 Tinghui Shao1
Tinghui Shao1 Mingming Fang
Mingming Fang- 1Key Laboratory of Targeted Intervention of Cardiovascular Disease and Collaborative Innovation Center for Cardiovascular Translational Medicine, Department of Pathophysiology, Nanjing Medical University, Nanjing, China
- 2Center for Experimental Medicine, Jiangsu Health Vocational College, Nanjing, China
- 3Institute of Biomedical Research, Liaocheng University, Liaocheng, China
Cardiac fibrosis is a key pathophysiological process that contributes to heart failure. Cardiac resident fibroblasts, exposed to various stimuli, are able to trans-differentiate into myofibroblasts and mediate the pro-fibrogenic response in the heart. The present study aims to investigate the mechanism whereby transcription of chloride channel accessory 2 (Clca2) is regulated in cardiac fibroblast and its potential implication in fibroblast-myofibroblast transition (FMyT). We report that Clca2 expression was down-regulated in activated cardiac fibroblasts (myofibroblasts) compared to quiescent cardiac fibroblasts in two different animal models of cardiac fibrosis. Clca2 expression was also down-regulated by TGF-β, a potent inducer of FMyT. TGF-β repressed Clca2 expression at the transcriptional level likely via the E-box element between −516 and −224 of the Clca2 promoter. Further analysis revealed that Twist1 bound directly to the E-box element whereas Twist1 depletion abrogated TGF-β induced Clca2 trans-repression. Twist1-mediated Clca2 repression was accompanied by erasure of histone H3/H4 acetylation from the Clca2 promoter. Mechanistically Twist1 interacted with HDAC1 and recruited HDAC1 to the Clca2 promoter to repress Clca2 transcription. Finally, it was observed that Clca2 over-expression attenuated whereas Clca2 knockdown enhanced FMyT. In conclusion, our data demonstrate that a Twist1-HDAC1 complex represses Clca2 transcription in cardiac fibroblasts, which may contribute to FMyT and cardiac fibrosis.
Introduction
Cardiac fibrosis is generally considered an adaptive response to adversarial stimuli when the heart is exposed to various injuries. A specialized cell type termed “myofibroblast,” typically absent from the normal myocardium under physiological conditions, emerges following injuries and mediates the fibrogenic response (Tomasek et al., 2002). On the one hand, myofibroblasts are capable of muscle-like contraction owing to the acquisition of the expression of genes encoding contractile proteins (e.g., α-SMA). Contraction by myofibroblasts facilitates wound healing and prevents the incidence of cardiac rupture (Talman and Ruskoaho, 2016). On the other hand, myofibroblasts produce multiple extracellular matrix proteins (e.g., type I collagen, type III collagen, fibronectin) to promote ventricular remodeling and maintain myocardial integrity (van den Borne et al., 2010). After the recession of injurious stimuli, myofibroblasts are no longer needed and thus must be removed or resolved to terminate cardiac fibrogenesis. On the contrary, persistent presence of myofibroblasts in the heart or failure of resolution often leads to aberrant and averse cardiac remodeling and increased rigidity of the myocardium dampening heart function. In fact, cardiac fibrosis is frequently observed and associated with poor diagnosis in patients with heart failure (Gonzalez et al., 2018).
The origin of myofibroblasts in the stressed heart was a subject matter of great controversy and remained elusive prior to the development and utilization of genetic lineage tracing technique. It has been proposed that microvascular endothelial cells (Zeisberg et al., 2007), epicardial epithelial cells (Zhou et al., 2010), myelocytic fibrocyte (Mollmann et al., 2006), and perivascular mesenchymal cells (Kramann et al., 2015) may trans-differentiate into myofibroblasts in vitro and/or in vivo under different conditions. Landmark studies from the Molkentin laboratory (Kanisicak et al., 2016) and the Evans laboratory (Moore-Morris et al., 2014), aided by lineage tracing, have unequivocally demonstrated that cardiac resident fibroblasts are the predominant source of mature myofibroblasts and become the effector cell type of cardiac fibrosis following cardiac injury via fibroblast-myofibroblast transition (FMyT). Further analysis has revealed that cardiac myofibroblasts can be labeled by periostin (encoded by postn), a matricellular protein that can function as a ligand for integrins to promote cell migration (Stempien-Otero et al., 2016). One of the most convincing pieces of evidence that supports the pivotal role of myofibroblasts in cardiac fibrosis is provided by Kaur et al. (2016) who demonstrate that elimination of periostin-positive cells (mature myofibroblasts), by diphtheria toxin mediated killing, abrogates aberrant fibrogenic response and preserves heart function after myocardial infarction. Despite these advances, many transcriptional events taking place during FMyT remain to be investigated in detail.
Chloride channel accessory 2 (Clca2) belongs to the family of calcium sensitive chloride conductance proteins or regulators (Jentsch and Pusch, 2018). Clca2 plays versatile pathophysiological roles by regulating multiple distinct yet interconnected cellular processes including proliferation (Walia et al., 2009), differentiation (Ramena et al., 2016), migration (Sasaki et al., 2012), and apoptosis (Seltmann et al., 2018). Early characterization of Clca2 protein structure and expression pattern indicated that Clca2 might be a regulator of cystic fibrosis (Gruber et al., 1999). More recently, Walia et al. (2012) have reported that Clca2 expression can be down-regulated by TGF-β, one of the most potent inducer of tissue fibrogenesis, in epithelial cells. In mammalian cells, gene expression is acutely influenced by the epigenetic machinery. Epigenetics mechanisms are heritable phenotypic changes that do not involve alterations in the DNA sequence; these mechanisms play an important role in a wide spectrum of human diseases (Surguchov et al., 2017). These observations prompted us to investigate whether and, if so, how Clca2 expression might be regulated in the process of cardiac FMyT. We report here that Clca2 is transcriptionally repressed by a Twist1-HDAC1 epigenetic complex in cardiac fibroblasts by pro-fibrogenic stimuli. In addition, Clca2 is able to modulate TGF-β induced FMyT in vitro.
Materials and Methods
Animals
All animal protocols were reviewed and approved the intramural Ethics Committee on Humane Treatment of Laboratory Animals of Jiangsu Health Vocational College. The mice were maintained in an SPF environment with 12 h light/dark cycles and libitum access to food and water. Cardiac fibrosis was induced by permanent ligation of left-anterior descending (LAD) coronary artery or transverse aortic constriction (TAC) as previously described (Yang et al., 2017; Liu et al., 2021b, c).
Cell Culture, Plasmids, Transient Transfection, and Reporter Assay
Primary cardiac fibroblasts were isolated and maintained in DMEM supplemented with 10% FBS as previously described (Gao et al., 2020; Liu et al., 2020; He et al., 2021; Zhao et al., 2021). Mouse embryonic fibroblasts (MEFs) were isolated and maintained in DMEM supplemented with 10% FBS as previously described (Angrisani et al., 2021). Clca2 promoter-luciferase construct was made by amplifying genomic DNA spanning the proximal promoter and the first exon of Clca2 gene (−1100/ + 91) and ligating into a pGL3-basic vector (Promega). Truncation mutants were made using a QuikChange kit (Thermo Fisher Scientific, Waltham, MA, United States) and verified by direct sequencing. Small interfering RNAs were purchased from Dharmacon. Transient transfection was performed with Lipofectamine 2000. Cells were harvested 48 h after transfection and reporter activity was measured using a luciferase reporter assay system (Promega) as previously described (Kong et al., 2021a, c; Liu et al., 2021c; Zhang et al., 2021). MS-275 and MC-1568 were purchased from Selleck. Mouse recombinant TGF-β was purchased from R&D.
Protein Extraction, Immunoprecipitation and Western Blot
Whole cell lysates were obtained by re-suspending cell pellets in RIPA buffer (50 mM Tris pH7.4, 150 mM NaCl, 1% Triton X-100) with freshly added protease inhibitor (Roche) as previously described (Chen et al., 2020a,b,c; Wu X. et al., 2020; Yang et al., 2020b; Zhang et al., 2020; Chen B. et al., 2021; Dong et al., 2021). Nuclear proteins were extracted using the NE-PER Kit (Pierce) following manufacturer’s recommendation. Specific antibodies or pre-immune IgGs were added to and incubated with cell lysates overnight before being absorbed by Protein A/G-plus Agarose beads (Santa Cruz). Precipitated immune complex was released by boiling with 1X SDS electrophoresis sample buffer. Western blot analyses were performed with anti-Clca2 (Proteintech, 19273-1, 1:500), anti-α-SMA (Sigma, A2547, 1:8000), anti-collagen type I (Proteintech, 14695-1, 1:2000), anti-Twist1 (Proteintech, 25465-1, 1:500), anti-HDAC1 (Santa Cruz, sc-7872, 1:1000), anti-HDAC2 (Santa Cruz, sc-7899, 1:1000), anti-HDAC3 (Santa Cruz, sc-11417, 1:1000), anti-HDAC8 (Santa Cruz, sc-11405, 1:1000), anti-FLAG (Sigma, F1804, 1:5000), and anti-β-actin (Sigma, A2228, 1:4000) antibodies.
