 Elizabeth Riffo1Mario Palma1
Elizabeth Riffo1Mario Palma1 Matías I. Hepp2Diego Benítez-Riquelme1
Matías I. Hepp2Diego Benítez-Riquelme1 Vicente A. Torres3
Vicente A. Torres3 Ariel F. Castro1
Ariel F. Castro1 Roxana Pincheira1*
Roxana Pincheira1*- 1Laboratorio de Transducción de Señales y Cáncer, Departamento de Bioquímica y Biología Molecular, Facultad de Ciencias Biológicas, Universidad de Concepción, Concepción, Chile
- 2Laboratorio de Investigación en Ciencias Biomédicas, Departamento de Ciencias Básicas y Morfología, Facultad de Medicina, Universidad Católica de la Santísima Concepción, Concepción, Chile
- 3Millennium Institute on Immunology and Immunotherapy, ICOD, Facultad de Odontología, Universidad de Chile, Santiago, Chile
SALL2/Sall2 is a transcription factor associated with development, neuronal differentiation, and cancer. Interestingly, SALL2/Sall2 deficiency leads to failure of the optic fissure closure and neurite outgrowth, suggesting a positive role for SALL2/Sall2 in cell migration. However, in some cancer cells, SALL2 deficiency is associated with increased cell migration. To further investigate the role of Sall2 in the cell migration process, we used immortalized Sall2 knockout (Sall2−/−) and Sall2 wild-type (Sall2+/+) mouse embryonic fibroblasts (iMEFs). Our results indicated that Sall2 positively regulates cell migration, promoting cell detachment and focal adhesions turnover. Sall2 deficiency decreased cell motility and altered focal adhesion dynamics. Accordingly, restoring Sall2 expression in the Sall2−/− iMEFs by using a doxycycline-inducible Tet-On system recovered cell migratory capabilities and focal adhesion dynamics. In addition, Sall2 promoted the autophosphorylation of Focal Adhesion Kinase (FAK) at Y397 and increased integrin β1 mRNA and its protein expression at the cell surface. We demonstrated that SALL2 increases ITGB1 promoter activity and binds to conserved SALL2-binding sites at the proximal region of the ITGB1 promoter, validated by ChIP experiments. Furthermore, the overexpression of integrin β1 or its blockade generates a cell migration phenotype similar to that of Sall2+/+ or Sall2−/− cells, respectively. Altogether, our data showed that Sall2 promotes cell migration by modulating focal adhesion dynamics, and this phenotype is associated with SALL2/Sall2-transcriptional regulation of integrin β1 expression and FAK autophosphorylation. Since deregulation of cell migration promotes congenital abnormalities, tumor formation, and spread to other tissues, our findings suggest that the SALL2/Sall2-integrin β1 axis could be relevant for those processes.
Introduction
SALL2/Sall2 is a transcription factor member of the Spalt-like (SALL) family conserved in many organisms, from nematodes to humans. SALL2 has an N-terminal zinc finger domain of the C2HC type, a glutamine-rich region, and several double and triple zinc fingers of the C2H2 type throughout its structure (Sweetman and Münsterberg, 2006; De Celis and Barrio, 2009). The SALL2 gene contains two alternative promoters, P1 and P2 controlling the expression of two main isoforms, E1 and E1A, respectively (Ma et al., 2001). These isoforms differ in the first 25 amino acids at the N-terminal domain. The E1 isoform contains a nuclear localization sequence, and a conserved repressor motif that is not present in E1A (Lauberth and Rauchman, 2006), suggesting isoform-specific functions. The E1 isoform is restricted to specific tissues, such as the brain, kidney, thymus, testis, and colon, while E1A has ubiquitous expression (Ma et al., 2001; Hermosilla et al., 2017). SALL2 was associated with development, neuronal differentiation, and cancer (Hermosilla et al., 2017; Álvarez et al., 2021). SALL2 deregulates in various cancer types, but its role in the disease is not entirely understood. Studies using mouse embryonic fibroblasts (MEFs) derived from the Sall2 knockout model suggest that SALL2/Sall2 behaves as a tumor suppressor by negatively regulating cell cycle progression (Sato et al., 2003; Hermosilla et al., 2018) and by inducing cellular apoptosis during genotoxic stress (Escobar et al., 2015). Consistent with these findings, SALL2 is downregulated in ovarian, lung, and colon cancer (Li et al., 2004; Chai et al., 2011; Farkas et al., 2021). However, clinical reports show that SALL2 is upregulated in other cancer types, such as glioblastoma and synovial sarcoma, suggesting an oncogenic role (Li et al., 2002; Nielsen et al., 2003; Estilo et al., 2009; Suvà et al., 2014).
Like other members of the SALL family, SALL1 and SALL4 (Wolf et al., 2014; Yuan et al., 2016; Itou et al., 2017), SALL2 was associated with the cell migration process. SALL2 silencing promoted cell proliferation, migration, and invasion in A2780 ovarian cancer cells (Miao et al., 2017). In addition, overexpression of SALL2 in radioresistant esophageal squamous cell carcinoma (ESCC) decreased cell migration and chemosensitivity (Luo et al., 2017). These studies suggest that SALL2 inhibits cell migration in these cancer contexts. Conversely, SALL2/Sall2 deficiency was associated with coloboma, a congenital disability characterized by failure to close the optic fissure during the embryonic development of the eye, causing blindness (Kelberman and Schlaepfer, 2017). Additionally, SALL2 interacts with the neurotrophin receptor p75NTR. It is required for nerve growth factor (NGF)-dependent neurite outgrowth of pheochromocytoma of the rat adrenal medulla (PC12) cells and hippocampal primary neurons (Pincheira et al., 2009). Because cell movement is needed for both closure of the optic fissure and neurite outgrowth, they suggest a positive role for SALL2/Sall2 in the migratory process in a normal context.
Cell migration is a complex and highly regulated process relevant to embryonic development, wound healing, and tumor dissemination. It initiates with cell polarity, followed by the generation of membrane protrusions and focal adhesions (FAs) at the leading edge, disassembly of FAs at the cell rear, and then cell traction (Trepat et al., 2012; SenGupta et al., 2021). For cells to migrate, they must sense different environmental signals through adhesion receptors, namely integrins, forming specialized FAs complexes, which mediate responses to these cues (Huttenlocher and Horwitz, 2011; Conway and Jacquemet, 2019). Integrins are a family of heterodimeric cell surface receptors consisting of one α and one β subunit. There are 18 α-subunits and 8 β-subunits, which can combine into 24 different heterodimers that recognize overlapping but distinct sets of extracellular ligands (Humphries et al., 2006). Integrin binding specificity is determined by their extracellular domain that recognizes diverse matrix ligands, including fibronectin (e.g., α5β1, α4β1), collagen (e.g., α1β1, α2β1), and laminin (e.g., α2β1, α6β1). In leukocytes, integrins also bind to soluble ligands such as the complement component iC3b and other cells by binding to ICAMs (αLβ2, αMβ2) and VCAM-1 (α4β1) molecules (Huttenlocher and Horwitz, 2011; Fagerholm et al., 2019). After integrin ligand-binding, several adaptor proteins like talin, vinculin, and paxillin, bind to the integrin cluster and recruit proteins with tyrosine kinase activity, such as focal adhesion kinase (FAK) (Wehrle-Haller, 2012a; Marsico et al., 2018). Once recruited in the adhesion site, FAK is autophosphorylated at tyrosine 397, generating a chain of successive phosphorylation events that modulate focal adhesion (FA) assembly-disassembly dynamics (Hamadi et al., 2005; Mitra, 2005). Polarized cell migration is characterized by asymmetric adhesion dynamics, where nascent adhesions mature into FAs or disassemble. The FAs disassembly involves weakening or disrupting the integrin-ECM and/or integrin-cytoskeletal linkages, with mechanisms that involve calpain-mediated proteolysis of FA proteins, endocytosis, and recycling of integrins, and degradation of the extracellular matrix (Wozniak et al., 2004; Wehrle-Haller, 2012b). Because of the essential role of integrins in cell migration and invasion, their deregulation leads to severe consequences. The absence of β1 is lethal. The absence or defective functions of β2, which plays essential roles in immune responses and inflammation (Gahmberg et al., 2019; Sun et al., 2021), results in leukocyte adhesion deficiency (LAD) syndromes. Defects in the αIIbβ3 integrin may result in Glanzmann’s thrombasthenia. The absence of leukocyte α4β1, LFA-1, and Mac-1 results in increased leukocytosis and susceptibility to microbial infections (Gahmberg et al., 2019). Integrins’ altered expression patterns and activities are frequent in many cancers, which could further promote tumor metastasis via downstream signaling pathways (Hamidi and Ivaska, 2018).
The role of SALL2/Sall2 in cell migration is controversial and mechanistically poorly explored. We used immortalized mouse embryonic fibroblasts (iMEFs) derived from previously characterized Sall2 knockout (KO) mice (Sato et al., 2003; Hermosilla et al., 2018) further to investigate the role of Sall2 in cell migration. Contrary to the previous findings in ovarian and esophageal squamous cell carcinoma (ESCC) cancer cell lines, our studies showed that Sall2 promotes cell migration. Sall2 regulated membrane protrusions, cell detachment, FAs maturation, and disassembly. Mechanistically, Sall2 increased FAK autophosphorylation at Y397. It positively regulated integrin β1 expression, increasing integrin protein levels and its availability at the cell surface. Finally, we demonstrated that Sall2 increases integrin β1 mRNA level by directly binding to and transactivating the ITGB1 promoter. Thus, our study identified integrin β1 as a novel Sall2 target. Taken together, we show that Sall2 promotes cell migration by regulating FA dynamics and increasing integrin β1 expression.
Materials and Methods
Reagents
Nocodazole (NZ; M1404), fibronectin (FN; F0895), protease inhibitor cocktail (P8340), phosphatase inhibitor cocktail II (P5726), polybrene (TR-1003), KAPA SYBR FAST qPCR Master Mix (2X) kit (KK4601) and antibodies raised against SALL2 (HPA004162), α-Tubulin (T902), FLAG (M2, F1804) and γ-Tubulin (T6557) were obtained from Sigma-Aldrich (St. Louis, MO United States). Lipofectamine 2000 (11668019), Alexa Fluor 488, 555 and 643 conjugated secondary antibodies (goat anti-mouse), Alexa Fluor 488 phalloidin (A12379) and the PE-conjugated CD29 antibody (eBioscience, #12-0291-82) were purchased from Invitrogen (Carlsbad, MI, United States). FAK Inhibitor compound PF562,271 was from Laviana pharma corporation (JS, China). Doxycycline (#14422) was from Cayman chemical (Ann Arbor, MI, United States). Puromycin dihydrochloride (sc-108071), normal mouse IgG2A (sc-3878), Integrin β1 (M-106, sc-8978), Integrin β1 (A-4, sc-374429), Integrin α4 (B-2, sc-376334), Integrin α6 (F-6, sc-374057), Calpain 2 (E-10, sc-373966), FAK (H-1, sc-1688), PTEN (A2B1, sc-7974), Talin (C-9, sc-365875), Integrin β3 (N-20, sc-6627) and Vinculin (7F9, sc-73614) antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, United States). Phospho-FAK Tyr397 (#3283) was purchased from Cell Signaling Technology (Danvers, MA, United States). Histone H3 (Ab1791) antibody was obtained from Abcam (Cambridge, United Kingdom). Acetyl-Histone H4 (06-598) antibody was purchased from Millipore (Burlington, MA, United States). Horseradish peroxidase–conjugated secondary antibodies (donkey anti-goat; goat anti-rabbit and goat anti-mouse) and Hoechst 33342 were obtained from Bio-Rad (Hercules, CA, United States).
Plasmids
The SV40 large T antigen expression pBSSVD2005 was a gift from David Ron (Addgene plasmid # 21826; http://n2t.net/addgene:21826; RRID: Addgene_21826). The pCW57-MCS1-2A-MCS2 (Addgene plasmid # 71782; http://n2t.net/addgene:71782; RRID: Addgene_71782) and pCW57-MCS1-P2A-MCS2 (Hygro) (Addgene plasmid # 80922; http://n2t.net/addgene:80922; RRID:Addgene_80922) were a gift from Adam Karpf, (Barger et al., 2019). For lentiviral infection, the pCMV-dR8.2 dvpr (Addgene plasmid # 8455; http://n2t.net/addgene:8455; RRID: Addgene_8455) and pCMV-VSV-G (Addgene plasmid # 8454; http://n2t.net/addgene:8454; RRID: Addgene_8454) were a gift from Bob Weinberg (Stewart et al., 2003). The EFIa-iTGB1 plasmid was a gift from Joan Massague (Addgene plasmid # 115799; http://n2t.net/addgene:115799; RRID:Addgene_115799) (Er et al., 2018). The pCDNA.3 FLAG_SALL2 was previously generated using the pCMV2-FLAG_SALL2 plasmid (Pincheira et al., 2009). To generate pCW57 Tet-On FLAG mSall2_E1A plasmid, FLAG mSall2_E1A was PCR amplified from pCMV2(NH)-FLAG mSall2_E1A, previously generated using the full-length mouse Sall2 coding sequence synthetized by GeneScrip (Escobar et al., 2015), using the following oligonucleotides: forward, 5′-GCAGGTCGACATGGACTACAAAGACGATGAC -3′ and reverse, 5′-GCAGCCTAGGTCATGGCATGGTGGGGTCATCTTT -3′ and then was subcloned into pCW57-MCS1-2A-MCS2 using SalI and AvrII restriction sites. The ITGB1 promoter_pGL4.30 was kindly provided by Dr. Masakazu Toi (Kyoto University, Japan; MTA (2019) to RP, Universidad de Concepción, Chile).
