 Maria Miquel
Maria Miquel Shuman Zhang1,2†
Shuman Zhang1,2† Christian Pilarsky
Christian Pilarsky
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 27 October 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.748631
This article is part of the Research Topic Translational Insights into Pancreatic Ductal Adenocarcinoma View all 16 articles
Pancreatic ductal adenocarcinoma (PDAC) is a hostile solid malignancy coupled with an extremely high mortality rate. Metastatic disease is already found in most patients at the time of diagnosis, resulting in a 5-year survival rate below 5%. Improved comprehension of the mechanisms leading to metastasis is pivotal for the development of new targeted therapies. A key field to be improved are modeling strategies applied in assessing cancer progression, since traditional platforms fail in recapitulating the complexity of PDAC. Consequently, there is a compelling demand for new preclinical models that mirror tumor progression incorporating the pressure of the immune system, tumor microenvironment, as well as molecular aspects of PDAC. We suggest the incorporation of 3D organoids derived from genetically engineered mouse models or patients as promising new tools capable to transform PDAC pre-clinical modeling and access new frontiers in personalized medicine.
Pancreatic ductal adenocarcinoma (PDAC) has the worst 5-year relative survival rate in comparison to all other solid tumors and has been prognosed to become the second leading cause of cancer-related mortality in the United States by 2030 after lung cancer (Chu et al., 2017; McGuigan et al., 2018). More than 90% of pancreatic cancers are exocrine tumors, being the most frequent type, PDAC. Other tumors like neuroendocrine tumors (PNET) are often indolent and treatable (Antonello et al., 2009). The poor outcome is correlated to late diagnosis, a result of non-specific symptoms, poor specificity of tumor markers, and non-accessible sites for routine palpation. Further, the PDAC is associated with a high capacity of metastatic dissemination to adjacent organs already in small tumor sizes. Common sites of dissemination are the liver, with metastases present in 76–80% of patients, peritoneum (48%) and the lungs (45%; Yachida et al., 2012). Even though surgical resection of the primary tumor is the only treatment with curative intention, 85–90% of patients are not eligible due to the systemic nature of the disease and a lack of early diagnosis. Even in the less than 20% operable cases, where the primary tumor has been completely removed (R0) and no manifestation of metastasis at resection, 75% of the patients will die of metastatic relapse in then 5 years after being operated (Chu et al., 2017; McGuigan et al., 2018).
Pancreatic ductal adenocarcinoma is a complex genetic disease, mainly determined by oncogenic activation of Kirsten rat sarcoma virus (KRAS) and mutations in tumor suppressor genes such Cyclin Dependent Kinase Inhibitor 2A (CDKN2A), Transformation Related Protein 53 (TP53), Lysine Demethylase 6A (KDM6A), Breast Cancer Gene (BRCA1/2), and SMAD Family Member 4 (SMAD4; Yachida and Iacobuzio-Donahue, 2009). The signature mutations of PDAC were identified in precursor lesions namely pancreatic intraepithelial neoplasia (PanIN), mucinous cystic neoplasia (MCNs), and intraductal papillary mucinous neoplasm (IPMNs; Hruban et al., 2001). Activation of oncogenic Kras in pancreatic epithelial cells triggers initiation of PDAC in mouse models and when combined with Trp53, Cdkn2a, or Smad4 mutations PDAC progression is accelerated, recapitulating many characteristics of the human disease (Izeradjene et al., 2007; see section “Genetically engineered mouse models”).
Using bulk tumor samples, separate studies identified at least two subtypes of PDAC (Collisson et al., 2011; Moffitt et al., 2015; Bailey et al., 2016; Chan-Seng-Yue et al., 2020), differentiated by markers of epithelial differentiation state, being the less differentiated subtype (“basal-like,” “squamous,” or “quasi-mesenchymal”) the one correlating with worse prognosis compared to the better differentiated subtypes (“classical” or “progenitor”; Dreyer et al., 2021a).
In primary patient-derived cell lines and bulky tumors of the various PDAC cohorts, a replication stress signature linked with the squamous subtype was identified. This is linked with functional impairments in replication of DNA and might also be utilized as biomarkers and give alternative therapeutics choices to standard care platinum chemotherapy for patients with DNA replication abnormalities (Dreyer et al., 2021b). The squamous subtype has also been defined by a distinct metabolic phenotype due to loss of genes that specify endodermal lineage, Hepatocyte Nuclear Factor 4 Alpha (HNF4A), and GATA Binding Protein 6 (GATA6). This subtype is therefore more sensitive to Glycogen synthase kinase 3 beta (GSK3β) inhibition except for a subgroup with distinct chromatin accessibility which acquires rapid drug resistance (Brunton et al., 2020).
Employing laser capture microdissection and RNA sequencing on PDAC epithelia and adjacent stroma defined two stromal subtypes differing in the immune-associated and extracellular matrix-associated processes. This study showed that across the same tumors, epithelial and stromal subtypes were partially linked [Extracellular Matrix (ECM) rich stroma was associated with Basal-like epithelium and Immune-rich stroma was found more often in association with Classical epithelia], showing potential dependence in the evolution of the tissue compartments in PDAC (Maurer et al., 2019).
Another study, based on the methylation patterns of the tumor genomes, defined two different origins of adenocarcinomas. One type of tumor is formed directly from ductal cells lining the ductal system of the pancreas, whereas the other develops from glandular cells and is less aggressive (Espinet et al., 2021).
Despite the current classification consensus, Juiz et al. showed that Basal-like and Classical cells coexist in PDAC as described by single-cell analysis on pancreatic cancer organoids derived from biopsies indicating that both subtypes can coexist in the same patient (Juiz et al., 2020).
Even though clinical decision-making based on histopathological criteria is widely established in several cancer types, subtypes of PDAC currently do not guide treatment decisions (Collisson et al., 2019). The only treatment with curative intent is surgery, which can be preceded by a neoadjuvant treatment and followed by adjuvant therapies such as gemcitabine monotherapy. However, recurrence rates in operated patients are still high and long-term survival is limited (Bijlsma, 2021).
The current standard of care for metastatic PDAC includes highly toxic chemotherapeutic cocktails with limited specificities. Gemcitabine has become a widely used drug for advanced and metastatic PDAC since it was reported (Burris et al., 1997), despite its low influence on patient survival. There are two gold-standard combination regimens for metastatic PDAC: 5-fluorouracil/leucovorin with irinotecan and oxaliplatin (FOLFIRINOX; Conroy et al., 2011), and gemcitabine with nab-paclitaxel since 2011 (Peixoto et al., 2017). A detailed review of PDAC chemotherapy can be found elsewhere: (Zeng et al., 2019; Singh and O’Reilly, 2020). According to developments and advances in other cancer types, it is expected that improvements in PDAC treatment are likely to come from the combination of classical cytotoxics with novel targeted agents against PDAC. Important matters in hand related to new therapeutic approaches include immunotherapy, DNA damage repair strategies, targeting the stroma, as well as cancer-cell metabolism (Dreyer et al., 2021b).
Current targeted therapies in PDAC undergoing Clinical Trials are divided into three approaches (Table 1). Firstly, inhibition of dysregulated oncogenes such as KRAS, c-MYC, Neurotrophic tyrosine receptor kinase, Neuregulin 1, and related molecules. Since these options have not led to an improvement of patient survival, alternative strategies are being developed to target these oncogenes, namely modification of mutant residues by small molecules, simultaneously inhibiting multiple molecules or pathways, and RNA interference. Secondly, reactivate tumor suppressors or modulate related molecules such as TP53, CDKN2A, SMAD4, KDM6A, and BRCA1/2. In addition to genetic event-guided treatment, immunotherapies such as antibody-drug conjugates, chimeric antigen receptor T cells (CAR-T), and immune checkpoint inhibitors also indicate the potential to target tumors precisely. Nonetheless, targeted therapies have been largely unsuccessful in PDAC. Currently, the only targeted therapeutic agent approved for PDAC is Erlotinib, which only slightly prolongs survival in metastatic disease (Moore et al., 2007; Sinn et al., 2017) but showed negative results in the adjuvant setting (Bijlsma et al., 2017). A possible reason for the unsuccessful outcomes of targeted agents in PDAC is incorrect patient selection. Interestingly, when tumor samples from several of the CONKO-005 trial participants were re-analyzed, a subgroup of patients with a combination of SMAD4 loss and low Mitogen-Activated Protein Kinase 9 (MAPK9) expression benefited from the addition of Erlotinib (Hoyer et al., 2021). Therefore, not only novel targeted therapies are needed but also integration with genomic profiling along with a full understanding of the tumor microenvironment and immunology (Qian et al., 2020). It is expected that in the future, comprehensive tumor analysis should become an essential part of diagnostic routines and guide treatment choice.
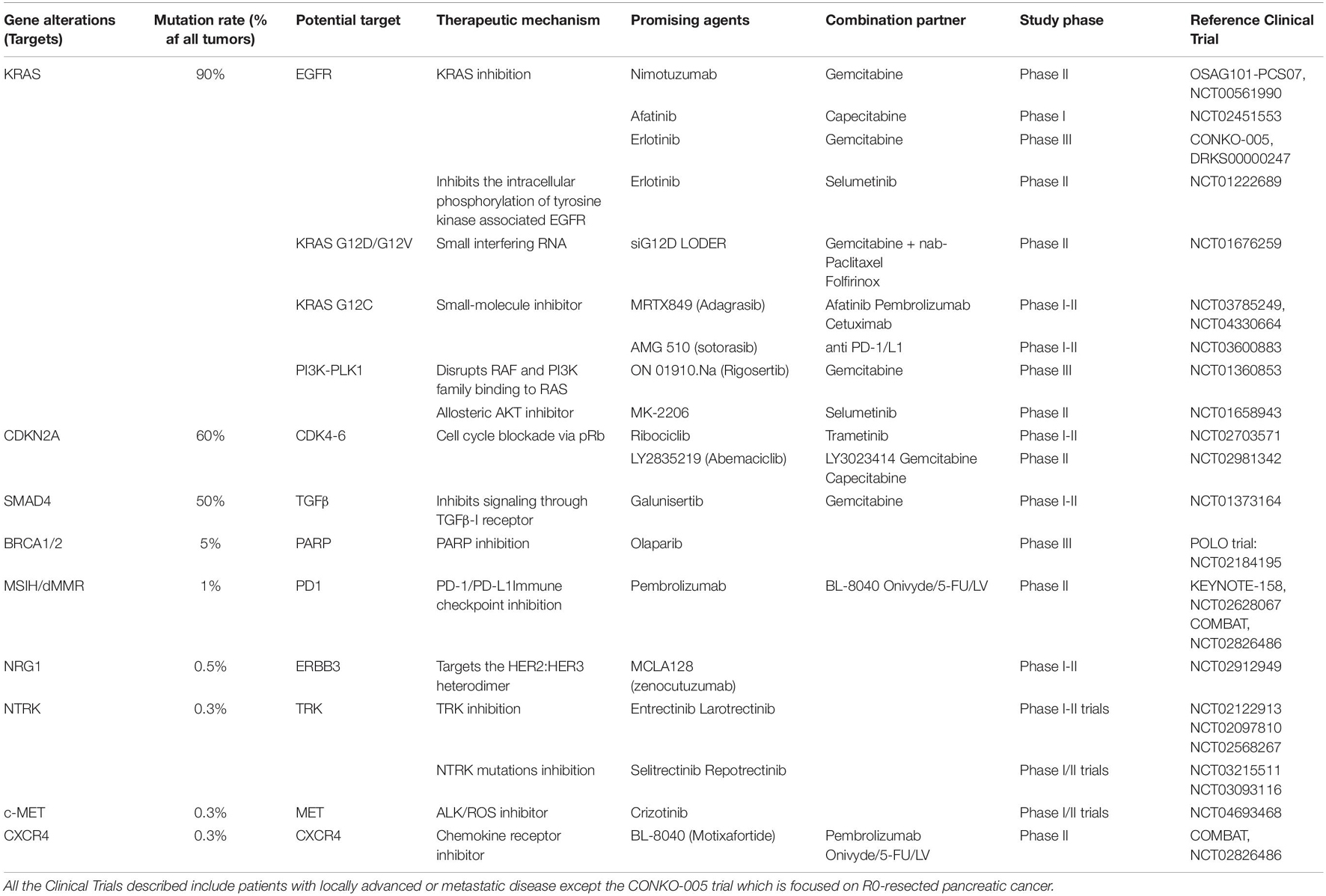
Table 1. Potential therapeutic targets in PDAC undergoing Clinical Trials.
Since most PDAC patients succumb due to metastatic cancer, this accentuates the crucial need to develop novel therapies that target, not only the primary tumor, but also the vulnerabilities of metastatic cells (Singh and O’Reilly, 2020). The last results of the COMBAT/KEYNOTE-202 Trial showed that Triple combination of motixafortide, pembrolizumab and chemotherapy are safe, well tolerated and showed signs of efficacy in a population with poor prognosis and aggressive PDAC (Bockorny et al., 2021). Promising new anticancer compounds are tested pre-clinically into in vitro and in vivo models. However, 90% of those fail when moving to clinical trials (Lai et al., 2019), showing the need for more consistent and representative models for drug testing, recapitulating better genetics, immunology, physiology, and metabolism from the human disease. Recent studies have highlighted the use of patient-derived organoids (PDOs) as a personalized model suitable for High throughput screening (HTS) which might help overcome some of the current model limitations (Tiriac et al., 2019).
Pancreatic ductal adenocarcinoma is characterized by dense desmoplasia, which can compose up to 80% of the whole tumor volume and low tumor cellularity, while metastases in the liver have less stroma and more tumor cellularity than primary tumors resulting in less overall survival (Rucki, 2014).
A complex network of inflammatory cells, fibroblasts, ECM and vasculature maintain tissue homeostasis in the stroma of normal epithelial tissues. In Contrast, around pancreatic cancer tissues, neoplastic cells corrupt the stroma to form a tumor-promoting environment which, at the same turn, promotes cancer cell proliferation and migration, and provides a reservoir for growth factors and cytokines.
The three dominant entities in the PDAC stroma are the vasculature, ECM, and cancer-associated fibroblasts (CAFs). Molecular subtypes of pancreatic CAFs have been described (Öhlund et al., 2017), most remarkably myofibroblastic CAFs and inflammatory CAFs, which have been speculated to participate in active crosstalk with cancer cells and pro-tumor and antitumor properties, respectively. Tumor progression is linked with disruption of the basement membrane integrity and desmoplastic reaction with enhanced production of type I collagen (Öhlund et al., 2009; Shields et al., 2012). Moffitt et al. (2015) by digitally separating tumor, normal and stromal gene expression, defined “normal” and “activated” stromal subtypes, which are independently prognostic of PDAC. Using a Hedgehog inhibitor, the decrement of stroma was beneficial in mouse models due to the blockade of stromal growth factors and elimination of the barrier for therapeutic delivery (Olive et al., 2009). Despite those observations, several strategies to target the ECM have been pursued in the last years but have so far failed to show an increase in patient survival (Hosein et al., 2020; Tomás-Bort et al., 2020). Experimentally manipulating stromal matrix content led to lower tissue stiffness, and increased tumor growth, resulting in decreased overall survival. Similarly, a multitude of anti-angiogenesis agents have been unsuccessful in late-stage clinical trials of PDAC probably due to hypovascularity (Hosein et al., 2020).
Since desmoplasia promotes hypovascularity and immunosuppression it results in a hypoxic environment due to limited oxygen diffusion through the tumor (Tuveson and Neoptolemos, 2012). In-depth research has shown that hypoxia modulates tumor biology promoting malignancy through hypoxia inducible factors (HIFs), which should be considered for targeted therapy (Shah et al., 2020). Hypoxia signaling also affects stromal cells, enhancing activation of macrophages, CAFs, stem cells, and secretion of specific ECM factors to produce widespread stroma of PDAC (Keith et al., 2012).
Metastasis is characterized by a sequential process initiated by the invasion of carcinoma cells into the basement membrane into the neighboring stroma, followed by invasion and survival into circulation (blood, lymph), extravasation into the parenchyma of distant tissues, and lastly by the reestablishment of foci of neoplastic cells at remote sites, even after a period of dormancy (Massagué and Obenauf, 2016; Massagué et al., 2017; Figure 1). Accordingly, a central step in the metastatic process is the gain of migratory and invasive phenotype. This demands pancreatic cancer cells to switch many of their epithelial characteristics for mesenchymal traits through a cellular program named epithelial to mesenchymal transition (EMT). The opposite process, mesenchymal to epithelial transition (MET), happens when colonies are re-established at the secondary organ (Beuran et al., 2015).
