 Inna Serganova1,2
Inna Serganova1,2 Sanjukta Chakraborty1
Sanjukta Chakraborty1 Samuel Yamshon1
Samuel Yamshon1 Yusuke Isshiki1Ryan Bucktrout1
Yusuke Isshiki1Ryan Bucktrout1 Ari Melnick1
Ari Melnick1 Wendy Béguelin1
Wendy Béguelin1 Roberta Zappasodi1,2,3,4*
Roberta Zappasodi1,2,3,4*- 1Division of Hematology and Medical Oncology, Department of Medicine, Weill Cornell Medical College, New York, NY, United States
- 2Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 3Immunology and Microbial Pathogenesis Program, Weill Cornell Graduate School of Medical Sciences, New York, NY, United States
- 4Parker Institute for Cancer Immunotherapy, San Francisco, CA, United States
B-cell non-Hodgkin lymphomas (B-NHLs) are highly heterogenous by genetic, phenotypic, and clinical appearance. Next-generation sequencing technologies and multi-dimensional data analyses have further refined the way these diseases can be more precisely classified by specific genomic, epigenomic, and transcriptomic characteristics. The molecular and genetic heterogeneity of B-NHLs may contribute to the poor outcome of some of these diseases, suggesting that more personalized precision-medicine approaches are needed for improved therapeutic efficacy. The germinal center (GC) B-cell like diffuse large B-cell lymphomas (GCB-DLBCLs) and follicular lymphomas (FLs) share specific epigenetic programs. These diseases often remain difficult to treat and surprisingly do not respond advanced immunotherapies, despite arising in secondary lymphoid organs at sites of antigen recognition. Epigenetic dysregulation is a hallmark of GCB-DLBCLs and FLs, with gain-of-function (GOF) mutations in the histone methyltransferase EZH2, loss-of-function (LOF) mutations in histone acetyl transferases CREBBP and EP300, and the histone methyltransferase KMT2D representing the most prevalent genetic lesions driving these diseases. These mutations have the common effect to disrupt the interactions between lymphoma cells and the immune microenvironment, via decreased antigen presentation and responsiveness to IFN-γ and CD40 signaling pathways. This indicates that immune evasion is a key step in GC B-cell lymphomagenesis. EZH2 inhibitors are now approved for the treatment of FL and selective HDAC3 inhibitors counteracting the effects of CREBBP LOF mutations are under development. These treatments can help restore the immune control of GCB lymphomas, and may represent optimal candidate agents for more effective combination with immunotherapies. Here, we review recent progress in understanding the impact of mutant chromatin modifiers on immune evasion in GCB lymphomas. We provide new insights on how the epigenetic program of these diseases may be regulated at the level of metabolism, discussing the role of metabolic intermediates as cofactors of epigenetic enzymes. In addition, lymphoma metabolic adaptation can negatively influence the immune microenvironment, further contributing to the development of immune cold tumors, poorly infiltrated by effector immune cells. Based on these findings, we discuss relevant candidate epigenetic/metabolic/immune targets for rational combination therapies to investigate as more effective precision-medicine approaches for GCB lymphomas.
Introduction
Despite the clinical success of immune checkpoint blockade (ICB) therapy in solid tumors (Zappasodi et al., 2018a; Zappasodi et al., 2018b; Ribas and Wolchok, 2018), B-cell lymphomas remain largely refractory to these treatments, with the exception of Hodgkin’s lymphoma, where PD-L1 (programmed death-ligand 1) constitutes a direct tumor target (Zappasodi et al., 2015; Ansell, 2019; Armand et al., 2021a). Moreover, while the majority of patients with relapsed/refractory B-cell non-Hodgkin lymphomas (B-NHLs) respond to CAR (chimeric antigen receptor) T-cell therapy, the duration of these responses is limited in many cases (Schuster et al., 2018; Hirayama et al., 2019). Hence, there is a need to identify and overcome the barriers that prevent successful immunotherapy in B-NHL patients. Although B-NHLs are diseases of the immune system and pose a completely different immunologic scenario compared to solid tumors, these malignancies arise at sites of antigen recognition where immunotherapies that precisely disengage T-cell effector functions are expected to work (Zappasodi et al., 2015; Zappasodi et al., 2018a). In fact, in the rare cases of B-NHLs where immunotherapy successfully elicits protective anti-tumor immune responses, tumor remissions can be long-lasting (Lesokhin et al., 2016; Fuca et al., 2021). Overall, this underscores the potential of immunotherapy to treat lymphomas and, at the same time, our poor understanding of the mechanisms that limit or prevent its efficacy in these diseases.
Recent efforts to improve the genetic classification of diffuse large B-cell lymphomas (DLBCLs)—the most common lymphoid malignancy in adults (Swerdlow et al., 2016)—have revealed the effect of specific driver epigenetic mutations to alter expression of T-cell immune co-receptors and/or downstream signaling molecules (Chapuy et al., 2018; Wright et al., 2020). These genetic features occur more frequently in germinal center (GC) subtypes of DLBCL and are also shared by follicular lymphoma (FL)—the second most frequent form of B-NHLs, which is also of GC origin (Morin et al., 2010; Carbone et al., 2019) (abbreviated thereafter as GCB lymphomas). Specifically, gain-of-function (GOF) mutations in the histone methyltransferase EZH2 (enhancer of zeste homolog 2) or loss-of-function (LOF) mutations in the histone acetyl transferase CREBBP (cAMP-response element binding protein (CREB) binding protein) or EP300 (E1A binding protein P300) or histone methyltransferase KMT2D (lysine methyltransferase 2D), which occur in 30–40% of these diseases, contribute to the repression of antigen presentation, IFN-γ response genes, or CD40 signaling in lymphoma cells (Mlynarczyk et al., 2019). These results suggest that escape from T-cell recognition and killing is inherent part of the GCB lymphoma oncogenic program and may be controlled at an epigenetic level in these diseases.
Epigenetic and metabolic reprogramming are usually deeply linked in cancer cells. The influence of tumor-intrinsic oncogenic signaling and tumor microenvironmental factors on the availability of metabolites that are substrates or inhibitors of epigenetic enzymes is well described (Kinnaird et al., 2016; Izzo et al., 2021). In addition, altered expression or activity of chromatin-modifying enzymes can impact directly and indirectly on cellular metabolism.
Here, we review the bidirectional relationship between epigenetics and metabolism in GCB lymphomas and its impact on the immune microenvironment. First, we focus on genetic and epigenetic characteristics of GCB DLBCLs and FLs, highlighting the most common alterations in EZH2, CREBBP, and/or KMT2D epigenetic modifiers and their function in histone modifications and chromatin remodeling. We then discuss principles linking the activity of chromatin-modifying enzymes and lymphoma metabolism and the impact of these mechanisms in anti-lymphoma immunity and disease progression. Lastly, we discuss current therapeutic interventions that could be harnessed in combination to target this metabolic-epigenetic crosstalk and potentially improve the response of GCB lymphomas to immunotherapy.
Molecular Features Supporting GC B-Cell Development and Lymphomagenesis
During their lifetime, B cells undergo a stepwise process including activation, proliferation, differentiation, and antibody secretion, which is controlled by a specific network of intracellular signaling pathways and transcription factors (TFs) deeply regulated at epigenetic level and in response to microenvironmental stimuli. GCs comprise two histologically distinct regions: the dark zone (DZ), with very proliferative GC B cells in which immunoglobulin (Ig) class-switch recombination (CSR) and somatic hypermutation (SHM) occur, and the light zone (LZ), where non-dividing GC B cells with appropriate B cell receptors (BCRs) interact with follicular dendritic cells (FDCs) and follicular helper CD4+ T cells (TFH) cells to receive proper help for further cycling into the DZ or maturation into plasma cells (PCs) (Mesin et al., 2016). Normal GC B-cell development depends on the cooperation of epigenetic and non-coding elements to control expression of multiple genes. Recent genome-wide studies allowed to map changes in the chromatin landscape, DNA methylome, 3-dimensional interactome, and coding and non-coding transcriptomes of normal and malignant B cells (Mlynarczyk et al., 2019). DNA methylation changes more frequently occur at gene body and remote upstream regions than promoter regions, although demethylation of key B-cell TF binding sites correlates with expression of those TFs and their transcriptional programs (Andrews and Payton, 2019). In GC B cells and GCB lymphomas, activation-induced cytosine deaminase (AID), which drives somatic hypermutation, also mediates DNA hypomethylation and increased methylome heterogeneity in regions associated with essential B-cell lineage genes (Dominguez et al., 2015; Teater et al., 2018). Recent data revealed that activating and repressive histone marks, chromatin accessibility, and gene expression determine defined regulatory landscape transitions in normal development of human or murine naïve B cells toward PCs (Kania et al., 2017; Kania et al., 2019). Perturbation of this program can lead to lymphomagenesis. As an example relevant for GCB lymphomagenesis, conditional deletion of the histone acetyltransferase CREBBP perturbs B-cell development and accelerates the development of lymphoma in BCL2-and MYC-driven mouse models (García-Ramírez et al., 2017; Hashwah et al., 2017; Jiang et al., 2017; Zhang et al., 2017). This is largely because of the role of CREBBP in the GC reaction to counteract the repressive effects of BCL6 by H3K27 acetylation at enhancers of BCL6 target genes, thus leading to GC exit. EZH2 histone methyltransferase—another commonly mutated epigenetic modifier in GCB lymphomas—is required for the formation and maintenance of GC reaction (Béguelin et al., 2013; Béguelin et al., 2016). Thus, several chromatin modifiers regulate key B-cell TFs to temporally regulate developmental transcriptional programs, and, when mutated, are lymphomagenic.
By genetic profiling of patient-derived DLBCLs, epigenetic regulator genes (EZH2, CREBBP, and/or KMT2D) were shown to help classify a subset of DLBCLs into a specific “cluster 3” (C3) (Chapuy et al., 2018) or “EZB” (EZH2 mutation and BCL2 translocation) (Wright et al., 2020) disease subtype. KMT2D is the most frequently mutated epigenetic regulator gene in DLBCL, with its mutation occurring in 24 and 28% of all DLBCL or GCB-DLBCL cases, respectively (Pasqualucci et al., 2011a; Reddy et al., 2017). Mutations in EZH2 and CREBBP are also enriched in GCB-DLBCL (12 and 16%) compared with all DLBCL cases (6 vs. 11%, respectively) (Pasqualucci et al., 2011a; Reddy et al., 2017). These genetic subclassifications have critically improved our ability to stratify patients with different prognosis after standard therapy with rituximab (anti-CD20) + CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone) chemotherapy (R-CHOP). According to transcriptional profiling, DLBCLs were classified into cell of origin (COO) categories, where GCB-DLBCLs were found to generally associate with a more favorable outcome compared with activated-B-cell (ABC)-DLBCLs. However, C3/EZB GCB-DLBCLs show significantly worse prognosis compared to other GCB-DLBCLs and progression-free survival (PFS) as short as the worst prognostic subtypes of ABC-DLBCL (Chapuy et al., 2018). The survival disadvantage of EZB/C3 DLBCLs may be at least partially explained by the fact that the mutational characteristics of double hit (DHIT) lymphomas, an aggressive subtype of DLBCL characterized by MYC and BCL2 translocation, are enriched in this category (Ennishi et al., 2019a; Sha et al., 2019; Wright et al., 2020). Indeed, the inferior survival of EZB DLBCLs was only observed in patients with EZB DLBCL expressing DHIT genetic signatures (Wright et al., 2020). MYC overexpression also affects lymphoma immunophenotype, transcriptional characteristics and metabolic conditions. C3/EZB DLBCLs without MYC alterations (EZB-MYC−) generally show LZ GCB cell-like gene expression profiles, in contrast to C3/EZB DLBCLs with MYC rearrangements (EZB-MYC+), which are enriched in DZ signatures, very likely because MYC expression promotes DZ re-entry and proliferation (Béguelin et al., 2020; Wright et al., 2020). Notably, the proliferative phenotype of EZB-MYC+ DLBCLs is coupled to highly glycolytic metabolism and sustained protein and lipid synthesis in contrast to EZB-MYC- and other types of DLBCL (Wright et al., 2020).
The mutational landscape of FL is close to that of C3/EZB DLBCLs and the incidence of mutations in epigenetic modifier genes are more frequent in FL than in DLBCLs (mutant KMT2D, >60%; mutant CREBBP, >50%; mutant EZH2, >15%) (Okosun et al., 2014; Pasqualucci et al., 2014; Green et al., 2015; Krysiak et al., 2017). Similar to C3/EZB DLBCLs, acquisition of MYC translocation, amplification or activating mutations is associated with aggressive histology in FL, predisposing to transformation to aggressive DLBCL (Pasqualucci et al., 2014). Thus, mutational characteristics of GCB-DLBCL and FL are similar and mutations in epigenetic modifier genes may play essential roles for the development and progression of these diseases.
Metabolic Features of Normal and Tumor GC B Cells
GC B cells, especially in the DZ, are highly proliferative and need to activate specific transduction programs to meet high energetic and biosynthetic demands. These dynamic processes are possible thanks to the great metabolic plasticity of B cells in the GC. Naive B cells are metabolically quiescent and require low levels of catabolic metabolism to sustain energy homeostasis. Following activation, B cells re-shape their metabolic program to meet the energetic and biosynthetic demands for proliferation (Jellusova, 2020). GC B cells use different carbon energy sources and metabolic pathways depending on their stage in the GC reaction process (Choi and Morel, 2020). Studies in mice showed that in comparison with naïve/resting B cells, GC B cells upregulate glucose consumption, together with upregulation of gene signatures for glycolysis, TCA (tricarboxylic acid) cycle and OxPhos (oxidative phosphorylation). These cells present increases in mitochondrial mass and HIF-1α accumulation (Jellusova et al., 2017; Jellusova and Rickert, 2017). Inhibition of glycolysis with hexokinase inhibitors or 2-deoxy-D-glucose (2-DG) significantly decreases the percentage of GC B cells, without affecting the overall percentage of B cells, CD4+PD1+ TFH cells or the CD4+:CD8+ T-cell ratio in GCs, pointing to the preferential dependency of GC B cells on glycolysis (Jellusova et al., 2017). However, without detailed direct metabolic analyses in vivo, the specific metabolic demand and related metabolic pathways of GC B cells are difficult to precisely determine. Studying these parameters in vitro is limiting and has led to conflicting results. For example, MS (mass spectroscopy) of spleen-derived CD19+B220+CD4−CD8− B cells cultured in 13C6-glucose detected reductions in total glycolytic metabolites, except for 3-phosphoglycerate (3-PG) upon B-cell activation. Moreover, lactate level accumulation was not observed in these studies (Waters et al., 2018). The isotopologue distribution in glycolytic metabolites suggested that glucose fluxed through the glycolytic pathway without accumulation of lactate, probably routed into alternative metabolic pathways (Waters et al., 2018). Recently, Weisel et al.(Weisel et al., 2020), using freshly isolated primary GC B cells, have shown that these cells are poorly glycolytic and consume higher oxygen amounts than resting naïve B cells or activated T cells. Specifically, GC B cells were found to oxidize both endogenous and exogenous fatty acids through high expression of the fatty-acid transporter CD36 (Weisel et al., 2020). These studies highlight a complex relationship between metabolic and activation states of B cells during the GC response. Interestingly, the transcriptional repressor Bcl6, which deeply controls the GC B-cell program, was identified among the genes specifically regulated in adipocytes, suggesting a role for BCL6 in lipid metabolism. Correspondingly, Bcl6-deficient mice were found to exhibit multiple features of dysregulated lipid metabolism (LaPensee et al., 2014).
According to the described metabolic features of normal GC B cells, a bioinformatics study in DLBCLs revealed that 30% of these tumors rely on OxPhos. In this work, DLBCLs were divided into three subgroups, based on COO and genetic basis for transformation: 1) OxPhos, 2) B-cell receptor (BCR)/proliferation, and 3) “host response” (HR) subsets (Monti et al., 2005). In comparison with BCR-DLBCLs, OxPhos-DLBCLs were found to display enhanced mitochondrial energy transduction, greater incorporation of nutrient-derived carbons into the TCA cycle, and increased glutathione levels. Moreover, perturbation of the fatty acid oxidation (FAO) program and glutathione synthesis proved selectively toxic to this tumor subset, providing evidence for distinct metabolic dependencies and underlying pro-survival mechanisms in DLBCLs (Caro et al., 2012). The differential utilization of fatty-acid-derived carbons and glucose in OxPhos vs. non-OxPhos DLBCLs correlated with the absence or presence of functional BCR signaling, respectively, presenting an example of heterogeneity in nutrient use within the same disease entity. Besides, the utilization of palmitate-derived acetyl-CoA for ATP production and citrate synthesis in OxPhos-DLBCLs suggests that FAO and fatty acid synthesis may coincide in these cells and the inhibition of the mitochondrial FAO program can compromise the survival of OxPhos-DLBCLs. Notably, OxPhos-DLBCLs have shown specific resistance to pan-HDAC inhibitors (HDACis) linked to upregulation of antioxidant pathways after HDAC inhibition, indicating that lymphoma metabolic subtypes may predispose to differential responses to epigenetic therapies (Mensah et al., 2021).
DLBCLs are also considerably dependent on the mitochondrial lysine deacetylase enzyme SIRT3, which belongs to the NAD+ dependent deacetylase family sirtuins and regulates anaplerotic glutaminolysis to fuel the TCA cycle and ensures elevated production of biosynthetic precursors needed for rapidly growing lymphoma cells (Li M. et al., 2019). In addition, SIRT3 promotes mitochondrial metabolism and reduces reactive oxygen species via multiple mechanisms. Interestingly, reliance on SIRT3 in DLBCL is independent of the COO (Alizadeh et al., 2000) or the OxPhos or BCR categories (Monti et al., 2005).
Principles Linking Metabolism to Epigenetics
The activity of epigenetic modifiers is influenced by cellular metabolism and the availability of metabolic products, which in tumors can depend on oncogenic alterations and/or on the tumor microenvironment (TME) (Table 1). DNA and histone methyltransferase and demethylases, histone acetyltransferases (HATs), and deacetylases (HDACs) utilize as substrates and co-factors metabolites derived from serine-glycine-one carbon metabolism, methionine, TCA cycle, ß-oxidation, glycolysis, and hexosamine biosynthesis (Allis and Jenuwein, 2016; Greer and Shi, 2012). More specifically, S-adenosyl methionine (SAM) from the one-carbon metabolism pathway, acetyl-coenzyme A (acetyl-CoA) from TCA-derived citrate, NAD+ from glycolysis or electron transport chain, α-ketoglutarate (αKG) from the TCA cycle, uridine diphosphate N-acetylglucosmaine and other metabolic intermediates in these pathways serve as substrates for chromatin-modifying enzymes (Figure 1). Acetyl-CoA, which is the donor for histone acetylation reactions, and SAM, which is the universal donor for all epigenetic methylation reactions involving DNA and histones, are generated through glucose, amino acid, fatty acid and vitamin metabolism are rate limiting substrates for these chromatin modifying reactions (Kim and Costello, 2017; Su et al., 2016). Therefore, these epigenetic modifications depend on the availability of substrates derived from specific metabolic pathways (Figure 1). For example, enzymes involved in histone and DNA methylation and demethylation can be regulated by both methionine metabolism (generating SAM) and TCA cycle (generating αKG as co-substrate for JmjC histone demethylases), thus linking epigenomic changes to the metabolic state of a cell (Figure 1). DNA methylation alters chromatin structure and regulates gene expressions by converting cytosine into 5-methylcytosine (5 mC). Changes in histone methylation at lysine (K) or arginine (R) amino acid residues can either activate or repress transcription. Histone methylation status can range from mono-/di-/tri-methylation or full demethylation, creating a diverse array of methylation patterns. Both histone and DNA methylation require SAM as the high-energy methyl donor, preferably localized in the nucleus (Figure 1). Acetyl-CoA, which is synthetized from glucose oxidation to pyruvate through pyruvate dehydrogenase (PDH), or from fatty acid ß-oxidation and acetate, metabolically sustains ATP production under aerobic condition and several biosynthetic processes. When not needed for these downstream metabolic processes, acetyl-CoA can diffuse from cytoplasm to the nucleus or can be locally produced in the nucleus (Nitsch et al., 2021), thus becoming available as substrate for HATs to modify histone tails (Figure 1), which is one of the major determinants of chromatin epigenetic state impacting on gene expression.

TABLE 1. Main metabolic substrates and co-factors of epigenetic enzymes.
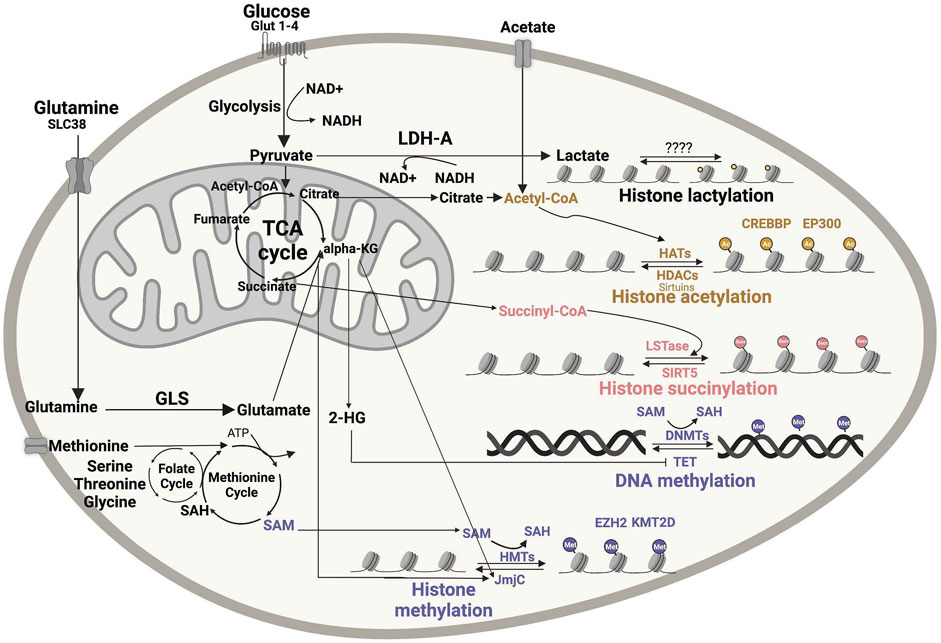
FIGURE 1. Impact of metabolic intermediates on cellular epigenomic. Numerous nutrients are metabolized to produce intermediates that can be used as substrates or modulators of enzymes involved in chromatin remodeling. Acetyl-CoA, lactate, succinyl-CoA and SAM are the major metabolic bioproducts involved in epigenetic reactions, including histone acetylation, histone methylation, succinylation, lactylation and DNA methylation. DNMT, DNA methyltransferases; GLS, glutaminase; 2-HG, 2-hydroxyglutarate; a-KG, α-ketoglutarate; SAH, S-adenosyl homocysteine; SAM, S-adenosyl methionine dehydrogenase 1; HAT, histone acetyltransferase; HDAC, histone deacetylases; HMT, histone methyltransferase; JmjC, Jumonji N/C-terminal domains; LDH-A, lactate dehydrogenase A; LSTase, lysine succinyltransferase; NAM, nicotinamide; SIRT, sirtuin; TCA, tricarboxylic acid; EZH2- Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit, KMT2D- Histone-lysine N-methyltransferase 2D. Figures were created using BioRender.com.
