 Jiabi Qian1,2,3†
Jiabi Qian1,2,3† Zifeng Li4†
Zifeng Li4† Kunlin Pei2
Kunlin Pei2 Ziping Li1
Ziping Li1 Chunjie Li1Muxia Yan2
Chunjie Li1Muxia Yan2 Maoxiang Qian3*
Maoxiang Qian3* Yuanbin Song5*
Yuanbin Song5* Hui Zhang2*
Hui Zhang2* Yingyi He2*
Yingyi He2*- 1Guangzhou Women and Children’s Medical Center, Institute of Pediatrics, Guangzhou, China
- 2Department of Hematology/Oncology, Guangzhou Women and Children’s Medical Center, Guangzhou, China
- 3Department of Hematology and Oncology, The Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism, Institute of Pediatrics, Institutes of Biomedical Sciences, Children’s Hospital of Fudan University, Ministry of Science and Technology, Fudan University, Shanghai, China
- 4Department of Hematology and Oncology, National Children’s Medical Center, Children’s Hospital of Fudan University, Shanghai, China
- 5Department of Hematologic Oncology, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, China
Through the advancements in recent decades, childhood acute lymphoblastic leukemia (ALL) is gradually becoming a highly curable disease. However, the truth is there remaining relapse in ∼15% of ALL cases with dismal outcomes. RAS mutations, in particular NRAS mutations, were predominant mutations affecting relapse susceptibility. KRAS mutations targeting has been successfully exploited, while NRAS mutation targeting remains to be explored due to its complicated and compensatory mechanisms. Using targeted sequencing, we profiled RAS mutations in 333 primary and 18 relapsed ALL patients and examined their impact on ALL leukemogenesis, therapeutic potential, and treatment outcome. Cumulative analysis showed that RAS mutations were associated with a higher relapse incidence in children with ALL. In vitro cellular assays revealed that about one-third of the NRAS mutations significantly transformed Ba/F3 cells as measured by IL3-independent growth. Meanwhile, we applied a high-throughput drug screening method to characterize variable mutation-related candidate targeted agents and uncovered that leukemogenic-NRAS mutations might respond to MEK, autophagy, Akt, EGFR signaling, Polo−like Kinase, Src signaling, and TGF−β receptor inhibition depending on the mutation profile.
Introduction
Translational genomic research and risk stratification-directed therapy have gradually made childhood acute lymphoblastic leukemia (ALL) a highly curable cancers (Vora et al., 2013; Pui et al., 2018), with over 90% leukemia-free survival in developed countries. However, about 15–20% children with ALL eventually relapse with dismal outcome (Mullighan et al., 2008; Ding et al., 2012; Bhojwani and Pui, 2013; Meyer et al., 2013; Pierro et al., 2017; Brown and Ferrando, 2018). Among the genetic alterations, RAS mutations, in particular NRAS mutations, are over presented in children with ALL (Ma et al., 2015). Studies have shown that the prevalence of NRAS mutations varies from 15 to 34% in children with ALL (Case et al., 2008; Irving et al., 2014; Ma et al., 2015). Impressively, Ma et al. has reported that NRAS mutations conferred susceptibilities on B cell ALL (B-ALL) relapse (Ma et al., 2015). Consequently, the oncogenic mutations in the NRAS represented crucial therapeutic targets (Ward et al., 2012). Therefore, it’s highly needed to explore the translational potential of NRAS mutations in pediatric ALL.
RAS GTPase (HRAS, KRAS and NRAS) family members play a critical role in human malignancies via regulating cell growth, differentiation, survival, motility, and adhesion through transmitting signals to activate downstream signaling cascades, including the RAF-MEK-ERK and PI3K-AKT pathways (Karnoub and Weinberg, 2008; Stephen et al., 2014; Burgess et al., 2017). In this regard, NRAS mutations have been found to be able to lead to constitutive activation, which in turn activate its downstream signaling pathways, including mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)-AKT, and others (i.e., RalGDS, and janus kinase (JAK) - signal transducer and activator of transcription (STAT)) (Brunet et al., 1999; Cox and Der, 2003; Downward, 2003; Xu et al., 2007; Wang et al., 2013; Kong et al., 2014; Zhang and Cheong, 2016; Bery et al., 2018). In the therapeutic targeting facet, much attentions have been paid to the breakthrough of KRASG12C targeting by several small molecules, such as AMG-510, MRTX849, and ARS-1620 (Janes et al., 2018; Canon et al., 2019; Hallin et al., 2020). Moreover, the KRASG12C targeting has been successfully translated into clinics with very promising results (Lito et al., 2016; Hallin et al., 2020). However, effective NRAS targeting remains to be explored.
It’s well established that NRAS stimulates proliferation through activating RAS-RAF-MAPK-ERK signaling pathway. Unfortunately, trials using ERK or MEK inhibitors to treated leukemic patients with NRAS mutations do not generate satisfactory results as expected. For example, Jain et al. has reported that three AML patients with NRAS mutations fail to respond to the MAPK inhibitor (selumetinib [AZD6244]) (Jain et al., 2014). Similarly, the reported NRAS-targeting agents have failed to demonstrate the satisfying outcomes. Furthermore, multiple in vitro and in vivo evidences has shown that NRAS mutated myeloma and/or leukemic cells are resistant to KRASG12C-targeted small molecules (Welsch et al., 2017; Janes et al., 2018; Canon et al., 2019; Hallin et al., 2020), indicating the specificity of NRAS targeting. Taken together, all these evidence has pointed out that the complex NRAS downstream signals and their compensatory effect might be the bottle-neck of precise targeting (Posch et al., 2013; Samatar and Poulikakos, 2014).
To this end, we retrospectively evaluated the impact of RAS mutations on children with ALL enrolled onto CCCG-ALL-2015 clinical trial and tested the contributions of NRAS mutations on ALL leukemogenesis and drug response. Furthermore, we utilized high-throughput drug screening (HDS) method to explore the candidates for NRAS targeting.
Methods
Patients
Newly Diagnosed (N = 333) and relapsed (N = 18) B-ALL patients enrolled onto CCCG-ALL-2015 clinical trial were included for this study. Ethical approval was obtained from the ethics committee at Guangzhou Women and Children’s Medical Centre 2015020936, 2017102307, and 2020-04500). Informed consent was provided by the patients’ legal guardians, or patients themselves if they were over 8 years old according to the Helsinki Declaration, and their related clinical information was collected for this study. The survival and relapse analyses were performed using Cox proportional hazards regression model.
Reagents and Cell Lines
All the reagents used in this study were listed in the Supplementary Table S1. The HEK-293T cells were purchased from the American Type Culture Collection (ATCC, United States), and Ba/F3 cells were gifted by Jun Yang at St. Jude Children’s Research Hospital (Xu et al., 2015). The HEK-293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen, United Kingdom) supplemented with 10% fetal bovine serum, and the Ba/F3 cells were maintained in RPMI1640 supplemented with 10% fetal bovine serum and 10 ng/ml recombinant mouse interleukin 3 (IL3) (PeproTech EC, London, United Kingdom).
