 Meng-Yi Luo1,2†Jian-Hui Su1,2†Shao-Xin Gong3Na Liang4Wen-Qian Huang1,2Wei Chen1,2
Meng-Yi Luo1,2†Jian-Hui Su1,2†Shao-Xin Gong3Na Liang4Wen-Qian Huang1,2Wei Chen1,2 Ai-Ping Wang1,2*Ying Tian1*
Ai-Ping Wang1,2*Ying Tian1*- 1Institute of Clinical Research, Affiliated Nanhua Hospital, University of South China, Hengyang, China
- 2Hengyang Key Laboratory of Neurodegeneration and Cognitive Impairment, Department of Physiology, Institute of Neuroscience Research, Hengyang Medical College, University of South China, Hengyang, China
- 3Department of Pathology, First Affiliated Hospital, University of South China, Hengyang, China
- 4Department of Anesthesiology, Affiliated Nanhua Hospital, University of South China, Hengyang, China
The dynamic balance of cardiomyocytes and neurons is essential to maintain the normal physiological functions of heart and brain. If excessive cells die in tissues, serious Cardio-Cerebrovascular Diseases would occur, namely, hypertension, myocardial infarction, and ischemic stroke. The regulation of cell death plays a role in promoting or alleviating Cardio-Cerebrovascular Diseases. Ferroptosis is an iron-dependent new type of cell death that has been proved to occur in a variety of diseases. In our review, we focus on the critical role of ferroptosis and its regulatory mechanisms involved in Cardio-Cerebrovascular Diseases, and discuss the important function of ferroptosis-related inhibitors in order to propose potential implications for the prevention and treatment of Cardio-Cerebrovascular Diseases.
1 Introduction
Cardio-Cerebrovascular Diseases (CCVDs) are the collective terms for cardiovascular and cerebrovascular diseases, which refer to ischemic or hemorrhagic diseases of heart, brain and systemic tissues caused by hyperlipidemia, hyperglycemia and hypertension (Vasan et al., 2001; McEvoy 2012), including atherosclerosis (AS), myocardial infarction (MI), heart failure (HF), diabetic cardiomyopathy (DCM), hypertension, ischemic stroke (IS), and so on. According to epidemiological surveys, CCVDs are very common among middle-aged people over 50, and have become the top one killer of people (Dalen et al., 2014). Patients with CCVDs are gradually becoming younger (Singh et al., 2015), accounting for almost one-third of global deaths every year (2015). CCVDs are characterized by high prevalence, disability and mortality rates (Zhang et al., 2011). Even with the application of the most advanced and perfect treatments available, more than 50% of survivors with cardio-cerebrovascular accidents may still be unable to take care of themselves completely (DeVon et al., 2017). However, the existing methods are not ideal for the prevention and treatment of CCVDs, and there is a lack of effective therapeutic strategies to improve neurological function. Therefore, in order to prevent and treat the disease and improve prognosis, there is an urgent need to explore the pathogenesis of CCVDs, as well as new therapeutic strategies. During these years, the role of cell death has been emerging in CCVDs (Lu et al., 2021).
Cell death (CD) is essential for maintaining homeostasis and basic biological functions within tissues, and its changes are of great importance in disease pathology. Different types of CD have their own manifestations, and many types of CD are defined by differences in morphological and biochemical characteristics (Galluzzi et al., 2012). Apoptosis is one of the first types of CD to be recognized, and is essential for maintaining homeostasis in the cardio-cerebrovascular internal environment (Favaloro et al., 2012). Both decreased and increased apoptosis can lead to pathological changes. The maintenance of normal structure and function of the cardio-cerebrovascular system requires a balance between the production and death of cells (including cardiomyocytes, endothelial cells, vascular smooth muscle cells, and neurons) in the tissues and organs (Zhaolin et al., 2019). Excess CD often leads to dysfunction. Consequently, we began to ponder over whether there are any other ways of death (i.e., necroptosis, pyroptosis, and ferroptosis) that play an important role in CCVDs as apoptosis?
Based on the functional differences, CD can be divided into accidental cell death (ACD) and regulated cell death (RCD). RCD concerns signal cascade reactions involving effector molecules, and has unique biochemical characteristics, morphological features and immunological consequences. For the past few years, many studies have explored the role of a new type of RCD-ferroptosis in CCVDs (Gao et al., 2015; Tuo et al., 2017). Ferroptosis, first proposed by Stockwell’s group in 2012, is an iron-dependent modality of RCD and characterized by lipid peroxide accumulation, which leads to oxidative damage to cell membranes, and differs from other forms of RCD in several ways (Dixon et al., 2012). Necroptosis is a type of RCD with necrotic features including cell membrane rupture and organelle swelling, and finally would cause inflammatory response. The activation of mixed lineage kinase domain-like protein (MLKL) by receptor-interacting protein 3 (RIPK3) is a key regulatory pathway of necroptosis, which disrupts cell membrane integrity and causes inflammation. Pyroptosis is a kind of RCD activated by inflammasomes, which is accompanied by cell swelling, cell membrane lysis and the release of inflammatory factors. Autophagy refers to a process that the concave membrane structure wraps senescent organelles and substances and then forms autophagosomes. Under the action of acidic lysosomal enzymes, the wrapped substances are degraded to achieve intracellular environmental homeostasis and organelle renewal. Autophagy plays an important role in neuro-toxicity and neuronal death induced by hypoxia-ischemia. The four RCD types differ largely in morphology, biochemistry and genetics. Ferroptosis is a way to regulate CD caused by iron overload and reactive oxygen species (ROS)-dependent lipid peroxides accumulation, with structural changes focused on mitochondria (Shojaie et al., 2020). Necroptosis and pyroptosis are characterized by the formation of plasma membrane pores via caspase-1, which leads to the pro-inflammatory cytokines release and cell lysis, and thus controls the inflammatory response. It is a profound inflammatory pattern of RCD associated with the innate immune system (Orning et al., 2018). Autophagy is an intracellular waste degradation pathway that is activated in response to cellular stress and usually mediates protective rather than cytotoxic effects (Galluzzi et al., 2018). Except for the differences, what remains the same is that they are all tightly regulated by intracellular signaling pathways. However, ferroptosis plays a key role in the pathogenesis of several CCVDs, and studies have shown that ferroptosis is closely related to AS, MI, HF, DCM, hypertension, and IS (Wu et al., 2021a). More importantly, in many cases, inhibiting ferroptosis through different mechanisms has been proved to have a protective effect on CCVDs (Magtanong and Dixon 2018; Fang et al., 2019). Thus, ferroptosis is a potential therapeutic target. In summary, the discovery of ferroptosis expands our understanding of CD in CCVDs, and targeting ferroptosis will provide a new strategy and broaden thoughts for the prevention and treatment of CCVDs.