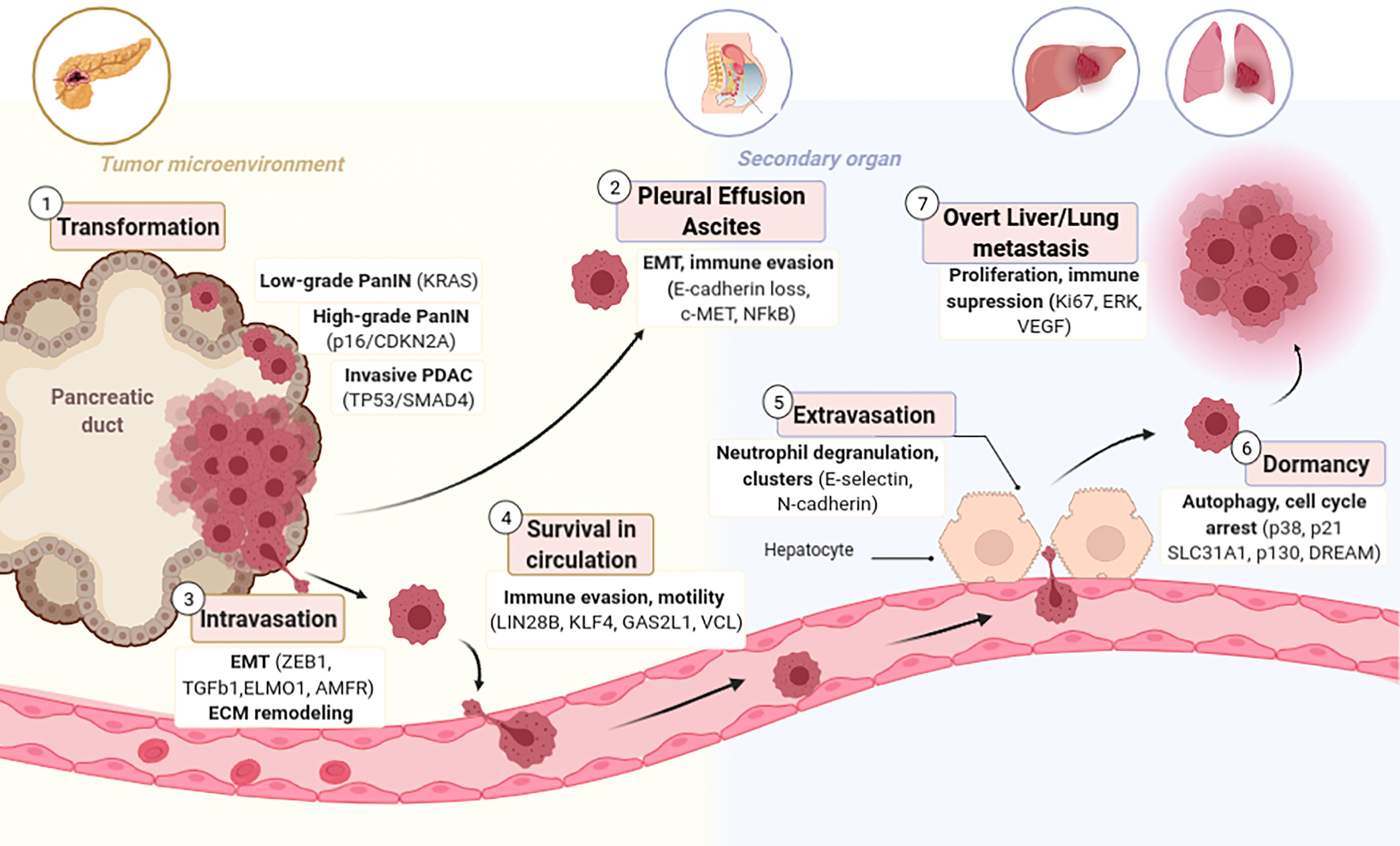
Figure 1. Stages of Metastatic Progression with candidate genes responsible per each stage. (1) Normal epithelial pancreatic ductal cells acquire an aggressive phenotype through serial mutations that transform them firstly to PanIN and lately to PDAC. (2) Transformed cells are capable of detaching and colonizing the Peritoneum forming Ascites or Pleural Effusion. (3) PDAC cells have enhanced motility due to EMT that allows them to invade blood or lymphatic vessels. (4) CTCs in circulation are abundant, but only few survive this pressure. (5) Several CTCs have tropism for the pre-metastatic niche (PMN) and are able to extravasate to a secondary organ where they might remain dormant (6) for several years and eventually relapse and form overt metastasis (7).
The decisive promoters of PDAC metastasis are not yet sufficiently understood, notably since the genetic composition of most metastases closely resembles the one of the complementary primary tumors (Campbell et al., 2010; Yachida et al., 2010; Makohon-Moore et al., 2017). The most studied drivers of metastasis in PDAC are: TP53 (Morton et al., 2010), SMAD4 (Ahmed et al., 2017), aberrant Wnt signaling (Yu et al., 2012), and aberrant ECM gene expression (Harris et al., 2017). Also, reduced expression in Liver Kinase B1 (LKB1) is correlated with liver metastasis, vascular invasion and thus, worse overall survival (Yang et al., 2015, 1). It has been suggested that Transforming growth factor beta (TGF-β) promotes invasion and migration via the initiation of EMT (Padua and Massagué, 2009; Aiello et al., 2017). Molecular perturbations coupled with this transition combine the loss of epithelial markers such as cytokeratins, E-cadherin, occludin, and claudin, with the gain of mesenchymal markers namely N-cadherin, fibronectin and vimentin (Maier et al., 2010), loss of cell-cell contacts and polarity leading to the gain of a mesenchymal migratory behavior (Thiery, 2002). Further, cells that have transitioned to the mesenchymal state embrace a spindle-like shape instead of a columnar one and have elevated invasiveness, migratory capacity, enhanced resistance to apoptosis and increased production of ECM factors (Rhim et al., 2012). It has been described that the PDAC EMT program is defined by an intermediate cell state “partial EMT” consisting of the maintenance of an epithelial program only at the protein level (Jolly, 2015). Partial EMT cells can migrate individually or as clusters while complete EMT cells mainly migrate in isolation (Duda et al., 2010; Grigore et al., 2016; Saitoh, 2018). The various mechanisms of dissemination (single cancer cells or clusters) seem to affect the metastatic capacity of cancer cells, since single cells do not have such a high metastatic capacity as tumor clusters (Friedl et al., 2012; Cheung and Ewald, 2016). Tumor clusters can be heterogeneous and integrated of stromal cells, such as CAFs, co-migrating with cancer cells to remote sites (Wang et al., 2016). Overall, in PDAC, the EMT program has been proved to enhance tumor-initiating potential (Rasheed et al., 2010) and drug resistance (Zhou et al., 2017).
When epithelial cells undergo EMT and enter circulation become circulating tumor cells (CTCs; Figure 1). CTCs isolated from patient blood express a cell motility gene signature consisting of upregulation of EMT and motility inducing genes such as Vinculin, Engulfment and cell motility protein 1, Autocrine Motility Factor Receptor, TFGß1 or p38 (Sergeant et al., 2012). CTCs necessarily contain the precursors of distant metastatic foci; thus, they may be characterized to identify drivers of dissemination, correlate gene set metastatic signatures, and develop targeted therapies to the “seeds” of metastasis (Franses et al., 2020). Using primarily CTCs collected from Genetically engineered mouse models (GEMMs), Growth Arrest Specific 2 Like 1 has been identified as a potential biomarker of CTCs in PDAC (Zhu et al., 2020). Contrary to the previous acceptance that PanINs are not able to undergo EMT, it has been reported that also low-grade PanINs, harboring only activating KRAS mutations, show indication of cells that have exfoliated and become CTCs expressing CD24 and CD44 (Rhim et al., 2012). Inflammation enhances the amount of circulating pancreatic preneoplastic cells, supporting the association between inflammation and PDAC (Mazur and Siveke, 2012). Consistently, treatment with anti-inflammatory agents reduces the amount of circulating PanIN cells and diminishes the number of PanINs in tissue (Tuveson and Neoptolemos, 2012).
The anatomical position of the primary tumor is a crucial determinant in the formation of peritoneal metastasis (Yachida and Iacobuzio-Donahue, 2009; Baretti et al., 2019). In some cases, the exfoliated cells directly attach to and invade organs and tissues in the peritoneal cavity (Jayne, 2007; Yachida and Iacobuzio-Donahue, 2009; Avula et al., 2020). It has been reported that intraperitoneal metastases can also take place via blood vessels or lymphatic absorption through the hematogenous route (Jayne, 2007; Ge et al., 2017). Peritoneal spread of disease is found in around a third of patients with PDAC, which may lead to ascites accumulation in up to 20–30% cases or pleural effusion in around 15% of patients (Golan et al., 2017). Symptomatic retention of fluid with viable cells usually occurs late in the course of the disease, at the clinically treatment resistant phase (Baretti et al., 2019). A detailed analysis of molecular pathways leading to Peritoneal metastasis is reviewed in: (Avula et al., 2020). Briefly, E-cadherin loss, especially in a KRAS mutated background, and HGF/c-Met [hepatocyte growth factor (HGF)/mesenchymal–epithelial transition factor (c-MET)] pathway lead to EMT and cell detachment from the pancreas (Furuyama et al., 2000; Takiguchi et al., 2017; Sato et al., 2020). It is still a challenge to effectively treat peritoneal metastasis, thus further efforts in revealing its mechanism should be addressed in the future. Although there is extensive literature describing ovarian and gastric cancer cell immune evasion through the transition of the peritoneal cavity, this issue has not been exhaustively studied in PDAC.
Several studies prove that the hostile milieu of the liver is particularly preconditioned early to favor the engraftment and growth of disseminated tumor cells (DTCs), so-called pre-metastatic niche (PMN) formation. The formation of PMNs is directed by an intricate series of mutual interactions among the TME and tumor cells, along with the exploitation of recruited and resident cells in secondary target organs. Extracellular vesicles and soluble factors are secreted by the primary tumor or premalignant lesions, even before the initiation of PDAC dissemination. They help to form a supportive niche in the liver by providing vascular docking sites for CTCs enhancing vascular permeability, remodeling the ECM and gathering immunosuppressive inflammatory cells (Houg and Bijlsma, 2018). Hepatic metastases show unique characteristics, such as increased proliferation (Ki67), M2 macrophage infiltration, 3p21.1 loss, downregulation of EMT, and metabolic rewiring (Yang et al., 2021). Reichert et al. (2018) demonstrate, using multiple mouse models, that liver metastases highly depend on P120CTN-mediated stabilization of membranous E-cadherin, while the lung seems permissive to colonization by cells that are not MET-capable. Interestingly, PDAC patients with recurrent lung metastases show significantly better overall survival compared with patients with metastasis at other sites (Yamashita et al., 2015).
Next-generation genome sequencing of untreated pancreatic primary tumors and the corresponding patient metastasis showed that cells triggering distant metastasis are genetically indistinguishable with the different metastatic locus bearing the same driver gene mutations (Makohon-Moore et al., 2017). This implies that post-transcriptional or transcriptional modifications are pivotal to support the intricated series of biological bottlenecks that must be surpassed for PDAC to metastasize (Fidler, 2003; Lambert et al., 2017). DTCs display clonal diversification according to the location of the metastatic foci. Lineage tracing studies revealed that metastases in the lung and liver tend to be monoclonal, while those in the peritoneum and diaphragm exhibit polyclonality due to a different via of dissemination (Kleeff et al., 2016; Knaack et al., 2018). These observations suggest that heterotypic interactions between tumor subclones as well as site-specific selective pressures are both central to influencing metastatic initiation and progression.
Since CTCs do not express β2-integrins, they form clusters with blood cells using them as a linker to attach the capillaries and extravasate to distant organs (Charles Jacob et al., 2021). Extravasation may be controlled by E-selectins, N-cadherin, or galectin-3 from endothelial cells (Yadavalli et al., 2017). Once CTCs extravasate to the secondary organ, they remain dormant with high resistance to current therapies (Sosa et al., 2014). TGF-β and BMPs stromal signals have been identified as promoters of tumor dormancy by enforcing quiescence and inhibiting self-renewal of DTCs (Gao et al., 2012). The perivascular niche has also been reported to induce cancer cell dormancy (Ghajar et al., 2013). In contrast, contexts rich in fibronectin or type 1 collagen inhibit dormancy (Massagué and Obenauf, 2016). A lack of stromal growth factors and an abundance of growth-inhibitory signals can favor metastatic dormancy in experimental models. DTCs are also kept in a dormant state due to constant pressure from the immune system. However, the acquisition of further mutations, inflammation, microenvironmental alterations, as well as immune and stromal signals can promote arouse of dormant cells inducing local relapse or metastases still after curative therapy (Aiello et al., 2017; Lenk et al., 2018; Park and Nam, 2020). Dormany explains why most patients that were resected with no margin experienced a relapse and died of metastatic disease. Thus, it is a future goal to fully understand the pathways leading to metastasis outgrowth and improve current adjuvant combinations to not only eliminate the primary tumor but also to eradicate the dormant foci to prevent relapse.
The formation of metastatic foci occurs with a transition from mesenchymal to epithelial phenotype, leading to enhanced proliferation and metastatic tumor deposit (Makohon-Moore et al., 2017). The aim of adjuvant chemotherapy is the prevention of metastatic relapse. Nonetheless, the current pharmacological armory employed attacks proliferating tumor cells instead of targeting metastasis. Avoiding metastasis in high-risk patients would be ideal rather than treating them. However, the few approved drugs targeting the stroma of metastasis (bisphosphonates, Zometa, anti-RANKL antibody, and denosumab) have not yielded an improvement in the adjuvant setting (Smith et al., 2015; Perrone et al., 2019). Hence, the current standard of care does not instruct any agent to prevent metastasis (Massagué et al., 2017). There are several undergoing clinical trials of targeted agents designed specifically with patients suffering from locally advanced disease or metastasis (Table 1) hoping to improve patients’ overall survival.
The early dissemination capacity of PDAC, even at the PanIN stage, explains why most patients have already found significant metastasis to the liver, lungs, peritoneum or lymph nodes at the time of diagnosis, and subsequent median survival is less than 1 year (Kleeff et al., 2016; American Cancer Society, 2021). Even though we hold an improved understanding of PDAC biology and progression, the translation for patient benefit has been slow. On this subject, a possible contributing factor may be the lack of solid trustworthy models of human PDAC for preclinical testing and research.
Compared to other models, 2D cell culture is the simplest, fastest and most economic form to study metastasis and invasion. There are 49 PDAC cell lines described in Cancer Cell Line Encyclopedia with specific mutations and from different tumor sites (Table 2). Many PDAC cell lines from patients and murine tumors accompanied by different mutations of KRAS, p53, p16, and SMAD4 are widely used in PDAC metastasis research (Sun et al., 2001).
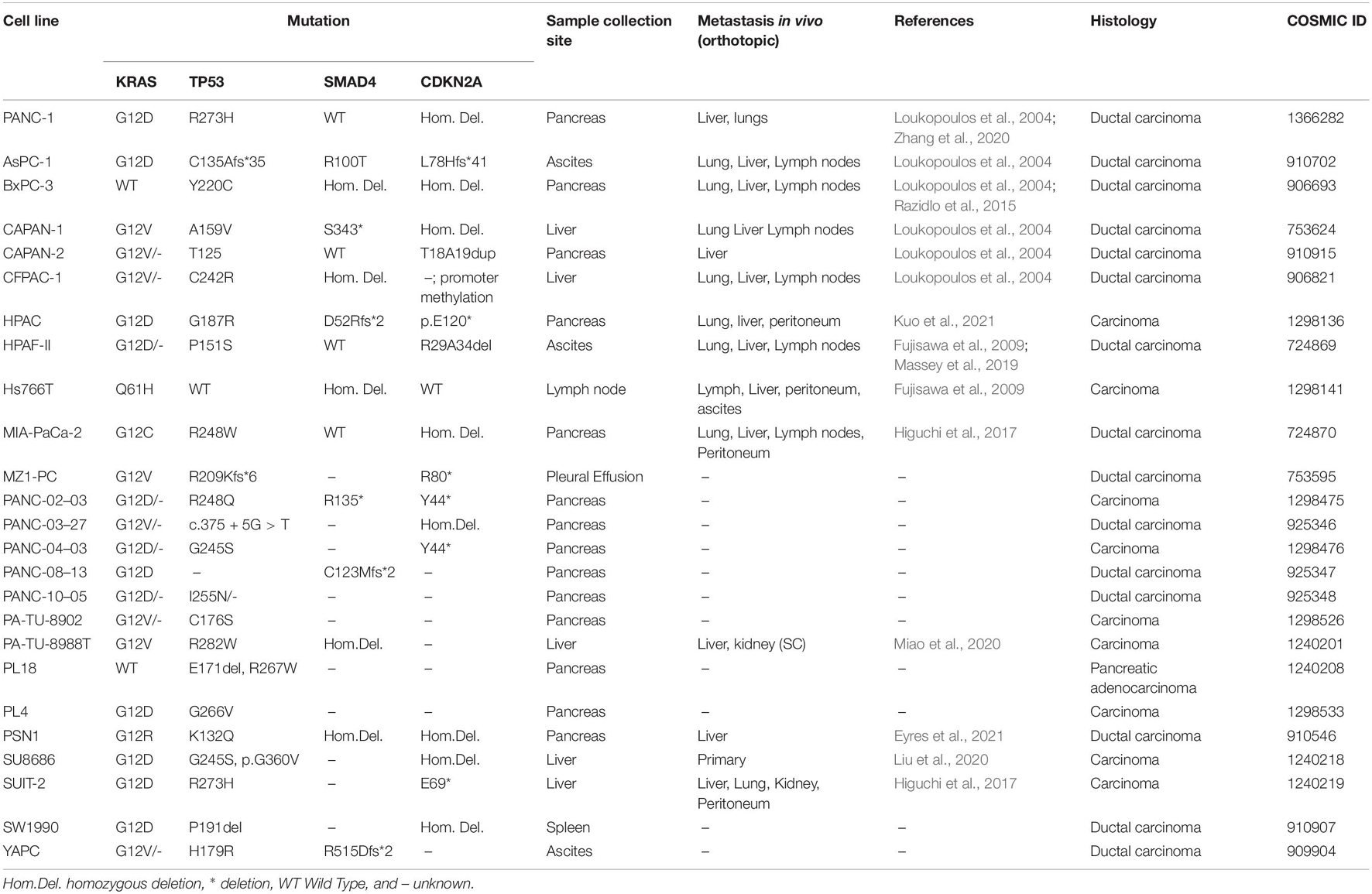
Table 2. Most common Human derived pancreatic cancer cell lines and metastatic activity in orthotopic transplantation model.