Targeted Next-Generation Sequencing and Validation
DNA was extracted among the diagnostic bone marrow and their matched saliva samples by Trizol (Thermofisher, United States) according to the manufacturer’s protocol. Targeted sequencing of hematological malignancies related genes (Supplementary Table S2) was completed at Kindstar Global (Beijing) Technology, Inc. As detailed, targeted gene capture and library construction for NGS were performed using NimbleGen Sequence Capture Arrays (Roche, Basel, Switzerland) according to the manufacturer’s protocol. Then, the NGS libraries were sent to generate 150-bp paired-end reads for sequencing on the Illumina HiSeq X10 platform (San Diego, CA, United States). Sequencing reads were aligned to the human reference genome (hg19) using Burrows–Wheeler Aligner (BWA-0.7.10). Duplicated reads were then marked and removed using Picard (picard-tools-2.17.0). Variant calls were performed using VarDictJava (1.5.8) (Lai et al., 2016) with pre-curated blacklist variant filters and custom Annovar scripts. Finally, the confident variants were then annotated and manually checked using IGV. Structural variants were called using Delly (Rausch et al., 2012; Hunger and Mullighan, 2015) and filtered using BreakTrans. In the meanwhile, we have retrieved and analyzed the RAS family mutation data from St. Jude PeCan Data Portal (McLeod et al., 2021).
Cytokine-independent Growth Assay in Ba/F3 Cells
The full-length NRAS cDNA was amplified and cloned into the cL20c-IRES-GFP lentiviral vector. NRAS mutations were generated using Q5 Site-Directed Mutagenesis Kit (New England Biolabs, United States) with primers listed in Supplementary Table S3. Lentiviral supernatants expressing NRAS mutants were generated by transient transfection of HEK-293T cells using Lipofectamine 3000 (Invitrogen, United Kingdom) following the manufacturer’s protocol. Ba/F3 cells were transduced with lentiviral supernatants expressing different NRAS mutants with MOI = 10, following with NRAS expressing cell sorting 48 h after lentiviral transduction by FACSAria II (BD, United States). Then, sorted Ba/F3 cells were washed three times with pre-cold PBS, seeded in the 96-well plate with 1×106/ml cell density, and maintained with full RPMI1640 media in the absence of murine IL3 cytokines. Cell viability was evaluated with Trypan blue using a TC10 automated cell counter (BIO-RAD) daily for at least 7 days.
High-Throughput Drug Screening Assay
High-throughput drug screening (HDS) was used to evaluate the cytotoxic effect of different candidate agents on NRASG12-transformed Ba/F3 cells (Supplementary Figure S1). Transformed Ba/F3 cells were grown in RPMI160 supplemented with 10% FBS and seeded in a 384-well plate (Corning, NY, United States) at a density of 1200 cells per well. The initial concentration of targeting drugs (Supplementary Table S4) was 10 μM and then serial diluted to generate the drug concentration series (10, 3.3, 1.1, 0.37, 0.12, 0.04, 0.013, 0.0045, 0.0015, and 0.0005 μM). The serial drug concentrations were added to the cells using an automated liquid handling system (PerkinElmer, MA, United States). Cell viability was assessed using CellTiter-Glo™ kits (Promega, WI, United States) after 72 h of drug exposure. The inhibition rate of each drug concentration was calculated after normalization using the formula below. The IC50 was calculated using GraphPad Prism v7.0 (GraphPad Software, Inc.). The HDS experiments were performed in triplicate and independently repeated three times.
Cell Counting Kit-8 (CCK-8) Assays
NRASG12-transformed Ba/F3 cells were seeded at a density of 2×105/ml in a 96-well plate, and treated with increasing doses of tested agents listed in Supplementary Table S1 for 72 h. The cell viability was tested using CCK-8 assay kit (Dojindo Molecular Technologies Inc., Japan) and colorimetric density was measured using a Multiscan MS spectrophotometer (Labsystems, Stockholm, Sweden). The experiments were performed in triplicate and repeated at least three times.
Western Blotting Assay
Ba/F3 cells with NRAS mutants were lysed in 1× lysis buffer (Cell Signaling Technology, United Kingdom). Proteins (20 mg) were electrophoresis on 10% PAGE gel (BIO-RAD) and then transferred onto PVDF membranes. After blocking membranes with 5% milk for 1 h at room temperature, the membranes were incubated with anti- Phospho- Erk1/2 antibody (Cell Signaling Technology, United Kingdom, 4370S, 1:1,000 dilution), anti- Erk1/2 antibody [Cell Signaling Technology, United Kingdom, 4696S, 1:1,000 dilution], anti- Phospho- Stat5 (Tyr694) antibody [Cell Signaling Technology, United Kingdom, 4322S, 1:1,000 dilution], and anti- Stat5 antibody (Cell Signaling Technology, United Kingdom, 94205S, 1:1,000 dilution). Tubulin was used as internal control. The blots were incubated with HRP-conjugated secondary antibodies for 1 h and visualized using the ECL system. All the antibodies we used were listed in Supplementary Table S1.
Statistical Analysis
All statistical analyses were performed using R (version 3.3.1) and GraphPad Prism v7.0 (GraphPad Software, Inc.). Kaplan–Meier survival analysis was performed and survival differences between groups were assessed with the log-rank test, assuming significance at p < 0.05. The other data values were presented as the mean ± SD. Statistical analysis methods were denoted in independent figure legends. p < 0.05 was considered statistically significant.
Results
RAS Family Alterations in Acute Lymphoblastic Leukemia Patients
Total 333 children with newly diagnosed ALL and 18 children with relapsed ALL from CCCG-ALL-2015 study at the Guangzhou Women and Children’s Medical Center were enrolled onto this study (Table 1; Figure 1A; Supplementary Figure S2). Targeted next-generation sequencing was performed to identify the ALL-related genetic alterations. We first analyzed the RAS mutation frequency and profile among newly diagnosed patients. As shown in Figure 1B, the frequency of KRAS, NRAS, and HRAS was 14.7, 9.9, and 0.6% respectively, while the frequency of K-, N-, and H- RAS mutation among relapsed patients was 27.8, 11.1, and 0% respectively (Figure 1B; Supplementary Tables S5–S7). Notably, KRAS mutation frequency in relapsed ALL was ∼1.9 folds higher than that of newly diagnosed ALL (27.8 vs. 14.7%; Figure 1B). In the PCGP cohort (McLeod et al., 2021), the mutation frequency of KRAS, NRAS, and HRAS were 13.9, 13.7, and 0% in HRAS among diagnostic samples and 25.5, 22.6, and 0% in relapsed samples (Supplementary Figure S3). To demonstrate the difference between B-ALL and T-cell ALL (T-ALL) as confirmed by flow cytometric immunophenotyping assay, we identified a higher RAS mutation frequency in newly diagnostic B-ALL patients than that in T-ALL patients (14.7 vs 0% in KRAS; 9.3 vs 6.7% in NRAS; 0.6 vs 0% in HRAS, Figure 1C). Because NRAS mutations were associated with B-ALL relapse, we then focused on exploring the NRAS mutation profiles in our study cohort. As shown in Figure 1D, most NRAS mutations located at G12, G13, and Q61 residues, with 52.5, 37.5, and 7.5% frequency, respectively. The NRAS mutations on other residues (G60, Y64, and A146) were very rare, which was in line with previous reports (Supplementary Figure S5A) (Prior et al., 2012). Similar pattern was also observed in KRAS mutations but not in HRAS mutations (Supplementary Figures S5B,C). To address the NRAS mutation profile, we retrieved the NRAS mutation data from pediatric Cancer Genome Project (PCGP) (McLeod et al., 2021) and identified a very similar pattern between our study cohort and PCGP study cohort. (upper panel, GWCMC study cohort; lower panel, PCGP study cohort; Figure 1E) (Hunger and Mullighan, 2015). To examine the association of RAS family mutations and ALL outcomes, we performed the survival analysis using Cox proportional hazards regression model. As shown in Supplementary Figure S4, we did not identify a significant lower overall survival (OS) was identified in ALL patients with RAS mutations (Hazard ratio [HR], 2.1, 95% CI, 0.6 to 6.8, p = 0.23, log-rank test). Similarly, the association of KRAS or NRAS mutations and ALL survival was not statistically significant, suggesting that RAS mutations might not impair the overall survival (Supplementary Figure S4). Next, we explored the effect of RAS mutations on ALL relapse and observed a higher risk of relapse among patients with RAS mutations than those with wild-type RAS (3-year cumulative relapse incidence: 18.7 ± 9.1% vs. 3.8 ± 1.3%, p = 0.0021, Gray test; Supplementary Figure S4). This pattern was observed in the KRAS mutation subgroup (p = 0.0012) but not in the NRAS-mutation subgroup (p = 0.18) (Supplementary Figure S4). Meanwhile, we did not identify an association of NRAS mutations with the therapeutic response as reflected by the minimal residual diseases (MRD) (Supplementary Table S5).