Isolation of primary MEFs and genotyping
Sall2 KO mice (Sato et al., 2003) were obtained by collaboration with Dr. Ruichi Nishinakamura (Kumamoto University, Japan; MTA to RP, Universidad de Concepción, Chile). Sall2+/+ and Sall2−/− mouse embryonic fibroblasts (MEFs) were isolated and genotyped as previously described (Escobar et al., 2015).
MEFs immortalization and lentiviral transduction
Primary Sall2+/+ and Sall2−/− MEFs (passage 4) were immortalized using simian virus 40 (SV40) large T antigen based on a modified protocol from Zhu et al. (Zhu et al., 1991). For transfection, we used Lipofectamine 2000 and 2 μg SV40 large T antigen expression vector. After cell transfection, we proceeded to select for low density. To complete the immortalization process, 5-6 post-transfection passages were carried out. The inducible reconstitution of Sall2 in immortalized Sall2−/− MEFs, was produced by transient transfection of pCMV-dR8.2 dvpr, pCMV-VSV-G, and pCW57 Tet-On FLAG mSall2_E1A into HEK293T cells in a 10 cm dish with Lipofectamine 2000 reagent according to the manufacturer’s instructions. Viral supernatants were collected at 24 h and passed through a 0.2 μm filter. Sall2−/− iMEFs were infected with viral supernatants containing polybrene at 8 μg/ml. For positive clone selection, a fresh medium containing 5 μg/ml of puromycin was used for 72 h. For Sall2_E1A induction, iMEFs were treated with doxycycline (1,000 ng/ml) for 24–48 h before each experiment. Like the generation of the inducible mSall2_E1A cellular model, the integrin β1 overexpression in Sall2−/− iMEFs was performed by transfecting HEK293T cells with the EFIa-iTGB1 plasmid or empty vector (negative control). For positive clone selection, a fresh medium containing 400 μg/ml of hygromycin was used for 96 h. After selection, the generation of the stable models was evaluated through western blot.
Cell culture
Sall2+/+ and Sall2−/− iMEFs, Tet-On Sall2 iMEFs, HEK293T (ATCC, Manassas, VA, United States; CRL-3216) and a previously generated SALL2 KO HEK293 cell model (Hermosilla et al., 2018) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Corning) supplemented with 10% (v/v) fetal bovine serum (FBS; Hyclone), 1% glutamine (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). The cell lines used in this study were regularly tested (6 months) for mycoplasma using EZ-PCR Mycoplasma Test Kit (Biological Industries).
Cell viability
iMEFs were seeded at 1 × 105 cells per 12-well plate. After 24 h, cells were serum-starved for 16 h. Survival rate was determined by using the trypan blue exclusion method. All experiments were performed in triplicate.
Wound healing assay
Cell culture dishes were previously covered with fibronectin (FN) 2 μg/ml, iMEFs were seeded at 4 × 104 cells per 24-well plate. After 48 h, cells were serum-starved overnight and wounded with a pipette tip. Phase contrast images were acquired at 0 and 16 h. Wound closure percentage was calculated by the change in wound area between 0 and 16 h. All experiments were performed in quintuplicate. For the integrin β1 blocking study, Sall2+/+ iMEFs were incubated at the time of wounding with 20 mg/L of anti-integrin β1 antibody (CD29) or non-immune IgG (mouse IgG2A). Non-immune IgG was used as a negative control. IncuCyte S3 system was used to perform a time-lapse video of the wound closure by iMEFs, every 30 min for a total period of 19 ½ h.
Transwell migration assay
Transwell inserts (Costar, 6.5-mm diameter, 8 um pore size) were previously covered with FN (2 μg/ml). iMEFs (2 × 104) were seeded into the upper chamber in serum-free medium, complete medium was placed into the bottom chamber. After 4 h, the cells remaining at the upper membrane surface were removed and migrating cells (on the lower membrane surface) were fixed with 10% v/v acetic acid, 10% v/v methanol solution, and stained with 0.4% (w/v) crystal violet for 10 min at room temperature. All experiments were performed in triplicate.
Immunofluorescence microscopy
iMEFs (4 × 104) were seeded on coverslips previously covered with FN (2 μg/ml). After 48 h, a wound was performed as an activator of migration. After wound completion (4 or 16 h), cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and incubated overnight at 4°C with primary antibodies (in blocking buffer, 3% bovine serum albumin in phosphate-buffered saline). After washing, fixed cells were incubated with Hoechst 33342 and Alexa fluor-conjugated secondary antibodies for 2 h. Images were obtained with LMS 780 spectral confocal system (Zeiss). For cell polarity analysis, the microtubule-organizing center (MTOC) was detected using γ-tubulin antibody. To measure the protrusion formation, actin filaments were stained with Alexa Fluor 488 phalloidin. For FAs analysis, vinculin protein was detected using a specific antibody. Identical exposure times and zoom (40x) were used for comparisons and quantification.
Cell detachment assay
iMEFs were seeded at 1 × 105 cells per 12-well plate. After 24 h, cells were incubated in ethylenediaminetetraacetic acid (EDTA) 0.05 mM at 37°C for 5, 10, and 15 min, respectively. As a control, untreated cells were used. Cells were fixed and stained with crystal violet for 10 min at room temperature. Then, crystal violet was extracted in 10% (v/v) acetic acid, and the absorbance at 590 nm was measured. The adherent cells were expressed as percentages, where 100% is A590, corresponding to the number of adherent cells before EDTA-detachment, and 0% is A590 of an empty well (no cells attached). All experiments were performed in triplicate.
Cell spreading
iMEFs (4 × 105) in suspension were allowed to attach and spread onto FN-coated 60-mm dishes (2 μg/ml) for the indicated periods (0, 10, 15, and 30 min). Samples were prepared for western blot analysis.
Focal adhesion dynamics analysis
For focal adhesion synchronization, iMEFs (2 × 104) were seeded on coverslips previously covered with FN (2 μg/ml). After 24 h, cells were treated with 10 μM nocodazole (NZ) in serum-free medium for 2 ½ h. NZ was washed-out with serum-free medium at 5, 10, 15, and 30 min. Subsequently, cells were fixed and prepared for immunofluorescence and stained with anti-vinculin antibody (Alexa 488). For inhibition of FAK autophosphorylation, PF562,271 was used at 1 μM concentration in conjunction with NZ treatment. The number of focal adhesions per cell was quantified at all time points. Identical exposure times and zoom (63x) were used for comparisons and quantification.
Western blot analysis
Proteins from cell lysates (30-50 μg of total protein) were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred for 16 h at 30 V to polyvinylidene fluoride (PVDF) membrane (Millipore; IPVH00010) using a wet transfer system. The PVDF membranes were blocked for 1 h at room temperature in 5% nonfat milk in TBS-T (TBS with 0.1% Tween) and incubated with primary antibody at an appropriate dilution at 4°C overnight in blocking buffer. After washing, the membranes were incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies diluted in TBS-T buffer. Immunolabeled proteins were visualized by ECL (RPN2209) or ECL prime (RPN2232) (Cytiva, Marlborough, MA, United States). For protein expression in vivo, proteins were extracted from brain tissues of 6–8 weeks old Sall2+/+ and Sall2−/− mice as previously described (Hermosilla et al., 2018).
Flow cytometry
For integrin β1 expression at the membrane surface, 2 × 105 iMEFs were washed, detached in 0.25% trypsin, and collected in 200 μL of cold sorting buffer (2% bovine serum albumin in phosphate-buffered saline). Then, the cells were incubated with 1 μg of PE-conjugated CD29 antibody in the dark for 30 min at 4°C. After incubation, cells were washed, resuspended in 500 μL of sorting buffer, and sorted using a BD FACSAria III cell sorter (BD Biosciences).
Bioinformatic analysis
Identification of putative SALL2/Sall2 sites was performed using a previously reported binding site matrix (consensus sequence GGG (T/C)GGG) (Gu et al., 2011) in Transcriptional Regulatory Element Database (TRED) (http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=analysisMatrixForm) with a cutoff score = 7.5 (Jiang et al., 2007). Sequences analyzed [–2000 bp from transcription start site (+1)] were obtained from Eukaryotic Promoter Database (EPD) (http://epd.vital-it.ch/). The comparative analysis of ITGB1 promoter derived from ChIP-seq data was performed as previously described (Farkas et al., 2021).
Real-time quantitative reverse transcription-PCR
Total RNA was extracted from cells with TRIzol reagent (Invitrogen; 15596026) according to the manufacturer’s instructions. RNA was treated with Turbo Dnase (Invitrogen; AM1907) to eliminate any residual DNA from the preparation. RNA (1 μg) was reverse transcribed using the Maloney murine leukemia virus reverse transcriptase (Invitrogen; 28025-01) and 0.25 μg of Anchored Oligo (dT) 20 Primer (Invitrogen; 12577-011). Real-time PCR was performed using KAPA SYBR FAST qPCR Master Mix (2X) kit and the AriaMX Real-Time PCR System (Agilent, Santa Clara, CA, United States) according to the manufacturer’s instructions. The thermal cycling variables used were as follows: 40 cycles at 95°C for 5 s and 60°C for 20 s. To control specificity of the amplified product, a melting-curve analysis was carried out. No amplification of unspecific product was observed. Amplification of RNA polymerase II (Polr2) was carried out for each sample as an endogenous control. Primers used for real-time reactions are summarized in Supplementary Table S1. The relative expression ratio of the Itgb1 gene was calculated using the standard curve method, using untreated Sall2+/+ iMEFs as reference.
Transient transfections and reporter gene assays
To evaluate ITGB1 promoter transcriptional activity, SALL2 KO HEK293 cells were transiently co-transfected with 1 μg of ITGB1 promoter, 0.125 μg of RSV-β-galactosidase (β-Gal), and 2 μg of FLAG_SALL2 (pCDNA.3 FLAG SALL2) or 1 μg of vector control per well. After 24 h, the transfected cells were lysed with reporter lysis 5X buffer (E4030; Promega, Madison, WI, United States) and centrifugated at 14000 × g to pellet cell debris. The supernatant was then assayed for luciferase and β-Gal activity using the manufacturer’s suggested protocols (Promega; E1483 and E4740, respectively). Luminescent reporter activity was measured using a Luminometer (Victor3; Perkin- Elmer). All transfections were normalized to β-Gal activity and performed in triplicate. Luciferase values were expressed as fold induction relative to the pGL3 vector control.
Chromatin immunoprecipitation assay
SALL2 KO HEK293 cells were seeded at 1 × 106 cells per 100-mm dish. After 24 h, cells were transiently transfected with 2 μg of pCDNA.3 FLAG_SALL2. Cell nuclei were sonicated in 300 μL of sonication buffer, using a Bioruptor plus sonicator (Diagenode) (40 times, 15 s on/15 s off each time, high potency), obtaining DNA lengths between 300 and 600 bp. Immunoprecipitations were carried out overnight at 4°C using 2 μg of FLAG (anti-FLAG antibody), 1 μg of H3 (anti-histone H3; Abcam), or 1 μg of acH4 (anti-histone H4 acetylated; Millipore) and 25 μg of chromatin. After crosslinking reversion, DNA was purified using DNA Clean & Concentrator™-25 kit (D4033, Zymo Research, Irvine, CA, United States). DNA was analyzed by PCR directed to ITGB1 promoter SALL2-specific proximal I and proximal II regions (-251/-127) and (-631/-297). The ITGB1 promoter region (-1986/-1878) was used as a negative control for SALL2 binding. Primers used for qPCR reactions are summarized in Supplementary Table S2. All PCR reactions were performed using KAPA SYBR FAST qPCR Master Mix (2X) kit containing 1 μL of input and 4 μL of IP samples.
Image quantification and statistical analysis
Confocal immunofluorescence and phase contrast images were manually analyzed using the ImageJ software. Data were analyzed using unpaired t-test performed with GraphPad Prism 9.3.1 software. Values averaged from at least three independent experiments were compared. A p-value of <0.05 was considered significant.
Results
Sall2 promotes cell migration
We investigated the role of Sall2 in cell migration by comparing the migration of iMEFs isolated from isogenic Sall2 knockout (Sall2−/−) and Sall2 wild-type (Sall2+/+) embryos immortalized with the large T antigen (Hermosilla et al., 2018). Figure 1A shows the expression of Sall2 in the Sall2+/+ iMEFs. We subjected iMEFs to an in vitro wound healing assay and evaluated cell migration 16 h post-scratching. As shown in Figure 1B and Supplementary movies S1, S2, Sall2−/− iMEFs exhibited a lower percentage of wound closure than the Sall2+/+ iMEFs (about 76% and 90%, respectively). There were no differences in cell viability between both genotypes at the time and in the conditions of the wound closure analysis (Supplementary Figure S1).
