 Fei Yuan
Fei Yuan Xiaoyu Cao
Xiaoyu Cao Yu-Hang Zhang
Yu-Hang Zhang Lei Chen
Lei Chen Tao Huang
Tao Huang ZhanDong Li
ZhanDong Li Yu-Dong Cai
Yu-Dong Cai- 1Department of Science and Technology, Binzhou Medical University Hospital, Binzhou, China
- 2Department of Neurology, Binzhou Medical University Hospital, Binzhou, China
- 3Channing Division of Network Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
- 4College of Information Engineering, Shanghai Maritime University, Shanghai, China
- 5Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China
- 6CAS Key Laboratory of Tissue Microenvironment and Tumor, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China
- 7College of Food Engineering, Jilin Engineering Normal University, Changchun, China
- 8School of Life Sciences, Shanghai University, Shanghai, China
Cancer driver gene is a type of gene with abnormal alterations that initiate or promote tumorigenesis. Driver genes can be used to reveal the fundamental pathological mechanisms of tumorigenesis. These genes may have pathological changes at different omics levels. Thus, identifying cancer driver genes involving two or more omics levels is essential. In this study, a computational investigation was conducted on lung cancer driver genes. Four omics levels, namely, epigenomics, genomics, transcriptomics, and post-transcriptomics, were involved. From the driver genes at each level, the Laplacian heat diffusion algorithm was executed on a protein–protein interaction network for discovering latent driver genes at this level. A following screen procedure was performed to extract essential driver genes, which contained three tests: permutation, association, and function tests, which can exclude false-positive genes and screen essential ones. Finally, the intersection operation was performed to obtain novel driver genes involving two omic levels. The analyses on obtained genes indicated that they were associated with fundamental pathological mechanisms of lung cancer at two corresponding omics levels.
Introduction
Driver gene is a commonly used description in oncology to describe genes with abnormal alterations that initiate or promote tumorigenesis (Pao and Girard, 2011; Tokheim et al., 2016). Identifying driver genes can help us reveal the fundamental pathological mechanisms of tumorigenesis (Pao and Girard, 2011). During tumorigenesis, genes may have pathological changes at different omics levels, including but not restricted to genomics (as DNA sequence alterations), epigenomics (as methylation status or other DNA modification status alterations), transcriptomics, and proteomics (Cancer Genome Atlas Research Network, 2014; Huang et al., 2015; Li et al., 2018). Using current biological techniques is time consuming but can detect all pathological alterations associated with tumorigenesis. However, not all alterations are associated with driver genes. Genes that are mutated or abnormally regulated during tumorigenesis but not associated with the initiation or progression of tumors are summarized as passenger genes (Pon and Marra, 2015). Most of the genes altered during tumorigenesis are actually passenger genes, which cannot help us understand or reveal the pathological mechanisms of cancer. Therefore, the identification of driver genes is one of the major research directions in oncology.
Based on traditional experiments, only hot spot genes, which have high mutation rate in sporadic cancer cases or have clear and obvious family hereditary histories, can be screened as candidates for cancer driver genes (Korenjak and Zavadil, 2019; Sears and Mazzone, 2020). For such pre-identified genes, driver potential needs to be validated using in vitro and in vivo experiments, which are quite time consuming and expensive (Chu et al., 2018; Sears and Mazzone, 2020), making it impossible and unreasonable for whole-omics wide screening. Therefore, to overcome the restrictions, scholars have introduced computational methods for discovering and pre-selecting candidate driver genes at whole omics level. Nowadays, identifying cancer driver genes using computational methods are not only effective but also reliable. At the beginning, computational methods are applied only for data at one omics level. With the development of algorithms and computational workflows, the integration of two or more omics levels of data to identify core cancer drivers has been realized (Turanli et al., 2018; Olivier et al., 2019). In 2016, Chen et al. (Chen et al., 2016) proposed shortest path-based method to identify novel lung cancer driver genes involving two omics levels. Later, Yuan and Lu employed another network algorithm, random walk with restart (RWR), to conduct the same investigation (Yuan and Lu, 2017). Several possible lung cancer driver genes involving two omics levels were proposed. This study continued the above two previous studies.
Cancer is one of the major threatening diseases for human health in the 21st century. As introduced above, identifying cancer biomarkers is one of the most effective way to explore the pathological mechanisms of tumorigenesis. However, revealing the potential biomarkers of all cancer subjects is difficult due to the limitation of data availability. In this study, we focused on one of the most common and deadly cancer subtype, namely, lung cancer. According to the statistics from GLOBOCAN estimates, in 2020, the general cumulative rates (0–75 years old) for lung cancer have been up to 3.78% in males and 1.77% in females, both of which are in the top of all cancer subtypes (rank 1 in males and rank 2 in females, following breast cancer) (Siegel et al., 2021). In 2021, more than 235,000 of new lung cancer cases and more than 130,000 of deaths from lung cancer are predicted in the United States alone (Siegel et al., 2021), confirming that lung cancer is a growing threat to humans in the 21st century.
Here, we integrated four levels of lung cancer omics data including epigenomics (methylation), genomics (gene variations), transcriptomics (gene expression), and post-transcriptomics regulation (microRNAs). Driver genes identified at each individual omics level were regarded as candidates for lung cancer drivers. Based on the candidates at each level, the Laplacian heat diffusion (LHD) (Carlin et al., 2017) algorithm was executed on a protein–protein interaction (PPI) network to identify raw driver genes. These genes were further filtered by a screen procedure, including permutation, association, and function tests, to exclude false-positive genes and select essential ones. Finally, intersection operation was carried out to identify driver genes involving at least two omics levels. Multiple genes were screened as potential multi-omics lung cancer drivers, and several genes were validated by recent publications via literature mining. The recognition of effective novel driver genes associated with lung cancer provided a novel approach for exploring the cancer pathological mechanisms of lung cancer and identifying clinical biomarkers.
Materials and Methods
Datasets
The driver genes at four omics levels, namely, epigenomics, genomics, transcriptomics, and post-transcriptomics regulation, were retrieved from a previous study (Chen et al., 2016). Genes were extracted from DNA methylation, somatic mutation, gene expression, and microRNA expression data, respectively, which were collected in TCGA (https://tcga-data.nci.nih.gov/docs/publications/luad_2014/) (Cancer Genome Atlas Research Network, 2014). The detailed data clean procedures can be found in Ref. (Chen et al., 2016). Finally, we obtained 153 driver genes at epigenomics level, 197 driver genes at genomics level, 1,373 driver genes at transcriptomics level, and 825 driver genes at post-transcriptomics level. Basing on these genes, we aimed to identify driver genes involving two levels.
