Positioning and Small Extracellular Vesicle Secretion in Arterial Stiffening and Calcification of Mice Lacking Mucolipin 1 Gene. Ijms 21 (5), 1713. doi:10.3390/ijms21051713
Bjørklund, G., Svanberg, E., Dadar, M., Card, D. J., Chirumbolo, S., Harrington, D. J., et al. (2020). The Role of Matrix Gla Protein (MGP) in Vascular Calcification. Cmc 27 (10), 1647–1660. doi:10.2174/0929867325666180716104159
Blaser, M. C., and Aikawa, E. (2018). Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 5, 187. doi:10.3389/fcvm.2018.00187
Bobryshev, Y., Orekhov, A., Sobenin, I., and Chistiakov, D. (2014). Role of Bone-type Tissue-Nonspecific Alkaline Phosphatase and PHOSPO1 in Vascular Calcification. Cpd 20 (37), 5821–5828. doi:10.2174/1381612820666140212193011
Bottini, M., Mebarek, S., Anderson, K. L., Strzelecka-Kiliszek, A., Bozycki, L., Simão, A. M. S., et al. (2018). Matrix Vesicles from Chondrocytes and Osteoblasts: Their Biogenesis, Properties, Functions and Biomimetic Models. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1862 (3), 532–546. doi:10.1016/j.bbagen.2017.11.005
Boulanger, C. M., Loyer, X., Rautou, P.-E., and Amabile, N. (2017). Extracellular Vesicles in Coronary Artery Disease. Nat. Rev. Cardiol. 14 (5), 259–272. doi:10.1038/nrcardio.2017.7
Brinton, T. J., Ali, Z. A., Hill, J. M., Meredith, I. T., Maehara, A., Illindala, U., et al. (2019). Feasibility of Shockwave Coronary Intravascular Lithotripsy for the Treatment of Calcified Coronary Stenoses. Circulation 139 (6), 834–836. doi:10.1161/CIRCULATIONAHA.118.036531
Cardoso, R. M. S., Rodrigues, S. C., Gomes, C. F., Duarte, F. V., Romao, M., Leal, E. C., et al. (2021). Development of an Optimized and Scalable Method for Isolation of Umbilical Cord Blood-Derived Small Extracellular Vesicles for Future Clinical Use. Stem Cell Transl Med 10 (6), 910–921. doi:10.1002/sctm.20-0376
Chaturvedi, P., Chen, N. X., O’Neill, K., McClintick, J. N., Moe, S. M., and Janga, S. C. (2015). Differential miRNA Expression in Cells and Matrix Vesicles in Vascular Smooth Muscle Cells from Rats with Kidney Disease. PLoS One 10 (6), e0131589. doi:10.1371/journal.pone.0131589
Chen, N. X., and Moe, S. M. (2015). Pathophysiology of Vascular Calcification. Curr. Osteoporos. Rep. 13 (6), 372–380. doi:10.1007/s11914-015-0293-9
Chen, N. X., O'Neill, K., Chen, X., Kiattisunthorn, K., Gattone, V. H., and Moe, S. M. (2013). Transglutaminase 2 Accelerates Vascular Calcification in Chronic Kidney Disease. Am. J. Nephrol. 37 (3), 191–198. doi:10.1159/000347031
Chen, N. X., O'Neill, K. D., and Moe, S. M. (2018). Matrix Vesicles Induce Calcification of Recipient Vascular Smooth Muscle Cells through Multiple Signaling Pathways. Kidney Int. 93 (2), 343–354. doi:10.1016/j.kint.2017.07.019
Chen, N. X., O’Neill, K. D., Dominguez, J. M., Moe, S. M., and Moe, S. M. (2021). Regulation of Reactive Oxygen Species in the Pathogenesis of Matrix Vesicles Induced Calcification of Recipient Vascular Smooth Muscle Cells. Vasc. Med. 26, 585–594. doi:10.1177/1358863X211024721
Chen, Q., Bei, J.-J., Liu, C., Feng, S.-B., Zhao, W.-B., Zhou, Z., et al. (2016). HMGB1 Induces Secretion of Matrix Vesicles by Macrophages to Enhance Ectopic Mineralization. PLoS One 11 (5), e0156686. doi:10.1371/journal.pone.0156686
Chen, Q., Wang, Z.-Y., Chen, L.-Y., and Hu, H.-Y. (2017). Roles of High Mobility Group Box 1 in Cardiovascular Calcification. Cell Physiol Biochem 42 (2), 427–440. doi:10.1159/000477591
Chen, Y., Wang, L., Zheng, M., Zhu, C., Wang, G., Xia, Y., et al. (2022). Engineered Extracellular Vesicles for Concurrent Anti-PDL1 Immunotherapy and Chemotherapy. Bioactive Mater. 9, 251–265. doi:10.1016/j.bioactmat.2021.07.012
