
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 12 October 2022
Sec. Developmental Epigenetics
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1022422
This article is part of the Research Topic Editors' Showcase 2021: Insights in Developmental Epigenetics View all 4 articles
 Michael Claxton1,2
Michael Claxton1,2 Michela Pulix1
Michela Pulix1 Michelle K. Y. Seah2
Michelle K. Y. Seah2 Ralph Bernardo1
Ralph Bernardo1 Peng Zhou1
Peng Zhou1 Sultan Aljuraysi1,3
Sultan Aljuraysi1,3 Triantafillos Liloglou4
Triantafillos Liloglou4 Philippe Arnaud5
Philippe Arnaud5 Gavin Kelsey6,7,8
Gavin Kelsey6,7,8 Daniel M. Messerschmidt2,9
Daniel M. Messerschmidt2,9 Antonius Plagge1*
Antonius Plagge1*Genomic imprinting is an epigenetic process through which genes are expressed in a parent-of-origin specific manner resulting in mono-allelic or strongly biased expression of one allele. For some genes, imprinted expression may be tissue-specific and reliant on CTCF-influenced enhancer-promoter interactions. The Peg13 imprinting cluster is associated with neurodevelopmental disorders and comprises canonical imprinted genes, which are conserved between mouse and human, as well as brain-specific imprinted genes in mouse. The latter consist of Trappc9, Chrac1 and Ago2, which have a maternal allelic expression bias of ∼75% in brain. Findings of such allelic expression biases on the tissue level raise the question of how they are reflected in individual cells and whether there is variability and mosaicism in allelic expression between individual cells of the tissue. Here we show that Trappc9 and Ago2 are not imprinted in hippocampus-derived neural stem cells (neurospheres), while Peg13 retains its strong bias of paternal allele expression. Upon analysis of single neural stem cells and in vitro differentiated neurons, we find not uniform, but variable states of allelic expression, especially for Trappc9 and Ago2. These ranged from mono-allelic paternal to equal bi-allelic to mono-allelic maternal, including biased bi-allelic transcriptional states. Even Peg13 expression deviated from its expected paternal allele bias in a small number of cells. Although the cell populations consisted of a mosaic of cells with different allelic expression states, as a whole they reflected bulk tissue data. Furthermore, in an attempt to identify potential brain-specific regulatory elements across the Trappc9 locus, we demonstrate tissue-specific and general silencer activities, which might contribute to the regulation of its imprinted expression bias.
Genomic imprinting has long been recognized as a paradigm of epigenetic regulation of a specific subset of ∼200 genes that have important roles in mammalian embryogenesis, regulation of nutrient supply and demand between mother and offspring, as well as brain development and neural functions (Ferguson-Smith, 2011; Peters, 2014; Tucci et al., 2019). Imprinted genes are defined by their parent-of-origin dependent mono-allelic or strongly biased allelic (>70%) expression. This expression bias towards a specific parental allele is a consequence of DNA methylation marks that are differentially established in either the male or female germ cells, respectively, at a defined set of CpG-rich islands (CGIs). Such germline differentially methylated regions (gDMRs) are maintained after fertilization in the somatic cells of the developing and adult offspring; they are only erased in its developing germline cells for re-setting according to its sex and transmission to the next generation (Plasschaert and Bartolomei, 2014). The germline DMRs often regulate the allelic expression of a cluster of neighboring genes and are, therefore, also called imprinting control regions (ICRs) (Ferguson-Smith, 2011; Peters, 2014; Tucci et al., 2019). In addition to DNA methylation, the mechanisms involved in the regulation of imprinted gene expression comprise histone modifications, non-coding RNAs and boundary or insulator elements that are recognized by CTCF, a methylation-sensitive DNA binding factor. CTCF binding at unmethylated sites within DMRs of imprinted genes has been shown to regulate access to tissue-specific enhancers and the formation of allelic topologically associated domains (TADs), thereby controlling the expression of neighboring genes in an allele-specific way (Bell and Felsenfeld, 2000; Lleres et al., 2019).
The imprinting status of most genes is conserved between human and mouse, although some genes do not show an allelic expression bias in one or the other species (Court et al., 2014; Tucci et al., 2019). Furthermore, while many imprinted genes have the same strong parental allele-specific expression bias in all tissues analyzed, others show tissue-specific imprinting effects, examples of which are Gnas in defined brain regions, endocrine glands and proximal renal tubules, Ube3a in neurons (but not glia and peripheral tissues), as well as Ago2, Trappc9 and Chrac1 in brain (Yamasaki et al., 2003; Weinstein et al., 2010; Mabb et al., 2011; Peters, 2014; Perez et al., 2015). The imprinting status of some genes can also be changed in specific cell types. Dlk1 is a paternally expressed gene with important functions in adipose tissue development, metabolic regulation and neurogenesis (Peters, 2014). However, its imprinting status is lost in neural stem cells (NSCs) of the subventricular zone and hippocampal dentate gyrus with bi-allelic expression of Dlk1 being required for postnatal and adult neurogenesis in these stem cell niches (Ferron et al., 2011; Montalban-Loro et al., 2021). The mechanism for this change in allelic expression status involves postnatal gain of methylation at the gDMR/ICR of the locus (Ferron et al., 2011).
The assessment of allelic expression biases of imprinted genes requires the generation of hybrid mice in crosses between different strains that carry a sufficient number of single nucleotide polymorphisms (SNPs) to be able to identify the parental alleles. Until recently, it was only possible to investigate the allelic expression of genes on the level of bulk primary cell culture or tissue lysates, which often contain different cell types. Although many imprinted genes show an almost exclusive parental bias of ∼90% and are considered mono-allelic in their expression, recent RNA-seq studies have revealed a number of genes with a weaker parental bias of ∼70%, which was used as a threshold for inclusion in the ‘imprinted gene’ category (Babak et al., 2015; Bonthuis et al., 2015; Crowley et al., 2015; Perez et al., 2015; Bouschet et al., 2016; Andergassen et al., 2017; Huang et al., 2017). These data raise questions about how such bulk tissue-level expression biases are reflected on a single cell level, and about the biological significance of such findings (Perez et al., 2016). Several single-cell expression scenarios can potentially lead to a parental allele-specific expression bias of ∼70% in tissues (Bonthuis et al., 2015; Perez et al., 2016), including 1) all cells show the same biased bi-allelic expression, 2) the tissue consists of a mixture of cells with mono-allelic and equal bi-allelic expression, 3) the tissue consists of an unbalanced mixture of cells with respectively mono-allelic paternal and mono-allelic maternal expression, which can be due to differential promoter usage as is the case for Grb10 in neurons versus glial cells and peripheral tissues (Yamasaki-Ishizaki et al., 2007; Sanz et al., 2008; Garfield et al., 2011). To address this question, two types of approaches have been applied recently. Using SNP-FISH in situ hybridization, which employs SNP-specific oligonucleotides, Ginart et al. (2016) were able to distinguish mono-versus bi-allelic expression of H19 and Igf2 in fixed fibroblasts and heart tissue via imaging. With a different in situ hybridization technique, using intronic RNAscope probes for nascent RNA in nuclei, Bonthuis et al. (2015) showed a mixture of cells with mono-allelic and bi-allelic expression of specific imprinted genes in brain sections. Novel single-cell RNA-seq methods have also been applied to analyze imprinted gene expression in single cortical neurons after labeling them with fluorescent proteins and FACS sorting (Laukoter et al., 2020). Their findings indicate some degree of variability of allelic expression in individual neurons, depending on the imprinted gene analyzed. While Meg3 and Snrpn showed the expected mono-allelic expression in almost all neurons, Inpp5f and Impact were mono-allelic paternally expressed in the majority of cells with smaller numbers of neurons displaying bi-allelic or even mono-allelic maternal expression (Laukoter et al., 2020). Thus, these initial studies indicate that the imprinted expression status of a gene, as determined on a tissue level, might not be reflected in all its cells.
The Peg13-Kcnk9-Trappc9 imprinting cluster (schematically shown in Figure 6A) on mouse chromosome 15/human chromosome 8 consists of several genes with neurodevelopmental functions; mutations of these cause disorders in both species. At the center of the locus, the non-coding RNA Peg13 is expressed from the paternal allele, starting at an unmethylated CGI promoter and DMR located within an intron of Trappc9 (Smith et al., 2003; Ruf et al., 2007). Germline-derived methylation silences Peg13 on the maternal allele. Complete deletion of Peg13 on the unmethylated paternal allele is lethal in mice, while the same mutation on the maternal allele does not cause a phenotype (Keshavarz and Tautz, 2021). A milder behavioral phenotype is observed with a mutation that truncates the non-coding RNA (Keshavarz and Tautz, 2021). The second gene of the cluster, Kcnk9, for which imprinting is conserved between human and mouse, encodes the two-pore domain potassium channel subunit Task3 (Court et al., 2014). Kcnk9 is expressed with a strong maternal allelic bias in adult brain (Ruf et al., 2007; Court et al., 2014; Cooper et al., 2020). Mutations of the gene cause Birk-Barel intellectual disability syndrome in humans and behavioral abnormalities in mice (Linden et al., 2007; Barel et al., 2008; Cooper et al., 2020). The three genes Trappc9, Chrac1 and Ago2 show an imprinted bias of expression from the maternal allele in mouse brain, but are not imprinted in human (Court et al., 2014; Babak et al., 2015; Bonthuis et al., 2015; Crowley et al., 2015; Perez et al., 2015; Bouschet et al., 2016; Andergassen et al., 2017; Huang et al., 2017). The mechanisms underlying the predominantly maternal expression of these three genes in mouse brain are currently unclear. Trappc9 encodes a subunit of the intracellular trafficking protein particle II complex (TrappII), mutations of which lead to a neurodevelopmental disorder in humans and mice, which includes symptoms of postnatal microcephaly, intellectual disability and speech impairment (Ke et al., 2020; Liang et al., 2020; Wilton et al., 2020; Aslanger et al., 2022). Ago2 forms a subunit of the RNA-induced silencing complex (RISC). Heterozygous mutations in humans result in a range of neurological phenotypes while homozygous mutation in mice is embryonic lethal (Liu et al., 2004; Lessel et al., 2020). Little is known about the chromatin accessibility factor Chrac1.