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) assays were performed essentially as described before (Wang et al., 2020; Liu et al., 2021a). In brief, chromatin in control and treated cells were cross-linked with 1% formaldehyde. Cells were incubated in lysis buffer (150 mM NaCl, 25 mM Tris pH 7.5, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) supplemented with protease inhibitor tablet and PMSF. DNA was fragmented into ∼200 bp pieces using a Branson 250 sonicator. Aliquots of lysates containing 200 μg of protein were used for each immunoprecipitation reaction with anti-Twist1 (Proteintech, 25465-1), anti-Slug (Cell Signaling Technology, 9585), anti-Zeb1 (Cell Signaling Technology, 3396), anti-Snail (Cell Signaling Technology, 3879), anti-anti-acetyl H3 (Millipore, 06-599), anti-acetyl H4 (Millipore, 06-598), anti-HDAC1 (Santa Cruz, sc-7872), or pre-immune IgG. For re-ChIP, immune complexes were eluted with the elution buffer (1% SDS, 100 mM NaCO3), diluted with the re-ChIP buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris pH 8.1), and subject to immunoprecipitation with a second antibody of interest.
RNA Isolation and Real-Time PCR
RNA was extracted with the RNeasy RNA isolation kit (Qiagen). Reverse transcriptase reactions were performed using a SuperScript First-strand Synthesis System (Invitrogen) as previously described (Dong et al., 2020; Hong et al., 2020; Wu T. et al., 2020; Yang et al., 2020a,b, 2021; Zhang et al., 2020; Kong et al., 2021b). Real-time PCR reactions were performed on an ABI Prism 7500 system. Ct values of target genes were normalized to the Ct values of housekeeping control gene (18s rRNA, 5′-CGCGGTTCTATTTTGTTGGT-3′ and 5′-TCGTCTTCGAAACTCCGACT-3′ for both human and mouse genes) using the ΔΔCt method and expressed as relative mRNA expression levels compared to the control group which is arbitrarily set as 1.
5-Ethynyl-2′-Deoxyuridine Incorporation Assay
5-ethynyl-2′-deoxyuridine (EdU) incorporation assay was performed in triplicate wells with a commercially available kit (Thermo Fisher Scientific) per vendor instruction. Briefly, the EdU solution was diluted with the culture media and added to the cells for an incubation period of 2 h at 37°C. After several washes with 1XPBS, the cells were then fixed with 4% formaldehyde and stained with Alexa FluorTM 488. The nucleus was counter-stained with DAPI. The images were visualized by fluorescence microscopy and analyzed with Image-Pro Plus (Media Cybernetics). For each group, at least six different fields were randomly chosen and the positively stained cells were counted and divided by the number of total cells. The data are expressed as relative EdU staining compared to the control group arbitrarily set as 1.
Statistical Analysis
One-way ANOVA with post hoc Scheff’e analyses were performed by SPSS software (IBM SPSS v18.0, Chicago, IL, United States). Unless otherwise specified, values of p < 0.05 were considered statistically significant.
Results
Chloride Channel Accessory 2 Expression Is Down-Regulated in Activated Cardiac Fibroblasts
When exposed to injurious stimuli, cardiac resident fibroblasts undergo trans-differentiation and become mature myofibroblasts to mediate the fibrogenic response. In order to compare Clca2 expression in quiescent cardiac fibroblasts and activated cardiac fibroblasts, C57B/6 mice were subjected to the LAD procedure to induce myocardial infarction; previous investigations have shown that FMyT peaks at 7 day after the surgery (Kanisicak et al., 2016). It was observed that compared to the sham-operated mice, expression levels of Acta2 (encoding α-SMA) and Col1a1 (encoding collagen type I), two typical myofibroblast markers, were significantly up-regulated in the primary cardiac fibroblasts isolated from the LAD-operated mice; on the contrary, Clca2 expression was down-regulated in the activated cardiac fibroblasts compared to the quiescent cardiac fibroblasts (Figure 1A). Western blotting confirmed that Clca2 protein levels were down-regulated as well (Figure 1B). In the second model of myocardial fibrosis, C57B/6 mice were subjected to the TAC procedure; FMyT typically peaks at 7 day after the surgery (Bursac, 2014). QPCR (Figure 1C) and Western blotting (Figure 1D) showed that Clca2 expression was lower in the activated cardiac fibroblasts isolated from the TAC mice than in the quiescent cardiac fibroblasts isolated from the sham mice, opposite to the changes in Acta2 expression and Col1a1 expression.
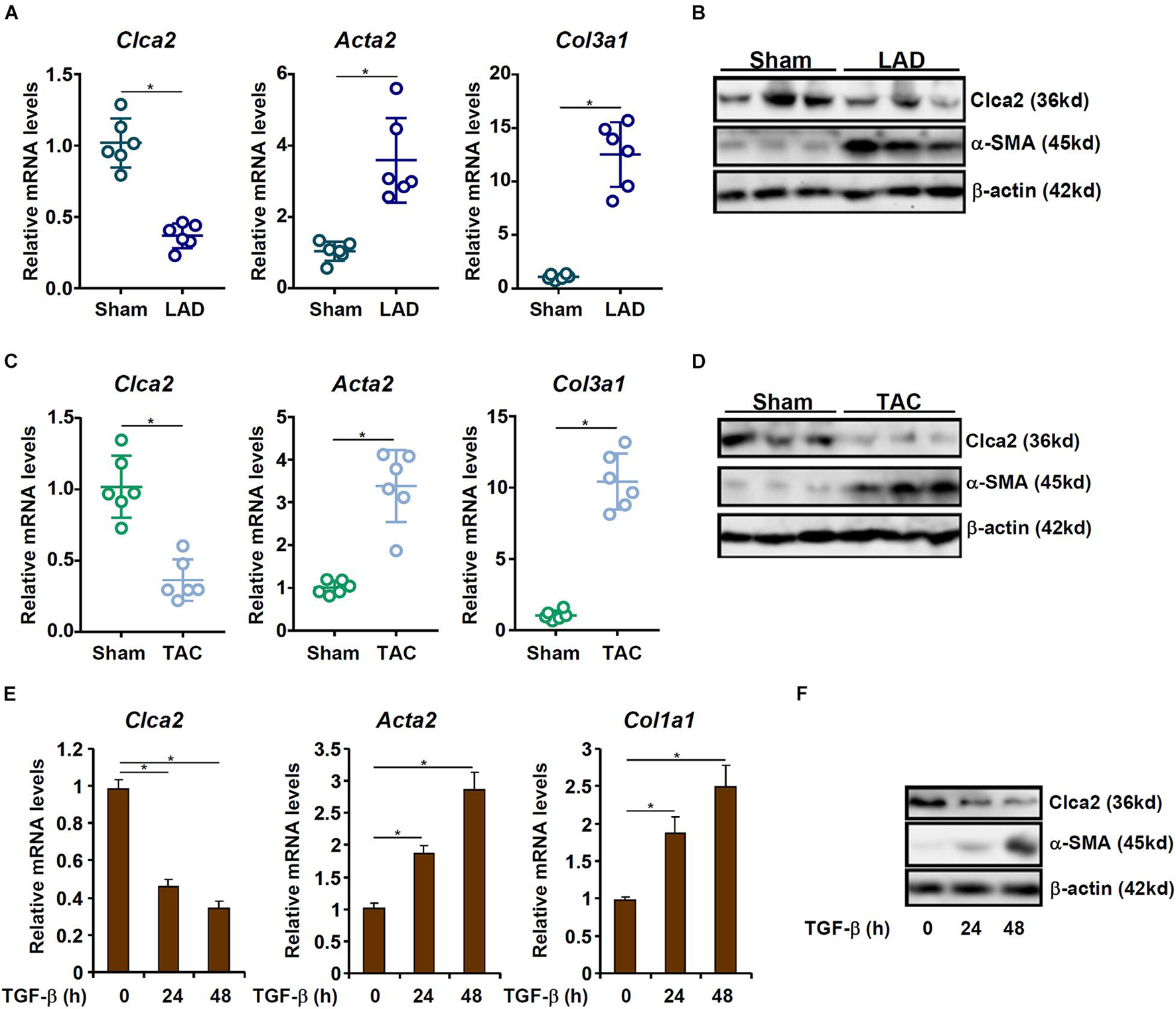
Figure 1. Chloride channel accessory 2 (Clca2) expression is down-regulated in activated cardiac fibroblasts. (A,B) C57B/6 mice were subjected to the LAD procedure to induce myocardial infarction. The mice were sacrificed 7 day after the surgery and primary cardiac fibroblasts were isolated. Clca2 expression was examined by qPCR and Western. N = 6 mice for each group. (C,D) C57B/6 mice were subjected to the TAC procedure to induce myocardial hypertrophy. The mice were sacrificed 7 day after the surgery and primary cardiac fibroblasts were isolated. Clca2 expression was examined by qPCR and Western. N = 6 mice for each group. (E,F) Primary cardiac fibroblasts were isolated from C57B/6 mice and treated with TGF-β (2 ng/ml). Cells were harvested at indicated time points and Clca2 expression was examined by qPCR and Western. Error bars represent SD (*p < 0.05, two-way Student’s t-test). All experiments were repeated three times and one representative experiment is shown.
