 Yuxin Guo
Yuxin Guo Ying Ju5
Ying Ju5 Dong Chen
Dong Chen- 1Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, Quzhou, China
- 2Key Laboratory of Computational Science and Application of Hainan Province, Haikou, China
- 3Key Laboratory of Data Science and Intelligence Education, Hainan Normal University, Ministry of Education, Haikou, China
- 4School of Mathematics and Statistics, Hainan Normal University, Haikou, China
- 5School of Informatics, Xiamen University, Xiamen, China
- 6College of Electrical and Information Engineering, Quzhou University, Quzhou, China
- 7Beidahuang Industry Group General Hospital, Harbin, China
Genes, the nucleotide sequences that encode a polypeptide chain or functional RNA, are the basic genetic unit controlling biological traits. They are the guarantee of the basic structures and functions in organisms, and they store information related to biological factors and processes such as blood type, gestation, growth, and apoptosis. The environment and genetics jointly affect important physiological processes such as reproduction, cell division, and protein synthesis. Genes are related to a wide range of phenomena including growth, decline, illness, aging, and death. During the evolution of organisms, there is a class of genes that exist in a conserved form in multiple species. These genes are often located on the dominant strand of DNA and tend to have higher expression levels. The protein encoded by it usually either performs very important functions or is responsible for maintaining and repairing these essential functions. Such genes are called persistent genes. Among them, the irreplaceable part of the body’s life activities is the essential gene. For example, when starch is the only source of energy, the genes related to starch digestion are essential genes. Without them, the organism will die because it cannot obtain enough energy to maintain basic functions. The function of the proteins encoded by these genes is thought to be fundamental to life. Nowadays, DNA can be extracted from blood, saliva, or tissue cells for genetic testing, and detailed genetic information can be obtained using the most advanced scientific instruments and technologies. The information gained from genetic testing is useful to assess the potential risks of disease, and to help determine the prognosis and development of diseases. Such information is also useful for developing personalized medication and providing targeted health guidance to improve the quality of life. Therefore, it is of great theoretical and practical significance to identify important and essential genes. In this paper, the research status of essential genes and the essential genome database of bacteria are reviewed, the computational prediction method of essential genes based on communication coding theory is expounded, and the significance and practical application value of essential genes are discussed.
Introduction
With the smooth progress of the Human Genome Project, more and more new genes have been discovered, and the study of gene function has become a major issue in the field of life sciences. Genes support the basic structure and performance of life. It stores all the information about the race, blood type, gestation, growth, apoptosis and other processes of life. The environment and genetics jointly determine important physiological processes such as reproduction, cell division, and protein synthesis. Genes are involved in diverse phenomena such as birth, growth, decline, illness, aging, and death (Yang et al., 2015; Hu et al., 2020; Cheng et al., 2021a). Except for some viruses whose genes are composed of ribonucleic acid (RNA) (Sun et al., 2013; Riaz and Li, 2019), in most organisms, genes are composed of deoxyribonucleic acid (DNA) arranged linearly on chromosomes. The existence of a simple life form requires at least 265 to 350 genes, whose functions are affected by internal factors and external environmental factors.
The impact of human genome research on medicine has gradually entered the clinic and changed the traditional medical model. Human genomic research has had a major impact on medicine. Genomic medicine will gradually be applied in clinical practice, and will change the traditional medical model (Liu et al., 2016a; Zhou et al., 2019; Zou, 2019; Chen et al., 2020a; Liu et al., 2020a; Li et al., 2021; Qi et al., 2021). Many believe that it will trigger the next medical revolution (Zeng et al., 2022). Genomic medicine aims to prevent diseases and apply specific gene therapies to address existing genetic defects. In-depth clinical genomic research heralds the arrival of a new era of medicine with a focus on drug optimization, and the prediction and prevention of diseases based on the genome of each patient. Genomic research has identified preferred targets for the development of new drugs (Grazziotin et al., 2015; Yan et al., 2019; Yu et al., 2020a; Zeng et al., 2020a; Liu et al., 2020b; Zhang et al., 2020; Zhuang et al., 2020; Yan et al., 2021a; Yan et al., 2021b; Deng et al., 2021; Shang et al., 2021).
So far, there are dozens of experimental methods to identify essential genes. The methods to realize essential gene identification are mainly divided into two kinds, one is to determine by means of wet experiment; The second is to use computational biology to predict the necessary genes identified by experiments. Wet experiments include: In 1995, Itaya used induced mutations to detect essential genes of Bacillus subtilis, Venter used global transposon mutations to identify essential genes of Mycoplasma genitalium, targeted gene knockout, transposon mutations, genetic imprinting, and scattered Shotgun method, RNA interference and CRISPR technology (Liu et al., 2016b; Fang et al., 2019a; Ru et al., 2019; Chen et al., 2021). And some researchers have made progress in different fields through the study of essential genes using different wet experiments. Such as: Uddin et al. conducted a comparative genomics analysis and docking studies on Acinetobacter baumannii, and predicted and analyzed its non-host essential genes to screen for new drug candidates (Cheng et al., 2018; Uddin et al., 2019; Wang et al., 2020). Wang et al. used the CRISPR (clustered regularly interspaced short palindromic repeats) system to construct a genome-wide single-guide RNA library and screened for genes essential for the survival and proliferation of human cancer cell lines, thus providing a cancer cell identification strategy (Lander et al., 2015). In the field of energy and chemical industries, Voshol et al. created a transposons library of Synechococcus elongatus PCC 7942, and then screened it to identify new target genes. The aim was to improve the production capacity of fatty acids and hydrocarbons, and the gene encoding the GTP-binding protein Era was identified as an essential gene in this process (Voshol et al., 2015; Hu et al., 2021a). Another research group suppressed genes in the chloroplasts of Chlamydomonas to study its essential signaling pathways, regulatory circuits, and gene functions (Rochaix and Ramundo, 2015; Fang et al., 2019b; Cheng et al., 2020; Wang et al., 2021a). Such analyses can shed light on the growth and photosynthetic processes of photosynthetic organisms, so that strategies can be developed to enhance the photosynthetic ability of engineered bacteria. Therefore, studies on essential genes have theoretical significance and also have practical application value. Therefore, it is of great theoretical significance and practical value to study essential genes.
