 Hongli Li
Hongli Li Celien Lismont
Celien Lismont Iulia Revenco
Iulia Revenco Mohamed A. F. Hussein
Mohamed A. F. Hussein Cláudio F. Costa
Cláudio F. Costa Marc Fransen
Marc Fransen- 1Laboratory of Peroxisome Biology and Intracellular Communication, Department of Cellular and Molecular Medicine, KU Leuven, Leuven, Belgium
- 2Department of Biochemistry, Faculty of Pharmacy, Assiut University, Asyut, Egypt
Peroxisomes harbor numerous enzymes that can produce or degrade hydrogen peroxide (H2O2). Depending on its local concentration and environment, this oxidant can function as a redox signaling molecule or cause stochastic oxidative damage. Currently, it is well-accepted that dysfunctional peroxisomes are selectively removed by the autophagy-lysosome pathway. This process, known as “pexophagy,” may serve a protective role in curbing peroxisome-derived oxidative stress. Peroxisomes also have the intrinsic ability to mediate and modulate H2O2-driven processes, including (selective) autophagy. However, the molecular mechanisms underlying these phenomena are multifaceted and have only recently begun to receive the attention they deserve. This review provides a comprehensive overview of what is known about the bidirectional relationship between peroxisomal H2O2 metabolism and (selective) autophagy. After introducing the general concepts of (selective) autophagy, we critically examine the emerging roles of H2O2 as one of the key modulators of the lysosome-dependent catabolic program. In addition, we explore possible relationships among peroxisome functioning, cellular H2O2 levels, and autophagic signaling in health and disease. Finally, we highlight the most important challenges that need to be tackled to understand how alterations in peroxisomal H2O2 metabolism contribute to autophagy-related disorders.
Introduction
Autophagy is a conserved catabolic program for the degradation of cytoplasmic components (e.g., dysfunctional organelles, protein aggregates, and non-specific portions of the cytoplasm) within the lysosome (Kaur and Debnath, 2015). Dysregulation of this process has been linked to pathologies such as neurodegeneration, cancer, and diabetes (Kenific and Debnath, 2015; Fang et al., 2019; Muralidharan et al., 2021). Depending on the delivery route of the cytoplasmic material to the lysosome interior, three primary types of autophagy have been recognized in mammalian cells: microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA) (Yim and Mizushima, 2020).
Proteins degraded by CMA contain a KFERQ-like motif that binds to HSC70, a cytosolic chaperone that delivers its cargo to the lysosomal surface for internalization and rapid degradation (Kaushik and Cuervo, 2018) (all protein acronyms are annotated as in the UniProtKB database and the full names can be retrieved in the Glossary). Approximately 30% of all soluble cytosolic proteins contain such a CMA-targeting motif (Chiang et al., 1989), and-after binding of the substrate-chaperone complex to the transmembrane protein LAMP2A (Cuervo and Dice, 2000)—the substrate proteins need to unfold before they cross the lysosomal membrane (Rout et al., 2014). These processes occur in cooperation with a set of cochaperones (Rout et al., 2014; Kaushik and Cuervo, 2018).
In microautophagy, the lysosomal or endosomal membrane is deformed to directly engulf cytosolic materials (Yim and Mizushima, 2020). At the morphological level, cargo can be engulfed through lysosomal protrusion, lysosomal invagination, or endosomal invagination (Oku and Sakai, 2018). At the mechanistic level, many aspects remain to be clarified (Mejlvang et al., 2018; Mesquita et al., 2021). However, this degradation route requires neither LAMP2A nor substrate unfolding (Yim and Mizushima, 2020).
Unlike the former two kinds of autophagy, macroautophagy involves the sequestration of cargo at a distinct site from lysosomes. During this process, a double-membrane structure (the phagophore) is formed (Klionsky et al., 2021). This structure wraps around the cytoplasmic target and creates, upon closure, a separate compartment (the autophagosome). This short-lived organelle subsequently fuses with lysosomes to deliver its content for degradation (Kriegenburg et al., 2018). Given that macroautophagy, hereafter referred to as “autophagy,” is widely recognized as the major removal pathway for organelles in mammalian cells (Klionsky et al., 2021), we focus here on the double-sided redox connection between peroxisomes and this type of autophagy: on one hand, as a major site of intracellular H2O2 metabolism, peroxisomes have the potential to turn on and tune autophagy, and this process may eliminate dysfunctional peroxisomes and protect the cell from oxidative damage; on the other hand, chronic impairment of peroxisome function may lead to an accumulation of H2O2 levels that inhibit autophagy, thereby driving a vicious cycle between peroxisome malfunction and cellular redox imbalance.
The Core Autophagy Machinery
The canonical autophagy process can be divided into multiple stages, including induction, phagophore nucleation and elongation, cargo sequestration, phagophore closure, autophagosome transport, and cargo degradation via fusion with lysosomes (Figure 1A) (Parzych and Klionsky, 2014; Yim and Mizushima, 2020; Melia et al., 2020). Many of these processes are executed by a dedicated cohort of autophagy-related (ATG) proteins (Feng et al., 2014). Here, we mainly focus on the molecular players involved in phagophore biogenesis (Figure 1B) (Kawabata and Yoshimori, 2020).
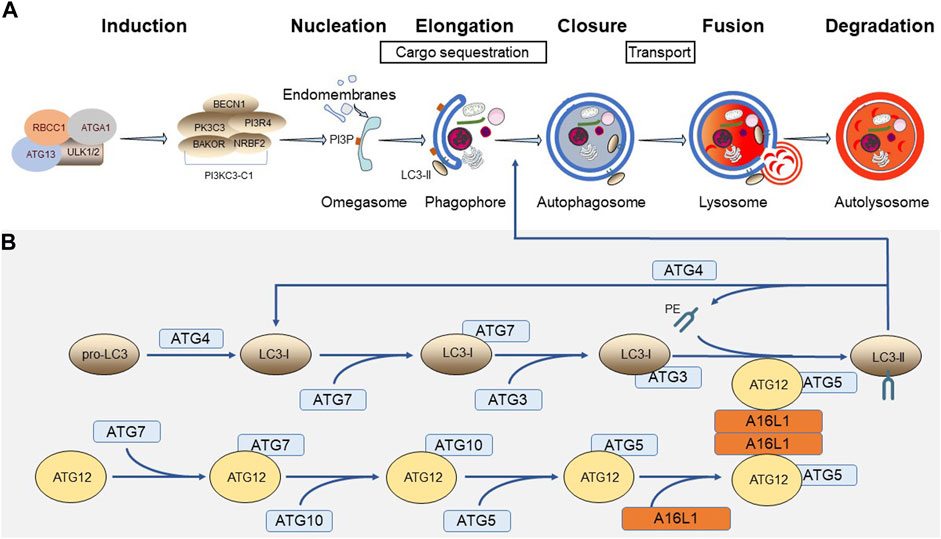
FIGURE 1. Distinct steps and core components of autophagy. (A) Different stages of canonical autophagy. (B) Ubiquitin-like conjugation systems involved in phagophore biogenesis.
The phagophore (or isolation membrane) is a small cup-shaped pre-autophagosomal structure that can originate from endomembranes (e.g., the ER membrane) containing phosphatidylinositol 3-phosphate (PI3P)-enriched subdomains, often referred to as omegasomes (Roberts and Ktistakis, 2013). PI3P is a phospholipid that is mainly generated by the class III phosphatidylinositol 3-kinase complex I (PI3KC3-C1), which is composed of a catalytic subunit (PK3C3), a PI3-kinase regulatory subunit (PI3R4), an allosteric modulator (BECN1), another regulator (NRBF2), and a phagophore targeting subunit (BAKOR) (Hurley and Young, 2017). Activation of the PI3KC3-C1 complex requires the serine/threonine protein kinase complex ULK. This complex, which integrates upstream nutrient and energy signals to coordinate autophagy induction, is made up of either ULK1 or ULK2 and 3 non-catalytic subunits (ATG13, RBCC1, and ATGA1) (Hurley and Young, 2017). Upon induction of autophagy, the ULK1 complex becomes active and translocates to the omegasome, where it exerts its function on PI3KC3-C1 (Mercer et al., 2018).
Elongation of the phagophore requires the concerted action of two ubiquitin-like conjugation systems. Here, it is worth noting that the ATG protein family includes two ubiquitin-like protein members, ATG8/MLP3/GBRL (here denoted as LC3) and ATG12 (Mohan and Wollert, 2018). Following translation, the pro-LC3 proteins first need to be cleaved into their LC3-I counterparts to expose a glycine residue at their C-terminus. This event, which is catalyzed by the cysteine protease ATG4, is necessary to become a substrate for ATG7, an E1-like enzyme that can activate both LC3-I and ATG12. ATG7 in turn transfers its activated ubiquitin-like substrates to ATG3 (in case of LC3-I) or ATG10 (in case of ATG12), two E2-like enzymes. Next, ATG12 is transferred onto ATG5 to form a complex with E3-like activity that, upon interaction with the A16L1 dimer (Lystad et al., 2019), is targeted to the autophagosomal membrane, where it stimulates the ATG3-mediated conjugation of LC3-I to phosphatidylethanolamine (PE) (Martens and Fracchiolla, 2020). This lipidated form of LC3, termed LC3-II, is the active form of LC3 and plays an important role in phagophore membrane expansion, cargo selection, and membrane closure (Li and Zhang, 2019). Note that ATG4 can also delipidate LC3-II to release this molecule from the autophagosomal membrane for reuse (Nakatogawa, 2013).
The closed autophagosome needs to be transported to and fused with the lysosome for digestion. These processes require cytoskeletal filaments, motor proteins, RABs, SNAREs, tethering factors, and lysosomal hydrolases. For a detailed overview of these factors, which are not further elaborated on here, the reader is referred to other dedicated reviews (Nakamura and Yoshimori, 2017; Yim and Mizushima, 2020; Zhao et al., 2021).
Cargo Receptors for Selective Autophagy
Depending on the nature of the substrate, autophagy can be classified into selective or non-selective (bulk) autophagy (Lamark and Johansen, 2021). In bulk autophagy, portions of the cytoplasm are randomly sequestered, degraded, and recycled to compensate for nutrient deficiencies. Selective autophagy, however, rather serves to eliminate functionally redundant or damaged cytoplasmic components, which are selectively sequestered and degraded as a stress response or quality control mechanism. Examples of selective autophagy processes in mammalian cells include aggrephagy (protein aggregates), ER-phagy (ER), ferritinophagy (ferritin), glycophagy (glycogen), lipophagy (lipid droplets), lysophagy (lysosomes), mitophagy (mitochondria), nucleophagy (nuclear fragments), pexophagy (peroxisomes), ribophagy (ribosomes), xenophagy (bacteria and viruses), and zymophagy (zymogen granules) (Gatica et al., 2018; Gubas and Dikic, 2021).
Both selective and bulk autophagy utilize the same core autophagy machinery. However, selective autophagy pathways require the additional action of specific autophagy receptors (SARs) including, among others, BNIP3, BNI3L, CACO2, FUND1, NBR1, OPTN, RETR1, and SQSTM (Figure 2A) (Gubas and Dikic, 2021). SARs can act either independently or cooperatively to bridge substrates to phagophores (Kirkin et al., 2009; Cemma et al., 2011). Therefore, they possess a cargo-binding domain as well as an LC3-interacting region. Given that autophagic substrates are frequently ubiquitinated, the cargo-binding domain is often a ubiquitin-binding domain (Kirkin and Rogov, 2019). Specific examples of such canonical SARs include SQSTM, NBR1, OPTN, and CACO2 (Kim et al., 2016). Importantly, the specificity, activity, and stability of most SARs are controlled by a diverse range of post-translational modifications (e.g., phosphorylation, ubiquitination, acetylation) and structural changes (e.g., oligomerization) (Gubas and Dikic, 2021), which may vary within different cell types or under specific environmental conditions.
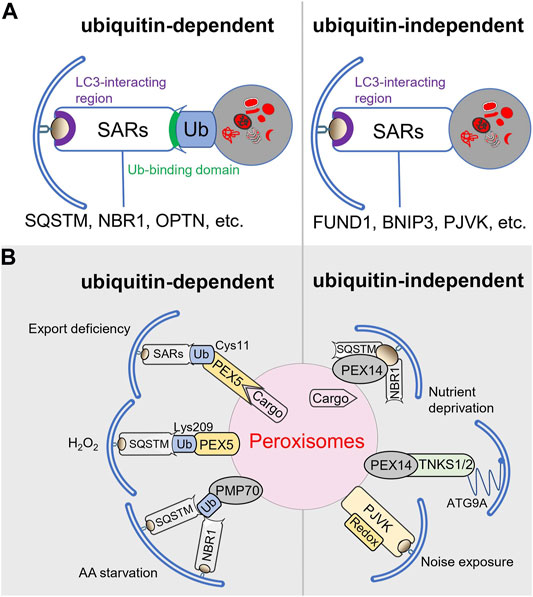
FIGURE 2. Selective autophagy pathways and pexophagy receptors. (A) Types of selective autophagy. (B) Cargo receptors involved in selective pexophagy.
