 Elena N. Huang
Elena N. Huang Henry Quach
Henry Quach Jin-A Lee
Jin-A Lee Joshua Dierolf1
Joshua Dierolf1 Theo J. Moraes
Theo J. Moraes Amy P. Wong
Amy P. Wong- 1Program in Developmental and Stem Cell Biology, Hospital for Sick Children, Toronto, ON, Canada
- 2Department of Laboratory Medicine & Pathobiology, University of Toronto, Toronto, ON, Canada
- 3Program in Translational Medicine, Hospital for Sick Children, Toronto, ON, Canada
The cystic fibrosis (CF) transmembrane conductance regulator (CFTR) protein is a cAMP-activated anion channel that is critical for regulating fluid and ion transport across the epithelium. This process is disrupted in CF epithelia, and patients harbouring CF-causing mutations experience reduced lung function as a result, associated with the increased rate of mortality. Much progress has been made in CF research leading to treatments that improve CFTR function, including small molecule modulators. However, clinical outcomes are not necessarily mutation-specific as individuals harboring the same genetic mutation may present with varying disease manifestations and responses to therapy. This suggests that the CFTR protein may have alternative functions that remain under-appreciated and yet can impact disease. In this mini review, we highlight some notable research implicating an important role of CFTR protein during early lung development and how mutant CFTR proteins may impact CF airway disease pathogenesis. We also discuss recent novel cell and animal models that can now be used to identify a developmental cause of CF lung disease.
Introduction
Cystic fibrosis (CF) is a lethal, monogenic disorder involving autosomal recessive mutations of the CF transmembrane conductance regulator (CFTR) gene encoding the CFTR protein. CFTR functions as an anion channel that regulates fluid transport across specialized epithelia. Loss of CFTR channel expression or function due to disease-causing mutations leads to defects in ion and fluid transport into the airway lumen (Bergeron and Cantin, 2019) which ultimately lead to persistent microbial presence causing chronic inflammation and irreversible lung damage (Ratjen et al., 2015). To date, there have been over 2,000 CFTR mutations identified, of which over 360 are considered disease-causing mutations1. Disease-causing CF mutations are classified into seven classes that contribute to disease severity (De Boeck, 2020). Class I protein production mutations produce defective mRNA that interfere with the production of the full-length mutant protein. The most common Class II (i.e., F508del) mutations are protein processing mutations that interfere with the polypeptide folding. Class III (i.e., G551D) are gating mutations that impair CFTR channel opening while Class IV and V mutations are conduction and insufficient production mutations that impair anion conduction or reduce protein expression on the surface of the cells, respectively. Finally, Class VI and VII affect surface protein stability or no expression of mRNA. CF is further complicated by phenotypic variances in patients even with the same genetic mutation and this may be a result of disease-causing gene modifiers (Strug et al., 2016) and environmental factors (Ramsay et al., 2016). Novel drugs are improving mutant CFTR protein function, however many challenges still remain. There are considerable variations in drug responses and current drugs are not effective for all CF mutations (Laselva et al., 2021). Multiple drug therapies are available for patients such as KalydecoTM for patients >4-months with at least one G551D mutant allele, and the latest life-saving drug TrikaftaTM approved for the majority of CF individuals with at least one F508del mutant allele in patients >6 years in the US and >12 years in Canada. Young children present with similar disease pathologies (Waters and Ratjen, 2015; Ramsey et al., 2017) to adults and delaying therapy may have negative implications for disease progression and severity. CF disease pathogenesis during development is not completely understood and requires additional investigation using relevant models for which simple knockout models (both animal and cell lines) cannot capture. This mini review aims to highlight evidence of early disease manifestations in the developing lung and the need to revisit the role of CFTR in fetal lung development, especially in the advent of more recent animal and relevant cell models which will provide insight into CFTR mutations and its impact on lung maturation and function.
The human lung emerges from the ventral foregut endoderm (Cardoso and Lü, 2006). Lung development is categorized into five distinct phases: embryonic, pseudoglandular, canalicular, saccular, and alveolar. The lung buds emerge in the embryonic phase. In the pseudoglandular phase, the primitive lung buds undergo branching morphogenesis coordinated by the crosstalk between the developing epithelial and surrounding mesenchymal cells. Early vessels emerge simultaneously to form the pulmonary vasculature. Differentiation of several lung cell types begins and is regulated through proximal-distal patterning (Lu et al., 2001). In the canalicular phase, further differentiation leads to formation of specialized epithelial cell subtypes and the appearance of the primitive alveolar cells. Extensive growth of the fetal lung with expansion of the air sacs, thinning of connective tissue, deposition of extracellular matrices, and the formation of surfactant proteins marks the saccular phase. Finally, secondary septations in the alveolar phase form complex alveoli that optimize gas exchange with a concomitant increase in surfactant production. The human lung continues to develop well past childhood years (Schittny, 2017), and by adulthood, it will consist of >60 cell types (Travaglini et al., 2020) that collectively play a vital role in breathing, gas exchange, acid-base balance, metabolism, and immunity. At this point, CFTR expression in the adult airways is restricted to the apical membrane of certain specialized cell types such as ciliated and alveolar epithelial cells (Regnier et al., 2008), secretory cells and submucosal glands (Engelhardt et al., 1992), and ionocytes (Montoro et al., 2018).
Spatio-temporal expression of CFTR in the developing lungs suggests it may play an important role in organ formation. High levels of CFTR mRNA are found in the developing pancreas, kidneys, liver, and gut (Amaral et al., 2020). CFTR protein is also broadly expressed in the developing human fetal airways with a progressive increase in expression by mid-gestation (∼75-fold higher than adult lung) (Marcorelles et al., 2007). Detectable as early as the pseudoglandular stage around 7 weeks, CFTR is found diffusely in the cytoplasm of multipotent progenitor cells (Trezise et al., 1993). After peaking at mid-gestation, CFTR expression declines, remains low and expressed on the apical membrane of a subset of bronchial epithelial cells (Trezise et al., 1993; Marcorelles et al., 2007; Saint-Criq and Gray, 2017). Interestingly, the decline in CFTR expression correlates with cellular differentiation and lineage commitment. A comparison of CFTR expression in lungs of healthy and CF fetuses showed a 3-week delay in CFTR expression in the latter group until 15 weeks gestation (Marcorelles et al., 2007). The morphogenetic impact of this delay remains unknown but evidence of congenital defects as a result of mutant CFTR and some proposed mechanisms of how CFTR may regulate developmental processes are suggested.
