 Shaoxian Wu1,2†
Shaoxian Wu1,2† Runzi Sun1,2†
Runzi Sun1,2† Bo Tan1
Bo Tan1 Bendong Chen2Wenyan Zhou2
Bendong Chen2Wenyan Zhou2 David Shihong Gao2Joshua Zhong2Hao Huang1
David Shihong Gao2Joshua Zhong2Hao Huang1 Jingting Jiang1*
Jingting Jiang1* Binfeng Lu2*
Binfeng Lu2*- 1Department of Tumor Biological Treatment, The Third Affiliated Hospital of Soochow University, Changzhou, China
- 2Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
In the era of immune checkpoint blockade cancer therapy, cytokines have become an attractive immune therapeutics to increase response rates. Interleukin 21 (IL21) as a single agent has been evaluated for cancer treatment with good clinical efficacy. However, the clinical application of IL21 is limited by a short half-life and concern about potential immune suppressive effect on dendritic cells. Here, we examined the antitumor function of a half-life extended IL21 alone and in combination with PD-1 blockade using preclinical mouse tumor models. We also determined the immune mechanisms of combination therapy. We found that combination therapy additively inhibited the growth of mouse tumors by increasing the effector function of type 1 lymphocytes. Combination therapy also increased the fraction of type 1 dendritic cells (DC1s) and M1 macrophages in the tumor microenvironment (TME). However, combination therapy also induced immune regulatory mechanisms, including the checkpoint molecules Tim-3, Lag-3, and CD39, as well as myeloid derived suppressor cells (MDSC). This study reveals the mechanisms of IL21/PD-1 cooperation and shed light on rational design of novel combination cancer immunotherapy.
Introduction
Recently, immune checkpoint blockade (ICB) has showed therapeutic efficacy and greatly prolonged survival in cancer patients. However, the response rates of ICB treatment are low for most carcinomas and new approach is needed to further improve cancer immune therapy (Chae et al., 2018; Li et al., 2018). ICB therapy has removed a major roadblock of cancer treatment by targeting molecules that hinder T cell-mediated immune responses (Hodi et al., 2010; Brahmer et al., 2012; Topalian et al., 2012). This new development has ushered in rich opportunities for using immune agonists as combination therapies. Cytokines drive T cell-mediated immune responses by enhancing proliferation, promoting type 1 differentiation, increasing the effector function, and directing the memory generation (Shourian et al., 2019; Zander et al., 2019; Xue et al., 2021). In contrast, molecules such as PD-1 impose “brakes” to an adaptive immune response. Therefore, the cytokine-based immunotherapy is in theory in concert with ICB therapy and promises to further improve clinical response rates.
Interleukin 21 (IL21) is an immune agonist that is an attractive cancer immunotherapeutic (Ma et al., 2003; Ugai et al., 2003; Di Carlo et al., 2004; Sivakumar et al., 2004; Spolski and Leonard, 2008; Xu et al., 2015; Lewis et al., 2017; Deng et al., 2020). Administration of IL21 in vivo directly increases the expression of effector molecules on CD8+T and NK cell, including granzyme B, perforin and IFN-γ (Kasaian et al., 2002; Brady et al., 2004; Zeng et al., 2005; White et al., 2007). Recently, IL21 was shown to promote the generation of memory stem CD8+T cells, thereby should promote a sustained antitumor immune response (Zhang et al., 2005; Klebanoff et al., 2011; Wölfl et al., 2011; Chen et al., 2018). IL21 also synergizes with IL-15 or IL-7 in vitro to promote the proliferation of central memory CD8+ T cells (Kasaian et al., 2002; Brady et al., 2004; Zeng et al., 2005). Other studies show that Th17 cells produce significantly higher levels of IL21 than either Th1 or Th2 cells and that IL21 is required for the generation of Th17 cells (Korn et al., 2007; Nurieva et al., 2007; Zhou et al., 2007). Th17 cells afford strong antitumor activities by stimulating CD8+T cells (Martin-Orozco et al., 2009). In addition to its direct effect on conventional T (Tconv) cells, IL21 inhibits the suppressive function of regulatory T cells (Tregs) and disrupts their homeostasis (Peluso et al., 2007; Clough et al., 2008; Attridge et al., 2012; Van Belle et al., 2012). Whether IL21 induces other immune regulatory pathways remain to be investigated. All in all, the existing data shows that IL21 strongly promotes the anti-tumor immune response. Indeed, administration of IL21 has shown strong antitumor efficacy in multiple preclinical mouse tumor models (Spolski and Leonard, 2008). Recent preclinical studies showed that recombinant IL21 synergizes with CTLA-4 and PD-1 blockade to inhibit cancer. These results validate the ability of IL21 to be combined with current ICB therapies (Lewis et al., 2017). Since many new checkpoint inhibitors are being evaluated in the clinics, it remains to be studied whether IL21 can be further combined with additional immune checkpoint inhibitors, such as anti-Lag-3 and anti-Tim-3 monoclonal antibodies (mAbs).
Recombinant IL21 has been tested as an antitumor agent in various clinical trials (Davis et al., 2007; Thompson et al., 2008; Davis et al., 2009; Schmidt et al., 2010; Petrella et al., 2012; Bhatia et al., 2014). The clinical program has advanced to phase II with promising antitumor activities and acceptable toxicity. However, the short half-life of IL21 reduces the in vivo levels of IL21 and requires frequent dosing, which limits its clinical application. In this study we first examined the therapeutic efficacy of a half-life-improved IL21 in preclinical mouse tumor models. We then investigated the immune mechanisms of combination therapy. Next, we determined whether IL21 induced the expression of additional immune checkpoint molecules, such as Lag-3, Tim-3, and CD39. Lastly, we examined whether targeting these molecules in triple and quadruple combinations therapies would further increase therapeutic efficacy.
Results
IL21R Was Differentially Expressed Among Tumor-Infiltrating Immune Cells
The cellular response to IL21-based therapy is dependent on the expression of its receptor IL21R. We examined IL21R expression on immune cells in the tumor microenvironment (TME). First, we analyzed published single-cell RNA-sequencing (scRNA-seq) data of mouse MC38 tumors (Figures 1A,B) (Zhang et al., 2020). IL21R was highly expressed on a broad range of tumor-infiltrating immune cells, including CD4+ Tconv cells, T regulatory cells (Tregs), CD8+ T cells, B cells, NK cells, macrophages, monocytes, and dendritic cells (DCs) but minimally expressed in neutrophils and mast cells (Figures 1C,D). We confirmed IL21R expression at the protein level by analyzing MC38 tumors with flow cytometry. IL21R was expressed on 41% of CD4+ Foxp3- T cells, 18% of Tregs, 38% of CD8+ T cells, 15% of NK cells, and 54% of B cells (Figures 1C,D and Supplementary Figure S1A). IL21R was also expressed on a smaller fraction of myeloid cells (16% of type 1 DCs (DC1s), 6% of type 2 DCs (DC2s), 3% of myeloid-derived suppressor cells (MDSCs), 8% of M1 macrophages, and 14% of M2 macrophages (Figures 1E,F). Interestingly, we observed that IL21R was expressed on more than 79% of T and B lymphocytes in lymph nodes and spleen (Supplementary Figures S1B,C). These data show that IL21 can directly act on multiple immune cell types in the TME and the secondary lymphoid system.
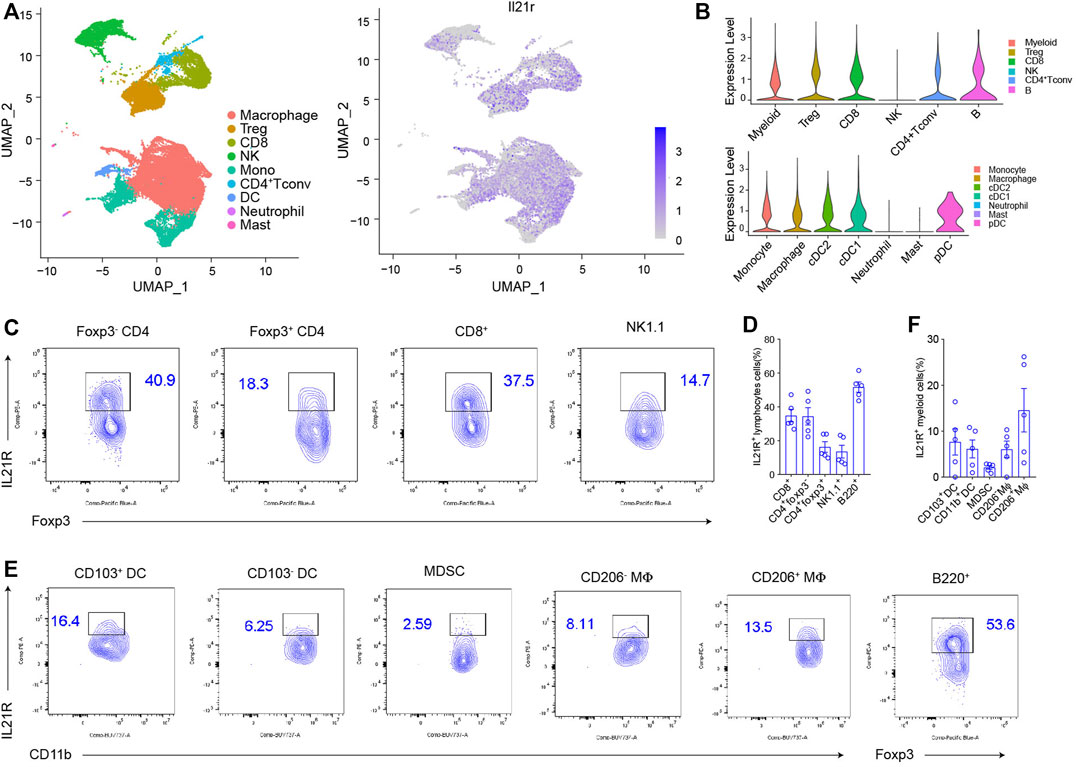
FIGURE 1. IL21R was differentially expressed among tumor-infiltrating immune cells (A) Cluster analysis. Immune cells from vaccinated MC38 mice were drawn by unified manifold approximation and projection (UMAP) dimensionality reduction of scRNA-seq data, and were colored by the expression of IL21R. (B) Violin plot shows the expression of IL21R in different immune cell subsets. Each dot represents a single cell. (C) Representative flow cytometry scatter plots of IL21R expression on tumor-infiltrating lymphocytes. (Foxp3-CD4+: CD45+CD4+Foxp3-, Foxp3+CD4+: CD45+CD4+Foxp3+, CD8+: CD45+CD8+, NK cell: CD45+CD8-CD4-NK1.1+, B cells: CD45+CD8-CD4-B220+). (D) Quantitative percentage of IL21R in various immune cells. (E) Representative flow cytometry analyses of IL21R expression on tumor-infiltrating myeloid cells (CD103+DC: CD45+Gr1-MHCII+CD24+CD103+, CD103-DC: CD45+Gr1-MHCII+CD24+CD103-CD11b+, MDSC: CD45+CD11b+Gr1+, CD206-Mφ: CD45+Gr1-MHCII+F4/80+CD206-Arg-1-, CD206+Mφ: CD45+Gr1-MHCII+F4/80+CD206+Arg-1+). (F) Quantitative percentage of IL21R in various immune cells.