The disadvantages of wet experiments are that they are expensive, time-consuming, inconsistent in their accuracy, and they can give different experimental results. In addition, wet experimental methods are not applicable to some bacteria. At the same time, as research progresses, it is necessary to obtain essential genes on a genome-wide scale in order to obtain as complete a set of data as possible. This presents a serious challenge to the determination of essential genes by wet assay (Yang et al., 2014; Tavasolian et al., 2020). Therefore, some scientists began to establish predictive models with higher accuracy based on the essential gene information and the biological characteristics of essential genes, combined with computer science and various mathematical algorithms, so as to realize the rapid identification of essential genes. Therefore, greatly reducing unnecessary time and capital consumption.
Essential Gene Database
Bacteria are abundant both in terms of the number of species and the size of populations. Only a small proportion of bacteria have been fully sequenced. Currently, the following databases provide essential genetic data:
1) NCBI: Holds genomic data for more than 35,000 bacteria. Essential/non-essential genes have been fully determined for only a few bacteria.
2) The Database of Essential Genes (DEG): This database has been developed and maintained by Tianjin University. The latest version (DEG 15.2) contains essential genetic information for 41 kinds of bacteria (48 groups) (Luo et al., 2014), 26 essential gene datasets for eukaryotes and one essential gene dataset for archaea.
3) CEG (Cluster of Essential Genes): Holds information about essential homologous gene clusters for 29 kinds of bacteria (Ye et al., 2013).
4) OGEE (Online Gene Essentiality Database): Holds information about essential genes for 39 kinds of bacteria (Chen et al., 2011).
5) PEC database: Holds information for essential genes in Escherichia coli, including relevant structural information and gene function information (Rivas et al., 2015).
Methods for Identifying Essential Genes
The study of essential genes can also obtain potential drug targets, which can be used to develop antibacterial drugs to resist the invasion of pathogenic microorganisms. Therefore, it is of great practical value to study the theoretical and computational prediction methods of essential genes. Theoretical prediction is a common method of comparative genomics, which is to understand the characteristics of genes to be tested by comparing with the structure of known genes. The disadvantage of this method is that it can not accurately determine all the necessary genes of a certain microorganism, and it is more difficult to analyze eukaryotes. Computational prediction is a potential computational prediction, which uses a variety of biological characteristics of the research object, combined with statistical and classification prediction algorithms, to predict essential genes. With the rapid development of technology, omics data such as protein structure and interaction are widely used in essential gene prediction analysis. Therefore, computational biology methods can be used to provide suitable candidate target genes. The main steps are as follows: First, algorithms and tools are developed to predict essential genes of pathogenic bacteria through computational modeling. This step yields a set of candidate essential genes. Second, an essential gene prediction model is constructed. The model includes information on conserved regions of genes, and this allows for the identification of essential genes showing homology among multiple species. Next, the sequences of the obtained essential genes are compared with the genome sequences of humans or other mammals, and those that are too similar are filtered out. The ones that remain are thus identified as candidate targets for the development of new therapeutic drugs. These genes can be targeted to design and screen effective therapeutic drugs. Compared with traditional drug design strategies, the computational model is more directed, so it can shorten the development time for new drugs (Yu et al., 2020b; Zeng et al., 2020b; Huo et al., 2020; Li et al., 2020; Cheng et al., 2021b; Wang et al., 2021b; Dong et al., 2021). For example, Paul et al. used a biological metabolic network of leishmaniasis and deletion mutations designed using bioinformatics methods to identify a collection of essential proteins for this disease (Ao et al., 2021; Hu et al., 2021b), and this method was more than five times more efficient than the method of randomly selecting potential drug targets (Stanly Paul et al., 2014; Chiu et al., 2020; Wang et al., 2020). As full genome sequences are obtained for more organisms, and with the continuing development of functional genomics research, more attention is being paid to the relationships among genes (Wang et al., 2019), proteins, and phenotypes (Kitano, 2002). Proteins are indispensable components of cellular structures, and participate in a wide range of processes that affect growth, physiology, and development (Eisenberg et al., 2000). In addition, proteins function in particular subcellular compartments (Huh et al., 2003).
Gene Computation and Prediction Methods Based on Communication Coding Theory
In 1995, Itaya studied the minimum number of chromosomal loci required for the survival of B. subtilis using an induced mutation method. Mutations of only six out of 79 randomly selected chromosomal loci prevented the formation of bacterial colonies. Thus, it was inferred that genes at these six loci were essential for the survival of B. subtilis (Itaya, 1995). Later, other researchers determined the essential genes of Mycoplasma genitalium and explored the composition of the minimum gene set with experimental methods (HutchisonPeterson et al., 1999). Ongoing research on the minimum gene set has continuously discovered new essential genes, thus opening the door to essential gene characterization and further in-depth research.
Hwang et al. studied the topological properties of essential and non-essential genes in protein interaction networks with Saccharomyces cerevisiae and E. coli as the research objects. Then, they predicted essential genes using machine learning methods and on the basis of protein interaction networks and sequence information (Hwang et al., 2009; Song et al., 2021). Deng et al. used machine learning methods, combined with three types of 8 characteristic parameters such as subcellular localization, phylogenetic information and expression levels, and co-expression networks to predict the essential genes of 4 species including E. coli. However, the effectiveness of this method relies on similar feature distributions in the research objects (Deng et al., 2011). Yang et al. studied human essential genes by constructing a classifier based on 28 protein interaction network topological features and 22 biological features (Yang et al., 2014). Yu et al. identified six fractal features of DNA and protein sequences of DEG bacteria, and classified sequences using naïve Bayes and random forest methods. Arun et al. studied the relationships among gene conservation, repetition, constitutive expression, and gene essentiality in E. coli (Arun et al., 2016). Although those studies made considerable progress, there were still some problems: few species were studied; the universality of prediction models and analysis results and prediction accuracy needed to be improved; and characteristic parameters that effectively describe essential genes needed to be identified.
Considering that information transmission and coding are similar between biological systems and modern communication systems, communication coding theory can be a useful tool for essential gene analysis and prediction of gene functions. The definition of coding problem in DNA computation was first proposed by Garzon and Deaton et al. in literature network, and then the complete definition of coding problem in DNA computation was given in 2004 after refining and summarizing (Garzon and Deaton, 2004).