Lung malformations and proinflammatory changes as a result of CFTR disruption have been found in fetal and newborn lungs. In rodents, malformations of the tracheal cartilaginous rings manifest in CFTR-deficient mice reduced breathing rate, suggestive of a role for CFTR in tracheal development (Bonvin et al., 2008). Moreover, in utero knockdown of CFTR in rats displayed classic CF morphologies of chronic inflammation, airway fibrosis and reactive airway disease (Cohen and Larson, 2005). In humans, abnormalities of the respiratory system caused by CF disease are observed in early childhood (Larson and Cohen, 2005). Studies of CF fetal lung tissue have demonstrated morphological abnormalities in tight junctions, dysfunction and absence of cilia in tracheal cells, and tracheal atrophy (Ornoy et al., 1987; Castellani et al., 2017). A study in newborn CF children found decreased tracheal lumen size (Meyerholz et al., 2010a). Although most CF infants are asymptomatic, 80.7% demonstrate abnormal lung changes and 77.2% exhibit detectable lung inflammation (Sly et al., 2009). Indeed, intensive CF early surveillance programs such as the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (ARESTCF) have shown that early lung disease in infants and young children can lead to irreversible lung damage. Therefore, optimal CF management may require early treatment but the potential risks of treatment-related side effects to the developing lung which remain unknown.
In utero correction of CFTR function can impact lung development and function. In utero delivery of CFTR has previously been shown to improve pulmonary under-development in a nitrofen-induced rat model of pulmonary hypoplasia (Larson and Cohen, 2006). Similarly, Sun et al. (2019) showed that a one-time antenatal administration of CFTR modulator rescued pancreatic and intestinal functions, reduced airway mucus production, and improved postnatal growth and survival of CF ferret kits. However, withdrawal of treatment at any point after birth reversed the phenotype resulting in bacterial infections and lung exacerbations similar to untreated animals. Together, both studies implicate CFTR in regulating lung morphogenesis and can impact postnatal lung function. Therefore, correcting prenatal defects may have a beneficial impact on reducing CF disease pathogenesis as has been suggested in the ARESTCF studies, and improve therapeutic outcomes for young CF children.
CFTR regulates fluid distension and mechanical stimulation of the developing lung. CFTR activation in the hypercalcemic fetal lung is facilitated by an extracellular calcium-sensing receptor (CaSR) that acts through activation of adenylate cyclase (Brennan et al., 2016). This secretion occurs during pseudoglandular branching and budding of airways and provides the lung with the distending pressure necessary for the expansion of the lungs. In utero CFTR gene transfer significantly affects the expression of proteins involved in smooth muscle contraction through the PI3 kinase and phospholipase-C pathways, modulating cytoskeletal tension and stretch induced differentiation of fetal lungs in a Rho kinase-independent manner (Cohen and Larson, 2006). In CF pig models, defects in CFTR-dependent anion transport and corresponding fluid secretion led to bud hypo-distension in the pseudoglandular airways that were unresponsive to CFTR agonists, forskolin/IBMX stimulation (Meyerholz et al., 2018). Altogether, these studies points to a mechanical role of CFTR in regulating growth of the developing fetal lung.
A putative role of CFTR in regulating epithelial differentiation has been previously shown (Brezillon et al., 1995; Dupuit et al., 1995). Mutant CFTR (F508del) localizes to the apical membrane of cells to a lesser degree (Kälin et al., 1999; Penque et al., 2000), suggesting that abnormal CFTR localization may be dependent on the differentiation state of the cell. In mice, in utero over-expression of CFTR increases bronchial cell differentiation and proliferation at the expense of alveolar development (Larson et al., 2000), resulting in death. In a rat model, transient in utero knockdown of CFTR altered alveolar Type II cell phenotype and associated surfactant homeostasis (Gad et al., 2009). An increase in CFTR mRNA in nasal epithelial cells has been shown to correlate with an increase in mucin and aquaporin expression (Jun et al., 2001). It has also been observed that genes associated with cilia biogenesis are down-regulated in F508del epithelia when compared to non-CF (Clarke et al., 2013). Overall, these studies implicate a role of CFTR in cellular differentiation. However, the mechanism(s) of how CFTR regulates differentiation during development is unclear.
Building evidence supports a direct regulatory role of CFTR protein in interacting with PDZ-containing signal transducers such as β-catenin (Liu et al., 2017), binding to cytoskeletal proteins such as zonula occludens-1 (ZO)-1, and regulating other ion channels (Benedetto et al., 2017; Figure 1) which can affect epithelial differentiation and proliferation. The canonical Wnt/β-catenin signaling plays a crucial role in normal lung development by regulating cellular differentiation and proliferation of uncommitted progenitor cells (Cardoso, 2001; De Langhe and Reynolds, 2008). Canonical Wnt activation through binding of Wnt ligands (i.e., Wnt3a) to its receptor Frizzled activates a cascade of signal transduction via the disheveled protein. This leads to the disruption of the Axin/APC/GSKβ degradation complex and in turn stabilizes β-catenin. Nuclear translocation of β-catenin binds to TCF/LEF transcription factors to transactivate Wnt target genes that regulate cellular proliferation and differentiation (MacDonald et al., 2009). An inverse relationship exists between CFTR and β-catenin activation as antisense-mediated knockdown of CFTR expression in the developing mouse lungs prolonged Wnt activity (Cohen et al., 2008). Overexpression of CFTR during lung development shortened Wnt activity and increased bronchial cell differentiation. Deep proteomic analysis of CF and wild-type cells revealed an association between wild-type CFTR with Wnt signaling components that was lost in mutant (F508del) CFTR (Pankow et al., 2015), further strengthening the observation of a direct relationship between CFTR and Wnt activity. Interestingly, CFTR/β-catenin interplay has also been demonstrated in other models such as kidney development (Zhang et al., 2017), intestinal inflammation (Liu et al., 2016), and pancreas development (Hyde et al., 1997).