Other metabolic intermediates, such as S-adenosylhomo-cysteine (SAH) and ß-hydroxyglutarate (2-HG), can instead affect the activity of chromatin-modifying enzymes by competitively inhibiting SAM and αKG substrate utilization, respectively. Notably, when not efficiently utilized or too abundant in the cell, SAM can regulate the folate cycle by directing it away from sustaining the methionine cycle, thus lowering SAM levels themselves. Overall, these observations indicate that the activity of epigenetic enzymes must tightly respond to changes in cellular metabolism (Reid et al., 2017).
Despite multiple studies suggesting a link between cellular metabolism and histone modifications, the integration of metabolic signals into chromatin changes via histone methylation and acetylation is challenging. Recent progress in MS and metabolic tracing approaches is now deepening our understanding of these mechanisms. By MS, it was recently found that histone modifications—especially acetylation—can be regulated both enzymatically and nonenzymatically (Simithy et al., 2017). Using metabolic tracing of [13C3] lactate, Zhang et al.(Zhang et al., 2019) identified lactylation as a new histone modification derived from lactate (Figure 1). Mentch et al.(Mentch et al., 2015) provided evidence that both SAM levels and the SAM/SAH ratio can be quantitatively altered through changes in the metabolic flux of the methionine cycle to affect a chromatin status. Overall, MS is proving an extremely useful tool to dissect the impact of metabolic pathways on epigenetic modifications (Lu et al., 2020). Similar technologies and assays applied to B-cell lymphomas that heavily rely on epigenetic reprogramming will provide new insights into the regulation of epigenetics through cellular metabolism in these diseases, with the potential to unveil novel vulnerabilities that can be targeted for therapy.
Role of the Immune Microenvironment in GCB Lymphomas
The role of the immune microenvironment in B-cell lymphoma pathogenesis is well recognized (Dave et al., 2004; Kotlov et al., 2021); however, the relative impact of different immune cell types on lymphoma immune escape and immunotherapy resistance is not entirely clear. For example, except for CD8+ T cells that seem to associate with better prognosis in B-NHL patients (Álvaro et al., 2006; Chang et al., 2007; Wahlin et al., 2007; Wahlin et al., 2010), other T-cell subsets, such as immunosuppressive regulatory T cells (Tregs) and T cells expressing various immune checkpoints variably associate with either positive or negative outcomes (Ansell et al., 2001; Carreras et al., 2006; Tzankov et al., 2008; Carreras et al., 2009; Farinha et al., 2010; Wahlin et al., 2010; Rajnai et al., 2012; Yang et al., 2012; Keane et al., 2013; Brady et al., 2014; Coutinho et al., 2015; Yang et al., 2015; Zhou et al., 2017; Greenbaum et al., 2019). B-cell lymphomas are unique given the fact that these tumor cells arise from professional antigen presenting cells (APCs)—a specialized subset of immune cells able to capture and optimally present antigens (Ags) to T cells through both MHC (major histocompatibility complex)-I and MHC-II routes (de Charette et al., 2016). This would suggest that these tumors are highly immunogenic in nature and may need to induce specific mechanisms of immune suppression to evade immunosurveillance. Aberrant oncogene expression in B cells can occur via genetic alterations during Ig gene rearrangements, but these potentially lymphomagenic cells are often recognized and eliminated by the immune system (Upadhyay et al., 2015). Nonetheless, depending on the sets of oncogenic mutations accumulated over time in altered B cells and their impact on direct pro-survival signals and immune evasion, the tumor can eventually manifest, indicating complete escape from the immune system. In this section we discuss the most common mechanisms of immune dysfunction and immunosuppression observed in GCB lymphomas, including DLBCLs and FLs (Figure 2).
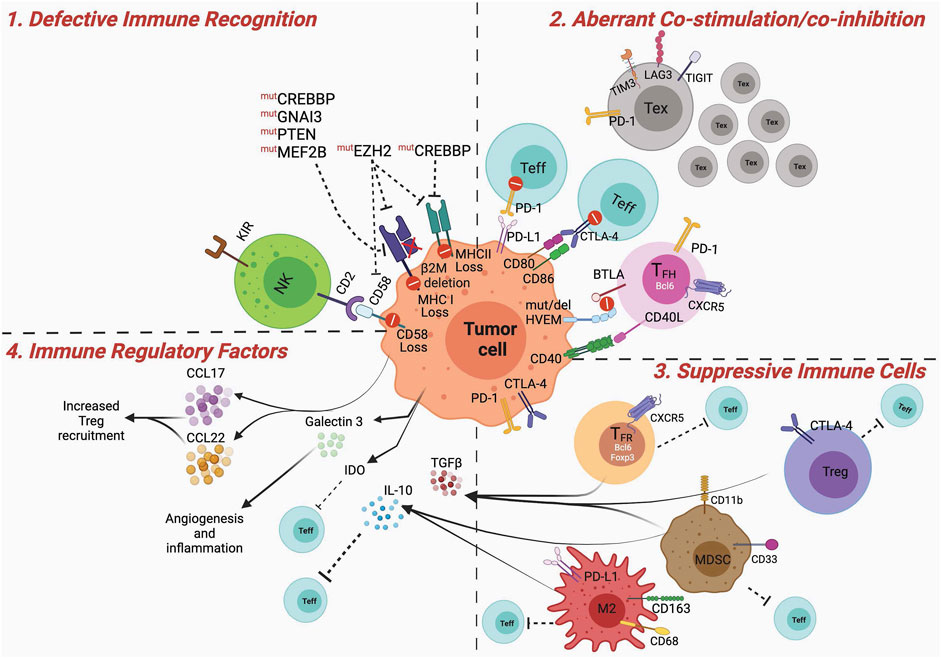
FIGURE 2. Dysfunctional immune microenvironment in GCB Lymphomas. Major mechanisms contributing to a dysfunctional and suppressive immune microenvironment in GCB lymphomas. 1) Defective immune recognition: MHC-I and MHC-II expression are often downregulated in GCB lymphoma cells, via mechanisms involving various genetic and epigenetic mutations, leading to poor antigen recognition. In addition, disruption of CD58/CD2 axis impedes tumor recognition by NK cells. 2) Aberrant co-stimulation: PD-1 and CTLA-4 can be expressed on tumor-infiltrating effector T cells (Teff) limiting or counteracting their activation via signals received by PD-L1 or inhibition of CD80/CD86 mediated co-stimulation. Tumor B cells can directly express PD-1 and CTLA-4 contributing to dampening T-cell activation. Mutation or deletion of HVEM on the lymphoma cells, renders them non-reactive to BTLA expressing TFH cells, and leads to aberrant expansion of lymphomagenic population (see text for more details). Lymphogenic B cells can proliferate independent of CD40/40L-mediated TFH cell help. Tumors are infiltrated by large numbers of exhausted T cells that, expressing TIM3, LAG3 and TIGIT, are subjected to sub-optimal co-stimulation and activation. 3) Suppressive immune cells: Tregs, TFR, M2 macrophages and MDSCs suppress activation of Teff cells. This can be mediated by receptor ligand interaction, such as PD-1:PD-L1, CTLA-4:CD80/CD86 or via soluble factors. 4) Immune regulatory factors: IL-10, TGFb, IDO secreted by MDSCs, macrophages, Tregs, TFR, or tumor cells induce immune suppression, by impeding optimal DC priming, promoting M2 polarization, or Treg differentiation. In addition, the release of chemoattractant (e.g. CCL17, CCL22) for suppressive immune cells can further contribute to the establishment of an immune suppressed lymphoma microenvironment. Figures were created using BioRender.com.
Defective Antigen Presentation
Downregulation or complete loss of Ag presentation machinery is observed in DLBCL (Challa-Malladi et al., 2011; Nijland et al., 2017) and MHC-I loss has been reported in 40–60% of DLBCL cases (Challa-Malladi et al., 2011; Rimsza et al., 2006; Rimsza et al., 2004). Genetic mutations or loss of beta2-microglobulin (β2M), which destabilize the assembly of the MHC class I, is a primary cause for MHC-I downregulation (Figure 2). β2M mutations are observed in 29% of DLBCL cases (Challa-Malladi et al., 2011), and cytoplasmic β2M isoforms were detected in 48.4% MHC-I-negative DLBCL cases (Nijland et al., 2017). In FL, ∼20% cases harbor MHC-I mutations (Fangazio et al., 2021) and β2M genetic alterations are generally rare (Green et al., 2015), but their frequency increases post histological transformation (Pasqualucci et al., 2014). Mutations in EZH2, GNA13, and MEF2B, as well as PTEN deletions, are significantly associated with MHC-I loss in DLBCLs (Figure 2) (Ennishi et al., 2019b). As discussed below in more detail, EZH2 activating mutations contribute to both MHC-I and MHC-II repression (Ennishi et al., 2019b). Around 40–50% of DLBCL cases have low MHC-II expression, which correlates with poor lymphocytes infiltration and shorter survival in DLBCL patients (Rosenwald et al., 2002; Roberts et al., 2006). MHC-II expression is under stringent epigenetic regulation. CREBBP activates the MHC-II-gene-expression regulator CIITA by catalyzing promoter/enhancer H3K27Ac. CREBBP LOF mutations prevent CIITA transcription in FLs and DLBCLs. MHC-II downregulation in DLBCL also results from overexpression of the TF FOXP1, which seems to be independent of CIITA mutation (Brown et al., 2016). Genetic alterations on chromosome 3p leading to FOXP1 overexpression are found in a small subset of DLBCLs (Koon et al., 2007). FOXP1 translocations are rare in DLBCLs and are often associated with extra-nodal localizations and high proliferative index (Haralambieva et al., 2006). MHC-II expression is reduced in FL cells at both transcription and protein levels, resulting in impaired Ag presentation (Green et al., 2015; Andor et al., 2019). Pharmacologic inhibition of EZH2 or thymidylate synthase (TS) were found to enhance MHC-I expression in human DLBCL cell lines (Dersh et al., 2021). TS contributes to the biosynthesis of thymidine and is important for DNA replication and repair. Interestingly, combined inhibition of EZH2 and TS displayed increased efficacy against DLBCL cells that are resistant to EZH2 inhibitors (Dersh et al., 2021).
Aberrant Immune Co-stimulation and Co-inhibition
Effective priming of T cells needs two signals from APCs: 1) recognition of the MHC-Ag complex by the T-cell receptor (TCR) and 2) co-stimulation by the interaction between CD80/CD86 on APCs and CD28 on T cells. CD80 and CD86 are B7 family members and are two of the most important mediators of this second signal post Ag recognition. Additional ligands for immune co-stimulatory and co-inhibitory receptors on T cells further fine tune T-cell activation and fate (Chen and Flies, 2013). In this regard, two major examples are CTLA-4 (cytotoxic T-lymphocyte associated protein 4)—a T-cell co-inhibitory receptor with higher affinity for the same CD28 ligands CD80/CD86 (Lee et al., 1998), and PD-1 (programmed cell death protein 1)—another crucial T-cell co-inhibitory receptor dampening the TCR and CD28 downstream signaling pathways upon engagement with its ligands PD-L1 and PD-L2, which can be expressed by tumor cells and immune cells, including APCs (Figure 2) (Pauken et al., 2021).
The role of CD80/CD86 is not yet fully clear in lymphoma, but their expression has been noted on tumor cells and/or on cells from the TME (Greaves and Gribben, 2013). 97% of FL cases and approximately 90% of DLBCL cases express CD80 (Dakappagari et al., 2012). Downregulation of CD80/CD86 has been associated with poor T-cell infiltration in DLBCL (Stopeck et al., 2000). As CD80 can directly interact with PD-L1 with either immune stimulatory or inhibitory outcomes (Pauken et al., 2021), the hierarchy and all possibility of these interactions in the various lymphoma TME need to be considered when assigning patients to ICB-based treatments.
CTLA-4 expression is detectable in both DLBCL and FL cells. Using human lymphoma cell line OCI-Ly3, Hermann et al. have shown that CTLA-4 expressed on these cells can interact with soluble CD86 and internalize it (Herrmann et al., 2017). This interaction can trigger the STAT3 (signal transducer and activator of transcription 3) pathway via phosphorylation of Tyk2 (tyrosin kinase 2) in B cells, with consequent induction of the immunoregulatory cytokines IL-10 and IL-6 (Herrmann et al., 2017), which facilitates immune evasion and supports tumor growth. In the same study, using a co-culture system, the authors showed that A20 lymphoma cells can internalize CD86 expressed on APCs via CTLA-4.
PD-1 is constitutively expressed on naïve B cells and is rapidly recruited to the immune synapse with BCR upon B-cell activation (Thibult et al., 2013). Various structural chromosomal alterations, including translocations involving the Ig heavy chain (IgH) locus or disruption of the 3’ region of the PD-L1 gene, can lead to aberrant PD-L1 expression in DLBCL (Georgiou et al., 2014; Kataoka et al., 2016). Interestingly, the Ig locus and CIITA are common partners of PD-L1 translocations in DLBCL (Steidl et al., 2011; Chapuy et al., 2016; Georgiou et al., 2016). However, the frequency of such events in GCB lymphomas is overall low, which may explain at least in part the lack of activity of PD-(L)-1 blockade in these diseases. In FL, a small fraction of neoplastic B cells (∼5%) and histiocytes express PD-L1 (Carreras et al., 2009; Myklebust et al., 2013).
T cells infiltrating lymphoma tissues compared to tonsil more frequently express PD-1 and display an exhausted phenotype, including co-expression of other T-cell inhibitory receptors, such as TIM3 (T-cell immunoglobulin (Ig) and mucin-domain-containing molecule 3) in FL (Yang et al., 2017), LAG3 (lymphocyte activating 3) in DLBCL (Roussel et al., 2021) and/or TIGIT (T-cell immunoreceptor with immunoglobulin and ITIM domains) in both FL and DLBCL (Figure 2) (Josefsson et al., 2019). PD-1 expression is detected in the TME of 39.5–68.6% of DLBCL cases (Song et al., 2019), but it has mixed clinical implications. While some studies observed that PD-1+ tumor-infiltrating lymphocytes (TILs) are associated with favorable clinical outcome in DLBCL (Muenst et al., 2010; Ahearne et al., 2014; Kiyasu et al., 2015; Fang et al., 2017), a recent study by Enemark et al. showed that PD-1 on intrafollicular T cell is a predictive biomarker for histological transformation of FL into DLBCL (Beck Enemark et al., 2021). In DLBCL, PD1+TIM3+CD8+T cells with an effector memory phenotype are observed inside CD20+ B-cell clusters (Roussel et al., 2021). In FL, TIM3 is expressed in ∼30–40% TILs (mainly CD8), with even greater expression in functionally exhausted PD-1low T-cell subsets (Yang et al., 2015). A recent study in two independent DLBCL cohorts uncovered similar trends in TIM3+LAG3+ TIL abundance in these diseases, and these cells were found to be an independent predictor of poor survival (Autio et al., 2021). Consistently, in FL patients, the presence of CD3+LAG-3+ as well as TIM-3+LAG-3+ TILs correlates with poor survival (Yang et al., 2017). TIM3 and LAG3 have also found to be expressed on DLBCL cells themselves and high TIM3 expression in these tumor cells correlates with shorter survival in patients (Chen et al., 2019; Keane et al., 2020). Interestingly, in FL patients, exhausted-phenotype TIGIT+CD8+ T cells and highly suppressive TIGIT+ Tregs in the TME contribute to resistance to PD-1 blockade (Josefsson et al., 2018; Yang et al., 2020). While the TIGIT inhibitory ligand CD155 can be expressed in normal B cells, its expression in lymphoma B cells in associating with infiltrating TIGIT+ T cells has not been deeply investigated.
The HVEM(TNFRSF14):LIGHT/BTLA/CD160 axis is another relevant immune modulatory pathway in B-cell lymphoma. HVEM can deliver co-stimulatory or inhibitory signals depending on the interactions with LIGHT vs. BTLA or CD160, respectively. While HVEM is mostly expressed in T cells, it can be also found on B cells, and is mutated in a fraction of FLs and often lost in EZB DLBCLs (Mlynarczyk et al., 2019) and other subtypes of DLBCLs (Kennedy and Klein, 2019). This suggests that the HVEM pathway normally controls GC B cells. This effect appears to be mainly mediated by TFH, which express high levels of the inhibitory counter-receptor BTLA (B- and T-lymphocyte attenuator) (Figure 2) (Kashiwakuma et al., 2010). TFH cells can have a dual effect on lymphoma B cells, especially of GC origin, because of their physiologic function to provide pro-survival signals (e.g. via CD40L:CD40) only to B cells that have optimally re-arranged their Ig genes and to restrain the growth of the other suboptimal clones (Basso et al., 2004; Good-Jacobson et al., 2010). Loss of HVEM in GCB lymphoma cells inactivates a major mechanism through which TFH properly dose and direct their helper functions toward the most fit B cells. Two complementary preclinical studies showed that HVEM insufficiency in lymphoma cells increases the proportion of TFH and FDCs in the TME (Boice et al., 2016) while concurrently reducing the ability of tumor B cells to interact with TFH (Mintz et al., 2019), leading to overgrowth of HVEM−/− malignant cells. Increases in intratumor TFH that no longer control the HVEM-defective lymphoma clone can in turn support tumor growth through the secretion of IL-4 and other pro-lymphomagenic cytokines (Boice et al., 2016). In this context, ICOS+PD-1+ TFH cells and their signature cytokine IL-21 are associated with poor therapeutic outcome in B-NHLs (Brady et al., 2014). Interestingly, FL is also enriched in follicular regulatory T cells (TFR), which persist along with TFH cells after rituximab therapy and contribute to immune suppression and poor therapeutic outcome (Ochando and Braza, 2017). Mechanistically, it has been shown that mesenchymal stromal cells can induce Foxp3 in FL-associated TFH cells, converting them into TFR. Clarifying the biologic significance of the TFH/TFR plasticity and potential inter-differentiation in GCB lymphomas will be crucial to understand how to precisely target or repolarize them for improved lymphoma control.
Overall, these findings illustrate the complex rewiring of the immune interactions between lymphoma B cells and T cells through aberrant co-stimulation and co-inhibitory pathways, which contributes to dampening anti-tumor immunity and to lymphoma growth. Clarifying whether and how defined sets of driver mutations in lymphoma specifically reshape the immune microenvironment will add important information to the molecular classification of these diseases and for improved patients’ treatment with immunotherapy. This is particularly relevant for GCB lymphomas, where mutations in epigenetic modifiers have a direct impact on Ag presentation and T-cell co-stimulation. Understanding whether these effects directly impact on the differentiation of specific immune microenvironments will be important for the development of more successful precision immune-oncology treatments for these diseases.
Cellular and Soluble Mediators of Active Immunosuppression
Major immunosuppressive mediators in lymphoma microenvironment include Tregs, tumor associated macrophages (TAM), myeloid derived suppressor cells (MDSCs), immunoregulatory cytokines/chemokines and other soluble factors, such as products of aberrant tumor metabolism (Figure 2).
Foxp3+ Tregs can suppress T- and B-cell function. In DLBCL tissue compared to normal lymph nodes, the frequency of intra-tumoral Tregs increases ∼3 times (38%, vs. 12% of CD4 cells) (Mittal et al., 2008). The prognostic implication of Tregs in B-cell lymphomas is still not entirely clear. In FL, Foxp3+ cells measured by immunohistochemistry have been found to correlate with prognosis depending on their spatial distribution, with intrafollicular localization of Tregs being associated with poor survival and risk of transformation (Farinha et al., 2010). More recently, it was shown that the TCR repertoire of the Tregs and CD8+ T cells inversely correlated, suggesting an antigen specific suppression of CD8+ T-cell clonal expansion (Liu et al., 2015). In other studies in both DLBCLs (Farinha et al., 2010) and FLs (de Charette et al., 2016), instead, Foxp3+ cells have been found to be associated with improved outcome after chemotherapy with or without rituximab. This may be attributed to the possibility that Tregs can directly suppress lymphomagenic B cells. More direct functional studies are needed in this area, especially to refine the identity of Foxp3 cells that correlate with outcome in these patients.
TAMs can be grouped in two major categories: 1) CD163- M1 macrophages, which are pro-inflammatory and 2) CD163+ M2 macrophages that are anti-inflammatory and are preferentially recruited at the tumor site (Figure 2). In DLBCL, CD68+CD163+ M2 macrophages are associated with poor clinical outcome (Komohara et al., 2015; Guo et al., 2016) and more frequent extra-nodal involvement (Li Y.-L. et al., 2019). The predictive value of the macrophage marker CD68 alone in lymphoma is debatable, with some studies indicating no predictive value in DLBCL (Matsuki et al., 2019), and others observing correlation with poor prognosis (Cai et al., 2012; Nam et al., 2014; Riihijarvi et al., 2015). Intriguingly, TAMs in DLBCL show STAT3-mediated expression of PD-L1 and this signature was found to correlate with prolonged PFS (McCord et al., 2019). The prognostic impact of TAMs in FL is not fully clear and can depend on the type of treatment. In an early study with FL patients treated with combination chemotherapy followed by radiation, elevated TAMs predicted inferior survival (Farinha et al., 2005). However, these cells did not correlate with poor survival if the patients received rituximab (Taskinen et al., 2007). The GELA FL-2000 clinical trial also showed that high frequency of intra-tumoral macrophages correlated with poor survival only in the patients who received chemotherapy without rituximab (Canioni et al., 2008). To incorporate the microenvironment component in the prognosis algorithms, the Lunenburg Lymphoma Biomarker Consortium studied a homogeneously rituximab-chemotherapy-treated group of FL patients and found that low CD8+ T-cell percentages, the presence of CD163-expressing macrophages, EZH2 wild-type (WT) status and gain of chromosome 18 in the diagnostic tumor biopsies predict a poor prognosis in FL treated with R-CHOP (Stevens et al., 2017), pointing to an overall negative impact of macrophages in the outcome of FL patients in these conditions.
MDSCs can be divided into two groups: polymorphonuclear (PMN-MDSC) and monocytic (M-MDSC) (Zhou et al., 2018; Tcyganov et al., 2018). Typically, in humans, MDSCs are identified by myeloid cell markers CD11b+, CD33+, HLA-DR low/−, and lineage-specific antigen Lin-negative (Figure 2). MDSCs can attenuate anti-tumor cytotoxic T lymphocyte (CTL) responses via metabolic competition, and generation of oxidative stress (Gabrilovich et al., 2012). A recent study by Wang et al. described a higher proportion of functionally suppressive M-MDSCs in DLBCL patients, which correlated with disease stage (Wang Z. et al., 2021). In an earlier preclinical study using the A20 lymphoma model, the authors showed that MDSCs can activate Tregs, thus reinforcing local immunosuppression (Serafini et al., 2008). Whether this mechanism occurs in human lymphoma remains to be established.
