 Lili Yu1,2
Lili Yu1,2 Gaoyang Zhu
Gaoyang Zhu Yang Xu
Yang Xu- 1Key Laboratory of Cancer Prevention and Intervention, Department of Medical Oncology, Ministry of Education, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Cancer Center, Zhejiang University, Hangzhou, China
- 3Cardiovascular Key Lab of Zhejiang Province, Department of Cardiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, China
- 4Shunde Hospital, Southern Medical University (The First People’s Hospital of Shunde), Foshan, China
- 5Division of Biological Sciences, University of California, San Diego, La Jolla, CA, United States
Metabolism plays critical roles in maintaining the homeostasis of cells. Metabolic abnormalities are often considered as one of the main driving forces for cancer progression, providing energy and substrates of biosynthesis to support neoplastic proliferation effectively. The tumor suppressor p53 is well known for its roles in inducing cell cycle arrest, apoptosis, senescence and ferroptosis. Recently, emerging evidence has shown that p53 is also actively involved in the reprogramming of cellular metabolism. In this review, we focus on recent advances in our understanding of the interplay between p53 and metabolism of glucose, fatty acid as well as amino acid, and discuss how the deregulation of p53 in these processes could lead to cancer.
Introduction
p53, encoded by the TP53 gene, is a critical tumor suppressor that is required to prevent the oncogenic transformation of cells. Of note, TP53 is the most frequently mutated gene in human cancers, and in most cases, TP53 mutation is associated with poor prognosis (Levine, 2020; Olivier et al., 2010). Mutant p53 (Mutp53) proteins not only lose tumor suppressive functions, but also frequently acquire various gain-of-functions (GOF) that promote tumorigenesis. Under normal conditions, p53 is maintained in an inactive and unstable form through the interaction between p53 and its E3 ligase MDM2 and negative regulator MDMX (Fu et al., 2020). Under various stress conditions, p53 is stabilized and activated by post-translational modifications such as phosphorylation, acetylation, sumoylation, disrupting the interaction between p53 and Mdm2 and Mdmx (Fu, et al., 2020).
As a transcriptional factor, p53 directly activates and suppresses the transcription of hundreds of genes, many of which play key roles in cell cycle, apoptosis, and senescence (Vousden and Prives, 2009). For a long time, the roles of p53 in cell cycle arrest, apoptosis and senescence have been considered the major mechanisms to mediate its tumor suppressive activities (Vousden and Prives, 2009). However, the disruption of p53-dependent cell cycle arrest, apoptosis and senescence is not sufficient to induce cancer (Fu et al., 2020). Instead, various studies in mouse models such as the p53 (3KR/3KR) knock-in mouse model have highlighted its metabolic roles in inhibiting cancer progression (Li et al., 2012).
Reprogramming of cellular metabolism is one of the “hallmarks of cancer”, and is considered one of the main driving forces for tumorigenesis (Hanahan and Weinberg, 2011). In order to effectively support neoplastic proliferation, cancer cells increase their uptake of nutrients, especially glucose and amino acids, and adapt themselves to ensure their maximum utilization of the metabolic intermediates of glycolysis and oxidative phosphorylation for biosynthesis and NADPH production (Pavlova and Thompson, 2016). Numerous reports indicate that p53 is playing extensive and complex roles in regulating various metabolic pathways, and the gain of function mutants of p53 promotes the oncogenic metabolic reprogramming that induces drug resistance and metastasis.