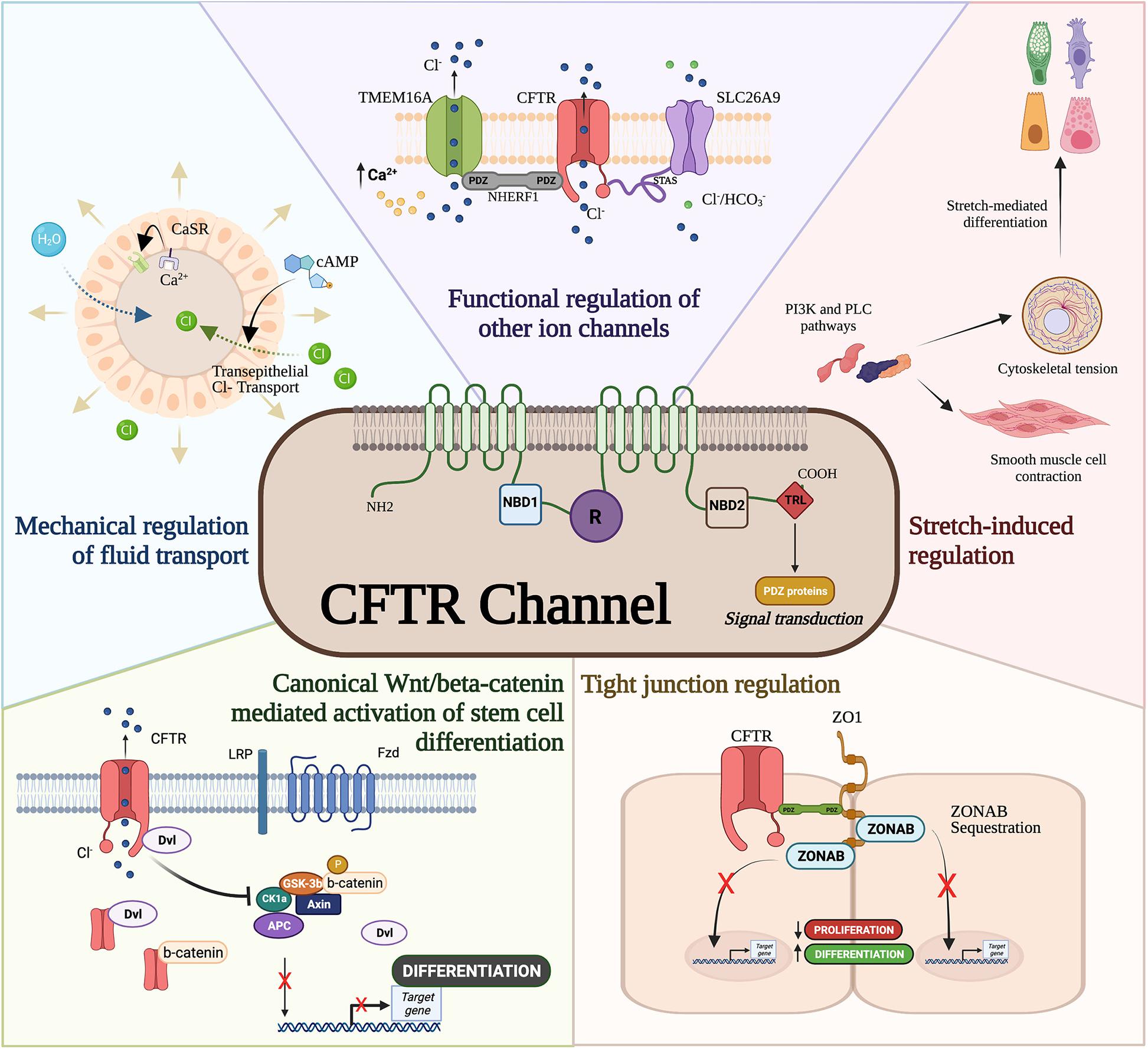
Figure 1. Cystic fibrosis transmembrane conductance regulator (CFTR) developmental interactome. The CFTR channel has been associated with developmental processes and regulatory pathways. CFTR mediates mechanical forces for airway development, functionally regulates other ion channels, drives differentiation through ZO-1, interferes with canonical Wnt/β-catenin activation of stem cell differentiation, and modulates smooth muscle cell contraction and cytoskeletal tension.
The last three amino acids in the C-terminal domain of the CFTR protein contain a PDZ domain required for apical membrane polarization (Moyer et al., 1999). While apical localization of CFTR is important for epithelial differentiation (Hollande et al., 1998; Castillon et al., 2002) and suppression of epithelial-mesenchymal transition (Corvol et al., 2018), CFTR’s PDZ binding domain is known to directly and indirectly regulate developmental pathways. Direct binding of CFTR to ZO-1 sequesters the ZO-1 associated nucleic acid binding (ZONAB) protein in tight junctions obstructing ZONAB nuclear translocation where it can activate genes associated with proliferation and differentiation (Ruan et al., 2014). CFTR/ZO-1 interaction has also been shown to form a complex with ezrin to induce ciliogenesis and secretory cell differentiation in dedifferentiated airway cells (Castillon et al., 2002). Furthermore, F508del CFTR mutations are capable of inhibiting intracellular transport activity of the SLC26A9 ion channel (Bertrand et al., 2017) through PDZ binding domains to the scaffolding protein, Na+/H+ exchanger regulatory factor isoform 1 (NHERF1). Interestingly, CFTR-NHERF1 interactions has been also shown to regulate Transmembrane member 16A (TMEM16A) (Benedetto et al., 2017), a calcium activated chloride channel previously shown to regulate fetal airway epithelial differentiation (He et al., 2020). Therefore, whether directly or indirectly, CFTR appears to be important in epithelial differentiation.
New animal and advanced cell models have emerged to elucidate the role of CFTR in lung development and cellular differentiation. This will provide important clues into why CF infants exhibit pulmonary abnormalities such as airway inflammation (Armstrong et al., 1997; Verhaeghe et al., 2007), functional (Beardsmore et al., 1988) and structural (Long et al., 2004) abnormalities, tracheal epithelial atrophy, and absence of cilia (Ornoy et al., 1987), and increased susceptibility to lower respiratory tract infections (Hiatt et al., 1999; Deschamp et al., 2019). While mouse models have provided important insight into the developmental pathways driving lung morphogenesis (Morrisey and Hogan, 2010), CF knockout mouse models do not recapitulate lung pathologies seen in humans (Guilbault et al., 2007). Here we focus on two animal models, ferrets and pigs, that do develop similar lung CF pathologies and highlight the emerging use of induced pluripotent stem cell (iPSC) in modeling airway development (Figure 2).