Cheruvanky, A., Zhou, H., Pisitkun, T., Kopp, J. B., Knepper, M. A., Yuen, P. S. T., et al. (2007). Rapid Isolation of Urinary Exosomal Biomarkers Using a Nanomembrane Ultrafiltration Concentrator. Am. J. Physiology-Renal Physiol. 292 (5), F1657–F1661. doi:10.1152/ajprenal.00434.2006
Cozzolino, M., Ciceri, P., Galassi, A., Mangano, M., Carugo, S., Capelli, I., et al. (2019). The Key Role of Phosphate on Vascular Calcification. Toxins 11 (4), 213. doi:10.3390/toxins11040213
Cui, L., Houston, D. A., Farquharson, C., and MacRae, V. E. (2016). Characterisation of Matrix Vesicles in Skeletal and Soft Tissue Mineralisation. Bone 87, 147–158. doi:10.1016/j.bone.2016.04.007
Cui, L., Rashdan, N. A., Zhu, D., Milne, E. M., Ajuh, P., Milne, G., et al. (2017). End Stage Renal Disease-Induced Hypercalcemia May Promote Aortic Valve Calcification via Annexin VI Enrichment of Valve Interstitial Cell Derived-Matrix Vesicles. J. Cel Physiol 232 (11), 2985–2995. doi:10.1002/jcp.25935
Cui, L., Zhou, Q., Zheng, X., Sun, B., and Zhao, S. (2020). Mitoquinone Attenuates Vascular Calcification by Suppressing Oxidative Stress and Reducing Apoptosis of Vascular Smooth Muscle Cells via the Keap1/Nrf2 Pathway. Free Radic. Biol. Med. 161, 23–31. doi:10.1016/j.freeradbiomed.2020.09.028
Dai, X.-Y., Zhao, M.-M., Cai, Y., Guan, Q.-C., Zhao, Y., Guan, Y., et al. (2013). Phosphate-induced Autophagy Counteracts Vascular Calcification by Reducing Matrix Vesicle Release. Kidney Int. 83 (6), 1042–1051. doi:10.1038/ki.2012.482
de Abreu, R. C., Fernandes, H., da Costa Martins, P. A., Sahoo, S., Emanueli, C., and Ferreira, L. (2020). Native and Bioengineered Extracellular Vesicles for Cardiovascular Therapeutics. Nat. Rev. Cardiol. 17 (11), 685–697. doi:10.1038/s41569-020-0389-5
Demer, L. L., and Tintut, Y. (2014). Inflammatory, Metabolic, and Genetic Mechanisms of Vascular Calcification. Atvb 34 (4), 715–723. doi:10.1161/ATVBAHA.113.302070
Drüeke, T. B., and Floege, J. (2020). Cardiovascular Complications of Chronic Kidney Disease: Pioneering Studies. Kidney Int. 98 (3), 522–526. doi:10.1016/j.kint.2020.07.001
Florea, A., Morgenroth, A., Bucerius, J., Schurgers, L. J., and Mottaghy, F. M. (2020). Locking and Loading the Bullet against Micro-calcification. Eur. J. Prev. Cardiol. 28, 1370–1375. doi:10.1177/2047487320911138
Freise, C., Querfeld, U., Ludwig, A., Hamm, B., Schnorr, J., and Taupitz, M. (2021). Uraemic Extracellular Vesicles Augment Osteogenic Transdifferentiation of Vascular Smooth Muscle Cells via Enhanced AKT Signalling and PiT‐1 Expression. J. Cel Mol Med 25 (12), 5602–5614. doi:10.1111/jcmm.16572
Furmanik, M., Chatrou, M., van Gorp, R., Akbulut, A., Willems, B., Schmidt, H., et al. (2020). Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 127 (7), 911–927. doi:10.1161/CIRCRESAHA.119.316159
Furmanik, M., van Gorp, R., Whitehead, M., Ahmad, S., Bordoloi, J., Kapustin, A., et al. (2021). Endoplasmic Reticulum Stress Mediates Vascular Smooth Muscle Cell Calcification via Increased Release of Grp78 (Glucose-Regulated Protein, 78 kDa)-Loaded Extracellular Vesicles. Atvb 41 (2), 898–914. doi:10.1161/ATVBAHA.120.315506
Gaurav, I., Thakur, A., Iyaswamy, A., Wang, X., Chen, X., and Yang, Z. (2021). Factors Affecting Extracellular Vesicles Based Drug Delivery Systems. Molecules 26 (6), 1544. doi:10.3390/molecules26061544
Gebraad, A., Kornilov, R., Kaur, S., Miettinen, S., Haimi, S., Peltoniemi, H., et al. (2018). Monocyte‐derived Extracellular Vesicles Stimulate Cytokine Secretion and Gene Expression of Matrix Metalloproteinases by Mesenchymal Stem/stromal Cells. FEBS J. 285 (12), 2337–2359. doi:10.1111/febs.14485