In this review, we focus on recent advances in the research of p53 and its GOF mutants in regulating oncogenic metabolic alterations, aiming to provide insights into the targeted therapy of human cancers with metabolic regulation regiments.
p53 and Glucose Metabolism
Numerous studies have shown that p53 plays complex roles in regulating glucose metabolism. Unlike normal cells, tumor cells use glucose mainly through glycolysis rather than oxidative phosphorylation (OXPHOS) to meet their energy and biosynthetic demand even under aerobic conditions, which is known as “Warburg effect” (Warburg et al., 1927). In many cases, p53 performs the tumor suppressive functions to inhibit aerobic glycolysis and promote OXPHOS.
p53 represses the transcription of glucose transporters GLUT1, GLUT3, and GLUT4 to reduce glucose uptake, which is the first rate-limiting event in glycolysis (Kawauchi et al., 2008; Schwartzenberg-Bar-Yoseph et al., 2004). p53 also transcriptionally induces TP53 Induced Glycolysis Regulatory Phosphatase (TIGAR) and inhibits 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3 and PFKFB4), resulting in reduced intracellular levels of fructose-2,6-bisphosphate (F-2,6-BP), which functions as allosteric activator of phosphofructokinase (PFK), the rate-limiting enzyme catalyzing the conversion from F6P to F-1,6-BP (Bensaad et al., 2006; Franklin et al., 2016; Liu et al., 2020; Ros et al., 2017). Moreover, p53 was also reported to inhibit other glycolytic enzymes such as hexokinase 2 (HK2) and phosphoglycerate mutase 1 (PGAM1) (Kondoh et al., 2005; Wang et al., 2014). These findings support the notion that wild-type p53 suppresses glycolysis.
To further tilt the balance from glycolysis to OXPHOS, p53 also promotes cellular OXPHOS by various complementary mechanisms. p53 is able to inhibit the expression of pyruvate dehydrogenase kinase 2 (PDK2), a negative regulator of pyruvate dehydrogenase (PDH) that converts pyruvate to acetyl-CoA, leading to increased OXPHOS (Contractor and Harris, 2012). In addition, p53 induces the expression of Synthesis of Cytochrome C Oxidase 2 (SCO2), thereby promoting the synthesis of cytochrome C oxidase complex that catalyzes the major step of OXPHOS (Matoba et al., 2006). It was also reported that the induction of ferredoxin reductase (FDXR) by p53 promotes electron transfer from NADPH to cytochrome p450 (Liu and Chen, 2002). Moreover, p53 could promote mitochondrial biogenesis, support mitochondrial fission, maintain mitochondrial genome integrity, and ensure the quality control and turnover of mitochondria, thereby guarantees the proper function of mitochondria (Lacroix et al., 2020). Besides, the pentose phosphate pathway (PPP) is also reported to be repressed by p53 through its direct binding to glucose-6-phosphate dehydrogenase (G6PD), which is the first and rate-limiting enzyme of PPP. Consequently, p53 suppresses the production of NADPH as well as precursors for nucleotide biosynthesis (Jiang et al., 2011).
In contrast to the above reviewed canonical functions, the complexity of the roles of p53 in glucose metabolism remains to be elucidated. In this context, p53 could play an oncogenic role by dominantly suppressing OXPHOS. For example, in contrast to many types of human cancers such as lung cancer, wide-type p53 is often retained in hepatocellular carcinomas (HCC), where it induces PUMA expression to disrupt the oligomerization and function of mitochondrial pyruvate carrier (MPC) through direct PUMA-MPC interaction, thereby inhibits the mitochondrial pyruvate uptake and promotes glycolysis of HCC (Kim et al., 2019). These findings underscore the complexity of wild-type p53, indicating that the impact of p53 on glucose metabolism in cancer cells is complex and cell context dependent.
p53, Lipid Metabolism and Ferroptosis
It has become increasingly clear that cancer cells gain the unique ability to synthesize fatty acids essential for cellular growth and survival (Beloribi-Djefaflia et al., 2016). Another non-canonical function of p53 is the capability to regulate lipid metabolism. p53 is thought to promote catabolism of fatty acids while simultaneously inhibit fatty acid synthesis. In addition to its inhibition of Glucose-6-phosphate dehydrogenase (G6PD) and pentose phosphate pathway (PPP) that is important for DNA synthesis and lipid synthesis (Jiang, et al., 2011), p53 can transcriptionally upregulate aromatase that is involved in lipid metabolism (Wang et al., 2013). Increased lipid accumulation in the livers of p53−/− mice is mitigated by the transgenic expression of aromatase, indicating important roles of p53-aromatase pathway in lipid metabolism (Wang, et al., 2013).
