
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 19 October 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1007641
This article is part of the Research TopicCerebellar Development and Medulloblastoma: New Players and Therapeutic OptionsView all 6 articles
 Veronica Marabitti1
Veronica Marabitti1 Manuela Giansanti1
Manuela Giansanti1 Francesca De Mitri1
Francesca De Mitri1 Francesca Gatto2
Francesca Gatto2 Angela Mastronuzzi1
Angela Mastronuzzi1 Francesca Nazio1*
Francesca Nazio1*Tumor-specific alterations in metabolism have been recognized to sustain the production of ATP and macromolecules needed for cell growth, division and survival in many cancer types. However, metabolic heterogeneity poses a challenge for the establishment of effective anticancer therapies that exploit metabolic vulnerabilities. Medulloblastoma (MB) is one of the most heterogeneous malignant pediatric brain tumors, divided into four molecular subgroups (Wingless, Sonic Hedgehog, Group 3 and Group 4). Recent progresses in genomics, single-cell sequencing, and novel tumor models have updated the classification and stratification of MB, highlighting the complex intratumoral cellular diversity of this cancer. In this review, we emphasize the mechanisms through which MB cells rewire their metabolism and energy production networks to support and empower rapid growth, survival under stressful conditions, invasion, metastasis, and resistance to therapy. Additionally, we discuss the potential clinical benefits of currently available drugs that could target energy metabolism to suppress MB progression and increase the efficacy of the current MB therapies.
Medulloblastoma (MB) is a malignant brain tumor arising mainly in childhood, including four molecular subgroups–Wingless (WNT), Sonic Hedgehog (SHH), Group 3 and Group 4– that have been defined through transcriptome, methylome and genome profiling analyses (Northcott et al., 2017). MB generally arises in the posterior fossa and it is thought to initiate from an unusual neural stem/progenitor cell during early brain development. In one-third of patients, tumor cells spread to the leptomeninges via the cerebrospinal fluid, resulting in the formation of metastasis. The current standard of care includes maximal safe resection, radiotherapy and chemotherapy, according to patients’ stratification; patients frequently fail first-line therapy and are still often incurable (Packer et al., 2006; Jakacki et al., 2012; Veneroni et al., 2017; Juraschka and Taylor, 2019). Intertumoral heterogeneity in MB has long been recognized from a histopathological and a molecular perspective (Cavalli et al., 2017; Morrissy et al., 2017; Juraschka and Taylor, 2019) and very recently from the composition of the tumor immune microenvironment (Bockmayr et al., 2018; Grabovska et al., 2020). Lately, single-cell sequencing studies have also revealed the presence of a profound clonal and spatial intratumoral heterogeneity that is critical for MB progression, relapse and treatment resistance (Tech et al., 2017; Hovestadt et al., 2019; Ocasio et al., 2019; Zhang et al., 2019; Luo et al., 2021). Given the marked heterogeneity among MBs, personalizing therapy to a patient’s tumor subtype and individual genetic, epigenetic and transcriptomic characteristics constitute the next frontier in oncology. Tumor heterogeneity is, at least in part, justified by the presence of cancer stem cells (CSCs) that represent a small subgroup of cells in the tumor bulk with stem-like features, recognized to be responsible for tumor onset, maintenance, and relapse after therapy. CSCs have been identified in many types of brain tumors, including glioblastoma (GBM) and MB; they are able to self-renew under clonal conditions, and differentiate into neuron- and glia-like cells as well as into atypical cells with mixed phenotypes (Singh et al., 2004). Many findings suggest that enriched, treatment-resistant subpopulations of CSCs are involved in driving MB tumor recurrence and metastasis subsequent to standard treatment (Sun et al., 2017; Bakhshinyan et al., 2021).
Metabolic adaptation is believed to be one of the hallmarks of tumor cells and several factors (both cell-intrinsic, such as oncogenes and tumor suppressor genes, and cell-extrinsic such as nutrient availability or hypoxia) contribute towards driving metabolic reprogramming. Normal cells usually convert glucose into pyruvate that has two possible fates mainly depending on the availability of oxygen. Under aerobic conditions, the glycolytic product pyruvate is mostly oxidized in the tricarboxylic acid cycle (TCA cycle; also known as the Krebs cycle) to produce H2O, CO2 and secondary metabolites; these act as electron donors in the oxygen-dependent mitochondrial oxidative phosphorylation (OXPHOS), a process which is coupled with high ATP production rate. Under hypoxic conditions, pyruvate undergoes anaerobic fermentation, giving lactate and concomitantly producing two molecules of ATP per molecule of glucose. The aim of the latter two processes (OXPHOS and lactic acid fermentation) is the recycling of co-enzymes for further glycolysis and other cellular enzymatic activities. On the contrary, cancer cells preferentially rely on glycolysis and lactic acid fermentation rather than OXPHOS to produce ATP, even in the presence of O2 (Warburg, 1956; Racker and Spector, 1981). This metabolic reprogramming is known as the Warburg effect or “aerobic glycolysis”.
Besides glycolysis, fatty acid (FA) biosynthesis and glutamine metabolism are two other important metabolic pathways altered in both cancer cells and CSCs. Indeed, cancer cells use the process of de novo lipogenesis to convert carbohydrates into FAs and lipids and alterations in this process are associated with increased proliferation and spreading (Koundouros and Poulogiannis, 2020). Lastly, according to recent studies, CSCs rely on the metabolism of the amino acid glutamine, which provides the carbon and the amino-nitrogen needed for the biosynthesis of amino acids, nucleotides, and lipids.
It is important to highlight that cellular heterogeneity inside the tumors may result in different metabolic requirements, and consequently, in different responses to metabolic therapies. In particular, the maintenance of stem cell functions is, at least in part, regulated by metabolic alterations that initially consist of the activation of glycolysis and the inhibition of OXPHOS. Despite the majority of data suggesting that CSCs are primarily glycolytic and that they are usually characterized by the upregulation of glycolytic enzymes, glucose uptake and lactate production, mounting evidence also shows their capability to exploit the mitochondrial oxidative metabolism.
This review summarizes the latest knowledge regarding MB-specific metabolic alterations at the cellular and the molecular level (Figure 1). Unraveling the metabolic networks of both tumor cells and CSCs in MB could be helpful for designing new targeted therapies that interfere with energy cell metabolism, with the potential to minimize relapse and resistance after treatments.
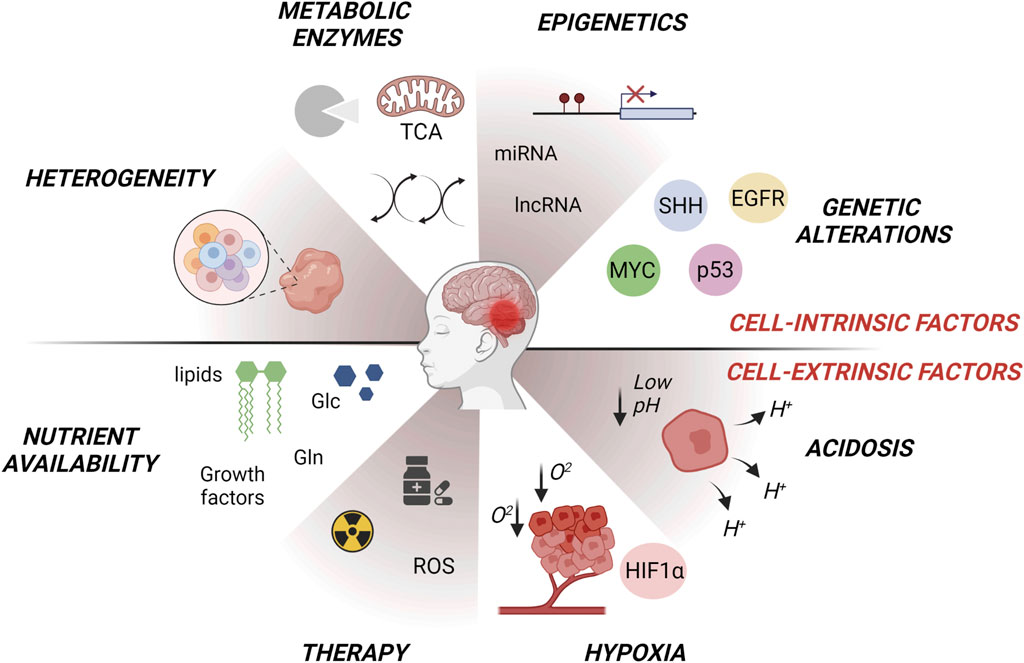
FIGURE 1. Intrinsic and extrinsic factors affecting metabolic changes in MB subgroups (WNT, SHH, Group3, Group4). Tumor intrinsic factors include heterogeneous oncogenic signaling and/or genetic alterations in metabolic enzymes that modulate gene expression through a variety of mechanisms such as signal transduction and epigenetic reprogramming. Tumor extrinsic factors consist of microenvironmental availability of nutrients such as glutamine or amino acids, oxygen and changes in extracellular pH. Alterations in metabolism influence glycolysis, lipogenesis, OXPHOS, oxygen reactive species (ROS) production, glutamine-dependent signaling and tumor heterogeneity, giving rise to metabolic adaptations. Figure is created in “BioRender.com”.
In the last decades, several works have focused on the role of metabolic rewiring in brain tumors such as GBM (Tardito et al., 2015; Bi et al., 2020), whereas our comprehension about metabolic features underlying MB pathogenesis and progression still remains limited. It has been shown that metabolic changes during cancer development are similar to the ones that occur during neural development: neurogenesis, indeed, requires an increase in spatiotemporal-regulated proliferation as it occurs in cancer. Many studies have shown that SHH-driven MB metabolic features largely mirror cerebellar granule precursors (CGPCs) phenotype. Aberrant SHH pathway activation induces an upregulation of aerobic glycolysis and lipogenesis, while decreasing fatty acid oxidation levels, as it occurs in CGPCs (Tech et al., 2015; Tech and Gershon, 2015). Nonetheless, since the SHH MB subgroup accounts for approximately one-third of MB diagnoses, it is of paramount importance to expand our knowledge about the other MB subgroups that likely arise from different tumor-initiating populations (Hovestadt et al., 2020). A large-scale bioinformatic analysis using data of 530 patients from the Medulloblastoma Advanced Genomic International Consortium (MAGIC) project has identified cancer metabolism-related pathways as new prognostic factors in all MB subgroups (Park et al., 2019). This work has allowed the identification of subgroup-specific metabolic signatures that could be targetable to improve the clinical outcome of MB patients. Park and others demonstrated that several prognostic genes involved in the glycolytic pathway (such as ENO1, encoding enolase 1) and lipogenesis (such as FASN and SCD encoding fatty acid synthase and stearoyl-CoA desaturase, respectively) are upregulated in three MB subgroups (SHH, Group 3, and Group 4). Specifically, glycolysis and glutaminolysis have been identified as prognostic or highly expressed in both SHH and Group 3 subgroups, suggesting that ‘Warburg effect’ may drive MB energy metabolism. One carbon (folate) cycle or serine synthesis (that are commonly used for nucleotide synthesis, methylation and reductive metabolism supporting the high proliferative rate of cancer cells), are mainly prognostic in both SHH and Group 4 subgroups. In addition, MYC-driven Group 3 MB are marked by pentose phosphate pathway (PPP) upregulation, which is frequently boosted by cancer cells to provide nucleic acids and nicotinamide-adenine dinucleotide phosphate (NADPH) for cell growth and survival under stressful conditions (Park et al., 2019). Yet, more research is needed to determine whether and how the identified metabolic signatures cross-talk with the genetic heterogeneity across MB subgroups.
Glucose metabolism and aerobic glycolysis. Glucose metabolism provides the fuel for physiological brain function. In neurons, glucose enters through the sodium-independent facilitative transporters GLUT1 and GLUT3 (encoded by SLC2A1 and SLC2A3 genes respectively), which are differentially distributed across the brain. GLUT1 is upregulated in embryonal tumors including MB (Loda et al., 2000), with a prominent expression in MYC-driven Group 3 MB (Park et al., 2019; Pham et al., 2022). GLUT1 overexpression determines a higher glucose influx into the cell that is avidly used for cytoplasmic glycolysis, supporting cell survival in low oxygen environments (Al Tameemi et al., 2019). As soon as glucose enter the cells, hexokinase converts glucose into glucose-6-phosphate (G6P) which is consumed by multiple pathways such as glycolysis, OXPHOS, PPP, hexosamine biosynthetic pathway (HBP) or amino acid biosynthesis. By using (18F)-fluorodeoxyglucose positron emission tomography (PET), a glycolytic metabolic phenotype has been reported in MB patients (Gururangan et al., 2004), and key genes involved in glycolysis are found to be significantly overexpressed in MYC-driven MB mouse models compared to SHH MB or non-neoplastic cerebellar tissue (Tao et al., 2019). Furthermore, both SHH MB and proliferating cells of the postnatal brain (i.e., hippocampus, subventricular zone, cerebellar external granule layer) preferentially express the glycolytic enzymes Hexokinase 2 (HK2) and the M2 isoform of pyruvate kinase (PKM2) (Gershon et al., 2013a, 2013b; Di Magno et al., 2014; Tech et al., 2017). HK2 and PKM2 are both key glycolysis rate-limiting enzymes, also in the presence of normal oxygen rate. Intriguingly, there are four mammalian hexokinases that vary in their tissue distribution and affinity for glucose; among them, HK2 drives aerobic glycolysis in MB (Tech et al., 2017). Moreover, pyruvate kinases catalyze the latest step of glycolysis: the M1 isoform (PKM1) is typically present in differentiated neurons, whereas high PKM2 expression characterizes all MB subgroups (Tech et al., 2017).
