 Tsung-Lin Cheng1,2
Tsung-Lin Cheng1,2 Chung-Hwan Chen2,3,4,5
Chung-Hwan Chen2,3,4,5 Meng-Hsing Wu6
Meng-Hsing Wu6 Chao-Han Lai7,8
Chao-Han Lai7,8 Ko-Hung Lee9Sheng-Hsiang Lin10Ai-Li Shiau11Chao-Liang Wu7
Ko-Hung Lee9Sheng-Hsiang Lin10Ai-Li Shiau11Chao-Liang Wu7 Lin Kang6*
Lin Kang6*- 1Department of Physiology, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
- 2Regenerative Medicine and Cell Therapy Research Center, Kaohsiung Medical University, Kaohsiung, Taiwan
- 3Orthopaedic Research Center, Kaohsiung Medical University, Kaohsiung, Taiwan
- 4Department of Orthopedics, Kaohsiung Municipal Ta-Tung Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
- 5Division of Adult Reconstruction Surgery, Department of Orthopedics, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
- 6Department of Obstetrics and Gynecology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan
- 7Department of Biochemistry and Molecular Biology, College of Medicine, National Cheng Kung University, Tainan, Taiwan
- 8Department of Surgery, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan
- 9An-an Women and Children Clinic, Tainan, Taiwan
- 10College of Medicine, Institute of Clinical Medicine, National Cheng Kung University, Tainan, Taiwan
- 11Department of Microbiology and Immunology, College of Medicine, National Cheng Kung University, Tainan, Taiwan
Fibrinogen-like 1 (FGL1) is involved in liver injury and liver regeneration, but its role in placenta and preeclampsia (PE) remains unclear. We assessed FGL1 expression in serum and placenta from L-NAME-induced PE-like mouse and in women with (n = 38) and without (n = 42) PE. For the mouse study, pregnant C57Bl/6 mouse (n = 6/group) were subcutaneously administered L-NAME with or without FGL1 once daily starting on days 7–14 of pregnancy and were sacrificed on gestational day (GD) 20. Maternal body weight, blood pressure, and urinary protein were assessed during GDs 8–20. The weight and length of the placenta and fetus were assessed. The placental structure was evaluated using hematoxylin staining. In the human study, the sera of the pregnant women during the late trimester were assessed with enzyme-linked immunosorbent assays (ELISAs). FGL1 expression in human trophoblast cell lines under L-NAME stimulation was measured using Western blotting and immunofluorescence staining. The detected FGL1 protein levels in serum and placenta were both significantly upregulated in patients and mouse with PE compared with those in the non-PE groups. FGL1 treatment decreased maternal hypertension and proteinuria, decreased fetal weight in mouse with PE, downregulated proinflammatory cytokine (interleukin-1b and interleukin-6) levels, and maintained the balance between antiangiogenic (fms-like tyrosine kinase-1) and proangiogenic (placental growth factor) substances in the placenta. L-NAME-upregulated FGL1 expression was inhibited following overexpression of FoxO3a. In summary, FoxO3a reduction is a potential pathophysiological mechanism leading to upregulated placental FGL1 expression that may play a pivotal role in preventing PE progression.
Introduction
Preeclampsia (PE) is one of the major issues in maternal-fetal medicine. According to the American College of Obstetricians and Gynecologists (ACOG) practice bulletin, the criteria for the diagnosis of PE are new-onset hypertension (systolic/diastolic pressure ≥140/90 mmHg) and proteinuria (a 24-h urine specimen with >300 mg protein) after gestational week 20 (Author Anonymous, 2019). PE occurs in approximately 4.6% of all pregnancies and causes substantial maternal and fetal morbidity and mortality worldwide (Abalos et al., 2013; Jeyabalan, 2013), and it affects approximately 1–2% of pregnancies in Taiwan (You et al., 2018). Several pathophysiological abnormalities, such as abnormal trophoblast differentiation and invasion, placental and endothelial dysfunction, immune maladaptation, and an exaggerated systemic inflammatory response, have been proposed to explain the pathogenesis of PE (Sibai et al., 2005; Sibai, 2008). However, the underlying pathophysiology remains unclear.
PE can occur in the presence of trophoblast tissue with hydatidiform moles even in the absence of a fetus and abate with the delivery of the placenta (Lain and Roberts, 2002). This finding suggests that the placenta should be the root cause of PE, and analysis of the placenta should help to elucidate the pathophysiology of PE. Clinical and experimental findings indicate that reduced placental perfusion is a unique feature that may result in PE, and PE is commonly found with diseases involving microvascular disorders (e.g., hypertension, diabetes mellitus) or with large placentas (e.g., hydatidiform moles, multiple gestations) (Roberts and Gammill, 2005). Fibrinogen-like protein 1 (FGL1) is a protein that contains the fibrinogen-related domain in its C-terminal portion (Hara et al., 2001). Proteins structurally related to FGL1 (angiopoietins, fibrinogen, tenascins) have been implicated in multiple cellular processes, including angiogenesis, proliferation, apoptosis and extracellular matrix modulation (Procopio et al., 1999; Kim et al., 2000; Sahni and Francis, 2000; El-Karef et al., 2007), suggesting a potential role for placental FGL1 in these processes during PE progression.
FGL1, also known as HFREP1 or hepassocin, was initially identified as an enriched transcript in hepatocellular carcinoma and rodent liver regeneration (Yamamoto et al., 1993; Hara et al., 2001). The forkhead box class O (FoxO) family comprises ubiquitously expressed transcription factors, and a predicted FoxO3 binding site was found in the FGL1 gene promoter (Liu et al., 2018). Increased plasma FGL1 levels disrupt insulin signaling to induce insulin resistance and type 2 diabetes through an extracellular signal-regulated kinase 1/2-dependent pathway. Accordingly, FGL1 knockdown improved insulin resistance in both high-fat-diet-fed mouse and ob/ob mouse (Wu et al., 2016). Our previous study indicated that FGL-1 was mainly expressed by placental trophoblasts and functions to increase trophoblast proliferation (Kang et al., 2020). However, limited data are available regarding the role and regulatory mechanisms of FGL1 in PE and PE-associated disorders.
Treating pregnant rodents with NG-nitroarginine methyl ester (L-NAME), a nitric oxide synthase (NOS) inhibitor, is an accepted method of inducing PE, and the experimental outcomes are similar to those in human PE, including hypertension, proteinuria, renal damage, and intrauterine growth restriction (Yallampalli and Garfield, 1993; Molnar et al., 1994; Buhimschi et al., 1995; Tsukimori et al., 2008). In this study, trophoblast cell lines, an L-NAME-induced PE-like mouse model, and human placental samples were used to evaluate the role of FGL1 in the progression of PE and the possible related regulatory mechanisms. Because FGL1 has been implicated in liver injury as a mitogenic growth factor (Cao et al., 2011), reduces the expression of proapoptotic factors (Li et al., 2010), and facilitates damaged liver tissue regeneration (Yan et al., 2002), we hypothesized that upregulated FGL1 expression may function as a complementary modulator to reduce the progression of PE and verified this hypothesis by treating L-NAME-induced PE-like mouse with recombinant FGL1.
Materials and Methods
Materials
Mouse anti-human FGL1 monoclonal antibody, goat anti-mouse FGL1 polyclonal antibody, purified recombinant human FGL1 (the similarity of FGL1 between humans and mouse is greater than 80%), and an FGL1 enzyme-linked immunosorbent assay (ELISA) kit were obtained from R&D Systems (Minneapolis, MN, United States). Antibodies recognizing actin, cytokeratin 7 (CK7), IL-6, and PlGF were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, United States), and 4′,6-diamidino-2-phenylindole hydrochloride (DAPI) was purchased from ATT Bioquest (CA, United States). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH, AM4300) was purchased from Thermo Fisher Scientific (Waltham, MA, United States). Rabbit anti-human FoxO3a monoclonal antibody was purchased from Cell Signaling Technology (Danvers, MA, United States). The FGL1 expression plasmid (pCMV6- FoxO3a) was purchased from Origene (Rockville, MD). All remaining reagents, unless otherwise specified, were obtained from Invitrogen (Carlsbad, CA, United States).
