
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Cell Dev. Biol., 07 October 2022
Sec. Cell Growth and Division
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1012820
This article is part of the Research TopicLive Cell Imaging: Cell and Developmental Research Bridging Education, Optical Engineering, Industry, Software, Shared FacilitiesView all 11 articles
 Jayson J. Smith1,2,3†
Jayson J. Smith1,2,3† Isabel W. Kenny3,4†
Isabel W. Kenny3,4† Carsten Wolff3,5
Carsten Wolff3,5 Rachel Cray5
Rachel Cray5 Abhishek Kumar3,5
Abhishek Kumar3,5 David R. Sherwood3,4*
David R. Sherwood3,4* David Q. Matus3,6*‡
David Q. Matus3,6*‡Light sheet fluorescence microscopy (LSFM) has become a method of choice for live imaging because of its fast acquisition and reduced photobleaching and phototoxicity. Despite the strengths and growing availability of LSFM systems, no generalized LSFM mounting protocol has been adapted for live imaging of post-embryonic stages of C. elegans. A major challenge has been to develop methods to limit animal movement using a mounting media that matches the refractive index of the optical system. Here, we describe a simple mounting and immobilization protocol using a refractive-index matched UV-curable hydrogel within fluorinated ethylene propylene (FEP) tubes for efficient and reliable imaging of larval and adult C. elegans stages.
Light sheet fluorescence microscopy (LSFM) affords several advantages for live imaging of biological samples over standard epifluorescence or confocal microscopy. Whereas wide-field microscopy illuminates an entire specimen for imaging, LSFM achieves reduced phototoxicity, photobleaching, and background signal by restricting the proportion of the sample that is illuminated during acquisition. Relative to wide-field imaging, point-scanning confocal methods reduce out of focus sample illumination in the X-Y dimension by only exciting a single point in the sample at a time. To cover the whole region of interest the laser repeatedly sweeps across the sample and for each point scanned the entire Z depth is illuminated. Thus, out of focus photobleaching and phototoxicity occurs in the Z-dimension (Fischer et al., 2011). In contrast to a confocal point-scanning microscope where out of focus light is rejected by discarding unwanted emitted photons, LSFM systems generate a light sheet that selectively illuminates a narrow z-range of the sample in the desired focal plane at a given time (Fischer et al., 2011; Albert-Smet et al., 2019). This eliminates out of focus photobleaching and permits the collection of the entire fluorescence signal of a section of the sample at one time point, dramatically increasing acquisition speeds (Fischer et al., 2011). Another advantage of LSFM is the ability to acquire multi-view image data via multidirectional illumination, sample rotation, or a combination of both techniques (Huisken and Stainier 2009; Schmid and Huisken 2015). To overcome loss of resolution at increased tissue depths, many LSFMs are equipped with the ability to simultaneously image an individual sample from multiple sides, which can then be computationally deconvolved and reconstructed to render a single image of isotropic resolution. These technical advantages have made LSFM a popular imaging method for visualization of complex three-dimensional cells and tissues over developmental time (Keller et al., 2008; Liu et al., 2018).
Most LSFMs are equipped with two or more perpendicular illumination and detection objectives with the sample centered under or between the objectives. This unique orientation of objectives relative to the sample impedes the use of traditional flat microscopy slide mounts for the majority of LSFM systems. Samples for LSFMs are thus often embedded in a cylinder of low-melt agarose that hangs vertically between the objectives. In cases where the agarose is not dense enough to maintain its form, rigid fluorinated ethylene propylene (FEP) tubes can be used to surround the agarose cylinder to stabilize and support the agar (Kaufmann et al., 2012; Girstmair et al., 2016; Steuwe et al., 2020). The refractive indices of low-melt agarose (1.33) and FEP tubes (1.34) are well matched to the refractive index of water (1.33) and this sample mounting method works well for many organisms.
The C. elegans embryo has been particularly helpful in advancing the use of LSFM. For example, C. elegans embryogenesis was used to demonstrate the enhanced spatiotemporal resolution that is achieved using lattice light-sheet microscopy (Chen et al., 2014). Similarly, the C. elegans embryo facilitated showing the effectiveness of four-dimensional (4D) live imaging with the Dual Inverted Selective Plane Illumination Microscope (diSPIM) system (Kumar et al., 2014). LSFM has also advanced our understanding C. elegans embryogenesis (Chardès et al., 2014; Duncan et al., 2019), such as helping to reveal how the rigid egg shell contributes to asymmetrical cell divisions (Fickentscher and Weiss 2017), how circuit structures are organized within the nerve ring (the C. elegans brain) (Moyle et al., 2021), and how the zinc finger protein PIE-1 concentration gradient is established and maintained in the zygote (Benelli et al., 2020).
Although LSFM can also be used to capture embryogenesis in mice (Ichikawa et al., 2014; Udan et al., 2014) and zebrafish (Keller et al., 2008; Kaufmann et al., 2012; Icha et al., 2016; Pang et al., 2020), the increased tissue size and thickness, tissue pigmentation, and lack of transparency limits post-embryonic imaging in these animal models. In contrast, the small size and transparency of C. elegans larvae and adults makes them ideal to examine post-embryonic developmental and physiological processes. C. elegans is also amenable to high-resolution live imaging of genetically encoded fluorophores fused to proteins to follow protein dynamics and assess gene expression levels and patterns (Tsuyama et al., 2013; Yoshida et al., 2017; Heppert et al., 2018; Mita et al., 2019; Keeley et al., 2020). Genetically encoded fluorophores can also be conjugated to biosensors, which have been used to quantitatively monitor cell cycle state (Adikes et al., 2020) and ATP in C. elegans larvae (Garde et al., 2022). C. elegans can also be easily stained with vital dyes (Hermann et al., 2005; Schultz and Gumienny 2012; Kelley et al., 2019).
Despite the advantages of LSFM in C. elegans for live imaging, LSFM use in larvae and adults has been limited by the difficulty of sample mounting. Because of its ease of use and good optical properties, low melt agar has been used extensively to mount larger organisms such as zebrafish for LSFM (Kaufmann et al., 2012; Icha et al., 2016; Pang et al., 2020). More recently, it has been reported to work as a mounting media for C. elegans (Rieckher et al., 2018). However, small organisms such as C. elegans are capable of burrowing into soft agar (Burnett et al., 2018), which might be one reason that the longest time lapse reported for this method was 20 min. In addition, the 2% low-melt agarose required to immobilize worms has a gelling temperature of 24–28°C (Icha et al., 2016; Hirsinger and Steventon 2017), which extends beyond the 25°C thermal tolerance of C. elegans (Stiernagle 2006). To avoid high temperatures, photo-activated polyethylene glycol (PEG) hydrogels have been used to physically immobilize C. elegans for live imaging (Burnett et al., 2018). Yet, the refractive indices of these hydrogels are often not well-matched for the imaging media or the organism. Here we present a simple protocol for preparing and mounting post-embryonic C. elegans for LSFM imaging using a combination of the refractive index matched, ultraviolet (UV)-activated adhesive hydrogel BIO-133 (Han et al., 2021) and FEP tube encasement. This approach has the advantage of much longer imaging sessions (several hours) and avoids potential negative physiological effects on the worm due to heat stress. We show how this protocol can be used to time-lapse image PVD neuron dendritic branching and pruning. We also demonstrate how this protocol is applicable to imaging a variety of proteins and structures, including extracellular matrix proteins (type IV collagen and laminin), the nuclear envelope, and the distal tip cell (DTC). We expect the adoption of these methods will enable better live-imaging studies of important dynamic cell and developmental processes, such as germ stem cell biology, cell migration, cell division, and cell invasion (Sherwood and Plastino, 2018; Gordon et al., 2020; Smith et al., 2022). Furthermore, this protocol is generalizable and applicable to other organisms with little or no modifications.
