 Rebekka Kühn1
Rebekka Kühn1 Peter Canoll
Peter Canoll Gunnar Hargus
Gunnar Hargus
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 10 November 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.766773
This article is part of the Research Topic Stem Cell Models of Human Neurological Diseases View all 9 articles
Neurodegenerative dementias are the most common group of neurodegenerative diseases affecting more than 40 million people worldwide. One of these diseases is frontotemporal dementia (FTD), an early onset dementia and one of the leading causes of dementia in people under the age of 60. FTD is a heterogeneous group of neurodegenerative disorders with pathological accumulation of particular proteins in neurons and glial cells including the microtubule-associated protein tau, which is deposited in its hyperphosphorylated form in about half of all patients with FTD. As for other patients with dementia, there is currently no cure for patients with FTD and thus several lines of research focus on the characterization of underlying pathogenic mechanisms with the goal to identify therapeutic targets. In this review, we provide an overview of reported disease phenotypes in induced pluripotent stem cell (iPSC)-derived neurons and glial cells from patients with tau-associated FTD with the aim to highlight recent progress in this fast-moving field of iPSC disease modeling. We put a particular focus on genetic forms of the disease that are linked to mutations in the gene encoding tau and summarize mutation-associated changes in FTD patient cells related to tau splicing and tau phosphorylation, microtubule function and cell metabolism as well as calcium homeostasis and cellular stress. In addition, we discuss challenges and limitations but also opportunities using differentiated patient-derived iPSCs for disease modeling and biomedical research on neurodegenerative diseases including FTD.
Human induced pluripotent stem cells (iPSCs) have been widely used for research on neurological disorders including neurodegenerative diseases. iPSCs are generated from differentiated somatic cells, usually fibroblasts or peripheral blood mononuclear cells, by overexpression of the reprogramming factors Oct4, Klf4, Sox2 and c-Myc (Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Figure 1). They share the main characteristics of embryonic stem cells (ESCs) such as the ability for unlimited self-renewal and the potential to differentiate into cells of all three germ layers: ectoderm, mesoderm, and endoderm (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). Thus, iPSCs provide powerful resources to study the differentiation of pluripotent cells into specialized cells of interest, such as functional neurons (Chambers et al., 2009; Shi et al., 2012; Reinhardt et al., 2013a), astrocytes (Serio et al., 2013; Hallmann et al., 2017; Guttikonda et al., 2021), oligodendrocytes (Stacpoole et al., 2013; Ehrlich et al., 2017) or microglia (Muffat et al., 2016; McQuade et al., 2018; Haenseler and Rajendran, 2019).
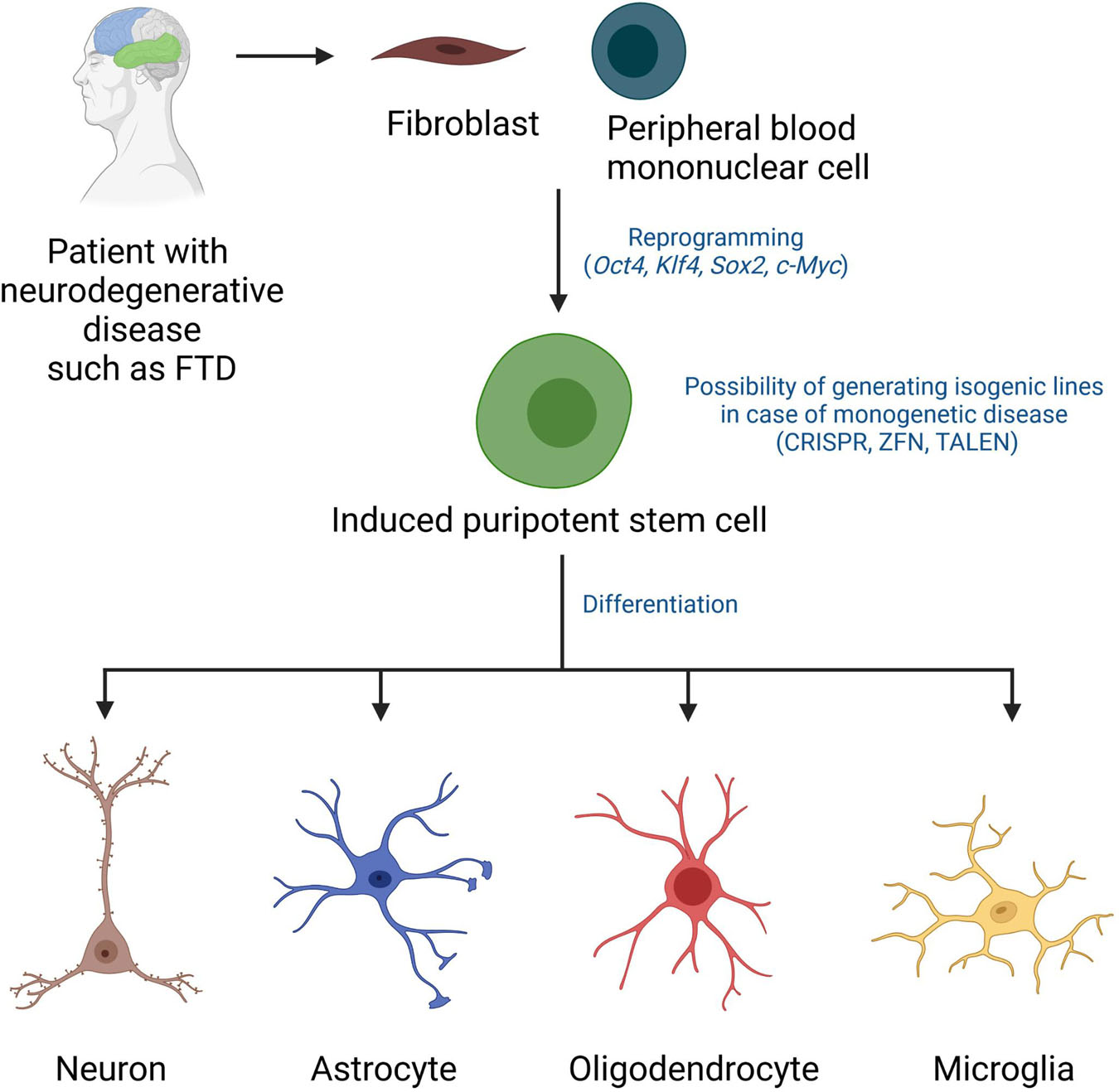
Figure 1. Disease modeling using patient-derived induced pluripotent stem cells. iPSCs can be derived from fibroblasts or peripheral blood mononuclear cells of patients using established reprogramming technologies involving overexpression of the transcription factors Oct4, Klf4, Sox2, c-Myc. Gene editing has been applied in various studies to generate isogenic lines. iPSCs from patients and control cells (isogenic iPSCs or healthy donor-derived iPSCs) can be guided to differentiate into neurons, astrocytes, oligodendrocytes, and microglia using optimized differentiation protocols and they can then be tested in various disease modeling and/or drug screening assays. CRISPR, clustered regularly interspaced short palindromic repeats; ZFN, zinc finger nucleases; TALEN, transcription activator-like effector nucleases.
Induced pluripotent stem cells also represent a unique cell source for high throughput toxicology, vulnerability and drug screens since high numbers of specialized cells can be readily generated from iPSCs, while the isolation of certain living human cells ex vivo (e.g., neurons or glial cells from the human brain) is difficult to accomplish due to technical limitations, lack of tissue availability and ethical concerns. Notably, since iPSCs can be derived from biopsies or blood samples of patients, they serve as unique tools to model patient-specific disease processes in vitro with the goal to identify mechanisms of disease development, to identify therapeutic targets and to develop customized therapies (Hargus et al., 2014). As such, iPSC models have been established to study pathology in various neurodegenerative disorders such as Alzheimer’s disease (AD) (Penney et al., 2020), Parkinson’s disease (Soldner et al., 2009; Hargus et al., 2010; Reinhardt et al., 2013b), Huntington’s disease (Csobonyeiova et al., 2020) or frontotemporal dementia (FTD) (Hargus et al., 2014; Lines et al., 2020).
Frontotemporal dementia represents 5–15% of all cases with dementia and is the second most common group of early onset dementia after AD with an incidence of 2.7–4.1 cases per 100,000 people annually (Knopman and Roberts, 2011; Rademakers et al., 2012). Patients with FTD typically present with progressive degeneration of the frontal and anterior temporal lobes but additional anatomical structures such as the basal ganglia and several brain stem areas are also affected to various degrees. As a consequence, the presentation of FTD as a clinical syndrome is heterogenous and different clinical variants of FTD exist (Neary et al., 1998; McKhann et al., 2001). Patients with the behavioral variant of FTD (bvFTD) present with changes in personality and executive dysfunction including disinhibition, impulsivity, apathy, loss of empathy, repetitive behaviors and dietary changes. A subset of FTD patients is diagnosed with primary progressive aphasia (PPA), a language abnormality which is associated with difficulties in the retrieval of words (semantic variant of PPA; svPPA) or with an impairment of language production (non-fluent variant of PPA; nfvPPA). Oftentimes, features of bvFTD and PPA overlap with disease progression. FTD is also often paralleled by movement abnormalities such as progressive supranuclear palsy (PSP), corticobasal syndrome (CBS), and/or amyotrophic lateral sclerosis (ALS; FTD-ALS) (Olney et al., 2017).
Due to the involvement of the frontal and temporal lobes, FTD is also referred to as frontotemporal lobar degeneration (FTLD) (Figure 2). Autopsy studies have revealed mislocalization and pathological accumulation of particular intracellular proteins in the brains of patients. These proteins include the microtubule-associated protein tau, the transactive response DNA binding protein molecular weight 43 (TDP-43), fused in sarcoma (FUS), Ewing’s sarcoma (EWS) or TATA-binding protein-associated factor 15 (TAF15) (Rademakers et al., 2012; Mackenzie and Neumann, 2016). These findings led to further subclassification of FTLD into FTLD-tau with deposition of hyperphosphorylated tau (∼45% of all FTLD patients), FTLD-TDP with deposition of TDP-43 (∼50–55% of all FTLD patients) and FTLD-FET with deposition of FET proteins (FUS, EWS, and TAF15; ∼6–8% of all FTLD patients) (Figure 2). A small minority of cases (below 1%) is grouped as FTLD-UPS with deposition of ubiquitinated proteins negative for TDP-43, tau and FET proteins (Figure 2A).
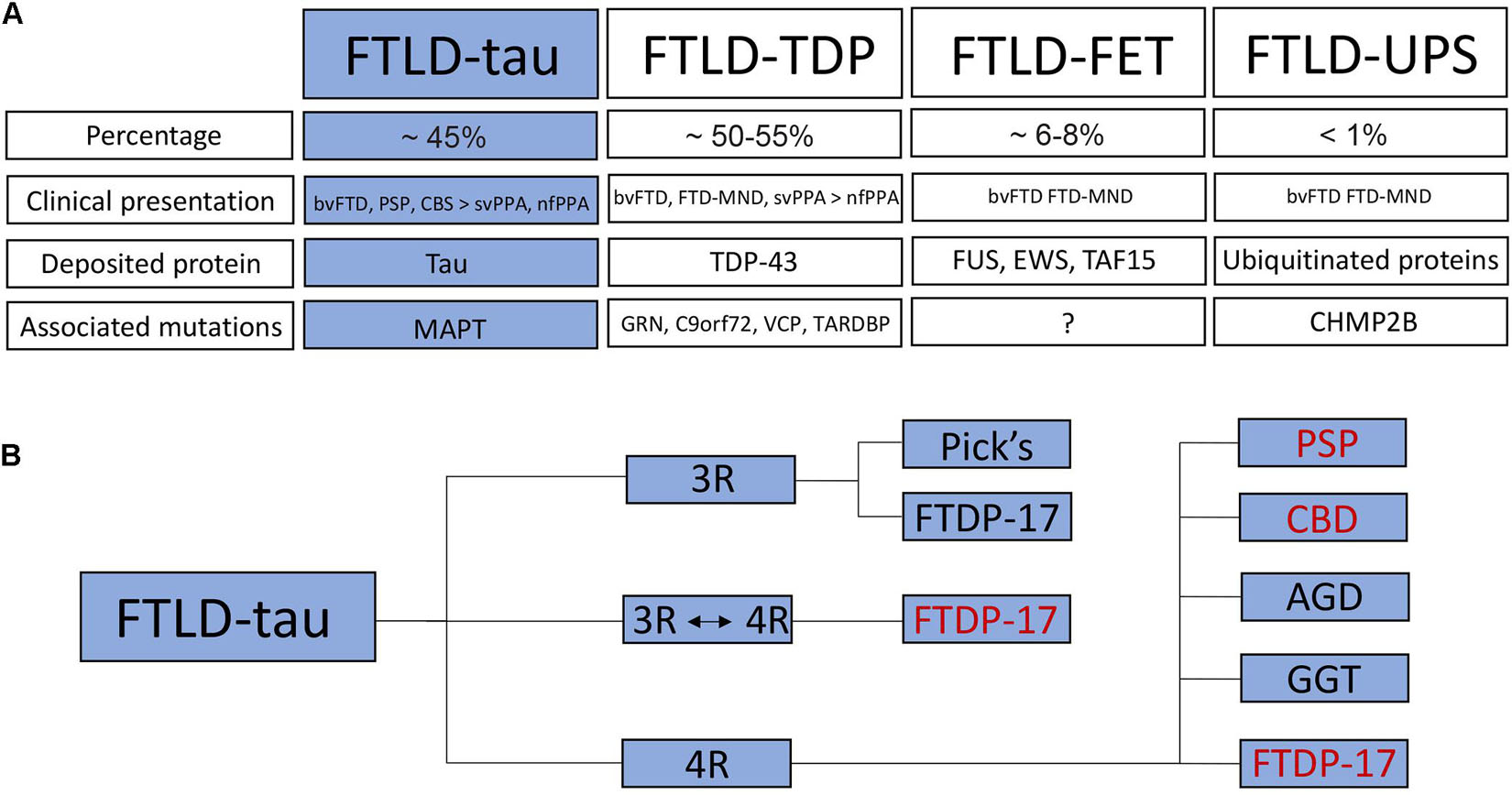
Figure 2. Classification of frontotemporal dementia / frontotemporal lobar degeneration. (A) FTLD is subdivided into FTLD-tau with deposition of hyperphosphorylated tau, FTLD-TDP with TDP-43 pathology, FTLD-FET with deposition of FET proteins as well as FTLD-UPS with deposition of ubiquitinated proteins. The percentage, clinical presentation, deposited proteins and associated mutations are listed for each FTLD subtype. (B) FTLD-tau (highlighted in blue) is further subdivided into 3R and 4R tauopathies as well as a small group of patients with unaltered 3R:4R ratio. Human iPSC models have been established for some FTLD-tau subtypes (in red). bvFTD, behavioral variant of frontotemporal dementia; PSP, progressive supranuclear palsy; CBS, corticobasal syndrome; svPPA, semantic variant of primary progressive aphasia; nfPPA, non-fluent variant of PPA; FTD-MND, FTD with motor neuron disease; TDP-43, transactive response DNA binding protein molecular weight 43; FUS, fused in sarcoma; EWS, Ewing‘s sarcoma; TAF15, TATA-binding protein-associated factor 15; GRN, granulin; C9orf72, chromosome 9 open reading frame 72; VCP, valosin-containing protein; TARDBP, TAR DNA-binding protein 43; CHMP2B, charged multivesicular body protein 2B; Pick’s, Pick’s disease; FTDP-17, Frontotemporal Dementia and Parkinsonism linked to chromosome 17; CBD, corticobasal degeneration; AGD, argyrophilic grain disease; GGT, globular glial tauopathy.
