 Wenchang Xu
Wenchang Xu Xinqi Liu
Xinqi Liu Wenjuan Han
Wenjuan Han Ling Zhao
Ling Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
EDITORIAL article
Front. Cell Dev. Biol. , 09 September 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1008907
This article is part of the Research Topic Genetic features contributing to Eye development and Disease View all 11 articles
Editorial on the Research Topic
Genetic features contributing to eye development and disease
Growing evidence has shown that genetic factors play crucial roles in the disorder of eye development and the progression of ocular diseases (Singh and Tyagi, 2018). Ocular disorders with complex inheritance are responsible for most blindness, however, there are currently no cures for many of these conditions (Singh and Tyagi, 2018; Chen et al., 2021). A better understanding of the genetic underpinnings of ocular diseases could facilitate the accurate diagnosis, counseling and treatment of these diseases.
The eye consists of three main types of tissues: 1) refracting tissues that focus incoming light onto light-sensitive tissues (including the pupil, iris, lens, ciliary muscle, cornea, vitreous and aqueous fluid), 2) light-sensitive tissues that convert detected light into electrical signals and transmit them to the brain (including the retina and optic nerve), and 3) support tissues that provide the architectural support for the shape of the eyeball (including the sclera, conjunctiva and uvea) (Rocher, 2010). These parts in the eyes must work together to produce a clear vision.
This topic summarizes ten original research articles that explored the genetic effects and mechanisms of genetic factors contributing to eye development and disease from diverse aspects, providing new insights into treating eye diseases (Table 1). According to the research object, these studies can be divided into three categories: novel causal and susceptibility genes in eye diseases, genetically engineered animal models for eye diseases, and novel concepts or innovative approaches for eye development and diseases.
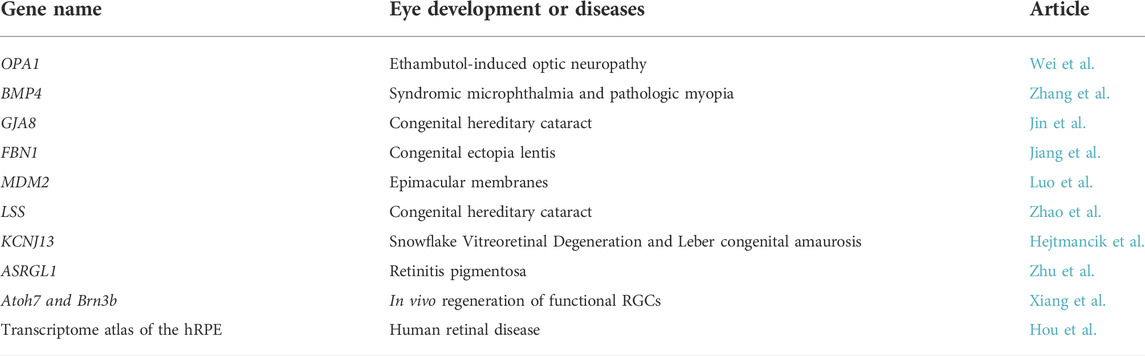
TABLE 1. Genes studied in the article collection.
Inherited eye diseases affect approximately one in 1,000 people worldwide, but the molecular mechanisms underlying most of them remain unclear (Mejecase et al., 2020). Identifying novel causal and susceptibility genes allows a better understanding of the disorders and offers new clues for better-targeted disease management.
Wei et al. identified mitochondrial mutations (OPA1 and LHON-mtDNA) in nearly half of patients with ethambutol-induced optic neuropathy (EON), a well-recognized ocular complication associated with ethambutol treatment in tuberculosis patients. Since some patients with EON have severe and permanent visual loss even without the known risk factors, their findings that mitochondrial genetic variations are major predisposing factors for the development of EON provided a better understanding of EON and additional support for genetic counseling.
Zhang et al. found variants in BMP4 contribute to a novel phenotype of pathologic myopia rather than syndromic microphthalmia that have been reported in a previous study (Reis et al., 2011). The observations that mutations in the same gene could cause both syndromic microphthalmia and pathologic myopia suggested bidirectional roles of BMP4 in early ocular development and provided new insight into the disease mechanism.
Jin et al. identified a novel connexin 50 mutation P88L in patients with congenital cataract and analyzed the function of this mutation. Congenital hereditary cataract is a heterogeneous disorder and the most common cause of childhood blindness (Berry et al., 2020). Their findings expand the spectrum of pathogenic connexin 50 mutations in congenital cataract and provide additional support for clinical diagnosis and genetic counseling.