Human Study
From February 3, 2017, to July 31, 2020, in the Department of Obstetrics and Gynecology of NCKUH, we prospectively collected 38 cases of PE meeting the ACOG criteria (2019) and 42 healthy controls. PE was defined as the development of hypertension in a previously normotensive pregnant woman after 20 weeks of gestation, accompanied by new-onset proteinuria. Hypertension was defined as systolic blood pressure ≥140 mmHg or diastolic blood pressure ≥90 mmHg on at least two occasions and 4–6 h apart. Proteinuria was defined as ≥300 mg protein excretion in the 24-h urine collection or a protein concentration of ≥300 mg/L in urine (≥1 + on dipstick). Plasma samples of all participants were obtained at the time of admission for delivery. Human placentas used in this study were obtained at the end of gestation, immediately after vaginal delivery or during cesarean section. The tissues were harvested by penetrating the center of the placenta (umbilical cord insertion) and snap-frozen in liquid nitrogen, and others were fixed-embedded in paraffin for immunohistochemistry. The decidua regions were performed to RT-PCR and Western blotting. During blood pressure measurement, patients were resting in the supine position in a quiet environment, and measurements were obtained in a fasting state between 08:00 and 10:00 AM. None of the healthy controls had clinical signs of PE or other medical or pregnancy complications. The exclusion criteria were as follows: multiple pregnancies, premature rupture of membranes, chorioamnionitis, chronic hypertension, diabetes mellitus, autoimmune disorders, and fetal abnormalities.
Cell Culture and Plasmid Transfection
The human trophoblast cell lines 3A-subE (ATCC CRL-1584) and HTR-8/SVneo (ATCC CRL-3271) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, United States), cultured in growth media [high-glucose DMEM containing 10% heat-inactivated fetal bovine serum (FBS), 1% antibiotics (penicillin and streptomycin; Invitrogen), and L-glutamine (292 mg/L)] and maintained at 37°C with 5% CO2. For transient transfections, Lipofectamine 2000 reagent (Invitrogen) was used according to the manufacturer’s instructions. The cells were collected for further experiments after transfection with plasmids (2 mg) for 48 h. Cells (2 × 105 cells/well) were seeded in 6-well plates, and after incubation with the indicated dose of L-NAME (0, 100, 1,000 μM) for 24 h, the cells were further analysed.
Experimental Animals and the Mouse Model of PE
Adult pregnant mouse (body weight: 21–23 g), the C57BL/6 inbred strains, were purchased from the BioLASCO Animal Center (Taipei, Taiwan, ROC) and received in our animal facilities on day 7 of pregnancy (day 0 = day of positive sperm smear). All animals were given free access to food and water. For the PE-like mouse model, L-NAME (75 mg/kg/day) was subcutaneously administered with or without recombinant FGL1 (1 μg/kg/day) on days 8–14 of pregnancy (FGL1 and non-FGL1 groups, respectively) based on a previous report (Motta et al., 2015). Both the control and treatment groups of mouse were age-matched, and saline was administered to the control groups. Blood pressure was noninvasively measured during pregnancy on gestational days (GDs) 8–20. The mouse were placed in metabolic cages for urine collection on GDs 12–15 and 15–18. The entire placenta without yolk sac and uterine layers were collected at GDs 19 for RNA isolation and Western blotting. The animal care and experimental procedures conformed to the ARRIVE guidelines (Kilkenny et al., 2010) and were approved by the Institutional Animal Care and User Committee (150213) at NCKU, Tainan, Taiwan.
RNA Isolation and Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction
Total RNA from mouse placentas was extracted using the RBC Total RNA Kit (Life Biomedical Limited) according to the manufacturer’s instructions. Next, 2 μg of extracted RNA was reverse-transcribed to cDNA using MuLV reverse transcriptase, 1 mM dNTPs, and 0.5 mg/ml oligo (dT12–18) as previously described (Cheng et al., 2009). cDNA was amplified through qRT-PCR using Fast Quant Green Master Mix with Low ROX (Protech Technology Enterprise, WI, United States) with 0.5 μM primers according to the manufacturer’s protocol. The sequences of the primers are as follows: FGL1 (F: 5′-AGCTGCCTGTGTTATTTCCTCTCA-3′; R: 5′-CCAGGGAGCCATTTTATTTATCCAA-3′) (Demchev et al., 2013), interleukin (IL)-1b (F: 5′-CACCTCTCAAGCAGAGCACAG -3′; R: 5′-GGGTTCCATGGTGAAGTCAAC-3′), IL-6 (F: 5′-TCCTACCCCAACTTCCAATGCTC-3′; R: 5′-TTGGATGGTCTTGGTCCTTAGCC-3′) (Iwashita et al., 2014), placenta growth factor (PlGF) (F: 5′-TCTGCTGGGAACAACTCAACA-3′; R: 5′-GTGAGACACCTCATCAGGGTA-3′) (Fu et al., 2020), FMS-like tyrosine kinase 1 (Flt-1) (F: 5′-AGGAGATGCTCCTCCCAAA-3′; R: 5′-GTGCAGGGATCCTCCAAAT-3′) (Zhou et al., 2007), and GAPDH (F: 5′- TGCACCACCAACTGCTTAGC-3′; R: 5′- GGCATGGACTGTGGTCATGAG-3′) (Xie et al., 2012). GAPDH gene expression was used as an internal control, and the
Histochemical Analysis
Histochemical analysis of 5-μm-thick sections was performed as described previously (Kang et al., 2020). Slides were counterstained using hematoxylin and eosin (Gill No. 1, Sigma-Aldrich), dehydrated in an ascending series of ethanol, cleared in xylene, and mounted with Cytoseal (Thermo Fisher Scientific). The center of the mouse placenta disc was marked with ink and sections were cut through the center. Photographs were obtained using an Olympus DP72 camera mounted on a BX61 Olympus microscope. The images were quantified using ImageJ software (National Institutes of Health) to analyze the spongiotrophoblast area.
Immunohistochemistry
Cells were fixed in 2% (w/v) formaldehyde and blocked with 10% (v/v) goat serum. Sections were dewaxed and rehydrated, followed by antigen retrieval. Peroxidase method: Sections exposed to the primary antibody for 30 min at room temperature, washed in phosphate-buffered saline (PBS), incubated for 30 min with rabbit-anti-mouse IgG conjugated with horseradish-peroxidase, then staining was achieved by incubating with 3,3′-diaminobenzidine (DAB; Sigma). Fluorescence method: One percent (w/v) Sudan black in 70% (v/v) ethanol was used for 1 min to block autofluorescence. Goat serum (10%, v/v) was applied for 1 h at room temperature for nonimmune blockade, followed by overnight incubation with mouse FGL1 polyclonal antibody (Sigma, diluted 1:100) at 4°C. Goat anti-mouse secondary antibody conjugated with Alexa Fluor-488 (Invitrogen) was applied at a 1:200 dilution with DAPI nuclear counterstaining for 1 h at room temperature. The sections were mounted using Shandon Immu-Mount (Thermo Fisher Scientific). For the negative control, the primary antibody was omitted to control for autofluorescence. Slides were stored at 4°C in the dark until photographed. ImageJ was used to assess the fluorescent images to quantify the mean fluorescence intensity or area.
ELISA
FGL1 (pg/ml) was measured in diluted human and mouse serum using an ELISA kit (R&D Systems) per the manufacturer’s protocol.
Blood Pressure Measurement
Systolic and diastolic blood pressures were measured daily in conscious mouse by tail-cuff plethysmography using a BP-2000 blood pressure analysis system (Visitech Systems, Apex, NC, United States) in animals that had been prewarmed in a metal chamber maintained at 30°C. Each day, values obtained from three consecutive measurements were averaged. The mean arterial pressure [MAP; (2 × diastolic + systolic)/3] was calculated as previously described (Domanski et al., 1999).
Total Urinary Proteins
Urinary protein concentrations were measured using Folin’s phenol reagent method (Lowry et al., 1951). Urine samples and bovine serum albumin standards were assayed in triplicate and read at 650 nm with an NIR filter in a spectrophotometer (model 6/20, Coleman). Sample urinary protein levels (mg/ml) were extrapolated from a standard curve derived from bovine serum albumin standards.
Western Blot Analysis
Tissues or cells were homogenized and lysed with a buffer containing 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, and a protease inhibitor mixture (PMSF, aprotinin, and sodium orthovanadate). Total protein lysate (50 μg per lane) was separated on SDS-PAGE gels (10% running and 4% stacking), transferred to PVDF membranes and analyzed using the designated primary antibodies (1:1000 dilution). After the membranes were probed with an appropriate secondary antibody (1:2000 dilution), signals were detected using an enhanced chemiluminescence reagent (Amersham Pharmacia Biotech, United Kingdom) under a LAS3000 imaging system (Fujifilm, Tokyo, Japan). The band intensity was quantified using ImageJ software.
