 José B. Sáenz
José B. Sáenz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 12 November 2021
Sec. Cell Death and Survival
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.741574
This article is part of the Research Topic Autoimmunity, Infection and Cancer, an Inflammatory Relationship with Intimate Implication to Cancer Prevention and Immunotherapy View all 9 articles
The human stomach functions as both a digestive and innate immune organ. Its main product, acid, rapidly breaks down ingested products and equally serves as a highly effective microbial filter. The gastric epithelium has evolved mechanisms to appropriately handle the myriad of injurious substances, both exogenous and endogenous, to maintain the epithelial barrier and restore homeostasis. The most significant chronic insult that the stomach must face is Helicobacter pylori (Hp), a stomach-adapted bacterium that can colonize the stomach and induce chronic inflammatory and pre-neoplastic changes. The progression from chronic inflammation to dysplasia relies on the decades-long interplay between this oncobacterium and its gastric host. This review summarizes the functional and molecular regionalization of the stomach at homeostasis and details how chronic inflammation can lead to characteristic alterations in these developmental demarcations, both at the topographic and glandular levels. More importantly, this review illustrates our current understanding of the epithelial mechanisms that underlie the pre-malignant gastric landscape, how Hp adapts to and exploits these changes, and the clinical implications of identifying these changes in order to stratify patients at risk of developing gastric cancer, a leading cause of cancer-related deaths worldwide.
The human stomach represents a unique digestive organ, capable of generating a remarkably acidic environment that serves essential physiologic and metabolic functions. While the stomach is also the source of multiple hormones that regulate satiety and hunger (Murphy and Bloom, 2006; Stengel and Tache, 2012; Stimac et al., 2014), for example, its endocrine function is dwarfed by its distinct exocrine ability to secrete acid. Acid defines the stomach and its purpose in the digestive tract (O'Connor and O'Morain, 2014; Schubert, 2015). From a nutritional standpoint, the ability of the human stomach to consistently maintain a pH less than 2 translates to an effective solubilization of ingested food and the reduction of cations, such as Fe3+ to Fe2+, for downstream intestinal absorption (Ems et al., 2021).
Perhaps the most fundamental and crucial function of acid is anti-microbial. Gastric acid serves as the primary line of defense against the myriad of microbes that are either ingested or resident to the oropharynx (Giannella et al., 1972). The potent neutralizing ability of gastric acid had long been known and was the impetus for many of the seminal studies that informed our current understanding of gastric physiology (Fourcroy, 1791). Moreover, the gastric mucosa’s tolerance to endogenous acid implies that the gastric epithelium has evolved mechanisms to avoid digesting itself (Wallace, 2008) and to appropriately respond to peptic injury through epithelial restitution (Lacy and Ito, 1984; Sorbye et al., 1988; Paimela et al., 1993), mucus production (Ichikawa, 2011; McColl, 2012), and prostaglandin synthesis (Robert et al., 1979; Konturek et al., 1982; Peskar, 2001).
One could argue that the large vat of hydrochloric acid that is the stomach is unrivaled as a microbial filter. Compared to the more distal intestine, where bacterial densities can reach levels as high as 108 cfu/ml in the cecum, for example (Marteau et al., 2001), the stomach is a bacterial desert, relatively speaking, with bacterial concentrations nearly one million times less dense (Giannella et al., 1972). Indeed, gastric acid eliminates over 99.9% of the micro-organisms that it encounters (Giannella et al., 1972) and represents an insurmountable barrier to chronic colonization for the vast majority of swallowed pathogens. The clinical consequences of raising intra-gastric pH, either through a loss of acid production or pharmacologic inhibition of acid secretion, have been intensely studied, particularly after the introduction of histamine receptor antagonists and, more recently, proton pump inhibitors (PPI) (Sanduleanu et al., 2001; Leonard et al., 2007; Janarthanan et al., 2012; Freedberg et al., 2014; McDonald et al., 2015). While the effects of long-term pharmacologic acid inhibition have been linked to a variety of pathophysiologic processes, ranging from chronic kidney disease to dementia, most of the published literature has relied on retrospective and observational studies that have failed to provide adequate evidence for causation (Vaezi et al., 2017). Of note, acid inhibition has been associated with an increased risk of enteric infections (Leonard et al., 2007; Janarthanan et al., 2012; McDonald et al., 2015), most notably nosocomial Clostridium difficile infection (McDonald et al., 2015; Tariq et al., 2017). In addition, gastric acidity has also been shown to promote intestinal microbial homeostasis, with PPI use demonstrating more destabilizing effects than antibiotics (Imhann et al., 2016). It seems biologically plausible that a reduction in one of the gastrointestinal tract’s most potent innate immune defenses could lead to gut dysbiosis, though well controlled prospective trials are still lacking (Villafuerte-Galvez and Kelly, 2018).
Broadly speaking, the stomach can be divided into two functionally distinct regions (Ban, 2013). The gastric corpus (or body) accounts for approximately two-thirds of the gastric surface area and is primarily responsible for generating gastric acid. The acid-producing, or oxyntic, glands of the corpus are lined by various epithelial cell types but are primarily defined by two distinct lineages, the parietal cell and the chief cell (Saenz and Mills, 2018). Oxyntic glands generate acidic gastric juice, primarily composed of hydrochloric acid, bicarbonate, and proteolytic enzymes, that aid in digesting food and creating a microbial barrier. The parietal cell secretes hydrochloric acid via its H+/K+ ATPase (Engevik et al., 2020), as well as intrinsic factor, which aids in absorption of vitamin B12 (Alpers and Russell-Jones, 2013), while the more basal chief cell secretes zymogenic proteolytic enzymes, including pepsinogen, (Raufman, 1992; Schubert, 2009), prochymosin (Richter et al., 1998), and gastric lipase (Moreau et al., 1989), which require acid activation. Together, these two epithelial lineages histologically and functionally define oxyntic mucosa (Rugge et al., 2008). In contrast, the more distal gastric antrum primarily functions to produce mucus, to regulate gastric acid secretion, and to propagate gastric peristalsis (O'Connor and O'Morain, 2014). These mucus-secreting glands differ in terms of morphology and composition compared to oxyntic glands of the corpus (Ban, 2013; Saenz and Mills, 2018). Antral glands are generally defined by a relative paucity of parietal cells and chief cells and by the presence of G cells (Watson et al., 2006), whose main product, gastrin, regulates acid secretion by parietal cells of the corpus and stimulates growth of oxyntic mucosa (Walsh, 1990). The differences in glandular composition of these two gastric compartments not only underlie their distinct functions in the stomach but also help to define some of the histologic changes that occur during chronic gastric injury (Rugge et al., 2008), which will be subsequently discussed in more detail. Importantly, the morphologic distinction between corpus and antral glands is less pronounced in humans (Choi et al., 2014) than in experimental animal models, such as mice.
It is worth noting that the functional organization of the stomach (i.e., acid-secreting corpus vs. mucus-producing antrum) can be explained by gastric specification during development (Kim and Shivdasani, 2016; Willet and Mills, 2016). We must consider that most of the developmental mechanisms behind human gastric corpus and antrum specification have been inferred from studies in mammalian and non-mammalian experimental models, and obvious limitations exist when extrapolating these findings to the human stomach. Regardless, one method of characterizing the distinct development of the corpus and antrum is by the regional pattern of transcription factor expression (Gracz and Magness, 2011; Kim and Shivdasani, 2016). In the mouse, the regions of the stomach that will become the gastric corpus and gastric antrum can both be characterized by the transcription factors Sox2 (Que et al., 2007; Gracz and Magness, 2011; Willet and Mills, 2016) and Gata4 (Jacobsen et al., 2002; Willet and Mills, 2016; Thompson et al., 2018), but the gastric antrum can be further defined by Pdx1 expression, which is absent from the corpus (Offield et al., 1996; Nomura et al., 2005; Fujitani et al., 2006). While this is an oversimplified method of distinguishing corpus from antrum, it can serve as a molecular framework for distinguishing antral and corpus progenitors. The phenotypic effects that result from inhibiting or mis-expressing these transcription factors is beyond the scope of this review and has been succinctly discussed elsewhere (Kim and Shivdasani, 2016; Willet and Mills, 2016; McCracken K. W. and Wells J. M., 2017).
Importantly, the spatiotemporal expression of local epithelial and mesenchymal signals (McCracken K. W. and Wells J. M., 2017) and their effects on lineage specification within corpus and antral glands can now be more easily recapitulated using gastric organoid culture models (Dedhia et al., 2016). Gastric organoids generated de novo from human pluripotent stem cells have been engineered to form differentiated antral (McCracken et al., 2014) or corpus organoids (McCracken et al., 2017), and this has expanded our understanding of the signaling pathways that drive corpus versus antral regionalization. For example, these studies have implicated HNF1B, expressed in the early endoderm, in antral specification and identified novel corpus-specific markers, such as IRX1/2/3/5 and PITX1 (McCracken et al., 2017). In addition, transient inhibition of MEK signaling enhanced expression of genes specific to mature parietal cells (McCracken et al., 2017), a defining cell type in the corpus. Finally, and perhaps most interestingly, temporal activation of WNT/β-catenin signaling promoted the development of corpus-like organoids at the expense of antral organoids (McCracken et al., 2014; Broda et al., 2019). Taken together, these findings would point to independent and inter-dependent developmental pathways for the functional regionalization of the stomach.
The distinct developmental pathways that drive corpus and antrum specification become particularly relevant during chronic gastric injury, when these developmentally regulated morphologic and molecular distinctions can become blurred. We refer to these epithelial changes as metaplasia, which is broadly defined as a change of cells to a form that does not normally occur in the tissue in which it is found (Kumar et al., 2005). Indeed, chronic inflammation in the corpus can lead to morphologic changes such that corpus glands begin to histologically resemble antral glands, which are not normally present in the majority of the gastric corpus. These glandular changes in the corpus have taken on various names, including pseudopyloric metaplasia (Hebbel, 1949; Magnus, 1954) and pyloric metaplasia (Morrissey et al., 1983; Steer, 1984; Chang et al., 2001) to describe antral-like glands observed adjacent to healing ulcers in the corpus (or duodenum) or to more generally denote the extension of antral-type glands into the corpus in the setting of chronic Hp infection (Goldenring, 2018). In either case, the defining features of these “antralized” corpus glands include the gradual loss of parietal cells and chief cells, resulting in atrophy, as well as the expression of metaplastic markers, some of which are inherent to antral glands (Xia et al., 2004b; Rubio et al., 2009). In the context of chronic Hp infection, the “antralization” of the corpus refers to the proximal progression of a front of inflammation from the antrum into the corpus, leaving in its wake a span of antral-like glands (Xia et al., 2000; Xia et al., 2004a). From a clinical standpoint, this pattern of antralization is not arbitrary: various longitudinal endoscopic studies suggest that the pattern of antralization proceeds from the corpus-antrum transition proximally into the corpus along the lesser curvature (Graham, 2014). More importantly, this pattern of inflammation carries an increased oncogenic risk (Cassaro et al., 2000) and forms the basis for endoscopically assessing pre-cancerous risk through gastric mapping, as defined by the Sydney system (Guarner et al., 2003).
It remains unclear why the antralization of the corpus follows a predictable expansion from the corpus-antrum transition and into the proximal corpus along the lesser curvature (Figures 1A–C). One could argue that, in the setting of chronic Hp infection, several foci of colonization could develop within the corpus and each give rise to a distinct nidus of inflammation from which atrophied (and perhaps antralized) glands can arise and/or radially expand in the setting of chronic inflammation (Figures 1D–F), though the evidence to date does not support this latter pattern. That the pattern of antralization instead emerges from the corpus-antrum border and extends along the lesser curvature, as opposed to the greater curvature, for example, suggests multiple possibilities that are not mutually exclusive. The first possibility is a programmed pattern of inflammation that develops irrespective of the insult: in the setting of chronic inflammation, the glands along the lesser curvature may adopt a more antral-like morphology. These glands may signal to neighboring glands to become more antral or to divide by gland fission, resulting in a gradual, gland-by-gland expansion. This possibility implies that antrum begets antrum and that the development of an antralized gland relies on an adjacent antralized gland. Another possibility relates to the span of antral glands that normally extends along the lesser curvature and into the fundus, with pyloric-type glands accounting for up to approximately 30% of the glands near the fundus in older individuals (Kimura, 1972). It is worth noting that the extension of the antrum into the corpus may occur with aging (Goldenring, 2018), though it appears to be accelerated and carry increased oncogenic potential in Hp-infected individuals (Umeda et al., 1971; Xia et al., 2002). It remains to be seen whether this strip of antral mucosa along the lesser curvature is a developmental remnant (Davis et al., 2017) and whether it serves as an area of heightened risk from which inflammation can arise (Busada et al., 2019).
