 Driti Ashok
Driti Ashok Laura Polcik
Laura Polcik Svenja Dannewitz Prosseda
Svenja Dannewitz Prosseda Tanja Nicole Hartmann
Tanja Nicole Hartmann- 1Department of Internal Medicine I, Faculty of Medicine and Medical Center, University of Freiburg, Freiburg, Germany
- 2University of Freiburg, Faculty of Biology, Freiburg, Germany
The bone marrow microenvironment provides critical cues for hematopoietic stem cell (HSC) self-renewal and differentiation and contributes to their malignant conversion. The microenvironment comprises a complex mixture of multiple cell types, soluble factors, and extracellular matrix in specialized regions termed ‘niches.’ Positioning of the various cellular players within these niches depends on their repertoire of adhesion molecules and chemotactic signaling, involving integrins and chemokine receptors and the corresponding intracellular players such as kinases and GTPases. The mechanical role of adhesion is to control the strength and morphology of the cell-cell and cell-extracellular matrix contacts and thereby the energy needed for the optimal localization of cells to their surroundings. While it is clear that biomechanical adhesive bonds are energetically expensive, the crosstalk between cell adhesion and metabolic pathways in the normal and malignant microenvironment is far from understood. The metabolic profile of the various cell types within the niche includes key molecules such as AMPK, glucose, mTOR, and HIF-1α. Here, we describe our most recent understanding of how the interplay between adhesion and these metabolic components is indispensable for bone marrow niche stability. In parallel, we compare the altered crosstalk of different cell types within the bone marrow niches in hematological malignancies and propose potential therapeutic associations.
Introduction
Hematopoiesis is a homeostatic process that starts with hematopoietic stem cells (HSCs) and ultimately leads to the formation of mature blood cells of the myeloid and lymphoid lineages. This process is supported by various cell types and soluble factors within the bone marrow (BM) microenvironment. Hematopoietic malignancies arise when the balance within the BM is disturbed, e.g., by DNA damage causing genetically abnormal hematopoietic cells (Hanahan and Weinberg, 2011; Bakker and Passegué, 2013) and converting HSCs into their malignant counterparts, the so-called leukemia stem cells (LSCs) or leukemia-initiating cells (LICs). These malignant cells further alter their microenvironment at the expense of normal hematopoiesis to facilitate their own growth and neoplastic processes (Wang and Zhong, 2018).
Retention within the BM is required to allow survival, proliferation, and differentiation of HSCs. For this purpose, HSCs express adhesion molecules such as integrins, selectins, and CD44 that bind their specific ligands presented by other cells, e.g., stromal cells (Klamer and Voermans, 2014; De Grandis et al., 2016; Levesque and Winkler, 2016; Mitroulis et al., 2020). Some of these molecules regulate HSC maintenance and retention and serve as biomarkers to determine the extent of malignant transformation (Kulkarni and Kale, 2020), e.g., the integrin very late antigen-4 (VLA-4, α4β1, see later chapters).
Normal hematopoiesis and malignant transformation are shaped by bioenergetics, biosynthesis, and the redox balance (Boroughs and Deberardinis, 2015; Deberardinis and Chandel, 2016; Pavlova and Thompson, 2016). Malignancies are often accompanied by hypoxia and altered ATP levels, which reflect the increased nutrient demand that is characteristic of malignant cells (Kim et al., 2006; Hardie, 2008). Adhesive processes are energetically expensive and thus modulated by bioenergetics. However, the crosstalk between cell adhesion and metabolic pathways in the normal and malignant microenvironment is far from understood.
This review describes how various hematopoietic and non-hematopoietic cell types interact within the normal and malignant microenvironment. We delineate the various factors that modulate integrin-dependent adhesion within the BM and the consequences of an altered adhesion-metabolism interplay for BM microenvironment stability.
Bone Marrow Compartmentalization: Cellular Structure and Adhesive Factors
Two types of niches exist in the BM, namely, the endosteal and the vascular niche. The endosteal niche promotes HSC (Lin− CD34+ CD38−) quiescence due to its unique composition (Nilsson et al., 1997; Wilson et al., 2007; Casanova-Acebes et al., 2013). It is composed of spongeous bone and specialized cells such as osteoblasts and osteoclasts (Figure 1A) (Nilsson et al., 1997; Wilson et al., 2007) and is enriched with a network of arterioles and sinusoids. Hematopoietic stem and progenitor cells (HSPCs) surround the surface of the endosteum. Along the sinusoidal belt, endothelial cells and mesenchymal progenitors are primed to give rise to osteoblasts (Nombela-Arrieta et al., 2013). Osteoblasts are important players within the endosteal niche (Xie et al., 2009), although osteoblast depletion in transgenic mice did not affect HSC numbers (Visnjic et al., 2001; Bowers et al., 2015; Yu et al., 2015), yet altered their cycling capacity (Bowers et al., 2015; Yu et al., 2015). Additionally, osteoblast depletion reduced pre-pro- and pro-B cell numbers in the BM (Zhu et al., 2007; Yu et al., 2015).
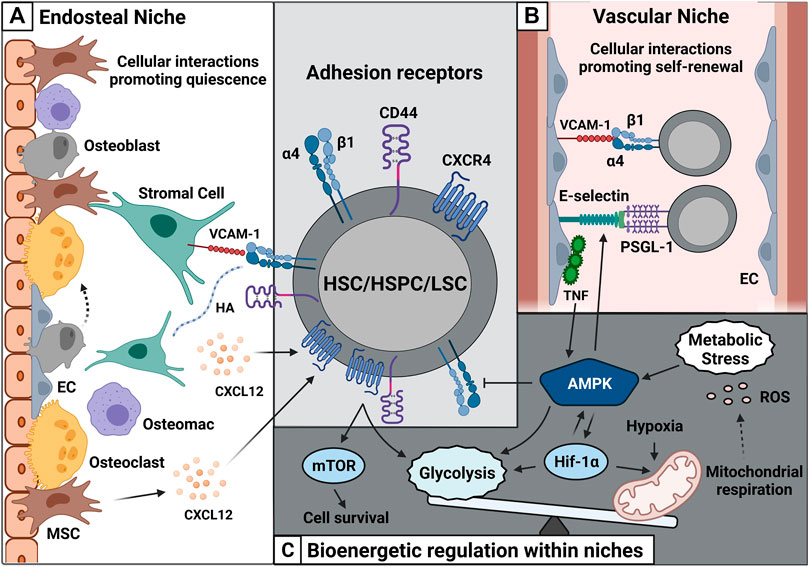
FIGURE 1. Cell-cell interactions within the BM microenvironment are shaped by adhesion and metabolism. (A) HSC/HSPC/LSC express adhesion receptors such as α4β1, CD44, and CXCR4. These receptors interact with their specific ligands such as VCAM-1, HA, and CXCL12 on non-hematopoietic cells such as stromal cells, MSCs and osteoblasts and contribute HSC quiescence. (B) The vascular niche made up of sinusoids contains numerous ECs. These ECs express large amounts of E-selectin and VCAM-1 and promote HSC/HSPC/LSC self-renewal. (C) Enhanced CXCR4-CD44 cooperativity can promote leukemic cell survival via mTOR and elevates glycolytic influx. Metabolic stress further increases AMPK activity promoting glycolysis and Hif-1α expression. High amounts of Hif-1α create a hypoxic environment followed by release of ROS. Abbreviations: HSC/HSPC, hematopoietic stem cell/hematopoietic stem and progenitor cell; LSC, leukemic stem cell; MSC, mesenchymal stem cell; EC, endothelial cell; VCAM-1, vascular cell-adhesion molecule-1; HA, hyaluronic acid; CXCL12, C-X-C Motif Chemokine Ligand 12; PSGL-1, P-selectin glycoprotein ligand-1; CXCR4, CXC receptor 4; mTOR, mammalian target of rapamycin complex; AMPK, adenosine-5′-monophosphate-activated protein kinase; Hif-1α, hypoxia-inducible factor α; ROS, reactive oxygen species.