In this study, we analyzed the allelic expression of these genes in bulk tissue and neurosphere lysates comparatively to single NSCs and differentiated neurons. We especially focused on Peg13, Trappc9 and Ago2 as examples of strongly and moderately biased imprinted genes. We found variability of allelic expression in individual cells, which was more pronounced for Trappc9 and Ago2 than for Peg13. All categories of expression from mono-allelic maternal to equal bi-allelic to mono-allelic paternal, as well as biased bi-allelic states, were identified for Trappc9 and Ago2 in single cells. For Peg13, a majority of cells showed the expected mono-allelic paternal or paternally biased bi-allelic expression, but a small number of cells deviated from this status, displaying equal bi-allelic or even a maternally biased expression. However, considering the whole population of single cells analyzed, we find that the imprinted gene expression status approximates the findings from bulk tissue lysates. Additionally, we determined the transcriptional start site of Trappc9 and investigated potential transcript variants as well as regulatory regions located within the locus, which led to the identification of sequence elements with a silencing function in primary neurons and/or fibroblasts.
Mouse strains C57BL/6J and Cast/EiJ were bred and maintained in the Babraham Institute Biological Support Unit. Ambient temperature was ∼19–21°C and relative humidity 52%. Lighting was provided on a 12 h light: 12 h dark cycle including 15 min “dawn” and “dusk” periods of subdued lighting. After weaning, mice were transferred to individually ventilated cages with 1–5 mice per cage. Mice were fed CRM (P) VP diet (Special Diet Services) ad libitum and received seeds (e.g., sunflower, millet) at the time of cage-cleaning as part of their environmental enrichment. Breeding and maintenance of these strains were performed under licenses issued by the Home Office (United Kingdom) in accordance with the Animals (Scientific Procedures) Act 1986 and were approved by the Animal Welfare and Ethical Review Body at the Babraham Institute. Tissues were collected from newborn or adult mice and either frozen for molecular biology or processed for cell culture. Frozen tissues from C57BL/6J and Mus musculus molossinus JF1 hybrid mice were kindly provided by Dr Philippe Arnaud, Université Clermont Auvergne, France.
Neurosphere culture was performed as described previously (Ferron et al., 2007; Chojnacki and Weiss, 2008) with slight modifications. Briefly, hippocampi were dissected from newborn mouse brain in ice-cold neurosphere growth medium (DMEM/F12 (Gibco) supplemented with 0.6% w/v glucose, 0.1% NaHCO3, 5 mM HEPES, 2 mM L-GIn, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 1x B27 (Gibco), 10 ng/ml FGF-2 (Peprotech), 20 ng/ml EGF (Peprotech), 4 mg/ml BSA (Sigma) and then transferred into Accutase (Gibco) for dissociation into a single-cell suspension by gentle trituration. Following centrifugation at 200 g for 5 min, cells were resuspended in growth medium and plated at a density of 3,000 cells/cm2 in suspension cell culture dishes (Corning). Neurospheres were allowed to grow for 6–8 days with intermittent medium supplementation before passaging via Accutase dissociation. Cells were re-plated at a lower density of 1,500–2,000 cells/cm2. Neurospheres could be stored in liquid nitrogen after freezing in growth medium with 10% DMSO. For bulk or single-cell gene expression analysis, neurospheres at early passage numbers (P3–P5) were used. Neurospheres were differentiated into neurons at the point of passaging by seeding a single cell suspension on Poly-L-Lysine (Sigma) coated dishes in differentiation medium (growth medium without EGF, FGF-2 and BSA, but containing 1% FBS). For single-neuron analysis, selection against replicating glial cells was started after 2 days of culture with 2 μM Cytosine β-D-arabinofuranoside (AraC) (Sigma) as described in the next paragraph.
Primary hippocampal neurons were cultured as described (Beaudoin et al., 2012; Ioannou et al., 2019) with modifications. Hippocampi were dissected from newborn mouse brain in ice-cold dissection medium [HBSS (Sigma) supplemented with 0.1% w/v glucose, 10 mM Hepes pH 7.4, 1% Na-pyruvate] and the tissue dissociated by adding an equal volume of 2x Papain stock solution (Worthington) at 37°C for 20 min. The supernatant was removed carefully, the tissue gently washed with plating medium [MEM (Gibco) supplemented with 0.45% glucose, 10% FBS, 1% Na-pyruvate, 2 mM Glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin] and then carefully triturated in fresh plating medium. The dissociated tissue was rinsed through a 70-μm cell strainer (Corning) and the collected cells centrifuged at 200 g for 5 min. Cells were resuspended in neuronal medium [Neurobasal medium (Gibco) supplemented with 2 mM glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 1x B27 (Gibco)] and plated in Poly-L-Lysine (Sigma) coated dishes at a density of 60,000 cells/cm2. Medium was replaced the following day, and on day two selection against replicating non-neuronal cells was started with neuronal medium containing 2 μM AraC. Half the medium was replaced with fresh neuronal medium every other day to dilute out the AraC.
Mouse embryonic fibroblasts were prepared as described (Matise et al., 2000) and cultured from frozen stocks in Hepes-buffered DMEM (Sigma) supplemented with 10% FBS, 2 mM Glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin.
The promoter-reporter gene plasmids were transfected into fibroblasts and into primary hippocampal neurons after 7 days of culture using Lipofectamine 2000 (Invitrogen). The firefly luciferase-based test constructs were mixed with a Renilla luciferase control plasmid (pGL4.74, Promega) at a 100:1 ratio to normalize for transfection efficiency. Cells were lysed 48 h after transfection and luciferase activities were measured using the Dual-Luciferase® Reporter Assay System (Promega) on a Glomax Multi Detection System (Promega).
RNA was isolated from neurospheres and tissues using TRIzol™ reagent (Invitrogen) or RNeasy Plus Mini kit (Qiagen). Samples were treated with DNAse I to remove any traces of DNA before cDNA was synthesized with ProtoScript® II Reverse Transcriptase (New England Biolabs) or SuperScript III™ Reverse Transcriptase (Invitrogen) using random hexamer primers, if not otherwise stated. PCR was performed using GoTaq® Hot Start Polymerase (Promega) or Q5™ High-Fidelity DNA Polymerase (New England Biolabs). For the bulk neurosphere and tissue gene expression analysis we used 1 μg of total RNA in reverse transcription reactions, which were then diluted 5-fold for endpoint PCR (initial denaturation 98°C, 30 s; 30 cycles of 98°C, 50–72°C annealing (primer dependent); 72°C, 30 s; final extension 72°C, 2 min) to obtain enough products for pyrosequencing analysis. 5′-RACE experiments were undertaken with ExactSTART™ Eukaryotic mRNA 5′&3′ RACE Kit (Epicentre/Illumina) on adult mouse brain RNA. The 5′-RACE cDNA was then amplified with a Trappc9-specific reverse primer (Pr_05RV; Supplementary Table S1) and a kit-supplied RACE 5′-linker primer, followed by cloning of PCR products into TOPO®-plasmids (Invitrogen) and sequencing.
SNPs in cDNA from tissues and neurospheres of hybrid mice, as well as genomic DNA for methylation analysis after bisulfite treatment, were sequenced using a PyroMark® Q96 ID instrument (Qiagen). PCR and sequencing primers (Supplementary Table S1) were designed using PyroMark Assay Design Software 2.0. Biotinylated PCR products were immobilized on streptavidin-coated beads for cleanup and sequenced using PyroMark® Gold Q96 reagents (Qiagen) following the manufacturer’s protocols.
Single C57BL/6J × Cast/EiJ neurosphere cells were obtained via dissociation with Accutase, dilution in growth medium and either FACS sorting or manual isolation via capillary action under a microscope using a protocol originally developed for oocytes (Lorthongpanich et al., 2013; Cheow et al., 2015; Cheow et al., 2016). Single neurons from differentiated neurospheres were collected after 7 days of culture in differentiation medium, which included 5 days of AraC treatment. Neurons were dissociated from culture dishes using Trypsin/EDTA (Sigma), diluted in differentiation medium and single cells isolated manually via capillary action under a microscope. Single cells were transferred into PCR tubes containing 5 μl of lysis buffer [CellsDirect Resuspension and Lysis Buffer, 10:1 (Invitrogen)] and incubated at 75°C for 10 min (Lorthongpanich et al., 2013; Cheow et al., 2015; Cheow et al., 2016). cDNA was synthesized at 37°C by adding an equal volume of a 2x reverse transcription master mix using MultiScribe™ Reverse Transcriptase (Invitrogen) and hexamer primers. This was followed by protease treatment (Qiagen Protease) as described (Lorthongpanich et al., 2013; Cheow et al., 2015; Cheow et al., 2016) to remove chromatin-associated proteins from genomic DNA. Next, we perform single-cell restriction analysis of methylation (SCRAM), using BstUI, to digest unmethylated CpG sites of genomic DNA. This provides the additional option of analyzing DNA methylation of CGIs in the single cells (Lorthongpanich et al., 2013; Cheow et al., 2015; Cheow et al., 2016). A final Proteinase K digest was carried out before undertaking multiplex PCR pre-amplification using a pool of primers for all target genes with the following conditions: initial denaturation 95°C, 10 min; 30 cycles of 95°C, 30 s; 60°C annealing/extension, 4 min. After pre-amplification, unincorporated primers were removed from the reactions by adding exonuclease I (Lorthongpanich et al., 2013; Cheow et al., 2015; Cheow et al., 2016). Pre-amplification reactions were then diluted 10-fold and aliquots were used to amplify individual target genes via nested-primer qPCR using 3 μl of diluted template and PowerUp SYBR Green 2x Master mix (Invitrogen) under the following conditions: initial denaturation 95°C, 10 min; 30 cycles of 95°C, 15 s; 60°C annealing/extension 1 min; melt curve analysis 60–95°C ramp 5 s/degree. Primers for target genes are listed in Supplementary Table S1. Where applicable, these qPCR products were purified using MinElute PCR® purification kit (Qiagen) and Sanger-sequenced for cDNA SNP expression analysis.
