 Zachary Fralish
Zachary Fralish Ethan M. Lotz
Ethan M. Lotz Taylor Chavez
Taylor Chavez Alastair Khodabukus
Alastair Khodabukus Nenad Bursac
Nenad Bursac- Department of Biomedical Engineering, Pratt School of Engineering, Duke University, Durham, NC, United States
The neuromuscular junction (NMJ) is a specialized cholinergic synaptic interface between a motor neuron and a skeletal muscle fiber that translates presynaptic electrical impulses into motor function. NMJ formation and maintenance require tightly regulated signaling and cellular communication among motor neurons, myogenic cells, and Schwann cells. Neuromuscular diseases (NMDs) can result in loss of NMJ function and motor input leading to paralysis or even death. Although small animal models have been instrumental in advancing our understanding of the NMJ structure and function, the complexities of studying this multi-tissue system in vivo and poor clinical outcomes of candidate therapies developed in small animal models has driven the need for in vitro models of functional human NMJ to complement animal studies. In this review, we discuss prevailing models of NMDs and highlight the current progress and ongoing challenges in developing human iPSC-derived (hiPSC) 3D cell culture models of functional NMJs. We first review in vivo development of motor neurons, skeletal muscle, Schwann cells, and the NMJ alongside current methods for directing the differentiation of relevant cell types from hiPSCs. We further compare the efficacy of modeling NMDs in animals and human cell culture systems in the context of five NMDs: amyotrophic lateral sclerosis, myasthenia gravis, Duchenne muscular dystrophy, myotonic dystrophy, and Pompe disease. Finally, we discuss further work necessary for hiPSC-derived NMJ models to function as effective personalized NMD platforms.
Introduction
Neuromuscular diseases (NMDs) are a broadly defined group of disorders that lead to progressive impairment of motor function. The NMDs primarily involve dysfunction of motor neurons (MNs), skeletal muscle (SkM), or their synaptic connection, the neuromuscular junction (NMJ). Different NMDs have distinct tissue origins such as MNs [e.g., amyotrophic lateral sclerosis (ALS) either from direct loss or retrograde degeneration of MNs], muscle [e.g., Duchenne muscular dystrophy (DMD) or myotonic dystrophy (DM)], NMJs [e.g., myasthenia gravis (MG) or congenital myasthenic syndromes (CMS)], or a combination thereof (e.g., Pompe disease). Regardless of the origin, the structural and/or functional deficit in a targeted tissue will resonate throughout the entire motor unit, leading to multiple shared symptoms among different NMDs. NMDs are estimated to affect 160 per 100,000 people worldwide (Deenen et al., 2015); however, despite this high prevalence, outcomes are often fatal as few curative treatments are available. Therefore, comprehensive biomimetic and clinically predictive in vitro and in vivo NMD models are essential for accelerating our understanding of the underlying disease mechanisms and development of effective therapeutics (Babin et al., 2014; Aartsma-Rus and van Putten, 2019). Animal models have been invaluable to our current understanding of NMDs as they capture important clinical features of the disease. Still, currently available animal models do not fully recapitulate the diverse range of disease phenotypes nor disease severity due to the complex genetic and non-genetic nature of human NMDs.
In recent years, high clinical and genetic heterogeneity of NMDs has prompted the considerations of personalized approaches to study and treat these devastating diseases. Historically, investigations of human NMDs have been hindered due to difficulty imaging NMJs in vivo, limited capability to isolate neural stem cells, and the postmitotic nature of adult MNs complicating in vitro studies. Over the past decade, advances in human induced pluripotent stem cell (hiPSC) technology, have provided a novel source of human somatic cells for pre-clinical research. hiPSCs can be generated from ethical and accessible sources, such as the skin and blood, and function as a potentially unlimited, patient-specific source of traditionally inaccessible cells such as MNs and cardiomyocytes. Additionally, hiPSC-derived cells can be integrated into two- (2D) and three-dimensional (3D) culture systems to enable novel studies of human development, disease, and pharmacology. hiPSC-based derivations of MNs and SkM, in particular, provide easily accessible, highly expandable sources of patient-specific NMD-relevant tissues. Notably, these platforms complement animal models creating an efficient and predictive system for patient specific NMD modeling and drug development. Incorporation of hiPSC-derived MNs and SkM into 3D cultures and organ-on-a-chip systems adds necessary structural complexity and genetic and environmental control over the cell-specific behavior. These tissue-engineered motor units recapitulate the nature of functional NMJs offering potential for improved mechanistic understanding of complex NMDs (Osaki et al., 2018; Bakooshli et al., 2019; Vila et al., 2019; Faustino Martins et al., 2020; Rimington et al., 2021).
In this review, we first explore the individual components of the NMJ including how they interact and contribute to NMJ functionality. We then compare current animal and in vitro hiPSC models of NMDs, focusing on ALS, MG, DMD, DM, and Pompe disease. We end by discussing the future of NMD modeling and strategies to address limitations in creating in vitro functional motor units that would allow predictive, patient-specific studies and treatment of NMDs.
Development
Understanding embryonic development of NMJs and their physiological roles is important to critically analyze methods to derive relevant cell types from hiPSCs and to compare biomimetic nature and effectiveness of the current and future tissue-engineered NMD models. Therefore, we provide an overview of MN, SkM, and Schwann cell (SC) development and draw parallels to current methods to differentiate these cells from hiPSCs. We finalize this section by describing NMJ development and function.
Motor Neuron Development
Over the past decade, methods to differentiate hiPSCs into neuronal cells has rapidly progressed due to increased understanding of early neural development and commitment of neuroprogenitor cells to highly specialized neural subtypes, including MNs (Deenen et al., 2015). MNs are found throughout the CNS and can be divided into upper MNs (UMNs) or lower MNs (LMNs), which, despite their shared nomenclature, are developmentally and genetically distinct. Different NMDs, such as primary lateral sclerosis, progressive muscular atrophy, or ALS, can target either or both of UMNs and LMNs (Liewluck and Saperstein, 2015) making their distinction an important consideration when modeling NMDs. UMNs originate from the pre-motor and primary motor regions of the cerebral cortex. Their axons form glutamatergic connections with LMNs located in the brainstem and ventral horn of the spinal cord. Axons of LMNs project beyond the CNS forming cholinergic synapses with multiple tissue types to control a wide variety of physiological processes. As a result of these regional differences, the genetic and molecular events leading to MN development diverge early. Therefore, it is important to consider the development of the CNS in its entirety and understand the molecular mechanisms underlying MN diversity to develop physiologically relevant models for NMDs.
Cells of the developing vertebrate nervous system are derived from the ectoderm which forms during gastrulation (Figures 1A–C). Inhibition of TGFβ and FGF initiates neurulation causing the ectoderm to fold inward generating three new regions: (1) neural tube, (2) neural crest, and (3) external ectoderm (Ozair et al., 2013). Each region contains cell progenies restricted to a limited number of distinct fates, and those found in the neural tube are destined to form the brain and spinal cord. Signaling molecules from the mesodermal notochord coordinate the formation of the neural tube in the proper spatial orientation along the rostral–caudal and dorsal–ventral axes (Muhr et al., 1999). After neurulation, morphogen production and its subsequent spatio-temporal organization along the two axes causes axial patterning of the neural tube responsible for regional specification of neural subtypes (Wichterle et al., 2002; Li et al., 2005).
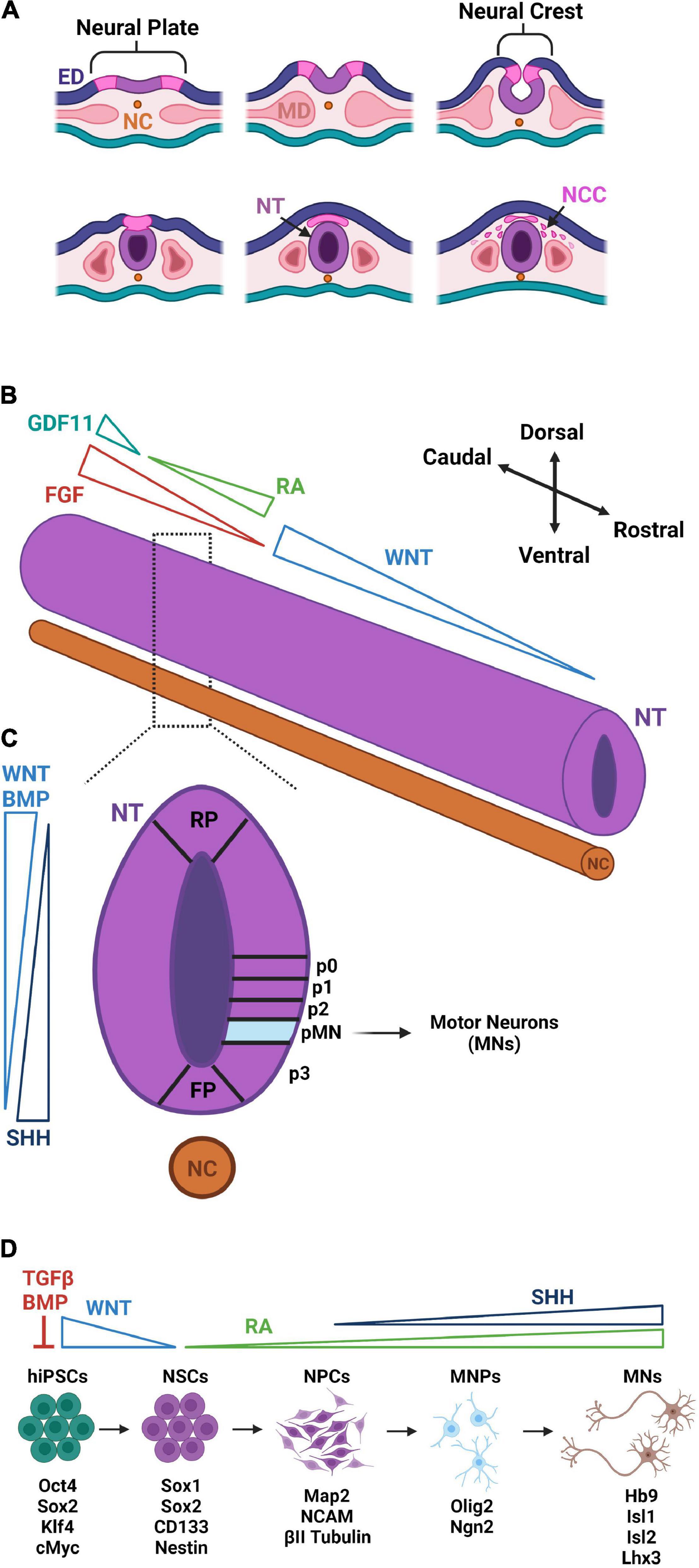
Figure 1. Early development and hiPSC-based differentiation of motor neurons. (A) After the notochord (NC) signals inward folding of the ectoderm at the neural plate, the neural crest is brought together forming the neural tube (NT). The neural crest then forms neural crest cells (NCCs) which differentiate in the peripheral nervous system. The cells of mesoderm (MD) differentiate into somites and, eventually, the musculoskeletal system. (B) Along the rostral–caudal axis of the NT, WNT gradients dictate regionalization of the brain and RA/FGF/GDF11 gradients dictate segmentation of the spinal cord. (C) Along the dorsal–ventral axis of the NT, antithetical WNT/BMP [derived from the roof plate (RP)] and SHH gradients [derived from the floor plate (FP)] dictate patterning and the formation of the five ventral progenitor cell domains (p0, p1, p2, pMN, and p3). The pMN domain is the source of subsequent MN specification. (D) hiPSC-derivations of MNs begin with dual SMAD inhibition of TGFβ and BMP pathways to trigger neural stem cell (NSC) differentiation. WNT and RA signaling direct NSC differentiation into neural progenitor cells (NPCs) of the spinal cord region. With the addition of SHH signaling, NPCs further differentiate to Olig2-expressing MN progenitors (MNPs). Suppression of Olig2 and upregulation of Ngn2 commit MNPs to a post-mitotic MN lineage that express Hb9, Isl1, Isl2, and Lhx3. Distinct colors are used to denote approximate correspondence between stages of hiPSC differentiation in panel (D) and embryonic development in panel (C).
Regionalization is first specified in the brain with cells assuming a rostral forebrain identity in the absence of morphogens. These cells continue to develop into the neurons comprising the telencephalic region of the brain, which houses the cerebral cortex where mature UMNs reside (Watanabe et al., 2005). The remaining cells are driven caudally in response to a WNT gradient established by dorsal roof plate cells. This gradient dictates caudal forebrain, midbrain, and hindbrain identities (Nordstrom et al., 2002). LMNs with a distinct spinal character require further caudalization. A major contributing factor to this LMN caudalization is retinoic acid (RA) signaling that leads to rostral identities of the cervical and upper thoracic spinal segments. Presomitic cells of the surrounding paraxial mesoderm convert retinaldehyde to RA via their expression of aldehyde dehydrogenase 1 A2 (ALDH1A2) (Liu et al., 2001). Decreased ALDH1A2 expression is found caudally and corresponds to decreased RA signaling (Liu et al., 2001) and increased FGF signaling, which govern the caudalization of neural precursors to identities of thoracic and lumbar spinal segments (Irioka et al., 2005). High expression of FGF alongside an increasing gradient of GDF11 dictate pattern a sacral spinal identity associated with the most caudal region of the developing spinal cord (Diez del Corral and Storey, 2004). Differential expression of HOX-family genes corresponds to cervical (HOX4 – HOX6), thoracic (HOX8 and HOX9), and lumbar (HOX10 and HOX11) positional identities along the spinal cord (Dasen and Jessell, 2009).
Neural precursors are also subject to dorsal–ventral patterning concurrent to rostral–caudal patterning. WNTs and BMPs derived from roof plate cells mediate dorsal patterning (Son et al., 2011). In contrast, exposure to increasing concentrations of sonic hedgehog (SHH) secreted by floor plate cells drives ventral patterning (Ericson et al., 1996). Dorsal–ventral patterning for the development of UMNs remains poorly understood. On the other hand, ventral positioning of LMN progenitors is known to require a coordinated balance between the antagonizing effects of BMPs/WNTs and SHH (Jessell, 2000). The ventral spinal cord consists of five domains that further restrict neural progenitors to a specific lineage. Interestingly, the MN progenitor (pMN) domain required for LMN specification is also required for oligodendrocyte specification (Ravanelli and Appel, 2015). OLIG2 expression is the earliest marker used to identify progenitors committed to the pMN domain (Ravanelli and Appel, 2015). Over time, oligodendrocyte progenitors will continue to express OLIG2 while committed MN progenitors will begin to express NGN2 which represses OLIG2 expression (Ravanelli and Appel, 2015). Continued expression of NGN2 induces HB9, signifying the formation of a post-mitotic MN (Lee et al., 2009).
Lower motor neurons at this stage are referred to as having a general character until organized into distinct motor columns that correspond to targeted regions of innervation. These regions include the median motor column (MMC), which innervates axial SkM, spinal accessory column (SAC), which innervates the branchial SkM of the face and neck, phrenic motor column (PMC), which innervates the SkM of the diaphragm, lateral motor column (LMC), which innervates appendage muscle, hypaxial motor column (HMC), which innervates intercostal and abdominal SkM, and preganglionic motor column (PGC), which synapse onto ganglionic neurons of the autonomic nervous system (ANS) (Nicolopoulos-Stournaras and Iles, 1983). Each motor column is organized along the rostro-caudal axis and is identified by a unique gene signature (Francius and Clotman, 2014).
Generation of Motor Neurons From Pluripotent Stem Cells
Over the past decade, a variety of protocols have been used to derive MNs from hiPSCs. These methods vary in length and efficiency; however, they are designed on the genetic and molecular principles of embryonic development (Figure 1D). Historically, neural induction was performed in suspended aggregates of hiPSCs, called embryoid bodies (EBs), in serum-free media without exogenous morphogens. Under these conditions, differentiating hiPSCs undergo spontaneous FGF and BMP inhibition, which naturally guides their differentiation to a neural fate (LaVaute et al., 2009). These early protocols were long and inefficient, often producing high experimental variability. Dramatic improvements came with the discovery that early inhibition of BMP and TGFβ signaling through SMAD inhibition selectively blocks the formation of mesodermal and endodermal cell fates leading to higher percentages of PAX6 and SOX2 expressing neural progenitors at earlier time points (Chambers et al., 2009). Based upon this discovery, dual-SMAD inhibition is now standard practice for the neuralization of hiPSCs in both monolayer cultures as well as EBs. The small molecule, SB431542, is the most utilized TGFβ inhibitor, and is commonly paired with a small molecule inhibitor of BMP (LDN193189, DMH1, or dorsomorphin) or recombinant Noggin, a naturally occurring BMP inhibitor.
After neuralization, neural precursors are committed to MN progenitors by following common patterning principles of caudalization and ventralization. MN differentiation efficiency and culture length have been improved by optimizing the concentrations and timing of patterning morphogens like WNTs, BMPs, RA, FGFs, and SHH (Nordstrom et al., 2002). Although experimental reproducibility has been improved, the variability and length of derivation protocols has remained a challenge. Specifically, the general application of RA and SHH for MN differentiation has been inefficient with yields ranging between 30 and 60% over a culture duration of 21–40 days (Hu and Zhang, 2009; Hester et al., 2011). Early activation of WNTs with CHIR99021 (CHIR) was shown to significantly improve MN differentiation efficiency and speed resulting in an 80% yield in 14 days (Maury et al., 2015). Moreover, when CHIR was continuously added throughout the differentiation, 90% of cells became mature MNs within 12 days (Du et al., 2015). This continued activation of WNTs stabilized excessive ventralization, maintaining a higher population of cells in the pMN domain and reducing the population of NKX2.2 expressing interneuron progenitors of the p3 domain (Du et al., 2015). While expedited protocols for MN generation may benefit cell manufacturing and screening, how accurately “fast-tracked” methods recapitulate adult MN cell physiology and maturation remains unclear.
Skeletal Muscle Development
Development of skeletal muscle (SkM) begins with the paraxial mesoderm (Wachtler, 1992). The paraxial mesoderm (PM) forms in the primitive streak/blastopore during gastrulation and is comprised of two bilateral strips of presomitic mesoderm (PSM) flanking the neural tube and notochord (White et al., 2005; Figure 2A). Cells acquiring the PM fate require suppression of BMP signaling in vivo (Winnier et al., 1995). In the posterior compartments of the PM, there is an unsegmented progenitor zone comprised of neuromesodermal progenitors (NMP) (Tzouanacou et al., 2009) and other progenitor cells which give rise to the paraxial mesoderm, neural tube derivatives, lateral plate derivatives, and notochord (Takemoto et al., 2011; Garriock et al., 2015). Cells within the progenitor zone develop into skeletal muscle progenitor cells as a result of WNT and FGF signaling gradients which target transcription factors essential for PSM specification and patterning such as brachyury (T), Tbx6, and Msgn1 (Ciruna and Rossant, 2001; Nowotschin et al., 2012). Differentiated cells acquire the identity of mesoderm progenitor cells (MPCs) within the most posterior region of the PSM (Chalamalasetty et al., 2014).
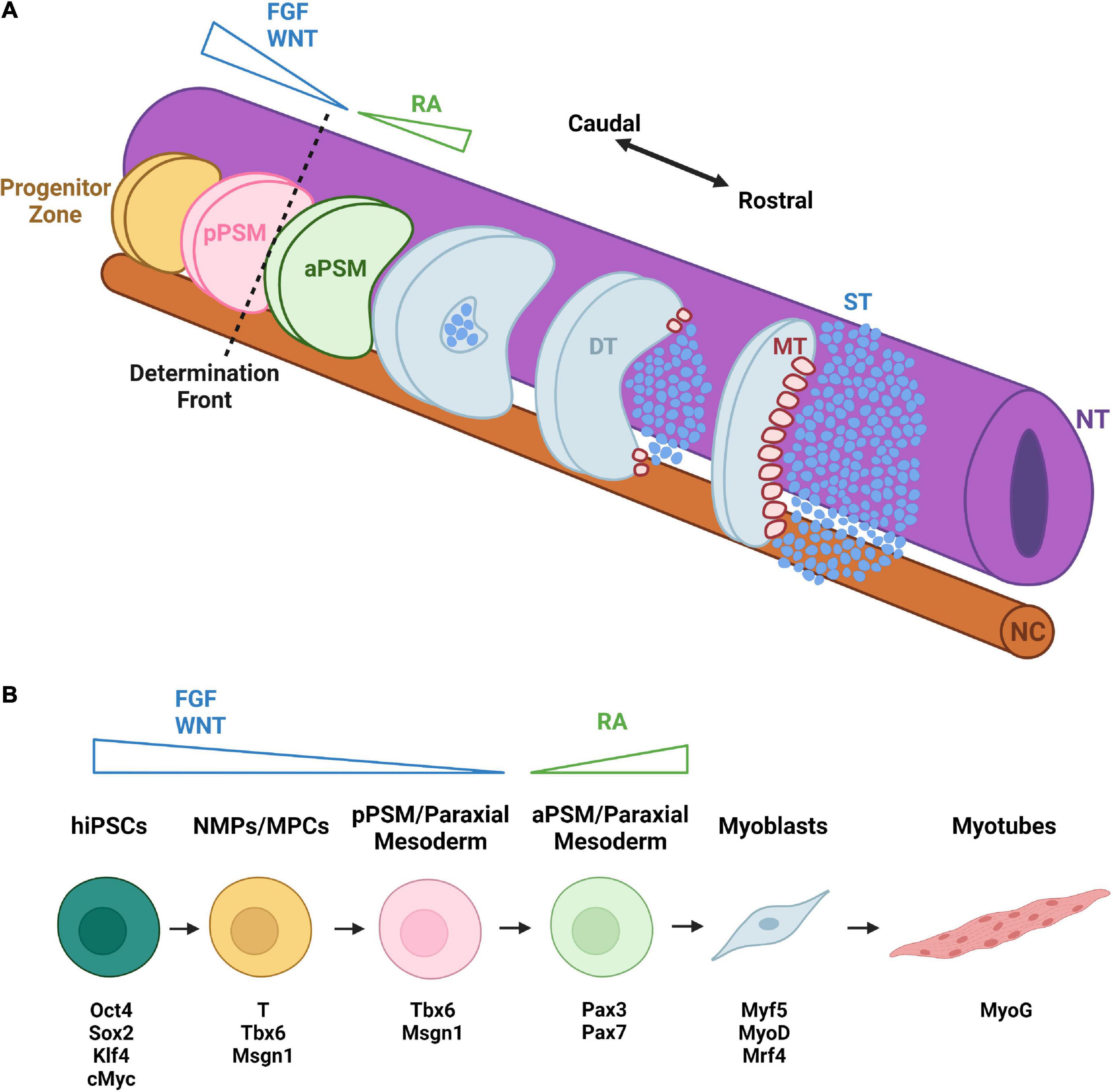
Figure 2. Early development and hiPSC-based differentiation of skeletal muscle. (A) Caudal-rostral development of SkM occurs bilaterally along the neural tube (NT) and notochord (NC). From the progenitor zone, cells migrate to the posterior presomitic mesoderm (pPSM) with a decreasing gradient of FGF and WNT signals. They then cross the determination front to enter the anterior presomitic mesoderm (aPSM). With increasing retinoic acid (RA) gradient, somite formation begins. As cells continue to travel rostrally, the dermomyotome (DT), sclerotome (ST), and myotome (MT) form, initiating primary myogenesis. (B) hiPSC differentiation to skeletal muscle begins with WNT and FGF activation, inducing a shift into neuromesodermal progenitors (NMPs) and then muscle progenitor cells (MPCs) expressing the transcription factors T, Tbx6, and Msgn1. Subsequent loss of T expression leads to formation of paraxial mesoderm cells resembling skeletal muscle progenitors of pPSM. Through RA activation, these muscle progenitors begin to express the muscle stem cell markers Pax3 and Pax7 and eventually differentiate into myoblasts expressing early myogenic markers Myf5, MyoD, and Mrf4. The myoblasts can fuse into myotubes that express the late muscle differentiation marker MyoG. Distinct colors are used to denote approximate correspondence between stages of hiPSC differentiation in panel (B) and embryonic development in panel (A).
In the next stage of development, MPCs attain the posterior PSM (pPSM) fate characterized by the downregulation of T and expression of Msgn1 and Tbx6 (Chalamalasetty et al., 2014). In the posterior two-thirds of the PSM, MPCs and pPSMs experience oscillations of the segmentation clock (pulses of Notch, FGF, and WNT signaling) to control the production of somites (Dubrulle et al., 2001; Aulehla et al., 2003). As skeletal muscle cells continue to develop, reach the determination front then enter the anterior third of the PSM (Dubrulle et al., 2001). At the determination front, the oscillations of the segmentation clock cease, Msgn1 is downregulated, and Pax3, Mesp2, Foxc1/2, and Meox1/2 genes are upregulated (Kume et al., 2001; Mankoo et al., 2003). Within the anterior PSM, retinoic acid (RA) counteracts the WNT and FGF signaling (Sakai et al., 2001). Furthermore, a posterior fissure forms at the junction between Mesp2+ and Mesp2– cells to create new somites (Dubrulle et al., 2001; Aulehla et al., 2003). Shortly after somites are formed, they become subdivided into ventral mesenchymal sclerotome and dorsal epithelial dermomyotome, the latter of which contains SkM (alongside dermis and brown fat) progenitors and maintains Pax3 expression (Lepper and Fan, 2010; Sanchez-Gurmaches and Guertin, 2014). Soon after its formation, primary myogenesis begins when dorsally located dermomyotomal cells lose Pax3 expression and upregulate myogenic factor Myf5 (Ott et al., 1991). These early myogenic cells delaminate from the dermomyotome and contribute to the formation of the first embryonic muscles—myotomes (Denetclaw et al., 1997). Myogenesis then officially commences with primary myogenesis and the generation of primary myofibers that serve as the foundation for adult muscle formation.
During secondary myogenesis, myogenic progenitors expressing transcription factor Pax7 sustain muscle growth by fusing among themselves or to existing primary myofibers generating β-enolase expressing secondary or fetal myofibers (Fougerousse et al., 2001). A subset of these Pax7+ progenitors localize under the basal lamina where the eventually become satellite cells which contribute to the repair and regeneration of damaged muscle fibers in adults (Dumont et al., 2015a). During muscle fiber maturation, expression of embryonic myosin heavy chain changes to adult myosin heavy chain isoforms with oxidative, slow twitch or glycolytic, fast twitch fiber phenotypes (Khodabukus, 2021). Additionally, actin and myosin assemble into sarcomeres, sarcomeres assemble into myofibrils, NMJs are formed at the sarcolemma, and triads are established from a network of tubules for facilitated neural excitation (Pourquié et al., 2018). Secondary and later stages of myogenesis are controlled by TGFβ (Gu et al., 2016), hepatocyte growth factor (HGF) (Bladt et al., 1995), WNT (van der Velden et al., 2006), and insulin-like growth factor (IGF) (Chargé and Rudnicki, 2004) signaling. The final phases of myogenesis are controlled by transcription factors including Myf5, MyoD, Myomaker, and MyoG to facilitate the fusion of muscle progenitor cells, hypertrophy of myofibers, and innervation by MNs to generate fully functional SkM (Zhang et al., 2020).
Generation of Skeletal Muscle From Pluripotent Stem Cells
The derivations of SkM from hiPSCs typically fall under one of two major approaches (Kodaka et al., 2017; Jiwlawat et al., 2018). The first approach involves transgene-based approaches whereby hiPSCs are directly reprogrammed into myogenic progenitor cells through overexpression of muscle specific transcription factors (Kodaka et al., 2017). The second approach involves transgene-free methods whereby developmental myogenesis in hiPSCs is recapitulated through the administration of small molecules, such as FGF2 and GSK3β inhibitor, which activate or inhibit myogenic signaling pathways (Jiwlawat et al., 2018; Figure 2B).
The transgene-based approaches generate myogenic progenitors from hiPSCs or their mesodermal derivatives by transient or constitutive overexpression of master regulators of myogenesis, such as PAX7 (Darabi et al., 2012; Rao et al., 2018) or MYOD1 (Abujarour et al., 2014; Albini and Puri, 2014; Maffioletti et al., 2015). Overexpression of exogenous myogenic genes has been accomplished through mRNA transfection (Warren et al., 2010), as well as transduction with adenoviral (Goudenege et al., 2012) or lentiviral (Albini and Puri, 2014; Maffioletti et al., 2015; Rao et al., 2018) vectors. Through these transdifferentiation methods, as many as 90% of cells commit to a myogenic identity and can differentiate into SkM progenitor cells (Tanaka et al., 2013; Abujarour et al., 2014; Rao et al., 2018). Use of fluorescence reporter genes co-expressed with transcription factors can allow further cell purification by fluorescence-activated cell sorting (FACS) (Darabi et al., 2012; Rao et al., 2018). Alternatively, activation of endogenous transcription factors (e.g., Pax7) using CRISPR/Cas9 methodology can lead to stable epigenetic reprogramming of hiPSCs and generation of myogenic progenitor cells (Kwon et al., 2020). The resulting SkM progenitors derived using transgene-based approaches survive and function when implanted in immunocompromised mice (Darabi et al., 2012; Kwon et al., 2020), while 3D engineered tissues generated from these cells can become functional muscle with the ability to survive and function in vivo (Rao et al., 2018). Nevertheless, these differentiation methods do not reflect normal development and despite the ability to obtain large numbers of human myogenic progenitors, regulatory concerns regarding genetic modification of cells may limit their potential therapeutic use (Jiwlawat et al., 2018).
A second approach, known as directed differentiation, mimics myogenic development through sequential addition of small molecules to activate or suppress specific signaling pathways. For example, CHIR-99021 activates WNT signaling through GSK3β inhibition, LDN-193189 inhibits BMP signaling, and HGF and IGF1 activate their respective signaling pathways (Chal et al., 2016). Despite no genetic modification and reliance on natural developmental ques, directed differentiation protocols require significantly longer culture time and exhibit considerably lower yields and higher heterogeneity of myogenic cells compared to transgene-based methods (Kodaka et al., 2017; Jiwlawat et al., 2018). Purity of myogenic progenitors can be increased by sorting for cell surface markers such as CDH13 (Nalbandian et al., 2021), FGFR4 (Nalbandian et al., 2021), ERBB3 (Hicks et al., 2018), and NGFR (Hicks et al., 2018), however, use of FACS further decreases cell yield. Recent protocols for expansion and cryopreservation of FACS-sorted hiPSC-derived myogenic progenitors may offer means to obtain clinically relevant cell quantities (van der Wal et al., 2018).