2 Contribution of Ferroptosis in Cardio-Cerebrovascular Disease
As the maintenance of cardio-cerebrovascular homeostasis depends on CD and cell renewal, excessive cell loss will contribute to the occurrence of many CCVDs (Lu et al., 2020). Hence, ferroptosis may participate in the occurrence, development and prognosis of diseases through the damage to normal tissues and organs or the loss of specific functions.
Studies have stated that heart tissue is easy to accumulate free iron, and high dietary iron intake increases the risk of CCVDs. Specific inhibitors of ferroptosis have been proved to effectively reduce heart damage and have cardio-protective effects (Fang et al., 2020). Cell damage in the infarct region of the brain is an inevitable upshot of focal cerebral ischemia (Masaldan et al., 2019; Datta et al., 2020), and delaying CD by targeting ferroptosis can save dysfunctional neurons and reduce the size of infarct region. Accordingly, inhibiting ferroptosis can effectively prevent and alleviate the progression of CCVDs.
2.1 The Role of Ferroptosis in Atherosclerosis
AS is a chronic progressive vascular disease characterized by disturbances in lipid metabolism (Weber and Noels 2011) and narrowing of the arterial lumen through the formation of atherosclerotic plaques in the arterial wall (Veseli et al., 2017; Moore and Tabas 2011). Endothelial dysfunction and inflammation are closely associated with the development of AS (Gimbrone and García-Cardeña 2016; Li et al., 2021b). Lipid peroxidation has been shown to be associated with endothelial dysfunction and inflammatory responses, and plays a key role in the pathogenesis of AS (Hammad et al., 2009; Gimbrone and García-Cardeña 2016). Firstly, in human and animal models, iron levels and inflammatory mediators in atherosclerotic lesions were significantly elevated compared to healthy arterial tissues (Ross 1999; Sullivan 2009). On the one hand, iron overload led to endothelial dysfunction by enhancing the oxidative and inflammatory responses in endothelial cells (ECs) (Gimbrone and García-Cardeña 2016; Xu 2019). On the other hand, iron overload induced ferroptosis in foam cells which led to plaque instability, and ferroptosis in foam cells increased IL-1 and IL-8 expression, promoting inflammatory responses (Su et al., 2021). Secondly, in ApoE-/- AS mice fed with high-fat diet, inhibition of ferroptosis by Ferrostatin-1 (Fer-1) protected against thoracic aortic lipid peroxidation and inhibited exacerbation of AS (Bai et al., 2020). In addition, inhibition of ferroptosis inhibited lipid peroxidation induced by oxidized low density lipoprotein (ox-LDL) and endothelial dysfunction in mouse aortic endothelial cells (MAECs) in vitro. Fer-1 also down-regulated the expression of adhesion molecules and up-regulated eNOS expression (Bai et al., 2020) (Figure 1). Similarly, lowering iron levels in atherosclerotic tissue with the iron chelator Deferoxamine (DFO) could inhibit ferroptosis and inflammation, and mitigate the development of AS (Zhang et al., 2010). Finally, other studies found that serum PDSS2 and nuclear factor erythroid-2-related factor 2 (Nrf2) levels were significantly reduced in AS patients (Yang et al., 2021). PDSS2 is a key enzyme for the synthesis of coenzyme Q10 (CoQ10) (Li Y. et al., 2018). Over-expression of PDSS2 in human coronary artery endothelial cells (HCAECs) inhibited ROS release and iron content to suppress ferroptosis and promote the proliferation of HCAECs. PDSS2 also attenuated ferroptosis in HCAECs by activating the antioxidant factor NF-E2-related factor 2 (Nrf2) (Yang et al., 2021). Nrf2 is the primary transcription factor that regulates antioxidant responses and inhibits ferroptosis in various cell types by protecting cells from lethal ROS stress (Gorrini et al., 2013; Dodson et al., 2019). MiRNA17-92 (miR-17-92) is a multifunctional oncogenic miRNA cluster that plays an important role in tumor angiogenesis and tissue development (Mendell 2008). Over-expression of miR-17-92 in HUVECs can protect ECs from erastin-induced ferroptosis by targeting the zinc lipoprotein (A20)-acyl-CoA synthetase long-chain family member 4 (ACSL4) axis that could protect glutathione peroxidase 4 (GPX4) and inhibit ROS production (Xiao et al., 2019). Macrophages, as one of the important immune cells in AS, play an vital role in the inflammatory response and foam cell formation (Xie et al., 2020). The current study shows that iron levels in plaque tissue, particularly in macrophages, are significantly elevated and are associated with AS (Thong, Selley, and Watt 1996; Bi et al., 2021). It has not been proved yet whether ferroptosis occurs in macrophages during AS. Due to lipid peroxidation, plaque hemorrhage and iron deposition are markers of progressive plaque in humans, and ferroptosis in cells may persist (Martinet et al., 2019).