MDSCs, M2 macrophages and Tregs can secrete IL-10 (Shen et al., 2016), which inhibits T-cell function, contributing to a suppressive lymphoma microenvironment (Figure 2). Another mechanism through which immunoregulatory myeloid cells limit T-cell function is by depleting critical nutrients for activated, proliferating T cells which are metabolically demanding. Tryptophan is an essential amino acid, critical for T-cell function, which can be degraded by indoleamine 1,2,3-dioxygenase (IDO). IDO is overexpressed in MDSCs and was also found to be upregulated in lymphoma cells (Elpek et al., 2007). It has been shown that intrasplenic injection of lymphoma cells in mice leads to Treg recruitment and that this effect is counteracted by IDO1 inhibition (Curti et al., 2007). TGFβ is another well-established immunosuppressive cytokine responsible for suppression of CD8+ effector T cells in the TME (Figure 2). However, the role of TGFβ in lymphoma is debated, as studies suggest that activation of this pathway might confer survival advantage to both DLBCL (Merdan et al., 2021) and FL patients (Labidi et al., 2010). In addition, in FL patients, elevated serum levels of IL-1R1, IL-6, IL-7, IL-10, IL-13, TNF-α, and vascular endothelial growth factor (VEGF) were identified (Labidi et al., 2010). Elevated serum VEGF and the glycolytic enzyme lactate dehydrogenase (LDH) are associated with shorter PFS in FL (Labidi et al., 2010). Locally in the TME, it was shown that TFH can induce FL cells to release the chemokines CCL17 and CCL22, which can in turn recruit Tregs and more IL-4-producing T cells to sustain tumor growth and immunosuppression (Rawal et al., 2013). Galectin 3 is another relevant soluble factor that can contribute to local immunosuppression in DLBCL and FL. A study by D'Haene et al. showed that galectin-3 is expressed in 50% of the DLBCL cases and 12.5% of FL cases (D'Haene et al., 2005). Galectin-3 mediates pro-tumor inflammatory process and is important in recruitment of macrophage and angiogenesis—which could potentially contribute to immune evasion collectively.
Impact of Major Epigenetic Modifiers in GCB Lymphoma Metabolism and Immune Evasion
Critical epigenetic modifiers commonly altered in GCB-DLBCL and FL modulate the way B cells interact with immune cells in GCs and require metabolic substrates as co-factors, pointing to a key role of these alterations in immune evasion of GCB lymphomas, which may be supported by specific metabolic processes (Figure 2).
EZH2
EZH2 is responsible for the enzymatic activity of polycomb repressive complex 2 (PRC2) which catalyzes histone 3 lysine 27 trimethylation (H3K27me3) at gene promoters and represses target gene expression (Di Croce and Helin, 2013). EZH2 is essential to maintain GC reactions by inhibiting PC differentiation and cell-cycle checkpoint genes in cooperation with BCL6, and EZH2 loss impairs GC formation (Béguelin et al., 2013; Caganova et al., 2013; Béguelin et al., 2016; Béguelin et al., 2017). Importantly, more than 90% of EZH2 mutations in DLBCL and FL occur at the Y641 residue located in the catalytic SET domain, which result in the GOF of EZH2 catalytic activity (Morin et al., 2010). Genetically engineered mice to specifically express the Ezh2Y641F point mutation in B cells develop GCB-like lymphomas in cooperation with BCL2 or BCL6 overexpression (Béguelin et al., 2013; Béguelin et al., 2016). Since EZH2 mutations in DLBCL and FL enhance EZH2 catalytic activity and EZH2 is essential for the development of GC B cells, EZH2 targeted therapy is a precision approach against GCB lymphomas. Indeed, pharmacologic inhibition of EZH2 is highly effective for the treatment of murine Ezh2-mutant B-cell lymphomas as well as EZH2-mutant-patient-derived xenograft (PDX) models in vivo (Béguelin et al., 2013; Béguelin et al., 2016; Scholze et al., 2020). As we describe in detail in the last section of this review, tazemetostat is the first FDA-approved EZH2 inhibitor for FL patients, which has shown activity especially in patients with EZH2-mutant FL (Morschhauser et al., 2020).
EZH2 methyltransferase activity can be regulated by SAM levels and EZH2 can self-sustain its own methylation activity by promoting SAM synthesis (Dann et al., 2015) (Figure 1). Other metabolites can post-translationally modify EZH2 (phosphorylation, O-GlcNAcylation, acetylation, methylation, ubiquitination) leading to function and stability alterations of PRCs (Li et al., 2020). In other type of cancers, EZH2 has been shown to suppress several metabolic activities, including branched amino acid (BCAA) metabolism, TCA cycle (IDH1), mTOR (mammalian target of rapamycin) signaling, and glutamine metabolism (Dann et al., 2015; Gu et al., 2019). EZH2 can also serve as a sensor of glycolytic metabolism in the TME through the miRNA-EZH2-Notch signaling pathway and this pathway is in turn regulated by glucose metabolism in the TME (Zhao et al., 2016). Overall, these observations indicate not only that EZH2 activity impacts on cell metabolism, but also that cell metabolism can influence EZH2 function. These effects deserve precise investigation in lymphoma, especially in EZH2-mutant cases.
EZH2 activating mutations in GCB lymphomas significantly alters the immune microenvironment. EZH2Y641F promotes abnormal expansion of centrocytes in GCs by preventing apoptosis and activation of the MYC pathway, which is crucial for recycling into the DZ (Béguelin et al., 2020). EZH2Y641F-mutant centrocytes downregulate TFH-interacting molecules such as SLAM, ICAM-1, ICAM-2, and Ly108 and are less dependent on the CD40/CD40L pro-survival signals induced by TFH (Béguelin et al., 2020). This gives a survival advantage to mutant centrocytes in a competitive microenvironment with WT centrocytes. EZH2 activating mutations in DLBCL patients are also strongly associated with loss of both MHC-I and MHC-II molecules (Ennishi et al., 2019b), which facilitates immune evasion. A recent study using genome-wide CRISPR screening in DLBCL cell lines has identified critical positive and negative regulators of MHC-I expression, among which EZH2 is the most crucial one in GCB-DLBCL (Dersh et al., 2021). Furthermore, EZH2 GOF is closely associated with epigenetic silencing of CD58 expression on lymphoma cells, thus blocking the interaction with cytotoxic effector CD2+ T and NK cells and interrupting another avenue of immune control (Figure 2) (Otsuka et al., 2020).
KMT2D
KMT2D is a part of the COMPASS-like complex which regulates gene enhancer functions through histone 3 lysine 4 mono- and di-methylation (H3K4me) for active gene transcription (Ford and Dingwall, 2015; Froimchuk et al., 2017). Among patients with GCB-DLBCL and FL, most KMT2D mutations are frameshift or nonsense mutations which result in KMT2D LOF (Zhang et al., 2015). Kmt2d-deficiency impairs B-cell differentiation and induces expansion of GC B cells in mice, suggesting that KMT2D is required to terminate GC reaction and promote PC differentiation (Ortega-Molina et al., 2015; Zhang et al., 2015). KMT2D LOF mutations accelerate B-cell lymphomagenesis in cooperation with Bcl2 in mice (Ortega-Molina et al., 2015; Zhang et al., 2015). KMT2D-target enhancers are repressed by BCL6 during GC reactions through the recruitment of LSD1, a histone demethylase at H3K4 (Hatzi et al., 2013). LSD1 loss in GC B cells impairs GC formation and prevents BCL6-driven lymphomagenesis through de-repression of BCL6 target genes (Hatzi et al., 2019). Although LSD1 knockdown inhibits the proliferation of DLBCL cell lines in vitro, pharmacologic inhibition of the enzymatic activity of LSD1 only shows modest effects against DLBCL in vivo (Hatzi et al., 2019). Since LSD1 is also responsible to recruit CoREST complex which induces a repressive chromatin state through the activity of HDAC1/2 in the complex, inhibition of the catalytic activity of LSD1 might not be enough to restore the expressions of B-cell differentiation genes (Shi et al., 2005; Yang et al., 2006). Therefore, LSD1 degraders rather than inhibitors of LSD1 enzymatic activity might be suitable for precision therapy of KMT2D-mutant GCB-lymphomas. KDM5 is another histone lysine-specific demethylase, which demethylates H3K4me1 to H3K4me0 and H3K4me3/me2 to H3K4me1. Notably, KDM5 inhibition has been shown to alleviate loss of H3K4 activating methylation marks in KMT2D-mutant lymphomas and may constitute a viable therapeutic strategy for KMT2D-mutant GC lymphomas (Heward J. et al., 2021).
The impact of KMT2D LOF in B-cell lymphoma metabolic rewiring has not been explored yet. However, in lung cancer, where KMT2D was found to be the most highly inactivated epigenetic modifier, KMT2D-inactivating mutations induce aberrant metabolic reprogramming via increased expression of glycolytic genes (Alam et al., 2020). Mechanistically, KMT2D was found to upregulate the circadian rhythm repressor PER2 which plays an important role in tumor suppression (Fu et al., 2002). Several glycolytic genes (e.g., Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1) were identified as target genes of PER2. Therefore, KMT2D-mediated Per2 activation represents a previously unknown tumor-suppressive mechanism that links an epigenetic tumor suppressor to a circadian rhythm regulator with direct metabolic implications. Accordingly, pharmacologic inhibition of glycolysis reduces tumorigenicity of human lung cancer cells bearing KMT2D-inactivating mutations, suggesting that KMT2D deficiency may present a therapeutic vulnerability to glycolytic inhibitors (Ding et al., 2008; Alam et al., 2020). The link between KMT2D and glycolysis may be relevant and worth to explore in detail in lymphoma, as glycolysis measured by expression of aldolase A and GAPDH was associated with significantly shorter transformation-free survival in FL patients (Monrad et al., 2020). High expression of aldolase A and GAPDH may indicate increased metabolic turnover, and these enzymes may be useful biomarkers in primary FL for predicting the risk of subsequent lymphoma transformation. It will be important to determine the extent to which KMT2D inactivating mutations support the glycolytic switch in GCB lymphomas.
KMT2D LOF mutations are associated with altered immune signatures in DLBCL. In a recent study by You et al., KMT2D non-synonymous mutations have been shown to correlate with an overall increase in mutational burden in DLBCL, which intriguingly corresponded with low intra-tumoral T-cell infiltration in GCB DLBCL patients with WT P53 (You et al., 2021). Similarly, in solid cancers, KMT2D LOF mutations have been recently found to contribute to DNA damage, increased mutational burden and activation of transposable elements, which in this setting are associated with increased infiltration of effector immune cells, such as CD8 T cells, NK cells and M1 macrophages and decreased infiltration of Tregs and immature macrophages and better response to ICB activity (Wang et al., 2020; Liu et al., 2021). This highlights potential distinct effects of this epigenetic modifier depending on the disease setting, underscoring the importance to study these mechanisms more carefully and specifically for rational design of more effective combination immunotherapies for lymphoma patients.
CREBBP and EP300
CREBBP catalyzes histone 3 lysine 27 acetylation (H3K27Ac) at enhancers, which activates transcription through the recruitment of DNA-binding TFs and other co-activators (Green, 2018). CREBBP activates PC differentiation genes, such as PRDM1 and IRF4, which are required to terminate the GC reaction (Jiang et al., 2017; Zhang et al., 2017). Mutations of CREBBP in DLBCL and FL are concentrated in the acetyltransferase catalytic domain, with hot spot mutations at the R1446 residue (Pasqualucci et al., 2011b). These mutations reduce CREBBP acetyltransferase activity and promote transcriptional repression of target genes (Pasqualucci et al., 2011b). Conditional Crebbp loss in B cells induces focal depletion of H3K27Ac at enhancers and accelerates B-cell lymphoma development in cooperation with Bcl2 in mice (García-Ramírez et al., 2017; Jiang et al., 2017; Zhang et al., 2017). During GC reactions, the enhancers regulated by CREBBP are generally repressed by BCL6 through recruitment of SMRT/NCOR complexes (Hatzi et al., 2013; Jiang et al., 2017). SMRT/NCOR complexes contain HDAC3 which antagonizes the function of CREBBP through H3K27 deacetylation, and HDAC3 loss in GCB-DLBCL cells restores H3K27Ac marks and enhances BCL6-SMRT target gene expression (Hatzi et al., 2013; Jiang et al., 2017; Mondello et al., 2020). These findings suggest that HDAC3 pharmacologic inhibition can be a promising therapeutic strategy for CREBBP-mutant GCB lymphomas, promoting GC B-cell exit and differentiation into PCs (Mondello et al., 2020).
In addition, EP300, which is also responsible for H3K27Ac and whose LOF mutations are found in GCB-DLBCL and FL (Cerchietti et al., 2010), partially compensates for the function of CREBBP, and EP300 may be critical for CREBBP-deficient B-cell survival (Meyer et al., 2019). Therefore, EP300 targeted therapy may be another precision approach against CREBBP-mutated GCB-lymphoma.
CREBBP and EP300 HAT activity may be modulated by the availability of acetyl-CoA substrates deriving from cellular metabolic processes (Figure 1). While poor evidence of these mechanisms is currently available for lymphoma, initial studies in hepatocellular carcinoma suggest that p300/CBP epigenetically induces expression of glycolysis-related enzymes (Cai et al., 2021), which may sustain HAT activity through increased acetyl-CoA levels.
In FL, CREBBP mutations are founder events, occur early and contribute to immune escape by downregulating MHC-II expression (Green et al., 2015; García-Ramírez et al., 2017), which is crucial for GC B-cell differentiation (Allen et al., 2007) and tumor-Ag presentation (Khodadoust et al., 2017). In DLBCL, it has been shown that CREBBP/EP300 mutations are also associated with the recruitment CD68+ and CD163+ M2 macrophages to the tumor site (Huang et al., 2021). The skewed M2 polarization in CREBBP/EP300-mutant DLBCLs was attributed to aberrant regulation of the FBXW7-NOTCH-CCL2/CSF1 axis (Huang et al., 2021). Moreover, CREBBP/EP300-mutant DLBCL patients were found to have higher serum levels of the immunosuppressive cytokine IL-10 compared to the pro-inflammatory cytokine IL-1β (Huang et al., 2021), suggesting potential systemic immune suppression in these patients. CREBBP mutations were also associated with upregulation of colony stimulating factor 1 (CSF1) and B7H4, both of which are linked to immunosuppressive myeloid cells. These findings suggest that the reprogrammed myeloid compartment in CREBBP/EP300-mutant lymphomas can be considered to identify novel therapeutic targets for patients bearing these diseases.
Emerging Therapies for B-Cell Lymphomas
EZH2 Inhibitors
Currently tazemetostat is the only the EZH2 inhibitor (EZH2i) FDA-approved for the treatment of FL and remains under investigation in DLBCL. In 2020, tazemetostat was approved for use in patients with EZH2-mutant FL who are relapsed or refractory (R/R) following at least 2 prior therapies, and for patients with WT EZH2 and R/R FL following 2 prior therapies and without other treatment options (Morin et al., 2021), based on phase-I and phase-II results (NCT01897571) showing efficacy in these populations (Italiano et al., 2018). The initial phase-I dose-escalation study included both patients with solid tumors and B-cell lymphomas, including 13 patients with DLBCL and 7 with FL (Italiano et al., 2018). Of these 20 patients, 7 responded and 3 had a complete response (CR). Notable toxicity included grade 3 or greater thrombocytopenia, anemia, hyperbilirubinemia and transaminitis with a significant number of patients experiencing grade 2 fatigue, anorexia, nausea/vomiting, and muscle spasms as well (Italiano et al., 2018). Following the efficacy in DLBCL and FL patients in the phase-I study, the phase-II portion of the trial recruited patients with R/R DLBCL and FL and treated with 800 mg of tazemetostat twice daily. 99 patients with FL were recruited, including 45 patients with EZH2-mutant FL and 44 with WT EZH2, with median age and prior lines of therapy similar between the two groups (Morschhauser et al., 2020). Overall response rate (ORR) and CR rate were higher in EZH2-mutant vs. WT cohort (69 and 13% vs. 35 and 4%, respectively). Responses were observed also among patients with progression of disease within 24 months of last therapy—an important negative prognostic factor in FL, albeit more frequently again in the EZH2-mutant vs. WT patient population (64 vs. 25% ORR, respectively) (Morschhauser et al., 2020). However, median PFS and duration of response (DOR) were similar between patients with EZH2-mutant vs. WT FL (13.8 and 10.9 vs. 11.1 and 13.0, respectively) (Morschhauser et al., 2020). Overall, these results showed efficacy in both EZH2-mutant and WT FL patients, with similar DOR and PFS between the two cohorts, despite higher response rate in EZH2-mutant patients, and similar toxicity profile compared to the phase-I trial (Morschhauser et al., 2020). Therefore, tazemetostat represents a valuable option with a reasonable toxicity profile and clinical efficacy for FL patients that are refractory to multiple prior lines, including those with WT EZH2. Preliminary results from the DLBCL portion of the trial (patient n = 226; EZH2 mutant, n = 36) were disappointing. ORR to tazemetostat monotherapy was similarly low (17%) in patients with either EZH2-mutant or WT tumors (3% CR for EZH2-mutant and 9% CR for EZH2-WT). Among 69 EZH2-WT patients that were treated with tazemetostat plus prednisolone, ORR was just 9%, with 1% CR, and median PFS and DOR not yet reached (Ribrag et al., 2018). Major toxicities included thrombocytopenia, anemia, neutropenia, nausea/vomiting, and fatigue (Ribrag et al., 2018). The variable results with tazemetostat in both EZH2-mutant and WT lymphoma patients underscore the need to identify reliable predictors of response that can help 1) allocate to this treatment the patients that are more likely to respond and 2) anticipate resistance. Systematic analysis of epigenetic, immune and metabolic profiles of the tumors that respond in comparison with tumors that do not respond will provide fundamental information in this direction.
While clinical trials have thus far evaluated tazemetostat mainly as a monotherapy in B-cell lymphomas, several rational drug combinations are currently being studied. A phase-I trial of tazemetostat plus R-CHOP in untreated DLBCL was published in 2020, with a phase-II trial planned to add patients with untreated FL (NCT02889523) (Sarkozy et al., 2020). In R/R FL, trials combining tazemetostat with lenalidomide plus rituximab regimen (NCT04224493) or rituximab (NCT04762160) are currently under way. Preclinical studies have also demonstrated synergy between tazemetostat and venetoclax (Bcl2 inhibitor) against DLBCL, with phase-I trials currently in development (Scholze et al., 2020). Due to its effects on the TME and on T cells, tazemetostat has also been proposed as an adjunct to several immunotherapies, which will be discussed in greater detail below. In addition to tazemetostat, other EZH2is are in development for GCB lymphomas. The highly selective EZH2i GSK2816126 showed disappointing results in a phase-I trial (Yap et al., 2019), while others, including CPI-0209 and SHR2554 (NCT04104776, NCT03603951) are still in clinical testing. Dual EZH1/2is are also currently being evaluated, including CPI-1205 (Harb et al., 2018), and valemetostat (NCT04842877). Clarifying the relative contribution of inhibiting EZH1 together with EZH2 in lymphoma remains an important aspect to determine (Yamagishi et al., 2019). Overall, the future appears to be bright for the potential of EZH2is in GCB lymphomas, with clinical efficacy of tazemetostat already apparent and many novel combinations and new agents in development.
HDAC Inhibitors
HDAC is have been considered as a form of epigenetic therapy, although there is little evidence to suggest that their anti-lymphoma effects are related to epigenetic regulation. Several such agents have been studied in clinical trials (Cao et al., 2018). The first clinically evaluated HDACi was vorinostat—a pan-HDACi that has now been studied in the phase-II setting for both newly diagnosed and R/R DLBCL and FL, both as monotherapy and in combination with other agents. In R/R DLBCL, vorinostat monotherapy proved very disappointing with underwhelming 6% ORR as well as high incidence of grade 3–4 thrombocytopenia and neutropenia (Crump et al., 2008). Vorinostat has also been combined with rituximab, cyclophosphamide, etoposide, and prednisone (R-CVEP) in R/R DLBCL with an ORR of 57% (35% CR) and a median PFS of 9.2 months with high rates of grade 3–4 hematologic toxicities (Smith et al., 2019). In untreated DLBCL, vorinostat was combined with R-CHOP but did not meet a predefined efficacy improvement over standard R-CHOP(Persky et al., 2018). In R/R FL, vorinostat monotherapy has shown efficacy with two phase-II studies demonstrating ORRs of 47% (Kirschbaum et al., 2011) and 49% (Ogura et al., 2014) with high rates of grade 3 and 4 neutropenia and thrombocytopenia in both studies. Another trial combined vorinostat with rituximab in R/R FL demonstrated similar 50% ORR and toxicity profile, with a 2-years PFS of 61% (Chen et al., 2015). Belinostat, another pan-HDACi, has also been studied in R/R DLBCL or transformed FL as monotherapy, but like vorinostat, it showed dismal response rates with just 11% ORR, although toxicity was more manageable than with vorinostat (Puvvada et al., 2016). Mocetinostat is an isotype-specific HDACi that was studied as monotherapy in R/R DLBCL and FL. ORR in both diseases was low at 19% in DLBCL and 12% in FL, with the most common grade 3 and 4 adverse events being neutropenia, thrombocytopenia, and fatigue (Chen et al., 2020). Another pan-HDAC inhibitor abexinostat, with a different pharmacokinetic profile, although showing promising results in FL and DLBCL patients, with ORRs of 56%, and 31% respectively, induced frequent ≥ grade 3 adverse events (Ribrag et al., 2017). Overall, the clinical activity with unselected HDACis have been limited by the pleiotropic and toxic effects, especially hematologic adverse events.
Based on the specific role of HDAC3 in lymphomagenesis, highly specific HDAC3is may offer a better option to both improve clinical efficacy and reduce toxicity from off-target effects. Specific HDAC3is have been challenging to develop and remain in early-stage pre-clinical development for phase I clinical trials (Mondello et al., 2020). HDAC3 inhibition offers the potential to block the BCL6-HDAC3 complex and restore the key pathways in cell cycle and differentiation inhibited by BCL6 in CREBBP-mutant lymphomas, including Ag presentation as well as BCR, NF-kB, and interferon signaling (Mondello et al., 2020). A selective HDAC3i developed by the Broad Institute (BRD3308/OKI422) has demonstrated promising activity both in vitro and in vivo. HDAC3 inhibition increased H3K27Ac, transcription of B-cell-terminal-differentiation genes, MHC-II expression, and inhibited cell proliferation of lymphoma cell lines even in the absence of a CREBBP mutation, although these effects were more marked in CREBBP-mutant lines (Mondello et al., 2020). Importantly, HDAC3-specific inhibition induces greater restoration of MHC-II expression compared to pan-HDACis, with particularly robust effects in CREBBP-mutant cell lines, and reduced toxicity against T cells (Mondello et al., 2020). In PDX models, HDAC3 inhibition with BRD3308 reduced tumor growth, and induced upregulation of BCL6 target genes and MHC-II expression in the setting of both CREBBP-mutant and CREBBP-WT diseases (Mondello et al., 2020). Notably, treatment with BRD3308 appeared to improve T-cell mediated tumor recognition and killing, when TILs were co-cultured with DLBCL cells pretreated with BRD3308 vs. vehicle (Mondello et al., 2020). These preclinical experiments suggest that HDAC3-specific inhibition may be an effective therapy for GCB lymphomas, particularly those with CREBBP-mutations, by reducing the dominance of BCL6 on transcription programs and improving both terminal differentiation and immunogenicity of tumor cells. In particular, the effects of HDAC3 inhibition in MHC-II expression and T-cell activation speak to its potential as a partner for immunotherapies, such as CAR T cells, ICB, or bispecific antibodies.