Genge, B. R., Wu, L. N. Y., and Wuthier, R. E. (2007). In Vitro Modeling of Matrix Vesicle Nucleation. J. Biol. Chem. 282 (36), 26035–26045. doi:10.1074/jbc.M701057200
Ghislat, G., and Knecht, E. (2012). New Ca2+-dependent Regulators of Autophagosome Maturation. Communicative Integr. Biol. 5 (4), 308–311. doi:10.4161/cib.20076
Ghosh, S., Luo, D., He, W., Chen, J., Su, X., and Huang, H. (2020). Diabetes and Calcification: The Potential Role of Anti-diabetic Drugs on Vascular Calcification Regression. Pharmacol. Res. 158, 104861. doi:10.1016/j.phrs.2020.104861
Goettsch, C., Hutcheson, J. D., and Aikawa, E. (2013). MicroRNA in Cardiovascular Calcification. Circ. Res. 112 (7), 1073–1084. doi:10.1161/CIRCRESAHA.113.300937
Goettsch, C., Hutcheson, J. D., Aikawa, M., Iwata, H., Pham, T., Nykjaer, A., et al. (2016). Sortilin Mediates Vascular Calcification via its Recruitment into Extracellular Vesicles. J. Clin. Invest. 126 (4), 1323–1336. doi:10.1172/JCI80851
Goettsch, C., Iwata, H., Hutcheson, J. D., O’Donnell, C. J., Chapurlat, R., Cook, N. R., et al. (2017). Serum Sortilin Associates with Aortic Calcification and Cardiovascular Risk in Men. Atvb 37 (5), 1005–1011. doi:10.1161/ATVBAHA.116.308932
Goody, P. R., Hosen, M. R., Christmann, D., Niepmann, S. T., Zietzer, A., Adam, M., et al. (2020). Aortic Valve Stenosis. Atvb 40 (4), 885–900. doi:10.1161/ATVBAHA.119.313067
Greening, D. W., Xu, R., Ji, H., Tauro, B. J., and Simpson, R. J. (2015). A Protocol for Exosome Isolation and Characterization: Evaluation of Ultracentrifugation, Density-Gradient Separation, and Immunoaffinity Capture Methods. Methods Mol. Biol. 1295, 179–209. doi:10.1007/978-1-4939-2550-6_15
Guo, Y., Bao, S., Guo, W., Diao, Z., Wang, L., Han, X., et al. (2019). Bone Marrow Mesenchymal Stem Cell-Derived Exosomes Alleviate High Phosphorus-Induced Vascular Smooth Muscle Cells Calcification by Modifying microRNA Profiles. Funct. Integr. Genomics 19 (4), 633–643. doi:10.1007/s10142-019-00669-0
Gurunathan, S., Kang, M.-H., Jeyaraj, M., Qasim, M., and Kim, J.-H. (2019). Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 8 (4), 307. doi:10.3390/cells8040307
Hannafon, B. N., Trigoso, Y. D., Calloway, C. L., Zhao, Y. D., Lum, D. H., Welm, A. L., et al. (2016). Plasma Exosome microRNAs Are Indicative of Breast Cancer. Breast Cancer Res. 18 (1), 90. doi:10.1186/s13058-016-0753-x
Hénaut, L., Sanchez-Nino, M. D., Aldamiz-Echevarría Castillo, G., Sanz, A. B., and Ortiz, A. (2016). Targeting Local Vascular and Systemic Consequences of Inflammation on Vascular and Cardiac Valve Calcification. Expert Opin. Ther. Targets 20 (1), 89–105. doi:10.1517/14728222.2015.1081685
Herrmann, J., Babic, M., Tölle, M., van der Giet, M., and Schuchardt, M. (2020). Research Models for Studying Vascular Calcification. Ijms 21 (6), 2204. doi:10.3390/ijms21062204
Hjortnaes, J., Shapero, K., Goettsch, C., Hutcheson, J. D., Keegan, J., Kluin, J., et al. (2015). Valvular Interstitial Cells Suppress Calcification of Valvular Endothelial Cells. Atherosclerosis 242 (1), 251–260. doi:10.1016/j.atherosclerosis.2015.07.008
Hodroge, A., Trécherel, E., Cornu, M., Darwiche, W., Mansour, A., Ait-Mohand, K., et al. (2017). Oligogalacturonic Acid Inhibits Vascular Calcification by Two Mechanisms. Atvb 37 (7), 1391–1401. doi:10.1161/ATVBAHA.117.309513
Hoshino, A., Kim, H. S., Bojmar, L., Gyan, K. E., Cioffi, M., Hernandez, J., et al. (2020). Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 182 (4), 1044–1061. doi:10.1016/j.cell.2020.07.009
Houben, E., Neradova, A., Schurgers, L. J., and Vervloet, M. (2016). The Influence of Phosphate, Calcium and Magnesium on Matrix Gla-Protein and Vascular Calcification: a Systematic Review. G Ital. Nefrol 33 (6), 1.