TGF-β is one of most potent inducer of FMyT and myocardial fibrosis (Davis and Molkentin, 2014). When quiescent cardiac fibroblasts were treated with TGF-β, it was found that both Acta2 and Col1a1 were progressively up-regulated whereas Clca2 expression was concomitantly down-regulated (Figures 1E,F).
TWIST1 Mediates Chloride Channel Accessory 2 Trans-Repression in Cardiac Fibroblasts
We next determined whether down-regulation of Clca2 expression by TGF-β occurred at the transcriptional level. A series of Clca2 promoter-luciferase reporter constructs were transfected into mouse embryonic fibroblasts (MEFs) followed by TGF-β treatment. As shown in Figure 2A, TGF-β treatment decreased the activity of the full-length Clca2 promoter (−1100/ + 91) suggesting that TGF-β could indeed repress Clca2 transcription. However, when deletions introduced to the full-length Clca2 promoter extended beyond −516, TGF-β treatment could no longer repress the Clca2 promoter activity. A closer examination revealed a conserved E-box (CAGGTG) located between −516 and −224 of the Clca2 promoter; mutation of the E-box completely abrogated the response to TGF-β treatment (Figure 2B).
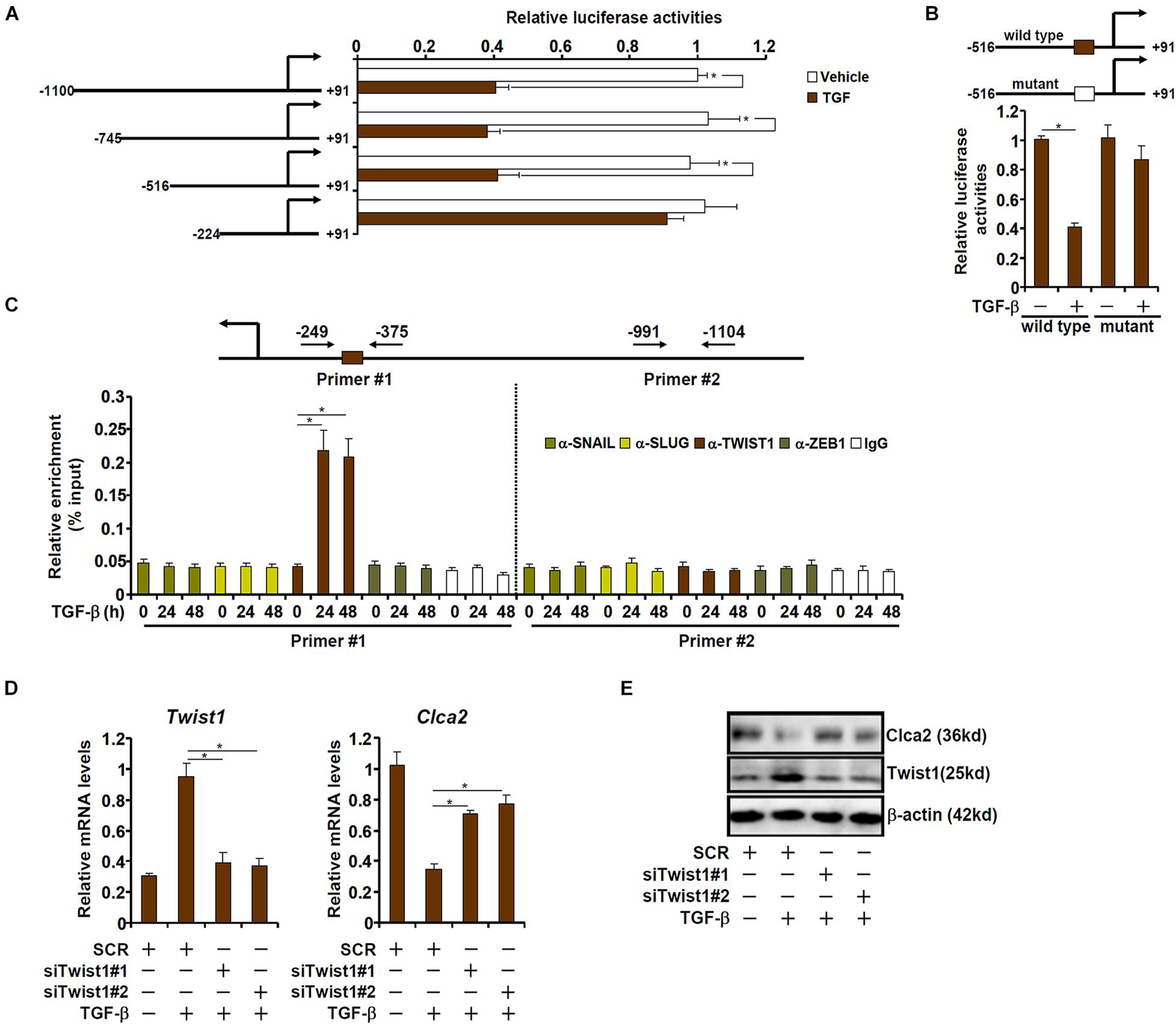
Figure 2. TWIST1 mediates Clca2 trans-repression in cardiac fibroblasts. (A) Different Clca2 promoter-luciferase constructs were transfected into mouse embryonic fibroblasts (MEFs) followed by treatment with TGF-β. Luciferase activities were normalized by protein concentration and GFP fluorescence. (B) Wild type and mutant Clca2 promoter-luciferase constructs were transfected into MEFs followed by treatment with TGF-β. Luciferase activities were normalized by protein concentration and GFP fluorescence. (C) Primary cardiac fibroblasts were treated with TGF-β (2 ng/ml) and were harvested at indicated time points. ChIP assays were performed with indicated antibodies. (D,E) Primary cardiac fibroblasts were transfected with indicated siRNAs followed by treatment with TGF-β (2 ng/ml). Gene expression was examined by qPCR and Western. Error bars represent SD (*p < 0.05, two-way Student’s t-test). All experiments were repeated three times and one representative experiment is shown.
The E-box binding family of zinc finger transcription repressors include Snail, Slug, Twist1, and Zeb1 (Kalluri and Weinberg, 2009). ChIP assay was performed to determine which one of these transcription factors (TFs). As shown in Figure 2C, Twist1, but not Snail, Slug, or Zeb1, occupied the proximal Clca2 promoter containing the E-box in response to TGF-β treatment; none of the TFs were detected on the distal Clca2 promoter. To further validate the role of Twist1 in Clca2 trans-repression, endogenous Twist1 was depleted with two independent pairs of siRNAs. Twist1 knockdown partially restored Clca2 expression in the presence of TGF-β in cardiac fibroblasts (Figures 2D,E).