Our objective was to develop a procedure for immobilizing larvae and adult C. elegans for two-to-three-hour long LSFM time lapse imaging sessions. To accomplish this, we developed a mounting strategy that combines anesthesia, the recently developed BIO-133 UV-activated adhesive hydrogel (Han et al., 2021) and animal encasement in an FEP tube (Figure 1). This mounting method allows liquid perfusion of the worms for long term live imaging (upper limit of 3 h to avoid physiological changes that occur from starvation) and is refractive index-matched to water to minimize the light interface resulting in optimal resolution during imaging. Furthermore, this mounting protocol can be adapted to work with LSFM systems equipped with either universal stage sample mounts (Figures 1A,B) or vertical mounts (Figures 1A–C). To validate our mounting protocol, we used the diSPIM (Kumar et al., 2014) to time-lapse image the PVD neurons using a strain harboring endogenously yellow fluorescent protein (YFP) tagged RAB-10 (strain wy1001[zf1::yfp::rab-10]) and a membrane tethered GFP expressed in the PVD and OLL neurons (wyIs592 [ser-2prom3p::myr-GFP]). Rab-10 is a small GTPase involved in post-Golgi vesicle trafficking and is a reporter for the Golgi and early endosome vesicles in the PVD neurons (Figure 2A) (Zou et al., 2015). The multi-dendritic mechanosensory PVD neurons exist as a pair, PVDL and PVDR. Each PVD neuron sits on one side of the animal and has a single axon that extends ventrally to the nerve cord (Figure 2A, bottom). PVD dendritic branching is predictable and developmentally regulated. Specifically, early in the L2 larval stage, the PVD extends three processes—one ventrally, one anteriorly, and one posteriorly. Beginning in late L2, the anterior and posterior processes send out short extensions that will elaborate into dendritic trees that compose the non-overlapping, anteroposterior repeating structural units of the PVDs referred to as “menorahs” (Figure 2B, top) (Oren-Suissa et al., 2010). The branches of these menorah structures cover most of the body, except for the neck and head, and are labeled in the proximal-distal and chronological order in which they occur: primary (1°), secondary (2°), tertiary (3°), or quaternary (4°) (Figure 2B, bottom) (Smith et al., 2010). Focusing on the PVDs allowed us to validate the efficacy of this protocol with respect to anterior, midbody, and posterior immobilization as well as imaging clarity throughout LSFM-based live cell imaging. Additionally, PVD development has been the subject of previous confocal-based time lapse studies (Zou et al., 2015) and thus provided us with a point of comparison in the validation of this protocol with respect to stereotyped subcellular dynamics and structural development in a two-to-three-hour timeframe (Chen and Pan 2021; Wang et al., 2021).

FIGURE 1. Schematic summary of C. elegans post-embryonic BIO-133 mounting strategies for LSFM imaging. (A) A schematic summary of steps #1–14 of the FEP-BIO-133 mounting protocol for time-lapse imaging of post-embryonic C. elegans on light sheet fluorescence microscopes, including animal anesthesia (top left, steps #1–4), transfer to BIO-133 (top right, steps #5–8), BIO-133 encapsulation (bottom left, step #9), and sample withdrawal into the FEP tube (bottom right, steps #10–14). Protocol steps #1–14 can be used for mounting samples on LSFMs configured with either a universal stage mount or a vertically-mounted sample. Pause points #1-2 in the procedure are indicated where they occur in the protocol. (B) A schematic summary of FEP tube-sample orientation (top, steps #15–16), UV-curation and bonding of FEP tube to Petri dish sample imaging chamber (middle, steps #17–18) and sample mounting (bottom, Steps #19–20) for LSFM systems equipped with a universal stage mount. After steps #1–14 (A), proceed to steps #15–20. Pause point #3 is indicated. (C) A schematic depicting preparation for a vertically-mounted sample, including sample chamber flooding (top, steps #21–22), UV-curation and loading of the FEP tube into the sample holder (middle, steps #23–24) and rotating the FEP tube to achieve optimal sample orientation (bottom, step #25). After steps #1–14 (A), skip steps #15–20 and proceed to steps #21–25. Pause point #3 is indicated. Created with BioRender.com.
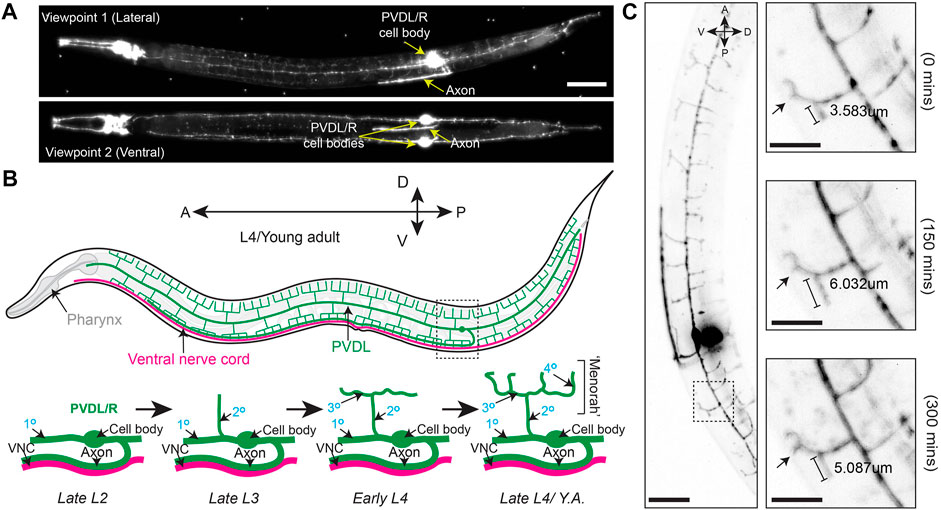
FIGURE 2. Branching and elongation of PVD neuron dendrites during a 5 h time-lapse on a DiSPIM. (A) LSFM Z-projections of an L4 hermaphrodite expressing yfp::rab-10 (acquired with 40x NA 0.8 water-dipping lenses, z-step = 1 μm, 200uW/cm2 488 nm laser power and 20 ms exposure) mounted using protocol Steps #1–20 on a diSPIM configured with a universal stage mount. Viewpoints were captured with imaging objectives oriented at 90° to simultaneously view the lateral and ventral aspects of the animal. Scale bar is 25 µm (B) (Top) A depiction of the fully elaborated PVD neurons in a young adult hermaphrodite animal. (Bottom) The developmental progression of PVD arborization focusing on the region indicated by the dashed box above. By late L2, the PVD neurons have extended their axons ventrally to contact the nerve cord and the primary (1°) dendrites have elongated along the anterior-posterior axis of the animal. The secondary (2°) dendrites branch dorsally and ventrally from the 1° dendrites by late L3. In early L4, the tertiary (3°) dendrites branch anteroposteriorly from the 2° dendrites, which is followed by the emergence of quaternary (4°) dendrites beginning in the late L4. (C) (Left) Timestamp from the beginning of a LSFM time-lapse in an L4 hermaphrodite expressing yfp:rab-10 as in (A) (Right) Time series of 3° and 4° dendritic dynamics over the course of a 300 min LSFM timelapse (acquired with the same parameters described in A). Scale bar 25 μm, 10 µm for inset.
We first performed time-lapse imaging of the posterior region of the PVD neuron in an L4 larval stage animal using 2-min acquisition intervals, a z-step size of 1 µm and z-range of 23 µm (Supplementary Movie S1). This allowed examination of PVD dendritic morphogenesis. We observed tertiary dendritic branch elongation (Figure 2C, bracket) as well as the growth of a quaternary branch (Figure 2C, arrow) (Smith et al., 2010; Albeg et al., 2011).