Hutcheson, J. D., Goettsch, C., Pham, T., Iwashita, M., Aikawa, M., Singh, S. A., et al. (2014). Enrichment of Calcifying Extracellular Vesicles Using Density-Based Ultracentrifugation Protocol. J. Extracellular Vesicles 3, 25129. doi:10.3402/jev.v3.25129
Icer, M. A., and Yıldıran, H. (2021). Effects of Fetuin-A with Diverse Functions and Multiple Mechanisms on Human Health. Clin. Biochem. 88, 1–10. doi:10.1016/j.clinbiochem.2020.11.004
Iordachescu, A., Hulley, P., and Grover, L. M. (2018). A Novel Method for the Collection of Nanoscopic Vesicles from an Organotypic Culture Model. RSC Adv. 8 (14), 7622–7632. doi:10.1039/c7ra12511a
Itel, F., Skovhus Thomsen, J., and Städler, B. (2018). Matrix Vesicles-Containing Microreactors as Support for Bonelike Osteoblasts to Enhance Biomineralization. ACS Appl. Mater. Inter. 10 (36), 30180–30190. doi:10.1021/acsami.8b10886
Itoh, S., Mizuno, K., Aikawa, M., and Aikawa, E. (2018). Dimerization of Sortilin Regulates its Trafficking to Extracellular Vesicles. J. Biol. Chem. 293 (12), 4532–4544. doi:10.1074/jbc.RA117.000732
Jain, M., Dev, R., Doddapattar, P., Kon, S., Dhanesha, N., and Chauhan, A. K. (2021). Integrin α9 Regulates Smooth Muscle Cell Phenotype Switching and Vascular Remodeling. JCI Insight 6 (10), 1. doi:10.1172/jci.insight.147134
Jing, L., Li, L., Ren, X., Sun, Z., Bao, Z., Yuan, G., et al. (2020). Role of Sortilin and Matrix Vesicles in Nϵ-Carboxymethyl-Lysine-Induced Diabetic Atherosclerotic Calcification. Dmso 13, 4141–4151. doi:10.2147/DMSO.S273029
Jing, L., Li, L., Sun, Z., Bao, Z., Shao, C., Yan, J., et al. (2019). Role of Matrix Vesicles in Bone-Vascular Cross-Talk. J. Cardiovasc. Pharmacol. 74 (5), 372–378. doi:10.1097/FJC.0000000000000720
Johnson, K. A., Polewski, M., and Terkeltaub, R. A. (2008). Transglutaminase 2 Is central to Induction of the Arterial Calcification Program by Smooth Muscle Cells. Circ. Res. 102 (5), 529–537. doi:10.1161/CIRCRESAHA.107.154260
Kapustin, A. N., Chatrou, M. L. L., Drozdov, I., Zheng, Y., Davidson, S. M., Soong, D., et al. (2015). Vascular Smooth Muscle Cell Calcification Is Mediated by Regulated Exosome Secretion. Circ. Res. 116 (8), 1312–1323. doi:10.1161/CIRCRESAHA.116.305012
Kapustin, A. N., Davies, J. D., Reynolds, J. L., McNair, R., Jones, G. T., Sidibe, A., et al. (2011). Calcium Regulates Key Components of Vascular Smooth Muscle Cell-Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 109 (1), e1–12. doi:10.1161/CIRCRESAHA.110.238808
Kapustin, A. N., and Shanahan, C. M. (2012). Calcium Regulation of Vascular Smooth Muscle Cell-Derived Matrix Vesicles. Trends Cardiovasc. Med. 22 (5), 133–137. doi:10.1016/j.tcm.2012.07.009
Kapustin, A. N., and Shanahan, C. M. (2016). Emerging Roles for Vascular Smooth Muscle Cell Exosomes in Calcification and Coagulation. J. Physiol. 594 (11), 2905–2914. doi:10.1113/JP271340
Kawakami, R., Katsuki, S., Travers, R., Romero, D. C., Becker-Greene, D., Passos, L. S. A., et al. (2020). S100A9-RAGE Axis Accelerates Formation of Macrophage-Mediated Extracellular Vesicle Microcalcification in Diabetes Mellitus. Atvb 40 (8), 1838–1853. doi:10.1161/ATVBAHA.118.314087
Khan, S. R., Canales, B. K., and Dominguez-Gutierrez, P. R. (2021). Randall's Plaque and Calcium Oxalate Stone Formation: Role for Immunity and Inflammation. Nat. Rev. Nephrol. 17 (6), 417–433. doi:10.1038/s41581-020-00392-1
Khavandgar, Z., Roman, H., Li, J., Lee, S., Vali, H., Brinckmann, J., et al. (2014). Elastin Haploinsufficiency Impedes the Progression of Arterial Calcification in MGP-Deficient Mice. J. Bone Miner Res. 29 (2), 327–337. doi:10.1002/jbmr.2039