TWIST1 Represses Chloride Channel Accessory 2 Transcription by Promoting Histone Deacetylation
Transcriptional repression is usually associated with erasure of histone acetylation surrounding the promoter region (Jenuwein and Allis, 2001). As shown in Figure 3A, TGF-β treatment led to disappearance of acetylated histone H3 and acetylated histone H4 from the proximal, but not the distal, Clca2 promoter; Twist1 knockdown normalized histone acetylation, suggesting that histone deacetylases (HDACs) might be involved in Twist1 mediated Clca2 trans-repression. HDACs can be categorized into three classes: class I and class II HDACs primarily catalyze histone deacetylation whereas class III HDACs (the sirtuins) primarily catalyze non-histone lysine deacetylation (Yang and Seto, 2008). Pre-treatment with a pan-class I HDAC inhibitor (MS-275), but not a pan-class II HDAC inhibitor (MC-1568), blocked TGF-β induced Clca2 repression (Figures 3B,C), indicating that class I HDAC might be involved in Clca2 trans-repression.
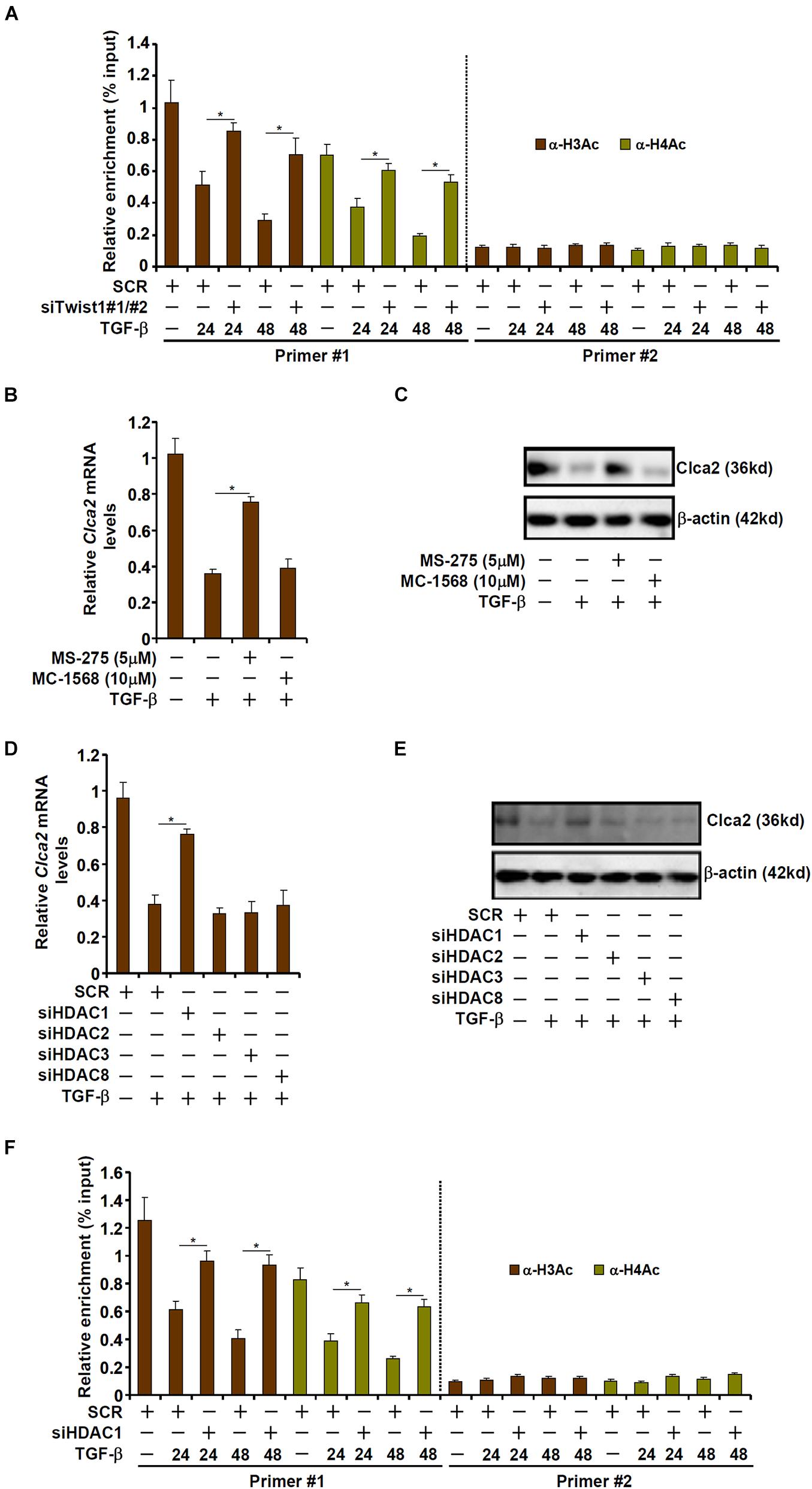
Figure 3. TWIST1 represses Clca2 transcription by promoting histone deacetylation. (A) Primary murine cardiac fibroblasts were transfected with indicated siRNAs by treatment with TGF-β (2 ng/ml). ChIP assays were performed with indicated antibodies. (B,C) Primary murine cardiac fibroblasts were treated with TGF-β (2 ng/ml) in the presence or absence of different HDAC inhibitors. Clca2 expression was examined by qPCR and Western. (D,E) Primary murine cardiac fibroblasts were transfected with indicated siRNAs by treatment with TGF-β (2 ng/ml). Clca2 expression was examined by qPCR and Western. (F) Primary cardiac murine fibroblasts were transfected with indicated siRNAs by treatment with TGF-β (2 ng/ml). ChIP assays were performed with indicated antibodies. Error bars represent SD (*p < 0.05, two-way Student’s t-test). All experiments were repeated three times and one representative experiment is shown.
Class I HDACs include HDAC1, HDAC2, HDAC3, and HDAC8. When individual class I HDACs were depleted with siRNAs, it was discovered that only HDAC1 knockdown significantly attenuated Clca2 repression by TGF-β treatment (Figures 3D,E). Consistently, HDAC1 knockdown largely normalized histone acetylation levels surrounding the Clca2 promoter (Figure 3F).
TWIST1 Interacts With and Recruits HDAC1 to Repress Chloride Channel Accessory 2 Transcription
We next investigated the possibility that Twist1 recruits HDAC1 to repress Clca2 transcription. ChIP assay showed that occupancies of HDAC1 on the Clca2 promoter were enhanced following TGF-β treatment with a kinetics similar to Twist1; Twist1 depletion blocked HDAC1 binding to the Clca2 promoter (Figure 4A). Co-immunoprecipitation confirmed that Twist1 and HDAC1 could interact with each other in cardiac fibroblasts (Figure 4B). Importantly, Re-ChIP assay showed that the Twist1-HDAC1 interaction was significantly cemented by TGF-β treatment on the Clca2 promoter (Figure 4C). In addition, whereas HDAC1 over-expression dose-dependently repressed the Clca2 promoter activity in reporter assay the mutant Clca2 promoter without the intact E-box was completely refractory to HDAC1 over-expression (Figure 4D).
