
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 11 January 2023
Sec. Cancer Cell Biology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1115903
This article is part of the Research Topic Epigenetics and Carcinogenesis View all 5 articles
 Tiina Viita
Tiina Viita Jacques Côté*
Jacques Côté*Acetylation of lysine residues on histone tails is an important post-translational modification (PTM) that regulates chromatin dynamics to allow gene transcription as well as DNA replication and repair. Histone acetyltransferases (HATs) are often found in large multi-subunit complexes and can also modify specific lysine residues in non-histone substrates. Interestingly, the presence of various histone PTM recognizing domains (reader domains) in these complexes ensures their specific localization, enabling the epigenetic crosstalk and context-specific activity. In this review, we will cover the biochemical and functional properties of the MOZ-BRPF1 acetyltransferase complex, underlining its role in normal biological processes as well as in disease progression. We will discuss how epigenetic reader domains within the MOZ-BRPF1 complex affect its chromatin localization and the histone acetyltransferase specificity of the complex. We will also summarize how MOZ-BRPF1 is linked to development via controlling cell stemness and how mutations or changes in expression levels of MOZ/BRPF1 can lead to developmental disorders or cancer. As a last touch, we will review the latest drug candidates for these two proteins and discuss the therapeutic possibilities.
The human genome comprises of approximately three billion nucleotides of DNA and for this reason the DNA must be highly compacted inside the nucleus (International Human Genome Sequencing Consortium, 2004). This is primarily done by wrapping DNA around histone proteins, to form the basic unit of chromatin, i.e., the nucleosome. DNA holds the genetic information needed for gene expression, which is essential for cell functions. As the DNA is tightly wrapped around histones in the form of chromatin, dynamic changes in the chromatin structure are necessary for effective gene expression, DNA repair as well as DNA replication. Epigenetic factors such as ATP-dependent chromatin remodeling complexes and histone modifying complexes modulate the compaction of genomic loci, allowing activation and repression of genes at given regions on the chromosome. The histone modifying complexes are responsible of histone post-transitional modifications (PTMs), which are deposited on the histone tails by specific enzymes (known as writers) and mark specific loci in the genome. Local specific combinations of these PTMs form epigenetic signatures that can be recognized by effector proteins (readers). As most PTMs are reversible, various enzymes (erasers) can also remove these marks. Chromatin modifying complexes are large multifunctional protein complexes and the presence of reader modules in these complexes ensures their specific localization, enabling the epigenetic crosstalk and context-specific activity [reviewed in (Talbert and Henikoff, 2021; Millan-Zambrano et al., 2022)].
One group of chromatin modifying complexes are histone acetyltransferases (HATs), mainly responsible of acetylating histone tails while some of them also have non-histone targets (Steunou et al., 2014). Acetylation of histone lysine residue removes the positive charge of this residue, which weakens the interactions between histones and DNA as well as between nucleosomes, leading to a more open chromatin structure. Open chromatin is more accessible to the transcription machinery; therefore, acetylation correlates with transcriptionally active genes. Acetylated lysine residues can also be recognized by proteins containing various reader domains, which makes them binding sites for other regulatory factors, including other chromatin modifying or remodeling complexes. Beside transcription, histone acetylation is also linked with DNA repair (Murr et al., 2006; Lashgari et al., 2022) and it has been shown to regulate the activity of DNA replication origins (Lubelsky et al., 2014). Different HATs can be categorized to different families based on their homologies: the Gcn5-related acetyltransferase (GNAT) family [reviewed in (Vetting et al., 2005)], the p300/CBP family [reviewed in (Dancy and Cole, 2015)], and the MYST family (named after its founding members, MOZ, Ybf2/Sas3, Sas2, and Tip60) [reviewed in (Avvakumov and Cote, 2007)]. In this review, we will focus on the 4-subunit MOZ-BRPF1 acetyltransferase complex, which belongs to MYST acetyltransferase family. We will summarize how different binding motifs inside the subunits of the complex drive the chromatin localization and biological functions of this histone modifying complex. We will also discuss how this complex has been linked to development as well as disease and hypothesize the therapeutic possibilities of drug candidates that target its structure/function.
Monocytic leukemia zinc finger protein (MOZ, also known as KAT6A) is a MYST family acetyltransferase, which was first discovered as a fusion partner of CBP in acute myeloid leukemia (AML) patients (Borrow et al., 1996). It is closely related to MORF (monocytic leukemia zinc finger protein related factor, also known as KAT6B), which shares 60% homology (Champagne et al., 1999). MOZ/MORF enzymes are found part of 4-subunit complexes that include ING4/5 (inhibitor of growth 4 or 5), MEAF6 (mammalian Esa1-associated factor-6) and BRPF1 (bromodomain and PHD finger-containing protein 1), the latter acting as scaffolding subunit binding simultaneously to MOZ or MORF on one hand, and ING4/5 and MEAF6 on the other (Doyon et al., 2006; Ullah et al., 2008; Klein et al., 2014; Feng et al., 2016; Klein et al., 2019). Although MOZ and MORF have high similarity and the same binding partners, they seem to drive different biological processes. MORF is required for neurogenesis and cerebral formation (Thomas et al., 2000; Merson et al., 2006; Sheikh et al., 2012), whereas knockout studies in mice have shown that MOZ is essential for normal hematopoiesis (Katsumoto et al., 2006; Thomas et al., 2006). These functional differences between MOZ and MORF may be due to distinct spatiotemporal gene expression, non-histone targets or regulation by post-translational modifications.
MOZ and MORF proteins harbor various domains [NEMM, double plant homeodomain (PHD) finger [DPF], MYST, glutamate/aspartate-rich and serine/methionine-rich domain] (Figure 1A) (Champagne et al., 1999; Champagne et al., 2001). The C-terminal region of MOZ/MORF contains the glutamate/aspartate-rich and serine/methionine-rich domains. These domains have been linked with transcription activation (Champagne et al., 1999; Champagne et al., 2001). This is supported by studies showing the serine/methionine-rich domain can bind and activate transcription factors RUNX1, RUNX2 and TP53 (Kitabayashi et al., 2001a; Pelletier et al., 2002; Rokudai et al., 2009). Moreover, these two domains are often lost in leukemic protein fusions with MOZ/MORF produced by chromosomal translocations (Borrow et al., 1996; Carapeti et al., 1998; Chaffanet et al., 2000), which indicates that lack of these domains may deregulate the MOZ-dependent gene expression network, leading to cancer. The NEMM (N-terminal part of Enok, MOZ or MORF) domain is also found in Enok, the homologous protein in Drosophila melanogaster (Huang et al., 2016). This domain had not been well characterized while showing similarities with the winged helix of linker histones H1 and H5 and being implicated in the nuclear localization of MOZ (Kitabayashi et al., 2001a). Overexpression studies with truncated constructs have suggested that the N-terminal region (1–84) of MOZ can interact with RNA polymerase 2 (RNAPII) (Miyamoto et al., 2020). Most recently, this domain was shown to recognize unmethylated CpG islands on DNA, in vitro and in vivo, and to be required for recruitment of MOZ/MORF and its HAT activity on the genome (Becht et al., 2023). Winged helix 1 (WH1) is critical for this function, recognizing unmethylated CpGs, and cooperates with winged helix 2 (WH2) to bind nucleosomes.

FIGURE 1. Multiple chromatin binding domains in the 4-subunit MOZ-BRPF1 complex inform about its biological properties and importance in transcription activation. (A) Protein domains of the MOZ-BRPF1 complex; MOZ, BRPF1, ING4/5 and MEAF6. Domains labelled as follows: NEMM, N-terminal part of Enok, MOZ and MORF; PHD, plant homeodomain-linked zinc finger; MYST, MYST histone acetyltransferase domain; Zn, zinc knuckle; I and II, Epc-homology region I and II; Bromo, bromo domain; PWWP, proline-tryptophan-tryptophan-proline-containing domain; LZ leucine zipper. Numbers correspond to total residues that each protein possesses. MOZ paralog MORF is indicated in parenthesis as it has the same domain structures/arrangement. (B) Predicted MOZ-BRPF1 complex orientation. Scaffolding subunit BRPF1 connects MOZ to ING4/5 and MEAF6. Experimentally determined histone marks/chromatin and DNA binding sites of the MOZ-BRPF1 complex are indicated. Red dots represent methylated histones and blue dots acetylated histones. Read arrow indicates acetyltransferase substrates of the complex. TF, transcription factor. (C) The MOZ-BRPF1 complex in gene activation. The complex binds to active transcription sites via acetylated H4 and/or methylated H3K4 and H3K36 and/or unmethylated CpG islands (left) or via transcription factors (TF) (right). After binding to specific chromatin regions, MOZ acetylates targeted histone lysines, which leads to opening of chromatin and induced gene expression. Individual histones in nucleosomes are indicated with different colors (upper left corner). DNA is represented by the black line Created with BioRender.com.
The most studied domains of MOZ/MORF are the DPF and MYST domains in the N-terminal part of the proteins. The DPF domain was first found to interact with the N-terminal part of histone H3 tail and helps the protein to localize to chromatin. Surprisingly, DPF interaction with the H3 tail strengthens when either H3K9 or H3K14 are acetylated but is crippled if H3K4 is modified/methylated (Ali et al., 2012) (Figure 1B). It seems that H3R2 must also be unmodified for DPF to bind H3K14ac (Qiu et al., 2012). Further structural studies have shown that DPF with the help of the MYST domain induces an α-helical conformation of H3K4-T11, which revealed a unique way of H3 recognition (Dreveny et al., 2014). It was suggested that this induced helical structure explains how MOZ recognizes the H3K4 methylation status and facilitates its own binding to selected chromatin regions. More recently, biochemical, structural and ChIP data argued that DPF allows a specific cross-talk in vivo, recognizing H3K14ac deposited by another HAT, HBO1/KAT7, to induce local H3K23 acetylation by MOZ/MORF (Klein et al., 2019). Importantly, DPF can associate with different forms of lysine acylation on histones and has a strong preference for H3K14 crotonylation (H3K14cr) (Tan et al., 2011; Xiong et al., 2016; Klein et al., 2017).
The first in vitro HAT assays with the MYST domain of MOZ showed acetylation of free histones H3, H4 and H2A, but it was not able by itself to acetylate nucleosomes (Kitabayashi et al., 2001a; Champagne et al., 2001) (Figure 1B). Structural studies with MOZ MYST domain revealed that it contains more divergent N- and C-terminal regions and possesses helix-turn-helix (HTH) DNA-binding motifs that can enhance catalytic activity (Holbert et al., 2007). Interestingly, a small region at the C-terminus of the MYST domain is needed to associate with BRPF1, the scaffolding platform of the 4-subunit complex (Ullah et al., 2008). Binding to BRPF1 enables MOZ to acetylate histone H3 tails within nucleosomes. BRPF1 also enhanced the transcription coactivator activity of MOZ, which indicates that complex formation helps MOZ to bind to specific genomic regions to activate transcription. As mentioned above, MOZ/MORF acetylates H3K23 and can be influenced by DPF recognition of H3K14 acylation (Klein et al., 2019). Interestingly, a recent study suggests that MOZ could prefer to propionylate this histone residue (Yan et al., 2017; Yan et al., 2020).
It has been suggested that a small region after the MYST domain of MOZ could bind to ENL, a common MLL protein fusion partner in mixed linage leukemia (Miyamoto et al., 2020). As this interaction was observed in chromatin fractions, it remains to be verified if ENL binds directly to MOZ or if they only occupy the same chromatin regions, as native purification from soluble nuclear extracts did not detect such interaction (Feng et al., 2016; Klein et al., 2019). Additionally, it has been suggested that MOZ would directly interact with MLL itself, so ENL could also associate with MOZ via MLL interaction (Paggetti et al., 2010; Miyamoto et al., 2020), or that ENL acts downstream of MOZ through recognition by its YEATS domain of H3K9ac deposited by the HAT (Yan et al., 2022). It is important to note that NuA3, the homologous HAT complex in budding yeast, was shown to associate with TAF14, a YEATS domain-containing protein that also recognizes H3K9ac (John et al., 2000; Shanle et al., 2015).