Under aerobic conditions, pyruvate could be converted into lactate by the enzyme lactate dehydrogenase (LDH), and this reaction is coupled with NAD+ regeneration. Lactate can be exported outside the cell and contributes to both intra- and extracellular acidification. High lactate levels have been primarily associated with Group 3/4 compared to SHH MB (Blüml et al., 2016), and MB Group 3 orthotopic mouse models also exhibit high lactate levels (Pham et al., 2022). Moreover, lactate dehydrogenase A (LDHA) levels are found overexpressed in the most aggressive MB subgroups (Valvona and Fillmore, 2018). Of interest, monocarboxylate transporter 1 (MCT1, encoded by the SLC16A1 gene), which mediates the import and export of lactate, pyruvate and ketone bodies across the plasma membrane, is upregulated in MB tumors and cell lines (Li et al., 2009). Supporting these findings, miRNA 124 (miR124), an important regulator of the temporal progression of neurogenesis in mice, targets MCT1 and it is downregulated in MB tumors (Li et al., 2009; Silber et al., 2013). On the other hand, c-MYC is reported to be a positive modulator of MCT1 (Coller et al., 2000), suggesting that c-MYC amplification or overexpression could contribute to enhance MCT1 expression levels in MB. These findings raise the possibility to target the glycolytic pathway as a novel therapeutic strategy against MB.
Interestingly, a comparative analysis of metabolic profiles of MB cells in three different environments -in vitro, in flank and in orthotopic xenografts- suggests that orthotopic MYC-amplified MB tumors have increased levels of glucosamine-6-phosphate compared to normal brain, proposing dependence on HBP (Pham et al., 2022). Similarly, RNA-sequencing data derived by the “Children’s Brain Tumor Network/Kids First Pediatric Brain Tumor Atlas'' indicate an increase in HBP enzymes expression in MB tumors compared to other pediatric brain tumors (i.e., glioma, ependymoma) (Pham et al., 2022).
Glutamine addiction. Glutamine is a nonessential amino acid (NEAA) implicated in different processes of cell proliferation, acting as carbon and nitrogen source for the biosynthesis of many other molecules such as nucleotides, other nonessential amino acids (alanine, aspartate, asparagine, glycine, etc), glutathione (GSH) and metabolic TCA intermediates (Zhang et al., 2017; Yoo et al., 2020). Glutamine as a precursor for the neurotransmitter glutamate plays a crucial role during the development of the nervous system. In normal conditions, glutamine is metabolized in the cytosol for the biosynthesis of nucleotides and amino acids and can also be transported into the mitochondria for glutaminolysis. The latter is a two-step process: the first reaction is the hydrolysis of glutamine in glutamate that is catabolized by glutaminases (GLS) 1/2, also known as glutamine synthetases; then glutamate is deaminated by the glutamate dehydrogenases (GDH) to produce α-ketoglutarate (α-KG) that supplies the TCA cycle. Additionally, glutamate undergoes expulsion into cytoplasm for generating GSH and other NAAs (Yoo et al., 2020). However, as also noted, glutaminase II pathway converts glutamine directly into α-KG by glutamine transaminase (GTK, and by ω-amidase). The latter has recently been reported as the main pathway implicated in the consumption of glutamine in the brain as well in primary Group 3 cells and MB mouse models (Pham et al., 2022).
Additional implications relevant to glutamine metabolism in MB have emerged. A role for p73 as a regulator of glutamine metabolism has been reported in MB (Velletri et al., 2013; Niklison-Chirou et al., 2017). P73 is a member of the p53-family, which is frequently overexpressed in different tumors, including MB (Zitterbart et al., 2007). In the most aggressive subgroups of MB, Niklison-Chirou and others reported high levels of TAp73, the p73 form that maintains the transactivation domain. TAp73 was previously recognized as a positive modulator of energetic metabolism by enhancing PPP (Jiang et al., 2013), inducing cytochrome c oxidase (Rufini et al., 2012), and activating the serine biosynthesis (Amelio et al., 2014). Additionally, high glutamine uptake and glutaminase activity are found to be upregulated in cisplatin-resistant MB cell line DAOY (Ge et al., 2022). Interestingly, the NEAT1 (non-coding RNA nuclear paraspeckle assembly transcript 1)/miR-23a-3p/GLS axis has emerged as a master regulator of glutamine metabolism in MB by regulating GLS activity and mitochondrial glutaminolysis. In both MB patients and in cisplatin-resistant DAOY cells, miR-23a-3p was downregulated, while the expression levels of NEAT1 were upregulated (Ge et al., 2022). Since cancer tissue of origin influences cellular metabolic reprogramming, the dependence of cancer cells on glutamine remains difficult to elucidate. For instance, GBM showed a high demand of glutamine, which is reported to be synthesized by GBM cells themselves and in part released by astrocytes in the cerebrospinal fluid (CSF). GBM cells convert glutamate to glutamine to sustain amino acid and nucleotide de novo biosynthesis, thus providing metabolites for TCA and OXPHOS (Tardito et al., 2015). A recent comprehensive metabolic study investigated the metabolic profiles of MYC-amplified MB Group 3 cell lines and different mouse models using labeled glucose. A high uptake of glucose in normal brain and orthotopic xenografts compared to in vitro cell cultures was found, suggesting that MYC-amplified MB use glucose as carbon source for the glutamate synthesis (deriving by TCA intermediates), depending on the tumor environmental conditions (Pham et al., 2022). These findings corroborate the hypothesis that high-risk MB tumors do not acquire glutamine by the TME but are able to synthesize it de novo using glucose.
As reported above, addiction to glutamine is exploited by cancer cells to produce GSH as antioxidant defense (Bansal and Celeste Simon, 2018) and several works now focus on the efficacy of targeting GLS to improve cancer therapy. High levels of GLS were associated with low reactive oxygen species (ROS) levels due to increased antioxidant response in cancer. Interestingly, a novel glutamine antagonist JHU395 was shown to inhibit GLS and to selectively kill MYC-driven MB, supporting the idea that ROS increase underlies GLS inhibition-induced cytotoxicity (Pham et al., 2021). C-MYC is a critical regulator of glutamine uptake by regulating the glutamine transporters SLC1A5 (also known as ASCT2). A work from Genovesi et al., identified SLC1A5 in a set of potentially druggable-targets in non-WNT restricted MB through a network-based system-pharmacogenomics approach (Ghasemi et al., 2022). Since c-MYC deregulation is frequent in non-WNT highly aggressive and metastatic MB subtypes, it is possible that SLC1A5 inhibition could induce high-risk MB regression by interfering with both glutamine catabolism and ROS scavenging, boosting ROS levels beyond the tolerable threshold. It is reasonable to hypothesize that targeting glutamine metabolism may impact on MB survival by both limiting the availability of critical metabolites and heightening oxidative stress in MYC-deregulated aggressive MB.
Using in-vivo Magnetic Resonance Spectroscopy (MRS), glutamate concentration has emerged as a survival prognostic marker of MB and elevated glutamate levels are reported in high-risk MB cases (Wilson et al., 2014). Glutamate decarboxylases (GAD1/2) are the enzyme responsible for the conversion of glutamate into the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Grone and Maruska, 2016). An increase in both GABA uptake and GABA metabolism is found in brain metastases derived from breast cancer, and an increase of NADH levels in the microenvironment is linked to this event (Neman et al., 2014).
Recent findings revealed GAD2 overexpression in primary MB Group 3 D425 cell line when cultured in CSF-mimicking condition, indicating de novo GABA biosynthesis. On the contrary, GABA catabolism is mediated by 4-aminobutyrate aminotransferase (ABAT, or GABA transaminase) as a shunt of energy and glutamine under nutrient deprivation and environmental stresses (Araújo et al., 2010; Feehily and Karatzas, 2013). Of interest, while differentiated neurons show both high OXPHOS rate and ABAT expression, neural stem cells and MB tumors exhibit low ABAT levels; notably, ABAT levels are lower in the metastatic G3/G4 subgroups compared to the less aggressive WNT/SHH subtypes. Intriguingly, during dissemination, MB cells exploit GABA metabolism to survive in the nutrient-poor environment of CSF, facilitating metastasis formation; this suggests a crucial role for ABAT expression fluctuation to promote leptomeningeal dissemination (Martirosian et al., 2021).
Lipogenesis. Lipid metabolism has been well-characterized in SHH MB, as SHH signaling suppresses fatty acid oxidation and exacerbates lipogenesis and aerobic glycolysis (Tech et al., 2015; Tech and Gershon, 2015). Mechanistically, the crucial lipid synthesis enzymes FASN and Acetyl-Coenzyme A (Acetyl-CoA) Carboxylase (Acc1) are upregulated by SHH MB, whereas lipid catabolism enzymes Acyl-Coenzyme A Oxidase 1 (Acox1) and Medium Chain Acyl-CoA Dehydrogenase (MCAD) are downregulated in the same MB subgroup (Bhatia et al., 2011). Accordingly, high lipid levels were found in SHH MB tissues compared to Group 3/4 (Blüml et al., 2016). The SHH signaling also drives lipid metabolic changes through the retinoblastoma protein (Rb)/E2F1 tumor suppressor complex inactivation in both CGNPs and SHH-driven MB (Bhatia et al., 2011). Under this condition, E2F1 up-regulates the nuclear nutrient sensor Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ), which positively regulates the expression of glycolytic enzymes HK2 and PKM2 (Bhatia et al., 2011, 2012).
A double regulation mechanism connecting SHH signaling and lipogenesis has recently emerged. Although SHH signaling drives lipogenesis, endogenous lipids can lead to SHH hyperactivation through the binding and the modulation of the SHH-activating key component Smoothened (SMO) protein (Deshpande et al., 2019; Daggubati et al., 2021). Endogenous sterol and oxysterol lipids (i.e., HSD11β2 and DHCR7) are indeed reported to activate SMO after ribosome biogenesis impairment, which consequently induces ER stress and unfolded protein response activation (Raleigh et al., 2018; Daggubati et al., 2021). Cholesterol biosynthesis is also found enhanced in SHH MB tumors as it promotes SMO activation (Gordon et al., 2018). Of interest, treatment with the current Food and Drug Administration (FDA)-approved SMO antagonist (i.e. Vismodegib and sonidegib) gives rise to drug resistance development in SHH MB patients due to several mechanisms, including SMO re-activating mutations. Statin treatment by simvastatin reduces SHH MB cells proliferation and SHH-derived MB young mice tumor growth (Fan et al., 2021), as single agents or combined with SMO antagonists (Gordon et al., 2018; Fan et al., 2021). These findings suggest that targeting lipid metabolism in association with current chemotherapy could represent a promising strategy to overcome SHH MB recurrence. However, further studies are required to delve into the role of fatty acid metabolism in all MB subgroups.
Genetic-driven metabolic adaptation. As previously described, genetic diversity is one of the major drivers of metabolic adaptation in MB. For this reason, mutational signatures specific to each subgroup may underlie metabolic heterogeneity across MB subgroups. Mutations affecting oncogenes or tumor suppressor genes could act as cell-intrinsic factors regulating metabolic functions in cancer cells to support the synthesis of specific metabolites in a context-dependent manner.
MYC. The oncogene MYC is the most studied gene driving MB progression and metastasization and it is frequently amplified or overexpressed in MB, especially in high-risk Group 3 (McManamy et al., 2007; Pfister et al., 2009; Cho et al., 2011; Northcott et al., 2012a, 2012b). However, since MYC was frequently found upregulated also in patients with good prognosis, the relationship between MYC expression and MB clinical outcome remains controversial. That is why it is becoming increasingly evident that MYC per se cannot be considered a good prognostic factor (Roussel and Robinson, 2013). These discrepancies might be explained by the presence of several determinants, both intrinsic (e.g. mutational co-dependencies, metabolic plasticity) or environmental (e.g. tumor microenvironment), that can influence MYC-related prognosis in MB patients. A large body of evidence supports the idea that MYC can profoundly shape the metabolic state of cancer cells by regulating a plethora of processes such as glutamine synthesis, glycolysis and lipogenesis as well as mitochondrial biogenesis and metabolism. Although inhibiting MYC proteins could ideally be a powerful approach to eradicate MB, this strategy results in low effectiveness in clinical settings, due to the highly flexible structure of MYC proteins and to the lack of drug binding pockets. A more promising therapeutic avenue encompasses the possibility of targeting pathways that are activated downstream of this oncoprotein; in this regard, MYC-dependent metabolic adaptation is a very attractive phenomenon with several potential novel targets for MB (Figure 2). As it occurs in other cancers, MYC stimulates the Warburg effect in MB by directly trans-activating the expression of key glycolytic genes (Tao et al., 2019). Several crucial metabolism-related genes are bona fide MYC targets both in healthy and in transformed cells. It has been demonstrated that MYC overexpression in astrocyte precursors is able to induce neoplastic formation, phenocopying Group 3 MB in mice. RNA-sequencing analyses of MYC-driven Group 3 mouse models revealed an upregulation of pro-glycolytic MYC-targets such as LDHA, PKM2, HK2 and Pyruvate dehydrogenase kinase 1 (PDK1) compared to normal cerebellum or SHH MB (Tao et al., 2019). This evidence supports the idea that MYC is a prominent positive regulator of the Warburg effect in MB. Interrogation of Gilbertson MB public dataset (Tumor MB-Gilbertson- 76–MAS5.0–u133p2 dataset) showed that LDHA mRNA is upregulated in WNT and Group 3 MB (Valvona and Fillmore, 2018) compared to non-neoplastic cerebellum and spinal cord tissue. LDHA was also found over-expressed in vitro both at the transcriptional and at the protein level in DAOY (SHH), UW402 (Group 3) and Res256 (subgroup non-attributed) MB cell lines (Valvona and Fillmore, 2018). LDHA inhibition through treatment with oxamate, a structural analogue of pyruvate, reduces MB glycolysis in favor of OXPHOS and blocks cell migration in vitro (Valvona and Fillmore, 2018). A very interesting question is whether and how and the intra-tumor heterogeneous expression of MYC contributes to MB progression and relapse. To this aim, a recent work has identified a functional cross-talk between MYC amplified and non-amplified cells within MB both in vitro and in vivo (Qin et al., 2022). By single-cell transcriptomics and mass-spectrometry-based metabolite quantification, the authors identified a prominent role for inter-clonal communication through secreted factors in the determination of MB aggressiveness (Figure 2). On one hand MYC-driven MB clones increased metastasizing capacity and leptomeningeal dissemination through LDHA release, while non-MYC-driven cells secreted Dickkopf WNT signaling pathway inhibitor three to drive neo-angiogenesis (Qin et al., 2022). The authors also found that LDHA secretion from MYC-driven cells in the tumor stroma is able to induce the conversion of lactate in non-MYC-driven cells, increasing their migratory propensity as a by-stander effect; this strengthens the notion that cell-to-cell communication may reinforce cancer aggressiveness. Moreover, pharmacological targeting of LDHA with GSK 2837808A reduces MB cell invasion in vitro prompting a highly relevant therapeutic opportunity associated with LDHA targeting (Qin et al., 2022).