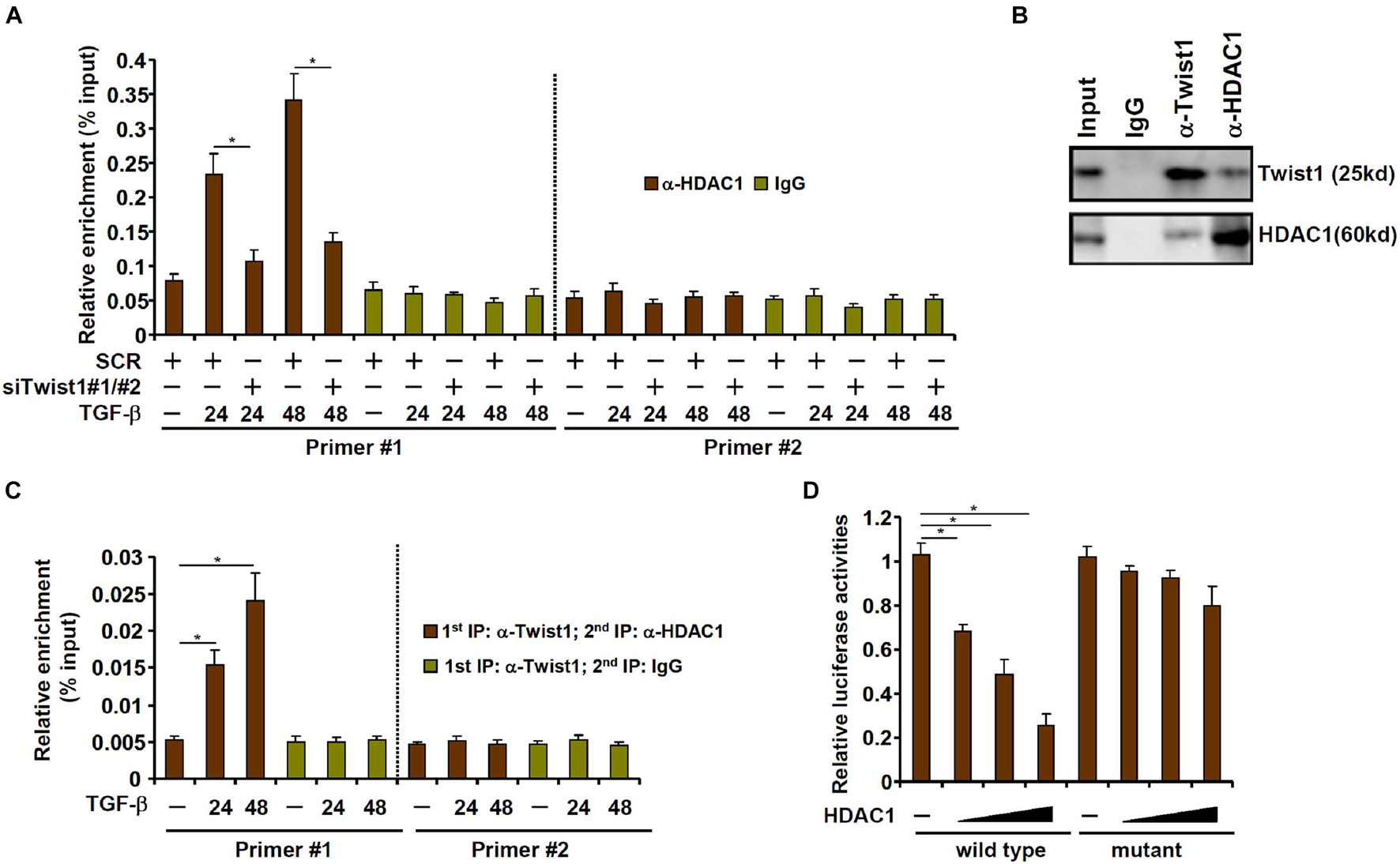
Figure 4. TWIST1 interacts with and recruits HDAC1 to repress Clca2 transcription. (A) Primary cardiac murine fibroblasts were transfected with indicated siRNAs by treatment with TGF-β (2 ng/ml). ChIP assays were performed with indicated antibodies. (B) Immunoprecipitation was performed with whole cell lysates from primary murine cardiac fibroblasts. (C) Primary murine cardiac fibroblasts were treated with or without TGF-β (2 ng/ml) for 48 h. Re-ChIP was performed with indicated antibodies. (D) Wild type and mutant Clca2 promoter-luciferase constructs were transfected into MEFs with increasing doses of HDAC1 followed by treatment with TGF-β. Luciferase activities were normalized by protein concentration and GFP fluorescence. Error bars represent SD (*p < 0.05, two-way Student’s t-test). All experiments were repeated three times and one representative experiment is shown.
Chloride Channel Accessory 2 Regulates Activation of Cardiac Fibroblasts
Finally, an attempt was made to place the finding that Clca2 transcription was epigenetically repressed during cardiac fibroblast activation in a pathophysiological perspective. To this end, primary murine cardiac fibroblasts were transduced with adenovirus carrying a Clca2 expression vector (Ad-FLAG-Clca2) or an empty vector (Ad-EV). Ad-FLAG-Clca2 transduction significantly boosted Clca2 expression in cardiac fibroblasts (Figures 5A,B). More important, Clca2 over-expression significantly down-regulated the expression of myofibroblast marker genes at both mRNA (Figure 5C) and protein (Figure 5D) levels. In addition, Clca2 over-expression attenuated proliferation of cardiac fibroblasts as measured by EdU incorporation (Figure 5E).
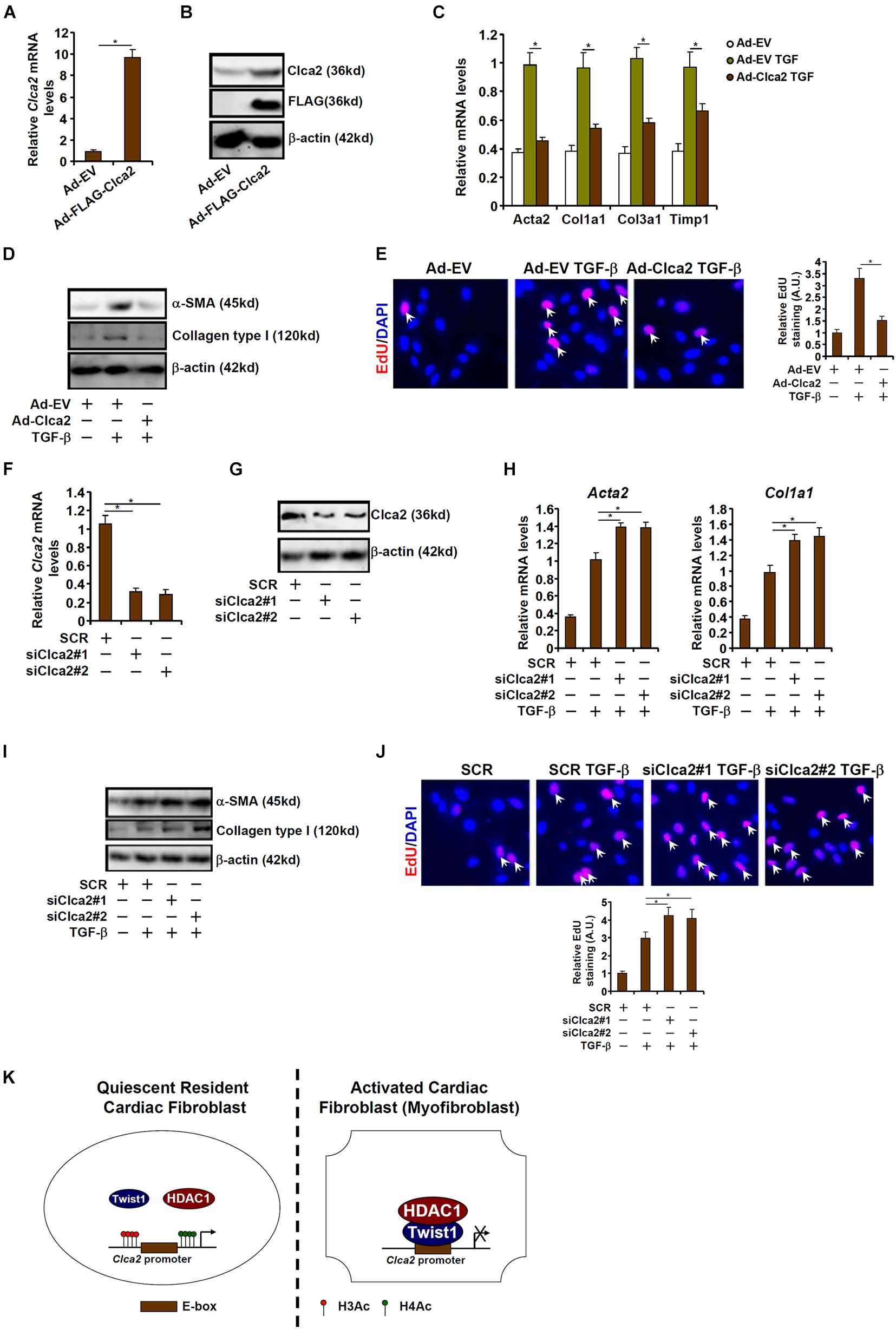
Figure 5. Chloride channel accessory 2 (Clca2) regulates activation of cardiac fibroblasts. (A–E) Primary murine cardiac fibroblasts were infected with adenovirus carrying a Clca2 expression vector (Ad-FLAG-Clca2) or an empty vector (Ad-EV) followed by treatment with TGF-β. Expression levels of Clca2 were examined by qPCR (A) and Western (B). Pro-fibrogenic genes were examined by qPCR (C) and Western (D). Cell proliferation was examined by EdU incorporation (E). (F–J) Primary murine cardiac fibroblasts were transfected with indicated siRNAs followed by treatment with TGF-β. Expression levels of Clca2 were examined by qPCR (F) and Western (G). Pro-fibrogenic genes were examined by qPCR (H) and Western (I). Cell proliferation was examined by EdU incorporation (J). Error bars represent SD (*p < 0.05, two-way Student’s t-test). All experiments were repeated three times and one representative experiment is shown. (K) A schematic model.