As mentioned above, BRPF1 is the scaffolding subunit of MOZ/MORF acetyltransferase complexes binding simultaneously MOZ or MORF, ING4/5 and MEAF6 (Figure 1A) (Ullah et al., 2008; Klein et al., 2014; Feng et al., 2016; Klein et al., 2019). This protein also has multiple functional domains such as a PHD-Zn knuckle-PHD (PZP) domain, a bromodomain and a chromo/Tudor-related Pro-Tyr-Tyr-Pro (PWWP) domain.
BRPF1 has been shown to able to bind both MOZ/MORF and HBO1 through its N-terminal region (domain I) (Ullah et al., 2008; Lalonde et al., 2013) but purification in native conditions indicates that the vast majority of BRPF1 is associated with MOZ/MORF in vivo (Feng et al., 2016). A small region between the PZP and bromodomain is required for association of ING4/5 and MEAF6 (Ullah et al., 2008; Lalonde et al., 2014) (domain II) (Figure 1A). Other domains of BRPF1 are epigenetic reader domains, which allow the protein to interact with chromatin. While BRPF1 PWWP domain is not important for acetyltransferase activity (Lalonde et al., 2013), it can recognize the H3K36me3 histone mark that is present on transcribed regions (Vezzoli et al., 2010) and seems required for BRPF1 to associate with chromatin regions in vivo (Laue et al., 2008). BRPF1 bromodomain can recognize histone acetylation marks including H2AK5ac, H4K12ac, H4K8ac, H4K5ac, and H3K14ac (Poplawski et al., 2014) (Figure 1B). It seems to have highest affinity towards H2AK5ac, followed by H4K12ac and H3K14ac. The PZP domain in BRPF1 is highly conserved in BRPF2/3 and JADE1/2/3 proteins associated with HBO1 complexes (Saksouk et al., 2009; Mishima et al., 2011; Avvakumov et al., 2012; Feng et al., 2016). Biochemical and structural studies with BRPF1 PZP domain indicate that both PHDs are required for the MOZ-BRPF1-ING5-MEAF6 complex to bind histone H3 in vivo and acetylate chromatin substrates in vitro. PHD2 assists PHD1 by allowing tight binding to a nucleosome with extra-nucleosomal linker DNA, while PHD1 locks in histone H3 N-terminus (Lalonde et al., 2013; Klein et al., 2016; Klein et al., 2020). Interestingly, the extreme N-terminus of BRPF1 also seems to have a role in the acetylation of chromatin substrates, driving the histone H3 tail specificity (Lalonde et al., 2013).
Although ING4/5 and MEAF6 do not directly bind to MOZ (Figure 1A), they were found to increase and specify the acetyltransferase activity of MOZ when compared to the MOZ-BRPF1 dimer (Ullah et al., 2008). ING4/5 proteins also contain a PHD domain specific for the H3K4me3 mark found around the transcription start sites of genes in vitro and in vivo (Shi et al., 2006; Champagne et al., 2008; Lalonde et al., 2013). Furthermore, the HAT activity of the ING5-containing MOZ/MORF complexes is greatly stimulated on H3K4me3 peptides when compared with unmodified or H3K9me peptides (Lalonde et al., 2013). This suggests that the PHD domain of ING4/5 not only guides the MOZ/MORF-BRPF1 complexes to active transcription sites, but it also enhances the acetyltransferase activity of the complexes at these sites.
MEAF6 function is much less understood while conserved from yeast to humans, essential for cell viability (depmap.org) and stoichiometric subunit of MOZ, MORF, HBO1 and NuA4/TIP60 HAT complexes. However, its role in the MOZ-BRPF1 complex does not seem to be as important as ING4/5 (Doyon et al., 2006; Ullah et al., 2008). Structural studies showed that MEAF6 associates with ING proteins and domain II of the scaffold subunit through three intertwined helices (Xu et al., 2016). Further studies are needed to understand the function of this small subunit in HAT complexes containing ING subunits.
The biochemical properties of the MOZ-BRPF1 acetyltransferase complex have shown that it has multiple chromatin binding/reader domains and several mechanisms for its recruitment to chromatin have been proposed. For example, ING4/5 and BRPF1 seem to recruit the complex to active transcription sites via binding to H3K4me3 (Lalonde et al., 2013) or H3K36me3 (Vezzoli et al., 2010), whereas the C-terminal part of MOZ has been shown to associate with different transcription factors (Kitabayashi et al., 2001a; Pelletier et al., 2002; Rokudai et al., 2009; Aikawa et al., 2010) possibly targeting the complex to a small subset of specific genes (Figure 1C). Being able to recognize specific histone PTMs indicates that localization of the MOZ-BRPF1 complex is regulated by other histone modifying complexes, e.g. HBO1-dependent H3K14ac recognized by MOZ DPF reader domain (Klein et al., 2019). This diversity of possible recruitment/regulatory mechanisms has made it difficult to fully understand the biological functions of this complex. Therefore, it has been important to conduct knockdown and knockout experiments of individual complex partners in vivo to pinpoint their actual role in biological processes. Careful examination of the biological processes will also help to identify how these complexes might behave in different pathological situations like cancer.
The first MOZ knockout studies were done in zebrafish, where MOZ was found to be an important regulator of developmental HOX genes (Miller et al., 2004; Crump et al., 2006) (Figure 2). HOX gene clusters are known to encode a family of transcriptional regulators that elicit distinct developmental programs along the head-to-tail axis of animals (Mallo and Alonso, 2013). Depletion of MOZ leads to defects in the zebrafish facial skeleton and lowers the expression levels of HOXB genes, which are known to be crucial regulators of head formation in zebrafish (Miller et al., 2004; Crump et al., 2006). Acetylation of histones by MOZ seems to be important in HOX gene regulation as inhibition of histone deacetylases (HDACs) rescues the phenotype. Mechanistic studies pinpointed that MOZ supports the skeletal development in the head (Crump et al., 2006). BRPF1 knockout studies in zebrafish follow the same trend as MOZ, which suggests that BRPF1 does not have MOZ-independent functions in skeletal development (Laue et al., 2008; Hibiya et al., 2009). This mechanism seems to be conserved as defects in craniofacial and caudal skeletons were also found in MOZ KO mice (Hibiya et al., 2009; Voss et al., 2009). In mice, MOZ seems to have a global effect on Hox gene expression and specification of the anterior expression boundary of Hox genes (Hoxc8/9). MOZ depletion diminishes the H3K9 acetylation mark of specific Hox genes in chromatin immunoprecipitation (ChIP) assays, without affecting the global levels of H3K9Ac (Voss et al., 2009). Thus, the MOZ-BRPF1 complex seems to regulate the transcription of HOX genes via H3K9 acetylation. Interestingly, ING5 association to HOX genes seems to be dependent on MOZ, suggesting that the complex is recruited via MOZ. It has also been shown that MOZ and BMI1, which is part of polycomb repressive complex 1 (PRC1), work simultaneously during the regulation of Hox genes in axial skeleton development (Sheikh et al., 2015a). Deletion of Moz or Bmi1 lead to opposing defects in axial skeleton development. However, if both are mutated, the mice shows a morphologically normal axial skeleton. Further studies showed that these two complexes indeed have opposing roles as PRC1 is required to prevent premature activation of Hox genes, while MOZ-BRPF1 is needed to activate and maintain normal levels of Hox gene expression after the initial activation phase. The role of MOZ in craniofacial development has been studied with full and conditional Moz knockout (KO) mice (Voss et al., 2012; Vanyai et al., 2019). Studies with the conditional KO showed that MOZ occupies and regulates H3K9 acetylation levels at distal-less homeobox (DLX) transcription factor-encoding Dlx5 gene loci and lowered expression of all DLX family genes and their targets (Vanyai et al., 2019). While conditional KO embryos showed downregulation of DLX transcription factors, there was upregulation of Runx2 and other bone-specific downstream effectors possibly leading to premature osteoblast lineage differentiation, causing craniofacial anomalies. Both DLX5 and MOZ have been previously linked with the RUNX2 transcription factor (Pelletier et al., 2002; Lee et al., 2003), leading Vanyai et al. (2019) to suggest a molecular mechanism where MOZ together with DLX5 regulate osteoblast development via suppression of RUNX2. Studies with the KO mice also showed that MOZ is involved in the development of the palate, facial structures, and is also linked to defects in the thymus and the cardiovascular system (Voss et al., 2012). Interestingly, the defects in the cardiovascular system closely resemble those seen in the DiGeorge syndrome (DGS). DGS is caused by mutations in the T-box 1 (TBX1) gene and further studies concluded that MOZ regulates TBX1, TXB2 and TXB5 expression via maintaining the H3K9ac levels at their loci. MOZ was shown to activate TBX1 and TBX5 in mesodermal cells during heart development (Vanyai et al., 2015).
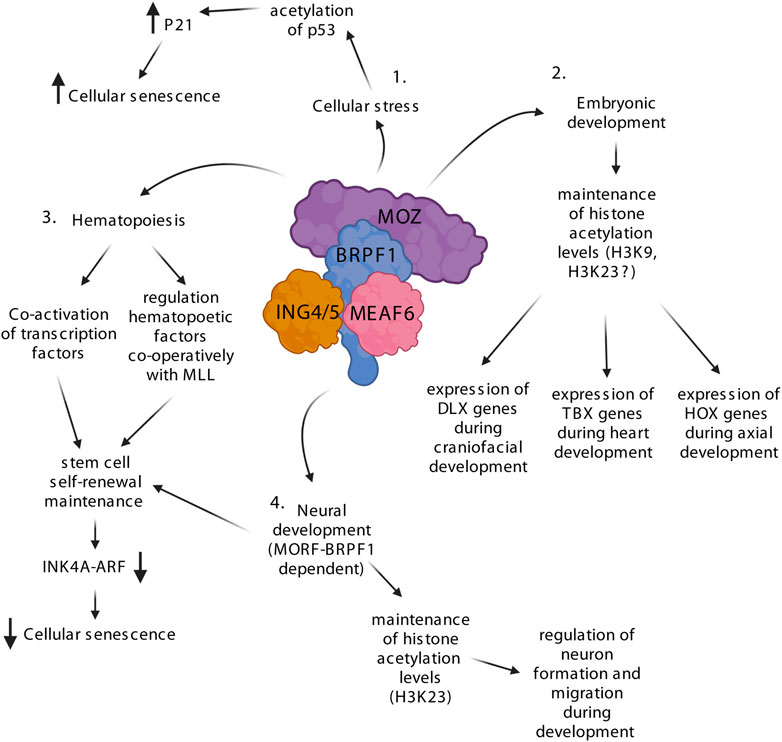
FIGURE 2. The MOZ-BRPF1 complex regulates various steps of development via controlling H3 acetylation levels. 1) Upon cellular stress, MOZ drives cellular senescence by interacting with PML-p53, which enhances p53 acetylation leading to expression of the p21 senescence factor. 2) MOZ is involved in embryogenesis by controlling the expression of multiple essential transcription factor families such as homeobox (HOX), T-box (TBX) and distal-less homeobox (DLX). Knockout experiments with MOZ show a decrease of H3K9ac levels at these genes, suggesting that the MOZ-BRPF1 complex regulates H3K9ac during development. H3K23ac is labeled with a question mark since its levels have not been fully addressed during MOZ KO studies. 3) MOZ and BRPF1 are crucial regulators of hematopoietic and neuronal stem cells and precursors. The MOZ-BRPF1 complex inhibits premature senescence of stem cells by downregulating the INK4a/ARF pathway. 4. Depletion of BRPF1 leads to decreased formation and motion of neurons causing neurodevelopmental defects. Loss of H3K23ac in the dorsal cortex of BRPF1 KO mice indicates that MOZ-BRPF1 or MORF-BRPF1 regulate this acetylation mark during neuronal development. Created with BioRender.com.