Recent transcriptomic analyses have shown that hiPSC-derived myogenic progenitors are developmentally immature and arrested between embryonic and fetal muscle stem cell stages (Xi et al., 2020; Nayak et al., 2021). Nevertheless, they can successfully fuse into myotubes that exhibit key functional behaviors of SkM, including generation of calcium transients and contractile force and robust response to acetylcholine (Skoglund et al., 2014; Rao et al., 2018), albeit at lower levels compared to primary human myotubes (Rao et al., 2018). Further advances in maturity of hiPSC-derived SkM cells will lead to improved modeling of human NMDs in vitro.
Schwann Cell Development
Schwann cells are varied group of glial cells that produce protective myelin sheaths and support NMJ function, remodeling, and regeneration (Son et al., 1996). SCs undergo three main transitions during development: (1) from migrating neural crest cells (NCCs) to SC precursors (SCPs), (2) SCPs to immature SCs, and (3) immature SCs to a mature myelinating or non-myelinating SCs (Jessen and Mirsky, 2005). These transitions and SC survival are dependent upon morphogens secreted from axons with which SCPs and SCs continuously associate (Jessen and Mirsky, 1999). However, the molecular mechanisms that regulate neural crest cell differentiation into SCs have not been fully elucidated.
Early in the process of neurulation, dorsally located NCCs segregate from the neural tube and migrate in ventrally (Jessen and Mirsky, 2005). The basic helix-loop-helix (bHLH) transcription factor Sox10 is expressed early by all NCCs (Kuhlbrodt et al., 1998; Woodhoo and Sommer, 2008). While high expression of Sox10 persists in glial and melanocyte NCC derivatives of the peripheral nervous system (PNS), its expression is downregulated in other NCC derivatives (Kuhlbrodt et al., 1998; Woodhoo and Sommer, 2008). The continued high expression of Sox10 is dependent on the expression of Pax3 (Kioussi et al., 1995; Blanchard et al., 1996; Doddrell et al., 2012), which is regulated in part by histone deacetylases 1 and 2 (HDAC1/2) (Jacob et al., 2011). Together, Sox10 and Pax3 induce the expression of key SC lineage genes including fatty acid binding protein 7 (Fabp7) and myelin protein zero (MP0) (Kioussi et al., 1995; Blanchard et al., 1996; Doddrell et al., 2012).
While Sox10 is necessary for SC specification, it is not sufficient. In the developing PNS, SCPs migrate alongside MN axons extending to targeted regions of innervation (Jessen and Mirsky, 1999). Moreover, migrating SCPs are dependent upon signals from these axons, such as Neuregulin-1 (NRG1), for appropriate development and survival (Jessen and Mirsky, 1999). In NCC cultures, NRG1 suppresses neuronal differentiation and promotes glial specification (Shah et al., 1994). NRG1 binds ErbB2/3, an obligate heteromeric receptor tyrosine kinase pair, on SCPs to activate key downstream signal transduction cascades that are essential for both proliferation and directed migration (Newbern and Birchmeier, 2010).
Immature SCs develop after SCPs cease migration and populate axons that are still projecting to their targeted region of innervation, while acquiring a set of properties that clearly distinguish them from SCPs. Specifically, they cease migration, become dependent on autocrine signaling for survival, and deposit an organized basal lamina (Jessen and Mirsky, 2005). Additionally, increased notch signaling is a critical mediator of the SCP transition to immature SCs, with a loss of notch signaling preventing immature SC formation (Woodhoo et al., 2009).
Generation of Schwann Cells From Pluripotent Stem Cells
Methods to differentiate hiPSCs into SCs have mimicked developmental process by first generating neural crest-derived SC precursors from hiPSCs. Neural crest stem cells have been derived from hiPSCs via FACS selection of p75+ cells derived from EBs cultured in stromal-cell-conditioned media, FGF2, and B-27 supplement (Liu et al., 2012). These cells were subsequently differentiated into a nearly pure population of SCs expressing glial fibrillary acidic protein, S100, and p75 through culture in mesenchymal stem cell medium supplemented with Neuregulin-1 for 40 days. A similar method using EB formation and FGF2 treatment was later developed that shortened the time of induction to 6 days (Huang et al., 2017).
To overcome the challenges with low reproducibility and throughput in EB cultures, methods for directed differentiation of hiPSCs to SCs have been developed. Specifically, sequential treatment of naïve hiPSCs with TGF-β and GSK-3β inhibitors followed by NRG1 produced SC precursors in 18 days (Kim et al., 2017). These precursors were further differentiated through treatment with NRG1, retinoic acid, platelet-derived growth factor-BB (PDGF-BB), and forskolin into SCs. This method shortened the total differentiation time from approximately 41 days (Huang et al., 2017) to approximately 32 days (Kim et al., 2017). However, a more recent protocol has been developed for the derivation of direct Schwann-cell precursors (SCPs) from SOX10-reporting hiPSCs that only required a total differentiation time of 21 days and allowed for in vitro culture up to 80 days with maintained expression of the SC proteins S100b, glial fibrillary acidic protein (GFAP), and galactosylceramidase (Mukherjee-Clavin et al., 2019). With all these methods, differentiated SCs show increased expression of SC-specific markers such as GFAP and S100β.
In addition to protein expression, hiPSC-derived SCs have been studied for their secretion rates of neurotrophic factors (Huang et al., 2017; Kim et al., 2017), ability to myelinate primary neurons (Liu et al., 2012; Kim et al., 2017), and ability to accelerate nerve healing within rodent sciatic nerve injury models (Huang et al., 2017; Kim et al., 2017). hiPSC-derived SCs have additionally been used in coculture with hiPSC-derived neurons to allow for stronger neuronal outgrowth within a 3D tissue-engineered skin model (Muller et al., 2018). Recent studies have further confirmed critical physiological roles of SCs in the stabilization and maintenance of NMJs in vitro (Singh and Vazquez, 2019; Martins et al., 2020), forming a foundation for the future development of novel biomimetic NMD models.
Neuromuscular Junction Development
The NMJ is a chemical synapse formed between MNs and SkM that allows the transmission of motor commands from the CNS (Figure 3). MNs communicate with SkM through the release of acetylcholine (ACh) into the synaptic cleft of the NMJ. ACh receptors (AChRs) located on muscle fibers are activated and depolarize the muscle cell which triggers calcium release from the sarcoplasmic reticulum initiating a contraction (Fambrough, 1979). Reciprocal signaling between MNs and SkM is important for the formation and maintenance of NMJs as highlighted by the coordination required for complex movements and sensory-motor feedback.
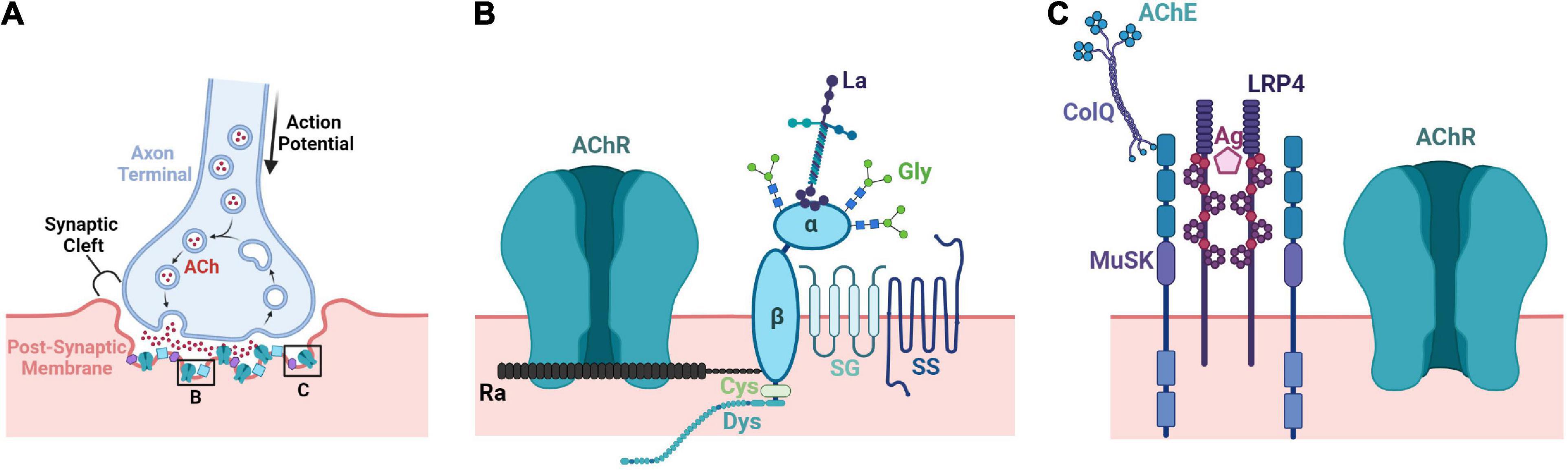
Figure 3. Structural and molecular architecture of the neuromuscular junction. (A) The NMJ is comprised of three components: (1) the axonal terminal of an MN (pre-synapse), (2) the basal lamina of the synapse (synaptic cleft), and (3) the sarcolemma (membrane) of a muscle fiber (post-synapse). Following the conduction of an action potential to the axon terminal, Ca2+ influx occurs at the presynaptic terminal releasing ACh-containing vesicles into the synaptic cleft. Released ACh can then bind to AChRs on the sarcolemma creating an endplate potential and eventually muscle contraction. (B) AChE secreted by the muscle binds to ColQ and inactivates residual ACh within the synapse. ColQ binds to MuSK to help stabilize the synapse. The synaptogenic proteoglycan agrin secreted by MNs binds to LRP4 to facilitate formation of the NMJ. (C) AChRs are stabilized by dystrophin-associated glycan (DAG) complexes. The AChR-clustering protein rapsyn connects AChRs to the DAG complex and dystrophin anchors the complex to the SkM cytoskeleton. Lamins and glycans additionally connect the complex to the ECM while the sarcoglycan and sarcospan stabilize the DAG complex within the membrane. ACh, acetylcholine; AChE, acetylcholine esterase; ColQ, collagen Q; MuSK, muscle-specific tyrosine kinase receptor; Ag, agrin; LRP4, low density lipoprotein receptor 4; AChR, acetylcholine receptor; AChR, acetylcholine receptor; La, laminin; α/β, α/β dystroglycan; Gly, glycans; SG, sarcoglycan; SS, sarcospan; Ra, rapsyn; Cys, cysteine; Dys, dystrophin.
During development, immature SCs migrate with MNs toward the periphery (Sugiura and Lin, 2011) and differentiate into either axonal SCs that myelinate axon extensions or terminal SCs that support the NMJ formation. Terminal SCs proliferate extensively around the NMJ (Hirata et al., 1997) and then cover or “cap” the nerve terminal with their processes (Court et al., 2008). Muscular innervation is preceded by the localization of small aneural AChR clusters to the central region of muscle fibers in a process called prepatterning. During this process, the MN terminal releases agrin which binds to the muscle-specific kinase (MuSK) co-receptor, low-density lipoprotein receptor related protein 4 (LRP4), promoting activation and transphosphorylation of MuSK. SC processes contact pre-patterned AChR clusters prior to the nerve and cover more of the postsynaptic membrane than axonal terminals during early synapse formation (Flanagan-Steet et al., 2005). Additionally, SCs express active agrin and encourage aggregation of AChRs on muscle fibers (Yang et al., 2001). In mice, SC loss results in MN defasciculation, but MNs still project toward muscle targets implying that SCs are not required for initial nerve–muscle contacts (Woldeyesus et al., 1999; Lin et al., 2000). However, further growth and maintenance of this early synapses is halted in SC absence (Riethmacher et al., 1997), suggesting that SCs are critical for NMJ homeostasis.
Eventually, innervation of muscle fibers induces the formation of larger, neural AChR clusters forming stable NMJs in the middle region of muscle fibers. Mice with a mutated agrin gene (agrin–/–) are unable to form NMJs; however, these mice can form aneural AChR clusters on muscle fibers prior to innervation (Lin et al., 2001). Conversely, aneural clusters are not formed in MuSK–/– mice, and their muscle fibers demonstrate a uniform distribution of AChRs with a broader region of innervation containing highly branched MN terminals. Neuronal agrin does not induce AChR clusters in MuSK–/– muscle cells (Glass et al., 1996); however, agrin sensitivity can be restored through expression of wild-type MuSK (Zhou et al., 1999). Interestingly, synapse formation can be rescued in agrin–/– mice with ectopic MuSK expression (Kim and Burden, 2008). Together, this suggests the importance of MuSK for aneural AChR clustering and prepatterning prior to innervation while agrin is also needed for neural AChR clustering and NMJ formation. Additionally, SC processes influence nerve terminal growth and are required for both the formation and maintenance of developing NMJs (Reddy et al., 2003).
Neuromuscular junction formation is also influenced by several extracellular components. For example, MuSK has a cysteine-rich domain (CRD) that shares homology with the WNT receptor, Frizzled. As a result, WNT proteins bind and activate MuSK prior to innervation, when neural agrin is absent (Barik et al., 2016). This signaling can regulate axon guidance as well as induce aneural cluster formation (Li et al., 2018). Moreover, both canonical and non-canonical WNT pathways are affected in transgenic mice with MuSK CRD deletions (Messeant et al., 2017). Many components of the extracellular matrix (ECM) have important regulatory roles in myogenesis and synaptogenesis. Within the synaptic basal lamina, ECM molecules help guide the process of innervation and are crucial to formation of post-synaptic density as well as organization and maintenance of functional appositions of the pre- and post-synaptic elements. The dystrophin-associated glycoprotein complex (DGC), through its α-dystroglycan subunit, organizes a functional scaffold in the basal lamina including perlecan, acetylcholinesterase/ColQ, and laminin that stabilizes AChR clusters (Jacobson et al., 2001). The DGC additionally connects networks of laminins and collagens to one another by nidogen and anchors them to the sarcolemma through the sarcoglycan-sarcospan subcomplex and intracellular cytoskeleton through dystrophin (Fox et al., 1991; Jacobson et al., 2001). The laminin β2 chain plays a role in synapse maturation by binding and clustering voltage-dependent calcium channels (VDCC) in the active zone of the NMJ. A reduced number of active zones and pre-synaptic release of ACh is observed in mice lacking laminin β2 (Rogers and Nishimune, 2017). Mice lacking ColQ, collagen XIII, collagen IV, or collagen VI also exhibit immature nerve terminals and/or NMJs (Sigoillot et al., 2016; Cescon et al., 2018; Zainul et al., 2018).
After forming functional NMJs with their target, MNs impinge on muscle fiber structural and functional diversity. A single muscle is composed of several fiber types that are innervated by specific classes of MNs. MNs are subdivided into three groups based on the type of muscle fiber they innervate: (1) alpha MNs, which innervate force generating extrafusal fibers, (2) gamma MNs, which innervate the proprioceptive intrafusal fibers, and (3) beta MNs, which innervate both extrafusal and intrafusal fibers. Alpha MNs are the most abundant of these classes and are categorized as SFR (slow-twitch, fatigue-resistant), FFR (fast-twitch, fatigue-resistant), and FF (fast-twitch, fatigable) reflecting the type of extrafusal muscle fiber they innervate (Totosy de Zepetnek et al., 1992). MNs are intrinsically competent to recognize and connect to either fast or slow muscle fibers (Landmesser, 2001). Slow MNs start to specifically express the synaptic vesicle glycoprotein 2a (SV2A) (Chakkalakal et al., 2010) as well as the estrogen-related receptor beta (ESRRB) (Enjin et al., 2010) soon after birth. Conversely, fast MNs specifically express the calcitonin-related polypeptide alpha (CALCA) and the chondrolectin (CHODL) (Enjin et al., 2010). The Notch ligand delta-like homolog1 (DLK1) has also been identified as a necessary regulator of fast MNs (Muller et al., 2014). Understanding of the influence of innervation by specific MN types upon SkM phenotype could provide important insight into certain NMDs that preferentially target specific muscle fiber types.
Current Models for Studies of Neuromuscular Junction Function and Disease
Animal Models
Various animal models have been broadly utilized to advance our understanding of the formation, function, and malfunction of NMJs during the development and progression of NMDs. Moreover, they have been used to study the pathophysiology and develop pharmacotherapies for NMDs. Specifically, Caenorhabditis elegans (Sleigh and Sattelle, 2010), zebrafish (Babin et al., 2014), Drosophila (Shields et al., 2017), and mice (Hsieh-Li et al., 2000) have been extensively employed to investigate the precise anatomy and function of NMJs. Their ease of genetic manipulation, tractable anatomy, relatively rapid growth, and low cost have contributed to their extensive use (Dawson et al., 2018). The mouse NMJ has been particularly useful due to its large size and accessibility, facilitating microscopic studies by immunofluorescence histology and functional analyses by electrophysiology (Webster, 2018). The imaging studies, in particular, have enabled enhanced understanding of how localization and density of pre-synaptic, post-synaptic, and synaptic proteins are rearranged or lost in disease states.
However, experimental results in animal models may have limited translational value due to distinct anatomical differences between animal and human NMJs. For example, the murine NMJ and human NMJ exhibit substantially different proteomes and the larger, more pretzeled murine NMJ readily remodels with age whereas the smaller, more fragmented human NMJ is mostly conserved (Jones et al., 2017). Interestingly, the density of the active portion of the human NMJ is greater compared to its mouse counterpart (Jones et al., 2017). Additionally, disease phenotypes in animals can vary widely from those in humans in terms of progression, severity, and etiology (Vainzof et al., 2008). High levels of inbreeding limit genetic diversity within common animal models and controlled environments prevent genetic drift, while removing common viral and microbial agents that can influence human NMD pathogenesis (Dawson et al., 2018). The inability of animals to fully capture the genotypic heterogeneity and allelic variations observed across human individuals has hindered the clinical success of NMD drugs validated through animal models (Vainzof et al., 2008). Only a small fraction of drugs that enter clinical trials are approved as many result in unanticipated drug responses and toxicities (Vainzof et al., 2008). This situation has prompted development of in vitro human models of NMJ and NMDs that could allow studies of disease and pharmacological effects in a personalized and clinically more relevant fashion.
Two-Dimensional in vitro Models
When developing in vitro models of NMJ, it is important to both consider its anatomical structure and enable relevant biological and functional studies. Initial rodent models of NMJ entailed mixed 2D co-cultures of myotubes with dissociated MNs (Kengaku et al., 1991) or spinal cord explants (Askanas et al., 1987), either plated simultaneously or sequentially (Figure 4A). Axonal projections in these co-cultures extended from the MNs to form NMJs with the myotubes that in turn exhibited functional post-synaptic potentials. Development of in vitro human models of the NMJ (Guo et al., 2011; Demestre et al., 2015; Yoshida et al., 2015) have additionally opened doors to personalized modeling of NMDs. For example, hiPSCs from spinal muscular atrophy (SMA) patients exhibited impaired AChR clustering which was ameliorated with valproic acid and antisense oligonucleotide treatment (Yoshida et al., 2015). The main advantages of these 2D culture models were relative simplicity and use of a flat substrate allowing for efficient and direct analysis of cell morphology and pathological features.
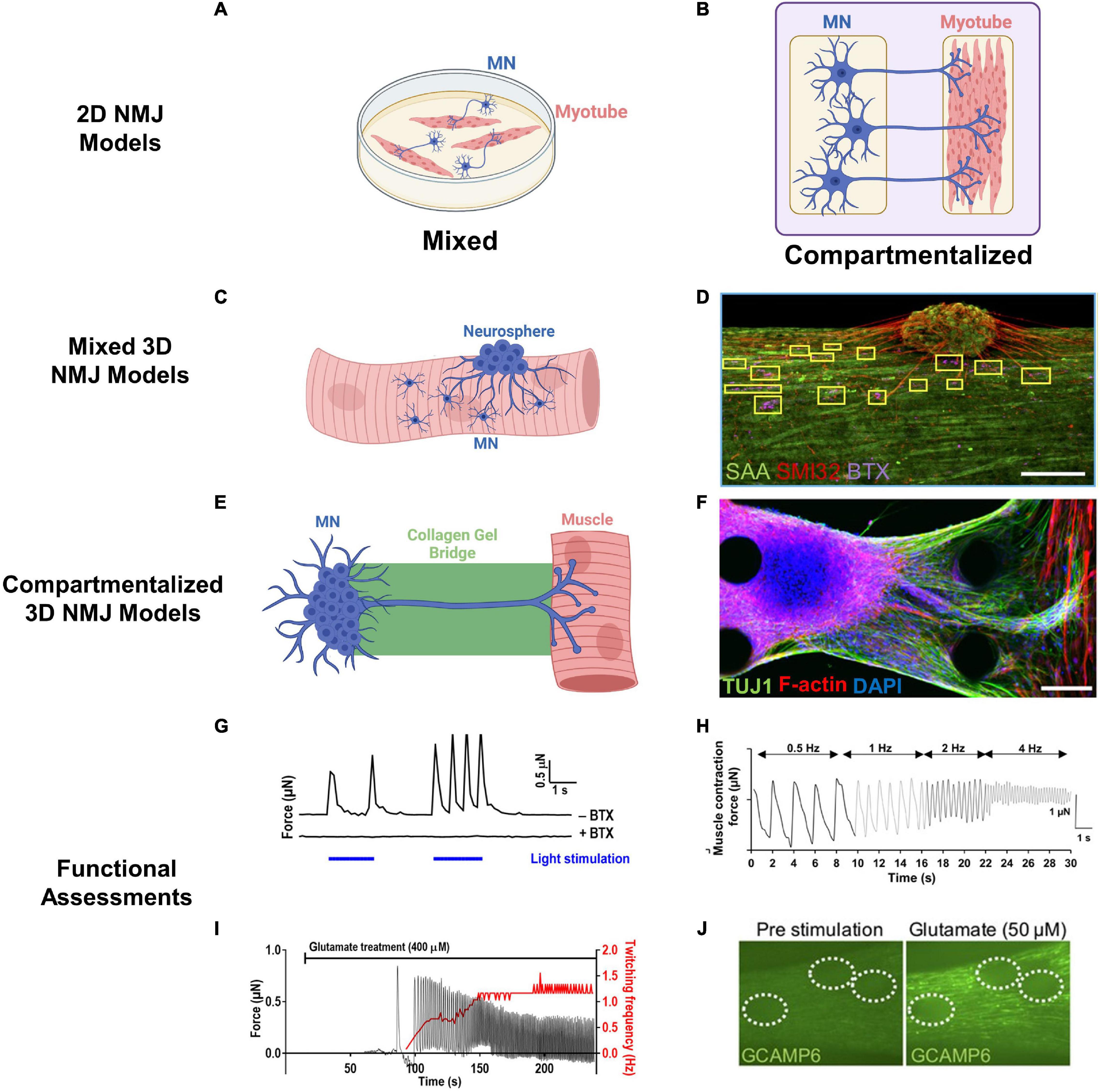
Figure 4. Engineered NMJ models. (A,B) Schematics of 2D NMJ co-culture models in mixed (A) and compartmentalized (B) configuration. (C) Mixed 3D NMJ co-culture systems incorporate MNs or neurospheres into SkM during 3D tissue formation. (D) Representative mixed 3D NMJ model with immunofluorescent staining of muscle sarcomeres (SAA), neurite extensions (SMI32), and acetylcholine receptors (BTX). Scale bar, 200 μm (Bakooshli et al., 2019; Copyright 2019, eLIFE). (E) Compartmentalized 3D NMJ co-culture systems culture MNs and SkM in separate compartments bridged by an extracellular matrix gel to facilitate axonal outreach and SkM innervation. (F) Representative compartmentalized NMJ model with immunofluorescent staining of neurite extensions (TUJ1) and myotubes [filamentous (F)-actin]. Scale bars, 100 μm (Osaki et al., 2018; Copyright 2018, Science Advances). (G) Example of optogenetic control of 3D NMJ models through blue light illumination (blue bars) of ChR2H134R-HBG3-MN neurospheres inducing contraction in muscle (y-axis) as measured by pillar displacement within a microfluidic system. Administration of α-bungarotoxin (BTX) prevented MN-induced contractions (Uzel et al., 2016; Copyright 2016, Science Advances). (H) Example of muscle contraction induced by electrical stimulation of MNs at varying frequencies (0.5–4 Hz) (Osaki et al., 2018; Copyright 2018, Science Advances). (I) Representative recording of contractile force (y-axis) in 3D SkM-MN co-culture induced by glutamate stimulation of neurospheres (Uzel et al., 2016; Copyright 2016, Science Advances). (J) Representative recording of glutamate-induced Ca2+ transients in 3D SkM-MN co-culture with muscle-specific expression of GCaMP6 reporter. MN neurospheres are encircled by dashed lines (Bakooshli et al., 2019; Copyright 2019, eLIFE).
However, AChR clustering in mixed 2D co-cultures exhibits poor co-localization of pre- and post-synaptic structures compared to native NMJs, hindering the ability to recapitulate the intricacies of specific NMDs (Das et al., 2010; Umbach et al., 2012). Specifically, without proper spatial cues, myoblasts in mixed 2D cultures fuse into randomly oriented and branched myotubes limiting formation of elongated myofibrils and mature sarcomeres (Bettadapur et al., 2016). These myotubes will often delaminate after few days of culture as they start to generate more mechanical stress against the underlying substrate, thus not providing sufficient time for proper NMJ maturation (Wang et al., 2012; Sun et al., 2013). Anatomical considerations also hinder physiological relevance of these models. In situ, the soma of the MNs reside in the spinal cord with only the axons projecting and physically interacting with the myofibers. Plating MNs on top of myotubes in mixed 2D co-cultures is therefore anatomically incorrect and may alter the physiology of one or both cell types. Additionally, measuring SkM force generation is not possible on most conventional culture substrates and individual analysis of each cell type, both functional and molecular, is often impractical.
To overcome these technical challenges, researchers have developed several types of tunable culture surfaces and microfabricated devices to engineer more biomimetic NMJs with improved anatomical organization. Use of topographical cues, such as polylactic acid (PLA) or polycaprolactone (PCL) nanofibers, served to align murine myoblasts and improve NMJ formation with co-cultured rat embryonic spinal MNs (Luo et al., 2018; Das et al., 2020). Micropatterning of alternating soft and stiff extracellular matrix strips increased expression of the NMJ markers MuSK and LRP4, improved myoblast fusion, and augmented AChR cluster size when rat primary MNs were co-cultured with both human and mouse SkM cells (Happe et al., 2017). Micropatterning techniques have also been shown to promote hiPSC-derived MN survival (Burbulla et al., 2016) and align human myoblasts (Ebrahimi et al., 2018).
Furthermore, incorporation of SCs has improved viability and survival of MNs in long-term 2D cultures in vitro while supporting longer, myelin ensheathed axonal projections in rodent models in vivo (Haastert et al., 2005; Honkanen et al., 2007; Paivalainen et al., 2008; Viader et al., 2011; Hyung et al., 2015). Within human cell lines, increased myotube number, length, and viability were observed in both SC/SkM cocultures and SC/SkM/MN tricultures highlighting the synergistic relationships among these cell types (Singh and Vazquez, 2019). Additionally, self-organization of hiPSC-derived NMJs has been accomplished following simultaneous generation of MNs, SkM, and SCs from a bipotent NMP population (Lin et al., 2019) fated to form both spinal neuroectodermal and associated musculoskeletal mesodermal cell derivatives (Gouti et al., 2017). Within this system, contractile and electrophysiological activity driven by functional NMJs was supported by the presence of terminal SCs and myelinated axons.
Additionally, development of compartmentalized 2D NMJ models has allowed for MN somas and myotubes to be spatially separated increasing the biomimetic nature of these co-cultures (Figure 4B). In the first example of a compartmentalized NMJ model, neurons derived from murine embryonic stem cells and fused C2C12 myoblasts were cultured separately in a microfluidic device and connected only through axon extensions (Park et al., 2013). Similar compartmentalized 2D models have been used to study rodent synaptic formation (Tong et al., 2014), AChR clustering (Southam et al., 2013), and MN-induced calcium transients in myotubes (Ionescu et al., 2016). A compartmentalized 2D NMJ model between primary embryonic rat MNs and myotubes inside an automated device allowed measurements of MN-initiated muscle contractile force through cantilever displacement (Smith et al., 2013). Similar studies testing effects of bungarotoxin, BOTOX®, and curare were performed in a human co-culture platform where video recording analysis was used to measure amplitude and frequency of MN-induced myotube contractions (Santhanam et al., 2018). Additionally, lentiviral transduction of human MNs with channelrhodopsins has enabled a more precise, light-mediated control over MN activity in NMJ co-cultures (Steinbeck et al., 2016). Use of single-donor hiPSC-derived SkM cells and MNs has further enabled patient-specific disease modeling (Guo et al., 2020a) with capability to assess NMJ function in response to electrical stimulation of MNs. While these 2D compartmentalized platforms partly recapitulate in vivo organization through physical separation of MNs and SkM cells, they lack the structural 3D complexity of the native innervated muscle.
Three-Dimensional in vitro Models
The lack of the 3D cell–cell and cell–ECM interactions in 2D NMJ models has prompted the development of 3D in vitro models of innervated SkM (Figures 4C–F), which despite being more expensive, time-consuming, and lower throughput than 2D cultures, are expected to provide a more physiologically relevant platform for NMD studies. The first example of a mixed 3D NMJ model incorporated fetal rodent nerve explants within 3D SkM constructs resulting in the formation of functional NMJs and expression of more mature myosin heavy chain (MHC) isoforms (Larkin et al., 2006). A similar mixing technique using neonatal rat myoblasts and embryonic ventral horn neurons improved myotube cytoskeletal organization and augmented force production of engineered SkM (Martin et al., 2015). Direct co-culture of mouse-derived MN spheroids and SkM allowed for contraction following glutamic acid activation of MNs that could be inhibited by curare treatment, a NMJ antagonist (Morimoto et al., 2013). However, compared to native NMJs, AChRs clustering remained relatively diffuse in these systems (Morimoto et al., 2013). These first-generation 3D NMJ models demonstrated the utility of 3D platforms to generate functional NMJs but their non-compartmentalize nature and use of embryonic rodent cells limit their utility for studies of human NMDs.