Furthermore, HSC survival depends on the interaction with the vascular network, comprising arteries that enter the BM and subdivide into numerous arterioles. The arterioles traverse the BM along with specialized capillaries, the so-called sinusoids (Figure 1B) (Kopp et al., 2005). These vessels lack a continuous basal lamina, and their open pores facilitate the passing of white blood cells from the BM while promoting hematopoietic cell proliferation and differentiation. The term ‘vascular niche’ describes this unique environment. Its self-renewing HSC pool is closely associated with BM endothelial cells along sinusoidal vessels towards the center of the BM (Figure 1B). This was confirmed by the knockout of the glycoprotein 130 (gp130) receptor in ECs, leading to reduced HSC numbers (Yao et al., 2005). The survival of ECs from Tie-2 gfp mice was co-dependent on HSCs in vitro, even in the presence of growth factors (Li et al., 2004).
Adhesive Factors and Metabolic Components in Leukemia
The interaction of HSC with their niche requires communication via cell surface receptors, that mediate their adhesion to this environment. Particularly CD44, CXCR4 and the integrin α4β1 (VLA-4) play important roles in regulating HSPC trafficking, self-renewal, proliferation, and differentiation. These molecules extensively interact with each other and influence each other’s function, with the integrin α4β1 being activated by CD44 and CXCR4. The CD44 family comprises different variants of glycoproteins with extensive posttranslational modifications. This regulates the binding of CD44 to its ligands and promotes heterodimerization with other surface molecules, such as α4β1, thereby modulating integrin function (Yu et al., 2021; (Gutjahr et al., 2021). Chemokines such as the CXCR4 ligand CXCL12 also serve as integrin activators. Binding of CXCL12 to CXCR4 activates a rapid cascade of intracellular signaling events that eventually direct the integrin conformation to a high affinity state. This high affinity state allows a strong adhesion of the cells to the substrate.
Once the crosstalk of these three receptors is altered, leukemogenesis is driven. For example, CD44 cooperativity with α4β1 triggers adhesion of leukemic cells to stromal cells (Gutjahr et al., 2021), inducing prosurvival signaling pathways. Similarly, CD44 interacts with CXCR4, which can promote chemoresistance in AML (Yu et al., 2021).
CXCL12 is secreted by all stromal cells in the BM. CXCL12 abundant reticular (CAR) cells/Leptin receptor-positive cells (LepR+) (Seike et al., 2021) are a subset of VCAM-1+ reticular cells that promote HSC activity (Omatsu et al., 2010). Cell-specific depletion of CXCL12 diminished CAR cells, followed by a marked decrease in HSC proliferation (Omatsu et al., 2010).
Furthermore, CXCL12 knockout in endothelial cells leads to modest loss of self-renewing activity (Greenbaum et al., 2013). Similarly, the co-culture of ECs with HSCs provided a growth advantage, increasing HSC numbers (Butler et al., 2010). In leukemic conditions, such as T-cell acute lymphoblastic leukemia, CXCL12 knockout in ECs suppressed tumor development, indicating a dependency of the tumor on the vascular environment (Pitt et al., 2015). In addition, CXCL12 expression on osteoblasts is decisive for BM lymphoid progenitor populations and their reconstitution capacity (Greenbaum et al., 2013); Zhu et al., 2007). Furthermore, differentiated osteoblasts regulate osteoclast differentiation and function via cytokines such as macrophage-colony stimulating factor (M-CSF), osteoprotegerin (OPG), and OPG ligand (OPGL) (Yasuda et al., 1998). Osteoclasts are F4/80 low or negative macrophages that mediate bone resorption at the endosteal surface (Figure 1A) (Lacey et al., 1998). Additionally, the endosteum contains F4/80 (high) expressing macrophages called osteomacs that contribute to bone remodeling (Chang et al., 2008). Osteomacs surround osteoblasts (Chang et al., 2008; Winkler et al., 2010), CXCL12-abundant reticular cells (Casanova-Acebes et al., 2013), and Nestin+ mesenchymal stem cells (MSCs). Osteomacs and Nestin+ MSCs synergize in maintaining a high-adhesive niche. Upon osteomac depletion, Nestin+ cells display reduced levels of the adhesive components CXCL12, VCAM-1, and Angpt-1 (Chow et al., 2011), which are essential for HSC retention within the BM. In malignancy, Nestin+ MSCs may facilitate this crosstalk as they are known to support leukemogenesis.
CXCL12 enhances the survival capacity of AML cells and supports chemoresistance (Yu et al., 2021). It also induces phosphorylation of mTOR (Braun et al., 2016), which has been linked to increased glycolysis and translation of HIF-1α (Figure 2) (Konopleva et al., 2009). The siRNA-mediated knockdown of CXCL12 in AML cell lines and primary AML cells reduces glucose levels and mTOR expression (Braun et al., 2016). This phenomenon was recapitulated using the CXC receptor 4 (CXCR4) inhibitor plerixafor (AMD3100) (Uy et al., 2012). Notably, CXCR4 cooperates with the glycoprotein CD44 in AML, thereby enhancing resistance to the BCL-2 inhibitor venetoclax (Yu et al., 2021). This cooperation conferred a phenotype attributed to cancer stem cells (Cd44high, CXCR4high) (Yu et al., 2021) followed by high expression of embryonic stem cell core transcription factors such as Sox2, Oct4, and Nanog (Figure 2).
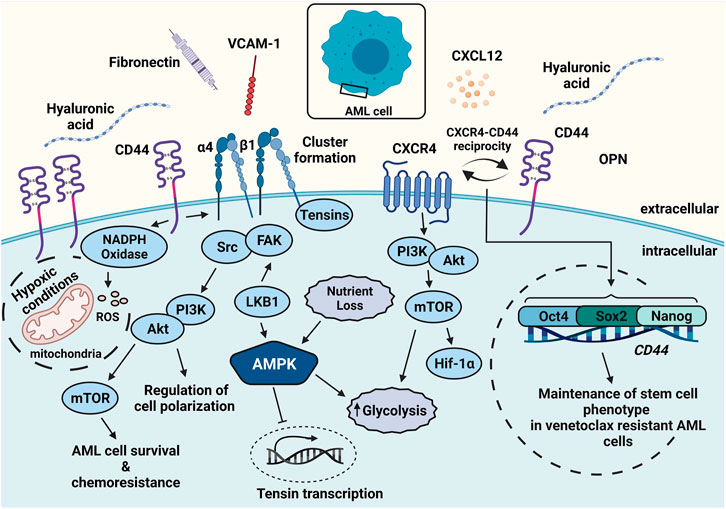
FIGURE 2. Leukemic cell survival can be augmented by adhesion and metabolic components. AML cells express high amounts of CD44, CXCR4, and α4β1. Ligands such as HA, CXCL12, and VCAM-1 secreted by stromal cells activate the receptors and render chemoresistance. CD44-mediated activation and clustering of α4β1 leads to adhesive hubs and promotes mTOR activity. CXCL12 interaction with CXCR4 can be followed by enhanced glycolysis. CXCR4 interaction with CD44 confers a cancer stem cell phenotype via Oct4, Sox2, and Nanog expression. Elevated CD44 expression increases ROS levels via NADPH oxidase and further promotes malignancy. Abbreviations: FAK, focal adhesion kinase.
In fact, CD44 is one of the first-described cancer stem cell markers and key homing receptors (Naor et al., 2002; Jin et al., 2006). Alternative splicing of the Cd44 gene gives rise to numerous variants (CD44v) (Naor et al., 2002; Jin et al., 2006). Normal cells expressing the standard isoform (CD44s) usually require activation and the induction of CD44 isoforms to bind their respective ligands, particularly hyaluronic acid (HA) and osteopontin. In contrast, tumor cells constitutively express CD44v and can bind ligands without further stimulation (Naor et al., 2002).