As MOZ was first found as a fusion partner of CBP produced by chromosomal translocation found in AML patients (Borrow et al., 1996), it suggested that it may be involved in hematopoiesis (Figure 2). Indeed, two independent knockout studies in mice showed that MOZ depletion impairs hematopoiesis and causes embryonic lethality around E14.5 (Katsumoto et al., 2006) or just before birth (Thomas et al., 2006), depending on the knockout mice strain. Different phenotypic analyses revealed that, although the precursors for all hematopoietic lineages and hematopoietic stem cells (HSCs) were present in MOZ KO mice, their amount and functionalities were dramatically reduced (Katsumoto et al., 2006; Thomas et al., 2006). Similar defects were found in mice carrying a point mutation in the catalytic site of MOZ, indicating that the acetyltransferase activity is required for efficient hematopoiesis (Perez-Campo et al., 2009). In parallel, BRPF1 has an important role in MOZ-mediated acetylation in HSCs as its depletion leads to dramatic reduction of H3 acetylation in bone marrow (You et al., 2016). MOZ has also been shown to be needed for B cell proliferation (Sheikh et al., 2015b). Moreover, conditional MOZ KO mice have reduced immune response and alterations in memory development of B cells (Good-Jacobson et al., 2014) and T cells (Newman et al., 2016). The role of MOZ in hematopoiesis is also supported by earlier biochemical studies showing that it can interact and activate hematopoietic transcription factors RUNX1 and PU.1 (Kitabayashi et al., 2001a; Bristow and Shore, 2003; Katsumoto et al., 2006). Mechanistically, MOZ seems to regulate important hematopoietic factors such as HOX genes, MIP-1α, c-Mpl, Meis1 and c-Kit as they are downregulated in KO/KD mice (Katsumoto et al., 2006; Sheikh et al., 2015b).
MOZ has been implicated with histone methyltransferase MLL in the regulation of HOX genes in hematopoietic cells (Paggetti et al., 2010). MLL is known to be important for HSC self-renewal (McMahon et al., 2007) and it regulates HOX genes by methylating H3K4, which promotes active transcription (Milne et al., 2002). In human cord blood cells, MOZ and MLL bind to the same promoter regions of various HOX genes and deletion of one or the other leads to lower levels of acetylated H3 as well as methylated H3K4 (Paggetti et al., 2010). These findings suggest a positive feedback loop where MOZ-mediated H3 acetylation favors MLL-dependent H3K4 methylation and vice versa, leading to increased transcription. This is supported in part by the fact that pre-acetylated H3 is a better substrate for methylation by MLL in vitro (Milne et al., 2002; Jain et al., 2022). Since most of the downregulated factors, including MLL, are linked to self-renewal and repopulating potential of HSCs, MOZ has likely a more profound role in the expansion and proliferation of early hematopoietic precursors. Indeed, MOZ can prevent HSCs and mouse embryonic fibroblasts (MEFs) from entering senescence via regulation of the INK4a/ARF pathway (Perez-Campo et al., 2014; Sheikh et al., 2015c). Upon MOZ depletion, the INK4a/ARF pathway is upregulated and studies in MEFs showed that MOZ maintains H3K9ac levels and expression of the INK4a/ARF pathway inhibitors CDC6, EZH2 and E2F2 (Sheikh et al., 2015c). In a related context, MOZ has already been linked to stress-induced cellular senescence, as it regulates expression of p21 and increases premature senescence through acetylation of TP53 (Rokudai et al., 2009; Rokudai et al., 2013). Thus, MOZ seems to either activate or prevent cellular senescence depending on the context. In addition, MOZ has also been implicated in maintaining the quiescence of adult HSCs (Sheikh et al., 2016; Sheikh et al., 2017), a mechanism essential to prevent HSCs exhaustion that could lead to long-term loss of stem cells. MLL and MOZ likely work again together in regulating the HSC quiescence as MLL is required to self-renew and maintain a non-cycling population of adult HSCs (Jude et al., 2007).
The knockout studies in zebrafish demonstrated that depletion of either MOZ or BRPF1 leads to similar defects and decreased in HOX gene expression, suggesting strictly overlapping roles in that organism (Miller et al., 2004; Crump et al., 2006; Laue et al., 2008; Hibiya et al., 2009). However, BRPF1 KO mice suffer more severe developmental issues and earlier embryonic lethality around E9.5 (You et al., 2014). During embryonic development, high BRPF1 expression is seen in the neuroepithelial cells of the neural tube and the headfold (You et al., 2014) while MOZ is expressed at low levels in these regions (Thomas et al., 2006). These findings indicate that BRPF1 could have MOZ independent functions during development. Indeed, BRPF1 KO mice embryos show various defects that are not seen in the MOZ KO (You et al., 2015a). Early embryonic lethality of the BRPF1-depleted mice can be explained by the blood vessel defects in placenta, which interrupt the nutrient and gas exchange between the embryonic and maternal circulation system. Also, the neurodevelopmental defects in the BRPF1 KO embryos are severe as the neural tube remains open (Figure 2). Immunostaining showed that formation and migration of neurons are compromised in the mutant embryos. Moreover, these defects are due to cell proliferation issues rather than apoptosis or DNA damage. Abnormal neuronal migration and cell cycle progression is also observed during brain and hippocampus development when BRPF1 is specifically depleted from the forebrain (bKO) (You et al., 2015b; You et al., 2015c). Behavioral tests with the BRPF1 bKO indicate that the mice suffer neocortical development problems, which commonly result in poor muscle motor coordination and learning disabilities. These findings indicate that BRPF1 has a crucial role in neuronal development. MOZ and MORF have been shown to regulate neuronal stem cell renewal (Sheikh et al., 2012; Perez-Campo et al., 2014) and loss of MORF causes neurological defects in mice (Thomas et al., 2000). Therefore, Since BRPF1 forms independent HAT complexes with MOZ and MORF, and possibly a small population of HBO1, its loss impacts the function of both (Doyon et al., 2006; Ullah et al., 2008; Lalonde et al., 2013; Feng et al., 2016; Klein et al., 2019). Based on biochemical studies BRPF1-MOZ/MORF prefers to acetylate H3K23, whereas BRPF1-HBO1 complex could acetylate H3K14 (Kueh et al., 2011; Lalonde et al., 2013; Feng et al., 2016; Yan et al., 2017; Klein et al., 2019). Accordingly, bone marrow cells of BRPF1 KO mice show lower levels of H3 acetylation at lysines 9, 14, and 23 (You et al., 2016). Although a small population of HBO1 could associate with BRPF1, it strongly prefers BRPF2/3 as binding partners, and loss of BRPF2 or BRPF3 have been linked with total loss of H3K14ac (Mishima et al., 2011; Feng et al., 2016; Yan et al., 2016). Moreover, studies with BRPF1 KO and heterozygote knockdown mice have shown total loss or reduced levels of H3K23ac in multiple tissues, indicating that BRPF1 solely governs specific H3K23 acetylation via MOZ/MORF in different tissues and cells (Yan et al., 2017). Therefore, loss of BRPF1 affects embryogenesis via preventing MOZ and MORF to acetylate H3K23.
In summary, while both MOZ and BRPF1 are important factors during embryogenesis (Figure 2), loss of BRPF1 creates more severe defects because of its function to enable chromatin binding and acetylation by both HAT enzymes. While MOZ binding to specific transcription factors allows the recruitment of the MOZ-BRPF1 complex to specific genomic loci, BRPF1 and ING4/5 are needed for efficient DNA and histone binding on local chromatin and acetyltransferase activity. A reduction in H3K9ac at specific target loci is observed in MOZ KO mice (Voss et al., 2009; Sheikh et al., 2015c; Vanyai et al., 2019). This is in contrast to biochemical and KD experiments in cells indicating that H3K23 is the major acetylation target of MOZ (Feng et al., 2016; Lv et al., 2017; Yan et al., 2017; Klein et al., 2019). Most recently, reduced H3K23 propionylation was also reported in MOZ KO embryo, indicating that MOZ can also performed other forms of lysine acylation in vivo (Yan et al., 2020), but H3K9 was not measured. Interestingly, antibody staining showed high H3K23ac in the E13.5 embryo liver and heart, whereas H3K23 propionylation (H3K23pr) was high in liver and cerebrocortical neuroepithelium. Although these two histone marks are chemically similar and linked with active transcription (Sabari et al., 2017), further studies are needed to address the role of them during development. Importantly, H3K23ac is by far one of the most abundant acetylation site in mammals [along H3K14ac and H4K16ac (Zheng et al., 2013)] and tumor suppressor TRIM24 has been shown to specifically bind H3K23ac with its bromodomain (Tsai et al., 2010). Therefore, it will be interesting to investigate its function alongside MOZ-BRPF1 complex in development. Additionally, studies are also needed to precisely investigate how and when MOZ and MORF may cooperate or play redundant roles during development.
Interestingly, patients with neurodevelopmental disorders have been shown to have mutations in MOZ (Arboleda et al., 2015; Tham et al., 2015; Kennedy et al., 2019) and BRPF1 (Mattioli et al., 2017; Yan et al., 2017; Naseer et al., 2020) (Figure 3A). Neurodevelopmental disorders impair or alter the growth and development of the brain and the central nervous system (Parenti et al., 2020). Transcriptional regulators or chromatin remodelers are often seen to be mutated in patients with neurodevelopmental disorders. Mutations in MOZ are usually heterozygous de novo mutations and they cause a disorder called the KAT6A syndrome (also known as Arboleda-Tham syndrome) (Arboleda et al., 2015; Tham et al., 2015). The core features of the syndrome are impaired intellectual development, significant speech/language deficit, heart defects, hypotonia, distinct facial features and gastrointestinal complications. Mechanistically, it was shown that H3K9ac is decreased in the fibroblasts collected from the patients, which is consistent with the KO/KD mice studies (Voss et al., 2009; Sheikh et al., 2015b). Truncating, frameshift, and non-sense mutations are the most common mutations in KAT6A syndrome patients, but missense mutations and mutations occurring at splice sites have also been mapped in some patients (Tham et al., 2015; Millan et al., 2016; Kennedy et al., 2019). Mutations in the MYST domain are rare as they are mostly found in those exons that encode the glutamate/aspartate-rich and serine/methionine-rich domains (Tham et al., 2015; Kennedy et al., 2019). Remarkably, patients with distal exon (16–18) mutations exhibit more severe symptoms than those with mutations in more proximal exons 1–15 (Kennedy et al., 2019). These findings indicate that the C-terminal part of MOZ, which is responsible for binding various transcription factors (Kitabayashi et al., 2001a; Pelletier et al., 2002; Katsumoto et al., 2006; Rokudai et al., 2009; Rokudai et al., 2013), is crucial for proper development in humans. Interestingly, while these monoallelic mutations in MOZ cause developmental anomalies (Tham et al., 2015; Kennedy et al., 2019), similar phenotypes are not observed in mice heterozygous MOZ KO (Voss et al., 2012; Vanyai et al., 2015; Vanyai et al., 2019). This suggests that the truncated MOZ mutant acts as dominant-negative. Another thing that needs to be pointed out is that the patients do not suffer from hematological abnormalities, although MOZ has an especially important role regulating the hematopoietic stem cells in mice (Katsumoto et al., 2006; Thomas et al., 2006). These issues require further studies to be fully understood. Interestingly, mutations in MORF, the MOZ paralog, have been linked to distinct developmental disorders with broad clinical spectrum called KAT6B disorders, which include the Genitopatellar syndrome and the Say-Barber-Biesecker-Young-Simpson syndrome (Zhang et al., 2020).