Alternatively, Clca2 expression was depleted with two separate pairs of siRNAs (Figures 5F,G). Clca2 knockdown further enhanced TGF-β induced expression of myofibroblast marker genes (Figures 5H,I) and augmented cell proliferation (Figure 5J). Together, these data suggest that Clca2 might regulate FMyT in vitro.
Discussion
Recent investigations have provided irrefutable evidence to support resident fibroblasts as the primary source of myofibroblasts contributing to cardiac fibrosis (Travers et al., 2016). Dynamic transcriptomic changes highlight the transition from quiescent cardiac fibroblasts to mature myofibroblasts (Krstevski et al., 2020). We show here that Twist1 is responsible for Clca2 trans-repression in activated cardiac fibroblasts by directly binding to the E-box element located on the Clca2 promoter (Figure 5K). Consistent with our observation, Al-Hattab et al. (2018) have previously reported that Twist1 transcription can be activated by TGF-β in cardiac fibroblasts, which is mediated by scleraxis. Of note, several studies have found that Twist1 can be placed among signature markers for cardiac fibroblasts (Zhou et al., 2010; Liu et al., 2016; Han et al., 2021). Whether or not Twist1 can directly regulate cardiac fibrosis remains to be determined. On the one hand, pharmaceutical inhibition and fibroblast-specific deletion of Twist1 have been shown to mitigate liver fibrosis (Dong et al., 2020) and skin fibrosis (Palumbo-Zerr et al., 2017), respectively, supporting Twist1 as a promoter of tissue fibrosis. On the other hand, Twist1 residing in the parenchymal cells, the mesenchymal cells, or infiltrating immune cells is able to rein in tissue injury and antagonize tissue fibrosis (Tan et al., 2017; Ren et al., 2019, 2021). Therefore, more studies should be conducted to test the feasibility of targeting Twist1 as a therapeutic strategy against aberrant cardiac fibrosis.
Our data indicate that Twist1 represses Clca2 transcription via, at least in part, by recruiting the histone deacetylase HDAC1. Curiously, our observation is in contrast to a previous study by Xu et al. (2006) where it was demonstrated that HDAC1, recruited by the RFX1, directly binds to the collagen type I promoter (Col1a2) and represses Col1a2 transcription in lung fibroblasts in response to IFN-γ treatment. It is likely that HDAC1 may exert differential effects on tissue fibrogenesis depending on the stimuli and the cell type. Global deletion of HDAC1 in mice results in early developmental arrest and embryonic lethality precluding the analysis of cardiac fibrosis in adult animals (Montgomery et al., 2007). More recently, Renaud et al. (2015) have shown that administration of a pan-HDAC inhibitor (HDACi) attenuates cardiac fibrosis in mice subjected to pressure overload although the mechanism is less clear but possibly can be attributable to HDAC1-mediated repression of miR-133a, an anti-fibrotic non-regulatory RNA. Of note, HDAC1-null MEFs display weakened proliferation compared to wild type MEFs (Yamaguchi et al., 2010), suggesting that HDAC1 deficiency may prevent cardiac fibrosis by limiting the expansion of myofibroblasts in vivo (Yamaguchi et al., 2010). Future studies employing fibroblast/myofibroblast conditional transgenic animals should clarify the role of HDAC1 in cardiac fibrosis.
We present data to show that manipulating Clca2 expression in cardiac fibroblasts influences FMyT in vitro. The underlying mechanism, however, awaits further investigation. Previous studies have shown that a variety of chloride channels may contribute to myofibroblast maturation via the MAPK-p38 signaling pathway (Shukla et al., 2014) or the PI3K-Akt signaling pathway (Sun et al., 2016) or the PKC signaling pathway (El Chemaly et al., 2014). Alternatively, chloride intracellular channel 4 (CLIC4) has been shown to promote TGF-β induced FMyT by inducing a dominant negative SMAD7 splicing isoform (Shukla et al., 2016). Despite the fact that several studies have provided evidence to show that chloride channel inhibitors/blockers can potentially attenuate the activation of cardiac fibroblasts (El Chemaly et al., 2014; Tian et al., 2018; Chen P. H. et al., 2021), no consensus seems to exist regarding the underlying mechanisms. It is therefore imperative for future investigators to focus on delineating the mode of action for Clca2 in the process of FMyT so that the plethora of data, including the ones presented here, can be exploited in the development of novel therapeutic solutions to treat adverse cardiac remodeling and heart failure.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Ethics Committee on Humane Treatment of Laboratory Animals of Jiangsu Health Vocational College.
Author Contributions
MF conceived the project and secured funding and provided supervision. TS, YX, and MF designed the experiments. TS and YX performed the experiments, collected the data, and analyzed the data. All authors contributed to the drafting and editing of manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81870302 and 81800520).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Hattab, D. S., Safi, H. A., Nagalingam, R. S., Bagchi, R. A., Stecy, M. T., and Czubryt, M. P. (2018). Scleraxis regulates Twist1 and Snai1 expression in the epithelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 315, H658–H668. doi: 10.1152/ajpheart.00092.2018
Angrisani, A., Di Fiore, A., Di Trani, C. A., Fonte, S., Petroni, M., Lospinoso Severini, L., et al. (2021). Specific Protein 1 and p53 interplay modulates the expression of the KCTD-containing Cullin3 adaptor suppressor of Hedgehog 2. Front. Cell Dev. Biol. 9:638508. doi: 10.3389/fcell.2021.638508
Bursac, N. (2014). Cardiac fibroblasts in pressure overload hypertrophy: the enemy within? J. Clin. Invest. 124, 2850–2853. doi: 10.1172/JCI76628
Chen, B., Fan, Z., Sun, L., Chen, J., Feng, Y., Fan, X., et al. (2020a). Epigenetic activation of the small GTPase TCL contributes to colorectal cancer cell migration and invasion. Oncogenesis 9:86. doi: 10.1038/s41389-020-00269-9
Chen, B., Yuan, Y., Sun, L., Chen, J., Yang, M., Yin, Y., et al. (2020b). MKL1 mediates TGF-β Induced RhoJ transcription to promote breast cancer cell migration and invasion. Front. Cell Dev. Biol. 8:832. doi: 10.3389/fcell.2020.00832
Chen, B., Zhao, Q., Xu, T., Yu, L., Zhuo, L., Yang, Y., et al. (2020c). BRG1 activates PR65A transcription to regulate NO bioavailability in vascular endothelial cell. Front. Cell Dev. Biol. 8:774. doi: 10.3389/fcell.2020.00774
Chen, B., Zhu, Y., Chen, J., Feng, Y., and Xu, Y. (2021). Activation of TCL transcription by lysine demethylase KDM4B in colorectal cancer cells. Front. Cell Dev. Biol. 9:617549. doi: 10.3389/fcell.2021.617549
Chen, P. H., Chung, C. C., Lin, Y. F., Kao, Y. H., and Chen, Y. J. (2021). Lithium reduces migration and collagen synthesis activity in human cardiac fibroblasts by inhibiting store-operated Ca(2+) entry. Int. J. Mol. Sci. 22:842. doi: 10.3390/ijms22020842
Davis, J., and Molkentin, J. D. (2014). Myofibroblasts: trust your heart and let fate decide. J. Mol. Cell Cardiol. 70, 9–18. doi: 10.1016/j.yjmcc.2013.10.019