Cargo Receptors for Pexophagy
To maintain peroxisome functionality, the superfluous and dysfunctional organelles need to be selectively removed through activation of a specialized form of autophagy, called pexophagy (Nordgren et al., 2013). Currently, there is good evidence that, in mammalian cells, pexophagy can occur through both ubiquitin-dependent and -independent mechanisms (Figure 2B) (Li and Wang, 2021).
Evidence that ubiquitin can indeed function as a self-removal signal for peroxisomes was obtained from an elegant proof-of-concept study showing that heterologous expression of peroxisomal membrane protein (PMPs)-ubiquitin fusion proteins in COS-7 or HeLa cells triggered pexophagy on condition that the ubiquitin moiety was facing the cytosol (Kim et al., 2008). Currently, the autophagy receptors SQSTM and NBR1 are both recognized to participate in this process (Kim et al., 2008; Deosaran et al., 2013; Germain and Kim, 2020). However, given that 1) exogenous expression of NBR1, but not SQSTM, promotes peroxisome clustering and lysosome targeting, 2) SQSTM increases the efficiency of NBR1-mediated pexophagy, and 3) SQSTM is not required for pexophagy upon NBR1 overexpression, their precise roles may differ (Deosaran et al., 2013). In addition, it cannot be ruled out that the relative contribution of NBR1 and SQSTM (or any other SAR) to pexophagy varies depending on the initial stimulus. In this context, it is worthwhile noting that in oxidative stress-induced pexophagy the contribution of SQSTM appears to be more important than that of NBR1 (Zhang et al., 2013; Jo et al., 2020b). Also, for a long time, it was unclear which endogenously ubiquitinated protein was serving as a prime recruitment factor for the SARs involved in pexophagy. Currently, it is common knowledge that PEX5, the cycling import receptor for peroxisomal matrix proteins, plays an active role in this process (Subramani, 2015; Li and Wang, 2021). This association is mainly based on the combined observations that 1) after delivery of its cargo into the peroxisome lumen, the protein is monoubiquitinated on a conserved cysteine residue (Cys11 in human PEX5) in order to be extracted from the peroxisomal membrane by the receptor export machinery (Carvalho et al., 2007; Francisco et al., 2017), 2) peroxisome-associated PEX5 can also be ubiquitinated at Lys209 in response to H2O2 treatment (Zhang et al., 2015), and 3) conditions resulting in an accumulation of (mono)ubiquitinated PEX5 on the peroxisomal membrane trigger peroxisome removal (Nordgren et al., 2015; Zhang et al., 2015; Lee et al., 2018). Importantly, these findings do not exclude that other ubiquitinated peroxisome-associated proteins may also be involved. Here, it should be noted that, besides peroxisome-associated PEX5, also PMP70 is ubiquitinated during amino acid starvation (Sargent et al., 2016). In addition, it is currently accepted that, under basal conditions, the ubiquitination state of PMPs is maintained at a low level by the peroxisome-associated pool of USP30, a ubiquitin-specific protease (Marcassa et al., 2018; Riccio et al., 2019).
Pexophagy can also take place in a ubiquitin-independent manner. Indeed, LC3-II can be recruited to the peroxisomal membrane through direct or indirect interactions with PEX14 or PJVK (Hara-Kuge and Fujiki, 2008; Jiang et al., 2015a; Li et al., 2017; Defourny et al., 2019). PEX14 is a peroxisomal membrane protein that normally functions as a docking factor for cargo-loaded PEX5 (Fransen et al., 1998), and PJVK is a redox-sensitive peroxisome-associated protein involved in sound-induced peroxisome proliferation in auditory hair cells (Defourny et al., 2019). Under conditions of starvation, where PEX5 is mostly cargo-unloaded, PEX14 can interact with LC3-II, thereby promoting peroxisome degradation (Hara-Kuge and Fujiki, 2008). In a follow-up study, the same authors reported that, upon starvation, PEX14 can also interact with NBR1 and SQSTM1, thereby (most likely) triggering conformational changes in PEX14 and increasing its affinity for LC3-II (Jiang et al., 2015b). Finally, large-scale protein-protein interaction studies have uncovered that PEX14 can also interact with TNKS1 and TNKS2, which were subsequently demonstrated to localize to peroxisomes and promote pexophagy under amino acid starvation conditions (Li et al., 2017). Given that TNKS1 and TNKS2 can also interact with ATG9A, an autophagosomal protein that promotes phagophore membrane growth (Nishimura and Tooze, 2020), it was suggested that the PEX14-TNKS/2-ATG9A complex may function as a non-canonical pexophagy receptor upon nutrient starvation (Li et al., 2017).
Triggers and Signaling Pathways Controlling (Selective) Autophagy
Autophagy serves a critical role in stress response and quality control networks, and imbalances in this process have been recognized as an important contributor to disease states such as neurodegeneration, cardiac ischemia-reperfusion, liver disease, Crohn’s disease, and infections (Murrow and Debnath, 2013). Major cellular stresses that can be linked to autophagy include nutrient and growth factor deprivation, hypoxia, ER stress, DNA damage, and oxidative stress. In the following subsections, we briefly outline how each of these factors can promote autophagy. For more detailed information, we refer the reader to other reviews.
Nutrient Deprivation
The basic processes underlying autophagy are controlled by complex signaling pathways. Key regulators include the mechanistic target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AAPK), two kinases whose activities respectively inhibit and stimulate autophagy through a coordinated phosphorylation of ULK1 (Egan et al., 2011; Kim et al., 2011): mTORC1 integrates signals from growth factors, nutrients, oxygen levels, and energy status; and AAPK is a sensor and regulator of cellular energy status (Alers et al., 2012). Both mTORC1 and AAPK are master regulators of cell metabolism, thereby linking autophagy to this process (Murrow and Debnath, 2013; Deleyto-Seldas and Efeyan, 2021). However, their actions are in general antagonistic. For example, nutrient deprivation, a condition well-known to induce autophagy, inhibits and stimulates the activities of mTORC1 and AAPK, respectively (Russell et al., 2014). A starvation-induced activation of AAPK also results in phosphorylation and activation of TSC2, a GTPase activating protein that functions as a key negative regulator of mTORC1 through the TSC complex-RHEB signaling axis (Inoki et al., 2003).
Hypoxia
Hypoxia, a condition in which oxygen availability is limited, is a significant contributor to cell damage in many acute (e.g., ischemic stroke) and chronic (e.g., pulmonary hypertension) disease processes (Lee et al., 2019). Hypoxia can induce autophagy through activation of multiple oxygen-sensitive signaling pathways (Fang et al., 2015). One such pathway involves hypoxia-inducible factors (HIFs). Under normoxic conditions, transcription factors belonging to the HIF protein family are rapidly degraded by the ubiquitin-proteasome system. However, under hypoxic conditions, these proteins are stabilized and translocated to the nucleus to initiate the transcription of genes involved in cellular adaptation and survival, including a set of genes essential for autophagy (e.g., ATG5, ATG7, ATG9A, BECN1, BNIP3, and BNI3L) (Daskalaki et al., 2018). In addition, HIF1 can act as regulator of autophagy by altering the expression levels of genes involved in glucose metabolism (Kierans and Taylor, 2020). Finally, autophagy can also be induced in a HIF-independent manner through hypoxic stress-induced activation of 1) the MK08 signaling pathway (Frazier et al., 2007), 2) the AAPK/TSC2 pathway (Papandreaou et al., 2008), or 3) the unfolded protein response in the ER (Rouschop et al., 2010; Yang et al., 2019). Note that, under normoxic conditions, ER stress can lead to either autophagy stimulation or inhibition (Rashid et al., 2015).
DNA Integrity
A third factor that can induce autophagy is diminished DNA integrity caused by, for example, UV-sunlight or metabolically-derived reactive oxygen species (ROS) (Juretschke and Beli, 2021). Such insults trigger a set of DNA damage response signaling pathways that lead to activation of PARP1, FOXO3, ATM, and P53: 1) PARP1 is a predominantly nuclear enzyme that converts NAD+ into poly(ADP-ribose), and hyperactivation of this enzyme causes NAD+ and ATP depletion, a condition promoting AAPK-mediated autophagy activation (Czarny et al., 2015); 2) FOXO3 is a transcription factor known to control the expression levels of multiple autophagy-related genes, including MLP3B and BNIP3, and binding of FOXO3 to the protein kinase ATM triggers autophosphorylation and activation of the latter protein (Rodriguez-Rocha et al., 2011); 3) activation of ATM triggers the initiation of a phosphorylation cascade that regulates the activity of various downstream targets, including AAPK and P53 (Rodriguez-Rocha et al., 2011); and 4) phosphorylation of P53, a multifunctional transcription factor, results in a transcriptional upregulation of TSC2 and PTEN, a negative regulator of PI3K signaling (Rodriguez-Rocha et al., 2011). In the end, all these events contribute to suppression of mTORC1 activity.
Oxidative Stress
Nutrient and growth factor deprivation, hypoxia, ER stress, and DNA damage can all be linked to perturbations in the cellular redox balance, another autophagy-modulating factor (Li et al., 2015; Sedlackova and Korolchuk, 2020). Indeed, physiologically relevant oxidants such as H2O2 can oxidatively modify proteins that are directly or indirectly involved in autophagy regulation and execution, thereby potentially effecting their localization, binding affinities, and/or activities (Sedlackova and Korolchuk, 2020). For example, exposure of cells to H2O2 can 1) upregulate the transcriptional expression of BECN, BNIP3, BNI3L, MLP3C, and SQSTM through activation of HIF1, P53, FOXO3, NFKB, and NF2L2 (Li et al., 2015), 2) activate PI3K signaling through inactivation of PTEN (Koundouros and Poulogiannis, 2018), 3) suppress mTORC1 activity through activation of ATM and AAPK (Wible and Bratton, 2018), and 4) oxidize and inhibit ATG4 (Wible and Bratton, 2018). For the underlying molecular mechanisms and physiological consequences, we refer the reader to Redox Regulation of Autophagy. Importantly, although it is generally thought that oxidative stress always induces autophagy as part of a cellular safeguard mechanism to limit oxidative injury, also other critical factors (e.g., the amount of ROS, nutrient availability, etc.) determine whether autophagy is effectively induced or suppressed under conditions of oxidative stress (Kma and Baruah, 2021).
Triggers and Signaling Pathways Controlling Pexophagy
Over the past decade, multiple studies have shown that pexophagy can be triggered by various stress stimuli including amino acid depletion (Li et al., 2017; Dutta et al., 2021), oxidative stress (Zhang et al., 2013; Jo et al., 2015; Zhang et al., 2015; Tripathi et al., 2016; Lee et al., 2018; Defourny et al., 2019; Jo et al., 2020b; Daussy et al., 2020; Dutta et al., 2021), hypoxia (Walter et al., 2014; Mu et al., 2020), viral infection (Daussy et al., 2020), and dysfunctional peroxisome biogenesis (Yamashita et al., 2014; Nordgren et al., 2015; Dahabieh et al., 2021; Wei et al., 2021). In the following subsections, these triggers will be discussed in more detail (Figure 3).
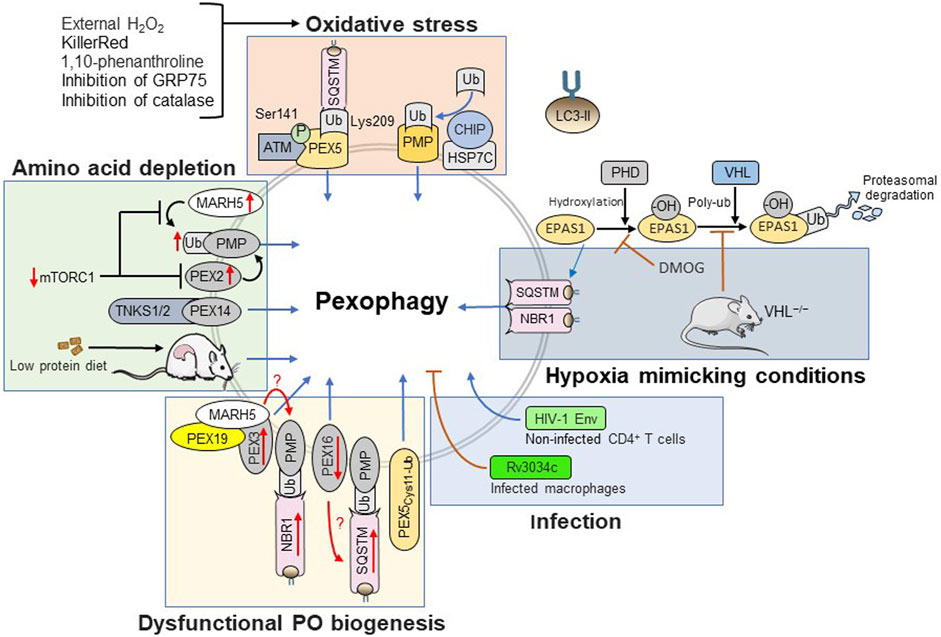
FIGURE 3. Triggers and signaling pathways controlling pexophagy.