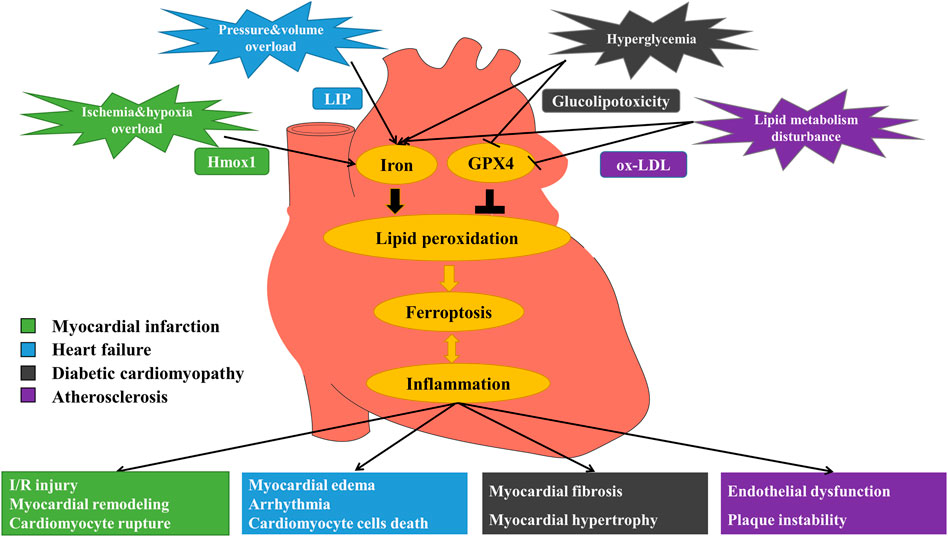
FIGURE 1. Mechanism of ferroptosis in myocardial infarction (MI), heart failure (HF), diabetic cardiomyopathy (DCM) and atherosclerosis (AS). MI: Under the stimulation of ischemia and hypoxia, excessive free iron releases via up-regulation of heme oxygenase 1 (Hmox1), promoting ferroptosis and eventually causing I/R injury, myocardial remodeling and myocardial cell rupture. HF: Stimulated by pressure and volume overload, labile iron pool (LIP) formation increases, leading to iron overload and the occurrence of ferroptosis, and then causing myocardial edema, arrhythmia and cardiomyocyte cell death. DCM: In the circumstance of hyperglycemia, free iron increases and glucolipotoxicity induces ferroptosis, triggering myocardial fibrosis, and myocardial hypertrophy. AS: When there exists lipid metabolism disturbance, iron overload and oxidized low-density lipoprotein (ox-LDL) induce lipid peroxidation that triggers ferroptosis, finally causing endothelial dysfunction and plaque instability.
In conclusion, ferroptosis is associated with the multiple vascular cytopathic processes that contribute to the development and progression of AS. Ferroptosis promotes the development of AS through inflammation, endothelial cell dysfunction, and the formation of foam cells, using ferroptosis inhibitors in AS could reverse these effects, which may provide new targets and strategies for the prevention and treatment of AS.
2.2 The Role of Ferroptosis in Myocardial Infarction
MI refers to severe and persistent myocardial ischemia caused by sharp decrease or interruption of arterial blood flow, resulting in myocardial ischemic necrosis. Refractory MI has gradually become one of the most serious diseases in the world (Zhang, Yu, et al., 2020). In MI, complete coronary artery occlusion is accompanied by a variety of structurally and functionally adverse consequences, the most irreversible of which is myocardial cell death.
Some studies have revealed that inhibiting myocardial cell death can reduce the size of MI (Del Re et al., 2019). It has been proved that Fe2+, erastin and RAS-selective lethal 3 (RSL3) can induce cardiomyocyte cell death (Kobayashi et al., 2018); and in the early and middle stages of MI, antioxidant GPX4 is detected at a decreased level (Wang H. et al., 2020). The content of Fe2+ is strictly regulated, and when its level is out of balance, excess ferrous will provide electron-promoting lipid peroxidation via Fenton reaction, resulting in an increased production of ROS and inducing ferroptosis (Kajarabille and Latunde-Dada 2019; Anandhan et al., 2020). Erastin and RSL3 are specific inducers of ferroptosis by inhibiting System Xc− and GPX4, respectively. In the acute myocardial infarction (AMI) model, the expression of divalent metal-ion transport 1 (DMT1) was found to be significantly up-regulated. DMT1, a member of solute carrier family 11 member 2 (SLC11A2), plays a role in iron transport, and its expression would be increased if exposed to high-iron environment. Over-expression of DMT1 can promote hypoxia/reperfusion-induced ferroptosis in cardiomyocyte cells, while its knockdown can suppress CD (Song et al., 2021). Besides, cardiomyocytes depend heavily on mitochondrial respiration to gain ATP as an energy source, thus mitochondrial dysfunction is an important cause of cardiomyocyte cell death (Patel and Karch 2020). In the animal model, iron chelator Deferiprone (DFP) improves myocardial mitochondrial function, and Liproxstatin-1 (Lip-1), a specific inhibitor of ferroptosis, also protects the integrity of mitochondrial structure (Ravingerová et al., 2020; Wu et al., 2021a). The evidence above suggests that ferroptosis is involved in the occurrence of MI, and the inhibition of ferroptosis can reduce the loss of cardiomyocytes and alleviate the progression of MI.
In terms of mechanism, ferroptosis promotes MI mainly through three pathways. During ferroptosis, heme oxygenase 1 (Hmox1) is up-regulated with increased free iron release from cardiomyocytes, and then left ventricular remodeling would be triggered, causing severely impaired function of left ventricular in MI patients (Baba et al., 2018). Besides, oxidative stress (OS) in cardiomyocytes would be increased, aggravating ischemia/reperfusion (I/R) injury (Song et al., 2021) (Figure 1). At the same time, ferroptosis is accompanied by excessive ROS production, resulting in depolarization of the mitochondrial membrane potential and opening of the mitochondrial permeability transition pore (MPTP), further leading to cell rupture (Ravingerová et al., 2020). Also, ferroptosis contributes to the accentuated myocardial remodeling and more severe dysfunction after MI by triggering inflammatory responses (Huang and Frangogiannis 2018). Therefore, blocking ferroptosis-related targets is expected to treat MI.