Dong, W., Kong, M., Zhu, Y., Shao, Y., Wu, D., Lu, J., et al. (2020). Activation of TWIST transcription by chromatin remodeling Protein BRG1 contributes to liver fibrosis in mice. Front. Cell Dev. Biol. 8:340. doi: 10.3389/fcell.2020.00340
Dong, W., Zhu, Y., Zhang, Y., Fan, Z., Zhang, Z., Fan, X., et al. (2021). BRG1 Links TLR4 trans-activation to LPS-Induced SREBP1a expression and liver injury. Front. Cell Dev. Biol. 9:617073. doi: 10.3389/fcell.2021.617073
El Chemaly, A., Norez, C., Magaud, C., Bescond, J., Chatelier, A., Fares, N., et al. (2014). ANO1 contributes to angiotensin-II-activated Ca2+-dependent Cl- current in human atrial fibroblasts. J. Mol. Cell Cardiol. 68, 12–19. doi: 10.1016/j.yjmcc.2013.12.027
Gao, Q. Y., Zhang, H. F., Tao, J., Chen, Z. T., Liu, C. Y., Liu, W. H., et al. (2020). Mitochondrial fission and mitophagy reciprocally orchestrate cardiac fibroblasts activation. Front. Cell Dev. Biol. 8:629397. doi: 10.3389/fcell.2020.629397
Gonzalez, A., Schelbert, E. B., Diez, J., and Butler, J. (2018). myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J. Am. Coll. Cardiol. 71, 1696–1706. doi: 10.1016/j.jacc.2018.02.021
Gruber, A. D., Schreur, K. D., Ji, H. L., Fuller, C. M., and Pauli, B. U. (1999). Molecular cloning and transmembrane structure of hCLCA2 from human lung, trachea, and mammary gland. Am. J. Physiol. 276, C1261–C1270. doi: 10.1152/ajpcell.1999.276.6.C1261
Han, J. K., Shin, Y., Sohn, M. H., Choi, S. B., Shin, D., You, Y., et al. (2021). Direct conversion of adult human fibroblasts into functional endothelial cells using defined factors. Biomaterials 272:120781. doi: 10.1016/j.biomaterials.2021.120781
He, S., Lu, Y., Guo, Y., Li, S., Lu, X., Shao, S., et al. (2021). Kruppel-Like Factor 15 modulates CXCL1/CXCR2 signaling-mediated inflammatory response contributing to angiotensin II-induced cardiac remodeling. Front. Cell Dev. Biol. 9:644954. doi: 10.3389/fcell.2021.644954
Hong, W., Kong, M., Qi, M., Bai, H., Fan, Z., Zhang, Z., et al. (2020). BRG1 mediates nephronectin activation in hepatocytes to Promote T lymphocyte infiltration in ConA-Induced hepatitis. Front. Cell Dev. Biol. 8:587502. doi: 10.3389/fcell.2020.587502
Jentsch, T. J., and Pusch, M. (2018). CLC chloride channels and transporters: structure. Function, physiology, and disease. Physiol. Rev. 98, 1493–1590. doi: 10.1152/physrev.00047.2017
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi: 10.1172/JCI39104
Kanisicak, O., Khalil, H., Ivey, M. J., Karch, J., Maliken, B. D., Correll, R. N., et al. (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7:12260. doi: 10.1038/ncomms12260
Kaur, H., Takefuji, M., Ngai, C. Y., Carvalho, J., Bayer, J., Wietelmann, A., et al. (2016). Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 118, 1906–1917. doi: 10.1161/CIRCRESAHA.116.308643
Kong, M., Dong, W., Xu, H., Fan, Z., Miao, X., Guo, Y., et al. (2021a). Choline kinase alpha is a novel transcriptional target of the brg1 in hepatocyte: implication in liver regeneration. Front. Cell Dev. Biol. 9:705302. doi: 10.3389/fcell.2021.705302
Kong, M., Dong, W., Zhu, Y., Fan, Z., Miao, X., Guo, Y., et al. (2021b). Redox-sensitive activation of CCL7 by BRG1 in hepatocytes during liver injury. Redox Biol. 46:102079. doi: 10.1016/j.redox.2021.102079
Kong, M., Zhu, Y., Shao, J., Fan, Z., and Xu, Y. (2021c). The chromatin remodeling protein BRG1 Regulates SREBP maturation by activating SCAP transcription in hepatocytes. Front. Cell Dev. Biol. 9:622866. doi: 10.3389/fcell.2021.622866
Kramann, R., Schneider, R. K., DiRocco, D. P., Machado, F., Fleig, S., Bondzie, P. A., et al. (2015). Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16, 51–66. doi: 10.1016/j.stem.2014.11.004
Krstevski, C., Cohen, C. D., Dona, M. S. I., and Pinto, A. R. (2020). New perspectives of the cardiac cellular landscape: mapping cellular mediators of cardiac fibrosis using single-cell transcriptomics. Biochem. Soc. Trans. 48, 2483–2493. doi: 10.1042/BST20191255
Liu, C., Han, Y., Gu, X., Li, M., Du, Y., Feng, N., et al. (2021a). Paeonol promotes Opa1-mediated mitochondrial fusion via activating the CK2alpha-Stat3 pathway in diabetic cardiomyopathy. Redox Biol. 46:102098. doi: 10.1016/j.redox.2021.102098
Liu, L., Zhao, Q., Kong, M., Mao, L., Yang, Y., and Xu, Y. (2021b). Myocardin-related transcription factor A (MRTF-A) regulates integrin beta 2 transcription to promote macrophage infiltration and cardiac hypertrophy in mice. Cardiovasc. Res. cvab110. doi: 10.1093/cvr/cvab110
Liu, L., Zhao, Q., Lin, L., Yang, G., Yu, L., Zhuo, L., et al. (2021c). Myeloid MKL1 disseminates cues to promote cardiac hypertrophy in mice. Front. Cell Dev. Biol. 9:583492. doi: 10.3389/fcell.2021.583492
Liu, C., Hu, T., Cai, Z., Xie, Q., Yuan, Y., Li, N., et al. (2020). Nucleotide-binding oligomerization domain-like receptor 3 deficiency attenuated isoproterenol-induced cardiac fibrosis via reactive oxygen species/high mobility group Box 1 Protein axis. Front. Cell Dev. Biol. 8:713. doi: 10.3389/fcell.2020.00713
Liu, Z., Chen, O., Zheng, M., Wang, L., Zhou, Y., Yin, C., et al. (2016). Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res. 16, 507–518. doi: 10.1016/j.scr.2016.02.037
Mollmann, H., Nef, H. M., Kostin, S., von Kalle, C., Pilz, I., Weber, M., et al. (2006). Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc. Res. 71, 661–671. doi: 10.1016/j.cardiores.2006.06.013
Montgomery, R. L., Davis, C. A., Potthoff, M. J., Haberland, M., Fielitz, J., Qi, X., et al. (2007). Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790–1802. doi: 10.1101/gad.1563807
Moore-Morris, T., Guimaraes-Camboa, N., Banerjee, I., Zambon, A. C., Kisseleva, T., Velayoudon, A., et al. (2014). Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Invest. 124, 2921–2934. doi: 10.1172/JCI74783
Palumbo-Zerr, K., Soare, A., Zerr, P., Liebl, A., Mancuso, R., Tomcik, M., et al. (2017). Composition of TWIST1 dimers regulates fibroblast activation and tissue fibrosis. Ann. Rheum Dis. 76, 244–251. doi: 10.1136/annrheumdis-2015-208470
Ramena, G., Yin, Y., Yu, Y., Walia, V., and Elble, R. C. (2016). CLCA2 Interactor EVA1 is required for mammary epithelial cell differentiation. PLoS One 11:e0147489. doi: 10.1371/journal.pone.0147489
Ren, J., Xu, Y., Lu, X., Wang, L., Ide, S., Hall, G., et al. (2021). Twist1 in podocytes ameliorates podocyte injury and proteinuria by limiting CCL2-dependent macrophage infiltration. JCI Insight 6:e148109. doi: 10.1172/jci.insight.148109
Ren, J., Zhang, J., Rudemiller, N. P., Griffiths, R., Wen, Y., Lu, X., et al. (2019). Twist1 in infiltrating macrophages attenuates kidney fibrosis via matrix metallopeptidase 13-Mediated matrix degradation. J. Am. Soc. Nephrol. 30, 1674–1685. doi: 10.1681/ASN.2018121253
Renaud, L., Harris, L. G., Mani, S. K., Kasiganesan, H., Chou, J. C., Baicu, C. F., et al. (2015). HDACs regulate miR-133a expression in pressure overload-induced cardiac fibrosis. Circ. Heart Fail. 8, 1094–1104. doi: 10.1161/CIRCHEARTFAILURE.114.001781
Sasaki, Y., Koyama, R., Maruyama, R., Hirano, T., Tamura, M., Sugisaka, J., et al. (2012). CLCA2, a target of the p53 family, negatively regulates cancer cell migration and invasion. Cancer Biol. Ther. 13, 1512–1521. doi: 10.4161/cbt.22280