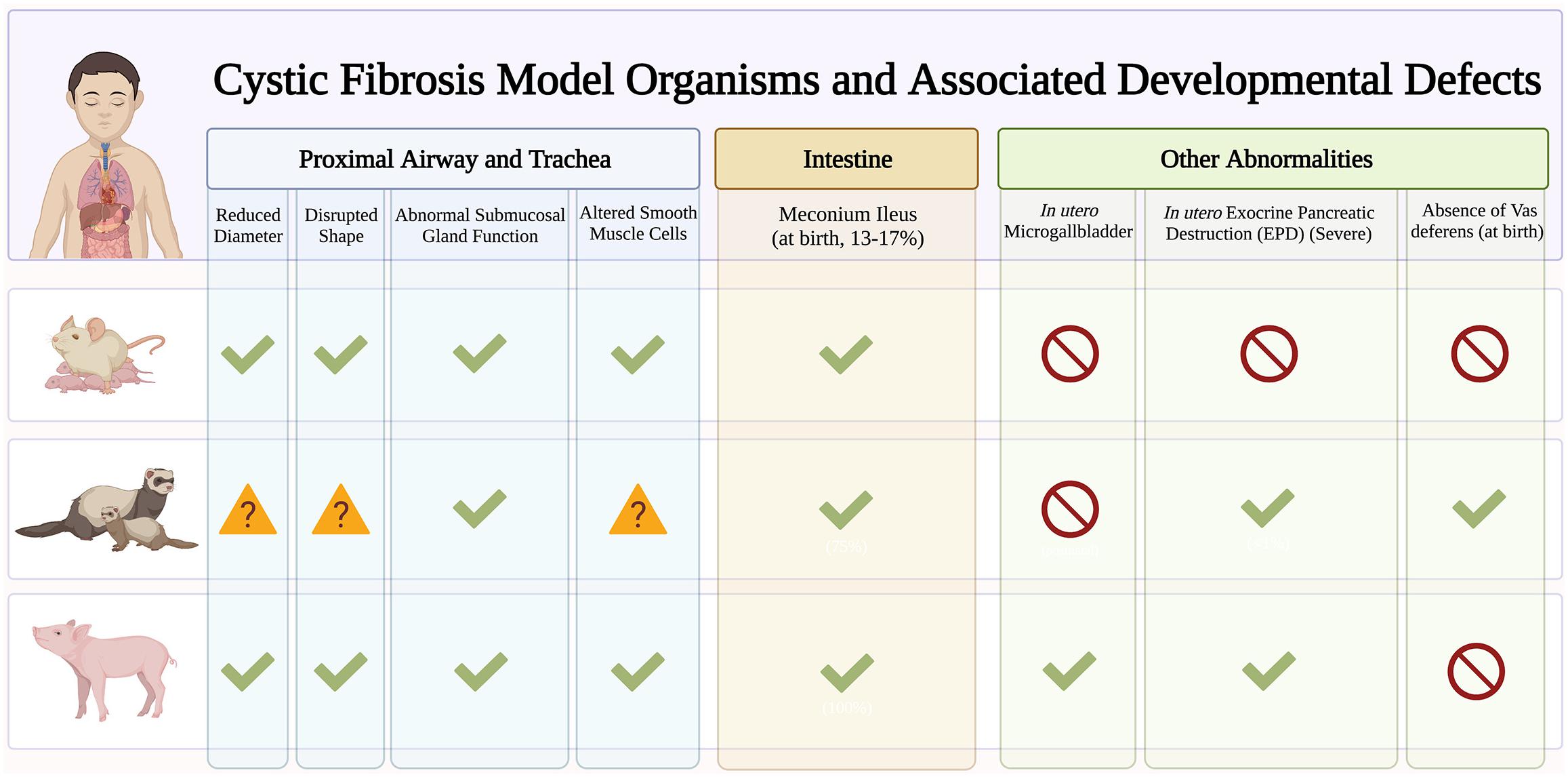
Figure 2. Early developmental pathology in cystic fibrosis (CF) animal models. Early manifestations of CF disease are recapitulated in different CF animal models. Checkmarks indicate presence of symptoms. Absence of symptoms is indicated by the red circle symbol. The question mark symbols indicate further investigation needed.
The ferret CF model exhibits multiorgan pathologies similar to human disease. These include pancreatic insufficiency, intestinal obstruction, stunted growth, and increased susceptibility to lung infections (Yan et al., 2015). As such, ferrets are great models to understand basic CF epithelial electrophysiology (Fisher et al., 2013) and assess small molecule and gene-based therapies (Cmielewski et al., 2014; Sun et al., 2019). Knockout ferrets develop severe lung and intestinal disease within the first week of life (Sun et al., 2010) leading to significant mortality soon after birth (Sun et al., 2014). Emerging ferret models with human mutations such as the G551D are useful for studying biological problems in a mutation-dependent manner (Semaniakou et al., 2019). With a short (42 day) gestational period, ferrets are great models to characterize the effects of modulator treatment in utero. In fact, in utero treatment with CFTR modulators improve postnatal CF kit lung functions which suggests fetal CFTR function is important in limiting pathogenic changes affecting postnatal function.
Pig models are suitable for studying lung development and diseases since they share many anatomical features with humans. CF pigs harboring F508del mutations have been generated that exhibit the same disease phenotype as humans (Rogers et al., 2008; Ostedgaard et al., 2011). CF piglets exhibit tracheal malformations similar to those seen in CF infants, including reduced lumen diameter and circularity (Meyerholz et al., 2010a, 2018) that contribute to abnormal particle distribution (Awadalla et al., 2014). CF piglets also exhibit no signs of inflammation at birth, but quickly develop lung disease and infections, resembling human disease (Stoltz et al., 2010). Human pancreatic disease such as small exocrine pancreas and inflammation are also observed in CF piglets. Interestingly, CF pancreatic disease begins in utero with activation of inflammatory and remodeling pathways (Abu-El-Haija et al., 2012). However, one important caveat is that CF pigs are prone to meconium ileus that often necessitates euthanasia despite surgical efforts (Meyerholz et al., 2010b; Guillon et al., 2015), and this may preclude the use of CF pigs in studying the long-term effects of CFTR in development.
Regulatory concerns over the use of human fetal tissues for research and the scarcity of fetal tissues with rare congenital disorders, is a major obstacle to understanding human lung development. Therefore, iPSC has become an attractive surrogate for fundamental discoveries in human development. These cells are artificially reprogrammed cells from somatic cells that phenocopy and function like embryonic stem cells (Takahashi and Yamanaka, 2006). These iPSC offer several advantages over cell lines and primary epithelial cells: (1) iPSC are an unlimited source of patient-derived cells that harbor the patient genotype allowing the study of patient-specific disease and therapies, (2) iPSC harboring various classes of CFTR mutations can be used to study mutation-dependent effects, (3) iPSC differentiation models stepwise developmental changes and therefore differentiation to fetal phenotypes offer an incredible opportunity to study mechanisms of development, (4) iPSC from one individual can generate multiple tissue/cell types and therefore offer a great source of cells to understand disease in tissue-specific context.