Genomic DNA was isolated from tissues through Proteinase K (100 μg/ml) digest in lysis buffer (100 mM Tris, 5 mM EDTA, 200 mM NaCl, 0.2% SDS, pH 8.5) at 55°C overnight, followed by Phenol/Chloroform extraction, Ethanol precipitation and resuspension in TE buffer. Neurosphere DNA was obtained using TRIzol™ reagent (Invitrogen) in a follow-on step after initial RNA isolation via Phenol/Ethanol extraction, precipitation and resuspension in TE. For DNA methylation analysis of CpG sites, bisulfite conversion of unmethylated cytosines was carried out using the EZ DNA Methylation-Gold™ kit (Zymo Research) according to manufacturer instructions. The bisulfite-treated and purified DNA was then used for PCR amplification of CGI fragments, followed either by methylation analysis via direct pyrosequencing or cloning of PCR products into TOPO®-vectors (Invitrogen) and Sanger sequencing of individually cloned plasmid samples. Sanger sequencing results were further analyzed using the free online tool “QUantification tool for Methylation Analysis” (QUMA; http://quma.cdb.riken.jp/).
Potential brain-specific gene regulatory elements were identified from histone modification and CTCF ChIP assay data, as well as DNAse I and ATAC-seq hypersensitivity data for the newborn (P0) mouse brain in comparison to peripheral tissues. These data were extracted from the databases ENCODE3 (https://www.encodeproject.org/), UCSC Genome Browser (https://genome-euro.ucsc.edu/) and ENSEMBL (https://www.ensembl.org/Mus_musculus/Info/Index). Seven candidate brain-specific regulatory elements across the Trappc9-Peg13 locus were found; their genomic positions (mouse GRCm38/mm10 genome version) and features are listed in Supplementary Figure S6. Similarly, the Trappc9 promoter characteristics at exon 1 were noted. To test the functionality of these regulatory elements in transfected cells, promoter-reporter gene plasmids were generated. The regulatory elements were amplified from C57BL/6J genomic DNA using Q5™ High-Fidelity DNA Polymerase (New England Biolabs), cloned into TOPO®-plasmids (Invitrogen) and sequenced for confirmation. Similarly, four Trappc9 promoter fragments of different lengths were cloned (positions 73,061,805–73,060,204 bp, 73,061,805–73,060,975 bp, 73,061,418–73,060,204 bp and 73,061,418–73,060,975 bp in GRC38/mm10). The firefly luciferase-encoding pGL4.23 [luc2/minP] vector (Promega) was used to generate reporter-gene constructs. First, the endogenous minimal promoter was removed via HindIII and NcoI digest and then replaced with a Trappc9 promoter fragment. The four Trappc9 promoter plasmids were tested in a preliminary reporter gene assay for their activity. The promoter fragment 73,061,418–73,060,975 bp, which avoids an upstream dinucleotide repeat sequence stretch and ends before the exon 1 splice donor site, showed the highest activity and was used in further experiments in combination with the identified regulatory elements. The regulatory elements were cloned into the pGL vector at the BamHI site downstream of the Luc2 reporter gene to reflect the same relative orientation to the Trappc9 promoter as in the genome. Only the Reg-E element was cloned upstream of the Trappc9 promoter as shown in Figure 6B.
The data for the promoter-reporter gene assays were analyzed using GraphPad Prism v.9.3 software. Data were analyzed for outliers using the ROUT method. The datasets were then analyzed for normality using the Shapiro-Wilkinson test. For non-parametric datasets, Mann-Whitney U-tests were performed in comparisons of the Reg-element datasets to the basic Trappc9 promoter dataset. For parametric datasets, unpaired t-tests were performed.
The core imprinted gene of this cluster on mouse chromosome 15, Peg13, is located within intron 17 of Trappc9 and is transcribed into a non-coding RNA (Smith et al., 2003; Wang et al., 2008). For Trappc9 itself, several alternatively spliced and truncated transcript variants have been described and annotated on ENSEMBL, including alternative first exons (Figure 1A). Furthermore, while some transcript variants were found to be predominantly expressed from the maternal allele in brain tissue, the truncated variant 203, which ends shortly after Peg13 in intron 17, was identified as a paternal allele-specific transcript in RNA-seq studies (Gregg et al., 2010; Hsu et al., 2018). We set out to confirm these variants using RT-PCR across specific Trappc9 exons. We readily detected the full-length transcript 202 in brain and other tissues of newborn mice but were not able to confirm the alternative first exon of the 201 transcript (Figure 1B). Exon 2 of the 202 transcript contains a conserved translational start codon across mammalian species. To further investigate potential alternative transcriptional start sites, we undertook 5′-RACE PCR and sequencing, which revealed several transcriptional start sites within the exon 1 5′-UTR of variant 202 (Figure 1C), but we did not detect the alternative exon 1 of variant 201. Sequencing of PCR products also showed alternative splicing of exon 5, which was missing in some kidney and spleen cDNAs, in line with ENSEMBL annotations. Additionally, we investigated the alternative, truncated Trappc9 splice forms 206 and 203, which terminate in intron 17 upstream and downstream of Peg13, respectively (Figure 1A, Supplementary Figure S1). We were unable to detect any cDNA containing the shared exon 17 in combination with 3′-UTR exons of variants 206 and 203 in tissues (Supplementary Figure S1), or containing shared exon 16 in single neurosphere cells.
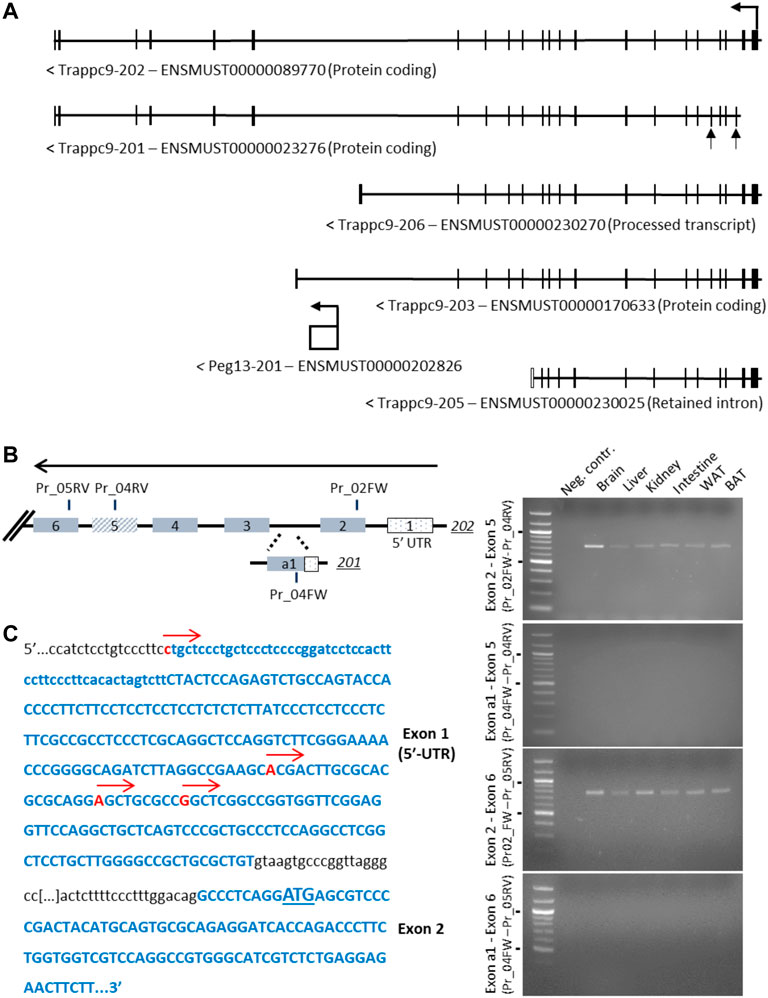
FIGURE 1. Analysis of alternative 5′-exons and transcriptional start sites of Trappc9. (A) Scheme of transcript variants 202, 201, 206, 203 and 205 as indicated on ENSEMBL. The alternative exons of variant 201, a1 and 5, are indicated by arrows. (B) Scheme of exons and primers used in RT-PCR (right) on total C57BL/6J RNA from multiple tissues using primer combinations specific for transcript 202 exon 2 or transcript 201 exon a1 with shared downstream primers in exons 5 and 6 (indicated in the scheme with FW and RV annotations). While expression of transcript 202 exons 2–5 and 2–6 (expected: 740 bp and 780 bp, respectively) was confirmed in multiple tissues, no expression involving transcript 201 exon a1 was detectable (expected: 318 bp and 358 bp, respectively). Size markers of 500 and 1,000 bp are indicated. (C) Sequencing of 5′-RACE RCR products using a downstream primer in exon 6 confirmed multiple transcription start sites in 202 exon 1 (indicated by red arrows), while no start sites in the region of 201 exon a1 could be detected. The first codon is underlined in exon 2.
In conclusion, we have experimentally verified the transcriptional start sites of Trappc9 located in exon 1 of the 202 variant as well as alternative splicing of exon 5 but found no evidence of an alternative promoter or truncated transcript variants.
Apart from Peg13, which constitutes a canonical imprinted gene with mono-allelic paternal expression in a wide range of tissues, the other genes of the cluster have been characterized via RNA-seq as tissue-specifically imprinted with biased expression from the maternal allele in mouse brain (Babak et al., 2015; Perez et al., 2015; Andergassen et al., 2017). We set out to validate these findings through SNP pyrosequencing and additionally included samples of primary NSC (neurosphere) cultures from newborn mice (Supplementary Figures S2A,B), since some imprinted genes have been shown to become bi-allelically expressed specifically in postnatal and adult NSCs (Ferron et al., 2011; Montalban-Loro et al., 2021). We used tissues and cultured hippocampal neurospheres from reciprocal crosses of C57BL/6J and Mus musculus castaneus (Cast/EiJ) newborn F1 hybrids. While Peg13 showed the expected, almost exclusive (80%–90%) expression from the paternal allele in brain, kidney and neurospheres, the other genes of this cluster displayed tissue-specific imprinted expression (Figure 2). Trappc9 and Ago2 were predominantly (70%–80%) transcribed from the maternal allele in brain, but showed equal bi-allelic expression in kidney, while Kcnk9 was expressed almost exclusively (>90%) from the maternal allele in both tissues. Due to unavailability of SNPs between C57BL/6J and Cast/EiJ strains, we analyzed Chrac1 in reciprocal crosses of C57BL/6J and Mus musculus molossinus (JF1) F1 tissue samples and found it to be bi-allelically expressed with only a small bias (<70%) towards the maternal allele in newborn brain (Supplementary Figure S3). We also confirmed brain-specific imprinted expression of Trappc9 in these hybrids (Supplementary Figure S3). Unexpectedly and in contrast to brain tissue as a whole, Ago2 and Trappc9 were not imprinted in the NSC cultures, but showed equal bi-allelic or only slightly biased (<70%) expression (Figure 2), which is reminiscent of Dlk1 (Ferron et al., 2011; Montalban-Loro et al., 2021). Kcnk9 allelic expression in neurospheres was inconclusive and prone to strain-specific biases (Figure 2).