2.3 The Role of Ferroptosis in Heart Failure
HF is not an independent disease, but the final stage of the development in most primary Cardiovascular Diseases, contributing to high morbidity and mortality worldwide and a huge social and economic burden on the healthcare system (Wu et al., 2021b). Keeping the dynamic balance of cardiomyocytes plays a key role in maintaining the normal physiological function of heart (Ma et al., 2020), and the loss of cardiomyocytes caused by CD would induce and aggravate the development of HF.
According to related reports on HF, myocardial hemorrhage can cause the increase of labile iron pool (LIP) production and trigger the release of free iron into myocardium, being one of the important pathogenesis of HF (Qiu et al., 2020). LIP is a pool of weakly chelated iron with redox activity and is an ionic source available for Fenton reaction (Jamnongkan et al., 2017). Higher than normal levels of LIP, can disrupt the balance of redox and consequently promote ferroptosis in cardiomyocytes (Lou et al., 2021). There is a study has found that the contents of LIP and lipid peroxides were increased in the rat model of HF induced by pressure overload, suggesting that HF is associated with the lipid peroxidation process which is highly related to iron-dependent metabolism. In addition, ferroptosis in cardiomyocytes also occurred in doxorubicin (DOX)-induced cardiotoxic mice model. In the HF model, it showed that GPX4 and ferritin heavy chain1 (FTH1) expression levels were down-regulated (Chen et al., 2019). GPX4 is considered to be the key enzyme of ferroptosis and its genetic inactivation or pharmacological inhibition would cause the accumulation of lipid peroxidation products, which is a crucial hallmark of ferroptosis (Stockwell et al., 2020). FTH1 is a ferroptosis-related protein that is reduced in cells where ferroptosis occurs. Previous studies have demonstrated that inhibition of mitochondrial death pathway could protect cardiac tissues from HF (Abdelwahid et al., 2017), while the opening of mitochondrial voltage-dependent anion channel (VDAC)2/3 induced by erastin could increase the uptake of labile iron and the generation of ROS, leading to ferroptosis in cells (Wang Y. et al., 2020). Iron chelator Deferasirox (DFX) has been proved to reduce the iron content of cardiac tissue in HF, thereby playing a role in cardiac protection. Besides, other specific inhibitors of ferroptosis also have the ability to alleviate disease progression in various clinical models, such as heart disease models (Fang et al., 2020). Hence, it can be determined that ferroptosis increases the risk of HF by accelerating the damage and loss of cardiomyocytes.
As an important cause of cardiomyocyte cell death during HF, the mechanisms of ferroptosis are mainly through the following four aspects. To start with, in the process of ferroptosis, Hmox1 is activated, which promotes the increase of LIP and destroys cell homeostatic capacity, thus compromising the integrity of cardiomyocytes (Kakhlon and Cabantchik 2002). Secondly, since cardiomyocytes are easily affected by free iron overload, the release of excess free iron during ferroptosis would result in up-regulation of transferrin receptor (TFR) and down-regulation of ferritin, promoting cardiomyocyte cell death through mitochondrial VDAC2/3 or membrane lipid peroxidation through Fenton reaction to induce myocardial edema and dysfunction (Liu et al., 2018). Afterwards, excessive ROS would interfere with Ca2+ homeostasis in cardiomyocytes, affecting multiple ion transporters responsible for myocardial electrical activity and bringing about diastolic and systolic dysfunction and arrhythmia. Lastly, ferroptosis can trigger systemic inflammatory response, activate multiple pro-inflammatory signaling, induce cardiomyocyte hypertrophy, fibrosis and death, and eventually lead to chronic adverse ventricular remodeling (Shirazi et al., 2017; Huang and Frangogiannis 2018) (Figure 1). The level of pro-inflammatory cytokines is closely related to the severity and poor prognosis of chronic HF (Thackeray et al., 2018). To sum up, one of the key pathogenic factors of lethal HF is the irreversible dysfunction and death of terminally differentiated cardiomyocytes (Fang et al., 2019). Inhibition of ferroptosis has notable clinical significance to prevent cardiomyocyte inflammation, hypertrophy and death, and to maintain normal cardiac function.
2.4 The Role of Ferroptosis in Diabetic Cardiomyopathy
Diabetes is a pro-inflammatory disease (De Blasio et al., 2020). CCVDs are the leading cause of death in patients with diabetes. DCM is a chronic and irreversible cardiac complication (Dillmann 2019), and is difficult to prevent from progressing to significant HF (Zhang J. et al., 2020). Therefore, there is a need to find new therapeutic approaches to treat DCM more effectively. The pathogenesis of DCM is multifactorial and complex, including hyperglycemia, fatty acids, OS, inflammation, endoplasmic reticulum stress (ERS), myocardial fibrosis and hypertrophy (Bugger and Abel 2014). Many of the molecular mechanisms that increase fibrosis and myocardial inflammation can activate ferroptosis signaling pathway. Fatty acid oxidation or nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in diabetes produces elevated levels of ROS, leading to myocardial death, inflammation and fibrosis that impair cardiac structure and function (Parim et al., 2019). OS due to an imbalance in ROS production and antioxidant capacity has been suggested as a common mechanism leading to inflammation and DCM (Liu et al., 2014; Lv et al., 2019). Lipid peroxide accumulation caused by ROS and fatty acid oxidation is a major cause of ferroptosis (Cao and Dixon 2016), leading to myocardial death, inflammation, fibrosis, and damage to cardiac structure and function (Li W. et al., 2017). Elevated ROS and OS are common features of inflammation (Lv et al., 2019), altering inflammatory phenotype in DCM, including increased expression of cell adhesion molecules, increased macrophage and leukocyte infiltration, and increased expression of inflammatory cytokines (Westermann et al., 2007; Singh et al., 2008; Rajesh et al., 2012). Biopsy showed that the apoptosis of a diabetic heart was 85 times higher than that of the nondiabetic heart, indicating that cardiomyocytes in diabetes were sensitive to suffer CD (e.g. ferroptosis) (Cai and Kang 2003). Essentially, CD is the end of cardiomyocytes during DCM (Chen et al., 2020a). It has been shown that in a DCM mouse model, glucolipotoxicity effects resulting from chronic diabetes, would promote lipid peroxidation; while inactivating Nrf2-mediated antioxidant defenses and impairing Nrf2-coordinated iron metabolism (Chien et al., 2020) would promote the expression of ferroptosis and exacerbate the progression of DCM (Zang et al., 2020). More and more research suggests that effective treatment of DCM requires a specific endogenous antioxidant defense system, rather than non-selective scavenging of ROS that may impair physiological redox signaling (Chen et al., 2014). Many natural and synthetic Nrf2 activators ameliorate OS and iron metabolism disorders, reduce lipid peroxidation and inflammation, showing good therapeutic effects in animal models with DCM (Hu et al., 2018; Wang et al., 2018). These findings all indicate that ferroptosis promotes the development of DCM through abnormal iron metabolism, lipid peroxidation, ROS production, and enhanced cardiomyocyte inflammation (Figure 1). Therefore, regulation of ferroptosis in cardiomyocyte and attenuation of myocardial inflammation in DCM by Nrf2 may provide new therapeutic options for DCM.