Consequently, recent efforts have focused on the development of human 3D NMJ co-cultures. For example, addition of hiPSC-MN clusters into 3D SkM/hydrogel suspensions or to pre-formed SkM tissues allowed generation of mixed 3D NMJ models (Figures 4C,D) where consequences of functional connectivity between MNs and muscle fibers were studied by recording calcium transients or contractile force generation (Osaki et al., 2018; Bakooshli et al., 2019; Rimington et al., 2021). Compared to 2D monolayers, 3D MN spheroid co-culture with SkM increased axon length and expression of SMI32, a marker of MN maturity (Rimington et al., 2021), while the presence of MNs improved the overall structure and function of myotubes (Bakooshli et al., 2019; Rimington et al., 2021), revealing the mutually beneficial effects of MNs and SkM within 3D co-culture systems. Interestingly, functional innervation was achieved following 2 weeks of culture within 3D, but not in comparable 2D NMJ co-cultures, and expression of the mature AChR ε-subunit was observed only in 3D NMJ co-cultures (Bakooshli et al., 2019). Beyond mixed 3D NMJ models, compartmentalized microdevices (Figures 4E,F) have been developed to spatially separate MN spheroids and engineered SkM and connect them via axon-permissive channels to more appropriately mimic in vivo muscle innervation (Uzel et al., 2016; Osaki et al., 2018; Vila et al., 2021). Through this compartmentalization, visualization of 3D neurite outgrowth and engineered SkM innervation is greatly simplified, similar to studies in 2D compartmentalized co-cultures.
Incorporation of SCs can further improve longevity and biomimetic organization of 3D NMJ models as shown in rodent co-cultures, where MN–SC interactions led to extended, myelinated axonal projections of MNs with improved viability (Gingras et al., 2008; Hyung et al., 2021), while optical stimulation of murine MNs acted reciprocally on SCs to enhance the myelination process, leading to the formation of thicker myelin sheaths (Hyung et al., 2019). Similarly, in human organoids, NMJs identified by accumulation of αBTX clusters in muscle fibers encompassed both myelinated axons and capping terminal SCs and were shown to be functional by curare-induced block of muscle activity (Faustino Martins et al., 2020).
Motor neuron activation within 3D NMJ models has been achieved through addition of the neurotransmitter glutamate (Osaki et al., 2018; Bakooshli et al., 2019) or its mimic N-Methyl-D-aspartate (NMDA), optogenetic control (Osaki et al., 2018; Vila et al., 2019, 2021), or direct electrical stimulation (Osaki et al., 2018; Rimington et al., 2021). Glutamate stimulates MNs (Figure 4I) through binding to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic (AMPA), kainic acid (KA), and NMDA receptors while NMDA specifically targets NMDA receptors (Newcomer et al., 2000). High doses of glutamate or NMDA can be used to study excitotoxicity (over-activation of glutamate receptors) while lower doses can access NMJ model sensitivity (Liu et al., 2007). Optogenetic control in NMJ models (Figure 4G) relies on genetic modification of MNs to express light-sensitive channels, such as channelrhodopsin, that induce an action potential and subsequent muscle contraction in response to blue light. This method, though utilizing genetically altered MNs, allows for spatiotemporal and noninvasive control over motor units. Direct electrical stimulation, while impractical in mixed co-culture systems as it would excite both MNs and SkM cells, can be implemented in compartmentalized systems to stimulate MNs (Figure 4H).
To assess NMJ functionality, recordings of calcium transients have been used as an indicator of MN-induced muscle excitation and gCaMP6 (Bakooshli et al., 2019), a genetically encoded calcium indicator, has been used to visualize calcium flow through muscles (Figure 4J). Furthermore, MN-innervated engineered SkM tissues can be cultured on microfabricated pillars, displacement of which can be imaged to assess muscle contractions induced via glutamate or light-stimulated MN activity (Uzel et al., 2016; Vila et al., 2019, 2021; Afshar et al., 2020). In addition to indirect functional measurements by video recordings, contractile force generation in mixed 3D NMJ co-cultures can be directly measured by a force transducer (Martin et al., 2015; Rizzuto et al., 2017; Rimington et al., 2021), which allows for assessment of the muscle force-length relationship and could be used for detailed functional studies in compartmentalized 3D NMJ models, similar to those performed in native nerve-muscle preparations (Martin et al., 2015; Rizzuto et al., 2017). Finally, transfer of MN activity to SkM can be blocked through a variety of AChR inhibitors including α-bungarotoxin (Osaki et al., 2018; Vila et al., 2019) and tubocurarine (Bakooshli et al., 2019; Rimington et al., 2021) to further validate NMJ functionality.
Neuromuscular Diseases
Neuromuscular diseases originate from various pathophysiological mechanisms, exhibit diverse symptoms, and differentially affect the NMJ (Figure 5). As such, they have historically been divided into subcategories and viewed through either a neurogenic or myogenic lens. However, increasing evidence for the important roles of cellular crosstalk in NMD pathogenesis suggest that modeling of the entire motor unit is necessary for proper studies of NMDs. In the following section, we present five NMD examples with diverse causes and manifestations highlighting the pathogenic roles of both MNs and SkM. First, we discuss ALS, a genetic disorder viewed to primarily affect MNs. Second, we consider MG, an autoimmune disorder focused upon the NMJ. Third, we examine DMD, a muscular dystrophy resulting from loss or truncation of the sarcolemmal protein dystrophin. Fourth, we review DM, a muscular dystrophy arising from toxic RNA repeats. Fifth, we discuss Pompe disease, a glycogen storage disorder affecting multiple cell types. For each NMD, we highlight most representative in vitro (Table 1) and in vivo models and offer perspective on future progress needed to advance NMD modeling toward translational applications.
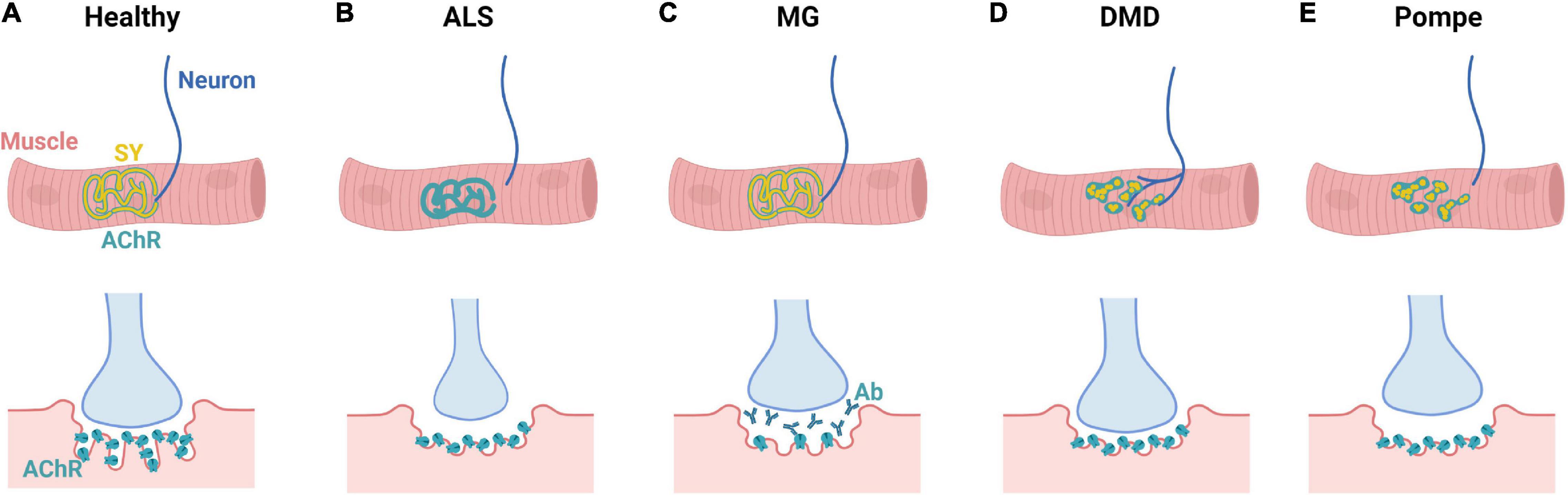
Figure 5. NMD-specific changes in NMJ morphology. (A) Mature human NMJs present with a characteristic “pretzel” shape and contain both synaptophysin (SY) and acetylcholine receptors (AChRs). NMJ endplates exhibit distinct membrane folds that extend into the muscle cell cytoplasm. (B) In ALS, marked decrease in synaptophysin and NMJ size occurs alongside denervation. Additionally, ALS patients present with smaller nerve terminals and flattened synaptic clefts. (C) In MG, NMJ morphology is generally maintained; however, there is synaptic accumulation of autoantibodies (Ab), decrease in AChRs, and shortening of synaptic clefts. (D) In DMD, there is marked axonal branching and NMJ fragmentation alongside shortening of the synaptic cleft. (E) In Pompe disease, there is denervation alongside NMJ fragmentation and increased rates of MN apoptosis.
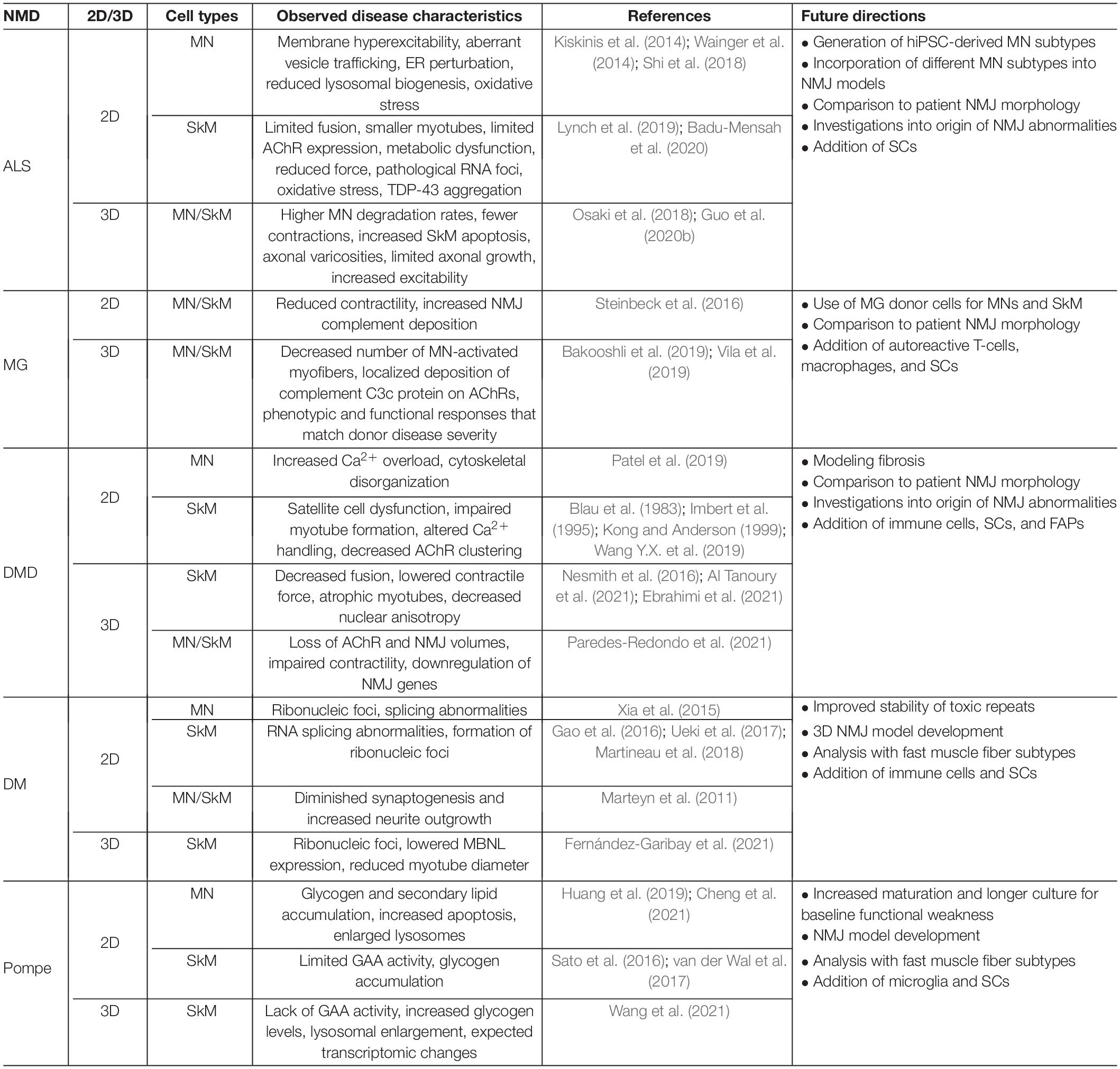
Table 1. Selected in vitro human models of NMD.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is late-onset, progressive NMD caused by SkM and MN wasting resulting in paralysis, respiratory failure, and death (Brown and Al-Chalabi, 2017). It is characterized by muscle stiffness and spasticity, but many patients also exhibit cognitive and behavioral changes (Oskarsson et al., 2018). Unfortunately, no ALS-specific biomarkers are currently known resulting in lengthy diagnosis periods and delayed treatments (Oskarsson et al., 2018). While no curative therapy is currently available, approved drugs, such as Edaravone and Riluzole, limit disease progression and may lengthen patient survival up to several months (Jaiswal, 2019). Approximately 90–95% of patients have sporadic ALS and 5–10% have familial disease, with no clear clinical or pathological differences between the groups (Loeffler et al., 2016). Over 100 genes have been attributed to familial ALS with the most commonly affected genes being C9ORF72, SOD1, TARDBP, and FUS, typically in combination (Wroe et al., 2008). The precise molecular mechanisms of ALS are unknown; however, many contributing factors have been proposed including protein aggregation (Ross and Poirier, 2004), excitotoxicity (Rothstein, 1995), aberrant nucleocytoplasmic or endosomal transport (Zhang et al., 2015), dysfunctional RNA metabolism (Strong, 2010), oxidative stress (Barber and Shaw, 2010), and axonal deformations (Bilsland et al., 2010). Although MN degradation is characteristic of ALS, denervation of the NMJ occurs first (Tremblay et al., 2017). Interestingly, ALS exhibits a preferential degradation of MNs with early loss of fast-fatigable MNs followed by fast fatigue-resistant, and then slow MNs (Tremblay et al., 2017). NMJs in ALS patients exhibit endplate fragmentation (Bjornskov et al., 1975), smaller endplates and nerve terminals (Tsujihata et al., 1984), flattened synaptic clefts (Yoshihara et al., 1998), and reduced mitochondrial presence within the nerve terminal (Tsujihata et al., 1984). Some small nerve terminals have been observed over distorted endplates, suggesting the possibility for NMJ regeneration (Yoshihara et al., 1998).
While ALS research has primarily focused upon MN pathology, studies with animal models have underlined the importance of pre-symptomatic SkM changes including atrophy and denervation (Loeffler et al., 2016). Prior to disease onset in SOD1 mice, there is an upregulation in muscle developmental genes (De Oliveira et al., 2014), a decrease in CDK5 (myogenic marker) (Park and Vincent, 2008), sarcoplasmic accumulation of neuronal NOS (Suzuki et al., 2010), and a loss in muscle volume (Kraft et al., 2007). Muscle-specific overexpression of SOD1 in mice has caused oxidative stress and muscular wasting without motor defects (Dobrowolny et al., 2008), while neuron-specific expression of SOD1 did not result in neuron abnormalities (Lino et al., 2002). Satellite cells from pre-symptomatic mice exhibit upregulated Pax7 expression (Manzano et al., 2011) and reduced proliferative capacity (Manzano et al., 2013). ALS patients exhibit a similar dysfunction in satellite cell proliferation (Scaramozza et al., 2014), indicating intrinsic muscle pathology in ALS as satellite cells are not directly innervated. Additionally, electrophysiological postsynaptic alterations presented in SOD1 mice prior to 6 weeks of age (Rocha et al., 2013) and decreased expression of choline acetyltransferase (ChAT) and vesicular acetylcholine transporter resulted in cholinergic dysfunction prior to MN degradation (Casas et al., 2016). In these mice, early NMJ dysfunction is accompanied by Ca2+ and reactive oxygen species accumulation, mitochondrial failure, and impaired transport within axons (Fischer-Hayes et al., 2013; Pollari et al., 2014). Clinically, pathophysiological axonal excitability is more pronounced within distal axonal branches (Nakata et al., 2006) and muscle denervation occurs before spinal cord MN degradation (Pollari et al., 2014). Based upon these observations, the “dying back” hypothesis of ALS suggests that this disease progresses through a retrograde degeneration of MNs from the periphery (Moloney et al., 2014) contrasting with the “dying forward” hypothesis that suggests glutamate excitotoxicity from cortical MNs advances forward to the periphery (Eisen et al., 1992). Additionally, astrocytes express most ALS related genes, and their dysregulation leads to neuroinflammation, oxidative stress, excitotoxicity, and protein aggregation further supporting the non-cell autonomous nature of ALS (Halpern et al., 2019).
Several animal models of ALS have been developed with a variety of mutations. Transgenic mice expressing mutant human SOD1 have been heavily studied and particularly useful in understanding pathophysiology of ALS. These mice progressively accumulate SOD1 within their muscles causing endoplasmic reticulum stress (Chen et al., 2015), recapitulate characteristic degradation of MNs and paralysis, and importantly demonstrate the non-cell autonomous nature of ALS (Nagai et al., 2007). Additionally, these mice exhibit altered gene expression related to muscle repair (De Oliveira et al., 2014), reduction in muscle volume (Marcuzzo et al., 2011), decreased proliferative capacity of satellite cells (Manzano et al., 2013), and a slow fiber-type shift (Hegedus et al., 2007). Studies with SOD1 mice have been useful to identify potential treatments with glial cell line-derived neurotrophic factor (GDNF) to rescue MN function through overexpression within the SkM (Li et al., 2007), stem cell-based delivery (Suzuki et al., 2007), and intramuscular injection of GDNF (Suzuki et al., 2008). Unfortunately, overexpression of healthy human SOD1 in these mice results in axonopathy undermining mutational importance within this disease model (Joyce et al., 2011) and spontaneous copy number deletions limit disease severity increasing variability within SOD1 mouse studies (Zwiegers et al., 2014). Additionally, rodent astrocytes exhibit significantly varied expression of many ALS-related genes, are less structurally and functionally diverse, and express a 10-fold decrease in glial fibrillary acidic protein-positive processes than human astrocytes, further limiting translational relevance of this model when studying the role of astrocytes in ALS (Oberheim et al., 2009).
To specifically study the effect of ALS on MN populations, hiPSC-based platforms have been widely employed. The first example of drug screening in ALS hiPSC-derived MNs used cells from patients with TARDBP mutations that exhibited decreased neurite length, which allowed identification of anacardic acid (a histone acetyltransferase inhibitor) as a potential ALS therapeutic agent (Egawa et al., 2012). These platforms have been expanded to the other ALS-associated mutations and have included assessments of membrane hyperexcitability (Wainger et al., 2014), vesicle trafficking (Shi et al., 2018), ER perturbation (Kiskinis et al., 2014), lysosomal biogenesis (Shi et al., 2018), and oxidative stress (Kiskinis et al., 2014). To model sporadic ALS in hiPSC-MNs, several models were combined to recapitulate heterogeneous neuronal degeneration, protein aggregation, and cell death and identify ropinirole as a potential therapeutic candidate (Fujimori et al., 2018). However, these models utilize rather immature neurons to model a disease that presents in mid to late life and only consider cell-autonomous effects of ALS. Accelerated aging through molecular manipulation, such as progerin overexpression (Miller et al., 2013), may improve the clinical relevance of these models.
To investigate non-cell autonomous contributions to ALS in a cell-specific manner, hiPSC-derived SkM and astrocyte models of ALS have been developed. Initial hiPSC-derived ALS SkM cells induced through both MyoD overexpression (Lenzi et al., 2016) and small molecule differentiation (Swartz et al., 2016) exhibited typical maturation patterns with limited pathologic alterations. Recently, however, a SkM model derived from SOD1-mutant ALS patient hiPSCs exhibited delayed and lower rates of fusion, smaller myotube size, limited AChR expression, metabolic dysfunction, and significantly reduced force production compared to healthy cells (Badu-Mensah et al., 2020). Additionally, a model using hiPSC-derived myotubes from C9ORF72 mutant patients exhibited pathological RNA foci, dipeptide repeat proteins, oxidative stress, and TDP-43 aggregation (Lynch et al., 2019). Beyond cultured hiPSC-SkM cells, hiPSC-derived astrocytes from ALS patients exhibit decreased expression of LC3-II causing p62 accumulation and modulated autophagy in HEK293T cells treated with astrocyte conditioned media (Madill et al., 2017). Additionally, C9ORF72 mutant hiPSC-derived astrocytes exhibited pathological RNA foci and dipeptide repeat proteins while causing MNs to undergo progressive action potential loss upon co-culture (Zhao et al., 2020). Through CRISPR-based removal of the C9ORF72 repeat, these phenotypes were reverted indicating both cell-autonomous astrocyte pathology and non-cell autonomous MN pathophysiology attributed to astrocytes (Zhao et al., 2020). Metabolically, C9ORF72-mutated astrocytes exhibit increased oxidative stress and senescence while secreting paracrine factors to induce oxidative stress in healthy MNs (Birger et al., 2019).
To combine these cell types in vitro, a compartmentalized ALS-on-a-chip model was developed to co-culture engineered 3D SkM tissues with heterogeneous (MNs and astrocytes) hiPSC-derived neural spheroids within a microfluidic device (Osaki et al., 2018). Functional NMJs were formed by axonal outgrowth from spheroids into SkM and light was used to stimulate the channelrhodopsin-2–expressing MNs to induce muscle contraction (Osaki et al., 2018). Engineered ALS motor units within this system exhibited higher degradation rates, induced fewer muscle contractions, and increased SkM apoptosis. These features were reversed through treatments with rapamycin and/or bosutinib as potential therapeutic agents (Osaki et al., 2018). A microfluidic compartmentalized 2D co-culture system made of hiPSC-MNs derived from three ALS mutant lines and primary wild-type myotubes exhibited axonal varicosities, limited axonal growth, and increased excitability (Guo et al., 2020b). Functional NMJs with ALS MNs were decreased in number and fidelity and showed increased fatigue index, while the Deanna protocol nutritional supplementation was found to correct these deficits in all lines (Guo et al., 2020b).
To improve upon our understanding of the underlying mechanisms of ALS pathology and to develop curative therapies, it will be critical to complement studies in both animal models and in vitro systems. ALS animal models with a wide variety of mutations have allowed for significant advancement in our understanding of the disease. However, limited disease severity and phenotypes, especially within supporting cells such as astrocytes (Oberheim et al., 2009), are likely to undermine clinical success of ALS therapeutics validated in animal models. Increasing disease severity in mice by additional knockdown of SOD1 in astrocytes, assessing the impact of different genetic backgrounds, and increased physical activity via treadmill running or swimming may yield more translationally relevant in vivo ALS models. For in vitro studies, current ALS-on-a-chip models (Osaki et al., 2018; Guo et al., 2020b) hold great potential for clinically relevant disease modeling and predictive drug screening as they exhibit therapeutically reversible ALS phenotypes and utilize platforms that support high-throughput studies. However, these models have yet to be analyzed to assess if they appropriately recapitulate pathological decrease in synaptophysin concentration, marked denervation, shrunken nerve terminals, and flattened synaptic clefts observed in ALS patients (Bjornskov et al., 1975; Tsujihata et al., 1984; Yoshihara et al., 1998). Further developments to in vitro ALS human models will be required to better recapitulate ALS pathology and disease progression. First, modular ALS platforms combined with non-invasive longitudinal and functional assessments can be utilized to help answer the fundamental question if ALS disease progression is due to direct loss of cortical MNs or their retrograde degeneration originating from muscle. Second, improved methodologies to generate hiPSC-derived MN subtypes will allow for further investigations into the preferential subtype-specific MN degradation observed in ALS (Tremblay et al., 2017). Third, incorporation of supportive cell types such as SCs will create a more realistic model of disease as SCs from ALS patients exhibit abnormal morphology with disorganized processes that extend into synaptic clefts (Bruneteau et al., 2015). In SOD1 mouse studies, similar disorganized processes were seen within SCs alongside upregulated galectin-3, a marker of phagocytosis (Martineau et al., 2020). These extensions may block the synaptic cleft, disrupting MN-SkM communication and contributing to ALS pathology, offering an interesting area for investigations within in vitro models.
Myasthenia Gravis
Myasthenia gravis is a rare autoimmune disorder characterized by accelerated fatigue within voluntary muscles, primarily in extraocular and facial muscles, proximal limbs, and neck extensors (Jayam Trouth et al., 2012). Weakness is highly variable and worsened by a variety of factors including heat, stress, and exercise (Jayam Trouth et al., 2012). MG is associated with accumulation of autoantibodies against NMJ proteins including muscle specific tyrosine kinase (MuSK), low-density lipoprotein receptor-related protein 4 (LRP4), and, most significantly, the nicotinic acetylcholine receptor (nAChR) (Nacu et al., 2015). The accumulation results from T cell-directed attack upon these postsynaptic membrane proteins (Ha and Richman, 2015). MG is often initially identified by ptosis and can be further classified into several subgroups based on autoantibody type and clinical features (Nacu et al., 2015). There are a wide range of treatments approved for MG including immunosuppression, anticholinesterase drugs, immunomodulation, and thymectomy (Farmakidis et al., 2018), but none provide curative outcomes.
Although the primary investigations of MG have focused on NMJ morphology and function, there have been reports of pathophysiological alterations in SkM. Human muscle atrophy was shown to present early in MG progression (Zouvelou et al., 2012) and accelerated atrophy was seen in type II fibers as compared to type I fibers (Wang et al., 2018). Additionally, accumulated anti-nAChR antibodies have been shown to modulate muscular IL-6 production altering mTOR signaling which may be responsible for MG-associated muscle fatigue (Maurer et al., 2015). Muscle biopsies from MG patients showed increased Pax7+ satellite cell pool, while isolated MG myoblasts exhibited increased proliferation and differentiation potential (Attia et al., 2017). A similar increase in satellite cell number, proliferation, and differentiation was observed in anti-nAChR mouse models of MG (Attia et al., 2017). Additionally, these mice exhibited delayed SkM maturation following development indicated by lower MyoG expression, reduced fiber size, and increased embryonic myosin heavy chain expression (Attia et al., 2017).
The role of autoantibodies in MG have been confirmed in multiple animal studies. Rabbits treated with anti-nAChR antibodies showed characteristic MG symptoms, confirming the autoimmune nature of MG (Patrick and Lindstrom, 1973). Similarly, administration of autoantibodies against LRP4 and MuSK in mouse models have reproduced MG symptoms indicating important roles of these receptors in disease (Wang et al., 2018). While useful in understanding NMJ dysfunction, acute induction of symptoms through autoantibody administration does not appropriately mimic the chronic progression of MG (Wang et al., 2018). Furthermore, immunized mice inconsistently exhibit clinically observable weakness, rarely present with extraocular muscle fatigue, and fail to recapitulate the dynamic nature of MG disease severity (Wang et al., 2018). Finally, there are additional auto-antibodies implicated in MG against proteins such as titin (Aarli, 2001), agrin (Gasperi et al., 2014), and cortactin (Gallardo et al., 2014) that are not considered in these models.
As MG is an autoimmune disorder, in vitro models of MG can be developed by adding patient serum to an existing NMJ platform. The first example of this utilized a 2D co-culture of optogenetically active hESC-derived MNs with primary myotubes to induce a reversible reduction in muscle contraction amplitude by treatment with IgG or active complement protein from MG patients (Steinbeck et al., 2016). In a 3D co-culture model of hiPSC-derived MNs and engineered SkM tissue, localized deposition of complement C3c protein was shown in AChRs of NMJs and treatment with MG IgG decreased number of MN-activated myofibers (Bakooshli et al., 2019). Additionally, automated optogenetic control was incorporated into a 3D MN-SkM culture system and differential responses to sera from donors were measured according to phenotype severity (Vila et al., 2019). Recently, hPSC-derived axial stem cells have been used to generate human neuromuscular organoids containing MNs, SCs, and SkM and study MG (Martins et al., 2020). This model showed reduced NMJ volumes alongside decreased rate and amplitude of contraction in response to MG sera. Although these models have recapitulated some aspects of MG through addition of patient sera, they are limited as they do not utilize MG patient-derived cells.
Overall, understanding the multi-organ nature of MG will require investigations in both animals and in vitro human model systems. While animal models are suitable for investigating the involvement of NMJs and immune system, they do not capture MG disease severity, resulting in failure of multiple MG treatments in clinical trials despite prior validation in animals (Mantegazza et al., 2016). Furthermore, direct analyses of heterocellular interactions important for MG pathology in animal models are hindered by the complexity of in vivo environments. On the other hand, in vitro models exhibit phenotypic and functional responses to patient sera that match disease severity in human donors (Vila et al., 2019) and can enable unique studies to elucidate the roles of cellular crosstalk in disease progression; however, they do not model systemic pathogenesis. Therefore, the next generation of in vitro MG models should incorporate immune cells to better recapitulate the autoimmune inflammatory environment. Specifically, incorporation of autoreactive T-cells would allow for investigations of the pathological development of autoantibodies beyond studying the effect of MG serum addition within existing platforms. In animal models of MG, macrophages can act as antigen-presenting cells and help produce self-AChR antibodies (Kinoshita et al., 1988) and as such, their incorporation into human in vitro systems (Juhas et al., 2018) would allow systematic studies of their roles in MG autoimmunity. Finally, further studies of SCs within in vitro models of MG should be performed to analyze their localization to the presynaptic membranes and potential neuroinflammatory roles (Ydens et al., 2013), including involvement in clinically observed pathological features such as disorganized axonal microorganelles, accumulation of Reich granules, and lipopigments (Kimura and Nezu, 1989).