Kiffer-Moreira, T., Yadav, M. C., Zhu, D., Narisawa, S., Sheen, C., Stec, B., et al. (2013). Pharmacological Inhibition of PHOSPHO1 Suppresses Vascular Smooth Muscle Cell Calcification. J. Bone Miner Res. 28 (1), 81–91. doi:10.1002/jbmr.1733
Kluszczynska, K., Czernek, L., Cypryk, W., Peczek, L., and Duchler, M. (2019). Methods for the Determination of the Purity of Exosomes. Curr. Pharm. Des. 25 (42), 4464–4485. doi:10.2174/1381612825666191206162712
Krohn, J. B., Hutcheson, J. D., Martínez-Martínez, E., and Aikawa, E. (2016a). Extracellular Vesicles in Cardiovascular Calcification: Expanding Current Paradigms. J. Physiol. 594 (11), 2895–2903. doi:10.1113/JP271338
Krohn, J. B., Hutcheson, J. D., Martínez-Martínez, E., Irvin, W. S., Bouten, C. V. C., Bertazzo, S., et al. (2016b). Discoidin Domain Receptor-1 Regulates Calcific Extracellular Vesicle Release in Vascular Smooth Muscle Cell Fibrocalcific Response via Transforming Growth Factor-β Signaling. Atvb 36 (3), 525–533. doi:10.1161/ATVBAHA.115.307009
Kubo, H. (2018). Extracellular Vesicles in Lung Disease. Chest 153 (1), 210–216. doi:10.1016/j.chest.2017.06.026
Kunitomi, Y., Hara, E. S., Okada, M., Nagaoka, N., Kuboki, T., Nakano, T., et al. (2019). Biomimetic Mineralization Using Matrix Vesicle Nanofragments. J. Biomed. Mater. Res. 107 (5), 1021–1030. doi:10.1002/jbm.a.36618
Lane, R. E., Korbie, D., Trau, M., and Hill, M. M. (2017). Purification Protocols for Extracellular Vesicles. Methods Mol. Biol. 1660, 111–130. doi:10.1007/978-1-4939-7253-1_10
Lee, K., Kim, H., and Jeong, D. (2014). Microtubule Stabilization Attenuates Vascular Calcification through the Inhibition of Osteogenic Signaling and Matrix Vesicle Release. Biochem. Biophysical Res. Commun. 451 (3), 436–441. doi:10.1016/j.bbrc.2014.08.007
Lee, K., Shao, H., Weissleder, R., and Lee, H. (2015). Acoustic Purification of Extracellular Microvesicles. ACS Nano 9 (3), 2321–2327. doi:10.1021/nn506538f
Li, Y., Sun, Z., Zhang, L., Yan, J., Shao, C., Jing, L., et al. (2020). Role of Macrophages in the Progression and Regression of Vascular Calcification. Front. Pharmacol. 11, 661. doi:10.3389/fphar.2020.00661
Lin, X., Zhu, T., Xu, F., Zhong, J.-Y., Li, F., Shan, S.-K., et al. (2020). Plasma Exosomes Derived from Patients with End-Stage Renal Disease and Renal Transplant Recipients Have Different Effects on Vascular Calcification. Front. Cel Dev. Biol. 8, 618228. doi:10.3389/fcell.2020.618228
Lin, Z., Rodriguez, N. E., Zhao, J., Ramey, A. N., Hyzy, S. L., Boyan, B. D., et al. (2016). Selective Enrichment of microRNAs in Extracellular Matrix Vesicles Produced by Growth Plate Chondrocytes. Bone 88, 47–55. doi:10.1016/j.bone.2016.03.018
Liu, L., Zeng, P., Yang, X., Duan, Y., Zhang, W., Ma, C., et al. (2018). Inhibition of Vascular Calcification. Arterioscler Thromb. Vasc. Biol. 38 (10), 2382–2395. doi:10.1161/ATVBAHA.118.311546
Lobb, R. J., Becker, M., Wen Wen, S., Wong, C. S. F., Wiegmans, A. P., Leimgruber, A., et al. (2015). Optimized Exosome Isolation Protocol for Cell Culture Supernatant and Human Plasma. J. Extracellular Vesicles 4, 27031. doi:10.3402/jev.v4.27031
Luong, T. T. D., Schelski, N., Boehme, B., Makridakis, M., Vlahou, A., Lang, F., et al. (2018). Fibulin-3 Attenuates Phosphate-Induced Vascular Smooth Muscle Cell Calcification by Inhibition of Oxidative Stress. Cel Physiol Biochem 46 (4), 1305–1316. doi:10.1159/000489144
Mathivanan, S., Ji, H., and Simpson, R. J. (2010). Exosomes: Extracellular Organelles Important in Intercellular Communication. J. Proteomics 73 (10), 1907–1920. doi:10.1016/j.jprot.2010.06.006
McCormick, M. M., Rahimi, F., Bobryshev, Y. V., Gaus, K., Zreiqat, H., Cai, H., et al. (2005). S100A8 and S100A9 in Human Arterial Wall. J. Biol. Chem. 280 (50), 41521–41529. doi:10.1074/jbc.M509442200