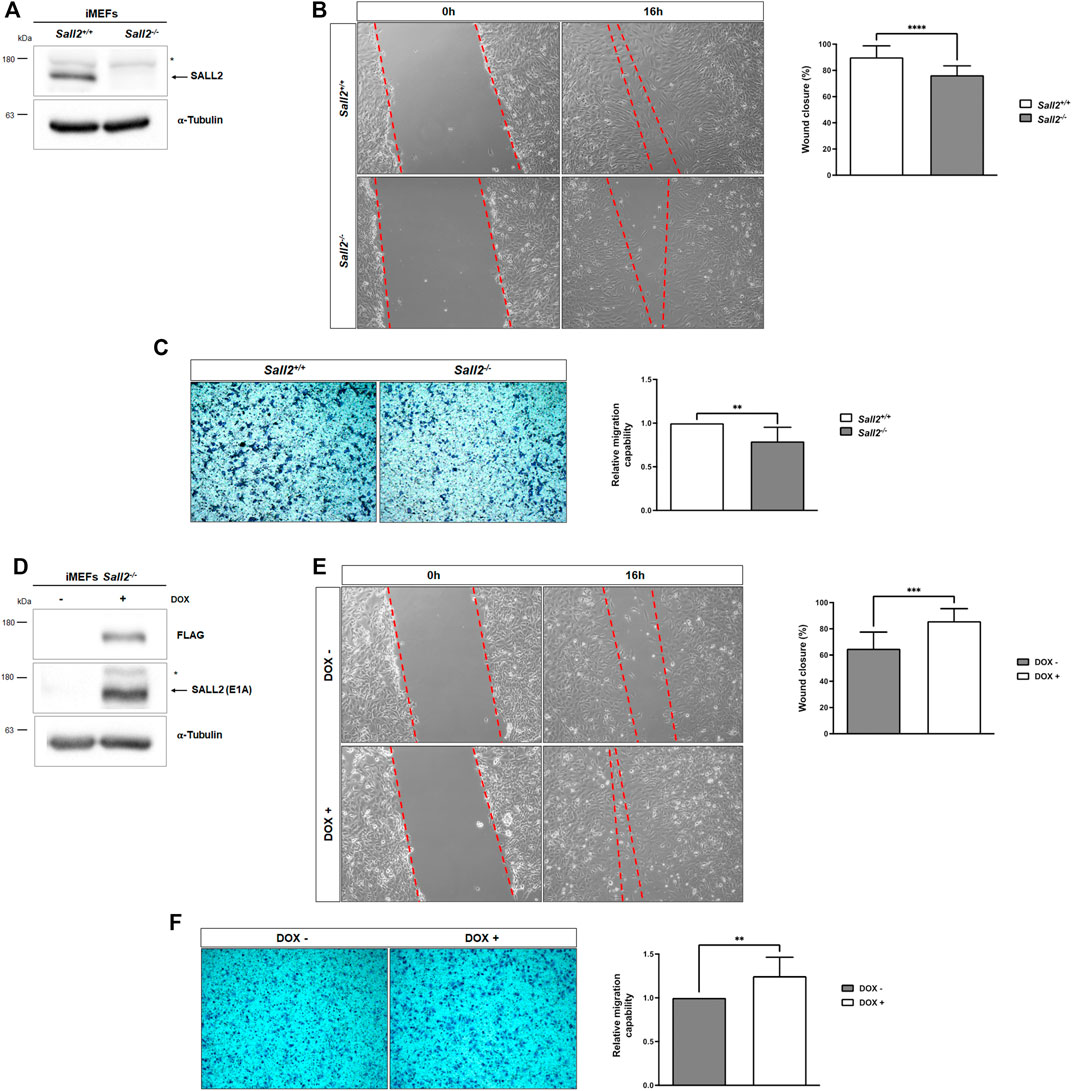
FIGURE 1. Sall2 promotes iMEFs migration. (A) Western blot analysis of endogenous Sall2 in Sall2+/+ and Sall2−/− iMEFs. The arrow indicates Sall2, and the asterisk corresponds to a nonspecific band. α-tubulin was used as the loading control. (B) Left, representative phase-contrast images (10x) at 0 and 16 h of in vitro wounding from Sall2+/+ and Sall2−/− iMEFs. Right, quantification of wound closure (as a percentage) at 16 h from images obtained in (B). (C) Left, representative microscopy images (10x) of transwell inserts from Sall2+/+ and Sall2−/− iMEFs after 4 h of cell migration. Right, quantification of the number of migrated cells from images obtained in (C). (D) Western blot analysis of mSall2 E1A isoform (FLAG) expression in the Tet-On Sall2 iMEFs inducible model (doxycycline (1,000 ng/ml, 48 h). (E) Left, representative phase-contrast images (10x) at 0 and 16 h of in vitro wounding from the Tet-On Sall2 iMEFs. Right, quantification of wound closure (as a percentage) at 16 h from images obtained in (E). (F) Left, representative microscopy images (10x) of transwell inserts from the Tet-On Sall2 iMEFs model after 4 h of cell migration. Right, quantification of the number of migrated cells from images obtained in (F). Data is expressed as mean ± SD from three independent experiments performed in quintuplicate and triplicated, respectively (**p = 0.001 to 0.01, ***p = 0.0001 to 0.001, ****p ≤ 0.0001; unpaired t-test).
We performed transwell migration assays further to analyze the effects of Sall2 in cell migration. As expected, Sall2−/− iMEFs exhibited fewer migratory cells than Sall2+/+ cells (Figure 1C) (about 21% less). To confirm the role of Sall2 in cell migration, we restored its expression in the Sall2−/− iMEFs. Since Sall2+/+ iMEFs mainly express the Sall2 E1A isoform (Hermosilla et al., 2018), we used a Tet-On system to generate a Sall2 E1A inducible cellular model (Tet-On Sall2 iMEFs) (Figure 1D). Consistent with the above results, induction of Sall2 expression by doxycycline increased cell migration (wound closure percentage from 65% to 86%, Figure 1E) and increased the number of migratory cells (about 20% more) (Figure 1F). Altogether, these results demonstrated that Sall2 promotes cell migration in iMEFs cells.
Sall2 decreases the lamellipodia size and promotes cell detachment
To determine how Sall2 promotes cell migration, we evaluated the requirement of Sall2 expression on the different steps involved in cellular movement, including cell polarity, membrane protrusion formation, and cell adhesion.
First, we assessed cell polarization. Like other studies in MEFs, we evaluated polarity establishment at 4 h after wounding (Grande-García et al., 2007; Tomar et al., 2009). The maintenance of cell polarity was evaluated at 16 h (Li and Gundersen, 2008), when the wound edges were still distinguishable, and migration was almost complete (Supplementary Figures S2B,E). Cell polarity was measured by analyzing the localization of the microtubule-organizing center (MTOC) relative to the nucleus following the induction of cell migration. The MTOC positioning was grouped into four grades, as previously described (Silverman-Gavrila et al., 2011), where grade 1 corresponds to a highly polarized state, grade 2 polarized, grade 3 non-polarized, and grade 4 to a highly non-polarized state (Supplementary Figure S2A). Our results showed no significant differences between Sall2 genotypes in cell polarization at 4 and 16 h (Supplementary Figures S2C,F). Similar results were observed when comparing highly polarized grade (G1) with medium polarized grade (G2). As shown in Supplementary Figures S2D,G, there were no significant differences between Sall2−/− and Sall2+/+ cells. All these results indicated that Sall2 is not required to induce cell polarity, polarization grade, and cell polarity maintenance over time.
We next evaluated whether Sall2 affects the membrane protrusions. We stained actin filaments after cell migration induction (Figure 2A) and analyzed the formation of lamellipodium and filopodium structures by quantifying their number, size, and polarization state. As shown in Figures 2B,C, Sall2−/− iMEFs depicted a higher lamellipodia area (mean = 352 versus 301 mm2) and exhibited a slightly lower filopodia number (mean = 23 versus 25) than Sall2+/+ cells. Still, there were no significant differences in the lamellipodia number, lamellipodia polarization state, filipodia length, and filipodia polarization state when comparing genotypes (Supplementary Figures S3A–D). Our data suggest that Sall2 mainly regulates the area of the lamellipodia.
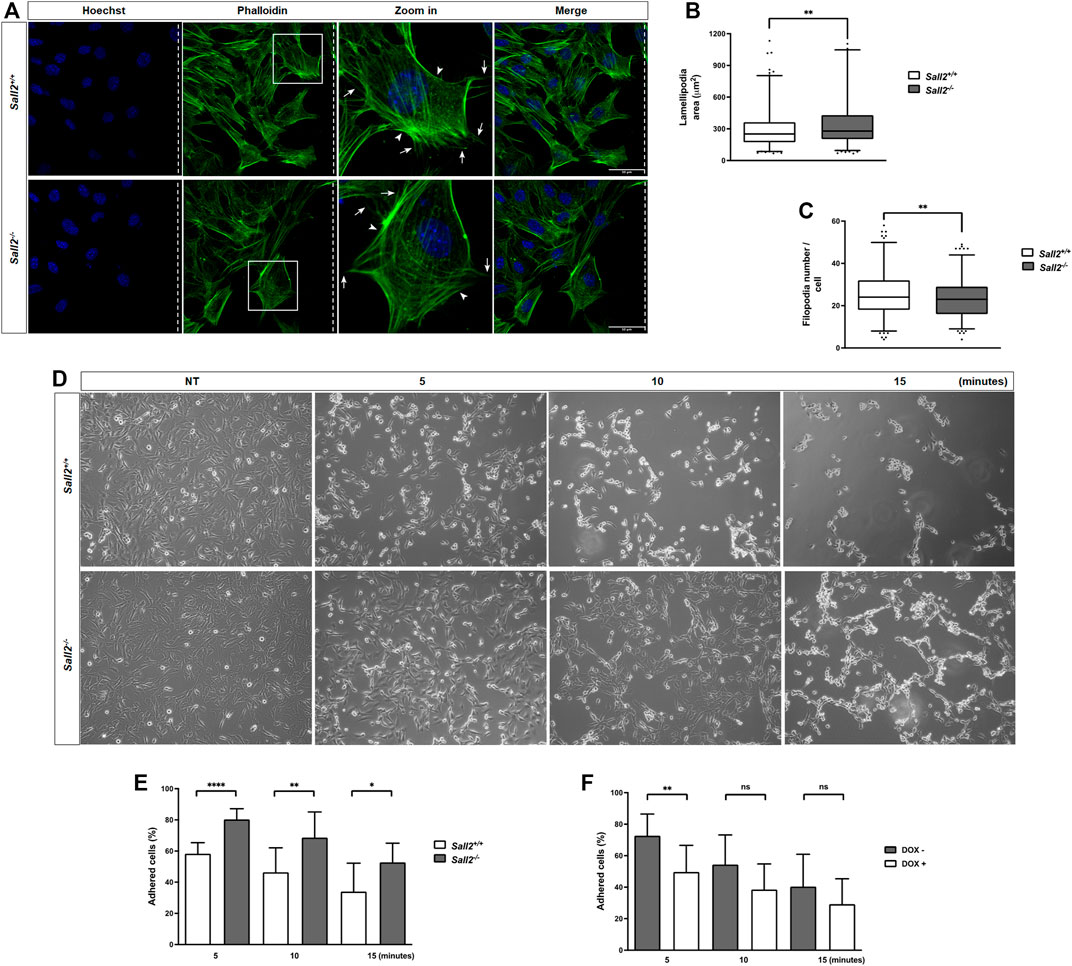
FIGURE 2. Sall2 decreases the lamellipodia size and promotes cell detachment. (A) Representative confocal images (40 ×) from Sall2+/+ and Sall2−/− iMEFs after cell migration induction at 16 h. Cells were fixed and stained with phalloidin (F- actin, green) and Hoechst (nuclei, blue). Dashed lines indicate the position of the wound, and arrows and arrowheads show the filopodia and lamellipodia, respectively. (B) Quantification of lamellipodia area from images obtained in (A). (C) Quantification of filopodia number from images obtained in (A). Data of cell protrusions analysis are shown as a box-and-whiskers plot from three independent experiments. The whiskers represent the 2.5th and 97.5th percentile, the box extends from the 25th to the 75th percentile and the line indicates the median. For each experiment at least 100 cells were analyzed per genotype. (D) Sall2+/+ and Sall2−/− iMEFs were treated with EDTA-containing solution for the indicated times, then fixed and evaluated by microscopy. Representative phase contrast images (10x) from each time point are shown. (E,F) Quantification of the percentage of adherent cells from Sall2+/+ and Sall2−/− iMEFs (E) and the Tet-On Sall2 iMEFs inducible model (F) performed as described in the MATERIALS AND METHODS section. Data of cell detachment assays are expressed as mean ± SD from three independent experiments performed in triplicate. (n.s, not significant, *p = 0.01 to 0.05, **p = 0.001 to 0.01, ****p ≤ 0.0001; unpaired t-test).
Additionally, we evaluated whether Sall2 regulates the cell adhesion process. We assessed cells binding to the extracellular matrix by comparing the detachment rate induced by adding EDTA. As shown in Figure 2D, iMEFs treated with an EDTA-containing solution resulted in a time-dependent detachment of cells. Interestingly, Sall2−/− cells exhibited a higher percentage of adherent cells (80% at 5 min, 69% at 10 min, and 53% at 15 min) than Sall2+/+ iMEFs (58% at 5 min, 46% at 10 min and 34% at 15 min) (Figure 2E). Accordingly, induction of Sall2 expression in the Tet-On Sall2 iMEFs significantly decreased the percentage of adherent cells at all times of EDTA treatment (47% at 5 min, 38% at 10 min, and 29% at 15 min), as observed in Sall2+/+ cells (Figure 2F). Our results support that Sall2 promotes cell detachment, suggesting a novel role of Sall2 in cell adhesion.
Sall2 decreases focal adhesions maturation and promotes focal adhesion dynamics
To further investigate the role of Sall2 in cell adhesion, we evaluated whether Sall2 regulates focal adhesions by quantifying their number, length, and fluorescence intensity. After induction of cell migration in Sall2+/+ and Sall2−/− iMEFs and the Tet-On Sall2 iMEFs model, we measured focal adhesion (FA) by immunocytochemistry of vinculin (Figures 3A,F), a protein involved in FA formation (Legate and Fässler, 2009; Wehrle-Haller, 2012a). As shown in Figure 3B, the FA number in Sall2−/− iMEFs was significantly higher than in Sall2+/+ cells (a mean of 15 versus 12 FA/cell). In addition, FA length (mean = 3.5 versus 2.7 mm) and FA fluorescence intensity (mean = 5,034 versus 4,088 a.u.) in Sall2−/− iMEFs were also significantly higher in comparison with Sall2+/+ cells (Figures 3C,D). Accordingly, Sall2 induction by doxycycline in Tet-On Sall2 iMEFs decreased the number, length, and fluorescence intensity of FAs, like the Sall2+/+ cells (Figures 3G–I). No significant differences in the polarization state of FAs between Sall2−/− and Sall2+/+ cells (Figure 3E) or after induction of Sall2 expression (Figure 3J) were observed. Since FA formation is a dynamic process where the FAs can disassemble or mature into stabilized complexes characterized by increased size (Gardel et al., 2010; Wehrle-Haller, 2012b), our results suggested that Sall2 negatively regulates the maturation of focal adhesions, as well as their number.
