 Manuela Sollazzo
Manuela Sollazzo Simona Paglia
Simona Paglia Simone Di Giacomo1†
Simone Di Giacomo1† Daniela Grifoni
Daniela Grifoni
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell Dev. Biol. , 10 January 2023
Sec. Cell Death and Survival
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1043630
Tumor cells exploit multiple mechanisms to evade apoptosis, hence the strategies aimed at reactivating cell death in cancer. However, recent studies are revealing that dying cells play remarkable pro-oncogenic roles. Among the mechanisms promoting cell death, cell competition, elicited by disparities in MYC activity in confronting cells, plays the primary role of assuring tissue robustness during development from Drosophila to mammals: cells with high MYC levels (winners) overproliferate while killing suboptimal neighbors (losers), whose death is essential to process completion. This mechanism is coopted by tumor cells in cancer initiation, where host cells succumb to high-MYC-expressing precancerous neighbors. Also in this case, inhibition of cell death restrains aberrant cell competition and rescues tissue structure. Inhibition of apoptosis may thus emerge as a good strategy to counteract cancer progression in competitive contexts; of note, we recently found a positive correlation between cell death amount at the tumor/stroma interface and MYC levels in human cancers. Here we used Drosophila to investigate the functional role of competition-dependent apoptosis in advanced cancers, observing dramatic changes in mass dimensions and composition following a boost in cell competition, rescued by apoptosis inhibition. This suggests the role of competition-dependent apoptosis be not confined to the early stages of tumorigenesis. We also show that apoptosis inhibition, beside restricting cancer mass, is sufficient to rescue tissue architecture and counteract cell migration in various cancer contexts, suggesting that a strong activation of the apoptotic pathways intensifies cancer burden by affecting distinct phenotypic traits at different stages of the disease.
Among the hallmark features of a cancer cell, evasion of apoptosis is the one which helps it survive the stressing conditions faced throughout its challenging life (Hanahan and Weinberg, 2011). Reactivation of cell death programs is thus common strategy in cancer treatment (Fulda, 2015). Apoptosis is usually thought as a mechanism through which a cell dictates its own demise but, in time, non-cell-autonomous processes involving apoptotic death have been emerging in literature (Labi and Erlacher, 2015). One of these processes, named “cell competition” (CC), was originally described in Drosophila as a phenomenon aimed at eliminating unfit cells during development (Morata and Ripoll, 1975), and following studies broadened the concept to a mechanism of surveillance through which viable but suboptimal cells could be detected and out-competed by fitter neighbors (Simpson, 1979; Moreno et al., 2002). In 2004, two studies found that MYC-upregulating cells acquired competitive properties in Drosophila epithelial tissues: cells expressing high MYC levels (winners) were able to induce apoptotic death of the wild-type neighbors (losers) and to overgrow at their expense, filling almost the entire tissue (de la Cova et al., 2004; Moreno and Basler, 2004). MYC-mediated CC (MMCC) is conserved in mammalian development: it has indeed been shown that mouse embryo is naturally composed of cells displaying different MYC activity: in time, high MYC-expressing cells colonize the whole tissue by killing and replacing cells with lower MYC levels (Claveria et al., 2013; Sancho et al., 2013). Since then, evidence of physiological MMCC was also found in adult tissues, such as the Drosophila ovary and eye (Rhiner et al., 2009; Merino et al., 2013) and the mammalian heart, where CC expands MYC-upregulating cardiomyocytes while silently replacing the wild-type counterparts without hindering normal heart functions (Villa del Campo et al., 2014; Villa del Campo et al., 2016). Conservation of MMCC across distant species is not surprising, as Drosophila MYC and mammalian c-MYC were found to substitute each other functions in several contexts (Schreiber-Agus et al., 1997; Gallant, 2013). In summary, CC requires the participation of different cell populations struggling for resources and space while growing in close proximity within a tissue (Johnston, 2014). This is a typical trait of cancer, where clonal expansion is promoted as a response to active selection (Bissel and Radisky, 2001; Laconi et al., 2008) MYC upregulation is long known to support human cancer emergence and progression (Gabay et al., 2014), and the identification of its essential function in CC has fostered a series of speculations about a role for this process in malignant growth (Baker, 2008; Moreno, 2008; Di Gregorio et al., 2016; Paglia et al., 2020). Studies performed in our and other labs indeed showed that, in Drosophila epithelia, cells with disrupted cell polarity upregulate MYC and behave as super-competitors, able to grow while killing neighboring wild-type cells (Froldi et al., 2010; Menéndez et al., 2010). In particular, loss-of-function mutations of the lgl (lethal giant larvae) tumor suppressor gene (TSG), which encodes a well-known polarity protein, have been found to promote MYC up-regulation and tumorigenesis in epithelial tissues (Grzeschik et al., 2007; Grzeschik et al., 2010; Grifoni et al., 2015). We previously showed that the human ortholog of Drosophila lgl, namely LLGL1 or HUGL-1, is functionally conserved (Grifoni et al., 2004), and our and other studies associated its transcriptional deregulation and/or protein delocalization with several types of cancers (Grifoni et al., 2004; Schimanski et al., 2005; Grifoni et al., 2007). In human epithelial cancers, polarity disruption is a hallmark of aggressivity (Dow and Humbert, 2007), and MMCC may contribute to trigger apoptosis of the less fit cells which, in turn, would fuel winners’ proliferation. In this sense, a recent study by Suijkerbuijk and others, where overgrowth of Drosophila intestinal adenomas due to CC was rescued by apoptosis inhibition, demonstrated the direct involvement of competition-dependent cell death in the early stages of carcinogenesis (Suijkerbuijk et al., 2016). The strict relationship between cell competition and apoptosis in human cancers has recently been found essential to tumor progression in a computational model, which has also highlighted how both contribute to cell resistance to conventional anti-cancer treatments (Pantziarka et al., 2019). Distinct oncogenic properties of apoptotic cells have recently been described in a mouse model of B cell lymphoma, where dying cells were found to promote angiogenesis and to alter the composition of the extracellular matrix, besides fueling tumor growth (Ford et al., 2015). In addition, several studies correlated the apoptotic index (AI) of tumors from distinct organs to a poor prognosis (De Jong et al., 2000; Naresh et al., 2001; Jalalinadoushan et al., 2004; Sun et al., 2006). Our recent analysis of polarity-deficient human cancers revealed that the abundance of apoptotic cells at the tumor/stroma interface positively correlated with MYC protein levels in the nearby tumor parenchyma also at late stages (Di Giacomo et al., 2017b), suggesting MMCC may help shape cancer evolution during malignant progression. To investigate MMCC functional relevance in advanced cancers, we induced clonal MYC overexpression in neoplastic epithelial tissues of Drosophila, observing a significant deviation both in terms of final cell composition and cancer size. Of note, we found this role depends on the presence of dying cells generated in the competitive islets. Then, we inhibited caspase activation in other Drosophila cancer models and again observed a restriction in cancer expansion, and besides, a normalization of tissue architecture and cell positioning. Altogether, our data indicate that massive activation of the apoptotic pathways can deeply impact a number of cancer-history traits, and any cell death-promoting interventions should take this relevant side effect into account.