Cell Proliferation Assay
HTR-8/SVneo cells were seeded in 96-well plates (5 × 103 cells/well) and cultured in DMEM containing 2% FBS for 12 h. Then, recombinant FGL1 (0, 50, 100, 200 ng/ml) was added to the culture medium, and the cells were cultured for another 48 h. Cell viability was subsequently analyzed by Cell Counting Kit-8 (CCK-8) colorimetric assays (Dojindo Molecular Technologies, Inc.). After treatment, 10 μl of CCK-8 was added to each well, and the incubation was continued at 37°C for 2 h in the dark. Then, the absorbance value at 450 nm was determined using a microplate reader (SpectraMAX 340; Molecular Devices, Sunnyvale, CA, United States).
Statistical Analysis
All cellular experiments were performed using triplicate samples and repeated at least three times. Statistical analyses were conducted using Prism 7.0 (GraphPad Software). Data are expressed as the mean ± SEM. The D’Agostino-Pearson normality test was used to assess the normality of the distribution. Student’s t test was used for comparisons between the two groups. One-way analysis of variance (ANOVA), followed by post hoc analysis (Tukey’s test), was used for comparisons among multiple groups. For comparisons among multiple groups and different time points, two-way ANOVA was used. p < 0.05 was considered statistically significant.
Results
FGL1 Expression was Upregulated in Both Mouse With Experimental PE and Human Patients
First, FGL1 gene and protein expression levels in the placentas from L-NAME-induced PE-like mouse were measured to determine the correlation between FGL1 and PE. qPCR, Western blotting, and immunofluorescence staining indicated that the FGL1 mRNA and protein expression levels in the placentas of the mouse with PE were significantly higher than those in the normal placentas (mRNA: 1.71 ± 0.07 vs. 1.0 ± 0.04-fold, p = 0.0001; Figure 1A; protein: 2.28 ± 0.32 vs. 1.01 ± 0.04-fold, p = 0.03; Figure 1B; immunofluorescence intensity: 70.94 ± 5.88 vs. 43.86 ± 4.18 a.u., p = 0.0038; Figure 1C). FGL1 is a secreted protein; ELISA revealed that the FGL1 concentration was higher in the serum of the mouse with PE than in that of the control mouse (355.8 ± 41.69 vs. 203.8 ± 33.25 pg/ml, p = 0.0172; Figure 1D). For the analysis of FGL1 levels in humans, pregnant women with PE and age-matched healthy pregnant women without PE (normotensive) were enrolled. Clinical definitions and exclusion criteria are described in the Methods section. Clinical characteristics of study subjects are shown in Table 1. As expected, patients with PE had a higher blood pressure and percent proteinuria but reduced placental weight, birth weight, and 5-min APGAR score than the healthy subjects. In our study population, the PE group had a significantly higher BMI and lower gestational age of sample collection. Trophoblast FGL1 expression in human placental sections (Figure 1E) and placental FGL1 protein levels (Figure 1F) were both significantly higher in the women with PE than in the controls. Similar results were obtained for the serum FGL1 concentration (196 ± 24.25 pg/ml, n = 38 vs. 120.4 ± 15.69 pg/ml, n = 44, p = 0.0088; Figure 1G). Taken together, the results suggest that the placenta trophoblasts in decidua should be one of the sources of FGL-1 production in both mice and humans and FGL1 upregulation correlates with PE.
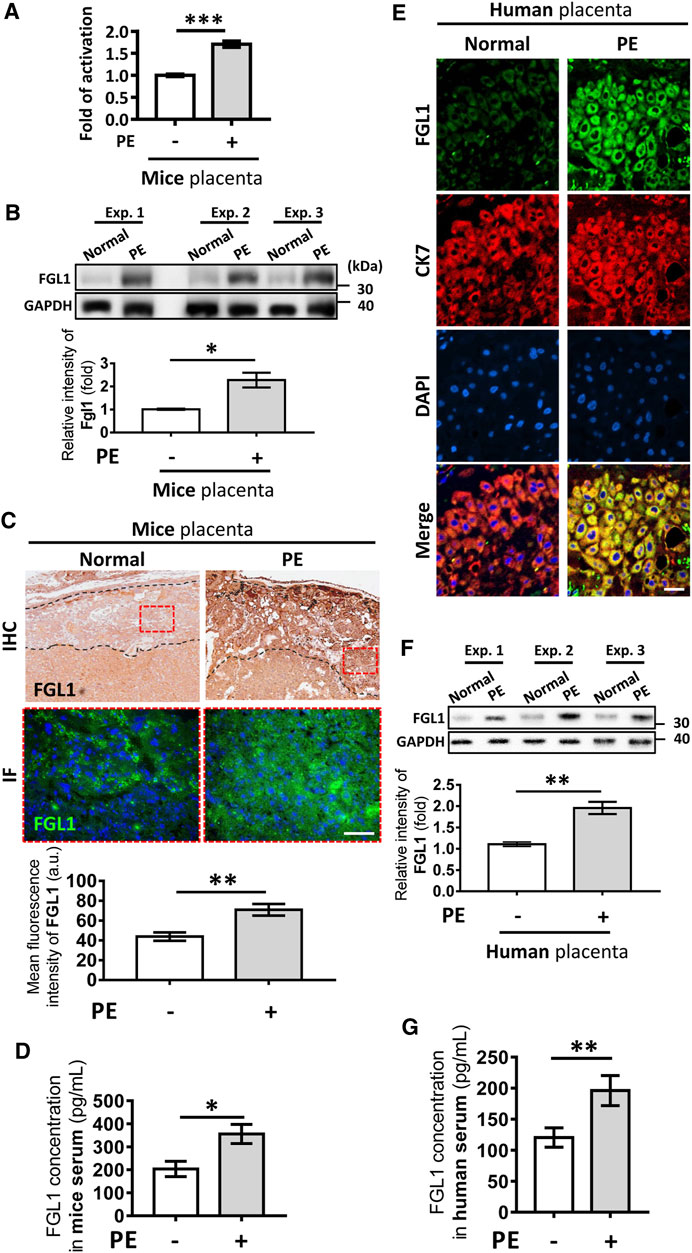
FIGURE 1. Expression of fibrinogen-like 1 (FGL1) in NG-nitroarginine methyl ester (L-NAME)-induced mouse with preeclampsia (PE) and human placenta and serum. FGL1 mRNA expression levels (A) and FGL1 protein expression levels (B) in the placenta of the mouse with PE and the controls were evaluated using RT-PCR and Western blotting, respectively. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (C) FGL1 protein expression in mouse placental sections was assessed using immunohistochemistry (IHC) and immunofluorescence (IF) staining. The dotted black area indicated the decidua; the dotted red rectangle indicated the picture area of IF. Brown and green indicates FGL1; blue indicates cell nuclei. Scale bar, 200 μm. Photographs were obtained from the placenta junctional zone. (D) Serum FGL1 concentration was analyzed in the PE and non-PE groups using enzyme-linked immunosorbent assays (ELISAs), n = 6/group. (E) Human placental FGL1 expression in the decidua was evaluated using immunofluorescence staining. Keratin-7 (CK7) is a trophoblast cell marker, and 4′,6-diamidino-2-phenylindole (DAPI) is an ideal nuclear counterstain. Scale bar, 10 μm. (F) Quantified protein expression level of FGL1 in human placenta. The experiment was repeated at least three times. (G) Serum FGL1 concentration in pregnant women with PE (n = 38) and those without PE (n = 42). *p < 0.05; **p < 0.01; ***p < 0.001 (Student’s t test).
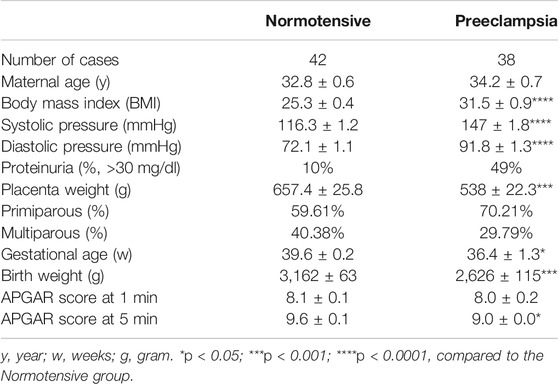
TABLE 1. Clinical characteristics of study subjects with and those without Preeclampsia.
