 Deepa S. Rajan1†Sukhleen Kour1†Tyler R. Fortuna1†Margot A. Cousin2,3Sarah S. Barnett4Zhiyv Niu4,5Dusica Babovic-Vuksanovic2,4,5Eric W. Klee2,3,4,5Brian Kirmse6Micheil Innes7Siri Lynne Rydning8Kaja K. Selmer9Magnus Dehli Vigeland10Anne Kjersti Erichsen11Andrea H. Nemeth12Francisca Millan13Catherine DeVile14Katherine Fawcett15,16Adrien Legendre17David Sims15Ricardo Parolin Schnekenberg12Lydie Burglen18,19Sandra Mercier26,27Somayeh Bakhtiari20,21Rosario Francisco-Velilla22Azman Embarc-Buh22Encarnacion Martinez-Salas22Kristen Wigby24,25Jerica Lenberg25Jennifer R. Friedman23,24,25Michael C. Kruer20,21Udai Bhan Pandey1*
Deepa S. Rajan1†Sukhleen Kour1†Tyler R. Fortuna1†Margot A. Cousin2,3Sarah S. Barnett4Zhiyv Niu4,5Dusica Babovic-Vuksanovic2,4,5Eric W. Klee2,3,4,5Brian Kirmse6Micheil Innes7Siri Lynne Rydning8Kaja K. Selmer9Magnus Dehli Vigeland10Anne Kjersti Erichsen11Andrea H. Nemeth12Francisca Millan13Catherine DeVile14Katherine Fawcett15,16Adrien Legendre17David Sims15Ricardo Parolin Schnekenberg12Lydie Burglen18,19Sandra Mercier26,27Somayeh Bakhtiari20,21Rosario Francisco-Velilla22Azman Embarc-Buh22Encarnacion Martinez-Salas22Kristen Wigby24,25Jerica Lenberg25Jennifer R. Friedman23,24,25Michael C. Kruer20,21Udai Bhan Pandey1*- 1Department of Pediatrics, Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center, Pittsburgh, PA, United States
- 2Department of Center for Individualized Medicine, Mayo Clinic, Rochester, MN, United States
- 3Department of Quantitative Health Sciences, Mayo Clinic, Rochester, MN, United States
- 4Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States
- 5Department of Clinical Genomics, Mayo Clinic, Rochester, MN, United States
- 6Division of Genetics, University of Mississippi Medical Center, Jackson, MS, United States
- 7Department of Medical Genetics and Alberta Children's Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 8Department of Neurology, Oslo University Hospital, Oslo, Norway
- 9Department of Research and Innovation, Division of Clinical Neuroscience, Oslo University Hospital and the University of Oslo, Oslo, Norway
- 10Department of Medical Genetics, Oslo University Hospital, and Institute of Clinical Medicine, University of Oslo, Oslo, Norway
- 11Department of Ophthalmology, Oslo University Hospital, Oslo, Norway
- 12Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom
- 13GeneDx, Gaithersburg, MD, United States
- 14Great Ormond Street Hospital, London, United Kingdom
- 15Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, United Kingdom
- 16Department of Health Sciences, University of Leicester, Leicester, United Kingdom
- 17Laboratoire de biologie médicale multisites Seqoia—FMG2025, Paris, France
- 18Centre de Référence des Malformations et Maladies Congénitales du Cervelet et Laboratoire de Neurogénétique Moléculaire, Département de Génétique, AP-HP. Sorbonne Université, Hôpital Trousseau, Paris, France
- 19Developmental Brain Disorders Laboratory, Imagine Institute, INSERM UMR 1163, Paris, France
- 20Barrow Neurological Institute, Phoenix Children’s Hospital, Phoenix, AZ, United States
- 21Departments of Child Health, Neurology, Cellular and Molecular Medicine and Program in Genetics, University of Arizona College of Medicine—Phoenix, Phoenix, AZ, United States
- 22Centro de Biologia Molecular Severo Ochoa, CSIC-UAM, Madrid, Spain
- 23Department of Neurosciences, University of California San Diego, San Diego, CA, United States
- 24Department of Pediatrics, University of California San Diego, San Diego, CA, United States
- 25Rady Children’s Institute for Genomic Medicine, San Diego, CA, United States
- 26CHU Nantes, Service de génétique médicale, Centre de Référence Anomalies du Développement et Syndromes Malformatifs, Nantes, France
- 27Nantes Université, CNRS, INSERM, l’institut du thorax, Nantes, France
The hereditary ataxias are a heterogenous group of disorders with an increasing number of causative genes being described. Due to the clinical and genetic heterogeneity seen in these conditions, the majority of such individuals endure a diagnostic odyssey or remain undiagnosed. Defining the molecular etiology can bring insights into the responsible molecular pathways and eventually the identification of therapeutic targets. Here, we describe the identification of biallelic variants in the GEMIN5 gene among seven unrelated families with nine affected individuals presenting with spastic ataxia and cerebellar atrophy. GEMIN5, an RNA-binding protein, has been shown to regulate transcription and translation machinery. GEMIN5 is a component of small nuclear ribonucleoprotein (snRNP) complexes and helps in the assembly of the spliceosome complexes. We found that biallelic GEMIN5 variants cause structural abnormalities in the encoded protein and reduce expression of snRNP complex proteins in patient cells compared with unaffected controls. Finally, knocking out endogenous Gemin5 in mice caused early embryonic lethality, suggesting that Gemin5 expression is crucial for normal development. Our work further expands on the phenotypic spectrum associated with GEMIN5-related disease and implicates the role of GEMIN5 among patients with spastic ataxia, cerebellar atrophy, and motor predominant developmental delay.
Introduction
Hereditary ataxias are a heterogenous group of neurological disorders affecting individuals of all age groups, and patients present with a disturbance in movement coordination. While cerebellar abnormalities are usually implicated, lesions elsewhere in the neuroaxis can lead to gait abnormalities that mimic the clinical phenotype. In addition, it is well recognized that many of the autosomal recessive cerebellar ataxias can present as spastic ataxias with pyramidal signs (Ruano et al., 2014). While traditionally these were clinically distinct entities, a spectrum of diseases ranging in clinical presentation from pure cerebellar ataxias to spastic paraplegias have been observed more recently (Synofzik and Schüle, 2017). The genetic causes of hereditary cerebellar ataxias and hereditary spastic ataxias are heterogenous, and often next-generation sequencing technologies are used to establish a molecular diagnosis (Németh et al., 2013). To date, more than 150 genes (OMIM categories include: 45 genes as autosomal dominant spinocerebellar ataxias, 29 autosomal recessive spinocerebellar ataxias, 8 spastic ataxias, and 73 spastic paraplegias) have been implicated in the ataxia paraplegia spectrum of diseases (McKusick, 2007). The identification of the molecular diagnosis in these patients is the first step toward understanding the molecular mechanism and is crucial for developing effective targeted therapeutic strategies.
GEMIN5 is an RNA-binding protein involved in regulating multiple aspects of transcriptional and translational processes (Francisco-Velilla et al., 2020; Martinez-Salas et al., 2020; Moreno-Morcillo et al., 2020). The GEMIN5 protein has 13 tryptophan–aspartic acid dipeptides (WD domains) at the N-terminus, followed by a dimerization domain, and a noncanonical RNA-binding site (RBS1) domain (Francisco-Velilla et al., 2020; Martinez-Salas et al., 2020; Moreno-Morcillo et al., 2020). GEMIN5 is predominantly a cytoplasmic protein with sparse expression in the nucleus, suggesting that this protein might be involved in regulating functions in both cellular compartments. The WD domain of GEMIN5 assists in the biogenesis of small nuclear ribonucleoproteins (snRNPs), the building blocks of splicing machinery, and aids in the formation of the snRNP complex along with the other complex proteins (SMN, Gemin2-8 and Unrip) (Lozano et al., 2018; Francisco-Velilla et al., 2020). Moreover, the noncanonical RNA-binding sites (RBS), located on the C-terminus, promote GEMIN5 to interact with various mRNAs and regulate their translation. Importantly, mutations affecting the survival motor neuron (SMN) protein have been linked with a neuromuscular disorder, spinal muscular atrophy (SMA), which has been linked with disruption of snRNP complex assembly (Melki et al., 1994; Lefebvre et al., 1995; Lefebvre et al., 1997; Burlet et al., 1998; Pellizzoni et al., 2002). The GEMIN5 protein has numerous functions as it incorporates into cytoplasmic stress granules, binds to ribosomes, and regulates global translation and SMN expression.
Here, we define a novel neurodevelopmental ataxia syndrome in patients with autosomal recessive variants in the GEMIN5 gene. We provide characterization of their neurological phenotype to include a spectrum of the cerebellar ataxia and spastic paraplegia phenotypes among seven unrelated families with nine affected individuals. We employ computational biology, biochemistry, and mouse genetics to examine the functional consequences of perturbing GEMIN5 protein in vitro and in vivo. Our data suggest that loss-of-function of GEMIN5 is detrimental in human patient cells and in mice.