Parental, larval and cellular genotypes used in the study can be found in Supplementary Table S1. All the listed stocks were built starting from original lines from the Bloomington Drosophila Stock Center (Indiana University), except for UAS-HAdm, made by P. Bellosta.
For the experiments as in Figures 1, 2, Supplementary Figure S2, S3 and S4, female larvae of the right genotypes were selected at 144 ± 2 h development at 25°C, transferred to a vial plugged with foam and immersed for 2 min in a water bath at 37°C. After the heat-shock, larvae were immediately transferred to fresh food and allowed to grow for additional 48 h at 25°C. Heat-shock duration was set so to obtain an average 5% of GFP+ cells in the final control masses, 48 h after clone induction. Q-VD-OPh (Sigma-Aldrich) was added to 500 μl standard food at 500 μM final concentration before larvae transfer. For the experiments as in Supplementary Figure S5, vials containing larvae at 48 ± 2 h development at 25°C were immersed for 20 min in a water bath at 37°C. After the heat-shock, control larvae were allowed to grow for additional 72 h at 25°C before dissection, while experimental larvae were allowed to grow for additional 24 h at 25°C, treated with Q-VD-OPh at the same concentration as above, and dissected after additional 48 h at 25°C. For the experiments as in Figures 3, 4, Supplementary Figure S6, S7, crosses were raised at 25°C.
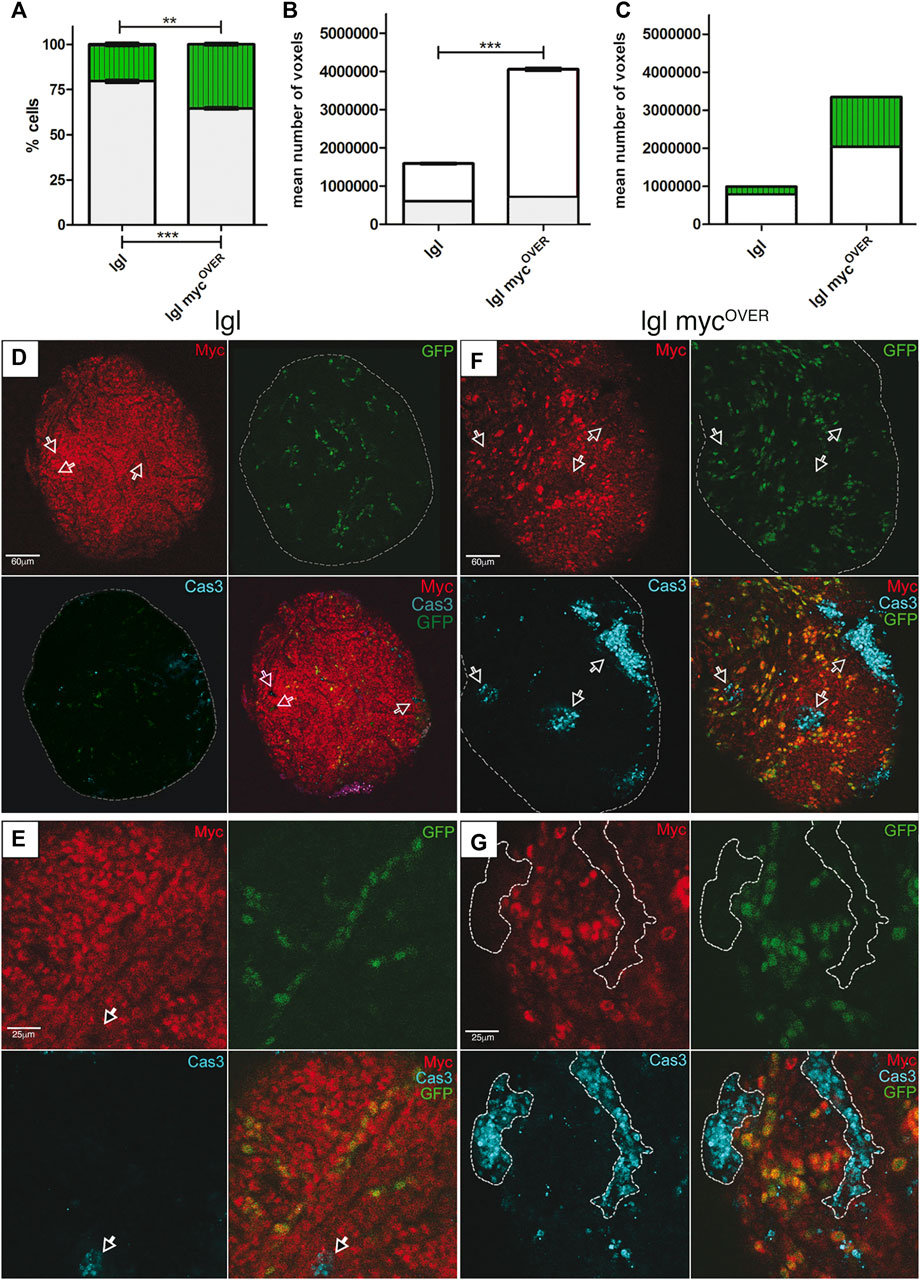
FIGURE 1. MYC-Mediated Cell Competition (MMCC) modifies tumor size and composition. (A) Percentage of GFP+ (green) and GFP− (grey) cells in lgl tumors collected at 8 days AEL after induction of neutral (n = 68) and mycOVER (n = 59) clones at 6 days AEL. See Supplementary Table 1 for plain genotypes. (B) Mean volume of the same samples as in A calculated from the images acquired immediately before dissociation (8 days AEL). The grey part of the bars represents the pre-induction volume (6 days AEL, n = 55 for neutral and n = 51 for mycOVER samples). (C) Relative volumes of GFP+ (green) and GFP− (white) cells in neutral and mycOVER samples after clone induction, calculated as described in the Methods section. Statistical significance and ±SEM are indicated. (D,E) MYC (red) and activated Cas3 (cyan) staining of lgl tumors collected at 8 days AEL in which GFP+, neutral clones were induced at 6 days AEL. The arrows highlight cells downregulating MYC positive to Cas3 signal. (F,G) MYC (red) and activated Cas3 (cyan) staining of lgl tumors collected at 8 days AEL in which GFP+, mycOVER clones were induced at 6 days AEL. The arrows in (F) and the dotted lines in (G) highlight cells with endogenous levels of MYC, positive to Cas3 signal. All the images represent disc cross-sections. Scale bars are indicated for each sample.