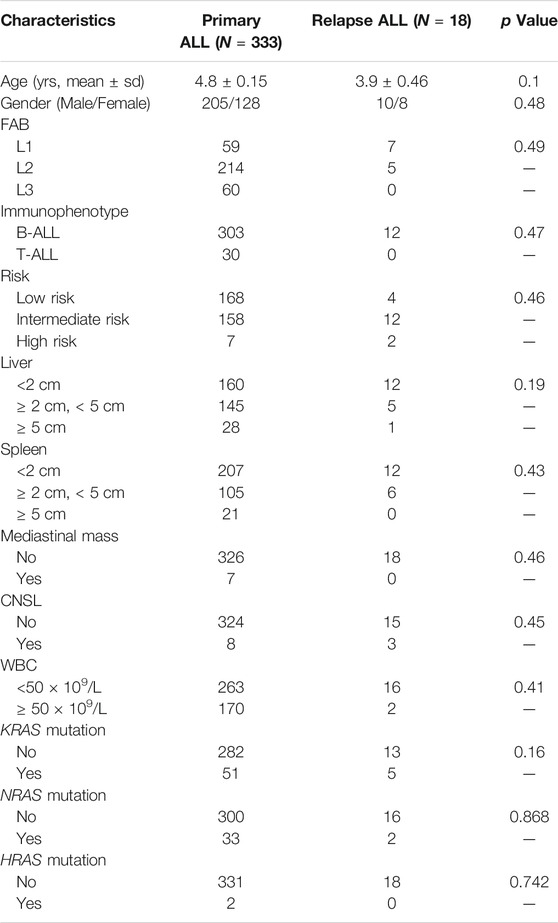
TABLE 1. Characteristics of enrolled patients from CCCG-ALL-2015 cohort.
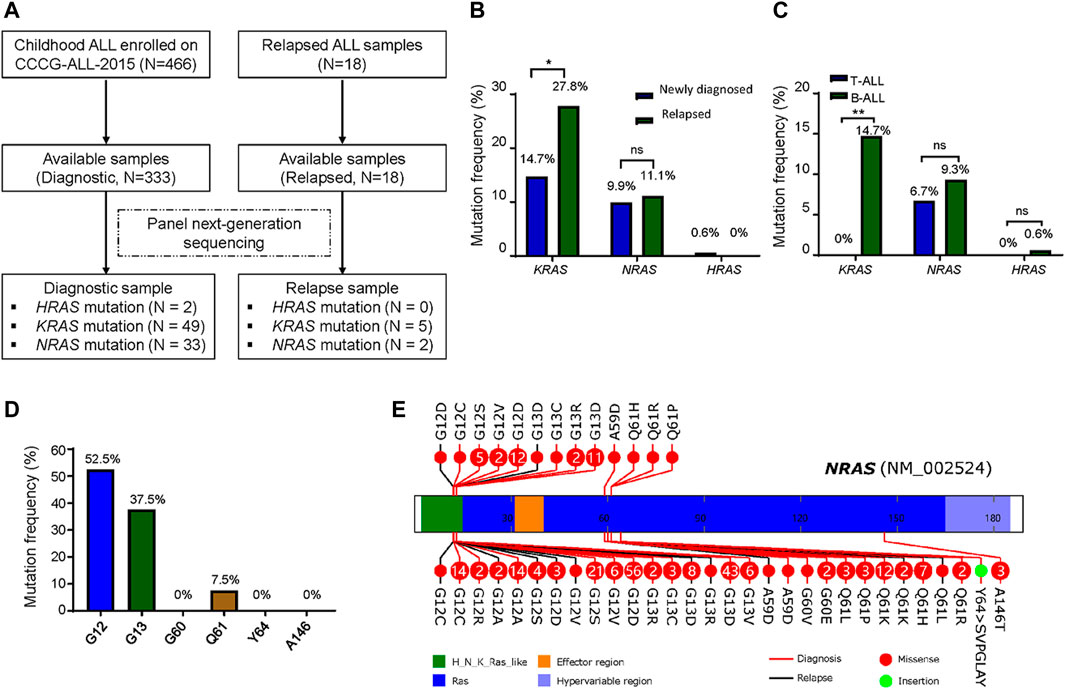
FIGURE 1. RAS mutations in patients with newly diagnosed and relapsed ALL. (A) Identification of HRAS, KRAS and NRAS mutations in patients with newly diagnosed and relapsed ALL enrolled onto CCCG-ALL-2015 clinical trial at Guangzhou Women and Children’s Medical Center. (B) RAS mutations frequency between newly diagnosed (N = 333) and relapsed (N = 18) cases. (C) RAS mutations frequency between diagnostic T-ALL and B-ALL. (D) NRAS mutations frequency including G12, G13, G60, Q61, Y64 and A146 residues in this study cohort. (E) NRAS mutation profile from our study cohort (upper panel) and PCGP study cohort. NRAS mutations spanned the full length of the gene (upper panel, our study cohort; lower panel, PCGP; red line, newly diagnosed ALL; black line, relapsed ALL; solid red circle, missense mutations; solid green circle, insertion mutations; number in this circle represents case number). McNemar chi-square test was performed to compare the frequency of KRAS, NRAS, and HRAS. p < 0.05 (*, <0.05; **, <0.01) was considered statistically significant.