DNA Methyltransferase Inhibitors
Another class of epigenetic drugs—DNA methyltransferase inhibitors (DNMTis)—has shown promising activity against B-cell lymphomas, reaching advanced clinical development. Demethylation at gene promoters induced by DNMTis can result in several beneficial anti-tumor effects: 1) re-expression of tumor suppressor genes, 2) reversal of chemotherapy resistance due to de-repression of SMAD1 and 3) upregulation of the Ag presentation machinery and IFN response genes, which can increase immune sensitivity, thus offering potential for combination with immunotherapy (Almstedt et al., 2010; Chiappinelli et al., 2015). In addition, demethylating agents may have a direct effect on T cells, limiting exhaustion during chronic antigen stimulation. Decitabine was found to prevent the development of exhaustion-associated epigenetic changes in T cells, and this synergized with T-cell reinvigoration upon PD-1 blockade in viral chronic infection mouse models, highlighting the rationale to combine ICB with DNMTis for the treatment of cancer (Ghoneim et al., 2017). Azacytidine and decitabine DNMTis are being tested in several trials in combination with other agents for B-NHLs (e.g. NCT03450343; NCT03579082; NCT01799083). Treatment of high-risk DLBCL patients with azacytidine resulted in reversal of SMAD1 hypermethylation and induction of its expression, which in turn enhanced the response to chemotherapy (Clozel et al., 2013). Accordingly, early promising results were obtained with sequential treatment of azacytidine followed by R-CHOP in high-risk B-cell lymphomas, with several clinical trials in progress including a phase II/III study (NCT04799275) (Clozel et al., 2013; Martin et al., 2021). The impact of these agents in anti-lymphoma immunity remains to be investigated.
Agents Targeting Lymphoma Metabolism
Understanding metabolic derangements in lymphomas reveals distinct therapeutic vulnerabilities, and one pathway that has shown promise in GCB lymphomas is PI3K, which regulates the PI3K, AKT, and mTOR pathway. Currently, four PI3K inhibitors (PI3Kis) are approved for use in FL and are undergoing study in DLBCL. Idelalisib was the first PI3Ki approved for FL and functions by specifically inhibiting the PI3Kδ isoform and is an oral drug. Following promising results in several phase-I studies, a phase-II trial enrolled patients with indolent lymphomas either refractory to rituximab or alkylating agents or relapsed within 6 months of these therapies (Gopal et al., 2014). While the study did not stratify results by disease subtype, 72 out of 125 patients on study had FL. ORR was 57% (6% CR), with a median PFS of 11 months in this very polyrefractory population (Gopal et al., 2014). The most common grade 3 or higher adverse events were neutropenia, transaminitis, and diarrhea, which may limit the use of this drug in certain cases. Based on this trial, idelalisib was approved for use R/R FL. Idelalisib has also been studied in DLBCL in combination with the Syk inhibitor entospletinib, but the combination was limited by toxicity due to pneumonitis (Barr et al., 2016). Copanlisib is a PI3Ki with activity against PI3Kα and PI3Kδ and is administered intravenously. Following promising phase-I-study results, a large phase-II trial administered copanlisib to patients with R/R indolent or aggressive lymphomas, including 16 FLs and 15 DLBCLs (Dreyling et al., 2017a). ORR in the indolent vs. aggressive subgroup was 44 and 27%, with a median PFS of 270 and 70 days, respectively. A subsequent large phase-II study of copanlisib in R/R indolent lymphomas (FL, n = 104) confirmed the promising results in this population, with 59% ORR (14% CR), 11.2 months median PFS and 22.6 months median DOR (Dreyling et al., 2017b). The most common grade 3–4 adverse events included hypertension, hyperglycemia, leukopenia, and neutropenia, wih nausea, fatigue, and diarrhea common as well. Based on these results, in 2017 copanlisib was approved in R/R FL who have received at least 2 prior therapies. Results of copanlisib monotherapy in DLBCL continued to be disappointing, with a subsequent phase-II study showing an ORR of only 19% (8% CR), although 32% of patients with ABC-DLBCL had a response (Lenz et al., 2020). Most recently, copanlisib has been combined with rituximab in a phase-III trial in patients with R/R indolent lymphomas, including FL, demonstrating 81% ORR (34% CR) in all indolent histologies vs. 48% ORR (15% CR) in the rituximab monotherapy arm. In the 184 FL patients included in the copanlisib-rituximab arm, median PFS was 22 vs. 18.7 months for the 91 FL patients in the rituximab arm (Matasar et al., 2021). However, nearly half of all patients had severe adverse events, including hyperglycemia and hypertension, while less than 20% patients in the single-agent rituximab arm had serious adverse events. Duvelisib is an oral agent that inhibits PI3Kδ and PI3Kγ that has been studied in a phase-II setting in R/R FL with at least 2 prior therapies. 83 patients with FL received duvelisib in a larger study including other indolent NHLs and achieved 42% ORR, including a CR (Flinn et al., 2019). Median PFS in the study as a whole was 9.5 months with a median OS of 29 months. The most common adverse events included diarrhea as well as neutropenia, anemia, and thrombocytopenia. The most recent PI3Ki to be approved is umbralisib, which is an oral agent that inhibits PI3Kδ as well as casein kinase-1 epsilon. In a large phase-II trial, patients with heavily pretreated indolent NHLs were treated with umbralisib monotherapy. In FL patients (n = 117), ORR was 43% with 3% CR rate, a median PFS of 10.6 months and median DOR of 11.1 months (Fowler et al., 2021). The most common toxicities were diarrhea, infection, nausea, neutropenia, transaminitis, and rash. Based on the results of these studies, umbralisib was approved for R/R FL following at least 3 prior lines of therapy. Notably, preclinical studies with a dual PI3K and HDAC inhibitor have revealed the potential of this combination strategy in DLBCLs irrespective or the COO (Mondello et al., 2017) and also in B-cell lymphoma refractory to Bruton’s tyrosine kinase inhibition (Guo et al., 2019).
Another metabolic pathway that may offer therapeutic vulnerability in B-cell lymphomas is the transport of metabolic products/substrates via the monocarboxylate transporter (MCT) family (MCT1, MCT2, MCT3, and MCT4). MCT is a family of transmemmbrane proteins that mediate the bi-directional transport of lactate, pyruvate, short-chain fatty acids and ketones (Halestrap, 2013). MCT1 is a monocarboxylate transporter associated with poor clinical outcomes in DLBCLs (Afonso et al., 2019). MCT1 is activated by the MEK signaling pathway, and both MEK inhbitors (MEKis) and direct MCT1 inhibitors (MCT1is) have been tested in phase-I trials in DLBCL. Selumetinib is a MEKi that was studied in patients with R/R DLBCL, but no patients had objective responses and the drug was poorly tolerated with most patients requiring dose de-escalations (Galanina et al., 2018). Early results of a phase-I study of the MCT1i AZD3965 in R/R DLBCL were recently presented at the ASCO annual meeting, showing CR in one out of 11 patients, with no other clinical responses noted (Halford et al., 2021). Based on these results the MCTi appears to work poorly as a monotherapy, although combinations with other drugs are being considered. Recent preclinical studies in mice have shown that in addition to rewiring the global metabolic activity of cancer cells, MCT1 inhibition can also impact on the TME including angiogenesis, metabolic symbiosis between cancer and stromal cells, and immune suppression (Romero-Garcia et al., 2016). For example, by using non-invasive proton nuclear magnetic resonance (1H NMR) spectroscopy (MRS), AZD3965 was found to inhibit tumor choline metabolism in vivo with the consequent increases in tumor-infiltrating NK cells and DCs in xenografted lymphoma models, where, however, only innate immune cells could be evaluated. In this study, AZD3965 treatment also showed to upregulate the immune checkpoint PD-L1 on NK cells, providing preliminary evidence for studying the impact of AZD3965 on anti-lymphoma immune responses and in combination with immune-modulating agents (Beloueche-Babari et al., 2020).
As an additional modality to counteract tumor metabolism, the anti-diabetic drug metformin—known to regulate blood glucose by different mechanisms—has started to be explored for B-NHL treatment. Retrospective analyses showed improved survival in diabetic DLBCL patients under metformin treatment during first-line chemotherapy. Moreover, metformin potentiated the anti-tumor activity of rituximab and chemotherapy in lymphoma models, suggesting potential therapeutic effects of metformin against these diseases (Singh et al., 2020).
T-Cell Targeting Immunotherapies in B-Cell Lymphomas
The development of immunotherapies for B-cell lymphomas including ICB, bispecific antibodies, and CAR T-cell therapies are an incredibly exciting area of innovation and studies in this space have exploded in the past few years. While the full description of this space would deserve a review article unto itself, here we highlight the most important immunotherapy trials in GCB lymphomas, before discussing the potential role of combining epigenetic and metabolic therapies with immunotherapy in these diseases.
Immune Checkpoint Blockade
In the past decade, ICB targeting PD-1, PD-L1, or CTLA-4 has proven effective against a variety of solid tumor malignancies as well as Hodgkin lymphoma, and there was optimism that ICB would be successful in GCB lymphomas as well. However, trials of ICB in both DLBCL and FL have proven disappointing thus far, underscoring the importance to understand the molecular determinants of this intrinsic immune resistance in these diseases for the design of more effective combination strategies.
Despite the promising results of the anti-PD-1 nivolumab in an initial phase-I study with R/R FL or DLBCL patients with ∼40% ORR and a very manageable toxicity profile (Lesokhin et al., 2016), subsequent phase II-trials showed markedly lower response rates, with 4 and 10% ORR and 2.2 and 1.9 months median PFS in R/R FL and DLBCL, respectively (Ansell et al., 2019; Armand et al., 2021b). Results were similarly poor in a trial with another PD-1 inhibitor, pembrolizumab, in R/R FL (ORR, 11% and median PFS, 3.4 months) (Ding et al., 2017). In R/R DLBCL patients, pembrolizumab was administered following autologous stem cell transplant, but did not meet its primary endpoint of PFS improvement relative to transplant alone (Frigault et al., 2020). The Keynote-013 study (NCT01953692), which has cohorts of pembrolizumab monotherapy in R/R DLBCL and FL, has not yet reported results. Multiagent therapy with pembrolizumab has been shown to increase its efficacy in this setting, with one study combining pembrolizumab and rituximab in R/R FL patients achieving 80% ORR, including 60% CR (Nastoupil et al., 2017).
Atezoliuzumab, a monoclonal antibody targeting PD-L1, has shown some clinical efficacy in combination with the anti-CD20 obinutuzumab in patients with R/R FL or DLBCL, with preliminary data from a phase-I study showing partial responses in one patient with FL and one with DLBCL out of 5 total evaluable patients (Till et al., 2015). Further studies have added lenalidomide to this regimen in FL and showed 85% ORR with a 72% CR rate at most recent update (Salles et al., 2018; Morschhauser et al., 2019). In the front line setting, atezolizumab combined with obinutuzumab and bendamustine for untreated FL showed 80% ORR with 67% CR by Lugano criteria, although 52% of the patients required treatment interruptions including one death due to atezolizumab-related cardiac arrest (Younes et al., 2017). Atezolizumab has also been combined with R-CHOP in untreated DLBCL, with patients receiving atezolizumab consolidation following induction with atezo-RCHOP, with an ORR of 87.5% (77.5% CR) and a 2-years PFS of 75%, although half of the patients discontinued consolidation prior to completion and half had grade 3–4 adverse events during consolidation (Younes et al., 2019).
Overall, these results indicate that PD-1/PD-L1 inhibitors have limited single-agent efficacy in GCB lymphomas, but there remains significant clinical potential when combined with other therapies, including epigenetic modulators and agents targeting tumor metabolism, as discussed below.
Chimeric Antigen Receptor (CAR) T Cells
CAR T-cell therapies represent an promising area of innovation in the treatment of B-cell lymphomas, in which patient’s T cells are transduced with a viral vector to form a CAR, comprising a tumor-targeting antibody portion linked to TCR intracellular signal transduction domains, which bypasses MHC restrictions for tumor-cell recognition and killing (Ansell and Lin, 2020). These cells are then re-infused into the patient following lymphodepletion. CD19-targeting CAR T cells have gained significant attention, showing long-term, durable efficacy in patients with poly-refractory lymphomas, which traditionally have a very poor prognosis (Crump et al., 2017), and three CD19-targeting CAR T-cell products are currently approved for DLBCL and FL patients. CAR T-cell therapy has its own set of unique toxicities, with cytokine release syndrome (CRS) and neurotoxicity being very common and deserving specific attention (Ansell and Lin, 2020).
Axicabtagene ciloleucel (axi-cel)—a CD19-targeting product which differs from the other approved CAR T-cell products for its CD28 co-stimulatory intracellular domain—was approved for both DLBCL and FL patients. The initial ZUMA-1 trial in R/R DLBCL following two or more lines of prior therapy reported 82% ORRs (54% CR) and long-term durable responses in a subset of patients with CR. Therapy was complicated by grade 3–4 CRS and neurotoxicity in 13 and 28% of patients, respectively (Locke et al., 2019). Subsequently ZUMA-5, which included 124 patients with R/R FL after 2 prior lines of therapy, demonstrated 94% ORR (80% CR) and grade 3–4 CRS and neurotoxicity in 7 and 19% of the patients, respectively (Jacobson et al., 2020). These results led to the approval of axi-cel in both R/R DLBCL and FL following two prior therapies. Axi-cel recently demonstrated preliminary efficacy in untreated double- or triple-hit DLBCL patients or patients with positive PET-CTs following 2 cycles of a rituximab and anthracycline containing regimen, with 93% ORR (80% CR) and grade 3–4 CRS and neurotoxicity in 20 and 27% of patients, respectively (Neelapu et al., 2020). While these results were certainly promising, long-term data will be needed to determine if axi-cel has a role in untreated DLBCL.
The other two CAR T-cell products are currently approved for DLBCL. Tisagenlecleucel (tisa-cel) is a CD3-4-1BB CAR construct that demonstrated 52% ORR (40% CR), with slightly higher rates of grade 3–4 CRS (22%) and similar rates of neurotoxicity (12%) in patients with R/R DLBCLs following two or more lines of therapy, leading to its approval in this setting (Schuster et al., 2019). Lisocabtagene maraleucel is also a CD3-4-1BB CAR T-cell product but it is given at a fixed CD4:CD8 ratio and showed 75% ORR (55% CR) in patients with R/R DLBCL following 2 or more prior therapies, leading to its approval in 2021 for this subgroup (Abramson et al., 2020).
While CAR T-cell therapy is an exciting area for clinical development for the treatment of B-cell lymphomas, there remain several crucial areas for improvement. One of the challenges of adoptive cell therapy is how to improve response duration. CAR T cells as well as effector T cells demand high metabolic support. The CAR structure impacts on CAR-T-cell-product metabolic profiles. CARs with 4-1BB co-stimulatory domains promote OxPhos of fatty acids, central-memory T-cell phenotypes with increased proliferation potential and persistence. CARs with CD28 co-stimulatory domains are more prone to aerobic glycolysis leading to CAR-T-cell products with effector-memory phenotypes (Kawalekar et al., 2016). Epigenetic modifiers can be employed to maintain the essential proportion of stem-cell memory pool in CAR T cells, which can in turn favor longer persistence in vivo. For example, treatment with decitabine during CAR-T-cell production was shown to improve CAR-T-cell functional phenotypes, persistence, tumor-homing ability and anti-tumor activity in B-NHL mouse models (Wang Y. et al., 2021). Lastly, prevention of CRS and neurotoxicity remains a critical problem and the use of CAR T-cell products in earlier lines of therapy also an open question that will require further study.
Bispecific T-Cell Engager (BiTE) Antibodies
BiTE antibodies offer another method for engaging the immune system with lymphoma by creating molecules with separate antigen binding sites targeting both tumor antigen (in the case of GCB lymphoma, CD19 or CD20) and a TCR-activating surface receptor, such as CD3. The toxicities of bispecific antibodies are different from that of traditional chemoimmunotherapy, with many patients developing CRS reminiscent of CAR T-cell toxicity (Ansell and Lin, 2020). BiTE therapy, unlike CAR T-cell therapy, does not require cells to be harvested from patients and is available directly as an off-the-shelf product.
Blinatumomab is a CD3/CD19 BiTE antibody that has gained FDA approval in acute lymphocytic leukemia but has also been studied in R/R DLBCL and FL patients. A phase-I trial recruiting patients with previously treated B-cell lymphomas showed 55% ORR (36% CR) in DLBCL and 80% ORR (40% CR) in FL patients (Goebeler et al., 2016).
Another BiTE product, mosunetuzumab, targets CD3 and CD20 and includes the Fc portion of the antibody to emulate human antibodies more closely. Mosunetuzumab is currently studied in previously treated B-cell lymphomas, and has demonstrated 33% ORR (21% CR) and 61% ORR (50% CR) in DLBCL and FL patients, respectively (Budde et al., 2018). Importantly, toxicity appeared to be more manageable compared to blinatumomab, with no grade 3–4 CRS. A recent update in R/R FL patients after 2 or more prior therapies reported 68% ORR (50% CR) in this patients population, which suggests that mosunetuzumab may be effective even in patients with poly-refractory disease (Assouline et al., 2020). Preliminary data were also published from patients with untreated DLBCL, who could not receive frontline chemotherapy due to age or comorbidities, indicating 55% ORR (46% CR) in this patient subset. Notably, toxicity was manageable with all CRS events being grade one. In a population with limited options, if chemotherapy cannot be given, mosunetuzumab may thus represent a valuable treatment (Olszewski et al., 2020).
Other bispecific antibodies currently in development include odronextuab—a CD3/CD20 IgG4 bispecific antibody—which has shown efficacy in previously treated B-cell lymphomas even in patients refractory to CAR T-cell therapy (Bannerji et al., 2020), and glofitamab—a CD3/CD20 BiTE with a 2:1 antigen configuration to allow for increased tumor antigen binding—which has been combined with obinutuzumab to reduce toxicity (Hutchings et al., 2020a). Epcoritamab—a CD3/CD20 bispecific antibody that is administered subcutaneously—has garnered significant excitement due to its ease of administration, with toxicity profile similar to other BiTE therapies and efficacy in both previously treated DLBCL (67% ORR, 33% CR; including 100% ORR and 50% CR in patients previously treated with CAR T-cell therapy) and FL (100% ORR, 25% CR) (Hutchings et al., 2020b). While clinical data from different BiTE therapies is emerging, the correct sequencing of therapies in more effective combination strategies and the patient populations that may benefit the most from them remain aspects to clarify and important areas of further study.
Rationale for Novel Immunotherapy-Based Combinations in B-Cell Lymphomas
The explosion in immunotherapy approaches offer potential exciting opportunities for GCB lymphomas patients and clinicians, but still have significant room for improvement. As described above, ICB thus far has shown poor efficacy as monotherapy, but improved efficacy in combination with other therapies and ideal partner drugs have yet to be determined. While CAR T-cell therapy has given durable responses in patients with high-risk, poly-refractory diseases, these patients are the minority and significant room for improvement in long-term efficacy remains (Locke et al., 2019). Similarly, long-term efficacy is an open question for BiTE therapy as well, and many drug partners are being explored in this space too.
Lymphoma resistance to immunotherapies is a critical hurdle and may be related to the immunosuppressed TME and immune escape mechanisms of these diseases. As described in detail above, MHC-II expression is decreased in many GCB lymphomas in association with a worse prognosis (Rimsza et al., 2004). Microenvironmental factors, such as the presence of Tregs, M2 macrophages, MDSCs or low levels of effector and cytotoxic T cells may further prevent anti-lymphoma immune responses (Good et al., 2019; Yan et al., 2019). These factors may be reversed or attenuated by epigenetic and/or metabolic therapy, allowing for increased efficacy of immunotherapies in GCB lymphomas. Moreover, agents targeting epigenetic modifiers and metabolic pathways may exert on-target/off-tumor effects leading to enhanced cytotoxic/functional profiles of anti-tumor T cells, either activated endogenously (e.g. via ICB or BiTE) or administered as cytotoxic T-cell products (e.g. CAR T cells) (Akbari et al., 2021).
EZH2 inhibition has the potential to improve the efficacy of immunotherapies in GCB lymphomas via several mechanisms. In addition to controlling immune-related genes in lymphoma cells, EZH2 is critical for the maintenance of Treg identity after activation, suggesting that EZH2 inhibition may also be able to reduce the suppressive role of Tregs (DuPage et al., 2015). Notably, CTLA-4 blockade with ipilimumab was found to increase EZH2 expression in T cells, including Tregs, and EZH2 inhibition was shown to improve the therapeutic response to anti–CTLA-4 through modulation of the Treg phenotype (Goswami et al., 2018; Wang et al., 2018). H3K27me3 accumulation also suppresses memory T-cell function and drives terminal T-cell differentiation (Gray et al., 2017), suggesting that EZH2 targeted therapy may be able to prolong T-cell functionality, thus providing the rationale for combination with T-cell targeting immunotherapies. EZH2 inactivation also contributes to the recruitment of CD4 and CD8 T cells into the TME by enhancing local Th1-type chemokine production (CXCL9, CXCL10), and loss of EZH2 has also been shown to increase Th2-type cytokine production (Tumes et al., 2013; Peng et al., 2015).
So far, data for combinations of tazemetostat with immunotherapies have been limited. A phase-I study with atezolizumab and twice daily oral tazemetostat until disease progression in previously treated DLBCL patients showed poor results. ORR was a dismal 16%, with 5% CR, although in 5 patients with EZH2-mutant disease, 3 responded and 1 had a CR. Although the lack of efficacy precluded this combination from further study, the responses in patients with EZH2-mutant lymphomas suggests that a precision-medicine approach targeting this specific patient population may be effective (Palomba et al., 2019).
HDAC3 and KDM5 inhibitors may similarly offer the potential to affect both lymphoma cells and the TME to improve the efficacy of immunotherapy. However, these combinations have only been tested in preclinical settings thus far. HDAC3is demonstrated the ability to induce transcription of BCL6 target genes and MHC-II expression even more so than pan-HDACis, while also increasing T-cell activation against tumor cells (Mondello et al., 2020). Similarly, KMD-5is restore expression of KMT2D- and CREBBP-regulated genes in KMT2D-mutant lymphomas, suggesting that these agents may have similar effects in the immune system (Heward J. A. et al., 2021). To add to the list of epigenetic strategies for combination therapies, co-targeting of EZH2 and HDACs has started to be investigated in preclinical models with promising results against EZH2 mutant lymphomas (Lue et al., 2019).
While these therapies remain in very early stages of development, their potential in combination with immunotherapies for the treatment of GCB lymphomas is obvious. Many important questions about targeting specific patient subpopulations and ideal drug combinations/sequences need to be answered for the design of effective epigenetic-immunotherapy approaches. In addition, these approaches may further benefit from incorporating agents targeting lymphoma metabolism, which are becoming clinically available. Considering the effects of cellular metabolism in the activity of epigenetic modifiers and immune microenvironment, starting to investigate rational combinations of metabolic and epigenetic therapies with immunotherapy is the logical next step.