2.5 The Role of Ferroptosis in Hypertension
Hypertension is one of the major risk factors of CCVDs (Zhang Y. et al., 2020). As blood pressure rises, the incidence of CCVDs increases (Pistoia et al., 2016). Brain is an early target from organ damage due to elevated changes in blood pressure (Sierra et al., 2011), and hypertensive brain injury is a serious complication of hypertension. Brain tissue is particularly vulnerable to OS and highly susceptible to oxygen radical damage because of its high oxygen consumption and lack of antioxidant enzymes. In addition, brain tissue is rich in unsaturated fatty acids, which are targets of lipid peroxidation (Dringen 2000; Nunomura et al., 2012; Zhang et al., 2018). Over these years, studies have demonstrated that OS mediates pathological changes in the brain and blood vessels (Dumitrescu et al., 2018), and ferroptosis is caused by OS and lipid peroxidation (Kerins and Ooi 2018; Morris et al., 2018) that are closely associated with brain injury and neuro-degenerative diseases. Studies have confirmed that ferroptosis is linked to a variety of central nervous system disorders, such as Parkinson's syndrome, epilepsy and stroke (Stockwell et al., 2017). Brain tissues in hypertensive brain-injured rats exhibited changes in ferroptosis-relevant indicators, such as decreased expression of GPX4, glutathione (GSH), increased iron content and malondialdehyde (MDA). This shows that hypertension leads to iron overload in the brain, and iron overload increases OS and lipid peroxidation, thereby causing ferroptosis (Yang et al., 2020) in neurons and ultimately leading to brain injury (Figure 2). However, how does hypertension lead to iron overload in the brain and what are specific mechanisms of ferroptosis in hypertensive brain injury are unknown. Therefore, whether inhibition of ferroptosis can play a protective role against hypertensive brain injury remains to be further investigated.
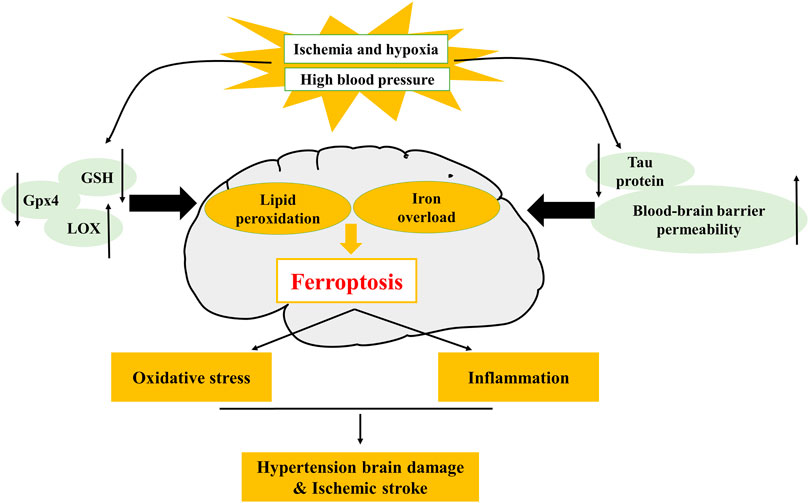
FIGURE 2. Mechanism of ferroptosis in hypertension and ischemic stroke. The decrease of glutathione peroxidase 4 (GPX4), glutathione (GSH), tau protein, and the increase of lipoxygenase (LOX), blood-brain barrier (BBB) permeability, can lead to the occurrence of ferroptosis. LOX is involved in hypertensive brain injury, tau protein and BBB permeability are involved in ischemic stroke.
2.6 The Role of Ferroptosis in Ischemic Stroke
IS refers to hemiplegia and disturbance of consciousness caused by cerebral infarction and clogged cerebral arteries, and is becoming a major global issue affecting human health, causing death and long-lasting disability. When the blood flows into a certain portion of brain and then is obstructed, IS would occur. This process is followed by hypoxia and nutritional deprivation, which leads to neuronal excitotoxicity and death (Shen et al., 2020). CD in the infarct region is the most inevitable consequence of focal cerebral ischemia (Datta et al., 2020). It was found that after cerebral injury, there were iron accumulation, metabolic disorder, increased expression of ferroptosis-related genes, GPX4 inactivation, ROS and pro-inflammatory factors generation (Li J. et al., 2020). The specific manifestations were iron overload, decreased GSH level and the enhancement of lipid peroxidation. Thusly, ferroptosis is considered to be an important pathway leading to IS.