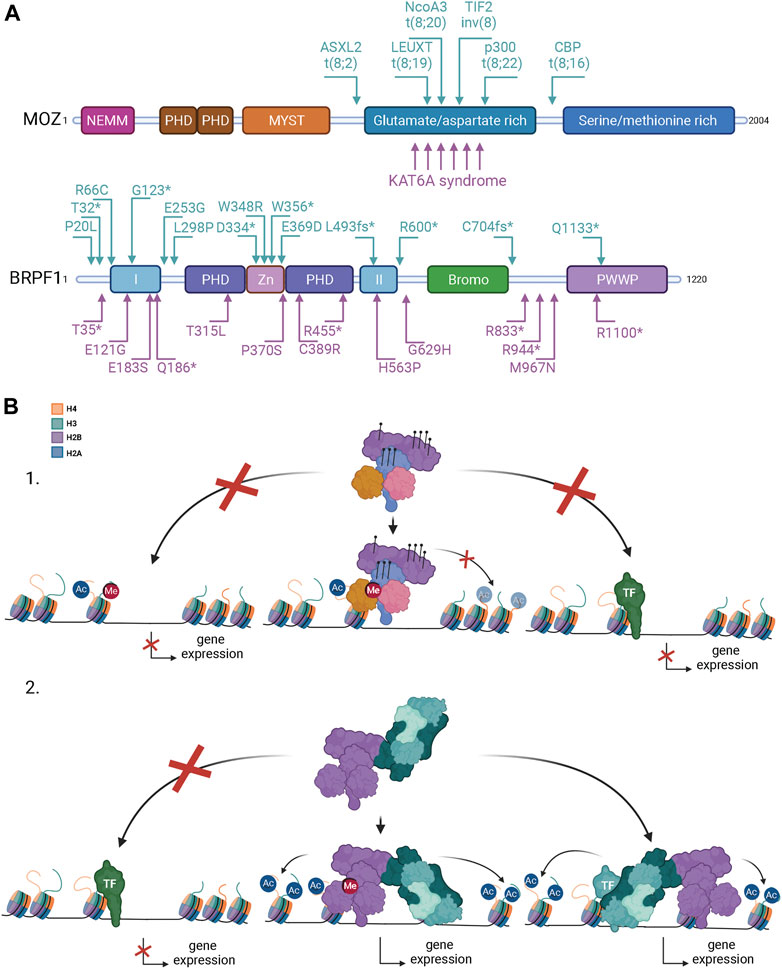
FIGURE 3. Mutations in MOZ and BRPF1 lead to developmental disorders and different forms of cancer. (A) MOZ is recurrently mutated in leukemia, non-hematologic malignancies, and developmental disorders with the common characteristics of intellectual disability and developmental delays. Arrows above the MOZ protein structure point to leukemia-associated translocations, the corresponding fusion partners being indicated. KAT6A syndrome refers to MOZ-specific intellectual disability disorders. For simplicity, only a few arrows below are used to illustrate mutation positions related to the KAT6A syndrome [refer to published reports for complete list of mutations (Arboleda et al., 2015; Tham et al., 2015; Kennedy et al., 2019)]. BRPF1 mutations are linked to developmental delays and cancer. Arrows above the BRPF1 protein structure indicate mutations associated with cancer (Huether et al., 2014; Kool et al., 2014; Aiello et al., 2019). Arrows below BRPF1 indicate mutations from patients with developmental disorders [modified from (Yan et al., 2020)]. Used abbreviations: CBP, CREB-binding protein; p300, E1A-associated p300 kDa protein; TIF2, transcription intermediary factor 2, ASXL2, additional sex combs-like 2; NcoA3, nuclear receptor co-activator three; leucine twenty homeobox, LEUTX. fs, reading frame shift; *, translational termination. Domain abbreviations for MOZ and BRPF1 are as in Figure 1. (B) Possible mechanisms how MOZ-BRPF1 mutations and translocations can change gene expression. 1) Mutations in the epigenetic reader domains of MOZ or BRPF1 can prevent chromatin binding on specific regions and alter gene expression (left). Mutations in the MOZ MYST domain or BRPF1 PZD domain can decrease or block the acetyltransferase activity of the complex leading to repressing chromatin structure unavailable for transcription (middle). Mutations in the C-terminal part of MOZ disrupts binding of specific (light green) transcription factors (TFs) blocking activation of genes (right). 2) Rearragements of the MOZ gene fuse the N-terminal of MOZ (indicated in A) with a fusion partner. Loss of MOZ C-terminus disrupts the complex binding to specific TFs (light green) blocking gene activation (left). The MOZ fusion protein can form a functional complex and binds to active transcription sites via BRPF1 and ING4/5 (middle). This can cause overexpression or silencing of target genes depending on the fusion partner. The MOZ complex can be recruited to fusion partner specific chromatin sites via TFs (teal) or other mechanisms leading to abnormal histone acetylation and gene expression in these sites (right) Created with BioRender.com.
BRPF1 mutations are linked to patients showing intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) (Mattioli et al., 2017; Yan et al., 2017; Demeulenaere et al., 2019; Pode-Shakked et al., 2019; Naseer et al., 2020). IDDDFP is an autosomal dominant neurodevelopmental disorder characterized by delayed motor and language development, intellectual disability, and dysmorphic facial features. The defects are milder but consistent with the BRPF1 KO studies in mice, underlining the importance of BRPF1 during neurodevelopment (You et al., 2015a; You et al., 2015b; You et al., 2015c). Similar to MOZ mutations, patients with BRPF1 mutations do not show hematological abnormalities (Mattioli et al., 2017; Yan et al., 2017; Demeulenaere et al., 2019; Pode-Shakked et al., 2019; Naseer et al., 2020; Yan et al., 2020), although loss of BRPF1 leads to acute bone-marrow failure in mice (You et al., 2016). In addition, while patients carry monoallelic mutations, no obvious phenotypes have been observed in heterozygous BRPF1 KO mice (You et al., 2015a; You et al., 2015b; You et al., 2015c). C-terminal truncating mutations are the most common ones and patients with truncations that disrupt MOZ/MORF binding or the PZP domain are considered the most severe (Mattioli et al., 2017; Yan et al., 2017; Demeulenaere et al., 2019; Pode-Shakked et al., 2019; Naseer et al., 2020; Yan et al., 2020). Biochemical and in vivo studies showed that disorder-linked BRPF1 mutations in the PZP domain block MOZ-dependent H3K23 acetylation (Mattioli et al., 2017; Yan et al., 2017). Slight decrease of H3K23ac was also seen in the histones extracted from patient fibroblasts, while global H3 acetylation levels is unchanged (Mattioli et al., 2017). Interestingly, lower H3K23ac levels are also observed in heterozygous BRPF1 KO mice, supporting the concept that heterozygous BRPF1 PZP mutations in humans can decrease H3K23ac levels mediated by MOZ and MORF (Yan et al., 2017).
All subunits of the MOZ-BRPF1 complex have been linked to tumorigenesis. ING4/5 are part of ING family of tumor suppressors also implicated in TP53 function (Dantas et al., 2019). Translocations within the MEAF6 gene are linked with endometrial stromal sarcoma (Antonescu et al., 2014; Micci et al., 2014). BRPF1 has been implicated as tumor suppressor in childhood leukemia (Huether et al., 2014), prostate cancer (Sole et al., 2020) and medulloblastoma (Kool et al., 2014). Recently, truncated BRPF1, which is found in human medulloblastoma patients, was shown to promote the medulloblastoma formation with SmoM2 from postmitotic neurons in adult mice (Aiello et al., 2019). Since the BRPF1 expression levels are significantly lower in medulloblastoma when compared to normal tissue, it supports a tumor suppressor function. In contrast, BRPF1 is frequently overexpressed in hepatocellular carcinoma (HCC) (Cheng et al., 2021). Inhibition and knockout studies with BRPF1 in HCC showed that its overexpression induces tumor migration and proliferation, acting like an oncogene in this specific cancer. Thus, the potential role of BRPF1 needs to be addressed individually between different cancers.
Although all subunits of the MOZ-BRPF1 complex have been linked to cancer, prevalent studies are related to the functions of MOZ in leukemia. In hematologic malignancies, MOZ has been frequently shown to form fusion proteins produced by chromosomal translocations (Figure 3A). Identified fusion partners for MOZ are CREB binding protein (CBP) (Borrow et al., 1996), E1A Binding Protein P300 (p300) (Chaffanet et al., 2000; Kitabayashi et al., 2001b), transcriptional intermediary binding factor 2 (TIF2) (Carapeti et al., 1998), additional sex combs-like 2 (ASXL2) (Imamura et al., 2003), nuclear receptor co-activator 3 (NcoA3) (Esteyries et al., 2008) and leucine Twenty Homeobox (LEUTX) (Chinen et al., 2014; Sramkova et al., 2020; Panagopoulos et al., 2021). MOZ fusions are mostly found in AML, which originates from primitive populations of HSCs and progenitor cells (Roussel et al., 2020). In normal hematopoiesis, these primitive populations represent a suppressed cell reservoir, which are needed for production of mature circulating blood cells. However, genetic alterations can either reignite the self-renewal capacity of the progenitors or induce uncontrolled self-renewal of the HSCs. Predominantly, these genetic alterations cause abnormal accumulation of non-differentiating cells (leukemic stem cells, LSCs) in the bone marrow, which is diagnosed as AML. Among these fusion proteins, MOZ-TIF2 has been studied the most, shown to have transforming activity in cultured cells and to induce AML in mice (Deguchi et al., 2003). MOZ-TIF2 transforming activity relies on the fusion protein ability to recruit CBP via TIF2 to MOZ specific binding sites. MOZ DNA binding capacity through its HTH motifs seemed to be crucial for the ability of MOZ-TIF2 to induce AML in mice (Deguchi et al., 2003). However, the double mutation Q654E/G657E in MOZ acetyl-CoA binding site prevents the formation of AML (Shima et al., 2014). This indicates that DNA binding as well as histone acetyltransferase activity are required for inducing AML in mice. In addition, the translocation hotspot of MOZ is in the glutamate/aspartate-rich domain after the MYST HAT domain (Figure 3A). Therefore, the MOZ fusion proteins retain the N-terminal moiety of the protein, containing NEMM, DPF and MYST domains. Biochemical studies have shown that the MOZ-TIF2 fusion can form the functional complex with BRPF1, MEAF6 and ING4/5 (Ullah et al., 2008). Association with BRPF1 enables transcription activation and induced colony formation by MOZ-TIF2 (Shima et al., 2014). This indicates that complex formation is required for leukemic activity of the fusion proteins (Figure 3B). MOZ-TIF2 uses the same INK4a/ARF pathway to escape senescence as MOZ (Largeot et al., 2016) and it can also deplete CBP from PML bodies to inhibit transcriptional activity of p53 and the retinoic acid receptor RAR (Kindle et al., 2005; Collins et al., 2006). Later studies have shown that MOZ-TIF2 can promote self-renewal of the committed progenitor cells, which allows preleukemic expansion of cells in the bone marrow facilitating the development of AML (Huntly et al., 2004). MOZ-TIF2 transforming potential can be partly suppressed by knocking down the hematopoietic transcription factor PU.1 (Aikawa et al., 2010). These results support the idea that MOZ fusion proteins can promote self-renewal to produce LSCs. Gene expression profiling of MOZ-CBP AML patients showed that this fusion produces a pattern of HOX expression characteristic of self-renewal, with upregulation of HOXA9, HOXA10, cofactor MEIS1 and marked downregulation of other homeobox genes (Camos et al., 2006). This profile partially resembles that of AML with MLL rearrangement, which may indicate that MLL and MOZ fusion proteins share gene targets for uncontrolled self-renewal. A recent study with MOZ and MLL fusions support this idea, showing that MOZ fusions and MLL can be recruited to unmethylated CpG-rich promoters to induce expression of leukemic genes (Miyamoto et al., 2020). In MLL-rearranged leukemia, MLL fusions rely on the AF4-ENL-P-TEFb complex (AEP/super elongation complexes) for transcriptional activation and DOT1L for transcriptional maintenance to fully transform hematopoietic progenitors to LSCs (Okuda et al., 2017). Miyamoto et al. (2020) claim that, in MOZ-rearranged leukemia, MLL is the recruiter of MOZ to specific genes as inhibition of MLL or DOT1L complex decreases the self-renewal capacity of leukemic stem cells produced by MOZ-TIF2. Mechanistically, the MOZ-CBP fusion seems to function in a slightly different manner, while both induce self-renewal capacity of cells (Kitabayashi et al., 2001a; Camos et al., 2006; Chan et al., 2007). While MOZ fusions drive leukemia by promoting self-renewal capacity of LSCs in different ways, implicating MLL, recent work demonstrates that MLL fusions themselves require endogenous MOZ function to induce AML, in part through ENL H3K9ac reader (Katsumoto et al., 2022; Yan et al., 2022). Cooperation between MOZ and MLL in oncogenic pathways has also been uncovered in gastrointestinal stromal tumors (Hemming et al., 2022).