CD44 variants v6, v8, v9, and v10 (numbering based on expressed exons) are found in normal peripheral blood mononuclear cells (Dougherty et al., 1991; Bendall et al., 2000), yet at a low abundance. In hematological malignancies, these variants are primarily associated with non-Hodgkin’s lymphoma, myeloma, and leukemia, while in solid tumors, they are associated with breast and colon cancer (Legras et al., 1998; Asosingh et al., 2000; Medina et al., 2010; Guo and Frenette, 2014). Multifaceted properties of CD44 have been deciphered as links between LSCs and their BM microenvironment (Jin et al., 2006). One of these many links is found in the metabolic shift in the microenvironment that involves elevated production of reactive oxygen species (ROS) due to oncogene activity (Figure 2), along with an increase in ROS-driving enzymes such as NADPH oxidase. CD44v8-10 elevates the synthesis of reduced state glutathione (GSH) via its cysteine-glutamate antiporter system (xcT) (Ishimoto et al., 2011). This phenomenon aids cancer cells in overcoming elevated ROS levels, e.g., in the context of aerobic glycolysis (Warburg effect) (Ishimoto et al., 2011). Blocking CD44 by monoclonal antibodies leads to the terminal differentiation of leukemic cells, thereby reducing their potential for engraftment (Jin et al., 2006). The role of CD44 in maintaining the stemness character of leukemic cells can be attributed to either its direct binding to ligands or to its cooperation with other surface receptors, which eventually shapes the niche (Goodison et al., 1999; Orian-Rousseau and Sleeman, 2014; Gutjahr et al., 2021; Yu et al., 2021). For example, HA binding to CD44 on malignant cells activates the integrin α4β1, enhancing strong adhesion to VCAM-1, FN, and laminin (Grassinger et al., 2009; Gutjahr et al., 2021). Strong adhesion is mechanically based on α4β1 clustering upon the CD44-α4β1 cooperation and promotes stromal cell-leukemia cell contacts that act as metabolically-relevant “adhesive hubs” (Figure 2).
The cooperation of CD44 with α4β1 is one example of the so-called inside-out signaling of integrins, which serves as a means to integrate extracellular cues in cellular responses. Several possible signaling pathways and processes are involved in inside-out signaling, which starts with the activation of cell surface receptors, e.g., cytokine-, chemokine-, or antigen receptors. The activated surface receptors then induce an intracellular signaling cascade involving phosphoinositide 3-kinase (PI3K) and PLC-γ, among others, which eventually culminates in a high-affinity integrin status (Harzschel et al., 2020). The PI3K/Akt signaling pathway is also relevant for stromal cell-mediated chemoprotection of leukemic cells and for α4β1 downstream signaling to metabolic pathways via the master regulator (mTOR) (Shishido et al., 2014; Guerra and Issinger, 2020; Harzschel et al., 2020). Focal adhesion kinase (FAK) interaction with integrins has proven essential for the metabolic regulation of tumor cells (Yin, 2011), leading to excessive glucose consumption via the PI3K/Akt pathway (Bisht and Dey, 2008; Paik et al., 2009). In AML, three-dimensional access to VCAM-1 using coated beads triggers high phosphorylation levels of Akt, ERK, and mTOR (Gutjahr et al., 2021), and this downstream signaling is intensified by the association of Src and FAK (Zhou et al., 2016; Gutjahr et al., 2021). FAK activation by integrins also triggers the insulin growth factor receptor-1 resulting in sustained PI3K activation (Andersson et al., 2009). This particularly facilitated the proliferation of leukemic cells. In solid tumors, reciprocal interaction between β1-integrins and glucose metabolism has also been reported. In breast cancer cells, glucose metabolism is promoted by β1-integrin signaling through Twist, a regulator of epithelial-mesenchymal transition (EMT) (Yang et al., 2016), while knockdown of Glut1 in breast cancer cells lowers integrin β1 levels followed by reduced Src and FAK expression (Oh et al., 2017). With metabolic deregulation being a hallmark of both solid and hematological malignancies, the involvement of integrins and the underlying mechanisms of cell adhesion changes warrant further investigation.
Hypoxia
Hypoxia regulates cell proliferation and survival via hypoxia-inducible factor 1-alpha (HIF-1α), mammalian target of rapamycin complex (mTORC), and adenosine-5′monophosphate-activated protein kinase (AMPK) (Kim et al., 2006; Hardie et al., 2012; Dibble and Manning, 2013).
The endosteal niche is widely believed to be hypoxic and shows oxygen levels of less than 2% (Spencer et al., 2014). This has been attributed to the quiescence of HSPCs that reside within the niche (Spencer et al., 2014). A report by Arrieta and others (Nombela-Arrieta et al., 2013) contradicted this traditional viewpoint. Using quantitative 3D imaging, the authors found that the hypoxic profile of HSPCs is independent of their cell cycle state and anatomical positions within the BM, implying that the tendency towards hypoxia could be driven by the cell populations within the BM rather than their location.
HIF-1α is one of the many critical factors driving glycolysis and mitochondrial respiration within the BM, with higher levels in hypoxic conditions (Figure 1B) (Papandreou et al., 2006). HSC survival is maintained by high rates of glycolysis (Simsek et al., 2010). For example, in quiescent HSCs, HIF-1α minimizes ROS generation and favors pyruvate dehydrogenase kinase expression (Takubo et al., 2013). When HIF-1α is knocked out in HSCs, glycolysis rates drop, and mitochondrial metabolism levels are elevated. This prevents pyruvate from entering the tricarboxylic acid cycle, thereby preventing mitochondrial respiration.
Integrin function is modulated by different oxygen levels in a partially HIF-dependent manner. For example, integrin α5β1-dependent upregulation of mast cell adhesion to fibronectin was observed in oxygen tensions as low as 1%. This upregulation was accompanied by increased Akt phosphorylation and sensitivity towards PI3K inhibition, indicating a hypoxia-driven integrin activation via the PI3K signaling pathway (Pastwinska et al., 2020).
AMPK
Glycolysis is influenced by AMPK (Almeida et al., 2004), which allows cancer cells to survive metabolic stress (Figure 1C) (Jiang and Nakada, 2016). This energy sensor and central regulator for cellular metabolism plays a crucial role in growth and metabolic programming and is activated during cellular stress such as nutrient deficiency. Notably, AMPK is the most prominent factor known to link integrins and metabolism. The metabolic signaling via AMPK extensively influences adhesion mechanisms, e.g., the AMPK-activating kinase LKB1 regulates focal adhesion kinase (FAK), thereby controlling cell migration and cell polarization via PI3K localization (Mihaylova and Shaw, 2011). LKB1 expression is also necessary to maintain HSC quiescence. Lkb1 abrogation resulted in enhanced cycling of HSCs and subsequent depletion (Nakada et al., 2010). AMPK phosphorylation of HSCs also required Lkb1.
AMPK-guided cell adhesion is regulated via the tensin family of proteins. Tensins bind β1-integrins and support their activation (Georgiadou et al., 2017) with the integrin adaptor talin. AMPK downregulates tensins and thereby negatively regulates β1-integrin activity. Factors that arrest gluconeogenesis, such as loss of nutrients, enhance AMPK activity. We propose that this results in reduced integrin-mediated adhesion in malignancies. However, AMPK levels have to be tightly balanced and thus both, enhanced and suppressed, AMPK activity can hinder cell migration (Nakano et al., 2010; Yan et al., 2015). This is molecularly based on dysregulation of the cytoskeletal arrangements during migration, with Rac activity at the front of the migrating cell being in dynamic balance with Rho A activity at the rear of the cell. Augmented AMPK phosphorylates its substrate, Pdlim5, higher phosphorylation levels of which down-regulates Rac1 expression at the leading edge of the cell (Yan et al., 2015). In contrast, suppressed AMPK activity alters microtubule polymerization (Nakano et al., 2010). While actomyosin filaments and the ECM mechanically regulate cell migration, the dual role of AMPK provides a possible explanation for the observed discrepancies in AMPK-mediated effects on integrin activity.