FIGURE 2. Allelic expression in tissues and primary hippocampal neurospheres of hybrid mice. Parental allelic expression was quantified via SNP pyrosequencing of cDNA from tissues or cultured neurospheres (NS) obtained from newborn mice. Average expression (n = 2–3) and example traces are shown. SNP IDs and exon locations are: Peg13 (rs238259968 and rs31423566 located in exon 1), Trappc9 (rs31440851 located in exon 2), Ago2 (rs232384843 located in exon 5) and Kcnk9 (rs225149059 located in exon 1). Points on the bar graphs represent individual pyrosequencing results from reciprocal crosses as shown in the legend. For Peg13 each data point shows the average of the two SNP values in the same sequence read. Representative pyrograms from whole brain samples are shown, indicating the SNP position in the sequence and the quantification of allelic expression.
We undertook pyrosequencing and/or Sanger sequencing of bisulfite-treated DNA to address DNA methylation states at the CGIs of the genes in neurospheres. The germline differentially methylated region (DMR) at Peg13 was maintained in NSCs with methylation observed on the maternal allele (Supplementary Figure S4A), in line with brain tissue observations (Ruf et al., 2007; Xie et al., 2012). The Trappc9 CGI1, located at the promoter/exon1, was unmethylated in NSCs, brain and kidney (Supplementary Figure S4B), while the CGI2 at exon 2 was fully methylated on both alleles in NSCs and brain (Supplementary Figure S4C). The promoter CGIs at Ago2, Chrac1 and Kcnk9 were also unmethylated in NSCs (Supplementary Figures S5A−C), which is in line with human brain data (Court et al., 2014) and their status as actively transcribed genes. This excludes any secondary DMRs at this imprinted gene cluster.
Overall, our data confirm brain-specific imprinting, i.e., preferential expression from the maternal allele, of Trappc9 and Ago2 in mouse, while maternal allelic expression of Kcnk9 occurs in brain and some peripheral tissues, e.g., kidney. Unexpectedly, Trappc9 and Ago2 have no allelic expression bias in hippocampal neurosphere cultures.
Analysis of imprinted gene expression on a bulk tissue level raises the question of whether the observed allelic bias is reflected in every cell of the lysate in the same way, or whether individual cells differ in their mono-/bi-allelic transcriptional status of the gene and, thus, deviate from the tissue average. Large-scale single-cell imprinted gene expression analysis is still in its infancy, but novel approaches indicate that not all cells of a tissue might show the same allelic expression status (Martini et al., 2022). To address this question specifically for the Peg13 imprinting cluster, we isolated single NSCs from C57BL/6J × Cast/EiJ neurospheres as well as single neurons differentiated from these in vitro. We then undertook qRT-PCR using the sc-GEM (single-cell analysis of genotype, expression and methylation) technique (Cheow et al., 2016) and Sanger sequencing to determine allelic SNP expression for Peg13, Trappc9 and Ago2. As a further confirmation of the NSC phenotype, we also verified marker gene expression (Hochgerner et al., 2018) in single neurosphere cells (Supplementary Figure S2C). Analyzing ∼50 NSCs and neurons, we found a variety of mono-allelic and bi-allelic expression states in individual cells.
For Peg13, 39% of the NSCs showed mono-allelic paternal expression and 40% of the cells had transcripts predominantly from the paternal allele, but additionally a small amount of maternal transcripts (Figures 3A–C). However, we also detected small numbers of cells with equal bi-allelic, predominantly maternal, or even mono-allelic maternal expression of Peg13 (Figures 3B,C), which indicates a surprising heterogeneity of imprinted expression between individual NSCs. Proportionately, the 79% of cells with mono-allelic and biased paternal expression approximate the bulk neurosphere expression bias of 87% paternal transcripts (Figure 2). In in vitro differentiated neurons, the cellular heterogeneity was reduced as almost all cells showed mono-allelic or paternal bias of Peg13 expression (Figure 3B), much in line with the paternal bias of 89% in brain tissue as a whole (Figure 2).
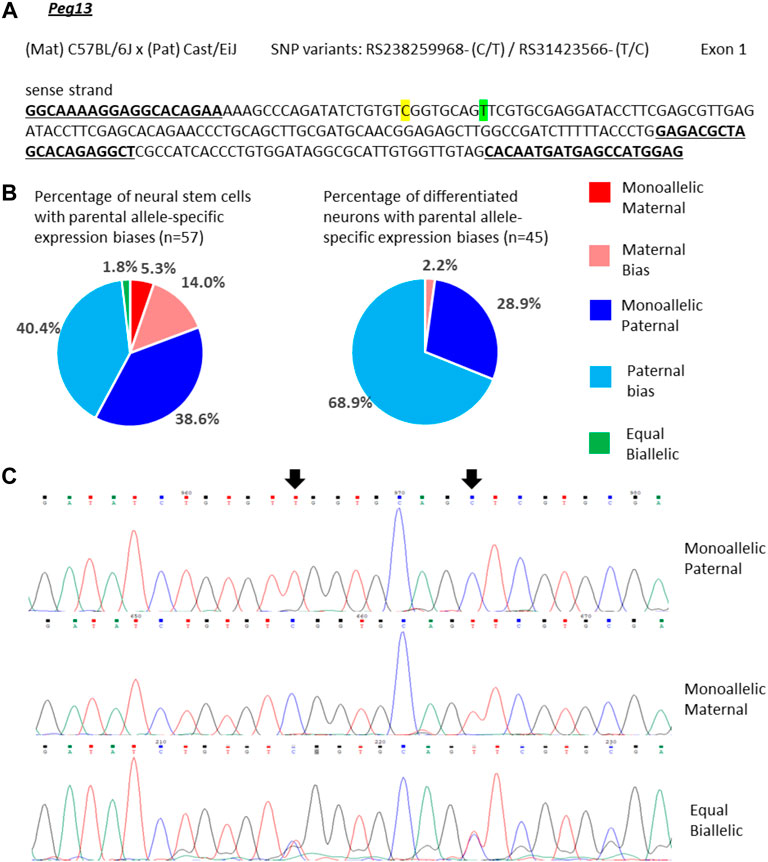
FIGURE 3. Variable single-cell allele-specific expression of Peg13 analyzed through Sanger sequencing of cDNA SNPs from neural stem (neurosphere) cells and their in vitro differentiated neurons. (A) The mouse cross, cDNA amplicon and SNP locations in exon 1 are shown. B6 SNP variants are indicated in color; primers are underlined. (B) Summary data showing the proportions of cells falling into the five categories of allelic expression. (C) Example single-cell sequence tracks for the three expression categories indicated. Sequence tracks for cells with maternal or paternal bias displayed a major SNP peak for those alleles with a minor overlapping SNP peak for the other allele, respectively. SNP positions are highlighted by arrows.
For Trappc9 the allelic expression varied considerably in single NSCs. We observed mono-allelic maternal or paternal Trappc9 expression in 10% of the cells respectively, while 29% displayed equal bi-allelic transcription (Figures 4A–C). Around half of the NSCs showed biased bi-allelic expression: 21% predominantly maternal, 31% predominantly paternal (Figure 4B). Considering all single-NSC categories in proportion, the data are in line with the bulk neurosphere analysis (Figure 2) as no clear overall parental allele bias was observed in either dataset. In differentiated neurons, the proportion of cells with equal bi-allelic expression was reduced to 11%, while another 11% showed mono-allelic maternal and 17% mono-allelic paternal Trappc9 expression (Figure 4B). Taking into account the proportions of neurons with biased bi-allelic expression (33% maternal, 28% paternal bias), the single-neuron dataset depicts an overall equal bi-allelic Trappc9 expression, which is in contrast to the maternal expression bias of 78% that was observed in whole-brain lysate (Figure 2). This discrepancy might be due to the brain lysate containing additional cell types (e.g., astrocytes, microglia, oligodendrocytes) and a wider range of neuron types from different brain sub-regions as compared to the in vitro differentiated neurons, which were derived from hippocampal neurospheres.
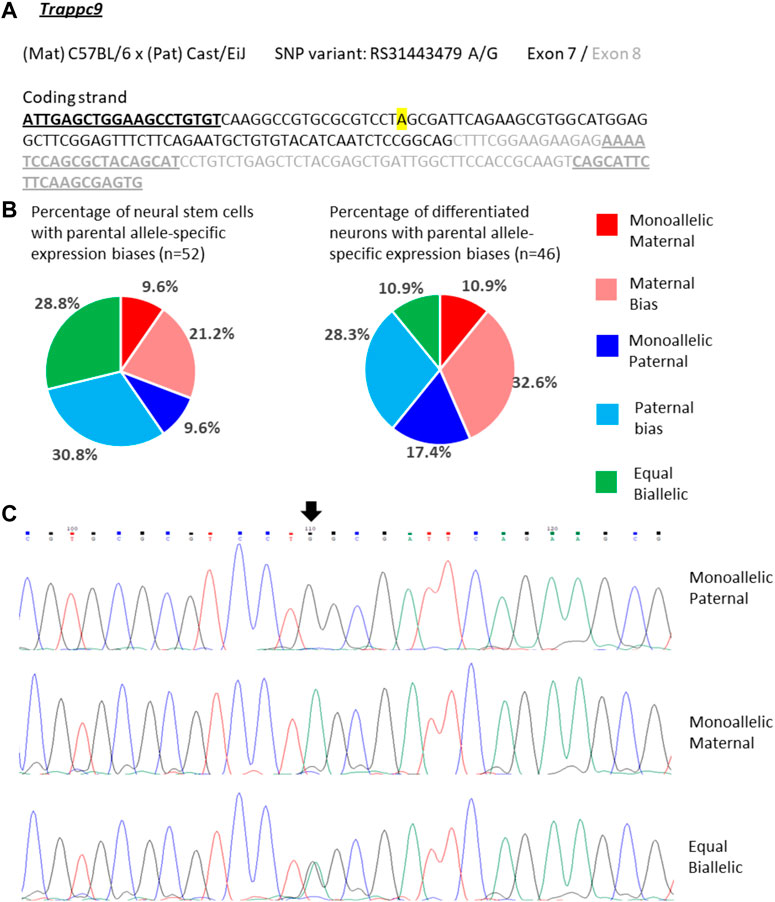
FIGURE 4. Variable single-cell allele-specific expression of Trappc9 analyzed through Sanger sequencing of a cDNA SNP from neural stem (neurosphere) cells and their in vitro differentiated neurons. (A) The mouse cross, cDNA amplicon and SNP location in exon 7 are shown. B6 SNP variant is indicated in color; primers are underlined. (B) Summary data showing the proportions of cells falling into the five categories of allelic expression. (C) Example single-cell sequence tracks for the three expression categories indicated. Sequence tracks for cells with maternal or paternal bias displayed a major SNP peak for those alleles with a minor overlapping SNP peak for the other allele, respectively. SNP position is highlighted by an arrow.