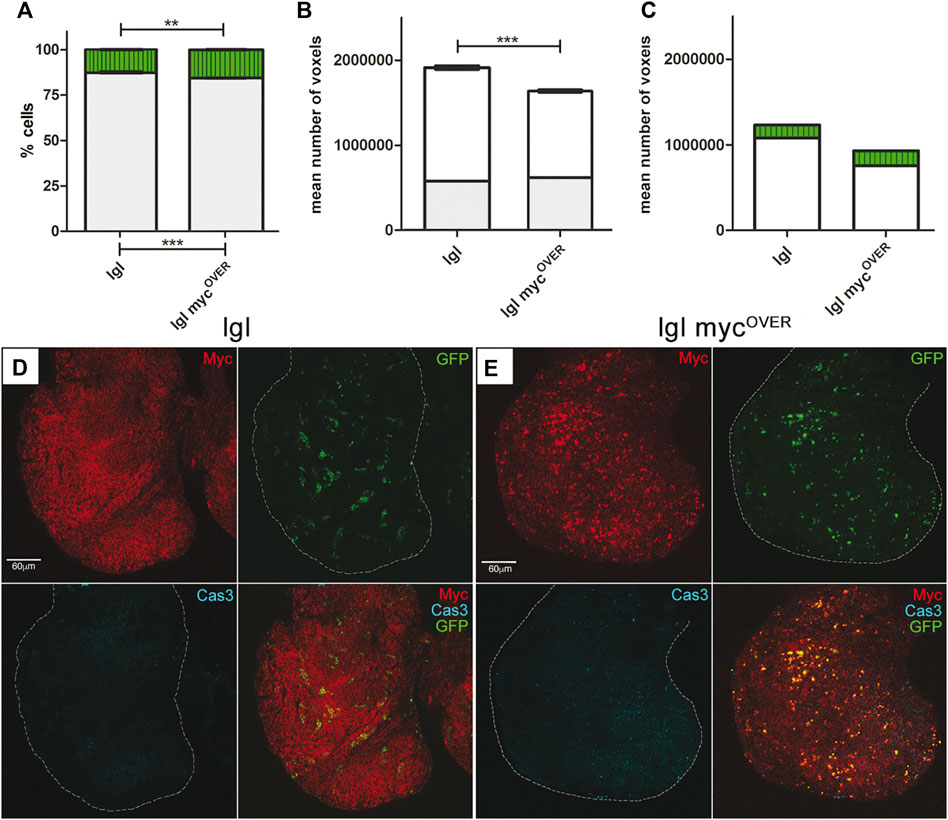
FIGURE 2. Apoptosis inhibition rescues both tumor size and composition. (A) Percentage of GFP+ (green) and GFP− (grey) cells in treated lgl tumors collected at 8 days AEL after induction of neutral (n = 64) and mycOVER (n = 62) clones at 6 days AEL. See Supplementary Table S1 for plain genotypes. (B) Mean volume of the same samples as in A calculated from the images acquired immediately before dissociation (8 days AEL). The grey part of the bars represents the pre-induction volume (6 days AEL, n = 58 for neutral and n = 55 for mycOVER samples). (C) Relative volumes of GFP+ (green) and GFP− (white) cells in neutral and mycOVER samples after clone induction and treatment, calculated as described in the Methods section. Statistical significance and ±SEM are indicated. (D) MYC (red) and activated Cas3 (cyan) staining of lgl treated tumors collected at 8 days AEL in which GFP+, lgl neutral clones were induced at 6 days AEL. (E) MYC (red) and activated Cas3 (cyan) staining of lgl treated tumors collected at 8 days AEL in which GFP+, lgl mycOVER clones were induced at 6 days AEL. All the images represent disc cross-sections. Scale bars are indicated for each sample.
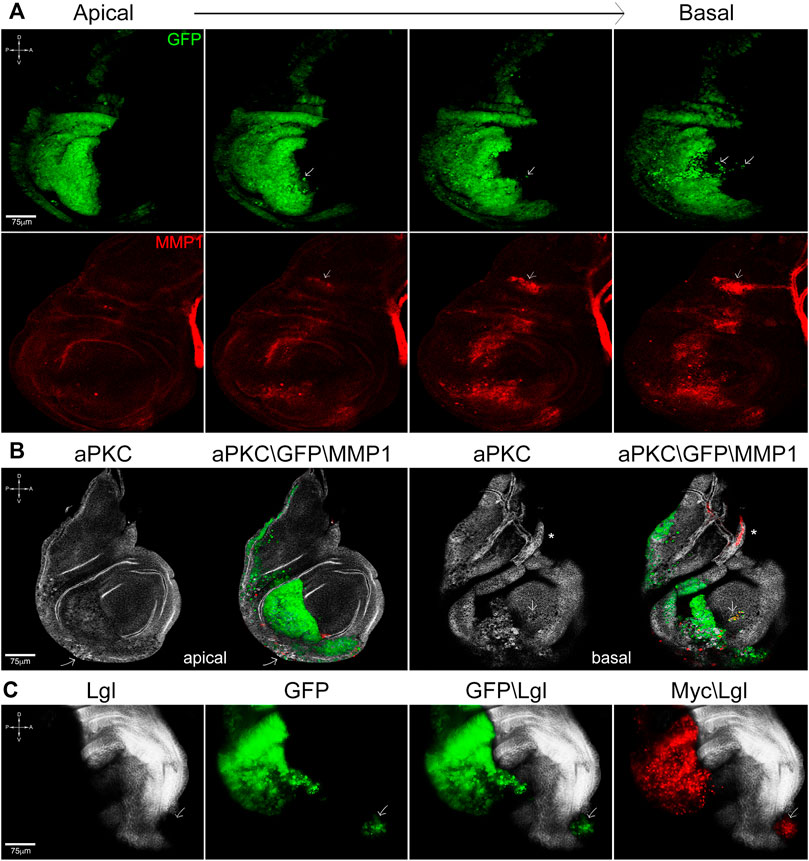
FIGURE 3. Cooperation between lgl knockdown and myc overexpression promotes cell migration and tissue disruption. (A) Apical-to-basal sections of en-lglKD, mycOVER wing discs showing GFP+ cells invading the anterior compartment (arrows) at the basal side of the disc epithelium. Cell migration is accompanied by MMP1 secretion (red), which also marks the air sac (arrow) and the tracheal tubes. (B) aPKC (white) and MMP1 (red) staining of en-lglKD, mycOVER wing discs showing loss of tissue structure and focal MMP1 secretion (arrow) at the apical face and noticeable cell migration at the basal face (arrow). The asterisk marks the tracheal structure. (C) Lgl (white) and Myc (red) staining of en-lglKD, mycOVER wing discs displaying local metastasis in the anterior compartment (arrow). See Supplementary Table S1 for plain genotypes. All the images represent disc cross-sections. Scale bars and A, p, D and V compartments are indicated for each sample.