Several migration and invasion assays can be performed in cell lines to study metastasis. A fast method to assess migration in 2D cell culture is the wound healing assay, where a scratch is performed in the middle of confluent monolayer cells, and the measurement of cell migration is quantified via microscopy. Using this method several groups were able to identify genes that promote motility namely Yes1 associated transcriptional regulator (YAP1), H19 imprinted maternally expressed transcript (H19) or C-X-C motif chemokine ligand 12 (CXCL12; Cecati et al., 2021; Gao et al., 2021; Luo et al., 2021). However, this method is not able for non-adherent cells and the scratch can cause cell damage. Alternatively, invasion can also be studied using inserts in which cells are seeded on top and invasive cells are able to pass through such as the Transwell or Boyden chamber. To reproduce ECM degradation, it is possible to add a layer of ECM in the inserts (Kramer et al., 2013). Several studies analyzed the invasive capacities of PDAC cell lines through several substrates. Quite unanimously, Capan-1 and Mia Paca-2 have the higher invasion rates followed by PANC-1 and BxPC-3 with slight variations between groups due to subtle differences in methodology (Deer et al., 2010). It is also possible to study the morphology, directionality and velocity of migrating cells using optical mobility assays such as the TAXIScan (Yamauchi et al., 2017). Using this method, it has been shown that only a few cells are able to invade the ECM but they up-regulate the expression of invasion-promoting pathways such as PI3K-AKT (Fujita et al., 2014).
Another benefit of 2D cultures is the capacity to co-culture cancer cells with stromal cells, and model the signals from the TME. For example, co-culturing cancer cells with Pancreatic stellate cells (PSCs) showed that cancer cells had enhanced EMT markers and migration (Kikuta et al., 2010). Recently, the development of microfluidic assays has allowed investigating the biophysical parameters in PDAC metastasis modeling the chemical gradients, flow/shear stress and the complex interactions between several cell types (Kramer et al., 2019). Using PDAC cell lines and combining the previously mentioned assays with CRISPR-Cas9 technique has revolutionized the field of metastasis research. Currently, there are available genome-wide or custom made sgRNA CRISPR libraries that have helped identify genes promoting migration, chemoresistance or radioresistance such as Histone Deacetylase 1, ATP binding cassette subfamily G member 2, Endoplasmic reticulum-associated protein degradation (Du et al., 2021; Ramaker et al., 2021).
Lack of germline DNA and missing clinical annotation are general problems when working with established cell lines. Since 2D cell lines are separated from tissue and cultured on a flat cell culture surface, they undergo several in vitro selection steps. They will divide abnormally, become flat, and lose their differentiated phenotype (Kapałczyńska et al., 2016). Thus, some cell types are not well-represented and the tumor heterogeneity is reduced. Recently, single-cell sequencing technologies showed that there is a high degree of heterogeneity in commonly used PDAC cell lines, induced by culturing identical PDAC cell lines in different laboratories. They also question the use of immortalized, non-transformed pancreatic lines as control lines in the experiments (Monberg et al., 2021). Despite their limitations, cell lines have been pivotal tools for screening in pre-clinical settings the genes promoting migration and survival in PDAC as well as chemo- or radioresistance.
Despite being time and cost-effective, 2D cell cultures do not represent the architecture and structural complexity of human tissues. 3D culture of normal cells and their neoplastic counterparts was introduced in the 1970s (Emerman and Pitelka, 1977). Since the development of the hanging drop technique, spheroids have been utilized to study morphogenesis together with the composition and architecture of malignant tissues (Kelm et al., 2003) using several techniques summarized in Table 3. In semisolid matrices resembling the basement membrane, cell-cell contacts and cell-matrix interactions allow epithelial cells to develop polarized structures. Abounding ECM components namely collagen, fibronectin and laminin are necessary to support these cultures. Interestingly, studies that have compared transcriptomes between 2D and 3D cultures have shown that cells are highly influenced by cell-matrix interactions. Several PDAC cell lines are able to grow in spheroids but it is unclear their ability to reflect the properties of the human tumor since they undergo deep transcriptomic transformations in transitioning from 2D to 3D (Monberg et al., 2021).
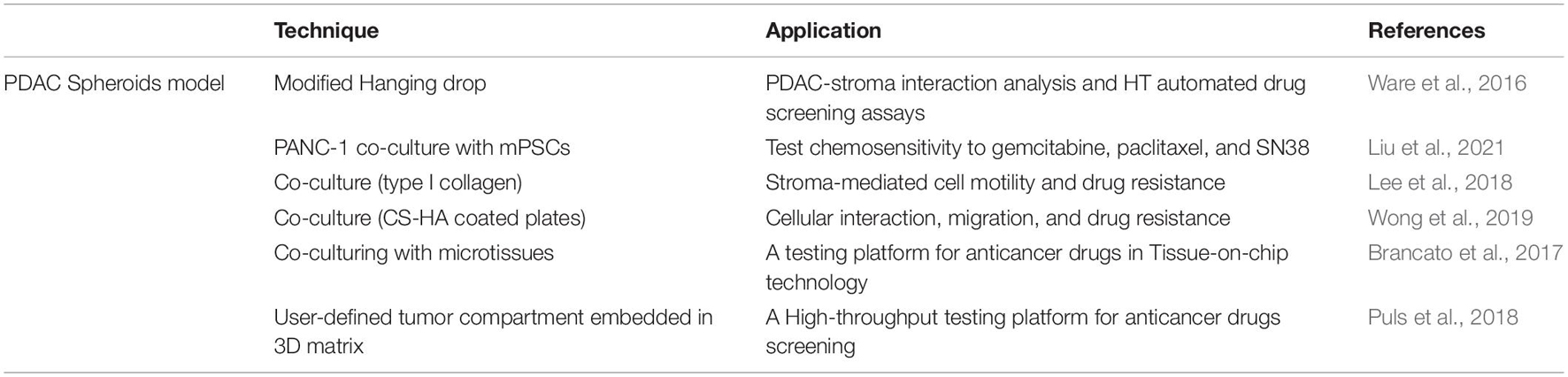
Table 3. Three-dimensional spheroid models for PDAC research and their applications in metastasis.
Pancreatic stellate cells are the main source of stromal fibrosis, interacting closely with cancer cells to produce a supportive environment that drives local and remote neoplastic development. By co-culturing PDAC spheroids with ECM components, it is possible to model PDAC-stroma interaction. Some groups have shown that PSC co-cultured spheroids reflect PDAC chemotherapeutic responses (Lee et al., 2018; Wong et al., 2019; Liu et al., 2021). For instance, PSC/PDAC spheroids showed enhanced resistance to gemcitabine in comparison to PDAC-only spheroids, while c-MET inhibitors crizotinib, tivantinib, and PHA-665752 were similarly effective in both models (Firuzi et al., 2019). Recently, other groups also proved the increased chemosensitivity from heterospecies spheroids to gemcitabine, paclitaxel and SN38. Interestingly, this group also showed that upon co-culture mPSC induces a shift from classical to a basal-like phenotype to PANC-1 spheroids showing the importance of TME in patient prognostic and metastasis development (Liu et al., 2021). CAFs are activated to myofibroblast and tumor-dependent lymphocyte infiltration is observed on co-culture reproducing the desmoplastic reaction of PDAC and providing a valuable tool for anticancer drug testing (Brancato et al., 2017; Tsai et al., 2018).
Immune cells interfere in treatment response and tumor progression (Qiu and Su, 2013). 3D approaches admit the inclusion of human immune cells in contrast to patient-derived xenografts (PDXs) which are established in immunodeficient mice. When T cells were added to monocyte and fibroblasts co-cultured with PDAC spheroids overexpressing Doublecortin-like kinase 1 -isoform 2, M2 monocytes were polarized via cytokine release which then inhibits CD4+ and CD8 + T cell activation and proliferation (Kuen et al., 2017; Chandrakesan et al., 2020). Since knockdown of DCLK-isoform2 resulted in enhanced CD8 + T cell activation and decreased pancreatic cancer cell viability—this study with spheroids suggested DCLK-isoform2 as a novel therapeutic target in PDAC (Chandrakesan et al., 2020). Importantly, another triple co-culture platform has been developed combining PANC-1, endothelial cells (HUVEC) and fibroblasts (MRC-5) which also mimicked the resistance to treatments observed in vivo to doxorubicin and gemcitabine hence proving the key role of a complex tumor microenvironment (Lazzari et al., 2018).
With additional therapies for stroma targeting and 3D patient models that replicate a patient’s specific TMEs, it is an exciting time for PDAC research. Several Clinical Trials target the tumor microenvironment of PDAC (Tomás-Bort et al., 2020). 3D cell cultures, and specially PSC/PDAC spheroids, are important tools for screening of cancer and stroma targeting drugs permitting a validation step preceding animal testing and reduce the number of animals required (Ishiguro et al., 2018). 3D modeling of cell culture may aid in drug discovery and biological treatment. While current 3D spheroid invasion models more precisely replicate tumor invasion compared to conventional 2D models, they have limitations such as low reproducibility and the difficulty to interact with high-throughput (HT) systems.
To overcome this limitation, Puls et al. developed a 3D tumor-tissue invasion model for HT phenotypic drug screening. In short, PDAC cell lines are embedded in an Oligomer in suitable plates for HTS where it is possible to monitor invasion into the surrounding tissue. When CAFs were added this highly enhanced PDAC invasion, as is expected to occur in vivo. Additionally, they showed that gemcitabine inhibited proliferation while not fully eradicating the tumor or blocking invasion. These results line up with those from PDAC xenograft models which show gemcitabine substantially arrests tumor growth and proliferation but does not induce apoptosis or reduction of remote metastases and invasion related markers (Puls et al., 2018). Although 3D spheroids have proven useful in cancer cell research, it is acknowledged that a passive environment does not adequately represent the cellular development of these cancer cells. The tumor cells grew throughout time without being suppressed by the drugs; however, it is not clear how much of the development was hindered or accelerated as a result of static media supply (Holub et al., 2020).
Inferring results from model systems to humans has been a major barrier in the drug discovery process. In the last decade, a surrogate in vitro 3D model for human and mice tissues, named organoids, has been refined. Unlike spheroids, organoids are not derived from cell lines but from primary cells. Moreover, organoids allow studies of tissue function since tissue-like structures are preserved (Marsee et al., 2021). Stem cells are isolated from mouse or human adult tissues and embedded in 3D matrices where they self-organize into epithelial structures (Tuveson and Clevers, 2019; Kim et al., 2020). They also maintain intra-tumor heterogeneity, cell polarity and interact with the ECM, resembling the molecular features of the original tumor. Not long ago, organoid cultures of pancreatic epithelium have allowed the culture of normal and neoplastic pancreatic epithelial cells for both humans and mice (Huch et al., 2013; Hindley et al., 2016; Boj et al., 2018).
To establish organoid cultures, it is necessary to mimic the homoeostatic surrounding of the normal tissue stem cells. For this purpose, cells are encapsulated in Matrigel, which contains the crucial components of the basement membrane, and complemented with the minimal essentials for sustainable growth of pancreatic epithelial cells left out mesenchyme. Since the majority of PDAC samples have high penetrance of KRAS activation (Yachida and Iacobuzio-Donahue, 2009), it is possible to apply selective pressure conditions withdrawing EGF or adding EGFR inhibitors to obtain a pure neoplastic culture.
Since organoids are genuine epithelial populations, they bypass the stromal suppression that primary tumors retain, allowing comparisons to normal ductal pancreatic cells (Boj et al., 2018). They can be established in several weeks even from small fine needle aspiration biopsies acquired from patients with advanced PDAC, enabling therapeutic testing and tumor response during treatment or disease progression. Employing a large cohort of PDOs, Tiriac et al. (2018) have developed a platform for testing single and targeted agents. They display, in retrospective case studies, that organoid response to therapeutic testing correlates with patient sensitivity to chemotherapy. By correlating the transcriptome and drug sensitivity profile of each organoid in the cohort, they defined transcriptomic signatures of chemosensitivity with prognostic clinical outcomes in treated cohorts of PDAC patients. Other authors found the same concordance when testing similar platforms (Huang et al., 2015; Romero-Calvo et al., 2019; Dreyer et al., 2021b). In PDOs obtained over multiple years in a metastatic PDAC patient, it was possible to show increased organoid resistance to chemotherapy in accord with treatment refractory cases (Tiriac et al., 2018). Organoid work has also shown that Beta-1,4-galactosyltransferase 1 (B4GALT1) promotes PDAC progression and chemoresistance via stabilization of CDK11p110 (Chen et al., 2021, 110). In the biomarker field, organoids showed that higher EV release is coupled to a high cell proliferation rate, promoted by Wnt pathway activation (Sándor et al., 2021).
Since their implementation, organoids have been able to demonstrate good genomic parallelization with the primary PDAC tumors (Tiriac et al., 2018; Gendoo et al., 2019; Romero-Calvo et al., 2019). Also, PDAC subtypes have been identified in independent cohorts of PDOs implying that the transcriptional programs are preserved. Seino et al. (2018) defined functional subtypes of PDAC and demonstrated an inverse correlation between and strict requirement for WNT-signaling and GATA6 expression (linked with classical subtype), thus implying that GATA6 acts as a key regulator of niche-dependency. This emphasizes the call for precision methods to select patients when considering Wnt pathway therapeutic approaches, for example with the porcupine clinical trials. In addition, organoids are genetically manageable for viral infection and transfection, allowing targeted evaluations of particular genes or genetic screening (Michels et al., 2020).
Co-cultures of organoids with PDAC stromal cells helped understand fibroblast heterogeneity and suggested new approaches for treating PDAC by blocking the fibroblasts that support the tumor and promoting tumor restraining fibroblasts (Öhlund et al., 2017; Tsai et al., 2018; Biffi et al., 2019). These co-culture conditions have also shown that CAFs modify the EMT phenotype and drive gemcitabine resistance induced by HGF derived from CAFs. Furthermore, high stromal expression of Paired related homeobox 1 (Prrx1), a transcription factor critical for activating CAFs, is displayed in the squamous subtype (Feldmann et al., 2021). Using organoids and mice, Walter et al. showed that MEK inhibition suppresses TGFβ-induced EMT and migration in vitro and eventually results in a greater decrease in CTCs in vivo (Walter et al., 2019). Further studies in PDAC metastasis have been achieved by creating organoid derived xenografts (ODX; section “Organoid derived xenografts”).
The recent findings prove that organoids should be a focal point of future studies of PDAC. Overall, organoids recapitulate the human disease much closer than spheroids or cell lines. They allow tissue function studies and co-culture. Since it is possible to culture the normal and neoplastic compartment, organoids are well suited for therapeutic testing and have intermediate scalability. However, organoids are still a complex model that requires a lot of technical training and represent a high cost to establish and maintain (Marsee et al., 2021). Despite showing correlation with patient transcriptomics subtypes and chemoresistance signatures, it has been shown that there are transcriptomic switches during ex vivo passage that may restrict their predictive abilities (Monberg et al., 2021), thus correct passage monitoring is required. To bring to the clinic fast organoid testing of PDAC patients, further work is required in accelerating organoid establishment and testing techniques of valuable compounds.
Recently, a consortium named PRECODE (European Commission, 2021) was established where several laboratories collaborate working on Pancreatic Cancer Research in Organoids in different fields helping to push forward the understanding of this disease and get closer to the development of an effective treatment for PDAC.
In vivo models are key to study alternative and innovative treatment approaches. Despite the great advantages of in vitro research, namely cost-efficiency and simplicity, these models are lacking a microenvironment, immune system and don’t represent tumor heterogeneity. Thus, in vivo models have been widely used to overcome these limitations for metastasis research and allow the understanding of the complex crosstalks involved in metastasis and defining its stages.
Transgenic models, using tissue or cell-type specific promoters, allow the ectopic and temporal expression of target genes in the mouse genome. Different pancreatic cancer lineage specific promoters have been employed in GEMMs like pancreatic and duodenal homeobox 1 (Pdx1), neurog3 (Ngn3), elastase (Ela), among others.