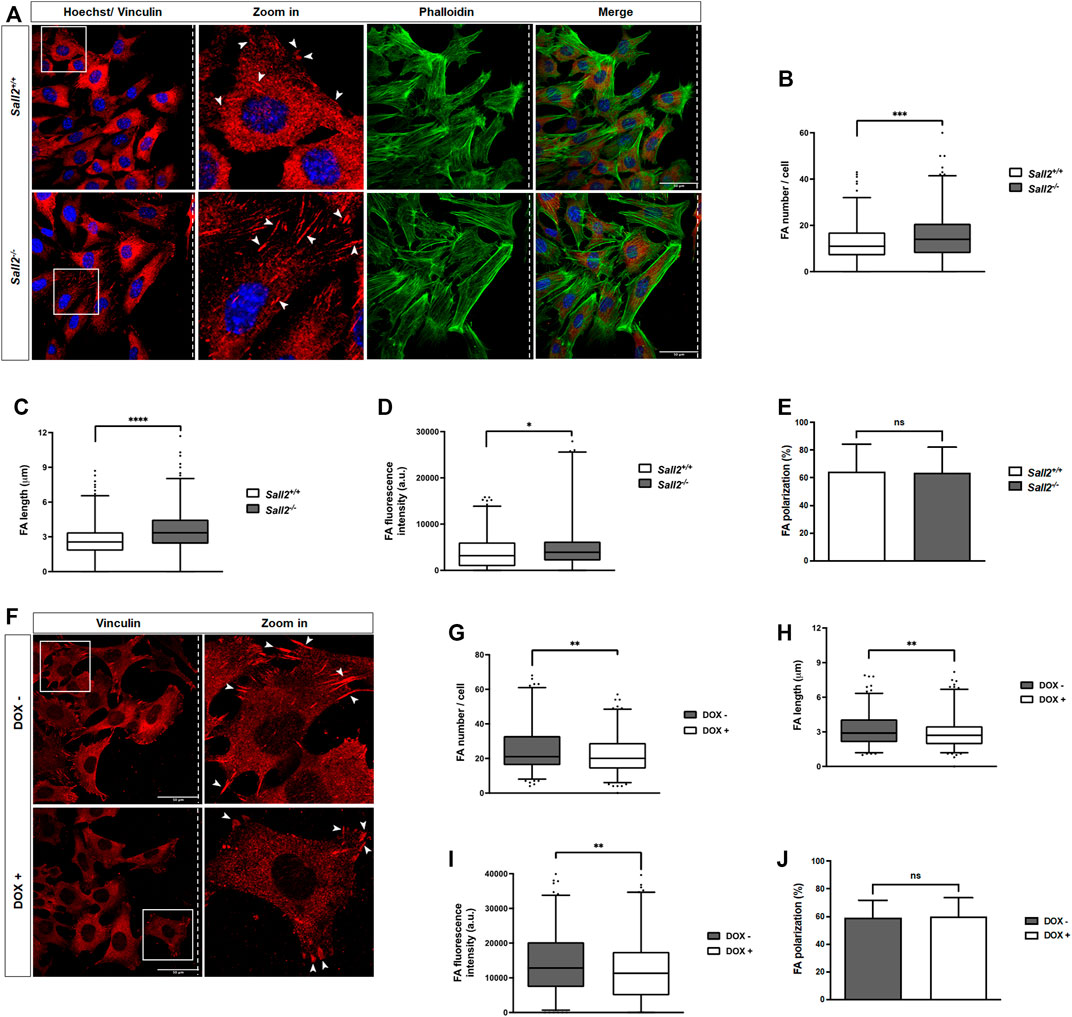
FIGURE 3. Sall2 decreases focal adhesions maturation. (A) Representative confocal images from Sall2+/+ and Sall2−/− iMEFs after cell migration induction at 16 h (40x). Cells were fixed, and FAs were stained with anti-vinculin antibody (red), phalloidin stained F- actin (green), and Hoechst stained the nuclei (blue). Dashed lines indicate the position of the wound, and the arrowheads show FA. (B–E) Quantification of FA number per cell (B), FA length (C), FA fluorescence intensity (D), and FA polarization (E) from Sall2+/+ and Sall2−/− cells. (F) Representative confocal images from the Sall2 Tet-On iMEFs inducible model after cell migration induction at 16 h (40x). (G–J) Quantification of FA number per cell (G), FA length (H), FA fluorescence intensity (I), and FA polarization (J) from the Tet-On Sall2 iMEFs. For each experiment at least 100 cells were analyzed per genotype. Data are shown as a box-and-whiskers plot from three independent experiments. The whiskers represent the 2.5th and 97.5th percentile, the box extends from the 25th to the 75th percentile, and the line indicates the median. (n.s, not significant, *p = 0.01 to 0.05, **p = 0.001 to 0.01, ***p = 0.0001 to 0.001, ****p ≤ 0.0001; unpaired t-test).
Considering these observations, we next evaluated whether Sall2 regulates FA dynamics. FAs were stabilized by microtubule depolymerization using nocodazole (Mendoza et al., 2013) at the concentration and time previously optimized (Figure 4A; Supplementary Figure S4). As expected, nocodazole increased the number of mature FAs compared with the non-treated cells ‘NT’. In contrast, nocodazole wash-out was followed by a progressive decrease of FA number, as observed in both genotypes (Figure 4B). Interestingly, Sall2−/− iMEFs exhibited delayed kinetics of FA disassembly (about 5 min) compared with Sall2+/+ cells (Figure 4C). In addition, Sall2 deficiency decreased the FA disassembly’s initial velocity (v0-D from 0.99 ± 0.1 to 0.55 ± 0.1). Conversely, we did not observe significant differences in FA assembly (kinetics and initial velocity of FA assembly, v0-A) when comparing Sall2 genotypes (Figure 4C). Accordingly, the induction of Sall2 expression significantly accelerated FAs disassembly (v0-D from 0.46 ± 0.1 to 0.73 ± 0.1), but not their assembly (Figure 4D). These results were supported by biochemical analysis of FAK autophosphorylation at Y397 (Hamadi et al., 2005). Phospho-Y397-FAK was evaluated after nocodazole release and during spreading on fibronectin at different time points (both approaches are widely used to assess changes in FAK activation, Nader et al., 2016; Erusappan et al., 2019). Phospho-Y397-FAK levels after 15 min of nocodazole wash-out were significantly lower in Sall2−/− iMEFs than in control cells (Figure 4E). Similarly, the phosphorylation of FAK at Y397 after 10 min of cell spreading was significantly lower in Sall2−/− iMEFs than in Sall2+/+ cells (Figure 4F). We further investigated the role of FAK activity in the Sall2-dependent regulation of FA dynamics. To this aim, Sall2+/+ iMEFs were treated with PF562,271, a specific inhibitor of FAK. As expected, PF562,271 decreased FAK- Y397 phosphorylation (Figure 4G). Like the Sall2−/− iMEFs phenotype, the treatment with PF562,271 decreased the FAs disassembly of Sall2+/+ iMEFs after 10 min of nocodazole wash-out. (Figure 4H). We observed a decrease in the initial velocity of FA disassembly (v0-D from 0.80 ± 0.1 to 0.30 ± 0.1) and assembly (v0-A from 0.80 ± 0.1 to 0.20 ± 0.1) (Figure 4I). Altogether, these results indicate that Sall2 promotes FAK activation and, consequently, the focal adhesion turnover.
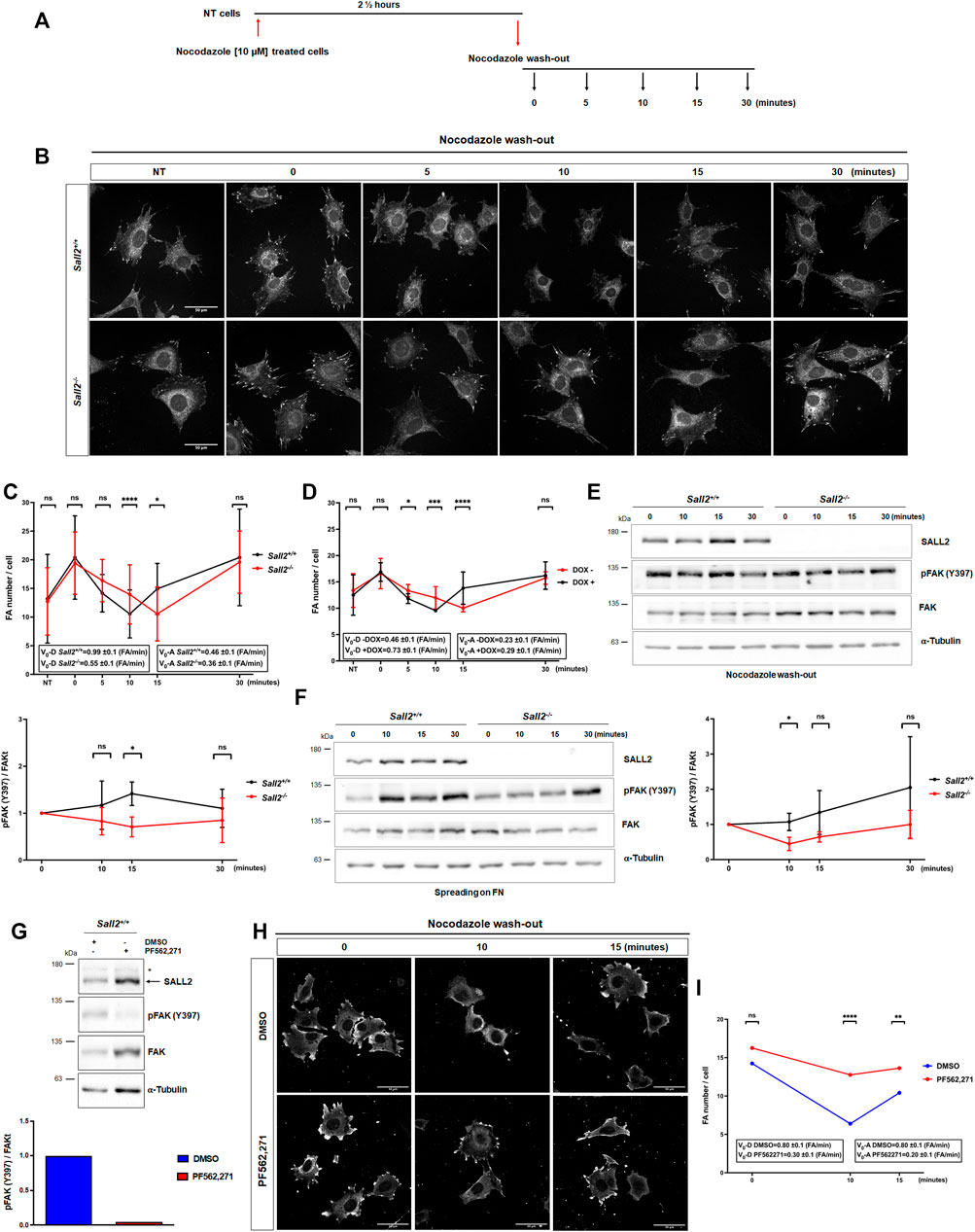
FIGURE 4. Sall2 promotes focal adhesion dynamics. (A) Schematic representation of nocodazole treatment and wash-out. (B) Representative confocal images (63x) from Sall2+/+ and Sall2−/− iMEFs incubated with 10 μM of nocodazole in serum-free medium. Nocodazole was washed-out at different times. Cells were fixed, and FAs were stained with anti-vinculin antibody (white). (C,D) Quantification of FA number per cell from Sall2+/+ and Sall2−/− iMEFs (C) and the Tet-On Sall2 iMEFs inducible model (D). Graphs included initial velocity (Vo-D) of FA disassembly and initial velocity (Vo-A) of FA assembly. For each experiment, at least 60 cells were analyzed per condition. (E) Left, FAK autophosphorylation (Y397) was evaluated by western blot at different times of nocodazole wash-out. Right, FAK autophosphorylation was quantified by densitometric analysis with respect to t = 0 min of nocodazole wash-out and normalized to total FAK and α-tubulin expression. (F) Left, FAK autophosphorylation (Y397) was evaluated by western blot at different times of spreading on fibronectin (FN). Right, FAK autophosphorylation was quantified by densitometric analysis with respect to t = 0 min of cell spreading and normalized to total FAK and α-tubulin expression. (G) Western blot of FAK autophosphorylation (Y397) and densitometric quantification from Sall2+/+ iMEFs treated with DMSO or 1 μM PF562,271. (H) Representative confocal images from Sall2+/+ iMEFs at different times of nocodazole wash-out treated with DMSO or 1 μM PF562,271. FAs were stained with anti-vinculin antibody (white). (I) Quantification of FA number per cell from Sall2+/+ iMEFs treated with DMSO or 1 μM PF562,271. Initial velocity (Vo-D) of FA disassembly and initial velocity (Vo-A) of FA assembly are indicated. For each experiment, at least 60 cells were analyzed per condition. Data are expressed as mean ± SD from three independent experiments (n.s, not significant, *p = 0.01 to 0.05, **p = 0.001 to 0.01, ***p = 0.0001 to 0.001, ****p ≤ 0.0001; unpaired t-test).