HSA-IL21 and PD-1 Blockade Combination Therapy Additively Inhibited Tumor Growth
In order to improve pharmacologic property of IL21, an anti-HSA nanoantibody was fused to IL21. The half-life of IL21 was extended less than 30 min to more than 15 h in the mouse (Zhong et al., 2020). We then tested the efficacy in vivo of HSA-IL21 administration alone and in combination with PD-1 blockade. HSA-IL21 alone significantly inhibited the growth of MC38 tumors at comparable levels to PD-1 blockade (Figure 2A). The weight of the mice stayed constant, showing that HSA-IL21 has no apparent toxicity (Figure 2B). Combination therapy completely stunted tumor growth (Figure 2A and Supplementary Figure S2A). Administration of recombinant IL21 at the same dose and frequency did not produce any antitumor effect (Supplementary Figures S2C,D).
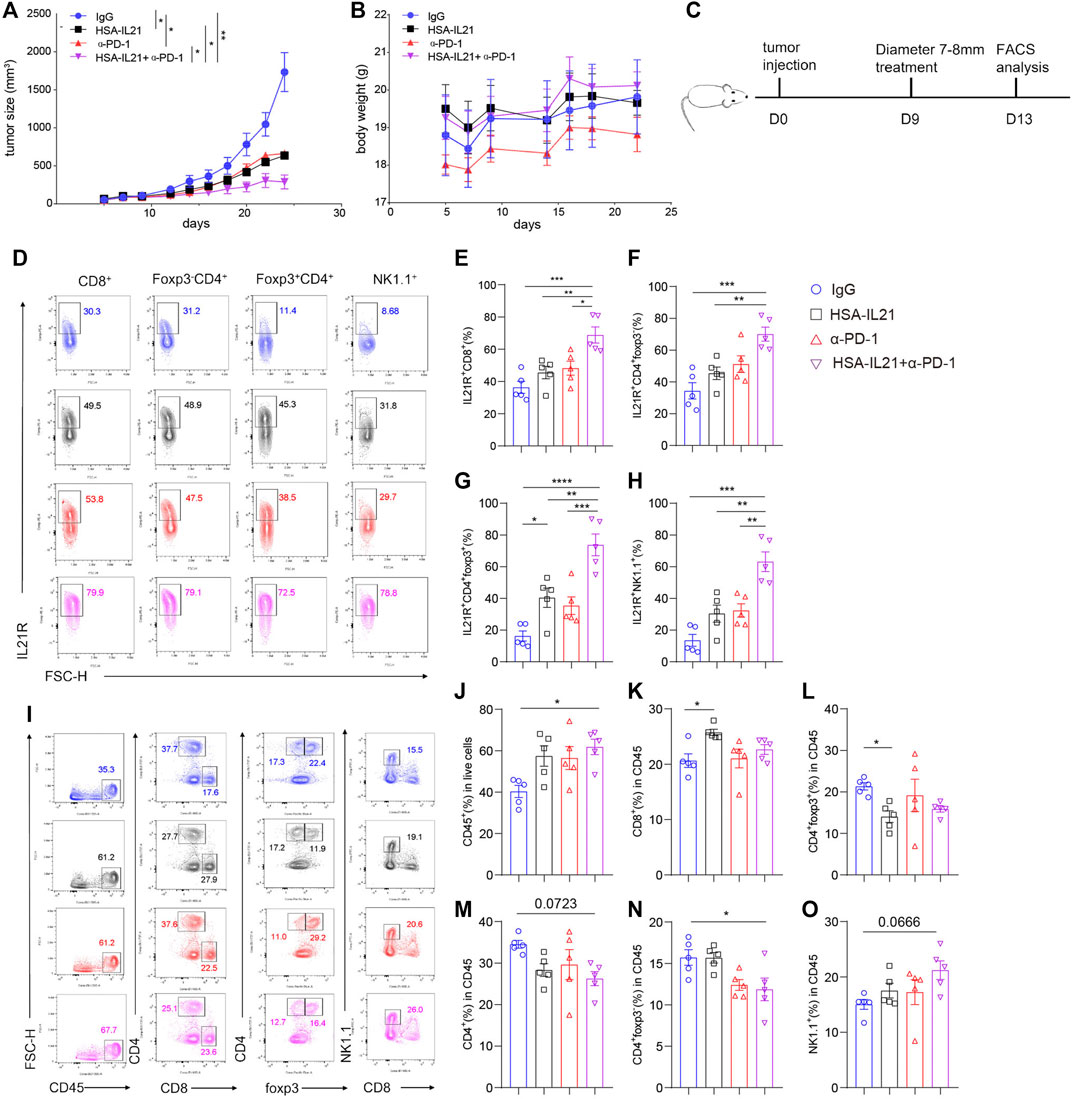
FIGURE 2. HSA-IL21 and PD-1 blockade combination therapy additively inhibited tumor growth. (A,B) MC38 tumor cells (1 × 106) were injected intradermally to B6 mice, monitored tumor volume (A) and body weight (B) of tumor-bearing mice treated with IgG, HAS-IL21, α-PD-1, HSA-IL21/α-PD-1. Data were presented as mean ± SEM, n = 4–5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA test were performed. (C) Graphic schematics of the mouse experiment. MC38 tumor cells (1 × 106) were injected intradermally to B6 mice, 9 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, HSA-IL21/α-PD-1.96h later, tumor-infiltrating lymphocytes were analyzed by flow cytometry. (D) Representative flow plots of IL21R expression on tumor-infiltrating CD8+, Foxp3-CD4+, Foxp3+CD4+ T and NK cells. (E–H) Quantitative analysis of IL21R expression depicted in (D). Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test were performed. (I) Representative flow plots showing the percentage of main immune populations. (J–O) Quantitative percentage of tumor infiltrating CD45+ lymphocytes, percentages of CD8+ T, CD4+ T cells, Foxp3+CD4+, Foxp3-CD4+ T and NK cells. Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test was performed.
To uncover the mechanisms of HSA-IL21/PD-1 blockade combination therapy, we studied the composition and functional states of immune cells in the TME using multi-color flow cytometry. Combination therapy additively increased the expression of IL21R on Foxp3- CD4+ T cells, Tregs, CD8+ T cells, and NK cells in the TME (Figures 2C–H). HSA-IL21 or PD-1 blockade alone increased the fraction of total immune cells (CD45+) out of all cells in the TME, though combination therapy did not cause any further increases (Figures 2I,J). Within the CD45+ immune cell compartment, HSA-IL21 treatment increased the fraction of CD8+ T cells and decreased the fraction of Treg cells (Figures 2K,L). In contrast, combined therapy decreased the fraction of CD4+ T cells (Figures 2M,N). Combination therapy shifted the T cell compartment towards the HSA-IL21 phenotype. The fraction of NK cells was not significantly altered by HSA-IL21 or PD-1 blockade alone. However, combination therapy significantly increased the fraction of NK cells (Figure 2O). These data show that HSA-IL21/PD-1 blockade combination therapy might act via IL21R to increase the immune response in the TME and thereby inhibit tumor growth.
Combination Therapy Enhances the Effector Function of Tumor-Infiltrating Lymphocytes
To determine whether combination therapy affects the effector function of tumor-infiltrating lymphocytes (TILs), we examined the effector and activation molecules GzmB, IFN-γ, and CD69 on CD8+, Foxp3-CD4+, Foxp3+CD4+ T cells and NK cells in the TME at 96 h after treatment by using multi-color flow cytometry (Figures 3A,I). The production of both IFN-γ and GzmB by CD8+ T cells and CD4+ T cells was additively increased by combination therapy (Figures 3B–H). Combination therapy also additively increased IFN-γ production on NK cells. GzmB production was enhanced by combination therapy or PD-1 blockade, but not HSA-IL21 alone (Figures 3G,H). The expression of CD69, a marker for activated and tissue resident T cells (Radulovic et al., 2013), was additively increased in Foxp3- CD4+ T cells, Tregs, CD8+ T cells, and NK cells following combination therapy. However, the expression of CD103, another marker for tissue resident T cells (Topham and Reilly, 2018), was unchanged. (Figures 3B,J–Q). Overall, HSA-IL21/PD-1 blockade combination therapy increases TIL effector function.