Development of differentiation protocols to generate lung cells have been established (Wong et al., 2012, 2015; Firth et al., 2014; Huang et al., 2014; Konishi et al., 2016; McCauley et al., 2017; Leibel et al., 2020). The differentiation process involves precise temporal and dose-dependent exposure of the cells to specific recombinant proteins and/or small molecules affecting key signaling pathways. Generally, the first major differentiation steps involve generation of definitive endoderm cells, followed by patterning to form the anterior ventral foregut endoderm that will form the earliest lung cells. Further differentiation generates fetal lung progenitors and requires key morphogens such as fibroblast growth factors, bone morphogenetic proteins and Wnts. Finally, exposure to air in vitro using air liquid interface cultures to mimic the microenvironment of the postnatal lung induces maturation and polarization of the epithelia. Recent sequencing technologies have mapped the faithful lung lineage differentiation of iPSC in modeling development compared to their in vivo tissue counterparts (Miller et al., 2019; Hurley et al., 2020; Hawkins et al., 2021). It has also been shown that iPSC-derived fetal lung cells transcriptionally match late pseudoglandular fetal lung epithelial cells (Ngan et al., 2021). Human iPSC-derived fetal lung cells provide an opportunity to determine the role of CFTR in pseudoglandular lung epithelial development. With iPSC harboring different CFTR mutations, the effects of CFTR mutations in lung epithelial lineage development and the long-term effects of modulator therapy in epithelial development can be studied. Overall, in vitro hPSC differentiations are powerful complementary models to study developmental mechanisms underlying genetic diseases.
Summary
Investigating the pleiotropic roles of CFTR during development and the impact of defective CFTR in utero is critical to understanding CF disease pathogenesis. Recent advances in cell and animal models will improve our understanding of an “old disease” from a developmental lens and potentially lead to the identification of novel therapies aimed at reversing or limiting disease progression.
Author Contributions
EH and HQ: conceptualization, methodology, validation, formal analysis, investigation, writing—original draft, writing—review and editing, and visualization. J-AL, JD, and TM: writing—review and editing. AW: resources, writing—review and editing, funding acquisition, and supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the SickKids Foundation-CIHR IHDCYH NI20-1070 and Stem Cell Network ECI-23 grants. EH is a recipient of the Lunenfeld Summer Studentship award (2021); JD is a recipient of the 2021 CRAFT (Centre for Research and Application in Fluidic Technologies) fellowship award.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All figures were prepared with BioRender.com.
Footnotes
References
Abu-El-Haija, M., Ramachandran, S., Meyerholz, D. K., Abu-El-Haija, M., Griffin, M., Giriyappa, R. L., et al. (2012). Pancreatic damage in fetal and newborn cystic fibrosis pigs involves the activation of inflammatory and remodelling pathways. Am. J. Pathol. 181, 499–507. doi: 10.1016/j.ajpath.2012.04.024
Amaral, M. D., Quaresma, M. C., and Pankonien, I. (2020). What role does CFTR play in development, differentiation, regeneration and cancer? Int. J. Mol. Sci. 21:E3133. doi: 10.3390/ijms21093133
Armstrong, D. S., Grimwood, K., Carlin, J. B., Carzino, R., Gutièrrez, J. P., Hull, J., et al. (1997). Lower airway inflammation in infants and young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 156, 1197–1204. doi: 10.1164/ajrccm.156.4.96-11058
Awadalla, M., Miyawaki, S., Abou Alaiwa, M. H., Adam, R. J., Bouzek, D. C., and Michalski, A. S., et al. (2014). Early airway structural changes in cystic fibrosis pigs as a determinant of particle distribution and deposition. Ann. Biomed. Eng. 42, 915–927. doi: 10.1007/s10439-013-0955-7
Beardsmore, C. S., Bar-Yishay, E., Maayan, C., Yahav, Y., Katznelson, D., and Godfrey, S. (1988). Lung function in infants with cystic fibrosis. Thorax 43, 545–551. doi: 10.1136/thx.43.7.545
Benedetto, R., Ousingsawat, J., Wanitchakool, P., Zhang, Y., Holtzman, M. J., Amaral, M., et al. (2017). Epithelial chloride transport by CFTR requires TMEM16A. Sci. Rep. 7:12397. doi: 10.1038/s41598-017-10910-0
Bergeron, C., and Cantin, A. M. (2019). Cystic fibrosis: pathophysiology of lung disease. Semin. Respir. Crit. Care Med. 40, 715–726. doi: 10.1055/s-0039-1694021
Bertrand, C. A., Mitra, S., Mishra, S. K., Wang, X., Zhao, Y., Pilewski, J. M., et al. (2017). The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L912–L925. doi: 10.1152/ajplung.00178.2016
Bonvin, E., Le Rouzic, P., Bernaudin, J.-F., Cottart, C.-H., Vandebrouck, C., Crié, A., et al. (2008). Congenital tracheal malformation in cystic fibrosis transmembrane conductance regulator-deficient mice. J. Physiol. 586, 3231–3243. doi: 10.1113/jphysiol.2008.150763
Brennan, S. C., Wilkinson, W. J., Tseng, H.-E., Finney, B., Monk, B., Dibble, H., et al. (2016). The extracellular calcium-sensing receptor regulates human fetal lung development via CFTR. Sci. Rep. 6:21975. doi: 10.1038/srep21975
Brezillon, S., Dupuit, F., Hinnrasky, J., Marchand, V., Kälin, N., Tümmler, B., et al. (1995). Decreased expression of the CFTR protein in remodeled human nasal epithelium from non-cystic fibrosis patients. Lab. Invest. 72, 191–200.
Cardoso, W. V. (2001). Molecular regulation of lung development. Ann. Rev. Physiol. 63, 471–494. doi: 10.1146/annurev.physiol.63.1.471
Cardoso, W. V., and Lü, J. (2006). Regulation of early lung morphogenesis: questions, facts and controversies. Development 133, 1611–1624. doi: 10.1242/dev.02310
Castellani, S., Favia, M., Guerra, L., Carbone, A., Abbattiscianni, A. C., Di Gioia, S., et al. (2017). Emerging relationship between CFTR, actin and tight junction organization in cystic fibrosis airway epithelium. Histol. Histopathol. 32, 445–459.