Congenital ectopia lentis (CEL), the second leading cause of pediatric lens surgery after congenital cataracts, could be caused by mutations in cbEGF-like domains of fibrillin-1 (FBN1) (Faivre et al., 2008). However, the correlations between genotype and phenotype of cbEGF-like mutations remain unknown. Jiang et al. focused on clinical manifestations of CEL in patients with different mutations in cbEGF-like domains of FBN1. And they clarified the correlations between genotype and phenotype for cbEGF-like mutations. Their work increases our knowledge of CEL and offers new clues for the targeted treatment of the disease.
Luo et al. presented work on the role of the mouse double minute 2 (MDM2) gene single nucleotide polymorphism (SNP) T309G in the development of epimacular membranes (EMMs), relatively common sight-threatening conditions characterized by fibrocellular proliferation along the surface of the internal limiting membrane (ILM) of the retina. They first reported that the MDM2 SNP309 G allele was a risk allele for EMM in a Chinese population. Their observation provides new insights into the molecular mechanism underlying these pathologies.
Since the experimental studies of many inherited eye disorders in human beings are limited, the availability of genetically engineered animal models is very valuable for exploring the pathogenic mechanisms of these conditions and developing translational therapies.
Zhao et al. generated a knock-in mouse model with lanosterol synthase (Lss) G589S mutation, which can recapitulate human congenital cataract resulting from G588S mutation in human LSS. Mice with homozygous Lss G589S mutation exhibited disrupted lens fiber differentiation at early-stage of lens development and down-regulated cholesterol synthesis signaling pathways. Their findings elucidate the important roles of LSS in lens development, contributing to a better understanding of LSS defects and disturbed sterol signaling in cataractogenesis and the development of therapies for cataracts.
Mutations in KCNJ13 are responsible for both snowflake vitreoretinal degeneration (SVD) and Leber congenital amaurosis (LCA) (Hejtmancik et al., 2008; Sergouniotis et al., 2011). Existing animal models have not been able to well verify causality and dissect the mechanisms and pathogenesis of these diseases. Hejtmancik et al. generated and characterized the Kcnj13 knockout mouse and RPE-specific conditional Kcnj13 knockout mice. Their work provides a potential mouse model system for elucidating the pathology of these diseases and developing gene therapy trials.
G178R in asparaginase and isoaspartyl peptidase 1 (ASRGL1) has been reported as a causing mutation for retinitis pigmentosa (RP), an inherited retinal degenerative disease for which there is currently no cure (Biswas et al., 2016). Since the pathological and molecular mechanisms of ASRGL1 in causing RP remains unknown, Zhu et al. developed Asrgl1 knockout mice and explored the function of Asrgl1 in the mammalian retina. Their findings provide a knockout mouse model for improving the understanding of RP disease mechanisms.
Novel concepts or innovative approaches could provide new insights into understanding the mechanism of eye development and diseases, and developing new diagnostic and treatment strategies.
Glaucoma is the most common cause of irreversible blindness worldwide and irreversible degeneration of retinal ganglion cells (RGCs) and the optic nerve are the common features shared by all forms of glaucoma (Jonas et al., 2017). In vivo RGC regeneration would be an ideal therapy but mammalian retinas are thought to lack regenerative capacity. Xiang et al. demonstrated that endogenous mouse Müller glia (MG) could be reprogramed into functional retinal ganglion cells (RGCs) in vivo by Math5 and Brn3b together, two crucial transcription factors (TFs) involved in retinal ganglion cell (RGC) generation and differentiation (Yang et al., 2003; Pan et al., 2008). Their study provides a promising new therapeutic approach for visual restoration in patients with glaucoma and other optic neuropathies.
Hou et al. profiled a single-cell transcriptome atlas of human RPE (hRPE) cells and provided a map of disease-related genes in the hRPE. They found two subpopulations, the macular RPE and peripheral RPE clusters which exhibited substantial differences in gene expression patterns and functions, play crucial roles in the potential treatment of retinal diseases. Their work provides important information for understanding the cellular mechanisms and treating pathological conditions of the hRPE associated with ocular diseases.