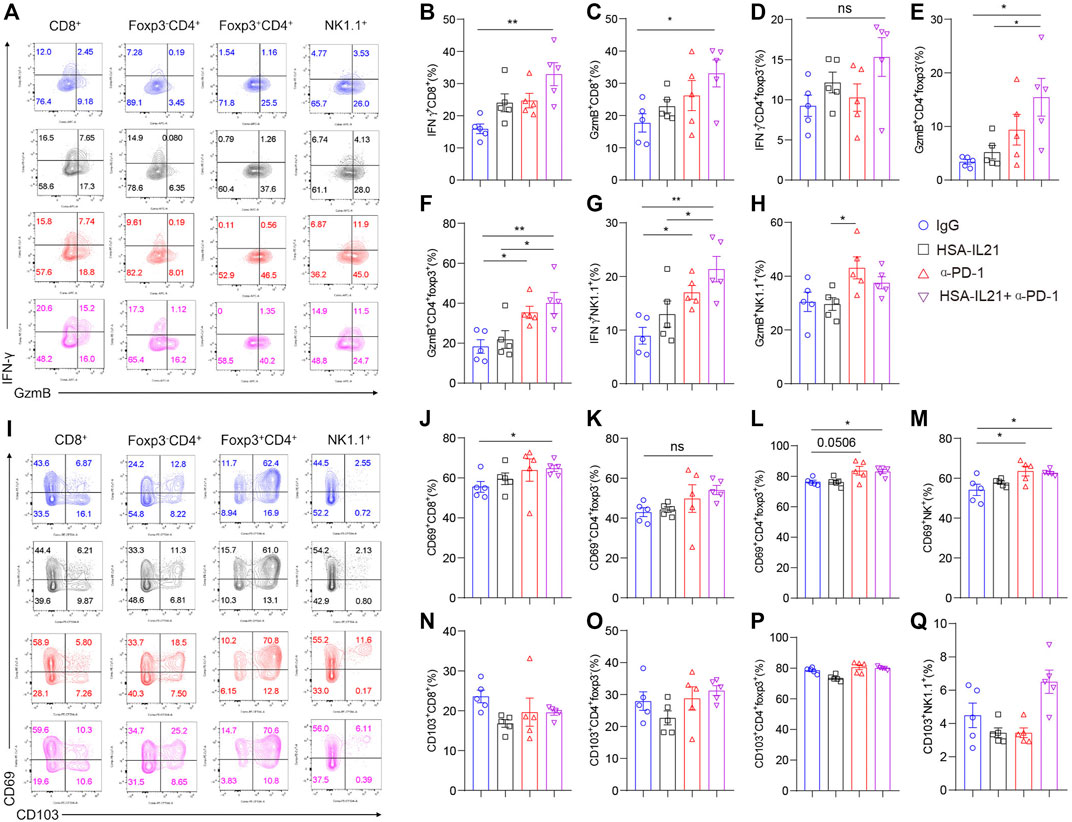
FIGURE 3. Combination therapy enhances the effector function of tumor-infiltrating lymphocytes. (A–Q) MC38 tumor cells (1×106) were injected intradermally to B6 mice, 9 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, HSA-IL21/α-PD-1, 96 h later, tumors were resected and analyzed by flow cytometry. (A and I) Representative flow plots showing IFN-γ, GzmB, CD69 and CD103 staining in CD8+, Foxp3-CD4+, Foxp3+CD4+ and NK cells. (B–H) Quantitative percentage of IFN-γ and GzmB expression in CD8+, Foxp3-CD4+, Foxp3+CD4+ T and NK cells. Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test was performed. (J–Q) Quantitative percentage of CD69 expression in CD8+, Foxp3-CD4+, Foxp3+CD4+ T and NK cells. Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA was performed.
Combination Therapy Induces Expression of Immune Checkpoint Molecules on Tumor-Infiltrating Lymphocytes
Since the expression of immune checkpoint molecules characterizes T and NK cell exhaustion and may limit the efficacy of combination therapy (Wherry, 2011; Wherry and Kurachi, 2015; Yang et al., 2020; Moesta et al., 2020; Bastid et al., 2015; Zhang et al., 2019; Sade-Feldman et al., 2019; Haas and Obenauf, 2019; Sun et al., 2021), we next looked at the expression of checkpoint molecules Tim-3, Lag-3, and CD39 by multi-color flow cytometry. The expression of all three inhibitory receptors was significantly enhanced on Foxp3-CD4+ T cells, Tregs, CD8+ T cells, and NK cells after combination therapy (Figures 4A–N). In addition, the fraction of Tim-3+CD39+ CD8+ T cells was significantly enhanced after combination treatment (Figures 4J,O–R). These results indicate that HSA-IL21/PD-1 blockade combination therapy may drive TILs into a hyperactivated state that is controlled by multiple immune checkpoint molecules (Yang et al., 2020).
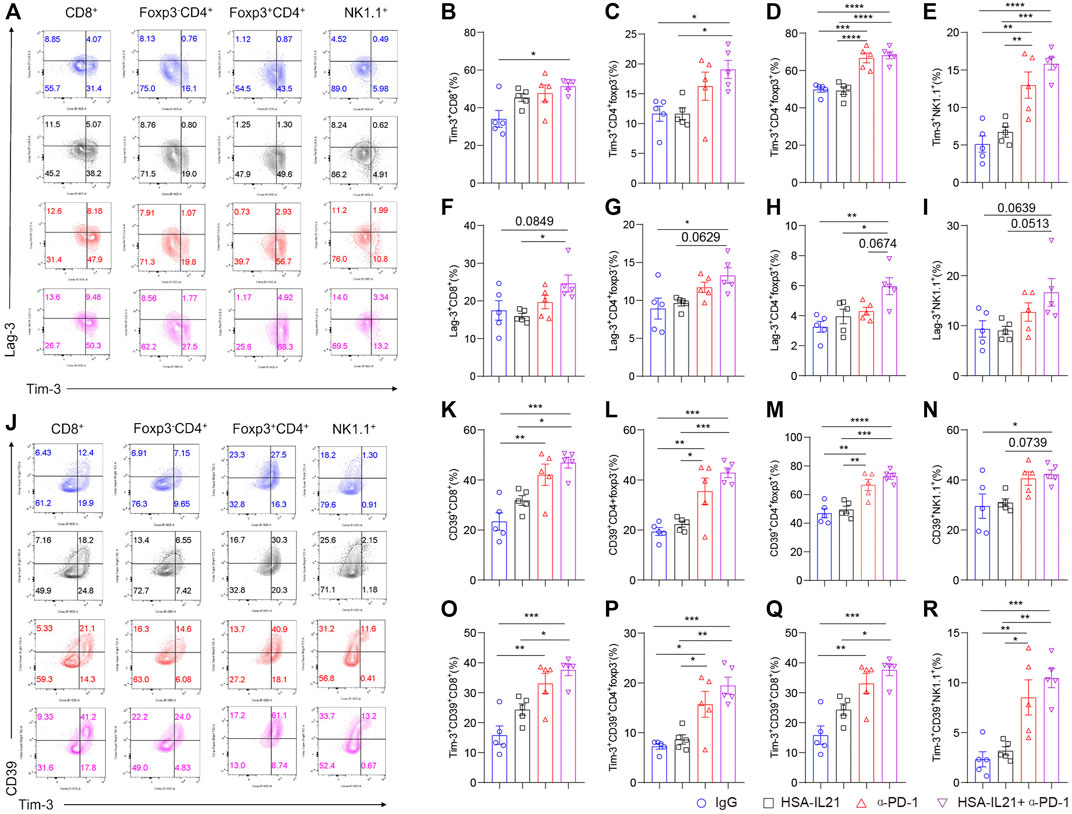
FIGURE 4. Combination therapy induces expression of immune checkpoint molecules on tumor-infiltrating lymphocytes. (A–I) MC38 tumor cells (1 × 106) were injected intradermally to B6 mice, 9 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, HSA-IL21/α-PD-1, 96 h later, tumors were resected and analyzed by flow cytometry. (A) Representative flow plots showing Tim-3 and Lag-3 staining in CD8+, Foxp3-CD4+, Foxp3+CD4+ T and NK cells. (B–I) Quantitative percentage of Tim-3 and Lag-3 expression in CD8+, Foxp3-CD4+, Foxp3+CD4+ T and NK cells. Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test was performed. (J) Representative flow plots showing Tim-3 and CD39 staining in CD8+, Foxp3-CD4+, Foxp3+CD4+ and NK cells. (K–R) Quantitative percentage of CD39 and CD39/Tim-3 double positive expression in CD8+, Foxp3-CD4+, Foxp3+CD4+ T and NK cells. Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test was performed.
Combination Therapy Promoted DC1 and M1 Cells and Decreased DC2 and M2 Cells in the TME
We next investigated whether combination therapy affected the myeloid compartment of the TME using multi-color flow cytometry. We found an increase in the fraction of DC1 and M1 macrophages in the immune compartment following combination therapy (Figures 5A–E). In contrast, the fraction of M2 macrophages and DC2 was decreased. These results are consistent with the observed increases in the CD8+ T cell-mediated immune response. Interestingly, we found that combination therapy increases the fraction of immunosuppressive MDSCs in the TME (Figures 5A,F,G). These data show that the myeloid compartment mainly promotes the anti-tumor lymphocyte response following combination therapy, but that some immunosuppression also occurs via MDSCs.