Network Construction
In this study, we used a powerful network algorithm, LHD algorithm (Carlin et al., 2017), to discover driver genes involving two omics levels. Thus, a network is necessary. PPI information is widely used to tackle protein- and gene-related problems (Ng et al., 2010; Chen et al., 2018; Zhang et al., 2019; Zhang and Chen, 2020; Liu et al., 2021; Pan et al., 2021a; Pan et al., 2021b; Zhu et al., 2021). Here, the human PPI information reported in STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, http://www.string-db.org/, Version 10.0) (Szklarczyk et al., 2014) was adopted to construct a PPI network. To this end, we downloaded the file “9606. protein.links.v10. txt.gz”, containing 4,274,001 PPIs that involve 19,247 human proteins. Each PPI consists of two proteins, encoded by Ensembl IDs, and one confidence score with range between 1 and 999. Such score indicates the strength of the PPI. In fact, such score is obtained by considering several aspects of proteins, including close neighborhood in (prokaryotic) genomes, gene fusion, occurrence across species, gene coexpression, literature description, etc. As elaborated in the website of STRING, PPIs in STRING are derived from genomic context predictions, high-throughput lab experiments, (conserved) co-expression, automated textmining, previous knowledge in databases. Thus, they can widely measure the associations of proteins. This is the great advantage compared with the PPIs reported in other public databases. The constructed PPI network defined 19,247 proteins as nodes, and two nodes were connected by an edge if and only if they can constitute a PPI. Evidently, each edge in the network represented a PPI. To further indicate the difference of PPIs, we assigned each edge with a weight, which was defined as the confidence score of the corresponding PPI. For easy description, such PPI network was denoted by N.
LHD-Based Method
Based on the PPI network N, an LHD-based method was designed to discover driver genes involving two omics levels. The method consisted of two stages. In the first stage, novel driver genes at each of four omics levels were identified by applying the LHD algorithm on the network N and performing a screen procedure. In the second stage, the novel driver genes at each level were refined to discover latent driver genes involving two omics levels.
LHD Algorithm
The LHD algorithm is a network diffusion algorithm (Carlin et al., 2017) that can deliver heat values on seed nodes to others in the network. Given a network N, let A be its adjacent matrix and D be the diagonal matrix, storing the degree of all vertices in the network. The Laplacian matrix L was defined as D-A, i.e., L = D-A. In addition, let S be the seed node set. The heat values on seed nodes are stored in a vector, denoted by
where t represents the time passed, and H(t) indicates the distribution of heat values at time t. Generally, as the time passes by, H(t) becomes stable. In reality, we tried several values of t and compared two consecutive vectors. If they were close enough, then the LHD algorithm was stopped. The final heat value distribution vector was selected as the outcome of the algorithm. Basing on this vector, we can extract the heat value of each node in the network. Nodes with high heat values were deemed to have strong associations with the seed nodes. By setting a proper threshold, important nodes can be selected.
For driver genes at one omics level, their encoding proteins were first obtained and fed into the LHD algorithm as seed nodes. The LHD algorithm was then performed on the PPI network N. According to the outcomes of the algorithm, nodes (proteins) assigned high heat values were selected as the raw driver genes at this level.
Screen Procedure
Some raw driver genes at each omics level can be obtained by executing the LHD algorithm. However, false-positive genes were inevitably included. A screen procedure was designed to control these genes.
Permutation Test
Heat value is very important to determine the selection of nodes (proteins). However, this value on some nodes (protein) was highly related to the structure of network N. Some nodes (proteins) more easily received high heat values, regardless of which nodes were seed nodes. Thus, the significance of heat value on each raw driver gene selected by the LHD algorithm should be further measured. For raw driver genes at each level, a permutation test was performed. In detail, we randomly generated 500 gene sets, which contained the same number of driver genes at this level. For each generated gene set, the LHD algorithm was executed on N with genes in this set as seed nodes. Finally, each raw driver gene was assigned a heat value. After all the randomly produced sets were considered, each raw driver gene was assigned 500 heat values. These values can be used to measure the significance of actual heat value that was obtained by driver genes at this level. A p-value was computed for each raw driver gene g as follows:
where Heat> denotes the number of randomly produced gene sets, on which the heat value of g is larger than its actual heat value. If a novel driver gene had a high p-value, then the heat values on several randomly generated gene sets were higher than its actual heat value, indicating that this heat value had no statistical significance. As such, this gene was not special for the driver genes at this level. Thus, we should select novel driver genes with low p-values. Given that 0.05 is always used as the cutoff to measure statistical significance, this study adopted it to filter novel driver genes at each level.
Association Test
By the permutation test, some false positive genes produced by the PPI network were excluded. To further select essential genes among remaining genes, we designed an association test. Several studies have reported that interacting proteins are more likely to share common functions. If the strength of the interaction was considered, then two proteins in a strong interaction were more likely to share common functions than those in a weak interaction. As mentioned in Network construction, the confidence score in STRING can measure the strength of an interaction and thus can be used to design the association test. For formulation, the confidence score on the interaction of proteins p1 and p2 was denoted by
Clearly, the gene with a high MAS was more important and should be kept. We can set a high threshold of MAS to select essential driver genes.
Function Test
To further extract essential driver genes at each level, we designed a third test, namely, function test. This test was based on two types of functional terms: 1) gene ontology and 2) KEGG pathway. In general, driver genes at some level may be annotated by some common functional terms. If the latent one exhibited similar functional terms to some driver genes, then it had a high probability to be a novel driver gene. We first used enrichment theory (Subramanian et al., 2005) to measure the relationship between one gene and all functional terms. In detail, for one gene g and one functional term f, let G be the set consisting of g and its interacting genes and F be the set containing genes annotated by f. The enrichment score between g and f was defined as the -log10 of the hypergeometric test p value of G and F, which can be computed by
where N stands for total number of human genes, M and n represent the number of genes in F and G, respectively, m denotes the number of common genes in F and G. The obtained values were collected in a vector, denoted by
where g1 and g2 represent two genes,
Similar to MAS, genes with high MFS values were more likely to be novel driver genes. By setting a proper threshold, essential genes can be obtained.
Intersection Operation
Based on the LHD algorithm and a screen procedure, some essential novel driver genes were obtained at each omics level. By taking the intersection operation, we identified some driver genes involving two levels. As four levels were considered in this study, we finally obtained six driver gene sets. Genes in each set were deemed to be driver genes involving two omics levels.
Results
In this study, an LHD-based method was proposed to identify novel driver genes involving two omics levels. The entire procedures are illustrated in Figure 1. The detailed results at each procedure are presented in this section.