Melki, I., Tessandier, N., Zufferey, A., and Boilard, E. (2017). Platelet Microvesicles in Health and Disease. Platelets 28 (3), 214–221. doi:10.1080/09537104.2016.1265924
Montañez-Barragán, A., Gómez-Barrera, I., Sanchez-Niño, M. D., Ucero, A. C., González-Espinoza, L., and Ortiz, A. (2014). Osteoprotegerin and Kidney Disease. J. Nephrol. 27 (6), 607–617. doi:10.1007/s40620-014-0092-x
Nabhan, J. F., Hu, R., Oh, R. S., Cohen, S. N., and Lu, Q. (2012). Formation and Release of Arrestin Domain-Containing Protein 1-mediated Microvesicles (ARMMs) at Plasma Membrane by Recruitment of TSG101 Protein. Proc. Natl. Acad. Sci. 109 (11), 4146–4151. doi:10.1073/pnas.1200448109
Nakahara, T., Dweck, M. R., Narula, N., Pisapia, D., Narula, J., and Strauss, H. W. (2017). Coronary Artery Calcification. JACC: Cardiovasc. Imaging 10 (5), 582–593. doi:10.1016/j.jcmg.2017.03.005
Nakai, W., Yoshida, T., Diez, D., Miyatake, Y., Nishibu, T., Imawaka, N., et al. (2016). A Novel Affinity-Based Method for the Isolation of Highly Purified Extracellular Vesicles. Sci. Rep. 6, 33935. doi:10.1038/srep33935
New, S. E. P., Goettsch, C., Aikawa, M., Marchini, J. F., Shibasaki, M., Yabusaki, K., et al. (2013). Macrophage-Derived Matrix Vesicles. Circ. Res. 113 (1), 72–77. doi:10.1161/CIRCRESAHA.113.301036
Nguyen, N. T., Nguyen, T. T., Da Ly, D., Xia, J.-B., Qi, X.-F., Lee, I.-K., et al. (2020). Oxidative Stress by Ca2+ Overload Is Critical for Phosphate-Induced Vascular Calcification. Am. J. Physiology-Heart Circulatory Physiol. 319 (6), H1302–H1312. doi:10.1152/ajpheart.00305.2020
Nitta, K., and Ogawa, T. (2015). Vascular Calcification in End-Stage Renal Disease Patients. Contrib. Nephrol. 185, 156–167. doi:10.1159/000380980
Omarjee, L., Nitschke, Y., Verschuere, S., Bourrat, E., Vignon, M. D., Navasiolava, N., et al. (2020). Severe Early‐onset Manifestations of Pseudoxanthoma Elasticum Resulting from the Cumulative Effects of Several Deleterious Mutations in ENPP1 , ABCC6 and HBB : Transient Improvement in Ectopic Calcification with Sodium Thiosulfate. Br. J. Dermatol. 183 (2), 367–372. doi:10.1111/bjd.18632
Pan, W., Liang, J., Tang, H., Fang, X., Wang, F., Ding, Y., et al. (2020). Differentially Expressed microRNA Profiles in Exosomes from Vascular Smooth Muscle Cells Associated with Coronary Artery Calcification. Int. J. Biochem. Cel Biol. 118, 105645. doi:10.1016/j.biocel.2019.105645
Perelló, J., Ferrer, M. D., Pérez, M., Kaesler, N., Brandenburg, V. M., Behets, G. J., et al. (2020). Mechanism of Action of SNF472, a Novel Calcification Inhibitor to Treat Vascular Calcification and Calciphylaxis. Br. J. Pharmacol. 177 (19), 4400–4415. doi:10.1111/bph.15163
Pescatore, L. A., Gamarra, L. F., and Liberman, M. (2019). Multifaceted Mechanisms of Vascular Calcification in Aging. Atvb 39 (7), 1307–1316. doi:10.1161/ATVBAHA.118.311576
Phadwal, K., Feng, D., Zhu, D., and MacRae, V. E. (2020). Autophagy as a Novel Therapeutic Target in Vascular Calcification. Pharmacol. Ther. 206, 107430. doi:10.1016/j.pharmthera.2019.107430
Proudfoot, D. (2019). Calcium Signaling and Tissue Calcification. Cold Spring Harb Perspect. Biol. 11 (10), a035303. doi:10.1101/cshperspect.a035303
Qi, Y., Dai, F., Gu, J., and Yao, W. (2019). Biomarkers in VSMC Phenotypic Modulation and Vascular Remodeling. Pharmazie 74 (12), 711–714. doi:10.1691/ph.2019.9743
Reynolds, J. L., Joannides, A. J., Skepper, J. N., McNair, R., Schurgers, L. J., Proudfoot, D., et al. (2004). Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: a Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. 15 (11), 2857–2867. doi:10.1097/01.ASN.0000141960.01035.28