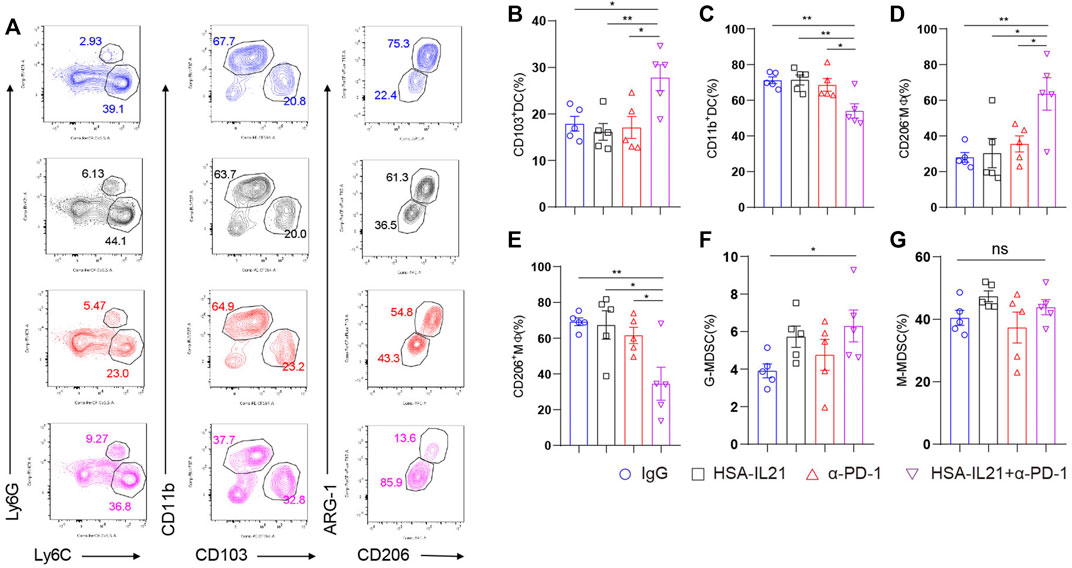
FIGURE 5. Combination therapy induced myeloid cells that promote the CD8+ T cell-mediated immune response against cancer. (A–G) MC38 tumor cells (1 × 106) were injected intradermally to B6 mice, 9 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, HSA-IL21/α-PD-1, 96h later, tumors were resected and analyzed by flow cytometry. Representative flow plots showing the percentage of myeloid populations. (B–G) Quantitative percentage of tumor infiltrating dendritic cells, macrophage, MDSC. Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test was performed.
Combination Therapy Promotes the Tumor-antigen-specific T Cell Response in Peripheral Lymphoid Organs
We also examined T cells in the spleen to determine changes in the peripheral lymphoid organs following combination therapy. We found no difference in the percentages of Foxp3- CD4+ T cells and CD8+ T cells. However, combination therapy additively decreased the fraction of naïve CD62L+ CD8+ and CD62L+ Foxp3- CD4+T cells (Figures 6A–C). The fraction of CD62L-CD44-CD8+ or CD62L-CD44-Foxp3-CD4+ T cells were increased in the combination therapy group (Figures 6A,D,E). There data suggest that the combined treatment with HSA-IL21 and PD-1 mAbs resulted in a systemic decrease in naive T cells. In order to further determine tumor-antigen-specific T cells, we performed the enzyme-linked immunosorbent spot (ELISpot) assay. We found that tumor antigen-specific effector T cells in the spleen were significantly increased upon combination therapy (Figure 6F). These data show that HSA-IL21/PD-1 mAbs combination therapy leads to both systemic T cell activation and an increase in the number of tumor-antigen-specific T cells in the peripheral lymphoid organs.
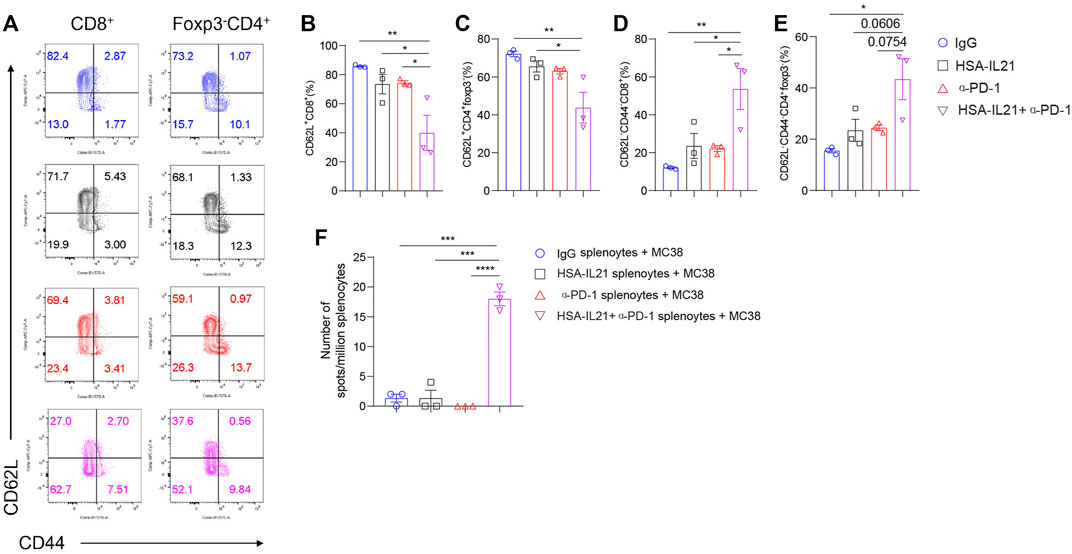
FIGURE 6. Combination therapy promotes the tumor-antigen-specific T cell response activation in peripheral lymphoid organs. (A–F) MC38 tumor cells (1 × 106) were injected intradermally to B6 mice, 9 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, HSA-IL21/α-PD-1, 96 h later, immune cells from spleen were analyzed by flow cytometry. Splenocytes were applied to IFN-γ ELISpot assay. (A) Representative flow plots showing CD62L and CD44 staining on CD8+ and Foxp3-CD4+T cells in spleen. Quantitative expression of CD44, CD62L expression on T cells gated on CD8+ (B–D) and Foxp3-CD4+T cells (E) in spleen. Data were presented as mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA test was performed. (F) Numbers of tumor-antigen specific IFN-γ producers in the splenocytes. Data were presented as mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA test was performed.
Combination Therapy Sustains Anti-Tumor Immune Responses in TME and Periphery
Given the strong anti-tumor immune response induced by combination therapy, we decided to test whether the therapy could sustain these responses overtime. We repeatedly administered of the combination therapy over four times at a 4-day interval, and performed multi-color flow cytometry of the TME (Figure 7). We found moderate increases in the fraction of total immune cells in the TME (Figures 7A,B). Within the immune compartment, we found a significant increase in the fraction of CD8+ T cells in the TME of the IL21 treatment group (Figures 7A,B,D,E). The fraction of CD4+ Foxp3- T cells and Tregs significantly decreased in all treatment groups (Figures 7A,C,F). We also found that combination therapy additively increased the fraction of IFN-γ+ CD4+ T cells, IFN-γ+ CD8+ T cells, GzmB+ CD8+ T cells, and GzmB+ NK cells (Figures 7G–K). The number of tumor antigen-specific T cells in the spleen were also increased upon treatment with combination therapy (Figure 7L). Collectively, these data demonstrate that combination therapy continually promotes the type 1 immune response over time and IL21 in particular greatly increases the tumor-antigen specific T cell in the periphery.