Sall2 promotes integrin β1 expression
To determine the underlying mechanism by which Sall2 promotes FA dynamics, we analyzed the expression of different proteins involved in FA assembly-disassembly, such as integrins, vinculin, talin, and calpain 2. The analysis was performed under the conditions where the highest differences in FAK activation were observed (15 min after nocodazole release and 10 min of cell spreading) and under normal growth conditions. Although Sall2+/+ cells depicted higher levels of FAK phosphorylation (Figures 4E,F), Sall2 deficiency was associated with higher FAK protein levels (Supplementary Figures S5B–G). These results suggested that Sall2-mediated FAK activation does not relate to positive regulation of FAK expression. In addition, we found that Sall2−/− iMEFs have significantly lower protein levels of integrin β1 than Sall2+/+ cells in all conditions analyzed (Figures 5A–C). Alpha subunits analysis showed slightly lower integrin α4 levels in Sall2−/− iMEFs than Sall2+/+ cells under normal growth conditions. However, we did not observe significative differences in the integrin α6 expression levels (Supplementary Figure S6). Additionally, we did not observe differences in the expression levels of vinculin, talin, calpain2 (Supplementary Figure S5), and integrin β3 (Supplementary Figure S6).
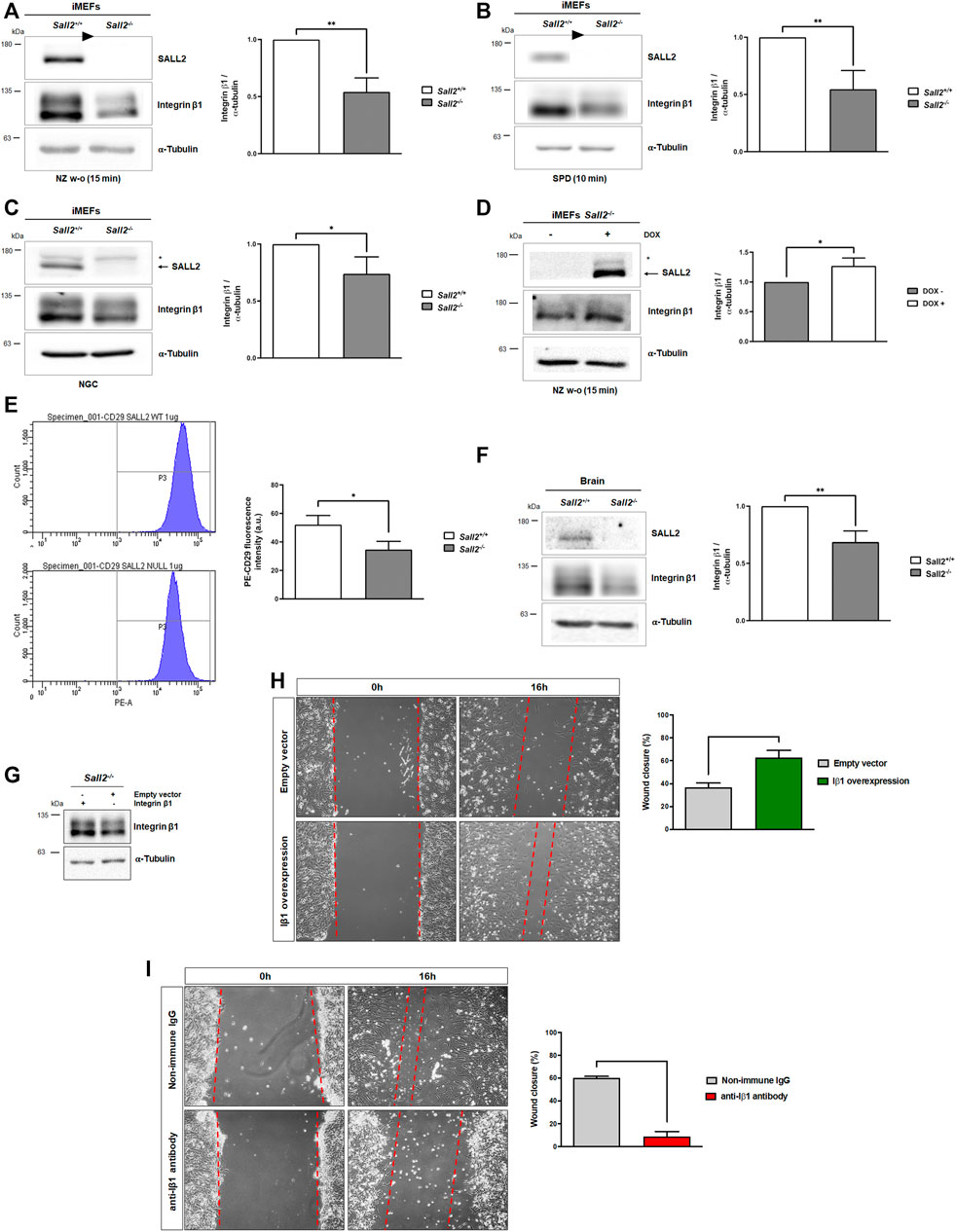
FIGURE 5. Sall2 promotes integrin β1 expression. (A–C) Representative blots of integrin β1 expression, and densitometric quantification, from Sall2+/+ and Sall2−/− iMEFs after 15 min of nocodazole wash-out treatment (NZ w-o) (A), 10 min of cell spreading (SPD) on FN (B), and in normal growth conditions (NGC) (C). (D) Representative blot of integrin β1 protein level, and densitometry, from the Tet-On Sall2 iMEFs inducible model after 15 min of NZ w-o. (E) Flow cytometry analysis of the cell surface expression of integrin β1 from Sall2+/+ and Sall2−/− iMEFs. (F) Left, a representative blot of brain tissues from 6 to 8-week-old Sall2+/+ and Sall2−/− mice. Right, integrin β1 densitometry from brain tissues. The arrow indicates Sall2, and the asterisk corresponds to a nonspecific band. The arrowheads in (A,B) indicate cropped unrelated columns and subsequent splicing of the blot. α-tubulin was used as loading control in western blot analysis. (G) Western blot analysis of integrin β1 overexpression in Sall2−/− iMEFs. (H) Left, representative phase-contrast images (10x) at 0 and 16 h of in vitro wounding from Sall2−/− iMEFs overexpressing integrin β1. Right, quantification of wound closure (as a percentage) at 16 h from images obtained in Left. (I) same as in H, but for Sall2+/+ iMEFs incubated with anti-β1 antibody. Data are expressed as mean ± SD from three independent experiments (*p = 0.01 to 0.05, **p = 0.001 to 0.01;unpaired t-test).
Considering the functional relevance of β-subunits in the recruitment of several adapters and signaling molecules to form focal adhesions (Schnittert et al., 2018), and since we did not observe changes in the integrin β3 protein levels, we focused our study on integrin β1. We evaluated how Sall2 induction affects integrin β1 levels after 15 min of nocodazole wash-out treatment, where the highest difference in integrin β1 expression was observed. We found that Sall2 expression significantly increases integrin β1 protein level, like the Sall2+/+ cells (Figure 5D). Additionally, we evaluated the integrin β1 expression at the cell surface of iMEFs by flow cytometry. As expected, Sall2−/− iMEFs exhibited a lower integrin β1 expression than Sall2+/+ cells (mean PE-CD29 fluorescence intensity = 35 versus 52 a.u.) (Figure 5E). Previously, we reported detectable Sall2 protein levels in mice tissues, including the brain, cerebellum, and spleen (Hermosilla et al., 2018). To evaluate the significance of the Sall2-dependent regulation of integrin β1 in vivo, we compared integrin β1 expression between brain tissues from isogenic Sall2+/+ and Sall2−/− mice. Consistent with our previous results (Figures 5A–D), integrin β1 protein levels were significantly lower in Sall2−/− than in Sall2+/+ tissues (Figure 5F).
To validate that the regulation of integrin β1 expression by Sall2 is directly involved in the cell migration phenotype, we performed functional studies with integrin β1 overexpression or the blocking of integrin β1 function. The integrin β1 overexpression in Sall2−/− iMEFs increased cell migration (wound closure percentage from 37% to 63%, Figures 5G,H), similar to that observed in Sall2+/+ cells. On the other hand, the integrin β1 blockage with an anti β1antibody in Sall2+/+ iMEFs showed a reduced wound closure percentage (from 60% to 9%, Figure 5I). Together, our results demonstrated that Sall2 positively regulates the integrin β1 expression and promotes cell migration through this regulation.
Sall2 transcriptionally regulates integrin β1
To investigate whether Sall2 regulates integrin β1 expression, we initially analyzed a previously published ChIP-seq (Farkas et al., 2021) and compared the ITGB1 promoter data between SALL2 wild-type and SALL2 knockout HEK293 cells. We identified SALL2-binding enrichment and associated peaks in the ITGB1 promoter region in the SALL2+/+ cells (Figure 6A). Additionally, using the Transcriptional Regulatory Element Database (TRED) (Jiang et al., 2007), we identified several putative binding sites in the mouse and human integrin β1 promoter (15 sites in the mouse and 30 sites in the human promoter), mainly located in the proximal promoter region, –500 bp to +1 (Figure 6B). Conservation analysis using BLASTn revealed 79% of identity between both species at the proximal region. Next, we evaluated the integrin β1 mRNA level by real-time PCR in the previous three cellular contexts. Figures 6C–E show that Sall2−/− iMEFs exhibited significantly lower integrin b1 mRNA than Sall2+/+ cells under nocodazole release, spreading on FN, and normal growth conditions. Like the protein analysis, the highest mRNA difference was observed under nocodazole release. Accordingly, induction of Sall2 expression increased the integrin β1 mRNA level after 15 min of nocodazole wash-out treatment (Figure 6F).
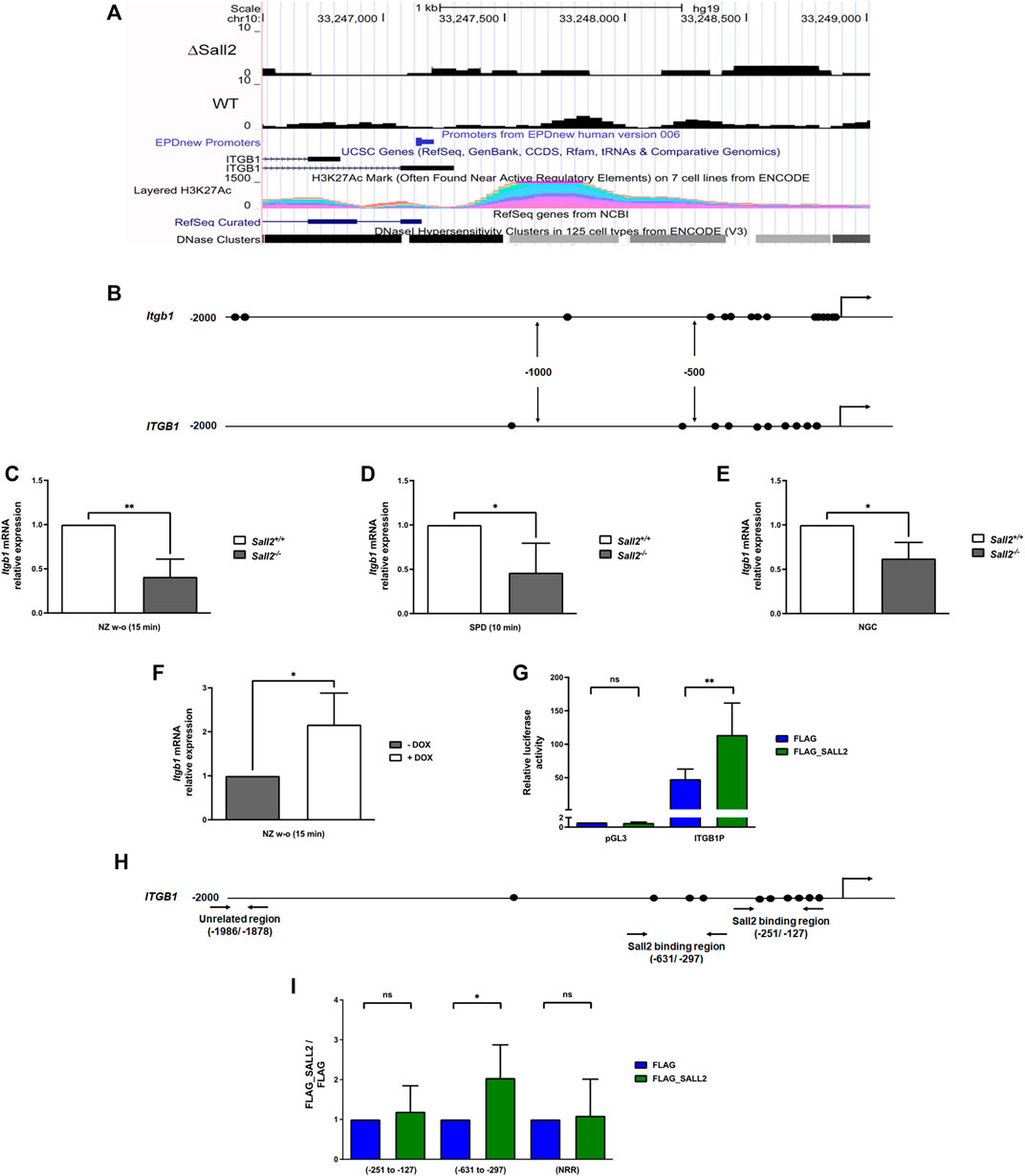
FIGURE 6. Sall2 transcriptionally regulates integrin β1. (A) Bigwig track from ITGB1 gene, according to reference from NCBI RefSeq genes and UCSC genes (ENCODE), from SALL2 wild-type (WT) and SALL2 knockout HEK293 cells (ΔSall2) ChIP-seq datasets. SALL2 binding enrichment and associated peaks in the promoter region, according with EPDnew Promoters ENCODE data are shown. H3K27Ac enhancer mark and DNase Clusters from ENCODE are also shown, plotted with the UCSC genome browser (https://genome.ucsc.edu/, hg19 track). (B) Schematic representation of mouse and human integrin β1 (Itgb1/ITGB1) promoters. The putative SALL2/Sall2 binding sites are represented by black ovals. The transcription start site (+1) is represented by arrows. (C–E) Integrin β1 mRNA level from Sall2+/+ and Sall2−/− iMEFs were determined by real-time PCR after 15 min of nocodazole wash-out treatment (NZ w-o) (C), 10 min of spreading (SPD) on FN (D) and under normal growth conditions (NGC) (E). (F) Integrin β1 mRNA levels from the Tet-On Sall2 iMEFs inducible model after 15 min of NZ w-o. RNA polymerase II was used as a normalizer. (G) ITGB1 promoter activity in the absence (FLAG vector) and presence of SALL2 (FLAG_SALL2) was performed as described in the MATERIALS AND METHODS. Luciferase activity was measured from cell lysates and normalized to β-galactosidase activity, and promoter activity was expressed as relative luciferase units (R.L.U). pGL3 vector served as control. (H) Schematic representation of human ITGB1 promoter. Horizontal arrows indicate the location of primers used for qPCR in site-specific ChIP assays. (I) Chromatin from SALL2 KO HEK293 cells transfected with FLAG_SALL2 was immunoprecipitated 24 h after transfection using FLAG antibody. Specific genomic regions of the human ITGB1 promoter and a nonrelated promoter region (NRR) were analyzed by real-time PCR. Graphs show quantification of the amplified DNA for each immunoprecipitation relative to FLAG. Data are expressed as mean ± SD from three independent experiments (n.s, not significant, *p = 0.01 to 0.05, **p = 0.001 to 0.01; unpaired t-test).