About 20–50% of patients with FTLD have a positive family history and autosomal-dominant patterns of inheritance have been linked to mutations in the following genes/loci associated with the following FTLD subtypes. These genes/loci are MAPT encoding tau causing FTLD-tau, GRN encoding progranulin causing FTLD-TDP, C9orf72 (chromosome 9 open reading frame 72) causing FTLD-TDP with or without motor neuron disease and representing the most common genetic alteration in ALS, VCP encoding the valosin-containing protein causing FTLD-TDP, and CHMP2B encoding the charged multivesicular body protein 2B causing FTLD-UPS. Rare cases of FTLD-TDP have been linked to a mutation in TARDBP encoding TDP-43 (Figure 2A), although these mutations are usually associated with ALS (Mackenzie and Neumann, 2016).
There are currently no effective disease-modifying therapies for patients with FTD and thus, research efforts aim at identifying mechanisms of pathogenesis and developing therapeutic strategies. Studies on postmortem brain tissue of patients have gained very important insights into the underlying pathology providing information on the involvement of the different brain areas and cell types as well as the deposition of aforementioned disease-associated pathological proteins. However, access to patient brain tissue is limited and postmortem brain studies capture a ‘snapshot’ of the final stage of the disease while data on early molecular events and an understanding of the development of the disease over time are difficult to extract. As an alternative, mouse models of FTD have been developed and could recapitulate some of the pathologic findings seen in patients’ brains (Lewis et al., 2000; Allen et al., 2002; Dawson et al., 2007; Roberson, 2012). However, important biological functions such as splicing of endogenous tau into 3R and 4R tau isoforms, a major dysregulated process in many patients with FTLD-tau as described below, cannot be modeled properly in adult mice since they express only 4R tau endogenous isoforms, limiting proper mechanistic studies and functional assays. Differences in biological processes between humans and mice like these may help to explain why drug trials that had shown promising results in studies on neurodegenerative diseases in mice then failed in human trials. In this context, patient-derived iPSCs may provide powerful tools.
In this review, we will provide an overview of human iPSC models of FTLD-tau. FTLD-tau forms with AD the large group of neurodegenerative tauopathies that are characterized by abundant deposition of hyperphosphorylated tau (p-tau) protein in various brain areas. While both AD and FTLD-tau show pronounced tau pathology, there are distinct differences in p-tau deposition between AD and FTLD-tau in regards to affected neuronal and glial cell types and in regards to the spatio-temporal distribution of p-tau aggregates in the brains. Also, in contrast to AD, the formation of β-amyloid-positive plaques is not a diagnostic feature of FTLD-tau. According to the most recent classification of FTLD, FTLD-tau includes non-AD tauopathies such as Pick’s disease (PiD), PSP, corticobasal degeneration (CBD), argyrophilic grain disease (AGD), globular glial tauopathy (GGT) as well as FTLD-tau linked to mutations in MAPT (Mackenzie and Neumann, 2016; Figure 2B).
Tau is a cytoplasmic protein that is mainly found in neurons but at lower levels also in glial cells such as astrocytes and oligodendrocytes. Tau is predominantly expressed in the axons of neurons where it binds to microtubules and controls microtubule polymerization and stabilization (Witman et al., 1976; Drechsel et al., 1992). Tau is essential for microtubule-associated functions such as axoplasmic transport of cargo molecules and organelles, axonal outgrowth, dendritic positioning and neuronal polarity (Kanaan et al., 2011; Noble et al., 2013). In addition, tau protects DNA from heat damage and oxidative stress and regulates neuronal excitability (Noble et al., 2013; Sohn et al., 2019). Tau is encoded by the MAPT gene on chromosome 17q21 that is composed of 16 exons (Figure 3). Alternative splicing of MAPT leads to the formation of six different tau isoforms that contain either 3 or 4 C-terminal microtubule-binding domains depending on alternative splicing of exon 10, which encodes one of these four microtubule-binding domains (3R and 4R tau isoforms) (Goedert et al., 1989). Additional splicing of exons 2 and 3 leads to the formation of three different 3R and three different 4R tau isoforms with either 0 (0N), 1 (1N), or 2 (2N) N-terminal repeats resulting in 0N3R, 1N3R, 2N3R, 0N4R, 1N4R, and 2N4R tau isoforms (Figure 3). In the healthy adult human brain, these 3R and 4R isoforms appear at equal levels (Kosik et al., 1989; Goedert and Jakes, 1990) but in many patients with FTLD-tau including PSP, CBD, AGD, and GGT and including many patients with MAPT mutations, this ratio is shifted toward 4R isoforms (Goedert and Spillantini, 2011; Ghetti et al., 2015; Rosler et al., 2019; Figure 2B). Patients with PiD and a few patients with MAPT mutations present with a decreased 4R:3R ratio, while no changes in the 4R:3R ratio are noted in some patients with FLTD-tau linked to mutant MAPT (Ghetti et al., 2015).
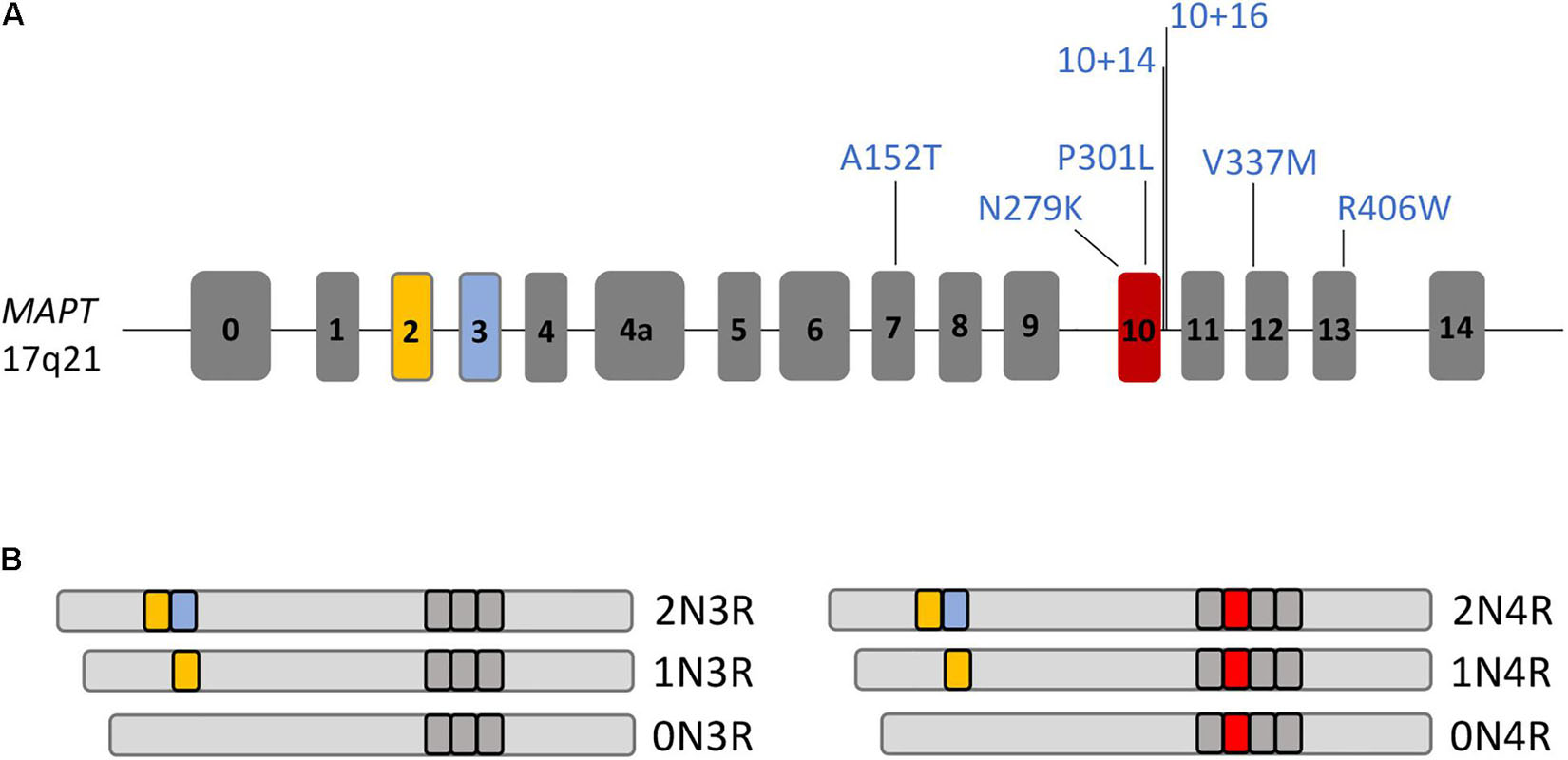
Figure 3. The MAPT gene and its different splice isoforms. (A) Drawing of the MAPT gene composed of 16 exons. MAPT mutations, for which human iPSC models with disease phenotypes have been described, are highlighted. (B) MAPT is spliced into 6 different isoforms in the adult human brain, 0N3R, 1N3R, 2N3R, 0N4R, 1N4R, or 2N4R tau, with presence or absence of exons 2, 3, and 10 in transcripts.
The formation of tau isoforms is developmentally regulated such that the fetal 0N3R isoform appears earliest during development, while the 2N4R tau isoform is the latest to be formed, resulting in the production of all six tau isoforms a few weeks after birth (Goedert et al., 1989). This developmental regulation of tau splicing is also reflected in differentiating human ESCs (Iovino et al., 2010; Sposito et al., 2015) and human iPSCs (Iovino et al., 2015; Sposito et al., 2015). Here, the fetal 0N3R tau isoform appears earliest during differentiation while other tau isoforms are absent. Further maturation over time results in the formation of first the 0N4R isoform with expression of all six isoforms in human iPSC-derived neurons after 150 days (Iovino et al., 2015) and 365 days (Sposito et al., 2015), respectively, of maturation in vitro. In addition, it was shown that tau protein is also secreted into the medium supernatant of human iPSC-derived neurons (Esteras et al., 2021). These findings highlight that human iPSCs are suitable for modeling FTLD-tau with the caveat that prolonged time windows of differentiation might be necessary to capture certain disease phenotypes in patient-derived neurons.
The vast majority of published studies on iPSC models of FTLD-tau is based on reprogrammed fibroblasts from patients with autosomal-dominant mutations in MAPT (Tables 1–4). MAPT mutations were first described in 1998 in affected individuals of 9 families with FTD with genetic linkage to chromosome 17q21 (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). MAPT mutations are autosomal-dominant, usually fully penetrant and represent about 20% of all mutations in familial FTLD (Moore et al., 2020). Male and females are equally affected (Moore et al., 2020). Sixty seven different MAPT mutations have been described so far (Moore et al., 2020) causing early onset FTD and Parkinsonian symptoms in many patients due to the involvement of the substantia nigra and functionally related structures, which resulted in the term Frontotemporal Dementia and Parkinsonism linked to chromosome 17 or FTDP-17 (Ghetti et al., 2015). Patients with MAPT mutations form the youngest group of FTD, with a mean age of symptom onset of 49.5 years and a mean disease duration of 9.3 years resulting in a mean age of death at 58.5 years (Moore et al., 2020). The majority of MAPT mutations are located in exons 9–13 and in the intron following exon 10 (Clavaguera et al., 2013). Among those, the most common mutations are the P301L, N279K and the IVS10 + 16C > T (henceforth termed 10 + 16) mutations that account for up to 60% of all FTDP-17 cases (Arendt et al., 2016; Moore et al., 2020). iPSC models of FTLD-tau revealed phenotypes linked to, besides others, tau pathology, microtubule function, cell metabolism, calcium homeostasis and dysregulated stress pathways. In the following chapters, we review pathologic changes in differentiated FTLD-tau patient iPSCs grouped by genetic background. A summary of disease phenotypes in different patient-derived neural cell types is provided in Figure 4.
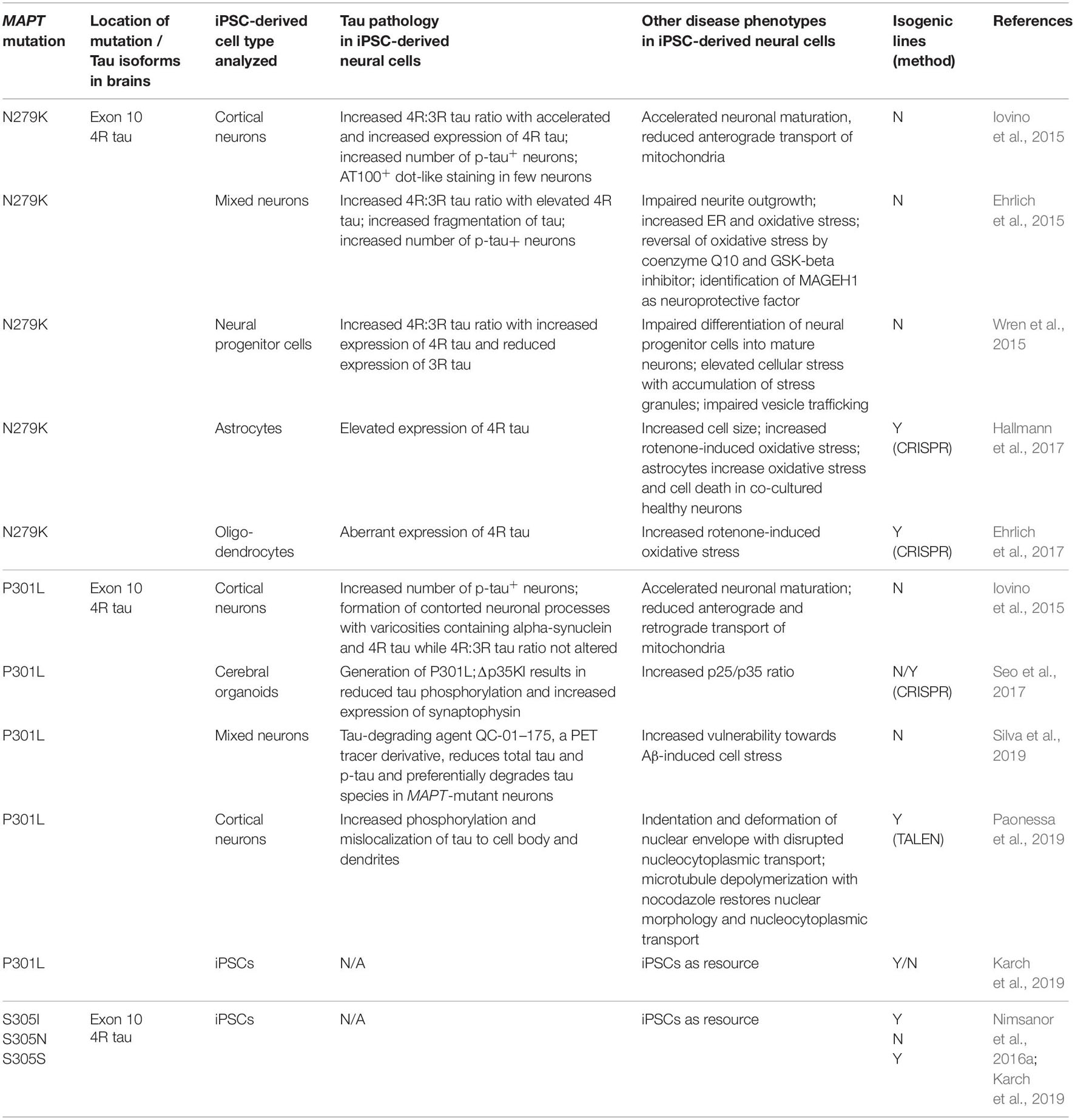
Table 1. Induced pluripotent stem cell models of FTLD-tau linked to MAPT mutations in exon 10.