Amino Acid Starvation
Free amino acids play a vital role in cellular metabolism. Examples include but are not limited to protein synthesis and energy metabolism. Amino acids activate mTORC1 signaling and amino acid starvation suppresses this pathway, thereby inducing autophagy and pexophagy (Germain and Kim, 2020; Deretic and Kroemer, 2021). On one hand, starvation-induced pexophagy can be linked to PEX2, a peroxisomal membrane-associated E3 ubiquitin ligase that is rapidly degraded by the proteasome under basal conditions (Sargent et al., 2016). Upon inhibition of mTORC1 (e.g., during amino acid starvation or upon rapamycin treatment), PEX2 is stabilized and promotes PMP ubiquitination, thereby triggering pexophagy (Sargent et al., 2016). On the other hand, starvation-induced pexophagy can be further enhanced by a PEX14-dependent recruitment of TNKS1/2 onto the peroxisomal membrane (Li et al., 2017). The in vivo observation that hepatic peroxisome content is dramatically decreased in rats fed with a low protein diet (Van Zutphen et al., 2014) is in line with the idea that there is indeed a causal link between amino acid starvation and pexophagy. However, direct effectors contributing to pexophagy upon mTORC1 inhibition remain to be identified. Nevertheless, in this regard, it must be noted that a very recent study demonstrated that 1) MARH5, an E3 ubiquitin ligase promoting mitophagy, can also be recruited to the peroxisomal membrane through interaction with the PMP import receptor PEX19 and its membrane docking protein PEX3, 2) recruitment of this E3 ligase to the peroxisomal membrane results in the ubiquitination of PMP70, and 3) MARH5 is playing an important role in mTORC1 inhibition-mediated pexophagy (Zheng et al., 2022). In addition, it is known that prolonged nutrient deprivation causes a significant decrease and increase in the intracellular GSH (Desideri et al., 2012) and H2O2 levels (Scherz-Shouval et al., 2007b; Chen et al., 2009), respectively.
Oxidative Stress
Over the last decade, multiple studies have suggested a strong link between oxidative stress and pexophagy, both at the cellular and organismal level. Examples include treatment of Chang liver cells with the chemical 1,10-phenanthroline (Jo et al., 2015), addition of H2O2 to HepG2 or HEK-293 cells (Zhang et al., 2013), treatment of mouse embryonic fibroblasts with 3-methyladenine (Ivashchenko et al., 2011), loss of GRP75 in neuroblastoma cells (Jo et al., 2020b), peroxisomal KillerRed-mediated ROS production in NIH/3T3 cells (Chen et al., 2020), suppression of catalase expression or activity in serum-starved HepG2 or RPE1 cells (Lee et al., 2018), and prolonged fasting of catalase-deficient mice (Dutta et al., 2021). Note that catalase is a major peroxisomal matrix protein that catalyzes the degradation of H2O2. Although the underlying molecular details have not yet been fully elucidated, different types of oxidative stressors can induce pexophagy through distinct mechanisms. For example, while it has been claimed that external H2O2 triggers pexophagy through activation of ATM (Zhang et al., 2015; Tripathi et al., 2016), this kinase appears to be dispensable for the removal of peroxisomes that are oxidatively damaged by activation of peroxisomal KillerRed (Chen et al., 2020). Here, it is important to point out that 1) activation of ATM not only suppresses mTORC1 activity (Wible and Bratton, 2018), but also triggers phosphorylation of PEX5 at Ser 141, an event that subsequently results in its ubiquitination at Lys 209 and the recruitment of SQSTM to the peroxisomal membrane (Zhang et al., 2015; Tripathi et al., 2016), and 2) intraperoxisomal ROS production by KillerRed triggers a HSP7C-mediated recruitment of CHIP, a ubiquitin E3 ligase, onto oxidatively stressed peroxisomes, thereby promoting their selective ubiquitination and autophagic degradation (Chen et al., 2020). Finally, it is worth noting that pexophagy in response to oxidative stress is no mammalian-specific phenomenon. For example, it has been reported that exposure of plant leaves to cadmium induces peroxisomal glycolate oxidase activity (McCarthy et al., 2001), which in turn leads to increased intraperoxisomal H2O2 levels and pexophagy induction (Calero and Muñoz et al., 2019).
Hypoxia
Given that peroxisomal respiration can be responsible for up to 20% of the oxygen consumption in tissues such a liver (De Duve and Baudhuin, 1966), it may not come as a surprise that limited oxygen availability (e.g., as a consequence of inadequate vascular networks in solid tumors) and hypoxia mimicking conditions (e.g., upon treatment of cells with HIF prolyl hydroxylase inhibitors such as DMOG) have been found to trigger pexophagy in an EPAS1-dependent manner, at least in certain cell types (Walter et al., 2014; Schönenberger et al., 2015; Mu et al., 2020). EPAS1 is a hypoxia-inducible transcription factor that, under normoxic conditions, is rapidly targeted for proteasomal degradation through hydroxylation by oxygen-sensing prolyl hydroxylases and subsequent recruitment of the von Hippel-Lindau (VHL) ubiquitination complex (Påhlman and Mohlin, 2018). In vivo evidence suggested that hypoxia-induced pexophagy involves both NBR1 and SQSTM (Walter et al., 2014). However, although there is evidence that hypoxia can induce ROS formation (Paddenberg et al., 2003; Rathore et al., 2008), the precise mechanisms underlying EPAS1-mediated pexophagy are unclear. Intriguingly, another study reported that peroxisomes are intact and even essential for growth of K562 and HEK-293 cells under hypoxia (Jain et al., 2020). Importantly, these researchers also demonstrated that this phenotype, which was attributed to the organelle’s role in maintenance of membrane fluidity, strongly depended on the medium lipid content and cell seeding conditions (Jain et al., 2020). As such, these seemingly conflicting data may be explained by differences in experimental conditions.
Viral and Bacterial Infections
Two recent studies have shown that pexophagy can also be modulated by pathogenesis-related proteins. One study demonstrated that the human immunodeficiency virus type 1 (HIV1) envelope glycoprotein (Env) has the potential to induce pexophagy in non-infected bystander CD4+ T cells (Daussy et al., 2020). The authors also demonstrated that HIV1 Env can provoke excessive ROS production, a condition that eventually leads to apoptosis thereby very likely contributing to the acquired immunodeficiency syndrome in HIV1-infected patients (Daussy et al., 2020). The other study showed that, upon infection of macrophages, Mycobacterium tuberculosis Rv3034c, a putative acetyltransferase, can 1) suppress pexophagy through phosphorylation of mTORC1, an event associated with the down-regulation of pexophagy-associated proteins (e.g., ATG5, NBR1, and SQSTM), and 2) activate peroxisome proliferator activated receptor-γ, a transcription factor that initiates the transcription of peroxisome biogenesis (e.g., PEX3, PEX5, and PEX19) and proliferation (e.g., PEX11B, FIS1, and DNM1L) factors (Ganguli et al., 2020). These changes are likely to favor redox homeostasis, thereby allowing the parasite to avoid ROS-mediated killing (Ganguli et al., 2020). Once again, the precise underlying molecular mechanisms remain unclear.
Dysfunctional Peroxisome Biogenesis
Another emerging pexophagy trigger is dysregulated PMP or matrix protein import. For example, the peroxisomal membrane proteins PEX3 and PEX16 are essential for PMP assembly, and both overexpression of PEX3 (Yamashita et al., 2014) or silencing of PEX16 (Wei et al., 2021) have been shown to induce pexophagy in an NBR1-and SQSTM-dependent manner, respectively. In addition, conditions leading to an accumulation of (mono)ubiquitinated PEX5 on the peroxisomal membrane (Nordgren et al., 2015; Park et al., 2021b; Dahabieh et al., 2021) can also trigger pexophagy. Note that the latter observation strongly indicates that peroxisome-associated monoubiquitinated PEX5 acts as a key surveillance factor for selective elimination of peroxisomes with a defective PEX5 export machinery (Nordgren et al., 2015; Law et al., 2017; Nazarko, 2017).
Redox Regulation of Autophagy
Currently, it is widely accepted that autophagy represents a prime mechanism of protection against oxidative damage (Ornatowski et al., 2020; Yun et al., 2020). In addition, autophagic activity is governed by complex redox-mediated signaling pathways that, depending on the context, exert positive or negative regulatory activities at the transcriptional and/or protein level (Scherz-Shouval et al., 2007a; Park et al., 2021a; Redza-Dutordoir and Averill-Bates, 2021; Zhou et al., 2021). In the following subsections, we first briefly explain the mechanisms behind H2O2 signaling. Next, we elaborate further on how autophagic activity can be directly (e.g., through oxidative modification of autophagy-related proteins) or indirectly (e.g., through oxidative modification of transcription factors or signaling proteins) modulated by H2O2, the major ROS in redox regulation of biological activities (Lismont et al., 2019b; Sies and Jones, 2020).
The Concept of H2O2 Signaling
A main mechanism by which H2O2 achieves specificity as signaling molecule is through direct oxidation of thiolate groups (RS−) in target proteins (Sies and Jones, 2020). These groups can react with H2O2 to form sulfenic acid (RSOH), an intermediate in inter- or intramolecular disulfide bond formation that–in the presence of high H2O2 concentrations–can be further oxidized to sulfinic (RSO2H) or sulfonic (RSO3H) acid. The latter modification is irreversible and causes permanent oxidative damage. Disulfide bond formation can act as a molecular switch to regulate the activity, localization, and stability of redox-sensitive proteins. Importantly, protein thiols with a low reactivity towards H2O2 can also form disulfide bonds through a redox relay mechanism whereby thiol peroxidases shuttle oxidative equivalents from H2O2 to other target proteins (Stöcker et al., 2018).
H2O2 as a Modulator of ATG Activity
Accumulating evidence points to H2O2 as a potent modulator of ATG activity (Lizama-Manibusan and McLaughlin, 2013). For example, it has been demonstrated that the human cysteine proteases ATG4A and ATG4B are direct targets for oxidation by H2O2, thereby rendering them enzymatically inactive through formation of inter- or intramolecular disulfide bridges (Scherz-Shouval et al., 2007b; Zheng et al., 2020). Given the dual function of ATG4 as pro-LC3 cleavage and LC3-II delipidating enzyme (Figure 1), these activities need to be tightly controlled to ensure LC3-I lipidation and autophagy progression when cells are exposed to oxidative insults. To cope with this dual role, it has been proposed that 1) the oxidative modification of ATG4 is mainly taking place at autophagosomes in close vicinity to H2O2-generating platforms such as mitochondria (Scherz-Shouval et al., 2007b), and 2) the ATG4-dependent cleavage of pro-LC3 into their LC3-I counterparts is more efficient than LC3-II deconjugation (and thus less impacted by partial inhibition of ATG4 activity) (Wible and Bratton, 2018). Nevertheless, although disulfide-bonded ATG4 can be efficiently reduced by the thioredoxin system, it can be expected that harsh or long-term exposure to oxidative stress will eventually fully inhibit ATG4 activity, thereby rather blocking than inducing LC3 lipid conjugation and autophagy (Wible and Bratton, 2018). Besides ATG4, also ATG3, ATG7, and ATG10 have been demonstrated to be redox-sensitive (Filomeni et al., 2010; Frudd et al., 2018). Under basal conditions, ATG3 and ATG7 form inactive thioester-bonded complexes with LC3; upon stimulation of autophagy, ATG3 and ATG7 become active and dissociate from LC3, thereby freeing their catalytic thiols; and under oxidative stress conditions, the non-LC3-shielded thiols in ATG3 and ATG7 form intermolecular disulfide linkages, thereby preventing LC3 lipidation, autophagosome maturation, and autophagy (Frudd et al., 2018). In analogy, it can be expected that the catalytic cysteine of ATG10, another E2-like enzyme, displays a redox-sensitive behavior (Filomeni et al., 2010). However, despite the observation that ATG10 is sensitive to oxidation by H2O2, this has apparently no impact on the conjugation of ATG5 to ATG12, at least not under the conditions tested (Frudd et al., 2018).
H2O2 as a Modulator of SARs Activity
Another important redox-regulated protein in autophagy is the autophagy receptor SQSTM. This protein can undergo self-polymerization through intermolecular disulfide bond formation, thereby facilitating cargo selection and degradation through high-avidity binding to LC3-II on nascent autophagic membranes (Cha-Molstad et al., 2018). The formation of such disulfide-linked conjugates is promoted by oxidative stress conditions, thereby activating prosurvival autophagy (Carroll et al., 2018). Whether or not the other SARs have the capacity to form similar disulfide-linked complexes, remains to be established.