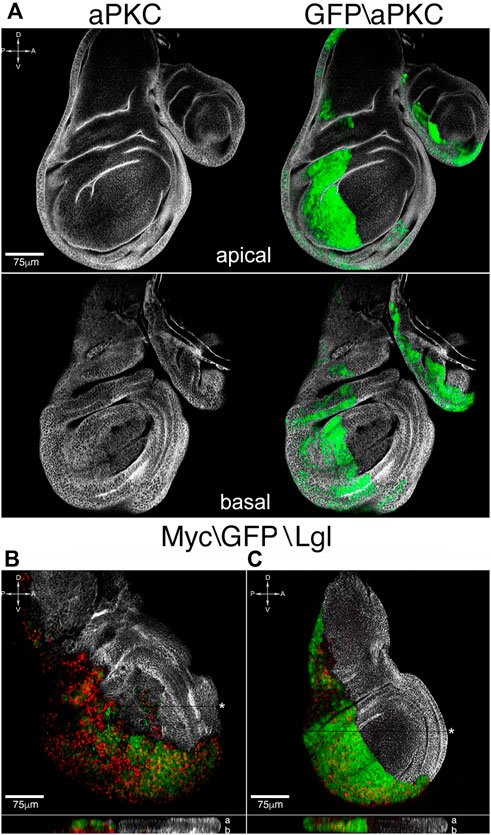
FIGURE 4. Apoptosis inhibition rescues tissue structure and cell migration. (A) Apical and basal sections of en-lglKD, mycOVER, dIAP1OVER wing discs showing normalization of tissue architecture and restriction border function. (B,C) Lgl (white) and Myc (red) staining of en-lglKD, mycOVER, dIAP1OVER wing discs (C) compared to en-lglKD, mycOVER counterparts (B). The reconstruction along the z axis shown at the bottom of each sample displays Myc-expressing nuclei going basal respect to GFP nuclei in (B), while they colocalize in (C). See Supplementary Table S1 for plain genotypes. All the images represent disc cross-sections; for xz images, the chosen section is indicated by a black stripe marked by an asterisk. In the z reconstructions, “a” and “b” stand for “apical” and “basal”. Scale bars and A, p, D and V compartments are indicated for each sample.
Figures 1, 2, Supplementary Figure S2, S3, S4: at 192 ± 2 h after egg laying (AEL), larvae displaying GFP+ cells were selected under a Nikon SMZ1000 fluorescence stereoscope, discarding those bearing clones in the fat body so to avoid possible systemic effects of Myc overexpression (Parisi et al., 2013). Collected larvae were dissected in cold PBS1X (Phosphate Buffer Saline, pH 7.5), overgrown wing discs were isolated paying attention to keep their shape unaltered, photographed (see after) and incubated with gentle agitation for 2.5 h in 1 ml PBT (4.5 mg/ml porcine trypsin-EDTA-Sigma-Aldrich-in PBS1X) prior to cell count. For the remaining figures, selected larvae were dissected at 120 ± 2 h AEL.
Discs from 192 ± 2 larvae were photographed each day before dissociation. 144 ± 2 h discs were also captured as a prior-to-treatment control. Major and minor axes were measured for each wing disc with ImageJ (NIH) and volumes were calculated approximating disc shape to a spheroid with depth = width. For cell volume, four different fields from confocal z stacks containing 55 ÷ 80 cells were measured for each sample. Cell perimeter was marked by aPKC staining. GFP+ and GFP− cell diameters were measured with ImageJ (NIH) and cell volumes were obtained approximating their shape to a sphere.
Vt = tumor volume, Vg = GFP+ cell volume; Vb = GFP− cell volume, Pg = GFP+ cell percentage, Pb = GFP− cell percentage. Total number is Ct = Vt (Vg×Pg + Vb×Pb), GFP+ and GFP− cell number is Cg = Ct×Pg and Cb = Ct×Pb. GFP+ and GFP− final volumes are: Cg×Vg and Cb×Vb. Volumetric values are reported in the graphs as mean number of voxels.
Tissue isolated from selected larvae were fixed and stained according to standard protocols. Primary antibodies: rabbit α-cleaved Caspase 3 (Cell Signaling #9961, 1:100), mouse α-dMyc (P. Bellosta, 1:5), rabbit α-aPKC (Santa Cruz Biotechnology #10800, 1:100), rat α-HA (Roche 11867423001, 1:100), mouse α-dIAP1 (B. Hay, 1:200), rabbit α-Lgl (D. Strand, 1:400), mouse α-MMP1 (DSHB, 1:50). Secondary antibodies: α-mouse 555 Alexa Fluor, 1:200 and α-rabbit or α-rat Cy5 DyLight (Jackson Laboratories), 1:500. Confocal images were processed as a whole with Adobe Photoshop software. All the images shown represent a single confocal stack if not otherwise specified.
Statistical significance was determined by using two-tailed, unpaired t-tests from at least three independent replicates and expressed as p values: p ≤ 0.05 = *, p ≤ 0.01 = **, p ≤ 0.001 = ***, p ≤ 0.0001 = ****. For the experiments as in Figures 1, 2A–C, sample size n) is given in the legend. For immunofluorescence analysis, 60÷90 discs were observed for each experiment and the most representative phenotypes were photographed. For the experiments as in Supplementary Figures S2 and S5C, caspase-positive and clone areas were calculated using the free ImageJ software from NIH, and sample size n) is indicated in the graph. Animals were randomized for drug feeding experiments. Results are presented as mean SEM in bar graphs which were created in GraphPad Prism 5. For graphs in 1B, 2B, SEM is indicated as the sum of the single SEM, following the error propagation rule.
It was recently demonstrated that cell competition is able to drive tumor initiation in different Drosophila organs (Eichenlaub et al., 2016; Suijkerbuijk et al., 2016), but what happens to cancer cells if some start to up-regulate MYC while contributing to an expanding tumor? What is the impact on cancer outcome? To answer these evolutionary questions, we developed an in vivo assay in Drosophila that allowed modulation of MYC levels in single cancer cells within a malignant mass growing in the animal. In Drosophila, the loss of function (LOF) of genes encoding polarity proteins, such as lgl, are known to trigger malignant growth in larval organs called the “wing discs”, which display aberrant gene expression (Bunker et al., 2015). lgl−/− epithelial organs display tissue disruption and neoplastic growth from 5 days after egg laying (AEL) onwards; the development of the mutant larvae lasts for about 10 days at 25 °C, with late individuals showing huge masses that fill their anterior half (Bilder, 2004). Therefore, to study the relevance of MMCC to cancer progression, we used a combination of the Flp-FRT and UAS-Gal4 binary systems (Caygill and Brand, 2016) to elicit the competitive behavior of lgl mutant cells through the induction of mycOVER GFP+ Flp-out clones at 6 days AEL, when the mutant wing disc shows evident structure aberrations (compare the wild-type and the lgl mutant discs in Supplementary Figure S1). All the parameters considered were compared with those obtained following induction of neutral GFP+ clones in the same experimental conditions. After two additional days of development, at 8 days AEL, we selected GFP+ larvae and, after sample imaging, we dissociated wing discs and counted GFP+ and GFP− cells. As can be seen in Figure 1A, GFP+ cell percentage (green) was much higher in the lgl mycOVER sample than in the lgl neutral sample (35,44% vs. 20,31%). This result showed that MYC clonal up-regulation in a tumor context drives the expansion of the MYC-overexpressing population. Concerning the final tumor mass, while before clone induction (at day 6) the two samples were comparable in size (Figure 1B, grey), at day 8 they appeared strikingly different: the average size of the lgl mycOVER samples was about 2.5 fold higher than that of the neutral samples (Figure 1B, white). With the aim to ascertain the post-induction contribution of GFP+ and GFP− cells to the final mass, we traced the cellular composition of the neutral and lgl mycOVER samples after clone induction, that is from day 6 to day 8 (See Materials and Methods for details). As can be observed in Figure 1C, both GFP+ and GFP− populations expanded in lgl mycOVER tumors with respect to the neutral controls: the GFP+ lgl mycOVER/lgl mass ratio was 6.5 and the GFP− lgl mycOVER/lgl mass ratio was 2.6. Therefore, the major variation was, as expected, ascribable to the GFP+ cells, but the GFP− cells were also found to contribute massively to the post-induction mass. This suggests that, in malignant tissues, some mutant cells may be competent to receive and transduce signals originating from the competitive niches, thus fueling non-cell-autonomous tumor growth.