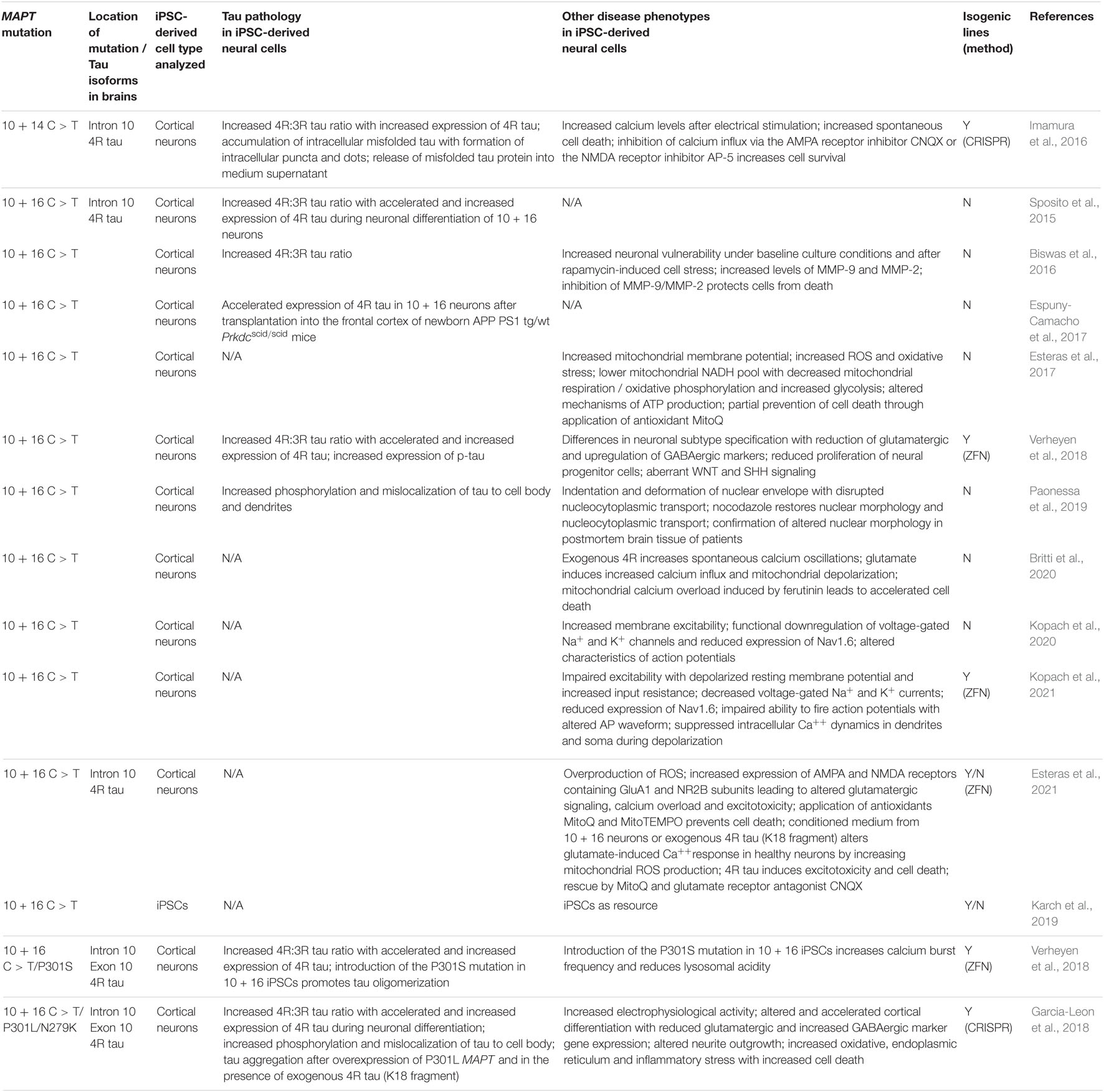
Table 2. Induced pluripotent stem cell models of FTLD-tau linked to MAPT mutations in intron 10.
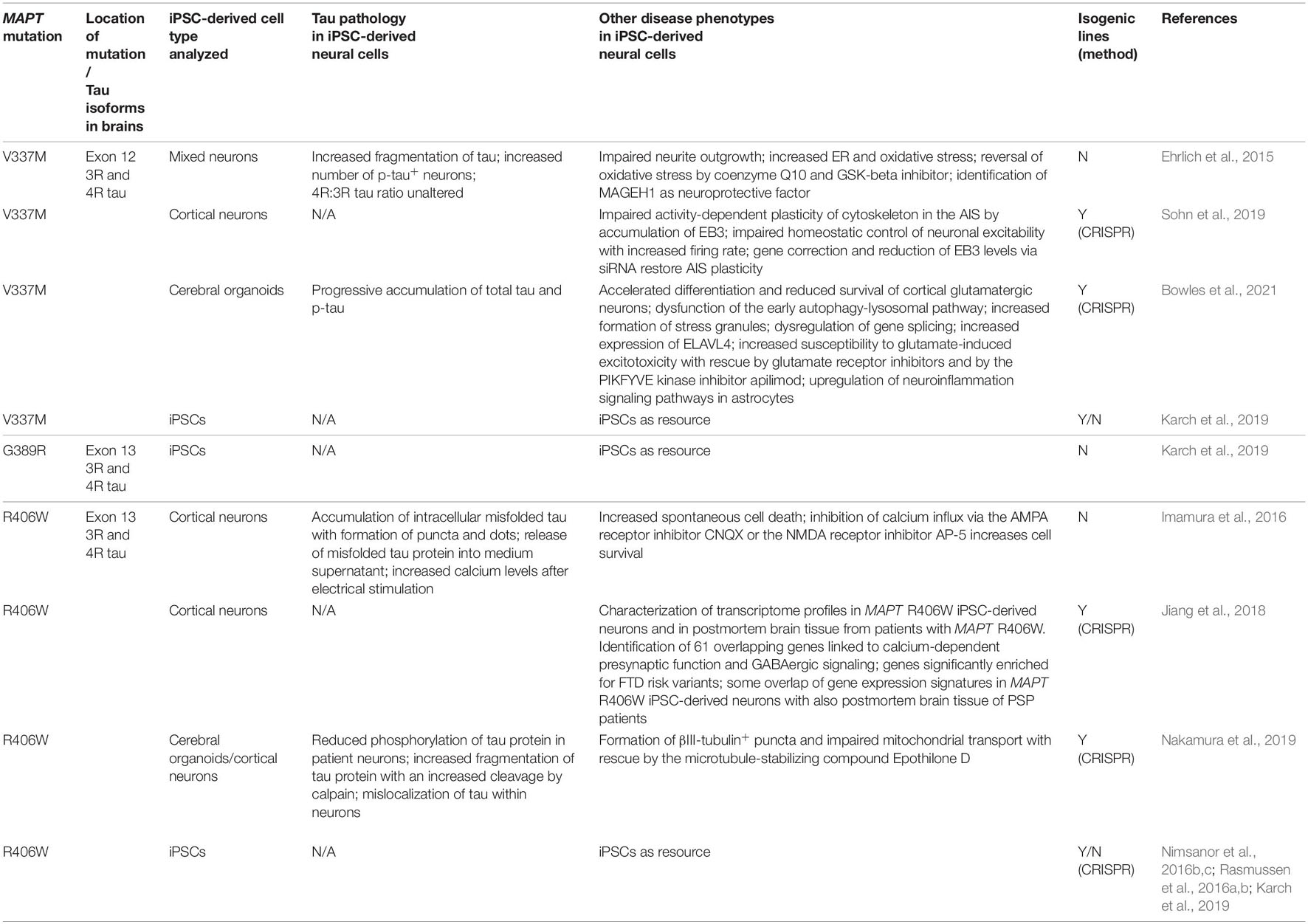
Table 3. Induced pluripotent stem cell models of FTLD-tau linked to MAPT mutations in exons 12 and 13.
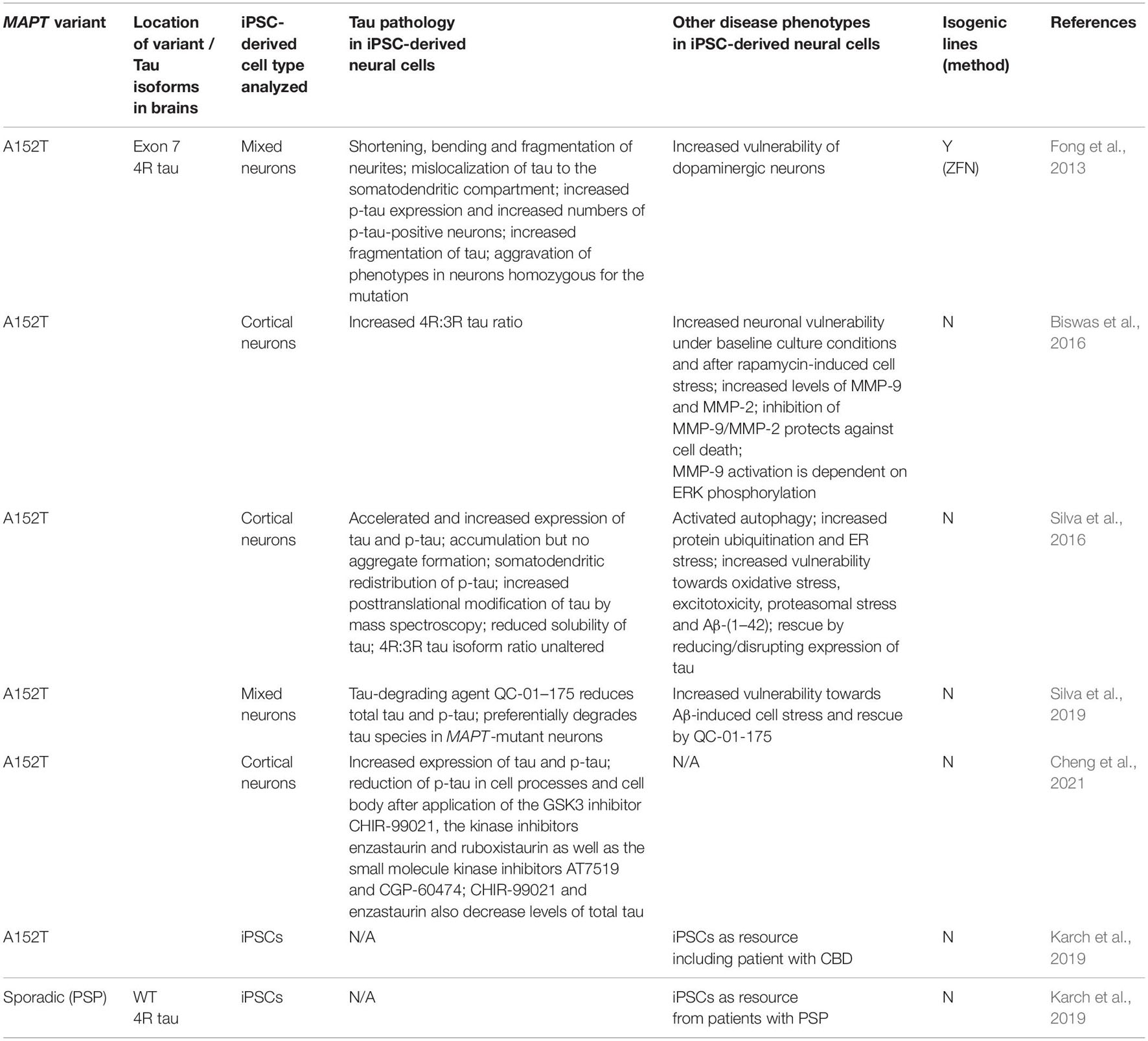
Table 4. Additional induced pluripotent stem cell models of FTLD-tau.