A reciprocal response from integrins is observed in regulating metabolic signaling. For instance, integrin-linked kinase (ILK) regulates factors upstream of YAP/TAZ, thereby promoting YAP/TAZ activity within the nucleus. This might be assisted by the TEAD family of transcription factors, which directly interact with AMPK (Ata and Antonescu, 2017). Indeed, the release of cyclic AMP, an activator of AMPK (Omar et al., 2009) supports the survival of B-lineage acute lymphoblastic leukemia cells. This suggests that regulating the cAMP pathway could serve as a therapeutic target in leukemia (Perez et al., 2016).
Selectins, CD44 and integrins strengthen the interaction between ECs and HSCs. During inflammatory conditions, selectins aid leukocytes in binding to endothelial cells and establish leukocyte rolling on the vasculature (Winkler et al., 2012). Constitutive E-selectin expression on ECs guides HSC activity (Schweitzer et al., 1996). Characterization of E-selectin knockout mice (Sele−/−) revealed slower HSC division, suggesting enhanced HSC quiescence and self-renewal capacity (Winkler et al., 2012). In BCR-ABL1-induced chronic myelogenous leukemia (CML)-like myeloproliferative neoplasia (MPN), Sele−/− mice showed lower engraftment rates of leukemia compared to the wild-type, suggesting a homing defect (Krause et al., 2014). Endothelial damage occurs during malignancy and via irradiation and chemotherapy (Lyu et al., 2019; Wijerathne et al., 2021). As a result, this dislodges the vascular endothelium, creating impaired vascular permeability and elevating vascular fibrosis. In TNF-stimulated ECs, overexpression of dominant-negative AMPK (AMPKα2−/−) resulted in higher levels of NF-ĸB and E-selectin. AMPK activation in endothelial cells by vasculoprotective agents, such as metformin and VEGF, triggers several biological effects beneficial to vascular homeostasis (Cheng et al., 2007). These involve suppression of hyperglycemia-induced generation of ROS (Kukidome et al., 2006), alleviation of free fatty acid-induced lipotoxicity (Mccarty, 2005), and inhibition of nuclear factor-κB activation by tumor necrosis factor-α (Hattori et al., 2006). AMPK activity triggers endothelial progenitor cell differentiation in vitro upon VEGF stimulation (Li et al., 2008). Highly phosphorylated AMPK was associated with high levels of VE-cadherin and the integrin ligand ICAM-1, while AMPK inhibition reduced expression of these markers, suggesting the involvement of AMPK in endothelial progenitor cell differentiation (Li et al., 2008). In AML, a dense vascular compartment is a hallmark of poor prognosis (Shih et al., 2009) and BMECs are known to support AML cell quiescence (Ding et al., 2012; Ding and Morrison, 2013). Treatment with the tubulin polymerization inhibitor combrestatin altered the shape of BMECs, depleting VCAM-1 and VE-cadherin in AML cells, leading to increased peripheral blood cycling of AML cells. Combrestatins trigger ROS production in AML cells, and in combination with cytarabine, decrease leukemic cells within the murine BM (Bosse et al., 2016).
Therapies Targeting the BM Microenvironment
Leukemia is marked by genetic and epigenetic alterations in hematopoietic cells and their respective progenitors (De Beauchamp et al., 2021). Myeloid leukemia (CML and AML) is particularly faced with the challenge of targeting cells that are treatment-resistant due to the large repertoire of mutations that arise, namely, point-mutations associated with BCR-ABL in CML or FLT3-TKD in AML (Corbin et al., 2011; Hamilton et al., 2012). Though widely implemented in clinical trials, tyrosine kinase inhibitors are not sufficient to eradicate minimal residual disease as they preferentially target differentiated cells (Corbin et al., 2011; Hamilton et al., 2012). An alternate route to target these malignant cells might be to interfere with metabolism. Preclinical data in CML showed that CD34+ cells display an oxidative phenotype marked by low levels of fatty acids such as oleic and linoleic acids and an increase in TCA metabolites, namely, glutamate and aspartate (Kuntz et al., 2017). Administration of the antibiotic tigecycline blocked levels of aspartate and, upon combination with the kinase inhibitor imatinib, arrested colony formation (Kuntz et al., 2017). Similarly, in CD44-overexpressing CD44high K562 cells, non-steroid anti-inflammatory drugs (NSAIDs) administration triggered autophagy followed by increased sensitivity to heat shock protein inhibitor 17-AAG (Moon et al., 2019). This was supported by the activation of AMPK, and thus, inhibition of Akt/mTOR/p70S6K/4EBP1 activity. Autophagic cell death eventually led to the downregulation of CD44, Oct4, and c-Myc in these cells (Moon et al., 2019), linking back adhesion and metabolism. Phase 2 clinical data suggests that 17-AAG (Tanespimycin) at a dosage of 340 mg/m2 in combination with bortezomib serves as a promising therapeutic option for patients with refractory multiple myeloma (Richardson et al., 2010). Regulation of metabolic components in AML was also studied by implementing dietary restriction coupled with the AMPK inhibitor Compound C, enhancing overall survival reducing the percentage of circulating AML cells in vivo (Saito et al., 2015). In addition, metformin administration stimulates the LKB1/AMPK cascade leading to mTORC declination in AML supported by shrinkage in tumor burden (Sujobert et al., 2015). Recent research suggests that resistance to chemotherapy in AML is mediated by metabolic alterations that are short-lived and sufficient to drive patients and patient-derived xenograft models into relapse (Van Gastel et al., 2020). Upon chemotherapeutic intervention specialized leukemic cells prevailed within the bone marrow. The metabolic signature of these cells comprised elevated levels of glutamine, further supporting pyrimidine synthesis. Pyrimidine synthesis, in turn, required a sustained aspartate supply from BM stromal cells. Blocking pyrimidines using Brequinar in combination with lower-dose chemotherapy cleared AML cells from the BM in PDX models (Van Gastel et al., 2020).
Multiple therapeutic options within the BM microenvironment have thus far targeted soluble factors and cell adhesion molecules. The CXCR4-CXCL12 axis was targeted using several individual inhibitors (Burger et al., 2005; Parameswaran et al., 2011; Uy et al., 2017); however, combination therapies proved more effective. For instance, the AML specific peptide E5 (Li et al., 2015) hindered the adhesion and migration of AML cells to the BM by interfering with the CXCR4-CXCL12 pathway. In combination with the chemotherapeutic drugs vincristine and cyclophosphamide, the leukemic burden in mice was reduced, and survival was prolonged (Li et al., 2015). Co-culture of leukemic cells with BM stromal cells confers resistance to the CD44 antibody A3D8 via the PI3K-Akt pathway (Chen et al., 2015). Administration of the PI3K-Akt inhibitor, LY294002, along with A3D8, partially resensitizes the AML cells (Chen et al., 2015). α4β1 inhibition reduces chemoresistance of AML and MM cell lines (Rashidi and Dipersio, 2016) and restores CD3-mediated cytotoxicity within the AML BM microenvironment (Nair-Gupta et al., 2020).
Conclusion
The importance of cell adhesion molecules within the BM is well-acclaimed and continuously advancing as the expression of certain molecules strongly correlates to malignancy. While our understanding of the crosstalk between integrins and these metabolites is improving, there remains a gap in its therapeutic application. Investigating the extent of chemo-sensitization within the BM upon combining adhesion molecules and metabolic inhibitors might bring novel insights into potential therapeutic alternatives.
Author Contributions
DA, LP, and TH conceived and wrote the manuscript. SD and TH edited the manuscript.
Funding
The authors of this review are supported by the José Carreras Leukemia Foundation DJCLS 04 R/2020, Deutsche Krebshilfe (DKH) 70113993, the Deutsche Forschungsgemeinshaft (DFG) 437764364, 419090910 and research support by AstraZeneca (AcertaPharma) to TH. This study received funding from AstraZeneca. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
BioRender was used to design figures and authors wish to thank the platform for the publication license.