While wild-type p53 can suppress lipid synthesis by regulating the activities or levels of downstream effectors/targets such as G6PD and aromatase (Jiang, et al., 2011; Wang, et al., 2013), numerous reports have demonstrated that mutant p53 can promote lipid synthesis by altering the activities of various transcription factors or signaling molecules such as p63, p73, Nrf2, and AMP-activated protein kinase (AMPK), which are involved in lipid metabolism (Do et al., 2012; Walerych et al., 2016; Xu et al., 2011). Several studies have shown that the upregulation of enzymes involved in the synthesis of fatty acids and cholesterol (mevalonate pathway) is required for tumor progression (Bathaie et al., 2017; Kuhajda et al., 1994; Ribas et al., 2016; Roongta et al., 2011; Zhan et al., 2008). The presence of p53 mutations correlates with high levels of enzymes involved in the mevalonate pathway in human breast cancer tissues (Freed-Pastor et al., 2012). Another study shows that ectopic expression of p53 mutants (p53R175H and p53P151S) inhibits AMPK activity and subsequently reduces phosphorylation of Acetyl-CoA carboxylase (ACC) under glucose and serum starvation in a p53-null head and neck squamous cell carcinoma (HNSCC) cell line UMSCC1 (Zhou et al., 2014).
Ferroptosis is a new form of programmed cell death characterized by the accumulation of iron-dependent lethal lipid peroxides (Dixon et al., 2012). p53 plays an important role in modulating ferroptotic responses by regulating the expression of its metabolic targets (Jiang et al., 2015). For example, recent studies have shown that ALOX12 is critical for p53-mediated ferroptosis (Chu et al., 2019). In addition, p53 induces ferroptosis partly through transcriptional activation of Glutaminase 2 (Jennis et al., 2016) and SAT1 (a polyamine catabolic enzyme) (Ou et al., 2016), and transcriptionally represses SLC7A11 (Jiang, et al., 2015). In addition, suppressor of cytokine signal transduction protein 1 (SOCS1) is required for p53-mediated expression of p53 target genes involved in ferroptosis. In this context, SOCS1 can reduce the expression of SLC7A11 to sensitize cells to ferroptosis (Saint-Germain et al., 2017). However, p53 behaves differently in a context dependent manner. While the basal p53 promotes ferroptosis, stress-induced p53 can inhibit ferroptosis (Tarangelo et al., 2018; Xie et al., 2017). For example, p53 inhibits ferroptosis by inhibiting dipeptidyl-peptidase-4 (DPP4) activity in human colorectal cancer cell lines (Xie, et al., 2017). Therefore, further studies would be needed to clarify the complex roles of p53 in ferroptosis.
p53 and Iron Metabolism
In addition to ferroptosis, p53 also modulates iron homeostasis. p53 expression is decreased upon the exposure to excessive levels of iron through heme-p53 interaction (Shen et al., 2014). Under iron-deprived conditions, HIF1α is activated to increase p53 protein stability and protein levels (An et al., 1998; Peyssonnaux et al., 2008; Peyssonnaux et al., 2007). In contrast, p53 is also found to be downregulated upon iron depletion via MDM2 (Dongiovanni et al., 2010). Therefore, the regulatory mechanisms of p53 by the iron concentration appear to be context dependent.
p53 can control the intracellular iron pool by modulating the expression of iron sensors. For example, p53 directly activates the expression of hepcidin, an iron-regulating hormone (Weizer-Stern et al., 2007). Another study suggests that p53 induces the expression of iron-sulfur cluster assembly proteins (ISCU) and protects cells from iron overload (Funauchi et al., 2015). p53 has been reported to modulate mitochondrial proteins that are involved in iron metabolism. For example, p53 mediates the expression of its target ferredoxin reductase (FDXR), and subsequently, modulates mitochondrial iron homeostasis through iron sulfur clusters (ISC) or heme synthesis (Sheftel et al., 2010).