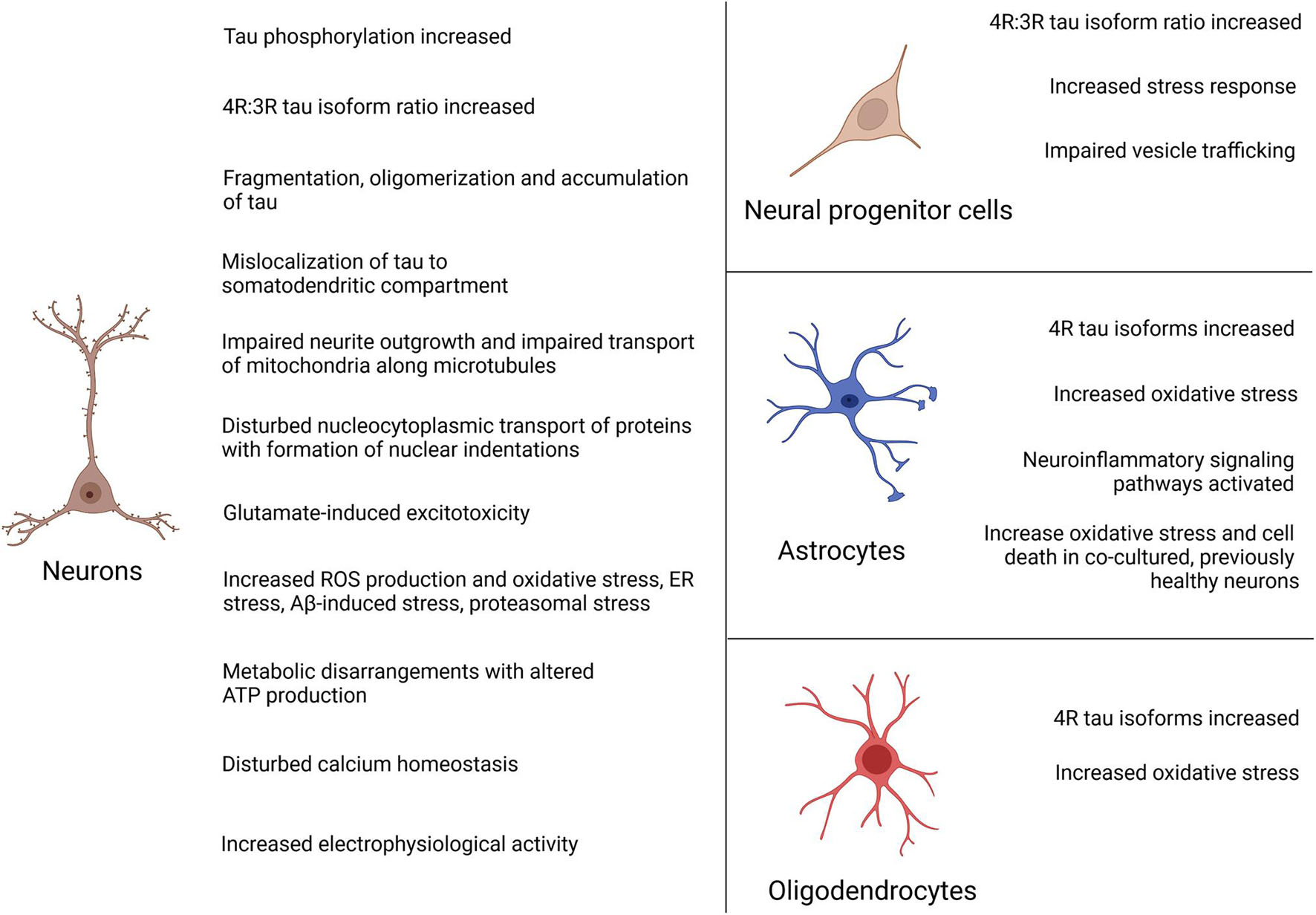
Figure 4. Summary of disease phenotypes in FTLD-tau iPSC-derived neural cells. Disease phenotypes have mainly been described in patient iPSC-derived neurons. In addition, pathologic changes have been reported in neural progenitor cells, astrocytes and oligodendrocytes.
Patients with the N279K mutation have the lowest age of onset of all familial forms of FTLD with a mean age at onset of 43.8 years (Moore et al., 2020). Tau pathology is seen in both neurons and glial cells. This mutation affects both peptide sequence and alternative splicing of the tau gene with increased production of 4R tau (Hasegawa et al., 1999; Kar et al., 2005).
Cultures composed of iPSC-derived neurons with the N279K mutation demonstrated an increased number of p-tau-positive neurons and an increased 4R:3R tau isoform ratio with an increased and accelerated expression of 4R tau (Iovino et al., 2015). This observation was paralleled by the formation of p-tau-positive dot-like aggregates in a subset of neurons (Iovino et al., 2015). Furthermore, an accelerated neuronal maturation and an impairment of intracellular transport along microtubules with significantly reduced anterograde transport of mitochondria was noted (Iovino et al., 2015).
Pronounced tau pathology in N279K patient iPSC-derived neurons was also observed in an independent study (Ehrlich et al., 2015) where patient neurons exhibited changes in the 4R:3R tau isoform ratio with elevated levels of 4R tau and an increased fragmentation of tau already 28 days after neuronal maturation of iPSC-derived neural progenitor cells (NPCs). These neuronal cultures also contained an increased number of p-tau-positive neurons and demonstrated an impairment of neurite outgrowth indicating disturbed microtubule function. Furthermore, N279K mutant neurons were much more susceptible to oxidative stress induced by rotenone, an inhibitor of complex 1 of the mitochondrial respiratory chain. This stress response could, at least in part, be rescued by the antioxidant coenzyme Q10 and the GSK-3 inhibitor CHIR99021, providing proof of the concept that stress responses are targetable and partially reversible in patient neurons. This study also described increased endoplasmic reticulum (ER) stress in patient iPSC-derived neurons and in postmortem patient brain tissue, and identified the cytoplasmic protein MAGEH1 as a neuroprotective molecule in patient neurons in vitro with an increased expression of MAGEH1 also in patient brains.
An increased stress response with an intracellular accumulation of stress granules, increased caspase-3/7 activity and an elevated ratio of 4R tau to 3R tau with an increased 4R tau but reduced 3R tau expression was already observed in iPSC-derived NPCs from patients with N279K MAPT (Wren et al., 2015). Interestingly, these N279K NPCs demonstrated an impaired ability to differentiate into neurons and showed disrupted endocytic trafficking with an accumulation and enlargement of exosomes and endosomes positive for Flotillin-1, a lipid raft marker, with a reduction of lysosomes (Wren et al., 2015). These observations were also confirmed in postmortem brain tissue from patients with mutant N279K MAPT, which showed increased levels of Flotillin-1 in the frontal and temporal cortex, indicating that abnormal vesicle trafficking is a pathogenic mechanism in FTLD-tau (Wren et al., 2015).
Induced pluripotent stem cells from patients with the N279K mutation were also differentiated into NPCs, at which stage CRISPR/Cas9 genome editing was performed to generate isogenic control cells (Hallmann et al., 2017). These patient and isogenic control cells were then differentiated into astrocytes by overexpression of Sox10 and application of pro-astrocytic growth and neurotrophic factors including ciliary neurotrophic factor (Hallmann et al., 2017). In the brains of many patients with mutant MAPT including N279K, astrocytes show pronounced tau pathology (Ghetti et al., 2015). Here, tufted astrocytes (Ghetti et al., 2015; Kovacs, 2020) are formed that demonstrate increased expression of 4R tau and abundant deposition of p-tau in the proximal segments of astrocytic processes. Both N279K patient and control iPSC-derived NPCs differentiated into astrocytes at comparable efficiencies and exhibited typical astrocyte-related properties including an uptake of glutamate from the cell culture medium and propagation of calcium waves across gap junctions (Hallmann et al., 2017). However, patient astrocytes appeared significantly larger, showed an aberrant expression of 4R tau and were much more vulnerable to oxidative stress compared to gene-corrected control cells (Hallmann et al., 2017). Interestingly, concentrations of rotenone that were needed to induce cell death in patient iPSC-derived astrocytes were 10 times higher than those needed to induce death in patient iPSC-derived neurons, reflecting differential vulnerabilities of these neural cells in the human brain. This iPSC-based study also demonstrated astrocyte-mediated non-cell autonomous mechanisms of neuronal degeneration in FTLD-tau. This was highlighted by co-culture of either patient or control astrocytes with healthy iPSC-derived control neurons that consequently acquired an increased susceptibility to oxidative stress in the presence of patient but not control astrocytes. These increased stress responses in previously healthy neurons were paralleled by an enrichment of stress-associated pathways in whole transcriptome analyses, further validating observed findings of cell non-autonomy (Hallmann et al., 2017).
In addition to neurons and astrocytes, oligodendrocytes degenerate in the brains of many patients with FTLD-tau linked to mutant MAPT (Ghetti et al., 2015). Hyperphosphorylated tau is deposited as coiled bodies in oligodendrocytes, most likely contributing to disease pathogenesis. Oligodendrocytes have been differentiated from human iPSCs through an NPC intermediate by overexpression of the transcription factors Sox10, Olig2, and Nkx6.2 and by the application of pro-glial factors and small molecules such as triiodo-L-thyronine (T3), platelet-derived growth factor (PDGF) and smoothened agonist (SAG) (Ehrlich et al., 2017). These oligodendrocytes had the ability to myelinate axons in vitro and to form myelin sheaths 16 weeks after the transplantation into the corpus callosum of newborn myelin basic protein (MBP)-deficient shiverer mice that lack the ability to produce myelin (Ehrlich et al., 2017). In this study, oligodendrocytes were also differentiated from N279K patient NPCs along with their isogenic control cells. These cells were not transplanted into shiverer mice but they showed pathologic changes in vitro with aberrant expression of 4R tau in differentiated patient oligodendrocytes. Furthermore, N279K patient oligodendrocytes were more susceptible to rotenone-induced oxidative stress with an increased expression of cleaved caspase-3 (Ehrlich et al., 2017).
The P301L MAPT mutation is the most common mutation in MAPT. This mutation is located in exon 10 and is a missense mutation (Kar et al., 2005) that makes tau more prone to phosphorylation (Alonso Adel et al., 2004). Furthermore, it is predicted to affect binding of the tau protein to microtubules (Kar et al., 2005; Iovino et al., 2015). Patients with the P301L mutation demonstrate abundant deposition of p-tau in neurons, astrocytes and oligodendrocytes, which is paralleled by increased 4R tau isoform expression (Mirra et al., 1999).
In one of the first reports on FTLD-tau iPSCs, stem cells were derived from two patients with the P301L MAPT mutation that demonstrated an accelerated neuronal maturation with formation of contorted neuronal processes with varicosities that, at least in a subset, contained alpha-synuclein and 4R tau without affecting the 4R:3R tau ratio (Iovino et al., 2015). Cultures composed of P301L neurons contained an increased number of p-tau-positive neurons and P301L neurons demonstrated a disturbed transport machinery along microtubules with reduced anterograde and retrograde transport of mitochondria (Iovino et al., 2015). Later, it was shown that increased phosphorylation of tau in P301L iPSC-derived neurons was accompanied by p-tau mislocalization to the cell body and dendrites (Paonessa et al., 2019). Such mislocalization of p-tau represents an early pathogenic event in tauopathies (Kowall and Kosik, 1987; Gotz et al., 1995) and was paralleled by indentation and deformation of the nuclear membrane in P301L neurons (Paonessa et al., 2019). These morphological changes were mediated by microtubules and resulted in disrupted protein transport across the nuclear membrane. Importantly, microtubule depolymerization with the small molecule nocodazole reduced these indentations, restored round nuclear morphology and rescued impaired nucleocytoplasmic transport in P301L neurons (Paonessa et al., 2019), further indicating that observed effects were directly mediated by microtubules.
Increased phosphorylation of the tau protein in P301L neural cells was mechanistically linked to p25, a proteolytic subunit of p35 and activator of cyclin-dependent kinase 5 (Cdk5) that, besides tau hyperphosphorylation, has been associated with tau aggregation, accumulation of β-amyloid, neuroinflammation and synaptic loss (Noble et al., 2003; Seo et al., 2017). In this study, P301L patient iPSC-derived cerebral organoids demonstrated increased tau phosphorylation that could be significantly reduced by disrupting of p25/Cdk5 activity via CRISPR/Cas9 editing of the p35 locus (Seo et al., 2017). This approach also resulted in an increased expression of synaptophysin in P301L organoids, overall demonstrating that disrupting p25/Cdk5 activity can ameliorate disease phenotypes in FTLD-tau (Seo et al., 2017). Tau phenotypes in P301L neurons could also be reversed with targeted protein degradation technology to transform the tau positron emission tomography (PET) tracer 18F-T807 into the tau degrader QC-01–175 (Silva et al., 2019). This study showed that P301L iPSC-derived neurons were more vulnerable to the aggregation-prone peptide Aβ(1-42) and that QC-01–175 reduced levels of total tau and levels of p-tau in P301L neurons (Silva et al., 2019). Notably, QC-01–175 preferentially degraded tau species in patient-derived neurons, while sparing tau in healthy controls, providing a potential therapeutic approach in neutralizing the neurotoxic effects of tau in tauopathies such as FTLD-tau (Silva et al., 2019).
Several mutations have been reported in intron 10 of MAPT located in the 5′-splice site of the intron following exon 10 (10 + 3, 10 + 11, 10 + 12, 10 + 13, 10 + 14, 10 + 16). These intronic mutations destabilize a stem-loop structure at the exon 10 5′-splice site intron junction resulting in a more frequent usage of the 5′-splice site with altered mRNA splicing of exon 10 and increased levels of 4R tau isoforms (Hutton et al., 1998; Grover et al., 1999).
The MAPT 10 + 16 mutation (Janssen et al., 2002; Lantos et al., 2002) is the most common of these intronic mutations and leads to a two- to six-fold increase of exon 10-containing MAPT mRNA (Connell et al., 2005). As a result, the brains of patients with the 10 + 16 mutation contain p-tau-positive inclusions in neurons, astrocytes and oligodendroglia that are composed of 4R tau (Ghetti et al., 2015).
In a first report on patient-derived iPSCs carrying the 10 + 16 mutation, expression of 4R tau was significantly accelerated during neuronal maturation resulting in an increased 4R:3R tau isoform ratio in these cells (Sposito et al., 2015). 10 + 16 patient neurons expressed the earliest 4R tau isoforms (0N4R) already at 100 days of differentiation, while only the fetal 3R tau isoform (0N3R) was expressed in cultured control neurons at this time (Sposito et al., 2015). Interestingly, 10 + 16 neurons showed an accelerated expression of 4R tau protein also after transplantation into a mouse model of AD (Espuny-Camacho et al., 2017). While no 4R tau expression was seen 2 months after injection of 10 + 16 NPCs into the frontal cortex of newborn APP PS1 tg/wt Prkdcscid/scid mice, grafted 10 + 16 neurons expressed 4R tau at 4 months (37% of grafted neurons) and at 6 months (84% of grafted neurons) after cell injection. In contrast, control human ESC-derived neurons did not express 4R tau until 6 months after transplantation into these mice (Espuny-Camacho et al., 2017). These findings demonstrated that accelerated tau splicing in 10 + 16 neurons occurred during similar time frames in vitro and in vivo. An accelerated expression of 4R tau in MAPT 10 + 16 iPSC-derived neurons with an increased 4R:3R tau ratio was confirmed in additional reports (Biswas et al., 2016; Verheyen et al., 2018).
It was also demonstrated that tau is hyperphosphorylated in 10 + 16 iPSC-derived neurons and that p-tau is mislocalized to the cell body and dendrites (Paonessa et al., 2019). The 10 + 16 neurons presented with nuclear indentations and nuclear deformities with disrupted nucleocytoplasmic transport of proteins, as similarly seen in iPSC-derived neurons carrying the P301L mutation. Likewise, these phenotypes were rescued by addition of the microtubule depolymerizing small molecule nocodazole (Paonessa et al., 2019) highlighting that these different MAPT mutations can lead to similar reversable morphological and functional nuclear phenotypes. Since the authors had access to postmortem brain tissue from several patients with the 10 + 16 mutation, they examined nuclear morphology in cortical brain samples from the frontal and temporal lobes. These studies identified an increased abundance of nuclei with nuclear lamina invaginations in deep cortical layers in these brain areas. Furthermore, invaginations were especially seen in neurons that contained high levels of p-tau and neurofibrillary tangles, validating the iPSC-model of FTLD-tau linked to MAPT 10 + 16 (Paonessa et al., 2019).