It has been proved that iron deposition was increased in the basal ganglia, thalami, periventricular and subcortical white matter areas after severe ischemic and hypoxic brain injury (Li J. et al., 2020). Following iron accumulation, the neuronal damage during reperfusion would be aggravated (Tuo et al., 2017). Moreover, cerebral ischemia decreased the level of tau protein (Lu et al., 2021). Tau protein has the ability of facilitating neuronal iron efflux (Tuo et al., 2017). If tau protein is pathologically reduced, it would cause iron ion transport disorder, thereby leading to ferroptosis in cells. Also, when blood-brain barrier (BBB) is disrupted by stroke and other brain diseases, it helps to transfer the systemic iron pool into neurons in the brain parenchyma, exacerbating ferroptosis (Datta et al., 2020; DeGregorio-Rocasolano et al., 2019). Additionally, iron chelators have been shown to improve the prognosis of IS in mammals (Wu et al., 2018). It was found that the administration of ferroptosis inhibitors partially protected neurons from death in the middle cerebral artery occlusion (MCAO) model (Weiland et al., 2019). Similarly, an increase in lipid peroxidation levels and a decrease in GSH levels could also be detected in the same above model (Li J. et al., 2020). GSH is the most abundant small-molecule antioxidant in cells (Bayır et al., 2020). As an effective substrate of GPX4 and an indispensable cofactor in GPX4 activation, GSH maintains GPX4 activity indirectly to inhibit the occurrence of ferroptosis. Selenocysteine peptides can transport selenium to the ventricle and improve the functional recovery after IS (Alim et al., 2019). Selenium is able to maintain the activity of GPX4 (Chen et al., 2020b) and improve neurological functions. Inactivation of GPX4 leads to neuronal death both in vivo and in vitro (Kenny et al., 2019). Cui et al. found that ACSL4 gene knockout could attenuate ischemic brain injury while over-expression aggravated IS (Cui et al., 2021). ACSL4 is not only a marker for ferroptosis sensitivity, but also an indispensable down-stream player in the ferroptosis process (Doll and Conrad, 2017; Seibt et al., 2019). Based on studies concerning ferroptosis and IS, there is growing body of evidence that ferroptosis would be induced during IS and then aggravate brain damage, suggesting that ferroptosis may be a potential target for preventing and treating IS.
Currently, it has been reported that multiple regulatory mechanisms of ferroptosis mediate neuronal death, including iron overload, OS and neuro-inflammation. During IS, tau protein decreases, generous free iron deposits, and then Fe2+ releases electrons, producing excessive ROS. At the same time, iron-dependent lipoxygenase (LOX) catalyzes polyunsaturated fatty acids (PUFAs) to induce OS, which further alters the permeability of BBB (Iadecola and Anrather 2011). At this point, the injured brain tissue rapidly produces pro-inflammatory cytokines, causing leukocyte infiltration and eventually inducing ferroptosis and exaggerating brain edema, neuron death and progress of IS (Jin et al., 2013) (Figure 2). Generally, iron accumulation induces OS, bringing about the increase of ROS levels and the imbalance of antioxidant balance and infiltrating pro-inflammatory mediators that further amplify cerebral inflammatory responses, which aggravate neuronal damage during reperfusion (Jin et al., 2013; Tuo et al., 2017). Ferroptosis inhibitor or iron chelation therapy can significantly lessen cerebral I/R injury and ameliorate the prognosis of patients with stroke. Targeting or combined with ferroptosis pathway to regulate the cerebral environment is a very advisable treatment at present, and the inhibition of ferroptosis is likely to provide a new strategy for the prevention of neuron death induced by IS.
2.7 The Role of Ferroptosis in Other Cerebrovascular Diseases
2.7.1 The Role of Ferroptosis in Intracerebral Hemorrhage
Intracerebral hemorrhage (ICH) refers to bleeding caused by non-traumatic vascular rupture in the brain parenchyma. Its incidence is relatively low, but only a part of patients could survive for more than 1 month, and most of them suffer from disability (Feigin et al., 2009; van Asch et al., 2010), which contribute to the high mortality of ICH. A large amount of studies have shown that the blood accumulates and flows into the surrounding brain and then compresses the tissue during ICH, which may be followed by multiple forms of CD, resulting collectively in neuronal death; at the same time, since the intracerebral hematoma releases hemoglobin (Hb)-based neurotoxin (Wang 2010), the neural damages secondary to perihematomal edema (PHE) that is caused by BBB disruption also occur in the brain parenchyma, becoming an important cause of morbidity and mortality in patients (Jeon et al., 2021).
The release of Hb is due to the accumulation and dissolution of iron-rich erythrocytes in the brain parenchyma, which consequently leads to iron toxicity and induces the production of ROS (Wu et al., 2011), interfering with normal function of cells and resulting in neuronal death (Zecca et al., 2004). Li et al. (Li et al., 2018) demonstrated for the first time that ferroptosis occurred in the collagenase-induced ICH mouse model, and ferroptosis was observed in neurons within the 1mm range of the edge from hematoma by transmission electron microscopy. Consistently, Zille et al. (Zille et al., 2017) also found that Hb could induce ferroptosis in primary cortical neurons. Ferroptosis is characterized by iron-dependent ROS accumulation and lipid peroxidation. The normal functions of antioxidant GPX4 and cystine transporter System Xc− are of great importance in controlling ferroptosis; while iron chelators, lipid peroxidation inhibitors and antioxidants can effectively inhibit the occurrence of ferroptosis. Iron deposition is a common indicator of ICH injury. Speer et al. (Speer et al., 2013) found that iron chelators were able to suppress ferroptosis by regulating hypoxia-inducible factor-1 (HIF-1) , thus playing a neuro-protective role in ICH. Chang et al. (Chang et al., 2014) showed that epicatechin could affect iron metabolism via regulating ferroptosis-related genes such as IREB2, protecting mice from ICH. In addition, GPX4 levels decreased in rat ICH models, while Fer-1, belonging to lipid peroxidation inhibitors, could block ferroptosis and improve secondary brain injury (Zhang et al., 2018). Similarly, mice treated with Lip-1 after ICH showed a more mild degree of neurologic deficits and less lesion volume (Li et al., 2017). N-acetyl cysteine (NAC) is an antioxidant that enhances GSH synthesis, increases cysteine level and System Xc− activity, thereby inhibiting ferroptosis in Hb-induced ICH model and improving the prognosis of ICH mice (Karuppagounder et al., 2018).