The Effect of NRAS Mutations on Acute Lymphoblastic Leukemia Leukemogenesis
The association of NRAS mutations with ALL relapse has been well studied by several groups. Thus, we next experimentally evaluated the role of NRAS mutations in ALL leukemogenesis, we cloned all NRAS mutants as we identified in Figure 1. We utilized a mouse hematopoietic progenitor Ba/F3 cell line with an IL3-dependent cell growth feature as a study model to test the leukemic transformation capacity of different NRAS mutations. As shown in Figure 2A, ectopic over-expression of NRASG12D but not wild-type NRAS or empty vector significantly induced Ba/F3 cells IL-3 independent growth (p < 0.0001). Using NRASG12D as a positive control, we next tested the leukemic transformation capacity of all NRAS mutations and found that nine of twenty mutations (NRASG12V, NRASG12R, NRASG12W, NRASG12C, NRASG13R, NRASQ61L, NRASQ61R, NRASQ61K, NRASY64>SVPGLAY) significantly potentiated Ba/F3 cells transformation after removing IL3 from culture media, with the comparable or stronger capacity to NRASG12D (Figure 2B). However, the other eleven NRAS mutant forms could not induce IL-3 independent growth. Since G12 residue is the mutation hot spot, we then used the saturated mutagenesis method to establish all nineteen G12 mutant forms and test their leukemic transformation capacity by the same strategy. Interestingly, not all NRAS G12 mutant forms could significantly activate or potentiate leukemogenesis (Figure 2C). Among of which, seven (36.8%) NRAS G12 mutants (NRASG12L, NRASG12T, NRASG12I, NRASG12K, NRASG12V, NRASG12Q, and NRASG12R) demonstrated stronger leukemogenic capacity than NRASG12D, and another seven (36.8%) mutant forms (NRASG12W, NRASG12C, NRASG12H, NRASG12E, NRASG12N, NRASG12M, and NRASG12A) showed comparable to or a little bit weaker capacity. The remaining four (26.4%) NRAS G12 mutants (NRASG12P, NRASG12Y, NRASG12S, and NRASG12F) could not transform Ba/F3 cells at all. Taken together, our data suggest that not all NRAS mutants have leukemogenic potentials or pathogenic effects (Supplementary Table S8).
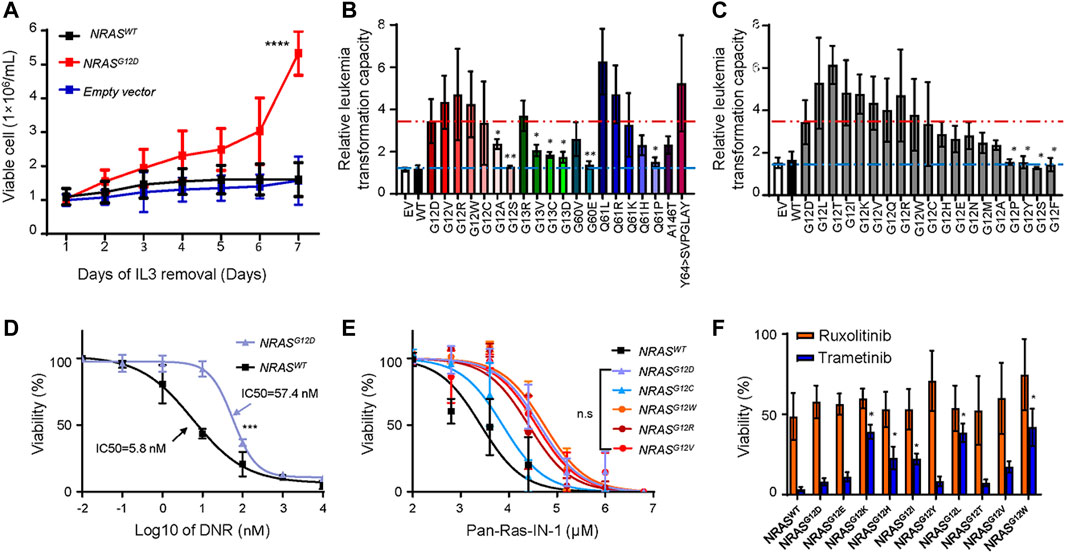
FIGURE 2. Transforming potentials and drug response of NRAS mutation s in Ba/F3 cell line. (A) IL-3 independent cell growth of Ba/F3 cells transduced with NRASG12D, NRASWT and empty vector. (B–C) Relative leukemogenic capacity between reported NRAS mutations (B) and G12 saturated mutant forms (C). Differences were calculated between NRASG12D and other NRAS mutants by student t test. Dark red dash line represents the NRASG12D transforming capacity as positive control, and light blue dash line represents non-transforming capacity. The relative capacity was calculated by the ratio of viable cells between day7 and day1. Cytotoxicity of DNR (D), Pan-Ras-IN-1 (E), trametinib and ruxolotinib (F) were examined in Ba/F3 cells with NRAS mutations (black line, NRASWT; purple line, NRASG12D; blue line, NRASG12C; orange line, NRASG12w; brown line, NRASG12R; red line, NRASG12V). The cell viability was measured after 72 h of drug exposure using an CCK-8 assay. All these experiments were performed in triplicate and independently repeated three times. Two-way ANOVA method was used to perform the statistical analysis for (D,E), and student t test was applied for (F)”. p < 0.05 (*, <0.05; **, <0.01; ****, <0.0001) was considered statistically significant.
Building upon the findings above, we further asked how to target ALL cells with NRAS mutations. To address this question, we first tried to answer that whether NRAS mutations conferred resistance to conventional and novel agents, such as daunorubicin (DNR) and tyrosine kinase inhibitors. We treated with NRASmut transformed Ba/F3 cells with DNR and found that NRASG12D ALL cells were more resistant to DNR than those with wild-type NRAS (IC50: 57.4 vs. 5.8 nM) (Figure 2D), which was in line with previous reports (Irving et al., 2014; Irving et al., 2016). Using a similar approach, we compared the effect of RAS inhibitors on NRASG12-transfected Ba/F3 cells. Ba/F3 cells transfected with distinct NRAS mutants (NRASG12D, NRASG12C, NRASG12W, NRASG12R, and NRASG12V) were more resistant to Pan-Ras-IN-1 (a pan-Ras inhibitor) variably than those with NRAS wild-type (Figure 2E). Similar results were observed for other RAS inhibitors, including Fendiline, ARS1620, and AMG510 (Supplementary Figure S6). As reported by Kirchberger et al. that MEK inhibition chemo-sensitized NRASG12D-mutated ALL cells to conventional therapeutic agents (i.e., DNR and dexamethasone) (Irving et al., 2016), we thus tested the MEK inhibition response among those NRASG12 mutants transformed Ba/F3 cells. Interestingly, we identified that Ba/F3 cells with NRASG12E, NRASG12T, and NRASG12Y mutation were as sensitive as Ba/F3 cells with NRASG12D mutation to trametinib treatment. Meanwhile, Ba/F3 cells with NRASG12K, NRASG12H, NRASG12I, NRASG12L, NRASG12V, and NRASG12W mutation just demonstrated a moderate response to trametinib treatment (Figure 2F). However, all tested NRASmut transformed Ba/F3 cells did not respond to ruxolitinib, a JAK2 inhibitor.