Conclusions and Perspectives
GCB lymphomas present significant heterogeneity, both across lymphoma samples and between patients. The degree of heterogeneity and derangement from the epigenetic status and dynamics of normal B-cells correlate with disease severity and patient survival (Abramson et al., 2020), adding more complexity to the genetic basis of B-cell lymphomas, with more than 150 genetic driver mutations identified (Chapuy et al., 2018). 85% of all DLBCL cases demonstrate alterations in at least one gene involved in epigenetic remodeling (García-Ramírez et al., 2017; Zhang et al., 2017), such as the histone methyltransferase KMT2D (Kawalekar et al., 2016), EZH2 (Elpek et al., 2007), and the HATs CREBBP and EP300 (Alam et al., 2020). In response to the genetic and epigenetic changes, GCB lymphomas have different ways to escape immune surveillance mechanisms. We described the most relevant ones: 1) defective immune recognition; 2) aberrant immune co-stimulation and co-inhibition; 3) cellular and 4) soluble mediators of active immunosuppression. Notably, EZH2 GOF and CREBBP LOF mutations correspond to downregulation of the Ag presentation machinery (MHC-I/MHC-II), and treatment with EZH2is can reverse these phenotypes (Morin et al., 2010). To add to the complex genetic-epigenetic-immune nature of these tumors, mechanisms of metabolic adaptation are also highly heterogeneous in GCB lymphomas and have the potential to influence the activity of epigenetic modifiers (Jellusova and Rickert, 2017). In earlier studies, the heterogeneous response of DLBCLs to R-CHOP was linked to glycolysis, with the glycolytic enzyme GAPDH being reported to predict for poor therapeutic outcome. Vice versa, GAPDHlow lymphomas were found to use other sources metabolic pathways, such as OxPhos and rely on mTORC1 signaling and glutaminolysis (Chiche et al., 2019).
Comprehensive genomic and transcriptomic analyses have allowed to detect subgroups of lymphomas with different metabolic profiles (Caro et al., 2012). However, studies of GCB lymphoma metabolic features are limited and mostly based on gene signatures analyses (Wang H. et al., 2021). Qualitative and quantitative information about preferential substrates and metabolic pathways in GCB lymphoma is required for a comprehensive and potentially actionable framework of the metabolic dysregulations in these diseases. Recently, metabolomic approached based on liquid and/or gas chromatography-mass spectrometry or nuclear magnetic resonance have been explored to identify possible biomarkers for characterization and early diagnosis of the different lymphoma subtypes (Ducker et al., 2017; Schwarzfischer et al., 2017; Barberini et al., 2019). Further deepening our understanding of lymphoma metabolism will be important, as different metabolic states could predict response to therapy and this information may reveal new targets for specific metabolic inhibitors.
The role of the immune microenvironment in GCB lymphomas and B-cell lymphomas overall is also not completely clear. Most findings are based on correlative analyses in patients, with contrasting results in many cases. A major limitation for these studies has been the paucity of faithful syngeneic pre-clinical models where to systematically investigate these mechanisms and the causal implications of specific microenvironmental factors in lymphomagenesis.
Recent advancements in the genetic/epigenetic classification of B-cell lymphomas have allowed to distinguish the oncogenic mutations driving specific disease subtypes more clearly (Chapuy et al., 2018; Mondello et al., 2020; Swenson et al., 2020; Wright et al., 2020; Reimann et al., 2021). This has tremendously advanced the generation of genetically engineered mouse models reproducing the same genetic mutations causing lymphomas in humans and the human diseases (Pasqualucci et al., 2011b; Béguelin et al., 2013; Béguelin et al., 2016; Béguelin et al., 2017; Béguelin et al., 2020). These now constitute an unprecedented resource that will foster and accelerate the study of lymphoma pathogenesis and treatment at different levels. The opportunity to study in vivo in a syngeneic setting rational combinations of lymphoma targeted therapies (e.g. epigenetic and metabolic therapies) with immunotherapies will provide fundamental and timely information for rapid clinical translation, including the identification of possible candidate biomarkers to follow in patients. In addition, we will be able to understand the impact of known lymphoma targeted therapies on the microenvironment, providing further rationale for combination with immunotherapy. This knowledge will likely help clarify some of the discordant or unexpected results obtained with some of these treatments in lymphoma patients and will guide the next chapter of precision immune-oncology treatments for these diseases.
Author Contributions
IS and RZ conceived the work. IS, SC, SY, YI, and RZ wrote the manuscript. WB, AM, and RZ provided critical revisions of the manuscript. IS, SC, and RB drafted figures and tables summarizing the main manuscript concepts.
Funding
IS is supported by NCI R50-CA221810. SY is supported by ASCO Conquer Cancer Foundation. YI is supported by Overseas Research Fellowship, Japan Society for The Promotion of Science. AM is supported by NCI R35 CA220499, R01 CA234561, P50 CA192937, Leukemia and Lymphoma Society (LLS) SCOR 7012-016, LLS SCOR 7021-20, LLS TRP 6605-20, LLS TRP6593-20. WB is supported by ASH Junior Faculty Scholar Award and LLS TRP 6641-22. RZ is supported by the NCI SPORE (P50-CA192937), LLS TRP 6643-22, and by the Parker Institute for Cancer Immunotherapy Bridge Fellows Award.
Conflict of Interest
AM has research funds from Janssen, Astra Zeneca, Epizyme, Daiichi Sankyo, and Sanofi, and has consulted for Astra Zeneca, Bristol Myers Squibb, Epizyme, Constellation, ExoTherapeutics, Daiichi Sankyo, and Janssen. W.B. is consulting for Eisai Co., Lrd. RZ is inventor on patent applications related to work on GITR, PD-1, and CTLA-4. RZ is scientific advisory board member of iTEOS Therapeutics, has consulted for Leap Therapeutics and receives grant support from Astra Zeneca and Bristol Myers Squibb.The other authors have no conflicts of interest to disclose.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abramson, J. S., Palomba, M. L., Gordon, L. I., Lunning, M. A., Wang, M., Arnason, J., et al. (2020). Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): a Multicentre Seamless Design Study. The Lancet 396 (10254), 839–852. doi:10.1016/s0140-6736(20)31366-0
Afonso, J., Pinto, T., Simões-Sousa, S., Schmitt, F., Longatto-Filho, A., Pinheiro, C., et al. (2019). Clinical Significance of Metabolism-Related Biomarkers in Non-hodgkin Lymphoma - MCT1 as Potential Target in Diffuse Large B Cell Lymphoma. Cell Oncol. 42 (3), 303–318. doi:10.1007/s13402-019-00426-2
Ahearne, M. J., Bhuller, K., Hew, R., Ibrahim, H., Naresh, K., and Wagner, S. D. (2014). Expression of PD-1 (CD279) and FoxP3 in Diffuse Large B-Cell Lymphoma. Virchows Arch. 465 (3), 351–358. doi:10.1007/s00428-014-1615-5
Akbari, B., Ghahri-Saremi, N., Soltantoyeh, T., Hadjati, J., Ghassemi, S., and Mirzaei, H. R. (2021). Epigenetic Strategies to Boost CAR T Cell Therapy. Mol. Ther. 29 (9), 2640–2659. doi:10.1016/j.ymthe.2021.08.003
Alam, H., Tang, M., Maitituoheti, M., Dhar, S. S., Kumar, M., Han, C. Y., et al. (2020). KMT2D Deficiency Impairs Super-enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 37 (4), 599–617. doi:10.1016/j.ccell.2020.03.005
Alizadeh, A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., et al. (2000). Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature 403 (6769), 503–511. doi:10.1038/35000501
Allen, C. D. C., Okada, T., and Cyster, J. G. (2007). Germinal-center Organization and Cellular Dynamics. Immunity 27 (2), 190–202. doi:10.1016/j.immuni.2007.07.009
Allis, C. D., and Jenuwein, T. (2016). The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 17 (8), 487–500. doi:10.1038/nrg.2016.59
Almstedt, M., Blagitko-Dorfs, N., Duque-Afonso, J., Karbach, J., Pfeifer, D., Jäger, E., et al. (2010). The DNA Demethylating Agent 5-Aza-2′-Deoxycytidine Induces Expression of NY-ESO-1 and Other Cancer/testis Antigens in Myeloid Leukemia Cells. Leuk. Res. 34 (7), 899–905. doi:10.1016/j.leukres.2010.02.004
Álvaro, T., Lejeune, M., Salvadó, M.-T., Lopez, C., Jaén, J., Bosch, R., et al. (2006). Immunohistochemical Patterns of Reactive Microenvironment Are Associated with Clinicobiologic Behavior in Follicular Lymphoma Patients. Jco 24 (34), 5350–5357. doi:10.1200/jco.2006.06.4766
Andor, N., Simonds, E. F., Czerwinski, D. K., Chen, J., Grimes, S. M., Wood-Bouwens, C., et al. (2019). Single-cell RNA-Seq of Follicular Lymphoma Reveals Malignant B-Cell Types and Coexpression of T-Cell Immune Checkpoints. Blood 133 (10), 1119–1129. doi:10.1182/blood-2018-08-862292
Andrews, J. M., and Payton, J. E. (2019). Epigenetic dynamics in normal and malignant B cells: die a hero or live to become a villain. Curr. Opin. Immunol. 57, 15–22. doi:10.1016/j.coi.2018.09.020
Ansell, S. M., and Lin, Y. (2020). Immunotherapy of Lymphomas. J. Clin. Invest. 130 (4), 1576–1585. doi:10.1172/jci129206
Ansell, S. M., Minnema, M. C., Johnson, P., Timmerman, J. M., Armand, P., Shipp, M. A., et al. (2019). Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. Jco 37 (6), 481–489. doi:10.1200/jco.18.00766
Ansell, S. M., Stenson, M., Habermann, T. M., Jelinek, D. F., and Witzig, T. E. (2001). Cd4+ T-Cell Immune Response to Large B-Cell Non-hodgkin's Lymphoma Predicts Patient Outcome. Jco 19 (3), 720–726. doi:10.1200/jco.2001.19.3.720
Ansell, S. M. (2019). The Highs and Lows of Immune-Checkpoint Blockade in Lymphoma. Cancer Immunol. Res. 7 (5), 696–700. doi:10.1158/2326-6066.cir-18-0890
Armand, P., Janssens, A., Gritti, G., Radford, J., Timmerman, J., Pinto, A., et al. (2021b). Efficacy and Safety Results from CheckMate 140, a Phase 2 Study of Nivolumab for Relapsed/refractory Follicular Lymphoma. Blood 137 (5), 637–645. doi:10.1182/blood.2019004753
Armand, P., Lesokhin, A., Borrello, I., Timmerman, J., Gutierrez, M., Zhu, L., et al. (2021a). A Phase 1b Study of Dual PD-1 and CTLA-4 or KIR Blockade in Patients with Relapsed/refractory Lymphoid Malignancies. Leukemia 35 (3), 777–786. doi:10.1038/s41375-020-0939-1
Assouline, S. E., Kim, W. S., Sehn, L. H., Schuster, S. J., Cheah, C. Y., Nastoupil, L. J., et al. (2020). Mosunetuzumab Shows Promising Efficacy in Patients with Multiply Relapsed Follicular Lymphoma: Updated Clinical Experience from a Phase I Dose-Escalation Trial. Blood 136 (Suppl. 1), 42–44. doi:10.1182/blood-2020-135839
Autio, M., Leivonen, S. K., Brück, O., Mustjoki, S., Mészáros Jørgensen, J., Karjalainen-Lindsberg, M. L., et al. (2021). Immune Cell Constitution in the Tumor Microenvironment Predicts the Outcome in Diffuse Large B-Cell Lymphoma. Haematologica 106 (3), 718–729. doi:10.3324/haematol.2019.243626
Bannerji, R., Allan, J. N., Arnason, J. E., Brown, J. R., Advani, R., Ansell, S. M., et al. (2020). Odronextamab (REGN1979), a Human CD20 X CD3 Bispecific Antibody, Induces Durable, Complete Responses in Patients with Highly Refractory B-Cell Non-hodgkin Lymphoma, Including Patients Refractory to CAR T Therapy. Blood 136 (Suppl. 1), 42–43. doi:10.1182/blood-2020-136659
Barberini, L., Noto, A., Fattuoni, C., Satta, G., Zucca, M., Cabras, M. G., et al. (2019). The Metabolomic Profile of Lymphoma Subtypes: A Pilot Study. Molecules 24 (13), 2367. doi:10.3390/molecules24132367
Barr, P. M., Saylors, G. B., Spurgeon, S. E., Cheson, B. D., Greenwald, D. R., O’Brien, S. M., et al. (2016). Phase 2 Study of Idelalisib and Entospletinib: Pneumonitis Limits Combination Therapy in Relapsed Refractory CLL and NHL. Blood 127 (20), 2411–2415. doi:10.1182/blood-2015-12-683516
Basso, K., Klein, U., Niu, H., Stolovitzky, G. A., Tu, Y., Califano, A., et al. (2004). Tracking CD40 Signaling during Germinal center Development. Blood 104 (13), 4088–4096. doi:10.1182/blood-2003-12-4291
Beck Enemark, M., Monrad, I., Madsen, C., Lystlund Lauridsen, K., Honoré, B., Plesner, T. L., et al. (2021). PD-1 Expression in Pre-treatment Follicular Lymphoma Predicts the Risk of Subsequent High-Grade Transformation. Ott Vol. 14, 481–489. doi:10.2147/ott.s289337
Béguelin, W., Popovic, R., Teater, M., Jiang, Y., Bunting, K. L., Rosen, M., et al. (2013). EZH2 Is Required for Germinal center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell 23 (5), 677–692. doi:10.1016/j.ccr.2013.04.011
Béguelin, W., Rivas, M. A., Calvo Fernández, M. T., Teater, M., Purwada, A., Redmond, D., et al. (2017). EZH2 Enables Germinal centre Formation through Epigenetic Silencing of CDKN1A and an Rb-E2f1 Feedback Loop. Nat. Commun. 8 (1), 877. doi:10.1038/s41467-017-01029-x
Béguelin, W., Teater, M., Gearhart, M. D., Calvo Fernández, M. T., Goldstein, R. L., Cárdenas, M. G., et al. (2016). EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 30 (2), 197–213. doi:10.1016/j.ccell.2016.07.006
Béguelin, W., Teater, M., Meydan, C., Hoehn, K. B., Phillip, J. M., Soshnev, A. A., et al. (2020). Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 37 (5), 655–673. doi:10.1016/j.ccell.2020.04.004
Beloueche-Babari, M., Casals Galobart, T., Delgado-Goni, T., Wantuch, S., Parkes, H. G., Tandy, D., et al. (2020). Monocarboxylate Transporter 1 Blockade with AZD3965 Inhibits Lipid Biosynthesis and Increases Tumour Immune Cell Infiltration. Br. J. Cancer 122 (6), 895–903. doi:10.1038/s41416-019-0717-x
Boice, M., Salloum, D., Mourcin, F., Sanghvi, V., Amin, R., Oricchio, E., et al. (2016). Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell 167 (2), 405–418. doi:10.1016/j.cell.2016.08.032
Brady, M. T., Hilchey, S. P., Hyrien, O., Spence, S. A., and Bernstein, S. H. (2014). Mesenchymal Stromal Cells Support the Viability and Differentiation of Follicular Lymphoma-Infiltrating Follicular Helper T-Cells. PLoS One 9 (5), e97597. doi:10.1371/journal.pone.0097597
Brown, P. J., Wong, K. K., Felce, S. L., Lyne, L., Spearman, H., Soilleux, E. J., et al. (2016). FOXP1 Suppresses Immune Response Signatures and MHC Class II Expression in Activated B-cell-like Diffuse Large B-Cell Lymphomas. Leukemia 30 (3), 605–616. doi:10.1038/leu.2015.299
Budde, L. E., Sehn, L. H., Assouline, S., Flinn, I. W., Isufi, I., Yoon, S. S., et al. (2018). Mosunetuzumab, a Full-Length Bispecific CD20/CD3 Antibody, Displays Clinical Activity in Relapsed/Refractory B-Cell Non-hodgkin Lymphoma (NHL): Interim Safety and Efficacy Results from a Phase 1 Study. Blood 132 (Suppl. 1), 399. doi:10.1182/blood-2018-99-118344
Caganova, M., Carrisi, C., Varano, G., Mainoldi, F., Zanardi, F., Germain, P.-L., et al. (2013). Germinal center Dysregulation by Histone Methyltransferase EZH2 Promotes Lymphomagenesis. J. Clin. Invest. 123 (12), 5009–5022. doi:10.1172/jci70626
Cai, L.-Y., Chen, S.-J., Xiao, S.-H., Sun, Q.-J., Ding, C.-H., Zheng, B.-N., et al. (2021). Targeting P300/CBP Attenuates Hepatocellular Carcinoma Progression through Epigenetic Regulation of Metabolism. Cancer Res. 81 (4), 860–872. doi:10.1158/0008-5472.can-20-1323
Cai, Q.-c., Liao, H., Lin, S.-x., Xia, Y., Wang, X.-x., Gao, Y., et al. (2012). High Expression of Tumor-Infiltrating Macrophages Correlates with Poor Prognosis in Patients with Diffuse Large B-Cell Lymphoma. Med. Oncol. 29 (4), 2317–2322. doi:10.1007/s12032-011-0123-6
Canioni, D., Salles, G., Mounier, N., Brousse, N., Keuppens, M., Morchhauser, F., et al. (2008). High Numbers of Tumor-Associated Macrophages Have an Adverse Prognostic Value that Can Be Circumvented by Rituximab in Patients with Follicular Lymphoma Enrolled onto the GELA-GOELAMS FL-2000 Trial. Jco 26 (3), 440–446. doi:10.1200/jco.2007.12.8298
Cao, F., Zwinderman, M., and Dekker, F. (2018). The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 23 (3), 551. doi:10.3390/molecules23030551
Carbone, A., Roulland, S., Gloghini, A., Younes, A., von Keudell, G., López-Guillermo, A., et al. (2019). Follicular Lymphoma. Nat. Rev. Dis. Primers 5 (1), 83. doi:10.1038/s41572-019-0132-x
Caro, P., Kishan, A. U., Norberg, E., Stanley, I. A., Chapuy, B., Ficarro, S. B., et al. (2012). Metabolic Signatures Uncover Distinct Targets in Molecular Subsets of Diffuse Large B Cell Lymphoma. Cancer Cell 22 (4), 547–560. doi:10.1016/j.ccr.2012.08.014
Carreras, J., Lopez-Guillermo, A., Fox, B. C., Colomo, L., Martinez, A., Roncador, G., et al. (2006). High Numbers of Tumor-Infiltrating FOXP3-Positive Regulatory T Cells Are Associated with Improved Overall Survival in Follicular Lymphoma. Blood 108 (9), 2957–2964. doi:10.1182/blood-2006-04-018218
Carreras, J., Lopez-Guillermo, A., Roncador, G., Villamor, N., Colomo, L., Martinez, A., et al. (2009). High Numbers of Tumor-Infiltrating Programmed Cell Death 1-positive Regulatory Lymphocytes Are Associated with Improved Overall Survival in Follicular Lymphoma. Jco 27 (9), 1470–1476. doi:10.1200/jco.2008.18.0513
Cerchietti, L. C., Hatzi, K., Caldas-Lopes, E., Yang, S. N., Figueroa, M. E., Morin, R. D., et al. (2010). BCL6 Repression of EP300 in Human Diffuse Large B Cell Lymphoma Cells Provides a Basis for Rational Combinatorial Therapy. J. Clin. Invest. 120 (12), 4569–4582. doi:10.1172/jci42869
Challa-Malladi, M., Lieu, Y. K., Califano, O., Holmes, A. B., Bhagat, G., Murty, V. V., et al. (2011). Combined Genetic Inactivation of β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell 20 (6), 728–740. doi:10.1016/j.ccr.2011.11.006
Chang, K. C., Huang, G. C., Jones, D., and Lin, Y. H. (2007). Distribution Patterns of Dendritic Cells and T Cells in Diffuse Large B-Cell Lymphomas Correlate with Prognoses. Clin. Cancer Res. 13 (22 Pt 1), 6666–6672. doi:10.1158/1078-0432.CCR-07-0504
Chapuy, B., Stewart, C., Dunford, A. J., Kim, J., Kamburov, A., Redd, R. A., et al. (2018). Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 24 (5), 679–690. doi:10.1038/s41591-018-0016-8
Chapuy, B., Roemer, M. G. M., Stewart, C., Tan, Y., Abo, R. P., Zhang, L., et al. (2016). Targetable Genetic Features of Primary Testicular and Primary central Nervous System Lymphomas. Blood 127 (7), 869–881. doi:10.1182/blood-2015-10-673236
Chen, B. J., Dashnamoorthy, R., Galera, P., Makarenko, V., Chang, H., Ghosh, S., et al. (2019). The Immune Checkpoint Molecules PD-1, PD-L1, TIM-3 and LAG-3 in Diffuse Large B-Cell Lymphoma. Oncotarget 10 (21), 2030–2040. doi:10.18632/oncotarget.26771
Chen, I.-C., Sethy, B., and Liou, J.-P. (2020). Recent Update of HDAC Inhibitors in Lymphoma. Front. Cel. Dev. Biol. 8, 576391. doi:10.3389/fcell.2020.576391
Chen, L., and Flies, D. B. (2013). Molecular Mechanisms of T Cell Co-stimulation and Co-inhibition. Nat. Rev. Immunol. 13 (4), 227–242. doi:10.1038/nri3405
Chen, R., Frankel, P., Popplewell, L., Siddiqi, T., Ruel, N., Rotter, A., et al. (2015). A Phase II Study of Vorinostat and Rituximab for Treatment of Newly Diagnosed and Relapsed/refractory Indolent Non-hodgkin Lymphoma. Haematologica 100 (3), 357–362. doi:10.3324/haematol.2014.117473
Chiappinelli, K. B., Strissel, P. L., Desrichard, A., Li, H., Henke, C., Akman, B., et al. (2015). Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162 (5), 974–986. doi:10.1016/j.cell.2015.07.011
Chiche, J., Reverso-Meinietti, J., Mouchotte, A., Rubio-Patiño, C., Mhaidly, R., Villa, E., et al. (2019). GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitors. Cel. Metab. 29 (6), 1243–1257. doi:10.1016/j.cmet.2019.02.002