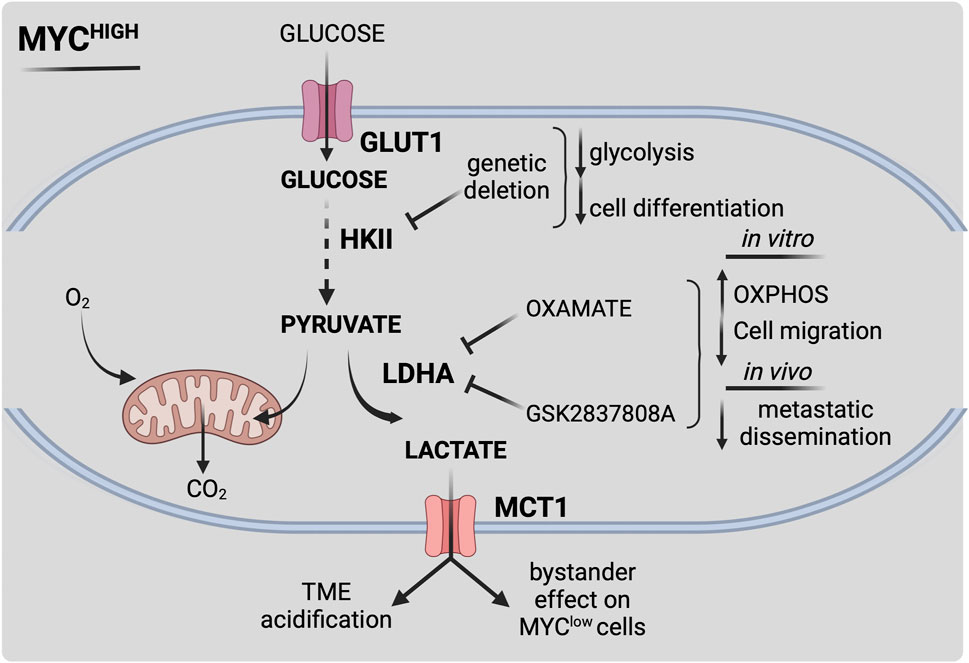
FIGURE 2. Schematic representation of MYC-dependent metabolic reprogramming in MB. MYC overexpression or amplification (MYChigh) drives metabolic reprogramming, leading to increased survival and therapy resistance. MYC-dependent alterations lead to enhanced aerobic glycolysis (i.e., Warburg effect) by a multiple-layered regulation. Activation of MYC stimulates glucose uptake by increasing transcription levels of glucose transporter GLUT1. MYC-dependent upregulation of hexokinase 2 (HKII) and pyruvate dehydrogenase kinase 1 (PDK1) fuels glucose flux within the cell. MYC highly contributes to extracellular acidification by acting on lactate synthesis and secretion through lactate dehydrogenase A (LDHA) and monocarboxylate transporter 1 (MCT1) expression, thus influencing MB microenvironment. Pharmacological targeting of MYC-dependent key metabolic reactions could be effective towards high-risk MYC-driven MB. Figure is created in “BioRender.com”.
A previously reported role for a c-MYC paralog, N-MYC, as an oncometabolic regulator has been established in SHH MB. N-MYC acts as a mediator of SHH/insulin/IGF/PI3K signaling pathways, leading to the final upregulation of HK2. Interference of N-MYC activity with the 10,058-F4 inhibitor leads to the impairment of SHH-induced HK2 upregulation in CGPCs (Gershon et al., 2013a; 2013b). Similarly, upon oncogenic SHH expression, HK2 deletion blocks glycolysis and induces cell differentiation while disrupting tumor growth (Gershon et al., 2013a; 2013b). These findings firstly corroborate the idea of cancer cells exploiting the energy metabolism of their developmental progenitors to maintain an undifferentiated state and a malignant potential. Moreover, they support the central role of MYC as master regulator of MB malignant phenotype and suggest its downstream effectors as potential novel targets and biomarkers for metabolic anticancer therapy.
P53. P53 is the most studied tumor suppressor, whose genetic inactivation has been extensively linked to the pathogenesis of the majority of human cancers. Beyond its well-known role as genome caretaker, p53 was also found to be a critical regulator of metabolic adaptation in several cancer types, including embryonal brain tumors (Marie and Shinjo, 2011; Xiong et al., 2020). At the same time, p53 is shown to be promptly activated by metabolic stresses. In human MBs, TP53 mutations are frequent in the WNT (16%) and SHH groups (21%), acting as promoter of cellular transformation, and are rare in Group 3 or 4 tumors (Zhukova et al., 2013). Although p53 genetic mutations are rare in MB with the poorest prognosis, a body of evidence suggests that p53 pathway is perturbed in high-risk MB. Even if the role of p53 is well-characterized in the SHH MB subgroup, its involvement in metabolic MB reprogramming is completely unexplored. Genomic data analysis from MB have shown that overexpression of the p53-antagonist gene WIP1 (wild-type p53-induced phosphatase one or protein phosphatase, magnesium-dependent 1, delta, PPM1D) is crucial to counteract p53-mediated apoptosis, thus mediating therapy resistance (Castellino et al., 2008; Buss et al., 2012, 2014; Akamandisa et al., 2016). Interestingly, WIP1 function can be regulated by phosphoglycerate mutase 1 (PGAM1), a glycolytic enzyme that is overexpressed in cancer tissues and that is able to block WIP1 cytoprotective function upon chemo- and radiotherapy in glioma (Ohba et al., 2020). These data highlight a key role for p53-related functions in the intricate crosstalk between cancer metabolic rewiring and therapy responsiveness, which could also be relevant for MB. An intriguing work studied the biological alterations of MB at relapse with respect to the primary tumors in patients and through in vivo studies (Hill et al., 2015). The authors strikingly found that combined MYC family amplifications and p53 pathway defects commonly emerged at relapse, and all patients in this group died of rapidly progressive post-relapse disease. This work contributed to elucidate an unprecedented role for novel co-occurring MYC/p53 mutations as determinants of dismal prognosis in MB (Hill et al., 2015). Since MYC amplification and p53 inactivation drive cell metabolism in the same direction, it is possible that MB stem cells acquire and exploit this second mutation to enhance their metabolic plasticity under the selective pressure of their microenvironment and drive metastases formation.
Many lines of evidence suggest that TIGAR (TP53-induced glycolysis and apoptosis regulator), a downstream target of p53, could fill the knowledge gap concerning P53 and metabolic plasticity in MB. TIGAR mainly functions as a glycolytic inhibitor by mediating the hydrolysis of fructose-1,6-diphosphate and fructose-2,6-diphosphate to inhibit glycolysis. While counteracting glycolysis, TIGAR facilitates PPP flux to produce NADPH and ribose, thereby promoting DNA repair, and reducing intracellular ROS (Lee et al., 2014). In recent years, TIGAR enzyme was shown to actively control cancer cell differentiation, mitochondrial function and autophagy (Geng et al., 2018) and it was found to be overexpressed in MB specimens (Li et al., 2012). Given its role in other tumors, TIGAR could antagonize p53 and promote PPP in a subpopulation of MB cells to sustain proliferation and antioxidant defenses during MB dissemination through the CSF. Moreover, it might be interesting to estimate TIGAR activity in MB and measure its metabolic-dependent contribution to therapy resistance.
EGFR. Very well-recognized oncogenic drivers are the members of the family receptor tyrosine kinase (RTKs) which actively support metabolic adaptation during cancer progression. Erb-B2 Receptor Tyrosine Kinase 2 (ERBB-2) is a member of the human epidermal growth factor receptor (EGFR) family and it is of particular relevance in MB; it is upregulated in approximately 40% of MB and its expression correlates with poor prognosis in patients (Bodey et al., 2005; Liu et al., 2014).
In several cancer settings, constitutive EGFR activation has been linked to increased glucose uptake, lactate secretion and glutamine utilization (Makinoshima et al., 2014; Sigismund et al., 2018). Additionally, it was shown that EGFR activation correlates with poor prognosis in MB patients and is a key determinant of MB migratory behavior (Rico-Varela et al., 2015). The contribution of EGFR activation to metabolic reprogramming in MB was however never explored directly. It was recently unveiled that EGFR oncogenic mutations can cause a switch from glycolysis towards the serine biosynthetic pathway (Jin et al., 2019), which was found enhanced in Group 4 MB (Park et al., 2019). In line with this observation, it could be relevant to explore whether this metabolic feature represents a specific metabolic vulnerability that could be exploited to hit EGFR-driven MB. In this respect, inhibition of 3-phosphoglycerate dehydrogenase (PHGDH), the rate limiting enzyme for de novo serine biosynthesis, turned out to be a promising therapeutic strategy to overcome resistance in metabolically-driven cancers (Rathore et al., 2020, 2021; McNamee et al., 2021; Zhao et al., 2021). Moreover, PHGDH inhibition was shown to be particularly effective in relapsing and therapy resistant tumors, as it was demonstrated for reduced brain metastasization (Ngo et al., 2020). It is plausible that upregulation of PHGDH-dependent serine/glycine biosynthesis supports EGFR-driven MB metabolic adaptation during leptomeningeal colonization and therefore PHGDH could be a potential target for metastatic MBs. Moreover, stratification of MB patients based on the expression of EGFR could help develop tailored metabolic therapies.
Epigenetic-driven metabolic adaptation. Amongst cell-intrinsic factors epigenetic alterations can shape the transcriptional program in order to allow cancer adaptation to the increased metabolic requirements. In MB, the interplay among cell metabolism and epigenetic regulation of gene expression is very poorly characterized. Mutations that typically arise in MB patients involve chromatin remodeling factors genes such as AT-Rich Interaction Domain 1A/2 (ARID1A, ARID2) and SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A, Member 4 (SMARCA4) (Morrissy et al., 2017). SMARCA4 mutations in WNT, Group 3 and Group 4 MB subgroups are reported and are found to counteract MYC activity in Group 3 MB (Morrissy et al., 2017; Ballabio et al., 2021). Moreover, in other cancers, SMARCA4 and ARID1A are found to repress the transcription of the sodium independent anionic amino acid transporter SLC7A11 (Koppula et al., 2018; Ogiwara et al., 2019; Song et al., 2020; Luo et al., 2021; Kito et al., 2022). Other than exporting glutamate outside the cell membrane, SLC7A11 imports cysteine, which is essential for GSH biosynthesis. Indeed, SMARCA4-dependent negative regulation of this transporter leads to reduction in intracellular cysteine required for antioxidant defense (Koppula et al., 2018; Song et al., 2020; Cermakova et al., 2021). Thus, loss-of-function mutations in the SMARCA4 gene could drive MB survival under oxidative stress conditions. Latest findings have unraveled new mechanisms linking epigenetics and metabolism in Group 4 MB. Badodi and others identified a cellular subset of Group 4 MB characterized by high expression of BMI1, a member of the Polycomb-Group Proteins, and low expression of the chromatin remodeler CHD7 (herein referred to as BMI1High; CHD7Low) both in vitro and in MB patients, respectively. This cellular population was shown to support MB growth (Badodi et al., 2017) through an overall decrease in mitochondrial function combined with upregulation of glycolysis (Badodi et al., 2021). Such mutated genetic background characterized by chromatin alterations is shown to favor the establishment of the Warburg effect as an adaptive strategy. Inositol hexakisphosphate (IP6) treatment is able to dramatically reduce glycolysis in the BMI1High; CHD7Low subpopulation without activating OXPHOS; it also synergizes with cisplatin to induce MB Group 4 clearance both in vitro and in vivo (Badodi et al., 2021). Given the role of both BMI1 and CHD7 in low oxygen cellular conditions, it is conceivable that this signature identifies a proportion of Group 4 MB with a specific advantage in hypoxic conditions due to the described metabolic adaptations. Another understudied layer of regulation of MB metabolic plasticity is represented by the plethora of non-coding RNAs affecting the expression of key metabolic genes, of which some are found deregulated in MB. An example of this epigenetic-dependent metabolic regulation in MB is provided by the discovery of a crosstalk between lncRNA NEAT1 and miRNA-23a-3p (miR-23a-3p) in the regulation of glutamine metabolism, which supports cisplatin resistance in MB cell lines (Ge et al., 2022). Specifically, NEAT1 is reported to promote therapy-resistance through the repression of miR-23a-3p expression, which is extremely low in MB samples. The authors have also demonstrated that miR-23a-3p exerts a tumor suppressive function in MB by targeting the gene encoding for GLS, thus attenuating glutamine metabolism. Therapeutic strategies enhancing miR-23a-3p expression should be validated in vivo to explore the possibility to target glutamine metabolism in cisplatin-refractory MB. Another miRNA that is implicated in MB metabolic reprogramming is microRNA 124 (miR124), whose function during neurogenesis was previously studied (Cheng et al., 2009). miR124 is significantly downregulated in MB (Li et al., 2009; Silber et al., 2013) and is found to control the expression of the main lactate transporter MCT1 (SLC16A1). Restoration of miR124 by ectopic expression induces MB cell death through the inhibition of lactate secretion by MCT1, probably due to cytotoxic intracellular acidosis.
Interplay between metabolic reprogramming and ROS production. In physiological conditions, ROS play a key role as a messenger in cell signal transduction and cell cycling while an increase in their production generates the so-called ‘oxidative stress’, that is widely considered as a fuel for tumorigenesis (Perillo et al., 2020; Cheung and Vousden, 2022). Cancer cells maintain low levels of ROS in two essential ways: by enhancing their antioxidant capacity (i.e. increase of NADPH and GSH) and/or by reducing the reliance on mitochondrial OXPHOS (Liemburg-Apers et al., 2015; Lee et al., 2018). It is now clear that altered bioenergetics affects ROS production, which is in turn able to regulate cancer metabolism through the control of key metabolic enzymes (Arfin et al., 2021). Little is known about the role of oxidative stress in the development and the evolution of MB. Given the ability of CSCs to maintain extremely low ROS levels to survive, unraveling the interplay between oxidative stress and MB stem cells (MBSCs) metabolism could be crucial for MB therapy. It has been shown that radioresistant MBSCs display reduced levels of mitochondrial ROS and a marked reduction of oxidative phosphorylation compared to their non-stem counterparts (Sun et al., 2017).