p53 and Amino Acid Metabolism
Amino acid metabolism has extensive effects on tumors, and it has been revealed that p53 functions to protect cells from metabolic stress and promote cellular survival. Cancer cells rely on glutamine for cellular proliferation after glucose depletion through a process named glutaminolysis, by which glutamine is converted to the intermediates of the TCA cycle (Pavlova and Thompson, 2016). p53 activates the expression of Glutaminase 2 (GLS2), a key enzyme in glutamine-based cellular energy production under glucose-deprivation conditions to support cancer cell growth. Glutamate also limits intracellular and extracellular oxidative stress to promote cell survival (Suzuki et al., 2010).
Under the conditions when both glucose and glutamine become limited, aspartate metabolism becomes very important for cellular energy production. p53 is reported to transactivate Solute Carrier Family 1 Member 3 (SLC1A3), an aspartate/glutamate transporter, under glutamine starvation conditions (Tajan et al., 2018). p53 can also promote cellular survival by the induction of high affinity amino acid transporter Solute Carrier Family 1 Member 3 (SLC1A3) (Tajan, et al., 2018). Another important player in tumor cell survival and proliferation is serine. p53 promotes serine synthesis by glutathionine (GSH) synthesis, eventually leading to overall cell survival (Maddocks et al., 2013). In summary, p53 promotes cellular survival by promoting energy production from amino acids under the condition of glucose deprivation.
Concluding Remarks
Tumor suppressor gene p53 is not only essential in cell cycle arrest or apoptosis, but also participates in various physiological functions. Here we take a closer look at the complexity of p53 function in regulating cellular metabolism. These findings together suggest that p53 could regulate various aspects of cellular metabolism via regulating different target gene expression or protein-protein interactions in a cellular and environmental context dependent manner (Figure 1). The roles of wild-type p53 in tumor metabolism are complex, and sometimes, could conflict with its status as a tumor suppressor. For example, some roles of p53 in suppressing OXPHOS and inducing amino acid based energy production can promote cancer cell survival and proliferation (Kim, et al., 2019; Suzuki, et al., 2010, Tajan et al., 201). While the full-length p53 mutants are found to be overexpressed in more than half of human cancers and apparently gain new oncogenic properties (Zhu et al., 2020), many questions remain unanswered for their roles in cellular metabolism. Further advancements in single-cell analysis and multi-omics analyses will provide more in-depth understanding of p53-related regulatory mechanisms.
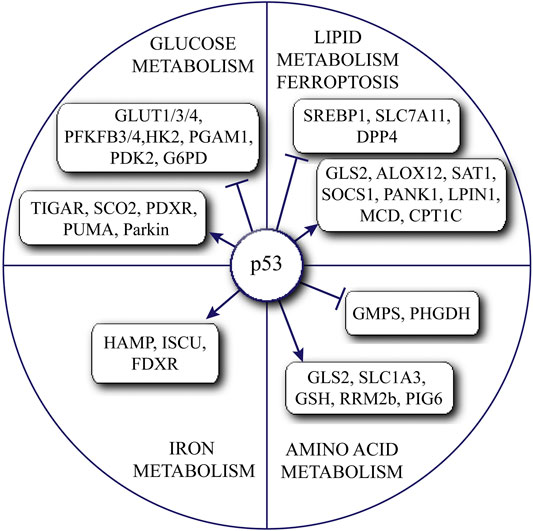
FIGURE 1. Summary of the complex roles of p53 in regulating various metabolic pathways.