Placental Trophoblasts are One of the Sources of FGL1 Under L-NAME Stimulation, and FGL1 Contributes to Promoting Trophoblast Proliferation
Our previous study indicated that FGL1 can be expressed in human trophoblasts (Kang et al., 2020). Here, we investigated whether L-NAME-enhanced placental FGL1 expression was correlated with trophoblasts. Two human trophoblast cell lines, 3A-sub E and HTR-8/SVneo, were stimulated with L-NAME at the indicated dose. As shown in Figure 2, Western blotting and immunofluorescence staining both indicated that FGL1 expression was dose-dependently upregulated under L-NAME stimulation, suggesting that L-NAME-induced progression of PE may also promote FGL1 expression in trophoblasts. We further found that exogenous FGL1 increased trophoblast proliferation (Figure 2E).
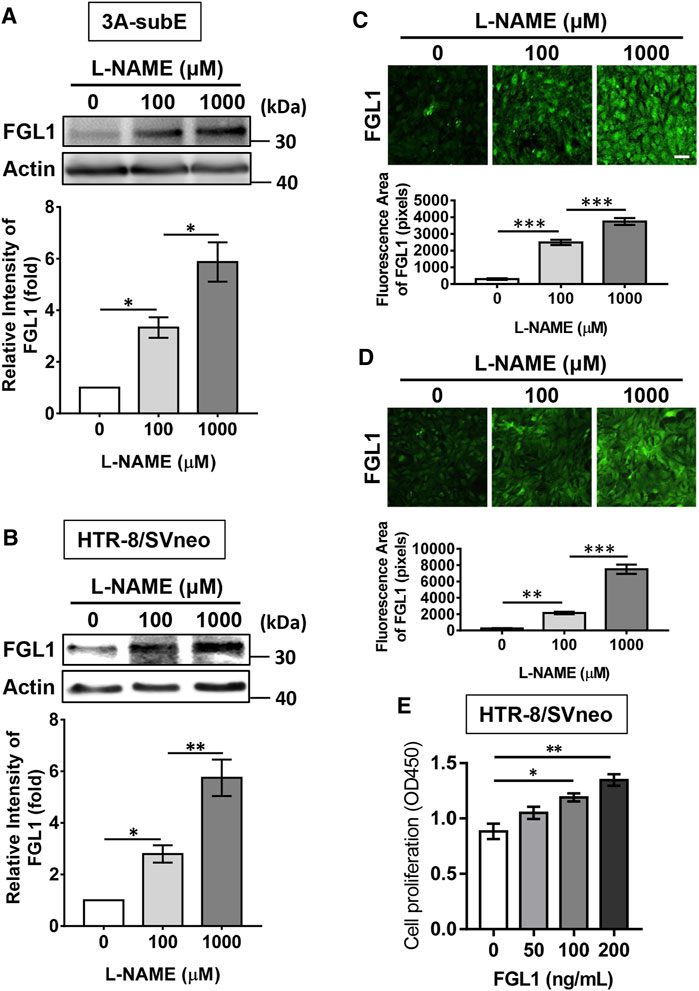
FIGURE 2. Effects of L-NAME stimulation on the expression of FGL1 in human placental trophoblast cell lines, and effects of FGL1 on the cell proliferation. Western blot analysis and immunofluorescence staining were performed to evaluate FGL1 expression in 3A-subE cells (A,C) and HTR-8/SVneo cells (B,D) incubated with the indicated dose of L-NAME for 24 h. Scale bar, 50 μm. (E) After FGL treatment at the indicated dose for 48 h, cell proliferation was assessed by CCK-8 assays. The experiments were repeated at least three times. *p < 0.05; **p < 0.01; ***p < 0.001 (Tukey’s test).
L-NAME-Induced Progression of PE in Mouse was Reduced by FGL1 Treatment
To determine whether the highly expressed FGL1 functioned as a protective factor against the progression of PE in mouse, we administered recombinant FGL1 to the mouse with PE. The body weight results revealed that the experimental group (PE + FGL1) had a minor improvement in body weight compared with the PE group during pregnancy (Figure 3A). Furthermore, FGL1 treatment significantly improved the MAP (Figure 3B), diastolic pressure and systolic pressure in the FGL1 group compared with the non-FGL1 group (diastolic: 58.61 ± 4.61 vs. 84.6 ± 3.95 mmHg, p < 0.0001; systolic: 112.28 ± 4.91 vs. 136.65 ± 1.73 mmHg, p = 0.0002; Figure 3C). The upregulated urinary protein expression under L-NAME stimulation at mid- and late pregnancy was significantly reduced by FGL1 treatment (mid: 14.93 ± 1.11 vs. 27.53 ± 1.57 mg/ml, p < 0.0001; late: 26.89 ± 1.47 vs. 38.93 ± 3.62 mg/ml, p = 0.0002; Figure 3D; n = 6/group). The results in this section suggest that upregulated FGL1 expression has critical antihypertensive and antiproteinuric effects in the mouse with PE, which may inhibit the progression of PE.
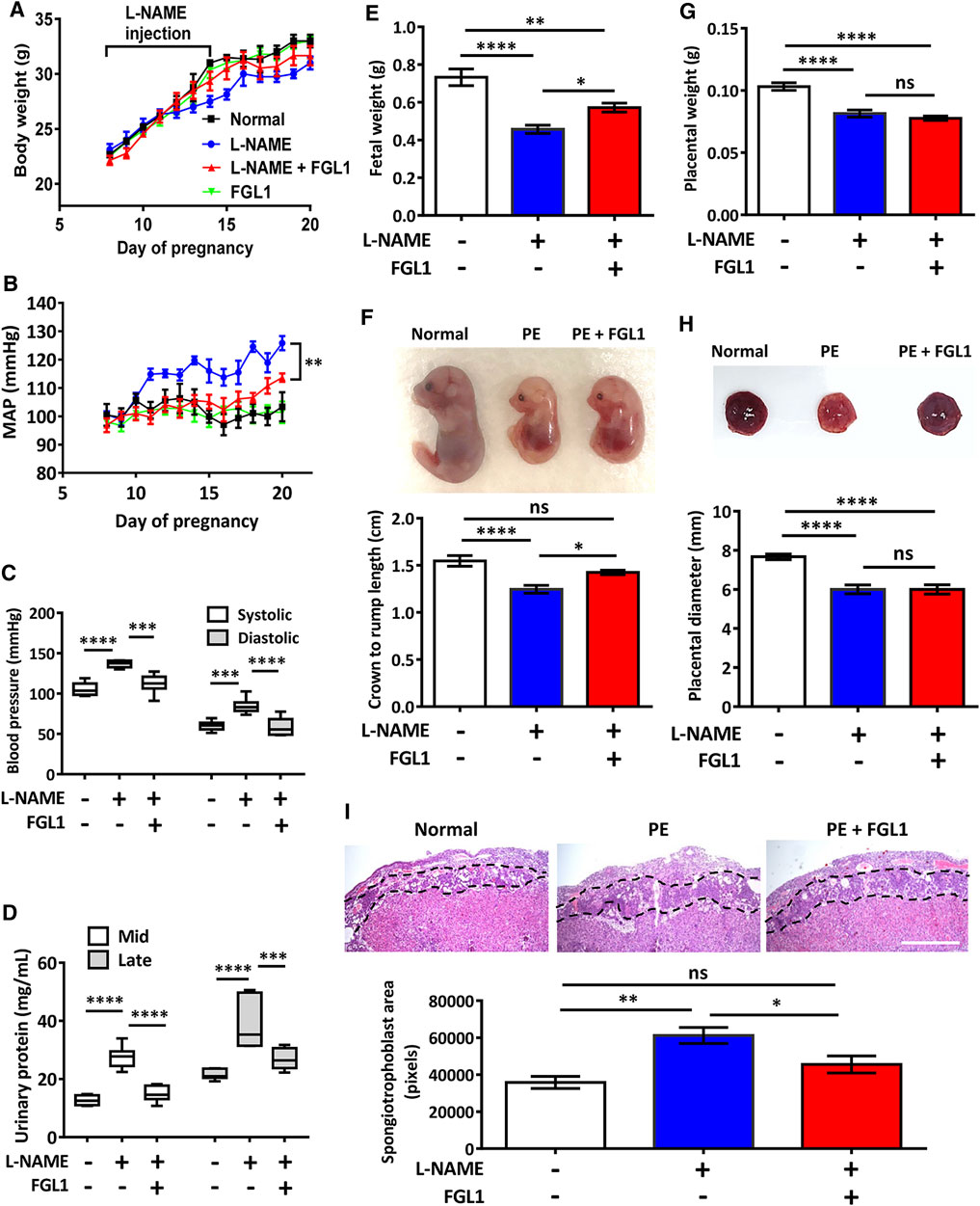
FIGURE 3. Effects of FGL1 treatment on the PE mouse. After the injection of L-NAME with or without FGL1 into pregnant mouse, representative parameters, including body weight (A), mean arterial pressure (MAP) (B), blood pressure (C), urinary protein (D), fetal weight (E), crown-to-rump length of the fetus (F), placenta weight (G), and placenta diameter (H), were analysed. Box and whisker plots for blood pressure and urinary protein. The box represents the median and interquartile range (IQR), and the whiskers were generated using Tukey’s method (values up to 75th percentile +1.5 IQR). (I) Hematoxylin-eosin staining of the mouse placenta. Dotted line indicates the spongiotrophoblast area. Mid, 12–15 days of pregnancy; late, 15–18 days of pregnancy. Scale bar, 10 mm. n = 6/group. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, no significance [(A, B): two-way ANOVA test; (C–I): Tukey’s test].