While several chemical and genetic approaches to generate PDAC in mice date back to the 1980s (Longnecker, 1984), it was the establishment of the KrasLSL.G12D mice (Johnson et al., 2001) in 2001 that permitted tissue-specific expression of oncogenic Kras under physiological control from the endogenous mouse locus. This model developed Lung cancer but not PDAC. After this, several GEMMs faithfully recapitulating the genetic, molecular, histological, and clinical hallmarks of human PDAC have been established (Table 4). A full review of GEMMs for PDAC is available in: (Westphalen and Olive, 2012).
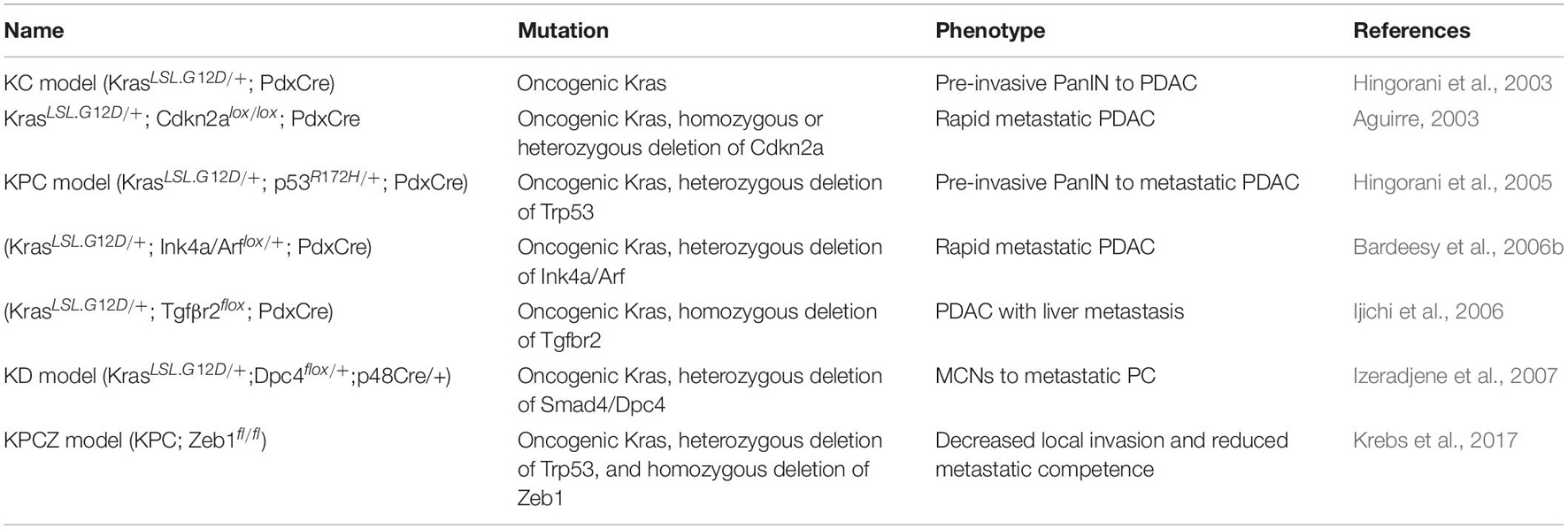
Table 4. Genetically engineered mouse models of pancreatic cancer summary of the most common GEMMs of PDAC driven by KrasG12D.
While transgenic mice are fast to develop and permit the expression of human genes (Qiu and Su, 2013), the expression occurs under foreign promoters. To circumvent these limitations, conditional models are used expressing the desired mutations within the endogenous locus by interbreeding mice carrying the mutant allele downstream of a “Lox-STOP-Lox” (LSL) cassette with Cre driver mice (Magnuson and Osipovich, 2013). In 2003 (Hingorani et al., 2003) by crossing PdxCre or p48Cre mice to KrasLSL.G12D mice the expression of KrasG12D was specifically targeted to the pancreas. In the KrasLSL.G12D/+; PdxCre model (KC model), mice are born with normal pancreas but develop PanIN at 8 weeks and slowly increase in grade, with a subset of those developing PDAC. The KC model proved that Kras mutations are enough to initiate PDAC formation in mice while targeted conditional mutations in Cdkn2A, Smad4, or p53 did not lead to PanIN or tumor development with PdxCre expression. However, this long latency, background tumors and sporadic progression to metastasis hampered the utility of the KC model for preclinical applications.
By combining oncogene activation and tumor suppressor inactivation, it has been successful to generate metastatic PDAC models resembling human disease. Aguirre (2003) showed that homozygous deletion of Cdkn2a in the context of Kras mutation in the pancreas (KrasLSL.G12D/+; Cdkn2alox/lox; and PdxCre) led to the rapid development of metastatic PDAC. Similarly, the loss of the Ink4a/ARF locus in Kras mutant mice promotes NF-kB, Notch signaling and metastasis (Bardeesy et al., 2006a). Conditional expression of p53R172H also accelerated KrasG12D pancreatic tumorigenesis. Although KrasLSL.G12D/+; p53R172H/+; and PdxCre mice (KPC mice) are born with histologically normal pancreas, they rapidly develop PanIN lesions, and die of PDAC in 5.5 months with ∼80% metastasis (Hingorani et al., 2005). This model showed indices of widespread genomic alterations, a characteristic that was previously missing in most GEMMs. Since KPC mice mirror the dynamics of the human TME, they are useful to study tumor-stroma interactions as well as disease progression and testing immunotherapies. Other models addressed the deletion of Smad4 in Kras mutant pancreatic cells but the histology of those tumors was more similar to MCNs or IPMNs (Bardeesy et al., 2006b; Izeradjene et al., 2007). Interestingly, homozygous deletion of transforming growth factor beta receptor 2 (Tgfbr2) combined with KrasG12D formed PDAC with metastasis with special tropism to the liver, suggesting that activated Ras signaling and hampered TGF-β signaling cooperate to advance PDAC progression (Ijichi et al., 2006).
Classical Cre-loxP GEMMs depend on a single Cre-mediated step of recombination to activate oncogenic Kras expression not allowing sequential multistep tumorigenesis and tumor heterogeneity, which are important hallmarks of PDAC. With a dual-recombinase system (Schönhuber et al., 2014), Krebs et al. (2017) generated the KPC; Zeb1fl/fl mice (termed KPCZ) model and discovered that the EMT-TF Zinc Finger E-Box Binding Homeobox 1 (Zeb1) is a crucial factor for driving metastasis.
Genetically engineered mouse models have enlightened the biology of PDAC, elucidated potential therapeutic and diagnostic targets, and accentuated the importance of the tumor stroma for pancreatic cancer immune evasion, maintenance, and drug resistance. Nonetheless, it is an expensive and labor-intensive model to generate and maintain. In addition, gene mutations are brought into the germline of mice, while they occur somatically and gradually in human tumors. Nonetheless, these limitations may be overcome by the use of CRISPR-Cas9 in mouse models. Recently, CRISPR-Cas9 technology has allowed more precise genome editing (Doudna and Charpentier, 2014; Platt et al., 2014). Using this method, Ideno et al. developed the Ptf1-Cre; LSL-Cas9 mouse model, which recapitulates human PDAC features such as PanIN or IPMN with potential advancement to PDAC (Ideno et al., 2019). Ischenko et al. (2021) used CRISPR-Cas9 to inactivate Kras in mice and demonstrated that in advanced tumors, Kras tumor growth dependence is diminished and is shown in the suppression of antitumor immunity.
All these models prove the crucial role of KRAS in the biology of pancreatic cancer; even though efforts to target KRAS directly have not been successful to date. Thus, Ras effector pathways namely Phosphatidylinositol 3-kinase (PI3K)- Protein Kinase B (AKT) and Raf- Mitogen-activated protein kinase kinase (MEK)- Extracellular signal-regulated kinase (ERK) have been investigated as potential surrogates (Mann et al., 2016). Following this line, by crossing to KC mice and analyzing transposon insertions in the resulting tumors, it has been described a large number of candidate genes that may promote tumor progression in KrasG12D initiated pancreatic tumors namely TGF-β and p16/CDKN2A. Genes implicated in chromatin remodeling were identified, including Ubiquitin Specific Peptidase 9 X-Linked (Usp9X), which plays an important role in the pathogenesis of PDAC (Pérez-Mancera et al., 2012; Mann et al., 2012). Several groups followed Usp9X and showed its association with worse prognosis in PDAC (Liu et al., 2017b; Pal et al., 2018).
The grade of aneuploidy in human tumors leads to a great variety of intertumoral gene modifications, with a completely different appearance as in mice. Overall, these species-related differences hamper the capacity of GEMMs to predict the true therapeutic response of PDAC patients in clinical trials. To overcome these limitations, transplantation models might be used.
Transplantation models consist of the engraftment of mouse or human cells/spheroids/organoids/tissues into recipient mice. This provides the benefit of tractability and a relatively lower and more predictable tumor latency than transgenic models. The transplantation can be orthotopic (in the pancreas), or heterotopic (subcutaneous, intraperitoneal, intravenous, intrasplenic, or intra-cardiac) according to where the cells are implanted. Cells engrafted via orthotopic transplantation may spread from the primary tumor to remote organ sites, hence permitting the modeling of the entire metastatic cascade; whereas when injected heterotopically into circulation it is possible to reproduce the steps of dissemination, extravasation, and colonization (Gómez-Cuadrado et al., 2017). Different sites of vascular injection define the site of colonization. For example, injection of cancer cells in the tail vein leads to the development of lung metastases since the cells are rapidly trapped in the microvasculature of the lung. Intrasplenic injection leads to the formation of micrometastasis in the liver. On the other hand, intracardiac injections allow systemic dissemination and are used to model brain or bone metastasis (Khanna, 2004). Moreover, transplantation models can be xenogeneic (xenograft) or syngeneic (allograft).
Allograft transplantation models are established by the transplantation of mice derived neoplastic cells and tumors into mice. They permit the study of metastatic dissemination with an intact immune system, and hence more closely recapitulate the TME. Allografts from isolated cancer cells or tumor pieces derived from GEMMs were characterized by a faster and more consistent development of primary tumors and up to 90% metastasis in the liver compared to GEMMs (Li et al., 2019). The abundance of metastasis in this model is probably a result of focal disease formation, closely mimicking the random mutations in KRAS present in human disease (Tseng et al., 2010).
In contrast to allograft models, xenografts require the implantation of human tumors or cancer cells into immunocompromised mice. Pancreatic cancer cell lines or spheroids are a frequent source for transplant. Nonetheless, as phenotypic and molecular properties may switch in culture, xenograft models of cancer cell lines do not always anticipate clinical responses (Garcia et al., 2020) and thus 3D models are a better alternative.
One of the solutions to address the many shortcomings of 2D cell lines is to establish cell line derived xenografts (CDX). PDAC cell lines are implanted into mouse models to research and test the efficacy of anti-cancer therapies in vivo and metastasis formation. Several studies produced allograft models with C57BL/6, such as TB 32047 (Lu et al., 2020), KPC cell line (Torres et al., 2013), or Pan02 (Jiang et al., 2014). In contrast to allograft models, human PDAC CDX are established by transplanting PDAC cell lines into immunocompromised mice. Resuspended cells in Matrigel for injection to establish an orthotopic mouse model of PDAC is a common method. The orthotopic injection of SUIT-2 cells into the pancreas can induce a process similar to the spread of human PDAC (Higuchi et al., 2017). 3–14 days after inoculation, Higuchi et al. observed intraperitoneal dissemination, extrapancreatic invasion, and further hematogenous organ metastases of SUIT-2 cells (Table 1).
The lung and liver are the most common sites of metastatic PDAC at diagnosis. Most CDX models are generated by subcutaneously injecting PDAC cells into immunodeficient mice. However, subcutaneous xenograft tumors rarely metastasize and thus orthotopic models are a better alternative (Table 1). PANC-1 and KP3, AsPC-1 and KP2 develop liver metastases while only AsPC-1 showed signs of lung metastases (Zhang and Du, 2019). Interestingly, multinucleated cells and spindle cells have been observed in liver and lung metastases playing an important part in metastasis formation. There is also a lung metastasis model by injecting PDAC cells via the tail vein (Kong et al., 2020). Metastatic tumors can be observed in the lungs and other organs after about a month.
Pancreatic ductal adenocarcinoma orthotopic metastasis mouse models are successfully established by injection 2D cells into pancreas. There have also been some studies that xenografted 3D spheroids from PDAC cells (Durymanov et al., 2019; Azmi et al., 2020). 3D spherical culture, as opposed to classic monolayer cell culture, more nearly replicate in vivo conditions inside a microenvironment, which can improve the defects of 2D culture (Liu et al., 2021). Furthermore, in contrast to their cell-based counterparts, spheroid-based xenografts (SDX) show increased expression of pro-fibrotic and pro-survival PDAC hallmarkers (Durymanov et al., 2019). Orthotopic implantation can progress pancreatic tumor to liver and lung metastasis tumor, which is similar to humans. But some PDAC cell lines are difficult to metastasize. Tail vein and splenic injection can easily perform metastasis, but it doesn’t produce a primary tumor. Secondly, immunodeficient mice successfully avoided rejection during xenotransplantation, but it also limits the study of metastasis progress since adaptation to the immune system plays an important role in the selection of metastatic mutations (Gonzalez et al., 2018). Another drawback of the CDX-SDX models is that they may not achieve the medical requirements of individualization and precision. Cell lines cannot accurately reflect the complexity of tumor heterogeneity and can only represent patients with certain types of cancer (Xu et al., 2018).
Orthotopic transplantation of human pancreatic tumor organoids into immunocompromised mice mimics the full spectrum PDAC progression, forming PanIN-like stages and advancing to invasive and metastatic PDAC (Boj et al., 2018). The only difference is that PanIN-like structures are not intraductal epithelial structures within the pancreas of the mouse. Conventional CDXs only repopulate the host environment but do not form any PanIN-like structure. In addition, ODXs closely recapitulate the dense collagen deposition present in human PDAC tissues and tumors from GEMMs, another feature missing in CDXs (Kim et al., 2009; Olive et al., 2009; Raimondi et al., 2020).
It is still unknown how engrafted PDAC organoids form PanIN-like structures. As organoid cultures can better retain tumor cell heterogeneity, it is possible that recovering several stem cells better mimics the different stages of disease progression upon implantation. In addition, it could be the organoid culture conditions that better reflect the cellular plasticity and epigenetic changes upon transplantation. The interaction of the matrix with pancreatic cancer cells in organoid cultures may help to switch into a PanIN-like biological stage (Hwang et al., 2016). Differentially from other xenograft models, ODXs offer a unique chance to study PDAC progression in vivo and early biomarkers. Undergoing research will evaluate the benefits of the ODXs for therapeutic and diagnostic development in comparison to classical PDAC models.
Patient-Derived Xenografts have been established as a rising tool to recapitulate tumor heterogeneity, genetics, and cancer microenvironment of PDAC. PDXs are used to identify new biomarkers, enhance therapeutic outcomes and also as tools for personalized treatments of PDAC patients.
Patient-derived xenografts from patient tumor tissue represent a more favorable alternative to CDX, SDX, or ODX since there is no in vitro selection. Patient tumor pieces are implanted into immunocompromised mice orthotopically or subcutaneously for propagation in vivo, followed by passage of tumor fragments in subsequent generations (Tentler et al., 2012). PDAC PDXs develop between one and 4 months after transplantation, with an engraftment rate between 20 and 80% (Garcia et al., 2020). They have been shown to conserve metastatic potential and histology of the original tumor (Hidalgo et al., 2014) and closely mirror drug responses in human patients (Voskoglou-Nomikos et al., 2003). This may represent the fact that PDXs are not composed of cancer cell populations separated from human tumors and adapted to culture conditions. Since PDX models are established from tumor fragments, the tumor neoplastic cell architecture is retained (Loukopoulos et al., 2004; Rubio-Viqueira and Hidalgo, 2009). Although the initial human stroma is gradually replaced with cells of the murine host, these models can recapitulate the complexity of the TME in PDAC (Duda et al., 2004). Also, successively passaged PDXs normally show consistent biological properties and homogeneous histological and molecular phenotypes (Aparicio et al., 2015). Since it is necessary to use immunocompromised mice when generating xenografts, this represents a major drawback to study metastasis, since the adaptive immune system plays a crucial role in the selection of metastatic variants (Gonzalez et al., 2018).
Chick chorioallantoic membrane (CAM) is a deeply vascularized extraembryonic membrane formed after embryonic day 5 rich in type IV collagen and laminin, which are similar to the human basement membrane. Chick embryos are not immune-competent until day 18 (Ribatti, 2016). More and more studies have confirmed that the CAM model can efficiently sustain tumor cell proliferation, making it a simple and rapid model for studying initial tumor development. It can reproduce all stages of tumor formation in a shorter amount of time as tumors can be detected after only 4 days of cancer cell injection (Komatsu et al., 2019). It has been shown that firefly luciferase-labeled primary PDAC cells can be engrafted onto the CAM with >80% success. A comparison of tumors harvested from the CAM with original human tissues by immunohistochemical staining showed similar positive staining for the PDAC markers cytokeratin, Cytokeratin 19, Cytokeratin 7, mucin-1, and Alcian blue. Importantly, the percentages of positive/negative cells within each model are very consistent (Rovithi et al., 2017). In addition to using fluorescently labeled cells, cell invasion can be analyzed by Alu PCR to estimate the presence of metastatic human cancer cells in the organs of the chick embryo. Li et al. extracted genomic DNA from chick CAM and liver, respectively, and Alu sequences in human cells were specifically detected by Alu PCR revealing that 31% of CAM and 0% of liver tissue were Alu positive in embryos with untreated Aspc-1 cells. After dexamethasone treatment, the invasion rate of Aspc-1 tumor cells to CAM and liver increased metastasis to 85% and 60%, respectively (Mira et al., 2002; Liu et al., 2017a).