Based on the above evidence, we can draw the view that ferroptosis is related to the neuronal death after ICH; inhibition of ferroptosis can effectively attenuate secondary injury after ICH and play a neuro-protective role.
2.7.2 The Role of Ferroptosis in Subarachnoid Hemorrhage
Subarachnoid hemorrhage (SAH) is a common acute cerebrovascular event in clinic, and its survivors will experience severe neurological disability. As a devastating subtype of stroke, the morbidity and mortality rates of SAH are pretty high (Li Y. et al., 2021). In recent years, the pathophysiological mechanisms of early brain injury (EBI) after SAH have become the hotspot of experimental and clinical research, and EBI is the key factor of poor prognosis of SAH. EBI is a complex and multifactorial process involving reduced cerebral blood flow, BBB disruption, lipid peroxidation and other pathological events. These pathological events exacerbate the injury, and finally lead to neuronal death which in turn leads to neurological dysfunction (Yan et al., 2015).
There are many modalities of CD in EBI. Li et al. (Li Y. et al., 2021) found that inhibiting ferroptosis could ameliorate CD in the brain and neuronal death in the cell model by improving BBB permeability and cerebral edema, thus alleviating EBI after SAH. ACSL4 is able to induce ferroptosis, and increasing its expression would exacerbate inflammation, OS and cognitive deficits after SAH (Qu et al., 2021); however, over-expression of GPX4, could reduce the brain water content and improve neurological behavior by suppressing ferroptosis (Gao et al., 2020). Similarly, in in vivo experiments, Lip-1, a ferroptosis inhibitor, decreased neuronal death and neuro-inflammation in EBI by protecting mitochondrial functions and reducing lipid peroxidation, thereby ameliorating cerebral edema and neurological deficits after SAH (Cao et al., 2021). To sum up, ferroptosis is involved in EBI, and inhibition of ferroptosis can attenuate the degree of EBI and have a neuro-protective effect after SAH (Dangol et al., 2019).
3 The Mechanism of Ferroptosis
Studies have indicated that ferroptosis may be induced by different physiological conditions and pathological stress. Among these factors, the dynamic balance of iron and the abnormality of lipid peroxidation are the most important to induce ferroptosis, while the functional state of System Xc− and the activity level of GPX4 are the key pathways to regulate ferroptosis. The relevant mechanisms and implications will be described in detail below (Figure 3).
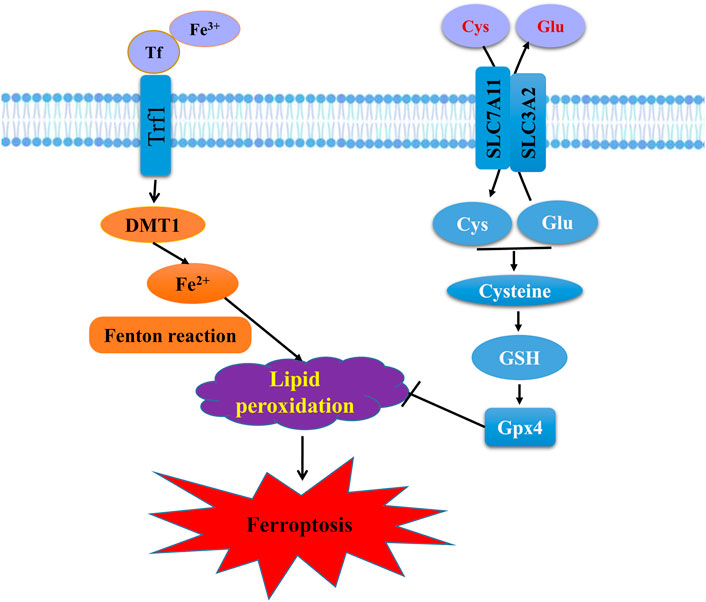
FIGURE 3. Schematic representation of the mechanism of ferroptosis. Ferroptosis is an iron-dependent form of regulated cell death mediated by iron overload and lipid peroxidation of cellular membranes. Fe3+ imported through the transferrin receptor is converted to Fe2+ in endosomes and released from endosome by divalent metal-ion transporter 1 (DMT1). Fenton reaction converts Fe2+ into Fe3+, which induces lipid peroxidation. GPX4 is the major endogenous mechanism to suppress lipid peroxidation. High extracellular concentrations of glutamate (Glu) inhibit system Xc−, which imports cystine (Cys) by exchanging intracellular Glu for extracellular Cys. Cys is subsequently converted to cysteine, which generates GSH, a cofactor for GPX4.
3.1 Inducing Factors of Ferroptosis
3.1.1 Iron (Fe2+) Metabolism
As an indispensable cofactor in many enzyme metabolism processes and a catalyst of redox-cycling reaction (Stoyanovsky et al., 2019), iron is involved in a variety of important physiological and biochemical processes in vivo. Studies have demonstrated that when iron accumulates in the body, it may be accompanied by OS, inflammation and CD (Masaldan et al., 2019). Since the homeostasis of iron in cells depends on the dynamic balance between its absorption, output and utilization, and thusly, the transport, metabolism and storage of iron should be strictly regulated. Excess iron would result in an increased production of ROS and induce ferroptosis (Kajarabille and Latunde-Dada 2019; Anandhan et al., 2020).
Over-expression of nuclear receptor coactivator 4 (NCOA4) or ATG protein can accelerate the degradation of ferritin, thus increase the level of free iron and aggravate the subsequent oxidative damage during ferroptosis (Liu J. et al., 2020). Traditional Chinese herbal extracts, such as artemisinin, also interfere with iron metabolism, increase Fe2+ concentration, and induce ferroptosis (Han et al., 2020). In contrast, Nrf2 is a stress-induced transcription factor responsible for maintaining cell metabolism, redox and protein balance, and controlling the expression of genes that counteract oxidative and electrophilic stress (Dodson et al., 2019). Once Nrf2 is activated, the storage of iron would increase, OS would be inhibited, and the occurrence of ferroptosis could be directly blocked.