Ago2 expression in single NSCs was similarly variable as Trappc9 expression. 10% of the NSCs displayed mono-allelic maternal, 16% mono-allelic paternal and 12% equal bi-allelic expression (Figures 5A–C). Most NSCs had a biased bi-allelic expression of Ago2 (29% predominantly maternal, 33% predominantly paternal). Overall, the proportions of NSCs falling into the various expression categories did not indicate a clear allele bias and are in line with the equal bi-allelic Ago2 expression found in bulk neurosphere samples (Figure 2). Compared to the NSCs, among the differentiated neurons more cells showed equal bi-allelic (21%), mono-allelic maternal (16%) and biased maternal (32%) expression (Figure 5B), while the proportions of neurons with mono-allelic paternal (9%) and biased paternal (23%) expression were reduced (Figure 5B). However, as with Trappc9, the in vitro differentiated neuron categories did not overall reflect the same strong bias of 75% maternal allele-specific Ago2 transcripts that were detected in whole-brain tissue (Figure 2).
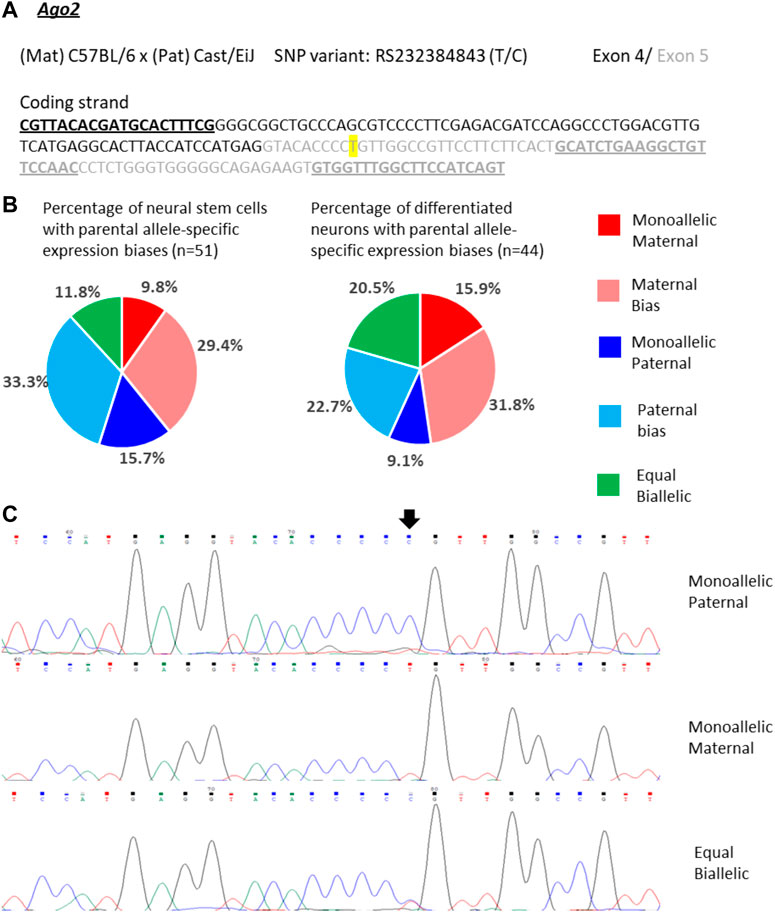
FIGURE 5. Variable single-cell allele-specific expression of Ago2 analyzed through Sanger sequencing of a cDNA SNP from neural stem (neurosphere) cells and their in vitro differentiated neurons. (A) The mouse cross, cDNA amplicon and SNP location in exon 5 are shown. B6 SNP variant is indicated in color; primers are underlined. (B) Summary data showing the proportions of cells falling into the five categories of allelic expression. (C) Example single-cell sequence tracks for the three expression categories indicated. Sequence tracks for cells with maternal or paternal bias displayed a major SNP peak for those alleles with a minor overlapping SNP peak for the other allele, respectively. SNP position is highlighted by an arrow.
Since the non-coding RNA Peg13 is transcribed from the core imprinting regulatory region (DMR) of the locus, findings of Peg13 expression other than mono-allelic paternal or paternally biased bi-allelic (Figures 3B,C) were unexpected. This raises the question of whether there is a specific pattern of allelic expression of the other imprinted genes of the locus associated with Peg13 transcription from the maternal allele. When analyzing the expression status of Trappc9 and Ago2 in those cells that showed equal bi-allelic, maternally biased or mono-allelic maternal expression of Peg13, we did not find any specific patterns or correlations (Supplementary Table S2). Some of these cells displayed the expected maternal bias of Trappc9 and/or Ago2, but other allelic biases, including mono-allelic expression states, were also observed and in varying combinations within individual cells.
In summary, our single-cell analysis of the three imprinted genes indicates a surprising variability of allelic expression states in individual cells, ranging from mono-allelic maternal to mono-allelic paternal transcription, even for the core imprinted gene of the locus, Peg13. Overall, Peg13 transcriptional states in the NSC and neuron populations matched the brain tissue level of allelic expression (∼89% paternal) very well as most cells displayed mono-allelic or strong paternal bias. Trappc9 and Ago2, which represent tissue-specifically imprinted genes with a maternal expression bias of ∼75% in brain, showed much more variability in their allelic transcriptional states in individual NSCs and neurons; all categories of allelic transcription were represented by substantial numbers of cells. Thus, our data do not support a model, in which a tissue-level imprinted gene expression status is reflected in each cell of the tissue in the same way. These findings might hint at a certain level of transcriptional noise or transient/random bursts of transcription, which might still be able to occur at alleles that are “silenced” by genomic imprinting (Varrault et al., 2020).
It is currently unclear how tissue-specific imprinting and maternal allele-biased expression of Trappc9 and Ago2 are regulated in the mouse brain. Furthermore, imprinting of the two genes is not conserved in humans (Court et al., 2014) and only homozygous mutations of TRAPPC9 cause a neurodevelopmental disorder, which is characterized by intellectual disability, speech impairment and microcephaly (Wilton et al., 2020; Aslanger et al., 2022). A potential mechanism could involve chromatin boundaries and CTCF-regulated access to tissue-specific enhancers as has been shown for the imprinted Igf2-H19 locus (Bell and Felsenfeld, 2000). Indeed, CTCF binding on the unmethylated paternal allele of the Peg13 DMR has been demonstrated in mouse brain and fibroblasts (Singh et al., 2011; Prickett et al., 2013) as well as in human brain (Court et al., 2014). In humans, CTCF regulates access of KCNK9 and PEG13 promoters to a brain-specific enhancer, most likely in a differential, allele-specific way (Court et al., 2014). However, since the imprinting status of the genes upstream of Peg13, i.e., Trappc9, Ago2 and Chrac1, differs between mouse and human brain, their regulation presumably involves enhancers that are not conserved or additional mechanisms. We, therefore, screened Encode3 mouse genome data for chromatin modifications and accessibility (Consortium et al., 2020; Gorkin et al., 2020) across the Trappc9-Peg13 locus in the UCSC Genome Browser and identified seven potential brain-specific regulatory elements with appropriate histone modification (H3K4 methylation, H3K27 acetylation, H3K9 acetylation), ATAC and DNAse I hypersensitivity marks (Supplementary Figure S6). To test these candidate elements in promoter-reporter gene assay, we first constructed a Luciferase plasmid that contained a 444 bp Trappc9 promoter fragment upstream of and including non-coding exon 1 (Figure 1C). The size of this promoter fragment is in line with standards from high-throughput testing of promoter-enhancer interactions in the mouse genome (Martinez-Ara et al., 2022). We positioned the candidate regulatory elements downstream or upstream of the promoter-reporter gene cassette, depending on their relative locations within the Trappc9 locus (Figures 6A,B). We performed reporter gene assays in cultures of mouse primary hippocampal neurons and embryonic fibroblasts. Compared to the promoter-only construct, regulatory elements Reg-B and Reg-E had significant silencing effects in fibroblasts, but not neurons, indicating a tissue-/cell type-specific function (Figure 6C). Reg-D displayed silencing activity in both neurons and fibroblasts. Unexpectedly, none of the regulatory elements showed enhancer activity in our assay conditions. All three silencing elements are located on the Trappc9-proximal side of the Peg13 DMR and CTCF-binding site (Figure 6A). These silencer elements might contribute to the regulation of tissue-specific expression of Trappc9, Chrac1 and/or Ago2 in vivo. Reg-D might also contribute specifically to the reduced transcription of their paternal alleles in brain, thereby generating an imprinted expression bias in this tissue, although any allele-specific mechanism remains to be elucidated.