Methods
Patient Recruitment
GEMIN5 variants were identified in probands by whole-exome sequencing in clinical diagnostic settings at different sites. We found a subset of our GEMIN5 patients through GeneMatcher (Wang et al., 2007). All patients were evaluated by a neurologist or geneticist at their respective referral centers. We reviewed the clinical information, neurological symptoms, examination, and radiological studies including brain MRI and other clinical evaluations in each patient. All patient information was deidentified. Informed consent was obtained from patients for publication at each site per local institution requirements by the authors.
Genetic Studies and Variant Assessment
Whole-exome sequencing (WES) was performed at different genetic centers using next-generation sequencing techniques, and all variants were confirmed via Sanger sequencing with standard methods.
Family 1: The trio was sequenced at the Yale Center for Genome Analysis (YCGA). Genomic DNA was captured using an IDT xGen exome kit followed by Illumina DNA sequencing. WES data were processed using two independent pipelines at the Yale School of Medicine and Phoenix Children’s Hospital. At each site, sequence reads were mapped to the reference genome (GRCh37) with BWA-MEM and further processed using GATK Best Practice workflows, which include duplication marking, indel realignment, and base quality recalibration. Single-nucleotide variants and small indels were called with GATK HaplotypeCaller and annotated using ANNOVAR, dbSNP (v138), 1000 Genomes (August 2015), NHLBI Exome Variant Server (EVS), and the Exome Aggregation Consortium v3 (ExAC). Rare deleterious missense variants and LOF variants (stop-gain, stop-loss, frameshift insertions/deletions, canonical splice site, and start-loss) were selected. MetaSVM and Combined Annotation Dependent Deletion (CADD v1.3) algorithms were used to predict deleteriousness of missense variants (MetaSVM-deleterious or CADD ≥20). Family 2: The trio was analyzed at the Genomic Sequencing Platform Seqoia (Paris) as follows: preparation of the libraries using NEBNext® Ultra II End repair/A-tailing module and Ligation module (New England Biolabs®), whole-genome sequencing on a NovaSeq 6000® (Illumina®) using 2 × 150 paired-end sequencing, high-quality reads mapping against the human reference genome (hg38), variant calling with GATK4 v4.1.7.0 (Broad Institute), and annotation with SnpEff (4.3t) and SnpSift (4.3t). Family 3: Extensive metabolic testing, microarray, spinocerebellar ataxia panel, and Friedrich’s ataxia panel were negative. Exome revealed GEMIN5: Chr 5 c.3046 C > T p. Arg 1016cys, c. 1452 dup p. Met485HisfsTer27. Family 4: The exome was done as per methodology described previously (Parolin Schnekenberg et al., 2015). Patients were recruited, and consent for participation in the study was obtained according to the Declaration of Helsinki and approved by the Central Oxford Research Ethics Committee and the Research and Development Department of the Oxford Radcliffe Hospitals NHS Trust, Oxford. All patients or their parents provided written consent for the study.
Family 5: Clinical exome sequencing was performed as described previously (Cousin et al., 2019). The proband and his parents provided written informed consent to a study approved by the Mayo Clinic Institutional Review Board. Family 5 sequence reads were mapped to the reference genome (GRCh37) with BWA-MEM and further processed using GATK Best Practice workflows, which include duplication marking, indel realignment, and base quality recalibration. Single-nucleotide variants and small indels were called with GATK HaplotypeCaller. The compound heterozygous variants originally identified and Sanger confirmed by the clinical laboratory for Family 5 were called with high confidence using the workflow implemented for Family 1. Family 6: Exome sequencing was done as per previously described protocol (Guillen Sacoto et al., 2020; Kour et al., 2021). All the variants are annotated by using the GEMIN5 NP_056280.2 reference transcript in GnomAD and the other databases to estimate the allelic frequency.
Family 7: Whole-exome sequencing (WES) was performed in the two affected siblings, using DNA from peripheral blood leukocytes. The exome was enriched using the SureSelectXT V5 exome kit (Agilent, Böblingen, Germany) and sequenced on a HiSeq2500 sequencer (Illumina, San Diego, CA, United States). High-quality reads were mapped against the human reference genome (hg19), and variants were called following the Genome Analysis Tool Kit version 3.3.0 best practice recommendations (McKenna et al., 2010; Ma et al., 2013). After annotation with Annovar, filtration and downstream analysis was done with FILTUS (Vigeland et al., 2016). The identified GEMIN5 variants were validated by Sanger sequencing.
The damaging index of GEMIN5 variants was determined by using various in silico prediction tools, such as Polyphen2, Provean, SNAP2, MUpro, PhD SNP, and SIFT (Cheng et al., 2006; Ng and Henikoff, 2006; Bromberg and Rost, 2007; Mi et al., 2010; Choi et al., 2012; Sim et al., 2012). Candidate variants were validated by Sanger sequencing and tested for cosegregation in all family members whenever samples were available (Supplementary Figure S1).
All the variants were annotated by using the GEMIN5 NP_056280.2 reference transcript to estimate the allelic frequency in GnomAD and the other databases. The effect of GEMIN5 variants on its structure and folding was predicted by using the molecular graphics tool PyMOL.
Immunoblotting
PBMCs were isolated from whole blood of the proband and the unaffected parents (family 5) by following the Mayo Clinic’s Biospecimen Accessioning and Processing (BAP) core’s standard protocol with a target count of 10 million cells. The isolated PBMCs were lysed in the buffer containing 150 mM NaCl, 50 mM NaF, 0.1% SDS, 1% NP40, 2 mM EDTA, 1% sodium deoxycholate, 1 mM DTT, 0.2 mM sodium orthovandate, and protease inhibitor (Roche) for 10 min on ice. The protein lysates were sonicated and centrifuged at 16,000 × g for 10 min to remove the debris. The protein concentration of each sample was determined by using Pierce BCA protein assay kit (Thermo-Scientific). Of the total protein, 40 µg was separated in 4%–12% NuPAGE gel (Novex/Life technologies) and transferred to a nitrocellulose membrane (Invitrogen). The membranes were blocked in 2.5% milk (BLOT- QuickBlocker™ EMD Millipore) in TBST followed by overnight incubation with primary antibody at 4°C. Membranes were washed, incubated with secondary antibody for 1 h at room temperature, and imaged on Odyssey® CLx (LI-COR Biosciences). The band intensities were measured using Image Studio™ (LI-COR Biosciences). All the primary and secondary antibodies were prepared in 2.5% blocking buffer.
The primary antibodies are as follows: Rabbit anti-GEMIN5 (1:1,000; Proteintech); Rabbit anti-GEMIN4 (1:2,000; Novas Biologicals); Rabbit anti-GEMIN2 (1:1,000; Invitrogen), Mouse anti-SMN (1:4,000; BD Biosciences); Mouse anti-α-tubulin (1:8,000; Sigma T5168).
The secondary antibodies are as follows: Goat anti-mouse Dylight 680 (1:10,000; LI-COR 925-68070); Goat anti-mouse Dylight 800 (1:10,000; Invitrogen SA5-10176); Goat anti-rabbit Dylight 680 (1:10,000; Invitrogen 35568); and Goat anti-rabbit Dylight 800 (1:10,000; Invitrogen SA5-35571).
Knockout Mice Creation
Creation of the Gemin5 Knockout mice in the C57BL/6N background was carried out at the MRC Harwell Institute through the International Mouse Phenotyping Consortium (IMPC). Using CRISPR/Cas9 and the EUCOMM/KOMP-CSD allele structure, a premature stop codon was introduced into exon 7 of the Mus muculus Gemin5 gene, resulting in a null GEMIN5 allele. Heterozygous Gemin5 knockout mice [C57BL/6NTac-Gemin5em1(IMPC)H/H] were confirmed via short-range PCR at the MRC Harwell Institute.
Results
Identification of Bi-Allelic GEMIN5 Variants
Here, we report nine patients with spastic ataxia and cerebellar atrophy (Figure 1). All patients had extensive metabolic and genetic testing, which was unrevealing before next-generation sequencing revealed biallelic variants in the GEMIN5 gene (Table 1). All variants were rare with allele frequencies ranging from 0 to 4.48e−3 as a heterozygote and showed autosomal recessive inheritance pattern (Table 3). The majority of these variants, with two exceptions, (p. Arg1016Cys and p. Pro594Arg), have not been reported in the GnomAD database in the homozygous state (Table 3). The p. Arg1016Cys was observed with a second allele disrupted by either a frameshift or a nonsense variant in three patients from three unrelated families. The p. Arg1016Cys variant is likely to affect the dimerization domain, which might result in downstream functional abnormality. Similarly, we found the p. Pro594Arg allele with another missense variant in two patients (Table 1). The variants identified were missense, frame shift, termination, and a predicted splice site variant. All missense variants affected conserved amino acid residues and were predicted to be pathogenic by various computational prediction tools (Table 4).