Castillon, N., Hinnrasky, J., Zahm, J.-M., Kaplan, H., Bonnet, N., Corlieu, P., et al. (2002). Polarized expression of cystic fibrosis transmembrane conductance regulator and associated epithelial proteins during the regeneration of human airway surface epithelium in three-dimensional culture. Lab. Invest. 82, 989–998. doi: 10.1097/01.LAB.0000022221.88025.43
Clarke, L. A., Sousa, L., Barreto, C., and Amaral, M. D. (2013). Changes in transcriptome of native nasal epithelium expressing F508del-CFTR and intersecting data from comparable studies. Respir. Res. 14:38. doi: 10.1186/1465-9921-14-38
Cmielewski, P., Farrow, N., Donnelley, M., McIntyre, C., Penny-Dimri, J., Kuchel, T., et al. (2014). Transduction of ferret airway epithelia using a pre-treatment and lentiviral gene vector. BMC Pulm. Med. 14:183. doi: 10.1186/1471-2466-14-183
Cohen, J. C., and Larson, J. E. (2005). Pathophysiologic consequences following inhibition of a CFTR-dependent developmental cascade in the lung. BMC Dev. Biol. 5:2. doi: 10.1186/1471-213X-5-2
Cohen, J. C., and Larson, J. E. (2006). Cystic fibrosis transmembrane conductance regulator (CFTR) dependent cytoskeletal tension during lung organogenesis. Dev. Dyn. 235, 2736–2748. doi: 10.1002/dvdy.20912
Cohen, J. C., Larson, J. E., Killeen, E., Love, D., and Takemaru, K.-I. (2008). CFTR and Wnt/beta-catenin signaling in lung development. BMC Dev. Biol. 8:70. doi: 10.1186/1471-213X-8-70
Corvol, H., Rousselet, N., Thompson, K. E., Berdah, L., Cottin, G., Foussigniere, T., et al. (2018). FAM13A is a modifier gene of cystic fibrosis lung phenotype regulating rhoa activity, actin cytoskeleton dynamics and epithelial-mesenchymal transition. J. Cyst. Fibros 17, 190–203. doi: 10.1016/j.jcf.2017.11.003
De Boeck, K. (2020). Cystic fibrosis in the year 2020: a disease with a new face. Acta Paediatr. 109, 893–899. doi: 10.1111/apa.15155
De Langhe, S. P., and Reynolds, S. D. (2008). Wnt signaling in lung organogenesis. Organogenesis 4, 100–108. doi: 10.4161/org.4.2.5856
Deschamp, A. R., Hatch, J. E., Slaven, J. E., Gebregziabher, N., Storch, G., Hall, G. L., et al. (2019). Early respiratory viral infections in infants with cystic fibrosis. J. Cyst. Fibros 18, 844–850. doi: 10.1016/j.jcf.2019.02.004
Dupuit, F., Kälin, N., Brézillon, S., Hinnrasky, J., Tümmler, B., and Puchelle, E. (1995). CFTR and differentiation markers expression in non-CF and delta F 508 homozygous CF nasal epithelium. J. Clin. Invest. 96, 1601–1611. doi: 10.1172/JCI118199
Engelhardt, J. F., Yankaskas, J. R., Ernst, S. A., Yang, Y., Marino, C. R., Boucher, R. C., et al. (1992). Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet. 2, 240–248. doi: 10.1038/ng1192-240
Firth, A. L., Dargitz, C. T., Qualls, S. J., Menon, T., Wright, R., Singer, O., et al. (2014). Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, E1723–E1730. doi: 10.1073/pnas.1403470111
Fisher, J. T., Tyler, S. R., Zhang, Y., Lee, B. J., Liu, X., Sun, X., et al. (2013). Bioelectric characterization of epithelia from neonatal CFTR knockout ferrets. Am. J. Respir. Cell Mol. Biol. 49, 837–844. doi: 10.1165/rcmb.2012-0433OC
Gad, A., Callender, D. L., Killeen, E., Hudak, J., Dlugosz, M. A., Larson, J. E., et al. (2009). Transient in utero disruption of cystic fibrosis transmembrane conductance regulator causes phenotypic changes in alveolar Type II cells in adult rats. BMC Cell Biol. 10:24. doi: 10.1186/1471-2121-10-24
Guilbault, C., Saeed, Z., Downey, G. P., and Radzioch, D. (2007). Cystic fibrosis mouse models. Am. J. Respir. Cell Mol. Biol. 36, 1–7. doi: 10.1165/rcmb.2006-0184TR
Guillon, A., Chevaleyre, C., Barc, C., Berri, M., Adriaensen, H., Lecompte, F., et al. (2015). Computed tomography (CT) scanning facilitates early identification of neonatal cystic fibrosis piglets. PLoS One 10:e0143459. doi: 10.1371/journal.pone.0143459
Hawkins, F. J., Suzuki, S., Beermann, M. L., Barillà, C., Wang, R., Villacorta-Martin, C., et al. (2021). Derivation of airway basal stem cells from human pluripotent stem cells. Cell Stem Cell 28, 79–95.e8. doi: 10.1016/j.stem.2020.09.017
He, M., Wu, B., Ye, W., Le, D. D., Sinclair, A. W., Padovano, V., et al. (2020). Chloride channels regulate differentiation and barrier functions of the mammalian airway. Elife 9:e53085. doi: 10.7554/eLife.53085
Hiatt, P. W., Grace, S. C., Kozinetz, C. A., Raboudi, S. H., Treece, D. G., Taber, L. H., et al. (1999). Effects of viral lower respiratory tract infection on lung function in infants with cystic fibrosis. Pediatrics 103, 619–626. doi: 10.1542/peds.103.3.619
Hollande, E., Fanjul, M., Chemin-Thomas, C., Devaux, C., Demolombe, S., Van Rietschoten, J., et al. (1998). Targeting of CFTR protein is linked to the polarization of human pancreatic duct cells in culture. Eur. J. Cell Biol. 76, 220–227. doi: 10.1016/S0171-9335(98)80037-X
Huang, S. X. L., Islam, M. N., O’Neill, J., Hu, Z., Yang, Y.-G., Chen, Y.-W., et al. (2014). Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 32, 84–91. doi: 10.1038/nbt.2754
Hurley, K., Ding, J., Villacorta-Martin, C., Herriges, M. J., Jacob, A., Vedaie, M., et al. (2020). Reconstructed single-cell fate trajectories define lineage plasticity windows during differentiation of human PSC-derived distal lung progenitors. Cell Stem Cell 26, 593–608.e8. doi: 10.1016/j.stem.2019.12.009
Hyde, K., Reid, C. J., Tebbutt, S. J., Weide, L., Hollingsworth, M. A., and Harris, A. (1997). The cystic fibrosis transmembrane conductance regulator as a marker of human pancreatic duct development. Gastroenterology 113, 914–919. doi: 10.1016/S0016-5085(97)70187-2