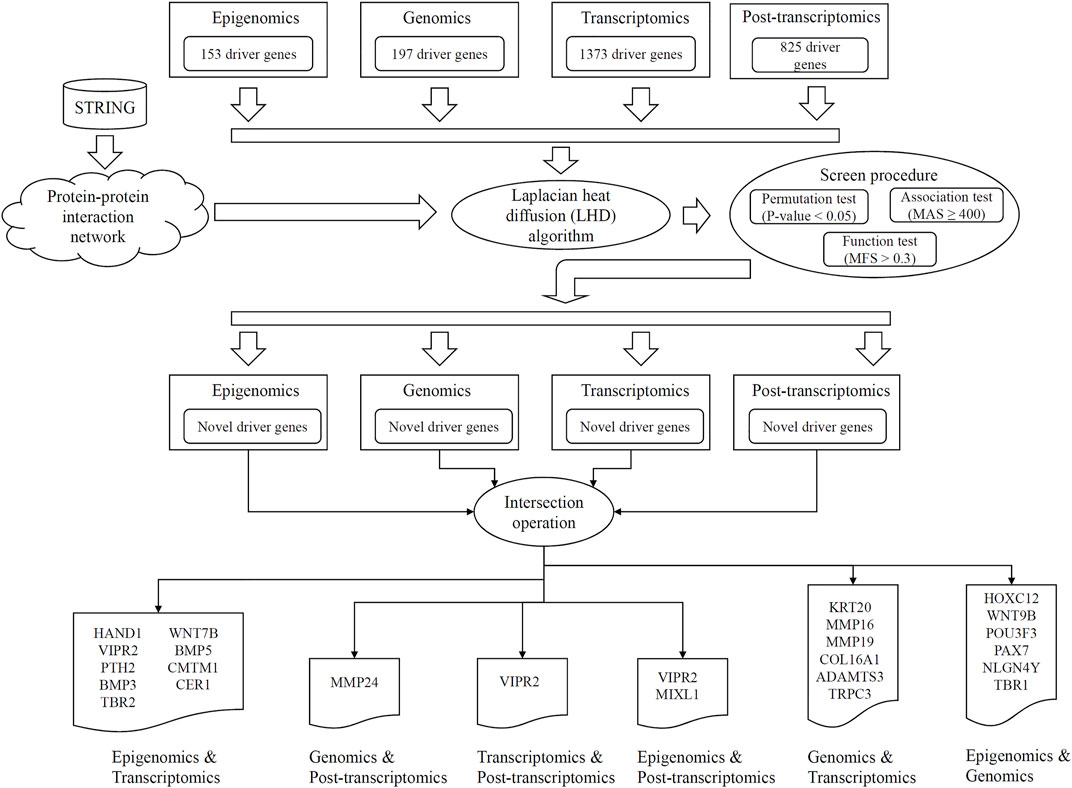
FIGURE 1. Entire procedures of LHD-based method for identification of multi-omics lung cancer driver genes. Based on driver genes at one omics level, the Laplacian heat diffusion (LHD) algorithm is executed on a protein–protein interaction network reported in STRING to identify raw driver genes. These genes are filtered by a screen procedure. The intersection operation is conducted to identify driver genes involving any two omics levels. Six groups are accessed, each of which contains driver genes involving two omics levels.
Latent Driver Genes at Each Level
In the first stage of LHD-based method, some latent driver genes were identified for each omics level. For epigenomics level, 153 validated driver genes were fed into the LHD algorithm, which was executed on the PPI network N. Each node was assigned a heat value. We selected the nodes with heat value no less than 10–5 and obtained 13,101 nodes. The heat values of the selected nodes are provided in Supplementary Table S1. A screen procedure was then performed to filter essential candidates. First, a permutation test was adopted to determine the statistical significance of heat value on each selected node, resulting in a p-value for each node (Supplementary Table S1). A total of 311 nodes with p-value less than 0.05 were selected. Second, an association test was executed to test the importance of the 311 remaining nodes, assigning an MAS to each node (Supplementary Table S1). The threshold of MAS was set as 400, resulting in 228 nodes. Finally, function test was used to evaluate each remaining node, which was assigned with an MFS (Supplementary Table S1). The threshold of MFS was set as 0.3. A total of 199 nodes were obtained, and their corresponding genes were selected as the latent driver genes at epigenomics level.
For the three other levels, the same procedures with common thresholds for all measurements were performed. All measurements on each node at the three levels are provided in Supplementary Tables S2–S4. The numbers of remaining nodes at each stage of LHD-based method are listed in Table 1. As a result, 84, 174, and 39 latent driver genes were accessed at levels of genomics, transcriptomics, and post-transcriptomics regulation, respectively.
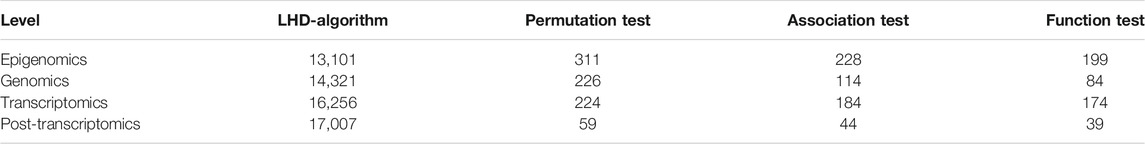
TABLE 1. Remaining latent driver genes at each stage of LHD-based method.
For latent driver genes at each omics level, we further investigated their associations with validated genes mentioned in Datasets. For each omics level, the PPIs between latent and validated genes were extracted. For each latent gene, we counted three values, which were defined as the number of validated genes that can interact with the latent gene with medium confidence (≥400), high confidence (≥700) and highest confidence (≥900). These values of all latent genes at four omics levels were indicated by four boxplots, as shown in Figure 2. It can be observed that each latent gene can interact with at least one validated gene with medium confidence and several latent genes can interact with validated genes with high or highest confidence. This fact indicated that the relationships between latent and validated genes were quite close, increasing the probabilities of latent genes to be actual driver genes.
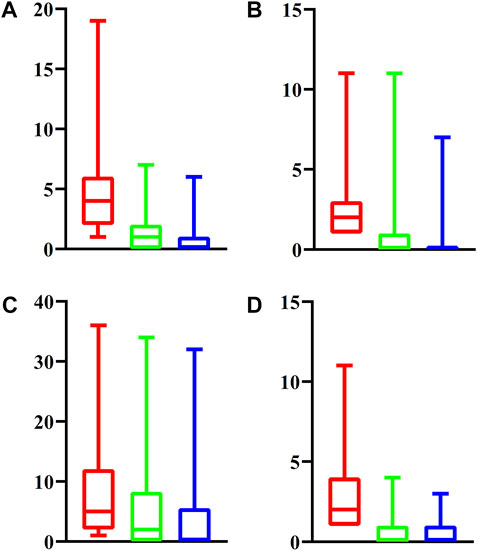
FIGURE 2. Boxplot to show the associations between latent driver genes and validated ones at each of four omic levels. (A) Epigenomics; (B) Genomics; (C) Transcriptomics; (D) Post-transcriptomics. The Y-axis represents the number of validated genes that can interact with latent genes with different strength. The red (green, blue, respectively) box denotes number of validated genes that can interact with latent genes with medium (high, highest, respectively) confidence.
Driver Genes Involving Two Levels
According to LHD-based method, we conducted the intersection operation of latent driver genes at two levels. Some driver genes involving two levels were obtained. The number of driver genes involving two levels is listed in Table 2, and the detailed genes for any two levels are also listed in Table 2. At least one driver gene was accessed for any two levels. Discussion presents an extensive discussion on some of the obtained genes.