References
Almeida, A., Moncada, S., and Bolaños, J. P. (2004). Nitric Oxide Switches on Glycolysis through the AMP Protein Kinase and 6-Phosphofructo-2-Kinase Pathway. Nat. Cel Biol 6, 45–51. doi:10.1038/ncb1080
Andersson, S., D’Arcy, P., Larsson, O., and Sehat, B. (2009). Focal Adhesion Kinase (FAK) Activates and Stabilizes IGF-1 Receptor. Biochem. Biophysical Res. Commun. 387, 36–41. doi:10.1016/j.bbrc.2009.06.088
Asosingh, K., Günthert, U., Bakkus, M. H. C., De Raeve, H., Goes, E., Van Riet, I., et al. (2000). In Vivo Induction of Insulin-like Growth Factor-I Receptor and CD44v6 Confers Homing and Adhesion to Murine Multiple Myeloma Cells. Cancer Res. 60, 3096–3104. https://cancerres.aacrjournals.org/content/60/11/3096.
Ata, R., and Antonescu, C. N. (2017). Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. Int. J. Mol. Sci. 18, 189. doi:10.3390/ijms18010189
Bakker, S. T., and Passegué, E. (2013). Resilient and Resourceful: Genome Maintenance Strategies in Hematopoietic Stem Cells. Exp. Hematol. 41, 915–923. doi:10.1016/j.exphem.2013.09.007
Bendall, L., Bradstock, K., and Gottlieb, D. (2000). Expression of CD44 Variant Exons in Acute Myeloid Leukemia Is More Common and More Complex Than that Observed in normal Blood, Bone Marrow or CD34+ Cells. Leukemia 14, 1239–1246. doi:10.1038/sj.leu.2401830
Bisht, B., and Dey, C. S. (2008). Focal Adhesion Kinase Contributes to Insulin-Induced Actin Reorganization into a Mesh Harboring Glucose Transporter-4 in Insulin Resistant Skeletal Muscle Cells. BMC Cel Biol 9, 48. doi:10.1186/1471-2121-9-48
Boroughs, L. K., and Deberardinis, R. J. (2015). Metabolic Pathways Promoting Cancer Cell Survival and Growth. Nat. Cel Biol 17, 351–359. doi:10.1038/ncb3124
Bosse, R. C., Wasserstrom, B., Meacham, A., Wise, E., Drusbosky, L., Walter, G. A., et al. (2016). Chemosensitizing AML Cells by Targeting Bone Marrow Endothelial Cells. Exp. Hematol. 44, 363–377. e365. doi:10.1016/j.exphem.2016.02.003
Bowers, M., Zhang, B., Ho, Y., Agarwal, P., Chen, C.-C., and Bhatia, R. (2015). Osteoblast Ablation Reduces normal Long-Term Hematopoietic Stem Cell Self-Renewal but Accelerates Leukemia Development. Blood 125, 2678–2688. doi:10.1182/blood-2014-06-582924
Braun, M., Qorraj, M., Büttner, M., Klein, F. A., Saul, D., Aigner, M., et al. (2016). CXCL12 Promotes Glycolytic Reprogramming in Acute Myeloid Leukemia Cells via the CXCR4/mTOR axis. Leukemia 30, 1788–1792. doi:10.1038/leu.2016.58
Burger, M., Hartmann, T., Krome, M., Rawluk, J., Tamamura, H., Fujii, N., et al. (2005). Small Peptide Inhibitors of the CXCR4 Chemokine Receptor (CD184) Antagonize the Activation, Migration, and Antiapoptotic Responses of CXCL12 in Chronic Lymphocytic Leukemia B Cells. Blood 106, 1824–1830. doi:10.1182/blood-2004-12-4918
Butler, J. M., Nolan, D. J., Vertes, E. L., Varnum-Finney, B., Kobayashi, H., Hooper, A. T., et al. (2010). Endothelial Cells Are Essential for the Self-Renewal and Repopulation of Notch-dependent Hematopoietic Stem Cells. Cell Stem Cell 6, 251–264. doi:10.1016/j.stem.2010.02.001
Casanova-Acebes, M., Pitaval, C., Weiss, L. A., Nombela-Arrieta, C., Chèvre, R., A-González, N., et al. (2013). Rhythmic Modulation of the Hematopoietic Niche through Neutrophil Clearance. Cell 153, 1025–1035. doi:10.1016/j.cell.2013.04.040
Chang, M. K., Raggatt, L.-J., Alexander, K. A., Kuliwaba, J. S., Fazzalari, N. L., Schroder, K., et al. (2008). Osteal Tissue Macrophages Are Intercalated throughout Human and Mouse Bone Lining Tissues and Regulate Osteoblast Function In Vitro and In Vivo. J. Immunol. 181, 1232–1244. doi:10.4049/jimmunol.181.2.1232
Chen, P., Huang, H., Wu, J., Lu, R., Wu, Y., Jiang, X., et al. (2015). Bone Marrow Stromal Cells Protect Acute Myeloid Leukemia Cells from Anti-CD44 Therapy Partly through Regulating PI3K/Akt-P27 Kip1 axis. Mol. Carcinog. 54, 1678–1685. doi:10.1002/mc.22239
Cheng, K. K. Y., Lam, K. S. L., Wang, Y., Huang, Y., Carling, D., Wu, D., et al. (2007). Adiponectin-induced Endothelial Nitric Oxide Synthase Activation and Nitric Oxide Production Are Mediated by APPL1 in Endothelial Cells. Diabetes 56, 1387–1394. doi:10.2337/db06-1580
Chow, A., Lucas, D., Hidalgo, A., Méndez-Ferrer, S., Hashimoto, D., Scheiermann, C., et al. (2011). Bone Marrow CD169+ Macrophages Promote the Retention of Hematopoietic Stem and Progenitor Cells in the Mesenchymal Stem Cell Niche. J. Exp. Med. 208, 261–271. doi:10.1084/jem.20101688
Corbin, A. S., Agarwal, A., Loriaux, M., Cortes, J., Deininger, M. W., and Druker, B. J. (2011). Human Chronic Myeloid Leukemia Stem Cells Are Insensitive to Imatinib Despite Inhibition of BCR-ABL Activity. J. Clin. Invest. 121, 396–409. doi:10.1172/jci35721
De Beauchamp, L., Himonas, E., and Helgason, G. V. (2021). Mitochondrial Metabolism as a Potential Therapeutic Target in Myeloid Leukaemia. Leukemia.
De Grandis, M., Lhoumeau, A.-C., Mancini, S. J. C., and Aurrand-Lions, M. (2016). Adhesion Receptors Involved in HSC and Early-B Cell Interactions with Bone Marrow Microenvironment. Cell. Mol. Life Sci. 73, 687–703. doi:10.1007/s00018-015-2064-2
Deberardinis, R. J., and Chandel, N. S. (2016). Fundamentals of Cancer Metabolism. Sci. Adv. 2, e1600200. doi:10.1126/sciadv.1600200
Dibble, C. C., and Manning, B. D. (2013). Signal Integration by mTORC1 Coordinates Nutrient Input with Biosynthetic Output. Nat. Cel Biol 15, 555–564. doi:10.1038/ncb2763
Ding, L., and Morrison, S. J. (2013). Haematopoietic Stem Cells and Early Lymphoid Progenitors Occupy Distinct Bone Marrow Niches. Nature 495, 231–235. doi:10.1038/nature11885
Ding, L., Saunders, T. L., Enikolopov, G., and Morrison, S. J. (2012). Endothelial and Perivascular Cells Maintain Haematopoietic Stem Cells. Nature 481, 457–462. doi:10.1038/nature10783
Dougherty, G. J., Landorp, P. M., Cooper, D. L., and Humphries, R. K. (1991). Molecular Cloning of CD44R1 and CD44R2, Two Novel Isoforms of the Human CD44 Lymphocyte "homing" Receptor Expressed by Hemopoietic Cells. J. Exp. Med. 174, 1–5. doi:10.1084/jem.174.1.1
Georgiadou, M., Lilja, J., Jacquemet, G., Guzmán, C., Rafaeva, M., Alibert, C., et al. (2017). AMPK Negatively Regulates Tensin-dependent Integrin Activity. J. Cel Biol 216, 1107–1121. doi:10.1083/jcb.201609066
Goodison, S., Urquidi, V., and Tarin, D. (1999). CD44 Cell Adhesion Molecules. Mol. Pathol. 52, 189–196. doi:10.1136/mp.52.4.189
Grassinger, J., Haylock, D. N., Storan, M. J., Haines, G. O., Williams, B., Whitty, G. A., et al. (2009). Thrombin-cleaved Osteopontin Regulates Hemopoietic Stem and Progenitor Cell Functions through Interactions with α9β1 and α4β1 Integrins. Blood 114, 49–59. doi:10.1182/blood-2009-01-197988
Greenbaum, A., Hsu, Y.-M. S., Day, R. B., Schuettpelz, L. G., Christopher, M. J., Borgerding, J. N., et al. (2013). CXCL12 in Early Mesenchymal Progenitors Is Required for Haematopoietic Stem-Cell Maintenance. Nature 495, 227–230. doi:10.1038/nature11926
Guerra, B., and Issinger, O. G. (2020). Role of Protein Kinase CK2 in Aberrant Lipid Metabolism in Cancer. Pharmaceuticals (Basel) 13, 292. doi:10.3390/ph13100292
Guo, W., and Frenette, P. S. (2014). Alternative CD44 Splicing in Intestinal Stem Cells and Tumorigenesis. Oncogene 33, 537–538. doi:10.1038/onc.2013.260
Gutjahr, J. C., Bayer, E., Yu, X., Laufer, J. M., Hopner, J. P., Tesanovic, S., et al. (2021). CD44 Engagement Enhances Acute Myeloid Leukemia Cell Adhesion to the Bone Marrow Microenvironment by Increasing VLA-4 Avidity. Haematologica 106, 2102–2113.