H2O2 as a Modulator of Transcriptional Autophagy Regulation
H2O2-induced posttranslational modifications can also modulate the stability, subcellular localization, and/or activity of many transcription factors (Marinho et al., 2014; Li et al., 2015). Here, we briefly summarize the main impact of H2O2 on HIF1A, P53, NF2L2, and FOXO, all of which have been implicated in autophagy regulation. For more details regarding the complex molecular mechanisms involved in the adaptive responses, we refer the reader to the references cited. HIF1A is the main driver of transcriptional responses to hypoxia, and H2O2-induced activation of HIF1A promotes the transcription of BNIP3 and BNI3L, thereby promoting selective mitophagy (Fan et al., 2019; Asgari et al., 2021). The tumor suppressor protein P53 can, depending on its intracellular location (e.g., cytoplasm versus nuclear) and the cellular environment (e.g., normal physiological conditions versus nutrient starvation or hypoxia), modulate autophagy at multiple levels and through diverse mechanisms (Hu et al., 2019). For example, nuclear-localized P53 can directly upregulate the expression levels of TSC2 and the β-scaffolding subunit of AAPK, thereby enhancing autophagy through inhibition of mTORC1 signaling; and cytoplasmic P53 can inhibit autophagosome formation through binding to RBCC1, an A16L1 interactor and component of the ULK complex (Hu et al., 2019). NF2L2 is a transcription factor that controls the expression of genes containing an antioxidant response element in their promoter, such as SQSTM (Puissant et al., 2012). H2O2 can enhance the expression, stability, and nuclear localization of NF2L2 through sequestration of oxidized KEAP1, a thiol-rich protein that promotes the continuous ubiquitin-mediated degradation of NF2L2 under basal conditions (Li et al., 2015; Yin et al., 2015). Finally, H2O2 can also activate FOXO transcription factors, which stimulate the transcription of LC3, BNIP3, and ATG12 (Sengupta et al., 2009; Li et al., 2015; Deng et al., 2021a).
H2O2 as a Modulator of Autophagy Signaling Pathways
Finally, H2O2 can also indirectly promote or inhibit autophagy via modulation of the AAPK, PI3K, and mitogen-activated protein kinase (MK) signaling pathways. Importantly, the outcome is context specific. For example, 1) exposure of HEK-293 cells to H2O2 results in S-glutathionylation of Cys299 and Cys304 (likely Cys297 and Cys302 in UniProt ID P54646) in AAPK2, a catalytic subunit of AAPK, and 2) these oxidative modifications stimulate AAPK activity through release of the autoinhibitory domain from its catalytic core, even under non-ATP depleting conditions (Zmijewski et al., 2010). On the other hand, oxidation of Cys130 and Cys174 has been reported to interfere with AAPK activity under energy starvation conditions, at least in mouse cardiomyocytes (Shao et al., 2014). For PI3K, it was shown that the α- and β-catalytic subunits respectively inhibit and promote autophagy in response to moderate and high levels of ROS (Kma and Baruah, 2021). The α-subunit inhibits autophagy through activation of AKT, a serine/threonine kinase that activates mTORC1 activity and arrests autophagic gene expression; and the β-subunit promotes autophagy through stimulating the activities of PI3KC3-C1 and FOXO (Kma and Baruah, 2021). High levels of ROS can also potentiate the PI3K/AKT pathway through inactivation of PTEN, a phosphatase that counteracts PI3K signaling through dephosphorylation of phosphatidylinositol (3,4,5)-trisphosphate to phosphatidylinositol (4,5)-bisphosphate (Koundouros and Poulogiannis, 2018).
MKs are a group of ROS-regulated serine-threonine protein kinases that play a role in diverse cellular processes (Son et al., 2011), including autophagy (Cagnol and Chambard, 2010; Zhou et al., 2015; He et al., 2018). This class of kinases can be grouped in three subclasses: the extracellular signal-regulated kinases, the c-jun N-terminal kinases, and the p38 kinases (Son et al., 2011; Sui et al., 2014). In general, these MKs can be activated by various oxidative stressors, including H2O2 (Son et al., 2011), and this subsequently triggers the initiation of complex signaling cascades that eventually modulate, among other processes, autophagic activity. For example, ROS-induced activation of the extracellular signal-regulated kinase pathway can induce adaptive and protective autophagy-associated responses in urinary protein-irritated renal tubular epithelial cells (Deng et al., 2021b); c-jun N-terminal kinase activation can enhance autophagy through 1) upregulation of LC3 (Sun et al., 2011) and DRAM1, a damage-regulated autophagy modulator (Lorin et al., 2010), and 2) the liberation of BECN1 from BCL2/B2CL1 (Zhou et al., 2011); and ROS-induced activation of p38 can induce the expression of various autophagy-related genes (McClung et al., 2010).
The Peroxisome-Autophagy Signaling Axes
Peroxisomes act as master regulators of cellular lipid and H2O2 metabolism (Van Veldhoven, 2010; Lismont et al., 2015), and emerging evidence hints changes in peroxisomal lipid or H2O2 metabolism have the potential to modulate autophagic activity (Figure 4). Specifically, peroxisomal β-oxidation-derived acetyl-CoA can downregulate autophagy by enhancing acetylation of the mTORC1 subunit RPTOR, a process driving mTORC1 activation (He et al., 2020); defects in peroxisomal β-oxidation can suppress autophagy through redox imbalances associated with an accumulation of very-long-chain fatty acids (VLCFAs) (Fourcade et al., 2015; Launay et al., 2015); and (peroxisome-derived) H2O2 can activate the peroxisomal pool of ATM (Tripathi et al., 2016), an event that enhances 1) autophagic flux through AAPK-TSC2-mediated suppression of mTORC1 activity, and 2) pexophagy through phosphorylation and subsequent ubiquitination of the peroxisome-associated pool of PEX5 (Zhang et al., 2013; Tripathi and Walker, 2016). At first sight, these findings appear somewhat paradoxical. However, this may highlight the complexity of the peroxisome-autophagy signaling axis and point to the importance of other factors. For example, it is well known that disturbances in peroxisomal fitness are intrinsically linked to mitochondrial redox imbalances (Fransen et al., 2017). In addition, we recently found that peroxisomes with a dysfunctional H2O2 metabolism are not necessarily predisposed to pexophagy, even though peroxisome-derived H2O2 has the potential to oxidize redox-sensitive cysteine residues in PEX5, PTEN, NFKB1, TF65, and FOXO3 (Lismont et al., 2019a), all proteins whose activities can be linked to pexophagy or autophagy regulation (Subramani, 2015; Füllgrabe et al., 2016; Koundouros and Poulogiannis, 2018). Importantly, given that H2O2 shows a Janus-faced effect on autophagy (see Redox Regulation of Autophagy), it remains to be investigated whether the observed oxidative modifications lead to autophagy stimulation or inhibition. In addition, it still must be clarified if and to which extent other redox-sensitive autophagy-related proteins can act as a target of peroxisome-derived H2O2.
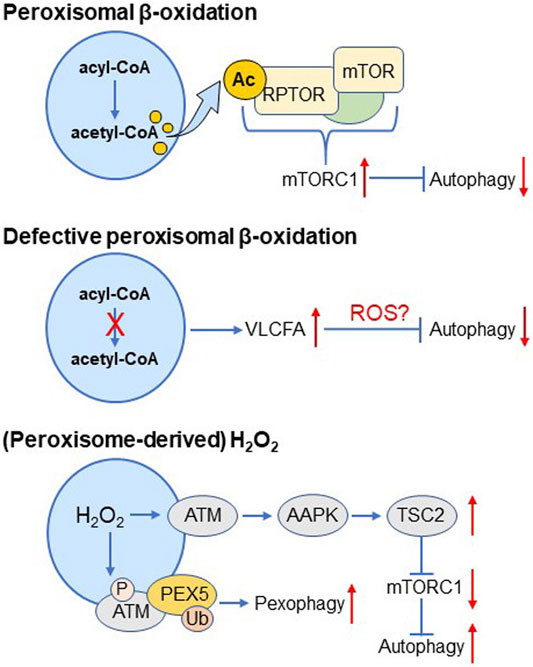
FIGURE 4. The peroxisome-autophagy signaling axes.
The Oxidative Stress-Pexophagy Signaling Axes
Oxidative stress is generally considered as one of the key mediators of cellular aging, a process that coincides with a decline in autophagic activity (Leidal et al., 2018) and a build-up of peroxisomes with a disturbed H2O2 metabolism (Legakis et al., 2002; Houri et al., 2020). Given that 1) in senescent cells, peroxisomes accumulate excessive amounts of PEX5 on their membranes (Legakis et al., 2002), 2) extraction of PEX5 from the peroxisomal membrane requires monoubiquitination of the protein at Cys11 (Carvalho et al., 2007), 3) Cys11 of human PEX5 functions as a redox switch that modulates the protein’s activity in response to intracellular oxidative stress (Apanasets et al., 2014), and 4) excessive peroxisomal H2O2 production decreases the intracellular levels of the (peroxisome-associated) PEX5Cys11-ubiquitin thioester conjugate (Lismont et al., 2019a), these findings point to an oxidative stress-induced decrease in PEX5-mediated pexophagy. On the other hand, dysfunctional autophagy/pexophagy will also lead to accumulation of SQSTM, a SAR linking autophagy and NF2L2 signaling through KEAP1 sequestration (Jiang et al., 2015a; Bartolini et al., 2018). This in turn enhances the expression of peroxisome proliferator-activated receptor-γ coactivator-1α, a protein whose expression is antioxidant response element-regulated (Gureev et al., 2019) and causes peroxisomal remodeling and biogenesis (Bagattin et al., 2010). As such, oxidative insults and defects in autophagy can lead to an accumulation of oxidatively burned-out peroxisomes, thereby fueling a vicious circle of oxidative injury (Vasko and Goligorsky, 2013).
Pexophagy and Disease
Pexophagy acts as a global regulator of peroxisome homeostasis and quality control and, as such, it does not come as a surprise that perturbations in this process have been linked to multiple disease conditions. Examples include the peroxisome biogenesis disorders (Nazarko, 2017), cancer (Walter et al., 2014; Dahabieh et al., 2018), lipopolysaccharide-induced acute kidney injury (Vasko, 2016), malnutrition-associated liver steatosis (van Zutphen et al., 2014), diabetes (Chu et al., 2020), noise-induced hearing loss (Defourny et al., 2019), HIV1 infections (Daussy et al., 2020), and neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease (Jo et al., 2020a). In the following paragraphs, we outline these examples in more detail.
In case of peroxisome biogenesis disorders, it was suggested that 1) in patients with mutations in genes coding for proteins constituting the ubiquitin-PEX5 export machinery, the disease phenotype is rather caused by excessive removal of peroxisomes than by defects in the peroxisomal matrix protein import machinery (Nazarko, 2017), and 2) low doses of autophagy inhibitors improve peroxisomal matrix protein import and peroxisome function without compromising cell viability (Law et al., 2017). Unfortunately, these findings could not be confirmed by others (Klouwer et al., 2021).
Both excessive and defective pexophagy have been linked to cancer (Dahabieh et al., 2018). For example, loss of peroxisomes due to enhanced pexophagy leads to metabolic alterations that have been suggested to promote a malignant phenotype in human clear cell renal cell carcinomas (Walter et al., 2014); and high expression levels of PEX6, PEX26 or MTOR, three negative regulators of pexophagy, have been associated with decreased patient survival in diffuse large B-cell lymphoma, lung cancer and melanoma cohorts. In the latter case, interference with the function of these proteins increased pexophagy and thwarted drug resistance in human melanoma and lymphoma cells (Dahabieh et al., 2021).
Pexophagy was also found to protect auditory hair cells against noise-induced oxidative damage (Defourny et al., 2019) and to attenuate lipopolysaccharide-induced acute kidney injury (Vasko et al., 2013; Vasko, 2016). In these conditions, the removal of dysfunctional peroxisomes may serve a quality control function to prevent ROS accumulation. On the other hand, enhanced peroxisome turnover because of HIV1 Env expression (Daussy et al., 2020) or mutations in GRP75 (Jo et al., 2020b) sensitize, respectively bystander CD4+ T lymphocytes and neuronal cells to oxidative injury, thereby potentially contributing to viral spreading and the progression of Parkinson’s disease. Finally, severe malnutrition-induced pexophagy contributes to hepatic mitochondrial dysfunction (Van Zutphen et al., 2014), and short-term inhibition of pexophagy benefits the health of pancreatic β-cells through elevation of ether phospholipid biosynthesis and by counteracting depletion of n-3 polyunsaturated fatty acids after fat-feeding (Chu et al., 2020).