In summary, CAM-assay is a flexible, cost-efficient, reproducible and rapid approach that can evaluate the metastatic capacity and aggressiveness of different PDAC cells within a short time in vivo. With the help of physiological and histological characteristics of PDAC, it is easy to assess the key features of tumor metastasis such as angiogenesis, intravasation and spontaneous metastasis. Therefore, CAM assay is an attractive metastasis model. Immune deficiency of chick embryos is up to 18 days, so the short observation periods (3–9 days) become an important limitation of CAM-assay. In addition, it cannot examine cancer-immune cell interactions. Another limitation to this system is that chick embryos are extensively vascularized organisms characterized by fast morphological changes (Lokman et al., 2020). CAM-assay for tumor research is also limited in its monitoring capabilities of tumor size. Because the tumor is encased by a radiopaque eggshell and has a modest structural size, it can only be monitored from above, posing a challenge to existing imaging modalities. Even though repetitive ultrasonography can monitor tumor growth and vascularization, it also relies on the experience of the experimenter in ultrasound (Eckrich et al., 2020).
Recently, the value of the zebrafish model has been appreciated. Teleost zebrafish (Danio rerio) shows large levels of physiologic and genetic analogies to mammals, closely resembling the clinical setting and allowing the natural history of the tumor to be monitored (Wu et al., 2017). Fishes are routinely maintained at 28°C, but the most favorable temperature for tumor cell proliferation is 37°C (Cabezas-Sainz et al., 2018). When engrafted zebrafish are raised at a compromise temperature (≤34°C), cancer cells do not proliferate at the same rate as when cultured in mice or humans. The Protein Kinase, DNA-Activated, Catalytic Subunit (prkdc) and Interleukin 2 receptor (il2rga) deficient zebrafish model can be raised at 37°C and can engraft a wide range of human malignancies (Yan et al., 2019). CRE/LOX technology and GAL4/UAS systems were combined to create the first kras-initiated PDAC model in zebrafish that highly recapitulates human PanIN development (Oh and Park, 2019). A xenograft model was established in zebrafish by transplanting human pancreatic cancer cells into the perivitelline cavity of 48 h post-fertilization zebrafish embryos (Guo et al., 2015). Subsequently, they observed that cells with kras mutations displayed significant proliferative and migratory behaviors invading the zebrafish vasculature system. Then xenotransplanted larvae were exposed to an inhibitor that targets the KRAS signaling pathway named U0126. There were fewer metastases in the bodies of the larvae in the following U0126 treatment group while the mock-treatment group displayed recurrent metastasis.
Zebrafish is a useful and economical in vivo animal model for speedy analysis of invasion and metastatic behavior. Zebrafish embryos are transparent, so it is easy to follow the invasion, circulation of tumor cells in blood vessels, migration and micro metastasis formation in real-time (Marques et al., 2009). The entire genome of zebrafish has been determined completely, and the genetic background is clear. Therefore, it can be used for large-scale genetic background screening (Gut et al., 2017). Zebrafish are highly reproductive, with a pair of zebrafish typically producing around 200 embryos (Hoo et al., 2016), and there is little difference between individuals, so they can be used for mass drug screening, such as anti-metastasis drugs (Nakayama et al., 2021). However, the most obvious shortcoming of zebrafish is that it is not a mammal. It is significantly different from humans directly and cannot fully simulate the type of human disease. In addition, antibodies against zebrafish protein are still lacking on the market compared to mice or humans (Hason and Bartùnìk, 2019). In the process of zebrafish xenograft, the temperature of embryo incubation, the different sites for implantation of tumor cells, the interaction between cells and host in embryo, and the changes of tumor microenvironment all affect the experimental results of cell proliferation, invasion and metastasis (Cabezas-Sáinz et al., 2020). Therefore, the technology of xenograft still needs to be improved in different aspects.
CRISPR/Cas9 research technology has been developing in recent years and has become a versatile tool for making changes to the genome of many organisms. Here describes some research on CRISPR technology in PDAC metastasis. It has been reported that Mucin 16 (MUC16) contributes to the metastasis of PDAC through FAK mediated Akt and ERK/MAPK pathway. MUC16 knock-out cells generated by CRISPR/Cas9 also exhibit reduced mesenchymal expression and enhanced epithelial expression in PDAC cells and inhibit cell metastasis (Muniyan et al., 2016). Vorvis et al. merged genetic and microRNA profiling analysis with CRISPR/Cas9 technology and identified that transcription factor Forkhead box A2 (FOXA2) is implicated in PDAC pathogenesis. Furthermore, suppression of FOXA2 levels by CRISPR/Cas9 in vitro resulted in the activation of the Plasminogen activator, urokinase receptor gene known to be implicated in invasive malignancy. These results were consistent when FOXA2 expression was blocked by siRNA (Vorvis et al., 2016). Core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase 1 was disrupted in human PDAC cells (T3M4 and CD18/HPAF) by CRISPR/Cas9 leading to enhanced O-glycosylation truncation on MUC16, which increases the formation of aggressive PDACs and metastasis in KPC mice. Stock, al. generated two cortactin knockout lines (PANC-1 and BxPC-3) using CRISPR/Cas9 technology to study the functional role of cortactin in PDAC (Chugh et al., 2018). In PDAC metastases, they detected more expression of cortactin and Tyr421-phosphorylation than the original tumor. Cortactin activation and the migratory ability of the PDAC cells both decreased significantly after treatment with an inhibitor of the Src family kinase. CRISPR/Cas9 technology is also applied to PDAC modeling and therapeutic research using a variety of animals. An individual mouse strain expressing Cas9 in the adult pancreas under a p48 promoter has been established to generate PDAC GEMMs of complicated genotypes with high efficiency (Ideno et al., 2019). By the use of an adeno-associated virus to transfer multiplexed RNA guidelines (sgRNAs) to an adult pancreas of p48-Cre; LSL-Cas9 mice, they produce a mutated Kras G12D allele using homology directed repair in combination with CRISPR-induced disruption of cooperative alleles [Trp53, AT-rich interaction domain 1A (Arid1A) and Lkb1].
Overall, CRISPR-Cas9 engineering provides new opportunities to model PDAC development. It allows the production of syngeneic and humanized mice which can help to eliminate transplant rejection by the host immune system without needing immunocompromised animals (Krempley and Yu, 2017). As a result, these models are particularly useful for researching immunotherapies, allowing to investigate several unanswered problems. For instance, it would be compelling to see if tumors with distinct histopathological features reported in the model have substantial variations in target allele frequency and/or if further mutations have accumulated and how this model’s total mutational and neoantigen load correlates to other GEMMs and human PDAC (Jørgensen and Hogg, 2016).
Several preclinical models for PDAC are accessible for basic and translational studies, which permit the description of the global genetic features of this disease (summarized in Figure 2).
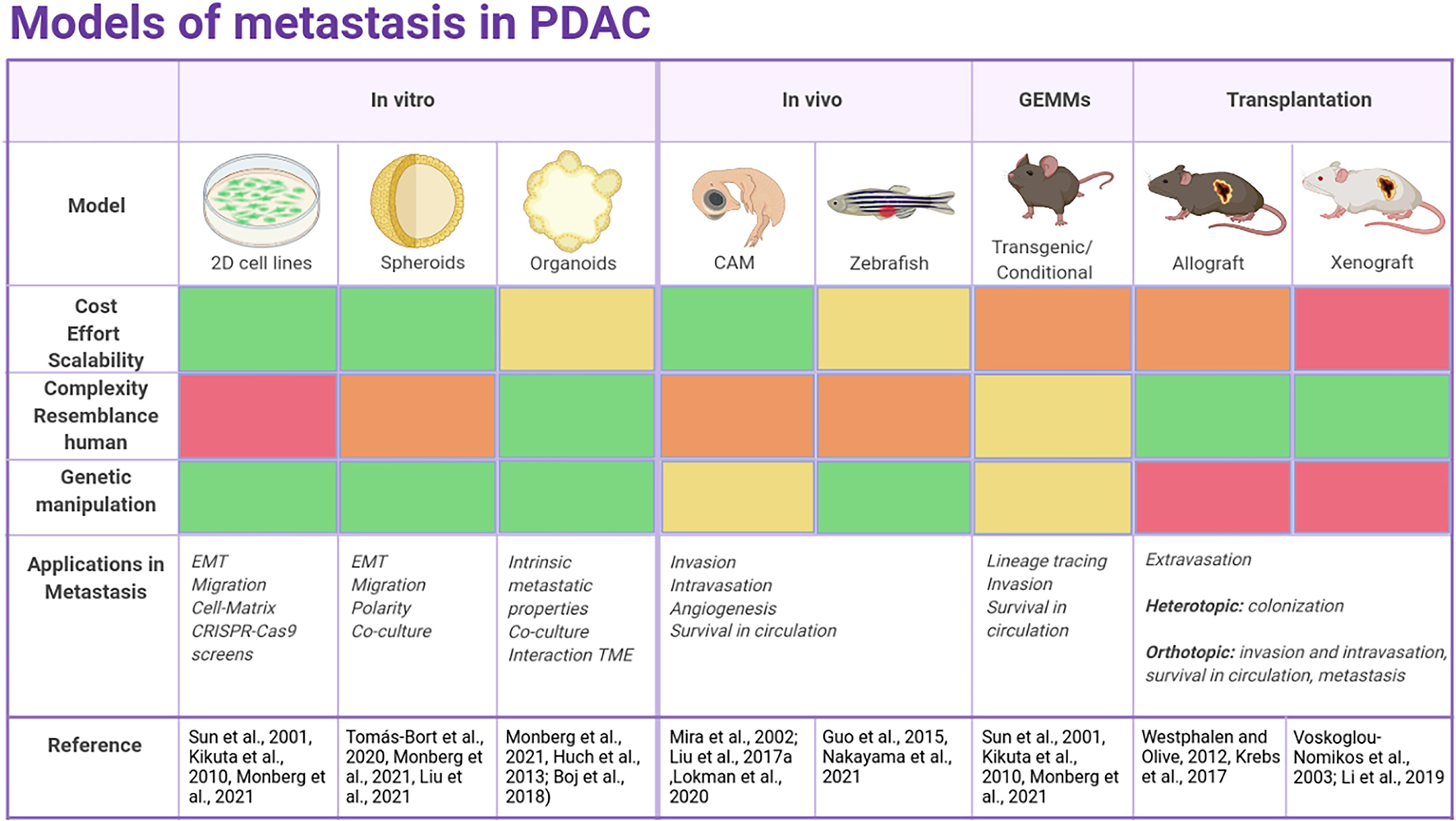
Figure 2. Overview of the current models to study metastasis in PDAC. The color scale indicates whether a model is more suitable (green) or less (red) for each purpose.
Pancreatic ductal adenocarcinoma is characterized by an early and fast metastatic process partially related to the location of the pancreas leading to peritoneal, liver or lung metastasis. Nonetheless, a deep molecular insight into the metastatic process of PDAC is still missing since very few studies have studied the mechanisms behind PDAC metastatic organotropism. This is pivotal since the location of the metastases determines the clinical outcome of the patient. Each PDAC preclinical model described above (2D cell lines, spheroids or organoids, GEMMs, and PDXs models) has pros and cons, and the model of choice will vary according to experimental goals.
The correct combination of currently available models is necessary for the development of more trustworthy therapeutic strategies against PDAC. A good strategy is a combination of the methods in each step of the experimental process. Namely, 2D cell lines, PDCL or spheroids are good tools for HTS, studying tumorigenesis and progression. Organoids offer similar benefits as cell lines, adding a step closer to personalized medicine. In vivo models are useful to model the TME and immune response, key players in PDAC transformation and progression. Murine models are ideal platforms for understanding PDAC progression, pathophysiology and testing therapeutic modalities. Once candidates are selected, more relevant models like ODXs or PDXs models are suitable for functional validation.
MM wrote the abstract, introduction, some models sections, discussion, and elaborated the figures of the manuscript. SZ wrote some models sections, elaborated some tables, and revised the manuscript. CP revised the manuscript, provided critical feedback, and helped shape the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful for the support from the EU (MSCA-ITN 861196, PRECODE; MM and CP) and the Chinese Scholarship Council (CSC Nr. 201806880012 SZ). CP wants to thank Ronald Belford Scott for his support. The present work was performed in (partial) fulfillment of the requirements for obtaining the degree Dr. rer. biol. hum of FAU by SZ.