To determine SALL2 activity at the ITGB1 promoter, we used SALL2 KO HEK293 cells co-transfected with a SALL2 expression plasmid and an ITGB1 promoter reporter previously described (Itou et al., 2017). SALL2 expression significantly increased the activity of the ITGB1 promoter (Figure 6G). By chromatin immunoprecipitation (ChIP) assays in SALL2 KO HEK293 cells, we demonstrated that SALL2 binds to the proximal promoter of ITGB1, specifically to the -631/-297 region (Figures 6H,I). In contrast, no binding of SALL2 was observed in a nonrelated promoter region (NRR) and the -251/-127 region of the ITGB1 promoter. The increment of SALL2 binding was correlated with an increase in histone H4 acetylation, a transcriptional activation marker (Supplementary Figure S7). These results support that integrin β1 is a novel Sall2 transcriptional target gene.
Discussion
Cell migration is an important event in several physiological processes. Deregulation of cell migration leads to misplaced cells and, consequently, abnormal functions, promoting several diseases such as congenital abnormalities, autoimmune syndromes, and primary tumor dissemination (Horwitz and Webb, 2003). A better understanding of the molecular factors that regulate cell migration could lead to novel therapeutic approaches.
In this study, we elucidated the role of the Sall2 transcription factor in the migration of immortalized mouse embryonic fibroblasts. Our studies demonstrated that Sall2 is required for optimal cell migration. Specifically, we found that Sall2 promotes cell detachment, FAK autophosphorylation, and FA dynamics. In addition, we demonstrated that Sall2 positively regulates integrin β1 expression by directly binding to the ITGB1 promoter.
Previous evidence is consistence with our findings, suggesting that SALL2/Sall2 promotes cell migration. SALL2 deficiency leads to optic fissure closure failure, causing blindness (Kelberman et al., 2014). In hippocampal neurons, SALL2 silencing significantly decreased neurite outgrowth (Pincheira et al., 2009). The stages before optic fissure closure, such as optic vesicle evagination, depend on the effective migration (Rembold et al., 2006). On the other hand, neurons must be highly polarized to extend their axon and dendrites (Li and Gundersen, 2008). Thus, these reported phenotypes associate the loss of SALL2/Sall2 function with a decrease in cell migration, reinforcing a positive role of SALL2/Sall2 in this process. Interestingly, two reports contradict this conclusion, associating SALL2 deficiency with increased cell migration in cancer. Depleting SALL2 in A2780 ovarian cancer cells increased migration and invasion and was associated with the activation of the PI3K/Akt signaling (Miao et al., 2017). Similarly, epigenetic silencing of SALL2 was associated with increased migration ability. Upregulation of SALL2 in ESCC cells decreased the growth and migration of radioresistant cells, which is directly associated with acquiring more aggressive phenotypes (Luo et al., 2017). Considering that no previous studies show a direct role of SALL2/Sall2 in cell migration in a non-cancer context, our results in iMEFs are the first to demonstrate that Sall2 plays a positive role in promoting cell migration.
The apparent controversy on the role of SALL2/Sall2 in the migration of A2780 cancer (Miao et al., 2017) versus MEFs, a non-cancer cell model, might relate to several reasons. The A2780 ovarian cell line is associated with amoeboid migration, characterized by the ability to move and invade independently of proteolysis by conferring increased motility and invasion of cells (Beaufort et al., 2014). These cells harbor mutations in BRCA2, PIK3CA, PTEN, and ARID1A genes, among others, associated with tumor initiation (Beaufort et al., 2014). In contrast, MEFs are associated with a mesenchymal-like phenotype. Mesenchymal cells move slowly, exhibit multiple protrusions, and depend strongly on integrin-mediated adhesions to the extracellular matrix (Panková et al., 2010; Bear et al., 2014). Here, we used immortalized MEFs, which are widely helpful in assessing migratory and invasive capacities (Birch et al., 2016; Prieto et al., 2020). These cells are a simple genetic model characterized mainly by the inactivation of p53 and Rb proteins (Ali and DeCaprio, 2001). Thus, depending on the cellular context and the presence or absence of specific partners, SALL2/Sall2 could affect its transcriptional activity and cellular functions. For example, SALL2 is a tumor suppressor in ovarian cancers but promotes aggressiveness in the glioma context. SALL2 binds target genes, BAX, p16, and c-MYC in ovarian cancer through the canonical GC-rich motif (Gu et al., 2011). In contrast, it binds to AT-rich motifs related to its interaction with SOX2 protein in glioma, reprogramming the tumor-propagating potential of glioblastoma stem-like cells (Suvà et al., 2014).
SALL2 isoforms might also account for different cellular outcomes. Two SALL2 isoforms, E1 and E1A, have been characterized. They are differentially expressed and only differ at the N-terminal end. The E1 isoform contains a repressor domain relevant to its interaction with the Nucleosome Remodeling Deacetylase (NuRD) complex, which is not present in E1A (Lauberth and Rauchman, 2006). A short E1A SALL2 isoform, lacking DNA binding domain and most Zinc finger motifs, is also deregulated in cancer contexts (Farkas et al., 2021). We previously showed that the E1A is the predominant Sall2 isoform in iMEFs (Hermosilla et al., 2018), suggesting that this isoform is responsible for promoting iMEFs’ cell migration. The expression of a different SALL2 isoform in A2780 and ESCC cancer cells may explain the contradictory results but was not established. Therefore, identifying differential functions of SALL2/Sall2 isoforms may resolve its contradictory role in cell migration between cancer and non-cancer cells.
We demonstrated that SALL2 binds to and activates the ITGB1 promoter transcriptional activity, identifying ITGB1 as a novel SALL2 target. Accordingly, our studies showed that Sall2 promotes the increase of mRNA and protein levels of integrin β1, correlating with an increase in its availability at the cell surface. Of physiological relevance, we also showed that Sall2 correlated with integrin β1 expression in mouse brain tissues, suggesting a conserved role of Sall2 in integrin β1 regulation, at least in normal conditions. We demonstrated that the regulation of integrin β1 is one of the mechanisms involved in the Sall2-dependent promotion of cell migration in fibroblasts.
We observed a more marked effect on cell migration with fibronectin as substrate (data not shown). Changes in the expression of extracellular matrix proteins could affect fibroblast cell migration. MEFs synthesize several matrix proteins, including fibronectin, laminin, and type I and IV collagen. Fibronectin is the most expressed (Blancas et al., 2011) and associates with the integrin heterodimers α4β1, α5β1, α8β1, and αvβ1 (Humphries et al., 2006). Despite this, there is no previous evidence of extracellular matrix protein expression in the context of Sall2 gain and loss of function. We observed that Sall2 might also regulate integrin α4 protein levels (Supplementary Figure S6). Whether Sall2 regulates the α4β1 heterodimer requires further investigation.
The integrin β1 on the plasma membrane activates through a combination of inside-out and outside-in mechanisms, leading to integrin clustering, FA maturation, and integrin downstream signaling mechanisms (Kechagia et al., 2019). As part of the process, integrin β1 is crucial for recruiting FAK, inducing its autophosphorylation at Y397 and the consequent events that modulate FA dynamics involved in the migration of cells (Wennerberg et al., 2000). Although Sall2 deficient cells show higher basal FAK levels, they have significantly lower levels of phospho-Y397-FAK than the wild-type cells, suggesting that Sall2 promotes FAK autophosphorylation independently of its effect on protein levels. A Sall2-dependent transcriptional mechanism may explain the higher levels of FAK in the Sall2 deficient cells since several putative Sall2 binding sites were identified in the Ptk2 (FAK) promoter (Supplementary Figure S8). Previous studies have shown that Sall2 transcriptionally represses c-MYC (Sung et al., 2012), CCND1, and CCNE1 genes transcription (Hermosilla et al., 2018). Similarly, Sall2 could bind and repress Ptk2 promoter activity and FAK expression.
Phosphorylation of FAK at Y397 creates a motif recognized by various SH2 domain-containing proteins, including SRC-Family Kinases (SFKs), phospholipase Cγ (PLCγ), growth-factor-receptor-bound protein-7 (GRB7), the Shc adaptor protein, and the p85 subunit of phosphatidylinositol 3-kinase (PI3K) (Mitra et al., 2005). The FAK–SRC signaling complex acts to recruit and/or phosphorylate several proteins, such as p130Cas and paxillin (Mitra et al., 2005). Although there is no evidence of Sall2 affecting some of these signaling components, the decrease in the autophosphorylation of FAK in the Sall2 deficient cells agreed with their FA dynamic phenotype. FAK autophosphorylation is an important event for its regulation by other kinases and, thus, the regulation of FA dynamics (Hamadi et al., 2005). Accordingly, Sall2 deficiency led to a substantial delay in the kinetics of FA disassembly and, consequently, to a slower initial velocity of FA disassembly. This result is consistent with the significantly higher FA length and fluorescence intensity observed in the Sall2−/− than in the Sall2+/+ iMEFs, suggesting a lower state of FA maturation in the wild-type cells contributing to a more dynamic FA turnover and migratory phenotype (Gardel et al., 2010). On the other hand, Sall2 deficiency results in increased FA number and cell adhesion, slowing cell movement. Consistent with their more adherent phenotype, Sall2−/− iMEFs depicted a larger lamellipodia area because the more extensive the cell membrane, the higher the number of FA and hence augmented cell adhesion (McGrath, 2007; Gauthier et al., 2012).
Interestingly, autophosphorylation and activation of Pyk2/FAK are essential for propagating signals that control neurite formation in PC12 and SH-SY5Y cells (Ivankovic-Dikic et al., 2000). Likewise, Sall2 is required for neurite outgrowth of PC12 cells and rat hippocampal neurons (Pincheira et al., 2009). We observed that Sall2 slightly increased the number of filopodia, an important structure for the directional response of the cells (Blanchoin et al., 2014). Filopodia participate in several cellular processes, including cell migration, neurite outgrowth, and wound healing (Mattila and Lappalainen, 2008). Whether the effect of Sall2 in the filopodia number relates to the role of Sall2 at the neuronal level is unknown. However, FAK not only plays a crucial role in cell migration. It also promotes cell survival, regulates transcription (Kleinschmidt and Schlaepfer, 2017), and activates the PI3K signaling pathway (Chen et al., 1996; Reif et al., 2003; Kim and Gumbiner, 2015). Thus, promoting FAK autophosphorylation by Sall2 becomes relevant and requires further investigation. Sall2’s effect on FAK activity and filopodia number could be common mechanisms for its role in neurite outgrowth and migration of iMEFs.
An additional mechanism explaining the role of Sall2 in cell migration could relate to the regulation of PTEN. It is well-known that PTEN regulates many cellular processes. They include cell polarity and migration through its lipid phosphatase activity, which antagonizes PI3K (phosphoinositide 3-kinase) signaling (Worby and Dixon, 2014). It also can regulate cell motility independently of its lipid phosphatase activity (Yamada and Araki, 2001). PTEN dephosphorylates FAK and inhibits cell migration, spreading, and focal adhesion formation of U-87MG cells (Tamura et al., 1998). Studies in breast cancer cells demonstrate that SALL2 silencing induced the AKT/mTOR pathway activation via the downregulation of PTEN. The mechanism involves the positive regulation of PTEN through the direct binding of SALL2 to the canonical GC-rich motif in the PTEN promoter (Ye et al., 2019). Contrary to the cancer context, we observed higher levels of PTEN in Sall2−/− cells (Supplementary Figure S9), which supports PTEN as an alternative or complementary mechanism in the Sall2-mediated FAK activation. This possibility requires further studies.