To further test the compatibility of this mounting protocol with other LSFMs, we imaged multiple fluorescently tagged strains on the Zeiss Lightsheet seven from two different acquisition angles. Compared to the diSPIM, which is equipped with a universal stage, the Lightsheet 7 has a vertical tube mount, which enables sample rotation during the acquisition for multi-view imaging. Using tiling and a small step size (0.30 µm), we imaged endogenously tagged type IV collagen (EMB-9:mRuby2, Figure 3A), endogenously tagged laminin (LAM-2:mNG, Figure 3B), endogenously tagged nucleoporin (NDC-1:mNG, Figure 3C), and a cell-specific transgene expressing membrane bound GFP in the somatic distal tip cells of the germline (lag-2p:GFP, Figure 3D). Using a 20X, 1.0 NA objective, we observed fine morphological and cellular structures. For example, we resolved the ring of type IV collagen at the edge of the spermatheca in young adult animals (Figure 3A′’), the laminin network surrounding the epithelial cells of the L4 stage spermatheca (Figure 3B′), the distribution of nucleoporin in L4 stage germ cells (Figure 3C′), and the elaborations of the distal tip cell in the young adult stage that enwrap the germ stem cell niche (Figure 3D′). Applying Multiview-registration [Fiji plugin BigStitcher (Hörl et al., 2019)] during image processing, we were also able to create an isotropic image of type IV collagen by combining two different 180° images of the same worm (Supplementary Movie S2).
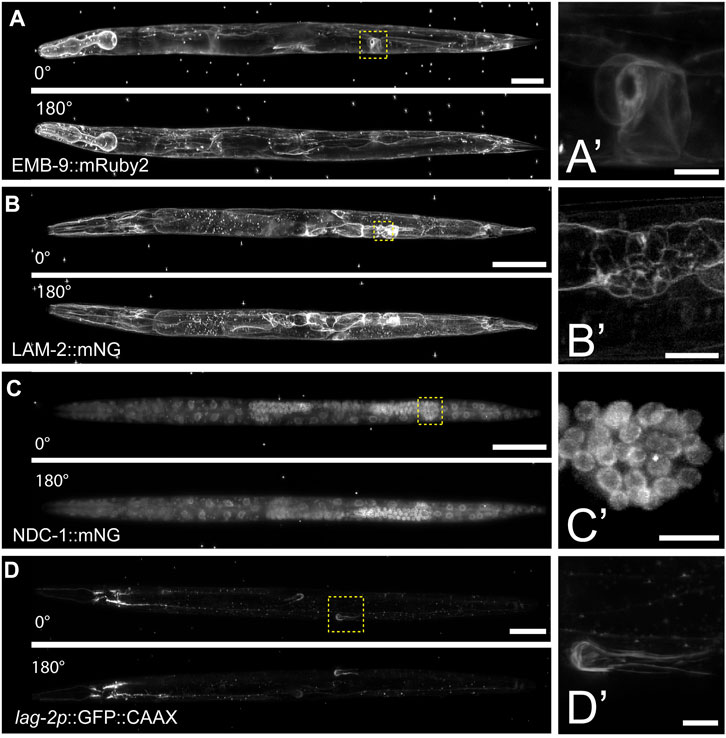
FIGURE 3. Multiview imaging of endogenously-tagged proteins in C. elegans young adults and larvae on a Zeiss Lightsheet 7 with a vertical mount. (A) Projected fluorescent images from two viewpoints on the Zeiss L7 showing endogenously-tagged type IV collagen (EMB-9:mRuby2) in a young adult hermaphrodite. The images were acquired from two angles 180° apart using a 20x NA 1.0 water dipping lens (z-step = 0.30 µm, 561 nm laser 2% power and 30 ms exposure). (B) Two projected images from LSFM sectioning of endogenously-tagged laminin (LAM-2:mNG) in an L4 hermaphrodite. The images were acquired from two angles 180° apart using a 20x NA 1.0 water dipping lens (z-step = 0.30 µm, 488 nm laser 1.5% power and 30 ms exposure). (C) Two projected images showing endogenously-tagged nucleoporin (NDC-1:mNG) in an L4 hermaphrodite. The images were acquired from two angles 180° apart using a 20x NA 1.0 water dipping lens (z-step = 0.30 µm, 488 nm laser 2% power and 50 ms exposure). (D) Two projected images showing distal tip cell (DTC) specific expression of membrane-tethered GFP in an adult hermaphrodite. The images were acquired from two angles 180° apart using a 20x NA 1.0 water dipping lens (z-step = 0.30 µm, 488 nm laser 7% power and 40 ms exposure). Scale bar for all images is 50 μm, 10 µm for inset. (A′–D′) Magnified insets of regions in the yellow dashed boxes in (A–D).
Key resources
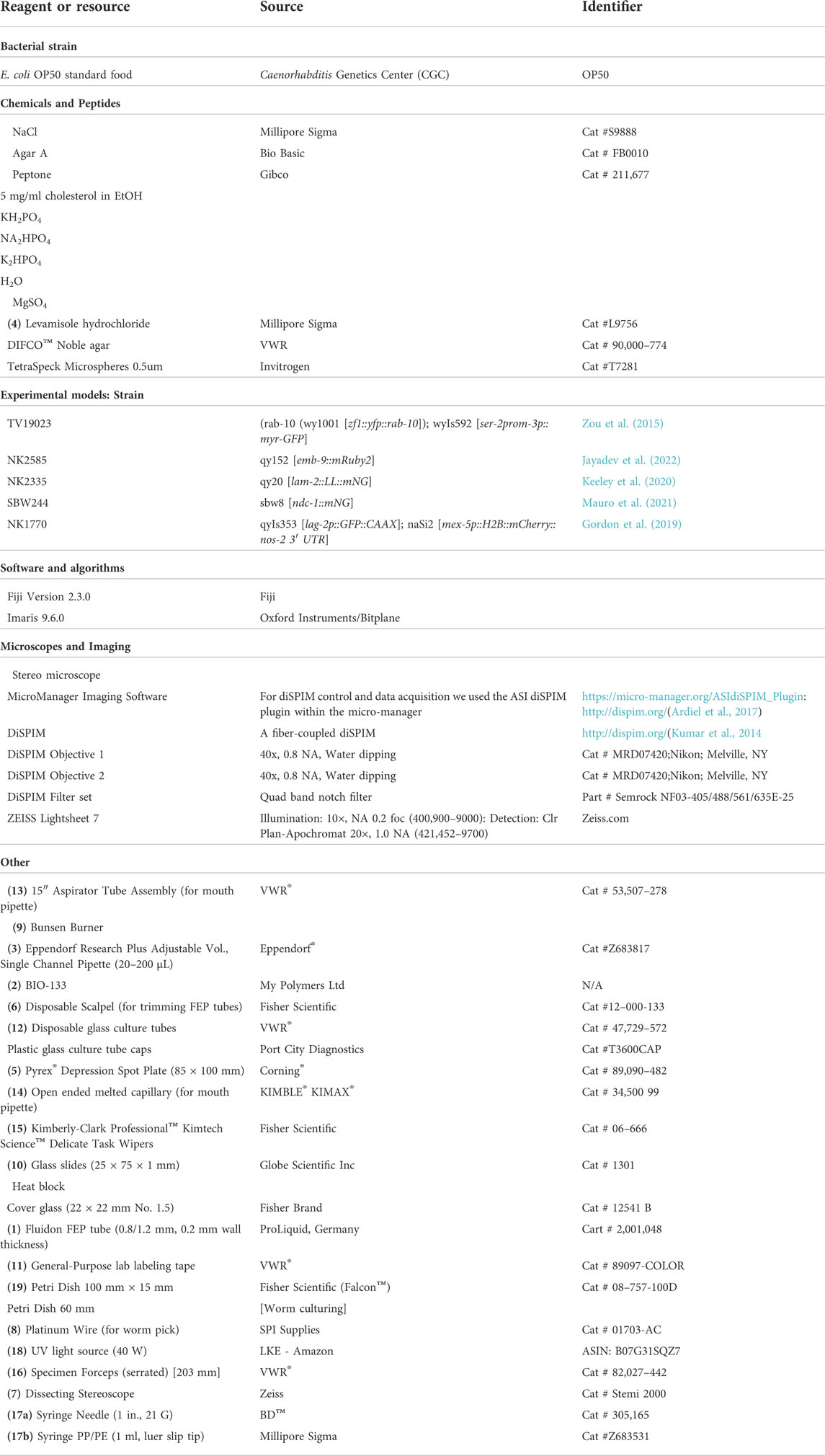
M9 buffer.