Hamilton, A., Helgason, G. V., Schemionek, M., Zhang, B., Myssina, S., Allan, E. K., et al. (2012). Chronic Myeloid Leukemia Stem Cells Are Not Dependent on Bcr-Abl Kinase Activity for Their Survival. J. Am. Soc. Hematol. 119, 1501–1510. doi:10.1182/blood-2010-12-326843
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of Cancer: the Next Generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hardie, D. G. (2008). AMPK: a Key Regulator of Energy Balance in the Single Cell and the Whole Organism. Int. J. Obes. 32, S7–S12. doi:10.1038/ijo.2008.116
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a Nutrient and Energy Sensor that Maintains Energy Homeostasis. Nat. Rev. Mol. Cel Biol 13, 251–262. doi:10.1038/nrm3311
Härzschel, A., Zucchetto, A., Gattei, V., and Hartmann, T. N. (2020). VLA-4 Expression and Activation in B Cell Malignancies: Functional and Clinical Aspects. Int. J. Mol. Sci. 21, 2206. doi:10.3390/ijms21062206
Hattori, Y., Suzuki, K., Hattori, S., and Kasai, K. (2006). Metformin Inhibits Cytokine-Induced Nuclear Factor κB Activation via AMP-Activated Protein Kinase Activation in Vascular Endothelial Cells. Hypertension 47, 1183–1188. doi:10.1161/01.hyp.0000221429.94591.72
Ishimoto, T., Nagano, O., Yae, T., Tamada, M., Motohara, T., Oshima, H., et al. (2011). CD44 Variant Regulates Redox Status in Cancer Cells by Stabilizing the xCT Subunit of System Xc− and Thereby Promotes Tumor Growth. Cancer Cell 19, 387–400. doi:10.1016/j.ccr.2011.01.038
Jiang, Y., and Nakada, D. (2016). Cell Intrinsic and Extrinsic Regulation of Leukemia Cell Metabolism. Int. J. Hematol. 103, 607–616. doi:10.1007/s12185-016-1958-6
Jin, L., Hope, K. J., Zhai, Q., Smadja-Joffe, F., and Dick, J. E. (2006). Targeting of CD44 Eradicates Human Acute Myeloid Leukemic Stem Cells. Nat. Med. 12, 1167–1174. doi:10.1038/nm1483
Kim, J.-w., Tchernyshyov, I., Semenza, G. L., and Dang, C. V. (2006). HIF-1-mediated Expression of Pyruvate Dehydrogenase Kinase: a Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cel Metab. 3, 177–185. doi:10.1016/j.cmet.2006.02.002
Klamer, S., and Voermans, C. (2014). The Role of Novel and Known Extracellular Matrix and Adhesion Molecules in the Homeostatic and Regenerative Bone Marrow Microenvironment. Cell Adhes. Migration 8, 563–577. doi:10.4161/19336918.2014.968501
Konopleva, M., Tabe, Y., Zeng, Z., and Andreeff, M. (2009). Therapeutic Targeting of Microenvironmental Interactions in Leukemia: Mechanisms and Approaches. Drug Resist. Updates 12, 103–113. doi:10.1016/j.drup.2009.06.001
Kopp, H.-G., Avecilla, S. T., Hooper, A. T., and Rafii, S. (2005). The Bone Marrow Vascular Niche: home of HSC Differentiation and Mobilization. Physiology 20, 349–356. doi:10.1152/physiol.00025.2005
Krause, D. S., Lazarides, K., Lewis, J. B., Von Andrian, U. H., and Van Etten, R. A. (2014). Selectins and Their Ligands Are Required for Homing and Engraftment of BCR-Abl1+ Leukemic Stem Cells in the Bone Marrow Niche. Blood 123, 1361–1371. doi:10.1182/blood-2013-11-538694
Kukidome, D., Nishikawa, T., Sonoda, K., Imoto, K., Fujisawa, K., Yano, M., et al. (2006). Activation of AMP-Activated Protein Kinase Reduces Hyperglycemia-Induced Mitochondrial Reactive Oxygen Species Production and Promotes Mitochondrial Biogenesis in Human Umbilical Vein Endothelial Cells. Diabetes 55, 120–127. doi:10.2337/diabetes.55.01.06.db05-0943
Kulkarni, R., and Kale, V. (2020). Physiological Cues Involved in the Regulation of Adhesion Mechanisms in Hematopoietic Stem Cell Fate Decision. Front. Cel Dev. Biol. 8, 611. doi:10.3389/fcell.2020.00611
Kuntz, E. M., Baquero, P., Michie, A. M., Dunn, K., Tardito, S., Holyoake, T. L., et al. (2017). Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat. Med. 23, 1234–1240. doi:10.1038/nm.4399
Lacey, D. L., Timms, E., Tan, H.-L., Kelley, M. J., Dunstan, C. R., Burgess, T., et al. (1998). Osteoprotegerin Ligand Is a Cytokine that Regulates Osteoclast Differentiation and Activation. Cell 93, 165–176. doi:10.1016/s0092-8674(00)81569-x
Legras, S., Günthert, U., Stauder, R., Curt, F., Oliferenko, S., Kluin-Nelemans, H. C., et al. (1998). A strong Expression of CD44-6v Correlates with Shorter Survival of Patients with Acute Myeloid Leukemia. Blood 91, 3401–3413. doi:10.1182/blood.v91.9.3401.3401_3401_3413
Levesque, J.-P., and Winkler, I. G. (2016). Cell Adhesion Molecules in Normal and Malignant Hematopoiesis: from Bench to Bedside. Curr. Stem Cel Rep 2, 356–367. doi:10.1007/s40778-016-0066-0
Li, W., Johnson, S. A., Shelley, W. C., and Yoder, M. C. (2004). Hematopoietic Stem Cell Repopulating Ability Can Be Maintained In Vitro by Some Primary Endothelial Cells. Exp. Hematol. 32, 1226–1237. doi:10.1016/j.exphem.2004.09.001
Li, X., Guo, H., Duan, H., Yang, Y., Meng, J., Liu, J., et al. (2015). Improving Chemotherapeutic Efficiency in Acute Myeloid Leukemia Treatments by Chemically Synthesized Peptide Interfering with CXCR4/CXCL12 axis. Sci. Rep. 5, 16228. doi:10.1038/srep16228
Li, X., Han, Y., Pang, W., Li, C., Xie, X., Shyy, J. Y.-J., et al. (2008). AMP-activated Protein Kinase Promotes the Differentiation of Endothelial Progenitor Cells. Arteriosclerosis, Thromb. Vasc. Biol. 28, 1789–1795. doi:10.1161/atvbaha.108.172452
Lyu, Z.-S., Yao, W.-L., Wen, Q., Zhao, H.-Y., Tang, F.-F., Wang, Y., et al. (2019). Glycolysis Restoration Attenuates Damaged Bone Marrow Endothelial Cells. Blood 134, 2491. doi:10.1182/blood-2019-122794