The steps involved in essential gene prediction based on the theory of communication coding are as follows: First, a genetic sequence analysis model is constructed based on communication coding theory. The model includes sequence analysis based on simple coding models (such as block codes, convolutional codes) and cascaded and mixed coding models. In error correction coding, convolutional codes (based on channel coding methods but with better performance), have been proven to be as effective as block codes in theory and in practice. The next step is to extract the characteristic parameters that describe essential genes. The combination of the theoretical model of communication coding for analyzing genetic sequences and genetic database information can reveal correlations between information units in DNA sequences at different scales in the coding sense, and extract the characteristic parameters of essential genes. Then, based on the features of the coding meaning and the omics data, the necessary gene calculation and prediction are carried out and combined with the determined gene data in the database, the performance optimization parameters of the established model are evaluated. Finally, the noise or redundancy is removed from the acquired features, and the key features are screened out, which are called “essential features,” and the feature set formed by these “essential features” is called “minimum feature group.” The “minimum feature group” should be constructed within the range of existing features. This will simplify and optimize the model and the analytical process, and improve its analytical efficiency and universality while ensuring prediction accuracy.
Analysis and Design of Gene Computational Prediction Models Based on Communication Coding Theory
According to the research of gene prediction method based on communication coding theory in 3.1 summary, we use the following research steps to establish a prediction model:
1. Check published studies to understand the current research situation.
2. Collect and preprocess datasets from open databases such as DEG, NCBI, KEGG, and Uniprot, and clean and sort the collected data.
3. Model research and feature extraction. This step includes encoding - based sequence analysis model and feature extraction. 1) Sequence analysis model based on coding; If the genetic sequence is analyzed as a sequence with certain information and coding characteristics, the biological sequence (DNA, RNA, or amino acid sequence) needs to be expressed in a form that is suitable for analysis, regardless of which analytical model is used (Dao et al., 2021). In this context, researchers have proposed digital mapping methods, graphic expression methods, and geometric expression methods. Taking a DNA sequence as an example, the digital mapping method expresses the four bases A, G, C, and T as different values, in a form that is convenient for computer processing, including integer expression and plural expression (Rosen, 2006; Liu et al., 2021a). The graphic and geometric expression methods express each base as a vector in space, and then connect the corresponding vectors to map a curve in the space. These methods allow for visualization of the sequence structure, and the local and overall characteristics of the sequence. Visualization of sequences is also useful for sequence comparisons and sequence similarity analyses. Once the biological sequence has been expressed and visualized, models based on convolutional code and block code models can be designed, adjusted, and modified according to analytical results and biological significance. Models can be analyzed using cascaded codes and mixed codes on the basis of simple model analysis. 2) Extract features of essential genes: According to different observation scales, the basic information units are set as bases, codons, genes, or the whole genome. Then, the association between essential genes and information units is investigated. This results in the extraction of characteristic parameters that reflect the essentiality of genes in combination with the parameters of the coding model (code length, generator matrix, constraint length, and code distance).
4. Computationally predict essential genes: After completing the tasks in Step 3, essential genes are predicted using machine learning algorithms. The best model is selected through performance evaluation. Redundant features are removed and essential features are screened to identify the “minimum feature group.”
5. Conduct performance testing and feedback optimization: Test the analytical performance of the prediction model based on cross validation and other methods (Jiang et al., 2013; Xu et al., 2021), using the essential/non-essential genes listed in DEG and other databases as training samples and verification samples.
6. Develop software to implement analysis and prediction algorithms and visualize the results. Package the final model into an executable program module with a functional interactive webpage. Establish an online service platform to provide online data analysis services.
The process of the research method is illustrated as a roadmap in Figure 1.
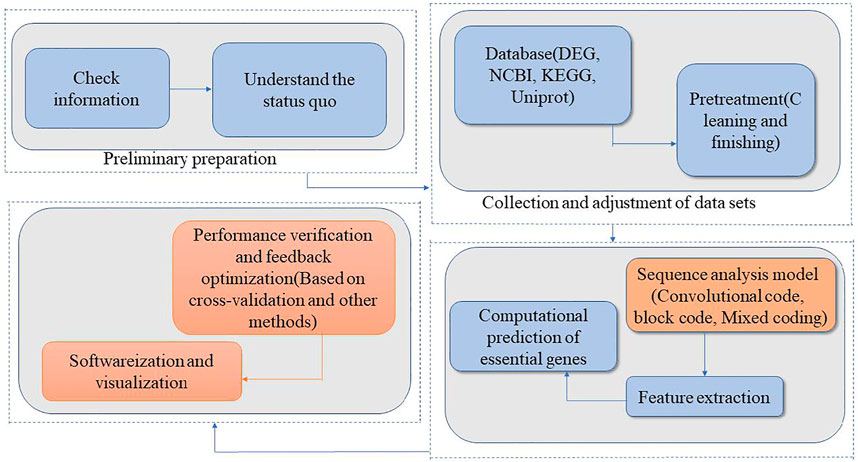
FIGURE 1. Technical roadmap of gene computational prediction model based on communication coding theory.
Key Protein Identification Method
A gene, the basic unit of heredity, is the DNA fragment required to produce a functional RNA or a polypeptide chain that forms into a functional protein. Therefore, genes and proteins are inextricably linked. For example, to explore the characteristics of essential genes, it is necessary to integrate and summarize the genomic metabolome, proteome and other omics data, and extract their combined data features, including metabolic pathways, evolutionary conservation, protein domains, and protein interactions (Wang et al., 2018; Zhang et al., 2018; Chen et al., 2019; Chen et al., 2020b; Yu et al., 2020c). Thus, it is important to identify proteins and collect all relevant information about their sequence, structure, localization, and function.