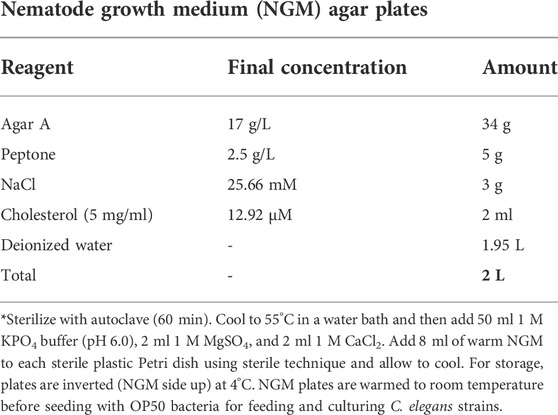
Nematode growth medium (NGM) agar plates.
Levamisole stock solution (anesthetic)
1. Prepare 200 mM levamisole stock solution in sterile water.
2. Aliquot 150 µL anesthetic stock solution into 1.5 ml Eppendorf tubes and store at -20°C.
4% (weight/volume) noble agar
1. Microwave 4% (weight/volume) noble agar in water to dissolve.
2. Aliquot 1 ml of the melted noble agar into disposable glass tubes and cover with foil or plastic cap. Store at room temperature for up to 3 months.
3. To use, melt noble agar in the glass tube over a Bunsen burner and add to heat block at 70°C to prevent solidification.
Steps 1–14 described below are shown in Figure 1A and Supplementary Video S1. Video tutorials for agar pad construction, worm anesthetization, and worm transfer can also be found elsewhere (Kelley et al., 2017). All necessary materials required to perform this procedure following preparation of M9 and NGM plates are shown in Supplementary Figure S1.
Total time: 45–65 min
C. elegans stage selection and anesthesia (timing: ∼30 min)
1. Synchronize worm cultures (Porta-de-la-Riva et al., 2012) (Time: 15 min) or pick appropriate staged animals for imaging. (Time: 2–3 min)
2. Add 50 µl anesthesia solution (5 mM Levamisole in M9) to a clean well in a glass depression dish.
Alternative to anesthesia: In addition to immobilization, the anesthetic relaxes the animals into a straight conformation, which facilitates consistent tissue geometry during imaging and permits Multiview registration. However, the use of anesthetic is not suitable for all experiments as levamisole is an acetylcholine receptor agonist that results in muscle contraction (Manjarrez and Mailler 2020). As an alternative, we found animals can be immobilized with cold temperatures by treatment at 5–7°C for ∼15 min prior BIO-133 UV-crosslinking.
3. Add 100 µl of BIO-133 to a clean well of the glass depression dish.
Detail for precision: BIO-133 is very viscous. Use a scalpel to trim the end of a pipette tip to transfer the hydrogel more easily.
(For Multiview registration) In an Eppendorf tube combine 80 µl BIO-133 and 20 µl of TetraSpeck Microspheres (1:2,000 dilution), vortex thoroughly to ensure beads are evenly dispersed in BIO-133. Once mixed, add 50–100 µl of BIO-133 to a clean well.
4. Transfer 20–50 animals to the anesthesia solution and wait for 12 min or until most of the animals have ceased moving. Larvae or adults should be straight and rod-like before proceeding to the next step. (Time: 15–20 min)
[Pause point 1]
Transferring C. elegans from anesthetic to BIO-133 (Timing: ∼15 min)
5. First swirl the glass depression dish to concentrate the anesthetized animals in the center of the well and then use the mouth pipette to remove most of the liquid anesthetic from the well to further concentrate the worm bodies. (Time: 1–3 min).
6. Prepare an agar pad on a glass slide (See Kelley et al., 2017 for details on agar pad construction) and allow to cool for 1 min (Time: 1–2 min)
7. Use a mouth pipette to transfer anesthetized animals from the well in the glass depression dish to the agar pad. (Time: 1 min)
8. Use a mouth pipette to remove anesthetic liquid from the agar pad until animals appear nearly dry (Supplementary Figure S2). Avoid removing anesthetized animals with the anesthetic solution. (Time: 1–3 min)
9. Using a worm pick, gather a droplet of BIO-133 at the end of the pick. Use the BIO-133 droplet to pick and then transfer worms from the nearly dry agar pad to the well of the glass depression dish that contains the BIO-133. Carefully and vigorously swirl the worms in the BIO-133 to separate individual animals and break up any liquid droplets or bubbles that form from the worm transfer. (Time: 2–5 min)
Detail for precision: Any transfer of the anesthetic or water to the BIO-133 solution will result in droplets forming in the adhesive, which will trap the animals, removing them from the hydrogel.
Detail for precision: Transferring individual animals rather than many larvae or adults on the pick at the same time will reduce the chances of aggregation.
[Pause point 2]
Loading BIO-133-encased C. elegans into FEP tube and polymerizing the mount (Timing: ∼20 min)
10. Attach the 21-G syringe needle to the 1 ml syringe barrel.
11. Use serrated forceps to slide the FEP tube onto the 21-G syringe needle.
Detail for precision: FEP tubes need to be rinsed and stored in double-distilled water prior to use (reference https://huiskenlab.com/sample-mounting/). Dry the outside of the tube with a Kimwipe and push air through the tube using the syringe plunger to dry the inside of the tube (Time: 1–3 min). Removing all water will reduce the number of droplets in the BIO-133.
Detail for precision: Depending on the length of the FEP tube, it may be necessary to use a disposable scalpel or razor blade to trim the tube into 2–5 cm lengths. Having a shorter segment of FEP tube reduces the time required to find the sample on a LSFM system by minimizing the area containing the sample. Shorter segments of FEP tubes also bond more easily to the bottom of the plastic Petri dish that will become the imaging chamber (See steps 14–16).
12. Place the open end of the FEP tube that is attached to the syringe into the BIO-133 adhesive solution. Using the syringe plunger, draw BIO-133 into the FEP tube until the tube is ¼ full. This primes the tube and ensures that C. elegans larvae and adults are positioned centrally, away from the edge of the FEP tube (Step 17). (Time: 1–3 min)
Detail for precision: Due to the high viscosity of the BIO-133 adhesive solution, there will be a delay between when you stop pulling the syringe plunger and when BIO-133 stops flowing into the FEP tube. If more than ¼ of the tube is filled with BIO-133 by the time the pressure is equalized, carefully expel the excess BIO-133 back into the well of the glass depression dish.