Patient-derived 10 + 16 neurons showed decreased survival at baseline culture conditions and demonstrated an increased vulnerability toward rapamycin-induced cell stress, suggesting that the mTOR signaling pathway was functionally altered in these cells (Biswas et al., 2016). Interestingly, increased rapamycin-induced vulnerability was, at least partially, rescued by blockage of the metalloproteinases MMP-9 and MMP-2, which were significantly upregulated and activated in 10 + 16 iPSC-derived neurons and which, by themselves, were able to induce cell death in human iPSC-derived control neurons (Biswas et al., 2016). These findings underscored a detrimental role of MMP-2 and MPP-9 on the survival of neurons, as previously described for MMP-9 for motor neurons in the context of ALS (Kaplan et al., 2014).
Additional studies demonstrated altered mitochondrial function in iPSC-derived neurons from patients with the 10 + 16 mutation (Esteras et al., 2017, 2021). 10 + 16 neurons carried an increased mitochondrial membrane potential leading to overproduction of cytosolic and mitochondrial reactive oxygen species (ROS). This in turn resulted in increased oxidative stress, as indicated by elevated lipid peroxidation, and it led to increased cell death that could be partially prevented by the application of the antioxidant compound MitoQ (Esteras et al., 2017). Interestingly, ATP production was also altered in 10 + 16 neurons. In fact, glycolysis was the main source of ATP in 10 + 16 neurons and ATP production in 10 + 16 neurons was much more dependent on glycolysis as energy fuel when compared to healthy control neurons. On the other hand, 10 + 16 neurons showed lower production of ATP by oxidative phosphorylation, indicating significant disturbances of metabolic programs in these cells (Esteras et al., 2017).
10 + 16 neurons also showed altered electrophysiological properties. Compared to control neurons, 10 + 16 patient-derived neurons demonstrated an increased membrane excitability after about 150 days of differentiation with functional downregulation of voltage-gated Na+ and K+ channels and reduced expression of the sodium channel Nav1.6, which is known to contribute to the initiation and propagation of action potentials (APs) (Kopach et al., 2020). In line with these observations, recordings revealed altered characteristics of action potentials (AP) in these cells. 10 + 16 neurons exhibited a depolarized AP threshold, a reduced amplitude and altered shape of the induced AP spike and the need for an almost two-fold stronger current that was needed to trigger neurons to fire an AP (Kopach et al., 2020). Furthermore, 10 + 16 patient-derived cortical neurons showed increased expression of AMPA and NMDA receptors containing GluA1 and NR2B subunits leading to altered glutamatergic signaling, calcium overload and excitotoxicity (Esteras et al., 2021). The application of mitochondrial antioxidants MitoQ and MitoTEMPO to the cell culture media led to recovery of impaired Ca++ signaling and to prevention of cell death (Esteras et al., 2021) as described for MitoQ before (Esteras et al., 2017).
Interestingly, conditioned medium from patient iPSC-derived 10 + 16 neurons as well as exogenous 4R tau (provided as K18 fragment comprising the four-repeat region of tau) altered glutamate-induced Ca++ response when added to cultures of healthy control iPSC-derived neurons (Esteras et al., 2021). This 4R tau treatment led to increased mitochondrial ROS production, induced excitotoxicity and eventually cell death which could be prevented by adding the antioxidant MitoQ or the glutamate AMPA receptor inhibitor CNQX to cultured neurons (Esteras et al., 2021). In addition, when added to patient iPSC-derived 10 + 16 neurons, exogenous 4R tau (K18 fragment) also increased calcium oscillations and glutamate elevated calcium influx with an increased mitochondrial depolarization (Britti et al., 2020). Increased calcium concentrations in patient neurons had detrimental effects, since mitochondrial calcium overload – induced by ferutinin – led to increased vulnerabiltiy and accelerated cell death in patient 10 + 16 neurons when compared to control neurons (Britti et al., 2020). These iPSC-based studies highlight that disturbed calcium homeostasis plays a critical role in the pathogenesis of FTLD-tau-linked MAPT 10 + 16.
For different sets of studies, zinc finger nuclease (ZFN) technology was applied to introduce the 10 + 16 mutation in iPSCs from a healthy donor and disease phenotypes were analyzed in genetically engineered neurons in comparison to isogenic controls (Verheyen et al., 2018). 10 + 16 mutant cells were generated as heterozygous and homozygous versions, which showed similar disease phenotypes. Those included reduced proliferation of NPCs, an accelerated expression of 4R tau isoforms during neuronal differentiation with an increased 4R:3R tau ratio, an increased expression of p-tau, aberrant WNT and SHH signaling, and significant differences in neuronal subtype specification with a reduction of glutamatergic markers and an upregulation of GABAergic markers in differentiated cortical neurons. In addition, 10 + 16 neurons showed an upregulation of TARDBP encoding the TDP-43 protein and a downregulation of APOE and GRN. Many of these phenotypes were also observed in neurons, that had been differentiated in parallel and for confirmation from a patient carrying MAPT 10 + 16 (Verheyen et al., 2018).
Interestingly, the authors of this paper also introduced a homozygous P301S mutation in homozygous 10 + 16 iPSCs to further aggravate pathologic changes in differentiated neurons (Verheyen et al., 2018). The P301S mutation is a MAPT missense mutation in the same locus as the P301L mutation that leads to tau pathology in neurons and glial cells. Tau pathology linked to P301S encompasses an impaired binding of tau to microtubules, a reduced ability of tau to promote microtubule assembly and an increased expression of 4R tau isoforms (Bugiani et al., 1999; Kar et al., 2005). In line with these known observations, the introduction of the P301S mutation in 10 + 16 iPSCs promoted tau oligomerization, but no aggregation, in differentiated neurons in the presence of recombinant human P301L mutant K18 tau fibrils as seeding ground. These changes were not observed in the absence of the P301S mutation and they confirmed a aggregation-promoting function of P301S tau. These findings were paralleled by reduced acidity of lysosomes and increased apoptosis. Furthermore, live cell calcium imaging revealed a significantly increased calcium burst frequency in 10 + 16 neurons carrying the P301S mutation indicating that the P301S mutation induced changes in neuronal excitability, which may contribute to an increased vulnerability of these cells and which may be linked to seizure activity seen in patients with the P301S mutation (Sperfeld et al., 1999; Verheyen et al., 2018).
In follow-up studies, genetically engineered 10 + 16 iPSC lines lacking the P301S mutation were used to further analyze electrophysiological properties in differentiated 10 + 16 neurons in comparison to isogenic controls (Esteras et al., 2021; Kopach et al., 2021). The results of these studies reproduced many findings from patient-derived 10 + 16 neurons and included an impaired excitability with depolarized resting membrane potential and increased input resistance, decreased voltage-gated Na+ and K+ currents, a reduced expression of Nav1.6, an impaired ability to fire action potentials with altered AP waveforms as well as suppressed intracellular Ca++ dynamics in dendrites and soma during depolarization as visualized by multiphoton fluorescent imaging (Kopach et al., 2021). Furthermore, genetically engineered 10 + 16 neurons demonstrated an increased mitochondrial membrane potential and two-photon excitation imaging showed increased glutamate-induced calcium influx (Esteras et al., 2021). These findings were paralleled by increased cytosolic and mitochondrial ROS production as similarly seen in patient iPSC-derived 10 + 16 neurons (Esteras et al., 2017).
In an unrelated study, CRISPR-FokI and piggyBac transposase technology was applied to introduce heterozygous P301L, N279K, and 10 + 16 MAPT mutations in healthy control iPSCs (Garcia-Leon et al., 2018). Differentiating triple-MAPT-mutant neurons showed an accelerated expression of 4R tau isoforms with an increased 4R:3R tau ratio already at 32 days of differentiation versus 160 days in parental control neurons. The mutant neurons demonstrated increased phosphorylation and mislocalization of tau to the cell body. Interestingly, aggregation of tau was also detected in triple-MAPT-mutant neurons when transduced with an adeno-associated viral vector encoding P301L MAPT. Such tau aggregation was further enhanced in the presence of exogenous 4R tau (K18 fragment). Triple-MAPT-mutant neurons also displayed altered neurite outgrowth, they were electrophysiologically more active when co-cultured on murine primary astrocytes and they showed an altered and accelerated differentiation into cortical neurons with an elevated expression of GABAergic and deep cortical markers and a reduced expression of glutamatergic markers. Triple-MAPT-mutant neurons also showed an elevated stress response signature linked to oxidative stress, ER stress and inflammatory stress with increased spontaneous cell death in culture (Garcia-Leon et al., 2018).
This intronic mutation leads to tau pathology in both neurons and glial cells with increased production of 4R tau (Ghetti et al., 2015). In one published study, iPSCs were generated from a patient with the MAPT 10 + 14 mutation and isogenic control cells were derived using CRISPR/Cas9 technology (Imamura et al., 2016). As seen for neurons carrying the 10 + 16 mutation, 10 + 14 neurons exhibited an increased expression of 4R tau resulting in an increased 4R:3R tau ratio (Imamura et al., 2016). Notably, the authors describe accumulation of intracellular misfolded tau protein with formation of intracellular puncta and dots in 10 + 14 neurons. This misfolded tau was also released into the medium supernatant. 10 + 14 neurons presented with increased calcium levels after electrical stimulation and showed increased spontaneous cell death, which could be prevented by blockage of calcium influx via application of the AMPA receptor inhibitor CNQX or the NMDA receptor inhibitor AP-5 (Imamura et al., 2016), as similarly seen in 10 + 16 neurons. These findings highlight that similar mechanisms may contribute to increased cell vulnerability in patient neurons with different intronic MAPT mutations.
The V337M MAPT mutation is located in exon 12 and is a missense mutation (Spillantini et al., 1996; Kar et al., 2005; Spina et al., 2017). It leads to increased phosphorylation and aggregation of tau and to an impaired binding of tau to microtubules (Alonso Adel et al., 2004; Kar et al., 2005). The V337M mutation causes neuronal, but no glial tau pathology and does not alter the ratio of 3R and 4R tau isoforms in contrast to the aforementioned MAPT mutations in exon 10 and intron 10.
In line with this finding, V337M patient iPSC-derived neurons did not aberrantly express 4R tau isoforms and an altered 4R:3R tau ratio was not seen (Ehrlich et al., 2015). However, and similar to N279K patient neurons, V337M neurons demonstrated an increased fragmentation of tau protein and contained an increased number of p-tau-positive neurons. In addition, V337M mutant neurons had an impairment of neurite outgrowth and were much more susceptible to rotenone-induced oxidative stress, which was partially reversable through GSK-3 inhibition and through the application of coenzyme Q10 (Ehrlich et al., 2015). Interestingly, MAGEH1 was also identified in V337M patient neurons as a neuroprotective molecule, similar to N279K neurons, further supporting its beneficial role in FTLD-tau (Ehrlich et al., 2015).
Induced pluripotent stem cell-derived neurons with the V337M mutation also demonstrated structural and functional alterations of the axon initial segment (AIS), which represents an area within neurons with unique cytoskeletal organization that contains tau-binding molecules and that initiates action potentials (Sohn et al., 2019). In this study, V337M neurons were either derived from FTLD-tau patients with the V337M mutation or by the introduction of the V337M mutation into human iPSCs via CRISPR/Cas9 technology. V337M mutant neurons showed shortening of the AIS and neuronal dysfunction with increased neuronal excitability after depolarization that was directly linked to an increased amount of ‘end-binding protein 3’ (EB3), one of the tau-binding molecules within the AIS. In fact, these alterations were normalized by reducing the levels of EB3 via siRNA, which resulted in elongation of the AIS and in the repair of AIS plasticity, as evidenced by lack of an abnormally increased firing rate upon depolarization (Sohn et al., 2019). Of note, these phenotypes were also rescued by repairing the V337M mutation in patient cells with CRISPR/Cas9 technology, demonstrating that structural and functional AIS pathology was directly driven by the V337M MAPT mutation (Sohn et al., 2019).
In a recent study, an isogenic organoid model of MAPT V337M-associated FTLD-tau was established to study disease-associated changes and mechanisms of early neural degeneration in a three-dimensional context using single cell RNA sequencing among other techniques (Bowles et al., 2021). Patient and gene-corrected iPSC-derived organoids were followed over 6 months of differentiation demonstrating accelerated differentiation of cortical glutamatergic neurons with earlier expression of glutamatergic signalling pathways and synaptic genes at 2 months and a significant reduction of excitatory deep and upper layer cortical neurons but not of interneurons at 4 and 6 months of culture of V337M organoids. These findings demonstrated selective excitatory neuron vulnerability in these organoids (Bowles et al., 2021). V337M organoids also showed a progressive upregulation and accumulation of total tau and p-tau, they demonstrated dysfunction of the early autophagy-lysosomal pathway, and they exhibited increased formation of stress granules. While abnormal splicing of MAPT was not observed, V337M organoids showed splicing dysregulation in genes associated with synaptic signaling and stress granule markers, further demonstrating dysfunctional synaptic maturation and increased stress responses in V337M organoids. Furthermore, V337M organoids presented with an accelerated and increased expression of the RNA-binding protein ELAVL4 that is known to regulate RNA splicing and that was found to bind MAPT RNA and to co-localize with the stress granules in V337M neurons. V337M organoids also demonstrated an increased susceptibility to glutamate-induced excitotoxicity, which could be rescued by the application of glutamate receptor inhibitors and by the PIKFYVE kinase inhibitor apilimod, providing a potential therapeutic target. Interestingly, neuronal disease phenotypes in V337M organoids were paralleled by an astrocyte-mediated neuroinflammatory response with an upregulation of interleukin-6, interleukin-8 and neuroinflammation signaling pathways in V337M astrocytes (Bowles et al., 2021). These findings highlight that iPSC-derived organoids represent an elegant platform to study neuronal but also glial pathology and, potentially, any glia-mediated mechanisms of neuronal degeneration over prolonged periods of time.
This MAPT mutation is located in exon 13. Similar to the V337M mutation, it is a missense mutation that leads to increased phosphorylation, an impaired binding of tau to microtubules and increased aggregation of tau (Reed et al., 1997; Miyasaka et al., 2001; Alonso Adel et al., 2004; Kar et al., 2005). The pathology in the brains of patients is neuronal with formation of neurofibrillary tangles and with an unaltered 3R:4R tau isoform ratio (Ghetti et al., 2015). In addition, clinical and histopathological features of AD with formation of rare Aβ-positive neuritic plaques have been found in patients.