Duchenne Muscular Dystrophy
Duchenne muscular dystrophy, a genetic myopathy with the highest prevalence of 7.1 in 100,000 male births (Crisafulli et al., 2020), is a fatal X-linked disorder caused by mutations in the dystrophin gene (Yiu and Kornberg, 2015). Dystrophin is an integral member of the dystrophin glycoprotein complex (DGC) that transmits contractile forces from the sarcomere to the ECM and functions as a molecular shock absorber (Le et al., 2018). In mature myofibers, dystrophin deficiency leads to sarcolemmal instability (Weller et al., 1990), abnormal calcium homeostasis (Tutdibi et al., 1999), and muscle degeneration (Torres and Duchen, 1987). In SCs, dystrophin regulates muscle stem cell commitment via epigenetic modifications (Chang et al., 2018) and regulation of cell polarity (Dumont et al., 2015b; Wang Y.X. et al., 2019). Loss of dystrophin results in increased levels of myogenic progenitors with an impaired ability to commit to differentiation resulting in diminished regenerative ability (Dumont et al., 2015b; Wang Y.X. et al., 2019). Together, this impaired satellite cell function, impeded muscle regeneration, and constant cycles of muscle degeneration result in progressive muscle weakness, loss of ambulation, and ultimately death due to respiratory failure (Yiu and Kornberg, 2015).
In addition to roles within SkM, dystrophin regulates neuronal function and is vital for healthy NMJ maintenance (Tintignac et al., 2015). Dystrophin and other members of the DGC are enriched at post-synaptic folds of the NMJ (Waite et al., 2009) and stabilize nAChRs (Zaccaria et al., 1998). Dystrophic myofibers are associated with increased rates of NMJ branching, fracturing, and transmission failure (Pratt et al., 2013, 2015a), suggesting a role of dystrophin in NMJ remodeling and/or maintenance. The role of dystrophin at the pre-synaptic level of NMJ structure and function is less clear. Increased pre-synaptic nerve terminal branching, axon sprouting, and denervation have been observed in mdx mice (Pratt et al., 2015b; van der Pijl et al., 2016) and humans (Nagao et al., 2003). These structural changes seen in dystrophic NMJs result in functional deficits including altered EMG characteristics and decreased safety factor of neuromuscular transmission in both patients (Priez et al., 1992) and mice (van der Pijl et al., 2016). Clinically, NMJ dysfunction results in increased sensitivity and slowed recovery from neuromuscular blocking drugs such as rocuronium and mivacurium (Ihmsen et al., 2009), contraindicating their use as anesthetics in DMD patients (Breucking et al., 2000). Lastly, dystrophin also regulates neuronal development and function in the brain which most likely contributes to the increased incidence of neurological abnormalities including autism, attention deficit disorder, and learning disabilities in DMD patients (Ricotti et al., 2016).
The majority of our understanding of DMD has been derived from a range of preclinical animal models (Wells, 2018) with the most common model being the mdx mouse which has a naturally occurring nonsense point mutation in exon 23 preventing dystrophin protein expression (Manning and O’Malley, 2015). The mdx mice exhibit several expected disease features including fibrosis (Gregorevic et al., 2008), respiratory dysfunction (Burns et al., 2018), cardiomyopathy (Mareedu et al., 2021), metabolic dysfunction (Moore et al., 2020), and muscle weakness (Barton et al., 2005). However, the mdx phenotype is milder and has a slower progression compared to clinical symptoms in DMD patients (Dangain and Vrbova, 1984). Disease severity of the mdx mouse model can be increased by the knockout of utrophin, which undergoes compensatory upregulation in mdx mice to protect against membrane instability (Deconinck et al., 1997). Alternatively, disease severity can be increased by knockout of the telomerase gene (mdx/mTR) which shortens telomere length and induces a more severe SkM (Sacco et al., 2010) and cardiac (Mourkioti et al., 2013) pathology. Larger preclinical animal models including rats (Larcher et al., 2014), rabbits (Sui et al., 2018), dogs (Nghiem and Kornegay, 2019), and pigs (Selsby et al., 2015) show greater disease severity and lethality than the traditional mdx mouse model. However, ethical concerns (Yokota et al., 2012) and secondary complications such as inability to feed (Gaschen et al., 1999) have limited their use to date. From larger animals, the golden retriever muscular dystrophy model (GRMD) (Nghiem and Kornegay, 2019) has been studied the most, but financial, ethical, and animal number concerns limit its use (Wells, 2018). Nevertheless, these preclinical models played important roles in validating current standard of care glucocorticoid therapy (Hudecki et al., 1993) and eteplirsen, a novel antisense oligonucleotide treatment approved by the FDA in 2016 (Khan et al., 2019).
Despite some success, numerous candidate therapeutics identified in animal studies have failed to be effective in humans driving the development of improved model systems (Rybalka et al., 2020). A primary limitation of current animal models is their low genetic and epigenetic diversity. DMD in humans is caused by over 4700 different mutations, and disease severity and response to pharmacological agents is heavily influenced by a range of disease modifiers such as expression levels of latent TGFB binding protein 4 (LTBP4) (Flanigan et al., 2013) and osteopontin-1 (Kyriakides et al., 2011). Disease severity of the mdx mouse model has been improved by crossing the mdx mutation onto the DBA/2J background, which contains a pro-fibrotic polymorphism in LTBP4 (Heydemann et al., 2009), resulting in greater fibrosis and functional impairment (van Putten et al., 2019). In the past decade, the development of genome editing tools such as TALEN and CRISPR-Cas9 has enabled the generation of humanized mdx mouse models (Aartsma-Rus and van Putten, 2019). These mice specifically model human dystrophin mutations to enable preclinical validation of gene editing therapies using CRISPR-Cas9 (Min et al., 2019) or antisense oligonucleotide (Veltrop et al., 2018) technologies.
While animal models can be genetically modified to include aspects of patients’ genetic diversity, true personalized disease platforms require use of patients’ tissues or cells. In vitro 2D studies of mouse or human DMD myoblasts have been utilized to model satellite cell dysfunction (Wang Y.X. et al., 2019), impaired myotube formation (Blau et al., 1983), and altered Ca2+ handling (Imbert et al., 1995). Myotubes lacking dystrophin or other members of the DGC display decreased AChR clustering (Kong and Anderson, 1999), indicating that post-synaptic NMJ abnormalities can occur in the absence of neural cells. When engineered into 3D tissues, DMD primary and immortalized patient cells display decreased fusion and force of contraction, atrophic myotubes, and decreased nuclear anisotropy (Nesmith et al., 2016; Al Tanoury et al., 2021; Ebrahimi et al., 2021). Large scale personalized platforms amenable to pharmacological screens will require the use of patient hiPSCs due to ethical and proliferative limitations of muscle biopsy-derived primary cells and a need for non-muscle cells such as MNs (Wang J. et al., 2019). Encouragingly, two chemicals (ginsenoside and fenofibrate) identified to improve fusion rate in hiPSC-derived DMD myoblasts were both found to improve muscle structure and function in mdx mice (Sun et al., 2020). Similarly, prednisolone, the current standard of care for DMD patients, rescued fusion, force of contraction, and branching defects in hiPSC-derived DMD myotubes (Al Tanoury et al., 2021), further implying a potentially predictive nature of these in vitro assays. In addition to drug development, hiPSC-derived DMD cells from patients with a wide-range of mutations can be utilized for optimization and validation of gene therapies such as guide RNA design for CRISPR-Cas9 mediated genome editing (Min et al., 2019).
Furthermore, hiPSC-based disease models allow building complex tissues to enable studies of the multi-cellular crosstalk in DMD pathogenesis. For example, astrocytes (Patel et al., 2019) and glutamatergic sensory neurons (Ruggieri et al., 2019) generated from DMD hiPSCs displayed increased Ca2+ overload and cytoskeletal disorganization indicating that neuronal involvement can be also studied using DMD hiPSC derivatives. Multicellular 3D cultures comprised of hiPSC-derived myoblasts, neurons, endothelial cells, and fibroblasts were successfully generated from both healthy and DMD cells (Maffioletti et al., 2018; Mazaleyrat et al., 2020), but the roles of cellular crosstalk in NMJ function and dysfunction in these cultures remain to be studied. Interestingly, in a primary and immortalized cell line based human 3D co-culture system, endothelial cells were required for DMD fibroblasts to undergo fibrinogenesis, suggesting that complex multicellular 3D platforms may be required to study mechanisms of DMD with high fidelity (Bersini et al., 2018). Recently, a compartmentalized optogenetic neuromuscular DMD model was developed by culturing hiPSC-derived DMD and isogenic control myoblasts with MN spheroids derived from wild-type channelrhodopsin-expressing murine or human ESCs (Paredes-Redondo et al., 2021). In this model, pharmacological inhibition of TGFβ signaling induced partial restoration of AChR and NMJ volumes along with significant up-regulation of MuSK expression (Paredes-Redondo et al., 2021). Besides SkM DMD platforms, hiPSC-derived DMD cardiomyocytes exhibit impaired contractile function (Chemello et al., 2021), altered calcium-handling (Chemello et al., 2021), and mitochondrial dysfunction (Sun et al., 2020), and have been utilized for in vitro studies of drug (Lin et al., 2015) and gene (Kyrychenko et al., 2017; Long et al., 2018; Chemello et al., 2021) therapies for DMD.
Since DMD arises from a range of mutations and its progression strongly depends on a variety of disease modifiers, complementary studies in animal and in vitro models will be required for improved understanding of disease mechanisms and development of effective therapeutic approaches. While translational relevance of current animal models is limited by low genetic and epigenetic diversity, genome editing technologies such as CRISPR can now enable improved phenotypic and mutational representation in animals (Pickar-Oliver et al., 2021). Regardless of these improvements, use of patient-derived cells will be necessary for truly personalized disease modeling. Recent 3D NMJ DMD models have provided a useful platform for studying NMJ dysfunction in vitro (Paredes-Redondo et al., 2021), but fully patient-specific DMD NMJ models remain to be developed. In these models, it will be important to perform careful morphological analysis of axonal branching and NMJ fragmentation to evaluate how well patient phenotypes are recapitulated (Pratt et al., 2013, 2015a). Similar to ALS models, the DMD platforms can be utilized in modular fashion to determine roles of distinct cellular and environmental components in NMJ pathology. For example, SCs within the mdx model exhibit disorganized processes directed away from endplates which may block innervation, indicating a potential role of SCs in DMD pathology (Personius and Sawyer, 2005) which remains to be studied in vitro. DMD patient and mouse muscles are also characterized by pro-inflammatory immune cell infiltration, which results in increased levels of neutrophils, T cells, and macrophages. Replicating this pro-inflammatory milieu in vitro could shed novel mechanistic insights into the effects of inflammation on NMJ structure and function in DMD. While the altered inflammatory milieu is thought to stimulate FAP proliferation and fibrosis (Juban et al., 2018), current DMD models do not exhibit fibrotic changes, thus the incorporation of both inflammatory and FAP cells will likely be essential to fully replicate the advanced stages of disease.
Myotonic Dystrophy
Myotonic dystrophy is the most prevalent form of muscular dystrophy in adults and is classified into Type I (DM1) and II (DM2). DM1 results from a series of CTG repeats in the DM protein kinase (DMPK) gene, while DM2 results from a series of CCTG repeats in the Zinc Finger 9 (ZNF9) gene (Thornton, 2014). The major pathogenic consequence of these DNA tandem repeats is gain-of-function of the resulting mutant RNA that form hairpin-like structures which bind and sequester RNA-binding proteins (Brouwer et al., 2009). This results in the sequestering and dysregulation of splice factors such as musclebind-like (MBNL) and CUG-binding proteins (CUGBP) (Fernandez-Costa et al., 2011). Ultimately, this leads to alternative splicing of multiple mRNAs including members of the DGC (Nakamori et al., 2007) and t-tubule proteins (Fugier et al., 2011) which contributes to the characteristic progressive myopathy and myotonia in DM. DM1 and 2 have a greater impact on fast muscle fibers and are associated with variable muscle fiber diameter, fiber splitting, and fibrofatty replacement (Vihola et al., 2003). Histologically, DM2 can be distinguished by pyknotic nuclear clumps that occur before the onset of muscle weakness (Meola and Cardani, 2015).
Aberrant splicing is not limited to SkM but occurs in multiple organs including the cardiac and neurological systems (Lee and Cooper, 2009). In DM1, splicing factors in the MBNL family accumulate within ribonuclear foci within both pre-synaptic MNs and post-synaptic nuclei leading to NMJ instability (Wheeler et al., 2007). DM1 patients exhibit thinned axon and myelin sheets without denervation (Fardeau and Tome, 1980). Additionally, repetitive nerve stimulation and single fiber electromyography in DM1 patients show abnormal nerve conduction and pathological jitter suggesting NMJ instability (Bombelli et al., 2016). DM1 mouse models exhibiting pathophysiological levels of CTG repeats in the DM1 region show distal denervation of diaphragm NMJs, reduced AChRs on the post synaptic membrane, and loss of unmyelinated fibers (Panaite et al., 2008). Furthermore, variable levels of axonal neuropathy (17–46%), axonal loss, and myelin sheath thinning have been reported in both DM1 patients (Peric et al., 2013) and mice (Panaite et al., 2011). Functionally, these structural alterations lead to abnormal nerve conduction and pathological jitter in DM1 patients (Bombelli et al., 2016). Currently there are no curative therapeutics for either DM1 or DM2 (Pascual-Gilabert et al., 2021). However, nearly two dozen preclinical and clinical drug development programs are currently active encompassing repurposed drugs, gene therapy, oligonucleotide therapeutics, and novel chemical treatments (Pascual-Gilabert et al., 2021).
Several animal models have been developed to study DM1 disease mechanisms and investigate potential therapies. DM1 was first modeled in DMPK knockout mice which only developed mild myopathy (Jansen et al., 1996), mild cardiac conduction dysfunction (Berul et al., 1999), and failed to replicate the multisystemic patient phenotype (Jansen et al., 1996). Through the overexpression of DMPK with toxic CTG repeats, a stronger disease phenotype was developed with ribonuclear foci changes, SkM atrophy, slowed growth, weakness, and myotonia (Vignaud et al., 2010). Neurologically, these mice exhibit RNA toxicity within Bergmann glia and Purkinje cell hyperexcitability, and reduced motor coordination representative of DM1 patients (Sicot et al., 2017). However, they still exhibit mild splicing defects and disease phenotype compared to patients (Huguet et al., 2012). Alternatively, myotonia and alternative splicing defects can be induced by combined MBNL1 inactivation and expression of untranslated CUG (HSALR model), but this does not lead to muscle wasting or denervation (Wheeler et al., 2007). Overexpression of CUGBP1 causes more severe myopathy and cardiomyopathy but is limited by high mortality and breeding issues. Through no individual mouse models capture all disease features or fully recapitulate severity seen in patients, they have provided significant mechanistic insights into the genetic causes of specific disease phenotypes.
Due to the multisystemic nature of DM, multiple muscle and non-muscle cell lines have been utilized to study DM pathology (Matloka et al., 2018). HEK, HeLa, and C2 cells with CTG repeats inserted in the 3′UTR of a truncated DMPK gene have replicated splicing misregulations and ribonuclear foci phenotypes (Philips et al., 1998; Warf and Berglund, 2007). In vitro drug screens to ameliorate these phenotypes have been utilized to identify novel therapeutics for DM (Warf and Berglund, 2007; Konieczny et al., 2017). DM patient-derived primary myoblast cultures exhibit metabolic alterations (Renna et al., 2017), splicing abnormalities (Laustriat et al., 2015), and ribonucleic foci formation (Fardaei et al., 2002). Myotube cultures have also been generated from MyoD overexpression in DM fibroblasts (Kuyumcu-Martinez and Cooper, 2006; Ravel-Chapuis et al., 2012) and hiPSCs (Gao et al., 2016; Ueki et al., 2017; Martineau et al., 2018) from DM patients. Similar to primary myoblasts, these cells exhibited RNA splicing abnormalities and formation of ribonucleic foci. Recently, the first 3D in vitro human muscle model of DM1 was developed by encapsulating patient-derived fibroblasts overexpressing MyoD in micromolded gelatin methacryloyl-carboxymethyl cellulose methacrylate hydrogels (Fernández-Garibay et al., 2021). Furthermore, for studies of neuromuscular abnormalities, DM patient ESCs were differentiated into MNs and co-cultured together with healthy SkM (Marteyn et al., 2011) and found to exhibit diminished synaptogenesis and increased neurite outgrowth associated with low expression of genes in the SLITRK family.
Additionally, hiPSC lines derived from DM patients have been used to study pathological alterations in distinct cell types (Gao et al., 2016; Ueki et al., 2017; Martineau et al., 2018). The neurological component of DM1 has been studied in hiPSC-derived neurons and astroglia which display expected ribonucleic foci and splicing abnormalities. These models have been used to demonstrate proof-of-principle phenotypic reversal through genome editing (Xia et al., 2015). While all models demonstrate histological alterations, no in vitro studies have shown myogenic or neuronal functional deficits. Additionally, CTG repeats have been shown to be unstable in pluripotent cells (Du et al., 2013) and CTG repeats do not expand when naive hiPSCs are differentiated into cardiomyocytes, muscle, or neurons as seen in vivo. Recently, DM1 hiPSC-derived cardiomyocytes from DM1 patients with varied CTG repeat lengths exhibited toxic RNA foci and mis-spliced MBNL1/2 transcripts and showed two distinct ions channel (Na+ and Ca2+) perturbations (Poulin et al., 2021). This platform revealed the underlying mechanism of electrical cardiac alterations in DM1 and can be used in the future to validate potential therapeutics in a high throughput fashion by monitoring action potential propagation and ionic currents in the human DM1 cardiomyocytes.
Overall, translationally relevant DM modeling is complicated by the multifaceted influences of DM throughout the entire body, most notably within neurological and muscular tissues. Although mouse models have greatly expanded our understanding of this pathogenic RNA disease, no single mouse model has exhibited severity comparable to patients or encompassed the myriad of DM phenotypes present in vivo. For example, DM1 patients exhibit toxic RNA accumulation whereas HSALR mice do not, which may contribute to the lack of denervation-like features in these mice (Wheeler et al., 2007). Intercrossing mouse lines may improve recapitulation of DM pathology and even show pathological synergy between symptoms. However, further development of patient-derived in vitro NMJ models will be critical for the ability to directly analyze the human DM NMJ. First, further optimization of hiPSC culture and differentiation protocols to produce myogenic and neuronal cells with stable CTG repeats that exhibit functional deficits will be critical. Second, novel methods for differentiating specific muscle subtypes from hiPSCs will augment our ability to accurately model and study DM as it preferentially affects fast muscle fibers (Vihola et al., 2003). Third, in DM1 patients, SCs exhibit abnormal glycogen accumulation and crystalline structures within their processes (Borenstein et al., 1977) and may contribute to disease progression through unknown mechanisms which remain to be studied in NMJ-SC co-cultures. Fourth, DM1 patients and mice show increased pro-inflammatory gene signatures and upregulation of the IL-6 pathway (Nakamori et al., 2017) and tumor necrosis factor superfamily member 12 (TNFSF12) signaling (Yadava et al., 2015). Incorporation of immune cells in DM NMJ models would allow important studies of how inflammation may contribute DM progression.
Pompe Disease
Pompe disease, also known as glycogen storage disease type II (GSDII), is a rare metabolic autosomal recessive disorder that results from deficiency of acid α-glucosidase (GAA) (Reuser et al., 1995). Pompe disease is categorized into two major types based upon disease onset and GAA enzyme activity, although a continuous spectrum of phenotypes exists. Infantile-onset Pompe disease (IOPD) is caused by very low GAA enzyme activity and results in fatal cardiac, neurologic, hepatic, and muscular dysfunction between ages 1 and 2 (Lim et al., 2014). In contrast, late-onset Pompe disease (LOPD) involves higher GAA activity which results in slower disease progression (Chan et al., 2017). GAA breaks down lysosomal glycogen, and its dysfunction leads to intralysosomal glycogen accumulation in various tissues but most abundantly within skeletal and cardiac muscle (Reuser et al., 1995). The build-up of glycogen in striated muscle leads to lysosomal enlargement, vacuolation (Prater et al., 2013), autophagosome proliferation (Nascimbeni et al., 2015), and lipofuscin aggregation (Feeney et al., 2014). These alterations are hypothesized to disrupt cellular and sarcomere architecture resulting in progressive muscle weakening and ultimately respiratory or cardiac failure (Reuser et al., 1995). However, neuromuscular abnormalities such as increased neuromuscular jitter and variance in response latency are seen within Pompe patient muscle prior to muscle weakness (Stålberg and Trontelj, 1997). This suggests that neurological dysfunction could be a key driver of muscle weakness and myopathy in Pompe disease.
In Pompe patients and mice, glycogen accumulation occurs in the brain (Mancall et al., 1965; Lee et al., 2011), central nervous system (CNS) (Mancall et al., 1965; Martini et al., 2001; DeRuisseau et al., 2009), and MNs (Gambetti et al., 1971; DeRuisseau et al., 2009). Furthermore, Pompe MNs have three-fold higher soma size (Mancall et al., 1965) and are more apoptotic (Turner et al., 2016), resulting in decreased motor output and neuronal loss (DeRuisseau et al., 2009). Pompe mice also exhibit significant NMJ alterations due to both presynaptic changes, such as reduced myelin thickness and neurofilament proteins, and postsynaptic changes, such as NMJ fragmentation (Falk et al., 2015). These structural alterations result in impaired neural output including increased numbers of denervated NMJs (Falk et al., 2015), decreased burst amplitude (DeRuisseau et al., 2009), and increased spontaneous EMG activity (Hobson-Webb et al., 2011). In addition to MNs, structural alterations and glycogen accumulation are found in neuronal support cells including astroglia and SCs (Martin et al., 1973). These alterations can lead to secondary disease traits including cognitive declines (Spiridigliozzi et al., 2017), speech disorders (Muller et al., 2009), and sensorineural and/or conductive hearing loss (Prater et al., 2012) demonstrating the wide ranging impacts of GAA deficiency.
Pompe disease has been predominantly modeled in the GAA knockout mice (Raben et al., 2001) that recapitulate key disease features such as striated muscle and nervous tissue glycogen accumulation (DeRuisseau et al., 2009), lysosomal abnormalities (Doyle et al., 2019), neuropathology (DeRuisseau et al., 2009), cardiac defects (Han et al., 2016), and muscle weakness (Lee et al., 2018). However, despite a complete lack of GAA, these mice exhibit a LOPD phenotype with late onset, slow disease progression, and normal breathing in normoxic conditions (Han et al., 2016; Gatto et al., 2017). Recently, crossing the GAA KO mice to the DBA2/J background resulted in a much more severe disease phenotype including early lethality, respiratory defects during normoxia, and more severe cardiomyopathy (Colella et al., 2020). While the pro-fibrotic polymorphism in LTBP4 found in the DBA2/J background has not been implicated in Pompe disease, polymorphisms in angiotensin-converting enzyme (ACE) and alpha-actinin 3 (ACTN3) (De Filippi et al., 2014) impact disease onset within LOPD. The classical GAA KO mouse model has been essential for identifying and validating potential therapeutics (Doyle et al., 2019) and has allowed for deeper understanding of the roles of the mTOR pathway, lysosomal dysregulation, and autophagocytosis in Pompe disease progression (Han et al., 2016; Gatto et al., 2017). In addition to mouse models, a novel zebrafish model, in which GAA activity is significantly reduced but not totally absent, displays significant motor behavior and NMJ abnormalities (Bragato et al., 2020). When this model was used as a drug screening platform, 3-bromopyruvic acid (Bragato et al., 2020) and 3,4-diaminopyridine phosphate (Cinzia et al., 2021) were found to increase AChR abundance, improve NMJ structure, and recover typical movement patterns. A baboon model of Pompe disease is currently used as a large animal preclinical model which has shown utility for therapeutic evaluation (Rastall et al., 2016). However, considering nearly 600 reported mutations within Pompe patients and the impact of disease gene modifiers (De Filippi et al., 2014), these models are unable to fully recapitulate human genetic complexity of the disease (Fukuda et al., 2007).
Current clinical therapy for Pompe disease is enzyme replacement therapy (ERT) which systemically delivers the recombinant human GAA (rhGAA) to break-down accumulated glycogen. While ERT significantly prolongs patient lifespan and augments quality of life (Kishnani et al., 2009), it is limited by inefficient delivery to SkM tissues (van der Ploeg et al., 2010), neutralization by host antibodies (De Vries et al., 2017), high dose requirements (Chien and Hwu, 2007), variable patient response (Kishnani et al., 2010), and high cost ($300,000 per year) (Güngör et al., 2013). Importantly, rhGAA does not appear to impact disease phenotypes in neurons nor does it cross the blood-brain barrier to help treat other neurological disease symptoms. Furthermore, restoration of SkM GAA activity does not restore nerve-invoked contractile function in GAA KO mice suggesting therapies should target neuronal tissues (Falk et al., 2015). In support, neuron-specific gene therapy in Pompe mice improved motor coordination, decreased astrogliosis, and increased myelination (Lee et al., 2018). However, early administration of AAV9-hGAA (age 1 month) led to the greatest restoration of GAA activity and overall function, while late administration (age 15 months) was not effective, reflecting how this treatment was unable to reverse a deficit in NMJ function and force production despite removing muscular glycogen accumulation (Todd et al., 2015).
In addition to studies in animal models and patients, in vitro human cell culture systems play important roles in modeling genetic diversity and neuromuscular dysfunction in Pompe disease. Specifically, human myotube cultures accurately model clinical differences between IOPD and LOPD patients (Raben et al., 2010), with IOPD myotubes displaying lysosomal enlargement (Spampanato et al., 2013) and LOPD myotubes exhibiting autophagosome accumulation (Nascimbeni et al., 2012). Glycogen accumulation and lysosomal enlargement in primary and hiPSC-derived IOPD myotubes could be prevented by overexpression of transcription factor EB (TFEB), a regulator of autophagy and lysosomal biogenesis (Spampanato et al., 2013; Sato et al., 2016). These findings were translated to GAA KO mice, where AAV delivery of TFEB ameliorated muscle pathology and restored contractile function (Gatto et al., 2017). Recently, the first in vitro 3D model of human Pompe disease SkM was reported using primary muscle cells (Wang et al., 2021). This model exhibited the expected lack of GAA activity, increased glycogen levels, lysosomal enlargement, and transcriptomic changes characteristic of Pompe disease but displayed no innate functional weakness (Wang et al., 2021). However, functional deficits could be induced by causing lysosomal stress with chloroquine, enforcing glycogen utilization by glucose starvation, or glycogen phosphorylase inhibition (Wang et al., 2021). Compared to 2D models, this biomimetic platform better recapitulates the in vivo Pompe phenotype for studies of glycogen accumulation and responses to candidate pharmacological and gene therapies.
Like other myopathies, large-scale and patient-specific studies of Pompe disease will require the utilization of hiPSCs. Encouragingly, hiPSC-derived myotubes generated through MyoD overexpression (Sato et al., 2016) or directed differentiation exhibit expected reductions in GAA activity and glycogen accumulation (van der Wal et al., 2017). Their utility as drug screening platforms has been shown by the ability of recombinant GAA (Yoshida et al., 2017), lentivirus encoding GAA (Sato et al., 2016), and antisense oligonucleotide (van der Wal et al., 2017) treatments to recover GAA enzyme activity and normalize cellular glycogen levels. The CNS involvement in Pompe disease has been modeled in vitro with hiPSC-derived neural stem cells which exhibited glycogen and secondary lipid accumulation, increased apoptosis, and enlarged lysosomes (Huang et al., 2019; Cheng et al., 2021). Lysosome size and glycogen accumulation could be decreased with rhGAA, hydroxypropyl-β-cyclodextrin, antioxidants (δ-tocopherol and ebselen), or PI3-K inhibitors (wortmannin and PX-866) (Huang et al., 2019; Cheng et al., 2021). Importantly, ebselen was able to increase GAA activity in the brain of GAA KO mice suggesting that these in vitro platforms could identify compounds that target neuronal tissues in Pompe patients (Huang et al., 2019). However, modeling of Pompe NMJ structure and function in hiPSC platforms has not been reported to date.
Overall, Pompe disease causes systemic accumulation of glycogen leading to a myriad of symptoms including cognitive decline, muscular wasting, and NMJ dysfunction. While animal models have been critical for therapeutic developments and understanding Pompe pathology, they do not recreate human genetic complexity. Further complications arise from variations in clinical features between IOPD and LOPD patients underlining the importance of complementing animal studies with in vitro disease modeling platforms. Promisingly, hiPSC-derived Pompe disease myotubes exhibit expected reductions in GAA activity and glycogen accumulation and can be utilized for personalized drug screening (Sato et al., 2016; van der Wal et al., 2017; Yoshida et al., 2017). However, current in vitro models of 3D Pompe muscle do not model functional weakness in the absence of exogenous stressors such as those inducing lysosomal deficiency or glucose starvation (Wang et al., 2021). Functional weakness could potentially be gained by increasing experimental duration or muscle maturation as done in 2D micropatterned cultures that increased pathological LAMP1-positive lysosome accumulation in hiPSC-derived Pompe myotubes (Jiwlawat et al., 2019). Alternatively, the lack of baseline functional deficit in engineered Pompe muscles could point to the requisite NMJ involvement, warranting the development of hiPSC-derived NMJ models of Pompe disease. Additional incorporation of SCs and microglia would be of particular interest as these cells also show glycogen accumulation and cytoplasmic ballooning (Martin et al., 1973) and can contribute to Pompe pathogenesis. Since reduced myelin is seen in Pompe mice (Falk et al., 2015), effects of SC myelination should be also assessed in human in vitro models. Finally, ERT is more effective in fast than slow muscle fibers (Hawes et al., 2007), thus the ability to differentiate specific muscle fiber subtypes from Pompe hiPSCs would further augment translational relevance of these in vitro models.
Discussion
Decades of work using both in vitro and in vivo models of NMDs have led to important mechanistic insights and notable therapeutic advances. Here, we have compared the utility of these models for studying the NMJ structure and function and discussed the current state of disease modeling in the context of five specific NMDs. Animal models have been essential for our understanding of the clinical features of NMDs, but their limited genetic diversity and non-human physiology hinder their ability to fully recapitulate human NMD phenotypes, severity, and progression. Advances in genome editing technologies have facilitated generation of animal models with human mutations, providing a means to generate improved preclinical models for testing pharmacological and gene therapies for NMDs. Nevertheless, truly personalized disease modeling that accurately represents patient genetic and epigenetic diversity will require development of high-fidelity in vitro human NMJ platforms.