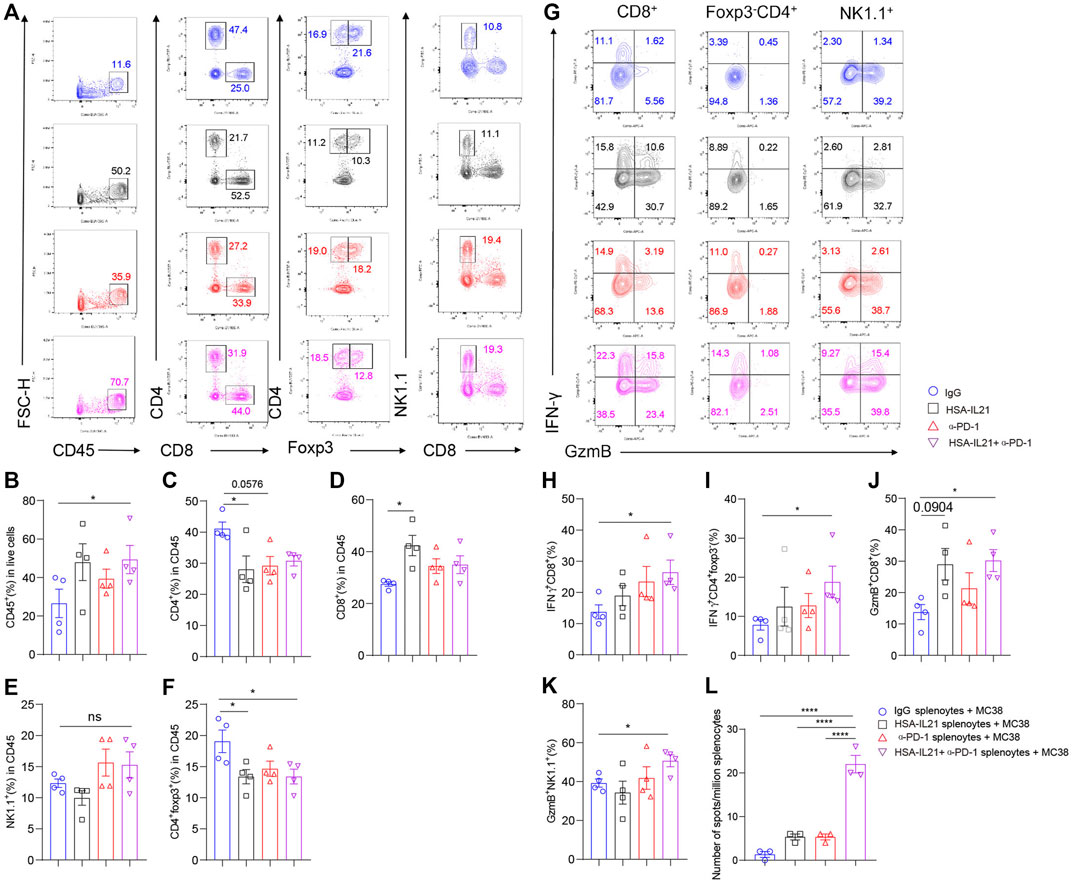
FIGURE 7. Combination therapy sustains anti-tumor immune responses in TME and periphery. (A–L) MC38 tumor cells (1 × 106) were injected intradermally to B6 mice, 5 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, HSA-IL21/α-PD-1 for total 4 times, after last treatment 48h, tumors were resected and analyzed by flow cytometry (n = 4). (A) Representative flow plots showing the percentage of main immune populations. (B–F) Quantitative percentage of tumor infiltrating CD45+ lymphocytes, percentages of CD8+, CD4+, Foxp3-CD4+, Foxp3+CD4+T cells and NK cells. Data were presented as mean ± SEM, n = 4, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test was performed. (G) Representative flow plots showing IFN-γ and GzmB staining in CD8+, Foxp3-CD4+ T cells and NK cells. (H–K) Quantitative percentage of IFN-γ and GzmB expression in CD8+, Foxp3-CD4+T cells and NK cells. Data were presented as mean ± SEM, n = 4, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test were performed. (L) Numbers of tumor-antigen specific IFN-γ producers in the splenocytes. Data were presented as mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA test was performed.
HSA-IL21 Combined With Tim-3, Lag-3, and PD-1 Blockade Additively Inhibits Tumor Growth
Given that HSA-IL21/PD-1 blockade combination therapy increases the expression of checkpoint molecules Tim-3 and Lag-3 expression on TILs, we next determined whether the efficacy the therapy could be improved by combination with ICB therapies targeting Tim-3 and Lag-3. Our results indicate that both HSA-IL21/PD-1/Tim-3 or Lag-3 blockade triple combinations produced a greater antitumor effect without apparent toxicity (Figures 8A–D). Moreover, HSA-IL21/PD-1/Tim-3/Lag-3 blockade quadruple therapy produced an even greater antitumor effect, again without apparent toxicity (Figures 8E,F and Supplementary Figures S3A–C). Our study shows that HSA-IL21 can be combined with multiple checkpoint inhibitors to improve current cancer immunotherapies.
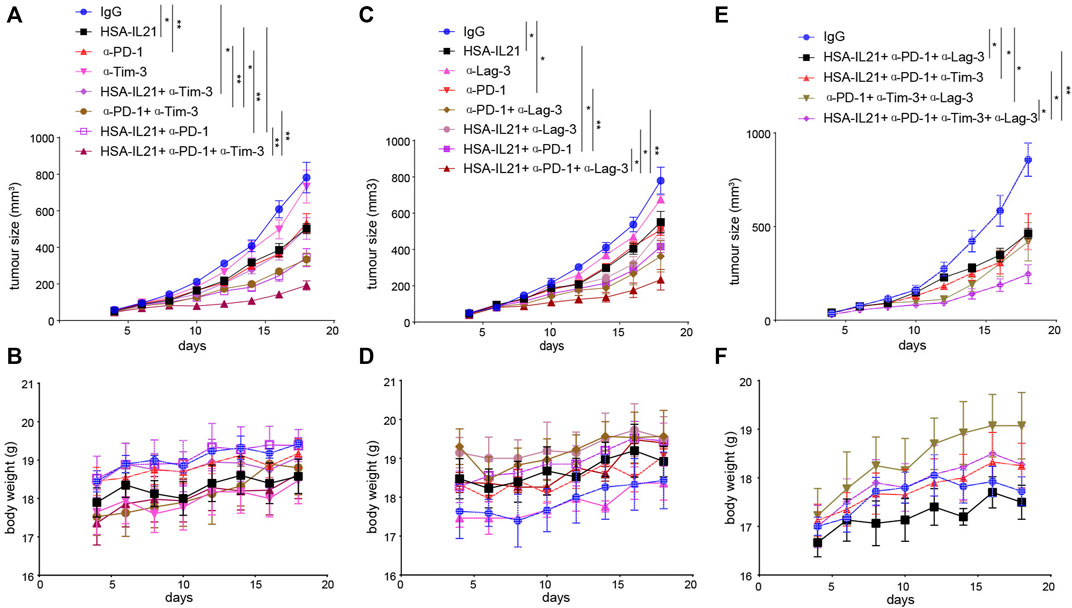
FIGURE 8. HSA-IL21 combined with Tim-3, Lag-3, and PD-1 blockade additively inhibits tumor growth. (A,B) MC38 tumor cells (1×106) were injected intradermally to B6 mice, 5 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, α-Tim-3, HSA-IL21/α-PD-1, HSA-IL21/α-Tim-3, α-PD-1/α-Tim-3, HSA-IL21/α-PD-1/α-Tim-3, for total 4 times. Tumor size (A) and body weight (B) were measured every 2 days. (n = 4–5). Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA test was performed. (C,D) MC38 tumor cells (1×106) were injected intradermally to B6 mice, 5 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21, α-PD-1, α-Lag-3, HSA-IL21/α-PD-1, HSA-IL21/α-Lag-3, α-PD-1/α-Lag-3, HSA-IL21/α-PD-1/α-Lag-3, for total 4 times. Tumor size(C) and body weight(D) were measured every 2 days. (n = 3–5). Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA test were performed. (E–F) MC38 tumor cells (1 × 106) were injected intradermally to B6 mice, 5 days after tumor inoculation, MC38 tumor-bearing mice were treated with IgG, HSA-IL21/α-PD-1/α-Tim-3, HSA-IL21/α-PD-1/α-Lag-3, α-PD-1/α-Tim-3/α-Lag-3, HSA-IL21/α-PD-1/α-Tim-3/α-Lag-3, for total 4 times. Tumor size (E) and body weight (F) were measured every 2 days. (n = 3–5). Data were presented as mean ± SEM, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA test was performed.
Discussion
Our work demonstrates that the half-life-extended HSA-IL21 retains the antitumor effect of WT IL21 and produces superior efficacy when combined with PD-1 blockade in vivo. The cellular mechanisms behind the additive effect of combination therapy involve increases in the fraction and effector functions of CD8, Th1 and NK cells, decreases in the fraction of Treg cells, increases in the fraction of DC1 and M1 macrophages, and increases in the number of tumor-antigen-specific T cells in the peripheral lymphoid organs. The additive effect provided by combination therapy shows that IL21 as a cancer immunotherapeutic is limited by the immune checkpoint molecule PD-1. We found that the effect of IL21 is also limited by multiple checkpoint molecules, including Tim-3 and Lag-3—combination therapies involving blockade of these molecules further improved therapeutic efficacy without causing severe toxicity. We found that the effect of IL21 is further limited by MDSCs. Our findings chart pathways for further improvement of IL21-based therapy.
We found that IL21 and PD-1 blockade alone or in combination act on T and NK cells. Combination therapy additively increases IL21R expression on T cells and NK cells in the TME. This finding suggests that IL21R might be an important molecular hub that integrates the signaling pathways of both IL21 and PD-1 blockade on TILs. We also showed that combination therapy synergistically increases the production of GzmB and IFN-γ by TILs. This is likely due to direct regulation of these genes by IL21 because a previous study showed that GzmB is an IL21 target gene (Shourian et al., 2019). Consistent with our conclusions, this study found that IL21 sustains the cytotoxic functions of CD8 T cells and increases their cytokine secretion capacities (Shourian et al., 2019). It has been shown that IL21 increases the number of central memory T cells and T memory stem cells in vitro (Kasaian et al., 2002; Brady et al., 2004; Zeng et al., 2005; Zhang et al., 2005; Klebanoff et al., 2011; Wölfl et al., 2011; Chen et al., 2018). Here, we found that IL21 increases the number of tumor-antigen-specific T cells in the spleen in vivo. These results show that IL21/PD-1 blockade combination can increase both effector function as well as the number of tumor antigen-specific T cells.
IL21/PD-1 blockade combination therapy activates TILs, but also induces the expression of immune checkpoint molecules Lag-3, Tim-3 and CD39. The strong stimulation provided by IL21 and PD-1 blockade on effector T cells may result in hyperactivation, a state that is characterized by the expression of multiple immune inhibitory receptors. These receptors prevent immune-mediated pathology, but limits the antitumor activity (Alvarez-Fernández et al., 2016; Haas and Obenauf, 2019; Sade-Feldman et al., 2019; Sun et al., 2021). We showed that triple combination therapy with Tim-3 or Lag-3, and quadruple therapy, further increases the efficacy of double therapy. Combination therapies should be tested in the clinic to further improve patient outcomes.