Protein is the material basis of life, is an organic macromolecule, is the basic organic matter that constitutes cells, and is the main undertaker of life activities (Wei et al., 2014; Wei et al., 2017; Guo et al., 2020; Tao et al., 2020; Wei et al., 2020; Guo et al., 2021). The human body contains many types of proteins with different properties and functions. All proteins have something in common: they are composed of 20 amino acids in different proportions, and they are constantly metabolized and renewed in the body. Many methods for identifying protein complexes based on protein interaction networks have been proposed. Some proteins are essential for the survival and reproduction of organisms, and for essential biological processes. Without them, organisms will have serious defects in their growth and development, or may even die. Thus, these are known as essential proteins. At the same time, research show many proteins perform specific biological functions only after they participate in the formation of protein complexes and interact with other proteins in the complex, suggesting that protein interactions are related to protein complexes. Therefore, this part takes the key proteins as the research object and uses the methods based on the subcellular protein interaction network and subcellular importance to identify the key proteins (Cheng et al., 2021a; Zulfiqar et al., 2021).
The centrality algorithm, a method used to identify key proteins in a protein-protein interaction network, is often used to quantify the contribution of a specific node in the graph and its impact on the network and it’s based on the law of central-lethality (Liu et al., 2020c; Zhai et al., 2020). Nodes with higher centrality values are usually regarded as key nodes in the network. Commonly used graph-based centrality algorithms include degree centrality, betweenness centrality, tight centrality, subgraph centrality, node cluster centrality, and local average connection centrality. However, this method cannot fully assess the criticality of proteins because it does not take temporal and spatial characteristics of protein interactions into account. Therefore, the subcellular location information can be used to construct a subcellular protein interaction network, and a key protein identification method based on the subcellular protein interaction network (LSED) is proposed. LSED is first based on a given global protein interaction network and protein The subcellular location information of each subcellular compartment is constructed to construct a subcellular compartment protein interaction network (PSLIN). Then, its credibility is calculated based on the size of the protein interaction network in each subcellular interval. Next, the centrality scores of the proteins in the subnets of the protein interaction network for each subcellular interval are calculated with a centrality method. Finally, the Localization-specific Centrality Score is calculated for each protein based on its centrality score in the protein interaction network for different subcellular intervals and the credibility of the network. By comparing the prediction accuracy of LSED method on PSLIN with that of centrality method on global protein interaction network, a multi-species average accuracy index (AKAcc) can be proposed, which can more comprehensively evaluate the prediction accuracy of various methods on multiple species. The key protein recognition methods mentioned in this paper are shown in Table 1.

TABLE 1. Key protein identification methods.
Because different subcellular intervals differ in importance, protein interactions in different subcellular intervals also differ in their importance. Therefore, the importance of protein interactions can be estimated based on the importance of subcellular intervals. A Centrality-based Independent Cascade (CIC) based on the importance of subcellular intervals has been proposed to detect essential proteins. For two interacting proteins u and v, the importance of the interaction (u,v) is defined as the maximum importance of the subcellular interval where the two proteins cooccur. A weighted protein interaction network is constructed based on the importance value of the interaction, and the CIC centrality method is used to estimate the criticality of proteins in this network. Thus, the CIC centrality score of a protein depends on the importance of its interactions in different subcellular intervals. Finally, the predictive performance of the CIC method is compared with those of other centrality algorithms. In addition, researchers conducted experiments on the protein interaction network of yeast, human, mouse and fruit fly to compare the prediction performance of CIC method with other centrality methods, including topology-based centrality methods and methods integrating other biological knowledge.
There are close relationships between protein complexes and essential proteins. Studies have shown that the criticality of a protein is not only determined by a single protein node, but often by the function of protein complexes. According to experimental data, essential proteins tend to aggregate in large amounts in certain complexes. Machine learning methods can be used to identify such protein complexes. At present, there is no strict mathematical expression or unified definition for the subgraph model corresponding to protein complexes and functional modules in interaction networks. However, some graph-clustering methods are effective for identifying protein complexes and functional modules, such as the graph-clustering algorithm RNSC, and density-based algorithms such as MCODE. Some studies have found that most proteins in the same protein complex have similar or identical functions, so some hierarchical clustering methods based on similarity or distance are also used to identify protein complexes. Hierarchical clustering algorithm can be divided into two categories, condensation algorithm and splitting algorithm. Among them, the G-N algorithm, a classic splitting algorithm, removes the edge with the highest betweenness and recalculates the betweenness of all edges. When at least two of the generated subgraphs meet the definition of the module after removing the edges, a corresponding phylogenetic tree can be drawn. And the HC-PIN method uses weighted edge clustering coefficient to do fast agglomerative clustering. The clustering idea is to merge the two clusters if there is an edge with the highest edge clustering coefficient between two clusters. At the same time, Table 2 shows various methods for identifying protein complexes.

TABLE 2. Identification methods of protein complexes.
Existing Problems and Research Prospects
In this paper, we have discussed some of the methods used to analyze and process biological sequence information, and introduced the use of coding theory in communication engineering to calculate and predict essential genes. However, some problems are yet to be solved. First, limited data are available for the preliminary analysis of essential genes. The DEG database contains information for a limited number of species, and this makes it difficult to test and validate analytical models. In the identification and function prediction of proteins, it is seldom possible to determine protein function based on a single experiment, because protein function is also affected by environmental factors. In addition, due to various biological, budgetary, or ethical factors, experiments cannot be performed on some organisms. Experiments conducted in vitro may not truly reflect the activity of proteins in the body. Moreover, the datasets in biological databases have problems such as noise, deviation, and missing data (Su et al., 2019; Liu et al., 2020d; Liu et al., 2021b; Su et al., 2021).
There are many commonalities between the transmission of genetic information and communication information. Therefore, methods based on communication coding theory can be applied to identify essential genes in bacteria and other organisms. This provides a new perspective and approach for analyzing and predicting essential genes. The information obtained in these analyses has applications in disease prevention, diagnosis, and treatment.
Author Contributions
Conceptualization, DC and LW; data collection or analysis, YG and YJ; validation, YG and YJ; writing—original draft preparation, YG and YJ; writing—review and editing, YG. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the National Natural Science Foundation of China (62072385), and the Special Science Foundation of Quzhou (2021D004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Thanks to the guidance of my tutor and the joint efforts of other authors, the success of this article is the result of everyone’s joint efforts.