In addition to leukemia, MOZ have been found abnormally expressed in other cancer types such as glioblastoma (Lv et al., 2017), medulloblastoma (Northcott et al., 2009), breast cancer (Yu et al., 2017), colorectal cancer (Mohammadi et al., 2019; Xie et al., 2020), uterine cervix cancer (Zack et al., 2013; Jiang et al., 2020), ovarian cancer (Zack et al., 2013; Liu et al., 2021), lung cancer, bladder cancer and several adenocarcinomas (Jiang et al., 2020). In most cancers MOZ expression is amplified. For example, lymphoma progression is highly dependent on MOZ expression, as depletion of one MOZ allele in mice can arrest transplantation-driven lymphoma development (Sheikh et al., 2015b). Recent studies on glioblastoma multiforme (GBM), ovarian, colorectal and breast cancers have dissected the molecular mechanisms used by MOZ to promote proliferation and growth of the tumors (Turner-Ivey et al., 2014; Lv et al., 2017; Yu et al., 2017; Lian et al., 2020; Xie et al., 2020; Liu et al., 2021). MOZ gene is frequently overexpressed in breast cancer samples, correlated with lower survival rates (Turner-Ivey et al., 2014; Yu et al., 2017; Jiang et al., 2020). MOZ is overexpressed in estrogen receptor-positive breast cancers, where it binds and regulates estrogen receptor α expression (Yu et al., 2017). SUM-52 breast cancer cells are known to have MOZ gene amplification (8p11-p12 amplicon), leading to its overexpression (Turner-Ivey et al., 2014). Downregulation of MOZ in these breast cancer cells inhibits tumor growth, supporting its function as an oncogene. Analysis of GBM primary specimens also linked MOZ overexpression with tumor progression and poor prognosis (Lv et al., 2017). MOZ depletion in GBM cell lines showed that high expression increases cell proliferation, migration, colony formation and tumor growth. Mechanistically, MOZ upregulates PIK3CA gene expression in GBM cells, activating the PI3K/ATK signaling pathway. MOZ binds to the PIK3CA promoter and acetylates H3K23, resulting in the recruitment of H3K23ac-specific reader/transcription factor TRIM24 (Lv et al., 2017). Interestingly, another study on colorectal cancer suggested a similar molecular mechanism but linked to YAP transcription/signaling (Xie et al., 2020). In colorectal cancer, MOZ cooperates with the DANCR long non-coding RNA (lncRNA) to bind and acetylate H3K23 at the YAP promoter, leading to the recruitment of TRIM24. Upregulation of YAP enhances proliferation and colony formation. YAP is part of the Hippo signaling pathway and regulates several biological processes including cell proliferation, organ size and cell fate (Pocaterra et al., 2020). Most recently, high expression of MOZ correlates with poor prognosis in ovarian cancer (Liu et al., 2021). MOZ was shown to promote tumor formation and drug resistance by acetylating the E3 ubiquitin ligase COP1. Normally, COP1 ubiquitinates β-catenin and regulates its degradation. Acetylation of COP1 impairs β-catenin ubiquitination, leading to its stabilization and abnormal activation of its signaling pathway, increasing proliferation and metastasis of ovarian cancer cells. Altogether, these studies have implicated MOZ in several crucial oncogenic pathways. It also becomes apparent that TRIM24 binding to the H3K23ac histone mark deposited by the MOZ-BRPF1 complex may become an important step in these oncogenic processes.
Nowadays epigenetics plays a central role in many types of diseases, including cardiovascular diseases, neurological diseases, metabolic disorders and cancer. Because of the reversible nature of epigenetic events, scientists have seen epigenetic factors as potential drug targets for therapies. MOZ and BRPF1 are no exceptions and highly specific inhibitors have recently been developed for both factors. Researchers had already designed potent HDAC inhibitors to manipulate histone acetylation in cancer patients but development of selective HAT inhibitors has been significantly more challenging (Wu et al., 2020). The MYST domain is extremely conserved between all family members, which has made it hard to design inhibitors specific for each HAT. Remarkably, two novel HAT inhibitors, WM-8014 and WM-1119, have been shown to be MOZ/MORF specific (Baell et al., 2018). Precise structural studies with MOZ acetyl-CoA binding pocket showed the inhibitors reversibly compete with acetyl-CoA for binding, thus preventing histone acetylation. In vitro inhibition studies revealed that both inhibitors were approximately 100 times more effective against MOZ and MORF when compared with other HATs. Moreover, they were the first HAT inhibitors showing nanomolar range of biochemical inhibition. In vivo studies showed that the inhibitors are able to prevent cancer growth in a zebrafish model of HCC as well as lymphoma in mice (Baell et al., 2018). Mechanistically, inhibition of MOZ activity leads to cellular senescence. These inhibitors have the potential to be powerful tools to study and treat MOZ-related cancers described in the previous section.
Epigenetic factors containing bromodomains are seen as potential drug targets because efficient and specific inhibition of bromodomain-containing BET proteins like Brd4 has been achieved and shown great anticancer effects in preclinical studies (Perez-Salvia and Esteller, 2017). BRPF1 is one of the three highly conserved BRPF family of proteins, harbouring a histone acetylation-binding bromodomain (Lloyd and Glass, 2018). Although the protein structures are similar, specific BRPF1 inhibitors have been developed. Benzoimidazolone based PFI-4, OF-1 and GSK6853 are BRPF1-specific inhibitors which prevent the protein to bind acetylated histones, affecting chromatin binding affinity (Bamborough et al., 2016; Meier et al., 2017). Studies with bone marrow cells and human primary monocytes showed that PFI-4 and OF-1 repress the transcription of osteoclastogenesis-related factors (Meier et al., 2017). The inhibitors do not affect normal cell growth or proliferation, which implies no high cytotoxic properties. Recent studies with GSK5959 (GSK6853 ortholog) have shown that BRPF1 inhibition can prevent HCC progression in cells and in mice, making these inhibitors possible therapeutic drugs for HCC treatment (Cheng et al., 2021). Another promising avenue is to develop synthetic lethality approaches targeting BRPF bromodomain in parallel to metabolism pathways producing acetyl-CoA, for example the GLUT1 glucose transporter as shown in breast cancer cells (Wu et al., 2019).
A large amount of information has been gathered on the biochemical properties of the MOZ histone acetyltransferase complex. But precise in vivo molecular mechanisms are still largely not clearly understood in terms of recruitment and the function of the abundant H3K23ac mark deposited by the complex. It has become clear that functional studies need to consider the whole protein complex instead of just focusing on the HAT enzyme, as BRPF1 is essential for its activity on chromatin and contains several reader domains for epigenetic marks. The MOZ-BRPF1 complex plays crucial roles in cellular homeostasis, development and different human pathologies. The efficient design of specific drugs targeting MOZ and BRPF1 provides great promise to more precisely dissect their function in vivo but also develop powerful therapeutic avenues to treat patients.
TV wrote the first draft of the review, which was then modified/completed by JC.
This work was supported by a Sigrid Juselius foundation fellowship grant and an Instrumentarium Science foundation grant to TV, and a Canadian Institutes of Health Research foundation grant to JC (FDN-143314). JC held the Canada Research Chair on Chromatin Biology and Molecular Epigenetics.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aiello, G., Ballabio, C., Ruggeri, R., Fagnocchi, L., Anderle, M., Morassut, I., et al. (2019). Truncated BRPF1 cooperates with smoothened to promote adult shh medulloblastoma. Cell Rep. 29, 4036–4052. doi:10.1016/j.celrep.2019.11.046
Aikawa, Y., Katsumoto, T., Zhang, P., Shima, H., Shino, M., Terui, K., et al. (2010). PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat. Med. 16, 580–585. doi:10.1038/nm.2122
Ali, M., Yan, K., Lalonde, M. E., Degerny, C., Rothbart, S. B., Strahl, B. D., et al. (2012). Tandem PHD fingers of MORF/MOZ acetyltransferases display selectivity for acetylated histone H3 and are required for the association with chromatin. J. Mol. Biol. 424, 328–338. doi:10.1016/j.jmb.2012.10.004
Antonescu, C. R., Sung, Y. S., Chen, C. L., Zhang, L., Chen, H. W., Singer, S., et al. (2014). Novel ZC3H7B-BCOR, MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoid tumors--molecular characterization shows genetic overlap with endometrial stromal sarcoma. Genes Chromosom. Cancer 53, 183–193. doi:10.1002/gcc.22132
Arboleda, V. A., Lee, H., Dorrani, N., Zadeh, N., Willis, M., Macmurdo, C. F., et al. (2015). De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am. J. Hum. Genet. 96, 498–506. doi:10.1016/j.ajhg.2015.01.017
Avvakumov, N., and Cote, J. (2007). The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 26, 5395–5407. doi:10.1038/sj.onc.1210608
Avvakumov, N., Lalonde, M. E., Saksouk, N., Paquet, E., Glass, K. C., Landry, A. J., et al. (2012). Conserved molecular interactions within the HBO1 acetyltransferase complexes regulate cell proliferation. Mol. Cell Biol. 32, 689–703. doi:10.1128/MCB.06455-11
Baell, J. B., Leaver, D. J., Hermans, S. J., Kelly, G. L., Brennan, M. S., Downer, N. L., et al. (2018). Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257. doi:10.1038/s41586-018-0387-5
Bamborough, P., Barnett, H. A., Becher, I., Bird, M. J., Chung, C. W., Craggs, P. D., et al. (2016). GSK6853, a chemical probe for inhibition of the BRPF1 bromodomain. ACS Med. Chem. Lett. 7, 552–557. doi:10.1021/acsmedchemlett.6b00092
Becht, D. C., Klein, B. J., Kanai, A., Jang, S. M., Cox, K. L., Zhou, B.-R., et al. (2023). MORF and MOZ acetyltransferases target unmethylated CpG islands through the winged helix domain. Nat. Commun. in press.