With the aim to analyze the dynamics occurring between the GFP+ and the GFP− cells during cancer expansion, samples underwent immunofluorescence (IF) assays for MYC and Cas3. In Figure 1D, a neoplastic organ is represented in which neutral clones have been induced. Irrespective of the GFP signal, we can observe sporadic cells positive to Cas3 signal (cyan), in which MYC staining (red) is lower compared to the surrounding cells (arrows). Figure 1E shows a closer view of this phenomenon: some cells expressing very low levels of MYC (arrow) are committed to die, as confirmed by Cas3 staining. These data suggest that MMCC may shape cancer evolution through continuous selection of cells with higher MYC levels, such as it happens in fly and mouse developing organs (de la Cova et al., 2004; Moreno and Basler, 2004; Claveria et al., 2013; Sancho et al., 2013). In the lgl mycOVER samples, widespread Cas3 activation was observed in cells with low MYC levels encircled by or adjacent to GFP+ cells with high MYC expression (Figure 1F, arrows). Figure 1G shows a close-up of this phenomenon: large clusters of GFP− cells (arrows) succumb when surrounded by high GFP+, high MYC-expressing neighbors. The difference in caspase activation between lgl and lgl mycOVER samples was highly significant, as shown in Supplementary Figure S2, where three distant sections/disc were analyzed for caspase-positive areas. Besides a huge expansion of the winner population, we reported an increase of the GFP− mass in the lgl mycOVER sample compared to the lgl neutral sample (Figure 1C, white). It is known that dying cells activate non-apoptotic caspase functions, resulting in the release of pro-proliferative factors both in Drosophila and mammals (Fuchs and Steller, 2005); in a cancer context this may translate into massive overgrowth. We indeed observed several GFP− cells expressing MYC levels comparable to those induced by the mycOVER transgene. To exclude these cells had lost the GFP construct due to genomic instability, whose relevance in fly tumors is still controversial (Dekanty et al., 2012; Sievers et al., 2014), we took advantage of the HA epitope tag fused to the myc cDNA in the mycOVER construct to reveal its presence in the sample. No GFP− MYC+ HA+ cells were however found in 20 discs analyzed. Supplementary Figure S3 shows that all the GFP−, high-MYC expressing cells (arrows and dotted lines) are negative to HA staining, confirming they are native cells. Therefore, these cells may respond to the growth signals emanating from the dying cells by directly or indirectly upregulating MYC.
Increasing evidence supports a pro-tumorigenic role of cell death in different contexts, played by apoptosis-dependent proliferation or other mechanisms promoted by non-apoptotic caspase functions (Dabrowska et al., 2016; Gregory et al., 2016; Diwanji and Bergmann 2019; Xu et al., 2022). It is supposed that even the dying cells generated in a competitive niche may contribute to tumor expansion, but functional demonstration is missing in advanced cancers. To investigate this aspect of cell competition, we used a pan-caspase inhibitor successfully employed in Drosophila (Monier et al., 2015) to prevent apoptotic cell death following clone induction in the same experimental samples as in the previous paragraphs. As can be seen in Figure 2A, the lgl mycOVER sample showed a very limited expansion of the GFP+ cells (+2.5%) respect to that observed in the untreated sample (+15%, compare to Figure 1A). As can be observed in Supplementary Figure S4, the average volumetric difference between treated and untreated entire imaginal discs is appreciable under an optical stereoscope. Moreover, lgl mycOVER samples were slightly undersized with respect to the neutral samples (Figure 2B) due to a reduction of both the GFP+ and the GFP− masses (compare Figure 2C and Figure 1C), confirming a role for the dying cells in fueling the growth of all the competent cells in the field. This phenomenon does not emerge in the neutral samples, where cell death is sporadic (Figure 1D). Figures 2D, E displays clone distribution (GFP) and MYC/Cas3 staining in cross-sections of neutral and lgl mycOVER samples respectively. As can be seen, MYC expression is not affected by the treatment but, despite the presence of mycOVER cells, apoptotic death is nearly absent. Finally, no native cells seemed to increase their MYC levels (not shown), confirming that MYC upregulation in GFP− cells resulted from signals coming from dying cells. Another trait we found affected by apoptosis inhibition was larval lifespan: respect to the 5 days AEL of a wild-type larva, lgl mutant animals show a prolonged larval life that lasts about 10 days, at the end of which they die with no sign of puparium formation (Bilder, 2004); in this case, both treated and untreated lgl larvae, as well as untreated lgl mycOVER animals, did not show any modification of this trait, while 12 out of 87 (13,8%) lgl mycOVER treated larvae were recovered from the vial walls, displaying a mature puparium and an arrhythmic heartbeat, which continued up to 5 days after treatment (Video 1). This was an unexpected effect of caspase restriction, suggesting it can also relieve the systemic consequences of cancer burden. Further analysis is requested to address this relevant issue. To broaden our findings, we inhibited caspase activation in another well-characterized cancer model, which uses the MARCM system (Lee and Luo, 2001) to combine lgl and oncogenic Ras mutations in single cells within the tissue. In this model of cooperative oncogenesis, where mutant cells are known to outcompete wild-type neighbors while colonizing the epithelium, it is feasible to induce cancer onset in a clonal fashion, as it is in mammals (Richardson and Portela, 2018). As can be seen in Supplementary Figure S5, the mutant clones grown in the developing wing disc (GFP+) are much smaller in samples treated with the caspase inhibitor (Supplementary Figure S5B) respect to those untreated (Supplementary Figure S5A). The difference is statistically significant, as shown in the graph (Supplementary Figure S5C). Altogether, these data suggest that activation of the apoptotic pathways is a primary event underlying mass expansion in competitive cancer contexts.