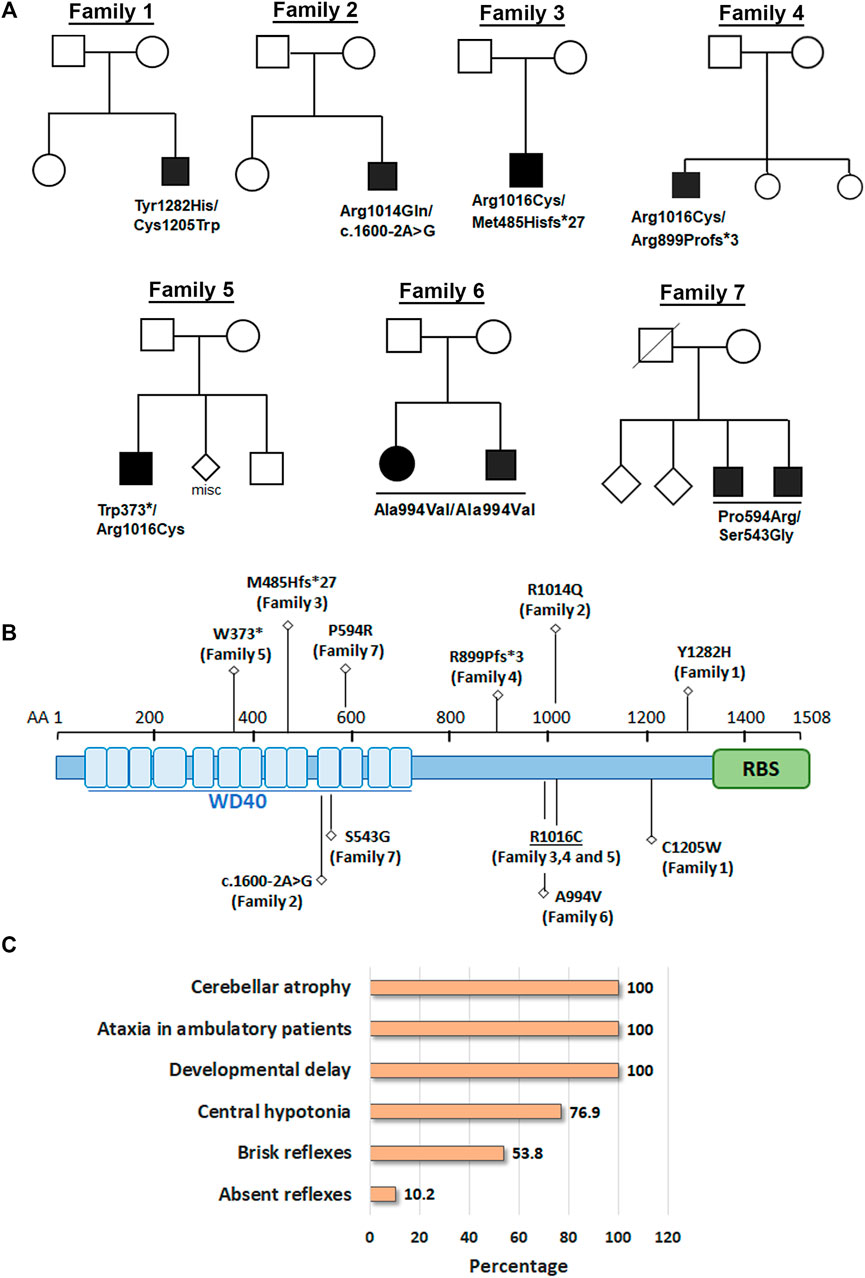
FIGURE 1. Biallelic variants in GEMIN5 leads to cerebellar atrophy and spastic ataxia neurodevelopmental disease. (A) Pedigrees of seven unrelated families with nine affected individuals are shown in the top panel. Variants in GEMIN5 are shown below the affected individuals. Affected individuals are shown as filled black circles/squires. (B) GEMIN5 protein domain structure. Type and position of the identified variants are shown along with the family numbers. (C) The frequency of clinical presentations in our patients with autosomal recessive variants in GEMIN5. The numerator and denominator in brackets indicate the number of affected probands and the number of probands assessed for the respective feature, respectively.
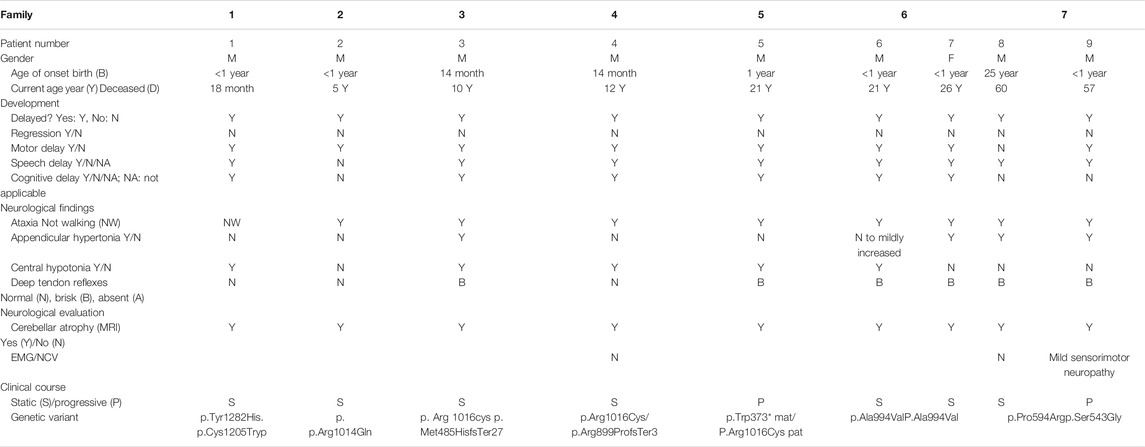
TABLE 1. Clinical presentations in patients with biallelic variants in GEMIN5.
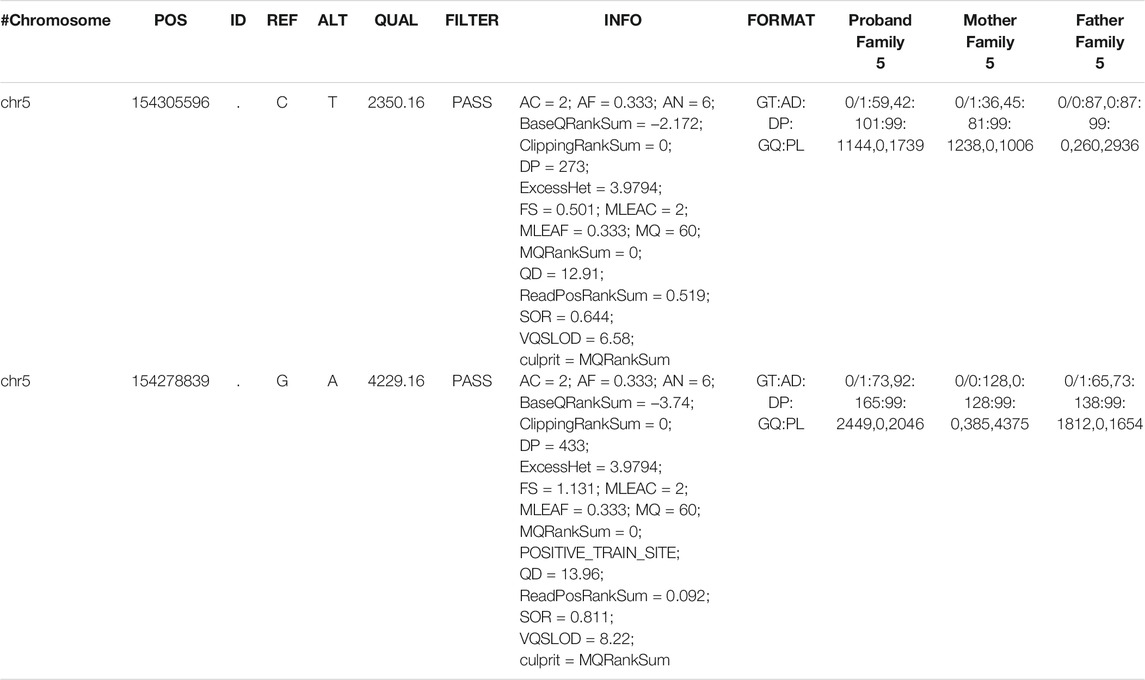
TABLE 2. Reanalysis of GEMIN5 variant (family 5).