Seltmann, K., Meyer, M., Sulcova, J., Kockmann, T., Wehkamp, U., Weidinger, S., et al. (2018). Humidity-regulated CLCA2 protects the epidermis from hyperosmotic stress. Sci. Transl. Med. 10:eaao4650. doi: 10.1126/scitranslmed.aao4650
Shukla, A., Edwards, R., Yang, Y., Hahn, A., Folkers, K., Ding, J., et al. (2014). CLIC4 regulates TGF-beta-dependent myofibroblast differentiation to produce a cancer stroma. Oncogene 33, 842–850. doi: 10.1038/onc.2013.18
Shukla, A., Yang, Y., Madanikia, S., Ho, Y., Li, M., Sanchez, V., et al. (2016). Elevating CLIC4 in multiple cell types reveals a TGF- dependent induction of a dominant negative Smad7 splice variant. PLoS One 11:e0161410. doi: 10.1371/journal.pone.0161410
Stempien-Otero, A., Kim, D. H., and Davis, J. (2016). Molecular networks underlying myofibroblast fate and fibrosis. J. Mol. Cell Cardiol. 97, 153–161. doi: 10.1016/j.yjmcc.2016.05.002
Sun, L., Dong, Y., Zhao, J., Yin, Y., and Zheng, Y. (2016). The CLC-2 chloride channel modulates ECM synthesis, differentiation, and migration of human conjunctival fibroblasts via the PI3K/Akt signaling pathway. Int. J. Mol. Sci. 17:910. doi: 10.3390/ijms17060910
Surguchov, A., Surgucheva, I., Sharma, M., Sharma, R., and Singh, V. (2017). Pore-forming proteins as mediators of novel epigenetic mechanism of epilepsy. Front. Neurol. 8:3. doi: 10.3389/fneur.2017.00003
Talman, V., and Ruskoaho, H. (2016). Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 365, 563–581. doi: 10.1007/s00441-016-2431-9
Tan, J., Tedrow, J. R., Nouraie, M., Dutta, J. A., Miller, D. T., Li, X., et al. (2017). Loss of Twist1 in the mesenchymal compartment promotes increased fibrosis in experimental lung injury by enhanced expression of CXCL12. J. Immunol. 198, 2269–2285. doi: 10.4049/jimmunol.1600610
Tian, X. Q., Ma, K. T., Wang, X. W., Wang, Y., Guo, Z. K., and Si, J. Q. (2018). Effects of the calcium-activated chloride channel inhibitors T16Ainh-A01 and CaCCinh-A01 on cardiac fibroblast function. Cell Physiol. Biochem. 49, 706–716. doi: 10.1159/000493036
Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C., and Brown, R. A. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363. doi: 10.1038/nrm809
Travers, J. G., Kamal, F. A., Robbins, J., Yutzey, K. E., and Blaxall, B. C. (2016). Cardiac fibrosis: the fibroblast awakens. Circ. Res. 118, 1021–1040. doi: 10.1161/CIRCRESAHA.115.306565
van den Borne, S. W., Diez, J., Blankesteijn, W. M., Verjans, J., Hofstra, L., and Narula, J. (2010). Myocardial remodeling after infarction: the role of myofibroblasts. Nat. Rev. Cardiol. 7, 30–37. doi: 10.1038/nrcardio.2009.199
Walia, V., Ding, M., Kumar, S., Nie, D., Premkumar, L. S., and Elble, R. C. (2009). hCLCA2 Is a p53-inducible inhibitor of breast cancer cell proliferation. Cancer Res. 69, 6624–6632. doi: 10.1158/0008-5472.CAN-08-4101
Walia, V., Yu, Y., Cao, D., Sun, M., McLean, J. R., Hollier, B. G., et al. (2012). Loss of breast epithelial marker hCLCA2 promotes epithelial-to-mesenchymal transition and indicates higher risk of metastasis. Oncogene 31, 2237–2246. doi: 10.1038/onc.2011.392
Wang, J. N., Yang, Q., Yang, C., Cai, Y. T., Xing, T., Gao, L., et al. (2020). Smad3 promotes AKI sensitivity in diabetic mice via interaction with p53 and induction of NOX4-dependent ROS production. Redox Biol. 32:101479. doi: 10.1016/j.redox.2020.101479
Wu, T., Wang, H., Xin, X., Yang, J., Hou, Y., Fang, M., et al. (2020). An MRTF-A-Sp1-PDE5 axis mediates Angiotensin-II-Induced cardiomyocyte hypertrophy. Front. Cell Dev. Biol. 8:839. doi: 10.3389/fcell.2020.00839
Wu, X., Dong, W., Zhang, T., Ren, H., Wang, J., Shang, L., et al. (2020). Epiregulin (EREG) and myocardin related transcription factor A (MRTF-A) form a feedforward loop to drive hepatic stellate cell activation. Front. Cell Dev. Biol. 8:591246. doi: 10.3389/fcell.2020.591246
Xu, Y., Sengupta, P. K., Seto, E., and Smith, B. D. (2006). Regulatory factor for X-box family proteins differentially interact with histone deacetylases to repress collagen alpha2(I) gene (COL1A2) expression. J. Biol. Chem. 281, 9260–9270.
Yamaguchi, T., Cubizolles, F., Zhang, Y., Reichert, N., Kohler, H., Seiser, C., et al. (2010). Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 24, 455–469. doi: 10.1101/gad.552310
Yang, G., Weng, X., Zhao, Y., Zhang, X., Hu, Y., Dai, X., et al. (2017). The histone H3K9 methyltransferase SUV39H links SIRT1 repression to myocardial infarction. Nat. Commun. 8:14941. doi: 10.1038/ncomms14941
Yang, X. J., and Seto, E. (2008). The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218. doi: 10.1038/nrm2346
Yang, Y., Li, Z., Guo, J., and Xu, Y. (2020a). Deacetylation of MRTF-A by SIRT1 defies senescence induced down-regulation of collagen type I in fibroblast cells. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165723. doi: 10.1016/j.bbadis.2020.165723
Yang, Y., Yang, G., Yu, L., Lin, L., Liu, L., Fang, M., et al. (2020b). An interplay between MRTF-A and the histone acetyltransferase TIP60 mediates hypoxia-reoxygenation induced iNOS transcription in macrophages. Front. Cell Dev. Biol. 8:484. doi: 10.3389/fcell.2020.00484
Yang, Y., Wang, H., Zhao, H., Miao, X., Guo, Y., Zhuo, L., et al. (2021). A GSK3-SRF axis mediates Angiotensin II induced endothelin transcription in vascular endothelial cells. Front. Cell Dev. Biol. 9:698254. doi: 10.3389/fcell.2021.698254
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., McMullen, J. R., Gustafsson, E., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961. doi: 10.1038/nm1613
Zhang, Y., Wang, H., Song, M., Xu, T., Chen, X., Li, T., et al. (2020). Brahma-related gene 1 deficiency in endothelial cells ameliorates vascular inflammatory responses in mice. Front. Cell Dev. Biol. 8:578790. doi: 10.3389/fcell.2020.578790
Zhang, Z., Chen, B., Zhu, Y., Zhang, T., Zhang, X., Yuan, Y., et al. (2021). The Jumonji domain-containing histone demethylase homolog 1D/lysine demethylase 7A (JHDM1D/KDM7A) is an epigenetic activator of RHOJ transcription in breast cancer cells. Front. Cell Dev. Biol. 9:664375. doi: 10.3389/fcell.2021.664375
Zhao, H., Zhang, Y., Xu, X., Sun, Q., Yang, C., Wang, H., et al. (2021). Sall4 and myocd empower direct cardiac reprogramming from adult cardiac fibroblasts after injury. Front. Cell Dev. Biol. 9:608367. doi: 10.3389/fcell.2021.608367
Keywords: transcriptional regulation, epigenetics, histone deacetylation, histone deacetylase, cardiac fibroblast, myocardial fibrosis
Citation: Shao T, Xue Y and Fang M (2021) Epigenetic Repression of Chloride Channel Accessory 2 Transcription in Cardiac Fibroblast: Implication in Cardiac Fibrosis. Front. Cell Dev. Biol. 9:771466. doi: 10.3389/fcell.2021.771466
Received: 06 September 2021; Accepted: 04 October 2021;
Published: 12 November 2021.
Edited by:
Andrei Surguchov, University of Kansas Medical Center, United StatesReviewed by:
Irina G. Sourgoutcheva, University of Kansas Medical Center, United StatesFabiana Passaro, University of Naples Federico II, Italy
Copyright © 2021 Shao, Xue and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingming Fang, ZGFmZWlmYW5nQDE2My5jb20=