Translational Potential of Differential NRAS Mutations on Acute Lymphoblastic Leukemia Therapeutics
The findings above suggested that ALL cells with NRAS mutation might differently respond to signaling inhibition. To address this question, we applied HDS strategy as an attempt to identify candidate agents that could preferentially target NRAS mutations. Among the 843 tested agents (Figure 3A; Supplementary Table S9), we observed that NRASG12D mutation well responded to MEK inhibition (GDC-0623, pimasertib, and TAK-733), which was in consistent with current clinical reports (Nakamura et al., 2013; Johnson et al., 2014; Kirchberger et al., 2018). Interestingly, NRASG12L and NRASG12N mutations also well responded to MEK inhibition. However, NRASG12C mutation well responded to autophagy inhibition and mTOR inhibition (WYE−354), and mix-lineage kinase inhibition (E−Necrosulfonamide), while NRASG12V and NRASG12T mutations responded to Akt inhibition (deguelin), EGFR inhibitor (mubritinib), Polo−like Kinase (PLK) inhibition (CFI−400945), Src inhibition (MCB−613), and TGF−β receptor inhibitor (LDN−212854). The distinctive drug response among NRAS mutations drove us mechanistically validate our findings above. We first utilized immunoblot assay to profile the impact of NRAS mutations on Erk, Jak2-Stat5 signaling pathway. As illustrated in Figures 3B,C, we found that NRASG12C, NRASG12K NRASG12E, NRASG12H and NRASG12N mutations did not activate Jak2-Stat5, or Erk signaling. NRASG12D strongly activated Erk signaling, while NRASG12I, NRASG12F, NRASG12W and NRASG12R, NRASG12S, NRASG12Y, NRASG12P, and NRASG12Q activated Jak2-Stat5 alone. Intriguingly, NRASG12T, NRASG12A, NRASG12L, NRASG12V, and NRASG12M co-stimulated Erk and Jak2-Stat5 signaling.
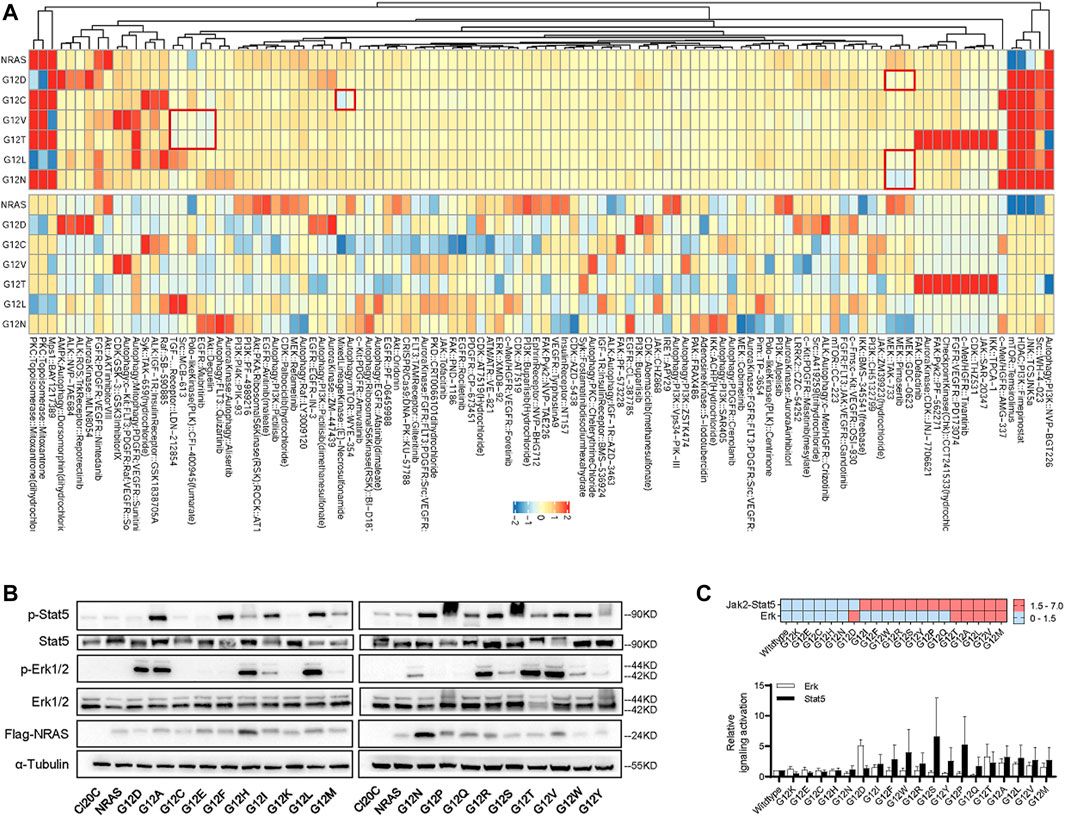
FIGURE 3. The impact of NRASG12 mutation on drug sensitivity. (A) Normalized heatmap of HDS results (log10 of IC50 concentration) among Ba/F3 cells with different NRAS mutation. All these experiments were performed in triplicate and independently repeated three times. (B) Western blotting assay of Ba/F3 cells transduced with NRASG12 mutants. (C) Heatmap (upper panel) and grey intensity plot (lower panel) to Erk and Jak2-Stat5 signaling pathways activation in Ba/F3 transduced with NRASG12 mutants.
Discussion
This study identified a group of RAS gene mutations with a high frequency in childhood ALL. Our data analysis showed that NRAS, KRAS, and HRAS mutations were almost mutually exclusive within our study cohort, with only eleven patients with KRAS and NRAS mutations concurrently. In consistent with reports from several other groups (Case et al., 2008; Davidsson et al., 2010; Irving et al., 2014; Oshima et al., 2016), we did not detect any changes in the frequency of RAS mutations based on gender or age. Irving et al. have identified that NRAS mutations were associated with an increased risk of progression within hyperdiploidy standard-risk patient group by analyzing cytogenetic data from 427 children with relapsed B-ALL (Irving et al., 2016). The impact of NRAS mutations on childhood ALL relapse in our study cohort was slightly different from other groups (Ma et al., 2015; Irving et al., 2016), which might be explained by several factors, including but not limited to patient demographics, socioeconomic status, clinical characteristics, and study sample size. Meanwhile, we found that the detectable genomic alteration in this cohort was only 36.55% (Supplementary Figure S1), suggesting whole transcriptome sequencing is highly needed to capture all genomic lesions.