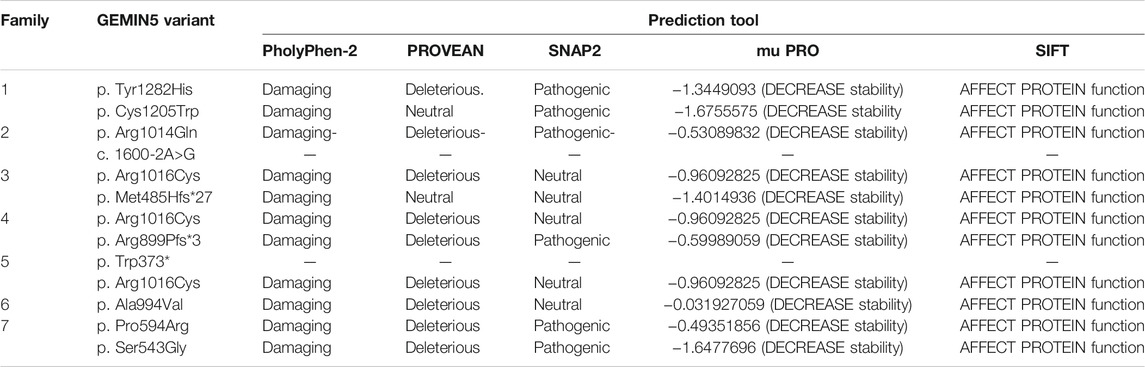
TABLE 3. Allelic frequencies of the variants identified among our GEMIN5 patients.

TABLE 4. In silico prediction of GEMIN5 variants.
Patients, 8/9, presented in the first 2 years of life with concerns for delayed motor development. No childhood motor or cognitive regression was observed in any of the patients. While all patients had signs of motor dysfunction, the neurological exam ranged from hypotonia and tremulousness to severe spastic ataxia. While central hypotonia was observed in some patients, appendicular tone ranged from normal to spastic. Interestingly, all patients presented with normal to brisk reflexes. All ambulatory patients were assessed by neurologists and were noted to be ataxic on clinical examination. The disease progression was variable among our patients, as some of the patients had a static course, while others showed a possibly slow, progressive ataxia. Magnetic resonance imaging (MRI) of the brain showed cerebellar atrophy in all patients (Figure 2).
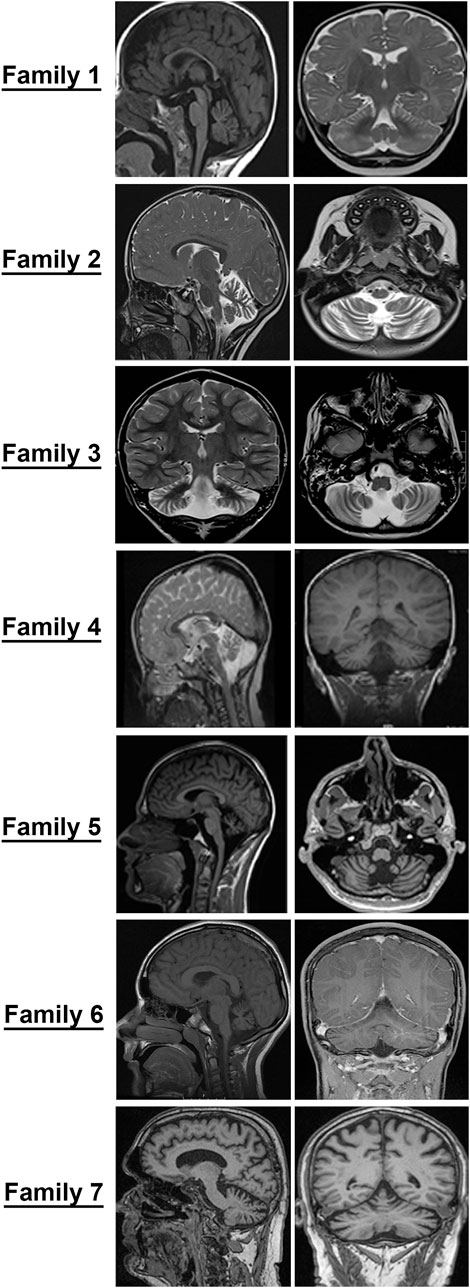
FIGURE 2. Cerebellar atrophy is a common feature among all individuals with GEMIN5-associated disease. Magnetic resonance imaging (MRI) of probands from families identified in the present study. Midsagittal, coronal, and axial T2/T1 images showing evidence of cerebellar atrophy, which was present in all patients.
Stratifying patients with GEMIN5-related disease from this study and our previously published cohort (Kour et al., 2021) by age of onset and severity revealed three main groups based on neurological involvement:
1. Infantile-onset severe global developmental delay with cerebellar atrophy on neuroimaging.
These patients presented in infancy with severe hypotonia and global developmental delay. Neuroimaging confirmed cerebellar atrophy.
2. Late infantile-onset developmental delay and ataxia syndrome with cerebellar atrophy on neuroimaging.
3. Juvenile/adult-onset spastic ataxia syndrome with cerebellar atrophy on neuroimaging.
Other clinical features seen in our patients include cataracts, strabismus, and nystagmus.
Overall, all variants of GEMIN5 appear to perturb GEMIN5 structure and function(s) and result in deleterious neurological symptoms.
Neuroradiological Features
The MRI images of all patients are shown in Figure 2. All patients showed diffuse cerebellar atrophy including vermian and hemispheric involvement of the cerebellum. Patient 5 had minimal brainstem and spinal atrophy as well. Most patients with repeat neuroimaging demonstrated a stable atrophy. At this time, it remains to be clarified if the loss of cerebellar volume is more of a hypoplasia rather than an atrophy and a natural history study with temporal follow-up of patients and imaging will be needed to better understand this.
GEMIN5 Variants Reduce the Levels of snRNP Complex Proteins in Patient Cells
GEMIN5 is a multidomain protein, which acts as a crucial anchor during the assembly of small nuclear ribonucleoproteins (snRNPs), the essential components of the spliceosome (Pellizzoni et al., 2002; Wan et al., 2005; Battle et al., 2006a; Battle et al., 2006b; Kolb et al., 2007; Yong et al., 2010). We asked if the patients carrying compound heterozygous variants of GEMIN5 perturb the levels of GEMIN5 and other SMN complex proteins. We isolated peripheral blood mononuclear cells (PBMCs) from patient 5 carrying p. Trp373* and p. Arg1016Cys variants of GEMIN5, as well as from the unaffected heterozygous parent (father) and performed Western blot (WB) with antibodies to GEMIN5, SMN, GEMIN4, and GEMIN2 to assess protein levels. We found a significant reduction in GEMIN5, SMN, GEMIN4, and GEMIN2 protein levels in the patient sample compared with the unaffected parent (Figure 3 and Supplementary Figure S2). These findings suggested that biallelic variants in GEMIN5 result in deleterious changes in the levels of snRNP complex proteins, which is likely contributing to various neurological defects observed in our patients.
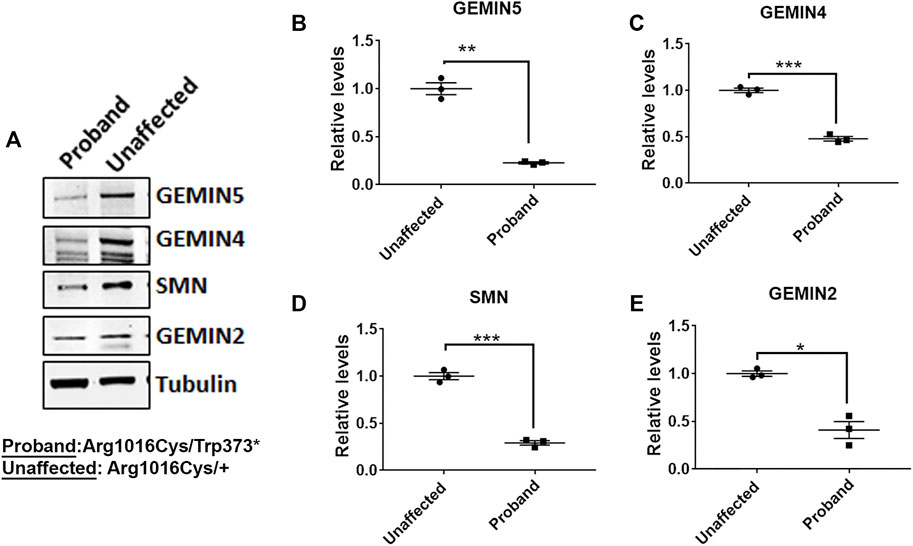
FIGURE 3. Individual with compound heterozygous variants in GEMIN5 demonstrates reduced levels of SMN complex proteins. (A) Representative immunoblot showing the protein levels of SMN, GEMIN4, and GEMIN2 in the patient carrying p.Trp373* and p.Arg1016Cys variants in GEMIN5. Protein lysates were prepared from the PBMCs isolated from the patient and the unaffected parent (Arg1016Cys/+). (B–E) Quantitative plot showing the reduced levels of GEMIN5 (B), GEMIN4 (C), SMN (D), and GEMIN2 (E) proteins in the patient relative to unaffected parent (father). The data represent mean ± SEM. p-Values (***<0.001, **<0.01, *<0.05) are calculated by unpaired t test (n = 3).