Jun, E. S., Kim, Y. S., Yoo, Roh, H. J., and Jung, J. S. (2001). Changes in expression of ion channels and aquaporins mRNA during differentiation in normal human nasal epithelial cells. Life Sci. 68, 827–840. doi: 10.1016/S0024-3205(00)00985-1
Kälin, N., Claass, A., Sommer, M., Puchelle, E., and Tümmler, B. (1999). DeltaF508 CFTR protein expression in tissues from patients with cystic fibrosis. J. Clin. Invest. 103, 1379–1389. doi: 10.1172/JCI5731
Konishi, S., Gotoh, S., Tateishi, K., Yamamoto, Y., Korogi, Y., Nagasaki, T., et al. (2016). Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep. 6, 18–25. doi: 10.1016/j.stemcr.2015.11.010
Larson, J. E., and Cohen, J. C. (2005). Developmental paradigm for early features of cystic fibrosis. Pediatr. Pulmonol. 40, 371–377. doi: 10.1002/ppul.20169
Larson, J. E., and Cohen, J. C. (2006). Improvement of pulmonary hypoplasia associated with congenital diaphragmatic hernia by in utero CFTR gene therapy. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L4–L10. doi: 10.1152/ajplung.00372.2005
Larson, J. E., Delcarpio, J. B., Farberman, M. M., Morrow, S. L., and Cohen, J. C. (2000). CFTR modulates lung secretory cell proliferation and differentiation. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L333–L341. doi: 10.1152/ajplung.2000.279.2.L333
Laselva, O., Ardelean, M. C., and Bear, C. E. (2021). Phenotyping rare CFTR mutations reveal functional expression defects restored by TRIKAFTATM. J. Pers. Med. 11:301. doi: 10.3390/jpm11040301
Leibel, S. L., McVicar, R. N., Winquist, A. M., Niles, W. D., and Snyder, E. Y. (2020). Generation of complete multi-cell type lung organoids from human embryonic and patient-specific induced pluripotent stem cells for infectious disease modeling and therapeutics validation. Curr. Protoc. Stem Cell Biol. 54:e118. doi: 10.1002/cpsc.118
Liu, K., Zhang, X., Zhang, J. T., Tsang, L. L., Jiang, X., and Chan, H. C. (2016). Defective CFTR- β-catenin interaction promotes NF-κB nuclear translocation and intestinal inflammation in cystic fibrosis. Oncotarget 7, 64030–64042. doi: 10.18632/oncotarget.11747
Liu, Z., Guo, J., Wang, Y., Weng, Z., Huang, B., Yu, M.-K., et al. (2017). CFTR-β-catenin interaction regulates mouse embryonic stem cell differentiation and embryonic development. Cell Death Differ. 24, 98–110. doi: 10.1038/cdd.2016.118
Long, F. R., Williams, R. S., and Castile, R. G. (2004). Structural airway abnormalities in infants and young children with cystic fibrosis. J. Pediatr. 144, 154–161. doi: 10.1016/j.jpeds.2003.09.026
Lu, M. M., Yang, H., Zhang, L., Shu, W., Blair, D. G., and Morrisey, E. E. (2001). The bone morphogenic protein antagonist gremlin regulates proximal-distal patterning of the lung. Dev. Dyn. 222, 667–680. doi: 10.1002/dvdy.1231
MacDonald, B. T., Tamai, K., and He, X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. doi: 10.1016/j.devcel.2009.06.016
Marcorelles, P., Montier, T., Gillet, D., Lagarde, N., and Ferec, C. (2007). Evolution of CFTR protein distribution in lung tissue from normal and CF human fetuses. Pediatr. Pulmonol. 42, 1032–1040. doi: 10.1002/ppul.20690
McCauley, K. B., Hawkins, F., Serra, M., Thomas, D. C., Jacob, A., and Kotton, D. N. (2017). Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt Signaling. Cell Stem Cell 20, 844–857.e6. doi: 10.1016/j.stem.2017.03.001
Meyerholz, D. K., Stoltz, D. A., Gansemer, N. D., Ernst, S. E., Cook, D. P., Strub, M. D., et al. (2018). Lack of cystic fibrosis transmembrane conductance regulator disrupts fetal airway development in pigs. Lab. Invest. 98, 825–838. doi: 10.1038/s41374-018-0026-7
Meyerholz, D. K., Stoltz, D. A., Namati, E., Ramachandran, S., Pezzulo, A. A., Smith, A. R., et al. (2010a). Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am. J. Respir. Crit. Care Med. 182, 1251–1261. doi: 10.1164/rccm.201004-0643OC
Meyerholz, D. K., Stoltz, D. A., Pezzulo, A. A., and Welsh, M. J. (2010b). Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am. J. Pathol. 176, 1377–1389. doi: 10.2353/ajpath.2010.090849
Miller, A. J., Dye, B. R., Ferrer-Torres, D., Hill, D. R., Overeem, A. W., Shea, L. D., et al. (2019). Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 14, 518–540. doi: 10.1038/s41596-018-0104-8
Montoro, D. T., Haber, A. L., Biton, M., Vinarsky, V., Lin, B., Birket, S. E., et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324. doi: 10.1038/s41586-018-0393-7
Morrisey, E. E., and Hogan, B. L. M. (2010). Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev. Cell 18, 8–23. doi: 10.1016/j.devcel.2009.12.010
Moyer, B. D., Denton, J., Karlson, K. H., Reynolds, D., Wang, S., Mickle, J. E., et al. (1999). A PDZ-interacting domain in CFTR is an apical membrane polarization signal. J. Clin. Invest. 104, 1353–1361. doi: 10.1172/JCI7453
Ngan, Z., Quach, H., Dierolf, J., Lee, J. A., Huang, E. N., and Wong, A. P. (2021). Modeling lung cell development using human pluripotent stem cells. bioRxiv [Preprint]. doi: 10.1101/2021.07.16.452691
Ornoy, A., Arnon, J., Katznelson, D., Granat, M., Caspi, B., and Chemke, J. (1987). Pathological confirmation of cystic fibrosis in the fetus following prenatal diagnosis. Am. J. Med. Genet. 28, 935–947. doi: 10.1002/ajmg.1320280420
Ostedgaard, L. S., Meyerholz, D. K., Chen, J.-H., Pezzulo, A. A., Karp, P. H., Rokhlina, T., et al. (2011). The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci. Transl. Med. 3:74ra24. doi: 10.1126/scitranslmed.3001868