Conclusion and Perspectives
Pexophagy is a complex cellular process that is tightly regulated at multiple levels and by distinct stimuli. The data presented in this review support the view that changes in the intracellular redox state have the potential to balance this process through activity modulation of autophagy-related proteins, transcription factors, kinases, phosphatases, and PEX5. An increasing number of studies started to examine the relationship between peroxisomal H2O2 emission and pexophagy, with a focus on the role of peroxisome-associated ubiquitin-PEX5. Major hurdles that have slowed down these studies include the lack of 1) easily accessible and reliable tools to monitor pexophagy in a dynamic manner, 2) compounds that rapidly and selectively trigger peroxisome degradation, and 3) study models that allow the modulation of peroxisomal H2O2 production in a time- and dose-dependent manner. Here, it is important to highlight that traditional platforms for studying pexophagy mainly focus on immunoblot and (immuno)cytochemistry analyses of key autophagy and peroxisome markers, which only provide a snapshot of a dynamic situation. In addition, unlike what is sometimes thought, amino acid starvation-induced pexophagy is a non-selective process, as also other kinds of cargo (e.g., portions of the cytosol, endoplasmic reticulum, and mitochondria) are sequestered during this type of “metabolic” autophagy (Deretic and Kroemer, 2021; and references therein). Furthermore, although there is mounting evidence that disturbances in peroxisomal H2O2 metabolism can trigger pexophagy, some frequently cited key experiments have been carried out by treating cells with external H2O2 (Zhang et al., 2013), a condition incomparable with intraperoxisomal H2O2 production (Lismont et al., 2021). As such, the recent development of 1) a genetically modified human cell line in which the intraperoxisomal production of H2O2 can be selectively modulated in a dose- and time-dependent manner (Lismont et al., 2019a), 2) a peroxisome-targeted variant of mKeima, a pH-sensitive red fluorescent protein suitable for imaging pexophagy in cellulo (Marcassa et al., 2018; Jo et al., 2020b), and 3) new fluorescent probes for in vitro and in vivo quantification of H2O2 (Ye et al., 2020), offers new opportunities to dynamically monitor (e.g., by flow cytometry) and study pexophagy flux in living cells in response to controlled fluctuations in peroxisomal H2O2 levels.
Despite the tremendous progress made in recent years, additional work is needed to better understand the peroxisome-autophagy redox connection and to sort out the exact nature of the mechanisms underlying the seemingly contradictory observations regarding the role of amino acid starvation, oxidative stress, and hypoxia in pexophagy regulation. Questions that deserve further research include but are not limited to: Which proteins with an established role in autophagy regulation are direct or indirect targets of peroxisome-derived H2O2? How do the corresponding oxidative modifications affect the activities of these proteins? Are the cellular responses induced dose-, time-, and cell type-specific? Do the in vitro studies recapitulate the in vivo situation? Obtaining answers to these questions will not only help us to unravel the molecular mechanisms underlying the Janus-role of pexophagy in health and disease, but also aid researchers to screen for pharmacological pexophagy regulators that can be used in a clinical setting to compensate for genetic and age-related changes in peroxisome homeostasis.
Author Contributions
HL and MF conceptualized and wrote the manuscript. HL generated the figures. HL, CL, IR, MAH, CC, and MF made a substantial intellectual contribution to the work, contributed to the editing, and approved the submitted version of the manuscript.
Funding
This work was supported by the KU Leuven (grant number: C14/18/088), the Research Foundation–Flanders (grant numbers G095315N and G091819N), and the European Union’s Horizon 2020 Research and Innovation Programme (Marie Skłodowska-Curie grant agreement No. 812968 (PERICO)). HL was a recipient of a doctoral fellowship from the China Scholarship Council (201906790005), CL was supported by postdoctoral fellowships from the KU Leuven (PDM/18/188) and the Research Foundation–Flanders (1213620N), and MAH was supported by a scholarship from the Ministry of Higher Education of the Arab Republic of Egypt.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors, MF.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge Servier Medical Art (https://smart.servier.com) for artistic templates.
References
Alers, S., Löffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cel. Biol. 32, 2–11. doi:10.1128/MCB.06159-11
Apanasets, O., Grou, C. P., Van Veldhoven, P. P., Brees, C., Wang, B., Nordgren, M., et al. (2014). PEX5, the Shuttling Import Receptor for Peroxisomal Matrix Proteins, Is a Redox-Sensitive Protein. Traffic 15, 94–103. doi:10.1111/tra.12129
Asgari, R., Yarani, R., Mohammadi, P., and Emami Aleagha, M. S. (2021). HIF-1α in the Crosstalk between Reactive Oxygen Species and Autophagy Process: a Review in Multiple Sclerosis. Cell. Mol. Neurobiol. in press. doi:10.1007/s10571-021-01111-5
Bagattin, A., Hugendubler, L., and Mueller, E. (2010). Transcriptional Coactivator PGC-1alpha Promotes Peroxisomal Remodeling and Biogenesis. Proc. Natl. Acad. Sci. USA 107, 20376–20381. doi:10.1073/pnas.1009176107
Bartolini, D., Dallaglio, K., Torquato, P., Piroddi, M., and Galli, F. (2018). Nrf2-p62 Autophagy Pathway and its Response to Oxidative Stress in Hepatocellular Carcinoma. Transl. Res. 193, 54–71. doi:10.1016/j.trsl.2017.11.007
Cagnol, S., and Chambard, J. C. (2010). ERK and Cell Death: Mechanisms of ERK-Induced Cell Death – Apoptosis, Autophagy and Senescence. FEBS J. 277, 2–21. doi:10.1111/j.1742-4658.2009.07366.x
Calero-Muñoz, N., Exposito-Rodriguez, M., Collado-Arenal, A. M., Rodríguez-Serrano, M., Laureano-Marín, A. M., Santamaría, M. E., et al. (2019). Cadmium Induces Reactive Oxygen Species-dependent Pexophagy in Arabidopsis Leaves. Plant Cel Environ 42, 2696–2714. doi:10.1111/pce.13597
Carroll, B., Otten, E. G., Manni, D., Stefanatos, R., Menzies, F. M., Smith, G. R., et al. (2018). Oxidation of SQSTM1/p62 Mediates the Link between Redox State and Protein Homeostasis. Nat. Commun. 9, 256. doi:10.1038/s41467-017-02746-z
Carvalho, A. F., Pinto, M. P., Grou, C. P., Alencastre, I. S., Fransen, M., Sá-Miranda, C., et al. (2007). Ubiquitination of Mammalian Pex5p, the Peroxisomal Import Receptor. J. Biol. Chem. 282, 31267–31272. doi:10.1074/jbc.M706325200
Cemma, M., Kim, P. K., and Brumell, J. H. (2011). The Ubiquitin-Binding Adaptor Proteins p62/SQSTM1 and NDP52 Are Recruited Independently to Bacteria-Associated Microdomains to Target Salmonella to the Autophagy Pathway. Autophagy 7, 341–345. doi:10.4161/auto.7.3.14046
Cha-Molstad, H., Lee, S. H., Kim, J. G., Sung, K. W., Hwang, J., Shim, S. M., et al. (2018). Regulation of Autophagic Proteolysis by the N-Recognin SQSTM1/p62 of the N-End Rule Pathway. Autophagy 14, 359–361. doi:10.1080/15548627.2017.1415190
Chen, B. H., Chang, Y. J., Lin, S., and Yang, W. Y. (2020). Hsc70/Stub1 Promotes the Removal of Individual Oxidatively Stressed Peroxisomes. Nat. Commun. 11, 5267. doi:10.1038/s41467-020-18942-3
Chen, Y., Azad, M. B., and Gibson, S. B. (2009). Superoxide Is the Major Reactive Oxygen Species Regulating Autophagy. Cell Death Differ. 16, 1040–1052. doi:10.1038/cdd.2009.49
Chiang, H. L., Terlecky, S. R., Plant, C. P., and Dice, J. F. (1989). A Role for a 70-kilodalton Heat Shock Protein in Lysosomal Degradation of Intracellular Proteins. Science 246, 382–385. doi:10.1126/science.2799391
Chu, K. Y., Mellet, N., Thai, L. M., Meikle, P. J., and Biden, T. J. (2020). Short-term Inhibition of Autophagy Benefits Pancreatic β-cells by Augmenting Ether Lipids and Peroxisomal Function, and by Countering Depletion of N-3 Polyunsaturated Fatty Acids after Fat-Feeding. Mol. Metab. 40, 101023. doi:10.1016/j.molmet.2020.101023
Cuervo, A. M., and Dice, J. F. (2000). Unique Properties of Lamp2a Compared to Other Lamp2 Isoforms. J. Cel Sci. 113, 4441–4450. doi:10.1242/jcs.113.24.4441
Czarny, P., Pawlowska, E., Bialkowska-Warzecha, J., Kaarniranta, K., and Blasiak, J. (2015). Autophagy in DNA Damage Response. Int. J. Mol. Sci. 16, 2641–2662. doi:10.3390/ijms16022641
Dahabieh, M. S., Di Pietro, E., Jangal, M., Goncalves, C., Witcher, M., Braverman, N. E., et al. (2018). Peroxisomes and Cancer: The Role of a Metabolic Specialist in a Disease of Aberrant Metabolism. Biochim. Biophys. Acta Rev. Cancer 1870, 103–121. doi:10.1016/j.bbcan.2018.07.004
Dahabieh, M. S., Huang, F., Goncalves, C., González, R. E. F., Prabhu, S., Bolt, A., et al. (2021). Silencing PEX26 as an Unconventional Mode to Kill Drug-Resistant Cancer Cells and Forestall Drug Resistance. Autophagy. in press. doi:10.1080/15548627.2021.1936932
Daskalaki, I., Gkikas, I., and Tavernarakis, N. (2018). Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cel Dev. Biol. 6, 104. doi:10.3389/fcell.2018.00104
Daussy, C. F., Galais, M., Pradel, B., Robert-Hebmann, V., Sagnier, S., Pattingre, S., et al. (2020). HIV-1 Env Induces Pexophagy and an Oxidative Stress Leading to Uninfected CD4+ T Cell Death. Autophagy 17, 2465–2474. doi:10.1080/15548627.2020.1831814
De Duve, C., and Baudhuin, P. (1966). Peroxisomes (Microbodies and Related Particles). Physiol. Rev. 46, 323–357. doi:10.1152/physrev.1966.46.2.323
Defourny, J., Aghaie, A., Perfettini, I., Avan, P., Delmaghan, i. S., and Petit, C. (2019). Pejvakin-mediated Pexophagy Protects Auditory Hair Cells against Noise-Induced Damage. Proc. Natl. Acad. Sci. USA 116, 8010–8017. doi:10.1073/pnas.1821844116