Choi, S.-C., and Morel, L. (2020). Immune Metabolism Regulation of the Germinal center Response. Exp. Mol. Med. 52 (3), 348–355. doi:10.1038/s12276-020-0392-2
Clozel, T., Yang, S., Elstrom, R. L., Tam, W., Martin, P., Kormaksson, M., et al. (2013). Mechanism-based Epigenetic Chemosensitization Therapy of Diffuse Large B-Cell Lymphoma. Cancer Discov. 3 (9), 1002–1019. doi:10.1158/2159-8290.cd-13-0117
Coutinho, R., Clear, A. J., Mazzola, E., Owen, A., Greaves, P., Wilson, A., et al. (2015). Revisiting the Immune Microenvironment of Diffuse Large B-Cell Lymphoma Using a Tissue Microarray and Immunohistochemistry: Robust Semi-automated Analysis Reveals CD3 and FoxP3 as Potential Predictors of Response to R-CHOP. Haematologica 100 (3), 363–369. doi:10.3324/haematol.2014.110189
Crump, M., Coiffier, B., Jacobsen, E. D., Sun, L., Ricker, J. L., Xie, H., et al. (2008). Phase II Trial of Oral Vorinostat (Suberoylanilide Hydroxamic Acid) in Relapsed Diffuse Large-B-Cell Lymphoma. Ann. Oncol. 19 (5), 964–969. doi:10.1093/annonc/mdn031
Crump, M., Neelapu, S. S., Farooq, U., Van Den Neste, E., Kuruvilla, J., Westin, J., et al. (2017). Outcomes in Refractory Diffuse Large B-Cell Lymphoma: Results from the International SCHOLAR-1 Study. Blood 130 (16), 1800–1808. doi:10.1182/blood-2017-03-769620
Curti, A., Pandolfi, S., Valzasina, B., Aluigi, M., Isidori, A., Ferri, E., et al. (2007). Modulation of Tryptophan Catabolism by Human Leukemic Cells Results in the Conversion of CD25− into CD25+ T Regulatory Cells. Blood 109 (7), 2871–2877. doi:10.1182/blood-2006-07-036863
D'Haene, N., Maris, C., Sandras, F., Dehou, M.-F., Remmelink, M., Decaestecker, C., et al. (2005). The Differential Expression of Galectin-1 and Galectin-3 in normal Lymphoid Tissue and Non-hodgkin's and Hodgkin's Lymphomas. Int. J. Immunopathol Pharmacol. 18 (3), 431–443. doi:10.1177/039463200501800304
Dakappagari, N., Ho, S. N., Gascoyne, R. D., Ranuio, J., Weng, A. P., and Tangri, S. (2012). CD80 (B7.1) Is Expressed on Both Malignant B Cells and Nonmalignant Stromal Cells in Non-hodgkin Lymphoma. Cytometry 82B (2), 112–119. doi:10.1002/cyto.b.20631
Dann, S. G., Ryskin, M., Barsotti, A. M., Golas, J., Shi, C., Miranda, M., et al. (2015). Reciprocal Regulation of Amino Acid Import and Epigenetic State through Lat1 and EZH 2. EMBO J. 34 (13), 1773–1785. doi:10.15252/embj.201488166
Dave, S. S., Wright, G., Tan, B., Rosenwald, A., Gascoyne, R. D., Chan, W. C., et al. (2004). Prediction of Survival in Follicular Lymphoma Based on Molecular Features of Tumor-Infiltrating Immune Cells. N. Engl. J. Med. 351 (21), 2159–2169. doi:10.1056/nejmoa041869
de Charette, M., Marabelle, A., and Houot, R. (2016). Turning Tumour Cells into Antigen Presenting Cells: The Next Step to Improve Cancer Immunotherapy? Eur. J. Cancer 68, 134–147. doi:10.1016/j.ejca.2016.09.010
Dersh, D., Phelan, J. D., Gumina, M. E., Wang, B., Arbuckle, J. H., Holly, J., et al. (2021). Genome-wide Screens Identify Lineage- and Tumor-specific Genes Modulating MHC-I- and MHC-II-Restricted Immunosurveillance of Human Lymphomas. Immunity 54 (1), 116–131. doi:10.1016/j.immuni.2020.11.002
Di Croce, L., and Helin, K. (2013). Transcriptional Regulation by Polycomb Group Proteins. Nat. Struct. Mol. Biol. 20 (10), 1147–1155. doi:10.1038/nsmb.2669
Ding, L., Getz, G., Wheeler, D. A., Mardis, E. R., McLellan, M. D., Cibulskis, K., et al. (2008). Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature 455 (7216), 1069–1075. doi:10.1038/nature07423
Ding, W., Laplant, B., Witzig, T. E., Johnston, P. B., Colgan, J. P., Rech, K. L., et al. (2017). PD-1 Blockade with Pembrolizumab in Relapsed Low Grade Non-hodgkin Lymphoma. Blood 130 (Suppl. 1), 4055. doi:10.1182/blood.V130.Suppl_1.4055.4055
Dominguez, P. M., Teater, M., Chambwe, N., Kormaksson, M., Redmond, D., Ishii, J., et al. (2015). DNA Methylation Dynamics of Germinal Center B Cells Are Mediated by AID. Cel. Rep. 12 (12), 2086–2098. doi:10.1016/j.celrep.2015.08.036
Dreyling, M., Morschhauser, F., Bouabdallah, K., Bron, D., Cunningham, D., Assouline, S. E., et al. (2017a). Phase II Study of Copanlisib, a PI3K Inhibitor, in Relapsed or Refractory, Indolent or Aggressive Lymphoma. Ann. Oncol. 28 (9), 2169–2178. doi:10.1093/annonc/mdx289
Dreyling, M., Santoro, A., Mollica, L., Leppä, S., Follows, G. A., Lenz, G., et al. (2017b). Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. Jco 35 (35), 3898–3905. doi:10.1200/jco.2017.75.4648
Ducker, G. S., Ghergurovich, J. M., Mainolfi, N., Suri, V., Jeong, S. K., Hsin-Jung Li, S., et al. (2017). Human SHMT Inhibitors Reveal Defective glycine Import as a Targetable Metabolic Vulnerability of Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 114 (43), 11404–11409. doi:10.1073/pnas.1706617114
DuPage, M., Chopra, G., Quiros, J., Rosenthal, W. L., Morar, M. M., Holohan, D., et al. (2015). The Chromatin-Modifying Enzyme Ezh2 Is Critical for the Maintenance of Regulatory T Cell Identity after Activation. Immunity 42 (2), 227–238. doi:10.1016/j.immuni.2015.01.007
Elpek, K. G., Lacelle, C., Singh, N. P., Yolcu, E. S., and Shirwan, H. (2007). CD4+CD25+ T Regulatory Cells Dominate Multiple Immune Evasion Mechanisms in Early but Not Late Phases of Tumor Development in a B Cell Lymphoma Model. J. Immunol. 178 (11), 6840–6848. doi:10.4049/jimmunol.178.11.6840
Ennishi, D., Jiang, A., Boyle, M., Collinge, B., Grande, B. M., Ben-Neriah, S., et al. (2019a). Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-cell-like Diffuse Large B-Cell Lymphoma. Jco 37 (3), 190–201. doi:10.1200/jco.18.01583
Ennishi, D., Takata, K., Béguelin, W., Duns, G., Mottok, A., Farinha, P., et al. (2019b). Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 9 (4), 546–563. doi:10.1158/2159-8290.cd-18-1090
Fang, X., Xiu, B., Yang, Z., Qiu, W., Zhang, L., Zhang, S., et al. (2017). The Expression and Clinical Relevance of PD-1, PD-L1, and TP63 in Patients with Diffuse Large B-Cell Lymphoma. Medicine (Baltimore) 96 (15), e6398. doi:10.1097/md.0000000000006398
Fangazio, M., Ladewig, E., Gomez, K., Garcia-Ibanez, L., Kumar, R., Teruya-Feldstein, J., et al. (2021). Genetic Mechanisms of HLA-I Loss and Immune Escape in Diffuse Large B Cell Lymphoma. Proc. Natl. Acad. Sci. U S A. 118 (22), e2104504118. doi:10.1073/pnas.2104504118
Farinha, P., Al-Tourah, A., Gill, K., Klasa, R., Connors, J. M., and Gascoyne, R. D. (2010). The Architectural Pattern of FOXP3-Positive T Cells in Follicular Lymphoma Is an Independent Predictor of Survival and Histologic Transformation. Blood 115 (2), 289–295. doi:10.1182/blood-2009-07-235598
Farinha, P., Masoudi, H., Skinnider, B. F., Shumansky, K., Spinelli, J. J., Gill, K., et al. (2005). Analysis of Multiple Biomarkers Shows that Lymphoma-Associated Macrophage (LAM) Content Is an Independent Predictor of Survival in Follicular Lymphoma (FL). Blood 106 (6), 2169–2174. doi:10.1182/blood-2005-04-1565
Flinn, I. W., Miller, C. B., Ardeshna, K. M., Tetreault, S., Assouline, S. E., Mayer, J., et al. (2019). DYNAMO: A Phase II Study of Duvelisib (IPI-145) in Patients with Refractory Indolent Non-hodgkin Lymphoma. Jco 37 (11), 912–922. doi:10.1200/jco.18.00915
Ford, D. J., and Dingwall, A. K. (2015). The Cancer COMPASS: Navigating the Functions of MLL Complexes in Cancer. Cancer Genet. 208 (5), 178–191. doi:10.1016/j.cancergen.2015.01.005
Fowler, N. H., Samaniego, F., Jurczak, W., Ghosh, N., Derenzini, E., Reeves, J. A., et al. (2021). Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients with Relapsed or Refractory Indolent Lymphoma. Jco 39 (15), 1609–1618. doi:10.1200/jco.20.03433
Frigault, M. J., Armand, P., Redd, R. A., Jeter, E., Merryman, R. W., Coleman, K. C., et al. (2020). PD-1 Blockade for Diffuse Large B-Cell Lymphoma after Autologous Stem Cell Transplantation. Blood Adv. 4 (1), 122–126. doi:10.1182/bloodadvances.2019000784
Froimchuk, E., Jang, Y., and Ge, K. (2017). Histone H3 Lysine 4 Methyltransferase KMT2D. Gene 627, 337–342. doi:10.1016/j.gene.2017.06.056
Fu, L., Pelicano, H., Liu, J., Huang, P., and Lee, C. C. (2002). The Circadian Gene Period2 Plays an Important Role in Tumor Suppression and DNA Damage Response In Vivo. Cell 111 (1), 41–50. doi:10.1016/s0092-8674(02)00961-3
Fuca, G., Ambrosini, M., Agnelli, L., Brich, S., Sgambelluri, F., Mortarini, R., et al. (2021). Fifteen-year Follow-Up of Relapsed Indolent Non-hodgkin Lymphoma Patients Vaccinated with Tumor-Loaded Dendritic Cells. J. Immunother. Cancer 9 (6), e002240. doi:10.1136/jitc-2020-002240
Gabrilovich, D. I., Ostrand-Rosenberg, S., and Bronte, V. (2012). Coordinated Regulation of Myeloid Cells by Tumours. Nat. Rev. Immunol. 12 (4), 253–268. doi:10.1038/nri3175
Galanina, N., Smith, S. M., Liao, C., Petrich, A., Libao, B., Gartenhaus, R., et al. (2018). University of Chicago Phase II Consortium Trial of Selumetinib (MEKi) Demonstrates Low Tolerability and Efficacy in Relapsed DLBCL. Br. J. Haematol. 181 (2), 264–267. doi:10.1111/bjh.14544
García-Ramírez, I., Tadros, S., González-Herrero, I., Martín-Lorenzo, A., Rodríguez-Hernández, G., Moore, D., et al. (2017). Crebbp Loss Cooperates with Bcl2 Overexpression to Promote Lymphoma in Mice. Blood 129 (19), 2645–2656. doi:10.1182/blood-2016-08-733469
Georgiou, G., Ippolito, G. C., Beausang, J., Busse, C. E., Wardemann, H., and Quake, S. R. (2014). The Promise and challenge of High-Throughput Sequencing of the Antibody Repertoire. Nat. Biotechnol. 32 (2), 158–168. doi:10.1038/nbt.2782
Georgiou, K., Chen, L., Berglund, M., Ren, W., de Miranda, N. F. C. C., Lisboa, S., et al. (2016). Genetic Basis of PD-L1 Overexpression in Diffuse Large B-Cell Lymphomas. Blood 127 (24), 3026–3034. doi:10.1182/blood-2015-12-686550
Ghoneim, H. E., Fan, Y., Moustaki, A., Abdelsamed, H. A., Dash, P., Dogra, P., et al. (2017). De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 170 (1), 142–157. doi:10.1016/j.cell.2017.06.007
Goebeler, M.-E., Knop, S., Viardot, A., Kufer, P., Topp, M. S., Einsele, H., et al. (2016). Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients with Relapsed/Refractory Non-hodgkin Lymphoma: Final Results from a Phase I Study. Jco 34 (10), 1104–1111. doi:10.1200/jco.2014.59.1586
Good, Z., Spiegel, J. Y., Sahaf, B., Malipatlolla, M. B., Frank, M. J., Baird, J., et al. (2019). Identification of Two CAR T-Cell Populations Associated with Complete Response or Progressive Disease in Adult Lymphoma Patients Treated with Axi-Cel. Blood 134 (Supplement_1), 779. doi:10.1182/blood-2019-130815
Good-Jacobson, K. L., Szumilas, C. G., Chen, L., Sharpe, A. H., Tomayko, M. M., and Shlomchik, M. J. (2010). PD-1 Regulates Germinal center B Cell Survival and the Formation and Affinity of Long-Lived Plasma Cells. Nat. Immunol. 11 (6), 535–542. doi:10.1038/ni.1877
Gopal, A. K., Kahl, B. S., de Vos, S., Wagner-Johnston, N. D., Schuster, S. J., Jurczak, W. J., et al. (2014). PI3Kδ Inhibition by Idelalisib in Patients with Relapsed Indolent Lymphoma. N. Engl. J. Med. 370 (11), 1008–1018. doi:10.1056/nejmoa1314583
Goswami, S., Apostolou, I., Zhang, J., Skepner, J., Anandhan, S., Zhang, X., et al. (2018). Modulation of EZH2 Expression in T Cells Improves Efficacy of Anti-CTLA-4 Therapy. J. Clin. Invest. 128 (9), 3813–3818. doi:10.1172/jci99760
Gray, S. M., Amezquita, R. A., Guan, T., Kleinstein, S. H., and Kaech, S. M. (2017). Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8 + T Cell Terminal Differentiation and Loss of Multipotency. Immunity 46 (4), 596–608. doi:10.1016/j.immuni.2017.03.012
Greaves, P., and Gribben, J. G. (2013). The Role of B7 Family Molecules in Hematologic Malignancy. Blood 121 (5), 734–744. doi:10.1182/blood-2012-10-385591
Green, M. R. (2018). Chromatin Modifying Gene Mutations in Follicular Lymphoma. Blood 131 (6), 595–604. doi:10.1182/blood-2017-08-737361
Green, M. R., Kihira, S., Liu, C. L., Nair, R. V., Salari, R., Gentles, A. J., et al. (2015). Mutations in Early Follicular Lymphoma Progenitors Are Associated with Suppressed Antigen Presentation. Proc. Natl. Acad. Sci. USA 112 (10), E1116–E1125. doi:10.1073/pnas.1501199112
Greenbaum, A., Gopal, A. K., Fromm, J. R., and Houghton, A. M. (2019). Diffuse Large B-Cell Lymphoma Is Infiltrated with Functional CD8+ T-Cells Lacking the Hallmarks of Exhaustion. Blood 134 (Supplement_1), 1518. doi:10.1182/blood-2019-126217
Greer, E. L., and Shi, Y. (2012). Histone Methylation: a Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 13 (5), 343–357. doi:10.1038/nrg3173
Gu, Z., Liu, Y., Cai, F., Patrick, M., Zmajkovic, J., Cao, H., et al. (2019). Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov. 9 (9), 1228–1247. doi:10.1158/2159-8290.cd-19-0152
Guo, B., Cen, H., Tan, X., and Ke, Q. (2016). Meta-analysis of the Prognostic and Clinical Value of Tumor-Associated Macrophages in Adult Classical Hodgkin Lymphoma. BMC Med. 14 (1), 159. doi:10.1186/s12916-016-0711-6
Guo, H., Zeng, D., Zhang, H., Bell, T., Yao, J., Liu, Y., et al. (2019). Dual Inhibition of PI3K Signaling and Histone Deacetylation Halts Proliferation and Induces Lethality in Mantle Cell Lymphoma. Oncogene 38 (11), 1802–1814. doi:10.1038/s41388-018-0550-3
Halestrap, A. P. (2013). The SLC16 Gene Family - Structure, Role and Regulation in Health and Disease. Mol. Aspects Med. 34 (2-3), 337–349. doi:10.1016/j.mam.2012.05.003
Halford, S. E. R., Walter, H., McKay, P., Townsend, W., Linton, K., Heinzmann, K., et al. (2021). Phase I Expansion Study of the First-In-Class Monocarboxylate Transporter 1 (MCT1) Inhibitor AZD3965 in Patients with Diffuse Large B-Cell Lymphoma (DLBCL) and Burkitt Lymphoma (BL). Jco 39 (15_Suppl. l), 3115. doi:10.1200/jco.2021.39.15_suppl.3115
Haralambieva, E., Adam, P., Ventura, R., Katzenberger, T., Kalla, J., Höller, S., et al. (2006). Genetic Rearrangement of FOXP1 Is Predominantly Detected in a Subset of Diffuse Large B-Cell Lymphomas with Extranodal Presentation. Leukemia 20 (7), 1300–1303. doi:10.1038/sj.leu.2404244
Harb, W., Abramson, J., Lunning, M., Goy, A., Maddocks, K., Lebedinsky, C., et al. (2018). A Phase 1 Study of CPI-1205, a Small Molecule Inhibitor of EZH2, Preliminary Safety in Patients with B-Cell Lymphomas. Ann. Oncol. 29, iii7. doi:10.1093/annonc/mdy048.001
Hashwah, H., Schmid, C. A., Kasser, S., Bertram, K., Stelling, A., Manz, M. G., et al. (2017). Inactivation of CREBBP Expands the Germinal center B Cell Compartment, Down-Regulates MHCII Expression and Promotes DLBCL Growth. Proc. Natl. Acad. Sci. USA 114 (36), 9701–9706. doi:10.1073/pnas.1619555114
Hatzi, K., Geng, H., Doane, A. S., Meydan, C., LaRiviere, R., Cardenas, M., et al. (2019). Histone Demethylase LSD1 Is Required for Germinal center Formation and BCL6-Driven Lymphomagenesis. Nat. Immunol. 20 (1), 86–96. doi:10.1038/s41590-018-0273-1
Hatzi, K., Jiang, Y., Huang, C., Garrett-Bakelman, F., Gearhart, M. D., Giannopoulou, E. G., et al. (2013). A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cel. Rep. 4 (3), 578–588. doi:10.1016/j.celrep.2013.06.016
Herrmann, A., Lahtz, C., Nagao, T., Song, J. Y., Chan, W. C., Lee, H., et al. (2017). CTLA4 Promotes Tyk2-STAT3-dependent B-Cell Oncogenicity. Cancer Res. 77 (18), 5118–5128. doi:10.1158/0008-5472.can-16-0342
Heward, J. A., Konali, L., D'Avola, A., Close, K., Yeomans, A., Philpott, M., et al. (2021). KDM5 Inhibition Offers a Novel Therapeutic Strategy for the Treatment of KMT2D Mutant Lymphomas. Blood 138, 370–381. doi:10.1182/blood.2020008743
Heward, J., Konali, L., D’Avola, A., Close, K., Yeomans, A., Philpott, M., et al. (2021). KDM5 Inhibition Offers a Novel Therapeutic Strategy for the Treatment of KMT2D Mutant Lymphomas. Blood 138 (5), 370–381. doi:10.1182/blood.2020008743
Hirayama, A. V., Gauthier, J., Hay, K. A., Voutsinas, J. M., Wu, Q., Pender, B. S., et al. (2019). High Rate of Durable Complete Remission in Follicular Lymphoma after CD19 CAR-T Cell Immunotherapy. Blood 134 (7), 636–640. doi:10.1182/blood.2019000905
Huang, Y.-H., Cai, K., Xu, P.-P., Wang, L., Huang, C.-X., Fang, Y., et al. (2021). CREBBP/EP300 Mutations Promoted Tumor Progression in Diffuse Large B-Cell Lymphoma through Altering Tumor-Associated Macrophage Polarization via FBXW7-NOTCH-Ccl2/csf1 axis. Sig. Transduct. Target. Ther. 6 (1), 10. doi:10.1038/s41392-020-00437-8
Hutchings, M., Carlo-Stella, C., Bachy, E., Offner, F. C., Morschhauser, F., Crump, M., et al. (2020a). Glofitamab Step-Up Dosing Induces High Response Rates in Patients with Hard-To-Treat Refractory or Relapsed Non-hodgkin Lymphoma. Blood 136 (Suppl. 1), 46–48. doi:10.1182/blood-2020-136044
Hutchings, M., Mous, R., Clausen, M. R., Johnson, P., Linton, K. M., Chamuleau, M. E. D., et al. (2020b). Subcutaneous Epcoritamab Induces Complete Responses with an Encouraging Safety Profile across Relapsed/Refractory B-Cell Non-hodgkin Lymphoma Subtypes, Including Patients with Prior CAR-T Therapy: Updated Dose Escalation Data. Blood 136 (Suppl. 1), 45–46. doi:10.1182/blood-2020-133820
Italiano, A., Soria, J.-C., Toulmonde, M., Michot, J.-M., Lucchesi, C., Varga, A., et al. (2018). Tazemetostat, an EZH2 Inhibitor, in Relapsed or Refractory B-Cell Non-hodgkin Lymphoma and Advanced Solid Tumours: a First-In-Human, Open-Label, Phase 1 Study. Lancet Oncol. 19 (5), 649–659. doi:10.1016/s1470-2045(18)30145-1
Izzo, L. T., Affronti, H. C., and Wellen, K. E. (2021). The Bidirectional Relationship between Cancer Epigenetics and Metabolism. Annu. Rev. Cancer Biol. 5, 235–257. doi:10.1146/annurev-cancerbio-070820-035832
Jacobson, C., Chavez, J. C., Sehgal, A. R., William, B. M., Munoz, J., Salles, G., et al. (2020). Primary Analysis of Zuma-5: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory (R/R) Indolent Non-hodgkin Lymphoma (iNHL). Blood 136 (Suppl. 1), 40–41. doi:10.1182/blood-2020-136834
Jellusova, J., and Rickert, R. C. (2017). A Brake for B Cell Proliferation: Appropriate Responses to Metabolic Stress Are Crucial to Maintain B Cell Viability and Prevent Malignant Outgrowth. Bioessays 39 (11), 1700079. doi:10.1002/bies.201700079
Jellusova, J., Cato, M. H., Apgar, J. R., Ramezani-Rad, P., Leung, C. R., Chen, C., et al. (2017). Gsk3 Is a Metabolic Checkpoint Regulator in B Cells. Nat. Immunol. 18 (3), 303–312. doi:10.1038/ni.3664
Jellusova, J. (2020). Metabolic Control of B Cell Immune Responses. Curr. Opin. Immunol. 63, 21–28. doi:10.1016/j.coi.2019.11.002
Jiang, Y., Ortega-Molina, A., Geng, H., Ying, H.-Y., Hatzi, K., Parsa, S., et al. (2017). CREBBP Inactivation Promotes the Development of HDAC3-dependent Lymphomas. Cancer Discov. 7 (1), 38–53. doi:10.1158/2159-8290.cd-16-0975
Josefsson, S. E., Beiske, K., Blaker, Y. N., Førsund, M. S., Holte, H., Østenstad, B., et al. (2019). TIGIT and PD-1 Mark Intratumoral T Cells with Reduced Effector Function in B-Cell Non-hodgkin Lymphoma. Cancer Immunol. Res. 7 (3), 355–362. doi:10.1158/2326-6066.cir-18-0351
Josefsson, S. E., Huse, K., Kolstad, A., Beiske, K., Pende, D., Steen, C. B., et al. (2018). T Cells Expressing Checkpoint Receptor TIGIT Are Enriched in Follicular Lymphoma Tumors and Characterized by Reversible Suppression of T-Cell Receptor Signaling. Clin. Cancer Res. 24 (4), 870–881. doi:10.1158/1078-0432.ccr-17-2337
Kania, A., Scharer, C. D., Price, M. J., Haines, R. R., Alexander-George, L. E., and Boss, J. (2019). H3K27me3-specific Demethylases Modulate B Cell Development and Differentiation. J. Immunol. 202 (1), 188.15.