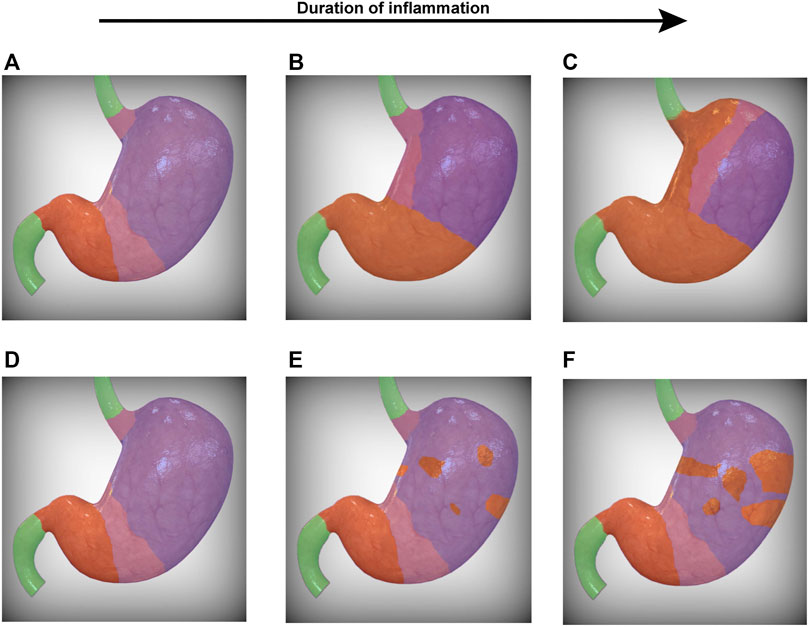
FIGURE 1. Progression of SPEM within the stomach. Two possibilities for the topographic progression of SPEM are presented. In (A), the corpus-antrum and gastroesophageal transition zones (pink) represent foci from which SPEM arises. In the setting of chronic inflammation, the transition zones converge along the lesser curvature (B), leaving in their wake gastric glands that morphologically resemble antrum (orange). The transition zone therefore represents a dynamic demarcation that serves as the leading edge for the progressive front of SPEM that ultimately extends into the greater curvature of the stomach (C). Alternatively, SPEM may arise from multiple foci within the corpus (E), gradually spreading radially and converging to form a lawn of SPEM (F). It should be noted that these possibilities are not mutually exclusive and may occur simultaneously. SPEM, spasmolytic polypeptide-expressing metaplasia. The human stomach images were modified from a stock image purchased from turboquid.com.
Why, then, do injured corpus glands morphologically resemble antral glands, and does this suggest that the conversion to a more antral phenotype represents a programmed, default injury response in the corpus (Saenz and Mills, 2018)? Does injured corpus epithelium take on molecular features of antral epithelium, or is the metaplastic epithelial gene signature in the corpus unique? It is worth noting that the term “antralization” of the corpus (Xia et al., 2004a; Rubio et al., 2009) may not be the most appropriate description for the epithelial changes taking place within the chronically inflamed corpus, though alternative terms like pyloric or pseudo-pyloric metaplasia may be equally misleading (Goldenring, 2018). Injury to corpus glands is often characterized by a loss of acid-secreting parietal cells and chief cells, resulting in glands that morphologically resemble antral glands (Xia et al., 2000), but the similarities appear to end there. Following either acute or chronic injury, the corpus epithelium undergoes reparative changes and an expression of novel markers that are not typically expressed within the corpus at homeostasis. These cellular changes are consistent with the definition of metaplasia, which makes the term pyloric or pseudopyloric metaplasia somewhat appropriate, though these metaplastic markers, which continue to be inventoried (Weis and Goldenring, 2009; Weis et al., 2013; Weis et al., 2014; Meyer and Goldenring, 2018), are not simply a re-expression of antral (or pyloric) genes. Notably, these metaplastic glands lack gastrin-producing cells, or G cells, a defining feature of antral (or pyloric) glands (Dockray, 1999). Indeed, certain genes upregulated in metaplastic corpus epithelium are highly expressed in antral glands at homeostasis, including DMBT1 (Sousa et al., 2012; Garay et al., 2017) and GKN3 (Menheniott et al., 2010; Bockerstett et al., 2020), but by the same token, various genes appear to be unique to metaplasia in the corpus, neither expressed in corpus nor antral glands at homeostasis. These include genes such as HE4 (Jeong et al., 2021; Nozaki et al., 2008; O'Neal et al., 2013), CD44v9 (Fan et al., 1996; Bertaux-Skeirik et al., 2015; Bertaux-Skeirik et al., 2017; Tsugawa et al., 2019), and CLU (Weis et al., 2013; Shimizu et al., 2016), among others (Weis et al., 2014). A more appropriate term to describe these glandular changes (and which will be used in this review to refer to these metaplastic changes) is spasmolytic polypeptide-expressing metaplasia (or SPEM), which is characterized by the expression of the antral spasmolytic polypeptide in the bases of corpus glands (Schmidt et al., 1999) and accurately describes the characteristic features of this metaplastic entity. Whether the expression of antralized or uniquely metaplastic genes increases the risk of neoplastic progression is largely correlative at this point. Regardless, chronically inflamed, metaplastic corpus glands adopt a hybrid expression pattern, demonstrating characteristics of antrum as well as features unique to the metaplastic corpus.
The developmental demarcations that anatomically divide the stomach therefore appear to be fluid during chronic injury. It has become increasingly evident that injured gastric epithelium also undergoes a cellular reprogramming that is characterized, in part, by the re-expression of markers typically associated with a more fetal (or primitive) state (Mills and Sansom, 2015). These cellular changes can occur in post-mitotic, differentiated cells, underscoring an inherent plasticity within gastric epithelium in the face of acute or chronic injury. More importantly, these post-mitotic cells can also re-enter the cell cycle, often in an attempt to repopulate glandular cells that have been injured or lost. This stepwise cellular reprogramming, termed paligenosis (Willet et al., 2018; Miao et al., 2020), has been proposed as a potential mechanism by which cells can appropriately deal with injury. It is worth noting, however, that this process has largely been described in the zymogenic chief cell of the corpus (Willet et al., 2018; Burclaff et al., 2019), and it remains to be seen whether other gastric epithelial lineages, either in the antrum or corpus, rely on a similar reprogramming sequence or whether this even occurs under more indolent and less proliferative injury conditions (e.g., chronic Hp infection).
The reparative and metaplastic potential within epithelial cells of the corpus seems to be more lineage-restricted (Bjerknes and Cheng, 2002; Saenz and Mills, 2018), and most studies to date have focused on the isthmal stem cell (Hayakawa et al., 2015a; Hayakawa et al., 2017a, b; Karam and Leblond, 1993) and zymogenic chief cell in mice (Nam et al., 2010; Goldenring et al., 2011; Mills and Goldenring, 2017) as being capable for heeding the call for repair. More recently, a single-cell RNA-sequencing (scRNA-seq) and trajectory analysis looking at possible cellular origins of metaplasia found mucous neck cells and chief cells as putative SPEM precursors (Bockerstett et al., 2020). It is likely that the relative contributions of different epithelial lineages to metaplasia along the corpus gland axis can vary and are not mutually exclusive, based on the acuity and severity of the metaplasia-inducing injury. Future studies should continue to focus on this epithelial plasticity.
On the other hand, the epithelial cells lining antral glands have long been thought of possessing varying degrees of inherent stemness (Qiao et al., 2007; Barker et al., 2010; Hayakawa et al., 2015b). Recent scRNA-seq profiling of human antral biopsies (Zhang et al., 2019) would suggest that the ability of epithelial cells to become metaplastic and give rise to gastric pre-neoplastic lesions may not be as stochastic as previously thought, however. Within human antral glands, differentiated epithelial lineages can not only express unique metaplastic/stem cell markers but can also transcriptionally activate distinct response pathways within a given pre-neoplastic stage. As expected, the proportion of differentiated antral mucous cells, characterized by MUC6 and TFF2 expression, significantly decreased during the progression to gastric cancer and correlated with a rise in cells expressing intestinal stem cell transcripts, including OLFM4, EPHB2, and/or SOX9. However, during chronic atrophic gastritis, for example, pit mucous cells of the antrum were more likely to transcriptionally activate components of the metallothione pathway, while the deeper gastric mucous cells were more likely to express components of the TNF pathway. Moreover, certain epithelial lineages were more likely to acquire metaplastic or pre-neoplastic transcriptional signatures than others. For example, OLFM4 expression in intestinal metaplasia lesions was primarily found within the gastric mucous cells of the antrum, located deep within antral glands, compared to the surface pit cells (Zhang et al., 2019). Taken together, these findings would suggest that cellular reprogramming during injury is not uniform across cell types and that the development of pre-neoplastic lesions, within the corpus and antrum, may be lineage-restricted.
It should be noted that an additional metaplastic phenotype can arise in chronically inflamed gastric mucosa. The appearance of intestinalized epithelium, referred to as gastric intestinal metaplasia (GIM), represents a distinct pre-neoplastic gastric lesion that is believed to confer increased oncogenic risk (Cassaro et al., 2000; de Vries et al., 2008; Tan and Yeoh, 2015). GIM is often portrayed as succeeding oxyntic atrophy in the Correa cascade (Correa and Piazuelo, 2012), but this has never been formally proven (Goldenring et al., 2010). Regardless, the presence of GIM is often a surrogate marker of chronic inflammation, especially when detected in the gastric corpus (Choi et al., 2015; Graham et al., 2019). Gastric metaplasia characterized by intestinalized epithelium (either small intestinal or colonic) remains a bit of a pathologic enigma. For one, gastric mucosa is not the only mucosa that adopts an intestinal phenotype following chronic inflammation, as intestinal metaplasia, referred to as Barrett’s metaplasia/esophagus, can also be seen in the chronically inflamed distal esophagus and also marks mucosa at risk of progressing to cancer (Souza et al., 2008; Peters et al., 2019). GIM exhibiting histologic features of small intestinal epithelium, often referred to as complete GIM, is defined by small intestinal-type mucosa containing goblet cells, a brush border, and eosinophilic enterocytes, whereas incomplete GIM demonstrates colonic-type epithelium with irregular mucin droplets and the lack of a brush border (Correa et al., 2010). This latter histologic entity is believed to carry significant prognostic weight, as patients with incomplete GIM have been felt to be at a heightened cancer risk compared to those with complete GIM (Gupta et al., 2020). A recent study of a 20-year follow-up of Colombian patients found that those with incomplete GIM at baseline had an odds ratio of greater than 13 for developing gastric cancer compared to those with complete GIM at baseline (Piazuelo et al., 2021). These differences in neoplastic risk would imply the importance of histologic sub-typing of GIM, though additional longitudinal studies are warranted (El-Zimaity et al., 2001; Altayar et al., 2020; Gupta et al., 2020). Complete GIM also represents a more differentiated intestinal epithelium, complete with a brush border and the expression of acidic mucins, while the colonic epithelium of incomplete GIM may represent less differentiated, sialomucin-predominant cells (Correa et al., 2010). How the histologic characterization of these metaplastic subtypes translates to differences in risk has not been investigated mechanistically, however. Regardless, this would suggest that the conversion (or reversion) of chronically injured epithelium to an intestinal phenotype could be part of a conserved, metaplastic, and pre-neoplastic response.