Deleyto-Seldas, N., and Efeyan, A. (2021). The mTOR-Autophagy axis and the Control of Metabolism. Front. Cel Dev. Biol. 9, 655731. doi:10.3389/fcell.2021.655731
Deng, A., Ma, L., Zhou, X., Wang, X., Wang, S., Chen, X., et al. (2021a). FoxO3 Transcription Factor Promotes Autophagy after Oxidative Stress Injury in HT22 Cells. Physiol. Pharmacol. 99, 627–634. doi:10.1139/cjpp-2020-0448
Deng, J. K., Zhang, X., Wu, H. L., Gan, Y., Ye, L., Zheng, H., et al. (2021b). ROS-ERK Pathway as Dual Mediators of Cellular Injury and Autophagy-Associated Adaptive Response in Urinary Protein-Irritated Renal Tubular Epithelial Cells. J. Diabetes Res. 2021, 6614848. doi:10.1155/2021/6614848
Deosaran, E., Larsen, K. B., Hua, R., Sargent, G., Wang, Y., Kim, S., et al. (2013). NBR1 Acts as an Autophagy Receptor for Peroxisomes. J. Cel Sci. 126, 939–952. doi:10.1242/jcs.114819
Deretic, V., and Kroemer, G. (2021). Autophagy in Metabolism and Quality Control: Opposing, Complementary or Interlinked Functions? Autophagy. in press. doi:10.1080/15548627.2021.1933742
Desideri, E., Filomeni, G., and Ciriolo, M. R. (2012). Glutathione Participates in the Modulation of Starvation-Induced Autophagy in Carcinoma Cells. Autophagy 8, 1769–1781. doi:10.4161/auto.22037
Dutta, R. K., Maharjan, Y., Lee, J. N., Park, C., Ho, Y. S., and Park, R. (2021). Catalase Deficiency Induces Reactive Oxygen Species Mediated Pexophagy and Cell Death in the Liver during Prolonged Fasting. Biofactors 47, 112–125. doi:10.1002/biof.1708
Egan, D., Kim, J., Shaw, R. J., and Guan, K. L. (2011). The Autophagy Initiating Kinase ULK1 Is Regulated via Opposing Phosphorylation by AMPK and mTOR. Autophagy 7, 643–644. doi:10.4161/auto.7.6.15123
Fan, P., Xie, X. H., Chen, C. H., Peng, X., Zhang, P., Yang, C., et al. (2019). Molecular Regulation Mechanisms and Interactions between Reactive Oxygen Species and Mitophagy. DNA Cel Biol. 38, 10–22. doi:10.1089/dna.2018.4348
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer's Disease. Nat. Neurosci. 22, 401–412. doi:10.1038/s41593-018-0332-9
Fang, Y., Tan, J., and Zhang, Q. (2015). Signaling Pathways and Mechanisms of Hypoxia-Induced Autophagy in the Animal Cells. Cell Biol. Int. 39, 891–898. doi:10.1002/cbin.10463
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The Machinery of Macroautophagy. Cell Res. 24, 24–41. doi:10.1038/cr.2013.168
Filomeni, G., Desideri, E., Cardaci, S., Rotilio, G., and Ciriolo, M. R. (2010). Under the ROS: Thiol Network Is the Principal Suspect for Autophagy Commitment. Autophagy 6, 999–1005. doi:10.4161/auto.6.7.12754
Fourcade, S., Ferrer, I., and Pujol, A. (2015). Oxidative Stress, Mitochondrial and Proteostasis Malfunction in Adrenoleukodystrophy: A Paradigm for Axonal Degeneration. Free Radic. Biol. Med. 88, 18–29. doi:10.1016/j.freeradbiomed.2015.05.041
Francisco, T., Rodrigues, T. A., Dias, A. F., Barros-Barbosa, A., Bicho, D., and Azevedo, J. E. (2017). Protein Transport into Peroxisomes: Knowns and Unknowns. Bioessays 39, 1700047. doi:10.1002/bies.201700047
Fransen, M., Lismont, C., and Walton, P. (2017). The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 18, 1126. doi:10.3390/ijms18061126
Fransen, M., Terlecky, S. R., and Subramani, S. (1998). Identification of a Human PTS1 Receptor Docking Protein Directly Required for Peroxisomal Protein Import. Proc. Natl. Acad. Sci. USA 95, 8087–8092. doi:10.1073/pnas.95.14.8087
Frazier, D. P., Wilson, A., Dougherty, C. J., Li, H., Bishopric, N. H., and Webster, K. A. (2007). PKC-alpha and TAK-1 Are Intermediates in the Activation of C-Jun NH2-terminal Kinase by Hypoxia-Reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 292, H1675–H1684. doi:10.1152/ajpheart.01132.2006
Frudd, K., Burgoyne, T., and Burgoyne, J. R. (2018). Oxidation of Atg3 and Atg7 Mediates Inhibition of Autophagy. Nat. Commun. 9, 95. doi:10.1038/s41467-017-02352-z
Füllgrabe, J., Ghislat, G., Cho, D. H., and Rubinsztein, D. C. (2016). Transcriptional Regulation of Mammalian Autophagy at a Glance. J. Cel Sci. 129, 3059–3066. doi:10.1242/jcs.188920
Ganguli, G., Pattanaik, K. P., Jagadeb, M., and Sonawane, A. (2020). Mycobacterium tuberculosis Rv3034c Regulates mTORC1 and PPAR-Gamma Dependant Pexophagy Mechanism to Control Redox Levels in Macrophages. Cell. Microbiol. 22, e13214. doi:10.1111/cmi.13214
Gatica, D., Lahiri, V., and Klionsky, D. J. (2018). Cargo Recognition and Degradation by Selective Autophagy. Nat. Cel Biol. 20, 233–242. doi:10.1038/s41556-018-0037-z
Germain, K., and Kim, P. K. (2020). Pexophagy: a Model for Selective Autophagy. Int. J. Mol. Sci. 21, 578. doi:10.3390/ijms21020578
Gubas, A., and Dikic, I. (2021). A Guide to the Regulation of Selective Autophagy Receptors. FEBS J. in press. doi:10.1111/febs.15824
Gureev, A. P., Shaforostova, E. A., and Popov, V. N. (2019). Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 10, 435. doi:10.3389/fgene.2019.00435
Hara-Kuge, S., and Fujiki, Y. (2008). The Peroxin Pex14p Is Involved in LC3-dependent Degradation of Mammalian Peroxisomes. Exp. Cel Res. 314, 3531–3541. doi:10.1016/j.yexcr.2008.09.015
He, A., Chen, X., Tan, M., Chen, Y., Lu, D., Zhang, X., et al. (2020). Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Mol. Cel. 79, 30–42. e4. doi:10.1016/j.molcel.2020.05.007
He, Y., She, H., Zhang, T., Xu, H., Cheng, L., Yepes, M., et al. (2018). p38 MAPK Inhibits Autophagy and Promotes Microglial Inflammatory Responses by Phosphorylating ULK1. J. Cel Biol. 217, 315–328. doi:10.1083/jcb.201701049
Houri, K., Mori, T., Onodera, Y., Tsujimoto, T., Takehara, T., Nakao, S., et al. (2020). miR-142 Induces Accumulation of Reactive Oxygen Species (ROS) by Inhibiting Pexophagy in Aged Bone Marrow Mesenchymal Stem Cells. Sci. Rep. 10, 3735. doi:10.1038/s41598-020-60346-2
Hu, W., Chen, S., Thorne, R. F., and Wu, M. (2019). TP53, TP53 Target Genes (DRAM, TIGAR), and Autophagy. Adv. Exp. Med. Biol. 1206, 127–149. doi:10.1007/978-981-15-0602-4_6
Hurley, J. H., and Young, L. N. (2017). Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 86, 225–244. doi:10.1146/annurev-biochem-061516-044820
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 115, 577–590. doi:10.1016/S0092-8674(03)00929-2
Ivashchenko, O., Van Veldhoven, P. P., Brees, C., Ho, Y. S., Terlecky, S. R., and Fransen, M. (2011). Intraperoxisomal Redox Balance in Mammalian Cells: Oxidative Stress and Interorganellar Cross-Talk. Mol. Biol. Cel. 22, 1440–1451. doi:10.1091/mbc.e10-11-0919
Jain, I. H., Calvo, S. E., Markhard, A. L., Skinner, O. S., To, T. L., Ast, T., et al. (2020). Genetic Screen for Cell Fitness in High or Low Oxygen Highlights Mitochondrial and Lipid Metabolism. Cell 181, 716–727. doi:10.1016/j.cell.2020.03.029
Jiang, L., Hara-Kuge, S., Yamashita, S. I., and Fujiki, Y. (2015a). Peroxin Pex14p Is the Key Component for Coordinated Autophagic Degradation of Mammalian Peroxisomes by Direct Binding to LC3-II. Genes Cells 20, 36–49. doi:10.1111/gtc.12198
Jiang, T., Harder, B., Rojo de la Vega, M., Wong, P. K., Chapman, E., and Zhang, D. D. (2015b). p62 Links Autophagy and Nrf2 Signaling. Free Radic. Biol. Med. 88, 199–204. doi:10.1016/j.freeradbiomed.2015.06.014
Jo, D. S., Bae, D. J., Park, S. J., Seo, H. M., Kim, H. B., Oh, J. S., et al. (2015). Pexophagy Is Induced by Increasing Peroxisomal Reactive Oxygen Species in 1,10-Phenanthroline-Treated Cells. Biochem. Biophys. Res. Commun. 467, 354–360. doi:10.1016/j.bbrc.2015.09.153
Jo, D. S., Park, N. Y., and Cho, D. H. (2020a). Peroxisome Quality Control and Dysregulated Lipid Metabolism in Neurodegenerative Diseases. Exp. Mol. Med. 52, 1486–1495. doi:10.1038/s12276-020-00503-9
Jo, D. S., Park, S. J., Kim, A. K., Park, N. Y., Kim, J. B., Bae, J. E., et al. (2020b). Loss of HSPA9 Induces Peroxisomal Degradation by Increasing Pexophagy. Autophagy 16, 1989–2003. doi:10.1080/15548627.2020.1712812
Juretschke, T., and Beli, P. (2021). Causes and Consequences of DNA Damage-Induced Autophagy. Matrix Biol. 100-101, 39–53. doi:10.1016/j.matbio.2021.02.004
Kaur, J., and Debnath, J. (2015). Autophagy at the Crossroads of Catabolism and Anabolism. Nat. Rev. Mol. Cel Biol. 16, 461–475. doi:10.1038/nrm4024
Kaushik, S., and Cuervo, A. M. (2018). The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cel Biol. 19, 365–381. doi:10.1038/s41580-018-0001-6
Kawabata, T., and Yoshimori, T. (2020). Autophagosome Biogenesis and Human Health. Cell Discov. 6, 33. doi:10.1038/s41421-020-0166-y
Kenific, C. M., and Debnath, J. (2015). Cellular and Metabolic Functions for Autophagy in Cancer Cells. Trends Cel Biol. 25, 37–45. doi:10.1016/j.tcb.2014.09.001
Kierans, S. J., and Taylor, C. T. (2021). Regulation of Glycolysis by the Hypoxia-Inducible Factor (HIF): Implications for Cellular Physiology. J. Physiol. 599, 23–37. doi:10.1113/JP280572
Kim, B. W., Kwon, D. H., and Song, H. K. (2016). Structure Biology of Selective Autophagy Receptors. BMB Rep. 49, 73–80. doi:10.5483/BMBRep.2016.49.2.265
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cel Biol. 13, 132–141. doi:10.1038/ncb2152
Kim, P. K., Hailey, D. W., Mullen, R. T., and Lippincott-Schwartz, J. (2008). Ubiquitin Signals Autophagic Degradation of Cytosolic Proteins and Peroxisomes. Proc. Natl. Acad. Sci. USA 105, 20567–20574. doi:10.1073/pnas.0810611105
Kirkin, V., and Rogov, V. V. (2019). A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cel. 76, 268–285. doi:10.1016/j.molcel.2019.09.005
Kirkin, V., Lamark, T., Sou, Y. S., Bjørkøy, G., Nunn, J. L., Bruun, J. A., et al. (2009). A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cel. 33, 505–516. doi:10.1016/j.molcel.2009.01.020
Klionsky, D. J., Abdel-Aziz, A. K., Abdelfatah, S., Abdellatif, M., Abdoli, A., Abel, S., et al. (2021). Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition). Autophagy 17, 1–382. doi:10.1080/15548627.2020.179728010.1080/15548627.2015.1100356