As described above for the MAPT 10 + 14 mutation, tau was misfolded in patient iPSC-derived MAPT R406W neurons. This misfolded tau protein was deposited within neurons as small puncta and dots and it was released into the supernatant of cultured neurons. Pathologic changes in MAPT R406W neurons were also linked to disturbed calcium homeostasis, as these neurons exhibited increased calcium levels after electrical stimulation and demonstrated increased spontaneous cell death linked to elevated calcium levels. In fact, blockage of calcium influx by aforementioned glutamate receptor inhibitors CNQX or AP-5 could prevent cell death in MAPT R406W neuronal cultures (Imamura et al., 2016).
An integrative system biology approach was applied to characterize transcriptome profiles in iPSC-derived neurons from a carrier of the MAPT R406W mutation versus those in isogenic control neurons and in comparison to postmortem brain tissue from FTLD-tau patients carrying the same MAPT R406W mutation (Jiang et al., 2018). The authors found 61 genes which were equally up- or downregulated in both patient iPSC-derived neurons and postmortem insular cortex. Functional annotation of these genes revealed enrichment for pathways involving calcium-dependent pre-synaptic function and GABAergic signaling with downregulation of various GABAergic receptors in both MAPT R406W iPSC-derived neurons and brains. Interestingly, improved gene-set enrichment analysis for GWAS (i-GSEA4GWAS) revealed that these 61 overlapping genes were significantly enriched for FTD risk variants and that SNPs within these 61 genes were more significantly associated with FTD risk than expected by chance (Jiang et al., 2018). In addition, the authors found that differential gene expression signatures in MAPT R406W iPSC-derived neurons also had some overlap with gene expression profiles from postmortem brain tissue of patients with PSP, a related disorder and another form of FTLD-tau, but not with gene expression signatures from postmortem brain tissue of patients with AD or FLD-TDP (Jiang et al., 2018).
Disease phenotypes in R406W neurons were also studied in neurons isolated from cerebral organoids. For this study, the authors dissociated cerebral organoids from R406W iPSCs and isogenic controls 30 days after initial differentiation and replated the neurons for analysis after an additional 30 days (Nakamura et al., 2019). In contrast to reports on neurons carrying other MAPT mutations, the R406W mutation led to reduced phosphorylation of tau in patient neurons with especially poor phosphorylation by GSK3β. Furthermore, patient R406W neurons exhibited increased fragmentation of tau protein with a particular increase in the amount of N-terminal tau fragments. Blocking experiments revealed that this increased fragmentation was driven by calpain cleavage. Tau was also mislocalized from axons to dendrites in patient neurons. In addition, axonal dystrophy with formation of βIII-tubulin-positive puncta and an impaired axonal transport of mitochondria along microtubles were seen in patient neurons. Both of these phenotypes could be rescued by adding the microtubule-stabilizing compound Epothilone D to the media, indicating disturbed but reversible microtubule function in R406W neurons (Nakamura et al., 2019).
Although its role is still under debate, the MAPT A152T variant significantly increases the risk for FTD and AD and it has been described in rare patients with PSP or CBD (Coppola et al., 2012; Kara et al., 2012; Biswas et al., 2016). This MAPT variant is located in exon 7 and it can cause tau pathologic changes with an increase of soluble tau oligomers and a decrease of binding of tau to microtubules (Coppola et al., 2012).
The first iPSCs with the A152T variant were derived by Fong et al. (2013), who also generated isogenic control iPSCs as well as homozygous, bi-allelic mutant iPSCs via ZFN-mediated gene-editing technology. Cultures composed of A152T neurons demonstrated an increased vulnerability of especially dopaminergic neurons as similarly seen in PSP, which can be caused, though rarely, by MAPT A152T (Coppola et al., 2012; Fong et al., 2013). Differentiated A152T neurons also showed pronounced tau pathology, which was further aggravated in neurons homozygous for the variant. Both heterozygous and homozygous A152T neurons exhibited shortening, bending and fragmentation of neurites with a punctate staining pattern of tau. Furthermore, mislocalization of tau to the somatodendritic compartment was observed in neurons that were homozygous for the A152T variant. A152T neuronal cultures showed increased p-tau expression and increased numbers of p-tau-positive neurons. Western blot studies revealed increased proteolysis of tau, which was in part mediated by caspase cleavage. Interestingly, several of these tau pathologic changes were also observed in postmortem brain tissue from a PSP patient carrying MAPT A152T (Fong et al., 2013).
An increased 4R:3R tau isoform ratio was described in A152T patient neuronal cultures (Biswas et al., 2016). In this study, A152T neurons presented with an increased vulnerability under normal culture conditions and in the presence of the mTOR inhibitor rapamycin, as similarly seen for neurons with the 10 + 16 mutation. Likewise, increased rapamycin-induced vulnerability in A152T neurons was, at least partially, reversed by blockage of MMP-9 and MMP-2, which were also significantly upregulated and activated in A152T iPSC-derived neurons (Biswas et al., 2016). This study demonstrated that MMP-9 activation in A152T neurons is dependent on ERK phosphorylation, since inhibition of the ERK pathway decreased MMP-9 expression in these cells. In addition, ectopic expression of 4R tau with the A152T variant activated the ERK pathway to increase MMP-9 expression in HEK293 cells. Taken together, these observations suggested an upstream role of 4R tau on ERK- and MMP-9-mediated vulnerability in patient-derived neurons carrying the A152T variant in MAPT (Biswas et al., 2016).
In an independent study, A152T neurons displayed an accelerated and increased expression of tau with unaltered 4R:3R isoform ratio and of p-tau leading to p-tau accumulation (Silva et al., 2016). While aggregation of tau and tangle formation was not seen, the tau protein in A152T neurons showed reduced solubility and somatodendritic redistribution. Mass spectroscopy confirmed increased posttranslational modification of tau and provided, for the first time, a quantification of allele-specific expression of tau protein in human iPSC-derived neurons. Here, A152T neurons expressed relatively higher levels of A152T tau (about 60%) than non-mutant tau (about 40%) and this ratio remained constant throughout prolonged differentiation. A152T neurons also demonstrated protein expression profiles linked to activated autophagy, increased protein ubiquitination and elevated ER stress and showed an increased vulnerability toward oxidative stress, glutamate-mediated excitotoxicity as well as proteasomal and Aβ-(1–42)-induced stress. Notably, these stress responses could be rescued by reducing or disrupting the expression tau in these neurons (Silva et al., 2016). Also, Aβ(1–42)-mediated stress could also almost completely be prevented by the application of aforementioned tau degrader QC-01–175, as similarly seen in P301L neurons, resulting in more than 70% clearance of tau and p-tau in A152T neurons via proteasomal degradation (Silva et al., 2019).
The same group later applied a high-content imaging assay on A152T and control cortical neurons to screen for kinases and small molecules to reduce tau and p-tau expression as well as their somatodendritic redistribution (Cheng et al., 2021). This study revealed that the GSK-3 inhibitor CHIR-99021 and the kinase inhibitors enzastaurin and ruboxistaurin reduced p-tau in cell processes and cell body in A152T and control neurons and that the small molecule kinase inhibitors AT7519 and CGP-60474, as part of a collection of 44 kinase inhibitors, had similar effects in a screen on A152T neurons. Furthermore, the application of CHIR-99021 and enzastaurin resulted not only in reduced phosphorylation of tau but also in a decrease of total tau in these cells suggesting clearance of tau by these compounds and a potentially beneficial function of kinase inhibitors in patient neurons (Cheng et al., 2021).
Several groups have published resource papers that report on the generation of iPSCs from patients with FTLD-tau without providing details on disease phenotypes in differentiated neural cells. The reports describe iPSC generation from a patient with the S305I MAPT mutation in exon 10 (Kovacs et al., 2008; Nimsanor et al., 2016a) and from a patient and a carrier with the R406W MAPT mutation in exon 13 (Rasmussen et al., 2016a, b) with generation of isogenic lines (Nimsanor et al., 2016b, c). A recent resource study of the TAU consortium reports on several iPSCs from patients with primary tauopathies, especially genetic forms of FTLD-tau, many of which are reviewed in this article (Karch et al., 2019). The authors also list iPSCs as resource from patients or carriers with the S305I (Kovacs et al., 2008), S305N (Hasegawa et al., 1999), and S305S (Stanford et al., 2000) MAPT mutations in exon 10, from patients or carriers with the G389R MAPT mutation in exon 13 (Murrell et al., 1999) as well as iPSCs from a CBD patient carrying the A152T mutation and from two PSP patients without MAPT mutations, providing additional opportunities for stem cell-based disease modeling. Table 5 provides a summary of alternative sources of FTLD-tau iPSCs.
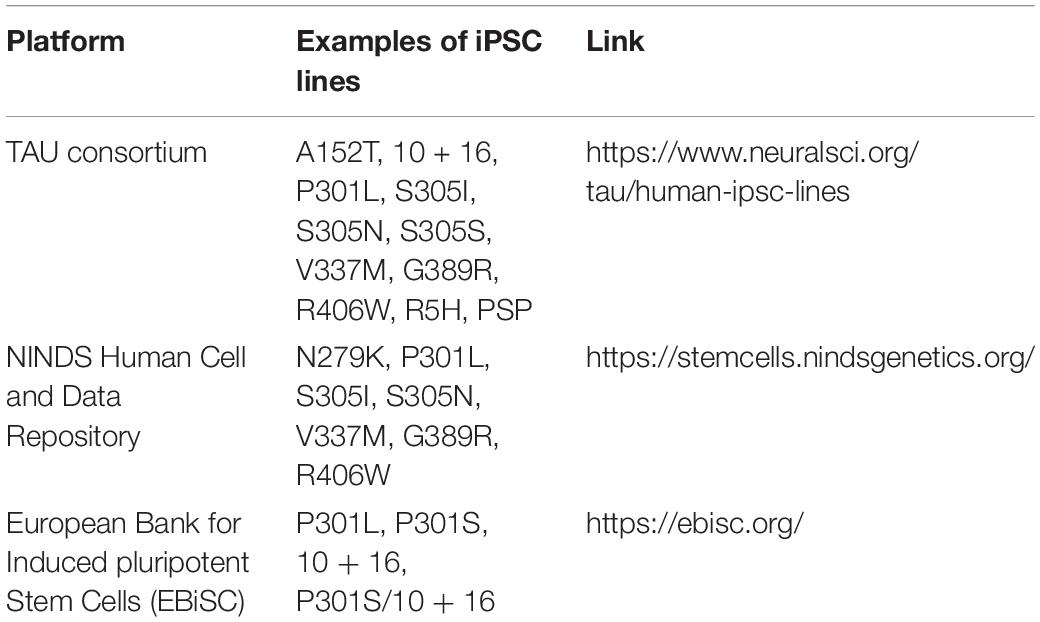
Table 5. Additional sources of FTLD-tau iPSC lines.
As reviewed in this article, iPSC technology allows to study FTLD-tau-associated changes in patient-derived neural cells. Most of the studies were performed on cortical neurons as the main affected cell type in FTLD-tau while few studies focused on other neural subtypes such as dopaminergic neurons, astrocytes or oligodendrocytes, which are also heavily involved by the disease. Main phenotypes included significant tau pathologic changes such as tau hyperphosphorylation, tau mis-splicing with increased and accelerated formation of 4R isoforms in mutations in exon 10 and intron 10, tau fragmentation, oligomerization and accumulation of misfolded tau as well mislocalization of tau to the somatodendritic compartment. They also encompassed disturbed microtubule function in patient neurons with impairment of neurite outgrowth, disturbed nucleocytoplasmic transport of proteins with formation of nuclear indentations as well as dysfunctional anterograde and retrograde transport of mitochondria along microtubules. Also, patient neurons presented with metabolic disarrangements with altered ATP production, an increased electrophysiological activity with formation of dysmorphic action potentials, significant calcium overload and increased glutamate-induced excitotoxicity, increased ROS production as well as pronounced vulnerability toward oxidative, Aβ-induced, proteasomal and ER stress.
Many of these disease phenotypes were reversable providing elegant platforms for high-throughput drug screening for compounds in pre-clinical settings to be able to potentially interfere at an early stage of the disease development process. In fact, human iPSC-derived drug screening platforms have been established (Wang et al., 2017; Cheng et al., 2021) and large numbers of patient-derived neural cells can readily be derived, cultured and tested in such dynamic cellular assays in vitro, in contrast to alternative approaches involving genetic mouse models or postmortem brain material from patients. Such drug screening assays may also involve the co-culture of patient iPSC-derived neurons with patient iPSC-derived glial cells such as astrocytes to test glia- or even neuron-mediated non-cell autonomous mechanism of disease development in neurodegenerative diseases, such as FTLD-tau. In this context, it should be noted that FTLD-tau patient iPSC-derived neurons, e.g., those carrying the 10 + 14 neurons or the R406W MAPT mutation, can secrete misfolded tau into the medium supernatant (Imamura et al., 2016) providing an opportunity study of tau seeding effects and mechanism of tau propagation in neuron-neuron or neuron-glia co-culture paradigms. Refinements of the reprogramming technology and application of genome editing strategies in patient-derived stem cells further improved iPSC-based disease modeling, such that ZFN, TALEN (transcription activator-like effector nucleases) and CRISPR/Cas9 technologies further reduced genetic heterogeneity and thus clonal variabilities for the differentiation of iPSCs into neural cell types of interest. These technologies have been applied in several of the FTLD-tau studies reviewed in this article (see also Tables 1–4).
While these findings highlight the importance of the iPSC technology in biomedical research, some limitations still have to be overcome. For instance, environmental factors are difficult to model and the overall number of patients in iPSC studies is generally low. Also, while the in vitro-differentiation of human iPSCs into neurons recapitulates neurogenesis in vivo with formation of all six tau isoforms over time, an extended differentiation is needed to express all tau isoforms in vitro, which is neither cost- nor time-efficient. As outlined here, many of the disease-associated changes in FTLD-tau neurons were already observed at early stages of differentiation with detection of similar pathologies in postmortem patient brain tissue in some of these studies. However, other typical disease-associated changes in patients’ brains such as the formation of p-tau-positive neurofibrillary tangles in neurons or p-tau-positive aggregates in glial cells have not been detected in human iPSC-derived cells. Thus, an acceleration of differentiation and an induction of premature aging may be necessary to shorten time of culture and to detect additional disease phenotypes in iPSC-derived neurons and glial cells. These approaches may involve the exposure to oxidative stress, introduction of DNA damage or impairment of DNA repair in differentiating cells (Miller et al., 2013), in part also addressing the limitation of lack of environmental cues. The culture of iPSC-derived cells as organoids (Seo et al., 2017; Bowles et al., 2021) and the transplantation of patient neural cells into rodent brains (Hargus et al., 2010) may provide additional alternatives since patient neurons could grow and communicate in a more physiological, three-dimensional context. The application of common immunohistochemical and biochemical tools as well as novel single cell technologies such as single cell proteomics or single cell RNA sequencing, as shown for MAPT V337M organoids (Bowles et al., 2021), may be very helpful in this context to detect previously unrecognized disease-associated changes. In addition, chimeric mice containing transplanted patient-derived neural cells could be used in pre-clinical settings to test for blood-brain-barrier-penetrance and in vivo-efficiency of compounds that had previously provided positive results in aforementioned high-throughput drug screening assays. Direct site-by-site comparisons of early versus aging-induced stages as well as comparisons with three-dimensional assays may be very helpful to further optimize iPSC-based disease modeling.