We showed that HSA-IL21/PD-1 blockade combination therapy increases the fraction of DC1 cells in the TME. Consistent with this finding, we observed an increase in the fraction and function of CD8+ T cells. Our results on DC1 contrast previous studies showing that IL21 has a potent inhibitory effect on DCs(Brandt et al., 2003; Wan et al., 2013). One study showed that addition of IL21 during generation of mouse bone marrow-derived DCs (BMDCs) reduces the expression of major histocompatibility complex II (MHCII) and the ability to induce antigen-specific CD4+ T cell proliferation (Brandt et al., 2003). IL21 added during lipopolysaccharide (LPS) stimulation inhibits DC activation and maturation, as well as the production of proinflammatory cytokines IL-1β, IL-12, IL-6, and tumor necrosis factor α (TNF-α). Another study showed that IL21 induces apoptosis of splenic conventional dendritic cells (cDCs) via induction of Bim (Wan et al., 2013). Consistent with these results, we also found that combination therapy decreases the fraction of DC2s in the TME. IL21 might act on DC2s in order to decrease the fraction of CD4+ Foxp3- T cells or Tregs in the TME. Our findings suggest that IL21 differentially acts on DC1s vs DC2s to promote the CD8+ T cell-mediated anti-tumor immune response.
Consistent with activation of CD8+, Th1 cells, and NK cells, we found that combination therapy increased the fraction of M1 macrophages and decreased the fraction of M2 macrophages in the TME. Again, we found increased the effector function of CD4+ Foxp3- T cells, CD8+ T cells, and NK cells. Combination therapy primarily shifts the immune response to a type 1 phenotype that is anti-tumor. However, we also observed increases in the fraction of immunosuppressive mMDSCs and nMDSCs out of all immune cells. The mechanism by which IL21 acts on MDSCs requires further work to be uncovered. Nonetheless, our findings highlight the opportunity to combine IL21 with immunotherapies that target MDSCs.
Recombinant IL21 as a single agent has been evaluated in clinical trials with encouraging efficacy. However, there are no published results about combination therapies using both IL21 and ICB in human cancer patients. The requirement for frequent dosing might be an obstacle to the success of IL21-based therapies in cancer patients. Our study demonstrates that the half-life extended HSA-IL21 has potential to be combined with existing ICB therapies as well as multiple additional checkpoint inhibitors in the clinic.
Materials and Methods
Mouse
C57BL/6J and BALB/c mice were purchased from The Jackson Laboratory and housed in the specific pathogen-free animal facility of University of Pittsburgh School of Medicine. All mice experiments have been approved by Institutional Animal Care and Use Committee of University of Pittsburgh.
Cell Culture and Tumor Model
MC38 tumor cell line was cultured in complete DMEM medium plus with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P.S). CT26 cancer cells are transfected with plasmid coding MSLN ORF, and selected by surface marker MSLN using fluorescenec-activated cell sorting (FACS). CT26-MSLN tumor cell line was cultured in complete DMEM medium plus with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P.S). For MC38 tumor model, C57BL/6 mice were injected with 1 million cells intradermally (i.d.). For CT26-MSLN tumor model, 1 million cells were injected intradermally (i.d.) into BALB/c mice. MC38 and CT26 bearing mice were randomized into four treatment cohorts: control IgG, HSA-IL21, α-PD-1 (clone J43, BioXCell) or HSA-IL21/α-PD-1. HSA-IL21 was injected by intraperitoneally (i.p.) 25 μg per mouse, α-PD-1 were injected by intraperitoneally (i.p.) 200 μg per mouse. All mice were administered on the 5th day after tumor inoculation. Tumor sizes were monitored every 2–3 days, and the tumor volume was calculated as L× W2/2.
Reagents and Antibodies
HSA-IL21 (the half-life extended IL21 was kindly provided by Anwita Biosciences, CA, USA), PD-1mAb (clone:J43), hamster IgG were purchased from Bioxcell company (catalog no. BE0091) for tumor therapy. For Flow cytometry, CD45 (clone: 30-F11), CD4 (clone: GK1.5), CD8 (clone: 53–6.7), NK1.1 (clone: PK136), B220 (clone: RA3-6B2), Foxp3 (MF-14), PD-1(29F.1A12), Tim-3 (clone: RMT3-23), Lag-3 (clone: C9B7W), CD39 (clone: 24DMS1), CD62L (clone: MEL-14), CD44 (clone: IM7), CD103 (clone: M290), CD69 (clone: H1.2F3), IFN-γ (clone: XMG1.2), GzmB (clone: QA16A02), CD11b (clone: M1/70), Ly6C (clone: HK1.4), Ly6G (clone: 1A8), Gr-1 (clone: RB6-8C5), CD24 (clone: M1/69), F4/80 (clone: BM8), CD11C (clone: N418), CD206 (clone: MR6F3), Arginase 1 (clone: A1exF5) were purchased from Biolegend ebioscience or BD Bioscience. Zombie NIR dye was purchased from Biolegend.
Processing of Tissues and Flow Cytometry
Mice were sacrificed, TDLN, spleen and tumors were removed. We placed the spleens and lymph nodes between the frosted surfaces of two glass slides and applied force to disrupt these organs to release immune cells, spleen was need to treated with ACK lysis buffer to remove red blood cells. Single-cell suspensions were filtered through a 40-μm cell strainer, washed, and resuspended in 1%FBS HBS for analysis. Tumor were cut into small pieces, digested in serum free RPMI with 0.25 mg/ml Liberase TL (Roche) and 0.33 mg/ml Dnase 1 (Sigma) in 37° for 30 min. Single-cell suspensions were filtered through a 40-μm cell strainer, washed, and resuspended in 1%FBS HBS for staining. For IFN-γ or Granzyme B staining, tumor cells were stimulated with leukocyte activation cocktail (Biolegend) for 6 h, then stained surface marker and intracellular markers. by the standard staining protocol described before (Chen et al., 2020). Flow cytometry analysis were applied to LSRII or Aurora (Cytek Biosciences) and analyzed by using Flowjo software (BD).
IFN-γ ELISpot Assay
15 μg/ml capture antibody anti-IFN-γ (clone:AN18, MabTech) was coated and incubated overnight at 4°. On the other day, 5*105 splenocytes were co-cultured with 5*104, 200Gy irradiated MC38 tumor cells at 37° incubator. Forty-8 hours later, Wash the plate 5 times with wash buffer (PBS/0.05% Tween 20) and incubated with 1.5 μg/ml biotinylated secondary antibody (clone: R4-6A2-biotin, MabTech) for 1 h, and then washed and developed with VECTASTAIN Elite ABC HRP Kit (Vector Labs) and incubated with AEC Peroxidase (HRP) Substrate Kit (Vector Labs). The plate was read and counted using the ImmunoSpot Analyzer (Cellular Technology).
Single-Cell RNA-Seq Data Processing
Zhang et al. (2020) scRNA-seq data of MC38 tumor downloaded from ENA website (ArrayExpress: E-MTAB-8832) was aligned and quantified using the Cellranger Software (Version 4.0.0) against the mm10 mouse reference genome. The preliminary filtered data generated from Cellranger were used for a Seurat object created by the R package Seurat (Version 3.2.3). Doublets were removed by DoubletFinder package. Further quality control was applied to cells based on four metrics step by step, including the total UMI count, number of detected genes and proportion of mitochondrial gene count per cell, and proportion of ribosomal gene count per cell. Specifically, cells with more than 50,000 UMI count and 10% mitochondrial gene count were filtered, as well as cells with more than 50% ribosomal gene count.
Integration of Multiple scRNA-Seq, Dimension Reduction and Unsupervised Clustering
Single cell data were processed for dimension reduction and unsupervised clustering by following the workflow in Seurat. In brief, 2,000 highly-variable genes were selected for downstream analysis by using FindVariableFeatures function with parameter “nfeatures = 2000.” Subsequently, IntegrateData function was used to integrate data and create a new matrix with 3,000 features, in which potential batch effect was regressed out. To reduce the dimensionality of the scRNA-seq dataset, principal component analysis (PCA) was performed on an scaled integrated data matrix. With ElbowPlot function of Seurat, top 40 PCs were used to perform the downstream analysis. The main cell clusters were identified with the FindClusters function offered by Seurat, with resolution set as default (res = 0.2). And then they were visualized with 2D UMAP plots. Conventional markers described in a previous study were used to categorize every cell into a known biological cell type.
Statistical Analysis
We used the one-way ANOVA test for comparisons between different treatment groups. Two-way ANOVA was used for comparing tumor growth curves. Statistical analyses were performed with Graphpad Prism.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by the Institution Animal Care and Use Committee at University of Pittsburgh.
Author Contributions
SW and RS designed the experiments, JZ and BL provided key reagents, SW, RS, WZ, and BC performed the experiments, SW, DG, BT, and HH analyzed the data. SW, JJ, and BL wrote the manuscript. All authors contributed to the article and approved.
Funding
This project is funded by a grant from Anwita Bioscences Inc. (to BL).