Kania, A., Scharer, C., and Boss, J. M. (2017). Inhibition of H3K27me3-specific Demethylases Promotes Plasma Cell Formation. J. Immunol. 198 (1), 195.21.
Kashiwakuma, D., Suto, A., Hiramatsu, Y., Ikeda, K., Takatori, H., Suzuki, K., et al. (2010). B and T Lymphocyte Attenuator Suppresses IL-21 Production from Follicular Th Cells and Subsequent Humoral Immune Responses. J.I. 185 (5), 2730–2736. doi:10.4049/jimmunol.0903839
Kataoka, K., Shiraishi, Y., Takeda, Y., Sakata, S., Matsumoto, M., Nagano, S., et al. (2016). Aberrant PD-L1 Expression through 3′-UTR Disruption in Multiple Cancers. Nature 534 (7607), 402–406. doi:10.1038/nature18294
Kawalekar, O. U., O’ Connor, R. S., Fraietta, J. A., Guo, L., McGettigan, S. E., Posey, A. D., et al. (2016). Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 44 (3), 712. doi:10.1016/j.immuni.2016.02.023
Keane, C., Gill, D., Vari, F., Cross, D., Griffiths, L., and Gandhi, M. (2013). CD4+Tumor Infiltrating Lymphocytes Are Prognostic and Independent of R-IPI in Patients with DLBCL Receiving R-CHOP Chemo-Immunotherapy. Am. J. Hematol. 88 (4), 273–276. doi:10.1002/ajh.23398
Keane, C., Law, S. C., Gould, C., Birch, S., Sabdia, M. B., Merida de Long, L., et al. (2020). LAG3: a Novel Immune Checkpoint Expressed by Multiple Lymphocyte Subsets in Diffuse Large B-Cell Lymphoma. Blood Adv. 4 (7), 1367–1377. doi:10.1182/bloodadvances.2019001390
Kennedy, R., and Klein, U. (2019). A T Cell-B Cell Tumor-Suppressive Axis in the Germinal Center. Immunity 51 (2), 204–206. doi:10.1016/j.immuni.2019.07.006
Khodadoust, M. S., Olsson, N., Wagar, L. E., Haabeth, O. A. W., Chen, B., Swaminathan, K., et al. (2017). Antigen Presentation Profiling Reveals Recognition of Lymphoma Immunoglobulin Neoantigens. Nature 543 (7647), 723–727. doi:10.1038/nature21433
Kim, M., and Costello, J. (2017). DNA Methylation: an Epigenetic Mark of Cellular Memory. Exp. Mol. Med. 49 (4), e322. doi:10.1038/emm.2017.10
Kinnaird, A., Zhao, S., Wellen, K. E., and Michelakis, E. D. (2016). Metabolic Control of Epigenetics in Cancer. Nat. Rev. Cancer 16 (11), 694–707. doi:10.1038/nrc.2016.82
Kirschbaum, M., Frankel, P., Popplewell, L., Zain, J., Delioukina, M., Pullarkat, V., et al. (2011). Phase II Study of Vorinostat for Treatment of Relapsed or Refractory Indolent Non-hodgkin's Lymphoma and Mantle Cell Lymphoma. Jco 29 (9), 1198–1203. doi:10.1200/jco.2010.32.1398
Kiyasu, J., Miyoshi, H., Hirata, A., Arakawa, F., Ichikawa, A., Niino, D., et al. (2015). Expression of Programmed Cell Death Ligand 1 Is Associated with Poor Overall Survival in Patients with Diffuse Large B-Cell Lymphoma. Blood 126 (19), 2193–2201. doi:10.1182/blood-2015-02-629600
Komohara, Y., Niino, D., Ohnishi, K., Ohshima, K., and Takeya, M. (2015). Role of Tumor-Associated Macrophages in Hematological Malignancies. Pathol. Int. 65 (4), 170–176. doi:10.1111/pin.12259
Koon, H. B., Ippolito, G. C., Banham, A. H., and Tucker, P. W. (2007). FOXP1: a Potential Therapeutic Target in Cancer. Expert Opin. Ther. Targets 11 (7), 955–965. doi:10.1517/14728222.11.7.955
Kotlov, N., Bagaev, A., Revuelta, M. V., Phillip, J. M., Cacciapuoti, M. T., Antysheva, Z., et al. (2021). Clinical and Biological Subtypes of B-Cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 11, 1468–1489. doi:10.1158/2159-8290.CD-20-0839
Krysiak, K., Gomez, F., White, B. S., Matlock, M., Miller, C. A., Trani, L., et al. (2017). Recurrent Somatic Mutations Affecting B-Cell Receptor Signaling Pathway Genes in Follicular Lymphoma. Blood 129 (4), 473–483. doi:10.1182/blood-2016-07-729954
Labidi, S. I., Ménétrier-Caux, C., Chabaud, S., Chassagne, C., Sebban, C., Gargi, T., et al. (2010). Serum Cytokines in Follicular Lymphoma. Correlation of TGF-β and VEGF with Survival. Ann. Hematol. 89 (1), 25–33. doi:10.1007/s00277-009-0777-8
LaPensee, C. R., Lin, G., Dent, A. L., and Schwartz, J. (2014). Deficiency of the Transcriptional Repressor B Cell Lymphoma 6 (Bcl6) Is Accompanied by Dysregulated Lipid Metabolism. PLoS One 9 (6), e97090. doi:10.1371/journal.pone.0097090
Lee, K.-M., Chuang, E., Griffin, M., Khattri, R., Hong, D. K., Zhang, W., et al. (1998). Molecular Basis of T Cell Inactivation by CTLA-4. Science 282 (5397), 2263–2266. doi:10.1126/science.282.5397.2263
Lenz, G., Hawkes, E., Verhoef, G., Haioun, C., Thye Lim, S., Seog Heo, D., et al. (2020). Single-agent Activity of Phosphatidylinositol 3-kinase Inhibition with Copanlisib in Patients with Molecularly Defined Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Leukemia 34 (8), 2184–2197. doi:10.1038/s41375-020-0743-y
Lesokhin, A. M., Ansell, S. M., Armand, P., Scott, E. C., Halwani, A., Gutierrez, M., et al. (2016). Nivolumab in Patients with Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. Jco 34 (23), 2698–2704. doi:10.1200/jco.2015.65.9789
Li, M., Chiang, Y.-L., Lyssiotis, C. A., Teater, M. R., Hong, J. Y., Shen, H., et al. (2019). Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 35 (6), 916–931. doi:10.1016/j.ccell.2019.05.002
Li, Y.-L., Shi, Z.-H., Wang, X., Gu, K.-S., and Zhai, Z.-M. (2019). Tumor-associated Macrophages Predict Prognosis in Diffuse Large B-Cell Lymphoma and Correlation with Peripheral Absolute Monocyte Count. BMC Cancer 19 (1), 1049. doi:10.1186/s12885-019-6208-x
Li, Z., Li, M., Wang, D., Hou, P., Chen, X., Chu, S., et al. (2020). Post-translational Modifications of EZH2 in Cancer. Cell Biosci. 10 (1), 143. doi:10.1186/s13578-020-00505-0
Liu, R., Niu, Y., Zhang, X., and Ma, T. (2021). Association of KMT2C/D Loss-Of-Function Mutations with Tumor Infiltrating Lymphocytes and Response to Immune Checkpoint Inhibitors in Solid Tumors. Jco 39 (15_Suppl. l), 2587. doi:10.1200/jco.2021.39.15_suppl.2587
Liu, X., Venkataraman, G., Lin, J., Kiyotani, K., Smith, S., Montoya, M., et al. (2015). Highly Clonal Regulatory T-Cell Population in Follicular Lymphoma - Inverse Correlation with the Diversity of CD8+T Cells. Oncoimmunology 4 (5), e1002728. doi:10.1080/2162402x.2014.1002728
Locke, F. L., Ghobadi, A., Jacobson, C. A., Miklos, D. B., Lekakis, L. J., Oluwole, O. O., et al. (2019). Long-term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): a Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol. 20 (1), 31–42. doi:10.1016/s1470-2045(18)30864-7
Lu, C., Coradin, M., Porter, E. G., and Garcia, B. A. (2020). Accelerating the Field of Epigenetic Histone Modification through Mass Spectrometry-Based Approaches. Mol. Cel. Proteomics 20, 100006. doi:10.1074/mcp.R120.002257
Lue, J. K., Prabhu, S. A., Liu, Y., Gonzalez, Y., Verma, A., Mundi, P. S., et al. (2019). Precision Targeting with EZH2 and HDAC Inhibitors in Epigenetically Dysregulated Lymphomas. Clin. Cancer Res. 25 (17), 5271–5283. doi:10.1158/1078-0432.ccr-18-3989
Martin, P., Bartlett, N. L., Chavez, J. C., Reagan, J. L., Smith, S. M., LaCasce, A. S., et al. (2021). Phase 1 Study of Oral Azacitidine (CC-486) Plus R-CHOP in Previously Untreated Intermediate- to High-Risk DLBCL. Blood. online ahead of print. doi:10.1182/blood.2021011679
Matasar, M. J., Capra, M., Özcan, M., Lv, F., Li, W., Yañez, E., et al. (2021). Copanlisib Plus Rituximab versus Placebo Plus Rituximab in Patients with Relapsed Indolent Non-hodgkin Lymphoma (CHRONOS-3): a Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 22 (5), 678–689. doi:10.1016/s1470-2045(21)00145-5
Matsuki, E., Bohn, O. L., El Jamal, S., Pichardo, J. D., Zelenetz, A. D., Younes, A., et al. (2019). Lymphocyte-to-Monocyte Ratio May Serve as a Better Prognostic Indicator Than Tumor-Associated Macrophages in DLBCL Treated with Rituximab. Appl. Immunohistochem. Mol. Morphol. 27 (8), 572–580. doi:10.1097/pai.0000000000000645
McCord, R., Bolen, C. R., Koeppen, H., Kadel, E. E., Oestergaard, M. Z., Nielsen, T., et al. (2019). PD-L1 and Tumor-Associated Macrophages in De Novo DLBCL. Blood Adv. 3 (4), 531–540. doi:10.1182/bloodadvances.2018020602
Mensah, A. A., Spriano, F., Sartori, G., Priebe, V., Cascione, L., Gaudio, E., et al. (2021). Study of the Antilymphoma Activity of Pracinostat Reveals Different Sensitivities of DLBCL Cells to HDAC Inhibitors. Blood Adv. 5 (10), 2467–2480. doi:10.1182/bloodadvances.2020003566
Mentch, S. J., Mehrmohamadi, M., Huang, L., Liu, X., Gupta, D., Mattocks, D., et al. (2015). Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cel. Metab. 22 (5), 861–873. doi:10.1016/j.cmet.2015.08.024
Merdan, S., Subramanian, K., Ayer, T., Van Weyenbergh, J., Chang, A., Koff, J. L., et al. (2021). Gene Expression Profiling-Based Risk Prediction and Profiles of Immune Infiltration in Diffuse Large B-Cell Lymphoma. Blood Cancer J. 11 (1), 2. doi:10.1038/s41408-020-00404-0
Mesin, L., Ersching, J., and Victora, G. D. (2016). Germinal Center B Cell Dynamics. Immunity 45 (3), 471–482. doi:10.1016/j.immuni.2016.09.001
Meyer, S. N., Scuoppo, C., Vlasevska, S., Bal, E., Holmes, A. B., Holloman, M., et al. (2019). Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity 51 (3), 535–547. doi:10.1016/j.immuni.2019.08.006
Mintz, M. A., Felce, J. H., Chou, M. Y., Mayya, V., Xu, Y., Shui, J.-W., et al. (2019). The HVEM-BTLA Axis Restrains T Cell Help to Germinal Center B Cells and Functions as a Cell-Extrinsic Suppressor in Lymphomagenesis. Immunity 51 (2), 310–323. doi:10.1016/j.immuni.2019.05.022
Mittal, S., Marshall, N. A., Duncan, L., Culligan, D. J., Barker, R. N., and Vickers, M. A. (2008). Local and Systemic Induction of CD4+CD25+ Regulatory T-Cell Population by Non-hodgkin Lymphoma. Blood 111 (11), 5359–5370. doi:10.1182/blood-2007-08-105395
Mlynarczyk, C., Fontán, L., and Melnick, A. (2019). Germinal center‐derived Lymphomas: The Darkest Side of Humoral Immunity. Immunol. Rev. 288 (1), 214–239. doi:10.1111/imr.12755
Mondello, P., Derenzini, E., Asgari, Z., Philip, J., Brea, E. J., Seshan, V., et al. (2017). Dual Inhibition of Histone Deacetylases and Phosphoinositide 3-kinase Enhances Therapeutic Activity against B Cell Lymphoma. Oncotarget 8 (8), 14017–14028. doi:10.18632/oncotarget.14876
Mondello, P., Tadros, S., Teater, M., Fontan, L., Chang, A. Y., Jain, N., et al. (2020). Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 10 (3), 440–459. doi:10.1158/2159-8290.cd-19-0116
Monrad, I., Madsen, C., Lauridsen, K. L., Honoré, B., Plesner, T. L., Hamilton-Dutoit, S., et al. (2020). Glycolytic Biomarkers Predict Transformation in Patients with Follicular Lymphoma. PLoS One 15 (5), e0233449. doi:10.1371/journal.pone.0233449
Monti, S., Savage, K. J., Kutok, J. L., Feuerhake, F., Kurtin, P., Mihm, M., et al. (2005). Molecular Profiling of Diffuse Large B-Cell Lymphoma Identifies Robust Subtypes Including One Characterized by Host Inflammatory Response. Blood 105 (5), 1851–1861. doi:10.1182/blood-2004-07-2947
Morin, R. D., Arthur, S. E., and Assouline, S. (2021). Treating Lymphoma Is Now a Bit EZ-Er. Blood Adv. 5 (8), 2256–2263. doi:10.1182/bloodadvances.2020002773
Morin, R. D., Johnson, N. A., Severson, T. M., Mungall, A. J., An, J., Goya, R., et al. (2010). Somatic Mutations Altering EZH2 (Tyr641) in Follicular and Diffuse Large B-Cell Lymphomas of Germinal-center Origin. Nat. Genet. 42 (2), 181–185. doi:10.1038/ng.518
Morschhauser, F., Ghosh, N., Lossos, I., Palomba, M. L., Mehta, A., Casasnovas, O., et al. (2019). Efficacy and Safety of Obinutuzumab + Lenalidomide + Atezolizumab in Patients with Relapsed or Refractory Follicular Lymphoma: Primary Analysis of a Phase 1b/2 Trial. Hematol. Oncol. 37 (S2), 113–114. doi:10.1002/hon.74_2629
Morschhauser, F., Tilly, H., Chaidos, A., McKay, P., Phillips, T., Assouline, S., et al. (2020). Tazemetostat for Patients with Relapsed or Refractory Follicular Lymphoma: an Open-Label, Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol. 21 (11), 1433–1442. doi:10.1016/s1470-2045(20)30441-1
Muenst, S., Hoeller, S., Willi, N., Dirnhofer, S., and Tzankov, A. (2010). Diagnostic and Prognostic Utility of PD-1 in B Cell Lymphomas. Dis. Markers 29 (1), 47–53. doi:10.1155/2010/404069
Myklebust, J. H., Irish, J. M., Brody, J., Czerwinski, D. K., Houot, R., Kohrt, H. E., et al. (2013). High PD-1 Expression and Suppressed Cytokine Signaling Distinguish T Cells Infiltrating Follicular Lymphoma Tumors from Peripheral T Cells. Blood 121 (8), 1367–1376. doi:10.1182/blood-2012-04-421826
Nam, S. J., Go, H., Paik, J. H., Kim, T. M., Heo, D. S., Kim, C. W., et al. (2014). An Increase of M2 Macrophages Predicts Poor Prognosis in Patients with Diffuse Large B-Cell Lymphoma Treated with Rituximab, Cyclophosphamide, Doxorubicin, Vincristine and Prednisone. Leuk. Lymphoma 55 (11), 2466–2476. doi:10.3109/10428194.2013.879713
Nastoupil, L. J., Westin, J. R., Fowler, N. H., Fanale, M. A., Samaniego, F., Oki, Y., et al. (2017). Response Rates with Pembrolizumab in Combination with Rituximab in Patients with Relapsed Follicular Lymphoma: Interim Results of an on Open-Label, Phase II Study. Jco 35 (15_Suppl. l), 7519. doi:10.1200/jco.2017.35.15_suppl.7519
Neelapu, S. S., Dickinson, M., Ulrickson, M. L., Oluwole, O. O., Herrera, A. F., Thieblemont, C., et al. (2020). Interim Analysis of ZUMA-12: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) as First-Line Therapy in Patients (Pts) with High-Risk Large B Cell Lymphoma (LBCL). Blood 136 (Suppl. 1), 49. doi:10.1182/blood-2020-134449
Nijland, M., Veenstra, R. N., Visser, L., Xu, C., Kushekhar, K., van Imhoff, G. W., et al. (2017). HLA Dependent Immune Escape Mechanisms in B-Cell Lymphomas: Implications for Immune Checkpoint Inhibitor Therapy? Oncoimmunology 6 (4), e1295202. doi:10.1080/2162402x.2017.1295202
Nitsch, S., Zorro Shahidian, L., and Schneider, R. (2021). Histone Acylations and Chromatin Dynamics: Concepts, Challenges, and Links to Metabolism. EMBO Rep. 22 (7), e52774. doi:10.15252/embr.202152774
Ochando, J., and Braza, M. S. (2017). T Follicular Helper Cells: a Potential Therapeutic Target in Follicular Lymphoma. Oncotarget 8 (67), 112116–112131. doi:10.18632/oncotarget.22788
Ogura, M., Ando, K., Suzuki, T., Ishizawa, K., Oh, S. Y., Itoh, K., et al. (2014). A Multicentre Phase II Study of Vorinostat in Patients with Relapsed or Refractory Indolent B‐cell non‐Hodgkin Lymphoma and Mantle Cell Lymphoma. Br. J. Haematol. 165 (6), 768–776. doi:10.1111/bjh.12819
Okosun, J., Bödör, C., Wang, J., Araf, S., Yang, C.-Y., Pan, C., et al. (2014). Integrated Genomic Analysis Identifies Recurrent Mutations and Evolution Patterns Driving the Initiation and Progression of Follicular Lymphoma. Nat. Genet. 46 (2), 176–181. doi:10.1038/ng.2856
Olszewski, A. J., Avigdor, A., Babu, S., Levi, I., Abadi, U., Holmes, H., et al. (2020). Single-Agent Mosunetuzumab Is a Promising Safe and Efficacious Chemotherapy-free Regimen for Elderly/Unfit Patients with Previously Untreated Diffuse Large B-Cell Lymphoma. Blood 136 (Suppl. 1), 43–45. doi:10.1182/blood-2020-136255
Ortega-Molina, A., Boss, I. W., Canela, A., Pan, H., Jiang, Y., Zhao, C., et al. (2015). The Histone Lysine Methyltransferase KMT2D Sustains a Gene Expression Program that Represses B Cell Lymphoma Development. Nat. Med. 21 (10), 1199–1208. doi:10.1038/nm.3943
Otsuka, Y., Nishikori, M., Arima, H., Izumi, K., Kitawaki, T., Hishizawa, M., et al. (2020). EZH2 Inhibitors Restore Epigenetically Silenced CD58 Expression in B-Cell Lymphomas. Mol. Immunol. 119, 35–45. doi:10.1016/j.molimm.2020.01.006
Palomba, M. L., Cartron, G., Popplewell, L., Ribrag, V., Westin, J., Chitra, S., et al. (2019). Safety and Clinical Activity of Atezolizumab in Combination with Tazemetostat in Relapsed or Refractory Diffuse Large B-Cell Lymphoma: Primary Analysis of a Phase 1b Study. Hematol. Oncol. 37 (S2), 517–519. doi:10.1002/hon.203_2631
Pasqualucci, L., Dominguez-Sola, D., Chiarenza, A., Fabbri, G., Grunn, A., Trifonov, V., et al. (2011b). Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 471 (7337), 189–195. doi:10.1038/nature09730
Pasqualucci, L., Khiabanian, H., Fangazio, M., Vasishtha, M., Messina, M., Holmes, A. B., et al. (2014). Genetics of Follicular Lymphoma Transformation. Cel. Rep. 6 (1), 130–140. doi:10.1016/j.celrep.2013.12.027
Pasqualucci, L., Trifonov, V., Fabbri, G., Ma, J., Rossi, D., Chiarenza, A., et al. (2011a). Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma. Nat. Genet. 43 (9), 830–837. doi:10.1038/ng.892
Pauken, K. E., Torchia, J. A., Chaudhri, A., Sharpe, A. H., and Freeman, G. J. (2021). Emerging Concepts in PD-1 Checkpoint Biology. Semin. Immunol. 52, 101480. doi:10.1016/j.smim.2021.101480
Peng, D., Kryczek, I., Nagarsheth, N., Zhao, L., Wei, S., Wang, W., et al. (2015). Epigenetic Silencing of TH1-type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature 527 (7577), 249–253. doi:10.1038/nature15520
Persky, D. O., Li, H., Rimsza, L. M., Barr, P. M., Popplewell, L. L., Bane, C. L., et al. (2018). A Phase I/II Trial of Vorinostat (SAHA) in Combination with Rituximab-CHOP in Patients with Newly Diagnosed Advanced Stage Diffuse Large B-Cell Lymphoma (DLBCL): SWOG S0806. Am. J. Hematol. 93 (4), 486–493. doi:10.1002/ajh.25010
Puvvada, S. D., Li, H., Rimsza, L. M., Bernstein, S. H., Fisher, R. I., LeBlanc, M., et al. (2016). A Phase II Study of Belinostat (PXD101) in Relapsed and Refractory Aggressive B-Cell Lymphomas: SWOG S0520. Leuk. Lymphoma 57 (10), 2359–2369. doi:10.3109/10428194.2015.1135431
Rajnai, H., Bödör, C., Balogh, Z., Gagyi, É., Csomor, J., Krenács, T., et al. (2012). Impact of the Reactive Microenvironment on the Bone Marrow Involvement of Follicular Lymphoma. Histopathology 60 (6B), E66–E75. doi:10.1111/j.1365-2559.2012.04187.x
Rawal, S., Chu, F., Zhang, M., Park, H. J., Nattamai, D., Kannan, S., et al. (2013). Cross Talk between Follicular Th Cells and Tumor Cells in Human Follicular Lymphoma Promotes Immune Evasion in the Tumor Microenvironment. J.I. 190 (12), 6681–6693. doi:10.4049/jimmunol.1201363
Reddy, A., Zhang, J., Davis, N. S., Moffitt, A. B., Love, C. L., Waldrop, A., et al. (2017). Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171 (2), 481–e15. doi:10.1016/j.cell.2017.09.027
Reid, M. A., Dai, Z., and Locasale, J. W. (2017). The Impact of Cellular Metabolism on Chromatin Dynamics and Epigenetics. Nat. Cel. Biol. 19 (11), 1298–1306. doi:10.1038/ncb3629
Reimann, M., Schrezenmeier, J., Richter-Pechanska, P., Dolnik, A., Hick, T. P., Schleich, K., et al. (2021). Adaptive T-Cell Immunity Controls Senescence-Prone MyD88- or CARD11-Mutant B-Cell Lymphomas. Blood 137 (20), 2785–2799. doi:10.1182/blood.2020005244