It was recently hypothesized that a metabolic deviation from glycolysis to the PPP pathway underlies CSCs ability to face oxidative stress and avoid redox imbalance (Ghanbari Movahed et al., 2019). Oxidative PPP pathway essentially converts the glycolytic intermediate G6P into ribulose-5-phosphate (R5P) with the production of two NADPH molecules to protect CSCs from free radicals. These metabolic steps are achieved through the enzymatic activity of glucose-6-phosphate dehydrogenase (G6PD) and 6GPD. A comprehensive metabolic profiling of MB identified PPP signature as a prognostic factor for Group 3 MB (Park et al., 2019). Moreover, the authors found a strong association between two key PPP genes, Transketolase (TKT) and Transaldolase 1 (TALDO1), and poor prognosis in Group 3 and Group 4 MB patients (Park et al., 2019). This evidence suggests the importance of capitalizing the ROS-induced metabolic deviation to PPP pathway to trigger CSCs death. To this aim, inhibition of G6PD and 6GPD might be applied in combination with ROS-inducing therapies to target MB stem cells. Anti-PPP agents are still not available in clinical settings; however, several works pinpoint a potent and promising antitumor effect both in vivo and in vitro for different cancer types (Mele et al., 2018). Moreover, treatment with G6PD inhibitors could counteract therapy resistance, mainly driven by CSCs (Hong et al., 2018). Otherwise, a possible pitfall of this approach is the insurgence of a resistance phenotype due to the activation of metabolic ways alternative to PPP. This case has been recently explored in melanoma cells, where the impairment of G6PD led to increased malic enzyme activity and glutamine consumption to face oxidative stress (Aurora et al., 2022).
An emerging target for selectively killing CSCs with a PPP-dependent phenotype is the nuclear factor erythroid 2-related factor 2 (NRF2), the master regulator of the antioxidant defense, whose function in cancer biology has been extensively explored. NRF2 is a transcription factor that manages the overall cellular response and metabolic adaptation to oxidative stress by controlling the expression of many genes such as G6PD and 6PGD. Under physiological conditions, the activity of NRF2 is inhibited by Kelch-Like ECH-Associated Protein 1 (Keap1), thus causing its cytoplasmic retention and its subsequent degradation. Upon oxidative stress, NRF2 dissociates from Keap1 and translocates into the nucleus where it trans-activates antioxidant responsive elements (ARE)-containing genes. A large body of evidence highlights a cytoprotective role for NRF2 in cancer progression and therapy resistance, including childhood brain tumors (Barrera et al., 2017; Godoy et al., 2020). Among neuroepithelial tumors, the World Health Organization (WHO) grade IV (the highest grade for CNS malignancies) MB shows the highest score of NRF2 staining (Barrera et al., 2017). In contrast to the significant amount of literature linking NRF2 with adult GBM, the current knowledge about its clinicopathological significance in MB is still poor. In a work from Tang et al., 2017 it has been demonstrated that NRF2 expression is enhanced in MB samples (n = 41) when compared to peritumoral control brain tissues (n = 27). Moreover, it was previously shown that pharmacological targeting of NRF2 with two NRF2-inducers (nifurtimox and tetrathiomolybdate) results in a synergic ROS-dependent antitumor response towards MB in vitro (Koto et al., 2011).
Otherwise, it is emerging that several anticancer drugs exert their cytotoxic function through the induction of intolerable levels of ROS. In line with this, it has been demonstrated that bortezomib, a proteasome inhibitor, which was previously found to selectively kill MB cells, induces cell death in a ROS-dependent manner (Ohshima-Hosoyama et al., 2011). Bortezomib treatment results in the stabilization of NOXA, a member of the Bcl-2 family, triggering apoptosis in response to high ROS levels in a p53-independent manner (Ohshima-Hosoyama et al., 2011; Hoerig et al., 2021). A recent molecular work from Li et al., has studied the effects of Liposomal Honokiol (Lip-HNK), a small bisphenol lignin delivered into liposomes, as a potential treatment against MB (Li et al., 2022). The authors found that HNK-dependent induction of apoptosis is mediated by ROS and thus reverted by treatment with the scavenger N-Acetylcysteine (NAC). In light of this data, it may be hypothesized that combinatorial approaches targeting both ROS production and specific metabolic adaptive mechanisms are needed to selectively kill MBSCs.
In addition to the complexity imposed by the oncogenotype and by cell-autonomous factors, environmental and cell-extrinsic factors are fundamental drivers of MB metabolism. During MB progression, cancer cells need to adapt their metabolism with respect to non-tumor cells to face the challenges posed by the harsh nature of the TME. Various metabolic stresses are encountered at different stages during MB development and relapse. TME is indeed generally hypoxic, acidic and with a distinct nutrient composition compared to non-tumor tissues, determining the first barrier that cancer cells must overcome in order to metastasize (Figure 3).
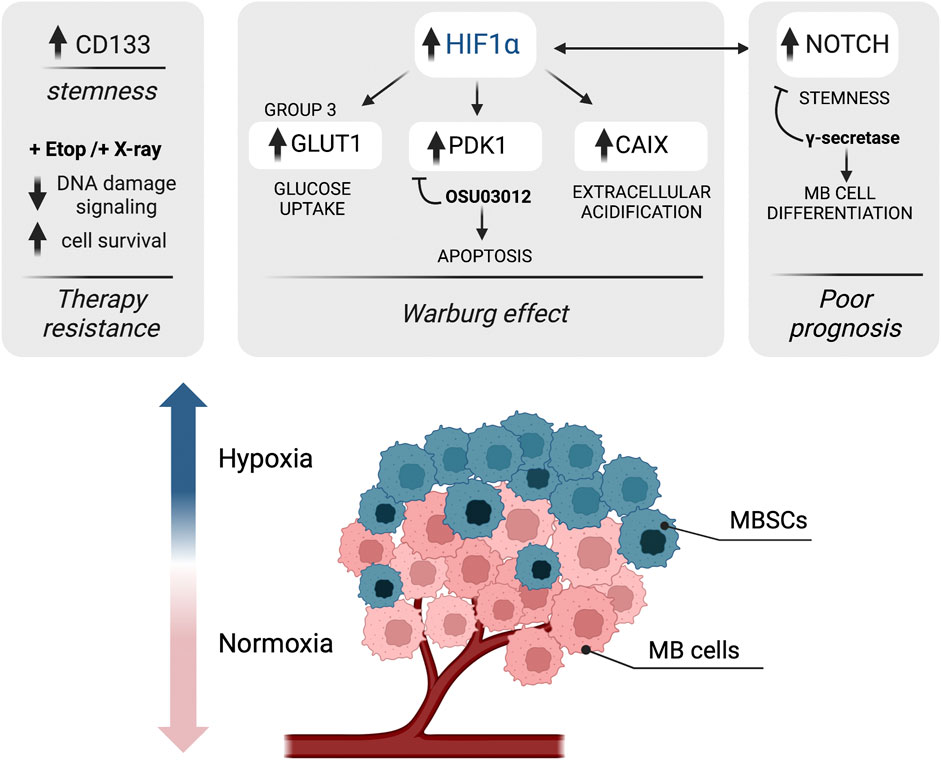
FIGURE 3. Schematic representation of the impact of hypoxia-associated metabolic adaptation in MB. Hypoxia is a major component of the tumor microenvironment that shapes tumor heterogeneity; the levels of oxygen in cancer cells decrease with increasing distance from the blood vessels. In hypoxic MB cells, hypoxia-inducible factor 1 alpha (HIF1α) supports the Warburg effect by upregulating the expression of genes involved in the glycolytic pathway, such as GLUT1, carbonic anhydrase IX (CAIX) and pyruvate dehydrogenase kinase 1 (PDK1). The use of PDK1 inhibitor OSU03012 induces mitochondrial-dependent apoptosis in MB. Moreover, hypoxic conditions were found to trigger upregulation of NOTCH, which correlates with a poor prognosis in MB. A connection between HIF1α and NOTCH signaling is also found, driving stemness and cancer stem cells (CSCs) expansion. Finally, hypoxia upregulates CD133 (staminal marker) expression in MB cells and contributes to therapy resistance (after both etoposide (Eto) and radiotherapy (X-ray) treatments) by attenuating DNA damage signaling. Figure is created in “BioRender.com”.
The role of hypoxia in metabolic shaping of MB. A common feature of solid tumors is the presence of regions with an imbalance in oxygen supply (Chouaib et al., 2016; Rankin and Giaccia, 2016). Cancer hypoxia is mainly caused by the uncontrolled proliferation of cancer cells that overcome the ability of the pre-existing blood vessels to satisfy the overall oxygen demand. Healthy cells are normally not viable at low concentrations of oxygen, which typically lead to cell death. On the contrary, cancer cells exploit genomic changes and modulate their entire metabolism to survive even at very low oxygen concentrations (pO2 ≤ 1%) (Al Tameemi et al., 2019) Hypoxia is clinically associated with a poor long-term prognosis and it is known to promote stemness and to block cell differentiation, thus favoring the selection and survival of CSCs within the TME. Due to the hard-to-practice techniques to measure pO2 in tumors and the lack of hypoxia-biomarkers, little is known about the extent of hypoxia in MB and other brain pediatric cancers.
Both in MB and glioma cell lines, it was demonstrated that in vitro induction of hypoxia increases the expression of the stem cell marker CD133 (Blazek et al., 2007). Moreover, low oxygen levels, particularly 2% oxygen, are required for in vitro expansion and survival of MB-derived precursor cells, whereas exposure to 20% oxygen induces tumor cell differentiation (Pistollato et al., 2010). Further, much evidence supports a role for hypoxia as an obstacle to therapy efficacy in brain tumors (Kitange et al., 2019; Mudassar et al., 2020). It was demonstrated that, after radiotherapy treatment under hypoxic conditions, MB cells display impaired DNA damage signaling by downregulating Nijmegen Breakage Syndrome 1 (NBS1) expression, thus increasing cell survival and limiting therapy effectiveness (Cowman et al., 2019). Understanding the mechanisms underlying MB adaptation to low oxygen is hugely important to develop and improve new targeted therapeutic strategies.
HIF1α as a central regulator of hypoxia-induced metabolic adaptation of MB. At a molecular level, the adaptation of tumor cells to hypoxia is mainly orchestrated by a family of transcription factors known as Hypoxia Inducible Factors (HIFs), which are highly sensitive to decreased cellular oxygen levels and are able to regulate a plethora of cellular pathways. HIF1α, the main hypoxia effector, is upregulated in MB and its genetic downregulation has been associated with reduced MB cell proliferation (Cruzeiro et al., 2018). In several cancer types, the stabilization of HIF1α leads to the upregulation of glycolytic metabolic pathways to produce ATP, strengthening the typical cancer Warburg effect. Induction of HIF1α expression also enhances lactate production which leads to tumor hypoxic acidosis, as it was shown for glioma cells (Wang et al., 2017). All of these metabolic changes are associated with increased aggressiveness, radio and chemo-therapy resistance and poor prognosis in several tumors. Multiple therapeutic options to inhibit HIF1α-dependent signaling have been developed in the last years. Amongst them, one of the most promising approaches for MB could be targeting HIF1α downstream targets, as they are found to be frequently upregulated (see below). Here, we briefly review the potential of targeting upregulated HIF1α targets in MB (Figure 3).
GLUT1. A downstream target of HIF1α that is known to contribute to hypoxia-induced metabolic adaptation of cancer cells is the pro-glycolytic factor GLUT1. HIF1α-dependent upregulation of GLUT1 supports the maintenance of stem-like features as it was shown in GBM, where GLUT1 blockade has been shown to inhibit self-renewal of CSCs (Shibuya et al., 2015). Inhibitors targeting GLUT1, such as BAY-876, also restored radiosensitivity in resistant breast cancer cells (Zhao et al., 2016). Due to its important role in cancer-related metabolic adaptation, GLUT1 is a promising therapeutic target for glycolytic and hypoxic tumors like MB.
PDK1. Pyruvate dehydrogenase kinase 1 (PDK1) is another target gene that has been reported to be transcriptionally regulated by HIF1α. PDK1 has been defined as a ‘glycolysis gatekeeper’ due to its ability to promote glycolysis by blocking the entry of pyruvate into the TCA cycle. HIF1α-dependent induction of PDK1 is associated with glycolytic reprogramming, enhanced cancer cell proliferation and stemness in several tumors (Du et al., 2016; Semba et al., 2016; Peng et al., 2018). Therefore, genetically or chemically targeting of PDK1 would result in decreased energy availability, leading to cancer cell death. In 2010 Baryawno et al. demonstrated that PDK1 is constitutively activated in immunohistochemical sections of human primary MB samples. The authors have also shown that targeting PDK1 with the small-molecule inhibitor OSU03012 suppresses MB growth by inducing mitochondrial-dependent apoptosis both in vitro and in vivo (Baryawno et al., 2010). Moreover, PDK1 inhibition can potentiate the anti-tumor effect of mTOR inhibitor CCI-779 in vivo MB models (Baryawno et al., 2010). These results suggest that targeting the HIF1α-PDK1 axis could improve the therapeutic benefit in patients with hypoxic and metabolically-adapted MB.
CAIX. Hypoxia-induced changes in cellular metabolism include accumulation of lactate, as the end-product of glycolysis, together with carbon dioxide, which results in the acidification of the extracellular environment. Microenvironmental pH maintenance upon hypoxia in tumors relies on the functional activation of carbonic anhydrase IX (CAIX), a cancer-related transmembrane enzyme catalyzing the reversible conversion of carbon dioxide to bicarbonate ion and proton. Through its ability to regulate intracellular and extracellular pH, CAIX expression supports cancer cells survival in hypoxia/acidosis conditions and facilitates cancer cell migration and metastasization in several tumors (Becker, 2019; Hui et al., 2022). CAIX was found to be highly expressed in MB, especially in tumor areas with low microvascular density (Pistollato et al., 2010) and in perinecrotic areas (Nordfors et al., 2010). Importantly, CAIX expression significantly correlated with poor prognosis in a panel of MB patient samples (Nordfors et al., 2010). In recent years, inhibitors of CAIX such as SLC-0111, were shown to sensitize cancer cells to chemotherapy and are currently under evaluation for the treatment of metastatic solid tumors (McDonald et al., 2020). Given the role of CAIX expression in MB, it is plausible that this approach is also highly effective towards this pediatric cancer.