Conflict of Interest
BL serves as a scientific adviser and owns stocks of Anwita Biosciences Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.779865/full#supplementary-material
Supplementary Figure S1 | Characterization of IL21R+ peripheral immune cells. (A) Representative flow cytometry gating strategy for identification of immune cell population. (Foxp3-CD4+: CD45+CD4+Foxp3-, Foxp3+CD4+: CD45+CD4+Foxp3+, CD8+: CD45+CD8+, NK cell: CD45+CD8-CD4-NK1.1+, B cells: CD45+CD8-CD4-B220+, CD103+DC: CD45+Gr1-MHCII+CD24+CD103+, CD103-DC: CD45+Gr1-MHCII+CD24+CD103-CD11b+, MDSC: CD45+CD11b+Gr1+, CD206-Mφ: CD45+Gr1-MHCII+F4/80+CD206-Arg-1-, CD206+Mφ: CD45+Gr1-MHCII+F4/80+CD206+Arg-1+, M-MDSC: CD45+CD11b+Ly6c+, G-MDSC: CD45+CD11b+Ly6G+). (B,C) Representative flow plots showing IL21R staining on CD8+, CD4+, Foxp3-CD4+, Foxp3+CD4+T cells and NK cells in TDLN (B) and spleen (C).
Supplementary Figure S2 | Administration of HSA-IL21 and PD-1 mAbs additively inhibited tumor growth. (A) CT26-MSLN tumor cells (1×106) were injected intradermally to BALB/c mice, monitored tumor volume of tumorbearing mice treated with IgG, IL21, HSA-IL21, α-PD-1, HSA-IL21/α-PD-1. (B) Individual tumor curves of mice depicted in Figure S2A. Data were presented as mean±SEM, n=5, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, two-way ANOVA test were performed. (C,D) MC38 tumor cells (1×106) were injected intradermally to B6 mice, monitored tumor volume (C) and individual tumor curves (D) of tumor-bearing mice treated with IgG, rIL21, HAS-IL21. Data were presented as mean±SEM, n=5, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, twoway ANOVA test were performed.
Supplementary Figure S3 | Administration with Tim-3 or Lag-3, PD-1 mAbs and HSA-IL21 additively inhibited the tumor growth. (A–C) Individual tumor curves of mice depicted in Figures 8A,C,E.
References
Alvarez-Fernández, C., Escribà-Garcia, L., Vidal, S., Sierra, J., and Briones, J. (2016). A Short CD3/CD28 Costimulation Combined with IL-21 Enhance the Generation of Human Memory Stem T Cells for Adoptive Immunotherapy. J. Transl. Med. 14 (1), 214. doi:10.1186/s12967-016-0973-y
Attridge, K., Wang, C. J., Wardzinski, L., Kenefeck, R., Chamberlain, J. L., Manzotti, C., et al. (2012). IL-21 Inhibits T Cell IL-2 Production and Impairs Treg Homeostasis. Blood 119 (20), 4656–4664. doi:10.1182/blood-2011-10-388546
Bastid, J., Regairaz, A., Bonnefoy, N., Déjou, C., Giustiniani, J., Laheurte, C., et al. (2015). Inhibition of CD39 Enzymatic Function at the Surface of Tumor Cells Alleviates Their Immunosuppressive Activity. Cancer Immunol. Res. 3 (3), 254–265. doi:10.1158/2326-6066.cir-14-0018
Bhatia, S., Curti, B., Ernstoff, M. S., Gordon, M., Heath, E. I., Miller, W. H., et al. (2014). Recombinant Interleukin-21 Plus Sorafenib for Metastatic Renal Cell Carcinoma: a Phase 1/2 Study. J. Immunother. Cancer 2, 2. doi:10.1186/2051-1426-2-2
Brady, J., Hayakawa, Y., Smyth, M. J., and Nutt, S. L. (2004). IL-21 Induces the Functional Maturation of Murine NK Cells. J. Immunol. 172 (4), 2048–2058. doi:10.4049/jimmunol.172.4.2048
Brahmer, J. R., Tykodi, S. S., Chow, L. Q. M., Hwu, W.-J., Topalian, S. L., Hwu, P., et al. (2012). Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 366 (26), 2455–2465. doi:10.1056/nejmoa1200694
Brandt, K., Bulfone-Paus, S., Foster, D. C., and Rückert, R. (2003). Interleukin-21 Inhibits Dendritic Cell Activation and Maturation. Blood 102 (12), 4090–4098. doi:10.1182/blood-2003-03-0669
Chae, Y. K., Arya, A., Iams, W., Cruz, M. R., Chandra, S., Choi, J., et al. (2018). Current Landscape and Future of Dual Anti-CTLA4 and PD-1/PD-L1 Blockade Immunotherapy in Cancer; Lessons Learned from Clinical Trials with Melanoma and Non-small Cell Lung Cancer (NSCLC). J. Immunother. Cancer 6 (1), 39. doi:10.1186/s40425-018-0349-3
Chen, L., Sun, R., Xu, J., Zhai, W., Zhang, D., Yang, M., et al. (2020). Tumor-Derived IL33 Promotes Tissue-Resident CD8+ T Cells and Is Required for Checkpoint Blockade Tumor Immunotherapy. Cancer Immunol. Res. 8 (11), 1381–1392. doi:10.1158/2326-6066.cir-19-1024
Chen, Y., Yu, F., Jiang, Y., Chen, J., Wu, K., Chen, X., et al. (2018). Adoptive Transfer of Interleukin-21-Stimulated Human CD8+ T Memory Stem Cells Efficiently Inhibits Tumor Growth. J. Immunother. 41 (6), 274–283. doi:10.1097/cji.0000000000000229
Clough, L. E., Wang, C. J., Schmidt, E. M., Booth, G., Hou, T. Z., Ryan, G. A., et al. (2008). Release from Regulatory T Cell-Mediated Suppression during the Onset of Tissue-specific Autoimmunity Is Associated with Elevated IL-21. J. Immunol. 180 (8), 5393–5401. doi:10.4049/jimmunol.180.8.5393
Davis, I. D., Brady, B., Kefford, R. F., Millward, M., Cebon, J., Skrumsager, B. K., et al. (2009). Clinical and Biological Efficacy of Recombinant Human Interleukin-21 in Patients with Stage IV Malignant Melanoma without Prior Treatment: A Phase IIa Trial. Clin. Cancer Res. 15 (6), 2123–2129. doi:10.1158/1078-0432.ccr-08-2663
Davis, I. D., Skrumsager, B. K., Cebon, J., Nicholaou, T., Barlow, J. W., Moller, N. P. H., et al. (2007). An Open-Label, Two-Arm, Phase I Trial of Recombinant Human Interleukin-21 in Patients with Metastatic Melanoma. Clin. Cancer Res. 13 (12), 3630–3636. doi:10.1158/1078-0432.ccr-07-0410
Deng, S., Sun, Z., Qiao, J., Liang, Y., Liu, L., Dong, C., et al. (2020). Targeting Tumors with IL-21 Reshapes the Tumor Microenvironment by Proliferating PD-1intTim-3-CD8+ T Cells. JCI Insight 5 (7), e132000. doi:10.1172/jci.insight.132000
Di Carlo, E., Comes, A., Orengo, A. M., Rosso, O., Meazza, R., Musiani, P., et al. (2004). IL-21 Induces Tumor Rejection by Specific CTL and IFN-γ-Dependent CXC Chemokines in Syngeneic Mice. J. Immunol. 172 (3), 1540–1547. doi:10.4049/jimmunol.172.3.1540
Haas, L., and Obenauf, A. C. (2019). Allies or Enemies-The Multifaceted Role of Myeloid Cells in the Tumor Microenvironment. Front. Immunol. 10, 2746. doi:10.3389/fimmu.2019.02746
Hodi, F. S., O'Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., et al. (2010). Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 363 (8), 711–723. doi:10.1056/nejmoa1003466
Kasaian, M. T., Whitters, M. J., Carter, L. L., Lowe, L. D., Jussif, J. M., Deng, B., et al. (2002). IL-21 Limits NK Cell Responses and Promotes Antigen-specific T Cell Activation. Immunity 16 (4), 559–569. doi:10.1016/s1074-7613(02)00295-9
Klebanoff, C. A., Gattinoni, L., Palmer, D. C., Muranski, P., Ji, Y., Hinrichs, C. S., et al. (2011). Determinants of Successful CD8+ T-Cell Adoptive Immunotherapy for Large Established Tumors in Mice. Clin. Cancer Res. 17 (16), 5343–5352. doi:10.1158/1078-0432.ccr-11-0503
Korn, T., Bettelli, E., Gao, W., Awasthi, A., Jäger, A., Strom, T. B., et al. (2007). IL-21 Initiates an Alternative Pathway to Induce Proinflammatory TH17 Cells. Nature 448 (7152), 484–487. doi:10.1038/nature05970
Lewis, K. E., Selby, M. J., Masters, G., Valle, J., Dito, G., Curtis, W. R., et al. (2017). Interleukin-21 Combined with PD-1 or CTLA-4 Blockade Enhances Antitumor Immunity in Mouse Tumor Models. Oncoimmunology 7 (1), e1377873. doi:10.1080/2162402x.2017.1377873
Li, X., Shao, C., Shi, Y., and Han, W. (2018). Lessons Learned from the Blockade of Immune Checkpoints in Cancer Immunotherapy. J. Hematol. Oncol. 11 (1), 31. doi:10.1186/s13045-018-0578-4
Ma, H.-L., Whitters, M. J., Konz, R. F., Senices, M., Young, D. A., Grusby, M. J., et al. (2003). IL-21 Activates Both Innate and Adaptive Immunity to Generate Potent Antitumor Responses that Require Perforin but Are Independent of IFN-γ. J. Immunol. 171 (2), 608–615. doi:10.4049/jimmunol.171.2.608