Schantl, A. E., Verhulst, A., Neven, E., Behets, G. J., D’Haese, P. C., Maillard, M., et al. (2020). Inhibition of Vascular Calcification by Inositol Phosphates Derivatized with Ethylene Glycol Oligomers. Nat. Commun. 11 (1), 721. doi:10.1038/s41467-019-14091-4
Shapiro, I. M., Landis, W. J., and Risbud, M. V. (2015). Matrix Vesicles: Are They Anchored Exosomes? Bone 79, 29–36. doi:10.1016/j.bone.2015.05.013
Shekar, C., and Budoff, M. (2018). Calcification of the Heart: Mechanisms and Therapeutic Avenues. Expert Rev. Cardiovasc. Ther. 16 (7), 527–536. doi:10.1080/14779072.2018.1484282
Shen, J., Zhang, N., Lin, Y.-N., Xiang, P., Liu, X.-b., Shan, P.-f., et al. (2018). Regulation of Vascular Calcification by Growth Hormone-Releasing Hormone and its Agonists. Circ. Res. 122 (10), 1395–1408. doi:10.1161/CIRCRESAHA.117.312418
Shi, W., Lu, J., Li, J., Qiu, M., Lu, Y., Gu, J., et al. (2020). Piperlongumine Attenuates High Calcium/Phosphate-Induced Arterial Calcification by Preserving P53/PTEN Signaling. Front. Cardiovasc. Med. 7, 625215. doi:10.3389/fcvm.2020.625215
Sohn, W., Kim, J., Kang, S. H., Yang, S. R., Cho, J.-Y., Cho, H. C., et al. (2015). Serum Exosomal microRNAs as Novel Biomarkers for Hepatocellular Carcinoma. Exp. Mol. Med. 47, e184. doi:10.1038/emm.2015.68
Ståhl, A.-l., Johansson, K., Mossberg, M., Kahn, R., and Karpman, D. (2019). Exosomes and Microvesicles in normal Physiology, Pathophysiology, and Renal Diseases. Pediatr. Nephrol. 34 (1), 11–30. doi:10.1007/s00467-017-3816-z
Sun, Z., Wang, Z., Li, L., Yan, J., Shao, C., Bao, Z., et al. (2019). RAGE/galectin-3 Yields Intraplaque Calcification Transformation via Sortilin. Acta Diabetol. 56 (4), 457–472. doi:10.1007/s00592-018-1273-1
Todorova, D., Simoncini, S., Lacroix, R., Sabatier, F., and Dignat-George, F. (2017). Extracellular Vesicles in Angiogenesis. Circ. Res. 120 (10), 1658–1673. doi:10.1161/CIRCRESAHA.117.309681
Tyson, K. L., Reynolds, J. L., McNair, R., Zhang, Q., Weissberg, P. L., and Shanahan, C. M. (2003). Osteo/chondrocytic Transcription Factors and Their Target Genes Exhibit Distinct Patterns of Expression in Human Arterial Calcification. Atvb 23 (3), 489–494. doi:10.1161/01.ATV.0000059406.92165.31
Veschi, E. A., Bolean, M., Strzelecka-Kiliszek, A., Bandorowicz-Pikula, J., Pikula, S., Granjon, T., et al. (2020). Localization of Annexin A6 in Matrix Vesicles during Physiological Mineralization. Ijms 21 (4), 1367. doi:10.3390/ijms21041367
Viegas, C. S. B., Costa, R. M., Santos, L., Videira, P. A., Silva, Z., Araújo, N., et al. (2017). Gla-rich Protein Function as an Anti-inflammatory Agent in Monocytes/macrophages: Implications for Calcification-Related Chronic Inflammatory Diseases. PLoS One 12 (5), e0177829. doi:10.1371/journal.pone.0177829
Viegas, C. S. B., Rafael, M. S., Enriquez, J. L., Teixeira, A., Vitorino, R., Luís, I. M., et al. (2015). Gla-rich Protein Acts as a Calcification Inhibitor in the Human Cardiovascular System. Atvb 35 (2), 399–408. doi:10.1161/ATVBAHA.114.304823
Viegas, C. S. B., Santos, L., Macedo, A. L., Matos, A. A., Silva, A. P., Neves, P. L., et al. (2018). Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification. Arterioscler Thromb. Vasc. Biol. 38 (3), 575–587. doi:10.1161/ATVBAHA.117.310578
Viegas, C. S. B., and Simes, D. C. (2018). “Inflammation and Calcification in the Vascular Tree; Insights into Atherosclerosis,” in Immunity and Inflammation in Health and Disease, 189–201. doi:10.1016/b978-0-12-805417-8.00015-9