Considering the unusual reliance of cancer cells on glycolysis, targeting tumor metabolic reprogramming has become a promising strategy for cancer treatment. In addition, the increased glycolysis contributes to higher levels of the acidic intermediates such as lactate and acidic tumor microenviroment, directly or indirectly suppress tumor immunity. Therefore, the activation of the roles of p53 in suppressing the metabolic reprogramming of cancer cells could become effective targeted therapy for human cancers. However, the development of such strategy requires attention to the complex and sometimes conflicting roles of p53 in cancer cells. The comprehensive understanding of various p53 regulated pathways will enable the precise activation of the p53-dependent pathways in suppressing tumor metabolism.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 81902506, 81930084), and the Key Research and Development Program of Guangdong Province (2019B020235003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
An, W. G., Kanekal, M., Simon, M. C., Maltepe, E., Blagosklonny, M. V., and Neckers, L. M. (1998). Stabilization of Wild-type P53 by Hypoxia-Inducible Factor 1α. Nature 392, 405–408. doi:10.1038/32925
Bathaie, S. Z., Ashrafi, M., Azizian, M., and Tamanoi, F. (2017). Mevalonate Pathway and Human Cancers. Curr. Mol. Pharmacol. 10, 77–85. doi:10.2174/1874467209666160112123205
Beloribi-Djefaflia, S., Vasseur, S., and Guillaumond, F. (2016). Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 5, e189. doi:10.1038/oncsis.2015.49
Bensaad, K., Tsuruta, A., Selak, M. A., Vidal, M. N. C., Nakano, K., Bartrons, R., et al. (2006). TIGAR, a P53-Inducible Regulator of Glycolysis and Apoptosis. Cell 126, 107–120. doi:10.1016/j.cell.2006.05.036
Chu, B., Kon, N., Chen, D., Li, T., Liu, T., Jiang, L., et al. (2019). ALOX12 Is Required for P53-Mediated Tumour Suppression through a Distinct Ferroptosis Pathway. Nat. Cel Biol 21, 579–591. doi:10.1038/s41556-019-0305-6
Contractor, T., and Harris, C. R. (2012). p53 Negatively Regulates Transcription of the Pyruvate Dehydrogenase Kinase Pdk2. Cancer Res. 72, 560–567. doi:10.1158/0008-5472.can-11-1215
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an Iron-dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Do, P. M., Varanasi, L., Fan, S., Li, C., Kubacka, I., Newman, V., et al. (2012). Mutant P53 Cooperates with ETS2 to Promote Etoposide Resistance. Genes Dev. 26, 830–845. doi:10.1101/gad.181685.111
Dongiovanni, P., Fracanzani, A. L., Cairo, G., Megazzini, C. P., Gatti, S., Rametta, R., et al. (2010). Iron-dependent Regulation of MDM2 Influences P53 Activity and Hepatic Carcinogenesis. Am. J. Pathol. 176, 1006–1017. doi:10.2353/ajpath.2010.090249
Franklin, D. A., He, Y., Leslie, P. L., Tikunov, A. P., Fenger, N., Macdonald, J. M., et al. (2016). p53 Coordinates DNA Repair with Nucleotide Synthesis by Suppressing PFKFB3 Expression and Promoting the Pentose Phosphate Pathway. Sci. Rep. 6, 38067. doi:10.1038/srep38067
Freed-Pastor, W. A., Mizuno, H., Zhao, X., Langerød, A., Moon, S.-H., Rodriguez-Barrueco, R., et al. (2012). Mutant P53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 148, 244–258. doi:10.1016/j.cell.2011.12.017