Next, the effects of FGL1 treatment on various parameters related to the fetus and placenta were investigated. Measurement of fetal weight and crown-to-rump length revealed that FGL1 treatment significantly reversed the L-NAME-reduced fetal growth (fetal weight: 0.57 ± 0.02 vs. 0.46 ± 0.02 g, p = 0.038; Figure 3E; crown-to-rump length: 1.43 ± 0.03 vs. 1.25 ± 0.04 cm, p = 0.033; Figure 3F; n = 12/group) but did not significantly affect placental weight or diameter (placental weight: 0.08 ± 0.002 vs. 0.08 ± 0.003 g, p = 0.564; Figure 3G; diameter: 6.0 ± 0.242 vs. 6.0 ± 0.229 cm, p > 0.99; Figure 3H; n = 12/group). In addition, hematoxylin-eosin staining performed to analyze the placental structure revealed that the L-NAME-increased spongiotrophoblast area was significantly reduced by FGL1 treatment (45,549 ± 4,585 vs. 61,232 ± 4,333 pixels, p = 0.046; Figure 3I; n = 6/group). These results indicate that FGL1 contributes to improving fetal growth and placental structure, thus retarding the progression of PE. Thus, upregulated FGL1 expression should function as a compensation factor to reduce the progression of PE.
FGL1 Downregulated Proinflammatory Cytokine Expression and Ameliorated the Imbalance of Angiogenic Molecules in Mouse With PE
Furthermore, qRT-PCR was used to elucidate how FGL1 treatment inhibited the progression of PE in mouse. The results revealed that FGL1 treatment significantly inhibited the expression of the proinflammatory cytokines IL-1b and IL-6 under L-NAME stimulation (IL-1b: 3.60 ± 0.75- vs. 6.34 ± 0.73-fold, p = 0.015, Figure 4A; IL-6: 3.26 ± 0.62- vs. 6.10 ± 0.87-fold, p = 0.014, Figure 4B; n = 6/group). The imbalanced expression of the angiogenic factors Flt-1 and PlGF was also improved in the FGL1 group compared with the non-FGL1 group (Flt-1: 0.93 ± 0.17- vs. 2.02 ± 0.19-fold, p = 0.0003, Figure 4C; PlGF: 0.96 ± 0.11- vs. 0.37 ± 0.08-fold, p = 0.0002, Figure 4D; Flt-1/PlGF ratio: 0.95 ± 0.09- vs. 7.29 ± 1.91-fold, p = 0.003, Figure 4E; n = 6/group). The protein levels of IL-6 and PlGF also showed consistent results (Figure 4F). These findings indicate that FGL1 inhibits the progression of PE by reducing the expression of proinflammatory cytokines and rescuing the dysregulated levels of angiogenic molecules.
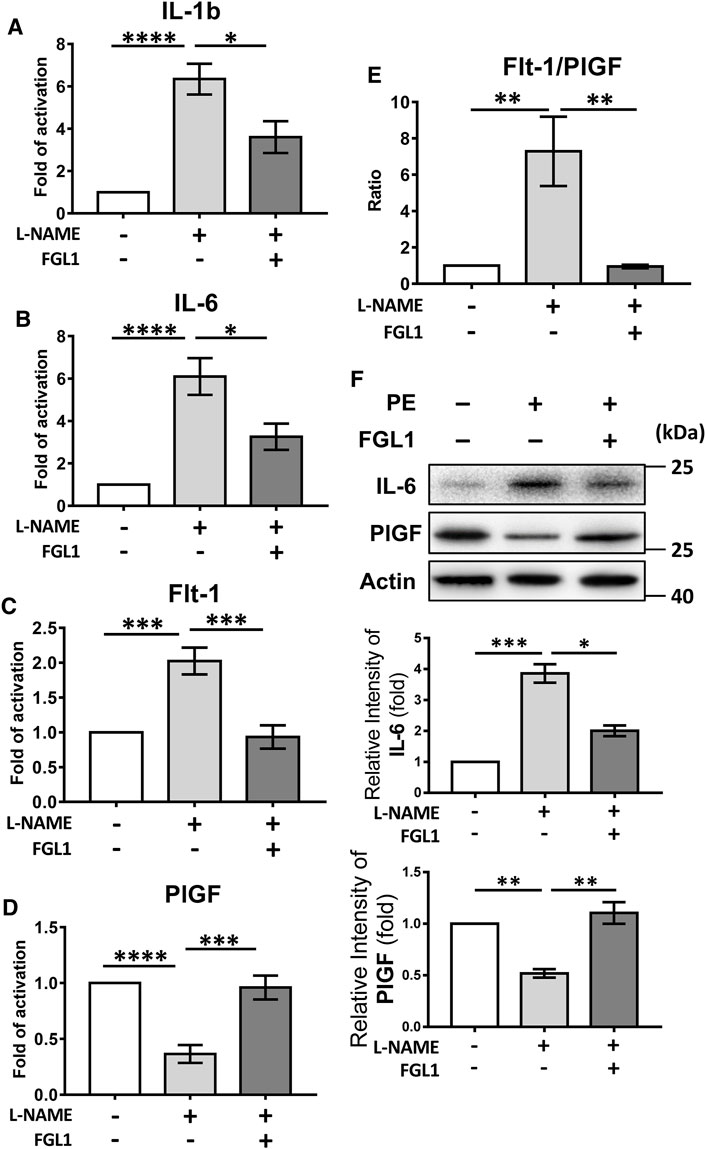
FIGURE 4. Effects of FGL1 treatment on proinflammatory cytokines and angiogenic markers in placentas from mouse with PE. After injection of L-NAME with or without FGL1, the placenta on the 18th day of pregnancy was subjected to quantitative real-time RT-PCR (qPCR) to evaluate the mRNA expression of proinflammatory cytokines and angiogenic markers. (A) Interleukin (IL)-1b. (B) IL-6. (C) FMS-like tyrosine kinase 1 (Flt-1). (D) Placental growth factor (PlGF). (E) Flt-1/PlGF ratio of mRNA level, n = 6/group. (F) Protein levels of IL-6 and PlGF. The experiments were repeated at least three times. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (Tukey’s test).
FoxO3a Overexpression Abolishes L-NAME-Induced Upregulation of FGL1
Finally, the intracellular mechanisms by which L-NAME moderates placental FGL expression and the cell function of FGL1 were investigated. Digital prediction by Qiagen (Hilden, Germany) indicated that the transcription factor binding site of FoxO3a is contained in the FGL1 promoter. L-NAME-induced upregulation of FGL1 was accompanied by a decrease in FoxO3a, and the FGL1 increase was abolished under FoxO3a overexpression (Figures 5A,B). Here, the results suggest that the overexpression of FoxO3a inhibits FGL1 expression in L-NAME-induced trophoblast cells.