Several reports have successfully linked genetic defects (i.e., RAS pathway alterations, drug-metabolism related genes [FPGS, NT5C2, NR3C1, and PRPS1], transcription factor [TP53, IKZF1, CREBBP]) with ALL relapse (Mullighan et al., 2011; Tzoneva et al., 2013; Mar et al., 2014; Song et al., 2020). Many study groups have reported that RAS mutations could be detected in ∼50% relapsed ALL patients (Irving et al., 2014; Malinowska-Ozdowy et al., 2015; Oshima et al., 2016; Tasian and Hunger, 2017; Ding, 2018; Jerchel et al., 2018), indicating the importance of RAS mutations in ALL relapse. Attractively, a recent study by Zhang et al. has shown that more than 50% of relapsed pediatric ALL patients have RAS pathway mutations (KRAS, NRAS, NF1, EPOR), further consolidating the role of RAS mutations in relapsed ALL. In this study, we included 333 newly-diagnosed and 18 relapsed B-ALL patients, which is the largest single institutional cohort in China to systemically explore the role of RAS mutations in childhood ALL. The prevalence of RAS mutations was 25.2 and 38.9% in newly-diagnosed and relapsed children with ALL in our study cohort, respectively (Figure 1), which was in line with recent reports (Irving et al., 2014; Ma et al., 2015). It has been reported that RAS mutations were more likely to be enriched in high-risk ALL group, including patients with early relapse (H-, K-, and N- RAS mutations) and with central nervous system (CNS) involvement (NRAS and KRAS mutations) (Reshmi et al., 2017; Takashima et al., 2018). Our report here demonstrated a correlation between KRAS mutations and ALL relapse (Supplementary Figure S4). However, we did not observe a significant association between RAS mutations and treatment outcome (i.e., early treatment response (defined by MRD), OS, CNS involvement, risk stratification). There may be several possibilities, including relatively small sample size, patient demographics, and treatment protocols. Additional studies or multi-institutional cooperation were warranted to further define their relationship.
It was reported that RAS activation in malignant hematopoietic cells induces multi-drug resistance (i.e., glucocorticoids and anthracyclines) in ALL therapy (McCubrey et al., 2007; Garza et al., 2009; Irving et al., 2014). Thus, it is critical to rescue the therapeutic response so as to improve the treatment outcome. Interestingly, we found that NRAS mutants differed in their ability to leukemic transformation, strongly indicating that not all NRAS mutations are driver mutations which can be potentially targeted (Figure 2). In combination with the in vitro cytotoxic and signaling activation results (Figures 2, 3), we believed that those leukemogenic NRAS mutants might contribute to leukemogenesis and therapeutic targeting via different mechanisms, which is supported by other groups. For example, Jerchel et al. have identified that NRAS mutation-related BCP-ALL may not activate the MAPK pathway (Jerchel et al., 2018). In contrast, Chan et al. have demonstrated NRAS mutation may promote B-cell leukemogenesis via STAT5 or MAPK (Chan et al., 2020), suggesting complicated mechanisms underlying the NRAS mutations in B-ALL. In this study, we confirmed the well response of NRASG12D to MEK inhibition by HDS assay and western blot (Figure 3). Interestingly, we had identified a distinctive signaling activation profile. It’s well established that NRASG12D activated ERK signaling and well responded to MEK inhibition. In the meanwhile, we also found that different NRASG12 mutant activated different down-steam signaling pathways (Figures 3B,C), which might partially explain the different drug response among NRAS G12 mutations (Figure 3A). Though we did not find that NRASG12N activated the ERK signaling with a similar pattern as NRASG12D did, NRASG12N surprisingly well responded to ERK inhibition (Figure 3A), suggesting some compensatory mechanisms might be existed. It’s noted that NRASG12C did not activate Jak2-Stat5 or Erk signaling as shown in the western blot, and the HDS assay showed that NRASG12C was resistant to MEK or JAK inhibition, again pointing to that one targeting strategy did not fit for all NRASG12 mutations. NRASG12T, and NRASG12V co-stimulated Erk and Jak2-Stat5 signaling, and demonstrated a similar drug responding pattern to Akt inhibition, autophagy inhibition, and TGF-β inhibition. Taken together, introducing proper NRAS targeting agents into current chemotherapy regimens might be of help in further improving current ALL treatment.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: National Genomics Data Center (NGDC) Genome Sequence Archive for Human (GSA-Human), https://ngdc.cncb.ac.cn/gsa-human, HRA000708
Ethics Statement
The studies involving human participants were reviewed and approved by This study was approved by the Institutional Review Board of Guangzhou Women and Children’s Medical Center (2015020936, 2017102307, and 2020-04500). . Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
JQ, MQ, YS, YH, and HZ designed the research; JQ, KP, FQ, ZL, and CL performed experiments; KP, YH, and MY collected the samples and recruited patients; JQ, KP, FQ, YS, MQ, YH, and HZ analyzed the results; and JQ, FQ, YS, MQ, YH, and HZ wrote the manuscript.
Funding
This work was supported by research grants from St. Baldrick’s Foundation International Scholar (581580), Natural Science Foundation of Guangdong Province (2015A030313460), and Guangzhou Women and Children’s Medical Center Internal Program (IP-2018-001, 5001-1600006, and 5001-1600008). HZ, YS, MQ, and CL were supported by the National Natural Science Foundation of China (82170152), (81800122), (81973997) and (32000392) respectively.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to show their gratitude and thanks to all of the patients for their kind participation. In the meanwhile, we would also like to thank Xujie Zhao at St. Jude Children’s Research Hospital for scientific editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.712484/full#supplementary-material
References
Bery, N., Cruz-Migoni, A., Bataille, C. J., Quevedo, C. E., Tulmin, H., Miller, A., et al. (2018). BRET-based RAS Biosensors that Show a Novel Small Molecule Is an Inhibitor of RAS-Effector Protein-Protein Interactions. Elife 7. doi:10.7554/elife.40515
Bhojwani, D., and Pui, C.-H. (2013). Relapsed Childhood Acute Lymphoblastic Leukaemia. Lancet Oncol. 14, e205–e217. doi:10.1016/s1470-2045(12)70580-6