Fu, X., Wu, S., Li, B., Xu, Y., and Liu, J. (2020). Functions of P53 in Pluripotent Stem Cells. Protein Cell 11, 71–78. doi:10.1007/s13238-019-00665-x
Funauchi, Y., Tanikawa, C., Yi Lo, P. H., Mori, J., Daigo, Y., Takano, A., et al. (2015). Regulation of Iron Homeostasis by the P53-ISCU Pathway. Sci. Rep. 5, 16497. doi:10.1038/srep16497
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of Cancer: the Next Generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Jennis, M., Kung, C.-P., Basu, S., Budina-Kolomets, A., Leu, J. I.-J., Khaku, S., et al. (2016). An African-specific Polymorphism in the TP53 Gene Impairs P53 Tumor Suppressor Function in a Mouse Model. Genes Dev. 30, 918–930. doi:10.1101/gad.275891.115
Jiang, L., Kon, N., Li, T., Wang, S.-J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 520, 57–62. doi:10.1038/nature14344
Jiang, P., Du, W., Wang, X., Mancuso, A., Gao, X., Wu, M., et al. (2011). p53 Regulates Biosynthesis through Direct Inactivation of Glucose-6-Phosphate Dehydrogenase. Nat. Cel Biol 13, 310–316. doi:10.1038/ncb2172
Kawauchi, K., Araki, K., Tobiume, K., and Tanaka, N. (2008). p53 Regulates Glucose Metabolism through an IKK-NF-Κb Pathway and Inhibits Cell Transformation. Nat. Cel Biol 10, 611–618. doi:10.1038/ncb1724
Kim, J., Yu, L., Chen, W., Xu, Y., Wu, M., Todorova, D., et al. (2019). Wild-Type P53 Promotes Cancer Metabolic Switch by Inducing PUMA-dependent Suppression of Oxidative Phosphorylation. Cancer Cell 35, 191–203. doi:10.1016/j.ccell.2018.12.012
Kondoh, H., Lleonart, M. E., Gil, J., Wang, J., Degan, P., Peters, G., et al. (2005). Glycolytic Enzymes Can Modulate Cellular Life Span. Cancer Res. 65, 177–185.
Kuhajda, F. P., Jenner, K., Wood, F. D., Hennigar, R. A., Jacobs, L. B., Dick, J. D., et al. (1994). Fatty Acid Synthesis: a Potential Selective Target for Antineoplastic Therapy. Proc. Natl. Acad. Sci. 91, 6379–6383. doi:10.1073/pnas.91.14.6379
Lacroix, M., Riscal, R., Arena, G., Linares, L. K., and Le Cam, L. (2020). Metabolic Functions of the Tumor Suppressor P53: Implications in normal Physiology, Metabolic Disorders, and Cancer. Mol. Metab. 33, 2–22. doi:10.1016/j.molmet.2019.10.002
Levine, A. J. (2020). p53: 800 Million Years of Evolution and 40 Years of Discovery. Nat. Rev. Cancer 20, 471–480. doi:10.1038/s41568-020-0262-1
Li, T., Kon, N., Jiang, L., Tan, M., Ludwig, T., Zhao, Y., et al. (2012). Tumor Suppression in the Absence of P53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell 149, 1269–1283. doi:10.1016/j.cell.2012.04.026
Liu, G., and Chen, X. (2002). The Ferredoxin Reductase Gene Is Regulated by the P53 Family and Sensitizes Cells to Oxidative Stress-Induced Apoptosis. Oncogene 21, 7195–7204. doi:10.1038/sj.onc.1205862
Liu, J., Liu, Z.-X., Wu, Q.-N., Lu, Y.-X., Wong, C.-W., Miao, L., et al. (2020). Long Noncoding RNA AGPG Regulates PFKFB3-Mediated Tumor Glycolytic Reprogramming. Nat. Commun. 11, 1507. doi:10.1038/s41467-020-15112-3
Maddocks, O. D. K., Berkers, C. R., Mason, S. M., Zheng, L., Blyth, K., Gottlieb, E., et al. (2013). Serine Starvation Induces Stress and P53-dependent Metabolic Remodelling in Cancer Cells. Nature 493, 542–546. doi:10.1038/nature11743