Aguirre, A. J. (2003). Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126. doi: 10.1101/gad.1158703
Ahmed, S., Bradshaw, A.-D., Gera, S., Dewan, M., and Xu, R. (2017). The TGF-β/Smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J. Clin. Med. 6:5. doi: 10.3390/jcm6010005
Aiello, N. M., Brabletz, T., Kang, Y., Nieto, M. A., Weinberg, R. A., and Stanger, B. Z. (2017). Upholding a role for EMT in pancreatic cancer metastasis. Nature 547, E7–E8. doi: 10.1038/nature22963
American Cancer Society (2021). Key Statistics for Pancreatic Cancer. Available online at: https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html (accessed July 8, 2021)
Antonello, D., Gobbo, S., Corbo, V., Sipos, B., Lemoine, N. R., and Scarpa, A. (2009). Update on the molecular pathogenesis of pancreatic tumors other than common ductal adenocarcinoma. Pancreatology 9, 25–33. doi: 10.1159/000178872
Aparicio, S., Hidalgo, M., and Kung, A. L. (2015). Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 15, 311–316. doi: 10.1038/nrc3944
Avula, L. R., Hagerty, B., and Alewine, C. (2020). Molecular mediators of peritoneal metastasis in pancreatic cancer. Cancer Metastasis Rev. 39, 1223–1243. doi: 10.1007/s10555-020-09924-4
Azmi, A. S., Khan, H. Y., Muqbil, I., Aboukameel, A., Neggers, J. E., Daelemans, D., et al. (2020). Preclinical assessment with clinical validation of selinexor with gemcitabine and nab-paclitaxel for the treatment of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 26, 1338–1348. doi: 10.1158/1078-0432.CCR-19-1728
Bailey, P., Chang, D. K., Nones, K., Johns, A. L., Patch, A. M., Gingras, M. C., et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. doi: 10.1038/nature16965
Bardeesy, N., Aguirre, A. J., Chu, G. C., Cheng, K.-H., Lopez, L. V., Hezel, A. F., et al. (2006a). Both p16Ink4a and the p19Arf-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. U.S.A. 103, 5947–5952. doi: 10.1073/pnas.0601273103
Bardeesy, N., Cheng, K.-H., Berger, J. H., Chu, G. C., Pahler, J., Olson, P., et al. (2006b). Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 20, 3130–3146. doi: 10.1101/gad.1478706
Baretti, M., Pulluri, B., Tsai, H.-L., Blackford, A. L., Wolfgang, C. L., Laheru, D., et al. (2019). The significance of ascites in patients with pancreatic ductal adenocarcinoma: a case-control study. Pancreas 48, 585–589. doi: 10.1097/MPA.0000000000001262
Beuran, M., Negoi, I., Paun, S., Ion, A. D., Bleotu, C., Negoi, R. I., et al. (2015). The epithelial to mesenchymal transition in pancreatic cancer: a systematic review. Pancreatology 15, 217–225. doi: 10.1016/j.pan.2015.02.011
Biffi, G., Oni, T. E., Spielman, B., Hao, Y., Elyada, E., Park, Y., et al. (2019). IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301. doi: 10.1158/2159-8290.CD-18-0710
Bijlsma, M. F. (2021). The case for a stratified application of targeted agents against pancreatic cancer. EBioMedicine 67:103344. doi: 10.1016/j.ebiom.2021.103344
Bijlsma, M. F., Sadanandam, A., Tan, P., and Vermeulen, L. (2017). Molecular subtypes in cancers of the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 14, 333–342. doi: 10.1038/nrgastro.2017.33
Bockorny, B., Macarulla, T., Semenisty, V., Borazanci, E., Feliu, J., Ponz-Sarvise, M., et al. (2021). Motixafortide and pembrolizumab combined to nanoliposomal irinotecan, fluorouracil and folinic acid in metastatic pancreatic cancer: the COMBAT/KEYNOTE-202 trial. Clin. Cancer Res., clincanres.CCR-21-0929-E.2021. doi: 10.1158/1078-0432.CCR-21-0929
Boj, S. F., Hwang, C. I., Baker, L. A., Chio, I. I. C., Engle, D. D., Corbo, V., et al. (2018). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. doi: 10.1016/j.cell.2014.12.021
Brancato, V., Comunanza, V., Imparato, G., Corà, D., Urciuolo, F., Noghero, A., et al. (2017). Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater. 49, 152–166. doi: 10.1016/j.actbio.2016.11.072
Brunton, H., Caligiuri, G., Cunningham, R., Upstill-Goddard, R., Bailey, U. M., Garner, I. M., et al. (2020). HNF4A and GATA6 loss reveals therapeutically actionable subtypes in pancreatic cancer. Cell Rep. 31:107625. doi: 10.1016/j.celrep.2020.107625
Burris, H. A., Moore, M. J., Andersen, J., Green, M. R., Rothenberg, M. L., Modiano, M. R., et al. (1997). Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15, 2403–2413. doi: 10.1200/JCO.1997.15.6.2403
Cabezas-Sainz, P., Guerra-Varela, J., Carreira, M. J., Mariscal, J., Roel, M., Rubiolo, J. A., et al. (2018). Improving zebrafish embryo xenotransplantation conditions by increasing incubation temperature and establishing a proliferation index with ZFtool. BMC Cancer 18:3. doi: 10.1186/s12885-017-3919-8
Cabezas-Sáinz, P., Pensado-López, A., Sáinz, B., and Sánchez, L. (2020). Modeling cancer using zebrafish xenografts: drawbacks for mimicking the human microenvironment. Cells 9:1978. doi: 10.3390/cells9091978
Campbell, P. J., Yachida, S., Mudie, L. J., Stephens, P. J., Pleasance, E. D., Stebbings, L. A., et al. (2010). The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113. doi: 10.1038/nature09460
Cecati, M., Giulietti, M., Righetti, A., Sabanovic, B., and Piva, F. (2021). Effects of CXCL12 isoforms in a pancreatic pre-tumour cellular model: microarray analysis. World J. Gastroenterol. 27, 1616–1629. doi: 10.3748/wjg.v27.i15.1616
Chandrakesan, P., Panneerselvam, J., May, R., Weygant, N., Qu, D., Berry, W. R., et al. (2020). DCLK1-isoform2 alternative splice variant promotes pancreatic tumor immunosuppressive M2-macrophage polarization. Mol. Cancer Ther. 19, 1539–1549. doi: 10.1158/1535-7163.MCT-19-0776
Chan-Seng-Yue, M., Kim, J. C., Wilson, G. W., Ng, K., Figueroa, E. F., O’Kane, G. M., et al. (2020). Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 52, 231–240. doi: 10.1038/s41588-019-0566-9
Charles Jacob, H., Charles Richard, J., Signorelli, R., Kashuv, T., Lavania, S., Vaish, U., et al. (2021). Modulation of early neutrophil granulation: the circulating tumor cell-extravesicular connection in pancreatic ductal adenocarcinoma. Cancers 13, 2727. doi: 10.3390/cancers13112727
Chen, Y., Su, L., Huang, C., Wu, S., Qiu, X., Zhao, X., et al. (2021). Galactosyltransferase B4GALT1 confers chemoresistance in pancreatic ductal adenocarcinomas by upregulating N-linked glycosylation of CDK11p110. Cancer Lett. 500, 228–243. doi: 10.1016/j.canlet.2020.12.006
Cheung, K. J., and Ewald, A. J. (2016). A collective route to metastasis: seeding by tumor cell clusters. Science 352, 167–169. doi: 10.1126/science.aaf6546
Chu, L. C., Goggins, M. G., and Fishman, E. K. (2017). Diagnosis and detection of pancreatic cancer. Cancer J. 23, 333–342. doi: 10.1097/PPO.0000000000000290
Chugh, S., Barkeer, S., Rachagani, S., Nimmakayala, R. K., Perumal, N., Pothuraju, R., et al. (2018). Disruption of C1galt1 gene promotes development and metastasis of pancreatic adenocarcinomas in mice. Gastroenterology 155, 1608–1624. doi: 10.1053/j.gastro.2018.08.007
Collisson, E. A., Bailey, P., Chang, D. K., and Biankin, A. V. (2019). Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 16, 207–220. doi: 10.1038/s41575-019-0109-y
Collisson, E. A., Sadanandam, A., Olson, P., Gibb, W. J., Truitt, M., Gu, S., et al. (2011). Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 17, 500–503. doi: 10.1038/nm.2344
Conroy, T., Desseigne, F., Ychou, M., Bouché, O., Guimbaud, R., Bécouarn, Y., et al. (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825. doi: 10.1056/NEJMoa1011923
Deer, E. L., González-Hernández, J., Coursen, J. D., Shea, J. E., Ngatia, J., Scaife, C. L., et al. (2010). Phenotype and genotype of pancreatic cancer cell lines. Pancreas 39, 425–435. doi: 10.1097/MPA.0b013e3181c15963
Doudna, J. A., and Charpentier, E. (2014). The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096
Dreyer, S. B., Rae, S., Bisset, K., Upstill-Goddard, R., Gemenetzis, G., Johns, A. L., et al. (2021a). The impact of molecular subtyping on pathological staging of pancreatic cancer. Ann. Surg. doi: 10.1097/SLA.0000000000005050 [Epub ahead of print].
Dreyer, S. B., Upstill-Goddard, R., Paulus-Hock, V., Paris, C., Lampraki, E.-M., Dray, E., et al. (2021b). Targeting DNA damage response and replication stress in pancreatic cancer. Gastroenterology 160, 362–377.e13. doi: 10.1053/j.gastro.2020.09.043
Du, R., Sullivan, D. K., Azizian, N. G., Liu, Y., and Li, Y. (2021). Inhibition of ERAD synergizes with FTS to eradicate pancreatic cancer cells. BMC Cancer 21:237. doi: 10.1186/s12885-021-07967-6
Duda, D. G., Duyverman, A. M. M. J., Kohno, M., Snuderl, M., Steller, E. J. A., Fukumura, D., et al. (2010). Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. U.S.A. 107, 21677–21682. doi: 10.1073/pnas.1016234107
Duda, D. G., Fukumura, D., Munn, L. L., Booth, M. F., Brown, E. B., Huang, P., et al. (2004). Differential transplantability of tumor-associated stromal cells. Cancer Res. 64, 5920–5924. doi: 10.1158/0008-5472.CAN-04-1268
Durymanov, M., Kroll, C., Permyakova, A., O’Neill, E., Sulaiman, R., Person, M., et al. (2019). Subcutaneous inoculation of 3D pancreatic cancer spheroids results in development of reproducible stroma-rich tumors. Transl. Oncol. 12, 180–189. doi: 10.1016/j.tranon.2018.10.003
Eckrich, J., Kugler, P., Buhr, C. R., Ernst, B. P., Mendler, S., Baumgart, J., et al. (2020). Monitoring of tumor growth and vascularization with repetitive ultrasonography in the chicken chorioallantoic-membrane-assay. Sci. Rep. 10:18585. doi: 10.1038/s41598-020-75660-y
Emerman, J. T., and Pitelka, D. R. (1977). Maintenance and induction of morphological differentiation in dissociated mammary epithelium on floating collagen membranes. In Vitro 13, 316–328. doi: 10.1007/BF02616178
Espinet, E., Gu, Z., Imbusch, C. D., Giese, N. A., Büscher, M., Safavi, M., et al. (2021). Aggressive PDACs show hypomethylation of repetitive elements and the execution of an intrinsic IFN program linked to a ductal cell of origin. Cancer Discov. 11, 638–659. doi: 10.1158/2159-8290.CD-20-1202
European Commission (2021). PancREatic Cancer OrganoiDs rEsearch Network | PRECODE Project | H2020 | CORDIS | European Commission. Available online at: https://cordis.europa.eu/project/id/861196 (accessed June 16, 2021).
Eyres, M., Lanfredini, S., Xu, H., Burns, A., Blake, A., Willenbrock, F., et al. (2021). TET2 drives 5hmc marking of GATA6 and epigenetically defines pancreatic ductal adenocarcinoma transcriptional subtypes. Gastroenterology 161, 653–668.e16. doi: 10.1053/j.gastro.2021.04.044
Feldmann, K., Maurer, C., Peschke, K., Teller, S., Schuck, K., Steiger, K., et al. (2021). Mesenchymal plasticity regulated by Prrx1 drives aggressive pancreatic cancer biology. Gastroenterology 160, 346–361.e24. doi: 10.1053/j.gastro.2020.09.010
Fidler, I. J. (2003). The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat. Rev. Cancer 3, 453–458. doi: 10.1038/nrc1098
Firuzi, O., Che, P. P., El Hassouni, B., Buijs, M., Coppola, S., Löhr, M., et al. (2019). Role of c-MET inhibitors in overcoming drug resistance in spheroid models of primary human pancreatic cancer and stellate cells. Cancers 11:638. doi: 10.3390/cancers11050638
Franses, J. W., Philipp, J., Missios, P., Bhan, I., Liu, A., Yashaswini, C., et al. (2020). Pancreatic circulating tumor cell profiling identifies LIN28B as a metastasis driver and drug target. Nat. Commun. 11:3303. doi: 10.1038/s41467-020-17150-3
Friedl, P., Locker, J., Sahai, E., and Segall, J. E. (2012). Classifying collective cancer cell invasion. Nat. Cell Biol. 14, 777–783. doi: 10.1038/ncb2548
Fujisawa, T., Joshi, B., Nakajima, A., and Puri, R. K. (2009). A novel role of interleukin-13 receptor 2 in pancreatic cancer invasion and metastasis. Cancer Res. 69, 8678–8685. doi: 10.1158/0008-5472.CAN-09-2100
Fujita, M., Imadome, K., Endo, S., Shoji, Y., Yamada, S., and Imai, T. (2014). Nitric oxide increases the invasion of pancreatic cancer cells via activation of the PI3K-AKT and RhoA pathways after carbon ion irradiation. FEBS Lett. 588, 3240–3250. doi: 10.1016/j.febslet.2014.07.006
Furuyama, H., Arii, S., Mori, A., and Imamura, M. (2000). Role of E-cadherin in peritoneal dissemination of the pancreatic cancer cell line, Panc-1, through regulation of cell to cell contact. Cancer Lett. 157, 201–209. doi: 10.1016/S0304-3835(00)00488-2
Gao, C., Quan, M.-Y., Chen, Q.-J., Yang, R., Wu, Y., Liu, J.-Y., et al. (2021). Yap1-2 isoform is the primary mediator in TGF-β1 induced EMT in pancreatic cancer. Front. Oncol. 11:649290. doi: 10.3389/fonc.2021.649290
Gao, H., Chakraborty, G., Lee-Lim, A. P., Mo, Q., Decker, M., Vonica, A., et al. (2012). The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779. doi: 10.1016/j.cell.2012.06.035
Garcia, P. L., Miller, A. L., and Yoon, K. J. (2020). Patient-derived xenograft models of pancreatic cancer: overview and comparison with other types of models. Cancers 12:1327. doi: 10.3390/cancers12051327
Ge, W., Chen, G., and Fan, X.-S. (2017). Pathway of peritoneal carcinomatosis maybe hematogenous metastasis rather than peritoneal seeding. Oncotarget 8, 41549–41554. doi: 10.18632/oncotarget.14607
Gendoo, D. M. A., Denroche, R. E., Zhang, A., Radulovich, N., Jang, G. H., Lemire, M., et al. (2019). Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput. Biol. 15:e1006596. doi: 10.1371/journal.pcbi.1006596
Ghajar, C. M., Peinado, H., Mori, H., Matei, I. R., Evason, K. J., Brazier, H., et al. (2013). The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 15, 807–817. doi: 10.1038/ncb2767
Golan, T., Stossel, C., Schvimer, M., Atias, D., Halperin, S., Buzhor, E., et al. (2017). Pancreatic cancer ascites xenograft-an expeditious model mirroring advanced therapeutic resistant disease. Oncotarget 8, 40778–40790. doi: 10.18632/oncotarget.17253
Gómez-Cuadrado, L., Tracey, N., Ma, R., Qian, B., and Brunton, V. G. (2017). Mouse models of metastasis: progress and prospects. Dis. Model. Mech. 10, 1061–1074. doi: 10.1242/dmm.030403
Gonzalez, H., Hagerling, C., and Werb, Z. (2018). Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284. doi: 10.1101/gad.314617.118
Grigore, A., Jolly, M., Jia, D., Farach-Carson, M., and Levine, H. (2016). Tumor budding: the name is EMT. Partial EMT. J. Clin. Med. 5:51. doi: 10.3390/jcm5050051
Guo, M., Wei, H., Hu, J., Sun, S., Long, J., and Wang, X. (2015). U0126 inhibits pancreatic cancer progression via the KRAS signaling pathway in a zebrafish xenotransplantation model. Oncol. Rep. 34, 699–706. doi: 10.3892/or.2015.4019
Gut, P., Reischauer, S., Stainier, D. Y. R., and Arnaout, R. (2017). Little fish, big data: zebrafish as a model for cardiovascular and metabolic disease. Physiol. Rev. 97, 889–938. doi: 10.1152/physrev.00038.2016
Harris, N. L. E., Vennin, C., Conway, J. R. W., Vine, K. L., Pinese, M., Cowley, M. J., et al. (2017). SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene 36, 4288–4298. doi: 10.1038/onc.2017.63
Hason, M., and Bartùnìk, P. (2019). Zebrafish models of cancer—new insights on modeling human cancer in a non-mammalian vertebrate. Genes 10:935. doi: 10.3390/genes10110935
Hidalgo, M., Amant, F., Biankin, A. V., Budinská, E., Byrne, A. T., Caldas, C., et al. (2014). Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 4, 998–1013. doi: 10.1158/2159-8290.CD-14-0001
Higuchi, T., Yokobori, T., Naito, T., Kakinuma, C., Hagiwara, S., Nishiyama, M., et al. (2017). Investigation into metastatic processes and the therapeutic effects of gemcitabine on human pancreatic cancer using an orthotopic SUIT-2 pancreatic cancer mouse model. Oncol. Lett. 15, 3091–3099. doi: 10.3892/ol.2017.7722
Hindley, C. J., Cordero-Espinoza, L., and Huch, M. (2016). Organoids from adult liver and pancreas: stem cell biology and biomedical utility. Dev. Biol. 420, 251–261. doi: 10.1016/j.ydbio.2016.06.039
Hingorani, S. R., Petricoin, E. F., Maitra, A., Rajapakse, V., King, C., Jacobetz, M. A., et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. doi: 10.1016/S1535-6108(03)00309-X