NOTCH signaling and Hypoxia. The interaction between HIF1α and NOTCH signaling plays an important role in promoting cell stemness (Gustafsson et al., 2005; Main et al., 2010). In normoxic conditions, NOTCH signaling is initiated when a cell-surface expressed ligand binds to the NOTCH receptor. This event triggers the cleavage of NOTCH intracellular domain (ICD) by ADAM metalloproteases and γ-secretase that translocates into the nucleus and induces the expression of target genes (Henrique and Schweisguth, 2019). Interestingly, HIF1α can directly interact with NOTCH ICD and allows its stabilization upon hypoxic conditions, sustaining NOTCH signaling in MB cells (Gustafsson et al., 2005; Pistollato et al., 2010). Inhibition of the NOTCH pathway by γ-secretase is sufficient to revert this effect, leading to MB cells differentiation upon hypoxia exposure (Pistollato et al., 2010). The implications of NOTCH signaling as an important driver of pluripotency and stemness in MB have been reported so far by several authors (Hallahan et al., 2004). NOTCH signaling pathway, indeed, promotes the development of both SHH and MYC-driven Group 3 MB, even though it is unable to initiate to MB in cerebellar precursors alone (Hallahan et al., 2004; Kahn et al., 2018; Ballabio et al., 2021). Additionally, SHH MB metastasization was found to be promoted by NOTCH pathway transcription factor Atonal Homolog 1 (ATOH1) (Grausam et al., 2017). Despite the role of NOTCH signaling in MB initiation, a recent analysis showed that NOTCH ligands and receptors are strongly associated with poor survival across MB patients, and NOTCH is most activated in both SHH and Group 3 samples (Northcott et al., 2017). Intriguingly, Mutvei and others found that HIF1α-dependent hyperactivation of NOTCH signaling induces a switch to HIF2α production in D341 cell line (MYC-amplified Group 3 MB) and not in DAOY cells (SHH MB) (Mutvei et al., 2018). Targeting the HIF1α-NOTCH axis could therefore affect hypoxia-driven oncogenic effects. Several NOTCH-targeting agents are now under clinical investigation for many cancer types and this approach might be beneficial especially for high-risk MB patients (Majumder et al., 2021).
Nutrient availability within the TME. Metabolic reprogramming arises as a consequence of the complex interplay between altered tumor properties and the tissue context that can fuel or hinder cancer proliferation. MB cells display an extensive crosstalk with both cellular (such as neurons, astrocytes and microglia) and non-cellular components of the brain extracellular matrix (ECM), which critically determine nutrient and growth factors supply. As reported above, it is widely accepted that MB can dramatically change its energy metabolism in response to nutrients availability in the TME. Although increasing attention has been focused on how environmental factors can shape MB evolution, the contribution of nutrient distribution to MB development and dissemination within primary and metastatic tumor sites remains yet unexplored. The case of pediatric brain tumors poses serious challenges due to the lack of experimental models that can faithfully reproduce the metabolic heterogeneity of both brain and leptomeningeal TME. In brain tumors, ECM is a main driver of metabolic adaptation; its composition and stiffness can determine the distribution of nutrients as well as the supply of oxygen to the tumor. Indeed, primary MB is surrounded by the ECM and mainly comes into contact with the interstitial matrix domain that is composed of loosely associated components (Van Ommeren et al., 2020). In contrast, leptomeningeal metastases are in contact with the basal lamina, which covers blood vessels and the pia mater and is mostly composed of a dense network of collagen, laminin and fibronectin. Therefore, in the leptomeningeal niche—the subarachnoid space—the concentration of growth factors and nutrients is dramatically different from the primary tumor (Van Ommeren et al., 2020). Moreover, during the metastasization process, MB cells must survive in a nutrient-deprived environment, especially when circulating through the CSF. With the exception of glutamine, CSF contains less glucose, amino acids and lipoproteins compared to blood. Recent studies are now focusing on the possible use of CSF as an indicator of MB disease status and also as a source of novel biomarkers. Metabolomic studies on MB CSF samples revealed high levels of metabolites that are typically secreted under hypoxia, such as α-KG, fumarate, hydroxypyruvate, malate and succinate as well as triacylglycerol (Lee et al., 2022). Moreover, recurrent MB patients-derived CSF samples show high concentrations of hypoxia-induced amino acids such as tryptophan, methionine, serine and lysine respectively (Reichl et al., 2020). These studies strengthen the concept that hypoxic and nutrient-deprived CSF represents a highly selective environment that may favor the adaptability of a small pool of metabolically flexible MB stem cells favoring tumor invasion and metastases.
Interplay among nutrient availability and autophagy. The nutrient sensing pathways in the microenvironment are controlled by two master regulators, the mammalian target of Rapamycin complex 1 (mTORC1) and the AMP-activated Kinase (AMPK), which antagonize each other to balance cancer metabolic demands. AMPK and mTORC1 exert their function through the regulation of autophagy, a fundamental process that allows the recycling of intracellular components through their lysosomal-dependent degradation. Autophagy is a key survival process induced by nutrient stress and upregulated in several types of cancers, including high-risk MB (Nazio et al., 2019, 2021; Paul et al., 2020; Gatto et al., 2021). It has recently been demonstrated that autophagy activation in Group 3 MB supports stemness and metastasization (Nazio et al., 2021). Moreover, targeting autophagy through pharmacological inhibition with chloroquine (CQ) results in sensitization of Group 3 MB and significantly improves survival in orthotopic mouse models (Nazio et al., 2021). Conversely, mTOR is found upregulated in a high proportion of MB samples (Folgiero et al., 2016) and several works address mTOR targeting as a therapeutic strategy for MB (Dimitrova and Arcaro, 2015; Aldaregia et al., 2018; Eckerdt et al., 2019), even if the rise of treatment-resistant phenotype is a rather controversial issue (Alammar et al., 2021). mTOR activity is mainly regulated by nutrient levels and is particularly sensitive to amino acids, such as tryptophan, leucine and arginine (Saxton and Sabatini, 2017). In amino acids-deprived conditions, mTOR activity is inhibited, which promotes autophagy-dependent catabolism, providing energy and supporting cell survival. Radiotracing experiments demonstrated that tryptophan is greedily metabolized in MB patients (Xin et al., 2020) and its depletion from the tumor microenvironment is associated with the activity of tryptophan catabolic enzyme indoleamine 2,3-dioxygenase 1 (IDO1). mTORC1 inhibition by rapamycin treatment was able to increase IDO1 expression (Folgiero et al., 2016), suggesting the existence of a crosstalk between tryptophan levels and mTOR in MB, which is critical for the effectiveness of mTOR targeting approaches.
Despite the lack of studies that compare tumor metabolism among MB subgroups, SHH MB tumors are the most well-characterized as Sonic Hedgehog, a mitogenic signal that directly regulates the bioenergetics of MB, drives metabolic dependence on aerobic glycolysis and lipogenesis rather on OXPHOS and fatty acids oxidation. However, low-carbohydrates ketogenic diets do not reduce tumor growth in allograft and spontaneous SHH-driven MB mouse models (Dang et al., 2015); thus, highlighting the presence of a complex scenario determining MB metabolic profiling and the possible pitfalls of therapeutic approaches targeting these changes. Indeed, human SHH MB tumors show high levels of OXPHOS and mitochondrial biogenesis (such as PGC1α) -associated genes (Łastowska et al., 2019), underlying the importance of mitochondrial-dependent metabolism that occur concomitantly to cytoplasmic metabolic reactions. It is not surprising that advanced solid tumors such as MB, after switching to glycolytic metabolism, remain dependent on mitochondria for key metabolic reactions such as glutaminolysis, fatty acid oxidation and for the synthesis of Krebs cycle intermediates that support tumor growth and spread. In light of this evidence, targeting the critical enzymes involved in metabolic adaptation to the microenvironment could be a valid approach to eradicate MB. However, progress in targeting cancer metabolism therapeutically in the past decade has been limited. Only a few metabolism-based drugs for cancer have been successfully developed, and some of them are currently being tested in clinical trials. Detailed descriptions about the currently available pharmacological compounds acting on metabolism together with the up-to-date clinical trials on tumors have been recently reviewed elsewhere (Lemberg et al., 2022; Stine et al., 2022). In this work, we have performed an unprecedented review of metabolism-related features of MB, detailing MB subgroup-specific metabolic alterations (Figure 4). Since intrinsical, genetic and clinical heterogeneity is a prominent feature of MB, it is of paramount importance to improve our knowledge about subgroup-specific molecular circuits adaptations to develop tailored target therapies for MB. Moreover, few metabolic drugs are already in clinical trials and it could be reasonable to promote repurposing of metabolism-based inhibitors for the treatment of MB. Here, we have summarized promising preclinical results of metabolic drugs tested directly against MB (Table 1). Importantly, even if the Warburg effect predominates MB metabolism, aerobic glycolysis and oxidative phosphorylation may coexist in MB and be differentially regulated throughout the malignant evolution of MB. However, the presence of intratumoral heterogeneity makes in vitro modeling of MB extremely difficult when studying metabolism. The use of 3D tumor organoids may reproduce MB heterogeneity but still lacks the high diversity of metabolites in the TME. Additionally, cell culture media contain nutrients such as glucose, amino acids and vitamins at levels that do not reflect those of human plasma or CSF and totally lack lipids and nucleotide precursors. Indeed, in vivo models better represent metabolic heterogeneity and TME, although mice diet, sex and environmental stressors are to be considered since they can have an impact on cancer metabolism. A very recent metabolomic analysis (Pham et al., 2022) shines a light on the discrepancies observed between in vitro MB cell cultures and orthotopic mouse xenografts in terms of metabolic phenotype, especially regarding glucose and glutamine uptake. This work showed how MYC-amplified orthotopic xenograft tumors display significantly higher glucose uptake and usage when compared to flank xenograft tumors and cells in culture (Pham et al., 2022). Together, this evidence suggests a crucial limitation of the metabolic profiling when conducted in non-native tumor environments. Beyond the experimental issue to in vitro model MB metabolism, comprehensive studies conducted both on patients and in mouse models have revealed a profound inter- and intra-variability based on the well-acknowledged subgroup MB classification. Therefore, staining for oncogenes and metabolic enzymes combined with assays measuring metabolic activities such as lactate production or glutamine uptake could help to stratify patients in order to develop personalized therapies. Moreover, the future entry of genomic, transcriptomic and metabolomic analyses into clinical routine will guide MB therapy integrating data considering both metabolic and genetic features.
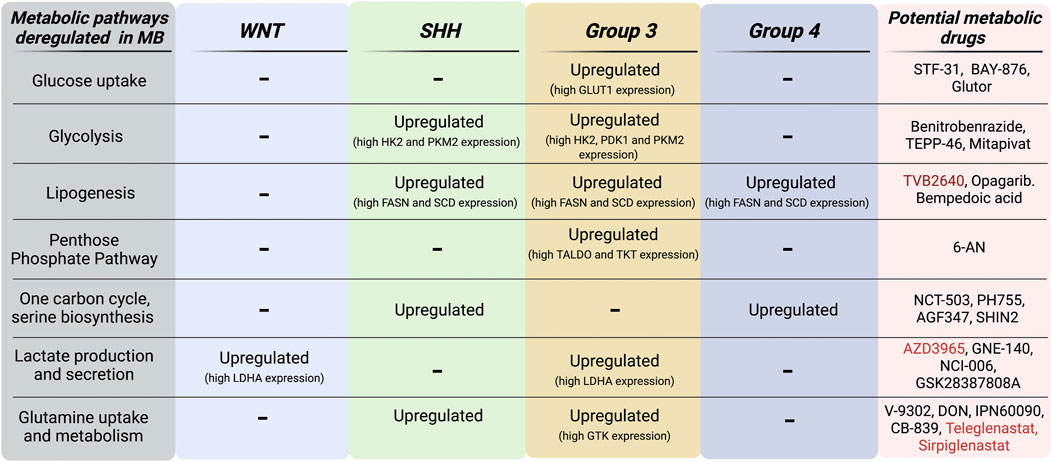
FIGURE 4. MB subgroup-specific alterations of metabolic pathways. Schematic representation of both metabolic pathways and genes that are found upregulated in MB subgroups. Metabolic inhibitors currently in preclinical (black) and clinical (red) development for oncology applications are listed in this scheme. Figure is created in “BioRender.com”.

TABLE 1. Preclinical studies targeting MB metabolism.