Detail for precision: To avoid introducing air bubbles into the FEP tube, do not remove the end of the tube from the BIO-133 until you have filled the final ¾ with anesthetized animals and BIO-133 (Step 14).
13. Slowly pull the plunger to draw 5–10 anesthetized animals into the primed FEP tube. (Time: 2–5 min)
Detail for precision: Due to the high viscosity of the BIO-133 adhesive solution, there will be a delay between pulling the syringe plunger and drawing anesthetized animals into the FEP tube. To avoid drawing BIO-133 and animals into the syringe barrel, stop pulling the syringe plunger when the FEP tube is ¾ full. Wait until the pressure equalizes, the FEP tube is full, and the worms stop flowing before removing the end of the FEP tube from the BIO-133 to avoid introducing air bubbles to the FEP tube.
Detail for precision: Position the opening of the FEP tube so that the animals will be drawn into the tube longitudinally. Draw one animal up at a time and avoid overlapping animals in the tube.
Detail for precision: Ensure that larvae and adults occupy the middle of the FEP tube since LSFM systems equipped with dip lenses will not be able to image animals that are too close to the ends of the FEP tube.
14. Remove the FEP tube from the BIO-133 and check the open end of the FEP tube and the end connected to the needle for air bubbles. The FEP tube should be filled with the adhesive solution, C. elegans larvae and adults, and free of air bubbles. Detach the FEP tube from the syringe with serrated forceps. (Time: 1–2 min)
IF USING A VERTICAL MOUNT, SKIP TO STEPS 21–25
(Steps 15–20 described below are shown in Figure 1B and Supplementary Video S2)
15. Place the FEP tube in the middle of the Petri dish. Add 2-3 drops of BIO-133 hydrogel to the FEP tube using a worm pick or pipette tip. BIO-133 will stabilize the FEP tube during and following UV-treatment. (Time: 1–2 min)
16. Use a stereo microscope to find the optimal orientation of the FEP tube such that your sample is as close as possible to the imaging objective. If multiple animals are mounted, roll the FEP tube in the uncured BIO-133 to achieve the orientation in which most animals are oriented properly (Figure 1B). (Time: 1–3 min)
17. Cure the mount with UV light for 2 min to crosslink the BIO-133 around the anesthetized animals and bond the sample-containing FEP tube to the plastic Petri dish imaging chamber. (Time: 2 min)
Installing the mount on an LSFM equipped with a universal stage and dipping lenses (Timing: ∼2 min)
18. After UV curing, the FEP tube should be stably attached to the surface of the plastic Petri dish and the sample should be encased in a rigid hydrogel in the FEP tube. Ensure that the FEP tube is securely attached to the Petri dish by lightly tapping it with forceps or a pipette tip. The tube should not budge or move at all before proceeding. (Time: 1 min)
19. Add mount to the universal stage on the LSFM system. Once the mount is resting on the universal stage, rotate the dish until your sample is optimally aligned with the imaging objectives (Figures 1B). Fasten the specimen clips to secure the Petri dish imaging chamber. (Time: 1 min)
20. Slowly fill the Petri dish imaging chamber with 45–50 ml room temperature M9 buffer (imaging medium), after which the dipping lens objectives can be lowered into the M9 for sample finding and subsequent imaging.
END OF PROCEDURE FOR LSFM WITH UNIVERSAL STAGE MOUNT
Installing the mount on an LSFM which requires a vertically mounted sample (Timing: ∼5 min)
(Steps 21–25 described below are shown in <b>Figure 1C</b> and step 23 (UV-curing) is shown in Supplementary Video S3)
21. Fill the LSFM media chamber with M9. (Time: 1 min)
Detail for Precision: M9 can be added to the media chamber prior to starting the protocol and does not need to be replaced between samples.
22. Wipe the FEP tube containing animals in BIO-133 with a Kimwipe to remove any BIO-133 from the outside of the tube. (Time: 1 min)
Detail for Precision: When possible, use forceps to handle the tube to keep the tube as clean as possible, as any smudges on the outside of the tube might impede the clarity of the imaging
23. Cure the mount with UV light for 2 min to crosslink the BIO-133 around the anesthetized animals; this can be done before or after detaching the FEP tube from the syringe needle. (Time: 2 min)
Detail for Precision: Use a stereomicroscope to locate the straight, centered, and non-overlapping animals within the FEP tube. (Time: 1 min)
24. Attach the tube in the sample holder, keeping in mind the positions of the animals as identified in step 24. If the animals are close to the end of the tube, place the opposite end of the tube in the sample holder. (Time: 1 min)
25. Place the sample holder with FEP tube back into the mount so that the FEP tube is submerged in M9 and ready for sample finding and imaging. (Time: 1 min)
This work introduces the advantages of LSFM live imaging to long term postembryonic C. elegans development, including faster acquisition speed and reduced phototoxicity and photobleaching. Prior to the development of this protocol, light-sheeting imaging of C. elegans had been limited to embryos, very short time-lapse imaging of larvae and adults, and fixed samples (Chardès et al., 2014; Chen et al., 2014; Liu et al., 2018; Rieckher et al., 2018; Breimann et al., 2019; Duncan et al., 2019). We anticipate that adult or larval encasement in BIO-133 within an FEP tube will enable continuous LSFM imaging for at least 2 h, a time span that is comparable to that typical of confocal time lapses (Kelley et al., 2017) and which approaches the physiological limit imposed by starvation (Schindler and Sherwood 2014). Unlike the confocal time-lapse mount, this protocol exposes animals to minimal amounts (up to 2 min) of direct UV light or low temperatures (7°C for the thermal immobilization method).
In addition to the extended time of imaging and optical properties of BIO-133, other major advantages of this protocol are the low material cost, accessibility of reagents and equipment (See Materials and Equipment table), and a similarity to established low-melt agarose-based LSFM mounting protocols currently used for C. elegans. The mounting strategy can be easily performed with resources already present in most C. elegans labs, except for a UV-light source, BIO-133, and FEP tubes. Compared to a previous method which uses low-melt agarose and a glass capillary tube to immobilize larvae and adults for LSFM imaging (Rieckher et al., 2018), this mounting strategy relies on similar procedural steps but avoids exposing animals to potentially harmful temperatures and provides a submersible mount that is compatible with most LSFM configurations. A minor drawback is that there are additional steps in the BIO-133 protocol relative to the low-melt agarose-based procedure. Namely, transfer of anesthetized animals to BIO-133 requires a drying process (steps 5–9) and the mount must be UV-cured (steps 15–18 or 21–23).
Compared to the short amount of time between preparing a traditional time lapse slide and imaging a sample on a point-scanning confocal system (Kelley et al., 2017), an additional limitation of this protocol is the length of time it takes to compose and cure the mount (∼30 min) before imaging. In this protocol animals are removed from food for a longer period before imaging, which reduces the time available for timelapse before starvation by ∼30 min compared to a slide-based time-lapse mount (Kelley et al., 2017). Furthermore, since the orientation of animals within the FEP tube is fixed after UV curing, it can take multiple mounting attempts to achieve optimal animal orientation. This protocol is therefore comparatively low throughput. This is a significant drawback to the investigation of developmental processes with sensitive timing, or if there is limited time available to use an LSFM system. To shorten the time to imaging, multiple LSFM time lapse mounts can be assembled in parallel.