It also remains unclear which injured cells give rise to intestinal cells in GIM. While there is more evidence for the cells that give rise to SPEM (Nam et al., 2010; Radyk et al., 2018), the cellular origin(s) and mechanisms of intestinalization in GIM are less clear. Some of the characteristic molecular features of human GIM have been described and shown to induce GIM when ectopically expressed in murine gastric mucosa. The homeobox gene Cdx2 is central to intestinal differentiation in the developing gut, as well as to the maintenance of differentiated intestinal cells (Barros et al., 2010; Barros et al., 2012). CDX2 expression under the embryonically transcribed Foxa3 promoter led to extensive intestinal metaplasia in the mouse stomach (Silberg et al., 2002), as did CDX2 expression under the parietal cell-specific Atp4b (β subunit of the H+/K+-ATPase) promoter (Mutoh et al., 2002). In addition, transgenic expression of the small intestinal transcription factor, CDX1, in parietal cells led to the replacement of oxyntic mucosa with intestinalized crypts with differentiated enterocytes, goblet cells, enteroendocrine cells, and Paneth cells. Interestingly, the location of Paneth cells was in the upper portion of the crypts, suggesting that cell-autonomous aspects of Paneth cell differentiation may not be fully recapitulated in this model. GIM in this mouse model was characterized by intestinalized cells scattered along the gastric gland axis, with the gland largely retaining gastric features and gastric epithelial lineages. Taken together, there appears to be evidence for CDX1/2, for example, in driving intestinal differentiation of undifferentiated cells of the foregut as well as inducing a trans-differentiation of adult parietal cells (Barros et al., 2012), but which of these cell fate mechanisms (if any) is pathologically relevant has yet to be determined. In the end, one could make an argument for both scenarios. On the one hand, injured gastric epithelium can take on a more fetal, progenitor-like state, and the expression of intestinal drivers, such as CDX1/2, in these reprogramming cells can direct them toward an intestinal cell type. On the other hand, intestinal cells can arise from the trans-differentiation of an adult gastric epithelial cell, similar to the trans-differentiation of chief cells to SPEM cells that has been reported (Weis and Goldenring, 2009; Meyer and Goldenring, 2018). One could speculate that all epithelial injury represents a transient reversion to a more primitive, progenitor state, and the spatiotemporal expression of certain lineage-defining features in this malleable population of cells dictates their fate.
At a more general level, what are the pathophysiologic implications of these changes in gastric landscape, both at the anatomic and glandular levels? Clinically, the anatomic extent of SPEM serves as a surrogate for the degree and duration of inflammation (Graham et al., 2019). It is generally assumed that the extent of atrophy and SPEM within the corpus portends a poorer clinical prognosis than atrophy restricted to the antrum (Hebbel, 1949; El-Zimaity, 2006). The corpus-restricted atrophy associated with autoimmune gastritis is a unique case (Neumann et al., 2013) and will not be discussed in this review. For patients with evidence of chronic inflammation, the pattern of atrophy and SPEM throughout the stomach is endoscopically mapped for purposes of risk stratification and surveillance. However, recent surveillance guidelines (Gupta et al., 2020) that rely on endoscopic mapping using the Sydney system (Dixon et al., 1996) focused on the histologic identification and mapping of GIM. Typically, these islands of GIM arise in a sea of atrophy and SPEM (Goldenring et al., 2010). GIM’s patchy distribution can make accurate mapping and surveillance all the more difficult (Crafa et al., 2018; Waddingham et al., 2018). Indeed, the presence of GIM in the gastric corpus is believed to confer increased oncogenic risk (Cassaro et al., 2000), but the guidelines for the timing and frequency of endoscopic surveillance of GIM in the United States remain vague (Gupta et al., 2020) and require further study.
Perhaps a more reasonable approach would be to consistently map the pattern of atrophy and SPEM, regardless of GIM. Whether GIM evolves from atrophic mucosa and independently confers increased oncogenic risk remain unresolved (Graham and Zou, 2018), but GIM in the corpus is almost invariably surrounded by atrophic mucosa (Goldenring et al., 2010). Importantly, the extent of atrophy has been shown to correlate with the risk of progressing to cancer (Liu et al., 2005). One Japanese study found that patients with severe (i.e., extensive) atrophy at index endoscopy were more likely to develop gastric cancer than patients with mild or moderate atrophy, within the time period studied (Shichijo et al., 2016). One could argue that endoscopically mapping the extent of corpus atrophy (or antralized corpus) would therefore be a more reliable and equally effective method for risk stratification than inconsistent sampling of patchy GIM (Kotelevets et al., 2021). Of note, this is commonly practiced in the Far East (Kishino et al., 2016; Song et al., 2017) using the Kimura-Takemoto classification (Kimura, 1969), which relies on the endoscopic pattern of atrophy to risk stratify patients. In addition, mucosal atrophy, unlike GIM, may still represent a reversible pathologic state (Rokkas et al., 2007; Lee et al., 2013), where Hp eradication can lead to a regression of atrophy or at the very least a halt in the progression to cancer. Identifying the extent of atrophic (or antralized) mucosa also seems more consistent with the pattern of field lineage changes that occur as a result of chronic inflammation and that follow a predictable, proximal extension from the antrum into the corpus along the lesser curvature, as discussed previously.
The extent of atrophy and SPEM has equally important implications in terms of understanding Hp colonization. Rather than focusing on how Hp effects metaplastic changes in the colonized stomach, one should consider how these metaplastic changes affect Hp pathogenesis. Hp has been shown to hone in on injured gastric epithelium, as seen in ulcerated mucosa. While injured mucosa may express chemoattractants for Hp (Aihara et al., 2014; Huang et al., 2015; Huang et al., 2016), ulcer margins are lined by SPEM glands that are believed to facilitate mucosal repair (Teal et al., 2020). Recent evidence suggests that Hp has a tropism for SPEM glands, and this is in part mediated by binding of the SabA adhesin to its cognate receptor, sialyl-Lewis X (sLex), which shows enhanced expression in metaplastic glands (Mahdavi et al., 2002; Saenz et al., 2019). More importantly, the expression of sLex extends deeper into the bases in SPEM glands (Saenz et al., 2019), allowing Hp to burrow deep within these glands. Teleologically, this could allow Hp not only to establish an acid-protected niche but also facilitate Hp’s interactions with proliferating, metaplastic cells near the gland base (Saenz and Mills, 2018). As Hp has been shown to inject genotoxins and induce distinct patterns of DNA damage (Hartung et al., 2015; Bauer et al., 2020), the interactions of Hp with actively dividing, metaplastic cells could be an under-appreciated method by which Hp promotes pre-neoplastic changes.
The pattern of atrophy and metaplasia throughout the corpus mimics the pattern of Hp’s spread throughout the stomach. The gradual migration of Hp from the antrum and into the corpus likely progresses along the lesser curvature (Van Zanten et al., 1999; Graham, 2014). It remains unclear, however, whether Hp is actively driving this pattern of SPEM through a gland-by-gland migration into the corpus, or whether Hp is simply following the pattern of SPEM glands, which are the result of chronic inflammation and for which Hp has an increased affinity (Mahdavi et al., 2002; Saenz et al., 2019). Regardless of this chicken vs egg scenario, it seems that Hp exploits the very metaplastic changes in the corpus that it has induced. This becomes particularly important when we consider the initial establishment of Hp infection and its clinical implications to the host. If we consider that Hp has a tropism for metaplastic glands, then we can speculate that metaplastic glands will be targeted and preferentially colonized upon initial infection.
Interestingly, at baseline and prior to the expansion of Hp-induced metaplasia, two anatomic and histologic junctions exist within the uninjured stomach and may represent hot spots for Hp colonization (Van Zanten et al., 1999). The first junction occurs at the transition from corpus to antrum, with a span of multiple glands with mixed features of both mucous and oxyntic glands (Odze and Goldblum, 2009; Stave et al., 1978). These glands show a decrease in parietal cell number per gland, as one moves distally toward the antrum (Grossman, 1960); Figure 2A). This likely represents a developmental transition in glandular composition from parietal- and chief cell-dense “pure” corpus glands to antral glands, which contain lower densities of these lineages. It also theoretically represents a transition in acidity (Engel et al., 1984; Schade et al., 1994), though local acid levels in this transition zone have not been accurately determined. Whether the spread of Hp into the corpus is simply a result of alterations in gastric acidity or changes in the metaplastic landscape remains an open question, but the transition zone likely serves a critical role both for the expansion of metaplasia in the host and for Hp pathogenesis (Van Zanten et al., 1999). Indeed, in Hp-infected patients with intact acid secretion, the pattern of gastritis is largely restricted to the antrum, where Hp likely avoids the inhospitable local acidity of the corpus and can more effectively replicate (Saenz and Mills, 2018). Over time, Hp-induced chronic inflammation can lead to a more proximal, corpus gastritis, ultimately leading to a loss of parietal cells and a hypo- or achlorhydric state, thereby allowing Hp to more effectively colonize the corpus (Bayerdorffer et al., 1989; Fiocca et al., 1992). The effect of lowering gastric acidity on Hp-induced gastritis was elegantly shown over 25 years ago, when Kuipers and others demonstrated that Hp-infected patients on chronic acid suppression, in the form of PPIs, were more likely to develop corpus gastritis than Hp-infected patients not on acid suppressants (Kuipers et al., 1996). This would argue that gastric acidity is a major determinant of Hp’s ability to colonize the corpus, as has been demonstrated in the murine stomach (Danon et al., 1995; Saenz et al., 2019). However, we would equally argue that, in addition to changes in gastric acidity that can accompany chronic inflammation, Hp can induce a characteristic metaplastic landscape, as discussed earlier, that it can exploit and follow in order to migrate out of the antrum and expand its niche. Indeed, this corpus-antrum transition zone has been shown to represent an area of consistent and persistent inflammation in infected patients (Genta et al., 1994b). We would agree with previous speculation (Van Zanten et al., 1999) that this transition zone serves as a nidus from which metaplasia, and subsequently Hp colonization, can expand proximally into the corpus.
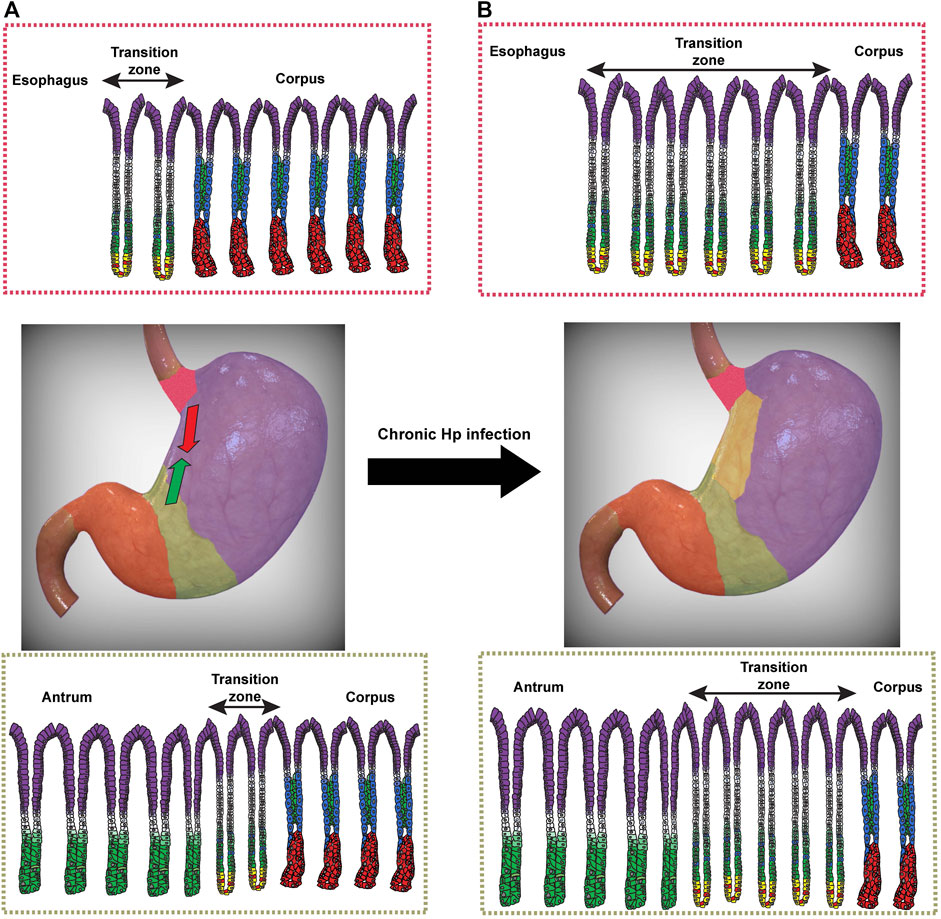
FIGURE 2. Expansion of transition zones during chronic Helicobacter pylori (Hp) infection. (A) Transition zones exist between the distal esophagus and glandular corpus (red) and between the corpus and antrum (green). At homeostasis, corpus glands are characterized by acid-secreting parietal cells (blue) lining the gland axis and zymogenic chief cells at the gland base (red). Pit cells (purple), mucous neck cells (green), and the proliferative isthmal stem cells (white) are also shown. At homeostasis, the cardia (top) contains a small number of glands within the transition zone (double-sided arrow) that show a mixed mucous/oxyntic morphology, with a relative paucity of parietal cells and an expansion of SPEM cells (yellow) with hybrid features of chief cells and mucous neck cells. A similar transition zone exists between the glands of the corpus and antrum (bottom). (B) Following chronic Hp infection, the number of glands with mixed mucous/oxyntic morphology increases, and the transition zones expand bi-directionally to converge along the lesser curvature. The human stomach images were modified from a stock image purchased from turboquid.com.