GEMIN5 Variants are Predicted to Cause Structural and Conformational Changes and Disrupt its Interaction With Other RNA/Proteins
GEMIN5 is a highly conserved protein across vertebrates, and mutations in GEMIN5 have been shown to cause a loss of function and loss of protein stability (Kour et al., 2021) (Table 4). Various in silico tools allow us to predict the impact of an amino acid substitution on the structure and function of a human protein. Using in silico tools, we found that the GEMIN5 variants are predicted to be deleterious in nature and lead to loss of protein function and stability (Table 4). Amino acid substitution at key sites within a protein due to a single-nucleotide variant (SNV) may result in various conformation changes, including remodeling or alteration of interaction network, salt bridges, and hydrogen bonds. These changes may perturb protein folding kinetics and can cause the destabilization of protein, impairing its subsequent function or interaction with other molecules (Dill et al., 1993; Teng et al., 2010). To determine the effect of GEMIN5 missense variants on the protein conformation and interaction with surrounding amino acids, we used PyMOL to predict the structural changes caused by five possibly deleterious substitutions—three GEMIN5 variants reported in our current study (p. Ser543Gly, p. Pro594Arg, and p. Arg1016Cys) and two GEMIN5 variants from our previous study (p. Gly683Asp and p. Asp704Glu) (Kour et al., 2021). All of the following variants are located within the WD40 repeat domains (PDB ID: 5H1J), except p. Arg1016Cys, which is located in the tetratricopeptide (TPR)-like dimerization domain (PDB ID: 6RNS) of the GEMIN5 protein (Figure 4). By limiting the affected area of mutagenesis to 5 Å, we measured changes in the orientation and conformation of surrounding amino acid by calculating their distance from the substituted GEMIN5 variant.
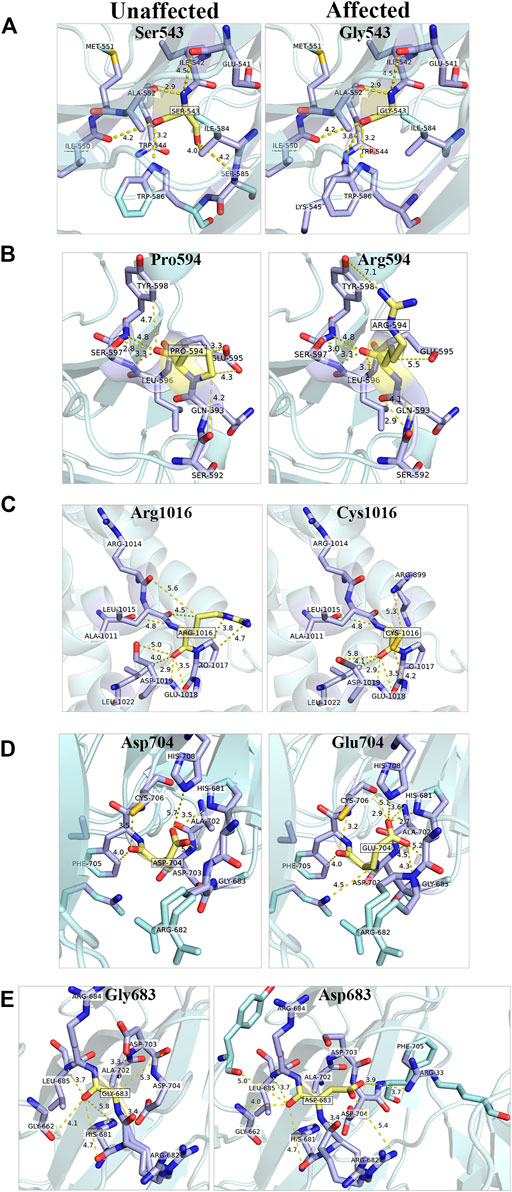
FIGURE 4. In silico prediction of structural changes caused by the GEMIN5 variants. Structural and interaction-based changes were measured around 5A region of GEMIN5 variants Ser543Gly (A), Pro594Arg (B), Asp704Glu (D), and Gly683Asp (E) in the WD40 domain (PDB ID: 5H1J) and Arg1016Cys (C) in the tetratricopeptide (TPR)-like domain (PDB ID: 6RNS) by using Pymol. The affected amino acids are colored yellow and their surrounding amino acid in light blue.
We found that the variant p. Ser543Gly in GEMIN5 may lead to changes in its angular parameters and distance from Ile584, Ser585, and Lys545, respectively, which could result in structural changes in anaphase-promoting complex subunit 4, the WD40 domain (540–588 aa) of GEMIN5 (Figure 4A). Similarly, we found that the conversion of Pro594, a nonpolar amino acid, to the positively charged arginine might affect its interactions with Glu595, Gln593, and Tyr598 (Figure 4B). Proline acts as a structural disruptor in alpha and beta helixes, which is a prerequisite for protein folding and structure. Therefore, its conversion to arginine, an amino acid found at the active centers of the protein and required for interactions with phosphorylated substrates, might result in the disruption of GEMIN5 structure and its interaction with other proteins.
PyMOL structure prediction suggested a possible change in the interaction and angular distance of 704Glu variant with His681, His708, Asp703, Gly683, and Arg682 (Figure 4D). Likewise, the p. Gly683Asp variant of GEMIN5, which is in a grove between two WD40 domains, induces an ionic charge that may result in the formation of additional bonds with Arg682 and Phe705 as well as change in the possible conformation due to its interaction with Arg33 (Figure 4E).
The p. Arg1016Cys variant of GEMIN5 lies in the TPR-like dimerization domain, which modulates the interaction of GEMIN5 with other proteins. Thus, the substitution of the highly polar arginine, which is typically found in the interface of the two proteins, to a small and neutrally charged cysteine, may affect its conformation and dimerization function. PyMOL predicted the possible changes in the distance and bonding with Arg1014, Pro1017, and Asp1019 (Figure 4C). Furthermore, the GEMIN5 variant p. Asp1019Cys has been reported to disrupt the dimerization properties of GEMIN5, suggesting the possible role and significance of Arg1016 and Asp1019 amino acids in protein–protein interactions. Overall, these findings suggest the possible deleterious effect of GEMIN5 variants on its structure conformation, interaction, and function.
Gemin5 is Essential for Development and Knocking out Gemin5 Causes Embryonic Lethality in Mice
To further understand the consequences of loss of function in Gemin5 in vivo, Gemin5 knockout (GEMIN5-DEL558) mice were created through the International Mouse Phenotyping Consortium (Dickinson et al., 2016; Mianné et al., 2017). To successfully generate the Gemin5 knockout mice, CRISPR/Cas9 was utilized to delete 558 nucleotides including exon 7 of the Mus musculus Gemin5 gene. This deletion of 558 nucleotides resulted in a frameshift and formation of a premature stop codon in the Gemin5 gene (Figure 5A). Heterozygous mice were validated for harboring one copy of Gemin5 WT and one copy of Gemin5 knockout by SR-PCR genotyping (Figures 5B, C). Heterozygous knockout mice were crossed together to assess the percentage of homozygous pups. No postnatal homozygous pups were observed at the P0 stage. Of the P0 pups, 63% were heterozygous for the Gemin5 knockout allele, while 37% of the pups harbored WT Gemin5 (n = 57).