Another potential limitation of this protocol is that BIO-133 must be cured with light of 300–400 nm in wavelength (Han et al., 2021) Animals must thus be exposed to UV-A light, which, while less damaging to DNA than UV-B or UV-C irradiation, is known to have a photoaging effect in C. elegans following prolonged exposure (more than 2 h) (Prasanth et al., 2016). Importantly, in this protocol we used a UV lamp which exposed the samples to UV-A light of 365 nm and 405 nm at an irradiance of approximately 6 mW/cm2 (Ford et al., 2021). At 2 minutes of exposure, animals received an estimated dose of UV-A light of 720 mJ/cm2, well-below the 15 J/cm2 that leads to C. elegans lethality (Ward et al., 2008). Although we did not monitor signs of UV-induced damage in animals treated with UV light, such as germ cell death, much higher doses of UV-A light are required to induce aging in the nematode (Prasanth et al., 2016) and all animals were viable following exposure and encapsulation in BIO-133.
Finally, we have not tested the diffusion mechanics of the activated BIO-133 hydrogel. It is possible that this protocol cannot be adapted for use in combination with diffusible cues and hormones (e.g., auxin for degron-based protein depletion) (Zhang et al., 2015; Martinez et al., 2020; Martinez and Matus 2020) or mitogens (Monsalve et al., 2019). However, pre-treatment with drugs or hormones prior to mounting animals may be sufficient to capture the desired effects, depending on the mechanics of the biological process or technique of interest. Since the ends of the FEP tubes are left open in the mount, the BIO-133 hydrogel matrix and sample should also be exposed to oxygen and media.
Here we describe a simple protocol for collecting high-quality post-embryonic LSFM time-lapse imaging data of larval and adult C. elegans. It is likely that this protocol can be adapted for the purposes of imaging other animal models, as the BIO-133 adhesive is biocompatible and FEP tubes are available in a variety of lengths and diameters. Though this method of immobilization and sample mounting provides novel opportunities for in vivo imaging of post-embryonic C. elegans, such as germ cell divisions, DTC migrations, sex myoblast migration, and anchor cell invasion, there remain a few shortcomings, such as the extended time it takes to prepare samples as discussed in the anticipated results section (Sherwood and Plastino 2018; Adikes et al., 2020; Gordon et al., 2020).
Among the many advantages to light-sheet microscopy mentioned above, this protocol enables multi-view image data via multidirectional illumination or sample rotation by providing access to the input image data necessary for 4D image reconstruction (Huisken and Stainier 2009; Schmid and Huisken 2015). Using 4D image reconstruction, we were able to discern the ring structure of type IV collagen in the spermathecal valve that opens to the uterus and laminin tightly covering the individual epithelial cells of the spermatheca. The BIO-133 can also be seeded with fluorescent beads (microspheres) as fiduciary markers (Preibisch et al., 2010; Wu et al., 2013) to improve multi-view image processing with greater temporal and spatial registration (Supplementary Movie S2). This protocol for C. elegans post-embryonic time-lapse imaging should be adaptable to any light sheet or confocal microscope that contains water dipping lenses and a universal stage mount or vertically mounted samples submerged in a sample chamber.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
JS, IK, and DM conceptualized the project. JS and IK designed the protocol, collected all data (with microscopy and image processing help from CW and AK), and wrote the manuscript. DM, DS edited and revised the manuscript. CW and AK provided additional comments on the manuscript. RC independently tested the protocol and provided helpful feedback.
IK and DS are supported by R35GM118049-06, R21OD028766, and R21OD032430. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). DM is supported by grant R01GM121597. The Embryology course was funded by NIH/NICHD grant R25HD094666, Burroughs Wellcome Fund 1021168 and the Company of Biologists. AK and RC are supported by the Chan Zuckerberg Initiative (CZI) Imaging Scientists Program, The Arnold and Mabel Beckman Foundation Lightsheet and Data Science Program, and startup funds provided to AK from the MBL. The ZEISS Lightsheet seven system at MBL was supported by the Howard Hughes Medical Institute.
The authors would like to thank J. Henry for help recording the videos and the Marine Biological Laboratory (MBL), the MBL Central Microscopy Facility, and the MBL Embryology course directors, staff, and students for providing the lab space and environment within which this protocol was developed. Michael Weber from the Flamingo team is greatly acknowledged for providing FEP tubes and support during preliminary imaging experiments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1012820/full#supplementary-material
Supplementary Movie S1 | Elaboration of the PVD neuron in the L4 midbody. A 5 h time-lapse of an L4 hermaphrodite expressing yfp::rab-10. The time-lapse was acquired on diSPIM with 40x NA 0.8 water-dipping objective lenses and images were collected every 2 min.
Supplementary Movie S2 | Using microspheres for enhanced spatiotemporal resolution. An isotropic image of endogenously-tagged type IV collagen (EMB-9:mRuby2) derived from the Multiview registration of images in Figure 3A.
Supplementary Video S1 | Step 1 to step 14.
Supplementary Video S2 | Step 15 to step 17.
Supplementary Video S3 | Step 23 (UV-curing mount for LSFMs which require vertically-mounted sample).
Adikes, R. C., Kohrman, A. Q., Martinez, M. A. Q., Palmisano, N. J., Smith, J. J., Medwig-Kinney, T. N., et al. (2020). Visualizing the metazoan proliferation-quiescence decision in vivo. eLife 9, e63265. doi:10.7554/eLife.63265
Albeg, A., Smith, C. J., Chatzigeorgiou, M., Feitelson, D. G., Hall, D. H., Schafer, W. R., et al. (2011). C. elegans multi-dendritic sensory neurons: Morphology and function. Mol. Cell. Neurosci. 46 (1), 308–317. doi:10.1016/j.mcn.2010.10.001
Albert-Smet, I., Marcos-Vidal, A., Vaquero, J. J., Desco, M., Muñoz-Barrutia, A., and Ripoll, J. (2019). Applications of light-sheet microscopy in microdevices. Front. Neuroanat. 13, 1. doi:10.3389/fnana.2019.00001
Ardiel, E. L., Kumar, A., Marbach, J., Christensen, R., Gupta, R., Duncan, W., et al. (2017). Visualizing calcium flux in freely moving nematode embryos. Biophys. J. 112 (9), 1975–1983. doi:10.1016/j.bpj.2017.02.035
Benelli, R., Struntz, P., Hofmann, D., and Weiss, M. (2020). Quantifying spatiotemporal gradient formation in early Caenorhabditis elegans embryos with lightsheet microscopy. J. Phys. D. Appl. Phys. 53 (29), 295401. doi:10.1088/1361-6463/ab8597