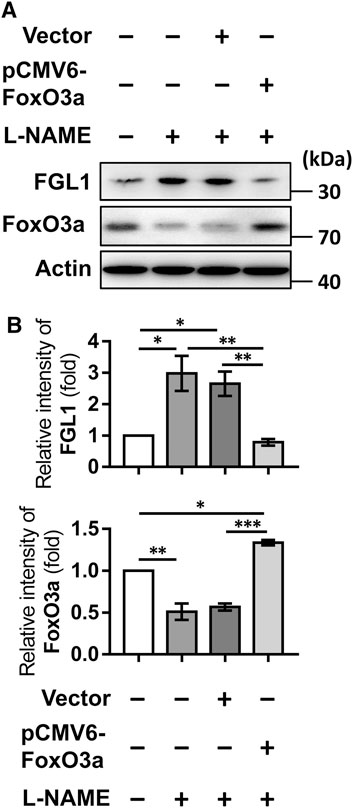
FIGURE 5. Overexpression of FoxO3a inhibits L-NAME-induced FGL1. (A) After transfection for 48 h and L-NAME (100 μM) treatment for 24 h, the effects of FoxO3a overexpression on L-NAME-upregulated FGL1 expression were evaluated by Western blotting. (B) Quantified protein expression levels of FGL1 and FoxO3a in HTR-8/SVneo cells. *p < 0.05; **p < 0.01; ***p < 0.001 (Tukey’s test).
Discussion
In this study, we found that the gene and protein levels of FGL1 were both significantly upregulated in the placenta and serum of mouse and humans with PE, indicating a correlation between PE and FGL1. In conclusion, this study highlighted the role of placental FGL1 in PE. Stress from reduced NOS activity and impaired NO formation/bioavailability increased placental FGL1 release in trophoblasts, at least in part, through FoxO3a signaling. FGL1 may function as a complementary factor to reduce the progression of PE by promoting trophoblast proliferation, downregulating proinflammatory cytokine expression and balancing placental perfusion (Figure 6).
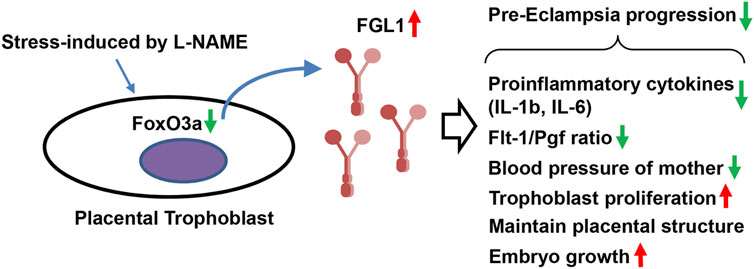
FIGURE 6. Schematic representation. Stress-upregulated FGL1 expression, through FoxO3a reduction, may function as a compensatory factor that contributes to reducing the progression of PE by mediating trophoblast proliferation, proinflammatory cytokines, and angiogenesis-related biomarkers.
The L-NAME-induced PE-like mouse model has been widely used for studying disease pathogenesis, such as blood vessel injury, inflammatory factors, and molecular mechanisms (Yallampalli and Garfield, 1993; Shu et al., 2018). Because the placenta is formed at GD 10.5 and is the root cause of PE (Lain and Roberts, 2002; Sones and Davisson, 2016), based on a previous report (Motta et al., 2015), we treated mouse with L-NAME and FGL1 on GDs 7–14. The results of blood pressure and urinary protein analysis showed that FGL1 treatment significantly reduced L-NAME-induced hypertension and proteinuria, which is consistent with our hypothesis.
Studies have suggested that L-NAME-induced placental hypoperfusion and ischemia are associated with severe PE (Wang et al., 2009). Our in vitro study on human trophoblast cell lines demonstrated that FGL1 expression can be dose-dependently enhanced by L-NAME stimulation. L-NAME is an NOS inhibitor (Fantel et al., 1997), implying that placental FGL1 expression may be mediated through reduced NOS activity and impaired NO formation/bioavailability.
Data on the role of placental FGL1 in PE have remained limited. FGL1 is known to regulate the expression of proliferative factors, promote liver regeneration, and repair liver damage (Yan et al., 2002; Li et al., 2010; Nayeb-Hashemi et al., 2015). FGL1 was suggested to be a biomarker for epithelial-mesenchymal transition and angiogenesis in specific lung adenocarcinomas (Bie et al., 2019). These findings can be extrapolated to indicate that upregulation of placental FGL1 expression is protective against the progression of PE. Consistent with this hypothesis, our investigation revealed that recombinant FGL1 administration to the mouse with PE significantly ameliorated maternal PE parameters, inducing, for example, decreases in blood pressure and urinary protein. FGL1 is a circulating factor that could be used as a biomarker for predicting disease activity and prognosis (Liu et al., 2020). However, to date, studies on FGL1 in the vasculature are lacking, and this issue may warrant further investigation.
The placenta is the root cause of the intrauterine growth restriction observed in PE (Roberts and Escudero, 2012). Insufficient invasion of trophoblast cells into the myometrial portions of the spiral arteries is crucial for the development of PE, leading to the failure of uteroplacental vessel adaptive changes (Kaufmann et al., 2003). Consistently, our results revealed that FGL1 treatment in L-NAME-stimulated mouse improved the proportion spongiotrophoblast layer and fetal growth but did not affect the L-NAME-reduced placental diameter. These observations indicate that the mechanisms of FGL1 in the placenta may involve the regulation of blood flow and angiogenesis, thus contributing to fetal growth.
FGL1 has been suggested as a novel biomarker for angiogenesis in patients with lung adenocarcinoma (Bie et al., 2019). Additionally, angiogenic imbalance is believed to contribute to PE. An increase in the levels of some antiangiogenic factors released by the placenta into the maternal circulation, such as Flt-1, and a decrease in the levels of proangiogenic factors, such as PlGF, cause angiogenic imbalance and contribute to the pathogenesis of PE (Dymara-Konopka et al., 2018). Accordingly, our data indicated that FGL1 treatment may retard the progression of PE by reducing the expression of proinflammatory cytokines and preventing the imbalance of angiogenic factors. Our findings may contribute to further knowledge regarding the role of placental FGL1 in PE.
Our study has some limitations. First, we did not have an FGL1 gene knockout mouse strain (FGL1-KO) (Bie et al., 2019) to evaluate the effects of FGL loss on the progression of PE. Second, although the results indicated the effects of FGL1 on the proliferation of trophoblasts, performing a toxicity study of FGL1 in trophoblasts would have provided further evidence for its use as a therapeutic agent. Third, though the results of our pilot study are consistent with an increase in placental expression and serum levels of FGL2 in patients with PE, some limitations must be kept in mind. The PE group in our study population had significantly higher BMI and lower gestational age of sample collection. However, using the L-NAME-induced PE-like mouse model, we confirmed that FGL1 treatment could reduce disease progression by decreasing proinflammatory cytokines and maintaining the balance of angiogenesis, which is a strength of our study. The results in this report indicated the benefits of FGL1 treatment on mice PE progression.
Accumulating evidence has shown that FoxO3a is involved in mediating various biological processes, including proliferation, apoptosis, protection against oxidative stress, and metabolism (van der Vos and Coffer, 2011). The polymorphisms of FoxO3 on rs2232365 are associated with the risk of late-onset PE in pregnant women (Pan et al., 2020). Increased expression levels of FoxO3a were found in patients with PE, and decreasing FoxO3a expression by siRNA reduced trophoblast apoptosis (Zhang Z. et al., 2020). FoxO3a plays a role in the regulation of trophoblast development and pregnancy complications, and a decreased expression of FoxO3a was found in the placenta of PE cases and was present in trophoblasts (Chen et al., 2021). Furthermore, FoxO3a posttranslational modifications were increased by accumulated reactive oxygen species (ROS) under oxidative stress (Zhang H. et al., 2020). Therefore, these results could be an indirect result of this cellular stress induced by L-NAME and more easily controlled with anti-free radical agents such as vitamins C and D. Here, our results showed that the FoxO3a protein level was downregulated in trophoblasts under NOS inhibition. In addition, the function of FGL1 in promoting trophoblast proliferation was similar to that in our previous report (Kang et al., 2020). However, the detailed mechanisms need to be further investigated.
In conclusion, FGL1 may function as a complementary factor to reduce the progression of PE by promoting trophoblast proliferation, downregulating proinflammatory cytokine expression and balancing placental perfusion, and it might also be a prognostic marker for PE. The effects of FGL1 are at least in part mediated through FoxO3a signaling.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the research was carried out according to The Code of Ethics of the World Medical Association (Declaration of Helsinki), informed consent was obtained, and the author’s institutional review board approved the study. This study was approved by the National Cheng Kung University Hospital (NCKUH) Institutional Review Board (No: A-ER-105-453; date of approval: 2017/02/03∼until now), and all participants provided written informed consent. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the animal care and experimental procedures conformed to the ARRIVE guidelines (Kilkenny et al., 2010) and were approved by the Institutional Animal Care and User Committee (150213) at NCKU, Tainan, Taiwan.