Recent advances in tissue-engineering methodologies have increased our in vitro modeling capabilities and furthered our understanding of human NMDs. hiPSC-derived models, in particular, hold promise for use in large-scale pharmaceutical testing, systematic analysis of disease mechanisms, and development of patient-specific treatments. However, additional progress will be needed to fully recapitulate NMD progression and complexity in vitro to allow for meaningful studies of underlying pathological mechanisms and drug responses (Figure 6). Specifically, hiPSC-derived NMJ models remain immature compared to primary NMJ models even after lengthy differentiation protocols. Therefore, improved differentiation methods will be necessary to not only replicate but accelerate developmental processes to obtain hiPSC-derived MNs and SkM cells and generate NMJs with mature structure and functionality akin to those of native motor units. Modifications of existing differentiation protocols to derive specific MN subtypes and SkM fiber types and stimulate NMJ maturation (Zhang et al., 2016) will allow development of in vitro models that can investigate why specific types of NMJs are distinctly affected by different NMDs and will enable targeted pharmacological testing of most affected tissues, such as type II muscle fibers in MG (Wang et al., 2018).
Figure 6. Future developments of hiPSC-derived NMD models. Optimization of soluble factor treatments will be needed to improve maturity of MNs, SkM, and NMJs toward their respective in vivo-mimetic phenotypes. Incorporation of biophysical stimulation during cell culture such as electrical stimulation, mechanical stretch, and media agitation are expected to further improve functional maturity of NMJs. Biomaterial design and microfabrication/microfluidics techniques can be applied to generate biomimetic microenvironments for cell growth via controlled presentation of developmentally relevant gradients in growth factors, topography, and stiffness. As the function of native NMJs relies on complex multi-cellular interactions, the ability to incorporate Schwann cells (SCs), endothelial cells (ECs), fibro-adipogenic progenitors (FAPs), and immune system cells (ICs) would improve modeling of in vivo states and our understanding of NMJ physiology and pathology. Incorporation of additional organ compartments sharing culture media with NMJs, such as intestine, gut, and liver will better emulate human drug responses, while adding bone, ligament, cartilage, and fat compartments will allow studies of the organ-organ crosstalk in NMDs. Finally, further miniaturization and standardization of culture systems along with development of non-invasive metabolic and functional readouts will enable establishment of versatile NMD platforms for high-throughput drug discovery research.
Replicating native, mature functional properties of NMJs will be crucial for accurate in vitro modeling of NMDs that often occur in adulthood. The immature state of hiPSC-derived cells remains a significant hurdle to generating predictive in vitro disease models or developing safe and effective regenerative therapies. Development of high-fidelity human NMJ models of NMDs is further complicated by the lack of detailed histological and functional descriptions of NMJs in NMD patients. Therefore, evaluation of in vitro human NMJ function and dysfunction as well as early formation relies largely on comparisons with murine models of development and disease. On the other hand, duration of human MN neurogenesis and maturation in vivo (Gogliotti et al., 2012; Stein et al., 2014) and in vitro (Johnson et al., 2007; Wainger et al., 2014) is substantially longer compared to murine counterparts, requiring longer-lasting and costly in vitro protocols to achieve mature MN states. Accelerating human MN maturation is commonly performed via small molecule inhibition of Notch. Disruption of Notch signaling accelerates neuronal differentiation by delaying the cell cycle transition from G1 to S phase increasing the commitment of progenitors toward neurogenesis (Crawford and Roelink, 2007; Borghese et al., 2010) and pre-MNs toward a post-mitotic, mature MN fate (Maury et al., 2015). While the small molecules that stimulate cell cycle exit have proven successful in accelerating MN maturation, their effects on the acquisition of distinct MN fates (e.g., MMC, SAC, PMC, LMC, HMC, PGC, ANS) are unexplored. As different MN subtypes emerge at distinct developmental stages in vivo, premature cell cycle exit could obstruct the ability to activate later MN subtype programs, which will require future investigations.
Similar to MNs, the immature state of current engineered SkM tissues lessens their physiological relevance and utility in disease modeling. A number of methods have been employed to enhance in vitro SkM maturation including dynamic culture (Juhas and Bursac, 2014), mechanical stretch (Powell et al., 2002), electrical stimulation (Khodabukus et al., 2019), growth factors (Ebrahimi et al., 2018), hormones (Butler-Browne et al., 1984), and small molecules (Selvaraj et al., 2019). Interestingly, some of these same interventions are also beneficial to both MN and NMJ formation, maturation, and regeneration. For example, electrical stimulation increases axon regeneration and recovery of motor function in sciatic nerve injury in Sprague-Dawley rats (Fu et al., 2020) and improves alignment and maturation of human myotubes in culture (Ahadian et al., 2012; Khodabukus et al., 2019). As functional innervation is required for muscle maturation and mature muscle contributes to maintaining functional NMJs, it will be important to develop methods that will improve overall maturation state in NMJ co-cultures. Additionally, myonuclei are typically localized at the periphery of muscle fibers, but specialized myonuclei are anchored below the mature postsynaptic membrane in vivo (Grady et al., 2005). Advances in single-nucleus RNA sequencing have revealed hundreds of novel genes expressed within postsynaptic myonuclei (Petrany et al., 2020). Genetic knockdown of a small subset of these genes revealed Gramd1b as a positive and Ufsp1 as a negative modulator of AchR clustering suggesting that novel insights into NMJ maturation and maintenance can be obtained from sequencing datasets. Similar studies of transcriptional specialization within modular neuromuscular cultures (consisting of multiple compartments and cell types) would improve our understanding of NMJ development leading to effective methods to increase NMJ maturation.
Ultimately, besides accelerated maturation of MNs and SkM cells, the incorporation of additional non-myogenic muscle-resident cell types, such as SCs, fibro-adipogenic progenitors (FAPs), and endothelial cells will be required to recreate native NMJ complexity. Here, heterocellular interactions can stimulate formation and advanced maturation of biomimetic NMJs to model both normal physiology and disease. For example, SCs are essential in the formation and maintenance of the adult NMJ (Ko and Robitaille, 2015) and their incorporation into NMJ models has supported MN and SkM viability, myelinated axons, and extended culture times (Singh and Vazquez, 2019; Hyung et al., 2021). Unfortunately, they have been underrepresented within NMD models despite their suggested disease-modifying roles in ALS (Bruneteau et al., 2015), MG (Kimura and Nezu, 1989), DMD (Personius and Sawyer, 2005), DM (Borenstein et al., 1977), and Pompe disease (Martin et al., 1973). Similarly, FAPs reside adjacent to NMJs, neighboring SCs and MNs, where they actively influence SCs and maintain NMJ integrity (Hogarth et al., 2019; Uezumi et al., 2021). Subpopulations of FAPs are dysregulated in many NMDs and accumulate within atrophied ALS mouse muscles (Gonzalez et al., 2017), exhibit varied gene signatures in response to denervation and cardiotoxin injury (Madaro et al., 2018), and activate IL-6-STAT3 signaling in response to denervation that may lead to fibrosis related to NMDs (Madaro et al., 2018). Vascular cells have critical roles in NMJ development, maintenance, and regeneration in vivo (Sawada et al., 2014) as well as secrete neurotrophic factors that support axonal growth in vitro (Grasman and Kaplan, 2017). Their roles in NMJ formation and function also remain to be explored and further elucidate their potential roles in NMDs.
Generation of advanced in vitro NMD models and platforms for predictive drug screening will also require the incorporation of cell of the immune system and cells involved in drug metabolism, respectively. The immune system is a critical regulator of both neurological (Prinz and Priller, 2017) and muscular (Farup et al., 2015) homeostasis, injury response, and pathology. The chronic injury cycles characteristic of multiple NMDs result in persistent and increased infiltration of monocytes/macrophages (Acharyya et al., 2007), T lymphocytes (Uzawa et al., 2021), and mast cells (Trias et al., 2017) at NMJs. These cells secrete multiple factors that induce a chronic pro-inflammatory environment and contribute to disease progression through several mechanisms that increase oxidative stress and alter stem cell function. Similarly, accurate modeling of autoimmune diseases such as MG will require incorporation of T and B lymphocytes to model chronic inflammation and autoantibody production (Uzawa et al., 2021). Realistic in vitro modeling of pharmacodynamics and expected drug concentrations in the bloodstream will necessitate incorporation of additional organ-on-chip (OOC) modules containing extramuscular tissues such as intestine, gut, liver, and fat. These multiplexed culture systems will be able to identify unexpected drug toxicities due to organ-organ crosstalk, such as that seen for bleomycin cardiomyocyte toxicity only in the presence of lung tissue (Skardal et al., 2017). Multiplexed OOC platforms will also enable studies of the more intimate tissue-tissue relationships between skeletal muscle and bone, ligament, or cartilage, which can be perturbed in NMDs as well as in various musculoskeletal degenerative diseases. In support of the feasibility of these complex systems, up to 10 unique OOC modules have been successfully interconnected to form a body-on-a-chip (BOC) platform (Novak et al., 2020). While additional tissue maturation within each OOC module is required, BOC platforms would have the unique potential to permit modular crosstalk studies allowing understanding of the NMD progression on a systemic level.
Beyond incorporation of additional cell types, further advancements in biomaterial design and microfabrication technologies will be critical for successful engineering of microphysiological systems that replicate in vivo 3D microenvironments of developing or diseased NMJs. For example, 3D hydrogels that incorporate the basement membrane proteins laminin and collagen IV have been shown to support increased contractile force generation in muscle-only (Hinds et al., 2011) and muscle-neuron (Vilmont et al., 2016) culture platforms and uniquely enable tau aggregation in organoid models of Alzheimer’s disease (Choi et al., 2014). Recent advancements in the design of smart biomaterials that allow spatiotemporal control of growth factor, topographical, and stiffness gradients (Darnell and Mooney, 2017; Kowalski et al., 2018), combined with use of microfluidic devices to establish tissue compartmentalization and gradients of soluble factors (Sun et al., 2019), offer opportunities to precisely influence the migration, proliferation, differentiation, and maturation of hiPSC derivatives through mimicry of the native ECM architecture and biophysical and biochemical cues. For example, 3D bioprinting (Kang et al., 2016; Zhang et al., 2018) of bioactive biomaterials to enable compartmentalized growth and differentiation of MNs and SkM (Osaki et al., 2018; Narayanan et al., 2021), along with microfluidic approaches to establish a local microenvironment supportive of NMJ formation and maturation via spatially defined delivery of auxiliary cells and agrin (Tourovskaia et al., 2008), could provide means to generate highly functional NMJs amenable to rigorous studies of NMD pathology.
Current in vitro NMJ models are mainly utilized for small-scale, stand-alone studies, or to supplement whole-organ and organism-level in vivo investigations with human cell- and tissue-specific data. Combining animal studies from a single genetic background with in vitro validations in hiPSC-based models from genetically diverse patients are likely to improve the predictive value of preclinical therapeutic tests. Eventually, however, advances in hiPSC technology along with miniaturization and standardization of microphysiological systems are expected to enable self-sufficient, high-throughput in vitro pharmacological screens with directly translational outcomes. Altogether, we anticipate that future advances in patient-specific hiPSC-based in vitro modeling of NMDs will be instrumental for gaining deeper understanding of human NMD pathophysiology and will lead to streamlined developments of pharmacotherapies for these devastating disorders.
Author Contributions
ZF, EL, and TC wrote the manuscript. TC and ZF generated figures for the manuscript. AK and NB edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants AR055226 and AR070543 from National Institute of Arthritis and Musculoskeletal and Skin Disease, grant UG3TR002142 from the NIH Common Fund for the Microphysiological Systems Initiative, grant U01EB028901 from National Institute of Biomedical Imaging and Bioengineering, and the Jain foundation. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aarli, J. A. (2001). Titin, thymoma, and myasthenia gravis. Arch. Neurol. 58, 869–870. doi: 10.1001/archneur.58.6.869
Aartsma-Rus, A., and van Putten, M. (2019). The use of genetically humanized animal models for personalized medicine approaches. Dis. Models Mech. 13:dmm041673. doi: 10.1242/dmm.041673
Abujarour, R., Bennett, M., Valamehr, B., Lee, T. T., Robinson, M., Robbins, D., et al. (2014). Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl. Med. 3, 149–160. doi: 10.5966/sctm.2013-0095
Acharyya, S., Villalta, S. A., Bakkar, N., Bupha-Intr, T., Janssen, P. M., Carathers, M., et al. (2007). Interplay of IKK/NF-κB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Invest. 117, 889–901. doi: 10.1172/JCI30556
Afshar, M. E., Abraha, H. Y., Bakooshli, M. A., Davoudi, S., Thavandiran, N., Tung, K., et al. (2020). A 96-well culture platform enables longitudinal analyses of engineered human skeletal muscle microtissue strength. Sci. Rep. 10:6918. doi: 10.1038/s41598-020-62837-8
Ahadian, S., Ramon-Azcon, J., Ostrovidov, S., Camci-Unal, G., Hosseini, V., Kaji, H., et al. (2012). Interdigitated array of Pt electrodes for electrical stimulation and engineering of aligned muscle tissue. Lab Chip 12, 3491–3503. doi: 10.1039/c2lc40479f
Al Tanoury, Z., Zimmerman, J. F., Rao, J., Sieiro, D., Mcnamara, H. M., Cherrier, T., et al. (2021). Prednisolone rescues Duchenne muscular dystrophy phenotypes in human pluripotent stem cell–derived skeletal muscle in vitro. Proc. Natl. Acad. Sci. U.S.A. 118:e2022960118. doi: 10.1073/pnas.2022960118
Albini, S., and Puri, P. L. (2014). Generation of myospheres from hESCs by epigenetic reprogramming. J. Vis. Exp. JoVE 88:51243. doi: 10.3791/51243
Askanas, V., Kwan, H., Alvarez, R. B., Engel, W. K., Kobayashi, T., Martinuzzi, A., et al. (1987). De novo neuromuscular junction formation on human muscle fibres cultured in monolayer and innervated by foetal rat spinal cord: ultrastructural and ultrastructural–cytochemical studies. J. Neurocytol. 16, 523–537. doi: 10.1007/BF01668506
Attia, M., Maurer, M., Robinet, M., Le Grand, F., Fadel, E., Le Panse, R., et al. (2017). Muscle satellite cells are functionally impaired in myasthenia gravis: consequences on muscle regeneration. Acta Neuropathol. 134, 869–888. doi: 10.1007/s00401-017-1754-2
Aulehla, A., Wehrle, C., Brand-Saberi, B., Kemler, R., Gossler, A., Kanzler, B., et al. (2003). Wnt3a plays a major role in the segmentation clock controlling somitogenesis. Dev. Cell 4, 395–406. doi: 10.1016/S1534-5807(03)00055-8
Babin, P. J., Goizet, C., and Raldua, D. (2014). Zebrafish models of human motor neuron diseases: advantages and limitations. Prog. Neurobiol. 118, 36–58. doi: 10.1016/j.pneurobio.2014.03.001
Badu-Mensah, A., Guo, X., Mcaleer, C. W., Rumsey, J. W., and Hickman, J. J. (2020). Functional skeletal muscle model derived from SOD1-mutant ALS patient iPSCs recapitulates hallmarks of disease progression. Sci. Rep. 10:14302. doi: 10.1038/s41598-020-70510-3
Bakooshli, M. A., Lippmann, E. S., Mulcahy, B., Iyer, N., Nguyen, C. T., Tung, K., et al. (2019). A 3D culture model of innervated human skeletal muscle enables studies of the adult neuromuscular junction. Elife 8:e44530. doi: 10.7554/eLife.44530.033
Barber, S. C., and Shaw, P. J. (2010). Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 48, 629–641. doi: 10.1016/j.freeradbiomed.2009.11.018
Barik, A., Li, L., Sathyamurthy, A., Xiong, W. C., and Mei, L. (2016). Schwann cells in neuromuscular junction formation and maintenance. J. Neurosci. 36, 9770–9781. doi: 10.1523/JNEUROSCI.0174-16.2016
Barton, E. R., Morris, L., Kawana, M., Bish, L. T., and Toursel, T. (2005). Systemic administration of L-arginine benefits mdx skeletal muscle function. Muscle Nerve 32, 751–760. doi: 10.1002/mus.20425
Bersini, S., Gilardi, M., Ugolini, G. S., Sansoni, V., Talo, G., Perego, S., et al. (2018). Engineering an environment for the study of fibrosis: a 3D human muscle model with endothelium specificity and endomysium. Cell Rep. 25, 3858–3868.e3854. doi: 10.1016/j.celrep.2018.11.092
Berul, C. I., Maguire, C. T., Aronovitz, M. J., Greenwood, J., Miller, C., Gehrmann, J., et al. (1999). DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Invest. 103, R1–R7. doi: 10.1172/JCI5346
Bettadapur, A., Suh, G. C., Geisse, N. A., Wang, E. R., Hua, C., Huber, H. A., et al. (2016). Prolonged culture of aligned skeletal myotubes on micromolded gelatin hydrogels. Sci. Rep. 6:28855. doi: 10.1038/srep28855
Bilsland, L. G., Sahai, E., Kelly, G., Golding, M., Greensmith, L., and Schiavo, G. (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 20523–20528. doi: 10.1073/pnas.1006869107
Birger, A., Ben-Dor, I., Ottolenghi, M., Turetsky, T., Gil, Y., Sweetat, S., et al. (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 50, 274–289. doi: 10.1016/j.ebiom.2019.11.026
Bjornskov, E. K., Dekker, N. P., Norris, F. H., and Stuart, M. E. (1975). End-plate morphology in amyotrophic lateral sclerosis. Arch. Neurol. 32, 711–712. doi: 10.1001/archneur.1975.00490520081016
Bladt, F., Riethmacher, D., Isenmann, S., Aguzzi, A., and Birchmeier, C. (1995). Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376, 768–771. doi: 10.1038/376768a0
Blanchard, A. D., Sinanan, A., Parmantier, E., Zwart, R., Broos, L., Meijer, D., et al. (1996). Oct-6 (SCIP/Tst-1) is expressed in Schwann cell precursors, embryonic Schwann cells, and postnatal myelinating Schwann cells: comparison with Oct-1, Krox-20, and Pax-3. J. Neurosci. Res. 46, 630–640. doi: 10.1002/(SICI)1097-4547(19961201)46:5<630::AID-JNR11>3.0.CO;2-0
Blau, H. M., Webster, C., and Pavlath, G. K. (1983). Defective myoblasts identified in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 80, 4856–4860. doi: 10.1073/pnas.80.15.4856
Bombelli, F., Lispi, L., Porrini, S. C., Giacanelli, M., Terracciano, C., Massa, R., et al. (2016). Neuromuscular transmission abnormalities in myotonic dystrophy type 1: a neurophysiological study. Clin. Neurol. Neurosurg. 150, 84–88. doi: 10.1016/j.clineuro.2016.08.020
Borenstein, S., Noël, P., Jacquy, J., and Flamentdurand, J. (1977). Myotonic dystrophy with nerve hypertrophy: report of a case with electrophysiological and ultrastructural study of the sural nerve. J. Neurol. Sci. 34, 87–99. doi: 10.1016/0022-510X(77)90094-6
Borghese, L., Dolezalova, D., Opitz, T., Haupt, S., Leinhaas, A., Steinfarz, B., et al. (2010). Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 28, 955–964. doi: 10.1002/stem.408
Bragato, C., Carra, S., Blasevich, F., Salerno, F., Brix, A., Bassi, A., et al. (2020). Glycogen storage in a zebrafish Pompe disease model is reduced by 3-BrPA treatment. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1866:165662. doi: 10.1016/j.bbadis.2020.165662
Breucking, E., Reimnitz, P., Schara, U., and Mortier, W. (2000). [Anesthetic complications. The incidence of severe anesthetic complications in patients and families with progressive muscular dystrophy of the Duchenne and Becker types]. Anaesthesist 49, 187–195. doi: 10.1007/s001010050813
Brouwer, J. R., Willemsen, R., and Oostra, B. A. (2009). Microsatellite repeat instability and neurological disease. Bioessays 31, 71–83. doi: 10.1002/bies.080122
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172. doi: 10.1056/NEJMra1603471
Bruneteau, G., Bauché, S., Gonzalez De Aguilar, J. L., Brochier, G., Mandjee, N., Tanguy, M. L., et al. (2015). Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2, 362–372. doi: 10.1002/acn3.179
Burbulla, L. F., Beaumont, K. G., Mrksich, M., and Krainc, D. (2016). Micropatterning facilitates the long-term growth and analysis of iPSC-derived individual human neurons and neuronal networks. Adv. Healthc. Mater. 5, 1894–1903. doi: 10.1002/adhm.201500900
Burns, D. P., Canavan, L., Rowland, J., O’flaherty, R., Brannock, M., Drummond, S. E., et al. (2018). Recovery of respiratory function in mdx mice co-treated with neutralizing interleukin-6 receptor antibodies and urocortin-2. J. Physiol. 596, 5175–5197. doi: 10.1113/JP276954
Butler-Browne, G. S., Herlicoviez, D., and Whalen, R. G. (1984). Effects of hypothyroidism on myosin isozyme transitions in developing rat muscle. FEBS Lett. 166, 71–75. doi: 10.1016/0014-5793(84)80047-2
Casas, C., Manzano, R., Vaz, R., Osta, R., and Brites, D. (2016). Synaptic failure: focus in an integrative view of ALS. Brain Plast. 1, 159–175. doi: 10.3233/BPL-140001
Cescon, M., Gregorio, I., Eiber, N., Borgia, D., Fusto, A., Sabatelli, P., et al. (2018). Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 136, 483–499. doi: 10.1007/s00401-018-1860-9
Chakkalakal, J. V., Nishimune, H., Ruas, J. L., Spiegelman, B. M., and Sanes, J. R. (2010). Retrograde influence of muscle fibers on their innervation revealed by a novel marker for slow motoneurons. Development 137, 3489–3499. doi: 10.1242/dev.053348
Chal, J., Al Tanoury, Z., Hestin, M., Gobert, B., Aivio, S., Hick, A., et al. (2016). Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 11, 1833–1850. doi: 10.1038/nprot.2016.110
Chalamalasetty, R. B., Garriock, R. J., Dunty, W. C., Kennedy, M. W., Jailwala, P., Si, H., et al. (2014). Mesogenin 1 is a master regulator of paraxial presomitic mesoderm differentiation. Development 141, 4285–4297. doi: 10.1242/dev.110908
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Chan, J., Desai, A. K., Kazi, Z. B., Corey, K., Austin, S., Hobson-Webb, L. D., et al. (2017). The emerging phenotype of late-onset Pompe disease: a systematic literature review. Mol. Genet. Metab. 120, 163–172. doi: 10.1016/j.ymgme.2016.12.004
Chang, N. C., Sincennes, M. C., Chevalier, F. P., Brun, C. E., Lacaria, M., Segales, J., et al. (2018). The dystrophin glycoprotein complex regulates the epigenetic activation of muscle stem cell commitment. Cell Stem Cell 22, 755–768 e756. doi: 10.1016/j.stem.2018.03.022
Chargé, S. B., and Rudnicki, M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238. doi: 10.1152/physrev.00019.2003
Chemello, F., Chai, A. C., Li, H., Rodriguez-Caycedo, C., Sanchez-Ortiz, E., Atmanli, A., et al. (2021). Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 7:eabg4910. doi: 10.1126/sciadv.abg4910
Chen, D., Wang, Y., and Chin, E. R. (2015). Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A∗ SOD1 amyotrophic lateral sclerosis mice. Front. Cell. Neurosci. 9:170. doi: 10.3389/fncel.2015.00170
Cheng, Y.-S., Yang, S., Hong, J., Li, R., Beers, J., Zou, J., et al. (2021). Modeling CNS involvement in pompe disease using neural stem cells generated from patient-derived induced pluripotent stem cells. Cells 10:8. doi: 10.3390/cells10010008
Chien, Y.-H., and Hwu, W.-L. (2007). A review of treatment of Pompe disease in infants. Biol. Targets Ther. 1, 195.
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278. doi: 10.1038/nature13800
Cinzia, B., Flavia, B., Gary, I., Renato, M., and Lorenzo, M. (2021). Therapeutic efficacy of 3, 4-Diaminopyridine phosphate on neuromuscular junction in Pompe disease. Biomed. Pharmacother. 137:111357. doi: 10.1016/j.biopha.2021.111357
Ciruna, B., and Rossant, J. (2001). FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev. Cell 1, 37–49. doi: 10.1016/S1534-5807(01)00017-X
Colella, P., Sellier, P., Gomez, M. J., Biferi, M. G., Tanniou, G., Guerchet, N., et al. (2020). Gene therapy with secreted acid alpha-glucosidase rescues Pompe disease in a novel mouse model with early-onset spinal cord and respiratory defects. EBioMedicine 61:103052. doi: 10.1016/j.ebiom.2020.103052
Court, F. A., Gillingwater, T. H., Melrose, S., Sherman, D. L., Greenshields, K. N., Morton, A. J., et al. (2008). Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J. Cell Sci. 121, 3901–3911. doi: 10.1242/jcs.031047
Crawford, T. Q., and Roelink, H. (2007). The notch response inhibitor DAPT enhances neuronal differentiation in embryonic stem cell-derived embryoid bodies independently of sonic hedgehog signaling. Dev. Dyn. 236, 886–892. doi: 10.1002/dvdy.21083
Crisafulli, S., Sultana, J., Fontana, A., Salvo, F., Messina, S., and Trifirò, G. (2020). Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J. Rare Dis. 15, 1–20. doi: 10.1186/s13023-020-01430-8
Dangain, J., and Vrbova, G. (1984). Muscle development in mdx mutant mice. Muscle Nerve 7, 700–704. doi: 10.1002/mus.880070903
Darabi, R., Arpke, R. W., Irion, S., Dimos, J. T., Grskovic, M., Kyba, M., et al. (2012). Human ES-and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10, 610–619. doi: 10.1016/j.stem.2012.02.015
Darnell, M., and Mooney, D. J. (2017). Leveraging advances in biology to design biomaterials. Nat. Mater. 16, 1178–1185. doi: 10.1038/nmat4991
Das, M., Rumsey, J. W., Bhargava, N., Stancescu, M., and Hickman, J. J. (2010). A defined long-term in vitro tissue engineered model of neuromuscular junctions. Biomaterials 31, 4880–4888. doi: 10.1016/j.biomaterials.2010.02.055
Das, S., Browne, K. D., Laimo, F. A., Maggiore, J. C., Hilman, M. C., Kaisaier, H., et al. (2020). Pre-innervated tissue-engineered muscle promotes a pro-regenerative microenvironment following volumetric muscle loss. Commun. Biol. 3:330. doi: 10.1038/s42003-020-1056-4
Dasen, J. S., and Jessell, T. M. (2009). Hox networks and the origins of motor neuron diversity. Curr. Top. Dev. Biol. 88, 169–200. doi: 10.1016/S0070-2153(09)88006-X
Dawson, T. M., Golde, T. E., and Lagier-Tourenne, C. (2018). Animal models of neurodegenerative diseases. Nat. Neurosci. 21, 1370–1379. doi: 10.1038/s41593-018-0236-8
De Filippi, P., Saeidi, K., Ravaglia, S., Dardis, A., Angelini, C., Mongini, T., et al. (2014). Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet J. Rare Dis. 9:102. doi: 10.1186/s13023-014-0102-z
De Oliveira, G. P., Maximino, J. R., Maschietto, M., Zanoteli, E., Puga, R. D., Lima, L., et al. (2014). Early gene expression changes in skeletal muscle from SOD1 g93a amyotrophic lateral sclerosis animal model. Cell. Mol. Neurobiol. 34, 451–462. doi: 10.1007/s10571-014-0029-x
De Vries, J. M., Kuperus, E., Hoogeveen-Westerveld, M., Kroos, M. A., Wens, S. C., Stok, M., et al. (2017). Pompe disease in adulthood: effects of antibody formation on enzyme replacement therapy. Genet. Med. 19, 90–97. doi: 10.1038/gim.2016.70
Deconinck, A. E., Rafael, J. A., Skinner, J. A., Brown, S. C., Potter, A. C., Metzinger, L., et al. (1997). Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90, 717–727. doi: 10.1016/S0092-8674(00)80532-2
Deenen, J. C., Horlings, C. G., Verschuuren, J. J., Verbeek, A. L., and Van Engelen, B. G. (2015). The Epidemiology of neuromuscular disorders: a comprehensive overview of the literature. J. Neuromuscul. Dis. 2, 73–85. doi: 10.3233/JND-140045
Demestre, M., Orth, M., Föhr, K., Achberger, K., Ludolph, A. C., Liebau, S., et al. (2015). Formation and characterisation of neuromuscular junctions between hiPSC derived motoneurons and myotubes. Stem Cell Res. 15, 328–336. doi: 10.1016/j.scr.2015.07.005
Denetclaw, W., Christ, B., and Ordahl, C. P. (1997). Location and growth of epaxial myotome precursor cells. Development 124, 1601–1610. doi: 10.1242/dev.124.8.1601
DeRuisseau, L. R., Fuller, D. D., Qiu, K., Deruisseau, K. C., Donnelly, W. H., Mah, C., et al. (2009). Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. U.S.A. 106, 9419–9424. doi: 10.1073/pnas.0902534106
Diez del Corral, R., and Storey, K. G. (2004). Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays 26, 857–869. doi: 10.1002/bies.20080
Dobrowolny, G., Aucello, M., Rizzuto, E., Beccafico, S., Mammucari, C., Bonconpagni, S., et al. (2008). Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436. doi: 10.1016/j.cmet.2008.09.002
Doddrell, R. D., Dun, X. P., Moate, R. M., Jessen, K. R., Mirsky, R., and Parkinson, D. B. (2012). Regulation of Schwann cell differentiation and proliferation by the Pax-3 transcription factor. Glia 60, 1269–1278. doi: 10.1002/glia.22346
Doyle, B. M., Turner, S. M., Sunshine, M. D., Doerfler, P. A., Poirier, A. E., Vaught, L. A., et al. (2019). Aav gene therapy utilizing glycosylation-independent lysosomal targeting tagged gaa in the hypoglossal motor system of pompe mice. Mol. Ther. Methods Clin. Dev. 15, 194–203. doi: 10.1016/j.omtm.2019.08.009
Du, J., Campau, E., Soragni, E., Jespersen, C., and Gottesfeld, J. M. (2013). Length-dependent CTG CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 22, 5276–5287. doi: 10.1093/hmg/ddt386
Du, Z. W., Chen, H., Liu, H., Lu, J., Qian, K., Huang, C. L., et al. (2015). Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 6:6626. doi: 10.1038/ncomms7626
Dubrulle, J., Mcgrew, M. J., and Pourquié, O. (2001). FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell 106, 219–232. doi: 10.1016/S0092-8674(01)00437-8
Dumont, N. A., Wang, Y. X., and Rudnicki, M. A. (2015a). Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 142, 1572–1581. doi: 10.1242/dev.114223
Dumont, N. A., Wang, Y. X., Von Maltzahn, J., Pasut, A., Bentzinger, C. F., Brun, C. E., et al. (2015b). Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 21, 1455–1463. doi: 10.1038/nm.3990
Ebrahimi, M., Lad, H., Fusto, A., Tiper, Y., Datye, A., Nguyen, C. T., et al. (2021). De novo revertant fiber formation and therapy testing in a 3D culture model of Duchenne muscular dystrophy skeletal muscle(). Acta Biomater. 132, 227–244. doi: 10.1016/j.actbio.2021.05.020
Ebrahimi, M., Ostrovidov, S., Salehi, S., Kim, S. B., Bae, H., and Khademhosseini, A. (2018). Enhanced skeletal muscle formation on microfluidic spun gelatin methacryloyl (GelMA) fibres using surface patterning and agrin treatment. J. Tissue Eng. Regen. Med. 12, 2151–2163. doi: 10.1002/term.2738
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4:145ra104. doi: 10.1126/scitranslmed.3004052
Eisen, A., Kim, S., and Pant, B. (1992). Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve 15, 219–224. doi: 10.1002/mus.880150215
Enjin, A., Rabe, N., Nakanishi, S. T., Vallstedt, A., Gezelius, H., Memic, F., et al. (2010). Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as markers for fast motor neurons and partition cells. J. Comp. Neurol. 518, 2284–2304. doi: 10.1002/cne.22332
Ericson, J., Morton, S., Kawakami, A., Roelink, H., and Jessell, T. M. (1996). Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 87, 661–673. doi: 10.1016/S0092-8674(00)81386-0
Falk, D. J., Todd, A. G., Lee, S., Soustek, M. S., Elmallah, M. K., Fuller, D. D., et al. (2015). Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum. Mol. Genet. 24, 625–636. doi: 10.1093/hmg/ddu476
Fambrough, D. M. (1979). Control of acetylcholine receptors in skeletal muscle. Physiol. Rev. 59, 165–227. doi: 10.1152/physrev.1979.59.1.165
Fardaei, M., Rogers, M. T., Thorpe, H. M., Larkin, K., Hamshere, M. G., Harper, P. S., et al. (2002). Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 11, 805–814. doi: 10.1093/hmg/11.7.805
Fardeau, M., and Tome, F. (1980). “Light and electron microscopic study of motor endplates in the adult and neo-natal forms of dystrophia myotonica,” in Proceedings of the INSEREM Symposium on Ontogenesis and Functional Mechanisms of Peripheral Synapses: Ontogenesis and Functional Mechanisms of Peripheral Synapses, (Amsterdam: Elsevier), 287–298.