Matoba, S., Kang, J.-G., Patino, W. D., Wragg, A., Boehm, M., Gavrilova, O., et al. (2006). p53 Regulates Mitochondrial Respiration. Science 312, 1650–1653. doi:10.1126/science.1126863
Olivier, M., Hollstein, M., and Hainaut, P. (2010). TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harbor Perspect. Biol. 2, a001008. doi:10.1101/cshperspect.a001008
Ou, Y., Wang, S.-J., Li, D., Chu, B., and Gu, W. (2016). Activation of SAT1 Engages Polyamine Metabolism with P53-Mediated Ferroptotic Responses. Proc. Natl. Acad. Sci. USA 113, E6806–E6812. doi:10.1073/pnas.1607152113
Pavlova, N. N., and Thompson, C. B. (2016). The Emerging Hallmarks of Cancer Metabolism. Cel Metab. 23, 27–47. doi:10.1016/j.cmet.2015.12.006
Peyssonnaux, C., Nizet, V., and Johnson, R. S. (2008). Role of the Hypoxia Inducible Factors HIF in Iron Metabolism. Cell Cycle 7, 28–32. doi:10.4161/cc.7.1.5145
Peyssonnaux, C., Zinkernagel, A. S., Schuepbach, R. A., Rankin, E., Vaulont, S., Haase, V. H., et al. (2007). Regulation of Iron Homeostasis by the Hypoxia-Inducible Transcription Factors (HIFs). J. Clin. Invest. 117, 1926–1932. doi:10.1172/jci31370
Ribas, V., García-Ruiz, C., and Fernández-Checa, J. C. (2016). Mitochondria, Cholesterol and Cancer Cell Metabolism. Clin. Transl Med. 5, 22. doi:10.1186/s40169-016-0106-5
Roongta, U. V., Pabalan, J. G., Wang, X., Ryseck, R.-P., Fargnoli, J., Henley, B. J., et al. (2011). Cancer Cell Dependence on Unsaturated Fatty Acids Implicates Stearoyl-CoA Desaturase as a Target for Cancer Therapy. Mol. Cancer Res. 9, 1551–1561. doi:10.1158/1541-7786.mcr-11-0126
Ros, S., Flöter, J., Kaymak, I., Da Costa, C., Houddane, A., Dubuis, S., et al. (2017). 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 Is Essential for P53-Null Cancer Cells. Oncogene 36, 3287–3299. doi:10.1038/onc.2016.477
Saint-Germain, E., Mignacca, L., Vernier, M., Bobbala, D., Ilangumaran, S., and Ferbeyre, G. (2017). SOCS1 Regulates Senescence and Ferroptosis by Modulating the Expression of P53 Target Genes. Aging 9, 2137–2162. doi:10.18632/aging.101306
Schwartzenberg-Bar-Yoseph, F., Armoni, M., and Karnieli, E. (2004). The Tumor Suppressor P53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Res. 64, 2627–2633. doi:10.1158/0008-5472.can-03-0846
Sheftel, A. D., Stehling, O., Pierik, A. J., Elsasser, H.-P., Muhlenhoff, U., Webert, H., et al. (2010). Humans Possess Two Mitochondrial Ferredoxins, Fdx1 and Fdx2, with Distinct Roles in Steroidogenesis, Heme, and Fe/S Cluster Biosynthesis. Proc. Natl. Acad. Sci. 107, 11775–11780. doi:10.1073/pnas.1004250107
Shen, J., Sheng, X., Chang, Z., Wu, Q., Wang, S., Xuan, Z., et al. (2014). Iron Metabolism Regulates P53 Signaling through Direct Heme-P53 Interaction and Modulation of P53 Localization, Stability, and Function. Cel Rep. 7, 180–193. doi:10.1016/j.celrep.2014.02.042
Suzuki, S., Tanaka, T., Poyurovsky, M. V., Nagano, H., Mayama, T., Ohkubo, S., et al. (2010). Phosphate-activated Glutaminase (GLS2), a P53-Inducible Regulator of Glutamine Metabolism and Reactive Oxygen Species. Proc. Natl. Acad. Sci. 107, 7461–7466. doi:10.1073/pnas.1002459107