Borrow, J., Stanton, V. P., Andresen, J. M., Becher, R., Behm, F. G., Chaganti, R. S., et al. (1996). The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat. Genet. 14, 33–41. doi:10.1038/ng0996-33
Bristow, C. A., and Shore, P. (2003). Transcriptional regulation of the human MIP-1alpha promoter by RUNX1 and MOZ. Nucleic Acids Res. 31, 2735–2744. doi:10.1093/nar/gkg401
Camos, M., Esteve, J., Jares, P., Colomer, D., Rozman, M., Villamor, N., et al. (2006). Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res. 66, 6947–6954. doi:10.1158/0008-5472.CAN-05-4601
Carapeti, M., Aguiar, R. C. T., Goldman, J. M., and Cross, N. C. P. (1998). A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 91, 3127–3133. doi:10.1182/blood.v91.9.3127.3127_3127_3133
Chaffanet, M., Gressin, L., Preudhomme, C., Soenen-Cornu, V., Birnbaum, D., and Pebusque, M. J. (2000). MOZ is fused to p300 in an acute monocytic leukemia with t(8;22). Genes Chromosom. Cancer 28, 138–144. doi:10.1002/(sici)1098-2264(200006)28:2<138:aid-gcc2>3.0.co;2-2
Champagne, K. S., Saksouk, N., Pena, P. V., Johnson, K., Ullah, M., Yang, X. J., et al. (2008). The crystal structure of the ING5 PHD finger in complex with an H3K4me3 histone peptide. Proteins 72, 1371–1376. doi:10.1002/prot.22140
Champagne, N., Bertos, N. R., Pelletier, N., Wang, A. H., Vezmar, M., Yang, Y., et al. (1999). Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. J. Biol. Chem. 274, 28528–28536. doi:10.1074/jbc.274.40.28528
Champagne, N., Pelletier, N., and Yang, X. J. (2001). The monocytic leukemia zinc finger protein MOZ is a histone acetyltransferase. Oncogene 20, 404–409. doi:10.1038/sj.onc.1204114
Chan, E. M., Chan, R. J., Comer, E. M., Goulet, R. J., Crean, C. D., Brown, Z. D., et al. (2007). MOZ and MOZ-CBP cooperate with NF-kappaB to activate transcription from NF-kappaB-dependent promoters. Exp. Hematol. 35, 1782–1792. doi:10.1016/j.exphem.2007.07.015
Cheng, C. L., Tsang, F. H., Wei, L., Chen, M., Chin, D. W., Shen, J., et al. (2021). Bromodomain-containing protein BRPF1 is a therapeutic target for liver cancer. Commun. Biol. 4, 888. doi:10.1038/s42003-021-02405-6
Chinen, Y., Taki, T., Tsutsumi, Y., Kobayashi, S., Matsumoto, Y., Sakamoto, N., et al. (2014). The leucine twenty homeobox (LEUTX) gene, which lacks a histone acetyltransferase domain, is fused to KAT6A in therapy-related acute myeloid leukemia with t(8; 19) (p11; q13). Genes, Chromosom. Cancer 53, 299–308. doi:10.1002/gcc.22140
Collins, H. M., Kindle, K. B., Matsuda, S., Ryan, C., Troke, P. J. F., Kalkhoven, E., et al. (2006). MOZ-TIF2 alters cofactor recruitment and histone modification at the RARbeta2 promoter: Differential effects of MOZ fusion proteins on CBP- and MOZ-dependent activators. J. Biol. Chem. 281, 17124–17133. doi:10.1074/jbc.M602633200
Crump, J. G., Swartz, M. E., Eberhart, J. K., and Kimmel, C. B. (2006). Moz-dependent Hox expression controls segment-specific fate maps of skeletal precursors in the face. Development 133, 2661–2669. doi:10.1242/dev.02435
Dancy, B. M., and Cole, P. A. (2015). Protein lysine acetylation by p300/CBP. Chem. Rev. 115, 2419–2452. doi:10.1021/cr500452k
Dantas, A., Al Shueili, B., Yang, Y., Nabbi, A., Fink, D., and Riabowol, K. (2019). Biological functions of the ING proteins. Cancers (Basel) 11, 1817. doi:10.3390/cancers11111817
Deguchi, K., Ayton, P. M., Carapeti, M., Kutok, J. L., Snyder, C. S., Williams, I. R., et al. (2003). MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell 3, 259–271. doi:10.1016/s1535-6108(03)00051-5
Demeulenaere, S., Beysen, D., De Veuster, I., Reyniers, E., Kooy, F., and Meuwissen, M. (2019). Novel BRPF1 mutation in a boy with intellectual disability, coloboma, facial nerve palsy and hypoplasia of the corpus callosum. Eur. J. Med. Genet. 62, 103691. doi:10.1016/j.ejmg.2019.103691
Doyon, Y., Cayrou, C., Ullah, M., Landry, A. J., Cote, V., Selleck, W., et al. (2006). ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 21, 51–64. doi:10.1016/j.molcel.2005.12.007
Dreveny, I., Deeves, S. E., Fulton, J., Yue, B., Messmer, M., Bhattacharya, A., et al. (2014). The double PHD finger domain of MOZ/MYST3 induces alpha-helical structure of the histone H3 tail to facilitate acetylation and methylation sampling and modification. Nucleic Acids Res. 42, 822–835. doi:10.1093/nar/gkt931
Esteyries, S., Perot, C., Adelaide, J., Imbert, M., Lagarde, A., Pautas, C., et al. (2008). NCOA3, a new fusion partner for MOZ/MYST3 in M5 acute myeloid leukemia. Leukemia 22, 663–665. doi:10.1038/sj.leu.2404930
Feng, Y., Vlassis, A., Roques, C., Lalonde, M. E., Gonzalez-Aguilera, C., Lambert, J. P., et al. (2016). BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 35, 176–192. doi:10.15252/embj.201591293
Good-Jacobson, K. L., Chen, Y., Voss, A. K., Smyth, G. K., Thomas, T., and Tarlinton, D. (2014). Regulation of germinal center responses and B-cell memory by the chromatin modifier MOZ. Proc. Natl. Acad. Sci. U. S. A. 111, 9585–9590. doi:10.1073/pnas.1402485111
Hemming, M. L., Benson, M. R., Loycano, M. A., Anderson, J. A., Andersen, J. L., Taddei, M. L., et al. (2022). MOZ and menin-MLL complexes are complementary regulators of chromatin association and transcriptional output in gastrointestinal stromal tumor. Cancer Discov. 12, 1804–1823. doi:10.1158/2159-8290.CD-21-0646
Hibiya, K., Katsumoto, T., Kondo, T., Kitabayashi, I., and Kudo, A. (2009). Brpf1, a subunit of the MOZ histone acetyl transferase complex, maintains expression of anterior and posterior Hox genes for proper patterning of craniofacial and caudal skeletons. Dev. Biol. 329, 176–190. doi:10.1016/j.ydbio.2009.02.021
Holbert, M. A., Sikorski, T., Carten, J., Snowflack, D., Hodawadekar, S., and Marmorstein, R. (2007). The human monocytic leukemia zinc finger histone acetyltransferase domain contains DNA-binding activity implicated in chromatin targeting. J. Biol. Chem. 282, 36603–36613. doi:10.1074/jbc.M705812200
Huang, F., Saraf, A., Florens, L., Kusch, T., Swanson, S. K., Szerszen, L. T., et al. (2016). The Enok acetyltransferase complex interacts with Elg1 and negatively regulates PCNA unloading to promote the G1/S transition. Genes Dev. 30, 1198–1210. doi:10.1101/gad.271429.115
Huether, R., Dong, L., Chen, X., Wu, G., Parker, M., Wei, L., et al. (2014). The landscape of somatic mutations in epigenetic regulators across 1, 000 paediatric cancer genomes. Nat. Commun. 5, 3630. doi:10.1038/ncomms4630
Huntly, B. J., Shigematsu, H., Deguchi, K., Lee, B. H., Mizuno, S., Duclos, N., et al. (2004). MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6, 587–596. doi:10.1016/j.ccr.2004.10.015
Imamura, T., Kakazu, N., Hibi, S., Morimoto, A., Fukushima, Y., Ijuin, I., et al. (2003). Rearrangement of the MOZ gene in pediatric therapy-related myelodysplastic syndrome with a novel chromosomal translocation t(2;8)(p23;p11). Genes Chromosom. Cancer 36, 413–419. doi:10.1002/gcc.10172
International Human Genome Sequencing Consortium (2004). Finishing the euchromatic sequence of the human genome. Nature 431, 931–945. doi:10.1038/nature03001
Jain, K., Marunde, M. R., Burg, J. M., Gloor, S. L., Joseph, F. M., Gillespie, Z. B., et al. (2022). An acetylation-mediated chromatin switch governs H3K4 methylation read-write capability. bioRxiv 2022, 482307. doi:10.1101/2022.02.28.482307
Jiang, Y., Guo, X., Liu, L., Rode, S., Wang, R., Liu, H., et al. (2020). Metagenomic characterization of lysine acetyltransferases in human cancer and their association with clinicopathologic features. Cancer Sci. 111, 1829–1839. doi:10.1111/cas.14385
John, S., Howe, L., Tafrov, S. T., Grant, P. A., Sternglanz, R., and Workman, J. L. (2000). The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30-containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)-FACT complex. Genes Dev. 14, 1196–1208. doi:10.1101/gad.14.10.1196
Jude, C. D., Climer, L., Xu, D., Artinger, E., Fisher, J. K., and Ernst, P. (2007). Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 1, 324–337. doi:10.1016/j.stem.2007.05.019
Katsumoto, T., Aikawa, Y., Iwama, A., Ueda, S., Ichikawa, H., Ochiya, T., et al. (2006). MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 20, 1321–1330. doi:10.1101/gad.1393106
Katsumoto, T., Ogawara, Y., Yamagata, K., Aikawa, Y., Goitsuka, R., Nakamura, T., et al. (2022). MOZ is critical for the development of MOZ/MLL fusion-induced leukemia through regulation of Hoxa9/Meis1 expression. Blood Adv. 6, 5527–5537. doi:10.1182/bloodadvances.2020003490
Kennedy, J., Goudie, D., Blair, E., Chandler, K., Joss, S., Mckay, V., et al. (2019). KAT6A syndrome: Genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 21, 850–860. doi:10.1038/s41436-018-0259-2
Kindle, K. B., Troke, P. J., Collins, H. M., Matsuda, S., Bossi, D., Bellodi, C., et al. (2005). MOZ-TIF2 inhibits transcription by nuclear receptors and p53 by impairment of CBP function. Mol. Cell Biol. 25, 988–1002. doi:10.1128/MCB.25.3.988-1002.2005
Kitabayashi, I., Aikawa, Y., Nguyen, L. A., Yokoyama, A., and Ohki, M. (2001a). Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 20, 7184–7196. doi:10.1093/emboj/20.24.7184
Kitabayashi, I., Aikawa, Y., Yokoyama, A., Hosoda, F., Nagai, M., Kakazu, N., et al. (2001b). Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 15, 89–94. doi:10.1038/sj.leu.2401983
Klein, B. J., Cox, K. L., Jang, S. M., Cote, J., Poirier, M. G., and Kutateladze, T. G. (2020). Molecular basis for the PZP domain of BRPF1 association with chromatin. Structure 28, 105–110. doi:10.1016/j.str.2019.10.014
Klein, B. J., Jang, S. M., Lachance, C., Mi, W., Lyu, J., Sakuraba, S., et al. (2019). Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation. Nat. Commun. 10, 4724. doi:10.1038/s41467-019-12551-5
Klein, B. J., Lalonde, M. E., Cote, J., Yang, X. J., and Kutateladze, T. G. (2014). Crosstalk between epigenetic readers regulates the MOZ/MORF HAT complexes. Epigenetics 9, 186–193. doi:10.4161/epi.26792
Klein, B. J., Muthurajan, U. M., Lalonde, M. E., Gibson, M. D., Andrews, F. H., Hepler, M., et al. (2016). Bivalent interaction of the PZP domain of BRPF1 with the nucleosome impacts chromatin dynamics and acetylation. Nucleic Acids Res. 44, 472–484. doi:10.1093/nar/gkv1321
Klein, B. J., Simithy, J., Wang, X., Ahn, J., Andrews, F. H., Zhang, Y., et al. (2017). Recognition of histone H3K14 acylation by MORF. Structure 25, 650–654. doi:10.1016/j.str.2017.02.003
Kool, M., Jones, D. T., Jager, N., Northcott, P. A., Pugh, T. J., Hovestadt, V., et al. (2014). Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25, 393–405. doi:10.1016/j.ccr.2014.02.004
Kueh, A. J., Dixon, M. P., Voss, A. K., and Thomas, T. (2011). HBO1 is required for H3K14 acetylation and normal transcriptional activity during embryonic development. Mol. Cell Biol. 31, 845–860. doi:10.1128/MCB.00159-10
Lalonde, M. E., Avvakumov, N., Glass, K. C., Joncas, F. H., Saksouk, N., Holliday, M., et al. (2013). Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev. 27, 2009–2024. doi:10.1101/gad.223396.113
Lalonde, M. E., Cheng, X., and Cote, J. (2014). Histone target selection within chromatin: An exemplary case of teamwork. Genes Dev. 28, 1029–1041. doi:10.1101/gad.236331.113
Largeot, A., Perez-Campo, F. M., Marinopoulou, E., Lie-a-Ling, M., Kouskoff, V., and Lacaud, G. (2016). Expression of the MOZ-TIF2 oncoprotein in mice represses senescence. Exp. Hematol. 44, 231–237. doi:10.1016/j.exphem.2015.12.006
Lashgari, A., Kougnassoukou Tchara, P. E., Lambert, J. P., and Cote, J. (2022). New insights into the DNA repair pathway choice with NuA4/TIP60. DNA Repair (Amst) 113, 103315. doi:10.1016/j.dnarep.2022.103315