Klouwer, F. C. C., Falkenberg, K. D., Ofman, R., Koster, J., van Gent, D., Ferdinandusse, S., et al. (2021). Autophagy Inhibitors Do Not Restore Peroxisomal Functions in Cells with the Most Common Peroxisome Biogenesis Defect. Front. Cel Dev. Biol. 9, 661298. doi:10.3389/fcell.2021.661298
Kma, L., and Baruah, T. J. (2021). The Interplay of ROS and the PI3K/Akt Pathway in Autophagy Regulation. Biotechnol. Appl. Biochem. 2021.in press. doi:10.1002/bab.2104
Koundouros, N., and Poulogiannis, G. (2018). Phosphoinositide 3-kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 8, 160. doi:10.3389/fonc.2018.00160
Kriegenburg, F., Ungermann, C., and Reggiori, F. (2018). Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 28, R512–R518. doi:10.1016/j.cub.2018.02.034
Lamark, T., and Johansen, T. (2021). Mechanisms of Selective Autophagy. Annu. Rev. Cel Dev. Biol. 37, 143–169. doi:10.1146/annurev-cellbio-120219-035530
Launay, N., Aguado, C., Fourcade, S., Ruiz, M., Grau, L., Riera, J., et al. (2015). Autophagy Induction Halts Axonal Degeneration in a Mouse Model of X-Adrenoleukodystrophy. Acta Neuropathol. 129, 399–415. doi:10.1007/s00401-014-1378-8
Law, K. B., Bronte-Tinkew, D., Di Pietro, E., Snowden, A., Jones, R. O., Moser, A., et al. (2017). The Peroxisomal AAA ATPase Complex Prevents Pexophagy and Development of Peroxisome Biogenesis Disorders. Autophagy 13, 868–884. doi:10.1080/15548627.2017.1291470
Lee, J. N., Dutta, R. K., Maharjan, Y., Liu, Z. Q., Lim, J. Y., Kim, S. J., et al. (2018). Catalase Inhibition Induces Pexophagy through ROS Accumulation. Biochem. Biophys. Res. Commun. 501, 696–702. doi:10.1016/j.bbrc.2018.05.050
Lee, J. W., Ko, J., Ju, C., and Eltzschig, H. K. (2019). Hypoxia Signaling in Human Diseases and Therapeutic Targets. Exp. Mol. Med. 51, 1–13. doi:10.1038/s12276-019-0235-1
Legakis, J. E., Koepke, J. I., Jedeszko, C., Barlaskar, F., Terlecky, L. J., Edwards, H. J., et al. (2002). Peroxisome Senescence in Human Fibroblasts. Mol. Biol. Cel. 13, 243–255. doi:10.1091/mbc.e02-06-0322
Leidal, A. M., Levine, B., and Debnath, J. (2018). Autophagy and the Cell Biology of Age-Related Disease. Nat. Cel Biol. 20, 1338–1348. doi:10.1038/s41556-018-0235-8
Li, J., and Wang, W. (2021). Mechanisms and Functions of Pexophagy in Mammalian Cells. Cells 10, 1094. doi:10.3390/cells10051094
Li, L., Tan, J., Miao, Y., Lei, P., and Zhang, Q. (2015). ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cel. Mol. Neurobiol. 35, 615–621. doi:10.1007/s10571-015-0166-x
Li, W., and Zhang, L. (2019). Regulation of ATG and Autophagy Initiation. Adv. Exp. Med. Biol. 1206, 41–65. doi:10.1007/978-981-15-0602-4_2
Li, X., Han, H., Zhou, M. T., Yang, B., Ta, A. P., Li, N., et al. (2017). Proteomic Analysis of the Human Tankyrase Protein Interaction Network Reveals its Role in Pexophagy. Cell Rep. 20, 737–749. doi:10.1016/j.celrep.2017.06.077
Lismont, C., Nordgren, M., Brees, C., Knoops, B., Van Veldhoven, P. P., and Fransen, M. (2019a). Peroxisomes as Modulators of Cellular Protein Thiol Oxidation: a New Model System. Antioxid. Redox Signal. 30, 22–39. doi:10.1089/ars.2017.6997
Lismont, C., Nordgren, M., Van Veldhoven, P. P., and Fransen, M. (2015). Redox Interplay between Mitochondria and Peroxisomes. Front. Cel Dev. Biol. 3, 35. doi:10.3389/fcell.2015.00035
Lismont, C., Revenco, I., and Fransen, M. (2019b). Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 20, 3673. doi:10.3390/ijms20153673
Lismont, C., Revenco, R., Li, H., Costa, C. F., Lenaerts, L., Hussein, M. A. F., et al. (2021). Peroxisome-derived Hydrogen Peroxide Can Modulate the Sulfenylation Profiles of Key Redox Signaling Proteins. Preprint. BioRxiv. doi:10.1101/2021.10.08.463647
Lizama-Manibusan, B., and McLaughlin, B. (2013). Redox Modification of Proteins as Essential Mediators of CNS Autophagy and Mitophagy. FEBS Lett. 587, 2291–2298. doi:10.1016/j.febslet.2013.06.007
Lorin, S., Pierron, G., Ryan, K. M., Codogno, P., and Djavaheri-Mergny, M. (2010). Evidence for the Interplay between JNK and P53-DRAM Signalling Pathways in the Regulation of Autophagy. Autophagy 6, 153–154. doi:10.4161/auto.6.1.10537
Lystad, A. H., Carlsson, S. R., and Simonsen, A. (2019). Toward the Function of Mammalian ATG12-ATG5-Atg16l1 Complex in Autophagy and Related Processes. Autophagy 15, 1485–1486. doi:10.1080/15548627.2019.1618100
Marcassa, E., Kallinos, A., Jardine, J., Rusilowicz-Jones, E. V., Martinez, A., Kuehl, S., et al. (2018). Dual Role of USP30 in Controlling Basal Pexophagy and Mitophagy. EMBO Rep. 19, e45595. doi:10.15252/embr.201745595
Marinho, H. S., Real, C., Cyrne, L., Soares, H., and Antunes, F. (2014). Hydrogen Peroxide Sensing, Signaling and Regulation of Transcription Factors. Redox Biol. 2, 535–562. doi:10.1016/j.redox.2014.02.006
Martens, S., and Fracchiolla, D. (2020). Activation and Targeting of ATG8 Protein Lipidation. Cel Discov. 6, 23. doi:10.1038/s41421-020-0155-1
McCarthy, I., Romero-Puertas, M. C., Palma, J. M., Sandalio, L. M., Corpas, F. J., Gómez, M., et al. (2001). Cadmium Induces Senescence Symptoms in Leaf Peroxisomes of Pea Plants. Plant Cel Environ 24, 1065–1073. doi:10.1046/j.1365-3040.2001.0075010.1046/j.1365-3040.2001.00750.x
McClung, J. M., Judge, A. R., Powers, S. K., and Yan, Z. (2010). p38 MAPK Links Oxidative Stress to Autophagy-Related Gene Expression in Cachectic Muscle Wasting. Am. J. Physiol. Cel Physiol. 298, C542–C549. doi:10.1152/ajpcell.00192.2009
Mejlvang, J., Olsvik, H., Svenning, S., Bruun, J. A., Abudu, Y. P., Larsen, K. B., et al. (2018). Starvation Induces Rapid Degradation of Selective Autophagy Receptors by Endosomal Microautophagy. J. Cel Biol. 217, 3640–3655. doi:10.1002/bies.20180000810.1083/jcb.201711002
Melia, T. J., Lystad, A. H., and Simonsen, A. (2020). Autophagosome Biogenesis: from Membrane Growth to Closure. J. Cel Biol. 219, e202002085. doi:10.1083/jcb.202002085
Mercer, T. J., Gubas, A., and Tooze, S. A. (2018). A Molecular Perspective of Mammalian Autophagosome Biogenesis. J. Biol. Chem. 293, 5386–5395. doi:10.1074/jbc.R117.810366
Mesquita, A., Glenn, J., and Jenny, A. (2021). Differential Activation of eMI by Distinct Forms of Cellular Stress. Autophagy 17, 1828–1840. doi:10.1080/15548627.2020.1783833
Mohan, J., and Wollert, T. (2018). Human Ubiquitin-like Proteins as central Coordinators in Autophagy. Interf. Focus 8, 20180025. doi:10.1098/rsfs.2018.0025
Mu, Y., Maharjan, Y., Dutta, R. K., Kim, H., Wei, X., Kim, J. H., et al. (2020). Dimethyloxaloylglycine Induces Pexophagy in a HIF-2alpha Dependent Manner Involving Autophagy Receptor P62. Biochem. Biophys. Res. Commun. 525, 46–52. doi:10.1016/j.bbrc.2020.02.051
Muralidharan, C., Conteh, A. M., Marasco, M. R., Crowder, J. J., Kuipers, J., de Boer, P., et al. (2021). Pancreatic Beta Cell Autophagy Is Impaired in Type 1 Diabetes. Diabetologia 64, 865–877. doi:10.1007/s00125-021-05387-6
Murrow, L., and Debnath, J. (2013). Autophagy as a Stress-Response and Quality-Control Mechanism: Implications for Cell Injury and Human Disease. Annu. Rev. Pathol. 8, 105–137. doi:10.1146/annurev-pathol-020712-163918
Nakamura, S., and Yoshimori, T. (2017). New Insights into Autophagosome-Lysosome Fusion. J. Cel Sci. 130, 1209–1216. doi:10.1242/jcs.196352
Nakatogawa, H. (2013). Two Ubiquitin-like Conjugation Systems that Mediate Membrane Formation during Autophagy. Essays Biochem. 55, 39–50. doi:10.1042/bse0550039
Nazarko, T. Y. (2017). Pexophagy Is Responsible for 65% of Cases of Peroxisome Biogenesis Disorders. Autophagy 13, 991–994. doi:10.1080/15548627.2017.1291480
Nishimura, T., and Tooze, S. A. (2020). Emerging Roles of ATG Proteins and Membrane Lipids in Autophagosome Formation. Cel Discov. 6, 32. doi:10.1038/s41421-020-0161-3
Nordgren, M., Francisco, T., Lismont, C., Hennebel, L., Brees, C., Wang, B., et al. (2015). Export-deficient Monoubiquitinated PEX5 Triggers Peroxisome Removal in SV40 Large T Antigen-Transformed Mouse Embryonic Fibroblasts. Autophagy 11, 1326–1340. doi:10.1080/15548627.2015.1061846
Nordgren, M., Wang, B., Apanasets, O., and Fransen, M. (2013). Peroxisome Degradation in Mammals: Mechanisms of Action, Recent Advances, and Perspectives. Front. Physiol. 4, 145. doi:10.3389/fphys.2013.00145
Oku, M., and Sakai, Y. (2018). Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 40, e1800008. doi:10.1002/bies.201800008
Ornatowski, W., Lu, Q., Yegambaram, M., Garcia, A. E., Zemskov, E. A., Maltepe, E., et al. (2020). Complex Interplay between Autophagy and Oxidative Stress in the Development of Pulmonary Disease. Redox Biol. 36, 101679. doi:10.1016/j.redox.2020.101679
Paddenberg, R., Ishaq, B., Goldenberg, A., Faulhammer, P., Rose, F., Weissmann, N., et al. (2003). Essential Role of Complex II of the Respiratory Chain in Hypoxia-Induced ROS Generation in the Pulmonary Vasculature. Am. J. Physiol. Lung Cel. Mol. Physiol. 284, L710–L719. doi:10.1152/ajplung.00149.2002
Påhlman, S., and Mohlin, S. (2018). Hypoxia and Hypoxia-Inducible Factors in Neuroblastoma. Cell Tissue Res. 372, 269–275. doi:10.1007/s00441-017-2701-1
Papandreou, I., Lim, A. L., Laderoute, K., and Denko, N. C. (2008). Hypoxia Signals Autophagy in Tumor Cells via AMPK Activity, Independent of HIF-1, BNIP3, and BNIP3L. Cel Death Differ. 15, 1572–1581. doi:10.1038/cdd.2008.84
Park, H., Kim, J., Shin, C., and Lee, S. (2021a). Intersection between Redox Homeostasis and Autophagy: Valuable Insights into Neurodegeneration. Antioxidants 10, 694. doi:10.3390/antiox10050694
Park, N. Y., Jo, D. S., Park, S. J., Lee, H., Bae, J. E., Hong, Y., et al. (2021b). Depletion of HNRNPA1 Induces Peroxisomal Autophagy by Regulating PEX1 Expression. Biochem. Biophys. Res. Commun. 545, 69–74. doi:10.1016/j.bbrc.2021.01.083
Parzych, K. R., and Klionsky, D. J. (2014). An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 20, 460–473. doi:10.1089/ars.2013.5371
Puissant, A., Fenouille, N., and Auberger, P. (2012). When Autophagy Meets Cancer through p62/SQSTM1. Am. J. Cancer Res. 2, 397–413.