TABLE 2. Driver genes involving two omics levelsa.
Comparison With Previous Results
Two previous studies (Chen et al., 2016; Yuan and Lu, 2017) reported some novel driver genes involving two omics levels. The comparison of driver genes indicated that the driver genes reported in the present study were completely different from those in previous two studies. The previous two studies adopted the shortest path-based and RWR-based methods, respectively, to discover novel driver genes, which had quite different principles and procedures; as such, the difference in the driver genes reported between the present study and previous studies was considered reasonable. On the other hand, each method has its limitations, and some driver genes may be omitted. The driver genes reported in this study can be essential supplements for the previous studies.
Discussion
Novel driver genes involving two of the four omics levels (epigenomics, genomics, transcriptomics, post-transcriptomics levels) were identified using LHD-based method. Six groups of latent multi-omics driver genes for lung cancer were screened. According to recent publications, several identified multi-omics driver genes involving two of the four omics levels can be confirmed to be associated with fundamental lung cancer tumorigenesis-associated pathological mechanisms. The detailed discussion on the genes identified in each group can be seen below.
Shared Genes Between Epigenomics and Post-transcriptomics Regulation
Two genes were identified to be lung cancer drivers at epigenomics (methylation) and post-transcriptomics regulation (microRNA) levels. The first gene is VIPR2 (ENSP00000262178). Early in 2017, researchers from Sungkyunkwan University confirmed that VIPR2 is associated with lung cancer at the DNA methylation level (Um et al., 2017). As for the microRNA level, no direct reports confirmed the correlation between lung cancer and VIPR2. However, such gene has been shown to be associated with pancreatic cancer tumorigenesis at the microRNA level (Naderi et al., 2014), indicating the specific role of the gene during tumorigenesis. Therefore, this gene could have regulatory effects on lung cancer at the microRNA level. For the next gene involving two omics levels, MIXL1 (ENSP00000355775), an epigenome analyses in 2019 on circulating tumor cells associated with metastasis revealed that circulating tumor cells in lung cancer has typical epigenomic alterations in MIXL1 (Gkountela et al., 2019). As for its regulatory effects at post-transcriptomics microRNA level, effective microRNAs from famous microRNA family, let-7 family, has shown to be associated with the dedifferentiation transformation of lung cells during embryonic development or malignant transformation (Navarro and Monzo, 2010). The gene MIXL1 is regulated by microRNAs from let-7 family during the initiation and proliferation of lung cancer stem cells (Navarro and Monzo, 2010). Therefore, such gene is a potential biomarker presenting abnormal microRNA level regulation during lung tumorigenesis.
Shared Genes Between Epigenomics and Genomics
Six genes were shown to be essential lung cancer drivers involving methylation and genomics levels. The first gene HOXC12 (ENSP00000243103) has been reported to be triggered by its pathological methylated and inactivated promoter during lung tumorigenesis, validating its specific driver role at the methylation level (Guerrero-Preston et al., 2014). At the genomics level, researchers from Iran in 2019 reported that a SNP in gene HOXC12 is associated with risk of multiple cancer subtypes, implying the specific lung cancer driver potentials of this gene (Hajjari and Rahnama, 2019). Genes such as WNT9B (ENSP00000290015) (Xu et al., 2019), POU3F3 (ENSP00000355001) (Zeng et al., 2020), and PAX7 (ENSP00000364524) (Rácz et al., 2000) are lung cancer biomarkers at different omics levels. As for their respective contribution at epigenomics and genomics levels, WNT9B (Lan et al., 2006; Farkas et al., 2014), POU3F3 (Li et al., 2014; Kumar et al., 2016), and PAX7 (Starzyńska et al., 2020) have all been shown to be associated with lung cancer at epigenomics and genomics levels independently. For the two remaining genes, NLGN4Y (ENSP00000342535) and TBR1 (ENSP00000374205), in 2019, researchers from University of Southampton summarized NLGN4Y as a multi-omics level driver for lung cancer at least at epigenomics and transcriptomics levels (Jeyananthan and Niranjan, 2019). As for the genomics level, variants in NLGN4Y can regulate cell proliferation in multiple pathogenesis, though not directly reported in lung cancer (Nardello et al., 2021). The potential regulatory effects of such gene on cell proliferation indicated that it may also be associated with lung cancer at the genomic level. Similar experimental works support the driver role of the TBR1 gene at genomics and epigenomics levels (Ischenko et al., 2014; Serth et al., 2020). Therefore, all predicted genes at epigenomic (methylation) and genomics levels have been validated to be potential lung cancer drivers.
Shared Genes Between Epigenomics and Transcriptomics
Nine genes have been shown to be associated with lung cancer and are potential lung cancer drivers at epigenomics and transcriptomics levels. Among the nine genes, VIPR2 (ENSP00000262178) has already been discussed above and shown to be regulated at epigenomics and post-transcriptomics level. According to the same supporting paper mentioned above, this gene can be regulated at the transcriptomics level (Um et al., 2017). Other genes, such as TBR2 (ENSP00000295743) and WNT7B (ENSP00000341032), which are the homologues of TBR1 and WNT9B, respectively, are also reasonable to be speculated as candidate regulators at epigenomics and transcriptomics levels. Considering the limitation of the manuscript’s length, we selected three candidates for detailed discussion: HAND1, BMP3, and CMTM1. According to recent publications, HAND1 is a typical DNA methylation biomarker of small-cell lung cancers (Kalari et al., 2013). At the transcriptomics level, HAND1 has been identified as a biomarker in a newly reported single-cell study (Yin et al., 2019). Apart from HAND1, BMP3 together with BMP5 has been considered potential multi-omics level lung cancer biomarkers according to recent publications. In 2015, researchers from Huazhong University of Science and Technology conducted transcriptomics level analyses on human lung squamous cell carcinoma, confirming the driver role of BMP3 and BMP5 at the transcriptomics levels (Deng et al., 2015). At the epigenomics level (methylation), an earlier study on colorectal cancer confirmed that the methylation alteration on BMP3 or BMP5 may trigger the malignant transformation of normal cells (Loh et al., 2008). Therefore, the two genes could be potential lung cancer biomarkers at multi-omics levels. The CMTM1 gene has also been reported by two independent studies to be associated with lung cancer at transcriptomics (Hou et al., 2020) and methylation (Shao et al., 2007) levels, respectively. Other genes, such as PTH2 (Kim et al., 1998), TBR2 (Kaowinn et al., 2017), WNT7B (Kirikoshi and Katoh, 2002), and CER1 (Semenova et al., 2016), either have similar effects shared with their homologues or have been independently reported to be associated with lung cancer at different omics levels.