Laue, K., Daujat, S., Crump, J. G., Plaster, N., Roehl, H. H., Tubingen Screen, C., et al. (2008). The multidomain protein Brpf1 binds histones and is required for Hox gene expression and segmental identity. Development 135, 1935–1946. doi:10.1242/dev.017160
Lee, M. H., Kim, Y. J., Kim, H. J., Park, H. D., Kang, A. R., Kyung, H. M., et al. (2003). BMP-2-induced Runx2 expression is mediated by Dlx5, and TGF-beta 1 opposes the BMP-2-induced osteoblast differentiation by suppression of Dlx5 expression. J. Biol. Chem. 278, 34387–34394. doi:10.1074/jbc.M211386200
Lian, J., Zhang, H., Wei, F., Li, Q., Lu, Y., Yu, B., et al. (2020). Long non-coding RNA DANCR promotes colorectal tumor growth by binding to lysine acetyltransferase 6A. Cell Signal 67, 109502. doi:10.1016/j.cellsig.2019.109502
Liu, W., Zhan, Z., Zhang, M., Sun, B., Shi, Q., Luo, F., et al. (2021). KAT6A, a novel regulator of beta-catenin, promotes tumorigenicity and chemoresistance in ovarian cancer by acetylating COP1. Theranostics 11, 6278–6292. doi:10.7150/thno.57455
Lloyd, J. T., and Glass, K. C. (2018). Biological function and histone recognition of family IV bromodomain-containing proteins. J. Cell Physiol. 233, 1877–1886. doi:10.1002/jcp.26010
Lubelsky, Y., Prinz, J. A., Denapoli, L., Li, Y., Belsky, J. A., and Macalpine, D. M. (2014). DNA replication and transcription programs respond to the same chromatin cues. Genome Res. 24, 1102–1114. doi:10.1101/gr.160010.113
Lv, D., Jia, F., Hou, Y., Sang, Y., Alvarez, A. A., Zhang, W., et al. (2017). Histone acetyltransferase KAT6A upregulates PI3K/AKT signaling through TRIM24 binding. Cancer Res. 77, 6190–6201. doi:10.1158/0008-5472.CAN-17-1388
Mallo, M., and Alonso, C. R. (2013). The regulation of Hox gene expression during animal development. Development 140, 3951–3963. doi:10.1242/dev.068346
Mattioli, F., Schaefer, E., Magee, A., Mark, P., Mancini, G. M., Dieterich, K., et al. (2017). Mutations in histone acetylase modifier BRPF1 cause an autosomal-dominant form of intellectual disability with associated ptosis. Am. J. Hum. Genet. 100, 105–116. doi:10.1016/j.ajhg.2016.11.010
Mcmahon, K. A., Hiew, S. Y., Hadjur, S., Veiga-Fernandes, H., Menzel, U., Price, A. J., et al. (2007). Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 1, 338–345. doi:10.1016/j.stem.2007.07.002
Meier, J. C., Tallant, C., Fedorov, O., Witwicka, H., Hwang, S. Y., Van Stiphout, R. G., et al. (2017). Selective targeting of bromodomains of the bromodomain-PHD fingers family impairs osteoclast differentiation. ACS Chem. Biol. 12, 2619–2630. doi:10.1021/acschembio.7b00481
Merson, T. D., Dixon, M. P., Collin, C., Rietze, R. L., Bartlett, P. F., Thomas, T., et al. (2006). The transcriptional coactivator Querkopf controls adult neurogenesis. J. Neurosci. 26, 11359–11370. doi:10.1523/JNEUROSCI.2247-06.2006
Micci, F., Gorunova, L., Gatius, S., Matias-Guiu, X., Davidson, B., Heim, S., et al. (2014). MEAF6/PHF1 is a recurrent gene fusion in endometrial stromal sarcoma. Cancer Lett. 347, 75–78. doi:10.1016/j.canlet.2014.01.030
Millan, F., Cho, M. T., Retterer, K., Monaghan, K. G., Bai, R., Vitazka, P., et al. (2016). Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. Am. J. Med. Genet. A 170, 1791–1798. doi:10.1002/ajmg.a.37670
Millan-Zambrano, G., Burton, A., Bannister, A. J., and Schneider, R. (2022). Histone post-translational modifications - cause and consequence of genome function. Nat. Rev. Genet. 23, 563–580. doi:10.1038/s41576-022-00468-7
Miller, C. T., Maves, L., and Kimmel, C. B. (2004). Moz regulates Hox expression and pharyngeal segmental identity in zebrafish. Development 131, 2443–2461. doi:10.1242/dev.01134
Milne, T. A., Briggs, S. D., Brock, H. W., Martin, M. E., Gibbs, D., Allis, C. D., et al. (2002). MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10, 1107–1117. doi:10.1016/s1097-2765(02)00741-4
Mishima, Y., Miyagi, S., Saraya, A., Negishi, M., Endoh, M., Endo, T. A., et al. (2011). The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 118, 2443–2453. doi:10.1182/blood-2011-01-331892
Miyamoto, R., Okuda, H., Kanai, A., Takahashi, S., Kawamura, T., Matsui, H., et al. (2020). Activation of CpG-rich promoters mediated by MLL drives MOZ-rearranged leukemia. Cell Rep. 32, 108200. doi:10.1016/j.celrep.2020.108200
Mohammadi, K., Safaralizadeh, R., Hosseinpour-Feizi, M., Dastmalchi, N., and Moaddab, Y. (2019). Investigation of the changes in the expression levels of MOZ gene in colorectal cancer tissues. J. Gastrointest. Oncol. 10, 68–73. doi:10.21037/jgo.2018.09.12
Murr, R., Loizou, J. I., Yang, Y. G., Cuenin, C., Li, H., Wang, Z. Q., et al. (2006). Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8, 91–99. doi:10.1038/ncb1343
Naseer, M. I., Abdulkareem, A. A., Guzman-Vega, F. J., Arold, S. T., Pushparaj, P. N., Chaudhary, A. G., et al. (2020). Novel missense variant in heterozygous state in the BRPF1 gene leading to intellectual developmental disorder with dysmorphic facies and ptosis. Front. Genet. 11, 368. doi:10.3389/fgene.2020.00368
Newman, D. M., Sakaguchi, S., Lun, A., Preston, S., Pellegrini, M., Khamina, K., et al. (2016). Acetylation of the Cd8 locus by KAT6A determines memory T cell diversity. Cell Rep. 16, 3311–3321. doi:10.1016/j.celrep.2016.08.056
Northcott, P. A., Nakahara, Y., Wu, X., Feuk, L., Ellison, D. W., Croul, S., et al. (2009). Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 41, 465–472. doi:10.1038/ng.336
Okuda, H., Stanojevic, B., Kanai, A., Kawamura, T., Takahashi, S., Matsui, H., et al. (2017). Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Invest. 127, 1918–1931. doi:10.1172/JCI91406
Paggetti, J., Largeot, A., Aucagne, R., Jacquel, A., Lagrange, B., Yang, X. J., et al. (2010). Crosstalk between leukemia-associated proteins MOZ and MLL regulates HOX gene expression in human cord blood CD34+ cells. Oncogene 29, 5019–5031. doi:10.1038/onc.2010.254
Panagopoulos, I., Andersen, K., Ramslien, L. F., Ikonomou, I. M., Micci, F., and Heim, S. (2021). Therapy-related myeloid leukemia with the translocation t(8;19)(p11;q13) leading to a kat6a-LEUTX fusion gene. Anticancer Res. 41, 1753–1760. doi:10.21873/anticanres.14940
Parenti, I., Rabaneda, L. G., Schoen, H., and Novarino, G. (2020). Neurodevelopmental disorders: From genetics to functional pathways. Trends Neurosci. 43, 608–621. doi:10.1016/j.tins.2020.05.004
Pelletier, N., Champagne, N., Stifani, S., and Yang, X. J. (2002). MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene 21, 2729–2740. doi:10.1038/sj.onc.1205367
Perez-Campo, F. M., Borrow, J., Kouskoff, V., and Lacaud, G. (2009). The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood 113, 4866–4874. doi:10.1182/blood-2008-04-152017
Perez-Campo, F. M., Costa, G., Lie, A. L. M., Stifani, S., Kouskoff, V., and Lacaud, G. (2014). MOZ-mediated repression of p16(INK) (4) (a) is critical for the self-renewal of neural and hematopoietic stem cells. Stem Cells 32, 1591–1601. doi:10.1002/stem.1606
Perez-Salvia, M., and Esteller, M. (2017). Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 12, 323–339. doi:10.1080/15592294.2016.1265710
Pocaterra, A., Romani, P., and Dupont, S. (2020). YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 133, jcs230425. doi:10.1242/jcs.230425
Pode-Shakked, N., Barel, O., Pode-Shakked, B., Eliyahu, A., Singer, A., Nayshool, O., et al. (2019). BRPF1-associated intellectual disability, ptosis, and facial dysmorphism in a multiplex family. Mol. Genet. Genomic Med. 7, e665. doi:10.1002/mgg3.665
Poplawski, A., Hu, K., Lee, W., Natesan, S., Peng, D., Carlson, S., et al. (2014). Molecular insights into the recognition of N-terminal histone modifications by the BRPF1 bromodomain. J. Mol. Biol. 426, 1661–1676. doi:10.1016/j.jmb.2013.12.007
Qiu, Y., Liu, L., Zhao, C., Han, C., Li, F., Zhang, J., et al. (2012). Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes Dev. 26, 1376–1391. doi:10.1101/gad.188359.112
Rokudai, S., Aikawa, Y., Tagata, Y., Tsuchida, N., Taya, Y., and Kitabayashi, I. (2009). Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J. Biol. Chem. 284, 237–244. doi:10.1074/jbc.M805101200
Rokudai, S., Laptenko, O., Arnal, S. M., Taya, Y., Kitabayashi, I., and Prives, C. (2013). MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. U. S. A. 110, 3895–3900. doi:10.1073/pnas.1300490110
Roussel, X., Daguindau, E., Berceanu, A., Desbrosses, Y., Warda, W., Neto Da Rocha, M., et al. (2020). Acute myeloid leukemia: From Biology to clinical practices through development and pre-clinical therapeutics. Front. Oncol. 10, 599933. doi:10.3389/fonc.2020.599933
Sabari, B. R., Zhang, D., Allis, C. D., and Zhao, Y. (2017). Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101. doi:10.1038/nrm.2016.140
Saksouk, N., Avvakumov, N., Champagne, K. S., Hung, T., Doyon, Y., Cayrou, C., et al. (2009). HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol. Cell 33, 257–265. doi:10.1016/j.molcel.2009.01.007
Shanle, E. K., Andrews, F. H., Meriesh, H., Mcdaniel, S. L., Dronamraju, R., Difiore, J. V., et al. (2015). Association of Taf14 with acetylated histone H3 directs gene transcription and the DNA damage response. Genes Dev. 29, 1795–1800. doi:10.1101/gad.269977.115
Sheikh, B. N., Dixon, M. P., Thomas, T., and Voss, A. K. (2012). Querkopf is a key marker of self-renewal and multipotency of adult neural stem cells. J. Cell Sci. 125, 295–309. doi:10.1242/jcs.077271
Sheikh, B. N., Downer, N. L., Phipson, B., Vanyai, H. K., Kueh, A. J., Mccarthy, D. J., et al. (2015a). MOZ and BMI1 play opposing roles during Hox gene activation in ES cells and in body segment identity specification in vivo. Proc. Natl. Acad. Sci. U. S. A. 112, 5437–5442. doi:10.1073/pnas.1422872112
Sheikh, B. N., Lee, S. C., El-Saafin, F., Vanyai, H. K., Hu, Y., Pang, S. H., et al. (2015b). MOZ regulates B-cell progenitors and, consequently, Moz haploinsufficiency dramatically retards MYC-induced lymphoma development. Blood 125, 1910–1921. doi:10.1182/blood-2014-08-594655
Sheikh, B. N., Metcalf, D., Voss, A. K., and Thomas, T. (2017). MOZ and BMI1 act synergistically to maintain hematopoietic stem cells. Exp. Hematol. 47, 83–97. doi:10.1016/j.exphem.2016.10.006
Sheikh, B. N., Phipson, B., El-Saafin, F., Vanyai, H. K., Downer, N. L., Bird, M. J., et al. (2015c). MOZ (MYST3, KAT6A) inhibits senescence via the INK4A-ARF pathway. Oncogene 34, 5807–5820. doi:10.1038/onc.2015.33
Sheikh, B. N., Yang, Y., Schreuder, J., Nilsson, S. K., Bilardi, R., Carotta, S., et al. (2016). MOZ (KAT6A) is essential for the maintenance of classically defined adult hematopoietic stem cells. Blood 128, 2307–2318. doi:10.1182/blood-2015-10-676072
Shi, X., Hong, T., Walter, K. L., Ewalt, M., Michishita, E., Hung, T., et al. (2006). ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99. doi:10.1038/nature04835
Shima, H., Yamagata, K., Aikawa, Y., Shino, M., Koseki, H., Shimada, H., et al. (2014). Bromodomain-PHD finger protein 1 is critical for leukemogenesis associated with MOZ-TIF2 fusion. Int. J. Hematol. 99, 21–31. doi:10.1007/s12185-013-1466-x
Sole, C., Goicoechea, I., Goni, A., Schramm, M., Armesto, M., Arestin, M., et al. (2020). The urinary transcriptome as a source of biomarkers for prostate cancer. Cancers (Basel) 12, 513. doi:10.3390/cancers12020513
Sramkova, L., Cermakova, J., Kutkova, K., Zemanova, Z., Pavlicek, P., Zuna, J., et al. (2020). Rapidly progressing acute myeloid leukemia with KAT6A-LEUTX fusion in a newborn. Pediatr. Blood Cancer 67, e28663. doi:10.1002/pbc.28663
Steunou, A.-L., Rossetto, D., and Côté, J. (2014). “Regulating chromatin by histone acetylation,” in Fundamentals of chromatin. Editors J. L. Workman,, and S. M. Abmayr 1 ed (Springer New York), 160–225.