FN conceptualized the work and, together with VM, MG and FM prepared a draft of the manuscript. FN, FG, and AM critically reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
FN’s lab is supported by grants from the Italian Ministry of Health #GR-2019-2369231 and from CureSearch for Children’s Cancer - Young Investigator Award in Pediatric Oncology Drug Development. VM is supported by a post-doctoral fellowship from Fondazione Umberto Veronesi. Figures are created in “BioRender.com” by FM and VM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akamandisa, M. P., Nahta, R., and Castellino, R. C. (2016). PPM1D/WIP1 in medulloblastoma. Oncoscience 3, 186–187. doi:10.18632/oncoscience.318
Al Tameemi, W., Dale, T. P., Al-Jumaily, R. M. K., and Forsyth, N. R. (2019). Hypoxia-modified cancer cell metabolism. Front. Cell. Dev. Biol. 7, 4–15. doi:10.3389/fcell.2019.00004
Alammar, H., Nassani, R., Alshehri, M. M., Aljohani, A. A., and Alrfaei, B. M. (2021). Deficiency in the treatment description of mTOR inhibitor resistance in medulloblastoma, a systematic review. Int. J. Mol. Sci. 23, 464. doi:10.3390/IJMS23010464
Aldaregia, J., Odriozola, A., Matheu, A., and Garcia, I. (2018). Targeting mTOR as a therapeutic approach in medulloblastoma. Int. J. Mol. Sci. 19, E1838. doi:10.3390/IJMS19071838
Amelio, I., Markert, E. K., Rufini, A., Antonov, A. V., Sayan, B. S., Tucci, P., et al. (2014). P73 regulates serine biosynthesis in cancer. Oncogene 33, 5039–5046. doi:10.1038/onc.2013.456
Araújo, W. L., Ishizaki, K., Nunes-Nesi, A., Larson, T. R., Tohge, T., Krahnert, I., et al. (2010). Identification of the 2-hydroxyglutarate and isovaleryl-CoA dehydrogenases as alternative electron donors linking lysine catabolism to the electron transport chain of Arabidopsis mitochondria. Plant Cell. 22, 1549–1563. doi:10.1105/tpc.110.075630
Arfin, S., Jha, N. K., Jha, S. K., Kesari, K. K., Ruokolainen, J., Roychoudhury, S., et al. (2021). Oxidative stress in cancer cell metabolism. Antioxidants (Basel), Switz. 10, 642. doi:10.3390/ANTIOX10050642
Aurora, A. B., Khivansara, V., Leach, A., Gill, J. G., Martin-Sandoval, M., Yang, C., et al. (2022). Loss of glucose 6-phosphate dehydrogenase function increases oxidative stress and glutaminolysis in metastasizing melanoma cells. Proc. Natl. Acad. Sci. U. S. A. 119, e2120617119. doi:10.1073/pnas.2120617119
Badodi, S., Dubuc, A., Zhang, X., Rosser, G., Da Cunha Jaeger, M., Kameda-Smith, M. M., et al. (2017). Convergence of BMI1 and CHD7 on ERK signaling in medulloblastoma. Cell. Rep. 21, 2772–2784. doi:10.1016/j.celrep.2017.11.021
Badodi, S., Pomella, N., Zhang, X., Rosser, G., Whittingham, J., Niklison-Chirou, M. V., et al. (2021). Inositol treatment inhibits medulloblastoma through suppression of epigenetic-driven metabolic adaptation. Nat. Commun. 12, 2148–2216. doi:10.1038/s41467-021-22379-7
Bakhshinyan, D., Adile, A. A., Liu, J., Gwynne, W. D., Suk, Y., Custers, S., et al. (2021). Temporal profiling of therapy resistance in human medulloblastoma identifies novel targetable drivers of recurrence. Sci. Adv. 7, eabi5568–13. doi:10.1126/sciadv.abi5568
Ballabio, C., Gianesello, M., Lago, C., Okonechnikov, K., Anderle, M., Aiello, G., et al. (2021). Notch1 switches progenitor competence in inducing medulloblastoma. Sci. Adv. 7, eabd2781. doi:10.1126/sciadv.abd2781
Bansal, A., and Celeste Simon, M. (2018). Glutathione metabolism in cancer progression and treatment resistance. J. Cell. Biol. 217, 2291–2298. doi:10.1083/jcb.201804161
Barrera, G., Daga, M., Cucci, A. M., and Pizzimenti, S. (2017). The potential therapeutic target Nrf2 in childhood brain tumors. J. Pediatr. Oncol. 5, 11–17.
Baryawno, N., Sveinbjörnsson, B., Eksborg, S., Chen, C. S., Kogner, P., and Johnsen, J. I. (2010). Small-molecule inhibitors of phosphatidylinositol 3-kinase/Akt signaling inhibit Wnt/beta-catenin pathway cross-talk and suppress medulloblastoma growth. Cancer Res. 70, 266–276. doi:10.1158/0008-5472.CAN-09-0578
Becker, H. M. (2019). Carbonic anhydrase IX and acid transport in cancer. Br. J. Cancer 1222 122, 157–167. doi:10.1038/s41416-019-0642-z
Bhatia, B., Hsieh, M., Kenney, A. M., and Nahlé, Z. (2011). Mitogenic Sonic hedgehog signaling drives E2F1-dependent lipogenesis in progenitor cells and medulloblastoma. Oncogene 30, 410–422. doi:10.1038/onc.2010.454
Bhatia, B., Potts, C. R., Guldal, C., Choi, S. P., Korshunov, A., Pfister, S., et al. (2012). Hedgehog-mediated regulation of PPARγ controls metabolic patterns in neural precursors and shh-driven medulloblastoma. Acta Neuropathol. 123, 587–600. doi:10.1007/s00401-012-0968-6
Bi, J., Chowdhry, S., Wu, S., Zhang, W., Masui, K., and Mischel, P. S. (2020). Altered cellular metabolism in gliomas — An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 20, 57–70. doi:10.1038/s41568-019-0226-5
Blazek, E. R., Foutch, J. L., and Maki, G. (2007). Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133− cells, and the CD133+ sector is enlarged by hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 67, 1–5. doi:10.1016/J.IJROBP.2006.09.037
Blüml, S., Margol, A. S., Sposto, R., Kennedy, R. J., Robison, N. J., Vali, M., et al. (2016). Molecular subgroups of medulloblastoma identification using noninvasive magnetic resonance spectroscopy. Neuro. Oncol. 18, 126–131. doi:10.1093/neuonc/nov097
Bockmayr, M., Mohme, M., Klauschen, F., Winkler, B., Budczies, J., Rutkowski, S., et al. (2018). Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 7, e1462430. doi:10.1080/2162402X.2018.1462430
Bodey, B., Kaiser, H. E., and Siegel, S. E. (2005). Epidermal growth factor receptor (EGFR) expression in childhood brain tumors. Vivo (Brooklyn) 19, 931–941.
Buss, M. C., Read, T. A., Schniederjan, M. J., Gandhi, K., and Castellino, R. C. (2012). HDM2 promotes WIP1-mediated medulloblastoma growth. Neuro. Oncol. 14, 440–458. doi:10.1093/neuonc/nos001
Buss, M. C., Remke, M., Lee, J., Gandhi, K., Schniederjan, M. J., Kool, M., et al. (2014). The WIP1 oncogene promotes progression and invasion of aggressive medulloblastoma variants. Oncogene 349 34, 1126–1140. doi:10.1038/onc.2014.37
Castellino, R. C., De Bortoli, M., Lu, X., Moon, S. H., Nguyen, T. A., Shepard, M. A., et al. (2008). Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J. Neurooncol. 86, 245–256. doi:10.1007/S11060-007-9470-8
Cavalli, F. M. G., Remke, M., Rampasek, L., Peacock, J., Shih, D. J. H., Luu, B., et al. (2017). Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 31, 737–754. e6. doi:10.1016/j.ccell.2017.05.005
Cermakova, K., Smith, E. A., Chan, Y. S., Cabrera, M. L., Chambers, C., Jarvis, M. I., et al. (2021). SMARCA4 regulates spatially restricted metabolic plasticity in 3D multicellular tissue, doi:10.1101/2021.03.21.436346
Cheng, L. C., Pastrana, E., Tavazoie, M., and Doetsch, F. (2009). MiR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 12, 399–408. doi:10.1038/nn.2294
Cheung, E. C., and Vousden, K. H. (2022). The role of ROS in tumour development and progression. Nat. Rev. Cancer 225 22, 280–297. doi:10.1038/s41568-021-00435-0
Cho, Y. J., Tsherniak, A., Tamayo, P., Santagata, S., Ligon, A., Greulich, H., et al. (2011). Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 29, 1424–1430. doi:10.1200/JCO.2010.28.5148
Chouaib, S., Noman, M. Z., Kosmatopoulos, K., and Curran, M. A. (2016). Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 364 36, 439–445. doi:10.1038/onc.2016.225
Coller, H. A., Grandori, C., Tamayo, P., Colbert, T., Lander, E. S., Eisenman, R. N., et al. (2000). Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. U. S. A. 97, 3260–3265. doi:10.1073/pnas.97.7.3260
Cowman, S., Fan, Y. N., Pizer, B., and Sée, V. (2019). Decrease of Nibrin expression in chronic hypoxia is associated with hypoxia-induced chemoresistance in some brain tumour cells. BMC Cancer 19, 300. doi:10.1186/S12885-019-5476-9
Cruzeiro, G. A. V., dos Reis, M. B., Silveira, V. S., Lira, R. C. P., Jr, C. G. C., Neder, L., et al. (2018). HIF1A is overexpressed in medulloblastoma and its inhibition reduces proliferation and increases EPAS1 and ATG16L1 methylation. Curr. Cancer Drug Targets 18, 287–294. doi:10.2174/1568009617666170315162525
Daggubati, V., Hochstelter, J., Bommireddy, A., Choudhury, A., Krup, A. L., Kaur, P., et al. (2021). Smoothened-activating lipids drive resistance to CDK4/6 inhibition in Hedgehog-associated medulloblastoma cells and preclinical models. J. Clin. Invest. 131, 141171. doi:10.1172/JCI141171
Dang, M. T., Wehrli, S., Dang, C. V., and Curran, T. (2015). The ketogenic diet does not affect growth of hedgehog pathway medulloblastoma in mice. PLoS One 10, e0133633. doi:10.1371/JOURNAL.PONE.0133633
Deshpande, I., Liang, J., Hedeen, D., Roberts, K. J., Zhang, Y., Ha, B., et al. (2019). Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 571, 284–288. doi:10.1038/s41586-019-1355-4
Di Magno, L., Manzi, D., D’Amico, D., Coni, S., Macone, A., Infante, P., et al. (2014). Druggable glycolytic requirement for Hedgehog-dependent neuronal and medulloblastoma growth. Cell. Cycle 13, 3404–3413. doi:10.4161/15384101.2014.952973
Dimitrova, V., and Arcaro, A. (2015). Targeting the PI3K/AKT/mTOR signaling pathway in medulloblastoma. Curr. Mol. Med. 15, 82–93. doi:10.2174/1566524015666150114115427
Du, J., Yang, M., Chen, S., Li, D., Chang, Z., and Dong, Z. (2016). PDK1 promotes tumor growth and metastasis in a spontaneous breast cancer model. Oncogene 35, 3314–3323. doi:10.1038/onc.2015.393
Eckerdt, F., Clymer, J., Bell, J. B., Beauchamp, E. M., Blyth, G. T., Goldman, S., et al. (2019). Pharmacological mTOR targeting enhances the antineoplastic effects of selective PI3Kα inhibition in medulloblastoma. Sci. Rep. 91 (9), 12822–12911. doi:10.1038/s41598-019-49299-3
Fan, Q., Gong, T., Zheng, C., Ng, J. M. Y., Chen, J., Myers, C., et al. (2021). Statins repress hedgehog signaling in medulloblastoma with no bone toxicities. Oncogene 40, 2258–2272. doi:10.1038/s41388-021-01701-z
Feehily, C., and Karatzas, K. A. G. (2013). Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 114, 11–24. doi:10.1111/j.1365-2672.2012.05434.x
Folgiero, V., Miele, E., Carai, A., Ferretti, E., Alfano, V., Po, A., et al. (2016). Ido1 involvement in mTOR pathway: A molecular mechanism of resistance to mTOR targeting in medulloblastoma. Oncotarget 7, 52900–52911. doi:10.18632/ONCOTARGET.9284
Gatto, F., Milletti, G., Carai, A., Mastronuzzi, A., and Nazio, F. (2021). Recent advances in understanding the role of autophagy in paediatric brain tumours. Diagnostics 202111, 481. doi:10.3390/DIAGNOSTICS11030481
Ge, J., Wang, B., Zhao, S., and Xu, J. (2022). Inhibition of lncRNA NEAT1 sensitizes medulloblastoma cells to cisplatin through modulating the miR-23a-3p-glutaminase (GLS) axis. Bioengineered 13, 7670–7682. doi:10.1080/21655979.2021.2008695
Geng, J., Yuan, X., Wei, M., Wu, J., and Qin, Z. H. (2018). The diverse role of TIGAR in cellular homeostasis and cancer. Free Radic. Res. 52, 1240–1249. doi:10.1080/10715762.2018.1489133
Genovesi, L. A., Millar, A., Tolson, E., Singleton, M., Hassall, E., Kojic, M., et al. (2021). Systems pharmacogenomics identifies novel targets and clinically actionable therapeutics for medulloblastoma. Genome Med. 13, 103. doi:10.1186/S13073-021-00920-Z
Gershon, T. R., Crowther, A. J., Liu, H., Miller, C. R., and Deshmukh, M. (2013a). Cerebellar granule neuron progenitors are the source of Hk2 in the postnatal cerebellum. Cancer Metab. 1, 15. doi:10.1186/2049-3002-1-15
Gershon, T. R., Crowther, A. J., Tikunov, A., Garcia, I., Annis, R., Yuan, H., et al. (2013b). Hexokinase-2-mediated aerobic glycolysis is integral to cerebellar neurogenesis and pathogenesis of medulloblastoma. Cancer Metab. 1, 2. doi:10.1186/2049-3002-1-2
Ghanbari Movahed, Z., Rastegari-Pouyani, M., Mohammadi, M., and Mansouri, K. (2019). Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 112, 108690. doi:10.1016/J.BIOPHA.2019.108690
Ghasemi, D. R., Fleischhack, G., Milde, T., and Pajtler, K. W. (2022). The current landscape of targeted clinical trials in non-WNT/non-SHH medulloblastoma. Cancers 14, 679. doi:10.3390/CANCERS14030679
Godoy, P. R. D. V., Pour Khavari, A., Rizzo, M., Sakamoto-Hojo, E. T., and Haghdoost, S. (2020). Targeting NRF2, regulator of antioxidant system, to sensitize glioblastoma neurosphere cells to radiation-induced oxidative stress. Oxid. Med. Cell. Longev. 2020, 2534643. doi:10.1155/2020/2534643