Hingorani, S. R., Wang, L., Multani, A. S., Combs, C., Deramaudt, T. B., Hruban, R. H., et al. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. doi: 10.1016/j.ccr.2005.04.023
Holub, A. R., Huo, A., Patel, K., Thakore, V., Chhibber, P., and Erogbogbo, F. (2020). Assessing advantages and drawbacks of rapidly generated ultra-large 3D breast cancer spheroids: studies with chemotherapeutics and nanoparticles. Int. J. Mol. Sci. 21:4413. doi: 10.3390/ijms21124413
Hoo, J. Y., Kumari, Y., Shaikh, M. F., Hue, S. M., and Goh, B. H. (2016). Zebrafish: a versatile animal model for fertility research. BioMed Res. Int. 2016:9732780. doi: 10.1155/2016/9732780
Hosein, A. N., Brekken, R. A., and Maitra, A. (2020). Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 17, 487–505. doi: 10.1038/s41575-020-0300-1
Houg, D. S., and Bijlsma, M. F. (2018). The hepatic pre-metastatic niche in pancreatic ductal adenocarcinoma. Mol. Cancer 17:95. doi: 10.1186/s12943-018-0842-9
Hoyer, K., Hablesreiter, R., Inoue, Y., Yoshida, K., Briest, F., Christen, F., et al. (2021). A genetically defined signature of responsiveness to erlotinib in early-stage pancreatic cancer patients: results from the CONKO-005 trial. EBioMedicine 66:103327. doi: 10.1016/j.ebiom.2021.103327
Hruban, R. H., Adsay, N. V., Albores–Saavedra, J., Compton, C., Garrett, E. S., Goodman, S. N., et al. (2001). Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 25, 579–586. doi: 10.1097/00000478-200105000-00003
Huang, L., Holtzinger, A., Jagan, I., Begora, M., Lohse, I., Ngai, N., et al. (2015). Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 21, 1364–1371. doi: 10.1038/nm.3973
Huch, M., Bonfanti, P., Boj, S. F., Sato, T., Loomans, C. J. M., Wetering, M. V. D., et al. (2013). Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 32, 2708–2721. doi: 10.1038/emboj.2013.204
Hwang, C., Boj, S. F., Clevers, H., and Tuveson, D. A. (2016). Preclinical models of pancreatic ductal adenocarcinoma. J. Pathol. 238, 197–204. doi: 10.1002/path.4651
Ideno, N., Yamaguchi, H., Okumura, T., Huang, J., Brun, M. J., Ho, M. L., et al. (2019). A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab. Invest. 99, 1233–1244. doi: 10.1038/s41374-018-0171-z
Ijichi, H., Chytil, A., Gorska, A. E., Aakre, M. E., Fujitani, Y., Fujitani, S., et al. (2006). Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 20, 3147–3160. doi: 10.1101/gad.1475506
Ischenko, I., D’Amico, S., Rao, M., Li, J., Hayman, M. J., Powers, S., et al. (2021). KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 12:1482. doi: 10.1038/s41467-021-21736-w
Ishiguro, S., Kawabata, A., Zulbaran-Rojas, A., Monson, K., Uppalapati, D., Ohta, N., et al. (2018). Co-treatment with a C1B5 peptide of protein kinase Cγ and a low dose of gemcitabine strongly attenuated pancreatic cancer growth in mice through T cell activation. Biochem. Biophys. Res. Commun. 495, 962–968. doi: 10.1016/j.bbrc.2017.11.102
Izeradjene, K., Combs, C., Best, M., Gopinathan, A., Wagner, A., Grady, W. M., et al. (2007). KrasG12D and smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 11, 229–243. doi: 10.1016/j.ccr.2007.01.017
Jayne, D. (2007). “Molecular biology of peritoneal carcinomatosis,” in Peritoneal Carcinomatosis, ed. W. P. Ceelen (Boston, MA: Springer US), 21–33. doi: 10.1007/978-0-387-48993-3_2
Jiang, Y.-J., Lee, C.-L., Wang, Q., Zhou, Z.-W., Yang, F., Jin, C., et al. (2014). Establishment of an orthotopic pancreatic cancer mouse model: cells suspended and injected in Matrigel. World J. Gastroenterol. 20, 9476–9485. doi: 10.3748/wjg.v20.i28.9476
Johnson, L., Mercer, K., Greenbaum, D., Bronson, R. T., Crowley, D., Tuveson, D. A., et al. (2001). Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116. doi: 10.1038/35074129
Jolly, M. K. (2015). Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 5:155. doi: 10.3389/fonc.2015.00155
Jørgensen, C., and Hogg, E. K. J. (2016). CRISPR/Cas9 engineering offers new opportunities to model pancreatic ductal adenocarcinoma development. Transl. Cancer Res. 5, S357–S359. doi: 10.21037/tcr.2016.08.01
Juiz, N., Elkaoutari, A., Bigonnet, M., Gayet, O., Roques, J., Nicolle, R., et al. (2020). Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 34, 12214–12228. doi: 10.1096/fj.202000363RR
Kapałczyńska, M., Kolenda, T., Przybyła, W., Zaja̧czkowska, M., Teresiak, A., Filas, V., et al. (2016). 2D and 3D cell cultures – a comparison of different types of cancer cell cultures. Arch. Med. Sci. 14, 910–919. doi: 10.5114/aoms.2016.63743
Keith, B., Johnson, R. S., and Simon, M. C. (2012). HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12, 9–22. doi: 10.1038/nrc3183
Kelm, J. M., Timmins, N. E., Brown, C. J., Fussenegger, M., and Nielsen, L. K. (2003). Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 83, 173–180. doi: 10.1002/bit.10655
Khanna, C. (2004). Modeling metastasis in vivo. Carcinogenesis 26, 513–523. doi: 10.1093/carcin/bgh261
Kikuta, K., Masamune, A., Watanabe, T., Ariga, H., Itoh, H., Hamada, S., et al. (2010). Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 403, 380–384. doi: 10.1016/j.bbrc.2010.11.040
Kim, J., Koo, B. K., and Knoblich, J. A. (2020). Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584. doi: 10.1038/s41580-020-0259-3
Kim, M. P., Evans, D. B., Wang, H., Abbruzzese, J. L., Fleming, J. B., and Gallick, G. E. (2009). Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 4, 1670–1680. doi: 10.1038/nprot.2009.171
Kleeff, J., Korc, M., Apte, M., Vecchia, C. L., Johnson, C. D., Biankin, A. V., et al. (2016). Pancreatic cancer. Nat. Rev. Dis. Primer 2, 1–23. doi: 10.1038/nrdp.2016.22
Knaack, H., Lenk, L., Philipp, L.-M., Miarka, L., Rahn, S., Viol, F., et al. (2018). Liver metastasis of pancreatic cancer: the hepatic microenvironment impacts differentiation and self-renewal capacity of pancreatic ductal epithelial cells. Oncotarget 9, 31771–31786. doi: 10.18632/oncotarget.25884
Komatsu, A., Higashi, Y., and Matsumoto, K. (2019). “Various CAM tumor models,” in The Enzymes, ed. F. Tamanoi (Amsterdam: Elsevier), 37–57. doi: 10.1016/bs.enz.2019.10.001
Kong, K., Guo, M., Liu, Y., and Zheng, J. (2020). Progress in animal models of pancreatic ductal adenocarcinoma. J. Cancer 11, 1555–1567. doi: 10.7150/jca.37529
Kramer, B., de Haan, L., Vermeer, M., Olivier, T., Hankemeier, T., Vulto, P., et al. (2019). Interstitial flow recapitulates gemcitabine chemoresistance in a 3D microfluidic pancreatic ductal adenocarcinoma model by induction of multidrug resistance proteins. Int. J. Mol. Sci. 20:4647. doi: 10.3390/ijms20184647
Kramer, N., Walzl, A., Unger, C., Rosner, M., Krupitza, G., Hengstschläger, M., et al. (2013). In vitro cell migration and invasion assays. Mutat. Res. Mutat. Res. 752, 10–24. doi: 10.1016/j.mrrev.2012.08.001
Krebs, A. M., Mitschke, J., Lasierra Losada, M., Schmalhofer, O., Boerries, M., Busch, H., et al. (2017). The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529. doi: 10.1038/ncb3513
Krempley, B. D., and Yu, K. H. (2017). Preclinical models of pancreatic ductal adenocarcinoma. Chin. Clin. Oncol. 6:25. doi: 10.21037/cco.2017.06.15
Kuen, J., Darowski, D., Kluge, T., and Majety, M. (2017). Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS One 12:e0182039. doi: 10.1371/journal.pone.0182039
Kuo, T.-C., Wu, M.-H., Yang, S.-H., Chen, S.-T., Hsu, T.-W., Jhuang, J.-Y., et al. (2021). C1GALT1 high expression is associated with poor survival of patients with pancreatic ductal adenocarcinoma and promotes cell invasiveness through integrin αv. Oncogene 40, 1242–1254. doi: 10.1038/s41388-020-01594-4
Lai, E., Puzzoni, M., Ziranu, P., Pretta, A., Impera, V., Mariani, S., et al. (2019). New therapeutic targets in pancreatic cancer. Cancer Treat. Rev. 81:101926. doi: 10.1016/j.ctrv.2019.101926
Lambert, A. W., Pattabiraman, D. R., and Weinberg, R. A. (2017). Emerging biological principles of metastasis. Cell 168, 670–691. doi: 10.1016/j.cell.2016.11.037
Lazzari, G., Nicolas, V., Matsusaki, M., Akashi, M., Couvreur, P., and Mura, S. (2018). Multicellular spheroid based on a triple co-culture: a novel 3D model to mimic pancreatic tumor complexity. Acta Biomater. 78, 296–307. doi: 10.1016/j.actbio.2018.08.008
Lee, J.-H., Kim, S.-K., Khawar, I. A., Jeong, S.-Y., Chung, S., and Kuh, H.-J. (2018). Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 37:4. doi: 10.1186/s13046-017-0654-6
Lenk, L., Pein, M., Will, O., Gomez, B., Viol, F., Hauser, C., et al. (2018). The hepatic microenvironment essentially determines tumor cell dormancy and metastatic outgrowth of pancreatic ductal adenocarcinoma. OncoImmunology 7:e1368603. doi: 10.1080/2162402X.2017.1368603
Li, J., Qian, W., Qin, T., Xiao, Y., Cheng, L., Cao, J., et al. (2019). Mouse-derived allografts: a complementary model to the KPC mice on researching pancreatic cancer in vivo. Comput. Struct. Biotechnol. J. 17, 498–506. doi: 10.1016/j.csbj.2019.03.016
Liu, C., Deng, S., Jin, K., Gong, Y., Cheng, H., Fan, Z., et al. (2020). Lewis antigen-negative pancreatic cancer: an aggressive subgroup. Int. J. Oncol. 56, 900–908. doi: 10.3892/ijo.2020.4989
Liu, L., Yao, D., Zhang, P., Ding, W., Zhang, X., Zhang, C., et al. (2017b). Deubiquitinase USP9X promotes cell migration, invasion and inhibits apoptosis of human pancreatic cancer. Oncol. Rep. 38, 3531–3537. doi: 10.3892/or.2017.6050
Liu, L., Aleksandrowicz, E., Schönsiegel, F., Gröner, D., Bauer, N., Nwaeburu, C. C., et al. (2017a). Dexamethasone mediates pancreatic cancer progression by glucocorticoid receptor, TGFβ and JNK/AP-1. Cell Death Dis. 8:e3064. doi: 10.1038/cddis.2017.455
Liu, X., Gündel, B., Li, X., Liu, J., Wright, A., Löhr, M., et al. (2021). 3D heterospecies spheroids of pancreatic stroma and cancer cells demonstrate key phenotypes of pancreatic ductal adenocarcinoma. Transl. Oncol. 14:101107. doi: 10.1016/j.tranon.2021.101107
Lokman, N. A., Ricciardelli, C., and Oehler, M. K. (2020). “Chick chorioallantoic membrane assay: a 3D animal model for cancer invasion and metastasis,” in Animal Biotechnology, (Amsterdam: Elsevier), 221–231. doi: 10.1016/B978-0-12-811710-1.00031-8
Longnecker, D. S. (1984). Lesions induced in rodent pancreas by azaserine and other pancreatic carcinogens. Environ. Health Perspect. 56, 245–251. doi: 10.1289/ehp.8456245
Loukopoulos, P., Kanetaka, K., Takamura, M., Shibata, T., Sakamoto, M., and Hirohashi, S. (2004). Orthotopic transplantation models of pancreatic adenocarcinoma derived from cell lines and primary tumors and displaying varying metastatic activity. Pancreas 29, 193–203. doi: 10.1097/00006676-200410000-00004
Lu, S., Zhang, Z., Du, P., Chard, L. S., Yan, W., El Khouri, M., et al. (2020). A virus-infected, reprogrammed somatic cell–derived tumor cell (VIReST) vaccination regime can prevent initiation and progression of pancreatic cancer. Clin. Cancer Res. 26, 465–476. doi: 10.1158/1078-0432.CCR-19-1395
Luo, Y., Yan, B., Liu, L., Yin, L., Ji, H., An, X., et al. (2021). Sulforaphane inhibits the expression of long noncoding RNA H19 and its target APOBEC3G and thereby pancreatic cancer progression. Cancers 13:827. doi: 10.3390/cancers13040827
Magnuson, M. A., and Osipovich, A. B. (2013). Pancreas-specific cre driver lines and considerations for their prudent use. Cell Metab. 18, 9–20. doi: 10.1016/j.cmet.2013.06.011
Maier, H. J., Wirth, T., and Beug, H. (2010). Epithelial-mesenchymal transition in pancreatic carcinoma. Cancers 2, 2058–2083. doi: 10.3390/cancers2042058
Makohon-Moore, A. P., Zhang, M., Reiter, J. G., Bozic, I., Allen, B., Kundu, D., et al. (2017). Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 49, 358–366. doi: 10.1038/ng.3764
Mann, K. M., Ward, J. M., Yew, C. C. K., Kovochich, A., Dawson, D. W., Black, M. A., et al. (2012). Sleeping beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A. 109, 5934–5941. doi: 10.1073/pnas.1202490109
Mann, K. M., Ying, H., Juan, J., Jenkins, N. A., and Copeland, N. G. (2016). KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 168, 29–42. doi: 10.1016/j.pharmthera.2016.09.003
Marques, I. J., Weiss, F. U., Vlecken, D. H., Nitsche, C., Bakkers, J., Lagendijk, A. K., et al. (2009). Metastatic behaviour of primary human tumours in a zebrafish xenotransplantation model. BMC Cancer 9:128. doi: 10.1186/1471-2407-9-128
Marsee, A., Roos, F. J. M., Verstegen, M. M. A., Gehart, H., de Koning, E., Lemaigre, F., et al. (2021). Building consensus on definition and nomenclature of hepatic, pancreatic, and biliary organoids. Cell Stem Cell 28, 816–832. doi: 10.1016/j.stem.2021.04.005
Massagué, J., and Obenauf, A. C. (2016). Metastatic colonization by circulating tumour cells. Nature 529, 298–306. doi: 10.1038/nature17038
Massagué, J., Batlle, E., and Gomis, R. R. (2017). Understanding the molecular mechanisms driving metastasis. Mol. Oncol. 11, 3–4. doi: 10.1002/1878-0261.12024
Massey, A. E., Sikander, M., Chauhan, N., Kumari, S., Setua, S., Shetty, A. B., et al. (2019). Next-generation paclitaxel-nanoparticle formulation for pancreatic cancer treatment. Nanomed. Nanotechnol. Biol. Med. 20:102027. doi: 10.1016/j.nano.2019.102027
Maurer, C., Holmstrom, S. R., He, J., Laise, P., Su, T., Ahmed, A., et al. (2019). Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 68, 1034–1043. doi: 10.1136/gutjnl-2018-317706
Mazur, P. K., and Siveke, J. T. (2012). Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut 61, 1488–1500. doi: 10.1136/gutjnl-2011-300756
McGuigan, A., Kelly, P., Turkington, R. C., Jones, C., Coleman, H. G., and McCain, R. S. (2018). Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 24, 4846–4861. doi: 10.3748/wjg.v24.i43.4846
Miao, J. X., Wang, J. Y., Li, H. Z., Guo, H. R., Dunmall, L. S. C., Zhang, Z. X., et al. (2020). Promising xenograft animal model recapitulating the features of human pancreatic cancer. World J. Gastroenterol. 26, 4802–4816. doi: 10.3748/WJG.V26.I32.4802
Michels, B. E., Mosa, M. H., Streibl, B. I., Zhan, T., Menche, C., Abou-El-Ardat, K., et al. (2020). Pooled in vitro and in vivo CRISPR-Cas9 screening identifies tumor suppressors in human colon organoids. Cell Stem Cell 26, 782–792.e7. doi: 10.1016/j.stem.2020.04.003
Mira, E., Lacalle, R. A., Gómez-Moutón, C., Leonardo, E., and Mañes, S. (2002). Quantitative determination of tumor cell intravasation in a real-time polymerase chain reaction-based assay. Clin. Exp. Metastasis 19, 313–318. doi: 10.1023/A:1015563031769
Moffitt, R. A., Marayati, R., Flate, E. L., Volmar, K. E., Loeza, S. G. H., Hoadley, K. A., et al. (2015). Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178. doi: 10.1038/ng.3398
Monberg, M. E., Geiger, H., Lee, J. J., Sharma, R., Semaan, A., Bernard, V., et al. (2021). Occult polyclonality of preclinical pancreatic cancer models drives in vitro evolution. Cancer Biol. doi: 10.1101/2021.04.13.439717
Moore, M. J., Goldstein, D., Hamm, J., Figer, A., Hecht, J. R., Gallinger, S., et al. (2007). Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of canada clinical trials group. J. Clin. Oncol. 25, 1960–1966. doi: 10.1200/JCO.2006.07.9525
Morton, J. P., Timpson, P., Karim, S. A., Ridgway, R. A., Athineos, D., Doyle, B., et al. (2010). Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. U.S.A. 107, 246–251. doi: 10.1073/pnas.0908428107
Muniyan, S., Haridas, D., Chugh, S., Rachagani, S., Lakshmanan, I., Gupta, S., et al. (2016). MUC16 contributes to the metastasis of pancreatic ductal adenocarcinoma through focal adhesion mediated signaling mechanism. Genes Cancer 7, 110–124. doi: 10.18632/genesandcancer.104
Nakayama, J., Tan, L., Goh, B. C., Wang, S., Makinoshima, H., and Gong, Z. (2021). A chemical screen based on an interruption of zebrafish gastrulation identifies the HTR2C inhibitor Pizotifen as a suppressor of EMT-mediated metastasis. Dev. Biol. doi: 10.1101/2021.03.04.434001