Interestingly, SALL4, the oncogenic member of the SALL family, is associated with increased cell migration and invasion in gastric and breast cancer cell lines (Yuan et al., 2016; Itou et al., 2017). SALL4 regulates integrins β1 and α6 expression in basal-like breast cancer cells, a cell type with high migratory properties (Itou et al., 2017). SALL4 mediates FAK activation, FA dynamics, and Rho inhibition to promote migration by a mechanism involving the SALL4/integrin α6β1 network (Itou et al., 2017). Unlike SALL4, SALL2/Sall2-dependent cell migration did not involve integrin a6. SALL4 binds DNA at the AT-Rich motif (Kong et al., 2021), but, as shown here, SALL2 binds to the canonical GC-Rich motif (Gu et al., 2011), suggesting a common regulation of ITGB1 by SALL proteins via differential promoter-binding sites. Studies indicate that the regulation of ITGB1 by specific SALL proteins might be context specific. For example, in breast cancer cells, SALL2 is downregulated, but SALL4 is upregulated (Alvarez et al., 2021). Thus, SALL4 is expected to have a primary role in ITGB1 regulation. On the other hand, in the glioma context, SALL2 and SALL4 are upregulated, potentially exerting a synergistic effect on the regulation of integrin β1 (Alvarez et al., 2021). In addition to SALL4 and SALL2, studies in breast cancer lines showed that SALL1 inhibition increases cell migration and correlates with decreased cadherin 1 expression. (Wolf et al., 2014). Our findings further support the role of the SALL family members in cell migration. However, how different contexts affect the involvement of SALL proteins in cell migration and integrins regulation requires further investigation.
In summary, our results propose that Sall2 promotes cell migration by modulation of focal adhesion dynamics in a non-cancer context. This effect might relate to the direct transcriptional regulation of the ITGB1 gene (Figure 7). Since deregulation of cell migration promotes congenital disabilities, autoimmune syndromes, and tumor formation and spreads to other tissues, our findings suggest that the Sall2-integrin β1 axis could lead to novel therapeutic approaches.
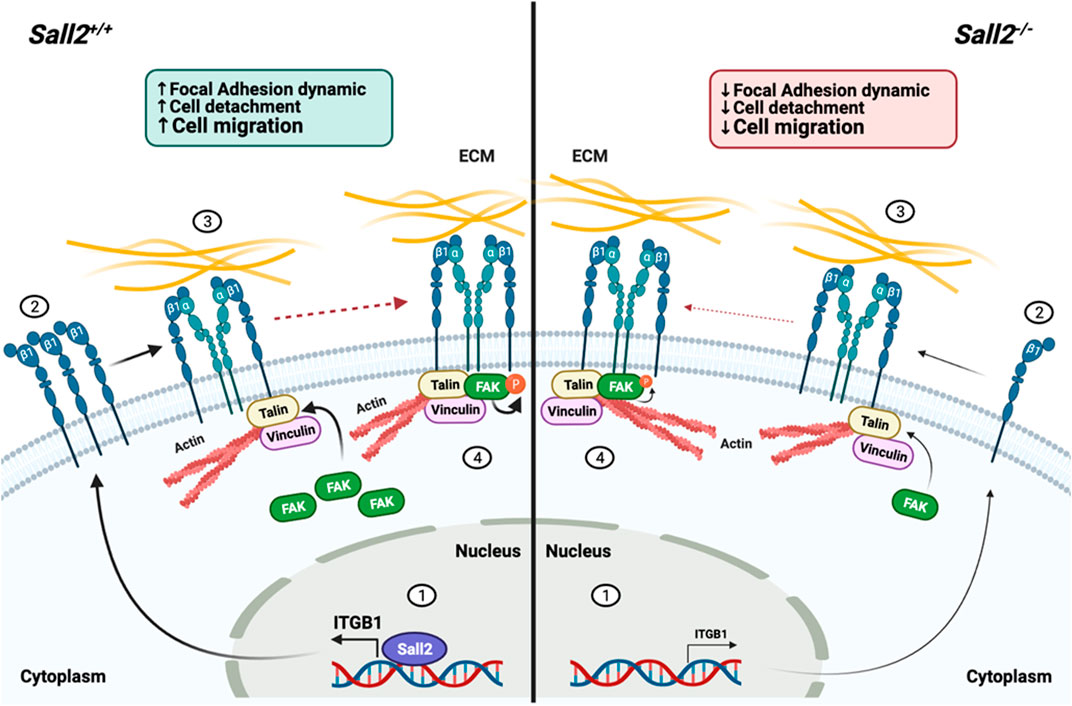
FIGURE 7. Proposed model of Sall2-dependent regulation of cell migration. In Sall2+/+ iMEFs cells, (1) Sall2 promotes the integrin β1 mRNA expression by directly binding to and transactivating its promoter, (2) increasing the integrin β1 protein levels and its expression at the membrane surface, contributing in part to the integrin β1 cluster formation. (3) After integrin ligand-binding, actin-associated proteins like talin and vinculin bind to the integrin β1 and recruit focal adhesion kinase (FAK). (4) Once recruited in the adhesion site, FAK is autophosphorylated modulating FA assembly-disassembly dynamics. Conversely, in Sall2−/− cells the same chain of events occurs but less favorably. In summary, Sall2 by promoting the FA dynamics favors cell detachment and subsequently cell migration.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/cfarkas/SALL2_conserved_network_analysis.
Ethics statement
The animal study was reviewed and approved by the Vicerrectoría de Investigación y Desarrollo Comité de Ética, Bioética y Bioseguridad. Universidad de Concepción.
Author contributions
ER and RP conceptualization. ER, MP, MH, and DB-R methodology. ER, MP, VAT, AFC, and RP investigation. VAT, AFC, and RP resources. ER and RP writing—original draft. ER, MP, MH, DB-R, AFC, VAT, and RP writing—review and editing. AFC, VAT, and RP supervision. All authors read and approved the final version of the manuscript.
Funding
This work was supported by Fondecyt Regular Grants #1151031, #1191172 to RP, Fondecyt Regular Grant #1201215 to AFC and Fondecyt Regular Grant #1220517 to VAT.
Acknowledgments
We thank David Ron and Adam Karpf for providing us the pBsSVD2005 and pCW57-MCS1-2A-MCS2, pCW57-MCS1-P2A-MCS2 plasmids, respectively. We thank Bob Weinberg for providing us with the pCMV-dR8.2 dvpr and pCMV-VSV-G plasmids. We thank Joan Massague for providing us the EFIa-iTGB1 plasmid. We thank Masakazu Toi for kindly providing us with the ITGB1 promoter_pGL4.30 vector. We thank to “Centro de Microscopía Avanzada (CMA Bio Bio),” Universidad de Concepción for immunofluorescence images, flow cytometry and time-lapse movies support. Finally, we thank the “Centro Regional de Estudios Avanzados para la Vida (CREAV),“ Universidad de Concepción for animal support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1031262/full#supplementary-material
Abbreviations
ChIP, chromatin immunoprecipitation; EDTA, ethylenediaminetetraacetic acid; EMC, extracellular matrix; ESCC, esophageal squamous cell carcinoma; FA, focal adhesion; FAK, focal adhesion kinase; FN, fibronectin; iMEFs, immortalized mouse embryonic fibroblasts; KO, knockout; MTOC, microtubule-organizing center; NGF, nerve growth factor; NZ, nocodazole; NuRD, Nucleosome Remodeling Deacetylase complex; PC12, pheochromocytoma of the rat adrenal medulla; SV40, simian virus 40.
References
Ali, S. H., and DeCaprio, J. A. (2001). Cellular transformation by SV40 large T antigen: Interaction with host proteins. Semin. Cancer Biol. 11 (1), 15–23. doi:10.1006/scbi.2000.0342
Álvarez, C., Quiroz, A., Benítez-Riquelme, D., Riffo, E., Castro, A. F., and Pincheira, R. (2021). SALL proteins; common and antagonistic roles in cancer. Cancers 13 (24), 6292. doi:10.3390/cancers13246292
Barger, C. J., Branick, C., Chee, L., and Karpf, A. R. (2019). Pan-cancer analyses reveal genomic features of FOXM1 overexpression in cancer. Cancers 11 (2), 251. doi:10.3390/cancers11020251
Bear, J. E., and Haugh, J. M. (2014). Directed migration of mesenchymal cells: Where signaling and the cytoskeleton meet. Curr. Opin. Cell Biol. 30, 74–82. doi:10.1016/j.ceb.2014.06.005
Beaufort, C. M., Helmijr, J. C., Piskorz, A. M., Hoogstraat, M., Ruigrok-Ritstier, K., Besselink, N., et al. (2014). Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PloS one 9 (9), e103988. doi:10.1371/journal.pone.0103988
Birch, J., Clarke, C. J., Campbell, A. D., Campbell, K., Mitchell, L., Liko, D., et al. (2016). The initiator methionine tRNA drives cell migration and invasion leading to increased metastatic potential in melanoma. Biol. Open 5 (10), 1371–1379. doi:10.1242/bio.019075
Blancas, A. A., Chen, C. S., Stolberg, S., and McCloskey, K. E. (2011). Adhesive forces in embryonic stem cell cultures. Cell adh. Migr. 5 (6), 472–479. doi:10.4161/cam.5.6.18270
Blanchoin, L., Boujemaa-Paterski, R., Sykes, C., and Plastino, J. (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 94 (1), 235–263. doi:10.1152/physrev.00018.2013
Chai, L. (2011). The role of HSAL (SALL) genes in proliferation and differentiation in normal hematopoiesis and leukemogenesis. Transfusion 51 (4), 87S–93S. doi:10.1111/j.1537-2995.2011.03371.x
Chen, H. C., Appeddu, P. A., Isoda, H., and Guan, J. L. (1996). Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 271 (42), 26329–26334. doi:10.1074/jbc.271.42.26329
Conway, J., and Jacquemet, G. (2019). Cell matrix adhesion in cell migration. Essays Biochem. 63 (5), 535–551. doi:10.1042/EBC20190012
De Celis, J. F., and Barrio, R. (2009). Regulation and function of Spalt proteins during animal development. Int. J. Dev. Biol. 53 (8-10), 1385–1398. doi:10.1387/ijdb.072408jd
Er, E. E., Valiente, M., Ganesh, K., Zou, Y., Agrawal, S., Hu, J., et al. (2018). Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 20 (8), 966–978. doi:10.1038/s41556-018-0138-8
Erusappan, P., Alam, J., Lu, N., Zeltz, C., and Gullberg, D. (2019). Integrin α11 cytoplasmic tail is required for FAK activation to initiate 3D cell invasion and ERK-mediated cell proliferation. Sci. Rep. 9 (1), 15283. doi:10.1038/s41598-019-51689-6
Escobar, D., Hepp, M. I., Farkas, C., Campos, T., Sodir, N. M., Morales, M., et al. (2015). Sall2 is required for proapoptotic Noxa expression and genotoxic stress-induced apoptosis by doxorubicin. Cell Death Dis. 6 (7), e1816. doi:10.1038/cddis.2015.165
Estilo, C. L., O-charoenrat, P., Talbot, S., Socci, N. D., Carlson, D. L., Ghossein, R., et al. (2009). Oral tongue cancer gene expression profiling: Identification of novel potential prognosticators by oligonucleotide microarray analysis. BMC cancer 9, 11. doi:10.1186/1471-2407-9-11
Fagerholm, S. C., Guenther, C., Llort Asens, M., Savinko, T., and Uotila, L. M. (2019). Beta2-Integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency disease. Front. Immunol. 10, 254. doi:10.3389/fimmu.2019.00254
Farkas, C., Quiroz, A., Alvarez, C., Hermosilla, V., Aylwin, C. F., Lomniczi, A., et al. (2021). Characterization of SALL2 gene isoforms and targets across cell types reveals highly conserved networks. Front. Genet. 12, 613808. doi:10.3389/fgene.2021.613808
Gahmberg, C. G., Grönholm, M., Madhavan, S., Jahan, F., Mikkola, E., Viazmina, L., et al. (2019). Regulation of cell adhesion: A collaborative effort of integrins, their ligands, cytoplasmic actors, and phosphorylation. Q. Rev. Biophys. 52, e10. doi:10.1017/S0033583519000088
Gardel, M. L., Schneider, I. C., Aratyn-Schaus, Y., and Waterman, C. M. (2010). Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333. doi:10.1146/annurev.cellbio.011209.122036
Gauthier, N. C., Masters, T. A., and Sheetz, M. P. (2012). Mechanical feedback between membrane tension and dynamics. Trends Cell Biol. 22 (10), 527–535. doi:10.1016/j.tcb.2012.07.005
Grande-García, A., Echarri, A., de Rooij, J., Alderson, N. B., Waterman-Storer, C. M., Valdivielso, J. M., et al. (2007). Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J. Cell Biol. 177 (4), 683–694. doi:10.1083/jcb.200701006
Gu, H., Li, D., Sung, C. K., Yim, H., Troke, P., and Benjamin, T. (2011). DNA-binding and regulatory properties of the transcription factor and putative tumor suppressor p150(Sal2). Biochim. Biophys. Acta 1809 (4-6), 276–283. doi:10.1016/j.bbagrm.2011.02.002
Hamadi, A., Bouali, M., Dontenwill, M., Stoeckel, H., Takeda, K., and Rondé, P. (2005). Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 118 (19), 4415–4425. doi:10.1242/jcs.02565
Hamidi, H., and Ivaska, J. (2018). Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 18 (9), 533–548. doi:10.1038/s41568-018-0038-z
Hermosilla, V. E., Hepp, M. I., Escobar, D., Farkas, C., Riffo, E. N., Castro, A. F., et al. (2017). Developmental SALL2 transcription factor: A new player in cancer. Carcinogenesis 38 (7), 680–690. doi:10.1093/carcin/bgx036
Hermosilla, V. E., Salgado, G., Riffo, E., Escobar, D., Hepp, M. I., Farkas, C., et al. (2018). SALL2 represses cyclins D1 and E1 expression and restrains G1/S cell cycle transition and cancer-related phenotypes. Mol. Oncol. 12 (7), 1026–1046. doi:10.1002/1878-0261.12308
Horwitz, R., and Webb, D. (2003). Cell migration. Curr. Biol. 13 (19), R756–R759. doi:10.1016/j.cub.2003.09.014