Gordon, R. E., Zhang, L., Peri, S., Kuo, Y. M., Du, F., Egleston, B. L., et al. (2018). Statins synergize with hedgehog pathway inhibitors for treatment of medulloblastoma. Clin. Cancer Res. 24, 1375–1388. doi:10.1158/1078-0432.CCR-17-2923
Grabovska, Y., Mackay, A., O’Hare, P., Crosier, S., Finetti, M., Schwalbe, E. C., et al. (2020). Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 11, 4324–4415. doi:10.1038/s41467-020-18070-y
Grausam, K. B., Dooyema, S. D. R., Bihannic, L., Premathilake, H., Morrissy, A. S., Forget, A., et al. (2017). ATOH1 promotes leptomeningeal dissemination and metastasis of sonic hedgehog subgroup medulloblastomas. Cancer Res. 77, 3766–3777. doi:10.1158/0008-5472.CAN-16-1836
Grone, B. P., and Maruska, K. P. (2016). Three distinct glutamate decarboxylase genes in vertebrates. Sci. Rep. 6, 30507–30509. doi:10.1038/srep30507
Gururangan, S., Hwang, E., Herndon, J. E., Fuchs, H., George, T., and Coleman, R. E. (2004). [18F]fluorodeoxyglucose-positron emission tomography in patients with medulloblastoma. Neurosurgery 55, 1280–1288. doi:10.1227/01.NEU.0000143027.41632.2B
Gustafsson, M. V., Zheng, X., Pereira, T., Gradin, K., Jin, S., Lundkvist, J., et al. (2005). Hypoxia requires Notch signaling to maintain the undifferentiated cell state. Dev. Cell. 9, 617–628. doi:10.1016/j.devcel.2005.09.010
Hallahan, A. R., Pritchard, J. I., Hansen, S., Benson, M., Stoeck, J., Hatton, B. A., et al. (2004). The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of Sonic Hedgehog-induced medulloblastomas. Cancer Res. 64, 7794–7800. doi:10.1158/0008-5472.CAN-04-1813
Hanaford, A. R., Alt, J., Rais, R., Wang, S. Z., Kaur, H., Thorek, D., et al. (2019). Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl. Oncol. 12 (10), 1314–1322. doi:10.1016/j.tranon.2019.05.013
Henrique, D., and Schweisguth, F. (2019). Mechanisms of notch signaling: A simple logic deployed in time and space. Development 146, dev172148. doi:10.1242/dev.172148
Hill, R. M., Kuijper, S., Lindsey, J. C., Petrie, K., Schwalbe, E. C., Barker, K., et al. (2015). Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell. 27, 72–84. doi:10.1016/J.CCELL.2014.11.002
Hoerig, C. M., Plant-Fox, A. S., Pulley, M. D., Di, K., and Bota, D. A. (2021). Exploring the role and clinical implications of proteasome inhibition in medulloblastoma. Pediatr. Blood Cancer 68, e29168. doi:10.1002/PBC.29168
Hong, W., Cai, P., Xu, C., Cao, D., Yu, W., Zhao, Z., et al. (2018). Inhibition of glucose-6-phosphate dehydrogenase reverses cisplatin resistance in lung cancer cells via the redox system. Front. Pharmacol. 9, 43. doi:10.3389/fphar.2018.00043
Hovestadt, V., Ayrault, O., Swartling, F. J., Robinson, G. W., Pfister, S. M., and Northcott, P. A. (2020). Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat. Rev. Cancer 20, 42–56. doi:10.1038/s41568-019-0223-8
Hovestadt, V., Smith, K. S., Bihannic, L., Filbin, M. G., Shaw, M. K. L., Baumgartner, A., et al. (2019). Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 572, 74–79. doi:10.1038/s41586-019-1434-6
Hui, C., Ong, C., Lee, D. Y., Lee, B., Li, H., Chun, J., et al. (2022). Hypoxia-regulated carbonic anhydrase IX (CAIX) protein is an independent prognostic indicator in triple negative breast cancer. Breast Cancer Res. 241 24, 38–11. doi:10.1186/S13058-022-01532-0
Jakacki, R. I., Burger, P. C., Zhou, T., Holmes, E. J., Kocak, M., Onar, A., et al. (2012). Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: A children’s oncology group phase I/II study. J. Clin. Oncol. 30, 2648–2653. doi:10.1200/JCO.2011.40.2792
Jiang, P., Du, W., and Yang, X. (2013). A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell. Cycle 12, 3720–3726. doi:10.4161/cc.27267
Jin, N., Bi, A., Lan, X., Xu, J., Wang, X., Liu, Y., et al. (2019). Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat. Commun. 10, 2701–2715. doi:10.1038/s41467-019-10427-2
Juraschka, K., and Taylor, M. D. (2019). Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 24, 353–363. doi:10.3171/2019.5.PEDS18381
Kahn, S. A., Wang, X., Nitta, R. T., Gholamin, S., Theruvath, J., Hutter, G., et al. (2018). Notch1 regulates the initiation of metastasis and self-renewal of Group 3 medulloblastoma. Nat. Commun. 9, 4121–4129. doi:10.1038/s41467-018-06564-9
Kitange, G., Shi, W., Lin, C.-M., Yu, C.-F., Huang, H.-Y., Chen, F.-H., et al. (2019). Distinct tumor microenvironment at tumor edge as a result of astrocyte activation is associated with therapeutic resistance for brain tumor. Front. Oncol. 1, 307. doi:10.3389/fonc.2019.00307
Kito, Y., Kawashima, K., Saigo, C., Hasegawa, M., Nomura, S., Mikamo, T., et al. (2022). Thorahcic SMARCA4-deficient undifferentiated tumors with ganglioneuroma and enchondroma: Implications for SLC7A11 and ARID1A expression: A case report. Diagn. Pathol. 17, 29–36. doi:10.1186/s13000-022-01205-8
Koppula, P., Zhang, Y., Zhuang, L., and Gan, B. (2018). Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. Lond. Engl. 38, 12. doi:10.1186/s40880-018-0288-x
Koto, K. S., Lescault, P., Brard, L., Kim, K., Singh, R. K., Bond, J., et al. (2011). Antitumor activity of nifurtimox is enhanced with tetrathiomolybdate in medulloblastoma. Int. J. Oncol. 38, 1329–1341. doi:10.3892/IJO.2011.971
Koundouros, N., and Poulogiannis, G. (2020). Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22. doi:10.1038/s41416-019-0650-z
Łastowska, M., Karkucińska-Więckowska, A., Waschek, J. A., and Niewiadomski, P. (2019). Differential expression of mitochondrial biogenesis markers in mouse and human SHH-Subtype Medulloblastoma. Cells 8, E216–E222. doi:10.3390/cells8030216
Lee, B., Mohamad, I., Pokhrel, R., Murad, R., Yuan, M., Stapleton, S., et al. (2022). Medulloblastoma cerebrospinal fluid reveals metabolites and lipids indicative of hypoxia and cancer-specific RNAs. Acta Neuropathol. Commun. 10, 25–14. doi:10.1186/s40478-022-01326-7
Lee, J., Cho, Y. S., Jung, H., and Choi, I. (2018). Pharmacological regulation of oxidative stress in stem cells. Oxid. Med. Cell. Longev. 2018, 4081890. doi:10.1155/2018/4081890
Lee, P., Vousden, K. H., and Cheung, E. C. (2014). TIGAR, TIGAR, burning bright. Cancer Metab. 2, 1. doi:10.1186/2049-3002-2-1
Lemberg, K. M., Gori, S. S., Tsukamoto, T., Rais, R., and Slusher, B. S. (2022). Clinical development of metabolic inhibitors for oncology. J. Clin. Invest. 132 (1), e148550. doi:10.1172/JCI148550
Li, K. K., Pang, J. C., Ching, A. K., Wong, C. K., Kong, X., Wang, Y., et al. (2009). miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum. Pathol. 40, 1234–1243. doi:10.1016/j.humpath.2009.02.003
Li, M., Lockwood, W., Zielenska, M., Northcott, P., Ra, Y. S., Bouffet, E., et al. (2012). Multiple CDK/CYCLIND genes are amplified in medulloblastoma and supratentorial primitive neuroectodermal brain tumor. Cancer Genet. 205, 220–231. doi:10.1016/J.CANCERGEN.2012.03.002
Li, S., Chen, J., Fan, Y., Wang, C., Wang, C., Zheng, X., et al. (2022). Liposomal Honokiol induces ROS-mediated apoptosis via regulation of ERK/p38-MAPK signaling and autophagic inhibition in human medulloblastoma. Signal Transduct. Target. Ther. 71 7, 49–53. doi:10.1038/s41392-021-00869-w
Liemburg-Apers, D. C., Willems, P. H. G. M., Koopman, W. J. H., and Grefte, S. (2015). Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 898 89, 1209–1226. doi:10.1007/S00204-015-1520-Y
Liu, W., Zhang, S., Zhang, L., Cui, Q., Wang, J., Gui, T., et al. (2014). A prognostic analysis of pediatrics central nervous system small cell tumors: Evaluation of EGFR family gene amplification and overexpression. Diagn. Pathol. 9, 132–210. doi:10.1186/1746-1596-9-132
Loda, M., Xu, X., Pession, A., Vortmeyer, A., and Giangaspero, F. (2000). Membranous expression of glucose transporter-1 protein (GLUT-1) in embryonal neoplasms of the central nervous system. Neuropathol. Appl. Neurobiol. 26, 91–97. doi:10.1046/J.1365-2990.2000.00225.X
Luo, W., Lin, G. N., Song, W., Zhang, Y., Lai, H., Zhang, M., et al. (2021). Single-cell spatial transcriptomic analysis reveals common and divergent features of developing postnatal granule cerebellar cells and medulloblastoma. BMC Biol. 19, 135–223. doi:10.1186/s12915-021-01071-8
Main, H., Lee, K. L., Yang, H., Haapa-Paananen, S., Edgren, H., Jin, S., et al. (2010). Interactions between Notch- and hypoxia-induced transcriptomes in embryonic stem cells. Exp. Cell. Res. 316, 1610–1624. doi:10.1016/J.YEXCR.2009.12.012
Majumder, S., Crabtree, J. S., Golde, T. E., Minter, L. M., Osborne, B. A., and Miele, L. (2021). Targeting notch in oncology: The path forward. Nat. Rev. Drug Discov. 20, 125–144. doi:10.1038/s41573-020-00091-3
Makinoshima, H., Takita, M., Matsumoto, S., Yagishita, A., Owada, S., Esumi, H., et al. (2014). Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J. Biol. Chem. 289, 20813–20823. doi:10.1074/jbc.M114.575464
Marie, S. K. N., and Shinjo, S. M. O. (2011). Metabolism and brain cancer. Clinics 66, 33–43. doi:10.1590/S1807-59322011001300005
Martirosian, V., Deshpande, K., Zhou, H., Shen, K., Smith, K., Northcott, P., et al. (2021). Medulloblastoma uses GABA transaminase to survive in the cerebrospinal fluid microenvironment and promote leptomeningeal dissemination. Cell. Rep. 35, 109302. doi:10.1016/j.celrep.2021.109302
McDonald, P. C., Chia, S., Bedard, P. L., Chu, Q., Lyle, M., Tang, L., et al. (2020). A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am. J. Clin. Oncol. 43, 484–490. doi:10.1097/COC.0000000000000691
McManamy, C. S., Pears, J., Weston, C. L., Hanzely, Z., Ironside, J. W., Taylor, R. E., et al. (2007). Nodule formation and desmoplasia in medulloblastomas - defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 17, 151–164. doi:10.1111/J.1750-3639.2007.00058.X
McNamee, M. J., Michod, D., and Niklison-Chirou, M. V. (2021). Can small molecular inhibitors that stop de novo serine synthesis be used in cancer treatment? Cell. Death Discov. 71 7, 87–95. doi:10.1038/s41420-021-00474-4
Mele, L., Paino, F., Papaccio, F., Regad, T., Boocock, D., Stiuso, P., et al. (2018). A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell. Death Dis. 9, 572. doi:10.1038/S41419-018-0635-5
Morrissy, A. S., Cavalli, F. M. G., Remke, M., Ramaswamy, V., Shih, D. J. H., Holgado, B. L., et al. (2017). Spatial heterogeneity in medulloblastoma. Nat. Genet. 49, 780–788. doi:10.1038/ng.3838
Mudassar, F., Shen, H., O’Neill, G., and Hau, E. (2020). Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res. 39, 208–217. doi:10.1186/s13046-020-01724-6
Mutvei, A. P., Landor, S. K. J., Fox, R., Braune, E. B., Tsoi, Y. L., Phoon, Y. P., et al. (2018). Notch signaling promotes a HIF2α-driven hypoxic response in multiple tumor cell types. Oncogene 37, 6083–6095. doi:10.1038/s41388-018-0400-3
Nazio, F., Bordi, M., Cianfanelli, V., Locatelli, F., and Cecconi, F. (2019). Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell. Death Differ. 26, 690–702. doi:10.1038/s41418-019-0292-y
Nazio, F., Po, A., Abballe, L., Ballabio, C., Diomedi Camassei, F., Bordi, M., et al. (2021). Targeting cancer stem cells in medulloblastoma by inhibiting AMBRA1 dual function in autophagy and STAT3 signalling. Acta Neuropathol. 1423 142, 537–564. doi:10.1007/S00401-021-02347-7
Neman, J., Termini, J., Wilczynski, S., Vaidehi, N., Choy, C., Kowolik, C. M., et al. (2014). Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. U. S. A. 111, 984–989. doi:10.1073/pnas.1322098111
Ngo, B., Kim, E., Osorio-Vasquez, V., Doll, S., Bustraan, S., Liang, R. J., et al. (2020). Limited environmental serine and Glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 10, 1352–1373. doi:10.1158/2159-8290.CD-19-1228
Niklison-Chirou, M. V., Erngren, I., Engskog, M., Haglöf, J., Picard, D., Remke, M., et al. (2017). TAp73 is a marker of glutamine addiction in medulloblastoma. Genes. Dev. 31, 1738–1753. doi:10.1101/gad.302349.117
Nordfors, K., Haapasalo, J., Korja, M., Niemelä, A., Laine, J., Parkkila, A. K., et al. (2010). The tumour-associated carbonic anhydrases CA II, CA IX and CA XII in a group of medulloblastomas and supratentorial primitive neuroectodermal tumours: An association of CA IX with poor prognosis. BMC Cancer 10, 148. doi:10.1186/1471-2407-10-148
Northcott, P. A., Buchhalter, I., Morrissy, A. S., Hovestadt, V., Weischenfeldt, J., Ehrenberger, T., et al. (2017). The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317. doi:10.1038/nature22973
Northcott, P. A., Jones, D. T. W., Kool, M., Robinson, G. W., Gilbertson, R. J., Cho, Y. J., et al. (2012a). Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 1212 12, 818–834. doi:10.1038/nrc3410