Pankow, S., Bamberger, C., Calzolari, D., Martínez-Bartolomé, S., Lavallée-Adam, M., Balch, W. E., et al. (2015). ΔF508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528, 510–516. doi: 10.1038/nature15729
Penque, D., Mendes, F., Beck, S., Farinha, C., Pacheco, P., Nogueira, P., et al. (2000). Cystic fibrosis F508del patients have apically localized CFTR in a reduced number of airway cells. Lab. Invest. 80, 857–868. doi: 10.1038/labinvest.3780090
Ramsay, K. A., Stockwell, R. E., Bell, S. C., and Kidd, T. J. (2016). Infection in cystic fibrosis: impact of the environment and climate. Expert Rev. Respir. Med. 10, 505–519. doi: 10.1586/17476348.2016.1162715
Ramsey, K., Ratjen, F., and Latzin, P. (2017). Elucidating progression of early cystic fibrosis lung disease. Eur. Respir. J. 50:1701916. doi: 10.1183/13993003.01916-2017
Ratjen, F., Bell, S. C., Rowe, S. M., Goss, C. H., Quittner, A. L., and Bush, A. (2015). Cystic fibrosis. Nat. Rev. Dis. Primers 1:15010. doi: 10.1038/nrdp.2015.10
Regnier, A., Dannhoffer, L., Blouquit-Laye, S., Bakari, M., Naline, E., and Chinet, T. (2008). Expression of cystic fibrosis transmembrane conductance regulator in the human distal lung. Hum. Pathol. 39, 368–376. doi: 10.1016/j.humpath.2007.06.020
Rogers, C. S., Hao, Y., Rokhlina, T., Samuel, M., Stoltz, D. A., Li, Y., et al. (2008). Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J. Clin. Invest. 118, 1571–1577. doi: 10.1172/JCI34773
Ruan, Y. C., Wang, Y., Da Silva, N., Kim, B., Diao, R. Y., Hill, E., et al. (2014). CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J. Cell Sci. 127, 4396–4408. doi: 10.1242/jcs.148098
Saint-Criq, V., and Gray, M. A. (2017). Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 74, 93–115. doi: 10.1007/s00018-016-2391-y
Schittny, J. C. (2017). Development of the lung. Cell Tissue Res. 367, 427–444. doi: 10.1007/s00441-016-2545-0
Semaniakou, A., Croll, R. P., and Chappe, V. (2019). Animal models in the pathophysiology of cystic fibrosis. Front. Pharmacol. 9:1475. doi: 10.3389/fphar.2018.01475
Sly, P. D., Brennan, S., Gangell, C., de Klerk, N., Murray, C., Mott, L., et al. (2009). Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir. Crit. Care Med. 180, 146–152. doi: 10.1164/rccm.200901-0069OC
Stoltz, D. A., Meyerholz, D. K., Pezzulo, A. A., Ramachandran, S., Rogan, M. P., Davis, G. J., et al. (2010). Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2:29ra31. doi: 10.1126/scitranslmed.3000928
Strug, L. J., Gonska, T., He, G., Keenan, K., Ip, W., Boëlle, P.-Y., et al. (2016). Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet. 25, 4590–4600. doi: 10.1093/hmg/ddw290
Sun, X., Olivier, A. K., Liang, B., Yi, Y., Sui, H., Evans, T. I. A., et al. (2014). Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator–knockout ferrets. Am. J. Respir. Cell Mol. Biol. 50, 502–512. doi: 10.1165/rcmb.2013-0261OC
Sun, X., Sui, H., Fisher, J. T., Yan, Z., Liu, X., Cho, H. J., et al. (2010). Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Invest. 120, 3149–3160. doi: 10.1172/JCI43052
Sun, X., Yi, Y., Yan, Z., Rosen, B. H., Liang, B., Winter, M. C., et al. (2019). In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med. 11:eaau7531. doi: 10.1126/scitranslmed.aau7531
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Travaglini, K. J., Nabhan, A. N., Penland, L., Sinha, R., Gillich, A., Sit, R. V., et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625. doi: 10.1038/s41586-020-2922-4
Trezise, A. E. O., Chambers, J. A., Wardle, C. J., Gould, S., and Harris, A. (1993). Expression of the cystic fibrosis gene in human foetal tissues. Hum. Mol. Genet. 2, 213–218. doi: 10.1093/hmg/2.3.213
Verhaeghe, C., Remouchamps, C., Hennuy, B., Vanderplasschen, A., Chariot, A., Tabruyn, S. P., et al. (2007). Role of IKK and ERK pathways in intrinsic inflammation of cystic fibrosis airways. Biochem. Pharmacol. 73, 1982–1994. doi: 10.1016/j.bcp.2007.03.019
Waters, V., and Ratjen, F. (2015). Pulmonary exacerbations in children with cystic fibrosis. Ann. Am. Thorac. Soc. 12(Suppl. 2), S200–S206.
Wong, A. P., Bear, C. E., Chin, S., Pasceri, P., Thompson, T. O., Huan, L.-J., et al. (2012). Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 30, 876–882. doi: 10.1038/nbt.2328
Wong, A. P., Chin, S., Xia, S., Garner, J., Bear, C. E., and Rossant, J. (2015). Efficient generation of functional CFTR-expressing airway epithelial cells from human pluripotent stem cells. Nat. Protoc. 10, 363–381. doi: 10.1038/nprot.2015.021
Yan, Z., Stewart, Z. A., Sinn, P. L., Olsen, J. C., Hu, J., McCray, P. B., et al. (2015). Ferret and pig models of cystic fibrosis: prospects and promise for gene therapy. Hum. Gene Ther. Clin. Dev. 26, 38–49. doi: 10.1089/humc.2014.154
Keywords: stem cells, disease modeling, lung development and pulmonary diseases, Wnt/β-catenin, cystic fibrosis
Citation: Huang EN, Quach H, Lee J-A, Dierolf J, Moraes TJ and Wong AP (2021) A Developmental Role of the Cystic Fibrosis Transmembrane Conductance Regulator in Cystic Fibrosis Lung Disease Pathogenesis. Front. Cell Dev. Biol. 9:742891. doi: 10.3389/fcell.2021.742891
Received: 19 July 2021; Accepted: 21 September 2021;
Published: 11 October 2021.
Edited by:
Cheng Yang, Fudan University, ChinaReviewed by:
Valerie Chappe, Dalhousie University, CanadaOscar Moran, Biophysics Institute, National Research Council (CNR), Italy
Copyright © 2021 Huang, Quach, Lee, Dierolf, Moraes and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy P. Wong, YXB3b25nQHNpY2traWRzLmNh
†These authors have contributed equally to this work