Shared Genes Between Genomics and Post-transcriptomics Regulation
Only one gene has been predicted to regulate lung cancer-associated pathological effects at genomics and post-transcriptomics levels. According to recent publications, MMP24 (ENSP00000246186) is associated with lung cancer at different omics levels (Fontenele et al., 2015; Wang et al., 2019; Wang et al., 2020). At genomics and post-transcriptomics levels, variants on MMP24 have been shown to be associated with lung cancer via a GWAS study in 2015 (Fontenele et al., 2015). In 2019, researchers from Tumor Hospital of Wuwei confirmed that microRNA-133a regulates MMP24 and further contributes to the tumorigenesis of lung cancer (Wang et al., 2019).
Shared Genes Between Transcriptomics and Post-transcriptomics Regulation
For genes that have been identified to be potential lung cancer drivers at transcriptomics and post-transcriptomics levels, only one gene, namely, VIPR2 (ENSP00000262178), was identified as multi-omics level regulator. As discussed above, this gene has shown to be effective at epigenomics, transcriptomics, and post-transcriptomics. Therefore, this gene could be a multi-omics regulator.
Shared Genes Between Genomics and Transcriptomics
Six genes have been identified to regulate lung tumorigenesis via genomics and transcriptomics levels. The first gene KRT20 (ENSP00000167588) has been shown to define the invasive characteristics of cancer cells at the transcriptomics level in multiple cancer subtypes (Eckstein et al., 2018), including lung cancer (Mollaoglu et al., 2018; Maly et al., 2019). As for the genomics level, variants in KRT20 are associated with lung cancer (Huang et al., 2015). Genes MMP16 (ENSP00000286614) and MMP19 (ENSP00000313437) belong to the matrix metalloproteinase family that is associated with multiple cancer subtypes, including lung cancer (Rudolph-Owen et al., 1998). As for their effects on genomics and transcriptomics levels, in the same publication, a summary of the effects of matrix metalloproteinases at multi-omics have been presented and demonstrated. TRPC3 (ENSP00000368966), as another novel driver gene associated with lung cancer at genomics and transcriptomics levels, has been validated to be potential genomic and transcriptomic driver for lung cancer. In 2016, a researcher from Guangzhou Medical University validated the effects of TRPC3 variants on lung cancer risk (Zhang et al., 2016). In 2021, such gene has been reported to be associated with malignant transformation at the transcriptomics level (Lin et al., 2021). The two remaining genes are COL16A1 (ENSP00000362776) and ADAMTS3 (ENSP00000286657). In 2016, COL16A1 has been shown to be associated with lung tumorigenesis via specific mutant patterns validated by in vitro A549 cell lines (Wang et al., 2016). As for the transcriptomics level, researchers from Medical University of South Carolina validated that at least in oral squamous cell carcinoma, the alteration of the gene expression of COL16A1 may promote tumor growth via interacting with the RNA-binding protein CELF1 (House et al., 2015). As for the last predicted gene ADAMTS3, according to recent publications, this gene has been confirmed to participate in lung tumorigenesis at genomics and transcriptomics levels (Vanni et al., 2016; Hannen et al., 2019).
Overall, several identified multi-omics lung cancer drivers were validated to be associated with fundamental pathological mechanisms. Therefore, LHD-based method was effective and accurate to identify lung cancer-associated tumor drivers and may help reveal the potential mechanisms of lung tumorigenesis.
Clinical Applications on New Driver Genes
Based on recent publications, some identified multi-omics lung cancer drivers have been already applied for clinical use on the diagnosis or treatment against lung cancer. According to recent publications, WNT9B, as the multi-omics biomarker for lung cancer at both epigenomics and genomics levels has been shown to act as an effective drug target (Stewart et al., 2014). The application of such drug can significantly improve the survival probability for lung cancer (Stewart et al., 2014). The next widely reported gene is MMP19, as a new driver gene at both genomics and transcriptomics levels. It has been widely reported to be associated with tumorigenesis, including gastric cancer (Shen et al., 2020), glioma (Luo et al., 2018) and lung cancer (Yu et al., 2014). As for its clinical application for lung cancer diagnosis and treatment, MMP19 has been reported to promote the metastasis of lung cancer and is associated with increased mortality of lung cancer as an active biomarker (Yu et al., 2014). TRPC3, as a driver gene at both genomics and transcriptomics levels have also been recognized as a promising clinical biomarker for lung cancer (Lastraioli et al., 2015), indicating the clinical significance of such multi-omics biomarker.
Conclusion
This study investigated lung cancer driver genes involving two omics levels. An LHD-based method was proposed to identify multi-omics lung cancer driver genes. Several genes, such as HOXC12, HAND1, VIPR2, KRT20, MMP24, and VIPR2, were discovered, and their special roles at different omics levels of lung cancer were confirmed. These findings may help improve the research progress on lung cancer.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://tcga-data.nci.nih.gov/docs/publications/luad_2014/.
Author Contributions
TH, ZL, and Y-DC designed the study. FY, XC, and LC performed the experiments. FY, XC, and Y-HZ analyzed the results. FY and Y-HZ wrote the manuscript. All authors contributed to the research and reviewed the manuscript.