Northcott, P. A., Shih, D. J. H., Remke, M., Cho, Y. J., Kool, M., Hawkins, C., et al. (2012b). Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 123, 615–626. doi:10.1007/S00401-011-0899-7
Ocasio, J., Babcock, B., Malawsky, D., Weir, S. J., Loo, L., Simon, J. M., et al. (2019). scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 10, 5829–5917. doi:10.1038/s41467-019-13657-6
Ogiwara, H., Takahashi, K., Sasaki, M., Kuroda, T., Yoshida, H., Watanabe, R., et al. (2019). Targeting the vulnerability of glutathione metabolism in arid1a-deficient cancers. Cancer Cell. 35, 177–190. e8. doi:10.1016/j.ccell.2018.12.009
Ohba, S., Johannessen, T. C. A., Chatla, K., Yang, X., Pieper, R. O., and Mukherjee, J. (2020). Phosphoglycerate mutase 1 activates DNA damage repair via regulation of WIP1 activity. Cell. Rep. 31, 107518. doi:10.1016/J.CELREP.2020.03.082
Ohshima-Hosoyama, S., Davare, M. A., Hosoyama, T., Nelon, L. D., and Keller, C. (2011). Bortezomib stabilizes NOXA and triggers ROS-associated apoptosis in medulloblastoma. J. Neurooncol. 105, 475–483. doi:10.1007/S11060-011-0619-0
Packer, R. J., Gajjar, A., Vezina, G., Rorke-Adams, L., Burger, P. C., Robertson, P. L., et al. (2006). Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J. Clin. Oncol. 24, 4202–4208. doi:10.1200/JCO.2006.06.4980
Park, A. K., Lee, J. Y., Cheong, H., Ramaswamy, V., Park, S. H., Kool, M., et al. (2019). Subgroup-specific prognostic signaling and metabolic pathways in pediatric medulloblastoma. BMC Cancer 19, 571–615. doi:10.1186/s12885-019-5742-x
Paul, R., Bharambe, H., and Shirsat, N. V. (2020). Autophagy inhibition impairs the invasion potential of medulloblastoma cells. Mol. Biol. Rep. 47, 5673–5680. doi:10.1007/S11033-020-05603-3
Peng, F., Wang, J. H., Fan, W. J., Meng, Y. T., Li, M. M., Li, T. T., et al. (2018). Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 37, 1062–1074. doi:10.1038/onc.2017.368
Perillo, B., Di Donato, M., Pezone, A., Di Zazzo, E., Giovannelli, P., Galasso, G., et al. (2020). ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 522 52, 192–203. doi:10.1038/s12276-020-0384-2
Pfister, S., Remke, M., Benner, A., Mendrzyk, F., Toedt, G., Felsberg, J., et al. (2009). Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J. Clin. Oncol. 27, 1627–1636. doi:10.1200/JCO.2008.17.9432
Pham, K., Hanaford, A. R., Poore, B. A., Maxwell, M. J., Sweeney, H., Parthasarathy, A., et al. (2022). Comprehensive metabolic profiling of MYC-amplified medulloblastoma tumors reveals key dependencies on amino acid, tricarboxylic acid and hexosamine pathways. Cancers (Basel) 14, 1311. doi:10.3390/cancers14051311
Pham, K., Maxwell, M. J., Sweeney, H., Alt, J., Rais, R., Eberhart, C. G., et al. (2021). Novel glutamine antagonist JHU395 suppresses MYC-driven medulloblastoma growth and induces apoptosis. J. Neuropathol. Exp. Neurol. 80, 336–344. doi:10.1093/jnen/nlab018
Pistollato, F., Rampazzo, E., Persano, L., Abbadi, S., Frasson, C., Denaro, L., et al. (2010). Interaction of hypoxia-inducible factor-1α and Notch signaling regulates medulloblastoma precursor proliferation and fate. Stem Cells 28, 1918–1929. doi:10.1002/STEM.518
Qin, N., Paisana, E., Langini, M., Picard, D., Malzkorn, B., Custódia, C., et al. (2022). Intratumoral heterogeneity of MYC drives medulloblastoma metastasis and angiogenesis. Neuro. Oncol. 24, 1509–1523. doi:10.1093/neuonc/noac068
Racker, E., and Spector, M. (1981). Warburg effect revisited: Merger of biochemistry and molecular biology. Sci. 80 213, 303–307. doi:10.1126/SCIENCE.6264596
Raleigh, D. R., Sever, N., Choksi, P. K., Sigg, M. A., Hines, K. M., Thompson, B. M., et al. (2018). Cilia-associated oxysterols activate smoothened. Mol. Cell. 72, 316–327. e5. doi:10.1016/j.molcel.2018.08.034
Rankin, E. B., and Giaccia, A. J. (2016). Hypoxic control of metastasis. Science 352, 175–180. doi:10.1126/science.aaf4405
Rathore, R., Caldwell, K. E., Schutt, C., Brashears, C. B., Prudner, B. C., Ehrhardt, W. R., et al. (2021). Metabolic compensation activates pro-survival mTORC1 signaling upon 3-phosphoglycerate dehydrogenase inhibition in osteosarcoma. Cell. Rep. 34, 108678. doi:10.1016/J.CELREP.2020.108678
Rathore, R., Schutt, C. R., and van Tine, B. A. (2020). PHGDH as a mechanism for resistance in metabolically-driven cancers. Cancer drug resist. (Alhambra, Calif.) 3, 762–774. doi:10.20517/CDR.2020.46
Reichl, B., Niederstaetter, L., Boegl, T., Neuditschko, B., Bileck, A., Gojo, J., et al. (2020). Determination of a tumor-promoting microenvironment in recurrent medulloblastoma: A multi-omics study of cerebrospinal fluid. Cancers (Basel) 12, 13500–E1414. doi:10.3390/cancers12061350
Rico-Varela, J., Singh, T., McCutcheon, S., and Vazquez, M. (2015). EGF as a new therapeutic target for medulloblastoma metastasis. Cell. Mol. Bioeng. 84 8, 553–565. doi:10.1007/S12195-015-0395-6
Roussel, M. F., and Robinson, G. W. (2013). Role of MYC in medulloblastoma. Cold Spring Harb. Perspect. Med. 3, a014308. doi:10.1101/CSHPERSPECT.A014308
Rufini, A., Niklison-Chirou, M. V., Inoue, S., Tomasini, R., Harris, I. S., Marino, A., et al. (2012). TAp73 depletion accelerates aging through metabolic dysregulation. Genes. Dev. 26, 2009–2014. doi:10.1101/gad.197640.112
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell. 168, 960–976. doi:10.1016/J.CELL.2017.02.004
Semba, H., Takeda, N., Isagawa, T., Sugiura, Y., Honda, K., Wake, M., et al. (2016). HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 7, 11635–11710. doi:10.1038/ncomms11635
Shibuya, K., Okada, M., Suzuki, S., Seino, M., Seino, S., Takeda, H., et al. (2015). Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 6, 651–661. doi:10.18632/ONCOTARGET.2892
Sigismund, S., Avanzato, D., and Lanzetti, L. (2018). Emerging functions of the EGFR in cancer. Mol. Oncol. 12, 3–20. doi:10.1002/1878-0261.12155
Silber, J., Hashizume, R., Felix, T., Hariono, S., Yu, M., Berger, M. S., et al. (2013). Expression of miR-124 inhibits growth of medulloblastoma cells. Neuro. Oncol. 15, 83–90. doi:10.1093/neuonc/nos281
Singh, Sheila K., Hawkins, Cynthia, Clarke, Ian D., Squire, J. A., Bayani, J., Hide, T., et al. (2004). Identification of human brain tumour initiating cells. Nature 432, 396–401. doi:10.1038/nature03128
Song, S., Nguyen, V., Schrank, T., Mulvaney, K., Walter, V., Wei, D., et al. (2020). Loss of SWI/SNF chromatin remodeling alters NRF2 signaling in non-small cell lung carcinoma. Mol. Cancer Res. 18, 1777–1788. doi:10.1158/1541-7786.MCR-20-0082
Stine, Z. E., Schug, Z. T., Salvino, J. M., and Dang, C. V. (2022). Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 21, 141–162. doi:10.1038/s41573-021-00339-6
Sun, L., Moritake, T., Ito, K., Matsumoto, Y., Yasui, H., Nakagawa, H., et al. (2017). Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets. PLoS One 12, 01761622–e176224. doi:10.1371/journal.pone.0176162
Tang, L., Deng, Y., Gao, M., Lin, X., Zhu, J., and Li, Y. (2017). Nrf-2 and HO-1 expression in medulloblastoma: A clinicopathological analysis. J. Biosci. Med. (Irvine). 05, 142–147. doi:10.4236/jbm.2017.53015
Tao, R., Murad, N., Xu, Z., Zhang, P., Okonechnikov, K., Kool, M., et al. (2019). MYC drives group 3 medulloblastoma through transformation of Sox2+ astrocyte progenitor cells. Cancer Res. 79, 1967–1980. doi:10.1158/0008-5472.CAN-18-1787
Tardito, S., Oudin, A., Ahmed, S. U., Fack, F., Keunen, O., Zheng, L., et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell. Biol. 17, 1556–1568. doi:10.1038/ncb3272
Tech, K., Deshmukh, M., and Gershon, T. R. (2015). Adaptations of energy metabolism during cerebellar neurogenesis are co-opted in medulloblastoma. Cancer Lett. 356, 268–272. doi:10.1016/j.canlet.2014.02.017
Tech, K., and Gershon, T. R. (2015). Energy metabolism in neurodevelopment and medulloblastoma. Transl. Pediatr. 4, 12–19. doi:10.3978/j.issn.2224-4336.2015.01.03
Tech, K., Tikunov, A. P., Farooq, H., Morrissy, A. S., Meidinger, J., Fish, T., et al. (2017). Pyruvate kinase inhibits proliferation during postnatal cerebellar neurogenesis and suppresses medulloblastoma formation. Cancer Res. 77, 3217–3230. doi:10.1158/0008-5472.CAN-16-3304
Valvona, C. J., and Fillmore, H. L. (2018). Oxamate, but not selective targeting of LDH-A, inhibits medulloblastoma cell glycolysis, growth and motility. Brain Sci. 8, E56. doi:10.3390/brainsci8040056
Van Ommeren, R., Garzia, L., Holgado, B. L., Ramaswamy, V., and Taylor, M. D. (2020). The molecular biology of medulloblastoma metastasis. Brain Pathol. 30, 691–702. doi:10.1111/bpa.12811
Velletri, T., Romeo, F., Tucci, P., Peschiaroli, A., Annicchiarico-Petruzzelli, M., Niklison-Chirou, M. V., et al. (2013). GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell. Cycle 12, 3564–3573. doi:10.4161/cc.26771
Veneroni, L., Boschetti, L., Barretta, F., Clerici, C. A., Simonetti, F., Schiavello, E., et al. (2017). Quality of life in long-term survivors treated for metastatic medulloblastoma with a hyperfractionated accelerated radiotherapy (HART) strategy. Child’s Nerv. Syst. 33, 1969–1976. doi:10.1007/s00381-017-3548-1
Wang, Q., Hu, B., Hu, X., Kim, H., Squatrito, M., Scarpace, L., et al. (2017). Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 32, 42–56. doi:10.1016/J.CCELL.2017.06.003
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309–314. doi:10.1126/science.123.3191.309
Wilson, M., Gill, S. K., MacPherson, L., English, M., Arvanitis, T. N., and Peet, A. C. (2014). Noninvasive detection of glutamate predicts survival in pediatric medulloblastoma. Clin. Cancer Res. 20, 4532–4539. doi:10.1158/1078-0432.CCR-13-2320
Xin, Y., Yue, X., Li, H., Li, Z., Cai, H., Choudhary, A. K., et al. (2020). PET imaging of medulloblastoma with an 18F-labeled tryptophan analogue in a transgenic mouse model. Sci. Rep. 10, 3800–3808. doi:10.1038/s41598-020-60728-6
Xiong, Y., Zhang, Y., Xiong, S., and Williams-Villalobo, A. E. (2020). A glance of p53 functions in brain development, neural stem cells, and brain cancer. Biol. (Basel) 9, 2855–E313. doi:10.3390/BIOLOGY9090285
Yoo, H. C., Yu, Y. C., Sung, Y., and Han, J. M. (2020). Glutamine reliance in cell metabolism. Exp. Mol. Med. 52, 1496–1516. doi:10.1038/s12276-020-00504-8
Zhang, J., Pavlova, N. N., and Thompson, C. B. (2017). Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 36, 1302–1315. doi:10.15252/embj.201696151
Zhang, L., He, X., Liu, X., Zhang, F., Huang, L. F., Potter, A. S., et al. (2019). Single-cell transcriptomics in medulloblastoma reveals tumor-initiating progenitors and oncogenic cascades during tumorigenesis and relapse. Cancer Cell. 36, 302–318. e7. doi:10.1016/j.ccell.2019.07.009
Zhao, F., Ming, J., Zhou, Y., and Fan, L. (2016). Inhibition of Glut1 by WZB117 sensitizes radioresistant breast cancer cells to irradiation. Cancer Chemother. Pharmacol. 77, 963–972. doi:10.1007/S00280-016-3007-9
Zhao, J. Y., Feng, K. R., Wang, F., Zhang, J. W., Cheng, J. F., Lin, G. Q., et al. (2021). A retrospective overview of PHGDH and its inhibitors for regulating cancer metabolism. Eur. J. Med. Chem. 217, 113379. doi:10.1016/J.EJMECH.2021.113379
Zhukova, N., Ramaswamy, V., Remke, M., Pfaff, E., Shih, D. J. H., Martin, D. C., et al. (2013). Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 31, 2927–2935. doi:10.1200/JCO.2012.48.5052
Zitterbart, K., Zavrelova, I., Kadlecova, J., Spesna, R., Kratochvilova, A., Pavelka, Z., et al. (2007). p73 expression in medulloblastoma: TAp73/DeltaNp73 transcript detection and possible association of p73alpha/DeltaNp73 immunoreactivity with survival. Acta Neuropathol. 114, 641–650. doi:10.1007/s00401-007-0298-2
Keywords: OXPHOS (oxidative phosphorylation), metabolism, warburg effect, glutamine/glutamate (GABA) cycle, ROS
Citation: Marabitti V, Giansanti M, De Mitri F, Gatto F, Mastronuzzi A and Nazio F (2022) Pathological implications of metabolic reprogramming and its therapeutic potential in medulloblastoma. Front. Cell Dev. Biol. 10:1007641. doi: 10.3389/fcell.2022.1007641
Received: 30 July 2022; Accepted: 05 October 2022;
Published: 19 October 2022.
Edited by:
Manuela Ceccarelli, National Research Council (CNR), ItalyReviewed by:
Timothy Gershon, University of North Carolina at Chapel Hill, United StatesCopyright © 2022 Marabitti, Giansanti, De Mitri, Gatto, Mastronuzzi and Nazio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Nazio, ZnJhbmNlc2NhLm5hemlvQG9wYmcubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.