Martin-Orozco, N., Muranski, P., Chung, Y., Yang, X. O., Yamazaki, T., Lu, S., et al. (2009). T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity 31 (5), 787–798. doi:10.1016/j.immuni.2009.09.014
Moesta, A. K., Li, X.-Y., and Smyth, M. J. (2020). Targeting CD39 in Cancer. Nat. Rev. Immunol. 20 (12), 739–755. doi:10.1038/s41577-020-0376-4
Nurieva, R., Yang, X. O., Martinez, G., Zhang, Y., Panopoulos, A. D., Ma, L., et al. (2007). Essential Autocrine Regulation by IL-21 in the Generation of Inflammatory T Cells. Nature 448 (7152), 480–483. doi:10.1038/nature05969
Peluso, I., Fantini, M. C., Fina, D., Caruso, R., Boirivant, M., MacDonald, T. T., et al. (2007). IL-21 Counteracts the Regulatory T Cell-Mediated Suppression of Human CD4+ T Lymphocytes. J. Immunol. 178 (2), 732–739. doi:10.4049/jimmunol.178.2.732
Petrella, T. M., Tozer, R., Belanger, K., Savage, K. J., Wong, R., Smylie, M., et al. (2012). Interleukin-21 Has Activity in Patients with Metastatic Melanoma: A Phase II Study. J. Clin. Oncol. 30 (27), 3396–3401. doi:10.1200/jco.2011.40.0655
Radulovic, K., Rossini, V., Manta, C., Holzmann, K., Kestler, H. A., and Niess, J. H. (2013). The Early Activation Marker CD69 Regulates the Expression of Chemokines and CD4 T Cell Accumulation in Intestine. PLoS One 8 (6), e65413. doi:10.1371/journal.pone.0065413
Sade-Feldman, M., Yizhak, K., Bjorgaard, S. L., Ray, J. P., de Boer, C. G., Jenkins, R. W., et al. (2019). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 176 (1-2), 404. doi:10.1016/j.cell.2018.12.034
Schmidt, H., Brown, J., Mouritzen, U., Selby, P., Fode, K., Svane, I. M., et al. (2010). Safety and Clinical Effect of Subcutaneous Human Interleukin-21 in Patients with Metastatic Melanoma or Renal Cell Carcinoma: A Phase I Trial. Clin. Cancer Res. 16 (21), 5312–5319. doi:10.1158/1078-0432.ccr-10-1809
Shourian, M., Beltra, J.-C., Bourdin, B., and Decaluwe, H. (2019). Common Gamma Chain Cytokines and CD8 T Cells in Cancer. Semin. Immunol. 42, 101307. doi:10.1016/j.smim.2019.101307
Sivakumar, P. V., Foster, D. C., and Clegg, C. H. (2004). Interleukin-21 Is a T-Helper Cytokine that Regulates Humoral Immunity and Cell-Mediated Anti-tumour Responses. Immunology 112 (2), 177–182. doi:10.1111/j.1365-2567.2004.01886.x
Spolski, R., and Leonard, W. J. (2008). Interleukin-21: Basic Biology and Implications for Cancer and Autoimmunity. Annu. Rev. Immunol. 26, 57–79. doi:10.1146/annurev.immunol.26.021607.090316
Sun, R., Wu, Y., Zhou, H., Wu, Y., Yang, Z., Gu, Y., et al. (2021). Eomes Impedes Durable Response to Tumor Immunotherapy by Inhibiting Stemness, Tissue Residency, and Promoting the Dysfunctional State of Intratumoral CD8+ T Cells. Front. Cel Dev. Biol. 9, 640224. doi:10.3389/fcell.2021.640224
Thompson, J. A., Curti, B. D., Redman, B. G., Bhatia, S., Weber, J. S., Agarwala, S. S., et al. (2008). Phase I Study of Recombinant Interleukin-21 in Patients with Metastatic Melanoma and Renal Cell Carcinoma. J. Clin. Oncol. 26 (12), 2034–2039. doi:10.1200/jco.2007.14.5193
Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott, D. F., et al. (2012). Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 366 (26), 2443–2454. doi:10.1056/nejmoa1200690
Topham, D. J., and Reilly, E. C. (2018). Tissue-Resident Memory CD8+ T Cells: From Phenotype to Function. Front. Immunol. 9, 515. doi:10.3389/fimmu.2018.00515
Ugai, S.-i., Shimozato, O., Kawamura, K., Wang, Y.-Q., Yamaguchi, T., Saisho, H., et al. (2003). Expression of the Interleukin-21 Gene in Murine colon Carcinoma Cells Generates Systemic Immunity in the Inoculated Hosts. Cancer Gene Ther. 10 (3), 187–192. doi:10.1038/sj.cgt.7700552
Van Belle, T. L., Nierkens, S., Arens, R., and von Herrath, M. G. (2012). Interleukin-21 Receptor-Mediated Signals Control Autoreactive T Cell Infiltration in Pancreatic Islets. Immunity 36 (6), 1060–1072. doi:10.1016/j.immuni.2012.04.005
Wan, C.-K., Oh, J., Li, P., West, E. E., Wong, E. A., Andraski, A. B., et al. (2013). The Cytokines IL-21 and GM-CSF Have Opposing Regulatory Roles in the Apoptosis of Conventional Dendritic Cells. Immunity 38 (3), 514–527. doi:10.1016/j.immuni.2013.02.011
Wherry, E. J., and Kurachi, M. (2015). Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 15 (8), 486–499. doi:10.1038/nri3862
White, L., Krishnan, S., Strbo, N., Liu, H., Kolber, M. A., Lichtenheld, M. G., et al. (2007). Differential Effects of IL-21 and IL-15 on Perforin Expression, Lysosomal Degranulation, and Proliferation in CD8 T Cells of Patients with Human Immunodeficiency Virus-1 (HIV). Blood 109 (9), 3873–3880. doi:10.1182/blood-2006-09-045278
Wölfl, M., Merker, K., Morbach, H., Van Gool, S. W., Eyrich, M., Greenberg, P. D., et al. (2011). Primed Tumor-Reactive Multifunctional CD62L+ Human CD8+ T Cells for Immunotherapy. Cancer Immunol. Immunother. 60 (2), 173–186. doi:10.1007/s00262-010-0928-8
Xu, M., Liu, M., Du, X., Li, S., Li, H., Li, X., et al. (2015). Intratumoral Delivery of IL-21 Overcomes Anti-Her2/Neu Resistance through Shifting Tumor-Associated Macrophages from M2 to M1 Phenotype. J. Immunol. 194 (10), 4997–5006. doi:10.4049/jimmunol.1402603
Xue, D., Hsu, E., Fu, Y.-X., and Peng, H. (2021). Next-generation Cytokines for Cancer Immunotherapy. Antib Ther. 4 (2), 123–133. doi:10.1093/abt/tbab014
Yang, M., Du, W., Yi, L., Wu, S., He, C., Zhai, W., et al. (2020). Checkpoint Molecules Coordinately Restrain Hyperactivated Effector T Cells in the Tumor Microenvironment. Oncoimmunology 9 (1), 1708064. doi:10.1080/2162402x.2019.1708064
Zander, R., Schauder, D., Xin, G., Nguyen, C., Wu, X., Zajac, A., et al. (2019). CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51 (6), 1028–1042. doi:10.1016/j.immuni.2019.10.009
Zeng, R., Spolski, R., Finkelstein, S. E., Oh, S., Kovanen, P. E., Hinrichs, C. S., et al. (2005). Synergy of IL-21 and IL-15 in Regulating CD8+ T Cell Expansion and Function. J. Exp. Med. 201 (1), 139–148. doi:10.1084/jem.20041057
Zhang, H., Vijayan, D., Li, X.-Y., Robson, S. C., Geetha, N., Teng, M. W. L., et al. (2019). The Role of NK Cells and CD39 in the Immunological Control of Tumor Metastases. Oncoimmunology 8 (6), e1593809. doi:10.1080/2162402x.2019.1593809
Zhang, L., Li, Z., Skrzypczynska, K. M., Fang, Q., Zhang, W., O’Brien, S. A., et al. (2020). Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181 (2), 442–459. doi:10.1016/j.cell.2020.03.048
Zhang, Y., Joe, G., Hexner, E., Zhu, J., and Emerson, S. G. (2005). Host-reactive CD8+ Memory Stem Cells in Graft-Versus-Host Disease. Nat. Med. 11 (12), 1299–1305. doi:10.1038/nm1326
Zhong, Z., Ye, F., Siegel, M., Huang, J., Liao, E., and Li, E. (2020). Albumin Binding Antibodies and Use Thereof. International Patent Publication Number: WO 2020/172528A1. Available at: https://patents.google.com/patent/WO2020172528A1/en Accessed August 27, 2020.
Keywords: interleukin 21, checkpoint inhibitors, tumor microenvironment, immunotherapy, mechanisms
Citation: Wu S, Sun R, Tan B, Chen B, Zhou W, Gao DS, Zhong J, Huang H, Jiang J and Lu B (2021) The Half-Life-Extended IL21 can Be Combined With Multiple Checkpoint Inhibitors for Tumor Immunotherapy. Front. Cell Dev. Biol. 9:779865. doi: 10.3389/fcell.2021.779865
Received: 20 September 2021; Accepted: 29 October 2021;
Published: 15 November 2021.
Edited by:
Qingqing Wang, Zhejiang University School of Medicine, ChinaReviewed by:
Weiguo Cui, Bloodcenter of Wisconsin, United StatesKarthik Dhatchinamoorthy, University of Massachusetts Medical School, United States
Zhinan Yin, Jinan University, China
Copyright © 2021 Wu, Sun, Tan, Chen, Zhou, Gao, Zhong, Huang, Jiang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingting Jiang, amlhbmdqaW5ndGluZ0BzdWRhLmVkdS5jbg==; Binfeng Lu, YmluZmVuZ0BwaXR0LmVkdQ==
†These authors have contributed equally to this work