Oh, S., and Park, J. T. (2019). Zebrafish model of KRAS -initiated pancreatic endocrine tumor. Anim. Cells Syst. 23, 209–218. doi: 10.1080/19768354.2019.1610058
Öhlund, D., Handly-Santana, A., Biffi, G., Elyada, E., Almeida, A. S., Ponz-Sarvise, M., et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596. doi: 10.1084/jem.20162024
Öhlund, D., Lundin, C., Ardnor, B., Öman, M., Naredi, P., and Sund, M. (2009). Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 101, 91–97. doi: 10.1038/sj.bjc.6605107
Olive, K. P., Jacobetz, M. A., Davidson, C. J., Gopinathan, A., McIntyre, D., Honess, D., et al. (2009). Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. doi: 10.1126/science.1171362
Padua, D., and Massagué, J. (2009). Roles of TGFβ in metastasis. Cell Res. 19, 89–102. doi: 10.1038/cr.2008.316
Pal, A., Dziubinski, M., Di Magliano, M. P., Simeone, D. M., Owens, S., Thomas, D., et al. (2018). Usp9x promotes survival in human pancreatic cancer and its inhibition suppresses pancreatic ductal adenocarcinoma in vivo tumor growth. Neoplasia 20, 152–164. doi: 10.1016/j.neo.2017.11.007
Park, S.-Y., and Nam, J.-S. (2020). The force awakens: metastatic dormant cancer cells. Exp. Mol. Med. 52, 569–581. doi: 10.1038/s12276-020-0423-z
Peixoto, R. D., Ho, M., Renouf, D. J., Lim, H. J., Gill, S., Ruan, J. Y., et al. (2017). Eligibility of metastatic pancreatic cancer patients for first-line palliative intent nab-paclitaxel plus gemcitabine versus FOLFIRINOX. Am. J. Clin. Oncol. 40, 507–511. doi: 10.1097/COC.0000000000000193
Pérez-Mancera, P. A., Rust, A. G., van der Weyden, L., Kristiansen, G., Li, A., Sarver, A. L., et al. (2012). The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270. doi: 10.1038/nature11114
Perrone, F., De Laurentiis, M., De Placido, S., Orditura, M., Cinieri, S., Riccardi, F., et al. (2019). Adjuvant zoledronic acid and letrozole plus ovarian function suppression in premenopausal breast cancer: HOBOE phase 3 randomised trial. Eur. J. Cancer 118, 178–186. doi: 10.1016/j.ejca.2019.05.004
Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. doi: 10.1016/j.cell.2014.09.014
Puls, T. J., Tan, X., Husain, M., Whittington, C. F., Fishel, M. L., and Voytik-Harbin, S. L. (2018). Development of a novel 3D tumor-tissue invasion model for high-throughput, high-content phenotypic drug screening. Sci. Rep. 8:13039. doi: 10.1038/s41598-018-31138-6
Qian, Y., Gong, Y., Fan, Z., Luo, G., Huang, Q., Deng, S., et al. (2020). Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 13:130. doi: 10.1186/s13045-020-00958-3
Qiu, W., and Su, G. H. (2013). Challenges and advances in mouse modeling for human pancreatic tumorigenesis and metastasis. Cancer Metastasis Rev. 32, 83–107. doi: 10.1007/s10555-012-9408-2
Raimondi, G., Mato-Berciano, A., Pascual-Sabater, S., Rovira-Rigau, M., Cuatrecasas, M., Fondevila, C., et al. (2020). Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine 56:102786. doi: 10.1016/j.ebiom.2020.102786
Ramaker, R. C., Hardigan, A. A., Gordon, E. R., Wright, C. A., Myers, R. M., and Cooper, S. J. (2021). Pooled CRISPR screening in pancreatic cancer cells implicates co-repressor complexes as a cause of multiple drug resistance via regulation of epithelial-to-mesenchymal transition. BMC Cancer 21:632. doi: 10.1186/s12885-021-08388-1
Rasheed, Z. A., Yang, J., Wang, Q., Kowalski, J., Freed, I., Murter, C., et al. (2010). Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst. 102, 340–351. doi: 10.1093/jnci/djp535
Razidlo, G. L., Magnine, C., Sletten, A. C., Hurley, R. M., Almada, L. L., Fernandez-Zapico, M. E., et al. (2015). Targeting pancreatic cancer metastasis by inhibition of Vav1, a driver of tumor cell invasion. Cancer Res. 75, 2907–2915. doi: 10.1158/0008-5472.CAN-14-3103
Reichert, M., Bakir, B., Moreira, L., Pitarresi, J. R., Feldmann, K., Simon, L., et al. (2018). Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev. Cell 45, 696–711.e8. doi: 10.1016/j.devcel.2018.05.025
Rhim, A. D., Mirek, E. T., Aiello, N. M., Maitra, A., Bailey, J. M., McAllister, F., et al. (2012). EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361. doi: 10.1016/j.cell.2011.11.025
Ribatti, D. (2016). The chick embryo chorioallantoic membrane (CAM). A multifaceted experimental model. Mech. Dev. 141, 70–77. doi: 10.1016/j.mod.2016.05.003
Romero-Calvo, I., Weber, C. R., Ray, M., Brown, M., Kirby, K., Nandi, R. K., et al. (2019). Human organoids share structural and genetic features with primary pancreatic adenocarcinoma tumors. Mol. Cancer Res. 17, 70–83. doi: 10.1158/1541-7786.MCR-18-0531
Rovithi, M., Avan, A., Funel, N., Leon, L. G., Gomez, V. E., Wurdinger, T., et al. (2017). Development of bioluminescent chick chorioallantoic membrane (CAM) models for primary pancreatic cancer cells: a platform for drug testing. Sci. Rep. 7:44686. doi: 10.1038/srep44686
Rubio-Viqueira, B., and Hidalgo, M. (2009). Direct in vivo xenograft tumor model for predicting chemotherapeutic drug response in cancer patients. Clin. Pharmacol. Ther. 85, 217–221. doi: 10.1038/clpt.2008.200
Rucki, A. A. (2014). Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies. World J. Gastroenterol. 20:2237. doi: 10.3748/wjg.v20.i9.2237
Saitoh, M. (2018). Involvement of partial EMT in cancer progression. J. Biochem. 164, 257–264. doi: 10.1093/jb/mvy047
Sándor, G. O., Soós, A. Á, Lörincz, P., Rojkó, L., Harkó, T., Bogyó, L., et al. (2021). Wnt activity and cell proliferation are coupled to extracellular vesicle release in multiple organoid models. Front. Cell Dev. Biol. 9:670825. doi: 10.3389/fcell.2021.670825
Sato, M., Matsumoto, M., Saiki, Y., Alam, M., Nishizawa, H., Rokugo, M., et al. (2020). BACH1 promotes pancreatic cancer metastasis by repressing epithelial genes and enhancing epithelial–mesenchymal transition. Cancer Res. 80, 1279–1292. doi: 10.1158/0008-5472.CAN-18-4099
Schönhuber, N., Seidler, B., Schuck, K., Veltkamp, C., Schachtler, C., Zukowska, M., et al. (2014). A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med. 20, 1340–1347. doi: 10.1038/nm.3646
Seino, T., Kawasaki, S., Shimokawa, M., Tamagawa, H., Toshimitsu, K., Fujii, M., et al. (2018). Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 22, 454–467.e6. doi: 10.1016/j.stem.2017.12.009
Sergeant, G., van Eijsden, R., Roskams, T., Van Duppen, V., and Topal, B. (2012). Pancreatic cancer circulating tumour cells express a cell motility gene signature that predicts survival after surgery. BMC Cancer 12, 527. doi: 10.1186/1471-2407-12-527
Shah, V. M., Sheppard, B. C., Sears, R. C., and Alani, A. W. G. (2020). Hypoxia: friend or foe for drug delivery in pancreatic cancer. Cancer Lett. 492, 63–70. doi: 10.1016/j.canlet.2020.07.041
Shields, M. A., Dangi-Garimella, S., Redig, A. J., and Munshi, H. G. (2012). Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 441, 541–552. doi: 10.1042/BJ20111240
Singh, R. R., and O’Reilly, E. M. (2020). New treatment strategies for metastatic pancreatic ductal adenocarcinoma. Drugs 80, 647–669. doi: 10.1007/s40265-020-01304-0
Sinn, M., Bahra, M., Liersch, T., Gellert, K., Messmann, H., Bechstein, W., et al. (2017). CONKO-005: adjuvant chemotherapy with gemcitabine plus erlotinib versus gemcitabine alone in patients after r0 resection of pancreatic cancer: a multicenter randomized phase III trial. J. Clin. Oncol. 35, 3330–3337. doi: 10.1200/JCO.2017.72.6463
Smith, M. R., Coleman, R. E., Klotz, L., Pittman, K., Milecki, P., Ng, S., et al. (2015). Denosumab for the prevention of skeletal complications in metastatic castration-resistant prostate cancer: comparison of skeletal-related events and symptomatic skeletal events. Ann. Oncol. 26, 368–374. doi: 10.1093/annonc/mdu519
Sosa, M. S., Bragado, P., and Aguirre-Ghiso, J. A. (2014). Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer 14, 611–622. doi: 10.1038/nrc3793
Sun, C., Yamato, T., Furukawa, T., Ohnishi, Y., Kijima, H., and Horii, A. (2001). Characterization of the mutations of the K-ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol. Rep. 8, 89–92. doi: 10.3892/or.8.1.89
Takiguchi, S., Inoue, K., Matsusue, K., Furukawa, M., Teramoto, N., and Iguchi, H. (2017). Crizotinib, a MET inhibitor, prevents peritoneal dissemination in pancreatic cancer. Int. J. Oncol. 51, 184–192. doi: 10.3892/ijo.2017.3992
Tentler, J. J., Tan, A. C., Weekes, C. D., Jimeno, A., Leong, S., Pitts, T. M., et al. (2012). Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 9, 338–350. doi: 10.1038/nrclinonc.2012.61
Thiery, J. P. (2002). Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454. doi: 10.1038/nrc822
Tiriac, H., Belleau, P., Engle, D. D., Plenker, D., Deschênes, A., Somerville, T. D. D., et al. (2018). Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 8, 1112–1129. doi: 10.1158/2159-8290.CD-18-0349
Tiriac, H., Plenker, D., Baker, L. A., and Tuveson, D. A. (2019). Organoid models for translational pancreatic cancer research. Curr. Opin. Genet. Dev. 54, 7–11. doi: 10.1016/j.gde.2019.02.003
Tomás-Bort, E., Kieler, M., Sharma, S., Candido, J. B., and Loessner, D. (2020). 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 10, 5074–5089. doi: 10.7150/thno.42441
Torres, M. P., Rachagani, S., Souchek, J. J., Mallya, K., Johansson, S. L., and Batra, S. K. (2013). Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: applications in diagnosis and therapy. PLoS One 8:e80580. doi: 10.1371/journal.pone.0080580
Tsai, S., McOlash, L., Palen, K., Johnson, B., Duris, C., Yang, Q., et al. (2018). Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 18:335. doi: 10.1186/s12885-018-4238-4
Tseng, W. W., Winer, D., Kenkel, J. A., Choi, O., Shain, A. H., Pollack, J. R., et al. (2010). Development of an orthotopic model of invasive pancreatic cancer in an immunocompetent murine host. Clin. Cancer Res. 16, 3684–3695. doi: 10.1158/1078-0432.CCR-09-2384
Tuveson, D. A., and Neoptolemos, J. P. (2012). Understanding metastasis in pancreatic cancer: a call for new clinical approaches. Cell 148, 21–23. doi: 10.1016/j.cell.2011.12.021
Tuveson, D., and Clevers, H. (2019). Cancer modeling meets human organoid technology. Science 364, 952–955. doi: 10.1126/science.aaw6985
Vorvis, C., Hatziapostolou, M., Mahurkar-Joshi, S., Koutsioumpa, M., Williams, J., Donahue, T. R., et al. (2016). Transcriptomic and CRISPR/Cas9 technologies reveal FOXA2 as a tumor suppressor gene in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G1124–G1137. doi: 10.1152/ajpgi.00035.2016
Voskoglou-Nomikos, T., Pater, J. L., and Seymour, L. (2003). Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 9, 4227–4239.
Walter, K., Tiwary, K., Trajkovic-Arsic, M., Hidalgo-Sastre, A., Dierichs, L., Liffers, S. T., et al. (2019). MEK inhibition targets cancer stem cells and impedes migration of pancreatic cancer cells In Vitro and In Vivo. Stem Cells Int. 2019, 1–11. doi: 10.1155/2019/8475389
Wang, X., Enomoto, A., Asai, N., Kato, T., and Takahashi, M. (2016). Collective invasion of cancer: perspectives from pathology and development: collective invasion of cancer. Pathol. Int. 66, 183–192. doi: 10.1111/pin.12391
Ware, M. J., Keshishian, V., Law, J. J., Ho, J. C., Favela, C. A., Rees, P., et al. (2016). Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials 108, 129–142. doi: 10.1016/j.biomaterials.2016.08.041
Westphalen, C. B., and Olive, K. P. (2012). Genetically engineered mouse models of pancreatic cancer. Cancer J. 18, 502–510. doi: 10.1097/PPO.0b013e31827ab4c4
Wong, C.-W., Han, H.-W., Tien, Y.-W., and Hsu, S. (2019). Biomaterial substrate-derived compact cellular spheroids mimicking the behavior of pancreatic cancer and microenvironment. Biomaterials 213:119202. doi: 10.1016/j.biomaterials.2019.05.013
Wu, J.-Q., Zhai, J., Li, C.-Y., Tan, A.-M., Wei, P., Shen, L.-Z., et al. (2017). Patient-derived xenograft in zebrafish embryos: a new platform for translational research in gastric cancer. J. Exp. Clin. Cancer Res. 36:160. doi: 10.1186/s13046-017-0631-0
Xu, C., Li, X., Liu, P., Li, M., and Luo, F. (2018). Patient-derived xenograft mouse models: a high fidelity tool for individualized medicine (Review). Oncol. Lett. 17, 3–10. doi: 10.3892/ol.2018.9583
Yachida, S., and Iacobuzio-Donahue, C. A. (2009). The pathology and genetics of metastatic pancreatic cancer. Arch. Pathol. Lab. Med. 133, 413–422. doi: 10.5858/133.3.413
Yachida, S., Jones, S., Bozic, I., Antal, T., Leary, R., Fu, B., et al. (2010). Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117. doi: 10.1038/nature09515
Yachida, S., White, C. M., Naito, Y., Zhong, Y., Brosnan, J. A., Macgregor-Das, A. M., et al. (2012). Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin. Cancer Res. 18, 6339–6347. doi: 10.1158/1078-0432.CCR-12-1215
Yadavalli, S., Jayaram, S., Manda, S. S., Madugundu, A. K., Nayakanti, D. S., Tan, T. Z., et al. (2017). Data-driven discovery of extravasation pathway in circulating tumor cells. Sci. Rep. 7:43710. doi: 10.1038/srep43710
Yamashita, K., Miyamoto, A., Hama, N., Asaoka, T., Maeda, S., Omiya, H., et al. (2015). Survival impact of pulmonary metastasis as recurrence of pancreatic ductal adenocarcinoma. Dig. Surg. 32, 464–471. doi: 10.1159/000439545
Yamauchi, A., Yamamura, M., Katase, N., Itadani, M., Okada, N., Kobiki, K., et al. (2017). Evaluation of pancreatic cancer cell migration with multiple parameters in vitro by using an optical real-time cell mobility assay device. BMC Cancer 17:234. doi: 10.1186/s12885-017-3218-4
Yan, C., Brunson, D. C., Tang, Q., Do, D., Iftimia, N. A., Moore, J. C., et al. (2019). Visualizing engrafted human cancer and therapy responses in immunodeficient zebrafish. Cell 177, 1903–1914.e14. doi: 10.1016/j.cell.2019.04.004
Yang, J., Lin, P., Yang, M., Liu, W., Fu, X., Liu, D., et al. (2021). Integrated genomic and transcriptomic analysis reveals unique characteristics of hepatic metastases and pro-metastatic role of complement C1q in pancreatic ductal adenocarcinoma. Genome Biol. 22:4. doi: 10.1186/s13059-020-02222-w
Yang, J.-Y., Jiang, S.-H., Liu, D.-J., Yang, X.-M., Huo, Y.-M., Li, J., et al. (2015). Decreased LKB1 predicts poor prognosis in pancreatic ductal adenocarcinoma. Sci. Rep. 5:10575. doi: 10.1038/srep10575
Yu, M., Ting, D. T., Stott, S. L., Wittner, B. S., Ozsolak, F., Paul, S., et al. (2012). RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 487, 510–513. doi: 10.1038/nature11217
Zeng, S., Pöttler, M., Lan, B., Grützmann, R., Pilarsky, C., and Yang, H. (2019). Chemoresistance in pancreatic cancer. Int. J. Mol. Sci. 20, 1–19. doi: 10.3390/ijms20184504
Zhang, G., and Du, Y.-C. N. (2019). “Orthotopic pancreatic tumor mouse models of liver metastasis,” in Pancreatic Cancer Methods in Molecular Biology, ed. G. H. Su (New York, NY: Springer New York), 309–320. doi: 10.1007/978-1-4939-8879-2_27
Zhang, J., Wang, W., Zhou, Y., Yang, J., Xu, J., Xu, Z., et al. (2020). Terphenyllin suppresses orthotopic pancreatic tumor growth and prevents metastasis in mice. Front. Pharmacol. 11, 457. doi: 10.3389/fphar.2020.00457
Zhou, P., Li, B., Liu, F., Zhang, M., Wang, Q., Liu, Y., et al. (2017). The epithelial to mesenchymal transition (EMT) and cancer stem cells: implication for treatment resistance in pancreatic cancer. Mol. Cancer 16:52. doi: 10.1186/s12943-017-0624-9
Keywords: metastasis, pancreatic cancer, organoids, metastasis models, PDAC – pancreatic ductal adenocarcinoma, GEMMs
Citation: Miquel M, Zhang S and Pilarsky C (2021) Pre-clinical Models of Metastasis in Pancreatic Cancer. Front. Cell Dev. Biol. 9:748631. doi: 10.3389/fcell.2021.748631
Received: 28 July 2021; Accepted: 20 September 2021;
Published: 27 October 2021.
Edited by:
Marc Stemmler, Friedrich-Alexander-Universität Erlangen-Nürnberg, GermanyReviewed by:
Baiwen Li, Shanghai General Hospital, ChinaCopyright © 2021 Miquel, Zhang and Pilarsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Pilarsky, Q2hyaXN0aWFuLlBpbGFyc2t5QHVrLWVybGFuZ2VuLmRl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.