These research articles on the topic show that elucidating the genetic underpinnings of ocular disorders leads to a better understanding of these diseases, which will contribute to clinical diagnosis, genetic counseling, early intervention and targeted treatment. The application and advancement of integrated multi-omics will further improve our knowledge of complex traits and provide new insights into the pathogenesis of ocular diseases. New genetic methodologies based on automated methods are expected to be developed for accurate and routine diagnosis of eye diseases that have highly diverse genetic causes and are difficult to identify. And gene and cell therapies will open new doors for the treatment of currently incurable eye disorders.
WX, XL and WH prepared the manuscript. LZ revised the manuscript and provided financial support. All authors read and approved the final manuscript.
This study was supported by funding from the National Natural Science Foundation of China (NSFC: 82000915; 81670894; 81721003), National Key Research and Development Program of China (2020YFA0112701), the Open Research Funds of the State Key Laboratory of Ophthalmology (2022KF04).
We would like to thank the support from the State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Berry, V., Georgiou, M., Fujinami, K., Quinlan, R., Moore, A., and Michaelides, M. (2020). Inherited cataracts: Molecular genetics, clinical features, disease mechanisms, and novel therapeutic approaches. Br. J. Ophthalmol. 104, 1331–1337. doi:10.1136/bjophthalmol-2019-315282
Biswas, P., Chavali, V. R., Agnello, G., Stone, E., Chakarova, C., Duncan, J. L., et al. (2016). A missense mutation in ASRGL1 is involved in causing autosomal recessive retinal degeneration. Hum. Mol. Genet. 25, 2483–2497. doi:10.1093/hmg/ddw113
Chen, H. Y., Lehmann, O. J., and Swaroop, A. (2021). Genetics and therapy for pediatric eye diseases. EBioMedicine 67, 103360. doi:10.1016/j.ebiom.2021.103360
Faivre, L., Collod-Beroud, G., Child, A., Callewaert, B., Loeys, B. L., Binquet, C., et al. (2008). Contribution of molecular analyses in diagnosing marfan syndrome and type I fibrillinopathies: An international study of 1009 probands. J. Med. Genet. 45, 384–390. doi:10.1136/jmg.2007.056382
Hejtmancik, J. F., Jiao, X., Li, A., Sergeev, Y. V., Ding, X., Sharma, A. K., et al. (2008). Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am. J. Hum. Genet. 82, 174–180. doi:10.1016/j.ajhg.2007.08.002
Jonas, J. B., Aung, T., Bourne, R. R., Bron, A. M., Ritch, R., and Panda-Jonas, S. (2017). Glaucoma. Lancet 390, 2183–2193. doi:10.1016/S0140-6736(17)31469-1
Mejecase, C., Malka, S., Guan, Z., Slater, A., Arno, G., and Moosajee, M. (2020). Practical guide to genetic screening for inherited eye diseases. Ther. Adv. Ophthalmol. 12, 2515841420954592. doi:10.1177/2515841420954592
Pan, L., Deng, M., Xie, X., and Gan, L. (2008). ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development 135, 1981–1990. doi:10.1242/dev.010751
Reis, L. M., Tyler, R. C., Schilter, K. F., Abdul-Rahman, O., Innis, J. W., Kozel, B. A., et al. (2011). BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 130, 495–504. doi:10.1007/s00439-011-0968-y
Sergouniotis, P. I., Davidson, A. E., Mackay, D. S., Li, Z., Yang, X., Plagnol, V., et al. (2011). Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am. J. Hum. Genet. 89, 183–190. doi:10.1016/j.ajhg.2011.06.002
Singh, M., and Tyagi, S. C. (2018). Genes and genetics in eye diseases: A genomic medicine approach for investigating hereditary and inflammatory ocular disorders. Int. J. Ophthalmol. 11, 117–134. doi:10.18240/ijo.2018.01.20
Keywords: eye diseases, eye development, causal genes, susceptibility genes, genetic mechanisms
Citation: Xu W, Liu X, Han W and Zhao L (2022) Editorial: Genetic features contributing to eye development and disease. Front. Cell Dev. Biol. 10:1008907. doi: 10.3389/fcell.2022.1008907
Received: 01 August 2022; Accepted: 22 August 2022;
Published: 09 September 2022.
Edited and reviewed by:
Ramani Ramchandran, Medical College of Wisconsin, United StatesCopyright © 2022 Xu, Liu, Han and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Zhao, emhhb2xpbmc2QG1haWwuc3lzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.