Breimann, L., Preusser, F., and Preibisch, S. (2019). Light-microscopy methods in C. elegans research. Curr. Opin. Syst. Biol. 13, 82–92. doi:10.1016/j.coisb.2018.11.004
Burnett, K., Edsinger, E., and Albrecht, D. R. (2018). Rapid and gentle hydrogel encapsulation of living organisms enables long-term microscopy over multiple hours. Commun. Biol. 1, 73. doi:10.1038/s42003-018-0079-6
Chardès, C., Mélénec, P., Bertrand, V., and Lenne, P.-F. (2014). Setting up a simple light sheet microscope for in toto imaging of C. elegans development. J. Vis. Exp. 87. doi:10.3791/51342
Chen, B.-C., Legant, W. R., Wang, K., Shao, L., Milkie, D. E., Davidson, M. W., et al. (2014). Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346 (6208), 1257998. doi:10.1126/science.1257998
Chen, C.-H., and Pan, C.-L. (2021). Live-cell imaging of PVD dendritic growth cone in post-embryonic C. elegans. Star. Protoc. 2 (2), 100402. doi:10.1016/j.xpro.2021.100402
Duncan, L. H., Moyle, M. W., Shao, L., Sengupta, T., Ikegami, R., Kumar, A., et al. (2019). Isotropic light-sheet microscopy and automated cell lineage analyses to catalogue Caenorhabditis elegans embryogenesis with subcellular resolution. J. Vis. Exp. 148. doi:10.3791/59533
Fickentscher, R., and Weiss, M. (2017). Physical determinants of asymmetric cell divisions in the early development of Caenorhabditis elegans. Sci. Rep. 7 (1), 9369. doi:10.1038/s41598-017-09690-4
Fischer, R. S., Wu, Y., Kanchanawong, P., Shroff, H., and Waterman, C. M. (2011). Microscopy in 3D: A biologist’s toolbox. Trends Cell Biol. 21 (12), 682–691. doi:10.1016/j.tcb.2011.09.008
Ford, H., Horsham, C., Urban, D., Tinker, R., and Hacker, E. (2021). Quantifying the ultraviolet radiation emitted by nail curing devices: A descriptive study. Australas. J. Dermatol. 62, e311–e313. doi:10.1111/ajd.13539
Garde, A., Kenny, I. W., Kelley, L. C., Chi, Q., Mutlu, A. S., Wang, M. C., et al. (2022). Localized glucose import, glycolytic processing, and mitochondria generate a focused ATP burst to power basement-membrane invasion. Dev. Cell 57 (6), 732–749. e7. doi:10.1016/j.devcel.2022.02.019
Girstmair, J., Zakrzewski, A., Lapraz, F., Handberg-Thorsager, M., Tomancak, P., Pitrone, P. G., et al. (2016). Light-sheet microscopy for everyone? Experience of building an OpenSPIM to study flatworm development. BMC Dev. Biol. 16 (1), 22. doi:10.1186/s12861-016-0122-0
Gordon, K. L., Payne, S. G., Linden-High, L. M., Pani, A. M., Goldstein, B., Hubbard, E. J. A., et al. (2019). Ectopic germ cells can induce niche-like enwrapment by neighboring body wall muscle. Curr. Biol. 29 (5), 823–833. e5. doi:10.1016/j.cub.2019.01.056
Gordon, K. L., Zussman, J. W., Li, X., Miller, C., and Sherwood, D. R. (2020). Stem cell niche exit in C. elegans via orientation and segregation of daughter cells by a cryptic cell outside the niche. eLife 9, e56383. doi:10.7554/eLife.56383
Han, X., Su, Y., White, H., O'Neill, K. M., Morgan, N. Y., Christensen, R., et al. (2021). A polymer index-matched to water enables diverse applications in fluorescence microscopy. Lab. Chip 21 (8), 1549–1562. doi:10.1039/d0lc01233e
Heppert, J. K., Pani, A. M., Roberts, A. M., Dickinson, D. J., and Goldstein, B. (2018). A CRISPR tagging-based screen reveals localized players in wnt-directed asymmetric cell division. Genetics 208 (3), 1147–1164. doi:10.1534/genetics.117.300487
Hermann, G. J., Schroeder, L. K., Hieb, C. A., Kershner, A. M., Rabbitts, B. M., Fonarev, P., et al. (2005). Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol. Biol. Cell 16 (7), 3273–3288. doi:10.1091/mbc.e05-01-0060
Hirsinger, E., and Steventon, B. (2017). A versatile mounting method for long term imaging of zebrafish development. J. Vis. Exp. 119. doi:10.3791/55210
Hörl, D., Rojas Rusak, F., Preusser, F., Tillberg, P., Randel, N., Chhetri, R. K., et al. (2019). BigStitcher: Reconstructing high-resolution image datasets of cleared and expanded samples. Nat. Methods 16 (9), 870–874. doi:10.1038/s41592-019-0501-0
Huisken, J., and Stainier, D. Y. R. (2009). Selective plane illumination microscopy techniques in developmental biology. Development 136 (12), 1963–1975. doi:10.1242/dev.022426
Icha, J., Schmied, C., Sidhaye, J., Tomancak, P., Preibisch, S., and Norden, C. (2016). Using light sheet fluorescence microscopy to image zebrafish eye development. J. Vis. Exp. (110), e53966. doi:10.3791/53966
Ichikawa, T., Nakazato, K., Keller, P. J., Kajiura-Kobayashi, H., Stelzer, E. H. K., Mochizuki, A., et al. (2014). Live imaging and quantitative analysis of gastrulation in mouse embryos using light-sheet microscopy and 3D tracking tools. Nat. Protoc. 9 (3), 575–585. doi:10.1038/nprot.2014.035
Jayadev, R., Morais, M. R. P. T., Ellingford, J. M., Srinivasan, S., Naylor, R. W., Lawless, C., et al. (2022). A basement membrane discovery pipeline uncovers network complexity, regulators, and human disease associations. Sci. Adv. 8 (20), eabn2265. doi:10.1126/sciadv.abn2265
Kaufmann, A., Mickoleit, M., Weber, M., and Huisken, J. (2012). Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development 139 (17), 3242–3247. doi:10.1242/dev.082586
Keeley, D. P., Hastie, E., Jayadev, R., Kelley, L. C., Chi, Q., Payne, S. G., et al. (2020). Comprehensive endogenous tagging of basement membrane components reveals dynamic movement within the matrix scaffolding. Dev. Cell 54 (1), 60–74. e7. doi:10.1016/j.devcel.2020.05.022
Keller, P. J., Schmidt, A. D., Wittbrodt, J., and Stelzer, E. H. K. (2008). Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science 322 (5904), 1065–1069. doi:10.1126/science.1162493
Kelley, L. C., Chi, Q., Cáceres, R., Hastie, E., Schindler, A. J., Jiang, Y., et al. (2019). Adaptive F-actin polymerization and localized ATP production drive basement membrane invasion in the absence of MMPs. Dev. Cell 48 (3), 313–328. e8. doi:10.1016/j.devcel.2018.12.018
Kelley, L. C., Wang, Z., Hagedorn, E. J., Shen, W., and Lei, S. (2017). Live-cell confocal microscopy and quantitative 4D image analysis of anchor-cell invasion through the basement membrane in Caenorhabditis elegans. Nat. Protoc. 12 (10), 2081–2096. doi:10.1038/nprot.2017.093
Kumar, A., Wu, Y., Christensen, R., Chandris, P., Gandler, W., McCreedy, E., et al. (2014). Dual-view plane illumination microscopy for rapid and spatially isotropic imaging. Nat. Protoc. 9 (11), 2555–2573. doi:10.1038/nprot.2014.172
Liu, T.-L., Upadhyayula, S., Milkie, D. E., Singh, V., Wang, K., Swinburne, I. A., et al. (2018). Observing the cell in its native state: Imaging subcellular dynamics in multicellular organisms. Science 360 (6386), eaaq1392. doi:10.1126/science.aaq1392
Manjarrez, J. R., and Mailler, R. (2020). Stress and timing associated with Caenorhabditis elegans immobilization methods. Heliyon 6 (7), e04263. doi:10.1016/j.heliyon.2020.e04263