The system used in the first set of experiments, while appropriate to study the impact of caspase inhibition on the progressive growth of advanced tumors, was not suitable to study its effect on other primary traits of cancer such as tissue disruption and cell migration, which start before the mass becomes overtly malignant. To analyze these traits, we used a distinct genetic system: briefly, the wing disc is divided into posterior (P) and anterior (A) compartments by a restriction border, which represents a robust barrier to cell intermingling during development; only mutations affecting cell fate can make them cross the P/A border (Wand and Dahmann, 2020). By using the engrailed (en) driver, active in P cells, we can modulate the expression of whatever gene, and the inclusion of a GFP transgene allows visualizing P cells shape and position in the tissue. We first analyzed the behavior of P cells promoted by lgl knockdown (lglKD) or myc overexpression (mycOVER) at 25°C. As can be seen in Supplementary Figure S6A, en-lglKD wing discs show sporadic tissue alterations, as shown by staining for the polarity determinant aPKC (white, asterisks), accompanied by focal protease secretion (Supplementary Figure S6A′, MMP1, red, asterisks). The arrows indicate the tracheal structure conveying oxygen to the disc cells, which physiologically expresses MMP1. GFP marks the P compartment in Supplementary Figure S6A″, which border is outlined in Supplementary Figure S6A and Supplementary Figure S6A’. en-mycOVER discs do not show signs of polarity disruption or protease secretion (Supplementary Figure S6B, B′), but display massive cell death instead, as it is known from the literature (Sollazzo et al., 2018; Supplementary Figure S6) and can be inferred by nuclear pyknosis in Supplementary Figure S6B” (arrows), where GFP marks the P compartment, which border is outlined in Supplementary Figure S6B and Supplementary Figure S6B’. When we combined the expression of the two transgenes, things changed radically: as can be appreciated in Figure 3A, which shows apical-to-basal sections of a representative en-lglKD, mycOVER wing disc, we observed GFP+ P cells trespassing on the A compartment (arrows), especially on the basal side of the disc, where the basement membrane favors motility by scaffolding cell adhesion and detachment (Vidal et al., 2007). The process was associated with MMP1 secretion (lower panel, red). The arrows point to the tracheal air sac which physiologically expresses MMP1. Concerning tissue structure, the polarity determinant aPKC appears to be severely affected both at the apical and basal sides of the disc (Figure 3B, white), where focal MMP1 secretion (red) supports cell motility (arrows). The asterisk indicates the tracheal structure. Migration of small clusters of cells, which was repeatedly observed in en-lglKD, mycOVER samples, generated local metastatic foci in 13 out of 123 wing discs analyzed (10.5%): an example of this behavior is found in Figure 3C, where the arrow points to a GFP+ lglKD (black) mycOVER (red) niche at the most anterior tip of the A compartment. In summary, while single lgl knockdown or myc overexpression failed in promoting carcinogenesis (Supplementary Figure S6), their combination favored cell migration and tissue disruption, possibly by coupling focal MMP1 secretion promoted by lglKD (Supplementary Figure S6A′) with growth signals due to massive cell death promoted by mycOVER (Supplementary Figure S6B″).
The anti-apoptotic protein dIAP1 is a central mediator of cell death in Drosophila, where it blocks the action of the initiator caspase Dronc (Vasudevan and Ryoo 2015). We carried out dIAP1 staining in en-lglKD mycOVER samples and saw a marked downregulation in the P compartment respect to the A counterpart (Supplementary Figure S7, red). We then induced dIAP1overexpression in the P cells and observed a rescue of tissue texture both at the apical and basal sides of the disc (Figure 4A, aPKC, white), together with an evident restriction of cell migration accompanied by a basal-to-apical shift of myc-expressing nuclei, that can be easily appreciated in the xz section shown at the bottom of the disc (compare the xz sections in Figure 4C and Figure 4B). In this case, apoptosis restriction was sufficient to block cell motility and rescue the correct disc shape and structure. Summing up the above findings, in this study we demonstrated that, in Drosophila epithelial tissues, distinct malignant traits occurring at different cancer stages depend on the activation of apoptotic programs and, given the strict conservation of the processes involved, we speculate comparable mechanisms are at work in mammalian cancers.
From the most recent studies in the field, it is emerging that the oncogenic potential of cancers growing under any kind of competitive pressure partly lies in the apoptotic death of the succumbing cells (Di Giacomo et al., 2017a). It is indeed known that the activation of caspases by cells entering the apoptotic program not only primes self-destruction, but also promotes the release of mitogenic signals, boosting proliferation of neighboring cells (Fuchs and Steller, 2015). A recent computational model of tumor growth identified cell competition and apoptosis as key drivers of cancer evolution (Pantziarka et al., 2019), and evidence about a pro-tumorigenic role of apoptosis, although not directly associated with cell competition, starts to surface in mammals (Labi and Erlacher, 2015). Using well-established Drosophila cancer models, here we showed that, under non-competitive conditions, epithelial tumors display sporadic cell death (Figures 1D,E), while its amount becomes relevant when the same cancers are forced to grow under competitive stress (Figures 1F, G). We also demonstrated the amazing overgrowth of cancer mass, together with the changes in cell composition (Figures 1A–C), mainly depends on caspase activation, as their inhibition was sufficient to take cancer size back to that of non-competitive tumors (Figure 2B). Then, we interrogated different fly cancer models to understand if apoptosis, beside accelerating cancer expansion, could be the driving force behind other aberrant cell behaviors in epithelial tumors. This analysis highlighted that cell migration, tissue disruption and local progression (Figure 3) are all counteracted by apoptosis inhibition (Figure 4), suggesting a prominent role for caspases in orchestrating a series of aberrant phenotypes during carcinogenesis. Altogether, our findings sound like a wake-up call: when chemotherapeutic drugs induce massive cell death in the tumor, will those dying cells activate non-apoptptic caspase functions and secrete factors that make low-proliferating cells (re)activate malignant pathways? A recent study found apoptosis to be a major player in cancer repopulation after chemotherapy in human cancer cells (Corsi et al., 2022), but antiblastic drugs are still the most rapid way to prevent organ failure in fast-growing cancers. Therefore, a fine genetic dissection of the intricate apoptotic pathways is mandatory to understand if the inhibition of the cascade at distinct levels may have different consequences on cancer evolution, so to develop target-specific therapies able to kill cancer cells without triggering secondary pro-tumorigenic effects.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
No animal studies are presented in the manuscript.
DG conceived the study; DG, SDG and MS designed the experiments; MS and SP performed the experiments; DG and SDG analyzed the data; DG wrote the paper.
The study was supported by AIRC (IG 12093 and IG 17252), Research Fellowships from the University of Bologna to SDG and MS and to DG FFO Grant 2021–2022 from Dept. Life, Health and Environmental Science, University of L’Aquila.