Induced pluripotent stem cell models of FTLD-tau with increased 4R tau expression, such as those linked to mutations in exon 10 and intron 10, require thorough assessment of 4R and 3R tau expression and their ratio in neural cells for model validation and to derive meaningful conclusions of translational value. While potentially challenging, given the predominant expression of fetal 3R tau in early iPSC-derived neurons, altered 4R:3R tau isoform ratios were reported in various stem cell models (see Tables 1–4 for details). It is possible that time frames of cell differentiation may have to be adjusted for certain differentiation protocols to assess the 4R:3R tau isoform ratio and to extract optimal results for model validation. Also, continual optimization of differentiation protocols for neuronal subtypes and glial cells as well as comparative analyses of phenotypes in different neural cell types, e.g., excitatory versus inhibitory cortical neurons or cortical versus subcortical/brainstem-type neurons and glial cells, are desirable to further strengthen iPSC-based modeling systems.
While disease phenotypes appeared to be robust in summarized studies, larger numbers of patient donors for iPSC generation would further support detection of smaller, yet critically important differences between groups. This is especially important when modeling sporadic forms of neurodegenerative diseases to account for donor- and aforementioned stem cell clone-related variabilities since isogenic control cells cannot be derived. In this context, the MAPT haplotype, either H1 or H2, of donors and stem cell lines could also play an important role since the H1 MAPT haplotype is associated with a higher risk for developing PSP, CBD, AD, and Parkinson’s disease (PD) (Kwok et al., 2004; Myers et al., 2005; Hoglinger et al., 2011; Kouri et al., 2015). Interestingly, allele-specific expression of MAPT transcripts with increased H1 MAPT transcripts has been reported in H1/H2 heterozygous iPSC-derived neurons from disease-free donors (Beevers et al., 2017). On the other hand, a comparative study on healthy-donor iPSC-derived neurons carrying either the H1/H1 or the H2/H2 MAPT haplotype did not reveal major haplotype-specific differences in tau expression but it showed significantly higher levels of conformationally altered MC1-positive insoluble tau in neurons carrying the H2/H2 haplotype, while neurons with the H1/H1 haplotype demonstrated an increased expression of alpha-synuclein that accumulates in neurons in PD (Strauss et al., 2021). Thus, genetic variants can modulate disease-associated processes, which is a very interesting aspect to explore in future iPSC-based studies on sporadic and familial forms of tauopathies including FTLD-tau.
Comparative studies with iPSCs from patients with other forms of FTD/FTLD such as FTLD-TDP may further identify mutual pathways of neurodegeneration such as pathways linked to neuroinflammation. Recent progress in iPSC research led to protocols that efficiently derive microglia from human iPSCs (Muffat et al., 2016; McQuade et al., 2018; Guttikonda et al., 2021). Thus, future studies could involve patient-derived microglia as pure cultures or as co-cultures with neurons and other glial cells as potential important modulators to study the role of neuroinflammation in FTLD-tau. Altogether, these various opportunities clearly highlight the strong value of iPSCs for research on neurodegenerative diseases including FTD/FTLD-tau.
RK, AM, PC, and GH contributed to the writing of this manuscript. All the authors contributed to the article and approved the submitted version.
This work was supported by grants from the NIH including R25 NS070697, R03NS112785, R21AG070414, and K08NS116166 as well as TAME-AD grant GT006988-19 to GH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank James Goldman for his input and critical reading of this manuscript. Figures 1, 4 were created with BioRender.com.
Allen, B., Ingram, E., Takao, M., Smith, M. J., Jakes, R., Virdee, K., et al. (2002). Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 22, 9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002
Alonso Adel, C., Mederlyova, A., Novak, M., Grundke-Iqbal, I., and Iqbal, K. (2004). Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 279, 34873–34881. doi: 10.1074/jbc.M405131200
Arendt, T., Stieler, J. T., and Holzer, M. (2016). Tau and tauopathies. Brain Res. Bull. 126, 238–292. doi: 10.1016/j.brainresbull.2016.08.018
Beevers, J. E., Lai, M. C., Collins, E., Booth, H. D. E., Zambon, F., Parkkinen, L., et al. (2017). MAPT genetic variation and neuronal maturity alter isoform expression affecting axonal transport in iPSC-derived dopamine neurons. Stem Cell Rep. 9, 587–599. doi: 10.1016/j.stemcr.2017.06.005
Biswas, M. H., Almeida, S., Lopez-Gonzalez, R., Mao, W., Zhang, Z., Karydas, A., et al. (2016). MMP-9 and MMP-2 contribute to neuronal cell death in iPSC models of frontotemporal dementia with MAPT mutations. Stem Cell Rep. 7, 316–324. doi: 10.1016/j.stemcr.2016.08.006
Bowles, K. R., Silva, M. C., Whitney, K., Bertucci, T., Berlind, J. E., Lai, J. D., et al. (2021). ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 184, 4547–4563 e4517. doi: 10.1016/j.cell.2021.07.003
Britti, E., Ros, J., Esteras, N., and Abramov, A. Y. (2020). Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 86:102150. doi: 10.1016/j.ceca.2019.102150
Bugiani, O., Murrell, J. R., Giaccone, G., Hasegawa, M., Ghigo, G., Tabaton, M., et al. (1999). Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J. Neuropathol. Exp. Neurol. 58, 667–677. doi: 10.1097/00005072-199906000-00011
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Cheng, C., Reis, S. A., Adams, E. T., Fass, D. M., Angus, S. P., Stuhlmiller, T. J., et al. (2021). High-content image-based analysis and proteomic profiling identifies Tau phosphorylation inhibitors in a human iPSC-derived glutamatergic neuronal model of tauopathy. Sci. Rep. 11: 17029. doi: 10.1038/s41598-021-96227-5
Clavaguera, F., Akatsu, H., Fraser, G., Crowther, R. A., Frank, S., Hench, J., et al. (2013). Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 9535–9540. doi: 10.1073/pnas.1301175110
Connell, J. W., Rodriguez-Martin, T., Gibb, G. M., Kahn, N. M., Grierson, A. J., Hanger, D. P., et al. (2005). Quantitative analysis of tau isoform transcripts in sporadic tauopathies. Brain Res. Mol. Brain Res. 137, 104–109. doi: 10.1016/j.molbrainres.2005.02.014
Coppola, G., Chinnathambi, S., Lee, J. J., Dombroski, B. A., Baker, M. C., Soto-Ortolaza, A. I., et al. (2012). Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum. Mol. Genet. 21, 3500–3512. doi: 10.1093/hmg/dds161
Csobonyeiova, M., Polak, S., and Danisovic, L. (2020). Recent overview of the use of iPSCs huntington’s disease modeling and therapy. Int. J. Mol. Sci. 21:2239. doi: 10.3390/ijms21062239
Dawson, H. N., Cantillana, V., Chen, L., and Vitek, M. P. (2007). The tau N279K exon 10 splicing mutation recapitulates frontotemporal dementia and parkinsonism linked to chromosome 17 tauopathy in a mouse model. J. Neurosci. 27, 9155–9168. doi: 10.1523/JNEUROSCI.5492-06.2007
Drechsel, D. N., Hyman, A. A., Cobb, M. H., and Kirschner, M. W. (1992). Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 3, 1141–1154. doi: 10.1091/mbc.3.10.1141
Ehrlich, M., Hallmann, A. L., Reinhardt, P., Arauzo-Bravo, M. J., Korr, S., Ropke, A., et al. (2015). Distinct neurodegenerative changes in an induced pluripotent stem cell model of frontotemporal dementia linked to mutant TAU protein. Stem Cell Rep. 5, 83–96. doi: 10.1016/j.stemcr.2015.06.001
Ehrlich, M., Mozafari, S., Glatza, M., Starost, L., Velychko, S., Hallmann, A. L., et al. (2017). Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. U.S.A. 114, E2243–E2252. doi: 10.1073/pnas.1614412114
Espuny-Camacho, I., Arranz, A. M., Fiers, M., Snellinx, A., Ando, K., Munck, S., et al. (2017). Hallmarks of Alzheimer’s disease in stem-cell-derived human neurons transplanted into mouse brain. Neuron 93, 1066–1081 e1068. doi: 10.1016/j.neuron.2017.02.001
Esteras, N., Kopach, O., Maiolino, M., Lariccia, V., Amoroso, S., Qamar, S., et al. (2021). Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimers Dement doi: 10.1002/alz.12394
Esteras, N., Rohrer, J. D., Hardy, J., Wray, S., and Abramov, A. Y. (2017). Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox. Biol. 12, 410–422. doi: 10.1016/j.redox.2017.03.008
Fong, H., Wang, C., Knoferle, J., Walker, D., Balestra, M. E., Tong, L. M., et al. (2013). Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 1, 226–234. doi: 10.1016/j.stemcr.2013.08.001
Garcia-Leon, J. A., Cabrera-Socorro, A., Eggermont, K., Swijsen, A., Terryn, J., Fazal, R., et al. (2018). Generation of a human induced pluripotent stem cell-based model for tauopathies combining three microtubule-associated protein TAU mutations which displays several phenotypes linked to neurodegeneration. Alzheimers Dement 14, 1261–1280. doi: 10.1016/j.jalz.2018.05.007
Ghetti, B., Oblak, A. L., Boeve, B. F., Johnson, K. A., Dickerson, B. C., and Goedert, M. (2015). Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46. doi: 10.1111/nan.12213
Goedert, M., and Jakes, R. (1990). Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x
Goedert, M., and Spillantini, M. G. (2011). Pathogenesis of the tauopathies. J. Mol. Neurosci. 45, 425–431. doi: 10.1007/s12031-011-9593-4
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x
Gotz, J., Probst, A., Spillantini, M. G., Schafer, T., Jakes, R., Burki, K., et al. (1995). Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 14, 1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x
Grover, A., Houlden, H., Baker, M., Adamson, J., Lewis, J., Prihar, G., et al. (1999). 5’ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J. Biol. Chem. 274, 15134–15143. doi: 10.1074/jbc.274.21.15134
Guttikonda, S. R., Sikkema, L., Tchieu, J., Saurat, N., Walsh, R. M., Harschnitz, O., et al. (2021). Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 24, 343–354. doi: 10.1038/s41593-020-00796-z
Haenseler, W., and Rajendran, L. (2019). Concise review: modeling neurodegenerative diseases with human pluripotent stem cell-derived microglia. Stem Cells 37, 724–730. doi: 10.1002/stem.2995
Hallmann, A. L., Arauzo-Bravo, M. J., Mavrommatis, L., Ehrlich, M., Ropke, A., Brockhaus, J., et al. (2017). Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci. Rep. 7:42991. doi: 10.1038/srep42991
Hargus, G., Cooper, O., Deleidi, M., Levy, A., Lee, K., Marlow, E., et al. (2010). Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U.S.A. 107, 15921–15926. doi: 10.1073/pnas.1010209107
Hargus, G., Ehrlich, M., Hallmann, A. L., and Kuhlmann, T. (2014). Human stem cell models of neurodegeneration: a novel approach to study mechanisms of disease development. Acta Neuropathol. 127, 151–173. doi: 10.1007/s00401-013-1222-6
Hasegawa, M., Smith, M. J., Iijima, M., Tabira, T., and Goedert, M. (1999). FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 443, 93–96. doi: 10.1016/S0014-5793(98)01696-2
Hoglinger, G. U., Melhem, N. M., Dickson, D. W., Sleiman, P. M., Wang, L. S., Klei, L., et al. (2011). Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 43, 699–705. doi: 10.1038/ng.859
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Imamura, K., Sahara, N., Kanaan, N. M., Tsukita, K., Kondo, T., Kutoku, Y., et al. (2016). Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 6:34904. doi: 10.1038/srep34904
Iovino, M., Agathou, S., Gonzalez-Rueda, A., Del Castillo Velasco-Herrera, M., Borroni, B., Alberici, A., et al. (2015). Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138, 3345–3359. doi: 10.1093/brain/awv222
Iovino, M., Patani, R., Watts, C., Chandran, S., and Spillantini, M. G. (2010). Human stem cell-derived neurons: a system to study human tau function and dysfunction. PLoS One 5:e13947. doi: 10.1371/journal.pone.0013947
Janssen, J. C., Warrington, E. K., Morris, H. R., Lantos, P., Brown, J., Revesz, T., et al. (2002). Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 58, 1161–1168. doi: 10.1212/WNL.58.8.1161
Jiang, S., Wen, N., Li, Z., Dube, U., Del Aguila, J., Budde, J., et al. (2018). Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl. Psychiatry 8:265. doi: 10.1038/s41398-018-0319-z
Kanaan, N. M., Morfini, G. A., LaPointe, N. E., Pigino, G. F., Patterson, K. R., Song, Y., et al. (2011). Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 31, 9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011
Kaplan, A., Spiller, K. J., Towne, C., Kanning, K. C., Choe, G. T., Geber, A., et al. (2014). Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 81, 333–348. doi: 10.1016/j.neuron.2013.12.009
Kar, A., Kuo, D., He, R., Zhou, J., and Wu, J. Y. (2005). Tau alternative splicing and frontotemporal dementia. Alzheimer Dis. Assoc. Disord. 19 Suppl 1, S29–S36. doi: 10.1097/01.wad.0000183082.76820.81
Kara, E., Ling, H., Pittman, A. M., Shaw, K., de Silva, R., Simone, R., et al. (2012). The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol. Aging 33, 2231.e7-2231.e14. doi: 10.1016/j.neurobiolaging.2012.04.006
Karch, C. M., Kao, A. W., Karydas, A., Onanuga, K., Martinez, R., Argouarch, A., et al. (2019). A comprehensive resource for induced pluripotent stem cells from patients with primary tauopathies. Stem Cell Rep. 13, 939–955. doi: 10.1016/j.stemcr.2019.09.006
Knopman, D. S., and Roberts, R. O. (2011). Estimating the number of persons with frontotemporal lobar degeneration in the US population. J. Mol. Neurosci. 45, 330–335. doi: 10.1007/s12031-011-9538-y