A similar transition zone exists between the distal esophagus and proximal stomach, often referred to as the cardia in humans (or limiting ridge in mice). Like with the corpus-antrum transition, there exists a span of mixed mucous/oxyntic glands as one moves from the distal esophagus to the gastric corpus (Figure 2A; Odze, 2005). The significance of the cardia in Hp pathogenesis remains largely understudied. Some studies have noted that the gastric cardia was one of the most consistently colonized regions of the stomach in Hp-infected patients, as determined endoscopically (Genta et al., 1994a; Liao et al., 2018). It remains to be seen whether the cardia indeed represents a preferential site for initial Hp colonization, like the corpus-antrum transition. Taken together, however, these transition zones could represent dynamic hot spots for the establishment of Hp infection. As Hp is able to delve deeper into glands with lower densities of parietal cells (Saenz et al., 2019), these transition zones could represent safe havens for Hp to colonize and replicate while avoiding the harsh acidic gastric lumen in the surface epithelium.
In addition to their contribution to Hp colonization and spread, the transition zones at the cardia and corpus-antrum border may also represent areas of increased oncogenic potential. Similar to the regional specification of the glandular stomach discussed previously, the delineation between the esophagus and the stomach at the gastroesophageal junction arises from a gradient of transcription factor expression as well as from communication between the mesenchyme and endoderm (Vogt and Panoskaltsis-Mortari, 2020). For example, Hoxa5 expression in the mesenchyme of murine embryos stimulates patterned expression of the transcription factors Sonic hedgehog (Shh) in esophageal epithelium and Indian Hedgehog (Ihh) in columnar gastric epithelium (McCracken K. W. and Wells J. M., 2017). In addition, the gastroesophageal junction is also defined by the coordinated development of muscle fibers of the lower esophagus that form the lower esophageal sphincter. The proper development and orientation of the smooth muscle fibers in the lower esophagus depend on Cdo, which encodes for a co-receptor for Hedgehog signaling. In the absence of Cdo, smooth muscle fascicles in the lower esophagus do not properly align (Romer et al., 2013).
Histologically, the gastric cardia is defined as the mucosa that is distal to the anatomic gastroesophageal junction and proximal to the characteristic oxyntic glands of the gastric corpus (Odze, 2005). There exists a debate on the origin and development of the gastric cardia, and it relies in part on whether the presence of mixed mucous/oxyntic glands is a result of a metaplastic conversion of pure corpus glands from chronic injury, such as gastroesophageal reflux disease (GERD) (Chandrasoma et al., 2000; Chandrasoma et al., 2003), or whether these mixed glands are normally present from birth (Kilgore et al., 2000; Glickman et al., 2002; Derdoy et al., 2003). While the intent of this review is not to engage in this debate, the correlation between the mixed glands that characterize this transition zone and the presence of cardia gastric tumors may not be coincidental.
Anatomically, the cardia can be exposed to various inflammatory insults, be it recurrent exposure to acid (Chandrasoma, 2005) and/or colonization by Hp (Genta et al., 1994a), as previously discussed. The etiology of inflammation of the gastric cardia, or carditis, can be distinguished histopathologically, with GERD-induced carditis showing more eosinophils in the inflammatory infiltrate and Hp-induced carditis demonstrating a more dense infiltrate of plasma cells, for example (Wieczorek et al., 2003; Odze, 2005). However, distinguishing whether a tumor in the cardia arose from metaplastic glands of the distal esophagus, gastric corpus, or cardia, can be more difficult (Wolf et al., 2001; Spechler, 2004). While various epidemiologic studies suggest that chronic Hp infection does not appear to increase the risk of cardia gastric cancer (Helicobacter and Cancer Collaborative, 2001; Abdi et al., 2019), tumorigenesis in the gastric cardia may be more nuanced. In the setting of chronic Hp infection, cancers arising in the cardia were positively associated with glandular atrophy and hypochlorhydria (Hansen et al., 2007), features consistent with non-cardia gastric cancer. But some studies have also suggested that the hypo- or achlorhydria that can develop from chronic Hp infection may protect against cancer of the cardia by reducing gastric acidity (Kamangar et al., 2006). In patients with chronic Hp infection and extensive atrophy involving the corpus, it is possible that atrophy and metaplasia begin in the cardia, an area of consistent Hp colonization and gastritis (Genta et al., 1994a). In the setting of chronic inflammation, this focus of atrophy and metaplasia migrates distally from the cardia into the corpus. That extensive atrophy can be seen in certain patients with cancer of the gastric cardia (Hansen et al., 2007) would simply illustrate the severity and duration of inflammation, but we should not lose sight of the fact that this atrophy and metaplasia may have originated in the cardia and that the glands of the cardia would have sustained the longest duration of inflammation and metaplastic injury. Whether the mixed mucous/oxyntic glands of the cardia are inherently at an increased risk of becoming metaplastic is unknown, but this transition zone appears to be at a unique risk for metaplastic injury and neoplasia.
The transition zone at the corpus-antrum junction may represent an area with similar metaplastic risk as the gastric cardia. Unlike cardia gastric cancer, the risk of non-cardia gastric cancer seems to be more strongly associated with chronic Hp infection and not directly linked to GERD (Axon, 2002; Moss and Malfertheiner, 2007). However, like the cardia, this junction is composed of a span of mixed mucous/oxyntic glands that are frequently colonized by Hp and where gastritis is consistently observed (Figure 2A; (Bayerdorffer et al., 1992; Satoh et al., 1991). As a result, this transition zone may also represent the initial site from which Hp and the metaplastic front can migrate proximally into the corpus. One could therefore imagine a bidirectional progression of metaplasia, moving (perhaps synchronously) distally from the cardia and proximally from the corpus-antrum junction and converging along the lesser curvature (Figure 2B). This pattern of atrophy and metaplasia carries an increased risk of progressing to cancer relative to other endoscopic patterns of metaplasia (Cassaro et al., 2000). This pattern of progression along the lesser curvature also represents the most direct route for the convergence of these two migrating, dynamic transition zones.
More importantly, the corpus-antrum transition zone, like the cardia, represents a region of sustained inflammation and metaplastic injury during chronic Hp infection. The duration of regional inflammation during chronic Hp infection should not be minimized, as these areas are the most prone to accumulating oncogenic mutations, either as a result of cellular proliferation during metaplastic reprogramming (Saenz and Mills, 2018) or from direct Hp-induced genotoxic injury (Hartung et al., 2015; Bauer et al., 2020). This may explain the fact that most non-cardia gastric adenocarcinomas are anatomically located in the antrum (You et al., 1992; Wanebo et al., 1993), even though the antrum occupies a significantly smaller surface area than the corpus. If Hp preferentially colonizes the antrum and the mixed mucous/oxyntic glands of the corpus-antrum junction when establishing an infection, then these regions would be exposed to inflammation longer than the corpus, which primarily experiences inflammation after Hp has migrated proximally (Bayerdorffer et al., 1989). Whether gastric cancers that arise solely in the corpus are related to initial Hp colonization and sustained gastritis in this region is not known. Regardless, the histologic characteristics of these transition zones, coupled with what we currently understand about Hp pathogenesis, could explain the patterns of atrophy, metaplasia, and cancer in chronically infected patients.
While we have enhanced our understanding of some of the topographic and glandular changes that result from chronic inflammation, we still have a rudimentary grasp of where gastric pre-neoplastic lesions first arise and why certain patterns of atrophy and metaplasia increase oncogenic risk. The transition zones (esophagus/gastric corpus and gastric corpus/antrum) may represent epicenters from which atrophy and SPEM develop and spread, but fundamental questions remain. What role(s), if any, do these poorly defined regions play in gastric tumorigenesis? Developmentally speaking, how are these transition zones formed and maintained, and are they inherently metaplastic? Do similar transition zones contribute to patterns of pathology and tumorigenesis seen in other chronically inflamed organs? As our appreciation of the mechanisms that give rise to atrophy and SPEM continue to evolve, we are likely to find that the functional and regional demarcations in the stomach, though developmentally determined, are fluid during injury and highlight an inherent plasticity in an organ that must constantly regenerate and adapt to injury.
The author confirms being the sole contributor to this work and has approved it for publication.
JBS is supported by a K08 award from the National Institute of Diabetes and Digestive and Kidney Diseases (5K08DK122116-02), a Research Scholar Award in Gastric Cancer from the American Gastroenterological Association, and a Pilot and Feasibility Award from the Digestive Diseases Research Core Center at the Washington University in St. Louis School of Medicine (P30-DK052574).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdi, E., Latifi‐Navid, S., Zahri, S., Yazdanbod, A., and Pourfarzi, F. (2019). Risk Factors Predisposing to Cardia Gastric Adenocarcinoma: Insights and New Perspectives. Cancer Med. 8, 6114–6126. doi:10.1002/cam4.2497
Aihara, E., Closson, C., Matthis, A. L., Schumacher, M. A., Engevik, A. C., Zavros, Y., et al. (2014). Motility and Chemotaxis Mediate the Preferential Colonization of Gastric Injury Sites by Helicobacter pylori. Plos Pathog. 10, e1004275. doi:10.1371/journal.ppat.1004275
Alpers, D. H., and Russell-Jones, G. (2013). Gastric Intrinsic Factor: the Gastric and Small Intestinal Stages of Cobalamin Absorption. A Personal Journey. Biochimie 95, 989–994. doi:10.1016/j.biochi.2012.12.006
Altayar, O., Davitkov, P., Shah, S. C., Gawron, A. J., Morgan, D. R., Turner, K., et al. (2020). AGA Technical Review on Gastric Intestinal Metaplasia-Epidemiology and Risk Factors. Gastroenterology 158, 732–744. doi:10.1053/j.gastro.2019.12.002
Axon, A. (2002). Gastric Cancer and Helicobacter pylori. Aliment. Pharmacol. Ther. 16 (Suppl. 4), 83–88. doi:10.1046/j.1365-2036.16.s4.14.x
Ban, S. (2013). “The Normal Stomach: Anatomy, Specimen Dissection and Histology Relevant to Pathological Practice,” in Morson and Dawson's Gastrointestinal Pathology. Editors N. A. Shepherd, B. F. Warren, G. T. Williams, J. K. Greenson, G. Y. Lauwers, and M. R. Novelli (Oxford, U.K.: Wiley-Blackwell).