Author Contributions
T-LC, C-HC, and LK contributed to the study conception, study design, data analysis, data interpretation, and manuscript writing; and M-HW, C-HL, K-HL, S-HL, A-LS, and C-LW contributed to the data interpretation and critical manuscript revision. All authors contributed to and approved the final manuscript.
Funding
This work was supported by grants from the Ministry of Science and Technology (MOST 106-2314-B-006-066 and MOST 108-2314-B-006-062 to LK), Executive Yuan, Taiwan; National Cheng Kung University Hospital (NCKUH-10604002, NCKUH-10705015, and NCKUH-10902008 to LK); College of Medicine, National Cheng Kung University, Tainan, Taiwan; and Kaohsiung Medical University (KMU-TC108A02, KMUH-DK(A)110003), Kaohsiung, Taiwan. The funders did not influence the design or data analysis of the study or contribute to the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Wei-Ting Chang for her assistance with this study.
References
Abalos, E., Cuesta, C., Grosso, A. L., Chou, D., and Say, L. (2013). Global and Regional Estimates of Preeclampsia and Eclampsia: a Systematic Review. Eur. J. Obstet. Gynecol. Reprod. Biol. 170, 1–7. doi:10.1016/j.ejogrb.2013.05.005
Author Anonymous, (2019). ACOG Practice Bulletin No. 202: Gestational Hypertension and Preeclampsia. Obstet. Gynecol. 133, e1. doi:10.1097/AOG.0000000000003018
Bie, F., Wang, G., Qu, X., Wang, Y., Huang, C., Wang, Y., et al. (2019). Loss of FGL1 Induces Epithelial-mesenchymal T-ransition and A-ngiogenesis in LKB1 M-utant L-ung A-denocarcinoma. Int. J. Oncol. 55, 697–707. doi:10.3892/ijo.2019.4838
Buhimschi, I., Yallampalli, C., Chwalisz, K., and Garfield, R. E. (1995). Pre-eclampsia-like Conditions Produced by Nitric Oxide Inhibition: Effects of L-Arginine, D-Arginine and Steroid Hormones. Hum. Reprod. 10, 2723–2730. doi:10.1093/oxfordjournals.humrep.a135775
Cao, M.-M., Xu, W.-X., Li, C.-Y., Cao, C.-Z., Wang, Z.-D., Yao, J.-W., et al. (2011). Hepassocin Regulates Cell Proliferation of the Human Hepatic Cells L02 and Hepatocarcinoma Cells through Different Mechanisms. J. Cell. Biochem. 112, 2882–2890. doi:10.1002/jcb.23202
Chen, H., Tang, X., Han, T. L., Zhu, J. N., Zhou, W., Baker, P. N., et al. (2021). Potential Role of FoxO3a in the Regulation of Trophoblast Development and Pregnancy Complications. J. Cell Mol Med 25, 4363–4372. doi:10.1111/jcmm.16499
Cheng, T.-L., Liao, C.-C., Tsai, W.-H., Lin, C.-C., Yeh, C.-W., Teng, C.-F., et al. (2009). Identification and Characterization of the Mitochondrial Targeting Sequence and Mechanism in Human Citrate Synthase. J. Cell. Biochem. 107, 1002–1015. doi:10.1002/jcb.22200
Demchev, V., Malana, G., Vangala, D., Stoll, J., Desai, A., Kang, H. W., et al. (2013). Targeted Deletion of Fibrinogen like Protein 1 Reveals a Novel Role in Energy Substrate Utilization. PLoS One 8, e58084. doi:10.1371/journal.pone.0058084
Domanski, M. J., Davis, B. R., Pfeffer, M. A., Kastantin, M., and Mitchell, G. F. (1999). Isolated Systolic Hypertension. Hypertension 34, 375–380. doi:10.1161/01.hyp.34.3.375
Dymara-Konopka, W., Laskowska, M., and Błażewicz, A. (2018). Angiogenic Imbalance as a Contributor of Preeclampsia. Cpb 19, 797–815. doi:10.2174/1389201019666180925115559
El-Karef, A., Yoshida, T., Gabazza, E., Nishioka, T., Inada, H., Sakakura, T., et al. (2007). Deficiency of Tenascin-C Attenuates Liver Fibrosis in Immune-Mediated Chronic Hepatitis in Mice. J. Pathol. 211, 86–94. doi:10.1002/path.2099
Fantel, A. G., Nekahi, N., Shepard, T. H., Cornel, L. M., Unis, A. S., and Lemire, R. J. (1997). The Teratogenicity of Nω-nitro-l-arginine Methyl Ester (L-NAME), a Nitric Oxide Synthase Inhibitor, in Rats. Reprod. Toxicol. 11, 709–717. doi:10.1016/s0890-6238(97)00033-6
Fu, R., Lv, W.-C., Xu, Y., Gong, M.-Y., Chen, X.-J., Jiang, N., et al. (2020). Endothelial ZEB1 Promotes Angiogenesis-dependent Bone Formation and Reverses Osteoporosis. Nat. Commun. 11, 460. doi:10.1038/s41467-019-14076-3
Hara, H., Yoshimura, H., Uchida, S., Toyoda, Y., Aoki, M., Sakai, Y., et al. (2001). Molecular Cloning and Functional Expression Analysis of a cDNA for Human Hepassocin, a Liver-specific Protein with Hepatocyte Mitogenic Activity. Biochim. Biophys. Acta (Bba) - Gene Struct. Expr. 1520, 45–53. doi:10.1016/s0167-4781(01)00249-4
Iwashita, Y., Zhang, E., Maruyama, J., Yokochi, A., Yamada, Y., Sawada, H., et al. (2014). Thrombomodulin Protects against Lung Damage Created by High Level of Oxygen with Large Tidal Volume Mechanical Ventilation in Rats. J. Intensive Care 2, 57. doi:10.1186/s40560-014-0057-0
Jeyabalan, A. (2013). Epidemiology of Preeclampsia: Impact of Obesity. Nutr. Rev. 71 (Suppl. 1), S18–S25. doi:10.1111/nure.12055
Kang, L., Li, H.-Y., Ou, H.-Y., Wu, P., Wang, S.-H., Chang, C.-J., et al. (2020). Role of Placental Fibrinogen-like Protein 1 in Gestational Diabetes. Translational Res. 218, 73–80. doi:10.1016/j.trsl.2020.01.001
Kaufmann, P., Black, S., and Huppertz, B. (2003). Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 69, 1–7. doi:10.1095/biolreprod.102.014977
Kilkenny, C., Browne, W., Cuthill, I. C., Emerson, M., Altman, D. G., and Group, N. C. R. R. G. W. (2010). Animal Research: Reporting In Vivo Experiments: the ARRIVE Guidelines. Br. J. Pharmacol. 160, 1577–1579. doi:10.1111/j.1476-5381.2010.00872.x
Kim, I., Kim, H.-G., Kim, H., Kim, H.-H., Park, S. K., Uhm, C.-S., et al. (2000). Hepatic Expression, Synthesis and Secretion of a Novel Fibrinogen/angiopoietin-Related Protein that Prevents Endothelial-Cell Apoptosis. Biochem. J. 346, 603–610. doi:10.1042/bj3460603
Lain, K. Y., and Roberts, J. M. (2002). Contemporary Concepts of the Pathogenesis and Management of Preeclampsia. JAMA 287, 3183–3186. doi:10.1001/jama.287.24.3183
Li, C. Y., Cao, C. Z., Xu, W. X., Cao, M. M., Yang, F., Dong, L., et al. (2010). Recombinant Human Hepassocin Stimulates Proliferation of Hepatocytes In Vivo and Improves Survival in Rats with Fulminant Hepatic Failure. Gut 59, 817–826. doi:10.1136/gut.2008.171124
Liu, S., Guo, Y., Lu, L., Lu, J., Ke, M., Xu, T., et al. (2020). Fibrinogen-Like Protein 1 Is a Novel Biomarker for Predicting Disease Activity and Prognosis of Rheumatoid Arthritis. Front. Immunol. 11, 579228. doi:10.3389/fimmu.2020.579228