References
Ao, C., Yu, L., and Zou, Q. (2021). Prediction of Bio-Sequence Modifications and the Associations with Diseases. Brief. Funct. genomics 20 (1), 1–18. doi:10.1093/bfgp/elaa023
Arun, P., Miryala, S. K., Chattopadhyay, S., Thiyyagura, K., Bawa, P., Bhattacharjee, M., et al. (2016). Identification and Functional Analysis of Essential, Conserved, Housekeeping and Duplicated Genes. FEBS Lett. 590, 1428–1437. doi:10.1002/1873-3468.12192
Chen, C., Zhang, Q., Ma, Q., and Yu, B. (2019). LightGBM-PPI: Predicting Protein-Protein Interactions through LightGBM with Multi-Information Fusion. Chemometrics Intell. Lab. Syst. 191, 54–64. doi:10.1016/j.chemolab.2019.06.003
Chen, C., Zhang, Q., Yu, B., Yu, Z., Lawrence, P. J., Ma, Q., et al. (2020). Improving Protein-Protein Interactions Prediction Accuracy Using XGBoost Feature Selection and Stacked Ensemble Classifier. Comput. Biol. Med. 123, 103899. doi:10.1016/j.compbiomed.2020.103899
Chen, S., Liu, Z., Li, M., Huang, Y., Wang, M., Zeng, W., et al. (2020). Potential Prognostic Predictors and Molecular Targets for Skin Melanoma Screened by Weighted Gene Co-expression Network Analysis. Curr. Gene Ther. 20 (1), 5–14. doi:10.2174/1566523220666200516170832
Chen, W.-H., Minguez, P., Lercher, M. J., and Bork, P. (2011). OGEE: an Online Gene Essentiality Database. Nucleic Acids Res. 40 (D1), D901–D906. doi:10.1093/nar/gkr986
Chen, Y., Ma, T., Yang, X., Wang, J., Song, B., and Zeng, X. (2021). MUFFIN: Multi-Scale Feature Fusion for Drug–Drug Interaction Prediction. Bioinformatics 37, 2651–2658. doi:10.1093/bioinformatics/btab169
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018). DincRNA: a Comprehensive Web-Based Bioinformatics Toolkit for Exploring Disease Associations and ncRNA Function. Bioinformatics 34 (11), 1953–1956. doi:10.1093/bioinformatics/bty002
Cheng, L., Qi, C., Yang, H., Lu, M., Cai, Y., Fu, T., et al. (2021). gutMGene: a Comprehensive Database for Target Genes of Gut Microbes and Microbial Metabolites. Nucleic Acids Res., gkab786. doi:10.1093/nar/gkab786
Cheng, L., Qi, C., Zhuang, H., Fu, T., and Zhang, X. (2020). gutMDisorder: a Comprehensive Database for Dysbiosis of the Gut Microbiota in Disorders and Interventions. Nucleic Acids Res. 48 (D1), D554–D560. doi:10.1093/nar/gkz843
Cheng, Y., Gong, Y., Liu, Y., Song, B., and Zou, Q. (2021). Molecular Design in Drug Discovery: a Comprehensive Review of Deep Generative Models. Brief. Bioinform. 22, bbab344. doi:10.1093/bib/bbab344
Chiu, T. P., Xin, B., Markarian, N., Wang, Y., and Rohs, R. (2020). TFBSshape: an Expanded Motif Database for DNA Shape Features of Transcription Factor Binding Sites. Nucleic Acids Res. 48 (D1), D246–D255. doi:10.1093/nar/gkz970
Dao, F. Y., Lv, H., Zhang, D., Zhang, Z. M., Liu, L., and Lin, H. (2021). DeepYY1: a Deep Learning Approach to Identify YY1-Mediated Chromatin Loops. Brief Bioinform 22 (4). bbaa356, doi:10.1093/bib/bbaa356
Deng, J., Deng, L., Su, S., Zhang, M., Lin, X., Wei, L., et al. (2011). Investigating the Predictability of Essential Genes across Distantly Related Organisms Using an Integrative Approach. Nucleic Acids Res. 39 (3), 795–807. doi:10.1093/nar/gkq784
Deng, L., Li, W., and Zhang, J. (2021). LDAH2V: Exploring Meta-Paths across Multiple Networks for lncRNA-Disease Association Prediction. Ieee/acm Trans. Comput. Biol. Bioinf. 18 (4), 1572–1581. doi:10.1109/tcbb.2019.2946257
Dong, J., Zhao, M., Liu, Y., Su, Y., and Zeng, X. (2021). Deep Learning in Retrosynthesis Planning: Datasets, Models and Tools. Brief. Bioinform., bbab391. doi:10.1093/bib/bbab391
Eisenberg, D., Marcotte, E. M., Xenarios, I., and Yeates, T. O. (2000). Protein Function in the post-genomic Era. Nature 405 (6788), 823–826. doi:10.1038/35015694
Fang, M., Lei, X., and Guo, L. (2019). A Survey on Computational Methods for Essential Proteins and Genes Prediction. Curr. Bioinformatics 14 (3), 211–225. doi:10.2174/1574893613666181112150422
Fang, S., Pan, J., Zhou, C., Tian, H., He, J., Shen, W., et al. (2019). Circular RNAs Serve as Novel Biomarkers and Therapeutic Targets in Cancers. Curr. Gene Ther. 19 (2), 125–133. doi:10.2174/1566523218666181109142756
Garzon, M. H., and Deaton, R. J. (2004). Codeword Design and Information Encoding in DNA Ensembles. Nat. Comput. 3 (3), 253–292. doi:10.1023/b:naco.0000036818.27537.c9
Grazziotin, A. L., Vidal, N. M., and Venancio, T. M. (2015). Uncovering Major Genomic Features of Essential Genes in Bacteria and a Methanogenic Archaea. Febs J. 282, 3395–3411. doi:10.1111/febs.13350
Guo, Y., Yan, K., Lv, H., and Liu, B. (2021). PreTP-EL: Prediction of Therapeutic Peptides Based on Ensemble Learning. Brief Bioinform 22, bbab358. doi:10.1093/bib/bbab358
Guo, Z., Wang, P., Liu, Z., and Zhao, Y. (2020). Discrimination of Thermophilic Proteins and Non-thermophilic Proteins Using Feature Dimension Reduction. Front. Bioeng. Biotechnol. 8, 584807. doi:10.3389/fbioe.2020.584807