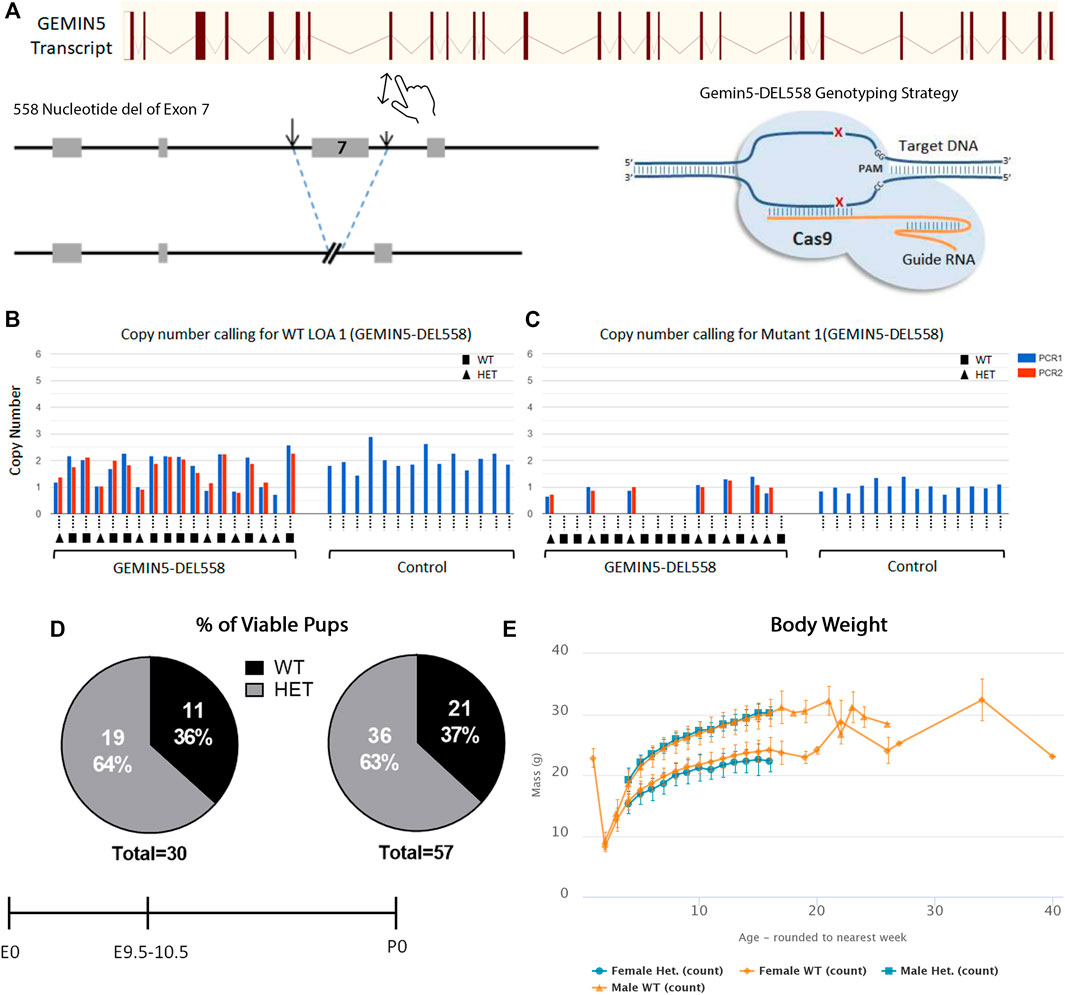
FIGURE 5. Knockout of Gemin5 leads to early embryonic lethality in mice. (A) Schematic representation of the wild-type allele of the mouse Gemin5 transcript. The Gemin5 KO mouse (GEMIN5-DEL558) was created by utilizing the EUCOMM/KOMP-CSD allele structure to delete 558 nucleotides from exon 7 of the Gemin5 gene, encompassing a critical exon to induce a premature stop codon and a null Gemin5 allele (Mianné et al., 2017). (B) SR-PCR genotyping detects the presence of the Gemin5 wild-type allele by short-range PCR in Gemin5 KO and control mice (copy number of 2). Samples are genotyped with a WT loss of allele (WT-LOA) assay. This is a FAM-labeled assay that is designed to detect the critical exon that has been floxed. If the animal contains 1 Gemin5 KO allele, the copy number of this assay should drop by 1 (copy number of 1). For autosomal genes that have been targeted, this means the following: WT = 2 copies of the LOA assay, HET = 1 copy of the LOA assay, HOM = 0 copy of the LOA assay. (C) SR-PCR genotyping detects the presence of the cassette lacZ, the Gemin5 wild-type allele, and the Gemin5 KO allele confirming heterozygotes by short-range PCR (copy number of 1). (D) Viability of Gemin5 KO mice at the embryonic stage of E9.5 and as first-born pups at P0. Homozygous Gemin5 KO mice are not viable up to the embryonic stage of E9.5. (E) A body composition (DEXA lean/fat) phenotypic assay was performed on 1,028 mice. The charts show the results of measuring lean mass in 8 Het females and 7 Het male mice compared with 514 female and 499 male controls. Heterozygote mice appear to have no change in body mass compared with controls.
To assess if pups were viable during embryonic development, mice were genotyped at embryonic day E9.5–10.5 across five separate litters (Figure 5D). No homozygous embryos were observed at E9.5–10.5, suggesting that Gemin5 is crucial for embryonic development in mice (n = 30). In addition, the body weight of heterozygous Gemin5 knockout mice was assessed over the course of 20 weeks. There was no significant change in the weight of heterozygous mice compared with wild-type control mice (n = 15 heterozygote mice, 1,013 control mice) (Figure 5E). Since we did not get any Gemin5 null animals at E9.5, we were unable to determine the impact of abolishing Gemin5 on brain development. These findings suggest that the homozygous loss of Gemin5 results in early embryonic lethality, while harboring one copy of defective Gemin5 does not appear to be detrimental and cause any obvious motor defects (Supplementary Figure S3).
Discussion
Here we expand on the neurological phenotypes observed among patients with biallelic pathogenic variants in the GEMIN5 gene. The clinical spectrum ranges from global developmental delay of early infantile onset with cerebellar atrophy to adult-onset spastic ataxia syndrome with cerebellar atrophy. Hereditary ataxias are a genetically heterogenous group of disorders with more than 150 genes being associated with this phenotype to date. With our cohort, we define an autosomal recessive hereditary ataxia syndrome associated with biallelic variants in GEMIN5. All but our early infantile patients had normal to brisk reflexes suggesting pyramidal tract involvement. The adult patients in our cohort were primarily followed in a spastic paraplegia clinic.
Compiling the neurological phenotype from patients we previously reported (Kour et al., 2021) together with our current cohort, 21 of the currently reported 39 patients showed brisk reflexes, 4 had absent reflexes, 9 were normal, and 5 were unknown. Central hypotonia was seen in 30 of the 39 patients, 7 had normal central tone, and 2 were unknown. Of the 39 patients, 18 had appendicular hypertonia. Cerebellar atrophy was characteristically seen in all patients on neuroimaging, suggesting that this is a key feature associated with GEMIN5 variants. We, hence, categorize the neurological phenotype associated with autosomal recessive variants in GEMIN5 as predominantly in the hereditary cerebellar ataxia–spastic paraplegia spectrum of disease. While the hereditary cerebellar ataxias and hereditary spastic paraplegias have been thought to be clinically distinct, epidemiological studies and recent advances in next-generation sequencing technologies have demonstrated that multiple genes are associated with the disease phenotypes and overlaps in clinical presentation as well as molecular pathways.
Furthermore, this GEMIN5 neurodevelopmental ataxia spectrum is characteristically distinct from the predominantly neuromuscular presentation of spinal muscular atrophy (SMA) caused by another member of the SMN protein complex. Of some overlap, pontocerebellar hypoplasia type 1 (PCH1) is a condition with pontocerebellar atrophy along with anterior horn cell involvement of the spinal cord associated with many autosomal recessive genes (Namavar et al., 2011; Wan et al., 2012; Rudnik-Schoneborn et al., 2013; Sánchez-Albisua et al., 2014; Lardelli et al., 2017; Ivanov et al., 2018; Nuovo et al., 2021). These diseases can present with a varying blend of neurological phenotypes including hypotonia to varying degrees of spasticity.
The GEMIN5 variants identified, so far, are missense, deletion, frameshift, nonsense, and splicing defects with autosomal recessive inheritance pattern. The variants identified in our current study, with two exceptions (p. Arg1016Cys and p. Pro594Arg), were never reported in any publicly available databases. The p. Arg1016Cys variant was identified with a frameshift or nonsense variant in three patients from three unrelated families (Table 1). This variant has been reported with allelic frequency of 4.48e−3 (heterozygote), and there are five individuals reported as homozygous. It is believed that the p. Arg1016Cys variant is a hypomorphic variant, and it is not sufficient to manifest any clinical symptoms unless the other allele is fully disrupted. Alternatively, the homozygous individuals in gnomAD may be at a presymptomatic stage.
GEMIN5 is expressed predominantly in the cytoplasm as well as in the nucleus, nucleoplasm, and gem bodies, suggesting functions in multiple cellular compartments (Pacheco et al., 2009; Piñeiro et al., 2015; Francisco-Velilla et al., 2016; Lozano et al., 2018; Martinez-Salas et al., 2020; Moreno-Morcillo et al., 2020). Animal model studies suggest that complete or partial reduction of the snRNP complex proteins are detrimental (Saida et al., 2021). Knocking out Smn, Gemin4, and Gemin5 is lethal in mice and Drosophila, suggesting that these proteins are essential for survival (Gates et al., 2004; Gavrilina et al., 2008; Meier et al., 2018). We have previously shown that genetic reduction of endogenous rigor mortis (fly homolog of human GEMIN5) leads to developmental delay, motor dysfunction, and reduced lifespan, similar to human patients with autosomal recessive variants in GEMIN5 (Kour et al., 2021). Our data in human patient cells (Figure 3) suggest that pathogenic variants in GEMIN5 significantly reduce the expression of snRNP complex assembly. Furthermore, knocking out Gemin5 in mice is embryonically lethal (Figure 5). Although these findings are interesting, we are unable to establish a link between cerebellar atrophy and Gemin5 loss of function due to early embryonic lethality in our knockout Gemin5 mice. We performed in silico analysis to determine the impact of GEMIN5 variants on the protein structure. We found that GEMIN5 missense variants cause significant alterations on protein conformation, such as remodeling the interaction network, peptide chain bending, and the ability to interact with other amino acids. Since the GEMIN5 protein has been shown to dimerize, it is likely that frameshift and termination variants might perturb the kinetics of protein folding and can cause the destabilization of proteins. These alterations might disrupt the ability of GEMIN5 to form a complex with snRNP proteins and exert physiological functions, which in turn lead to neurological symptoms observed in patients with GEMIN5-related disease. We recently showed that reduction of the levels of GEMIN5 (by mutations or shRNA knockdown) is sufficient to disrupt the snRNP assembly in mammalian cells and iPSC neurons (Kour et al., 2021). These findings support the idea that each component of the snRNP complex has an important role to play in maintaining cellular functions. Alternatively, it is possible that loss of normal GEMIN5 function might affect translation of mRNAs either in a tissue-specific manner or ubiquitously leading to defective protein synthesis and their regulatory pathways.