Ribas, A., and Wolchok, J. D. (2018). Cancer Immunotherapy Using Checkpoint Blockade. Science 359 (6382), 1350–1355. doi:10.1126/science.aar4060
Ribrag, V., Kim, W. S., Bouabdallah, R., Lim, S. T., Coiffier, B., Illes, A., et al. (2017). Safety and Efficacy of Abexinostat, a Pan-Histone Deacetylase Inhibitor, in Non-hodgkin Lymphoma and Chronic Lymphocytic Leukemia: Results of a Phase II Study. Haematologica 102 (5), 903–909. doi:10.3324/haematol.2016.154377
Ribrag, V., Morschhauser, F., McKay, P., Salles, G. A., Batlevi, C. L., Schmitt, A., et al. (2018). Interim Results from an Ongoing Phase 2 Multicenter Study of Tazemetostat, an EZH2 Inhibitor, in Patients with Relapsed or Refractory (R/R) Diffuse Large B-Cell Lymphoma (DLBCL). Blood 132 (Suppl. 1), 4196. doi:10.1182/blood-2018-99-113411
Riihijarvi, S., Fiskvik, I., Taskinen, M., Vajavaara, H., Tikkala, M., Yri, O., et al. (2015). Prognostic Influence of Macrophages in Patients with Diffuse Large B-Cell Lymphoma: a Correlative Study from a Nordic Phase II Trial. Haematologica 100 (2), 238–245. doi:10.3324/haematol.2014.113472
Rimsza, L. M., Roberts, R. A., Campo, E., Grogan, T. M., Bea, S., Salaverria, I., et al. (2006). Loss of Major Histocompatibility Class II Expression in Non-immune-privileged Site Diffuse Large B-Cell Lymphoma Is Highly Coordinated and Not Due to Chromosomal Deletions. Blood 107 (3), 1101–1107. doi:10.1182/blood-2005-04-1510
Rimsza, L. M., Roberts, R. A., Miller, T. P., Unger, J. M., LeBlanc, M., Braziel, R. M., et al. (2004). Loss of MHC Class II Gene and Protein Expression in Diffuse Large B-Cell Lymphoma Is Related to Decreased Tumor Immunosurveillance and Poor Patient Survival Regardless of Other Prognostic Factors: a Follow-Up Study from the Leukemia and Lymphoma Molecular Profiling Project. Blood 103 (11), 4251–4258. doi:10.1182/blood-2003-07-2365
Roberts, R. A., Wright, G., Rosenwald, A. R., Jaramillo, M. A., Grogan, T. M., Miller, T. P., et al. (2006). Loss of Major Histocompatibility Class II Gene and Protein Expression in Primary Mediastinal Large B-Cell Lymphoma Is Highly Coordinated and Related to Poor Patient Survival. Blood 108 (1), 311–318. doi:10.1182/blood-2005-11-4742
Romero-Garcia, S., Moreno-Altamirano, M. M. B., Prado-Garcia, H., and Sánchez-García, F. J. (2016). Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 7, 52. doi:10.3389/fimmu.2016.00052
Rosenwald, A., Wright, G., Chan, W. C., Connors, J. M., Campo, E., Fisher, R. I., et al. (2002). The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 346 (25), 1937–1947. doi:10.1056/nejmoa012914
Roussel, M., Le, K.-S., Granier, C., Llamas Gutierrez, F., Foucher, E., Le Gallou, S., et al. (2021). Functional Characterization of PD1+TIM3+ Tumor-Infiltrating T Cells in DLBCL and Effects of PD1 or TIM3 Blockade. Blood Adv. 5 (7), 1816–1829. doi:10.1182/bloodadvances.2020003080
Salles, G., Ghosh, N., Lossos, I. S., Palomba, M. L., Mehta, A., Casasnovas, O., et al. (2018). Atezolizumab in Combination with Obinutuzumab and Lenalidomide Demonstrates Favorable Activity and Manageable Toxicity in Patients with Relapsed/Refractory Follicular Lymphoma (FL): An Interim Analysis of a Phase Ib/II Trial. Blood 132 (Suppl. 1), 1603. doi:10.1182/blood-2018-99-114992
Sarkozy, C., Morschhauser, F., Dubois, S., Molina, T., Michot, J. M., Cullières-Dartigues, P., et al. (2020). A LYSA Phase Ib Study of Tazemetostat (EPZ-6438) Plus R-CHOP in Patients with Newly Diagnosed Diffuse Large B-Cell Lymphoma (DLBCL) with Poor Prognosis Features. Clin. Cancer Res. 26 (13), 3145–3153. doi:10.1158/1078-0432.ccr-19-3741
Scholze, H., Stephenson, R. E., Reynolds, R., Shah, S., Puri, R., Butler, S. D., et al. (2020). Combined EZH2 and Bcl-2 Inhibitors as Precision Therapy for Genetically Defined DLBCL Subtypes. Blood Adv. 4 (20), 5226–5231. doi:10.1182/bloodadvances.2020002580
Schuster, S. J., Bishop, M. R., Tam, C. S., Waller, E. K., Borchmann, P., McGuirk, J. P., et al. (2018). Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 380 (1), 45–56. doi:10.1056/NEJMoa1804980
Schuster, S. J., Bishop, M. R., Tam, C. S., Waller, E. K., Borchmann, P., McGuirk, J. P., et al. (2019). Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 380 (1), 45–56. doi:10.1056/nejmoa1804980
Schwarzfischer, P., Reinders, J., Dettmer, K., Kleo, K., Dimitrova, L., Hummel, M., et al. (2017). Comprehensive Metaboproteomics of Burkitt's and Diffuse Large B-Cell Lymphoma Cell Lines and Primary Tumor Tissues Reveals Distinct Differences in Pyruvate Content and Metabolism. J. Proteome Res. 16 (3), 1105–1120. doi:10.1021/acs.jproteome.6b00164
Serafini, P., Mgebroff, S., Noonan, K., and Borrello, I. (2008). Myeloid-derived Suppressor Cells Promote Cross-Tolerance in B-Cell Lymphoma by Expanding Regulatory T Cells. Cancer Res. 68 (13), 5439–5449. doi:10.1158/0008-5472.can-07-6621
Sha, C., Barrans, S., Cucco, F., Bentley, M. A., Care, M. A., Cummin, T., et al. (2019). Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group that Requires Different Approaches to Therapy. Jco 37 (3), 202–212. doi:10.1200/jco.18.01314
Shen, L., Li, H., Shi, Y., Wang, D., Gong, J., Xun, J., et al. (2016). M2 Tumour-Associated Macrophages Contribute to Tumour Progression via Legumain Remodelling the Extracellular Matrix in Diffuse Large B Cell Lymphoma. Sci. Rep. 6, 30347. doi:10.1038/srep30347
Shi, Y.-J., Matson, C., Lan, F., Iwase, S., Baba, T., and Shi, Y. (2005). Regulation of LSD1 Histone Demethylase Activity by its Associated Factors. Mol. Cel. 19 (6), 857–864. doi:10.1016/j.molcel.2005.08.027
Simithy, J., Sidoli, S., Yuan, Z.-F., Coradin, M., Bhanu, N. V., Marchione, D. M., et al. (2017). Characterization of Histone Acylations Links Chromatin Modifications with Metabolism. Nat. Commun. 8 (1), 1141. doi:10.1038/s41467-017-01384-9
Singh, A. R., Gu, J. J., Zhang, Q., Torka, P., Sundaram, S., Mavis, C., et al. (2020). Metformin Sensitizes Therapeutic Agents and Improves Outcome in Pre-clinical and Clinical Diffuse Large B-Cell Lymphoma. Cancer Metab. 8, 10. doi:10.1186/s40170-020-00213-w
Smith, M., Jegede, O., Parekh, S., Hanson, C. A., Martin, P., Till, B. G., et al. (2019). Minimal Residual Disease (MRD) Assessment in the ECOG1411 Randomized Phase 2 Trial of Front-Line Bendamustine-Rituximab (BR)-Based Induction Followed by Rituximab (R) ± Lenalidomide (L) Consolidation for Mantle Cell Lymphoma (MCL). Blood 134 (Supplement_1), 751. doi:10.1182/blood-2019-129323
Song, M. K., Park, B. B., and Uhm, J. (2019). Understanding Immune Evasion and Therapeutic Targeting Associated with PD-1/pd-L1 Pathway in Diffuse Large B-Cell Lymphoma. Int. J. Mol. Sci. 20 (6). doi:10.3390/ijms20061326
Steidl, C., Shah, S. P., Woolcock, B. W., Rui, L., Kawahara, M., Farinha, P., et al. (2011). MHC Class II Transactivator CIITA Is a Recurrent Gene Fusion Partner in Lymphoid Cancers. Nature 471 (7338), 377–381. doi:10.1038/nature09754
Stevens, W. B. C., Mendeville, M., Redd, R., Clear, A. J., Bladergroen, R., Calaminici, M., et al. (2017). Prognostic Relevance of CD163 and CD8 Combined with EZH2 and Gain of Chromosome 18 in Follicular Lymphoma: a Study by the Lunenburg Lymphoma Biomarker Consortium. Haematologica 102 (8), 1413–1423. doi:10.3324/haematol.2017.165415
Stopeck, A. T., Gessner, A., Miller, T. P., Hersh, E. M., Johnson, C. S., Cui, H., et al. (2000). Loss of B7.2 (CD86) and Intracellular Adhesion Molecule 1 (CD54) Expression is Associated With Decreased Tumor-Infiltrating T Lymphocytes in Diffuse Lymphoma. Clin. Cancer Res. 6 (10), 3904–3909.
Su, X., Wellen, K. E., and Rabinowitz, J. D. (2016). Metabolic Control of Methylation and Acetylation. Curr. Opin. Chem. Biol. 30, 52–60. doi:10.1016/j.cbpa.2015.10.030
Swenson, S. A., Gilbreath, T. J., Vahle, H., Hynes-Smith, R. W., Graham, J. H., Law, H. C.-H., et al. (2020). UBR5 HECT Domain Mutations Identified in Mantle Cell Lymphoma Control Maturation of B Cells. Blood 136 (3), 299–312. doi:10.1182/blood.2019002102
Swerdlow, S. H., Campo, E., Pileri, S. A., Harris, N. L., Stein, H., Siebert, R., et al. (2016). The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 127 (20), 2375–2390. doi:10.1182/blood-2016-01-643569
Taskinen, M., Karjalainen-Lindsberg, M.-L., Nyman, H., Eerola, L.-M., and Leppä, S. (2007). A High Tumor-Associated Macrophage Content Predicts Favorable Outcome in Follicular Lymphoma Patients Treated with Rituximab and Cyclophosphamide-Doxorubicin-Vincristine-Prednisone. Clin. Cancer Res. 13 (19), 5784–5789. doi:10.1158/1078-0432.ccr-07-0778
Tcyganov, E., Mastio, J., Chen, E., and Gabrilovich, D. I. (2018). Plasticity of Myeloid-Derived Suppressor Cells in Cancer. Curr. Opin. Immunol. 51, 76–82. doi:10.1016/j.coi.2018.03.009
Teater, M., Dominguez, P. M., Redmond, D., Chen, Z., Ennishi, D., Scott, D. W., et al. (2018). AICDA Drives Epigenetic Heterogeneity and Accelerates Germinal center-derived Lymphomagenesis. Nat. Commun. 9 (1), 222. doi:10.1038/s41467-017-02595-w
Thibult, M.-L., Mamessier, E., Gertner-Dardenne, J., Pastor, S., Just-Landi, S., Xerri, L., et al. (2013). PD-1 Is a Novel Regulator of Human B-Cell Activation. Int. Immunol. 25 (2), 129–137. doi:10.1093/intimm/dxs098
Till, B. G., Park, S. I., Popplewell, L. L., Goy, A., Penuel, E., Venstrom, J. M., et al. (2015). Safety and Clinical Activity of Atezolizumab (Anti-PDL1) in Combination with Obinutuzumab in Patients with Relapsed or Refractory Non-hodgkin Lymphoma. Blood 126 (23), 5104. doi:10.1182/blood.v126.23.5104.5104
Tumes, D. J., Onodera, A., Suzuki, A., Shinoda, K., Endo, Y., Iwamura, C., et al. (2013). The Polycomb Protein Ezh2 Regulates Differentiation and Plasticity of CD4+ T Helper Type 1 and Type 2 Cells. Immunity 39 (5), 819–832. doi:10.1016/j.immuni.2013.09.012
Tzankov, A., Meier, C., Hirschmann, P., Went, P., Pileri, S. A., and Dirnhofer, S. (2008). Correlation of High Numbers of Intratumoral FOXP3+ Regulatory T Cells with Improved Survival in Germinal center-like Diffuse Large B-Cell Lymphoma, Follicular Lymphoma and Classical Hodgkin's Lymphoma. Haematologica 93 (2), 193–200. doi:10.3324/haematol.11702
Upadhyay, R., Hammerich, L., Peng, P., Brown, B., Merad, M., and Brody, J. (2015). Lymphoma: Immune Evasion Strategies. Cancers 7 (2), 736–762. doi:10.3390/cancers7020736
Wahlin, B. E., Sander, B., Christensson, B., and Kimby, E. (2007). CD8+ T-Cell Content in Diagnostic Lymph Nodes Measured by Flow Cytometry Is a Predictor of Survival in Follicular Lymphoma. Clin. Cancer Res. 13 (2 Pt 1), 388–397. doi:10.1158/1078-0432.CCR-06-1734
Wahlin, B. E., Aggarwal, M., Montes-Moreno, S., Gonzalez, L. F., Roncador, G., Sanchez-Verde, L., et al. (2010). A Unifying Microenvironment Model in Follicular Lymphoma: Outcome Is Predicted by Programmed Death-1-Positive, Regulatory, Cytotoxic, and Helper T Cells and Macrophages. Clin. Cancer Res. 16 (2), 637–650. doi:10.1158/1078-0432.ccr-09-2487
Wang, D., Quiros, J., Mahuron, K., Pai, C.-C., Ranzani, V., Young, A., et al. (2018). Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cel. Rep. 23 (11), 3262–3274. doi:10.1016/j.celrep.2018.05.050
Wang, G., Chow, R. D., Zhu, L., Bai, Z., Ye, L., Zhang, F., et al. (2020). CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade. Cancer Discov. 10 (12), 1912–1933. doi:10.1158/2159-8290.cd-19-1448
Wang, H., Shao, R., Liu, W., Tang, H., and Lu, Y. (2021). Identification of a Prognostic Metabolic Gene Signature in Diffuse Large B‐cell Lymphoma. J. Cel. Mol. Med. 25 (14), 7066–7077. doi:10.1111/jcmm.16720
Wang, Y., Tong, C., Dai, H., Wu, Z., Han, X., Guo, Y., et al. (2021). Low-dose Decitabine Priming Endows CAR T Cells with Enhanced and Persistent Antitumour Potential via Epigenetic Reprogramming. Nat. Commun. 12 (1), 409. doi:10.1038/s41467-020-20696-x
Wang, Z., Jiang, R., Li, Q., Wang, H., Tao, Q., and Zhai, Z. (2021). Elevated M-MDSCs in Circulation Are Indicative of Poor Prognosis in Diffuse Large B-Cell Lymphoma Patients. J. Clin. Med. 10 (8), 1768. doi:10.3390/jcm10081768
Waters, L. R., Ahsan, F. M., Wolf, D. M., Shirihai, O., and Teitell, M. A. (2018). Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 5, 99–109. doi:10.1016/j.isci.2018.07.005
Weisel, F. J., Mullett, S. J., Elsner, R. A., Menk, A. V., Trivedi, N., Luo, W., et al. (2020). Germinal center B Cells Selectively Oxidize Fatty Acids for Energy while Conducting Minimal Glycolysis. Nat. Immunol. 21 (3), 331–342. doi:10.1038/s41590-020-0598-4
Wright, G. W., Huang, D. W., Phelan, J. D., Coulibaly, Z. A., Roulland, S., Young, R. M., et al. (2020). A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 37 (4), 551–568. doi:10.1016/j.ccell.2020.03.015
Yamagishi, M., Hori, M., Fujikawa, D., Ohsugi, T., Honma, D., Adachi, N., et al. (2019). Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cel. Rep. 29 (8), 2321–2337. doi:10.1016/j.celrep.2019.10.083
Yan, Z.-X., Li, L., Wang, W., OuYang, B.-S., Cheng, S., Wang, L., et al. (2019). Clinical Efficacy and Tumor Microenvironment Influence in a Dose-Escalation Study of Anti-CD19 Chimeric Antigen Receptor T Cells in Refractory B-Cell Non-hodgkin's Lymphoma. Clin. Cancer Res. 25 (23), 6995–7003. doi:10.1158/1078-0432.ccr-19-0101
Yang, M., Gocke, C. B., Luo, X., Borek, D., Tomchick, D. R., Machius, M., et al. (2006). Structural Basis for CoREST-dependent Demethylation of Nucleosomes by the Human LSD1 Histone Demethylase. Mol. Cel. 23 (3), 377–387. doi:10.1016/j.molcel.2006.07.012
Yang, Z.-Z., Grote, D. M., Ziesmer, S. C., Niki, T., Hirashima, M., Novak, A. J., et al. (2012). IL-12 Upregulates TIM-3 Expression and Induces T Cell Exhaustion in Patients with Follicular B Cell Non-hodgkin Lymphoma. J. Clin. Invest. 122 (4), 1271–1282. doi:10.1172/jci59806
Yang, Z.-Z., Grote, D. M., Ziesmer, S. C., Xiu, B., Novak, A. J., and Ansell, S. M. (2015). PD-1 Expression Defines Two Distinct T-Cell Sub-populations in Follicular Lymphoma that Differentially Impact Patient Survival. Blood Cancer J. 5, e281. doi:10.1038/bcj.2015.1
Yang, Z.-Z., Kim, H. J., Villasboas, J. C., Chen, Y.-P., Price-Troska, T., Jalali, S., et al. (2017). Expression of LAG-3 Defines Exhaustion of Intratumoral PD-1+ T Cells and Correlates with Poor Outcome in Follicular Lymphoma. Oncotarget 8 (37), 61425–61439. doi:10.18632/oncotarget.18251
Yang, Z.-Z., Kim, H. J., Wu, H., Jalali, S., Tang, X., Krull, J. E., et al. (2020). TIGIT Expression Is Associated with T-Cell Suppression and Exhaustion and Predicts Clinical Outcome and Anti-PD-1 Response in Follicular Lymphoma. Clin. Cancer Res. 26 (19), 5217–5231. doi:10.1158/1078-0432.ccr-20-0558
Yap, T. A., Winter, J. N., Giulino-Roth, L., Longley, J., Lopez, J., Michot, J.-M., et al. (2019). Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 25 (24), 7331–7339. doi:10.1158/1078-0432.ccr-18-4121
You, H., Xu-Monette, Z. Y., Wei, L., Nunns, H., Nagy, M. L., Bhagat, G., et al. (2021). Genomic Complexity Is Associated with Epigenetic Regulator Mutations and Poor Prognosis in Diffuse Large B-Cell Lymphoma. Oncoimmunology 10 (1), 1928365. doi:10.1080/2162402X.2021.1928365
Younes, A., Burke, J. M., Cheson, B. D., Diefenbach, C., Ferrari, S., Hahn, U. H., et al. (2019). Safety and Efficacy of Atezolizumab in Combination with Rituximab Plus CHOP in Previously Untreated Patients with Diffuse Large B-Cell Lymphoma (DLBCL): Updated Analysis of a Phase I/II Study. Blood 134 (Supplement_1), 2874. doi:10.1182/blood-2019-123368
Younes, A., John, B. M., Diefenbach, C. S., Ferrari, S., Kahn, C., Sharman, J. P., et al. (2017). Safety and Efficacy of Atezolizumab in Combination with Obinutuzumab and Bendamustine in Patients with Previously Untreated Follicular Lymphoma: An Interim Analysis. Blood 130 (Suppl. 1), 481. doi:10.1182/blood.V130.Suppl_1.481.481
Zappasodi, R., de Braud, F., and Di Nicola, M. (2015). Lymphoma Immunotherapy: Current Status. Front. Immunol. 6, 448. doi:10.3389/fimmu.2015.00448
Zappasodi, R., Merghoub, T., and Wolchok, J. D. (2018a). Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 33 (4), 581–598. doi:10.1016/j.ccell.2018.03.005
Zappasodi, R., Wolchok, J. D., and Merghoub, T. (2018b). Strategies for Predicting Response to Checkpoint Inhibitors. Curr. Hematol. Malig. Rep. 13, 383–395. doi:10.1007/s11899-018-0471-9
Zhang, D., Tang, Z., Huang, H., Zhou, G., Cui, C., Weng, Y., et al. (2019). Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 574 (7779), 575–580. doi:10.1038/s41586-019-1678-1
Zhang, J., Dominguez-Sola, D., Hussein, S., Lee, J.-E., Holmes, A. B., Bansal, M., et al. (2015). Disruption of KMT2D Perturbs Germinal center B Cell Development and Promotes Lymphomagenesis. Nat. Med. 21 (10), 1190–1198. doi:10.1038/nm.3940
Zhang, J., Vlasevska, S., Wells, V. A., Nataraj, S., Holmes, A. B., Duval, R., et al. (2017). The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-Cell Lymphoma. Cancer Discov. 7 (3), 322–337. doi:10.1158/2159-8290.cd-16-1417
Zhao, E., Maj, T., Kryczek, I., Li, W., Wu, K., et al. (2016). Cancer Mediates Effector T Cell Dysfunction by Targeting microRNAs and EZH2 via Glycolysis Restriction. Nat. Immunol. 17 (1), 95–103. doi:10.1038/ni.3313
Zhou, D.-M., Xu, Y.-X., Zhang, L.-Y., Sun, Y., Wang, Z.-Y., Yuan, Y.-Q., et al. (2017). The Role of Follicular T Helper Cells in Patients with Malignant Lymphoid Disease. Hematology 22 (7), 412–418. doi:10.1080/10245332.2017.1300623
Keywords: GCB-DLBCLs, epigenetics, metabolic intermediates, immune microenvironment, combination therapies
Citation: Serganova I, Chakraborty S, Yamshon S, Isshiki Y, Bucktrout R, Melnick A, Béguelin W and Zappasodi R (2022) Epigenetic, Metabolic, and Immune Crosstalk in Germinal-Center-Derived B-Cell Lymphomas: Unveiling New Vulnerabilities for Rational Combination Therapies. Front. Cell Dev. Biol. 9:805195. doi: 10.3389/fcell.2021.805195
Received: 30 October 2021; Accepted: 30 November 2021;
Published: 07 January 2022.
Edited by:
Rada Amin, University of Nebraska-Lincoln, United StatesReviewed by:
Jennifer Amengual, Columbia University, United StatesMomoko Nishikori, Kyoto University, Japan
Kim Good-Jacobson, Monash University, Australia
Copyright © 2022 Serganova, Chakraborty, Yamshon, Isshiki, Bucktrout, Melnick, Béguelin and Zappasodi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta Zappasodi, cm96NDAwMkBtZWQuY29ybmVsbC5lZHU=