Hu, Y., Qiu, S., and Cheng, L. (2021). Integration of Multiple-Omics Data to Analyze the Population-specific Differences for Coronary Artery Disease. Comput. Math. Methods Med. 2021, 7036592. doi:10.1155/2021/7036592
Hu, Y., Sun, J. Y., Zhang, Y., Zhang, H., Gao, S., Wang, T., et al. (2021). rs1990622 Variant Associates with Alzheimer's Disease and Regulates TMEM106B Expression in Human Brain Tissues. BMC Med. 19 (1), 11. doi:10.1186/s12916-020-01883-5
Hu, Y., Zhang, H., Liu, B., Gao, S., Wang, T., Han, Z., et al. (2020). rs34331204 Regulates TSPAN13 Expression and Contributes to Alzheimer's Disease with Sex Differences. Brain. 143 (11), e95. doi:10.1093/brain/awaa302
Huh, W.-K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., et al. (2003). Global Analysis of Protein Localization in Budding Yeast. Nature 425, 686–691. doi:10.1038/nature02026
Huo, Y., Xin, L., Kang, C., Wang, M., Ma, Q., and Yu, B. (2020). SGL-SVM: A Novel Method for Tumor Classification via Support Vector Machine with Sparse Group Lasso. J. Theor. Biol. 486, 110098. doi:10.1016/j.jtbi.2019.110098
Hutchison, , Peterson, S. N., Gill, S. R., Cline, R. T., White, O., Fraser, C. M., et al. (1999). Global Transposon Mutagenesis and a Minimal Mycoplasma Genome. Science 286 (5447), 2165–2169. doi:10.1126/science.286.5447.2165
Hwang, Y.-C., Lin, C.-C., Chang, J.-Y., Mori, H., Juan, H.-F., and Huang, H.-C. (2009). Predicting Essential Genes Based on Network and Sequence Analysis. Mol. Biosyst. 5 (12), 1672–1678. doi:10.1039/b900611g
Itaya, M. (1995). An Estimation of Minimal Genome Size Required for Life. Febs Lett. 362 (3), 257–260. doi:10.1016/0014-5793(95)00233-y
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting Human microRNA-Disease Associations Based on Support Vector Machine. Ijdmb 8 (3), 282–293. doi:10.1504/ijdmb.2013.056078
Kitano, H. (2002). Computational Systems Biology. Nature 420 (6912), 206–210. doi:10.1038/nature01254
Lander, E. S., Wang, T., Hughes, N. W., and Jenny, W. (2015). Identification and Characterization of Essential Genes in the Human Genome. Science 350, 1096–1101. doi:10.1126/science.aac7041
Li, F., Luo, M., Zhou, W., Li, J., Jin, X., Xu, Z., et al. (2020). Single Cell RNA and Immune Repertoire Profiling of COVID-19 Patients Reveal Novel Neutralizing Antibody. Protein Cell 12, 751–755. doi:10.1007/s13238-020-00807-6
Li, H.-L., Pang, Y.-H., and Liu, B. (2021). BioSeq-BLM: a Platform for Analyzing DNA, RNA and Protein Sequences Based on Biological Language Models. Nucleic Acids Res., gkab829. doi:10.1093/nar/gkab829
Liu, C., Wei, D., Xiang, J., Ren, F., Huang, L., Lang, J., et al. (2020). An Improved Anticancer Drug-Response Prediction Based on an Ensemble Method Integrating Matrix Completion and Ridge Regression. Mol. Ther. - Nucleic Acids 21, 676–686. doi:10.1016/j.omtn.2020.07.003
Liu, H., Zhang, W., Zou, B., Wang, J., Deng, Y., and Deng, L. (2020). DrugCombDB: a Comprehensive Database of Drug Combinations toward the Discovery of Combinatorial Therapy. Nucleic Acids Res. 48 (D1), D871–D881. doi:10.1093/nar/gkz1007
Liu, J., Liu, S., Liu, C., Zhang, Y., Pan, Y., Wang, Z., et al. (2021). Nabe: an Energetic Database of Amino Acid Mutations in Protein–Nucleic Acid Binding Interfaces. Database 2021, baab050. doi:10.1093/database/baab050
Liu, X., Hong, Z., Liu, J., Lin, Y., Rodríguez-Patón, A., Zou, Q., et al. (2020). Computational Methods for Identifying the Critical Nodes in Biological Networks. Brief. Bioinform. 21 (2), 486–497. doi:10.1093/bib/bbz011
Liu, X., Yang, J., Zhang, Y., Fang, Y., Wang, F., Wang, J., et al. (2016). A Systematic Study on Drug-Response Associated Genes Using Baseline Gene Expressions of the Cancer Cell Line Encyclopedia. Sci. Rep. 6, 22811. doi:10.1038/srep22811
Liu, Y., Chen, D., Su, R., Chen, W., and Wei, L. (2020). iRNA5hmC: The First Predictor to Identify RNA 5-Hydroxymethylcytosine Modifications Using Machine Learning. Front. Bioeng. Biotechnol. 8, 227. doi:10.3389/fbioe.2020.00227
Liu, Y., Zeng, X., He, Z., and Zou, Q. (2016). Inferring microRNA-Disease Associations by Random Walk on a Heterogeneous Network with Multiple Data Sources. Ieee/acm Trans. Comput. Biol. Bioinform 14 (4), 905–915. doi:10.1109/TCBB.2016.2550432
Liu, Y., Zhang, X., Zou, Q., and Zeng, X. (2021). Minirmd: Accurate and Fast Duplicate Removal Tool for Short Reads via Multiple Minimizers. Bioinformatics 37 (11), 1604–1606. doi:10.1093/bioinformatics/btaa915
Luo, H., Lin, Y., Gao, F., Zhang, C.-T., and Zhang, R. (2014). DEG 10, an Update of the Database of Essential Genes that Includes Both Protein-Coding Genes and Noncoding Genomic Elements: Table 1. Nucl. Acids Res. 42, D574–D580. doi:10.1093/nar/gkt1131