Barker, N., Huch, M., Kujala, P., van de Wetering, M., Snippert, H. J., van Es, J. H., et al. (2010). Lgr5+ve Stem Cells Drive Self-Renewal in the Stomach and Build Long-Lived Gastric Units In Vitro. Cell Stem Cell 6, 25–36. doi:10.1016/j.stem.2009.11.013
Barros, R., Camilo, V., Pereira, B., Freund, J.-N., David, L., and Almeida, R. (2010). Pathophysiology of Intestinal Metaplasia of the Stomach: Emphasis on CDX2 Regulation. Biochem. Soc. Trans. 38, 358–363. doi:10.1042/bst0380358
Barros, R., Freund, J.-N., David, L., and Almeida, R. (2012). Gastric Intestinal Metaplasia Revisited: Function and Regulation of CDX2. Trends Mol. Med. 18, 555–563. doi:10.1016/j.molmed.2012.07.006
Bauer, M., Nascakova, Z., Mihai, A.-I., Cheng, P. F., Levesque, M. P., Lampart, S., et al. (2020). The ALPK1/TIFA/NF-κB axis Links a Bacterial Carcinogen to R-Loop-Induced Replication Stress. Nat. Commun. 11, 5117. doi:10.1038/s41467-020-18857-z
Bayerdörffer, E., Lehn, N., Hatz, R., Mannes, G. A., Oertel, H., Sauerbruch, T., et al. (1992). Difference in Expression of Helicobacter pylori Gastritis in Antrum and Body. Gastroenterology 102, 1575–1582. doi:10.1016/0016-5085(92)91716-h
Bayerdorffer, E., Oertel, H., Lehn, N., Kasper, G., Mannes, G. A., Sauerbruch, T., et al. (1989). Topographic Association between Active Gastritis and Campylobacter pylori Colonisation. J. Clin. Pathol. 42, 834–839. doi:10.1136/jcp.42.8.834
Bertaux-Skeirik, N., Feng, R., Schumacher, M. A., Li, J., Mahe, M. M., Engevik, A. C., et al. (2015). CD44 Plays a Functional Role in Helicobacter Pylori-Induced Epithelial Cell Proliferation. Plos Pathog. 11, e1004663. doi:10.1371/journal.ppat.1004663
Bertaux-Skeirik, N., Wunderlich, M., Teal, E., Chakrabarti, J., Biesiada, J., Mahe, M., et al. (2017). CD44 Variant Isoform 9 Emerges in Response to Injury and Contributes to the Regeneration of the Gastric Epithelium. J. Pathol. 242, 463–475. doi:10.1002/path.4918
Bjerknes, M., and Cheng, H. (2002). Multipotential Stem Cells in Adult Mouse Gastric Epithelium. Am. J. Physiology-Gastrointestinal Liver Physiol. 283, G767–G777. doi:10.1152/ajpgi.00415.2001
Bockerstett, K. A., Lewis, S. A., Noto, C. N., Ford, E. L., Saenz, J. B., Jackson, N. M., et al. (2020). Single-Cell Transcriptional Analyses Identify Lineage-specific Epithelial Responses to Inflammation and Metaplastic Development in the Gastric Corpus. Gastroenterology 159, 2116–2129. doi:10.1053/j.gastro.2020.08.027
Broda, T. R., McCracken, K. W., and Wells, J. M. (2019). Generation of Human Antral and Fundic Gastric Organoids from Pluripotent Stem Cells. Nat. Protoc. 14, 28–50. doi:10.1038/s41596-018-0080-z
Burclaff, J., Willet, S., Saenz, J. B., and Mills, J. (2019). Proliferation and Differentiation of Gastric Mucous Neck and Chief Cells during Homeostasis and Injury-Induced Metaplasia. Gastroenterology 158, 598–609.
Busada, J. T., Ramamoorthy, S., Cain, D. W., Xu, X., Cook, D. N., and Cidlowski, J. A. (2019). Endogenous Glucocorticoids Prevent Gastric Metaplasia by Suppressing Spontaneous Inflammation. J. Clin. Invest. 129, 1345–1358. doi:10.1172/jci123233
Cassaro, M., Rugge, M., Gutierrez, O., Leandro, G., Graham, D. Y., and Genta, R. M. (2000). Topographic Patterns of Intestinal Metaplasia and Gastric Cancer. Am. J. Gastroenterol. 95, 1431–1438. doi:10.1111/j.1572-0241.2000.02074.x
Chandrasoma, P. (2005). Controversies of the Cardiac Mucosa and Barrett's Oesophagus. Histopathology 46, 361–373. doi:10.1111/j.1365-2559.2005.02088.x
Chandrasoma, P. T., Der, R., Ma, Y., Dalton, P., and Taira, M. (2000). Histology of the Gastroesophageal Junction. Am. J. Surg. Pathol. 24, 402–409. doi:10.1097/00000478-200003000-00009
Chandrasoma, P. T., Der, R., Ma, Y., Peters, J., and Demeester, T. (2003). Histologic Classification of Patients Based on Mapping Biopsies of the Gastroesophageal Junction. Am. J. Surg. Pathol. 27, 929–936. doi:10.1097/00000478-200307000-00008
Chang, C.-C., Pan, S., Lien, G.-S., Chen, S.-H., Cheng, C.-J., Liu, J.-D., et al. (2001). Investigation of the Extent of Gastric Metaplasia in the Duodenal Bulb by Using Methylene Blue Staining. J. Gastroenterol. Hepatol. 16, 729–733. doi:10.1046/j.1440-1746.2001.02521.x
Choi, C. E., Sonnenberg, A., Turner, K., and Genta, R. M. (2015). High Prevalence of Gastric Preneoplastic Lesions in East Asians and Hispanics in the USA. Dig. Dis. Sci. 60, 2070–2076. doi:10.1007/s10620-015-3591-2
Choi, E., Roland, J. T., Barlow, B. J., O'Neal, R., Rich, A. E., Nam, K. T., et al. (2014). Cell Lineage Distribution Atlas of the Human Stomach Reveals Heterogeneous Gland Populations in the Gastric Antrum. Gut 63, 1711–1720. doi:10.1136/gutjnl-2013-305964
Correa, P., Piazuelo, B. M., and Wilson, K. T. (2010). Pathology of Gastric Intestinal Metaplasia: Clinical Implications. Am. J. Gastroenterol. 105, 493–498. doi:10.1038/ajg.2009.728
Correa, P., and Piazuelo, M. B. (2012). The Gastric Precancerous Cascade. J. Dig. Dis. 13, 2–9. doi:10.1111/j.1751-2980.2011.00550.x
Crafa, P., Russo, M., Miraglia, C., Barchi, A., Moccia, F., Nouvenne, A., et al. (2018). From Sidney to OLGA: an Overview of Atrophic Gastritis. Acta Biomed. 89, 93–99. doi:10.23750/abm.v89i8-S.7946
Danon, S. J., O'Rourke, J. L., Moss, N. D., and Lee, A. (1995). The Importance of Local Acid Production in the Distribution of Helicobacter felis in the Mouse Stomach. Gastroenterology 108, 1386–1395. doi:10.1016/0016-5085(95)90686-x
Davis, A., Amin, N. M., Johnson, C., Bagley, K., Ghashghaei, H. T., and Nascone-Yoder, N. (2017). Stomach Curvature Is Generated by Left-Right Asymmetric Gut Morphogenesis. Development 144, 1477–1483. doi:10.1242/dev.143701
de Vries, A. C., van Grieken, N. C. T., Looman, C. W. N., Casparie, M. K., de Vries, E., Meijer, G. A., et al. (2008). Gastric Cancer Risk in Patients with Premalignant Gastric Lesions: a Nationwide Cohort Study in the Netherlands. Gastroenterology 134, 945–952. doi:10.1053/j.gastro.2008.01.071
Dedhia, P. H., Bertaux-Skeirik, N., Zavros, Y., and Spence, J. R. (2016). Organoid Models of Human Gastrointestinal Development and Disease. Gastroenterology 150, 1098–1112. doi:10.1053/j.gastro.2015.12.042
Derdoy, J. J., Bergwerk, A., Cohen, H., Kline, M., Monforte, H. L., and Thomas, D. W. (2003). The Gastric Cardia. Am. J. Surg. Pathol. 27, 499–504. doi:10.1097/00000478-200304000-00010
Dixon, M. F., Genta, R. M., Yardley, J. H., and Correa, P. (1996). Classification and Grading of Gastritis. Am. J. Surg. Pathol. 20, 1161–1181. doi:10.1097/00000478-199610000-00001
Dockray, G. J. (1999). Topical Review. Gastrin and Gastric Epithelial Physiology. J. Physiol. 518 ( Pt 2) (Pt 2), 315–324. doi:10.1111/j.1469-7793.1999.0315p.x
El-Zimaity, H. M. (2006). Gastric Atrophy, Diagnosing and Staging. Wjg 12, 5757–5762. doi:10.3748/wjg.v12.i36.5757
El-Zimaity, H. M. T., Ramchatesingh, J., Ali Saeed, M., and Graham, D. Y. (2001). Gastric Intestinal Metaplasia: Subtypes and Natural History. J. Clin. Pathol. 54, 679–683. doi:10.1136/jcp.54.9.679
Ems, T., St Lucia, K., and Huecker, M. R. (2021). “Biochemistry, Iron Absorption,”. StatPearls (Treasure Island (FL).
Engel, E., Peskoff, A., Kauffman, G. L., and Grossman, M. I. (1984). Analysis of Hydrogen Ion Concentration in the Gastric Gel Mucus Layer. Am. J. Physiology-Gastrointestinal Liver Physiol. 247, G321–G338. doi:10.1152/ajpgi.1984.247.4.g321
Engevik, A. C., Kaji, I., and Goldenring, J. R. (2020). The Physiology of the Gastric Parietal Cell. Physiol. Rev. 100, 573–602. doi:10.1152/physrev.00016.2019
Fan, X., Long, A., Goggins, M., Fan, X., Keeling, P. W., and Kelleher, D. (1996). Expression of CD44 and its Variants on Gastric Epithelial Cells of Patients with Helicobacter pylori Colonisation. Gut 38, 507–512. doi:10.1136/gut.38.4.507
Fiocca, R., Villani, L., Luinetti, O., Gianatti, A., Perego, M., Alvisi, C., et al. (1992). Helicobacter Colonization and Histopathological Profile of Chronic Gastritis in Patients with or without Dyspepsia, Mucosal Erosion and Peptic Ulcer: a Morphological Approach to the Study of Ulcerogenesis in Man. Vichows Archiv A. Pathol. Anat. 420, 489–498. doi:10.1007/bf01600253
Fourcroy, A. F. (1791. Elements D'Histoire Naturelle et de Chimie: Tome Premier Paris, France: Chez Cuchet.