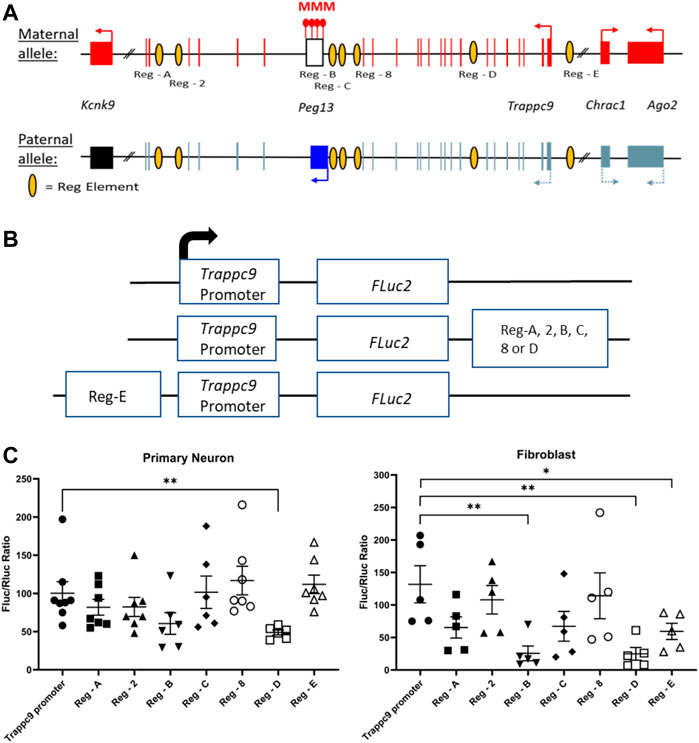
FIGURE 6. Promoter-reporter gene assays indicate silencer elements for Trappc9. (A) Scheme of the murine Peg13—Kcnk9—Trappc9 cluster of imprinted genes. For Trappc9 all introns are shown; for Chrac1, Ago2 and Kcnk9 introns have been omitted. Promoters, transcriptional activity and directions are indicated by arrows. Also indicated are the approximate locations of the candidate regulatory elements (listed in Supplementary Figure S6) across Trappc9 introns. Not to scale. (B) Schematic of the promoter/enhancer constructs used for transfection of primary cells. Enhancers were positioned downstream or upstream of the promoter-reporter gene cassette depending on their relative locations within the Trappc9 gene and in the same orientation. (C) Reporter gene activity of the constructs in cultures of primary hippocampal neurons from newborn mice and primary embryonic fibroblasts. Normalized activity of Firefly luciferase (FLuc) to co-transfected Renilla luciferase (RLuc) is shown. Mean values ± S.E.M. are indicated. *p ≤ 0.05; **p ≤ 0.01.
Our findings of allelic expression biases of these imprinting cluster genes in newborn mouse brain are in line with previous data from whole transcriptome studies, showing strong (∼90%) paternal and maternal preferences for Peg13 and Kcnk9, respectively, while Trappc9 and Ago2 displayed a more moderate (∼75%) preference for the maternal allele (Babak et al., 2015; Bonthuis et al., 2015; Crowley et al., 2015; Perez et al., 2015; Bouschet et al., 2016; Andergassen et al., 2017; Huang et al., 2017). Chrac1 fell below the threshold of 70% bias in our data and also showed a weaker maternal bias than Trappc9 and Ago2 in the studies by Crowley et al. (2015) and Perez et al. (2015). Brain-specificity of Trappc9 and Ago2 imprinting was also confirmed, since we found expression in kidney to be equal bi-allelic, in line with previous data. Unexpectedly, we did not detect an imprinted expression bias for Trappc9 and Ago2 in cultured hippocampal NSCs (neurospheres), where both genes showed equal bi-allelic expression in bulk sample analysis. By contrast, Peg13 retained its strong paternal expression bias in neurospheres. These findings are reminiscent of another imprinted gene, Dlk1, which loses its mono-allelic paternal expression and becomes bi-allelically expressed in postnatal NSCs of the ventricular zone and hippocampal subgranular zone (Ferron et al., 2011; Montalban-Loro et al., 2021). The change to bi-allelic expression of Dlk1 is associated with gain of methylation at its germline DMR and is a requirement for normal postnatal and adult neurogenesis. However, we did not find any change in methylation at the Peg13 germline DMR in neurospheres. Also, the promoter CGI of Trappc9 remained unmethylated on both alleles, while the second CGI at exon 2 retained its high levels of methylation in neurospheres. Similarly, the Ago2 promoter CGI remained unmethylated on both alleles in neurospheres. Thus, the regulation of allelic expression of Trappc9 and Ago2 in NSCs is likely to differ from that in differentiated neural cells and might involve changes in histone modifications, transcription factor binding and/or enhancer access. In any case, a relevance of Trappc9 expression in NSCs is implied by the finding of reduced numbers of Sox2-positive stem cells in the subventricular zone and hippocampal subgranular zone of knock-out mice (Usman et al., 2022), which might be linked to their microcephaly phenotype (Ke et al., 2020; Liang et al., 2020; Wilton et al., 2020; Aslanger et al., 2022).
Due to advances in technology, especially single-cell transcriptomics and highly sensitive in situ hybridization methods, it has now become possible to investigate imprinted gene expression on the cellular level (Varrault et al., 2020; Martini et al., 2022). Instead of a single-cell RNA-seq approach, we used the sc-GEM method (Lorthongpanich et al., 2013; Cheow et al., 2015; Cheow et al., 2016) for a more limited analysis of the genes of this imprinting cluster in single cultured NSCs and differentiated neurons. Our data for Trappc9 and Ago2 show a broad variability of allelic expression status in individual neurosphere cells. All categories of allelic expression, ranging from mono-allelic maternal to equal bi-allelic to mono-allelic paternal and intermediately biased bi-allelic states, were found in significant numbers of cells. Taking into account all single NSCs analyzed, the allelic expression of these two genes leveled out in line with the bulk neurosphere data, i.e., overall there was no allelic bias in the neural stem cell population. Our findings were similar in single neurons that were differentiated from the neurospheres; significant numbers of cells were found for each category of allelic expression. When comparing the cell population of NSCs with the neuronal population for Ago2 expression biases, a slight overall shift from paternal to maternal allelic biases was observed. For Trappc9, the proportion of cells with equal bi-allelic expression was reduced in the neuron population compared to the NSC population, but the proportions of cells with maternal and paternal expression biases, respectively, remained balanced. Neither of the two genes displayed an overall maternal expression preference in the population of single neurons, which is in contrast to the data obtained from brain lysates. However, our neuron culture is not fully representative of all the cell types that would be included in a brain tissue lysate, since we actively selected against dividing glial cells by adding Ara-C to the culture. Furthermore, since the neurons were differentiated from hippocampal NSCs, our culture is likely to contain only a limited range of neuronal cell types. Data from the Allen Brain Map (https://portal.brain-map.org/) in situ hybridization atlas and single-cell transcriptomics indicate medium levels of Trappc9 and Ago2 expression in many neurons of the cortex and hippocampus, with lower levels occurring in some types of neurons as well as astrocytes and oligodendrocytes. Limited histological analysis for Trappc9 by Ke et al. (2020) support these data.
On a more general note, we have no indication that our single-cell data are affected by potential technical issues, for example, allele drop-out during reverse transcription, and we have not found a way to test for such eventualities at extremely low numbers of RNA molecules. However, this would affect the weakly expressed allele, i.e., the paternal allele of Trappc9 or the maternal allele for Peg13 as judged from brain lysates, more than the strongly expressed allele and should lead to an increased number of cells with mono-allelic maternal expression of Trappc9, or exclusively cells with mono-allelic paternal expression of Peg13. There is no indication for such an effect in our data. On the contrary, our data show the opposite, i.e., a surprisingly large number of cells that display a biased paternal expression of Peg13 with weak expression of the maternal allele readily detectable, and even cells with predominantly maternal Peg13 expression. Such results would not be expected, if there were a significant rate of allelic drop-out of the weakly expressed allele. Furthermore, our findings of varying mono- or bi-allelic expression states of Ago2 in individual neurons is in line with in situ hybridization data obtained by Bonthuis et al. (2015), who analyzed nascent transcripts in nuclei of brain sections and determined that 46% of Ago2 expressing cells in the arcuate nucleus, and 63% in the dorsal raphe nucleus, showed mono-allelic expression with the remainder having two visible sites of nuclear transcription.
For Peg13, our single-cell data indicated a predominantly paternal expression bias in NSCs, and even more so in differentiated neurons, although there was a substantial proportion of cells with paternally biased bi-allelic (instead of mono-allelic) expression. A small number of cells, mainly NSCs, deviated from this expected bias and showed equal bi-allelic or even mono-allelic maternal expression. Although surprising, these findings are not unprecedented. A recent study of imprinted gene expression in single cortical cells identified similar variability and occasional deviations from expected biases (Laukoter et al., 2020). For example, Meg3 (also known as Gtl2), which usually has a strong maternal expression bias, was found to be bi-allelic in a small number of cortical cells. For two other imprinted genes with an expected paternal expression bias, i.e., Inpp5f and Impact, a substantial number of cortical cells deviated towards bi-allelic or predominantly maternal expression (Laukoter et al., 2020). The mechanisms and reasons behind such variable allelic expression states of imprinted genes in individual cells are largely unclear. The cases of Dlk1 gain of methylation and loss of imprinting in NSCs (Ferron et al., 2011; Montalban-Loro et al., 2021), or Grb10 alternative promoter usage on the maternal and paternal alleles (Yamasaki-Ishizaki et al., 2007; Sanz et al., 2008; Garfield et al., 2011) are unlikely models for our findings. On the other hand, random mono-allelic expression (RMAE) effects, especially transcriptional bursting (Reinius and Sandberg, 2015; Chess, 2016; Xu et al., 2017; Varrault et al., 2020), might affect imprinted genes and be the underlying reason for the variable allelic expression states we find in single cells. Such RMAE effects might be stochastic and dynamic, rather than permanent as in the case of random allelic exclusion of immunoglobulin genes, and might involve relatively short-lived CTCF-cohesin chromatin loops (Gabriele et al., 2022).
For the human PEG13-KCNK9 locus, CTCF-cohesin binding sites, chromatin looping and enhancer interactions with those two gene promoters have been described (Court et al., 2014). CTCF-binding on the unmethylated paternal allele of the Peg13 gDMR is conserved in mice (Singh et al., 2011; Prickett et al., 2013), but tissue-specific enhancer elements and chromatin looping might differ in this species and could underly the imprinted expression of Trappc9, Chrac1 and Ago2 in murine brain tissue. Our ENCODE3-based search for brain-specific regulatory elements considered active histone and open chromatin marks and resulted in several candidate regions, but unexpectedly these did not show enhancer function when tested in transfected primary neurons or fibroblasts. Instead, two elements had silencing activity specifically in fibroblasts, while a third element silenced reporter gene activity in both neurons and fibroblasts. Typically, active enhancers are associated with active chromatin marks, e.g., H3K27ac, H3K4me1, H3K9ac, and silencers are variably marked by H4K20me, H3K9me3 (typical for heterochromatin and methylated DNA) and/or H3K27me3, although chromatin at silencer regions is still expected to be open for binding of repressive transcription factors and, therefore, also associated with H3K79me2 and H3K36me3 marks (Pang and Snyder, 2020). However, there is currently no widely accepted consensus for a silencer chromatin signature and many silencer elements might be bifunctional elements acting through various mechanisms (Segert et al., 2021). Furthermore, a recent functional study of ENCODE3 candidate cis-regulatory elements (cCREs) found that the majority of annotated cCREs had no effect on transcription, while similar numbers of the remaining elements had enhancer or repressor activity, respectively, which was surprising given that cCREs are predicted to be enhancers (Martinez-Ara et al., 2022). Further experiments will be required to determine whether the silencer elements we identified within the Trappc9 locus might function in an allele-specific way and contribute to the brain-specific imprinted expression bias of this gene.