Funding
This research was funded by the Strategic Priority Research Program of Chinese Academy of Sciences (XDA26040304, XDB38050200), National Key R and D Program of China (2018YFC0910403), the Fund of the Key Laboratory of Tissue Microenvironment and Tumor of Chinese Academy of Sciences (202002), the science and technology project of Binzhou Medical University (BY2016KYQD22), the medicine and health science technology development program of Shandong Province (2018WS541).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.825272/full#supplementary-material
References
Cancer Genome Atlas Research Network (2014). Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 511, 543–550. doi:10.1038/nature13385
Carlin, D. E., Demchak, B., Pratt, D., Sage, E., and Ideker, T. (2017). Network Propagation in the Cytoscape Cyberinfrastructure. Plos Comput. Biol. 13, e1005598. doi:10.1371/journal.pcbi.1005598
Chen, L., Huang, T., Zhang, Y.-H., Jiang, Y., Zheng, M., and Cai, Y.-D. (2016). Identification of Novel Candidate Drivers Connecting Different Dysfunctional Levels for Lung Adenocarcinoma Using Protein-Protein Interactions and a Shortest Path Approach. Sci. Rep. 6, 29849. doi:10.1038/srep29849
Chen, L., Zhang, Y.-H., Zhang, Z., Huang, T., and Cai, Y.-D. (2018). Inferring Novel Tumor Suppressor Genes with a Protein-Protein Interaction Network and Network Diffusion Algorithms. Mol. Ther. Methods Clin. Dev. 10, 57–67. doi:10.1016/j.omtm.2018.06.007
Chu, G. C. W., Lazare, K., and Sullivan, F. (2018). Serum and Blood Based Biomarkers for Lung Cancer Screening: a Systematic Review. BMC Cancer 18, 181. doi:10.1186/s12885-018-4024-3
Deng, T., Lin, D., Zhang, M., Zhao, Q., Li, W., Zhong, B., et al. (2015). Differential Expression of Bone Morphogenetic Protein 5 in Human Lung Squamous Cell Carcinoma and Adenocarcinoma. Acta Biochim. Biophys. Sin. 47, 557–563. doi:10.1093/abbs/gmv037
Eckstein, M., Wirtz, R., Gross-Weege, M., Breyer, J., Otto, W., Stoehr, R., et al. (2018). mRNA-Expression of KRT5 and KRT20 Defines Distinct Prognostic Subgroups of Muscle-Invasive Urothelial Bladder Cancer Correlating with Histological Variants. Int. J. Mol. Sci. 19, 3396. doi:10.3390/ijms19113396
Farkas, S. A., Vodickova, L., Vodicka, P., Nilsson, T. K., and Vymetalkova, V. (2014). DNA Methylation Changes in Genes Frequently Mutated in Sporadic Colorectal Cancer and in the DNA Repair and Wnt/β-Catenin Signaling Pathway Genes. Epigenomics 6, 179–191. doi:10.2217/epi.14.7
Fontenele, E. G. P., Amaral de Moraes, M. E., Brasil d'Alva, C., Pinheiro, D. P., Aguiar Sales Pinheiro Landim, S., de Sousa Barros, F. A., et al. (2015). Association Study of GWAS-Derived Loci with Height in Brazilian Children: Importance of MAP3K3, MMP24 and IGF1R Polymorphisms for Height Variation. Horm. Res. Paediatr. 84, 248–253. doi:10.1159/000437324
Gkountela, S., Castro-Giner, F., Szczerba, B. M., Vetter, M., Landin, J., Scherrer, R., et al. (2019). Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 176, 98–112. doi:10.1016/j.cell.2018.11.046
Guerrero-Preston, R., Michailidi, C., Marchionni, L., Pickering, C. R., Frederick, M. J., Myers, J. N., et al. (2014). Key Tumor Suppressor Genes Inactivated by "greater Promoter" Methylation and Somatic Mutations in Head and Neck Cancer. Epigenetics 9, 1031–1046. doi:10.4161/epi.29025
Hajjari, M., and Rahnama, S. (2019). Association between SNPs of Long Non-coding RNA HOTAIR and Risk of Different Cancers. Front. Genet. 10, 113. doi:10.3389/fgene.2019.00113
Hannen, R., Selmansberger, M., Hauswald, M., Pagenstecher, A., Nist, A., Stiewe, T., et al. (2019). Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers 11, 921. doi:10.3390/cancers11070921
Hou, X., He, S., Zhang, D., Yang, C., Shi, Y., and Zhang, K. (2020). Expression and Clinical Significance of CMTM6 in Nonsmall Cell Lung Cancer. DNA Cel Biol. 39, 2265–2271. doi:10.1089/dna.2020.5564
House, R. P., Talwar, S., Hazard, E. S., Hill, E. G., and Palanisamy, V. (2015). RNA-binding Protein CELF1 Promotes Tumor Growth and Alters Gene Expression in Oral Squamous Cell Carcinoma. Oncotarget 6, 43620–43634. doi:10.18632/oncotarget.6204
Huang, T., Yang, J., and Cai, Y. D. (2015). Novel Candidate Key Drivers in the Integrative Network of Genes, microRNAs, Methylations, and Copy Number Variations in Squamous Cell Lung Carcinoma. Biomed. Res. Int. 2015, 358125. doi:10.1155/2015/358125
Ischenko, I., Liu, J., Petrenko, O., and Hayman, M. J. (2014). Transforming Growth Factor-Beta Signaling Network Regulates Plasticity and Lineage Commitment of Lung Cancer Cells. Cell Death Differ 21, 1218–1228. doi:10.1038/cdd.2014.38
Jeyananthan, P., and Niranjan, M. (2019). “Classification and Regression Analysis of Lung Tumors from Multi-Level Gene Expression Data,” in International Joint Conference on Neural Networks (IJCNN), Budapest, Hungary, 14-19 July 2019 (IEEE), 1–8. doi:10.1109/IJCNN.2019.8852282
Kalari, S., Jung, M., Kernstine, K. H., Takahashi, T., and Pfeifer, G. P. (2013). The DNA Methylation Landscape of Small Cell Lung Cancer Suggests a Differentiation Defect of Neuroendocrine Cells. Oncogene 32, 3559–3568. doi:10.1038/onc.2012.362
Kaowinn, S., Kim, J., Lee, J., Shin, D. H., Kang, C.-D., Kim, D.-K., et al. (2017). Cancer Upregulated Gene 2 Induces Epithelial-Mesenchymal Transition of Human Lung Cancer Cells via TGF-β Signaling. Oncotarget 8, 5092–5110. doi:10.18632/oncotarget.13867
Kim, S. K., Su, L.-K., Oh, Y., Kemp, B. L., Hong, W. K., and Mao, L. (1998). Alterations of PTEN/MMAC1, a Candidate Tumor Suppressor Gene, and its Homologue, PTH2, in Small Cell Lung Cancer Cell Lines. Oncogene 16, 89–93. doi:10.1038/sj.onc.1201512
Kirikoshi, H., and Katoh, M. (2002). Expression of WNT7A in Human normal Tissues and Cancer, and Regulation of WNT7A and WNT7B in Human Cancer. Int. J. Oncol. 21, 895–900. doi:10.3892/ijo.21.4.895
Korenjak, M., and Zavadil, J. (2019). Experimental Identification of Cancer Driver Alterations in the Era of pan‐cancer Genomics. Cancer Sci. 110, 3622–3629. doi:10.1111/cas.14210
Kumar, S., Rathkolb, B., Kemter, E., Sabrautzki, S., Michel, D., Adler, T., et al. (2016). Generation and Standardized, Systemic Phenotypic Analysis of Pou3f3L423P Mutant Mice. PLoS One 11, e0150472. doi:10.1371/journal.pone.0150472
Lan, Y., Ryan, R. C., Zhang, Z., Bullard, S. A., Bush, J. O., Maltby, K. M., et al. (2006). Expression ofWnt9b and Activation of Canonical Wnt Signaling during Midfacial Morphogenesis in Mice. Dev. Dyn. 235, 1448–1454. doi:10.1002/dvdy.20723
Lastraioli, E., Iorio, J., and Arcangeli, A. (2015). Ion Channel Expression as Promising Cancer Biomarker. Biochim. Biophys. Acta Biomembr. 1848, 2685–2702. doi:10.1016/j.bbamem.2014.12.016
Li, W., Zheng, J., Deng, J., You, Y., Wu, H., Li, N., et al. (2014). Increased Levels of the Long Intergenic Non-protein Coding RNA POU3F3 Promote DNA Methylation in Esophageal Squamous Cell Carcinoma Cells. Gastroenterology 146, 1714–1726. doi:10.1053/j.gastro.2014.03.002
Li, Y., Gu, J., Xu, F., Zhu, Q., Ge, D., and Lu, C. (2018). Transcriptomic and Functional Network Features of Lung Squamous Cell Carcinoma through Integrative Analysis of GEO and TCGA Data. Sci. Rep. 8, 15834. doi:10.1038/s41598-018-34160-w
Lin, D.-C., Zheng, S.-Y., Zhang, Z.-G., Luo, J.-H., Zhu, Z.-L., Li, L., et al. (2021). TRPC3 Promotes Tumorigenesis of Gastric Cancer via the CNB2/GSK3β/NFATc2 Signaling Pathway. Cancer Lett. 519, 211–225. doi:10.1016/j.canlet.2021.07.038
Liu, H., Hu, B., Chen, L., and Lu, L. (2021). Identifying Protein Subcellular Location with Embedding Features Learned from Networks. CP 18, 646–660. doi:10.2174/1570164617999201124142950
Loh, K., Chia, J. A., Greco, S., Cozzi, S.-J., Buttenshaw, R. L., Bond, C. E., et al. (2008). Bone Morphogenic Protein 3 Inactivation Is an Early and Frequent Event in Colorectal Cancer Development. Genes Chromosom. Cancer 47, 449–460. doi:10.1002/gcc.20552
Luo, Q., Luo, H., Chen, X., Yan, P., Fu, H., Huang, H., et al. (2018). The Expression of MMP19 and its Clinical Significance in Glioma. Int. J. Clin. Exp. Pathol. 11, 5407–5412.