Freedberg, D. E., Lebwohl, B., and Abrams, J. A. (2014). The Impact of Proton Pump Inhibitors on the Human Gastrointestinal Microbiome. Clin. Lab. Med. 34, 771–785. doi:10.1016/j.cll.2014.08.008
Fujitani, Y., Fujitani, S., Boyer, D. F., Gannon, M., Kawaguchi, Y., Ray, M., et al. (2006). Targeted Deletion of a Cis-Regulatory Region Reveals Differential Gene Dosage Requirements for Pdx1 in Foregut Organ Differentiation and Pancreas Formation. Genes Development 20, 253–266. doi:10.1101/gad.1360106
Garay, J., Piazuelo, M. B., Lopez-Carrillo, L., Leal, Y. A., Majumdar, S., Li, L., et al. (2017). Increased Expression of Deleted in Malignant Brain Tumors (DMBT1) Gene in Precancerous Gastric Lesions: Findings from Human and Animal Studies. Oncotarget 8, 47076–47089. doi:10.18632/oncotarget.16792
Genta, R. M., Huberman, R. M., and Graham, D. Y. (1994a). The Gastric Cardia in Helicobacter pylori Infection. Hum. Pathol. 25, 915–919. doi:10.1016/0046-8177(94)90011-6
Genta, R. M., Robason, G. O., and Graham, D. Y. (1994b). Simultaneous Visualization of Helicobacter pylori and Gastric Morphology: a New Stain. Hum. Pathol. 25, 221–226. doi:10.1016/0046-8177(94)90191-0
Giannella, R. A., Broitman, S. A., and Zamcheck, N. (1972). Gastric Acid Barrier to Ingested Microorganisms in Man: Studies In Vivo and In Vitro. Gut 13, 251–256. doi:10.1136/gut.13.4.251
Glickman, J. N., Fox, V., Antonioli, D. A., Wang, H. H., and Odze, R. D. (2002). Morphology of the Cardia and Significance of Carditis in Pediatric Patients. Am. J. Surg. Pathol. 26, 1032–1039. doi:10.1097/00000478-200208000-00008
Goldenring, J. R., Nam, K. T., and Mills, J. C. (2011). The Origin of Pre-neoplastic Metaplasia in the Stomach: Chief Cells Emerge from the Mist. Exp. Cel Res. 317, 2759–2764. doi:10.1016/j.yexcr.2011.08.017
Goldenring, J. R., Nam, K. T., Wang, T. C., Mills, J. C., and Wright, N. A. (2010). Spasmolytic Polypeptide-Expressing Metaplasia and Intestinal Metaplasia: Time for Reevaluation of Metaplasias and the Origins of Gastric Cancer. Gastroenterology 138, 22072210–2210. doi:10.1053/j.gastro.2010.04.023
Goldenring, J. R. (2018). Pyloric Metaplasia, Pseudopyloric Metaplasia, Ulcer‐associated Cell Lineage and Spasmolytic Polypeptide‐expressing Metaplasia: Reparative Lineages in the Gastrointestinal Mucosa. J. Pathol. 245, 132–137. doi:10.1002/path.5066
Gracz, A. D., and Magness, S. T. (2011). Sry-box (Sox) Transcription Factors in Gastrointestinal Physiology and Disease. Am. J. Physiology-Gastrointestinal Liver Physiol. 300, G503–G515. doi:10.1152/ajpgi.00489.2010
Graham, D. Y. (2014). History of Helicobacter Pylori, Duodenal Ulcer, Gastric Ulcer and Gastric Cancer. Wjg 20, 5191–5204. doi:10.3748/wjg.v20.i18.5191
Graham, D. Y., Rugge, M., and Genta, R. M. (2019). Diagnosis: Gastric Intestinal Metaplasia-What to Do Next?. Curr. Opin. Gastroenterol. 35, 535–543. doi:10.1097/mog.0000000000000576
Graham, D. Y., and Zou, W. Y. (2018). Guilt by Association: Intestinal Metaplasia Does Not Progress to Gastric Cancer. Curr. Opin. Gastroenterol. 34, 458–464. doi:10.1097/mog.0000000000000472
Grossman, M. I. (1960). The Pyloric Gland Area of the Stomach. Gastroenterology 38, 1–6. doi:10.1016/s0016-5085(60)80111-4
Guarner, J., Herrera-Goepfert, R., Mohar, A., Smith, C., Schofield, A., Halperin, D., et al. (2003). Diagnostic Yield of Gastric Biopsy Specimens when Screening for Preneoplastic Lesions. Hum. Pathol. 34, 28–31. doi:10.1053/hupa.2003.3
Gupta, S., Li, D., El Serag, H. B., Davitkov, P., Altayar, O., Sultan, S., et al. (2020). AGA Clinical Practice Guidelines on Management of Gastric Intestinal Metaplasia. Gastroenterology 158, 693–702. doi:10.1053/j.gastro.2019.12.003
Hansen, S., Vollset, S. E., Derakhshan, M. H., Fyfe, V., Melby, K. K., Aase, S., et al. (2007). Two Distinct Aetiologies of Cardia Cancer; Evidence from Premorbid Serological Markers of Gastric Atrophy and Helicobacter pylori Status. Gut 56, 918–925. doi:10.1136/gut.2006.114504
Hartung, M. L., Gruber, D. C., Koch, K. N., Grüter, L., Rehrauer, H., Tegtmeyer, N., et al. (2015). Helicobacter pylori -Induced DNA Strand Breaks Are Introduced by Nucleotide Excision Repair Endonucleases and Promote NF-Κb Target Gene Expression. Cell Rep. 13, 70–79. doi:10.1016/j.celrep.2015.08.074
Hayakawa, Y., Ariyama, H., Stancikova, J., Sakitani, K., Asfaha, S., Renz, B. W., et al. (2015a). Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 28, 800–814. doi:10.1016/j.ccell.2015.10.003
Hayakawa, Y., Fox, J. G., and Wang, T. C. (2017a). Isthmus Stem Cells Are the Origins of Metaplasia in the Gastric Corpus. Cell Mol. Gastroenterol. Hepatol. 4, 89–94. doi:10.1016/j.jcmgh.2017.02.009
Hayakawa, Y., Fox, J. G., and Wang, T. C. (2017b). The Origins of Gastric Cancer from Gastric Stem Cells: Lessons from Mouse Models. Cell Mol. Gastroenterol. Hepatol. 3, 331–338. doi:10.1016/j.jcmgh.2017.01.013
Hayakawa, Y., Jin, G., Wang, H., Chen, X., Westphalen, C. B., Asfaha, S., et al. (2015b). CCK2R Identifies and Regulates Gastric Antral Stem Cell States and Carcinogenesis. Gut 64, 544–553. doi:10.1136/gutjnl-2014-307190
Hebbel, R. (1949). The Topography of Chronic Gastritis in Otherwise Normal Stomachs. Am. J. Pathol. 25, 125–141.
Helicobacter and Cancer Collaborative Group, , and Cancer Collaborative, G. (2001). Gastric Cancer and Helicobacter pylori: a Combined Analysis of 12 Case Control Studies Nested within Prospective Cohorts. Gut 49, 347–353. doi:10.1136/gut.49.3.347
Huang, J. Y., Sweeney, E. G., Sigal, M., Zhang, H. C., Remington, S. J., Cantrell, M. A., et al. (2015). Chemodetection and Destruction of Host Urea Allows Helicobacter pylori to Locate the Epithelium. Cell Host & Microbe 18, 147–156. doi:10.1016/j.chom.2015.07.002
Huang, Y., Wang, Q.-l., Cheng, D.-d., Xu, W.-t., and Lu, N.-h. (2016). Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter pylori. Front. Cel. Infect. Microbiol. 6, 159. doi:10.3389/fcimb.2016.00159
Ichikawa, T. I. K. (2011). Protective Effects of Gastric Mucus, Gastritis, and Gastric Cancer - New Insights in Gastroprotection, Diagnosis, and Treatments. Rijeka, Croatia: InTech.
Imhann, F., Bonder, M. J., Vich Vila, A., Fu, J., Mujagic, Z., Vork, L., et al. (2016). Proton Pump Inhibitors Affect the Gut Microbiome. Gut 65, 740–748. doi:10.1136/gutjnl-2015-310376
Jacobsen, C. M., Narita, N., Bielinska, M., Syder, A. J., Gordon, J. I., and Wilson, D. B. (2002). Genetic Mosaic Analysis Reveals that GATA-4 Is Required for Proper Differentiation of Mouse Gastric Epithelium. Developmental Biol. 241, 34–46. doi:10.1006/dbio.2001.0424
Janarthanan, S., Ditah, I., Adler, D. G., and Ehrinpreis, M. N. (2012). Clostridium difficile -Associated Diarrhea and Proton Pump Inhibitor Therapy: A Meta-Analysis. Am. J. Gastroenterol. 107, 1001–1010. doi:10.1038/ajg.2012.179
Jeong, H., Lee, B., Kim, K. H., Cho, S. Y., Cho, Y., Park, J., et al. (2021). WFDC2 Promotes Spasmolytic Polypeptide-Expressing Metaplasia through the Upregulation of IL33 in Response to Injury. Gastroenterology 161, 953–967. doi:10.1053/j.gastro.2021.05.058
Kamangar, F., Dawsey, S. M., Blaser, M. J., Perez-Perez, G. I., Pietinen, P., Newschaffer, C. J., et al. (2006). Opposing Risks of Gastric Cardia and Noncardia Gastric Adenocarcinomas Associated with Helicobacter pylori Seropositivity. J. Natl. Cancer Inst. 98, 1445–1452. doi:10.1093/jnci/djj393
Karam, S. M., and Leblond, C. P. (1993). Dynamics of Epithelial Cells in the Corpus of the Mouse Stomach. I. Identification of Proliferative Cell Types and Pinpointing of the Stem Cell. Anat. Rec. 236, 259–279. doi:10.1002/ar.1092360202
Kilgore, S. P., Ormsby, A. H., Gramlich, T. L., Rice, T. W., Richter, J. E., Falk, G. W., et al. (2000). The Gastric Cardia: Fact or Fiction? Am. J. Gastroenterol. 95, 921–924. doi:10.1111/j.1572-0241.2000.01930.x
Kim, T.-H., and Shivdasani, R. A. (2016). Stomach Development, Stem Cells and Disease. Development 143, 554–565. doi:10.1242/dev.124891
Kimura, K. (1972). Chronological Transition of the Fundic-Pyloric Border Determined by Stepwise Biopsy of the Lesser and Greater Curvatures of the Stomach. Gastroenterology 63, 584–592. doi:10.1016/s0016-5085(19)33241-x
Kimura, K. T. T., and Takemoto, T. (1969). An Endoscopic Recognition of the Atrophic Border and its Significance in Chronic Gastritis. Endoscopy 1, 87–97. doi:10.1055/s-0028-1098086
Kishino, M., Nakamura, S., and Shiratori, K. (2016). Clinical and Endoscopic Features of Undifferentiated Gastric Cancer in Patients with Severe Atrophic Gastritis. Intern. Med. 55, 857–862. doi:10.2169/internalmedicine.55.4841
Konturek, S. J., Brzozowski, T., Piastucki, I., Radecki, T., Dembiński, A., and Dembińska-Kieć, A. (1982). Role of Locally Generated Prostaglandins in Adaptive Gastric Cytoprotection. Dig. Dis Sci 27, 967–971. doi:10.1007/bf01391740
Kotelevets, S. M., Chekh, S. A., and Chukov, S. Z. (2021). Updated Kimura-Takemoto Classification of Atrophic Gastritis. Wjcc 9, 3014–3023. doi:10.12998/wjcc.v9.i13.3014
Kuipers, E. J., Lundell, L., Klinkenberg-Knol, E. C., Havu, N., Festen, H. P. M., Liedman, B., et al. (1996). Atrophic Gastritis and Helicobacter pylori Infection in Patients with Reflux Esophagitis Treated with Omeprazole or Fundoplication. N. Engl. J. Med. 334, 1018–1022. doi:10.1056/nejm199604183341603
Kumar, V., Abbas, A. K., Fausto, N., Robbins, S. L., and Cotran, R. S. (2005). Robbins and Cotran Pathologic Basis of Disease. 7th edn. Philadelphia, PA: Elsevier Saunders.
Lacy, E. R., and Ito, S. (1984). Rapid Epithelial Restitution of the Rat Gastric Mucosa after Ethanol Injury. Lab. Invest. 51, 573–583.