Martinez, M. A. Q., Kinney, B. A., Medwig-Kinney, T. N., Ashley, G., Ragle, J. M., Johnson, L., et al. (2020). Rapid degradation of Caenorhabditis elegans proteins at single-cell resolution with a synthetic auxin. G3 10, 267–280. doi:10.1534/g3.119.400781
Martinez, M. A. Q., and Matus, D. Q. (2020). Auxin-mediated protein degradation in Caenorhabditis elegans. Bio. Protoc. 10 (8), e3589. doi:10.21769/BioProtoc.3589
Mauro, M. S., Celma, G., Zimyanin, V., Gibson, K. H., Redemann, S., and Bahmanyar, S. (2021). Ndc1 drives nuclear pore complex assembly independent of membrane biogenesis to promote nuclear formation and growth. eLife 11 e75513. doi:10.7554/eLife.75513
Mita, M., Ito, M., Harada, K., Sugawara, I., Ueda, H., Tsuboi, T., et al. (2019). Green fluorescent protein-based glucose indicators report glucose dynamics in living cells. Anal. Chem. 91 (7), 4821–4830. doi:10.1021/acs.analchem.9b00447
Monsalve, G. C., Yamamoto, K. R., and Ward, J. D. (2019). A new tool for inducible gene expression in Caenorhabditis elegans. Genetics 211 (2), 419–430. doi:10.1534/genetics.118.301705
Moyle, M. W., Barnes, K. M., Kuchroo, M., Gonopolskiy, A., Duncan, L. H., Sengupta, T., et al. (2021). Structural and developmental principles of neuropil assembly in C. elegans. Nature 591 (7848), 99–104. doi:10.1038/s41586-020-03169-5
Oren-Suissa, M., Hall, D. H., Treinin, M., Shemer, G., and Podbilewicz, B. (2010). The fusogen EFF-1 controls sculpting of mechanosensory dendrites. Science 328 (5983), 1285–1285. doi:10.1126/science.1189095
Pang, M., Bai, L., Zong, W., Wang, X., Bu, Y., Xiong, C., et al. (2020). Light-sheet fluorescence imaging charts the gastrula origin of vascular endothelial cells in early zebrafish embryos. Cell Discov. 6, 74. doi:10.1038/s41421-020-00204-7
Porta-de-la-Riva, M., Fontrodona, L., Villanueva, A., and Cerón, J. (2012). Basic Caenorhabditis elegans methods: Synchronization and observation. J. Vis. Exp. (64), e4019. doi:10.3791/4019
Prasanth, M. I., Santoshram, G. S., Bhaskar, J. P., and Balamurugan, K. (2016). Ultraviolet-A triggers photoaging in model nematode Caenorhabditis elegans in a DAF-16 dependent pathway. AGE 38 (27). doi:10.1007/s11357-016-9889-y
Preibisch, S., Saalfeld, S., Schindelin, J., and Tomancak, P. (2010). Software for bead-based registration of selective plane illumination microscopy data. Nat. Methods 7 (6), 418–419. doi:10.1038/nmeth0610-418
Rieckher, M., Psycharakis, S. E., Ancora, D., Zacharopoulos, A., Ripoll, J., Tavernarakis, N., et al. (2018). Demonstrating improved multiple transport-mean-free-path imaging capabilities of light sheet microscopy in the quantification of fluorescence dynamics. Biotechnol. J. 13 (1), 1700419. doi:10.1002/biot.201700419
Schindler, A. J., and Sherwood, D. R. (2014). Should I stay or should I go? Identification of novel nutritionally regulated developmental checkpoints in C. elegans. Worm 3 (4), e979658. doi:10.4161/21624054.2014.979658
Schmid, B., and Huisken, J. (2015). Real-time multi-view deconvolution. Bioinformatics 31 (20), 3398–3400. doi:10.1093/bioinformatics/btv387
Schultz, R. D., and Gumienny, T. L. (2012). Visualization of Caenorhabditis elegans cuticle structures using the lipophilic vital dye, DiI. J. Vis. Exp. (59), e3362. doi:10.3791/3362
Sherwood, D. R., and Plastino, J. (2018). Invading, leading and navigating cells in Caenorhabditis elegans: Insights into cell movement in vivo. Genetics 208 (1), 53–78. doi:10.1534/genetics.117.300082
Smith, C. J., Watson, J. D., Spencer, W. C., O'Brien, T., Cha, B., Albeg, A., et al. (2010). Time-lapse imaging and cell-specific expression profiling reveal dynamic branching and molecular determinants of a multi-dendritic nociceptor in C. elegans. Dev. Biol. 345 (1), 18–33. doi:10.1016/j.ydbio.2010.05.502
Smith, J. J., Xiao, Y., Parsan, N., Medwig-Kinney, T. N., Martinez, M. A. Q., Moore, F. E. Q., et al. (2022). The SWI/SNF chromatin remodeling assemblies BAF and PBAF differentially regulate cell cycle exit and cellular invasion in vivo. PLoS Genet. 18 (1), e1009981. doi:10.1371/journal.pgen.1009981
Steuwe, C., Vaeyens, M.-M., Jorge-Peñas, A., Cokelaere, C., Hofkens, J., Roeffaers, M. B. J., et al. (2020). Fast quantitative time lapse displacement imaging of endothelial cell invasion. Plos One 15 (1), e0227286. doi:10.1371/journal.pone.0227286
Stiernagle, T. (2006). Maintenance of C. elegans. Wormbook: The online Review of C. Elegans biology, 1–11.
Tsuyama, T., Kishikawa, J., Han, Y.-W., Harada, Y., Tsubouchi, A., Noji, H., et al. (2013). In vivo fluorescent adenosine 5’-triphosphate (ATP) imaging of Drosophila melanogaster and Caenorhabditis elegans by using a genetically encoded fluorescent ATP biosensor optimized for low temperatures. Anal. Chem. 85 (16), 7889–7896. doi:10.1021/ac4015325
Udan, R. S., Piazza, V. G., Hsu, C.-W., Hadjantonakis, A.-K., and Dickinson, M. E. (2014). Quantitative imaging of cell dynamics in mouse embryos using light-sheet microscopy. Development 141 (22), 4406–4414. doi:10.1242/dev.111021
Wang, X., Li, T., Hu, J., Feng, Z., Zhong, R., Nie, W., et al. (2021). In vivo imaging of a PVD neuron in Caenorhabditis elegans. Star. Protoc. 2 (1), 100309. doi:10.1016/j.xpro.2021.100309
Ward, A., Liu, J., Feng, Z., and Xu, X. Z. S. (2008). Light-sensitive neurons and channels mediate phototaxis in C. elegans. Nat. Neurosci. 11, 916–922. doi:10.1038/nn.2155
Wu, Y., Wawrzusin, P., Senseney, J., Fischer, R. S., Christensen, R., Santella, A., et al. (2013). Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nat. Biotechnol. 31 (11), 1032–1038. doi:10.1038/nbt.2713
Yoshida, T., Alfaqaan, S., Sasaoka, N., and Imamura, H. (2017). Application of FRET-based biosensor “ATeam” for visualization of ATP levels in the mitochondrial matrix of living mammalian cells. Methods Mol. Biol. 1567, 231–243. doi:10.1007/978-1-4939-6824-4_14
Zhang, L., Ward, J. D., Cheng, Z., and Dernburg, A. F. (2015). The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development 142 (24), 4374–4384. doi:10.1242/dev.129635
Keywords: C. elegans, light sheet fluorescence microscopy, BIO-133, postembryonic development, timelapse
Citation: Smith JJ, Kenny IW, Wolff C, Cray R, Kumar A, Sherwood DR and Matus DQ (2022) A light sheet fluorescence microscopy protocol for Caenorhabditis elegans larvae and adults. Front. Cell Dev. Biol. 10:1012820. doi: 10.3389/fcell.2022.1012820
Received: 05 August 2022; Accepted: 20 September 2022;
Published: 07 October 2022.
Edited by:
Konstantinos Palikaras, National and Kapodistrian University of Athens, GreeceReviewed by:
Matthias Rieckher, University of Cologne, GermanyCopyright © 2022 Smith, Kenny, Wolff, Cray, Kumar, Sherwood and Matus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David R. Sherwood, ZGF2aWQuc2hlcndvb2RAZHVrZS5lZHU=; David Q. Matus, ZGF2aWQubWF0dXNAc3Rvbnlicm9vay5lZHU=
†These authors have contributed equally to this work and share first authorship
‡Paid employee of: Arcadia Science
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.