Farmakidis, C., Pasnoor, M., Dimachkie, M. M., and Barohn, R. J. (2018). Treatment of myasthenia gravis. Neurol. Clin. 36, 311–337. doi: 10.1016/j.ncl.2018.01.011
Farup, J., Madaro, L., Puri, P., and Mikkelsen, U. (2015). Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis. 6:e1830. doi: 10.1038/cddis.2015.198
Faustino Martins, J. M., Fischer, C., Urzi, A., Vidal, R., Kunz, S., Ruffault, P. L., et al. (2020). Self-Organizing 3D human trunk neuromuscular organoids. Cell Stem Cell 27:498. doi: 10.1016/j.stem.2020.08.011
Feeney, E. J., Austin, S., Chien, Y.-H., Mandel, H., Schoser, B., Prater, S., et al. (2014). The value of muscle biopsies in Pompe disease: identifying lipofuscin inclusions in juvenile-and adult-onset patients. Acta Neuropathol. Commun. 2:2. doi: 10.1186/2051-5960-2-2
Fernández-Garibay, X., Ortega, M. A., Cerro-Herreros, E., Comelles, J., Martínez, E., Artero, R., et al. (2021). Bioengineered in vitro 3D model of myotonic dystrophy type 1 human skeletal muscle. Biofabrication 13:035035. doi: 10.1088/1758-5090/abf6ae
Fernandez-Costa, J. M., Llamusi, M. B., Garcia-Lopez, A., and Artero, R. (2011). Alternative splicing regulation by Muscleblind proteins: from development to disease. Biol. Rev. 86, 947–958. doi: 10.1111/j.1469-185X.2011.00180.x
Fischer-Hayes, L. R., Brotherton, T., and Glass, J. D. (2013). Axonal degeneration in the peripheral nervous system: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp. Neurol. 246, 6–13. doi: 10.1016/j.expneurol.2013.05.001
Flanagan-Steet, H., Fox, M. A., Meyer, D., and Sanes, J. R. (2005). Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development 132, 4471–4481. doi: 10.1242/dev.02044
Flanigan, K. M., Ceco, E., Lamar, K. M., Kaminoh, Y., Dunn, D. M., Mendell, J. R., et al. (2013). LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann. Neurol. 73, 481–488. doi: 10.1002/ana.23819
Fougerousse, F., Edom-Vovard, F., Merkulova, T., Ott, M. O., Durand, M., Butler-Browne, G., et al. (2001). The muscle-specific enolase is an early marker of human myogenesis. J. Muscle Res. Cell Motil. 22, 535–544. doi: 10.1023/A:1015008208007
Fox, J. W., Mayer, U., Nischt, R., Aumailley, M., Reinhardt, D., Wiedemann, H., et al. (1991). Recombinant nidogen consists of three globular domains and mediates binding of laminin to collagen type IV. EMBO J. 10, 3137–3146. doi: 10.1002/j.1460-2075.1991.tb04875.x
Francius, C., and Clotman, F. (2014). Generating spinal motor neuron diversity: a long quest for neuronal identity. Cell. Mol. Life Sci. 71, 813–829. doi: 10.1007/s00018-013-1398-x
Fu, T., Jiang, L., Peng, Y., Li, Z., Liu, S., Lu, J., et al. (2020). Electrical muscle stimulation accelerates functional recovery after nerve injury. Neuroscience 426, 179–188. doi: 10.1016/j.neuroscience.2019.10.052
Fugier, C., Klein, A. F., Hammer, C., Vassilopoulos, S., Ivarsson, Y., Toussaint, A., et al. (2011). Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 17, 720–725. doi: 10.1038/nm.2374
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi: 10.1038/s41591-018-0140-5
Fukuda, T., Roberts, A., Plotz, P. H., and Raben, N. (2007). Acid alpha-glucosidase deficiency (Pompe disease). Curr. Neurol. Neurosci. Rep. 7, 71–77. doi: 10.1007/s11910-007-0024-4
Gallardo, E., Martínez-Hernández, E., Titulaer, M. J., Huijbers, M. G., Martínez, M. A., Ramos, A., et al. (2014). Cortactin autoantibodies in myasthenia gravis. Autoimmun. Rev. 13, 1003–1007. doi: 10.1016/j.autrev.2014.08.039
Gambetti, P., Dimauro, S., and Baker, L. (1971). Nervous system in Pompe’s disease: ultrastructure and biochemistry. J. Neuropathol. Exp. Neurol. 30, 412–430. doi: 10.1097/00005072-197107000-00008
Gao, Y., Guo, X., Santostefano, K., Wang, Y., Reid, T., Zeng, D., et al. (2016). Genome therapy of myotonic dystrophy type 1 iPS cells for development of autologous stem cell therapy. Mol. Ther. 24, 1378–1387. doi: 10.1038/mt.2016.97
Garriock, R. J., Chalamalasetty, R. B., Kennedy, M. W., Canizales, L. C., Lewandoski, M., and Yamaguchi, T. P. (2015). Lineage tracing of neuromesodermal progenitors reveals novel Wnt-dependent roles in trunk progenitor cell maintenance and differentiation. Development 142, 1628–1638. doi: 10.1242/dev.111922
Gaschen, L., Lang, J., Lin, S., Adé-Damilano, M., Busato, A., Lombard, C. W., et al. (1999). Cardiomyopathy in dystrophin-deficient hypertrophic feline muscular dystrophy. J. Vet. Intern. Med. 13, 346–356. doi: 10.1111/j.1939-1676.1999.tb02193.x
Gasperi, C., Melms, A., Schoser, B., Zhang, Y., Meltoranta, J., Risson, V., et al. (2014). Anti-agrin autoantibodies in myasthenia gravis. Neurology 82, 1976–1983. doi: 10.1212/WNL.0000000000000478
Gatto, F., Rossi, B., Tarallo, A., Polishchuk, E., Polishchuk, R., Carrella, A., et al. (2017). AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci. Rep. 7:15089. doi: 10.1038/s41598-017-15352-2
Gingras, M., Beaulieu, M. M., Gagnon, V., Durham, H. D., and Berthod, F. (2008). In vitro study of axonal migration and myelination of motor neurons in a three-dimensional tissue-engineered model. Glia 56, 354–364. doi: 10.1002/glia.20617
Glass, D. J., Bowen, D. C., Stitt, T. N., Radziejewski, C., Bruno, J., Ryan, T. E., et al. (1996). Agrin acts via a MuSK receptor complex. Cell 85, 513–523. doi: 10.1016/S0092-8674(00)81252-0
Gogliotti, R. G., Quinlan, K. A., Barlow, C. B., Heier, C. R., Heckman, C. J., and Didonato, C. J. (2012). Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci. 32, 3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012
Gonzalez, D., Contreras, O., Rebolledo, D. L., Espinoza, J. P., Van Zundert, B., and Brandan, E. (2017). ALS skeletal muscle shows enhanced TGF-β signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS One 12:e0177649. doi: 10.1371/journal.pone.0177649
Goudenege, S., Lebel, C., Huot, N. B., Dufour, C., Fujii, I., Gekas, J., et al. (2012). Myoblasts derived from normal hESCs and dystrophic hiPSCs efficiently fuse with existing muscle fibers following transplantation. Mol. Ther. 20, 2153–2167. doi: 10.1038/mt.2012.188
Gouti, M., Delile, J., Stamataki, D., Wymeersch, F. J., Huang, Y., Kleinjung, J., et al. (2017). A gene regulatory network balances neural and mesoderm specification during vertebrate trunk development. Dev. Cell 41, 243–261.e247. doi: 10.1016/j.devcel.2017.04.002
Grady, R. M., Starr, D. A., Ackerman, G. L., Sanes, J. R., and Han, M. (2005). Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc. Natl. Acad. Sci. U.S.A. 102, 4359–4364. doi: 10.1073/pnas.0500711102
Grasman, J. M., and Kaplan, D. L. (2017). Human endothelial cells secrete neurotropic factors to direct axonal growth of peripheral nerves. Sci. Rep. 7:4092. doi: 10.1038/s41598-017-04460-8
Gregorevic, P., Blankinship, M. J., Allen, J. M., and Chamberlain, J. S. (2008). Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. 16, 657–664. doi: 10.1038/mt.2008.28
Gu, J.-M., Wang, D. J., Peterson, J. M., Shintaku, J., Liyanarachchi, S., Coppola, V., et al. (2016). An NF-κB-EphrinA5-dependent communication between NG2+ interstitial cells and myoblasts promotes muscle growth in neonates. Dev. Cell 36, 215–224. doi: 10.1016/j.devcel.2015.12.018
Güngör, D., Kruijshaar, M. E., Plug, I., D’agostino, R. B., Hagemans, M. L., Van Doorn, P. A., et al. (2013). Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J. Rare Dis. 8:49. doi: 10.1186/1750-1172-8-49
Guo, X., Badu-Mensah, A., Thomas, M. C., Mcaleer, C. W., and Hickman, J. J. (2020a). Characterization of functional human skeletal myotubes and neuromuscular junction derived—from the same induced pluripotent stem cell source. Bioengineering 7:133. doi: 10.3390/bioengineering7040133
Guo, X., Gonzalez, M., Stancescu, M., Vandenburgh, H. H., and Hickman, J. J. (2011). Neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle in a defined system. Biomaterials 32, 9602–9611. doi: 10.1016/j.biomaterials.2011.09.014
Guo, X., Smith, V., Jackson, M., Tran, M., Thomas, M., Patel, A., et al. (2020b). A human-based functional NMJ system for personalized ALS modeling and drug testing. Adv. Therap. 3:2000133. doi: 10.1002/adtp.202000133
Ha, J. C., and Richman, D. P. (2015). Myasthenia gravis and related disorders: pathology and molecular pathogenesis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1852, 651–657. doi: 10.1016/j.bbadis.2014.11.022
Haastert, K., Grosskreutz, J., Jaeckel, M., Laderer, C., Bufler, J., Grothe, C., et al. (2005). Rat embryonic motoneurons in long-term co-culture with Schwann cells–a system to investigate motoneuron diseases on a cellular level in vitro. J. Neurosci. Methods 142, 275–284. doi: 10.1016/j.jneumeth.2004.09.003
Halpern, M., Brennand, K. J., and Gregory, J. (2019). Examining the relationship between astrocyte dysfunction and neurodegeneration in ALS using hiPSCs. Neurobiol. Dis. 132:104562. doi: 10.1016/j.nbd.2019.104562
Han, S.-O., Li, S., and Koeberl, D. D. (2016). Salmeterol enhances the cardiac response to gene therapy in Pompe disease. Mol. Genet. Metab. 118, 35–40. doi: 10.1016/j.ymgme.2016.03.006
Happe, C. L., Tenerelli, K. P., Gromova, A. K., Kolb, F., and Engler, A. J. (2017). Mechanically patterned neuromuscular junctions-in-a-dish have improved functional maturation. Mol. Biol. Cell 28, 1950–1958. doi: 10.1091/mbc.e17-01-0046
Hawes, M. L., Kennedy, W., O’callaghan, M. W., and Thurberg, B. L. (2007). Differential muscular glycogen clearance after enzyme replacement therapy in a mouse model of Pompe disease. Mol. Genet. Metab. 91, 343–351. doi: 10.1016/j.ymgme.2007.04.018
Hegedus, J., Putman, C., and Gordon, T. (2007). Time course of preferential motor unit loss in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 28, 154–164. doi: 10.1016/j.nbd.2007.07.003
Hester, M. E., Murtha, M. J., Song, S., Rao, M., Miranda, C. J., Meyer, K., et al. (2011). Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. 19, 1905–1912. doi: 10.1038/mt.2011.135
Heydemann, A., Ceco, E., Lim, J. E., Hadhazy, M., Ryder, P., Moran, J. L., et al. (2009). Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Invest. 119, 3703–3712. doi: 10.1172/JCI39845
Hicks, M. R., Hiserodt, J., Paras, K., Fujiwara, W., Eskin, A., Jan, M., et al. (2018). ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat. Cell Biol. 20, 46–57. doi: 10.1038/s41556-017-0010-2
Hinds, S., Bian, W., Dennis, R. G., and Bursac, N. (2011). The role of extracellular matrix composition in structure and function of bioengineered skeletal muscle. Biomaterials 32, 3575–3583. doi: 10.1016/j.biomaterials.2011.01.062
Hirata, K., Zhou, C., Nakamura, K., and Kawabuchi, M. (1997). Postnatal development of Schwann cells at neuromuscular junctions, with special reference to synapse elimination. J. Neurocytol. 26, 799–809. doi: 10.1023/A:1018570500052
Hobson-Webb, L. D., Dearmey, S., and Kishnani, P. S. (2011). The clinical and electrodiagnostic characteristics of Pompe disease with post-enzyme replacement therapy findings. Clin. Neurophysiol. 122, 2312–2317. doi: 10.1016/j.clinph.2011.04.016
Hogarth, M. W., Defour, A., Lazarski, C., Gallardo, E., Manera, J. D., Partridge, T. A., et al. (2019). Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 10:2430. doi: 10.1038/s41467-019-10438-z
Honkanen, H., Lahti, O., Nissinen, M., Myllyla, R. M., Kangas, S., Paivalainen, S., et al. (2007). Isolation, purification and expansion of myelination-competent, neonatal mouse Schwann cells. Eur. J. Neurosci. 26, 953–964. doi: 10.1111/j.1460-9568.2007.05726.x
Hsieh-Li, H. M., Chang, J. G., Jong, Y. J., Wu, M. H., Wang, N. M., Tsai, C. H., et al. (2000). A mouse model for spinal muscular atrophy. Nat. Genet. 24, 66–70. doi: 10.1038/71709
Hu, B. Y., and Zhang, S. C. (2009). Differentiation of spinal motor neurons from pluripotent human stem cells. Nat. Protoc. 4, 1295–1304. doi: 10.1038/nprot.2009.127
Huang, C.-W., Huang, W.-C., Qiu, X., Da Silva, F. F. F., Wang, A., Patel, S., et al. (2017). The differentiation stage of transplanted stem cells modulates nerve regeneration. Sci. Rep. 7:17401. doi: 10.1038/s41598-017-17043-4
Huang, H.-P., Chiang, W., Stone, L., Kang, C.-K., Chuang, C.-Y., and Kuo, H.-C. (2019). Using human Pompe disease-induced pluripotent stem cell-derived neural cells to identify compounds with therapeutic potential. Hum. Mol. Genet. 28, 3880–3894. doi: 10.1093/hmg/ddz218
Hudecki, M. S., Pollina, C. M., Granchelli, J. A., Daly, M. K., Byrnes, T., Wang, J. C., et al. (1993). Strength and endurance in the therapeutic evaluation of prednisolone-treated MDX mice. Res. Commun. Chem. Pathol. Pharmacol. 79, 45–60.
Huguet, A., Medja, F., Nicole, A., Vignaud, A., Guiraud-Dogan, C., Ferry, A., et al. (2012). Molecular, physiological, and motor performance defects in DMSXL mice carrying> 1,000 CTG repeats from the human DM1 locus. PLoS Genet. 8:e1003043. doi: 10.1371/journal.pgen.1003043
Hyung, S., Lee, S. R., Kim, J., Kim, Y., Kim, S., Kim, H. N., et al. (2021). A 3D disease and regeneration model of peripheral nervous system-on-a-chip. Sci. Adv. 7:eabd9749. doi: 10.1126/sciadv.abd9749
Hyung, S., Lee, S. R., Kim, Y. J., Bang, S., Tahk, D., Park, J. C., et al. (2019). Optogenetic neuronal stimulation promotes axon outgrowth and myelination of motor neurons in a three-dimensional motor neuron-Schwann cell coculture model on a microfluidic biochip. Biotechnol. Bioeng 116, 2425–2438. doi: 10.1002/bit.27083
Hyung, S., Yoon Lee, B., Park, J. C., Kim, J., Hur, E. M., and Francis Suh, J. K. (2015). Coculture of primary motor neurons and schwann cells as a model for in vitro myelination. Sci. Rep. 5:15122.
Ihmsen, H., Schmidt, J., Schwilden, H., Schmitt, H. J., and Muenster, T. (2009). Influence of disease progression on the neuromuscular blocking effect of mivacurium in children and adolescents with Duchenne muscular dystrophy. Anesthesiology 110, 1016–1019. doi: 10.1097/ALN.0b013e31819daf31
Imbert, N., Cognard, C., Duport, G., Guillou, C., and Raymond, G. (1995). Abnormal calcium homeostasis in Duchenne muscular dystrophy myotubes contracting in vitro. Cell Calcium 18, 177–186. doi: 10.1016/0143-4160(95)90062-4
Ionescu, A., Zahavi, E. E., Gradus, T., Ben-Yaakov, K., and Perlson, E. (2016). Compartmental microfluidic system for studying muscle–neuron communication and neuromuscular junction maintenance. Eur. J. Cell Biol. 95, 69–88. doi: 10.1016/j.ejcb.2015.11.004
Irioka, T., Watanabe, K., Mizusawa, H., Mizuseki, K., and Sasai, Y. (2005). Distinct effects of caudalizing factors on regional specification of embryonic stem cell-derived neural precursors. Dev. Brain Res. 154, 63–70. doi: 10.1016/j.devbrainres.2004.10.004
Jacob, C., Christen, C. N., Pereira, J. A., Somandin, C., Baggiolini, A., Lotscher, P., et al. (2011). HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat. Neurosci. 14, 429–436. doi: 10.1038/nn.2762
Jacobson, C., Cote, P. D., Rossi, S. G., Rotundo, R. L., and Carbonetto, S. (2001). The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J. Cell Biol. 152, 435–450. doi: 10.1083/jcb.152.3.435
Jaiswal, M. K. (2019). Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 39, 733–748. doi: 10.1002/med.21528
Jansen, G., Groenen, P. J., Bächner, D., Jap, P. H., Coerwinkel, M., Oerlemans, F., et al. (1996). Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 13, 316–324. doi: 10.1038/ng0796-316
Jayam Trouth, A., Dabi, A., Solieman, N., Kurukumbi, M., and Kalyanam, J. (2012). Myasthenia gravis: a review. Autoimmune Dis. 2012:874680. doi: 10.1155/2012/874680
Jessell, T. M. (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20–29. doi: 10.1038/35049541
Jessen, K. R., and Mirsky, R. (1999). Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci. 22, 402–410. doi: 10.1016/S0166-2236(98)01391-5
Jessen, K. R., and Mirsky, R. (2005). The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6, 671–682. doi: 10.1038/nrn1746
Jiwlawat, N., Lynch, E., Jeffrey, J., Van Dyke, J. M., and Suzuki, M. (2018). Current progress and challenges for skeletal muscle differentiation from human pluripotent stem cells using transgene-free approaches. Stem Cells Int. 2018:6241681. doi: 10.1155/2018/6241681
Jiwlawat, N., Lynch, E. M., Napiwocki, B. N., Stempien, A., Ashton, R. S., Kamp, T. J., et al. (2019). Micropatterned substrates with physiological stiffness promote cell maturation and Pompe disease phenotype in human induced pluripotent stem cell-derived skeletal myocytes. Biotechnol. Bioeng. 116, 2377–2392. doi: 10.1002/bit.27075
Johnson, M. A., Weick, J. P., Pearce, R. A., and Zhang, S. C. (2007). Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J. Neurosci. 27, 3069–3077. doi: 10.1523/JNEUROSCI.4562-06.2007
Jones, R. A., Harrison, C., Eaton, S. L., Hurtado, M. L., Graham, L. C., Alkhammash, L., et al. (2017). Cellular and molecular anatomy of the human neuromuscular junction. Cell Rep. 21, 2348–2356. doi: 10.1016/j.celrep.2017.11.008
Joyce, P. I., Fratta, P., Fisher, E. M., and Acevedo-Arozena, A. (2011). SOD1 and TDP-43 animal models of amyotrophic lateral sclerosis: recent advances in understanding disease toward the development of clinical treatments. Mamm. Genome 22, 420–448. doi: 10.1007/s00335-011-9339-1
Juban, G., Saclier, M., Yacoub-Youssef, H., Kernou, A., Arnold, L., Boisson, C., et al. (2018). AMPK activation regulates LTBP4-dependent TGF-β1 secretion by pro-inflammatory macrophages and controls fibrosis in Duchenne muscular dystrophy. Cell Rep. 25, 2163–2176.e2166. doi: 10.1016/j.celrep.2018.10.077
Juhas, M., Abutaleb, N., Wang, J. T., Ye, J., Shaikh, Z., Sriworarat, C., et al. (2018). Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat. Biomed. Eng. 2, 942–954. doi: 10.1038/s41551-018-0290-2
Juhas, M., and Bursac, N. (2014). Roles of adherent myogenic cells and dynamic culture in engineered muscle function and maintenance of satellite cells. Biomaterials 35, 9438–9446. doi: 10.1016/j.biomaterials.2014.07.035
Kang, H.-W., Lee, S. J., Ko, I. K., Kengla, C., Yoo, J. J., and Atala, A. (2016). A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat. Biotechnol. 34, 312–319. doi: 10.1038/nbt.3413
Kengaku, M., Kawata, A., Kawashima, S., and Nakane, M. (1991). Role of fibronectin in the inhibitory effect of TGF-beta on choline acetyltransferase activity in co-cultures of spinal cord neurons and myotubes. Brain Res. Dev. Brain Res. 61, 281–284. doi: 10.1016/0165-3806(91)90144-8
Khan, N., Eliopoulos, H., Han, L., Kinane, T. B., Lowes, L. P., Mendell, J. R., et al. (2019). Eteplirsen treatment attenuates respiratory decline in ambulatory and non-ambulatory patients with duchenne muscular dystrophy. J. Neuromuscul. Dis. 6, 213–225. doi: 10.3233/JND-180351
Khodabukus, A. (2021). Tissue-Engineered skeletal muscle models to study muscle function, plasticity, and disease. Front. Physiol. 12:619710. doi: 10.3389/fphys.2021.619710
Khodabukus, A., Madden, L., Prabhu, N. K., Koves, T. R., Jackman, C. P., Muoio, D. M., et al. (2019). Electrical stimulation increases hypertrophy and metabolic flux in tissue-engineered human skeletal muscle. Biomaterials 198, 259–269. doi: 10.1016/j.biomaterials.2018.08.058
Kim, H.-S., Lee, J., Lee, D. Y., Kim, Y.-D., Kim, J. Y., Lim, H. J., et al. (2017). Schwann cell precursors from human pluripotent stem cells as a potential therapeutic target for myelin repair. Stem Cell Rep. 8, 1714–1726. doi: 10.1016/j.stemcr.2017.04.011
Kim, N., and Burden, S. J. (2008). MuSK controls where motor axons grow and form synapses. Nat. Neurosci. 11, 19–27. doi: 10.1038/nn2026
Kimura, S., and Nezu, A. (1989). Peripheral nerve involvement in myasthenia gravis. Brain Dev. 11, 429–432. doi: 10.1016/S0387-7604(89)80030-0
Kinoshita, I., Nakamura, T., Satoh, A., Matsuo, H., Seto, M., Tomita, I., et al. (1988). Role of the macrophage in the pathogenesis of experimental autoimmune myasthenia gravis. J. Neurol. Sci. 87, 49–59. doi: 10.1016/0022-510X(88)90053-6
Kioussi, C., Gross, M. K., and Gruss, P. (1995). Pax3: a paired domain gene as a regulator in PNS myelination. Neuron 15, 553–562. doi: 10.1016/0896-6273(95)90144-2
Kishnani, P. S., Corzo, D., Leslie, N. D., Gruskin, D., Van Der Ploeg, A., Clancy, J. P., et al. (2009). Early treatment with alglucosidase alfa prolongs long-term survival of infants with Pompe disease. Pediatr. Res. 66, 329–335. doi: 10.1203/PDR.0b013e3181b24e94
Kishnani, P. S., Goldenberg, P. C., Dearmey, S. L., Heller, J., Benjamin, D., Young, S., et al. (2010). Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 99, 26–33. doi: 10.1016/j.ymgme.2009.08.003
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. doi: 10.1016/j.stem.2014.03.004
Ko, C. P., and Robitaille, R. (2015). Perisynaptic schwann cells at the neuromuscular synapse: adaptable, multitasking glial cells. Cold Spring Harb. Perspect. Biol. 7:a020503. doi: 10.1101/cshperspect.a020503
Kodaka, Y., Rabu, G., and Asakura, A. (2017). Skeletal muscle cell induction from pluripotent stem cells. Stem Cells Int. 2017:1376151. doi: 10.1155/2017/1376151
Kong, J., and Anderson, J. E. (1999). Dystrophin is required for organizing large acetylcholine receptor aggregates. Brain Res. 839, 298–304. doi: 10.1016/S0006-8993(99)01737-0
Konieczny, P., Selma-Soriano, E., Rapisarda, A. S., Fernandez-Costa, J. M., Perez-Alonso, M., and Artero, R. (2017). Myotonic dystrophy: candidate small molecule therapeutics. Drug Discov. Today 22, 1740–1748. doi: 10.1016/j.drudis.2017.07.011
Kowalski, P. S., Bhattacharya, C., Afewerki, S., and Langer, R. (2018). Smart biomaterials: recent advances and future directions. ACS Biomater. Sci. Eng. 4, 3809–3817. doi: 10.1021/acsbiomaterials.8b00889
Kraft, A. D., Resch, J. M., Johnson, D. A., and Johnson, J. A. (2007). Activation of the Nrf2–ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 207, 107–117. doi: 10.1016/j.expneurol.2007.05.026
Kuhlbrodt, K., Herbarth, B., Sock, E., Hermans-Borgmeyer, I., and Wegner, M. (1998). Sox10, a novel transcriptional modulator in glial cells. J. Neurosci. 18, 237–250. doi: 10.1523/JNEUROSCI.18-01-00237.1998
Kume, T., Jiang, H., Topczewska, J. M., and Hogan, B. L. (2001). The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 15, 2470–2482. doi: 10.1101/gad.907301
Kuyumcu-Martinez, N. M., and Cooper, T. A. (2006). Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Altern. Splicing Dis. 44, 133–159. doi: 10.1007/978-3-540-34449-0_7
Kwon, J. B., Vankara, A., Ettyreddy, A. R., Bohning, J. D., and Gersbach, C. A. (2020). Myogenic progenitor cell lineage specification by CRISPR/Cas9-based transcriptional activators. Stem Cell Rep. 14, 755–769. doi: 10.1016/j.stemcr.2020.03.026
Kyriakides, T., Pegoraro, E., Hoffman, E. P., Piva, L., Cagnin, S., Lanfranchi, G., et al. (2011). SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy: predicting the severity of Duchenne muscular dystrophy: implications for treatment. Neurology 77:1858. author reply 1858-1859 doi: 10.1212/WNL.0b013e318239b9ae
Kyrychenko, V., Kyrychenko, S., Tiburcy, M., Shelton, J. M., Long, C., Schneider, J. W., et al. (2017). Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2:e95918. doi: 10.1172/jci.insight.95918
Landmesser, L. T. (2001). The acquisition of motoneuron subtype identity and motor circuit formation. Int. J. Dev. Neurosci. 19, 175–182. doi: 10.1016/S0736-5748(00)00090-3
Larcher, T., Lafoux, A., Tesson, L., Remy, S., Thepenier, V., Francois, V., et al. (2014). Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One 9:e110371. doi: 10.1371/journal.pone.0110371
Larkin, L. M., Van Der Meulen, J. H., Dennis, R. G., and Kennedy, J. B. (2006). Functional evaluation of nerve-skeletal muscle constructs engineered in vitro. In Vitro Cell. Dev. Biol. Anim. 42, 75–82. doi: 10.1290/0509064.1
Laustriat, D., Gide, J., Barrault, L., Chautard, E., Benoit, C., Auboeuf, D., et al. (2015). In vitro and in vivo modulation of alternative splicing by the biguanide metformin. Mol. Ther. Nucleic Acids 4:e262. doi: 10.1038/mtna.2015.35
LaVaute, T. M., Yoo, Y. D., Pankratz, M. T., Weick, J. P., Gerstner, J. R., and Zhang, S. C. (2009). Regulation of neural specification from human embryonic stem cells by BMP and FGF. Stem Cells 27, 1741–1749. doi: 10.1002/stem.99
Le, S., Yu, M., Hovan, L., Zhao, Z., Ervasti, J., and Yan, J. (2018). Dystrophin as a molecular shock absorber. ACS Nano 12, 12140–12148. doi: 10.1021/acsnano.8b05721
Lee, J. E., and Cooper, T. A. (2009). Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 37, 1281–1286. doi: 10.1042/BST0371281
Lee, K.-Z., Qiu, K., Sandhu, M. S., Elmallah, M. K., Falk, D. J., Lane, M. A., et al. (2011). Hypoglossal neuropathology and respiratory activity in pompe mice. Front. Physiol. 2:31. doi: 10.3389/fphys.2011.00031
Lee, N.-C., Hwu, W.-L., Muramatsu, S.-I., Falk, D. J., Byrne, B. J., Cheng, C.-H., et al. (2018). A neuron-specific gene therapy relieves motor deficits in Pompe disease mice. Mol. Neurobiol. 55, 5299–5309. doi: 10.1007/s12035-017-0763-4
Lee, S., Lee, B., Lee, J. W., and Lee, S. K. (2009). Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron 62, 641–654. doi: 10.1016/j.neuron.2009.04.025
Lenzi, J., Pagani, F., De Santis, R., Limatola, C., Bozzoni, I., Di Angelantonio, S., et al. (2016). Differentiation of control and ALS mutant human iPSCs into functional skeletal muscle cells, a tool for the study of neuromuscolar diseases. Stem Cell Res. 17, 140–147. doi: 10.1016/j.scr.2016.06.003
Lepper, C., and Fan, C. M. (2010). Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis 48, 424–436. doi: 10.1002/dvg.20630