Liu, Y., Ao, X., Ding, W., Ponnusamy, M., Wu, W., Hao, X., et al. (2018). Critical Role of FOXO3a in Carcinogenesis. Mol. Cancer 17, 104. doi:10.1186/s12943-018-0856-3
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408. doi:10.1006/meth.2001.1262
Lowry, O., Rosebrough, N., Farr, A. L., and Randall, R. (1951). Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 193, 265–275. doi:10.1016/s0021-9258(19)52451-6
Molnár, M., Sütö, T., Tóth, T., and Hertelendy, F. (1994). Prolonged Blockade of Nitric Oxide Synthesis in Gravid Rats Produces Sustained Hypertension, Proteinuria, Thrombocytopenia, and Intrauterine Growth Retardation. Am. J. Obstet. Gynecol. 170, 1458–1466. doi:10.1016/s0002-9378(94)70179-2
Motta, C., Grosso, C., Zanuzzi, C., Molinero, D., Picco, N., Bellingeri, R., et al. (2015). Effect of Sildenafil on Pre-eclampsia-like Mouse Model Induced by L-Name. Reprod. Dom Anim. 50, 611–616. doi:10.1111/rda.12536
Nayeb-Hashemi, H., Desai, A., Demchev, V., Bronson, R. T., Hornick, J. L., Cohen, D. E., et al. (2015). Targeted Disruption of Fibrinogen like Protein-1 Accelerates Hepatocellular Carcinoma Development. Biochem. Biophysical Res. Commun. 465, 167–173. doi:10.1016/j.bbrc.2015.07.078
Pan, X., Wei, B., Wang, H., Ma, L., Du, Z., and Chen, Y. (2020). Novel Association between FOXO3 Rs2232365 Polymorphism and Late-Onset Preeclampsia: a Case-Control Candidate Genetic Study. BMC Pregnancy Childbirth 20, 779. doi:10.1186/s12884-020-03479-6
Procopio, W. N., Pelavin, P. I., Lee, W. M. F., and Yeilding, N. M. (1999). Angiopoietin-1 and -2 Coiled Coil Domains Mediate Distinct Homo-Oligomerization Patterns, but Fibrinogen-like Domains Mediate Ligand Activity. J. Biol. Chem. 274, 30196–30201. doi:10.1074/jbc.274.42.30196
Roberts, J. M., and Escudero, C. (2012). The Placenta in Preeclampsia. Pregnancy Hypertens. Int. J. Women's Cardiovasc. Health 2, 72–83. doi:10.1016/j.preghy.2012.01.001
Roberts, J. M., and Gammill, H. S. (2005). Preeclampsia. Hypertension 46, 1243–1249. doi:10.1161/01.hyp.0000188408.49896.c5
Sahni, A., and Francis, C. W. (2000). Vascular Endothelial Growth Factor Binds to Fibrinogen and Fibrin and Stimulates Endothelial Cell Proliferation. Blood 96, 3772–3778. doi:10.1182/blood.v96.12.3772
Shu, W., Li, H., Gong, H., Zhang, M., Niu, X., Ma, Y., et al. (2018). Evaluation of Blood Vessel Injury, Oxidative Stress and Circulating Inflammatory Factors in an L-NAME-Induced Preeclampsia-like Rat Model. Exp. Ther. Med. 16, 585–594. doi:10.3892/etm.2018.6217
Sibai, B., Dekker, G., and Kupferminc, M. (2005). Pre-eclampsia. The Lancet 365, 785–799. doi:10.1016/s0140-6736(05)71003-5
Sibai, B. M. (2008). Hypertensive Disorders of Pregnancy: the United States Perspective. Curr. Opin. Obstet. Gynecol. 20, 102–106. doi:10.1097/gco.0b013e3282f73380
Sones, J. L., and Davisson, R. L. (2016). Preeclampsia, of Mice and Women. Physiol. Genomics 48, 565–572. doi:10.1152/physiolgenomics.00125.2015
Tsukimori, K., Komatsu, H., Fukushima, K., Kaku, T., Nakano, H., and Wake, N. (2008). Inhibition of Nitric Oxide Synthetase at Mid-gestation in Rats Is Associated with Increases in Arterial Pressure, Serum Tumor Necrosis Factor- , and Placental Apoptosis. Am. J. Hypertens. 21, 477–481. doi:10.1038/ajh.2007.80
van der Vos, K. E., and Coffer, P. J. (2011). The Extending Network of FOXO Transcriptional Target Genes. Antioxid. Redox Signaling 14, 579–592. doi:10.1089/ars.2010.3419
Wang, A., Rana, S., and Karumanchi, S. A. (2009). Preeclampsia: the Role of Angiogenic Factors in its Pathogenesis. Physiology 24, 147–158. doi:10.1152/physiol.00043.2008
Wu, H.-T., Ou, H.-Y., Hung, H.-C., Su, Y.-C., Lu, F.-H., Wu, J.-S., et al. (2016). A Novel Hepatokine, HFREP1, Plays a Crucial Role in the Development of Insulin Resistance and Type 2 Diabetes. Diabetologia 59, 1732–1742. doi:10.1007/s00125-016-3991-7
Xie, J., Cha, S.-K., An, S.-W., Kuro-o, M., Birnbaumer, L., and Huang, C.-L. (2012). Cardioprotection by Klotho through Downregulation of TRPC6 Channels in the Mouse Heart. Nat. Commun. 3, 1238. doi:10.1038/ncomms2240
Yallampalli, C., and Garfield, R. E. (1993). Inhibition of Nitric Oxide Synthesis in Rats during Pregnancy Produces Signs Similar to Those of Preeclampsia. Am. J. Obstet. Gynecol. 169, 1316–1320. doi:10.1016/0002-9378(93)90299-x
Yamamoto, T., Gotoh, M., Sasaki, H., Terada, M., Kitajima, M., and Hirohashi, S. (1993). Molecular Cloning and Initial Characterization of a Novel Fibrinogen-Related Gene, HFREP-1. Biochem. Biophysical Res. Commun. 193, 681–687. doi:10.1006/bbrc.1993.1678
Yan, J., Ying, H., Gu, F., He, J., Li, Y. L., Liu, H. M., et al. (2002). Cloning and Characterization of a Mouse Liver-specific Gene Mfrep-1, Upregulated in Liver Regeneration. Cell Res 12, 353–361. doi:10.1038/sj.cr.7290137
You, S.-H., Cheng, P.-J., Chung, T.-T., Kuo, C.-F., Wu, H.-M., and Chu, P.-H. (2018). Population-based Trends and Risk Factors of Early- and Late-Onset Preeclampsia in Taiwan 2001-2014. BMC Pregnancy Childbirth 18, 199. doi:10.1186/s12884-018-1845-7
Zhang, H., Lin, F., Zhao, J., and Wang, Z. (2020a). Expression Regulation and Physiological Role of Transcription Factor FOXO3a during Ovarian Follicular Development. Front. Physiol. 11, 595086. doi:10.3389/fphys.2020.595086
Zhang, Z., Huang, C., Wang, P., Gao, J., Liu, X., Li, Y., et al. (2020b). HIF-1α A-ffects T-rophoblastic A-poptosis I-nvolved in the O-nset of P-reeclampsia by R-egulating FOXO3a under H-ypoxic C-onditions. Mol. Med. Rep. 21, 2484–2492. doi:10.3892/mmr.2020.11050
Keywords: fibrinogen-like 1, preeclampsia, placenta, embryo (animal), proinflammatory cytokines
Citation: Cheng T-L, Chen C-H, Wu M-H, Lai C-H, Lee K-H, Lin S-H, Shiau A-L, Wu C-L and Kang L (2021) Upregulation of Fibrinogen-Like 1 Expression Contributes to Reducing the Progression of Preeclampsia. Front. Cell Dev. Biol. 9:757643. doi: 10.3389/fcell.2021.757643
Received: 13 August 2021; Accepted: 16 November 2021;
Published: 08 December 2021.
Edited by:
Rashmi Sood, Medical College of Wisconsin, United StatesReviewed by:
Piyali Chatterjee, Central Texas Veterans Health Care System, United StatesCamila Ferreira Bannwart Castro, Sao Paulo State University, Brazil
Copyright © 2021 Cheng, Chen, Wu, Lai, Lee, Lin, Shiau, Wu and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Kang, a2FuZ2xpbkBuY2t1LmVkdS50dw==