A number of Trappc9 transcript variants have been annotated on ENSEMBL, including two alternative promoters and truncated transcripts, one of which has been described as specifically expressed from the paternal allele in RNA-seq data (Gregg et al., 2010; Hsu et al., 2018). Our attempts to confirm such truncated transcript versions with primer combinations that span transcript-specific and shared exons were unsuccessful. Trappc9 and Peg13 are transcribed in the same direction and Peg13 is an unspliced long non-coding RNA located in intron 17 of Trappc9. Peg13 transcription from the paternal allele might extend further downstream than is currently known, similar to other long non-coding RNAs at imprinted loci, e.g., Nespas, or Meg3/Gtl2 (Ferguson-Smith, 2011; Peters, 2014). Therefore, allelic expression analysis of Trappc9 RNA downstream of the Peg13 start site requires careful consideration. We also found no evidence for a second promoter of Trappc9. The transcriptional start sites we identified at the first non-coding exon will create transcripts with a translational start site in exon 2, which encodes the well-conserved NH2-terminal end of the protein. This would be missing upon alternative promoter usage. From our data, we can exclude a second promoter, which is also of relevance in the context of the variable allelic expression of Trappc9 discussed above. We can exclude a second promoter as a possible explanation for the variable allelic expression in single cells.
Overall, the mechanisms of brain-specific imprinted expression of Trappc9 in mice remain to be fully elucidated, but this allelic bias is of biological relevance, since maternal transmission of a knock-out mutation of Trappc9 results in phenotypes similar to homozygous deletion and, vice versa, mice carrying a paternally transmitted mutation are not different from wild-types (Liang et al., 2020).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://zenodo.org/record/7002711#.YwSFdnbMKUk.
The animal study was reviewed and approved by the Animal Welfare and Ethical Review Body at the Babraham Institute.
MC, MP, MS, RB, PZ, SA, and TL conducted the experiments and collected and analyzed the data. PA, GK, and DM provided essential materials for the work. MC, SA, and AP carried out the statistical analysis. MC and AP wrote the manuscript. DM and AP conceived, designed and supervised the project and acquired funding. All authors reviewed and edited the manuscript before submission.
This project was funded by a Wellcome Trust grant (099795/Z/12/Z), a University of Liverpool—A*STAR Research Attachment Programme (ARAP, Singapore) PhD studentship and a PhD studentship from the King Saud University, Saudi Arabia. Work in G.K.’s lab was funded by the UK Biotechnology and Biological Sciences Research Council (BBS/E/B/000C0423) and Medical Research Council (MR/S000437/1). This work was supported by funding from the National Research Foundation, Singapore (NRFF-2015-05) to DMM’s lab.
We would like to thank all staff of the animal facility for their dedicated work. We would also like to thank Abigail Clark and Megan Green for contributions to early stages of the project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1022422/full#supplementary-material
Andergassen, D., Dotter, C. P., Wenzel, D., Sigl, V., Bammer, P. C., Muckenhuber, M., et al. (2017). Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. Elife 6, e25125. doi:10.7554/eLife.25125
Aslanger, A. D., Goncu, B., Duzenli, O. F., Yucesan, E., Sengenc, E., and Yesil, G. (2022). Biallelic loss of TRAPPC9 function links vesicle trafficking pathway to autosomal recessive intellectual disability. J. Hum. Genet. 67 (5), 279–284. doi:10.1038/s10038-021-01007-8
Babak, T., DeVeale, B., Tsang, E. K., Zhou, Y., Li, X., Smith, K. S., et al. (2015). Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet. 47 (5), 544–549. doi:10.1038/ng.3274
Barel, O., Shalev, S. A., Ofir, R., Cohen, A., Zlotogora, J., Shorer, Z., et al. (2008). Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am. J. Hum. Genet. 83 (2), 193–199. doi:10.1016/j.ajhg.2008.07.010
Beaudoin, G. M., Lee, S. H., Singh, D., Yuan, Y., Ng, Y. G., Reichardt, L. F., et al. (2012). Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 7 (9), 1741–1754. doi:10.1038/nprot.2012.099
Bell, A. C., and Felsenfeld, G. (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405 (6785), 482–485. doi:10.1038/35013100
Bonthuis, P. J., Huang, W. C., Stacher Horndli, C. N., Ferris, E., Cheng, T., and Gregg, C. (2015). Noncanonical genomic imprinting effects in offspring. Cell Rep. 12 (6), 979–991. doi:10.1016/j.celrep.2015.07.017
Bouschet, T., Dubois, E., Reynes, C., Kota, S. K., Rialle, S., Maupetit-Mehouas, S., et al. (2016). In vitro corticogenesis from embryonic stem cells recapitulates the in vivo epigenetic control of imprinted gene expression. Cereb. Cortex 27, 2418–2433. doi:10.1093/cercor/bhw102
Cheow, L. F., Courtois, E. T., Tan, Y., Viswanathan, R., Xing, Q., Tan, R. Z., et al. (2016). Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat. Methods 13 (10), 833–836. doi:10.1038/nmeth.3961
Cheow, L. F., Quake, S. R., Burkholder, W. F., and Messerschmidt, D. M. (2015). Multiplexed locus-specific analysis of DNA methylation in single cells. Nat. Protoc. 10 (4), 619–631. doi:10.1038/nprot.2015.041
Chess, A. (2016). Monoallelic gene expression in mammals. Annu. Rev. Genet. 50, 317–327. doi:10.1146/annurev-genet-120215-035120
Chojnacki, A., and Weiss, S. (2008). Production of neurons, astrocytes and oligodendrocytes from mammalian CNS stem cells. Nat. Protoc. 3 (6), 935–940. doi:10.1038/nprot.2008.55
Consortium, E. P., Moore, J. E., Purcaro, M. J., Pratt, H. E., Epstein, C. B., Shoresh, N., et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583 (7818), 699–710. doi:10.1038/s41586-020-2493-4
Cooper, A., Butto, T., Hammer, N., Jagannath, S., Fend-Guella, D. L., Akhtar, J., et al. (2020). Inhibition of histone deacetylation rescues phenotype in a mouse model of Birk-Barel intellectual disability syndrome. Nat. Commun. 11 (1), 480. doi:10.1038/s41467-019-13918-4
Court, F., Camprubi, C., Garcia, C. V., Guillaumet-Adkins, A., Sparago, A., Seruggia, D., et al. (2014). The PEG13-DMR and brain-specific enhancers dictate imprinted expression within the 8q24 intellectual disability risk locus. Epigenetics Chromatin 7 (1), 5. doi:10.1186/1756-8935-7-5
Crowley, J. J., Zhabotynsky, V., Sun, W., Huang, S., Pakatci, I. K., Kim, Y., et al. (2015). Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nat. Genet. 47 (4), 353–360. doi:10.1038/ng.3222
Ferguson-Smith, A. C. (2011). Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 12 (8), 565–575. doi:10.1038/nrg3032
Ferron, S. R., Andreu-Agullo, C., Mira, H., Sanchez, P., Marques-Torrejon, M. A., and Farinas, I. (2007). A combined ex/in vivo assay to detect effects of exogenously added factors in neural stem cells. Nat. Protoc. 2 (4), 849–859. doi:10.1038/nprot.2007.104
Ferron, S. R., Charalambous, M., Radford, E., McEwen, K., Wildner, H., Hind, E., et al. (2011). Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature 475 (7356), 381–385. doi:10.1038/nature10229
Gabriele, M., Brandao, H. B., Grosse-Holz, S., Jha, A., Dailey, G. M., Cattoglio, C., et al. (2022). Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376 (6592), 496–501. doi:10.1126/science.abn6583
Garfield, A. S., Cowley, M., Smith, F. M., Moorwood, K., Stewart-Cox, J. E., Gilroy, K., et al. (2011). Distinct physiological and behavioural functions for parental alleles of imprinted Grb10. Nature 469 (7331), 534–538. doi:10.1038/nature09651
Ginart, P., Kalish, J. M., Jiang, C. L., Yu, A. C., Bartolomei, M. S., and Raj, A. (2016). Visualizing allele-specific expression in single cells reveals epigenetic mosaicism in an H19 loss-of-imprinting mutant. Genes Dev. 30 (5), 567–578. doi:10.1101/gad.275958.115
Gorkin, D. U., Barozzi, I., Zhao, Y., Zhang, Y., Huang, H., Lee, A. Y., et al. (2020). An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583 (7818), 744–751. doi:10.1038/s41586-020-2093-3
Gregg, C., Zhang, J., Weissbourd, B., Luo, S., Schroth, G. P., Haig, D., et al. (2010). High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329 (5992), 643–648. doi:10.1126/science.1190830
Hochgerner, H., Zeisel, A., Lonnerberg, P., and Linnarsson, S. (2018). Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat. Neurosci. 21 (2), 290–299. doi:10.1038/s41593-017-0056-2
Hsu, C. L., Chou, C. H., Huang, S. C., Lin, C. Y., Lin, M. Y., Tung, C. C., et al. (2018). Analysis of experience-regulated transcriptome and imprintome during critical periods of mouse visual system development reveals spatiotemporal dynamics. Hum. Mol. Genet. 27 (6), 1039–1054. doi:10.1093/hmg/ddy023
Huang, W. C., Ferris, E., Cheng, T., Horndli, C. S., Gleason, K., Tamminga, C., et al. (2017). Diverse non-genetic, allele-specific expression effects shape genetic architecture at the cellular level in the mammalian brain. Neuron 93(5), 1094–1109 e1097. doi:10.1016/j.neuron.2017.01.033