Qi, C., Wang, C., Zhao, L., Zhu, Z., Wang, P., Zhang, S., et al. (2021). SCovid: Single-Cell Atlases for Exposing Molecular Characteristics of COVID-19 across 10 Human Tissues. Nucleic Acids Res., gkab881. doi:10.1093/nar/gkab881
Riaz, F., and Li, D. (2019). Non-coding RNA Associated Competitive Endogenous RNA Regulatory Network: Novel Therapeutic Approach in Liver Fibrosis. Curr. Gene Ther. 19 (5), 305–317. doi:10.2174/1566523219666191107113046
Rivas, M. A., Pirinen, M., Conrad, D. F., Lek, M., Tsang, E. K., Karczewski, K. J., et al. (2015). Effect of Predicted Protein-Truncating Genetic Variants on the Human Transcriptome. Science 348 (6235), 666–669. doi:10.1126/science.1261877
Rochaix, J.-D., and Ramundo, S. (2015). Conditional Repression of Essential Chloroplast Genes: Evidence for New Plastid Signaling Pathways. Biochim. Biophys. Acta (Bba) - Bioenerg. 1847 (9), 986–992. doi:10.1016/j.bbabio.2014.11.011
Rosen, G. (2006). Examining Coding Structure and Redundancy in DNA. IEEE Eng. Med. Biol. Mag. 25 (1), 62–68. doi:10.1109/memb.2006.1578665
Ru, X., Cao, P., Li, L., and Zou, Q. (2019). Selecting Essential MicroRNAs Using a Novel Voting Method. Mol. Ther. - Nucleic Acids 18, 16–23. doi:10.1016/j.omtn.2019.07.019
Shang, Y., Gao, L., Zou, Q., and Yu, L. (2021). Prediction of Drug-Target Interactions Based on Multi-Layer Network Representation Learning. Neurocomputing 434, 80–89. doi:10.1016/j.neucom.2020.12.068
Song, B., Li, F., Liu, Y., and Zeng, X. (2021). Deep Learning Methods for Biomedical Named Entity Recognition: a Survey and Qualitative Comparison. Brief. Bioinform. 22, bbab282. doi:10.1093/bib/bbab282
Stanly Paul, M. L., Kaur, A., Geete, A., and Elizabeth Sobhia, M. (2014). Essential Gene Identification and Drug Target Prioritization in Leishmania Species. Mol. Biosyst. 10 (5), 1184–1195. doi:10.1039/c3mb70440h
Su, R., Liu, X., Wei, L., and Zou, Q. (2019). Deep-Resp-Forest: A Deep forest Model to Predict Anti-cancer Drug Response. Methods 166, 91–102. doi:10.1016/j.ymeth.2019.02.009
Su, W., Liu, M.-L., Yang, Y.-H., Wang, J.-S., Li, S.-H., Lv, H., et al. (2021). PPD: A Manually Curated Database for Experimentally Verified Prokaryotic Promoters. J. Mol. Biol. 433 (11), 166860. doi:10.1016/j.jmb.2021.166860
Sun, H., Yang, J., Zhang, T., Long, L. P., Jia, K., Yang, G., et al. (2013). Using Sequence Data to Infer the Antigenicity of Influenza Virus. mBio 44 (4), e00230–13. doi:10.1128/mBio.00230-13
Tao, Z., Li, Y., Teng, Z., and Zhao, Y. (2020). A Method for Identifying Vesicle Transport Proteins Based on LibSVM and MRMD. Comput. Math. Methods Med. 2020, 8926750. doi:10.1155/2020/8926750
Tavasolian, F., Hosseini, A. Z., Soudi, S., and Naderi, M. (2020). miRNA-146a Improves Immunomodulatory Effects of MSC-Derived Exosomes in Rheumatoid Arthritis. Curr. Gene Ther. 20 (4), 297–312. doi:10.2174/1566523220666200916120708
Uddin, R., Masood, F., Azam, S. S., and Wadood, A. (2019). Identification of Putative Non-host Essential Genes and Novel Drug Targets against Acinetobacter Baumannii by In Silico Comparative Genome Analysis. Microb. Pathogenesis 128, 28–35. doi:10.1016/j.micpath.2018.12.015
Voshol, G. P., Meyer, V., and van den Hondel, C. A. (2015). GTP-binding Protein Era: a Novel Gene Target for Biofuel Production. BMC Biotechnol. 15 (1), 21. doi:10.1186/s12896-015-0132-1
Wang, J., Liu, X., Shen, S., Deng, L., and Liu, H. (2021). DeepDDS: Deep Graph Neural Network with Attention Mechanism to Predict Synergistic Drug Combinations. Brief. Bioinform., bbab390. doi:10.1093/bib/bbab390
Wang, J., Shi, Y., Wang, X., and Chang, H. (2020). A Drug Target Interaction Prediction Based on LINE-RF Learning. Curr. Bioinformatics 15 (7), 750–757. doi:10.2174/1574893615666191227092453
Wang, N., Zhang, J., and Liu, B. (2021). IDRBP-PPCT: Identifying Nucleic Acid-Binding Proteins Based on Position-specific Score Matrix and Position-specific Frequency Matrix Cross Transformation. Ieee/acm Trans. Comput. Biol. Bioinf. (99), 1. doi:10.1109/TCBB.2021.3069263
Wang, X., Yu, B., Ma, A., Chen, C., Liu, B., and Ma, Q. (2018). Protein-protein Interaction Sites Prediction by Ensemble Random Forests with Synthetic Minority Oversampling Technique. Bioinformatics 35 (14), 2395–2402. doi:10.1093/bioinformatics/bty995
Wang, Y., Yang, S., Zhao, J., Du, W., Liang, Y., Wang, C., et al. (2019). Using Machine Learning to Measure Relatedness between Genes: A Multi-Features Model. Sci. Rep. 9 (1), 4192. doi:10.1038/s41598-019-40780-7
Wei, L., Tang, J., and Zou, Q. (2017). SkipCPP-Pred: an Improved and Promising Sequence-Based Predictor for Predicting Cell-Penetrating Peptides. Bmc Genomics 18, 742. doi:10.1186/s12864-017-4128-1
Wei, L., He, W., Malik, A., Su, R., Cui, L., and Manavalan, B. (2020). Computational Prediction and Interpretation of Cell-specific Replication Origin Sites from Multiple Eukaryotes by Exploiting Stacking Framework. Brief. Bioinform. 22, bbaa275. doi:10.1093/bib/bbaa275