Talbert, P. B., and Henikoff, S. (2021). The yin and yang of histone marks in transcription. Annu. Rev. Genomics Hum. Genet. 22, 147–170. doi:10.1146/annurev-genom-120220-085159
Tan, M., Luo, H., Lee, S., Jin, F., Yang, J. S., Montellier, E., et al. (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028. doi:10.1016/j.cell.2011.08.008
Tham, E., Lindstrand, A., Santani, A., Malmgren, H., Nesbitt, A., Dubbs, H. A., et al. (2015). Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 96, 507–513. doi:10.1016/j.ajhg.2015.01.016
Thomas, T., Corcoran, L. M., Gugasyan, R., Dixon, M. P., Brodnicki, T., Nutt, S. L., et al. (2006). Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 20, 1175–1186. doi:10.1101/gad.1382606
Thomas, T., Voss, A. K., Chowdhury, K., and Gruss, P. (2000). Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development 127, 2537–2548. doi:10.1242/dev.127.12.2537
Tsai, W. W., Wang, Z., Yiu, T. T., Akdemir, K. C., Xia, W., Winter, S., et al. (2010). TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932. doi:10.1038/nature09542
Turner-Ivey, B., Guest, S. T., Irish, J. C., Kappler, C. S., Garrett-Mayer, E., Wilson, R. C., et al. (2014). KAT6A, a chromatin modifier from the 8p11-p12 amplicon is a candidate oncogene in luminal breast cancer. Neoplasia 16, 644–655. doi:10.1016/j.neo.2014.07.007
Ullah, M., Pelletier, N., Xiao, L., Zhao, S. P., Wang, K., Degerny, C., et al. (2008). Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell Biol. 28, 6828–6843. doi:10.1128/MCB.01297-08
Vanyai, H. K., Garnham, A., May, R. E., Mcrae, H. M., Collin, C., Wilcox, S., et al. (2019). MOZ directs the distal-less homeobox gene expression program during craniofacial development. Development 146, dev175042. doi:10.1242/dev.175042
Vanyai, H. K., Thomas, T., and Voss, A. K. (2015). Mesodermal expression of Moz is necessary for cardiac septum development. Dev. Biol. 403, 22–29. doi:10.1016/j.ydbio.2015.04.011
Vetting, M. W., Lp, S. D. C., Yu, M., Hegde, S. S., Magnet, S., Roderick, S. L., et al. (2005). Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226. doi:10.1016/j.abb.2004.09.003
Vezzoli, A., Bonadies, N., Allen, M. D., Freund, S. M., Santiveri, C. M., Kvinlaug, B. T., et al. (2010). Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 17, 617–619. doi:10.1038/nsmb.1797
Voss, A. K., Collin, C., Dixon, M. P., and Thomas, T. (2009). Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev. Cell 17, 674–686. doi:10.1016/j.devcel.2009.10.006
Voss, A. K., Vanyai, H. K., Collin, C., Dixon, M. P., Mclennan, T. J., Sheikh, B. N., et al. (2012). MOZ regulates the Tbx1 locus, and Moz mutation partially phenocopies DiGeorge syndrome. Dev. Cell 23, 652–663. doi:10.1016/j.devcel.2012.07.010
Wu, D., Qiu, Y., Jiao, Y., Qiu, Z., and Liu, D. (2020). Small molecules targeting HATs, HDACs, and BRDs in cancer therapy. Front. Oncol. 10, 560487. doi:10.3389/fonc.2020.560487
Wu, Q., Heidenreich, D., Zhou, S., Ackloo, S., Kramer, A., Nakka, K., et al. (2019). A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 10, 1915. doi:10.1038/s41467-019-09672-2
Xie, W., Zhang, Y., Wang, B., Hu, Y., Zhan, B., Wei, F., et al. (2020). Tripartite motif containing 24 regulates cell proliferation in colorectal cancer through YAP signaling. Cancer Med. 9, 6367–6376. doi:10.1002/cam4.3310
Xiong, X., Panchenko, T., Yang, S., Zhao, S., Yan, P., Zhang, W., et al. (2016). Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat. Chem. Biol. 12, 1111–1118. doi:10.1038/nchembio.2218
Xu, P., Li, C., Chen, Z., Jiang, S., Fan, S., Wang, J., et al. (2016). The NuA4 core complex acetylates nucleosomal histone H4 through a double recognition mechanism. Mol. Cell 63, 965–975. doi:10.1016/j.molcel.2016.07.024
Yan, F., Li, J., Milosevic, J., Petroni, R., Liu, S., Shi, Z., et al. (2022). KAT6A and ENL form an epigenetic transcriptional control module to drive critical leukemogenic gene-expression programs. Cancer Discov. 12, 792–811. doi:10.1158/2159-8290.CD-20-1459
Yan, K., Rousseau, J., Littlejohn, R. O., Kiss, C., Lehman, A., Rosenfeld, J. A., et al. (2017). Mutations in the chromatin regulator gene BRPF1 cause syndromic intellectual disability and deficient histone acetylation. Am. J. Hum. Genet. 100, 91–104. doi:10.1016/j.ajhg.2016.11.011
Yan, K., Rousseau, J., Machol, K., Cross, L. A., Agre, K. E., Gibson, C. F., et al. (2020). Deficient histone H3 propionylation by BRPF1-KAT6 complexes in neurodevelopmental disorders and cancer. Sci. Adv. 6, eaax0021. doi:10.1126/sciadv.aax0021
Yan, K., You, L., Degerny, C., Ghorbani, M., Liu, X., Chen, L., et al. (2016). The chromatin regulator BRPF3 preferentially activates the HBO1 acetyltransferase but is dispensable for mouse development and survival. J. Biol. Chem. 291, 2647–2663. doi:10.1074/jbc.M115.703041
You, L., Chen, L., Penney, J., Miao, D., and Yang, X. J. (2014). Expression atlas of the multivalent epigenetic regulator Brpf1 and its requirement for survival of mouse embryos. Epigenetics 9, 860–872. doi:10.4161/epi.28530
You, L., Li, L., Zou, J., Yan, K., Belle, J., Nijnik, A., et al. (2016). BRPF1 is essential for development of fetal hematopoietic stem cells. J. Clin. Invest. 126, 3247–3262. doi:10.1172/JCI80711
You, L., Yan, K., Zou, J., Zhao, H., Bertos, N. R., Park, M., et al. (2015b). The lysine acetyltransferase activator Brpf1 governs dentate gyrus development through neural stem cells and progenitors. PLoS Genet. 11, e1005034. doi:10.1371/journal.pgen.1005034
You, L., Yan, K., Zou, J., Zhao, H., Bertos, N. R., Park, M., et al. (2015a). The chromatin regulator Brpf1 regulates embryo development and cell proliferation. J. Biol. Chem. 290, 11349–11364. doi:10.1074/jbc.M115.643189
You, L., Zou, J., Zhao, H., Bertos, N. R., Park, M., Wang, E., et al. (2015c). Deficiency of the chromatin regulator BRPF1 causes abnormal brain development. J. Biol. Chem. 290, 7114–7129. doi:10.1074/jbc.M114.635250
Yu, L., Liang, Y., Cao, X., Wang, X., Gao, H., Lin, S. Y., et al. (2017). Identification of MYST3 as a novel epigenetic activator of ERα frequently amplified in breast cancer. Oncogene 36, 2910–2918. doi:10.1038/onc.2016.433
Zack, T. I., Schumacher, S. E., Carter, S. L., Cherniack, A. D., Saksena, G., Tabak, B., et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140. doi:10.1038/ng.2760
Zhang, L. X., Lemire, G., Gonzaga-Jauregui, C., Molidperee, S., Galaz-Montoya, C., Liu, D. S., et al. (2020). Further delineation of the clinical spectrum of KAT6B disorders and allelic series of pathogenic variants. Genet. Med. 22, 1338–1347. doi:10.1038/s41436-020-0811-8
Keywords: MOZ, KAT6A, BRPF1, epigenetics, development, cancer, MYST acetyltransferase, MORF
Citation: Viita T and Côté J (2023) The MOZ-BRPF1 acetyltransferase complex in epigenetic crosstalk linked to gene regulation, development, and human diseases. Front. Cell Dev. Biol. 10:1115903. doi: 10.3389/fcell.2022.1115903
Received: 04 December 2022; Accepted: 29 December 2022;
Published: 11 January 2023.
Edited by:
Fen Yang, Nanjing Medical University, ChinaReviewed by:
Hengbin Wang, Virginia Commonwealth University, United StatesCopyright © 2023 Viita and Côté. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacques Côté, amFjcXVlcy5jb3RlQGNyY2h1ZGVxdWViZWMudWxhdmFsLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.