Ioannou, M. S., Liu, Z., and Lippincott-Schwartz, J. (2019). A neuron-glia Co-culture system for studying intercellular lipid transport. Curr. Protoc. Cell Biol. 84 (1), e95. doi:10.1002/cpcb.95
Ke, Y., Weng, M., Chhetri, G., Usman, M., Li, Y., Yu, Q., et al. (2020). Trappc9 deficiency in mice impairs learning and memory by causing imbalance of dopamine D1 and D2 neurons. Sci. Adv. 6 (47), eabb7781. doi:10.1126/sciadv.abb7781
Keshavarz, M., and Tautz, D. (2021). The imprinted lncRNA Peg13 regulates sexual preference and the sex-specific brain transcriptome in mice. Proc. Natl. Acad. Sci. U. S. A. 118 (10), e2022172118. doi:10.1073/pnas.2022172118
Laukoter, S., Pauler, F. M., Beattie, R., Amberg, N., Hansen, A. H., Streicher, C., et al. (2020). Cell-type specificity of genomic imprinting in cerebral cortex. Neuron 107 (6), 1160–1179. e1169. doi:10.1016/j.neuron.2020.06.031
Lessel, D., Zeitler, D. M., Reijnders, M. R. F., Kazantsev, A., Hassani Nia, F., Bartholomaus, A., et al. (2020). Germline AGO2 mutations impair RNA interference and human neurological development. Nat. Commun. 11 (1), 5797. doi:10.1038/s41467-020-19572-5
Liang, Z. S., Cimino, I., Yalcin, B., Raghupathy, N., Vancollie, V. E., Ibarra-Soria, X., et al. (2020). Trappc9 deficiency causes parent-of-origin dependent microcephaly and obesity. PLoS Genet. 16 (9), e1008916. doi:10.1371/journal.pgen.1008916
Linden, A. M., Sandu, C., Aller, M. I., Vekovischeva, O. Y., Rosenberg, P. H., Wisden, W., et al. (2007). TASK-3 knockout mice exhibit exaggerated nocturnal activity, impairments in cognitive functions, and reduced sensitivity to inhalation anesthetics. J. Pharmacol. Exp. Ther. 323 (3), 924–934. doi:10.1124/jpet.107.129544
Liu, J., Carmell, M. A., Rivas, F. V., Marsden, C. G., Thomson, J. M., Song, J. J., et al. (2004). Argonaute2 is the catalytic engine of mammalian RNAi. Science 305 (5689), 1437–1441. doi:10.1126/science.1102513
Lleres, D., Moindrot, B., Pathak, R., Piras, V., Matelot, M., Pignard, B., et al. (2019). CTCF modulates allele-specific sub-TAD organization and imprinted gene activity at the mouse Dlk1-Dio3 and Igf2-H19 domains. Genome Biol. 20 (1), 272. doi:10.1186/s13059-019-1896-8
Lorthongpanich, C., Cheow, L. F., Balu, S., Quake, S. R., Knowles, B. B., Burkholder, W. F., et al. (2013). Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos. Science 341 (6150), 1110–1112. doi:10.1126/science.1240617
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34 (6), 293–303. doi:10.1016/j.tins.2011.04.001
Martinez-Ara, M., Comoglio, F., van Arensbergen, J., and van Steensel, B. (2022). Systematic analysis of intrinsic enhancer-promoter compatibility in the mouse genome. Mol. Cell 82 (13), 2519–2531.e6. e2516. doi:10.1016/j.molcel.2022.04.009
Martini, P., Sales, G., Diamante, L., Perrera, V., Colantuono, C., Riccardo, S., et al. (2022). BrewerIX enables allelic expression analysis of imprinted and X-linked genes from bulk and single-cell transcriptomes. Commun. Biol. 5 (1), 146. doi:10.1038/s42003-022-03087-4
Matise, M. P., Auerbach, W., and Joyner, A. L. (2000). “Production of targeted embryonic stem cell clones,” in Gene targeting - a practical approach. Editor A. L. Joyner. 2nd ed (New York: Oxford University Press), 101–132.
Montalban-Loro, R., Lassi, G., Lozano-Urena, A., Perez-Villalba, A., Jimenez-Villalba, E., Charalambous, M., et al. (2021). Dlk1 dosage regulates hippocampal neurogenesis and cognition. Proc. Natl. Acad. Sci. U. S. A. 118 (11), e2015505118. doi:10.1073/pnas.2015505118
Pang, B., and Snyder, M. P. (2020). Systematic identification of silencers in human cells. Nat. Genet. 52 (3), 254–263. doi:10.1038/s41588-020-0578-5
Perez, J. D., Rubinstein, N. D., and Dulac, C. (2016). New perspectives on genomic imprinting, an essential and multifaceted mode of epigenetic control in the developing and adult brain. Annu. Rev. Neurosci. 39, 347–384. doi:10.1146/annurev-neuro-061010-113708
Perez, J. D., Rubinstein, N. D., Fernandez, D. E., Santoro, S. W., Needleman, L. A., Ho-Shing, O., et al. (2015). Quantitative and functional interrogation of parent-of-origin allelic expression biases in the brain. Elife 4, e07860. doi:10.7554/eLife.07860
Peters, J. (2014). The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 15 (8), 517–530. doi:10.1038/nrg3766
Plasschaert, R. N., and Bartolomei, M. S. (2014). Genomic imprinting in development, growth, behavior and stem cells. Development 141 (9), 1805–1813. doi:10.1242/dev.101428
Prickett, A. R., Barkas, N., McCole, R. B., Hughes, S., Amante, S. M., Schulz, R., et al. (2013). Genome-wide and parental allele-specific analysis of CTCF and cohesin DNA binding in mouse brain reveals a tissue-specific binding pattern and an association with imprinted differentially methylated regions. Genome Res. 23 (10), 1624–1635. doi:10.1101/gr.150136.112
Reinius, B., and Sandberg, R. (2015). Random monoallelic expression of autosomal genes: Stochastic transcription and allele-level regulation. Nat. Rev. Genet. 16 (11), 653–664. doi:10.1038/nrg3888
Ruf, N., Bahring, S., Galetzka, D., Pliushch, G., Luft, F. C., Nurnberg, P., et al. (2007). Sequence-based bioinformatic prediction and QUASEP identify genomic imprinting of the KCNK9 potassium channel gene in mouse and human. Hum. Mol. Genet. 16 (21), 2591–2599. doi:10.1093/hmg/ddm216
Sanz, L. A., Chamberlain, S., Sabourin, J. C., Henckel, A., Magnuson, T., Hugnot, J. P., et al. (2008). A mono-allelic bivalent chromatin domain controls tissue-specific imprinting at Grb10. EMBO J. 27 (19), 2523–2532. doi:10.1038/emboj.2008.142
Segert, J. A., Gisselbrecht, S. S., and Bulyk, M. L. (2021). Transcriptional silencers: Driving gene expression with the brakes on. Trends Genet. 37 (6), 514–527. doi:10.1016/j.tig.2021.02.002
Singh, P., Wu, X., Lee, D. H., Li, A. X., Rauch, T. A., Pfeifer, G. P., et al. (2011). Chromosome-wide analysis of parental allele-specific chromatin and DNA methylation. Mol. Cell. Biol. 31 (8), 1757–1770. doi:10.1128/MCB.00961-10
Smith, R. J., Dean, W., Konfortova, G., and Kelsey, G. (2003). Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome Res. 13 (4), 558–569. doi:10.1101/gr.781503
Tucci, V., Isles, A. R., Kelsey, G., Ferguson-Smith, A. C., and Erice Imprinting, G. (2019). Genomic imprinting and physiological processes in mammals. Cell 176 (5), 952–965. doi:10.1016/j.cell.2019.01.043
Usman, M., Li, Y., Ke, Y., Chhetri, G., Islam, M. A., Wang, Z., et al. (2022). Trappc9 deficiency impairs the plasticity of stem cells. Int. J. Mol. Sci. 23 (9), 4900. doi:10.3390/ijms23094900
Varrault, A., Dubois, E., Le Digarcher, A., and Bouschet, T. (2020). Quantifying genomic imprinting at tissue and cell resolution in the brain. Epigenomes 4 (3), 21. doi:10.3390/epigenomes4030021
Wang, X., Sun, Q., McGrath, S. D., Mardis, E. R., Soloway, P. D., and Clark, A. G. (2008). Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One 3 (12), e3839. doi:10.1371/journal.pone.0003839
Weinstein, L. S., Xie, T., Qasem, A., Wang, J., and Chen, M. (2010). The role of GNAS and other imprinted genes in the development of obesity. Int. J. Obes. 34 (1), 6–17. doi:10.1038/ijo.2009.222
Wilton, K. M., Gunderson, L. B., Hasadsri, L., Wood, C. P., and Schimmenti, L. A. (2020). Profound intellectual disability caused by homozygous TRAPPC9 pathogenic variant in a man from Malta. Mol. Genet. Genomic Med. 8 (5), e1211. doi:10.1002/mgg3.1211
Xie, W., Barr, C. L., Kim, A., Yue, F., Lee, A. Y., Eubanks, J., et al. (2012). Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148 (4), 816–831. doi:10.1016/j.cell.2011.12.035
Xu, J., Carter, A. C., Gendrel, A. V., Attia, M., Loftus, J., Greenleaf, W. J., et al. (2017). Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat. Genet. 49 (3), 377–386. doi:10.1038/ng.3769
Yamasaki, K., Joh, K., Ohta, T., Masuzaki, H., Ishimaru, T., Mukai, T., et al. (2003). Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 12 (8), 837–847. doi:10.1093/hmg/ddg106
Keywords: genomic imprinting, allelic expression, single-cell analysis, neurosphere, neural stem cell, Peg13, Trappc9, Ago2
Citation: Claxton M, Pulix M, Seah MKY, Bernardo R, Zhou P, Aljuraysi S, Liloglou T, Arnaud P, Kelsey G, Messerschmidt DM and Plagge A (2022) Variable allelic expression of imprinted genes at the Peg13, Trappc9, Ago2 cluster in single neural cells. Front. Cell Dev. Biol. 10:1022422. doi: 10.3389/fcell.2022.1022422
Received: 18 August 2022; Accepted: 20 September 2022;
Published: 12 October 2022.
Edited by:
Rosalind M. John, Cardiff University, United KingdomReviewed by:
Anthony Isles, Cardiff University, United KingdomCopyright © 2022 Claxton, Pulix, Seah, Bernardo, Zhou, Aljuraysi, Liloglou, Arnaud, Kelsey, Messerschmidt and Plagge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonius Plagge, cGxhZ2dlQGxpdmVycG9vbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.