Brown, J. A., and Ferrando, A. (2018). Glucocorticoid Resistance in Acute Lymphoblastic Leukemia: BIM Finally. Cancer Cell 34, 869–871. doi:10.1016/j.ccell.2018.11.011
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., et al. (1999). Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 96, 857–868. doi:10.1016/s0092-8674(00)80595-4
Burgess, M. R., Hwang, E., Mroue, R., Bielski, C. M., Wandler, A. M., Huang, B. J., et al. (2017). KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 168, 817–829. doi:10.1016/j.cell.2017.01.020
Canon, J., Rex, K., Saiki, A. Y., Mohr, C., Cooke, K., Bagal, D., et al. (2019). The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-tumour Immunity. Nature 575, 217–223. doi:10.1038/s41586-019-1694-1
Case, M., Matheson, E., Minto, L., Hassan, R., Harrison, C. J., Bown, N., et al. (2008). Mutation of Genes Affecting the RAS Pathway Is Common in Childhood Acute Lymphoblastic Leukemia. Cancer Res. 68, 6803–6809. doi:10.1158/0008-5472.can-08-0101
Chan, L. N., Murakami, M. A., Robinson, M. E., Caeser, R., Sadras, T., Lee, J., et al. (2020). Signalling Input from Divergent Pathways Subverts B Cell Transformation. Nature 583, 845–851. doi:10.1038/s41586-020-2513-4
Cox, A. D., and Der, C. J. (2003). The Dark Side of Ras: Regulation of Apoptosis. Oncogene 22, 8999–9006. doi:10.1038/sj.onc.1207111
Davidsson, J., Paulsson, K., Lindgren, D., Lilljebjörn, H., Chaplin, T., Forestier, E., et al. (2010). Relapsed Childhood High Hyperdiploid Acute Lymphoblastic Leukemia: Presence of Preleukemic Ancestral Clones and the Secondary Nature of Microdeletions and RTK-RAS Mutations. Leukemia 24, 924–931. doi:10.1038/leu.2010.39
Ding, L., Ley, T. J., Larson, D. E., Miller, C. A., Koboldt, D. C., Welch, J. S., et al. (2012). Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 481, 506–510. doi:10.1038/nature10738
Ding, Z.-C. (2018). The Promise and Challenges of Chimeric Antigen Receptor T Cells in Relapsed B-Cell Acute Lymphoblastic Leukemia. Ann. Transl. Med. 6, 235. doi:10.21037/atm.2018.05.35
Downward, J. (2003). Targeting RAS Signalling Pathways in Cancer Therapy. Nat. Rev. Cancer 3, 11–22. doi:10.1038/nrc969
Garza, A. S., Miller, A. L., Johnson, B. H., and Thompson, E. B. (2009). Converting Cell Lines Representing Hematological Malignancies from Glucocorticoid-Resistant to Glucocorticoid-Sensitive: Signaling Pathway Interactions. Leuk. Res. 33, 717–727. doi:10.1016/j.leukres.2008.10.006
Hallin, J., Engstrom, L. D., Hargis, L., Calinisan, A., Aranda, R., Briere, D. M., et al. (2020). The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 10, 54–71. doi:10.1158/2159-8290.cd-19-1167
Hunger, S. P., and Mullighan, C. G. (2015). Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 373, 1541–1552. doi:10.1056/nejmra1400972
Irving, J. A. E., Enshaei, A., Parker, C. A., Sutton, R., Kuiper, R. P., Erhorn, A., et al. (2016). Integration of Genetic and Clinical Risk Factors Improves Prognostication in Relapsed Childhood B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 128, 911–922. doi:10.1182/blood-2016-03-704973
Irving, J., Matheson, E., Minto, L., Blair, H., Case, M., Halsey, C., et al. (2014). Ras Pathway Mutations Are Prevalent in Relapsed Childhood Acute Lymphoblastic Leukemia and Confer Sensitivity to MEK Inhibition. Blood 124, 3420–3430. doi:10.1182/blood-2014-04-531871
Jain, N., Curran, E., Iyengar, N. M., Diaz-Flores, E., Kunnavakkam, R., Popplewell, L., et al. (2014). Phase II Study of the Oral MEK Inhibitor Selumetinib in Advanced Acute Myelogenous Leukemia: a University of Chicago Phase II Consortium Trial. Clin. Cancer Res. 20, 490–498. doi:10.1158/1078-0432.ccr-13-1311
Janes, M. R., Zhang, J., Li, L.-S., Hansen, R., Peters, U., Guo, X., et al. (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-specific Inhibitor. Cell 172, 578–589. doi:10.1016/j.cell.2018.01.006
Jerchel, I. S., Hoogkamer, A. Q., Ariës, I. M., Steeghs, E. M. P., Boer, J. M., Besselink, N. J. M., et al. (2018). RAS Pathway Mutations as a Predictive Biomarker for Treatment Adaptation in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 32, 931–940. doi:10.1038/leu.2017.303
Johnson, D. B., Smalley, K. S. M., and Sosman, J. A. (2014). Molecular Pathways: Targeting NRAS in Melanoma and Acute Myelogenous Leukemia. Clin. Cancer Res. 20, 4186–4192. doi:10.1158/1078-0432.ccr-13-3270
Karnoub, A. E., and Weinberg, R. A. (2008). Ras Oncogenes: Split Personalities. Nat. Rev. Mol. Cel Biol 9, 517–531. doi:10.1038/nrm2438
Kirchberger, M. C., Ugurel, S., Mangana, J., Heppt, M. V., Eigentler, T. K., Berking, C., et al. (2018). MEK Inhibition May Increase Survival of NRAS-Mutated Melanoma Patients Treated with Checkpoint Blockade: Results of a Retrospective Multicentre Analysis of 364 Patients. Eur. J. Cancer 98, 10–16. doi:10.1016/j.ejca.2018.04.010
Kong, G., Wunderlich, M., Yang, D., Ranheim, E. A., Young, K. H., Wang, J., et al. (2014). Combined MEK and JAK Inhibition Abrogates Murine Myeloproliferative Neoplasm. J. Clin. Invest. 124, 2762–2773. doi:10.1172/jci74182
Lai, Z., Markovets, A., Ahdesmaki, M., Chapman, B., Hofmann, O., McEwen, R., et al. (2016). VarDict: a Novel and Versatile Variant Caller for Next-Generation Sequencing in Cancer Research. Nucleic Acids Res. 44, e108. doi:10.1093/nar/gkw227
Lito, P., Solomon, M., Li, L.-S., Hansen, R., and Rosen, N. (2016). Allele-specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science 351, 604–608. doi:10.1126/science.aad6204
Ma, X., Edmonson, M., Yergeau, D., Muzny, D. M., Hampton, O. A., Rusch, M., et al. (2015). Rise and Fall of Subclones from Diagnosis to Relapse in Pediatric B-Acute Lymphoblastic Leukaemia. Nat. Commun. 6, 6604. doi:10.1038/ncomms7604
Malinowska-Ozdowy, K., Frech, C., Schönegger, A., Eckert, C., Cazzaniga, G., Stanulla, M., et al. (2015). KRAS and CREBBP Mutations: a Relapse-Linked Malicious Liaison in Childhood High Hyperdiploid Acute Lymphoblastic Leukemia. Leukemia 29, 1656–1667. doi:10.1038/leu.2015.107
Mar, B. G., Bullinger, L. B., McLean, K. M., Grauman, P. V., Harris, M. H., Stevenson, K., et al. (2014). Mutations in Epigenetic Regulators Including SETD2 Are Gained during Relapse in Paediatric Acute Lymphoblastic Leukaemia. Nat. Commun. 5, 3469. doi:10.1038/ncomms4469
McCubrey, J. A., Steelman, L. S., Chappell, W. H., Abrams, S. L., Wong, E. W. T., Chang, F., et al. (2007). Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1773, 1263–1284. doi:10.1016/j.bbamcr.2006.10.001