Humphries, J. D., Byron, A., and Humphries, M. J. (2006). Integrin ligands at a glance. J. Cell Sci. 119 (19), 3901–3903. doi:10.1242/jcs.03098
Huttenlocher, A., and Horwitz, A. R. (2011). Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 3 (9), a005074. doi:10.1101/cshperspect.a005074
Itou, J., Tanaka, S., Li, W., Iida, A., Sehara-Fujisawa, A., Sato, F., et al. (2017). The Sal-like 4 - integrin α6β1 network promotes cell migration for metastasis via activation of focal adhesion dynamics in basal-like breast cancer cells. Biochim. Biophys. Acta. Mol. Cell Res. 1864 (1), 76–88. doi:10.1016/j.bbamcr.2016.10.012
Ivankovic-Dikic, I., Grönroos, E., Blaukat, A., Barth, B. U., and Dikic, I. (2000). Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat. Cell Biol. 2 (9), 574–581. doi:10.1038/35023515
Jiang, C., Xuan, Z., Zhao, F., and Zhang, M. Q. (2007). Tred: A transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. 35, D137–D140. doi:10.1093/nar/gkl1041
Kechagia, J. Z., Ivaska, J., and Roca-Cusachs, P. (2019). Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20 (8), 457–473. doi:10.1038/s41580-019-0134-2
Kelberman, D., Islam, L., Lakowski, J., Bacchelli, C., Chanudet, E., Lescai, F., et al. (2014). Mutation of SALL2 causes recessive ocular coloboma in humans and mice. Hum. Mol. Genet. 23 (10), 2511–2526. doi:10.1093/hmg/ddt643
Kim, N. G., and Gumbiner, B. M. (2015). Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 210 (3), 503–515. doi:10.1083/jcb.201501025
Kleinschmidt, E. G., and Schlaepfer, D. D. (2017). Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 45, 24–30. doi:10.1016/j.ceb.2017.01.003
Kong, N. R., Bassal, M. A., Tan, H. K., Kurland, J. V., Yong, K. J., Young, J. J., et al. (2021). Zinc finger protein SALL4 functions through an AT-rich motif to regulate gene expression. Cell Rep. 34 (1), 108574. doi:10.1016/j.celrep.2020.108574
Lauberth, S. M., and Rauchman, M. (2006). A conserved 12-amino acid motif in Sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J. Biol. Chem. 281 (33), 23922–23931. doi:10.1074/jbc.M513461200
Legate, K. R., and Fässler, R. (2009). Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J. Cell Sci. 122 (2), 187–198. doi:10.1242/jcs.041624
Li, C. M., Guo, M., Borczuk, A., Powell, C. A., Wei, M., Thaker, H. M., et al. (2002). Gene expression in Wilms' tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am. J. Pathol. 160 (6), 2181–2190. doi:10.1016/S0002-9440(10)61166-2
Li, D., Tian, Y., Ma, Y., and Benjamin, T. (2004). p150(Sal2) is a p53-independent regulator of p21(WAF1/CIP). Mol. Cell. Biol. 24 (9), 3885–3893. doi:10.1128/MCB.24.9.3885-3893.2004
Li, R., and Gundersen, G. G. (2008). Beyond polymer polarity: How the cytoskeleton builds a polarized cell. Nat. Rev. Mol. Cell Biol. 9 (11), 860–873. doi:10.1038/nrm2522
Luo, J., Wang, W., Tang, Y., Zhou, D., Gao, Y., Zhang, Q., et al. (2017). mRNA and methylation profiling of radioresistant esophageal cancer cells: The involvement of Sall2 in acquired aggressive phenotypes. J. Cancer 8 (4), 646–656. doi:10.7150/jca.15652
Ma, Y., Li, D., Chai, L., Luciani, A. M., Ford, D., Morgan, J., et al. (2001). Cloning and characterization of two promoters for the human HSAL2 gene and their transcriptional repression by the Wilms tumor suppressor gene product. J. Biol. Chem. 276 (51), 48223–48230. doi:10.1074/jbc.M106468200
Marsico, G., Russo, L., Quondamatteo, F., and Pandit, A. (2018). Glycosylation and integrin regulation in cancer. Trends Cancer 4 (8), 537–552. doi:10.1016/j.trecan.2018.05.009
Mattila, P. K., and Lappalainen, P. (2008). Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 9 (6), 446–454. doi:10.1038/nrm2406
McGrath, J. L. (2007). Cell spreading: The power to simplify. Curr. Biol. 17 (10), R357–R358. doi:10.1016/j.cub.2007.03.057
Mendoza, P., Ortiz, R., Díaz, J., Quest, A. F., Leyton, L., Stupack, D., et al. (2013). Rab5 activation promotes focal adhesion disassembly, migration and invasiveness in tumor cells. J. Cell Sci. 126 (17), 3835–3847. doi:10.1242/jcs.119727
Miao, F., Zhang, X., Cao, Y., Wang, Y., and Zhang, X. (2017). Effect of siRNA-silencing of SALL2 gene on growth, migration and invasion of human ovarian carcinoma A2780 cells. BMC cancer 17 (1), 838. doi:10.1186/s12885-017-3843-y
Mitra, S. K., Hanson, D. A., and Schlaepfer, D. D. (2005). Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6 (1), 56–68. doi:10.1038/nrm1549
Nader, G. P., Ezratty, E. J., and Gundersen, G. G. (2016). FAK, talin and PIPKIγ regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat. Cell Biol. 18 (5), 491–503. doi:10.1038/ncb3333
Nielsen, T. O., Hsu, F. D., O'Connell, J. X., Gilks, C. B., Sorensen, P. H., Linn, S., et al. (2003). Tissue microarray validation of epidermal growth factor receptor and SALL2 in synovial sarcoma with comparison to tumors of similar histology. Am. J. Pathol. 163 (4), 1449–1456. doi:10.1016/S0002-9440(10)63502-X
Panková, K., Rösel, D., Novotný, M., and Brábek, J. (2010). The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 67 (1), 63–71. doi:10.1007/s00018-009-0132-1
Pincheira, R., Baerwald, M., Dunbar, J. D., and Donner, D. B. (2009). Sall2 is a novel p75NTR-interacting protein that links NGF signalling to cell cycle progression and neurite outgrowth. EMBO J. 28 (3), 261–273. doi:10.1038/emboj.2008.274
Prieto, I., Alarcón, C. R., García-Gómez, R., Berdún, R., Urgel, T., Portero, M., et al. (2020). Metabolic adaptations in spontaneously immortalized PGC-1α knock-out mouse embryonic fibroblasts increase their oncogenic potential. Redox Biol. 29, 101396. doi:10.1016/j.redox.2019.101396
Reif, S., Lang, A., Lindquist, J. N., Yata, Y., Gabele, E., Scanga, A., et al. (2003). The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J. Biol. Chem. 278 (10), 8083–8090. doi:10.1074/jbc.M212927200
Rembold, M., Loosli, F., Adams, R. J., and Wittbrodt, J. (2006). Individual cell migration serves as the driving force for optic vesicle evagination. Sci. (New York, N.Y.) 313 (5790), 1130–1134. doi:10.1126/science.1127144
Sato, A., Matsumoto, Y., Koide, U., Kataoka, Y., Yoshida, N., Yokota, T., et al. (2003). Zinc finger protein sall2 is not essential for embryonic and kidney development. Mol. Cell. Biol. 23 (1), 62–69. doi:10.1128/MCB.23.1.62-69.2003
Schnittert, J., Bansal, R., Storm, G., and Prakash, J. (2018). Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 129, 37–53. doi:10.1016/j.addr.2018.01.020
SenGupta, S., Parent, C. A., and Bear, J. E. (2021). The principles of directed cell migration. Nat. Rev. Mol. Cell Biol. 22 (8), 529–547. doi:10.1038/s41580-021-00366-6
Silverman-Gavrila, R., Silverman-Gavrila, L., Hou, G., Zhang, M., Charlton, M., and Bendeck, M. P. (2011). Rear polarization of the microtubule-organizing center in neointimal smooth muscle cells depends on PKCα, ARPC5, and RHAMM. Am. J. Pathol. 178 (2), 895–910. doi:10.1016/j.ajpath.2010.10.001
Stewart, S. A., Dykxhoorn, D. M., Palliser, D., Mizuno, H., Yu, E. Y., An, D. S., et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA (New York, N.Y.) 9 (4), 493–501. doi:10.1261/rna.2192803
Sun, H., Hu, L., and Fan, Z. (2021). β2 integrin activation and signal transduction in leukocyte recruitment. Am. J. Physiol. Cell Physiol. 321 (2), C308–C316. doi:10.1152/ajpcell.00560.2020
Sung, C. K., Yim, H., Gu, H., Li, D., Andrews, E., Duraisamy, S., et al. (2012). The polyoma virus large T binding protein p150 is a transcriptional repressor of c-MYC. PLoS ONE 7 (9), e46486. doi:10.1371/journal.pone.0046486
Suvà, M. L., Rheinbay, E., Gillespie, S. M., Patel, A. P., Wakimoto, H., Rabkin, S. D., et al. (2014). Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157 (3), 580–594. doi:10.1016/j.cell.2014.02.030
Sweetman, D., and Münsterberg, A. (2006). The vertebrate spalt genes in development and disease. Dev. Biol. 293 (2), 285–293. doi:10.1016/j.ydbio.2006.02.009
Tamura, M., Gu, J., Matsumoto, K., Aota, S., Parsons, R., and Yamada, K. M. (1998). Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Sci. (New York, N.Y.) 280 (5369), 1614–1617. doi:10.1126/science.280.5369.1614
Tomar, A., Lim, S. T., Lim, Y., and Schlaepfer, D. D. (2009). A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J. Cell Sci. 122 (11), 1852–1862. doi:10.1242/jcs.046870
Trepat, X., Chen, Z., and Jacobson, K. (2012). Cell migration. Compr. Physiol. 2 (4), 2369–2392. doi:10.1002/cphy.c110012
Wehrle-Haller, B. (2012b). Assembly and disassembly of cell matrix adhesions. Curr. Opin. Cell Biol. 24 (5), 569–581. doi:10.1016/j.ceb.2012.06.010
Wehrle-Haller, B. (2012a). Structure and function of focal adhesions. Curr. Opin. Cell Biol. 24 (1), 116–124. doi:10.1016/j.ceb.2011.11.001
Wennerberg, K., Armulik, A., Sakai, T., Karlsson, M., Fässler, R., Schaefer, E. M., et al. (2000). The cytoplasmic tyrosines of integrin subunit beta1 are involved in focal adhesion kinase activation. Mol. Cell. Biol. 20 (15), 5758–5765. doi:10.1128/MCB.20.15.5758-5765.2000
Wolf, J., Müller-Decker, K., Flechtenmacher, C., Zhang, F., Shahmoradgoli, M., Mills, G. B., et al. (2014). An in vivo RNAi screen identifies SALL1 as a tumor suppressor in human breast cancer with a role in CDH1 regulation. Oncogene 33 (33), 4273–4278. doi:10.1038/onc.2013.515
Worby, C. A., and Dixon, J. E. (2014). Pten. Annu. Rev. Biochem. 83, 641–669. doi:10.1146/annurev-biochem-082411-113907
Wozniak, M. A., Modzelewska, K., Kwong, L., and Keely, P. J. (2004). Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 1692 (2-3), 103–119. doi:10.1016/j.bbamcr.2004.04.007
Yamada, K. M., and Araki, M. (2001). Tumor suppressor PTEN: Modulator of cell signaling, growth, migration and apoptosis. J. Cell Sci. 114 (13), 2375–2382. doi:10.1242/jcs.114.13.2375
Ye, L., Lin, C., Wang, X., Li, Q., Li, Y., Wang, M., et al. (2019). Epigenetic silencing of SALL2 confers tamoxifen resistance in breast cancer. EMBO Mol. Med. 11 (12), e10638. doi:10.15252/emmm.201910638
Yuan, X., Zhang, X., Zhang, W., Liang, W., Zhang, P., Shi, H., et al. (2016). SALL4 promotes gastric cancer progression through activating CD44 expression. Oncogenesis 5 (11), e268. doi:10.1038/oncsis.2016.69
Keywords: Sall2, cell migration, focal adhesion (FA), focal adhesion kinase (FAK), integrin β1, mouse embrionic fibroblasts (MEFs)
Citation: Riffo E, Palma M, Hepp MI, Benítez-Riquelme D, Torres VA, Castro AF and Pincheira R (2022) The Sall2 transcription factor promotes cell migration regulating focal adhesion turnover and integrin β1 expression. Front. Cell Dev. Biol. 10:1031262. doi: 10.3389/fcell.2022.1031262
Received: 29 August 2022; Accepted: 25 October 2022;
Published: 09 November 2022.
Edited by:
Claudia Tanja Mierke, Leipzig University, GermanyReviewed by:
Donna Peters, University of Wisconsin-Madison, United StatesVita Golubovskaya, ProMab Biotechnologies, United States
Copyright © 2022 Riffo, Palma, Hepp, Benítez-Riquelme, Torres, Castro and Pincheira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roxana Pincheira, cm9waW5jaGVpcmFAdWRlYy5jbA==