Kopach, O., Esteras, N., Wray, S., Abramov, A. Y., and Rusakov, D. A. (2021). Genetically engineered MAPT 10+16 mutation causes pathophysiological excitability of human iPSC-derived neurons related to 4R tau-induced dementia. Cell Death Dis. 12:716. doi: 10.1038/s41419-021-04007-w
Kopach, O., Esteras, N., Wray, S., Rusakov, D. A., and Abramov, A. Y. (2020). Maturation and phenotype of pathophysiological neuronal excitability of human cells in tau-related dementia. J. Cell Sci. 133:jcs241687. doi: 10.1242/jcs.241687
Kosik, K. S., Orecchio, L. D., Bakalis, S., and Neve, R. L. (1989). Developmentally regulated expression of specific tau sequences. Neuron 2, 1389–1397. doi: 10.1016/0896-6273(89)90077-9
Kouri, N., Ross, O. A., Dombroski, B., Younkin, C. S., Serie, D. J., Soto-Ortolaza, A., et al. (2015). Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 6:7247. doi: 10.1038/ncomms8247
Kovacs, G. G. (2020). Astroglia and Tau: new perspectives. Front. Aging Neurosci. 12:96. doi: 10.3389/fnagi.2020.00096
Kovacs, G. G., Pittman, A., Revesz, T., Luk, C., Lees, A., Kiss, E., et al. (2008). MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol. 116, 103–118. doi: 10.1007/s00401-007-0322-6
Kowall, N. W., and Kosik, K. S. (1987). Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann. Neurol. 22, 639–643. doi: 10.1002/ana.410220514
Kwok, J. B., Teber, E. T., Loy, C., Hallupp, M., Nicholson, G., Mellick, G. D., et al. (2004). Tau haplotypes regulate transcription and are associated with Parkinson’s disease. Ann. Neurol. 55, 329–334. doi: 10.1002/ana.10826
Lantos, P. L., Cairns, N. J., Khan, M. N., King, A., Revesz, T., Janssen, J. C., et al. (2002). Neuropathologic variation in frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 58, 1169–1175. doi: 10.1212/WNL.58.8.1169
Lewis, J., McGowan, E., Rockwood, J., Melrose, H., Nacharaju, P., Van Slegtenhorst, M., et al. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405. doi: 10.1038/78078
Lines, G., Casey, J. M., Preza, E., and Wray, S. (2020). Modelling frontotemporal dementia using patient-derived induced pluripotent stem cells. Mol. Cell Neurosci. 109:103553. doi: 10.1016/j.mcn.2020.103553
Mackenzie, I. R., and Neumann, M. (2016). Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J. Neurochem. 138 Suppl 1, 54–70. doi: 10.1111/jnc.13588
McKhann, G. M., Albert, M. S., Grossman, M., Miller, B., Dickson, D., Trojanowski, J. Q., et al. (2001). Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch. Neurol. 58, 1803–1809. doi: 10.1001/archneur.58.11.1803
McQuade, A., Coburn, M., Tu, C. H., Hasselmann, J., Davtyan, H., and Blurton-Jones, M. (2018). Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13:67. doi: 10.1186/s13024-018-0297-x
Miller, J. D., Ganat, Y. M., Kishinevsky, S., Bowman, R. L., Liu, B., Tu, E. Y., et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. doi: 10.1016/j.stem.2013.11.006
Mirra, S. S., Murrell, J. R., Gearing, M., Spillantini, M. G., Goedert, M., Crowther, R. A., et al. (1999). Tau pathology in a family with dementia and a P301L mutation in tau. J. Neuropathol. Exp. Neurol. 58, 335–345. doi: 10.1097/00005072-199904000-00004
Miyasaka, T., Morishima-Kawashima, M., Ravid, R., Heutink, P., van Swieten, J. C., Nagashima, K., et al. (2001). Molecular analysis of mutant and wild-type tau deposited in the brain affected by the FTDP-17 R406W mutation. Am. J. Pathol. 158, 373–379. doi: 10.1016/S0002-9440(10)63979-X
Moore, K. M., Nicholas, J., Grossman, M., McMillan, C. T., Irwin, D. J., Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 19, 145–156. doi: 10.1016/S1474-4422(19)30394-1
Muffat, J., Li, Y., Yuan, B., Mitalipova, M., Omer, A., Corcoran, S., et al. (2016). Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 22, 1358–1367. doi: 10.1038/nm.4189
Murrell, J. R., Spillantini, M. G., Zolo, P., Guazzelli, M., Smith, M. J., Hasegawa, M., et al. (1999). Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J. Neuropathol. Exp. Neurol. 58, 1207–1226. doi: 10.1097/00005072-199912000-00002
Myers, A. J., Kaleem, M., Marlowe, L., Pittman, A. M., Lees, A. J., Fung, H. C., et al. (2005). The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum. Mol. Genet. 14, 2399–2404. doi: 10.1093/hmg/ddi241
Nakamura, M., Shiozawa, S., Tsuboi, D., Amano, M., Watanabe, H., Maeda, S., et al. (2019). Pathological progression induced by the frontotemporal dementia-associated R406W Tau mutation in patient-derived iPSCs. Stem Cell Rep. 13, 684–699. doi: 10.1016/j.stemcr.2019.08.011
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi: 10.1212/WNL.51.6.1546
Nimsanor, N., Jorring, I., Rasmussen, M. A., Clausen, C., Mau-Holzmann, U. A., Kitiyanant, N., et al. (2016a). Induced pluripotent stem cells (iPSCs) derived from a symptomatic carrier of a S305I mutation in the microtubule-associated protein tau (MAPT)-gene causing frontotemporal dementia. Stem Cell Res. 17, 564–567. doi: 10.1016/j.scr.2016.10.006
Nimsanor, N., Poulsen, U., Rasmussen, M. A., Clausen, C., Mau-Holzmann, U. A., Nielsen, J. E., et al. (2016b). Generation of an isogenic, gene-corrected iPSC line from a pre-symptomatic 28-year-old woman with an R406W mutation in the microtubule associated protein tau (MAPT) gene. Stem Cell Res. 17, 600–602. doi: 10.1016/j.scr.2016.09.024
Nimsanor, N., Poulsen, U., Rasmussen, M. A., Clausen, C., Mau-Holzmann, U. A., Nielsen, J. E., et al. (2016c). Generation of an isogenic, gene-corrected iPSC line from a symptomatic 59-year-old female patient with frontotemporal dementia caused by an R406W mutation in the microtubule associated protein tau (MAPT) gene. Stem Cell Res. 17, 576–579. doi: 10.1016/j.scr.2016.09.020
Noble, W., Hanger, D. P., Miller, C. C., and Lovestone, S. (2013). The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 4:83. doi: 10.3389/fneur.2013.00083
Noble, W., Olm, V., Takata, K., Casey, E., Mary, O., Meyerson, J., et al. (2003). Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38, 555–565. doi: 10.1016/S0896-6273(03)00259-9
Olney, N. T., Spina, S., and Miller, B. L. (2017). Frontotemporal dementia. Neurol. Clin. 35, 339–374. doi: 10.1016/j.ncl.2017.01.008
Paonessa, F., Evans, L. D., Solanki, R., Larrieu, D., Wray, S., Hardy, J., et al. (2019). Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep. 26, 582–593 e585. doi: 10.1016/j.celrep.2018.12.085
Penney, J., Ralvenius, W. T., and Tsai, L. H. (2020). Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 25, 148–167. doi: 10.1038/s41380-019-0468-3
Poorkaj, P., Bird, T. D., Wijsman, E., Nemens, E., Garruto, R. M., Anderson, L., et al. (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825. doi: 10.1002/ana.410430617
Rademakers, R., Neumann, M., and Mackenzie, I. R. (2012). Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8, 423–434. doi: 10.1038/nrneurol.2012.117
Rasmussen, M. A., Hjermind, L. E., Hasholt, L. F., Waldemar, G., Nielsen, J. E., Clausen, C., et al. (2016a). Induced pluripotent stem cells (iPSCs) derived from a patient with frontotemporal dementia caused by a R406W mutation in microtubule-associated protein tau (MAPT). Stem Cell Res. 16, 75–78. doi: 10.1016/j.scr.2015.12.006
Rasmussen, M. A., Hjermind, L. E., Hasholt, L. F., Waldemar, G., Nielsen, J. E., Clausen, C., et al. (2016b). Induced pluripotent stem cells (iPSCs) derived from a pre-symptomatic carrier of a R406W mutation in microtubule-associated protein tau (MAPT) causing frontotemporal dementia. Stem Cell Res. 16, 105–109. doi: 10.1016/j.scr.2015.12.012
Reed, L. A., Grabowski, T. J., Schmidt, M. L., Morris, J. C., Goate, A., Solodkin, A., et al. (1997). Autosomal dominant dementia with widespread neurofibrillary tangles. Ann. Neurol. 42, 564–572. doi: 10.1002/ana.410420406
Reinhardt, P., Glatza, M., Hemmer, K., Tsytsyura, Y., Thiel, C. S., Hoing, S., et al. (2013a). Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One 8:e59252. doi: 10.1371/journal.pone.0059252
Reinhardt, P., Schmid, B., Burbulla, L. F., Schondorf, D. C., Wagner, L., Glatza, M., et al. (2013b). Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367. doi: 10.1016/j.stem.2013.01.008
Roberson, E. D. (2012). Mouse models of frontotemporal dementia. Ann. Neurol. 72, 837–849. doi: 10.1002/ana.23722
Rosler, T. W., Tayaranian Marvian, A., Brendel, M., Nykanen, N. P., Hollerhage, M., Schwarz, S. C., et al. (2019). Four-repeat tauopathies. Prog. Neurobiol. 180:101644. doi: 10.1016/j.pneurobio.2019.101644
Seo, J., Kritskiy, O., Watson, L. A., Barker, S. J., Dey, D., Raja, W. K., et al. (2017). Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. J. Neurosci. 37, 9917–9924. doi: 10.1523/JNEUROSCI.0621-17.2017
Serio, A., Bilican, B., Barmada, S. J., Ando, D. M., Zhao, C., Siller, R., et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U.S.A. 110, 4697–4702. doi: 10.1073/pnas.1300398110
Shi, Y., Kirwan, P., Smith, J., Robinson, H. P., and Livesey, F. J. (2012). Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 15, 477–486, S471. doi: 10.1038/nn.3041
Silva, M. C., Cheng, C., Mair, W., Almeida, S., Fong, H., Biswas, M. H., et al. (2016). Human iPSC-derived neuronal model of Tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Rep. 7, 325–340. doi: 10.1016/j.stemcr.2016.08.001
Silva, M. C., Ferguson, F. M., Cai, Q., Donovan, K. A., Nandi, G., Patnaik, D., et al. (2019). Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. elife 8:e45457. doi: 10.7554/eLife.45457.037
Sohn, P. D., Huang, C. T., Yan, R., Fan, L., Tracy, T. E., Camargo, C. M., et al. (2019). Pathogenic Tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104, 458–470 e455. doi: 10.1016/j.neuron.2019.08.008
Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G. W., Cook, E. G., et al. (2009). Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977. doi: 10.1016/j.cell.2009.02.013
Sperfeld, A. D., Collatz, M. B., Baier, H., Palmbach, M., Storch, A., Schwarz, J., et al. (1999). FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann. Neurol. 46, 708–715. doi: 10.1002/1531-8249(199911)46:5<708::AID-ANA5>3.0.CO;2-K
Spillantini, M. G., Crowther, R. A., and Goedert, M. (1996). Comparison of the neurofibrillary pathology in Alzheimer’s disease and familial presenile dementia with tangles. Acta Neuropathol. 92, 42–48. doi: 10.1007/s004010050487
Spillantini, M. G., Murrell, J. R., Goedert, M., Farlow, M. R., Klug, A., and Ghetti, B. (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U.S.A. 95, 7737–7741. doi: 10.1073/pnas.95.13.7737
Spina, S., Schonhaut, D. R., Boeve, B. F., Seeley, W. W., Ossenkoppele, R., O’Neil, J. P., et al. (2017). Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology 88, 758–766. doi: 10.1212/WNL.0000000000003636
Sposito, T., Preza, E., Mahoney, C. J., Seto-Salvia, N., Ryan, N. S., Morris, H. R., et al. (2015). Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum. Mol. Genet. 24, 5260–5269. doi: 10.1093/hmg/ddv246
Stacpoole, S. R., Spitzer, S., Bilican, B., Compston, A., Karadottir, R., Chandran, S., et al. (2013). High yields of oligodendrocyte lineage cells from human embryonic stem cells at physiological oxygen tensions for evaluation of translational biology. Stem Cell Rep. 1, 437–450. doi: 10.1016/j.stemcr.2013.09.006
Stanford, P. M., Halliday, G. M., Brooks, W. S., Kwok, J. B., Storey, C. E., Creasey, H., et al. (2000). Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain 123 (Pt 5), 880–893. doi: 10.1093/brain/123.5.880
Strauss, T., Marvian-Tayaranian, A., Sadikoglou, E., Dhingra, A., Wegner, F., Trumbach, D., et al. (2021). iPS cell-based model for MAPT haplotype as a risk factor for human tauopathies identifies no major differences in TAU expression. Front. Cell Dev. Biol. 9:726866. doi: 10.3389/fcell.2021.726866
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Verheyen, A., Diels, A., Reumers, J., Van Hoorde, K., Van den Wyngaert, I., van Outryve d’Ydewalle, C., et al. (2018). Genetically engineered iPSC-derived FTDP-17 MAPT neurons display mutation-specific neurodegenerative and neurodevelopmental phenotypes. Stem Cell Rep. 11, 363–379. doi: 10.1016/j.stemcr.2018.06.022
Wang, C., Ward, M. E., Chen, R., Liu, K., Tracy, T. E., Chen, X., et al. (2017). Scalable production of iPSC-derived human neurons to identify Tau-lowering compounds by high-content screening. Stem Cell Rep. 9, 1221–1233. doi: 10.1016/j.stemcr.2017.08.019
Witman, G. B., Cleveland, D. W., Weingarten, M. D., and Kirschner, M. W. (1976). Tubulin requires tau for growth onto microtubule initiating sites. Proc. Natl. Acad. Sci. U.S.A. 73, 4070–4074. doi: 10.1073/pnas.73.11.4070
Wren, M. C., Zhao, J., Liu, C. C., Murray, M. E., Atagi, Y., Davis, M. D., et al. (2015). Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol. Neurodegener. 10:46. doi: 10.1186/s13024-015-0042-7
Keywords: frontotemporal dementia (FTD), induced pluriopotent stem cells, tau, disease modeling, frontotemporal lobar degeneration (FTLD), neurodegenenerative diseases, tauopathy, neurodegeneration
Citation: Kühn R, Mahajan A, Canoll P and Hargus G (2021) Human Induced Pluripotent Stem Cell Models of Frontotemporal Dementia With Tau Pathology. Front. Cell Dev. Biol. 9:766773. doi: 10.3389/fcell.2021.766773
Received: 30 August 2021; Accepted: 27 September 2021;
Published: 10 November 2021.
Edited by:
Florian Wegner, Hannover Medical School, GermanyReviewed by:
Jared Sterneckert, Technische Universität Dresden, GermanyCopyright © 2021 Kühn, Mahajan, Canoll and Hargus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gunnar Hargus, Z2gyMzc0QGN1bWMuY29sdW1iaWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.