Li, L., Xiong, W. C., and Mei, L. (2018). Neuromuscular junction formation, aging, and disorders. Annu. Rev. Physiol. 80, 159–188. doi: 10.1146/annurev-physiol-022516-034255
Li, W., Brakefield, D., Pan, Y., Hunter, D., Myckatyn, T. M., and Parsadanian, A. (2007). Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp. Neurol. 203, 457–471. doi: 10.1016/j.expneurol.2006.08.028
Li, X. J., Du, Z. W., Zarnowska, E. D., Pankratz, M., Hansen, L. O., Pearce, R. A., et al. (2005). Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 23, 215–221. doi: 10.1038/nbt1063
Liewluck, T., and Saperstein, D. S. (2015). Progressive muscular atrophy. Neurol. Clin. 33, 761–773. doi: 10.1016/j.ncl.2015.07.005
Lim, J.-A., Li, L., and Raben, N. (2014). Pompe disease: from pathophysiology to therapy and back again. Front. Aging Neurosci. 6:177. doi: 10.3389/fnagi.2014.00177
Lin, B., Li, Y., Han, L., Kaplan, A. D., Ao, Y., Kalra, S., et al. (2015). Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model. Mech. 8, 457–466. doi: 10.1242/dmm.019505
Lin, C. Y., Yoshida, M., Li, L. T., Ikenaka, A., Oshima, S., Nakagawa, K., et al. (2019). iPSC-derived functional human neuromuscular junctions model the pathophysiology of neuromuscular diseases. JCI Insight 4:e124299. doi: 10.1172/jci.insight.124299
Lin, W., Burgess, R. W., Dominguez, B., Pfaff, S. L., Sanes, J. R., and Lee, K. F. (2001). Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410, 1057–1064. doi: 10.1038/35074025
Lin, W., Sanchez, H. B., Deerinck, T., Morris, J. K., Ellisman, M., and Lee, K.-F. (2000). Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 97, 1299–1304. doi: 10.1073/pnas.97.3.1299
Lino, M. M., Schneider, C., and Caroni, P. (2002). Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22, 4825–4832. doi: 10.1523/JNEUROSCI.22-12-04825.2002
Liu, J. P., Laufer, E., and Jessell, T. M. (2001). Assigning the positional identity of spinal motor neurons: rostrocaudal patterning of Hox-c expression by FGFs, Gdf11, and retinoids. Neuron 32, 997–1012. doi: 10.1016/S0896-6273(01)00544-X
Liu, Q., Spusta, S. C., Mi, R., Lassiter, R. N., Stark, M. R., Höke, A., et al. (2012). Human neural crest stem cells derived from human ESCs and induced pluripotent stem cells: induction, maintenance, and differentiation into functional schwann cells. Stem Cells Transl. Med. 1, 266–278. doi: 10.5966/sctm.2011-0042
Liu, Y., Wong, T. P., Aarts, M., Rooyakkers, A., Liu, L., Lai, T. W., et al. (2007). NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 27, 2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007
Loeffler, J. P., Picchiarelli, G., Dupuis, L., and Gonzalez De Aguilar, J. L. (2016). The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol. 26, 227–236. doi: 10.1111/bpa.12350
Long, C., Li, H., Tiburcy, M., Rodriguez-Caycedo, C., Kyrychenko, V., Zhou, H., et al. (2018). Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 4:eaa9004. doi: 10.1126/sciadv.aap9004
Luo, B., Tian, L., Chen, N., Ramakrishna, S., Thakor, N., and Yang, I. H. (2018). Electrospun nanofibers facilitate better alignment, differentiation, and long-term culture in an in vitro model of the neuromuscular junction (NMJ). Biomater. Sci. 6, 3262–3272. doi: 10.1039/C8BM00720A
Lynch, E., Semrad, T., Belsito, V. S., Fitzgibbons, C., Reilly, M., Hayakawa, K., et al. (2019). C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis. Model. Mech. 12:dmm039552. doi: 10.1242/dmm.039552
Madaro, L., Passafaro, M., Sala, D., Etxaniz, U., Lugarini, F., Proietti, D., et al. (2018). Denervation-activated STAT3–IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 20, 917–927. doi: 10.1038/s41556-018-0151-y
Madill, M., Mcdonagh, K., Ma, J., Vajda, A., Mcloughlin, P., O’brien, T., et al. (2017). Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 10:22. doi: 10.1186/s13041-017-0300-4
Maffioletti, S. M., Gerli, M. F., Ragazzi, M., Dastidar, S., Benedetti, S., Loperfido, M., et al. (2015). Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat. Protoc. 10, 941–958. doi: 10.1038/nprot.2015.057
Maffioletti, S. M., Sarcar, S., Henderson, A. B. H., Mannhardt, I., Pinton, L., Moyle, L. A., et al. (2018). Three-Dimensional human iPSC-Derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 23, 899–908. doi: 10.1016/j.celrep.2018.03.091
Mancall, E. L., Aponte, G. E., and Berry, R. G. (1965). Pompe’s disease (diffuse glycogenosis) with neuronal storage. J. Neuropathol. Exp. Neurol. 24, 85–96. doi: 10.1097/00005072-196501000-00008
Mankoo, B. S., Skuntz, S., Harrigan, I., Grigorieva, E., Candia, A., Wright, C. V., et al. (2003). The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development 130, 4655–4664. doi: 10.1242/dev.00687
Manning, J., and O’Malley, D. (2015). What has the mdx mouse model of Duchenne muscular dystrophy contributed to our understanding of this disease? J. Muscle Res. Cell Motil. 36, 155–167. doi: 10.1007/s10974-015-9406-4
Mantegazza, R., Cordiglieri, C., Consonni, A., and Baggi, F. (2016). Animal models of myasthenia gravis: utility and limitations. Int, J. Gen. Med. 9:53. doi: 10.2147/IJGM.S88552
Manzano, R., Toivonen, J. M., Calvo, A. C., Oliván, S., Zaragoza, P., Rodellar, C., et al. (2013). Altered in vitro proliferation of mouse SOD1-G93A skeletal muscle satellite cells. Neurodegener. Dis. 11, 153–164. doi: 10.1159/000338061
Manzano, R., Toivonen, J. M., Oliván, S., Calvo, A. C., Moreno-Igoa, M., Muñoz, M. J., et al. (2011). Altered expression of myogenic regulatory factors in the mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 8, 386–396. doi: 10.1159/000324159
Marcuzzo, S., Zucca, I., Mastropietro, A., De Rosbo, N. K., Cavalcante, P., Tartari, S., et al. (2011). Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: a longitudinal MRI study. Exp. Neurol. 231, 30–37. doi: 10.1016/j.expneurol.2011.05.007
Mareedu, S., Pachon, R., Thilagavathi, J., Fefelova, N., Balakrishnan, R., Niranjan, N., et al. (2021). Sarcolipin haploinsufficiency prevents dystrophic cardiomyopathy in mdx mice. Am. J. Physiol. Heart Circ. Physiol. 320, H200–H210. doi: 10.1152/ajpheart.00601.2020
Marteyn, A., Maury, Y., Gauthier, M. M., Lecuyer, C., Vernet, R., Denis, J. A., et al. (2011). Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell 8, 434–444. doi: 10.1016/j.stem.2011.02.004
Martin, J., De Barsy, T., Van Hoof, F., and Palladini, G. (1973). Pompe’s disease: an inborn lysosomal disorder with storage of glycogen. Acta Neuropathol. 23, 229–244. doi: 10.1007/BF00687878
Martin, N. R., Passey, S. L., Player, D. J., Mudera, V., Baar, K., Greensmith, L., et al. (2015). Neuromuscular junction formation in tissue-engineered skeletal muscle augments contractile function and improves cytoskeletal organization. Tissue Eng. Part A 21, 2595–2604. doi: 10.1089/ten.tea.2015.0146
Martineau, É, Arbour, D., Vallée, J., and Robitaille, R. (2020). Properties of Glial Cell at the Neuromuscular Junction are Incompatible with synaptic repair in the SOD1G37R ALS mouse model. J. Neurosci. 40, 7759–7777. doi: 10.1523/JNEUROSCI.1748-18.2020
Martineau, L., Racine, V., Benichou, S. A., and Puymirat, J. (2018). Lymphoblastoids cell lines–Derived iPSC line from a 26-year-old myotonic dystrophy type 1 patient carrying (CTG) 200 expansion in the DMPK gene: CHUQi001-A. Stem Cell Res. 26, 103–106. doi: 10.1016/j.scr.2017.12.010
Martini, C., Ciana, G., Benettoni, A., Katouzian, F., Severini, G., Bussani, R., et al. (2001). Intractable fever and cortical neuronal glycogen storage in glycogenosis type 2. Neurology 57, 906–908. doi: 10.1212/WNL.57.5.906
Martins, J.-M. F., Fischer, C., Urzi, A., Vidal, R., Kunz, S., Ruffault, P.-L., et al. (2020). Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell 26, 172–186.e176. doi: 10.1016/j.stem.2019.12.007
Matloka, M., Klein, A. F., Rau, F., and Furling, D. (2018). Cells of matter—in vitro models for myotonic dystrophy. Front. Neurol. 9:361. doi: 10.3389/fneur.2018.00361
Maurer, M., Bougoin, S., Feferman, T., Frenkian, M., Bismuth, J., Mouly, V., et al. (2015). IL-6 and Akt are involved in muscular pathogenesis in myasthenia gravis. Acta Neuropathol. Commun. 3, 1–14. doi: 10.1186/s40478-014-0179-6
Maury, Y., Come, J., Piskorowski, R. A., Salah-Mohellibi, N., Chevaleyre, V., Peschanski, M., et al. (2015). Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 33, 89–96. doi: 10.1038/nbt.3049
Mazaleyrat, K., Badja, C., Broucqsault, N., Chevalier, R., Laberthonniere, C., Dion, C., et al. (2020). Multilineage differentiation for formation of innervated skeletal muscle fibers from healthy and diseased human pluripotent stem cells. Cells 9:1531. doi: 10.3390/cells9061531
Meola, G., and Cardani, R. (2015). Myotonic dystrophy type 2: an update on clinical aspects, genetic and pathomolecular mechanism. J. Neuromuscul. Dis. 2, S59–S71. doi: 10.3233/JND-150088
Messeant, J., Ezan, J., Delers, P., Glebov, K., Marchiol, C., Lager, F., et al. (2017). Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development 144, 1712–1724. doi: 10.1242/dev.146167
Miller, J. D., Ganat, Y. M., Kishinevsky, S., Bowman, R. L., Liu, B., Tu, E. Y., et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. doi: 10.1016/j.stem.2013.11.006
Min, Y. L., Li, H., Rodriguez-Caycedo, C., Mireault, A. A., Huang, J., Shelton, J. M., et al. (2019). CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 5:eaav4324. doi: 10.1126/sciadv.aav4324
Moloney, E. B., De Winter, F., and Verhaagen, J. (2014). ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 8:252. doi: 10.3389/fnins.2014.00252
Moore, T. M., Lin, A. J., Strumwasser, A. R., Cory, K., Whitney, K., Ho, T., et al. (2020). Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol. 11:690. doi: 10.21203/rs.2.23961/v1
Morimoto, Y., Kato-Negishi, M., Onoe, H., and Takeuchi, S. (2013). Three-dimensional neuron-muscle constructs with neuromuscular junctions. Biomaterials 34, 9413–9419. doi: 10.1016/j.biomaterials.2013.08.062
Mourkioti, F., Kustan, J., Kraft, P., Day, J. W., Zhao, M. M., Kost-Alimova, M., et al. (2013). Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat. Cell Biol. 15, 895–904. doi: 10.1038/ncb2790
Muhr, J., Graziano, E., Wilson, S., Jessell, T. M., and Edlund, T. (1999). Convergent inductive signals specify midbrain, hindbrain, and spinal cord identity in gastrula stage chick embryos. Neuron 23, 689–702. doi: 10.1016/S0896-6273(01)80028-3
Mukherjee-Clavin, B., Mi, R., Kern, B., Choi, I. Y., Lim, H., Oh, Y., et al. (2019). Comparison of three congruent patient-specific cell types for the modelling of a human genetic Schwann-cell disorder. Nat. Biomed. Eng. 3, 571–582. doi: 10.1038/s41551-019-0381-8
Muller, C. W., Jones, H. N., O’grady, G., Suarez, A., Heller, J. H., and Kishnani, P. S. (2009). Language and speech function in children with infantile Pompe disease. J. Pediatr. Neurol. 7, 147–156.
Muller, D., Cherukuri, P., Henningfeld, K., Poh, C. H., Wittler, L., Grote, P., et al. (2014). Dlk1 promotes a fast motor neuron biophysical signature required for peak force execution. Science 343, 1264–1266. doi: 10.1126/science.1246448
Muller, Q., Beaudet, M.-J., De Serres-Bérard, T., Bellenfant, S., Flacher, V., and Berthod, F. (2018). Development of an innervated tissue-engineered skin with human sensory neurons and Schwann cells differentiated from iPS cells. Acta Biomater. 82, 93–101. doi: 10.1016/j.actbio.2018.10.011
Nacu, A., Andersen, J. B., Lisnic, V., Owe, J. F., and Gilhus, N. E. (2015). Complicating autoimmune diseases in myasthenia gravis: a review. Autoimmunity 48, 362–368. doi: 10.3109/08916934.2015.1030614
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nagao, M., Kato, S., Hayashi, H., and Misawa, H. (2003). Hyperproliferation of synapses on spinal motor neurons of Duchenne muscular dystrophy and myotonic dystrophy patients. Acta Neuropathol. 106, 557–560. doi: 10.1007/s00401-003-0759-1
Nakamori, M., Hamanaka, K., Thomas, J. D., Wang, E. T., Hayashi, Y. K., Takahashi, M. P., et al. (2017). Aberrant myokine signaling in congenital myotonic dystrophy. Cell Rep. 21, 1240–1252. doi: 10.1016/j.celrep.2017.10.018
Nakamori, M., Kimura, T., Fujimura, H., Takahashi, M. P., and Sakoda, S. (2007). Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve 36, 251–257. doi: 10.1002/mus.20809
Nakata, M., Kuwabara, S., Kanai, K., Misawa, S., Tamura, N., Sawai, S., et al. (2006). Distal excitability changes in motor axons in amyotrophic lateral sclerosis. Clin. Neurophysiol. 117, 1444–1448. doi: 10.1016/j.clinph.2006.04.005
Nalbandian, M., Zhao, M., Sasaki-Honda, M., Jonouchi, T., Lucena-Cacace, A., Mizusawa, T., et al. (2021). Characterization of hiPSC-derived muscle progenitors reveals distinctive markers for myogenic cell purification toward cell therapy. Stem Cell Rep. 16, 883–898. doi: 10.1016/j.stemcr.2021.03.004
Narayanan, N., Lengemann, P., Kim, K. H., Kuang, L., Sobreira, T., Hedrick, V., et al. (2021). Harnessing nerve–muscle cell interactions for biomaterials-based skeletal muscle regeneration. J. Biomed. Mater. Res. Part A 109, 289–299. doi: 10.1002/jbm.a.37022
Nascimbeni, A. C., Fanin, M., Masiero, E., Angelini, C., and Sandri, M. (2012). The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ. 19, 1698–1708. doi: 10.1038/cdd.2012.52
Nascimbeni, A. C., Fanin, M., Tasca, E., Angelini, C., and Sandri, M. (2015). Impaired autophagy affects acid α−glucosidase processing and enzyme replacement therapy efficacy in late-onset glycogen storage disease type II. Neuropathol. Appl. Neurobiol. 41, 672–675. doi: 10.1111/nan.12214
Nayak, P., Colas, A., Mercola, M., Varghese, S., and Subramaniam, S. (2021). Temporal mechanisms of myogenic specification in human induced pluripotent stem cells. Sci. Adv. 7:eabf7412. doi: 10.1126/sciadv.abf7412
Nesmith, A. P., Wagner, M. A., Pasqualini, F. S., O’connor, B. B., Pincus, M. J., August, P. R., et al. (2016). A human in vitro model of Duchenne muscular dystrophy muscle formation and contractility. J. Cell Biol. 215, 47–56. doi: 10.1083/jcb.201603111
Newbern, J., and Birchmeier, C. (2010). Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin. Cell Dev. Biol. 21, 922–928. doi: 10.1016/j.semcdb.2010.08.008
Newcomer, J. W., Farber, N. B., and Olney, J. W. (2000). NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2:219. doi: 10.31887/DCNS.2000.2.3/jnewcomer
Nghiem, P. P., and Kornegay, J. N. (2019). Gene therapies in canine models for Duchenne muscular dystrophy. Hum. Genet. 138, 483–489. doi: 10.1007/s00439-019-01976-z
Nicolopoulos-Stournaras, S., and Iles, J. F. (1983). Motor neuron columns in the lumbar spinal cord of the rat. J. Comp. Neurol. 217, 75–85. doi: 10.1002/cne.902170107
Nordstrom, U., Jessell, T. M., and Edlund, T. (2002). Progressive induction of caudal neural character by graded Wnt signaling. Nat. Neurosci. 5, 525–532. doi: 10.1038/nn0602-854
Novak, R., Ingram, M., Marquez, S., Das, D., Delahanty, A., Herland, A., et al. (2020). Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng. 4, 407–420.
Nowotschin, S., Ferrer-Vaquer, A., Concepcion, D., Papaioannou, V. E., and Hadjantonakis, A.-K. (2012). Interaction of Wnt3a, Msgn1 and Tbx6 in neural versus paraxial mesoderm lineage commitment and paraxial mesoderm differentiation in the mouse embryo. Dev. Biol. 367, 1–14. doi: 10.1016/j.ydbio.2012.04.012
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009
Osaki, T., Uzel, S. G., and Kamm, R. D. (2018). Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4:eaat5847. doi: 10.1126/sciadv.aat5847
Oskarsson, B., Gendron, T. F., and Staff, N. P. (2018). Amyotrophic lateral sclerosis: an update for 2018. Mayo Clin. Proc. 93, 1617–1628. doi: 10.1016/j.mayocp.2018.04.007
Ott, M.-O., Bober, E., Lyons, G., Arnold, H., and Buckingham, M. (1991). Early expression of the myogenic regulatory gene, myf-5, in precursor cells of skeletal muscle in the mouse embryo. Development 111, 1097–1107. doi: 10.1242/dev.111.4.1097
Ozair, M. Z., Kintner, C., and Brivanlou, A. H. (2013). Neural induction and early patterning in vertebrates. Wiley Interdiscip. Rev. Dev. Biol. 2, 479–498. doi: 10.1002/wdev.90
Paivalainen, S., Nissinen, M., Honkanen, H., Lahti, O., Kangas, S. M., Peltonen, J., et al. (2008). Myelination in mouse dorsal root ganglion/Schwann cell cocultures. Mol. Cell. Neurosci. 37, 568–578. doi: 10.1016/j.mcn.2007.12.005
Panaite, P.-A., Gantelet, E., Kraftsik, R., Gourdon, G., Kuntzer, T., and Barakat-Walter, I. (2008). Myotonic dystrophy transgenic mice exhibit pathologic abnormalities in diaphragm neuromuscular junctions and phrenic nerves. J. Neuropathol. Exp. Neurol. 67, 763–772. doi: 10.1097/NEN.0b013e318180ec64
Panaite, P.-A., Kielar, M., Kraftsik, R., Gourdon, G., Kuntzer, T., and Barakat-Walter, I. (2011). Peripheral neuropathy is linked to a severe form of myotonic dystrophy in transgenic mice. J. Neuropathol. Exp. Neurol. 70, 678–685. doi: 10.1097/NEN.0b013e3182260939
Paredes-Redondo, A., Harley, P., Maniati, E., Ryan, D., Louzada, S., Meng, J., et al. (2021). Optogenetic modeling of human neuromuscular circuits in Duchenne muscular dystrophy with CRISPR and pharmacological corrections. Sci. Adv. 7:eabi8787. doi: 10.1126/sciadv.abi8787
Park, H. S., Liu, S., Mcdonald, J., Thakor, N., and Yang, I. H. (2013). “Neuromuscular junction in a microfluidic device,” in Proceedings of the 2013 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), (Piscataway, NJ: IEEE), 2833–2835.
Park, K. H., and Vincent, I. (2008). Presymptomatic biochemical changes in hindlimb muscle of G93A human Cu/Zn superoxide dismutase 1 transgenic mouse model of amyotrophic lateral sclerosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1782, 462–468. doi: 10.1016/j.bbadis.2008.04.001
Pascual-Gilabert, M., López-Castel, A., and Artero, R. (2021). Myotonic dystrophy type 1 drug development: a pipeline toward the market. Drug Discov. Today 26, 1765–1772. doi: 10.1016/j.drudis.2021.03.024
Patel, A. M., Wierda, K., Thorrez, L., Van Putten, M., De Smedt, J., Ribeiro, L., et al. (2019). Dystrophin deficiency leads to dysfunctional glutamate clearance in iPSC derived astrocytes. Transl. Psychiatry 9:200. doi: 10.1038/s41398-019-0535-1
Patrick, J., and Lindstrom, J. (1973). Autoimmune response to acetylcholine receptor. Science 180, 871–872. doi: 10.1126/science.180.4088.871
Peric, S., Stojanovic, V. R., Nikolic, A., Kacar, A., Basta, I., Pavlovic, S., et al. (2013). Peripheral neuropathy in patients with myotonic dystrophy type 1. Neurol. Res. 35, 331–335. doi: 10.1179/1743132812Y.0000000144
Personius, K. E., and Sawyer, R. P. (2005). Terminal Schwann cell structure is altered in diaphragm of mdx mice. Muscle Nerve 32, 656–663. doi: 10.1002/mus.20405
Petrany, M. J., Swoboda, C. O., Sun, C., Chetal, K., Chen, X., Weirauch, M. T., et al. (2020). Single-nucleus RNA-seq identifies transcriptional heterogeneity in multinucleated skeletal myofibers. Nat. Commun. 11:6374. doi: 10.1038/s41467-020-20063-w
Philips, A. V., Timchenko, L. T., and Cooper, T. A. (1998). Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280, 737–741. doi: 10.1126/science.280.5364.737
Pickar-Oliver, A., Gough, V., Bohning, J. D., Liu, S., Robinson-Hamm, J. N., Daniels, H., et al. (2021). Full-length dystrophin restoration via targeted genomic integration by aav-crispr in a humanized mouse model of duchenne muscular dystrophy. Mol. Ther. (in press). doi: 10.1016/j.ymthe.2021.09.003
Pollari, E., Goldsteins, G., Bart, G., Koistinaho, J., and Giniatullin, R. (2014). The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8:131. doi: 10.3389/fncel.2014.00131
Poulin, H., Mercier, A., Djemai, M., Pouliot, V., Deschenes, I., Boutjdir, M., et al. (2021). iPSC-derived cardiomyocytes from patients with myotonic dystrophy type 1 have abnormal ion channel functions and slower conduction velocities. Sci. Rep. 11:2500. doi: 10.1038/s41598-021-82007-8
Pourquié, O., Al Tanoury, Z., and Chal, J. (2018). “Chapter Five – the long road to making muscle in vitro,” in Current Topics in Developmental Biology, ed. A. H. Brivanlou (Cambridge, MA: Academic Press), 123–142. doi: 10.1016/bs.ctdb.2018.03.003
Powell, C. A., Smiley, B. L., Mills, J., and Vandenburgh, H. H. (2002). Mechanical stimulation improves tissue-engineered human skeletal muscle. Am. J. Physiol. Cell Physiol. 283, C1557–C1565. doi: 10.1152/ajpcell.00595.2001
Prater, S. N., Banugaria, S. G., Dearmey, S. M., Botha, E. G., Stege, E. M., Case, L. E., et al. (2012). The emerging phenotype of long-term survivors with infantile Pompe disease. Genet. Med. 14, 800–810. doi: 10.1038/gim.2012.44
Prater, S. N., Patel, T. T., Buckley, A. F., Mandel, H., Vlodavski, E., Banugaria, S. G., et al. (2013). Skeletal muscle pathology of infantile Pompe disease during long-term enzyme replacement therapy. Orphanet J. Rare Dis. 8:90. doi: 10.1186/1750-1172-8-90
Pratt, S. J., Shah, S. B., Ward, C. W., Inacio, M. P., Stains, J. P., and Lovering, R. M. (2013). Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles. J. Physiol. 591, 559–570. doi: 10.1113/jphysiol.2012.241679
Pratt, S. J., Shah, S. B., Ward, C. W., Kerr, J. P., Stains, J. P., and Lovering, R. M. (2015a). Recovery of altered neuromuscular junction morphology and muscle function in mdx mice after injury. Cell. Mol. Life Sci. 72, 153–164. doi: 10.1007/s00018-014-1663-7
Pratt, S. J., Valencia, A. P., Le, G. K., Shah, S. B., and Lovering, R. M. (2015b). Pre-and postsynaptic changes in the neuromuscular junction in dystrophic mice. Front. Physiol. 6:252. doi: 10.3389/fphys.2015.00252
Priez, A., Duchene, J., and Goubel, F. (1992). Duchenne muscular dystrophy quantification: a multivariate analysis of surface EMG. Med. Biol. Eng. Comput. 30, 283–291. doi: 10.1007/BF02446966
Prinz, M., and Priller, J. (2017). The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 20, 136–144. doi: 10.1038/nn.4475
Raben, N., Lu, N., Nagaraju, K., Rivera, Y., Lee, A., Yan, B., et al. (2001). Conditional tissue-specific expression of the acid α-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Hum. Mol. Genet. 10, 2039–2047. doi: 10.1093/hmg/10.19.2039
Raben, N., Ralston, E., Chien, Y.-H., Baum, R., Schreiner, C., Hwu, W.-L., et al. (2010). Differences in the predominance of lysosomal and autophagic pathologies between infants and adults with Pompe disease: implications for therapy. Mol. Genet. Metab. 101, 324–331. doi: 10.1016/j.ymgme.2010.08.001
Rao, L., Qian, Y., Khodabukus, A., Ribar, T., and Bursac, N. (2018). Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat. Commun. 9:126. doi: 10.1038/s41467-017-02636-4
Rastall, D., Seregin, S., Aldhamen, Y., Kaiser, L., Mullins, C., Liou, A., et al. (2016). Long-term, high-level hepatic secretion of acid α-glucosidase for Pompe disease achieved in non-human primates using helper-dependent adenovirus. Gene Ther. 23, 743–752. doi: 10.1038/gt.2016.53
Ravanelli, A. M., and Appel, B. (2015). Motor neurons and oligodendrocytes arise from distinct cell lineages by progenitor recruitment. Genes Dev. 29, 2504–2515. doi: 10.1101/gad.271312.115
Ravel-Chapuis, A., Bélanger, G., Yadava, R. S., Mahadevan, M. S., Desgroseillers, L., Côté, J., et al. (2012). The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J. Cell Biol. 196, 699–712. doi: 10.1083/jcb.201108113
Reddy, L. V., Koirala, S., Sugiura, Y., Herrera, A. A., and Ko, C.-P. (2003). Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron 40, 563–580. doi: 10.1016/S0896-6273(03)00682-2
Renna, L. V., Bosè, F., Iachettini, S., Fossati, B., Saraceno, L., Milani, V., et al. (2017). Receptor and post-receptor abnormalities contribute to insulin resistance in myotonic dystrophy type 1 and type 2 skeletal muscle. PLoS One 12:e0184987. doi: 10.1371/journal.pone.0184987
Reuser, A. J., Kroos, M. A., Hermans, M. M., Bijvoet, A. G., Verbeet, M., Van Diggelen, O. A., et al. (1995). Glycogenosis type II (acid maltase deficiency). Muscle Nerve 18, S61–S69. doi: 10.1002/mus.880181414
Ricotti, V., Mandy, W. P., Scoto, M., Pane, M., Deconinck, N., Messina, S., et al. (2016). Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev. Med. Child Neurol. 58, 77–84. doi: 10.1111/dmcn.12922
Riethmacher, D., Sonnenberg-Riethmacher, E., Brinkmann, V., Yamaai, T., Lewin, G. R., and Birchmeier, C. (1997). Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389, 725–730. doi: 10.1038/39593
Rimington, R. P., Fleming, J. W., Capel, A. J., Wheeler, P. C., and Lewis, M. P. (2021). Bioengineered model of the human motor unit with physiologically functional neuromuscular junctions. Sci. Rep. 11:11695. doi: 10.1038/s41598-021-91203-5
Rizzuto, E., Pisu, S., Nicoletti, C., Del Prete, Z., and Musarò, A. (2017). Measuring neuromuscular junction functionality. JoVE J. Vis. Exp. 2017:e55227. doi: 10.3791/55227
Rocha, M. C., Pousinha, P. A., Correia, A. M., Sebastião, A. M., and Ribeiro, J. A. (2013). Early changes of neuromuscular transmission in the SOD1 (G93A) mice model of ALS start long before motor symptoms onset. PLoS One 8:e73846. doi: 10.1371/journal.pone.0073846
Rogers, R. S., and Nishimune, H. (2017). The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 57-58, 86–105. doi: 10.1016/j.matbio.2016.08.008
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17. doi: 10.1038/nm1066
Rothstein, J. D. (1995). Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clin. Neurosci. (New York, NY) 3, 348–359.