Villa-Bellosta, R. (2019). ATP-based Therapy Prevents Vascular Calcification and Extends Longevity in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Proc. Natl. Acad. Sci. USA 116 (47), 23698–23704. doi:10.1073/pnas.1910972116
Voelkl, J., Luong, T. T. D., Tuffaha, R., Musculus, K., Auer, T., Lian, X., et al. (2018a). SGK1 Induces Vascular Smooth Muscle Cell Calcification through NF-Κb Signaling. J. Clin. Invest. 128 (7), 3024–3040. doi:10.1172/JCI96477
Voelkl, J., Tuffaha, R., Luong, T. T. D., Zickler, D., Masyout, J., Feger, M., et al. (2018b). Zinc Inhibits Phosphate-Induced Vascular Calcification through TNFAIP3-Mediated Suppression of NF-Κb. Jasn 29 (6), 1636–1648. doi:10.1681/ASN.2017050492
Wang, Z., Wu, H.-j., Fine, D., Schmulen, J., Hu, Y., Godin, B., et al. (2013). Ciliated Micropillars for the Microfluidic-Based Isolation of Nanoscale Lipid Vesicles. Lab. Chip 13 (15), 2879–2882. doi:10.1039/c3lc41343h
Wei, W., Guo, X., Gu, L., Jia, J., Yang, M., Yuan, W., et al. (2021). Bone Marrow Mesenchymal Stem Cell Exosomes Suppress Phosphate-Induced Aortic Calcification via SIRT6-HMGB1 Deacetylation. Stem Cel Res Ther 12 (1), 235. doi:10.1186/s13287-021-02307-8
Willems, B. A., Furmanik, M., Caron, M. M. J., Chatrou, M. L. L., Kusters, D. H. M., Welting, T. J. M., et al. (2018). Ucma/GRP Inhibits Phosphate-Induced Vascular Smooth Muscle Cell Calcification via SMAD-dependent BMP Signalling. Sci. Rep. 8 (1), 4961. doi:10.1038/s41598-018-23353-y
Xiao, H., Chen, J., Duan, L., and Li, S. (2021). Role of Emerging Vitamin K-dependent P-roteins: Growth A-rrest-specific P-rotein 6, Gla-rich P-rotein and P-eriostin (Review). Int. J. Mol. Med. 47 (3), 2. doi:10.3892/ijmm.2020.4835
Xiao, X., Yang, C., Qu, S.-L., Shao, Y.-D., Zhou, C.-Y., Chao, R., et al. (2020). S100 Proteins in Atherosclerosis. Clinica Chim. Acta 502, 293–304. doi:10.1016/j.cca.2019.11.019
Xu, F., Zhong, J. Y., Lin, X., Shan, S. K., Guo, B., Zheng, M. H., et al. (2020). Melatonin Alleviates Vascular Calcification and Ageing through Exosomal miR‐204/miR‐211 Cluster in a Paracrine Manner. J. Pineal Res. 68 (3), e12631. doi:10.1111/jpi.12631
Xu, J., Camfield, R., and Gorski, S. M. (2018). The Interplay between Exosomes and Autophagy - Partners in Crime. J. Cel Sci. 131 (15), jcs215210. doi:10.1242/jcs.215210
Zazzeroni, L., Faggioli, G., and Pasquinelli, G. (2018). Mechanisms of Arterial Calcification: The Role of Matrix Vesicles. Eur. J. Vasc. Endovascular Surg. 55 (3), 425–432. doi:10.1016/j.ejvs.2017.12.009
Zeringer, E., Barta, T., Li, M., and Vlassov, A. V. (2015). Strategies for Isolation of Exosomes. Cold Spring Harb Protoc. 2015 (4), pdb.top074476–323. doi:10.1101/pdb.top074476
Zhang, C., Zhang, K., Huang, F., Feng, W., Chen, J., Zhang, H., et al. (2018). Exosomes, the Message Transporters in Vascular Calcification. J. Cel. Mol. Med. 22 (9), 4024–4033. doi:10.1111/jcmm.13692
Keywords: vascular calcification, matrix vesicles, extracellular vesicles, exosomes, vascular smooth muscle cells
Citation: Li T, Yu H, Zhang D, Feng T, Miao M, Li J and Liu X (2022) Matrix Vesicles as a Therapeutic Target for Vascular Calcification. Front. Cell Dev. Biol. 10:825622. doi: 10.3389/fcell.2022.825622
Received: 30 November 2021; Accepted: 04 January 2022;
Published: 21 January 2022.
Edited by:
Roland Wohlgemuth, Lodz University of Technology, PolandReviewed by:
Tzongshi Lu, Brigham and Women’s Hospital and Harvard Medical School, United StatesMiriam Zacchia, University of Campania Luigi Vanvitelli, Italy
Copyright © 2022 Li, Yu, Zhang, Feng, Miao, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianwei Li, amVycnlsaTY3OEB5YWhvby5jb20=; Xiaoheng Liu, bGl1eGlhb2hnQHNjdS5lZHUuY24=
†These authors have contributed equally to this work