Maly, V., Maly, O., Kolostova, K., and Bobek, V. (2019). Circulating Tumor Cells in Diagnosis and Treatment of Lung Cancer. In Vivo 33, 1027–1037. doi:10.21873/invivo.11571
Mollaoglu, G., Jones, A., Wait, S. J., Mukhopadhyay, A., Jeong, S., Arya, R., et al. (2018). The Lineage-Defining Transcription Factors SOX2 and NKX2-1 Determine Lung Cancer Cell Fate and Shape the Tumor Immune Microenvironment. Immunity 49, 764–779. doi:10.1016/j.immuni.2018.09.020
Naderi, E., Mostafaei, M., Pourshams, A., and Mohamadkhani, A. (2014). Network of microRNAs-mRNAs Interactions in Pancreatic Cancer. Biomed. Res. Int. 2014, 534821. doi:10.1155/2014/534821
Nardello, R., Antona, V., Mangano, G. D., Salpietro, V., Mangano, S., and Fontana, A. (2021). A Paradigmatic Autistic Phenotype Associated with Loss of PCDH11Y and NLGN4Y Genes. BMC Med. Genomics 14, 98. doi:10.1186/s12920-021-00934-x
Navarro, A., and Monzó, M. (2010). MicroRNAs in Human Embryonic and Cancer Stem Cells. Yonsei Med. J. 51, 622–632. doi:10.3349/ymj.2010.51.5.622
Ng, K.-L., Ciou, J.-S., and Huang, C.-H. (2010). Prediction of Protein Functions Based on Function-Function Correlation Relations. Comput. Biol. Med. 40, 300–305. doi:10.1016/j.compbiomed.2010.01.001
Olivier, M., Asmis, R., Hawkins, G. A., Howard, T. D., and Cox, L. A. (2019). The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 20, 4781. doi:10.3390/ijms20194781
Pan, X., Chen, L., Liu, M., Niu, Z., Huang, T., and Cai, Y. D. (2021a). Identifying Protein Subcellular Locations with Embeddings-Based Node2loc. IEEE/ACM Trans. Comput. Biol. Bioinform. doi:10.1109/tcbb.2021.3080386
Pan, X., Li, H., Zeng, T., Li, Z., Chen, L., Huang, T., et al. (2021b). Identification of Protein Subcellular Localization with Network and Functional Embeddings. Front. Genet. 11, 626500. doi:10.3389/fgene.2020.626500
Pao, W., and Girard, N. (2011). New Driver Mutations in Non-small-cell Lung Cancer. Lancet Oncol. 12, 175–180. doi:10.1016/s1470-2045(10)70087-5
Pon, J. R., and Marra, M. A. (2015). Driver and Passenger Mutations in Cancer. Annu. Rev. Pathol. Mech. Dis. 10, 25–50. doi:10.1146/annurev-pathol-012414-040312
Rácz, A., Brass, N., Höfer, M., Sybrecht, G. W., Remberger, K., and Meese, E. U. (2000). Gene Amplification at Chromosome 1pter-P33 Including the Genes PAX7 and ENO1 in Squamous Cell Lung Carcinoma. Int. J. Oncol. 17, 67–73. doi:10.3892/ijo.17.1.67
Rudolph-Owen, L. A., Chan, R., Muller, W. J., and Matrisian, L. M. (1998). The Matrix Metalloproteinase Matrilysin Influences Early-Stage Mammary Tumorigenesis. Cancer Res. 58, 5500–5506.