Lee, Y.-C., Chen, T. H.-H., Chiu, H.-M., Shun, C.-T., Chiang, H., Liu, T.-Y., et al. (2013). The Benefit of Mass Eradication of Helicobacter pylori Infection: a Community-Based Study of Gastric Cancer Prevention. Gut 62, 676–682. doi:10.1136/gutjnl-2012-302240
Leonard, J., Marshall, J. K., and Moayyedi, P. (2007). Systematic Review of the Risk of Enteric Infection in Patients Taking Acid Suppression. Am. J. Gastroenterol. 102, 2047–2056. doi:10.1111/j.1572-0241.2007.01275.x
Liao, P.-H., Lin, Y.-C., Chu, C.-H., Shih, S.-C., and Liou, T.-C. (2018). Colonization of Helicobacter pylori in the Gastric Cardia: A Comparison between the UFT300 and CLO Tests. JGH Open 2, 93–96. doi:10.1002/jgh3.12053
Liu, Y., Uemura, N., Xiao, S.-D., Tytgat, G. N. J., and Ten Kate, F. J. W. (2005). Agreement between Endoscopic and Histological Gastric Atrophy Scores. J. Gastroenterol. 40, 123–127. doi:10.1007/s00535-004-1511-x
Magnus, H. A. (1954). The Pathology of Peptic Ulceration. Postgrad. Med. J. 30, 131–136. doi:10.1136/pgmj.30.341.131
Mahdavi, J., Sondén, B., Hurtig, M., Olfat, F. O., Forsberg, L., Roche, N., et al. (2002). Helicobacter pylori SabA Adhesin in Persistent Infection and Chronic Inflammation. Science 297, 573–578. doi:10.1126/science.1069076
Marteau, P., Pochart, P., Doré, J., Béra-Maillet, C., Bernalier, A., and Corthier, G. (2001). Comparative Study of Bacterial Groups within the Human Cecal and Fecal Microbiota. Appl. Environ. Microbiol. 67, 4939–4942. doi:10.1128/aem.67.10.4939-4942.2001
McColl, K. E. L. (2012). The Elegance of the Gastric Mucosal Barrier: Designed by Nature for Nature. Gut 61, 787–788. doi:10.1136/gutjnl-2011-301612
McCracken, K. W., Aihara, E., Martin, B., Crawford, C. M., Broda, T., Treguier, J., et al. (2017). Wnt/β-catenin Promotes Gastric Fundus Specification in Mice and Humans. Nature 541, 182–187. doi:10.1038/nature21021
McCracken, K. W., Catá, E. M., Crawford, C. M., Sinagoga, K. L., Schumacher, M., Rockich, B. E., et al. (2014). Modelling Human Development and Disease in Pluripotent Stem-Cell-Derived Gastric Organoids. Nature 516, 400–404. doi:10.1038/nature13863
McCracken, K. W., and Wells, J. M. (2017a). Mechanisms of Embryonic Stomach Development. Semin. Cel Dev Biol. doi:10.1016/j.semcdb.2017.02.004
McCracken, K. W., and Wells, J. M. (2017b). Mechanisms of Embryonic Stomach Development. Semin. Cel Developmental Biol. 66, 36–42. doi:10.1016/j.semcdb.2017.02.004
McDonald, E. G., Milligan, J., Frenette, C., and Lee, T. C. (2015). Continuous Proton Pump Inhibitor Therapy and the Associated Risk of Recurrent Clostridium difficile Infection. JAMA Intern. Med. 175, 784–791. doi:10.1001/jamainternmed.2015.42
Menheniott, T. R., Peterson, A. J., O'Connor, L., Lee, K. S., Kalantzis, A., Kondova, I., et al. (2010). A Novel Gastrokine, Gkn3, marks Gastric Atrophy and Shows Evidence of Adaptive Gene Loss in Humans. Gastroenterology 138, 1823–1835. doi:10.1053/j.gastro.2010.01.050
Meyer, A. R., and Goldenring, J. R. (2018). Injury, Repair, Inflammation and Metaplasia in the Stomach. J. Physiol. 596, 3861–3867. doi:10.1113/jp275512
Miao, Z.-F., Lewis, M. A., Cho, C. J., Adkins-Threats, M., Park, D., Brown, J. W., et al. (2020). A Dedicated Evolutionarily Conserved Molecular Network Licenses Differentiated Cells to Return to the Cell Cycle. Developmental Cell 55, 178–194. doi:10.1016/j.devcel.2020.07.005
Mills, J. C., and Sansom, O. J. (2015). Reserve Stem Cells: Differentiated Cells Reprogram to Fuel Repair, Metaplasia, and Neoplasia in the Adult Gastrointestinal Tract. Sci. Signal. 8, re8. doi:10.1126/scisignal.aaa7540
Mills, J. C., and Goldenring, J. R. (2017). Metaplasia in the Stomach Arises from Gastric Chief Cells. Cell Mol. Gastroenterol. Hepatol. 4, 85–88. doi:10.1016/j.jcmgh.2017.03.006
Moreau, H., Bernadac, A., Gargouri, Y., Benkouka, F., Laugier, R., and Verger, R. (1989). Immunocytolocalization of Human Gastric Lipase in Chief Cells of the Fundic Mucosa. Histochemistry 91, 419–423. doi:10.1007/bf00493829
Morrissey, S. M., Ward, P. M., Jayaraj, A. P., Tovey, F. I., and Clark, C. G. (1983). Histochemical Changes in Mucus in Duodenal Ulceration. Gut 24, 909–913. doi:10.1136/gut.24.10.909
Moss, S. F., and Malfertheiner, P. (2007). Helicobacter and Gastric Malignancies. Helicobacter 12 Suppl 1 (Suppl. 1), 23–30. doi:10.1111/j.1523-5378.2007.00539.x
Murphy, K. G., and Bloom, S. R. (2006). Gut Hormones and the Regulation of Energy Homeostasis. Nature 444, 854–859. doi:10.1038/nature05484
Mutoh, H., Hakamata, Y., Sato, K., Eda, A., Yanaka, I., Honda, S., et al. (2002). Conversion of Gastric Mucosa to Intestinal Metaplasia in Cdx2-Expressing Transgenic Mice. Biochem. Biophysical Res. Commun. 294, 470–479. doi:10.1016/s0006-291x(02)00480-1
Nam, K. T., Lee, H. J., Sousa, J. F., Weis, V. G., O'Neal, R. L., Finke, P. E., et al. (2010). Mature Chief Cells Are Cryptic Progenitors for Metaplasia in the Stomach. Gastroenterology 139, 2028–2037. doi:10.1053/j.gastro.2010.09.005
Neumann, W. L., Coss, E., Rugge, M., and Genta, R. M. (2013). Autoimmune Atrophic Gastritis-Pathogenesis, Pathology and Management. Nat. Rev. Gastroenterol. Hepatol. 10, 529–541. doi:10.1038/nrgastro.2013.101
Nomura, S., Settle, S. H., Leys, C. M., Means, A. L., Peek, R. M., Leach, S. D., et al. (2005). Evidence for Repatterning of the Gastric Fundic Epithelium Associated with Ménétrier's Disease and TGFα Overexpression. Gastroenterology 128, 1292–1305. doi:10.1053/j.gastro.2005.03.019
Nozaki, K., Ogawa, M., Williams, J. A., Lafleur, B. J., Ng, V., Drapkin, R. I., et al. (2008). A Molecular Signature of Gastric Metaplasia Arising in Response to Acute Parietal Cell Loss. Gastroenterology 134, 511–522. doi:10.1053/j.gastro.2007.11.058
O'Connor, A., and O'Moráin, C. (2014). Digestive Function of the Stomach. Dig. Dis. 32, 186–191. doi:10.1159/000357848
O'Neal, R. L., Nam, K. T., LaFleur, B. J., Barlow, B., Nozaki, K., Lee, H.-J., et al. (2013). Human Epididymis Protein 4 Is Up-Regulated in Gastric and Pancreatic Adenocarcinomas. Hum. Pathol. 44, 734–742. doi:10.1016/j.humpath.2012.07.017
Odze, R. D., and Goldblum, J. R. (2009). Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas. Philadelphia, PA: Elsevier Health Sciences.
Odze, R. D. (2005). Pathology of the Gastroesophageal junction. Semin. Diagn. Pathol. 22, 256–265. doi:10.1053/j.semdp.2006.04.007
Offield, M. F., Jetton, T. L., Labosky, P. A., Ray, M., Stein, R. W., Magnuson, M. A., et al. (1996). PDX-1 Is Required for Pancreatic Outgrowth and Differentiation of the Rostral Duodenum. Development 122, 983–995. doi:10.1242/dev.122.3.983
Paimela, H., Goddard, P. J., Carter, K., Khakee, R., McNeil, P. L., Ito, S., et al. (1993). Restitution of Frog Gastric Mucosa In Vitro: Effect of Basic Fibroblast Growth Factor. Gastroenterology 104, 1337–1345. doi:10.1016/0016-5085(93)90342-a
Peskar, B. M. (2001). Role of Cyclooxygenase Isoforms in Gastric Mucosal Defence. J. Physiology-Paris 95, 3–9. doi:10.1016/s0928-4257(01)00003-1
Peters, Y., Al-Kaabi, A., Shaheen, N. J., Chak, A., Blum, A., Souza, R. F., et al. (2019). Barrett Oesophagus. Nat. Rev. Dis. Primers 5, 35. doi:10.1038/s41572-019-0086-z
Piazuelo, M. B., Bravo, L. E., Mera, R. M., Camargo, M. C., Bravo, J. C., Delgado, A. G., et al. (2021). The Colombian Chemoprevention Trial: 20-Year Follow-Up of a Cohort of Patients with Gastric Precancerous Lesions. Gastroenterology 160, 1106–1117. doi:10.1053/j.gastro.2020.11.017
Qiao, X. T., Ziel, J. W., McKimpson, W., Madison, B. B., Todisco, A., Merchant, J. L., et al. (2007). Prospective Identification of a Multilineage Progenitor in Murine Stomach Epithelium. Gastroenterology 133, 1989–1998. doi:10.1053/j.gastro.2007.09.031
Que, J., Okubo, T., Goldenring, J. R., Nam, K.-T., Kurotani, R., Morrisey, E. E., et al. (2007). Multiple Dose-dependent Roles for Sox2 in the Patterning and Differentiation of Anterior Foregut Endoderm. Development 134, 2521–2531. doi:10.1242/dev.003855
Radyk, M. D., Burclaff, J., Willet, S. G., and Mills, J. C. (2018). Metaplastic Cells in the Stomach Arise, Independently of Stem Cells, via Dedifferentiation or Transdifferentiation of Chief Cells. Gastroenterology 154, 839–843. doi:10.1053/j.gastro.2017.11.278
Raufman, J.-P. (1992). Gastric Chief Cells: Receptors and Signal-Transduction Mechanisms. Gastroenterology 102, 699–710. doi:10.1016/0016-5085(92)90124-h
Richter, C., Tanaka, T., and Yada, R. Y. (1998). Mechanism of Activation of the Gastric Aspartic Proteinases: Pepsinogen, Progastricsin and Prochymosin. Biochem. J. 335 (Pt 3), 481–490. doi:10.1042/bj3350481
Robert, A., Nezamis, J. E., Lancaster, C., and Hanchar, A. J. (1979). Cytoprotection by Prostaglandins in Rats. Gastroenterology 77, 433–443. doi:10.1016/0016-5085(79)90002-7
Rokkas, T., Pistiolas, D., Sechopoulos, P., Robotis, I., and Margantinis, G. (2007). The Long-Term Impact of Helicobacter pylori Eradication on Gastric Histology: a Systematic Review and Meta-Analysis. Helicobacter 12 (Suppl. 2), 32–38. doi:10.1111/j.1523-5378.2007.00563.x
Romer, A. I., Singh, J., Rattan, S., and Krauss, R. S. (2013). Smooth Muscle Fascicular Reorientation Is Required for Esophageal Morphogenesis and Dependent on Cdo. J. Cel Biol 201, 309–323. doi:10.1083/jcb.201301005
Rubio, C. A., Jaramillo, E., Suzuki, G., Lagergren, P., and Nesi, G. (2009). Antralization of the Gastric Mucosa of the Incisura Angularis and its Gastrin Expression. Int. J. Clin. Exp. Pathol. 2, 65–70.
Rugge, M., Correa, P., Di Mario, F., El-Omar, E., Fiocca, R., Geboes, K., et al. (2008). OLGA Staging for Gastritis: a Tutorial. Dig. Liver Dis. 40, 650–658. doi:10.1016/j.dld.2008.02.030
Sáenz, J. B., and Mills, J. C. (2018). Acid and the Basis for Cellular Plasticity and Reprogramming in Gastric Repair and Cancer. Nat. Rev. Gastroenterol. Hepatol. 15, 257–273. doi:10.1038/nrgastro.2018.5
Sáenz, J. B., Vargas, N., and Mills, J. C. (2019). Tropism for Spasmolytic Polypeptide-Expressing Metaplasia Allows Helicobacter pylori to Expand its Intragastric Niche. Gastroenterology 156, 160–174. doi:10.1053/j.gastro.2018.09.050
Sanduleanu, S., Jonkers, D., de Bruïne, A., Hameeteman, W., and Stockbrügger, R. W. (2001). Changes in Gastric Mucosa and Luminal Environment during Acid-Suppressive Therapy: a Review in Depth. Dig. Liver Dis. 33, 707–719. doi:10.1016/s1590-8658(01)80050-5
Satoh, K., Kimura, K., Yoshida, Y., Kasano, T., Kihira, K., and Taniguchi, Y. (1991). A Topographical Relationship between Helicobacter pylori and Gastritis: Quantitative Assessment of Helicobacter pylori in the Gastric Mucosa. Am. J. Gastroenterol. 86, 285–291.
Schade, C., Flemström, G., and Holm, L. (1994). Hydrogen Ion Concentration in the Mucus Layer on Top of Acid-Stimulated and -inhibited Rat Gastric Mucosa. Gastroenterology 107, 180–188. doi:10.1016/0016-5085(94)90075-2
Schmidt, P. H., Lee, J. R., Joshi, V., Playford, R. J., Poulsom, R., Wright, N. A., et al. (1999). Identification of a Metaplastic Cell Lineage Associated with Human Gastric Adenocarcinoma. Lab. Invest. 79, 639–646.