Ruggieri, S., Viggiano, L., Annese, T., Rubolino, C., Gerbino, A., De Zio, R., et al. (2019). DP71 and SERCA2 alteration in human neurons of a Duchenne muscular dystrophy patient. Stem Cell Res. Ther. 10:29. doi: 10.1186/s13287-018-1125-5
Rybalka, E., Timpani, C. A., Debruin, D. A., Bagaric, R. M., Campelj, D. G., and Hayes, A. (2020). The failed clinical story of myostatin inhibitors against Duchenne Muscular Dystrophy: Exploring the biology behind the battle. Cells 9:2657. doi: 10.3390/cells9122657
Sacco, A., Mourkioti, F., Tran, R., Choi, J., Llewellyn, M., Kraft, P., et al. (2010). Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143, 1059–1071. doi: 10.1016/j.cell.2010.11.039
Sakai, Y., Meno, C., Fujii, H., Nishino, J., Shiratori, H., Saijoh, Y., et al. (2001). The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 15, 213–225. doi: 10.1101/gad.851501
Sanchez-Gurmaches, J., and Guertin, D. A. (2014). Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat. Commun. 5:4099. doi: 10.1038/ncomms5099
Santhanam, N., Kumanchik, L., Guo, X., Sommerhage, F., Cai, Y., Jackson, M., et al. (2018). Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials 166, 64–78. doi: 10.1016/j.biomaterials.2018.02.047
Sato, Y., Kobayashi, H., Higuchi, T., Shimada, Y., Ida, H., and Ohashi, T. (2016). TFEB overexpression promotes glycogen clearance of Pompe disease iPSC-derived skeletal muscle. Mol. Ther. Methods Clin. Dev. 3:16054. doi: 10.1038/mtm.2016.54
Sawada, M., Matsumoto, M., and Sawamoto, K. (2014). Vascular regulation of adult neurogenesis under physiological and pathological conditions. Front. Neurosci. 8:53. doi: 10.3389/fnins.2014.00053
Scaramozza, A., Marchese, V., Papa, V., Salaroli, R., Soraru, G., Angelini, C., et al. (2014). Skeletal muscle satellite cells in amyotrophic lateral sclerosis. Ultrastruct. Pathol. 38, 295–302. doi: 10.3109/01913123.2014.937842
Selsby, J. T., Ross, J. W., Nonneman, D., and Hollinger, K. (2015). Porcine models of muscular dystrophy. ILAR J. 56, 116–126. doi: 10.1093/ilar/ilv015
Selvaraj, S., Mondragon-Gonzalez, R., Xu, B., Magli, A., Kim, H., Laine, J., et al. (2019). Screening identifies small molecules that enhance the maturation of human pluripotent stem cell-derived myotubes. Elife 8:e47970. doi: 10.7554/eLife.47970.030
Shah, N. M., Marchionni, M. A., Isaacs, I., Stroobant, P., and Anderson, D. J. (1994). Glial growth factor restricts mammalian neural crest stem cells to a glial fate. Cell 77, 349–360. doi: 10.1016/0092-8674(94)90150-3
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W.-H., Hung, S.-T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24:313. doi: 10.1038/nm.4490
Shields, M. C., Bowers, M. R., Fulcer, M. M., Bollig, M. K., Rock, P. J., Sutton, B. R., et al. (2017). Drosophila studies support a role for a presynaptic synaptotagmin mutation in a human congenital myasthenic syndrome. PLoS One 12:e0184817. doi: 10.1371/journal.pone.0184817
Sicot, G., Servais, L., Dinca, D. M., Leroy, A., Prigogine, C., Medja, F., et al. (2017). Downregulation of the glial GLT1 glutamate transporter and Purkinje cell dysfunction in a mouse model of myotonic dystrophy. Cell Rep. 19, 2718–2729. doi: 10.1016/j.celrep.2017.06.006
Sigoillot, S. M., Bourgeois, F., Karmouch, J., Molgo, J., Dobbertin, A., Chevalier, C., et al. (2016). Neuromuscular junction immaturity and muscle atrophy are hallmarks of the ColQ-deficient mouse, a model of congenital myasthenic syndrome with acetylcholinesterase deficiency. FASEB J. 30, 2382–2399. doi: 10.1096/fj.201500162
Singh, T., and Vazquez, M. (2019). Time-dependent addition of neuronal and Schwann cells increase myotube viability and length in an in vitro tri-culture model of the neuromuscular junction. Regener. Eng. Transl. Med. 5, 402–413. doi: 10.1007/s40883-019-00095-5
Skardal, A., Murphy, S. V., Devarasetty, M., Mead, I., Kang, H.-W., Seol, Y.-J., et al. (2017). Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 7:8837. doi: 10.1038/s41598-017-08879-x
Skoglund, G., Laine, J., Darabi, R., Fournier, E., Perlingeiro, R., and Tabti, N. (2014). Physiological and ultrastructural features of human induced pluripotent and embryonic stem cell-derived skeletal myocytes in vitro. Proc. Natl. Acad. Sci. U.S.A. 111, 8275–8280. doi: 10.1073/pnas.1322258111
Sleigh, J., and Sattelle, D. (2010). C. elegans models of neuromuscular diseases expedite translational research. Transl. Neurosci. 1, 214–227. doi: 10.2478/v10134-010-0032-9
Smith, A., Long, C., Pirozzi, K., and Hickman, J. (2013). A functional system for high-content screening of neuromuscular junctions in vitro. Technology 1, 37–48. doi: 10.1142/S2339547813500015
Son, E. Y., Ichida, J. K., Wainger, B. J., Toma, J. S., Rafuse, V. F., Woolf, C. J., et al. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218. doi: 10.1016/j.stem.2011.07.014
Son, Y.-J., Trachtenberg, J. T., and Thompson, W. J. (1996). Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci. 19, 280–285. doi: 10.1016/S0166-2236(96)10032-1
Southam, K. A., King, A. E., Blizzard, C. A., Mccormack, G. H., and Dickson, T. C. (2013). Microfluidic primary culture model of the lower motor neuron–neuromuscular junction circuit. J. Neurosci. Methods 218, 164–169. doi: 10.1016/j.jneumeth.2013.06.002
Spampanato, C., Feeney, E., Li, L., Cardone, M., Lim, J. A., Annunziata, F., et al. (2013). Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 5, 691–706. doi: 10.1002/emmm.201202176
Spiridigliozzi, G. A., Keeling, L. A., Stefanescu, M., Li, C., Austin, S., and Kishnani, P. S. (2017). Cognitive and academic outcomes in long-term survivors of infantile-onset Pompe disease: a longitudinal follow-up. Mol. Genet. Metab. 121, 127–137. doi: 10.1016/j.ymgme.2017.04.014
Stålberg, E., and Trontelj, J. V. (1997). The study of normal and abnormal neuromuscular transmission with single fibre electromyography. J. Neurosci. Methods 74, 145–154. doi: 10.1016/S0165-0270(97)02245-0
Stein, J. L., De La Torre-Ubieta, L., Tian, Y., Parikshak, N. N., Hernandez, I. A., Marchetto, M. C., et al. (2014). A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86. doi: 10.1016/j.neuron.2014.05.035
Steinbeck, J. A., Jaiswal, M. K., Calder, E. L., Kishinevsky, S., Weishaupt, A., Toyka, K. V., et al. (2016). Functional connectivity under optogenetic control allows modeling of human neuromuscular disease. Cell Stem Cell 18, 134–143. doi: 10.1016/j.stem.2015.10.002
Strong, M. J. (2010). The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 288, 1–12. doi: 10.1016/j.jns.2009.09.029
Sugiura, Y., and Lin, W. (2011). Neuron–glia interactions: the roles of Schwann cells in neuromuscular synapse formation and function. Biosci. Rep. 31, 295–302. doi: 10.1042/BSR20100107
Sui, T., Lau, Y. S., Liu, D., Liu, T., Xu, L., Gao, Y., et al. (2018). A novel rabbit model of Duchenne muscular dystrophy generated by CRISPR/Cas9. Dis. Model. Mech. 11:dmm032201. doi: 10.1242/dmm.032201
Sun, C., Choi, I. Y., Gonzalez, Y. I. R., Andersen, P., Talbot, C. C. Jr., Iyer, S. R., et al. (2020). Duchenne muscular dystrophy hiPSC-derived myoblast drug screen identifies compounds that ameliorate disease in mdx mice. JCI Insight 5:e134287. doi: 10.1172/jci.insight.134287
Sun, J., Ma, X., Chu, H. T., Feng, B., Tuan, R. S., and Jiang, Y. (2019). Biomaterials and advanced biofabrication techniques in hiPSCs based neuromyopathic disease modeling. Front. Bioeng. Biotechnol. 7:373. doi: 10.3389/fbioe.2019.00373
Sun, Y., Duffy, R., Lee, A., and Feinberg, A. W. (2013). Optimizing the structure and contractility of engineered skeletal muscle thin films. Acta Biomater. 9, 7885–7894. doi: 10.1016/j.actbio.2013.04.036
Suzuki, M., Mchugh, J., Tork, C., Shelley, B., Hayes, A., Bellantuono, I., et al. (2008). Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol. Ther. 16, 2002–2010. doi: 10.1038/mt.2008.197
Suzuki, M., Mchugh, J., Tork, C., Shelley, B., Klein, S. M., Aebischer, P., et al. (2007). GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One 2:e689. doi: 10.1371/journal.pone.0000689
Suzuki, N., Mizuno, H., Warita, H., Takeda, S. I., Itoyama, Y., and Aoki, M. (2010). Neuronal NOS is dislocated during muscle atrophy in amyotrophic lateral sclerosis. J. Neurol. Sci. 294, 95–101. doi: 10.1016/j.jns.2010.03.022
Swartz, E. W., Baek, J., Pribadi, M., Wojta, K. J., Almeida, S., Karydas, A., et al. (2016). A novel protocol for directed differentiation of C9orf72-associated human induced pluripotent stem cells into contractile skeletal myotubes. Stem Cells Transl. Med. 5, 1461–1472. doi: 10.5966/sctm.2015-0340
Takemoto, T., Uchikawa, M., Yoshida, M., Bell, D. M., Lovell-Badge, R., Papaioannou, V. E., et al. (2011). Tbx6-dependent Sox2 regulation determines neural or mesodermal fate in axial stem cells. Nature 470, 394–398. doi: 10.1038/nature09729
Tanaka, A., Woltjen, K., Miyake, K., Hotta, A., Ikeya, M., Yamamoto, T., et al. (2013). Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling Miyoshi Myopathy in vitro. PLoS One 8:e61540. doi: 10.1371/journal.pone.0061540
Thornton, C. A. (2014). Myotonic dystrophy. Neurol. Clin. 32, 705–719. doi: 10.1016/j.ncl.2014.04.011
Tintignac, L. A., Brenner, H.-R., and Rüegg, M. A. (2015). Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol. Rev. 95, 809–852. doi: 10.1152/physrev.00033.2014
Todd, A. G., Mcelroy, J. A., Grange, R. W., Fuller, D. D., Walter, G. A., Byrne, B. J., et al. (2015). Correcting neuromuscular deficits with gene therapy in pompe disease. Ann. Neurol. 78:222. doi: 10.1002/ana.24433
Tong, Z., Seira, O., Casas, C., Reginensi, D., Homs-Corbera, A., Samitier, J., et al. (2014). Engineering a functional neuro-muscular junction model in a chip. RSC Adv. 4, 54788–54797. doi: 10.1039/C4RA10219C
Torres, L., and Duchen, L. (1987). The mutant mdx: inherited myopathy in the mouse: morphological studies of nerves, muscles and end-plates. Brain 110, 269–299. doi: 10.1093/brain/110.2.269
Totosy de Zepetnek, J., Zung, H., Erdebil, S., and Gordon, T. (1992). Motor-unit categorization based on contractile and histochemical properties: a glycogen depletion analysis of normal and reinnervated rat tibialis anterior muscle. J. Neurophysiol. 67, 1404–1415. doi: 10.1152/jn.1992.67.5.1404
Tourovskaia, A., Li, N., and Folch, A. (2008). Localized acetylcholine receptor clustering dynamics in response to microfluidic focal stimulation with agrin. Biophys. J. 95, 3009–3016. doi: 10.1529/biophysj.107.128173
Tremblay, E., and Martineau, É, and Robitaille, R. (2017). Opposite synaptic alterations at the neuromuscular junction in an ALS mouse model: when motor units matter. J. Neurosci. 37, 8901–8918. doi: 10.1523/JNEUROSCI.3090-16.2017
Trias, E., Ibarburu, S., Barreto-Núñez, R., Varela, V., Moura, I. C., Dubreuil, P., et al. (2017). Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight 2:e95934. doi: 10.1172/jci.insight.95934
Tsujihata, M., Hazama, R., Yoshimura, T., Satoh, A., Mori, M., and Nagataki, S. (1984). The motor end-plate fine structure and ultrastructural localization of acetylcholine receptors in amyotrophic lateral sclerosis. Muscle Nerve 7, 243–249. doi: 10.1002/mus.880070310
Turner, S. M., Hoyt, A. K., Elmallah, M. K., Falk, D. J., Byrne, B. J., and Fuller, D. D. (2016). Neuropathology in respiratory-related motoneurons in young Pompe (Gaa−/−) mice. Respir. Physiol. Neurobiol. 227, 48–55. doi: 10.1016/j.resp.2016.02.007
Tutdibi, O., Brinkmeier, H., Rüdel, R., and Föhr, K. (1999). Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 515, 859–868. doi: 10.1111/j.1469-7793.1999.859ab.x
Tzouanacou, E., Wegener, A., Wymeersch, F. J., Wilson, V., and Nicolas, J.-F. (2009). Redefining the progression of lineage segregations during mammalian embryogenesis by clonal analysis. Dev. Cell 17, 365–376. doi: 10.1016/j.devcel.2009.08.002
Ueki, J., Nakamori, M., Nakamura, M., Nishikawa, M., Yoshida, Y., Tanaka, A., et al. (2017). Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep. 7:42522. doi: 10.1038/srep42522
Uezumi, A., Ikemoto-Uezumi, M., Zhou, H., Kurosawa, T., Yoshimoto, Y., Nakatani, M., et al. (2021). Mesenchymal Bmp3b expression maintains skeletal muscle integrity and decreases in age-related sarcopenia. J. Clin. Invest. 131:e139617. doi: 10.1172/JCI139617
Umbach, J. A., Adams, K. L., Gundersen, C. B., and Novitch, B. G. (2012). Functional neuromuscular junctions formed by embryonic stem cell-derived motor neurons. PLoS One 7:e36049. doi: 10.1371/journal.pone.0036049
Uzawa, A., Kuwabara, S., Suzuki, S., Imai, T., Murai, H., Ozawa, Y., et al. (2021). Roles of cytokines and T cells in the pathogenesis of myasthenia gravis. Clin. Exp. Immunol. 203, 366–374. doi: 10.1111/cei.13546
Uzel, S. G., Platt, R. J., Subramanian, V., Pearl, T. M., Rowlands, C. J., Chan, V., et al. (2016). Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci. Adv. 2:e1501429. doi: 10.1126/sciadv.1501429
Vainzof, M., Ayub-Guerrieri, D., Onofre, P. C., Martins, P. C., Lopes, V. F., Zilberztajn, D., et al. (2008). Animal models for genetic neuromuscular diseases. J. Mol. Neurosci. 34, 241–248. doi: 10.1007/s12031-007-9023-9
van der Pijl, E. M., Van Putten, M., Niks, E. H., Verschuuren, J. J., Aartsma-Rus, A., and Plomp, J. J. (2016). Characterization of neuromuscular synapse function abnormalities in multiple Duchenne muscular dystrophy mouse models. Eur. J. Neurosci. 43, 1623–1635. doi: 10.1111/ejn.13249
van der Ploeg, A. T., Clemens, P. R., Corzo, D., Escolar, D. M., Florence, J., Groeneveld, G. J., et al. (2010). A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 362, 1396–1406. doi: 10.1056/NEJMoa0909859
van der Velden, J. L., Langen, R. C., Kelders, M. C., Wouters, E. F., Janssen-Heininger, Y. M., and Schols, A. M. (2006). Inhibition of glycogen synthase kinase-3β activity is sufficient to stimulate myogenic differentiation. Am. J. Physiol. Cell Physiol. 290, C453–C462. doi: 10.1152/ajpcell.00068.2005
van der Wal, E., Bergsma, A. J., Van Gestel, T. J., Lm, S., Zaehres, H., Araúzo-Bravo, M. J., et al. (2017). GAA deficiency in Pompe disease is alleviated by exon inclusion in iPSC-derived skeletal muscle cells. Mol. Ther. Nucleic Acids 7, 101–115. doi: 10.1016/j.omtn.2017.03.002
van der Wal, E., Herrero-Hernandez, P., Wan, R., Broeders, M., In ’T Groen, S. L. M., Van Gestel, T. J. M., et al. (2018). Large-Scale expansion of human iPSC-derived skeletal muscle cells for disease modeling and cell-based therapeutic strategies. Stem Cell Rep. 10, 1975–1990. doi: 10.1016/j.stemcr.2018.04.002
van Putten, M., Putker, K., Overzier, M., Adamzek, W. A., Pasteuning-Vuhman, S., Plomp, J. J., et al. (2019). Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. FASEB J. 33, 8110–8124. doi: 10.1096/fj.201802488R
Veltrop, M., Van Vliet, L., Hulsker, M., Claassens, J., Brouwers, C., Breukel, C., et al. (2018). A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLoS One 13:e0193289. doi: 10.1371/journal.pone.0193289
Viader, A., Golden, J. P., Baloh, R. H., Schmidt, R. E., Hunter, D. A., and Milbrandt, J. (2011). Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J. Neurosci. 31, 10128–10140. doi: 10.1523/JNEUROSCI.0884-11.2011
Vignaud, A., Ferry, A., Huguet, A., Baraibar, M., Trollet, C., Hyzewicz, J., et al. (2010). Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin–proteasome pathway. Neuromuscul. Disord. 20, 319–325. doi: 10.1016/j.nmd.2010.03.006
Vihola, A., Bassez, G., Meola, G., Zhang, S., Haapasalo, H., Paetau, A., et al. (2003). Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology 60, 1854–1857. doi: 10.1212/01.WNL.0000065898.61358.09
Vila, O., Chavez, M., Ma, S. P., Yeager, K., Zholudeva, L. V., Colón-Mercado, J. M., et al. (2021). Bioengineered optogenetic model of human neuromuscular junction. Biomaterials 276:121033. doi: 10.1016/j.biomaterials.2021.121033
Vila, O. F., Uzel, S. G., Ma, S. P., Williams, D., Pak, J., Kamm, R. D., et al. (2019). Quantification of human neuromuscular function through optogenetics. Theranostics 9:1232. doi: 10.7150/thno.25735
Vilmont, V., Cadot, B., Ouanounou, G., and Gomes, E. R. (2016). A system for studying mechanisms of neuromuscular junction development and maintenance. Development 143, 2464–2477. doi: 10.1242/dev.130278
Wachtler, F. (1992). The basic embryology of skeletal muscle formation in vertebrates: the avian model. Semin. Dev. Biol. 3, 217–227.
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Waite, A., Tinsley, C. L., Locke, M., and Blake, D. J. (2009). The neurobiology of the dystrophin-associated glycoprotein complex. Ann. Med. 41, 344–359. doi: 10.1080/07853890802668522
Wang, J., Khodabukus, A., Rao, L., Vandusen, K., Abutaleb, N., and Bursac, N. (2019). Engineered skeletal muscles for disease modeling and drug discovery. Biomaterials 221:119416. doi: 10.1016/j.biomaterials.2019.119416
Wang, J., Xiao, Y., Zhang, K., Luo, B., and Shen, C. (2018). Introducing autoimmunity at the synapse by a novel animal model of experimental autoimmune myasthenia gravis. Neuroscience 374, 264–270. doi: 10.1016/j.neuroscience.2018.01.042
Wang, J., Zhou, C. J., Khodabukus, A., Tran, S., Han, S.-O., Carlson, A. L., et al. (2021). Three-dimensional tissue-engineered human skeletal muscle model of Pompe disease. Commun. Biol. 4:524. doi: 10.1038/s42003-021-02059-4
Wang, P. Y., Thissen, H., and Tsai, W. B. (2012). The roles of RGD and grooved topography in the adhesion, morphology, and differentiation of C2C12 skeletal myoblasts. Biotechnol. Bioeng. 109, 2104–2115. doi: 10.1002/bit.24452
Wang, Y. X., Feige, P., Brun, C. E., Hekmatnejad, B., Dumont, N. A., Renaud, J. M., et al. (2019). EGFR-Aurka signaling rescues polarity and regeneration defects in dystrophin-deficient muscle stem cells by increasing asymmetric divisions. Cell Stem Cell 24, 419–432.e416. doi: 10.1016/j.stem.2019.01.002
Warf, M. B., and Berglund, J. A. (2007). MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA 13, 2238–2251. doi: 10.1261/rna.610607
Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y.-H., Li, H., Lau, F., et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630. doi: 10.1016/j.stem.2010.08.012
Watanabe, K., Kamiya, D., Nishiyama, A., Katayama, T., Nozaki, S., Kawasaki, H., et al. (2005). Directed differentiation of telencephalic precursors from embryonic stem cells. Nat. Neurosci. 8, 288–296. doi: 10.1038/nn1402
Webster, R. G. (2018). Animal models of the neuromuscular junction, vitally informative for understanding function and the molecular mechanisms of congenital myasthenic syndromes. Int. J. Mol. Sci. 19:1326. doi: 10.3390/ijms19051326
Weller, B., Karpati, G., and Carpenter, S. (1990). Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J. Neurol. Sci. 100, 9–13. doi: 10.1016/0022-510X(90)90005-8
Wells, D. J. (2018). Tracking Progress: An Update on Animal Models for Duchenne Muscular Dystrophy. Cambridge: The Company of Biologists Ltd. doi: 10.1242/dmm.035774
Wheeler, T., Krym, M., and Thornton, C. (2007). Ribonuclear foci at the neuromuscular junction in myotonic dystrophy type 1. Neuromuscul. Disord. 17, 242–247. doi: 10.1016/j.nmd.2006.12.015
White, P. H., Farkas, D. R., and Chapman, D. L. (2005). Regulation of Tbx6 expression by Notch signaling. Genesis 42, 61–70. doi: 10.1002/gene.20124
Wichterle, H., Lieberam, I., Porter, J. A., and Jessell, T. M. (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. doi: 10.1016/S0092-8674(02)00835-8
Winnier, G., Blessing, M., Labosky, P. A., and Hogan, B. (1995). Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 9, 2105–2116. doi: 10.1101/gad.9.17.2105
Woldeyesus, M. T., Britsch, S., Riethmacher, D., Xu, L., Sonnenberg-Riethmacher, E., Abou-Rebyeh, F., et al. (1999). Peripheral nervous system defects in erbB2 mutants following genetic rescue of heart development. Genes Dev. 13, 2538–2548. doi: 10.1101/gad.13.19.2538
Woodhoo, A., Alonso, M. B., Droggiti, A., Turmaine, M., D’antonio, M., Parkinson, D. B., et al. (2009). Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat. Neurosci. 12, 839–847. doi: 10.1038/nn.2323
Woodhoo, A., and Sommer, L. (2008). Development of the Schwann cell lineage: from the neural crest to the myelinated nerve. Glia 56, 1481–1490. doi: 10.1002/glia.20723
Wroe, R., Wai-Ling Butler, A., Andersen, P. M., Powell, J. F., and Al-Chalabi, A. (2008). ALSOD: the amyotrophic lateral sclerosis online database. Amyotroph. Lateral Scler. 9, 249–250. doi: 10.1080/17482960802146106
Xi, H., Langerman, J., Sabri, S., Chien, P., Young, C. S., Younesi, S., et al. (2020). A human skeletal muscle atlas identifies the trajectories of stem and progenitor cells across development and from human pluripotent stem cells. Cell Stem Cell 27, 158–176.e110. doi: 10.1016/j.stem.2020.04.017
Xia, G., Gao, Y., Jin, S., Subramony, S., Terada, N., Ranum, L. P., et al. (2015). Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 33, 1829–1838. doi: 10.1002/stem.1970
Yadava, R. S., Foff, E. P., Yu, Q., Gladman, J. T., Kim, Y. K., Bhatt, K. S., et al. (2015). TWEAK/Fn14, a pathway and novel therapeutic target in myotonic dystrophy. Hum. Mol. Genet. 24, 2035–2048. doi: 10.1093/hmg/ddu617
Yang, J.-F., Cao, G., Koirala, S., Reddy, L. V., and Ko, C.-P. (2001). Schwann cells express active agrin and enhance aggregation of acetylcholine receptors on muscle fibers. J. Neurosci. 21, 9572–9584. doi: 10.1523/JNEUROSCI.21-24-09572.2001
Ydens, E., Lornet, G., Smits, V., Goethals, S., Timmerman, V., and Janssens, S. (2013). The neuroinflammatory role of Schwann cells in disease. Neurobiol. Dis. 55, 95–103. doi: 10.1016/j.nbd.2013.03.005
Yiu, E. M., and Kornberg, A. J. (2015). Duchenne muscular dystrophy. J. Paediatr. Child Health 51, 759–764. doi: 10.1111/jpc.12868
Yokota, T., Duddy, W., Echigoya, Y., and Kolski, H. (2012). Exon skipping for nonsense mutations in Duchenne muscular dystrophy: too many mutations, too few patients? Expert Opin. Biol. Ther. 12, 1141–1152. doi: 10.1517/14712598.2012.693469
Yoshida, M., Kitaoka, S., Egawa, N., Yamane, M., Ikeda, R., Tsukita, K., et al. (2015). Modeling the early phenotype at the neuromuscular junction of spinal muscular atrophy using patient-derived iPSCs. Stem Cell Rep. 4, 561–568. doi: 10.1016/j.stemcr.2015.02.010
Yoshida, T., Awaya, T., Jonouchi, T., Kimura, R., Kimura, S., Era, T., et al. (2017). A skeletal muscle model of infantile-onset Pompe disease with patient-specific iPS cells. Sci. Rep. 7:13473. doi: 10.1038/s41598-017-14063-y
Yoshihara, T., Ishii, T., Iwata, M., and Nomoto, M. (1998). Ultrastructural and histochemical study of the motor end plates of the intrinsic laryngeal muscles in amyotrophic lateral sclerosis. Ultrastruct. Pathol. 22, 121–126. doi: 10.3109/01913129809032266
Zaccaria, M. L., Egle De Stefano, M., Properzi, F., Gotti, C., Petrucci, T. C., and Paggi, P. (1998). Disassembly of the cholinergic postsynaptic apparatus induced by axotomy in mouse sympathetic neurons: the loss of dystrophin and β-dystroglycan immunoreactivity precedes that of the acetylcholine receptor. J. Neuropathol. Exp. Neurol. 57, 768–779. doi: 10.1097/00005072-199808000-00006
Zainul, Z., Heikkinen, A., Koivisto, H., Rautalahti, I., Kallio, M., Lin, S., et al. (2018). Collagen XIII is required for neuromuscular synapse regeneration and functional recovery after peripheral nerve injury. J. Neurosci. 38, 4243–4258. doi: 10.1523/JNEUROSCI.3119-17.2018
Zhang, B. G., Quigley, A. F., Bourke, J. L., Nowell, C. J., Myers, D. E., Choong, P. F., et al. (2016). Combination of agrin and laminin increase acetylcholine receptor clustering and enhance functional neuromuscular junction formation in vitro. Dev. Neurobiol. 76, 551–565. doi: 10.1002/dneu.22331
Zhang, H., Wen, J., Bigot, A., Chen, J., Shang, R., Mouly, V., et al. (2020). Human myotube formation is determined by MyoD–Myomixer/Myomaker axis. Sci. Adv. 6:eabc4062. doi: 10.1126/sciadv.abc4062
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, Q., Nguyen, P. D., Shi, S., Burrell, J. C., Cullen, D. K., and Le, A. D. (2018). 3D bio-printed scaffold-free nerve constructs with human gingiva-derived mesenchymal stem cells promote rat facial nerve regeneration. Sci. Rep. 8:6634. doi: 10.1038/s41598-018-24888-w
Zhao, C., Devlin, A. C., Chouhan, A. K., Selvaraj, B. T., Stavrou, M., Burr, K., et al. (2020). Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 68, 1046–1064. doi: 10.1002/glia.23761
Zhou, H., Glass, D. J., Yancopoulos, G. D., and Sanes, J. R. (1999). Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations. J. Cell Biol. 146, 1133–1146. doi: 10.1083/jcb.146.5.1133
Zouvelou, V., Rentzos, M., Toulas, P., and Evdokimidis, I. (2012). AchR-positive myasthenia gravis with MRI evidence of early muscle atrophy. J. Clin. Neurosci. 19, 918–919. doi: 10.1016/j.jocn.2011.09.022
Keywords: tissue engineering, induced pluripotent stem cells, disease modeling, neuromuscular junction, human skeletal muscle, muscular dystrophy, drug development, organ on a chip
Citation: Fralish Z, Lotz EM, Chavez T, Khodabukus A and Bursac N (2021) Neuromuscular Development and Disease: Learning From in vitro and in vivo Models. Front. Cell Dev. Biol. 9:764732. doi: 10.3389/fcell.2021.764732
Received: 25 August 2021; Accepted: 06 October 2021;
Published: 27 October 2021.
Edited by:
Alec S. T. Smith, University of Washington, United StatesReviewed by:
Gabsang Lee, Johns Hopkins University, United StatesDarren Player, University College London, United Kingdom
Copyright © 2021 Fralish, Lotz, Chavez, Khodabukus and Bursac. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nenad Bursac, bmVuYWQuYnVyc2FjQGR1a2UuZWR1