Given the distinct neurological presentation, we propose the term GEMIN5-related neurodevelopmental ataxia with cerebellar atrophy to describe the variability in clinical presentation associated with this gene.
Data Availability Statement
The datasets presented in this article are not readily available due to patient-related privacy regulations and institutional policies. Requests to access the datasets should be directed to udai.pandey@chp.edu
Ethics Statement
The studies involving human participants were reviewed and approved by The University of Pittsburgh Institutional Review Board (IRB). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by The University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) (Protocol #: 21069518).
Author Contributions
DR, SK, TF, and UP planned, performed and analyzed the data. All other authors were involved in collecting patient data, analysis, and interpreting the data. All authors read the manuscript and gave their consent to publish.
Funding
The C57BL/6NTac-Gemin5em1(IMPC)H/H mice and phenotypes were generated at the MRC Harwell Institute. The knockout mouse generation protocol has been approved as per MRC Harwell Institute guidelines. The MRC Harwell Institute generated the mice as part of the International Mouse Phenotyping Consortium (IMPC) and has received funding from the National Institutes for Health for generating and/or phenotyping (5UM1HG006348-08) the C57BL/6NTac-Gemin5em1(IMPC)H/H mice. The research reported in this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding and associated primary phenotypic information may be found at www.mousephenotype.org.
Conflict of Interest
FM is employed by GeneDx (MD, USA). JF conducts Clinical Trials with Biogen (Angelman’s Syndrome). JF’s spouse is Founder and Principal of Friedman Bioventure, which holds a variety of publicly traded and private biotechnology interests.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
UP and DR are thankful to the Children’s Neuroscience Institute, Children’s Hospital of Pittsburgh, for providing a pilot grant funding.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.783762/full#supplementary-material
References
Battle, D. J., Kasim, M., Yong, J., Lotti, F., Lau, C.-K., Mouaikel, J., et al. (2006). The SMN Complex: an Assembly Machine for RNPs. Cold Spring Harbor Symposia Quantitative Biol. 71, 313–320. doi:10.1101/sqb.2006.71.001
Battle, D. J., Lau, C.-K., Wan, L., Deng, H., Lotti, F., and Dreyfuss, G. (2006). The Gemin5 Protein of the SMN Complex Identifies snRNAs. Mol. Cel 23 (2), 273–279. doi:10.1016/j.molcel.2006.05.036
Bromberg, Y., and Rost, B. (2007). SNAP: Predict Effect of Non-synonymous Polymorphisms on Function. Nucleic Acids Res. 35 (11), 3823–3835. doi:10.1093/nar/gkm238
Burlet, P., Huber, C., Bertrandy, S., Ludosky, M. A., Zwaenepoel, I., Clermont, O., et al. (1998). The Distribution of SMN Protein Complex in Human Fetal Tissues and its Alteration in Spinal Muscular Atrophy. Hum. Mol. Genet. 7 (12), 1927–1933. doi:10.1093/hmg/7.12.1927
Cheng, J., Randall, A., and Baldi, P. (2006). Prediction of Protein Stability Changes for Single-Site Mutations Using Support Vector Machines. Proteins 62 (4), 1125–1132. doi:10.1002/prot.20810
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS One 7 (10), e46688. doi:10.1371/journal.pone.0046688
Cousin, M. A., Conboy, E., Wang, J.-S., Lenz, D., Schwab, T. L., Williams, M., et al. (2019). RINT1 Bi-allelic Variations Cause Infantile-Onset Recurrent Acute Liver Failure and Skeletal Abnormalities. Am. J. Hum. Genet. 105 (1), 108–121. doi:10.1016/j.ajhg.2019.05.011
Dickinson, M. E., Flenniken, A. M., Ji, X., Teboul, L., Wong, M. D., White, J. K., et al. (2016). High-throughput Discovery of Novel Developmental Phenotypes. Nature 537 (7621), 508–514. doi:10.1038/nature19356
Dill, K. A., Fiebig, K. M., and Chan, H. S. (1993). Cooperativity in Protein-Folding Kinetics. Proc. Natl. Acad. Sci. 90 (5), 1942–1946. doi:10.1073/pnas.90.5.1942
Francisco-Velilla, R., Embarc-Buh, A., Rangel-Guerrero, S., Basu, S., Kundu, S., and Martinez-Salas, E. (2020). RNA-protein Coevolution Study of Gemin5 Uncovers the Role of the PXSS Motif of RBS1 Domain for RNA Binding. RNA Biol. 17 (9), 1331–1341. doi:10.1080/15476286.2020.1762054
Francisco-Velilla, R., Fernandez-Chamorro, J., Ramajo, J., and Martinez-Salas, E. (2016). The RNA-Binding Protein Gemin5 Binds Directly to the Ribosome and Regulates Global Translation. Nucleic Acids Res. 44 (17), 8335–8351. doi:10.1093/nar/gkw702
Gates, J. J., Lam, J. A., Ortiz, C. S., Losson, R., and Thummel, C. S. (2004). Rigor Mortisencodes a Novel Nuclear Receptor Interacting Protein Required for Ecdysone Signaling duringDrosophilalarval Development. Development 131 (1), 25–36. doi:10.1242/dev.00920
Gavrilina, T. O., McGovern, V. L., Workman, E., Crawford, T. O., Gogliotti, R. G., DiDonato, C. J., et al. (2008). Neuronal SMN Expression Corrects Spinal Muscular Atrophy in Severe SMA Mice while Muscle-specific SMN Expression Has No Phenotypic Effect. Hum. Mol. Genet. 17 (8), 1063–1075. doi:10.1093/hmg/ddm379
Guillen Sacoto, M. J., Tchasovnikarova, I. A., Torti, E., Forster, C., Andrew, E. H., Anselm, I., et al. (2020). De Novo Variants in the ATPase Module of MORC2 Cause a Neurodevelopmental Disorder with Growth Retardation and Variable Craniofacial Dysmorphism. Am. J. Hum. Genet. 107 (2), 352–363. doi:10.1016/j.ajhg.2020.06.013
Ivanov, I., Atkinson, D., Litvinenko, I., Angelova, L., Andonova, S., Mumdjiev, H., et al. (2018). Pontocerebellar Hypoplasia Type 1 for the Neuropediatrician: Genotype-Phenotype Correlations and Diagnostic Guidelines Based on New Cases and Overview of the Literature. Eur. J. Paediatric Neurol. 22 (4), 674–681. doi:10.1016/j.ejpn.2018.03.011
Kolb, S. J., Battle, D. J., and Dreyfuss, G. (2007). Molecular Functions of the SMN Complex. J. Child. Neurol. 22 (8), 990–994. doi:10.1177/0883073807305666
Kour, S., Rajan, D. S., Fortuna, T. R., Anderson, E. N., Ward, C., Lee, Y., et al. (2021). Loss of Function Mutations in GEMIN5 Cause a Neurodevelopmental Disorder. Nat. Commun. 12 (1), 2558. doi:10.1038/s41467-021-22627-w
Lardelli, R. M., Schaffer, A. E., Eggens, V. R. C., Zaki, M. S., Grainger, S., Sathe, S., et al. (2017). Biallelic Mutations in the 3′ Exonuclease TOE1 Cause Pontocerebellar Hypoplasia and Uncover a Role in snRNA Processing. Nat. Genet. 49 (3), 457–464. doi:10.1038/ng.3762
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 80 (1), 155–165. doi:10.1016/0092-8674(95)90460-3
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., et al. (1997). Correlation between Severity and SMN Protein Level in Spinal Muscular Atrophy. Nat. Genet. 16 (3), 265–269. doi:10.1038/ng0797-265
Lozano, G., Francisco-Velilla, R., and Martinez-Salas, E. (2018). Deconstructing Internal Ribosome Entry Site Elements: an Update of Structural Motifs and Functional Divergences. Open Biol. 288 (11), 180155. doi:10.1098/rsob.180155
Ma, Q., Liu, B., Zhou, C., Yin, Y., Li, G., and Xu, Y. (2013). An Integrated Toolkit for Accurate Prediction and Analysis of Cis-Regulatory Motifs at a Genome Scale. Bioinformatics. 29 (18), 2261–2268. doi:10.1093/bioinformatics/btt397
Martinez-Salas, E., Embarc-Buh, A., and Francisco-Velilla, R. (2020). Emerging Roles of Gemin5: From snRNPs Assembly to Translation Control. Ijms 21 (11), 3868. doi:10.3390/ijms21113868