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. Ieee/acm Trans. Comput. Biol. Bioinf. 11 (1), 192–201. doi:10.1109/tcbb.2013.146
Xu, Z., Luo, M., Lin, W., Xue, G., Wang, P., Jin, X., et al. (2021). DLpTCR: an Ensemble Deep Learning Framework for Predicting Immunogenic Peptide Recognized by T Cell Receptor. Brief Bioinform 22, bbab335. doi:10.1093/bib/bbab335
Yan, K., Fang, X., Xu, Y., and Liu, B. (2019). Protein Fold Recognition Based on Multi-View Modeling. Bioinformatics 35 (17), 2982–2990. doi:10.1093/bioinformatics/btz040
Yan, K., Wen, J., Liu, J.-X., Xu, Y., and Liu, B. (2021). Protein Fold Recognition by Combining Support Vector Machines and Pairwise Sequence Similarity Scores. Ieee/acm Trans. Comput. Biol. Bioinf. 18, 2008–2016. doi:10.1109/TCBB.2020.2966450
Yan, K., Wen, J., Xu, Y., and Liu, B. (2021).Protein Fold Recognition Based on Auto-Weighted Multi-View Graph Embedding Learning Model, Proceeding of the IEEE/ACM Trans. Comput. Biol. and Bioinf., April 2020. IEEE, 1. doi:10.1109/TCBB.2020.2991268
Yang, J., Huang, T., Huang, T., Petralia, F., Long, Q., Zhang, B., et al. (2015). Synchronized Age-Related Gene Expression Changes across Multiple Tissues in Human and the Link to Complex Diseases. Sci. Rep. 5, 15145. doi:10.1038/srep15145
Yang, L., Wang, J., Wang, H., Lv, Y., Zuo, Y., Li, X., et al. (2014). Analysis and Identification of Essential Genes in Humans Using Topological Properties and Biological Information. Gene 551 (2), 138–151. doi:10.1016/j.gene.2014.08.046
Ye, Y. N., Hua, Z. G., Huang, J., Rao, N., and Guo, F. B. (2013). CEG: a Database of Essential Gene Clusters. BMC Genomics 14 (1), 1–10. doi:10.1186/1471-2164-14-769
Yu, B., Chen, C., Zhou, H., Liu, B., and Ma, Q. (2020). GTB-PPI: Predict Protein-Protein Interactions Based on L1-Regularized Logistic Regression and Gradient Tree Boosting. Genomics, Proteomics & Bioinformatics 18 (5), 582–592. doi:10.1016/j.gpb.2021.01.001
Yu, L., Xu, F., and Gao, L. (2020). Predict New Therapeutic Drugs for Hepatocellular Carcinoma Based on Gene Mutation and Expression. Front. Bioeng. Biotechnol. 8, 8. doi:10.3389/fbioe.2020.00008
Yu, L., Zhou, D., Gao, L., and Zha, Y. (2020). Prediction of Drug Response in Multilayer Networks Based on Fusion of Multiomics Data. Methods 192, 85–92. doi:10.1016/j.ymeth.2020.08.006
Zeng, X., Song, X., Ma, T., Pan, X., Zhou, Y., Hou, Y., et al. (2020). Repurpose Open Data to Discover Therapeutics for COVID-19 Using Deep Learning. J. Proteome Res. 19 (11), 4624–4636. doi:10.1021/acs.jproteome.0c00316
Zeng, X., Tu, X., Liu, Y., Fu, X., and Su, Y. (2022). Toward Better Drug Discovery with Knowledge Graph. Curr. Opin. Struct. Biol. 72, 114–126. doi:10.1016/j.sbi.2021.09.003
Zeng, X., Zhu, S., Hou, Y., Zhang, P., Li, L., Li, J., et al. (2020). Network-based Prediction of Drug-Target Interactions Using an Arbitrary-Order Proximity Embedded Deep forest. Bioinformatics 36 (9), 2805–2812. doi:10.1093/bioinformatics/btaa010
Zhai, Y., Chen, Y., Teng, Z., and Zhao, Y. (2020). Identifying Antioxidant Proteins by Using Amino Acid Composition and Protein-Protein Interactions. Front. Cel Dev. Biol. 8, 591487. doi:10.3389/fcell.2020.591487
Zhang, F., Ma, A., Wang, Z., Ma, Q., Liu, B., Huang, L., et al. (2018). A Central Edge Selection Based Overlapping Community Detection Algorithm for the Detection of Overlapping Structures in Protein-Protein Interaction Networks. Molecules 23 (10), 2633. doi:10.3390/molecules23102633
Zhang, S., Su, M., Sun, Z., Lu, H., and Zhang, Y. (2020). The Signature of Pharmaceutical Sensitivity Based on ctDNA Mutation in Eleven Cancers. Exp. Biol. Med. (Maywood) 245 (8), 720–732. doi:10.1177/1535370220906518
Zhou, L.-Y., Qin, Z., Zhu, Y.-H., He, Z.-Y., and Xu, T. (2019). Current RNA-Based Therapeutics in Clinical Trials. Curr. Gene Ther. 19 (3), 172–196. doi:10.2174/1566523219666190719100526
Zhuang, J., Dai, S., Zhang, L., Gao, P., Han, Y., Tian, G., et al. (2020). Identifying Breast Cancer-Induced Gene Perturbations and its Application in Guiding Drug Repurposing. Curr. Bioinformatics 15 (9), 1075–1089. doi:10.2174/1574893615666200203104214
Zou, Q. (2019). Latest Machine Learning Techniques for Biomedicine and Bioinformatics. Curr. Bioinformatics 14 (3), 176–177. doi:10.2174/157489361403190220112855
Keywords: communication coding, database, essential genes, machine learning, minimum feature group
Citation: Guo Y, Ju Y, Chen D and Wang L (2021) Research on the Computational Prediction of Essential Genes. Front. Cell Dev. Biol. 9:803608. doi: 10.3389/fcell.2021.803608
Received: 28 October 2021; Accepted: 22 November 2021;
Published: 06 December 2021.
Edited by:
Lei Deng, Central South University, ChinaCopyright © 2021 Guo, Ju, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Chen, cGVha2dyaW5Ab3V0bG9vay5jb20=; Lihong Wang, Mjk3NTI0MTYyNUBxcS5jb20=