Mccarty, M. F. (2005). AMPK Activation as a Strategy for Reversing the Endothelial Lipotoxicity Underlying the Increased Vascular Risk Associated with Insulin Resistance Syndrome. Med. Hypotheses 64, 1211–1215. doi:10.1016/j.mehy.2004.01.042
Medina, P. P., Nolde, M., and Slack, F. J. (2010). OncomiR Addiction in an In Vivo Model of microRNA-21-Induced Pre-B-cell Lymphoma. Nature 467, 86–90. doi:10.1038/nature09284
Mihaylova, M. M., and Shaw, R. J. (2011). The AMPK Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat. Cel Biol 13, 1016–1023. doi:10.1038/ncb2329
Mitroulis, I., Kalafati, L., Bornhäuser, M., Hajishengallis, G., and Chavakis, T. (2020). Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol. 11, 1540. doi:10.3389/fimmu.2020.01540
Moon, H.-J., Park, S.-Y., Lee, S.-H., Kang, C.-D., and Kim, S.-H. (2019). Nonsteroidal Anti-inflammatory Drugs Sensitize CD44-Overexpressing Cancer Cells to Hsp90 Inhibitor through Autophagy Activation. Oncol. Res. 27, 835–847. doi:10.3727/096504019x15517850319579
Nair-Gupta, P., Rudnick, S. I., Luistro, L., Smith, M., Mcdaid, R., Li, Y., et al. (2020). Blockade of VLA4 Sensitizes Leukemic and Myeloma Tumor Cells to CD3 Redirection in the Bone Marrow Microenvironment. Blood Cancer J. 10, 65. doi:10.1038/s41408-020-0331-4
Nakada, D., Saunders, T. L., and Morrison, S. J. (2010). Lkb1 Regulates Cell Cycle and Energy Metabolism in Haematopoietic Stem Cells. Nature 468, 653–658. doi:10.1038/nature09571
Nakano, A., Kato, H., Watanabe, T., Min, K.-D., Yamazaki, S., Asano, Y., et al. (2010). AMPK Controls the Speed of Microtubule Polymerization and Directional Cell Migration through CLIP-170 Phosphorylation. Nat. Cel Biol 12, 583–590. doi:10.1038/ncb2060
Naor, D., Nedvetzki, S., Golan, I., Melnik, L., and Faitelson, Y. (2002). CD44 in Cancer. Crit. Rev. Clin. Lab. Sci. 39, 527–579. doi:10.1080/10408360290795574
Nilsson, S. K., Dooner, M. S., Tiarks, C. Y., Weier, H.-U., and Quesenberry, P. J. (1997). Potential and Distribution of Transplanted Hematopoietic Stem Cells in a Nonablated Mouse Model. Blood 89, 4013–4020. doi:10.1182/blood.v89.11.4013
Nombela-Arrieta, C., Pivarnik, G., Winkel, B., Canty, K. J., Harley, B., Mahoney, J. E., et al. (2013). Quantitative Imaging of Haematopoietic Stem and Progenitor Cell Localization and Hypoxic Status in the Bone Marrow Microenvironment. Nat. Cel Biol 15, 533–543. doi:10.1038/ncb2730
Oh, S., Kim, H., Nam, K., and Shin, I. (2017). Glut1 Promotes Cell Proliferation, Migration and Invasion by Regulating Epidermal Growth Factor Receptor and Integrin Signaling in Triple-Negative Breast Cancer Cells. BMB Rep. 50, 132–137. doi:10.5483/bmbrep.2017.50.3.189
Omar, B., Zmuda-Trzebiatowska, E., Manganiello, V., Göransson, O., and Degerman, E. (2009). Regulation of AMP-Activated Protein Kinase by cAMP in Adipocytes: Roles for Phosphodiesterases, Protein Kinase B, Protein Kinase A, Epac and Lipolysis. Cell Signal. 21, 760–766. doi:10.1016/j.cellsig.2009.01.015
Omatsu, Y., Sugiyama, T., Kohara, H., Kondoh, G., Fujii, N., Kohno, K., et al. (2010). The Essential Functions of Adipo-Osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity 33, 387–399. doi:10.1016/j.immuni.2010.08.017
Orian-Rousseau, V., and Sleeman, J. (2014). CD44 Is a Multidomain Signaling Platform that Integrates Extracellular Matrix Cues with Growth Factor and Cytokine Signals. Adv. Cancer Res. 123, 231–254. doi:10.1016/b978-0-12-800092-2.00009-5
Paik, J.-Y., Ko, B.-H., Jung, K.-H., and Lee, K.-H. (2009). Fibronectin Stimulates Endothelial Cell 18F-FDG Uptake through Focal Adhesion Kinase-Mediated Phosphatidylinositol 3-kinase/Akt Signaling. J. Nucl. Med. 50, 618–624. doi:10.2967/jnumed.108.059386
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L., and Denko, N. C. (2006). HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cel Metab. 3, 187–197. doi:10.1016/j.cmet.2006.01.012
Parameswaran, R., Yu, M., Lim, M., Groffen, J., and Heisterkamp, N. (2011). Combination of Drug Therapy in Acute Lymphoblastic Leukemia with a CXCR4 Antagonist. Leukemia 25, 1314–1323. doi:10.1038/leu.2011.76
Pavlova, N. N., and Thompson, C. B. (2016). The Emerging Hallmarks of Cancer Metabolism. Cel Metab. 23, 27–47. doi:10.1016/j.cmet.2015.12.006
Pitt, L. A., Tikhonova, A. N., Hu, H., Trimarchi, T., King, B., Gong, Y., et al. (2015). CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 27, 755–768. doi:10.1016/j.ccell.2015.05.002
Rashidi, A., and Dipersio, J. F. (2016). Targeting the Leukemia-Stroma Interaction in Acute Myeloid Leukemia: Rationale and Latest Evidence. Ther. Adv. Hematol. 7, 40–51. doi:10.1177/2040620715619307
Richardson, P. G., Badros, A. Z., Jagannath, S., Tarantolo, S., Wolf, J. L., Albitar, M., et al. (2010). Tanespimycin with Bortezomib: Activity in Relapsed/refractory Patients with Multiple Myeloma. Br. J. Haematol. 150, 428–437. doi:10.1111/j.1365-2141.2010.08264.x
Saito, Y., Chapple, R. H., Lin, A., Kitano, A., and Nakada, D. (2015). AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 17, 585–596. doi:10.1016/j.stem.2015.08.019
Schweitzer, K. M., Dräger, A. M., Van Der Valk, P., Thijsen, S. F., Zevenbergen, A., Theijsmeijer, A. P., et al. (1996). Constitutive Expression of E-Selectin and Vascular Cell Adhesion Molecule-1 on Endothelial Cells of Hematopoietic Tissues. Am. J. Pathol. 148, 165–175. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1861610/?page=2.