Schubert, M. L. (2015). Functional Anatomy and Physiology of Gastric Secretion. Curr. Opin. Gastroenterol. 31, 479–485. doi:10.1097/mog.0000000000000213
Schubert, M. L. (2009). Gastric Exocrine and Endocrine Secretion. Curr. Opin. Gastroenterol. 25, 529–536. doi:10.1097/mog.0b013e328331b62a
Shichijo, S., Hirata, Y., Niikura, R., Hayakawa, Y., Yamada, A., Ushiku, T., et al. (2016). Histologic Intestinal Metaplasia and Endoscopic Atrophy Are Predictors of Gastric Cancer Development after Helicobacter pylori Eradication. Gastrointest. Endosc. 84, 618–624. doi:10.1016/j.gie.2016.03.791
Shimizu, T., Choi, E., Petersen, C. P., Noto, J. M., Romero-Gallo, J., Piazuelo, M. B., et al. (2016). Characterization of Progressive Metaplasia in the Gastric Corpus Mucosa of Mongolian Gerbils Infected with Helicobacter pylori. J. Pathol. 239, 399–410. doi:10.1002/path.4735
Silberg, D. G., Sullivan, J., Kang, E., Swain, G. P., Moffett, J., Sund, N. J., et al. (2002). Cdx2 Ectopic Expression Induces Gastric Intestinal Metaplasia in Transgenic Mice. Gastroenterology 122, 689–696. doi:10.1053/gast.2002.31902
Song, J. H., Kim, S. G., Jin, E. H., Lim, J. H., and Yang, S. Y. (2017). Risk Factors for Gastric Tumorigenesis in Underlying Gastric Mucosal Atrophy. Gut and Liver 11, 612–619. doi:10.5009/gnl16488
Sørbye, H., Svanes, C., Stangeland, L., Kvinnsland, S., and Svanes, K. (1988). Epithelial Restitution and Cellular Proliferation after Gastric Mucosal Damage Caused by Hypertonic NaCl in Rats. Virchows Arch. A. Pathol. Anat. Histopathol 413, 445–455. doi:10.1007/BF00716993
Sousa, J. F., Ham, A.-J. L., Whitwell, C., Nam, K. T., Lee, H.-J., Yang, H.-K., et al. (2012). Proteomic Profiling of Paraffin-Embedded Samples Identifies Metaplasia-specific and Early-Stage Gastric Cancer Biomarkers. Am. J. Pathol. 181, 1560–1572. doi:10.1016/j.ajpath.2012.07.027
Souza, R. F., Krishnan, K., and Spechler, S. J. (2008). Acid, Bile, and CDX: the ABCs of Making Barrett's Metaplasia. Am. J. Physiology-Gastrointestinal Liver Physiol. 295, G211–G218. doi:10.1152/ajpgi.90250.2008
Spechler, S. J. (2004). Intestinal Metaplasia at the Gastroesophageal junction. Gastroenterology 126, 567–575. doi:10.1053/j.gastro.2003.11.061
Stave, R., Brandtzaeg, P., Nygaard, K., and Fausa, O. (1978). The Transitional Body-Antrum Zone in Resected Human Stomachs. Scand. J. Gastroenterol. 13, 685–691. doi:10.3109/00365527809181782
Steer, H. W. (1984). Surface Morphology of the Gastroduodenal Mucosa in Duodenal Ulceration. Gut 25, 1203–1210. doi:10.1136/gut.25.11.1203
Stengel, A., and Taché, Y. (2012). Gastric Peptides and Their Regulation of Hunger and Satiety. Curr. Gastroenterol. Rep. 14, 480–488. doi:10.1007/s11894-012-0291-3
Štimac, D., Klobucar Majanovic, S., and Franjic, N. (2014). Stomach - Key Player in the Regulation of Metabolism. Dig. Dis. 32, 192–201. doi:10.1159/000357849
Tan, P., and Yeoh, K.-G. (2015). Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 149, 1153–1162. doi:10.1053/j.gastro.2015.05.059
Tariq, R., Singh, S., Gupta, A., Pardi, D. S., and Khanna, S. (2017). Association of Gastric Acid Suppression with Recurrent Clostridium difficile Infection. JAMA Intern. Med. 177, 784–791. doi:10.1001/jamainternmed.2017.0212
Teal, E., Dua-Awereh, M., Hirshorn, S. T., and Zavros, Y. (2020). Role of Metaplasia during Gastric Regeneration. Am. J. Physiology-Cell Physiol. 319, C947–C954. doi:10.1152/ajpcell.00415.2019
Thompson, C. A., DeLaForest, A., and Battle, M. A. (2018). Patterning the Gastrointestinal Epithelium to Confer Regional-specific Functions. Developmental Biol. 435, 97–108. doi:10.1016/j.ydbio.2018.01.006
Tsugawa, H., Kato, C., Mori, H., Matsuzaki, J., Kameyama, K., Saya, H., et al. (2019). Cancer Stem-Cell Marker CD44v9-Positive Cells Arise from Helicobacter pylori-Infected CAPZA1-Overexpressing Cells. Cell Mol. Gastroenterol. Hepatol. 8, 319–334. doi:10.1016/j.jcmgh.2019.05.008
Umeda, N., Mahood, W. H., Hermann, G. A., and Herrera, A. F. (1971). Distribution of Gastric Glands and Gastritis: a Topographic Study by Endoscopy. Gastrointest. Endosc. 18, 17–20. doi:10.1016/s0016-5107(71)74012-7
Vaezi, M. F., Yang, Y.-X., and Howden, C. W. (2017). Complications of Proton Pump Inhibitor Therapy. Gastroenterology 153, 35–48. doi:10.1053/j.gastro.2017.04.047
Veldhuyzen van Zanten, S. J. O., Dixon, M. F., and Lee, A. (1999). The Gastric Transitional Zones: Neglected Links between Gastroduodenal Pathology and Helicobacter Ecology. Gastroenterology 116, 1217–1229. doi:10.1016/s0016-5085(99)70025-9
Villafuerte-Gálvez, J. A., and Kelly, C. P. (2018). Proton Pump Inhibitors and Risk of Clostridium difficile Infection. Curr. Opin. Gastroenterol. 34, 11–18. doi:10.1097/mog.0000000000000414
Vogt, C. D., and Panoskaltsis‐Mortari, A. (2020). Tissue Engineering of the Gastroesophageal Junction. J. Tissue Eng. Regen. Med. 14, 855–868. doi:10.1002/term.3045
Waddingham, W., Graham, D., Banks, M., and Jansen, M. (2018). The Evolving Role of Endoscopy in the Diagnosis of Premalignant Gastric Lesions. F1000Res 7, 715. doi:10.12688/f1000research.12087.1
Wallace, J. L. (2008). Prostaglandins, NSAIDs, and Gastric Mucosal Protection: Why Doesn't the Stomach Digest Itself? Physiol. Rev. 88, 1547–1565. doi:10.1152/physrev.00004.2008
Walsh, J. H. (1990). Role of Gastrin as a Trophic Hormone. Digestion 47 (Suppl. 1), 11–52. doi:10.1159/000200509
Wanebo, H. J., Kennedy, B. J., Chmiel, J., Steele, G., Winchester, D., and Osteen, R. (1993). Cancer of the Stomach. Ann. Surg. 218, 583–592. doi:10.1097/00000658-199321850-00002
Watson, S. A., Grabowska, A. M., El-Zaatari, M., and Takhar, A. (2006). Gastrin - Active Participant or Bystander in Gastric Carcinogenesis? Nat. Rev. Cancer 6, 936–946. doi:10.1038/nrc2014
Weis, V. G., and Goldenring, J. R. (2009). Current Understanding of SPEM and its Standing in the Preneoplastic Process. Gastric Cancer 12, 189–197. doi:10.1007/s10120-009-0527-6
Weis, V. G., Petersen, C. P., Mills, J. C., Tuma, P. L., Whitehead, R. H., and Goldenring, J. R. (2014). Establishment of Novel In Vitro Mouse Chief Cell and SPEM Cultures Identifies MAL2 as a Marker of Metaplasia in the Stomach. Am. J. Physiology-Gastrointestinal Liver Physiol. 307, G777–G792. doi:10.1152/ajpgi.00169.2014
Weis, V. G., Sousa, J. F., LaFleur, B. J., Nam, K. T., Weis, J. A., Finke, P. E., et al. (2013). Heterogeneity in Mouse Spasmolytic Polypeptide-Expressing Metaplasia Lineages Identifies Markers of Metaplastic Progression. Gut 62, 1270–1279. doi:10.1136/gutjnl-2012-302401
Wieczorek, T. J., Wang, H. H., Antonioli, D. A., Glickman, J. N., and Odze, R. D. (2003). Pathologic Features of Reflux and Helicobacter Pylori-Associated Carditis. Am. J. Surg. Pathol. 27, 960–968. doi:10.1097/00000478-200307000-00011
Willet, S. G., Lewis, M. A., Miao, Z. F., Liu, D., Radyk, M. D., Cunningham, R. L., et al. (2018). Regenerative Proliferation of Differentiated Cells by mTORC1-dependent Paligenosis. EMBO J. 37. doi:10.15252/embj.201798311
Willet, S. G., and Mills, J. C. (2016). Stomach Organ and Cell Lineage Differentiation: from Embryogenesis to Adult Homeostasis. Cell Mol. Gastroenterol. Hepatol. 2, 546–559. doi:10.1016/j.jcmgh.2016.05.006
Wolf, C., Seldenrijk, C. A., Timmer, R., Breumelhof, R., and Smout, A. J. P. M. (2001). Does Carditis Have Two Different Etiologies? Dig. Dis. Sci. 46, 2424–2432. doi:10.1023/a:1012315617940
Xia, H. H.-X., Kalantar, J. S., Talley, N. J., Wyatt, J. M., Adams, S., Cheung, K., et al. (2000). Antral-type Mucosa in the Gastric Incisura, Body, and Fundus (Antralization): a Link between Helicobacter pylori Infection and Intestinal Metaplasia? Am. J. Gastroenterol. 95, 114–121. doi:10.1111/j.1572-0241.2000.01609.x
Xia, H. H.-X., Wong, B. C.-Y., Zhang, G.-S., Yang, Y., Wyatt, J. M., Adams, S., et al. (2004a). Antralization of Gastric Incisura Is Topographically Associated with Increased Gastric Epithelial Apoptosis and Proliferation, but Not with CagA Seropositivity. J. Gastroenterol. Hepatol. 19, 1257–1263. doi:10.1111/j.1440-1746.2004.03489.x
Xia, H. H.-X., Yang, Y., Lam, S. K., Wong, W. M., Leung, S. Y., Yuen, S. T., et al. (2004b). Aberrant Epithelial Expression of Trefoil Family Factor 2 and Mucin 6 in Helicobacter pylori Infected Gastric Antrum, Incisura, and Body and its Association with Antralisation. J. Clin. Pathol. 57, 861–866. doi:10.1136/jcp.2003.015487
Xia, H. H.-X., Zhang, G.-S., Talley, N. J., Wong, B. C. Y., Yang, Y., Henwood, C., et al. (2002). Topographic Association of Gastric Epithelial Expression of Ki-67, Bax, and Bcl-2 with Antralization in the Gastric Incisura, Body, and Fundus. Am. J. Gastroenterol. 97, 3023–3031. doi:10.1111/j.1572-0241.2002.07120.x
You, W.-c., Blot, W. J., Chang, Y.-s., Li, J.-y., Jin, M.-l., Zhao, Y., et al. (1992). Comparison of the Anatomic Distribution of Stomach Cancer and Precancerous Gastric Lesions. Jpn. J. Cancer Res. 83, 1150–1153. doi:10.1111/j.1349-7006.1992.tb02738.x
Keywords: metaplasia, Helicobacter pylori, atrophy, cancer, inflammation
Citation: Sáenz JB (2021) Follow the Metaplasia: Characteristics and Oncogenic Implications of Metaplasia’s Pattern of Spread Throughout the Stomach. Front. Cell Dev. Biol. 9:741574. doi: 10.3389/fcell.2021.741574
Received: 14 July 2021; Accepted: 29 October 2021;
Published: 12 November 2021.
Edited by:
Rich Dipaolo, Saint Louis University, United StatesReviewed by:
Massimoi Rugge, University of Padua, ItalyCopyright © 2021 Sáenz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José B. Sáenz, c2FlbnpqQHd1c3RsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.