McLeod, C., Gout, A. M., Zhou, X., Thrasher, A., Rahbarinia, D., Brady, S. W., et al. (2021). St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov. 11 (5), 1082–1099. doi:10.1158/2159-8290.CD-20-1230
Meyer, J. A., Wang, J., Hogan, L. E., Yang, J. J., Dandekar, S., Patel, J. P., et al. (2013). Relapse-specific Mutations in NT5C2 in Childhood Acute Lymphoblastic Leukemia. Nat. Genet. 45, 290–294. doi:10.1038/ng.2558
Mullighan, C. G., Phillips, L. A., Su, X., Ma, J., Miller, C. B., Shurtleff, S. A., et al. (2008). Genomic Analysis of the Clonal Origins of Relapsed Acute Lymphoblastic Leukemia. Science 322, 1377–1380. doi:10.1126/science.1164266
Mullighan, C. G., Zhang, J., Kasper, L. H., Lerach, S., Payne-Turner, D., Phillips, L. A., et al. (2011). CREBBP Mutations in Relapsed Acute Lymphoblastic Leukaemia. Nature 471, 235–239. doi:10.1038/nature09727
Nakamura, A., Arita, T., Tsuchiya, S., Donelan, J., Chouitar, J., Carideo, E., et al. (2013). Antitumor Activity of the Selective Pan-RAF Inhibitor TAK-632 in BRAF Inhibitor-Resistant Melanoma. Cancer Res. 73, 7043–7055. doi:10.1158/0008-5472.can-13-1825
Oshima, K., Khiabanian, H., da Silva-Almeida, A. C., Tzoneva, G., Abate, F., Ambesi-Impiombato, A., et al. (2016). Mutational Landscape, Clonal Evolution Patterns, and Role of RAS Mutations in Relapsed Acute Lymphoblastic Leukemia. Proc. Natl. Acad. Sci. USA 113, 11306–11311. doi:10.1073/pnas.1608420113
Pierro, J., Hogan, L. E., Bhatla, T., and Carroll, W. L. (2017). New Targeted Therapies for Relapsed Pediatric Acute Lymphoblastic Leukemia. Expert Rev. anticancer Ther. 17, 725–736. doi:10.1080/14737140.2017.1347507
Posch, C., Moslehi, H., Feeney, L., Green, G. A., Ebaee, A., Feichtenschlager, V., et al. (2013). Combined Targeting of MEK and PI3K/mTOR Effector Pathways Is Necessary to Effectively Inhibit NRAS Mutant Melanoma In Vitro and In Vivo. Proc. Natl. Acad. Sci. 110, 4015–4020. doi:10.1073/pnas.1216013110
Prior, I. A., Lewis, P. D., and Mattos, C. (2012). A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 72, 2457–2467. doi:10.1158/0008-5472.can-11-2612
Pui, C.-H., Yang, J. J., Bhakta, N., and Rodriguez-Galindo, C. (2018). Global Efforts toward the Cure of Childhood Acute Lymphoblastic Leukaemia. Lancet Child. Adolesc. Health 2, 440–454. doi:10.1016/s2352-4642(18)30066-x
Rausch, T., Zichner, T., Schlattl, A., Stutz, A. M., Benes, V., and Korbel, J. O. (2012). DELLY: Structural Variant Discovery by Integrated Paired-End and Split-Read Analysis. Bioinformatics 28, i333–i339. doi:10.1093/bioinformatics/bts378
Reshmi, S. C., Harvey, R. C., Roberts, K. G., Stonerock, E., Smith, A., Jenkins, H., et al. (2017). Targetable Kinase Gene Fusions in High-Risk B-ALL: a Study from the Children's Oncology Group. Children's Oncol. Group 129, 3352–3361. doi:10.1182/blood-2016-12-758979
Samatar, A. A., and Poulikakos, P. I. (2014). Targeting RAS-ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 13, 928–942. doi:10.1038/nrd4281
Song, M., Pebworth, M.-P., Yang, X., Abnousi, A., Fan, C., Wen, J., et al. (2020). Cell-type-specific 3D Epigenomes in the Developing Human Cortex. Nature 587, 644–649. doi:10.1038/s41586-020-2825-4
Stephen, A. G., Esposito, D., Bagni, R. K., and McCormick, F. (2014). Dragging Ras Back in the Ring. Cancer Cell 25, 272–281. doi:10.1016/j.ccr.2014.02.017
Takashima, Y., Sasaki, Y., Hayano, A., Homma, J., Fukai, J., Iwadate, Y., et al. (2018). Target Amplicon Exome-Sequencing Identifies Promising Diagnosis and Prognostic Markers Involved in RTK-RAS and PI3K-AKT Signaling as central Oncopathways in Primary central Nervous System Lymphoma. Oncotarget 9, 27471–27486. doi:10.18632/oncotarget.25463
Tasian, S. K., and Hunger, S. P. (2017). Genomic Characterization of Paediatric Acute Lymphoblastic Leukaemia: an Opportunity for Precision Medicine Therapeutics. Br. J. Haematol. 176, 867–882. doi:10.1111/bjh.14474
Tzoneva, G., Perez-Garcia, A., Carpenter, Z., Khiabanian, H., Tosello, V., Allegretta, M., et al. (2013). Activating Mutations in the NT5C2 Nucleotidase Gene Drive Chemotherapy Resistance in Relapsed ALL. Nat. Med. 19, 368–371. doi:10.1038/nm.3078
Vora, A., Goulden, N., Wade, R., Mitchell, C., Hancock, J., Hough, R., et al. (2013). Treatment Reduction for Children and Young Adults with Low-Risk Acute Lymphoblastic Leukaemia Defined by Minimal Residual Disease (UKALL 2003): a Randomised Controlled Trial. Lancet Oncol. 14, 199–209. doi:10.1016/s1470-2045(12)70600-9
Wang, J., Kong, G., Liu, Y., Du, J., Chang, Y.-I., Tey, S. R., et al. (2013). NrasG12D/+ Promotes Leukemogenesis by Aberrantly Regulating Hematopoietic Stem Cell Functions. Blood 121, 5203–5207. doi:10.1182/blood-2012-12-475863
Ward, A. F., Braun, B. S., and Shannon, K. M. (2012). Targeting Oncogenic Ras Signaling in Hematologic Malignancies. Blood 120, 3397–3406. doi:10.1182/blood-2012-05-378596
Welsch, M. E., Kaplan, A., Chambers, J. M., Stokes, M. E., Bos, P. H., Zask, A., et al. (2017). Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 168, 878–889. doi:10.1016/j.cell.2017.02.006
Xu, H., Zhang, H., Yang, W., Yadav, R., Morrison, A. C., Qian, M., et al. (2015). Inherited Coding Variants at the CDKN2A Locus Influence Susceptibility to Acute Lymphoblastic Leukaemia in Children. Nat. Commun. 6, 7553. doi:10.1038/ncomms8553
Xu, J., Shi, S., Matsumoto, N., Noda, M., and Kitayama, H. (2007). Identification of Rgl3 as a Potential Binding Partner for Rap-Family Small G-Proteins and Profilin II. Cell Signal. 19, 1575–1582. doi:10.1016/j.cellsig.2007.02.004
Keywords: NRAS proto-oncogene, acute lymphoblastic leukemia, signaling pathway activation, therapeutic targeting, leukemogenic potential
Citation: Qian J, Li Z, Pei K, Li Z, Li C, Yan M, Qian M, Song Y, Zhang H and He Y (2022) Effects of NRAS Mutations on Leukemogenesis and Targeting of Children With Acute Lymphoblastic Leukemia. Front. Cell Dev. Biol. 10:712484. doi: 10.3389/fcell.2022.712484
Received: 20 May 2021; Accepted: 04 January 2022;
Published: 08 February 2022.
Edited by:
Claudia Scotti, University of Pavia, ItalyReviewed by:
Sheik Pran Babu Sardar Pasha, University of California, Davis, United StatesPeng Xu, Soochow University, China
Copyright © 2022 Qian, Li, Pei, Li, Li, Yan, Qian, Song, Zhang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maoxiang Qian, bXhxaWFuQGZ1ZGFuLmVkdS5jbg==; Yuanbin Song, amltbXlzb25nQGZveG1haWwuY29t; Hui Zhang, emhhbmdodWlyamhAZ3djbWMub3Jn; Yingyi He, aHl5YmJzQDE2My5jb20=
†These authors have contributed equally to this work