Shih, T. T.-F., Hou, H.-A., Liu, C.-Y., Chen, B.-B., Tang, J.-L., Chen, H.-Y., et al. (2009). Bone Marrow Angiogenesis Magnetic Resonance Imaging in Patients with Acute Myeloid Leukemia: Peak Enhancement Ratio Is an Independent Predictor for Overall Survival. Blood J. Am. Soc. Hematol. 113, 3161–3167. doi:10.1182/blood-2008-08-173104
Shishido, S., Bã¶nig, H., and Kim, Y.-M. (2014). Role of Integrin Alpha4 in Drug Resistance of Leukemia. Front. Oncol. 4, 99. doi:10.3389/fonc.2014.00099
Simsek, T., Kocabas, F., Zheng, J., Deberardinis, R. J., Mahmoud, A. I., Olson, E. N., et al. (2010). The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 7, 380–390. doi:10.1016/j.stem.2010.07.011
Spencer, J. A., Ferraro, F., Roussakis, E., Klein, A., Wu, J., Runnels, J. M., et al. (2014). Direct Measurement of Local Oxygen Concentration in the Bone Marrow of Live Animals. Nature 508, 269–273. doi:10.1038/nature13034
Sujobert, P., Poulain, L., Paubelle, E., Zylbersztejn, F., Grenier, A., Lambert, M., et al. (2015). Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cel Rep. 11, 1446–1457. doi:10.1016/j.celrep.2015.04.063
Takubo, K., Nagamatsu, G., Kobayashi, C. I., Nakamura-Ishizu, A., Kobayashi, H., Ikeda, E., et al. (2013). Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 12, 49–61. doi:10.1016/j.stem.2012.10.011
Uy, G. L., Rettig, M. P., Motabi, I. H., Mcfarland, K., Trinkaus, K. M., Hladnik, L. M., et al. (2012). A Phase 1/2 Study of Chemosensitization with the CXCR4 Antagonist Plerixafor in Relapsed or Refractory Acute Myeloid Leukemia. Blood 119, 3917–3924. doi:10.1182/blood-2011-10-383406
Uy, G. L., Rettig, M. P., Stone, R. M., Konopleva, M. Y., Andreeff, M., Mcfarland, K., et al. (2017). A Phase 1/2 Study of Chemosensitization with Plerixafor Plus G-CSF in Relapsed or Refractory Acute Myeloid Leukemia. Blood Cancer J. 7, e542. doi:10.1038/bcj.2017.21
Van Gastel, N., Spinelli, J. B., Sharda, A., Schajnovitz, A., Baryawno, N., Rhee, C., et al. (2020). Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cel Metab. 32, 391–403. e396. doi:10.1016/j.cmet.2020.07.009
Visnjic, D., Kalajzic, I., Gronowicz, G., Aguila, H. L., Clark, S. H., Lichtler, A. C., et al. (2001). Conditional Ablation of the Osteoblast Lineage in Col2.3Δtk Transgenic Mice. J. Bone Miner Res. 16, 2222–2231. doi:10.1359/jbmr.2001.16.12.2222
Wang, A., and Zhong, H. (2018). Roles of the Bone Marrow Niche in Hematopoiesis, Leukemogenesis, and Chemotherapy Resistance in Acute Myeloid Leukemia. Hematology 23, 729–739. doi:10.1080/10245332.2018.1486064
Wijerathne, H., Langston, J. C., Yang, Q., Sun, S., Miyamoto, C., Kilpatrick, L. E., et al. (2021). Mechanisms of Radiation-Induced Endothelium Damage: Emerging Models and Technologies. Radiother. Oncol. 158, 21–32. doi:10.1016/j.radonc.2021.02.007
Wilson, A., Oser, G. M., Jaworski, M., Blanco-Bose, W. E., Laurenti, E., Adolphe, C., et al. (2007). Dormant and Self-Renewing Hematopoietic Stem Cells and Their Niches. Ann. N Y Acad. Sci. 1106, 64–75. doi:10.1196/annals.1392.021
Winkler, I. G., Barbier, V., Nowlan, B., Jacobsen, R. N., Forristal, C. E., Patton, J. T., et al. (2012). Vascular Niche E-Selectin Regulates Hematopoietic Stem Cell Dormancy, Self Renewal and Chemoresistance. Nat. Med. 18, 1651–1657. doi:10.1038/nm.2969
Winkler, I. G., Sims, N. A., Pettit, A. R., Barbier, V., Nowlan, B., Helwani, F., et al. (2010). Bone Marrow Macrophages Maintain Hematopoietic Stem Cell (HSC) Niches and Their Depletion Mobilizes HSCs. Blood 116, 4815–4828. doi:10.1182/blood-2009-11-253534
Xie, Y., Yin, T., Wiegraebe, W., He, X. C., Miller, D., Stark, D., et al. (2009). Detection of Functional Haematopoietic Stem Cell Niche Using Real-Time Imaging. Nature 457, 97–101. doi:10.1038/nature07639
Yan, Y., Tsukamoto, O., Nakano, A., Kato, H., Kioka, H., Ito, N., et al. (2015). Augmented AMPK Activity Inhibits Cell Migration by Phosphorylating the Novel Substrate Pdlim5. Nat. Commun. 6, 6137. doi:10.1038/ncomms7137
Yang, J., Hou, Y., Zhou, M., Wen, S., Zhou, J., Xu, L., et al. (2016). Twist Induces Epithelial-Mesenchymal Transition and Cell Motility in Breast Cancer via ITGB1-FAK/ILK Signaling axis and its Associated Downstream Network. Int. J. Biochem. Cel Biol. 71, 62–71. doi:10.1016/j.biocel.2015.12.004
Yao, L., Yokota, T., Xia, L., Kincade, P. W., and Mcever, R. P. (2005). Bone Marrow Dysfunction in Mice Lacking the Cytokine Receptor Gp130 in Endothelial Cells. Blood 106, 4093–4101. doi:10.1182/blood-2005-02-0671
Yasuda, H., Shima, N., Nakagawa, N., Yamaguchi, K., Kinosaki, M., Mochizuki, S.-i., et al. (1998). Osteoclast Differentiation Factor Is a Ligand for Osteoprotegerin/osteoclastogenesis-Inhibitory Factor and Is Identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. 95, 3597–3602. doi:10.1073/pnas.95.7.3597
Yin, B. (2011). Focal Adhesion Kinase as a Target in the Treatment of Hematological Malignancies. Leuk. Res. 35, 1416–1418. doi:10.1016/j.leukres.2011.04.017
Yu, V. W. C., Saez, B., Cook, C., Lotinun, S., Pardo-Saganta, A., Wang, Y.-H., et al. (2015). Specific Bone Cells Produce DLL4 to Generate Thymus-Seeding Progenitors from Bone Marrow. J. Exp. Med. 212, 759–774. doi:10.1084/jem.20141843
Yu, X., Munoz-Sagredo, L., Streule, K., Muschong, P., Bayer, E., Walter, R. J., et al. (2021). CD44 Loss of Function Sensitizes AML Cells to the BCL-2 Inhibitor Venetoclax by Decreasing CXCL12-Driven Survival Cues. Blood 138, 1067–1080. doi:10.1182/blood.2020006343
Zhou, H.-S., Zhou, H.-S., Z. Carter, B., Andreeff, M., Z. Carter, B., and Andreeff, M. (2016). Bone Marrow Niche-Mediated Survival of Leukemia Stem Cells in Acute Myeloid Leukemia: Yin and Yang. Cancer Biol. Med. 13, 248–259. doi:10.20892/j.issn.2095-3941.2016.0023
Keywords: Bone marrow, microenvironment, adhesion, metabolism, integrin, VLA-4, α4β1, AML
Citation: Ashok D, Polcik L, Dannewitz Prosseda S and Hartmann TN (2022) Insights Into Bone Marrow Niche Stability: An Adhesion and Metabolism Route. Front. Cell Dev. Biol. 9:798604. doi: 10.3389/fcell.2021.798604
Received: 20 October 2021; Accepted: 14 December 2021;
Published: 18 January 2022.
Edited by:
Zhichao Fan, UCONN Health, United StatesReviewed by:
Yi Zhang, Fudan University, ChinaTakashi Nagasawa, Osaka University, Japan
Ioannis Mitroulis, Democritus University of Thrace, Greece
Copyright © 2022 Ashok, Polcik, Dannewitz Prosseda and Hartmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanja Nicole Hartmann, dGFuamEuaGFydG1hbm5AdW5pa2xpbmlrLWZyZWlidXJnLmRl