Rashid, H. O., Yadav, R. K., Kim, H. R., and Chae, H. J. (2015). ER Stress: Autophagy Induction, Inhibition and Selection. Autophagy 11, 1956–1977. doi:10.1080/15548627.2015.1091141
Rathore, R., Zheng, Y. M., Niu, C. F., Liu, Q. H., Korde, A., Ho, Y. S., et al. (2008). Hypoxia Activates NADPH Oxidase to Increase [ROS]i and [Ca2+]i through the Mitochondrial ROS-PKCepsilon Signaling axis in Pulmonary Artery Smooth Muscle Cells. Free Radic. Biol. Med. 45, 1223–1231. doi:10.1016/j.freeradbiomed.2008.06.012
Redza-Dutordoir, M., and Averill-Bates, D. A. (2021). Interactions between Reactive Oxygen Species and Autophagy. Biochim. Biophys. Acta Mol. Cel. Res. 1868, 119041. doi:10.1016/j.bbamcr.2021.119041
Riccio, V., Demers, N., Hua, R., Vissa, M., Cheng, D. T., Strilchuk, A. W., et al. (2019). Deubiquitinating Enzyme USP30 Maintains Basal Peroxisome Abundance by Regulating Pexophagy. J. Cel Biol. 218, 798–807. doi:10.1083/jcb.201804172
Roberts, R., and Ktistakis, N. T. (2013). Omegasomes: PI3P Platforms that Manufacture Autophagosomes. Essays Biochem. 55, 17–27. doi:10.1042/bse0550017
Rodriguez-Rocha, H., Garcia-Garcia, A., Panayiotidis, M. I., and Franco, R. (2011). DNA Damage and Autophagy. Mutat. Res. 711, 158–166. doi:10.1016/j.mrfmmm.2011.03.007
Rouschop, K. M. A., van den Beucken, T., Dubois, L., Niessen, H., Bussink, J., Savelkouls, K., et al. (2010). The Unfolded Protein Response Protects Human Tumor Cells during Hypoxia through Regulation of the Autophagy Genes MAP1LC3B and ATG5. J. Clin. Invest. 120, 127–141. doi:10.1172/JCI40027
Rout, A. K., Strub, M. P., Piszczek, G., and Tjandra, N. (2014). Structure of Transmembrane Domain of Lysosome-Associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-Mediated Autophagy. J. Biol. Chem. 289, 35111–35123. doi:10.1074/jbc.M114.609446
Russell, R. C., Yuan, H. X., and Guan, K. L. (2014). Autophagy Regulation by Nutrient Signaling. Cel Res. 24, 42–57. doi:10.1038/cr.2013.166
Sargent, G., van Zutphen, T., Shatseva, T., Zhang, L., Giovanni, V. S., Bandsma, R., et al. (2016). PEX2 Is the E3 Ubiquitin Ligase Required for Pexophagy during Starvation. J. Cel Biol. 214, 677–690. doi:10.1083/jcb.201511034
Scherz-Shouval, R., Shvets, E., and Elazar, Z. (2007a). Oxidation as a post-translational Modification that Regulates Autophagy. Autophagy 3, 371–373. doi:10.4161/auto.4214
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007b). Reactive Oxygen Species Are Essential for Autophagy and Specifically Regulate the Activity of Atg4. EMBO J. 26, 1749–1760. doi:10.1038/sj.emboj.7601623
Schönenberger, M. J., Krek, W., and Kovacs, W. J. (2015). EPAS1/HIF-2α Is a Driver of Mammalian Pexophagy. Autophagy 11, 967–969. doi:10.1080/15548627.2015.1045180
Sedlackova, L., and Korolchuk, V. I. (2020). The Crosstalk of NAD, ROS and Autophagy in Cellular Health and Ageing. Biogerontology 21, 381–397. doi:10.1007/s10522-020-09864-0
Sengupta, A., Molkentin, J. D., and Yutzey, K. E. (2009). FoxO Transcription Factors Promote Autophagy in Cardiomyocytes. J. Biol. Chem. 284, 28319–28331. doi:10.1074/jbc.M109.024406
Shao, D., Oka, S., Liu, T., Zhai, P., Ago, T., Sciarretta, S., et al. (2014). A Redox-dependent Mechanism for Regulation of AMPK Activation by Thioredoxin1 during Energy Starvation. Cell Metab 19, 232–245. doi:10.1016/j.cmet.2013.12.013
Sies, H., and Jones, D. P. (2020). Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cel Biol. 21, 363–383. doi:10.1038/s41580-020-0230-3
Son, Y., Cheong, Y. K., Kim, N. H., Chung, H. T., Kang, D. G., and Pae, H. O. (2011). Mitogen-activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 792639. doi:10.1155/2011/792639
Stöcker, S., Van Laer, K., Mijuskovic, A., and Dick, T. P. (2018). The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal. 28, 558–573. doi:10.1089/ars.2017.7162
Subramani, S. (2015). A Mammalian Pexophagy Target. Nat. Cel Biol. 17, 1371–1373. doi:10.1038/ncb3253
Sui, X., Kong, N., Ye, L., Han, W., Zhou, J., Zhang, Q., et al. (2014). p38 and JNK MAPK Pathways Control the Balance of Apoptosis and Autophagy in Response to Chemotherapeutic Agents. Cancer Lett. 344, 174–179. doi:10.1016/j.canlet.2013.11.019
Sun, T., Li, D., Wang, L., Xia, L., Ma, J., Guan, Z., et al. (2011). c-Jun NH2-terminal Kinase Activation Is Essential for Up-Regulation of LC3 during Ceramide-Induced Autophagy in Human Nasopharyngeal Carcinoma Cells. J. Transl. Med. 9, 161. doi:10.1186/1479-5876-9-161
Tripathi, D. N., and Walker, C. L. (2016). The Peroxisome as a Cell Signaling Organelle. Curr. Opin. Cel Biol. 39, 109–112. doi:10.1016/j.ceb.2016.02.017
Tripathi, D. N., Zhang, J., Jing, J., Dere, R., and Walker, C. L. (2016). A New Role for ATM in Selective Autophagy of Peroxisomes (Pexophagy). Autophagy 12, 711–712. doi:10.1080/15548627.2015.1123375
Van Veldhoven, P. P. (2010). Biochemistry and Genetics of Inherited Disorders of Peroxisomal Fatty Acid Metabolism. J. Lipid Res. 51, 2863–2895. doi:10.1194/jlr.R005959
van Zutphen, T., Ciapaite, J., Bloks, J. W., Ackereley, C., Gerding, A., Jurdzinski, A., et al. (2014). Malnutrition-associated Liver Steatosis and ATP Depletion Is Caused by Peroxisomal and Mitochondrial Dysfunction. J. Hepatol. 65, 1198–1208. doi:10.1016/j.jhep.2016.05.046
Vasko, R., and Goligorsky, M. S. (2013). Dysfunctional Lysosomal Autophagy Leads to Peroxisomal Oxidative Burnout and Damage during Endotoxin-Induced Stress. Autophagy 9, 442–444. doi:10.4161/auto.23344
Vasko, R. (2016). Peroxisomes and Kidney Injury. Antioxid. Redox Signal. 25, 217–231. doi:10.1089/ars.2016.6666
Vasko, R., Ratliff, B. B., Bohr, S., Nadel, E., Chen, J., Xavier, S., et al. (2013). Endothelial Peroxisomal Dysfunction and Impaired Pexophagy Promotes Oxidative Damage in Lipopolysaccharide-Induced Acute Kidney Injury. Antioxid. Redox Signal. 19, 211–230. doi:10.1089/ars.2012.4768
Walter, K. M., Schönenberger, M. J., Trötzmüller, M., Horn, M., Elsässer, H. P., Moser, A. B., et al. (2014). Hif-2α Promotes Degradation of Mammalian Peroxisomes by Selective Autophagy. Cel Metab 20, 882–897. doi:10.1016/j.cmet.2014.09.017
Wei, X., Maharjan, Y., Dorotea, D., Dutta, R. K., Kim, D., Kim, H., et al. (2021). Knockdown of PEX16 Induces Autophagic Degradation of Peroxisomes. Int. J. Mol. Sci. 22, 7989. doi:10.3390/ijms22157989
Wible, D. J., and Bratton, S. B. (2018). Reciprocity in ROS and Autophagic Signaling. Curr. Opin. Toxicol. 7, 28–36. doi:10.1016/j.cotox.2017.10.006
Yamashita, S. I., Abe, K., Tatemichi, Y., and Fujiki, Y. (2014). The Membrane Peroxin PEX3 Induces Peroxisome-Ubiquitination-Linked Pexophagy. Autophagy 10, 1549–1564. doi:10.4161/auto.29329
Yang, Y., Ma, F., Liu, Z., Su, Q., Liu, Y., Liu, Z., et al. (2019). The ER-Localized Ca2+-Binding Protein Calreticulin Couples ER Stress to Autophagy by Associating with Microtubule-Associated Protein 1A/1B Light Chain 3. J. Biol. Chem. 294, 772–782. doi:10.1074/jbc.RA118.005166
Ye, S., Hu, J. J., Zhao, Q. A., and Yang, D. (2020). Fluorescent Probes for In Vitro and In Vivo Quantification of Hydrogen Peroxide. Chem. Sci. 11, 11989–11997. doi:10.1039/d0sc04888g
Yim, W. W. Y., and Mizushima, N. (2020). Lysosome Biology in Autophagy. Cel Discov 6, 6. doi:10.1038/s41421-020-0141-7
Yin, J., Duan, J., Cui, Z., Ren, W., Li, T., and Yin, Y. (2015). Hydrogen Peroxide-Induced Oxidative Stress Activates NF-kB and Nrf2/Keap1 Signals and Triggers Autophagy in Piglets. RSC Adv. 5, 15479. doi:10.1039/C4RA13557A
Yun, H. R., Jo, Y. H., Kim, J., Shin, Y., Kim, S. S., and Choi, T. G. (2020). Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 21, 3289. doi:10.3390/ijms21093289
Zhang, J., Kim, J., Alexander, A., Cai, S., Tripathi, D. N., Dere, R., et al. (2013). A Tuberous Sclerosis Complex Signalling Node at the Peroxisome Regulates mTORC1 and Autophagy in Response to ROS. Nat. Cel Biol. 15, 1186–1196. doi:10.1038/ncb2822
Zhang, J., Tripathi, D. N., Jing, J., Alexander, A., Kim, J., Powell, R. T., et al. (2015). ATM Functions at the Peroxisome to Induce Pexophagy in Response to ROS. Nat. Cel. Biol. 17, 1259–1269. doi:10.1038/ncb3230
Zhao, Y. G., Codogno, P., and Zhang, H. (2021). Machinery, Regulation and Pathophysiological Implications of Autophagosome Maturation. Nat. Rev. Mol. Cel Biol. 22, 733–750. doi:10.1038/s41580-021-00392-4
Zheng, J., Chen, X., Liu, Q., Zhong, G., and Zhuang, M. (2022). Ubiquitin Ligase MARCH5 Localizes to Peroxisomes to Regulate Pexophagy. J. Cel Biol. 221, e202103156. doi:10.1083/jcb.202103156
Zheng, X., Yang, Z., Gu, Q., Xia, F., Fu, Y., Liu, P., et al. (2020). The Protease Activity of Human ATG4B Is Regulated by Reversible Oxidative Modification. Autophagy 16, 1838–1850. doi:10.1080/15548627.2019.1709763
Zhou, F., Yang, Y., and Xing, D. (2011). Bcl-2 and Bcl-xL Play Important Roles in the Crosstalk between Autophagy and Apoptosis. FEBS J. 278, 403–413. doi:10.1111/j.1742-4658.2010.07965.x
Zhou, J., Li, X. Y., Liu, Y. J., Feng, J., Wu, Y., Shen, H. M., et al. (2021). Full-coverage Regulations of Autophagy by ROS: from Induction to Maturation. Autophagy. in press. doi:10.1080/15548627.2021.1984656
Zhou, Y. Y., Li, Y., Jiang, W. Q., and Zhou, L. F. (2015). MAPK/JNK Signalling: a Potential Autophagy Regulation Pathway. Biosci. Rep. 35, e00199. doi:10.1042/BSR20140141
Zmijewski, J. W., Banerjee, S., Bae, H., Friggeri, A., Lazarowski, E. R., and Abraham, E. (2010). Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-Activated Protein Kinase. J. Biol. Chem. 285, 33154–33164. doi:10.1074/jbc.M110.143685
Glossary
AAPK AMP-activated protein kinase
ATG autophagy-related protein
ATM ataxia telangiectasia mutated
BAKOR beclin 1-associated autophagy-related key regulator
BCL B-cell CLL/lymphoma
B2Cl Bcl-2-like protein
BECN beclin
BNIP ATM, ataxia telangiectasia mutated; BCL2/adenovirus E1B 19 kDa protein-interacting protein
BNI3L BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like
CACO calcium-binding and coiled-coil domain-containing protein
CHIP C-terminus of Hsc70-interacting protein
CMA chaperone-mediated autophagy
DMOG dimethyloxalylglycine
DNM1L dynamin-1-like protein
DRAM DNA damage-regulated autophagy modulator protein
Env envelope glycoprotein
EPAS endothelial PAS domain-containing protein
ER endoplasmic reticulum;
FIS fission protein
FOXO forkhead box protein
FUND FUN14 domain-containing protein
GRP stress-70 protein
HIF hypoxia-inducible factor
HIV1 human immunodeficiency virus type 1
HSP7C constitutive heat shock protein 70
HSC70 heat shock cognate 70 kDa protein
HSPA heat shock protein family A
KEAP Kelch-like ECH-associated protein
LAMP lysosome-associated membrane glycoprotein
LC3 microtubule associated protein 1 light chain 3
MARH5 membrane-associated RING-CH protein V
MK mitogen-activated protein kinase
MLP3 microtubule-associated proteins 1A/1B light chain
NBR next to BRCA1 gene protein
mTORC mechanistic target of rapamycin complex
NF2L2 nuclear factor erythroid 2-related factor
NFKB nuclear factor NF-kappa-B
NRBF nuclear receptor-binding factor
OPTN optineurin
P53 cellular tumor antigen p53
PARP poly (ADP-ribose) polymerase
PE phosphatidylethanolamine
PEX peroxin
PI3KC phosphatidyl 3-kinase complex
PI3P phosphatidylinositol 3-phosphate
PI3R4 phosphoinositide 3-kinase regulatory subunit
PJVK pejvakin
PK3C phosphatidylinositol 3-kinase catalytic subunit
PMP peroxisomal membrane protein
PTEN Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase
RAB RAS-associated binding
RBCC RB1-inducible coiled-coil protein
RETR reticulophagy regulator
RHEB RAS homologue enriched in brain
ROS reactive oxygen species
RPTOR regulatory-associated protein of mTOR. SAR, specific autophagy receptor
SNARE soluble N-ethylmaleimide-sensitive fusion protein attachment receptor
SQSTM sequestosome
TF transcription factor
TNKS poly (ADP-ribose) polymerase tankyrase
TSC tuberin
ULK UNC-51 like autophagy activating kinase
USP ubiquitin carboxyl-terminal hydrolase
VHL Von Hippel-Lindau
VLCFA very-long-chain fatty acid
Keywords: autophagy, disease, hydrogen peroxide, oxidative damage, peroxisomes, pexophagy, thiol-based redox signaling
Citation: Li H, Lismont C, Revenco I, Hussein MAF, Costa CF and Fransen M (2021) The Peroxisome-Autophagy Redox Connection: A Double-Edged Sword?. Front. Cell Dev. Biol. 9:814047. doi: 10.3389/fcell.2021.814047
Received: 12 November 2021; Accepted: 02 December 2021;
Published: 16 December 2021.
Edited by:
Michael Schrader, University of Exeter, United KingdomReviewed by:
Luisa M. Sandalio, Experimental Station of Zaidín (CSIC), SpainSuresh Subramani, University of California, San Diego, United States
Paul Walton, Western University, Canada
Copyright © 2021 Li, Lismont, Revenco, Hussein, Costa and Fransen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc Fransen, bWFyYy5mcmFuc2VuQGt1bGV1dmVuLmJl