Tajan, M., Hock, A. K., Blagih, J., Robertson, N. A., Labuschagne, C. F., Kruiswijk, F., et al. (2018). A Role for P53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cel Metab. 28, 721–736. e726. doi:10.1016/j.cmet.2018.07.005
Tarangelo, A., Magtanong, L., Bieging-Rolett, K. T., Li, Y., Ye, J., Attardi, L. D., et al. (2018). p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cel Rep. 22, 569–575. doi:10.1016/j.celrep.2017.12.077
Vousden, K. H., and Prives, C. (2009). Blinded by the Light: The Growing Complexity of P53. Cell 137, 413–431. doi:10.1016/j.cell.2009.04.037
Walerych, D., Lisek, K., Sommaggio, R., Piazza, S., Ciani, Y., Dalla, E., et al. (2016). Proteasome Machinery Is Instrumental in a Common Gain-Of-Function Program of the P53 Missense Mutants in Cancer. Nat. Cel Biol 18, 897–909. doi:10.1038/ncb3380
Wang, L., Xiong, H., Wu, F., Zhang, Y., Wang, J., Zhao, L., et al. (2014). Hexokinase 2-mediated Warburg Effect Is Required for PTEN- and P53-Deficiency-Driven Prostate Cancer Growth. Cel Rep. 8, 1461–1474. doi:10.1016/j.celrep.2014.07.053
Wang, X., Zhao, X., Gao, X., Mei, Y., and Wu, M. (2013). A New Role of P53 in Regulating Lipid Metabolism. J. Mol. Cel Biol 5, 147–150. doi:10.1093/jmcb/mjs064
Warburg, O., Wind, F., and Negelein, E. (1927). The Metabolism of Tumors in the Body. J. Gen. Physiol. 8, 519–530. doi:10.1085/jgp.8.6.519
Weizer-Stern, O., Adamsky, K., Margalit, O., Ashur-Fabian, O., Givol, D., Amariglio, N., et al. (2007). Hepcidin, a Key Regulator of Iron Metabolism, Is Transcriptionally Activated by P53. Br. J. Haematol. 138, 253–262. doi:10.1111/j.1365-2141.2007.06638.x
Xie, Y., Zhu, S., Song, X., Sun, X., Fan, Y., Liu, J., et al. (2017). The Tumor Suppressor P53 Limits Ferroptosis by Blocking DPP4 Activity. Cel Rep. 20, 1692–1704. doi:10.1016/j.celrep.2017.07.055
Xu, J., Reumers, J., Couceiro, J. R., De Smet, F., Gallardo, R., Rudyak, S., et al. (2011). Gain of Function of Mutant P53 by Coaggregation with Multiple Tumor Suppressors. Nat. Chem. Biol. 7, 285–295. doi:10.1038/nchembio.546
Zhan, Y., Ginanni, N., Tota, M. R., Wu, M., Bays, N. W., Richon, V. M., et al. (2008). Control of Cell Growth and Survival by Enzymes of the Fatty Acid Synthesis Pathway in HCT-116 colon Cancer Cells. Clin. Cancer Res. 14, 5735–5742. doi:10.1158/1078-0432.ccr-07-5074
Zhou, G., Wang, J., Zhao, M., Xie, T.-X., Tanaka, N., Sano, D., et al. (2014). Gain-of-function Mutant P53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Mol. Cel 54, 960–974. doi:10.1016/j.molcel.2014.04.024
Keywords: p53, glucose metabolism, lipid metabolism, ferroptosis, amino acid metabolism, iron metabolism
Citation: Yu L, Wu M, Zhu G and Xu Y (2022) Emerging Roles of the Tumor Suppressor p53 in Metabolism. Front. Cell Dev. Biol. 9:762742. doi: 10.3389/fcell.2021.762742
Received: 22 August 2021; Accepted: 27 December 2021;
Published: 18 January 2022.
Edited by:
Ji Hoon Jung, Kyung Hee University, South KoreaReviewed by:
Chia-Hsin (Lori) Chan, Roswell Park Comprehensive Cancer Center, United StatesGabriella D’Orazi, G. D’Annunzio University of Chieti-Pescara, Italy
Copyright © 2022 Yu, Wu, Zhu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaoyang Zhu, emh1Z3kxOTkwQDE2My5jb20=; Yang Xu, eHV5YW5nMjAyMEB6anUuZWR1LmNu