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
McKusick, V. A. (2007). Mendelian Inheritance in Man and its Online Version, OMIM. Am. J. Hum. Genet. 80 (4), 588–604. doi:10.1086/514346
Meier, I. D., Walker, M. P., and Matera, A. G. (2018). Gemin4 Is an Essential Gene in Mice, and its Overexpression in Human Cells Causes Relocalization of the SMN Complex to the Nucleoplasm. Biol. Open 7 (2). doi:10.1242/bio.032409
Melki, J., Lefebvre, S., Burglen, L., Burlet, P., Clermont, O., Millasseau, P., et al. (1994). De Novo and Inherited Deletions of the 5q13 Region in Spinal Muscular Atrophies. Science 264 (5164), 1474–1477. doi:10.1126/science.7910982
Mi, H., Dong, Q., Muruganujan, A., Gaudet, P., Lewis, S., and Thomas, P. D. (2010). PANTHER Version 7: Improved Phylogenetic Trees, Orthologs and Collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38 (Database issue), D204–D210. doi:10.1093/nar/gkp1019
Mianné, J., Codner, G. F., Caulder, A., Fell, R., Hutchison, M., King, R., et al. (2017). Analysing the Outcome of CRISPR-Aided Genome Editing in Embryos: Screening, Genotyping and Quality Control. Methods 121-122, 68–76. doi:10.1016/j.ymeth.2017.03.016
Moreno-Morcillo, M., Francisco-Velilla, R., Embarc-Buh, A., Fernández-Chamorro, J., Ramón-Maiques, S., and Martinez-Salas, E. (2020). Structural Basis for the Dimerization of Gemin5 and its Role in Protein Recruitment and Translation Control. Nucleic Acids Res. 48 (2), 788–801. doi:10.1093/nar/gkz1126
Namavar, Y., Barth, P. G., Poll-The, B. T., and Baas, F. (2011). Classification, Diagnosis and Potential Mechanisms in Pontocerebellar Hypoplasia. Orphanet J. Rare Dis. 6, 50. doi:10.1186/1750-1172-6-50
Németh, A. H., Kwasniewska, A. C., Lise, S., Parolin Schnekenberg, R., Becker, E. B. E., Bera, K. D., et al. (2013). Next Generation Sequencing for Molecular Diagnosis of Neurological Disorders Using Ataxias as a Model. Brain 136 (Pt 10), 3106–3118. doi:10.1093/brain/awt236
Ng, P. C., and Henikoff, S. (2006). Predicting the Effects of Amino Acid Substitutions on Protein Function. Annu. Rev. Genom. Hum. Genet. 7, 61–80. doi:10.1146/annurev.genom.7.080505.115630
Nuovo, S., Micalizzi, A., Romaniello, R., Arrigoni, F., Ginevrino, M., Casella, A., et al. (2021). Refining the Mutational Spectrum and Gene-Phenotype Correlates in Pontocerebellar Hypoplasia: Results of a Multicentric Study. J. Med. Genet. 5, jmedgenet–2020. doi:10.1136/jmedgenet-2020-107497
Pacheco, A., de Quinto, S. L., Ramajo, J., Fernández, N., and Martínez-Salas, E. (2009). A Novel Role for Gemin5 in mRNA Translation. Nucleic Acids Res. 37 (2), 582–590. doi:10.1093/nar/gkn979
Parolin Schnekenberg, R., Perkins, E. M., Miller, J. W., Davies, W. I. L., D’Adamo, M. C., Pessia, M., et al. (2015). De Novopoint Mutations in Patients Diagnosed with Ataxic Cerebral Palsy. Brain 138 (Pt 7), 1817–1832. doi:10.1093/brain/awv117
Pellizzoni, L., Yong, J., and Dreyfuss, G. (2002). Essential Role for the SMN Complex in the Specificity of snRNP Assembly. Science 29298 (5599), 1775–1779. doi:10.1126/science.1074962
Piñeiro, D., Fernandez-Chamorro, J., Francisco-Velilla, R., and Martinez-Salas, E. (2015). Gemin5: A Multitasking RNA-Binding Protein Involved in Translation Control. Biomolecules 5 (2), 528–544. doi:10.3390/biom5020528
Ruano, L., Melo, C., Silva, M. C., and Coutinho, P. (2014). The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: a Systematic Review of Prevalence Studies. Neuroepidemiology 42 (3), 174–183. doi:10.1159/000358801
Rudnik-Schoneborn, S., Senderek, J., Jen, J. C., Houge, G., Seeman, P., Puchmajerova, A., et al. (2013). Pontocerebellar Hypoplasia Type 1: Clinical Spectrum and Relevance of EXOSC3 Mutations. Neurology 80 (5), 438–446. doi:10.1212/WNL.0b013e31827f0f66
Saida, K., Tamaoki, J., Sasaki, M., Haniffa, M., Koshimizu, E., Sengoku, T., et al. (2021). Pathogenic Variants in the Survival of Motor Neurons Complex Gene GEMIN5 Cause Cerebellar Atrophy. Clin. Genet. 100 (6), 722–730. doi:10.1111/cge.14066
Sánchez-Albisua, I., Frölich, S., Barth, P. G., Steinlin, M., and Krägeloh-Mann, I. (2014). Natural Course of Pontocerebellar Hypoplasia Type 2A. Orphanet J. Rare Dis. 9, 70. doi:10.1186/1750-1172-9-70
Sim, N.-L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 40, W452–W457. doi:10.1093/nar/gks539
Synofzik, M., and Schüle, R. (2017). Overcoming the divide between Ataxias and Spastic Paraplegias: Shared Phenotypes, Genes, and Pathways. Mov Disord. 32 (3), 332–345. doi:10.1002/mds.26944
Teng, S., Srivastava, A. K., Schwartz, C. E., Alexov, E., and Wang, L. (2010). Structural Assessment of the Effects of Amino Acid Substitutions on Protein Stability and Protein Protein Interaction. Ijcbdd 3 (4), 334–349. doi:10.1504/IJCBDD.2010.038396
Vigeland, M. D., Gjøtterud, K. S., and Selmer, K. K. (2016). FILTUS: a Desktop GUI for Fast and Efficient Detection of Disease-Causing Variants, Including a Novel Autozygosity Detector. Bioinformatics 32 (10), 1592–1594. doi:10.1093/bioinformatics/btw046
Wan, J., Yourshaw, M., Mamsa, H., Rudnik-Schöneborn, S., Menezes, M. P., Hong, J. E., et al. (2012). Mutations in the RNA Exosome Component Gene EXOSC3 Cause Pontocerebellar Hypoplasia and Spinal Motor Neuron Degeneration. Nat. Genet. 44 (6), 704–708. doi:10.1038/ng.2254
Wan, L., Battle, D. J., Yong, J., Gubitz, A. K., Kolb, S. J., Wang, J., et al. (2005). The Survival of Motor Neurons Protein Determines the Capacity for snRNP Assembly: Biochemical Deficiency in Spinal Muscular Atrophy. Mol. Cel Biol 25 (13), 5543–5551. doi:10.1128/MCB.25.13.5543-5551.2005
Wang, J. Z., Du, Z., Payattakool, R., Yu, P. S., and Chen, C.-F. (2007). A New Method to Measure the Semantic Similarity of GO Terms. Bioinformatics 23 (10), 1274–1281. doi:10.1093/bioinformatics/btm087
Keywords: Gemin5, ataxia, cerebellar atrophy, developmental delay, neurodegeneration, cell death, development
Citation: Rajan DS, Kour S, Fortuna TR, Cousin MA, Barnett SS, Niu Z, Babovic-Vuksanovic D, Klee EW, Kirmse B, Innes M, Rydning SL, Selmer KK, Vigeland MD, Erichsen AK, Nemeth AH, Millan F, DeVile C, Fawcett K, Legendre A, Sims D, Schnekenberg RP, Burglen L, Mercier S, Bakhtiari S, Francisco-Velilla R, Embarc-Buh A, Martinez-Salas E, Wigby K, Lenberg J, Friedman JR, Kruer MC and Pandey UB (2022) Autosomal Recessive Cerebellar Atrophy and Spastic Ataxia in Patients With Pathogenic Biallelic Variants in GEMIN5. Front. Cell Dev. Biol. 10:783762. doi: 10.3389/fcell.2022.783762
Received: 20 October 2021; Accepted: 17 January 2022;
Published: 28 February 2022.
Edited by:
Jiyan Zhang, Independent researcher, Beijing, ChinaReviewed by:
Craig Blackstone, National Institute of Neurological Disorders and Stroke (NINDS), United StatesSokol V. Todi, Wayne State University, United States
Bing Lang, Central South University, China
Copyright © 2022 Rajan, Kour, Fortuna, Cousin, Barnett, Niu, Babovic-Vuksanovic, Klee, Kirmse, Innes, Rydning, Selmer, Vigeland, Erichsen, Nemeth, Millan, DeVile, Fawcett, Legendre, Sims, Schnekenberg, Burglen, Mercier, Bakhtiari, Francisco-Velilla, Embarc-Buh, Martinez-Salas, Wigby, Lenberg, Friedman, Kruer and Pandey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Udai Bhan Pandey, udai@pitt.edu
†These authors have contributed equally to this work