Sears, C. R., and Mazzone, P. J. (2020). Biomarkers in Lung Cancer. Clin. Chest Med. 41, 115–127. doi:10.1016/j.ccm.2019.10.004
Semenova, E. A., Kwon, M.-c., Monkhorst, K., Song, J.-Y., Bhaskaran, R., Krijgsman, O., et al. (2016). Transcription Factor NFIB Is a Driver of Small Cell Lung Cancer Progression in Mice and Marks Metastatic Disease in Patients. Cel Rep. 16, 631–643. doi:10.1016/j.celrep.2016.06.020
Serth, J., Peters, I., Dubrowinskaja, N., Reese, C., Albrecht, K., Klintschar, M., et al. (2020). Age-, Tumor-, and Metastatic Tissue-Associated DNA Hypermethylation of a T-Box Brain 1 Locus in Human Kidney Tissue. Clin. Epigenet 12, 33. doi:10.1186/s13148-020-0823-x
Shao, L., Cui, Y., Li, H., Liu, Y., Zhao, H., Wang, Y., et al. (2007). CMTM5 Exhibits Tumor Suppressor Activities and Is Frequently Silenced by Methylation in Carcinoma Cell Lines. Clin. Cancer Res. 13, 5756–5762. doi:10.1158/1078-0432.ccr-06-3082
Shen, D., Zhao, H., Zeng, P., Song, J., Yang, Y., Gu, X., et al. (2020). Circular RNA Hsa_circ_0005556 Accelerates Gastric Cancer Progression by Sponging miR-4270 to Increase MMP19 Expression. J. Gastric Cancer 20, 300. doi:10.5230/jgc.2020.20.e28
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2021). Cancer Statistics, 2021. CA Cancer J. Clin. 71, 7–33. doi:10.3322/caac.21654
Starzyńska, T., Karczmarski, J., Paziewska, A., Kulecka, M., Kuśnierz, K., Żeber-Lubecka, N., et al. (2020). Differences between Well-Differentiated Neuroendocrine Tumors and Ductal Adenocarcinomas of the Pancreas Assessed by Multi-Omics Profiling. Int. J. Mol. Sci. 21, 4470. doi:10.3390/ijms21124470
Stewart, D. J., Chang, D. W., Ye, Y., Spitz, M., Lu, C., Shu, X., et al. (2014). Wnt Signaling Pathway Pharmacogenetics in Non-small Cell Lung Cancer. Pharmacogenomics J. 14, 509–522. doi:10.1038/tpj.2014.21
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene Set Enrichment Analysis: a Knowledge-Based Approach for Interpreting Genome-wide Expression Profiles. Proc. Natl. Acad. Sci. 102, 15545–15550. doi:10.1073/pnas.0506580102
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2014). STRING V10: Protein-Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 43, D447–D452. doi:10.1093/nar/gku1003
Tokheim, C. J., Papadopoulos, N., Kinzler, K. W., Vogelstein, B., and Karchin, R. (2016). Evaluating the Evaluation of Cancer Driver Genes. Proc. Natl. Acad. Sci. USA 113, 14330–14335. doi:10.1073/pnas.1616440113
Turanli, B., Karagoz, K., Gulfidan, G., Sinha, R., Mardinoglu, A., and Arga, K. Y. (2018). A Network-Based Cancer Drug Discovery: From Integrated Multi-Omics Approaches to Precision Medicine. Curr. Pharm. Des. 24, 3778–3790. doi:10.2174/1381612824666181106095959
Um, S.-W., Kim, H. K., Kim, Y., Lee, B. B., Kim, D., Han, J., et al. (2017). Bronchial Biopsy Specimen as a Surrogate for DNA Methylation Analysis in Inoperable Lung Cancer. Clin. Epigenet 9, 131. doi:10.1186/s13148-017-0432-5
Vanni, I., Coco, S., Bonfiglio, S., Cittaro, D., Genova, C., Biello, F., et al. (2016). Whole Exome Sequencing of Independent Lung Adenocarcinoma, Lung Squamous Cell Carcinoma, and Malignant Peritoneal Mesothelioma. Medicine (Baltimore) 95, e5447. doi:10.1097/md.0000000000005447
Wang, Y., Yan, P., Liu, Z., Yang, X., Wang, Y., Shen, Z., et al. (2016). MEK Inhibitor Can Reverse the Resistance to Bevacizumab in A 549 Cells Harboring Kirsten Rat Sarcoma Oncogene Homolog Mutation. Thorac. Cancer 7, 279–287. doi:10.1111/1759-7714.12325
Wang, H., Zhang, Y., Zhang, Y., Liu, W., and Wang, J. (2019). Cryptotanshinone Inhibits Lung Cancer Invasion via microRNA-133a/matrix Metalloproteinase 14 Regulation. Oncol. Lett. 18, 2554–2559. doi:10.3892/ol.2019.10580
Wang, Y., Ding, X., Liu, B., Li, M., Chang, Y., Shen, H., et al. (2020). ETV4 Overexpression Promotes Progression of Non-small Cell Lung Cancer by Upregulating PXN and MMP1 Transcriptionally. Mol. Carcinog 59, 73–86. doi:10.1002/mc.23130
Xu, H., Jiao, X., Wu, Y., Li, S., Cao, L., and Dong, L. (2019). Exosomes Derived from PM2.5-treated Lung Cancer Cells Promote the Growth of Lung Cancer via the Wnt3a/β Catenin Pathway. Oncol. Rep. 41, 1180–1188. doi:10.3892/or.2018.6862
Yin, N., Liang, X., Liang, S., Liang, S., Yang, R., Hu, B., et al. (2019). Embryonic Stem Cell- and Transcriptomics-Based In Vitro Analyses Reveal that Bisphenols A, F and S Have Similar and Very Complex Potential Developmental Toxicities. Ecotoxicol. Environ. Saf. 176, 330–338. doi:10.1016/j.ecoenv.2019.03.115
Yu, G., Herazo-Maya, J. D., Nukui, T., Romkes, M., Parwani, A., Juan-Guardela, B. M., et al. (2014). Matrix Metalloproteinase-19 Promotes Metastatic BehaviorIn Vitroand Is Associated with Increased Mortality in Non-small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 190, 780–790. doi:10.1164/rccm.201310-1903oc
Yuan, F., and Lu, W. (2017). Prediction of Potential Drivers Connecting Different Dysfunctional Levels in Lung Adenocarcinoma via a Protein-Protein Interaction Network. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2284–2293. doi:10.1016/j.bbadis.2017.11.018
Zeng, Q., Dai, Y., Duan, C., Zeng, R., Zeng, Q., and Wei, C. (2020). Long Noncoding RNA POU3F3 Enhances Cancer Cell Proliferation, Migration and Invasion in Non-small Cell Lung Cancer (Adenocarcinoma) by Downregulating microRNA-30d-5p. BMC Pulm. Med. 20, 185. doi:10.1186/s12890-020-01218-3
Zhang, X., and Chen, L. (2020). Prediction of Membrane Protein Types by Fusing Protein-Protein Interaction and Protein Sequence Information. Biochim. Biophys. Acta Proteins Proteomics 1868, 140524. doi:10.1016/j.bbapap.2020.140524
Zhang, Z., Wang, J., He, J., Zeng, X., Chen, X., Xiong, M., et al. (2016). Identification of TRPCs Genetic Variants that Modify Risk for Lung Cancer Based on the Pathway and Two-Stage Study. Meta Gene 9, 191–196. doi:10.1016/j.mgene.2016.07.005
Zhang, X., Chen, L., Guo, Z.-H., and Liang, H. (2019). Identification of Human Membrane Protein Types by Incorporating Network Embedding Methods. IEEE Access 7, 140794–140805. doi:10.1109/access.2019.2944177
Keywords: lung cancer, driver gene, epigenomics, genomics, transcriptomics, posttranscriptomics, heat diffusion algorithm, protein-protein interaction network
Citation: Yuan F, Cao X, Zhang Y-H, Chen L, Huang T, Li Z and Cai Y-D (2022) Identification of Novel Lung Cancer Driver Genes Connecting Different Omics Levels With a Heat Diffusion Algorithm. Front. Cell Dev. Biol. 10:825272. doi: 10.3389/fcell.2022.825272
Received: 30 November 2021; Accepted: 06 January 2022;
Published: 26 January 2022.
Edited by:
Liang Cheng, Harbin Medical University, ChinaReviewed by:
Juntao Li, Henan Normal University, ChinaMeijing Li, Shanghai Maritime University, China
Copyright © 2022 Yuan, Cao, Zhang, Chen, Huang, Li and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Huang, dG9odWFuZ3Rhb0AxMjYuY29t; ZhanDong Li, bGl6ZDU5MUBqbGVudS5lZHUuY24=; Yu-Dong Cai, Y2FpX3l1ZEAxMjYuY29t
†These authors have contributed equally to this work