The authors want to thank Fabrizio Ferrè and Marilena Ignesti for scientific help, Bruce Hay and Paola Bellosta for reagents, the Bloomington Drosophila Stock Center for fly stocks and the Developmental Studies Hybridoma Bank at Iowa University for antibodies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1043630/full#supplementary-material
Baker, N. E., and Li, W. (2008). Cell competition and its possible relation to cancer. Cancer Res. 68, 5505–5507. doi:10.1158/0008-5472.CAN-07-6348
Bilder, D. (2004). Epithelial polarity and proliferation control: Links from the Drosophila neoplastic tumor suppressors. Genes Dev. 18, 1909–1925. doi:10.1101/gad.1211604
Bissell, M. J., and Radisky, D. (2001). Putting tumours in context. Nat. Rev. Cancer. 1, 46–54. doi:10.1038/35094059
Bunker, B. D., Nellimoottil, T. T., Boileau, R. M., Classen, A. K., and Bilder, D. (2015). The transcriptional response to tumorigenic polarity loss in Drosophila. eLife 4, e03189. doi:10.7554/eLife.03189
Caygill, E. E., and Brand, A. H. (2016). The GAL4 system: A versatile system for the manipulation and analysis of gene expression. Methods Mol. Biol. 1478, 33–52. doi:10.1007/978-1-4939-6371-3_2
Claveria, C., Giovinazzo, G., Sierra, R., and Torres, M. (2013). Myc-driven endogenous cell competition in the early mammalian embryo. Nature 500, 39–44. doi:10.1038/nature12389
Corsi, F., Capradossi, F., Pelliccia, A., Briganti, S., Bruni, E., Traversa, E., Torino, F., Reichle, A., and Ghibelli, L. (2022). Apoptosis as driver of therapy-induced cancer repopulation and acquired cell-resistance (crac): A simple in vitro model of phoenix rising in prostate cancer. Int. J. Mol. Sci. 23, 1152. doi:10.3390/ijms23031152
Dekanty, A., Barrio, L., Muzzopappa, M., Auer, H., and Milan, M. (2012). Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc. Nat. Acad. Sci. USA. 109, 20549–20554. doi:10.1073/pnas.1206675109
Dabrowska, C., Li, M., and Fan, Y. (2016). Apoptotic caspases in promoting cancer: Implications from their roles in development and tissue homeostasis. Adv. Exp. Med. Biol. 930, 89–112. doi:10.1007/978-3-319-39406-0_4
de Jong, J. S., van Diest, P. J., and Baak, J. P. A. (2000). Number of apoptotic cells as a prognostic marker in invasive breast cancer. Br. J. Cancer. 82, 368–373. doi:10.1054/bjoc.1999.0928
de la Cova, C., Abril, M., Bellosta, P., Gallant, P., and Johnston, L. A. (2004). Drosophila myc regulates organ size by inducing cell competition. Cell 117, 107–116. doi:10.1016/s0092-8674(04)00214-4
Di Giacomo, S., Sollazzo, M., de Biase, D., Ragazzi, M., Bellosta, P., Pession, A., et al. (2017b). Human cancer cells signal their competitive fitness through MYC activity. Sci. Rep. 7, 12568. doi:10.1038/s41598-017-13002-1
Di Giacomo, S., Sollazzo, M., Paglia, S., and Grifoni, D. (2017a). Myc, cell competition, and cell death in cancer: The inseparable triad. Genes 8, 120. doi:10.3390/genes8040120
Di Gregorio, A., Bowling, S., and Rodriguez, T. A. (2016). Cell competition and its role in the regulation of cell fitness from development to cancer. Dev. Cell. 38, 621–634. doi:10.1016/j.devcel.2016.08.012
Diwanji, N., and Bergmann, A. (2019). Two sides of the same coin - compensatory proliferation in regeneration and cancer. Adv. Exp. Med. Biol. 1167, 65–85. doi:10.1007/978-3-030-23629-8_4
Dow, L. E., and Humbert, P. O. (2007). Polarity regulators and the control of epithelial architecture, cell migration, and tumorigenesis. Int. Rev. Cytol. 262, 253–302. doi:10.1016/S0074-7696(07)62006-3
Eichenlaub, T., Cohen, S. M., and Herranz, H. (2016). Cell competition drives the formation of metastatic tumors in a Drosophila model of epithelial tumor formation. Curr. Biol. CB 26, 419–427. doi:10.1016/j.cub.2015.12.042
Ford, C. A., Petrova, S., Pound, J. D., Voss, J. J., Melville, L., Paterson, M., et al. (2015). Oncogenic properties of apoptotic tumor cells in aggressive b cell lymphoma. Curr. Biol. 25, 577–588. doi:10.1016/j.cub.2014.12.059
Froldi, F., Ziosi, M., Garoia, F., Pession, A., Grzeschik, N. A., Bellosta, P., et al. (2010). The lethal giant larvae tumour suppressor mutation requires dmyc oncoprotein to promote clonal malignancy. BMC Biol. 8, 33. doi:10.1186/1741-7007-8-33
Fuchs, Y., and Steller, H. (2015). Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell. Biol. 16, 329–344. doi:10.1038/nrm3999
Fulda, S. (2015). Targeting apoptosis for anticancer therapy. Semin. Cancer Biol. 31, 84–88. doi:10.1016/j.semcancer.2014.05.002
Gabay, M., Li, Y., and Felsher, D. W. (2014). Myc activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 4, a014241. doi:10.1101/cshperspect.a014241
Gallant, P. (2013). Myc function in drosophila. Cold Spring Harb. Perspect. Med. 3, a014324. doi:10.1101/cshperspect.a014324
Gregory, C. D., Ford, C. A., and Voss, J. J. (2016). Microenvironmental effects of cell death in malignant disease. Adv. Exp. Med. Biol. 930, 51–88. doi:10.1007/978-3-319-39406-0_3
Grifoni, D., Garoia, F., Bellosta, P., Parisi, F., De Biase, D., Collina, G., et al. (2007). Apkczeta cortical loading is associated with lgl cytoplasmic release and tumor growth in drosophila and human epithelia. Oncogene 26, 5960–5965. doi:10.1038/sj.onc.1210389
Grifoni, D., Garoia, F., Schimanski, C. C., Schmitz, G., Laurenti, E., Galle, P. R., et al. (2004). The human protein hugl-1 substitutes for drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene 23, 8688–8694. doi:10.1038/sj.onc.1208023
Grifoni, D., Sollazzo, M., Fontana, E., Froldi, F., and Pession, A. (2015). Multiple strategies of oxygen supply in drosophila malignancies identify tracheogenesis as a novel cancer hallmark. Sci. Rep. 5, 9061. doi:10.1038/srep09061
Grzeschik, N. A., Amin, N., Secombe, J., Brumby, A. M., and Richardson, H. E. (2007). Abnormalities in cell proliferation and apico-basal cell polarity are separable in drosophila lgl mutant clones in the developing eye. Dev. Biol. 311, 106–123. doi:10.1016/j.ydbio.2007.08.025
Grzeschik, N. A., Parsons, L. M., Allott, M. L., Harvey, K. F., and Richardson, H. E. (2010). Lgl, apkc, and crumbs regulate the salvador/warts/hippo pathway through two distinct mechanisms. Curr. Biol. 20, 573–581. doi:10.1016/j.cub.2010.01.055
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Jalalinadoushan, M., Peivareh, H., and Azizzadeh Delshad, A. (2004). Correlation between apoptosis and histological grade of transitional cell carcinoma of urinary bladder. Urol. J. 1, 177–179.
Johnston, L. A. (2014). Socializing with myc: Cell competition in development and as a model for premalignant cancer. Cold Spring Harb. Perspect. Med. 4, a014274. doi:10.1101/cshperspect.a014274
Labi, V., and Erlacher, M. (2015). How cell death shapes cancer. Cell Death Dis. 6, e1675. doi:10.1038/cddis.2015.20
Laconi, E., Doratiotto, S., and Vineis, P. (2008). The microenvironments of multistage carcinogenesis. Semin. Cancer Biol. 18, 322–329. doi:10.1016/j.semcancer.2008.03.019
Lee, T., and Luo, L. (2001). Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251–254. doi:10.1016/s0166-2236(00)01791-4
Menendez, J., Perez-Garijo, A., Calleja, M., and Morata, G. (2010). A tumor-suppressing mechanism in drosophila involving cell competition and the hippo pathway. Proc. Nat. Acad. Sci. USA. 107, 14651–14656. doi:10.1073/pnas.1009376107
Merino, M. M., Rhiner, C., Portela, M., and Moreno, E. (2013). Fitness fingerprints" mediate physiological culling of unwanted neurons in drosophila. Curr. Biol. 23, 1300–1309. doi:10.1016/j.cub.2013.05.053
Monier, B., Gettings, M., Gay, G., Mangeat, T., Schott, S., Guarner, A., et al. (2015). Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature 518, 245–248. doi:10.1038/nature14152
Morata, G., and Ripoll, P. (1975). Minutes: Mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 42, 211–221. doi:10.1016/0012-1606(75)90330-9
Moreno, E., and Basler, K. (2004). Dmyc transforms cells into super-competitors. Cell 117, 117–129. doi:10.1016/s0092-8674(04)00262-4
Moreno, E., Basler, K., and Morata, G. (2002). Cells compete for decapentaplegic survival factor to prevent apoptosis in drosophila wing development. Nature 416, 755–759. doi:10.1038/416755a
Moreno, E. (2008). Is cell competition relevant to cancer? Nat. Rev. Cancer. 8, 141–147. doi:10.1038/nrc2252
Naresh, K. N., Lakshminarayanan, K., Pai, S. A., and Borges, A. M. (2001). Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: A hypothesis to support this paradoxical association. Cancer 91, 578–584. doi:10.1002/1097-0142(20010201)91:3<578:aid-cncr1037>3.0.co;2-w
Paglia, S., Sollazzo, M., Di Giacomo, S., Strocchi, S., and Grifoni, D. (2020). Exploring MYC relevance to cancer biology from the perspective of cell competition. Semin. Cancer Biol. 63, 49–59. doi:10.1016/j.semcancer.2019.05.009
Pantziarka, P., Ghibelli, L., and Reichle, A. (2019). A computational model of tumor growth and anakoinosis. Front. Pharmacol. 10, 287. doi:10.3389/fphar.2019.00287
Parisi, F., Riccardo, S., Zola, S., Lora, C., Grifoni, D., Brown, L. M., et al. (2013). dMyc expression in the fat body affects DILP2 release and increases the expression of the fat desaturase Desat1 resulting in organismal growth. Dev. Biol. 379, 64–75. doi:10.1016/j.ydbio.2013.04.008
Rhiner, C., Diaz, B., Portela, M., Poyatos, J. F., Fernandez-Ruiz, I., Lopez-Gay, J. M., et al. (2009). Persistent competition among stem cells and their daughters in the drosophila ovary germline niche. Development 136, 995–1006. doi:10.1242/dev.033340
Sancho, M., Di-Gregorio, A., George, N., Pozzi, S., Sanchez, J. M., Pernaute, B., et al. (2013). Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev. Cell. 26, 19–30. doi:10.1016/j.devcel.2013.06.012
Schimanski, C. C., Schmitz, G., Kashyap, A., Bosserhoff, A. K., Bataille, F., Schafer, S. C., et al. (2005). Reduced expression of hugl-1, the human homologue of drosophila tumour suppressor gene lgl, contributes to progression of colorectal cancer. Oncogene 24, 3100–3109. doi:10.1038/sj.onc.1208520
Schreiber-Agus, N., Stein, D., Chen, K., Goltz, J. S., Stevens, L., and DePinho, R. A. (1997). Drosophila myc is oncogenic in mammalian cells and plays a role in the diminutive phenotype. Proc. Nat. Acad. Sci. USA. 94, 1235–1240. doi:10.1073/pnas.94.4.1235
Sievers, C., Comoglio, F., Seimiya, M., Merdes, G., and Paro, R. (2014). A deterministic analysis of genome integrity during neoplastic growth in Drosophila. PloS one 9, e87090. doi:10.1371/journal.pone.0087090
Simpson, P. (1979). Parameters of cell competition in the compartments of the wing disc of drosophila. Dev. Biol. 69, 182–193. doi:10.1016/0012-1606(79)90284-7
Sollazzo, M., Genchi, C., Paglia, S., Di Giacomo, S., Pessionm, A., de Biase, D., et al. (2018). High MYC levels favour multifocal carcinogenesis. Front. Genet. 9, 612. doi:10.3389/fgene.2018.00612
Suijkerbuijk, S. J., Kolahgar, G., Kucinski, I., and Piddini, E. (2016). Cell competition drives the growth of intestinal adenomas in drosophila. Curr. Biol. 26, 428–438. doi:10.1016/j.cub.2015.12.043
Sun, B., Sun, Y., Wang, J., Zhao, X., Wang, X., and Hao, X. (2006). Extent, relationship and prognostic significance of apoptosis and cell proliferation in synovial sarcoma. Eur. J. Canc. Prev. 15, 258–265. doi:10.1097/01.cej.0000198896.02185.68
Vasudevan, D., and Ryoo, H. D. (2015). Regulation of cell death by IAPs and their antagonists. Curr. Top. Dev. Biol. 114, 185–208. doi:10.1016/bs.ctdb.2015.07.026
Vidal, M., Warner, S., Read, R., and Cagan, R. L. (2007). Differing Src signaling levels have distinct outcomes in Drosophila. Cancer Res. 67, 10278–10285. doi:10.1158/0008-5472.CAN-07-1376
Villa del Campo, C., Claveria, C., Sierra, R., and Torres, M. (2014). Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep. 8, 1741–1751. doi:10.1016/j.celrep.2014.08.005
Villa Del Campo, C., Lioux, G., Carmona, R., Sierra, R., Munoz-Chapuli, R., Claveria, C., et al. (2016). Erratum: Myc overexpression enhances epicardial contribution to the developing heart and promotes extensive expansion of the cardiomyocyte population. Sci. Rep. 6, 37880–35366. doi:10.1038/srep37880
Wang, J., and Dahmann, C. (2020). Establishing compartment boundaries in Drosophila wing imaginal discs: An interplay between selector genes, signaling pathways and cell mechanics. Semin. Cell Dev. Biol. 107, 161–169. doi:10.1016/j.semcdb.2020.07.008
Xu, D. C., Wang, L., Yamada, K. M., and Baena-Lopez, L. A. (2022). Non-apoptotic activation of Drosophila caspase 2/9 modulates JNK signaling, the tumor microenvironment, and growth of wound-like tumors. Cell Rep. 39, 110718. doi:10.1016/j.celrep.2022.110718
Keywords: cell competition, cancer evolution, apoptosis, drosophila, lethal (2) giant larvae
Citation: Sollazzo M, Paglia S, Di Giacomo S and Grifoni D (2023) Apoptosis inhibition restrains primary malignant traits in different Drosophila cancer models. Front. Cell Dev. Biol. 10:1043630. doi: 10.3389/fcell.2022.1043630
Received: 13 September 2022; Accepted: 22 December 2022;
Published: 10 January 2023.
Edited by:
Jean-Philippe Chambon, Université de Montpellier, FranceReviewed by:
Luis Alberto Baena-Lopez, University of Oxford, United KingdomCopyright © 2023 Sollazzo, Paglia, Di Giacomo and Grifoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Grifoni, ZGFuaWVsYS5ncmlmb25pQHVuaXZhcS5pdA==
†Present address: Manuela Sollazzo, Mito B&B Lab, Department of Pharmacy and Biotechnology, University of Bologna, Bologna, Italy; Simone Di Giacomo, OaCP ResearchLAB, OaCP IE LTD, Cork, Ireland
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.