3.1.2 Lipid Peroxidation
Lipid peroxidation refers to the oxidation and degradation of lipids. In this process, free radicals “steal” electrons from lipids in the cell membrane, thereby leading to cell damage. In living cells, the most common initiator of lipid peroxidation is ROS. The lethal accumulation of ROS is a sign of ferroptosis (Feng and Stockwell 2018; Hirschhorn and Stockwell 2019; Kajarabille and Latunde-Dada 2019). When the content of ROS is too high and completely destroys the redox dynamic balance, lipid peroxidation will be induced by non-enzymatic and enzymatic pathways, and then result in ferroptosis (Su et al., 2019). Therefore, the abundance and location of intracellular oxidizable substrates also determine the degree of lipid peroxidation and ferroptosis (Han et al., 2020).
Lipoxygenases and phosphorylase kinase G2 are two key drivers of lipid peroxidation (Yang et al., 2016), which can directly damage phospholipids and intrigue ferroptosis. FINO2 is an organic substance containing internal peroxides (Li J. et al., 2020). It promotes widespread lipid peroxidation by directly inducing iron oxide and indirectly inhibiting GPX4, and preferentially initiates ferroptosis (Gaschler et al., 2018; Zhang X. et al., 2020). Hmox-1 can also accelerate erastin-induced ferroptosis by generating ROS. In addition, artemisinin increases ROS levels, and FIN56 binds and activates squalene synthase to accumulate ROS (Kang et al., 2019), leading to the depletion of endogenous antioxidant CoQ10 and then inducing ferroptosis by lipid peroxidation. However, ferroptosis suppressor protein 1 (FSP1) has an inhibitory effect on ferroptosis by reducing CoQ10, inhibiting lipid peroxidation and ferroptosis terminal driver pLPO.
P53, as a gene encoding transcription factor that controls the initiation of cell cycle, has dual regulatory effects on lipid peroxidation. On the one side, p53 promotes the expression of SAT1 (Qiu et al., 2020), and then increased SAT1 cooperates with ROS to lead to lipid peroxidation (Murphy 2016), further oxidizes PUFAs, and finally causes ferroptosis; on the other side, p53 directly inhibits the activity of DPP4 or induces the expression of CDKN1A/p21, to make it difficult for NOX1 to interact with DPP4 to form complexes, thus alleviating lipid peroxidation and ferroptosis (Tao et al., 2020).
3.2 The Up-Stream Regulatory Mechanisms of Ferroptosis
3.2.1 System Xc−
The cystine-glutamate antiporter System Xc− is a disulfide-linked heterodimer composed of a light chain XCT and a heavy chain 4F2, namely, substrate-specific subunit SLC7A11 and regulatory subunit SLC3A2 (Song et al., 2018; Kajarabille and Latunde-Dada 2019; Qiu et al., 2020), with the ability to maintain cell reduction environment. System Xc−, the most up-stream node in the ferroptosis signaling cascade (Sato et al., 2018; Seibt et al., 2019; Stockwell et al., 2020), synthesizes antioxidant GSH by up-taking cystine and exchanging intracellular glutamate at a molar ratio of 1:1 and then reducing cystine to cysteine (Seibt et al., 2019; Stockwell et al., 2020). The administration of System Xc− inhibitors can deplete intracellular cysteine, reduce GSH concentration, trigger OS, and increase cell sensitivity to ferroptosis (Han et al., 2020).
Small molecule erastin can directly and selectively target and inhibit the System Xc−, and irreversibly bind to and inactivate XCT, leading to continuous disorder of cystine uptake, a decrease in cellular antioxidant capacity, and eventually ROS accumulation and ferroptosis (Sato et al., 2018; Kajarabille and Latunde-Dada 2019; Su et al., 2019). Similarly, BECN1 directly blocks the activity of System Xc− and initiates ferroptosis by binding to SLC7A11, the core component of System Xc−. P53 down-regulates the expression of SLC7A11 to reduce cystine uptake and enhance the cell sensitivity to ferroptosis.
As an important antioxidant defense system in vivo, pharmacological inhibition on System Xc− can promote the occurrence of lipid peroxidation and ferroptosis. Enhancing the stability of SLC7A11 can inhibit ferroptosis, while the destabilization of SLC7A11 can increase the sensitivity to ferroptosis (Liu et al., 2019). It has been proved to be a feasible treatment strategy that inhibiting the occurrence and development of ferroptosis by regulating the activity of System Xc− or the expression level of SLC7A11.
3.2.2 Glutathione Peroxidase 4
GPX4 is a selenoprotein enzyme, and its activity is the cornerstone of antioxidant defense (Ursini and Maiorino 2020). GPX4 can interrupt the lipid peroxidation chain reaction by reducing complex hydroperoxides to the corresponding counterparts, to quench lipid peroxidation (Seibt st al., 2019).
RAS-selective lethal 3 (RSL3) is the first described GPX4 inhibitor (Seibt et al., 2019). RSL3 binds to the nucleophilic active site of GPX4, and then reduces the expression level of GPX4 most effectively through chloroacetamide moiety (Jelinek et al., 2018), thus inactivating GPX4, producing lethal ROS accumulation and inducing ferroptosis. ACSL4 is a fatty acid activating enzyme expressed in the outer membrane of mitochondria and endoplasmic reticulum. Its over-expression reduces the activity of GPX4, resulting in increased release of lactic dehydrogenase, decreased cell viability and increased ferroptosis markers. Erastin indirectly blocks the function of GPX4 by depleting GSH, then intrigues ferroptosis. FIN56 promotes the degradation of GPX4 (Belavgeni et al., 2020), that leads to ferroptosis in cells.
Selenium is an essential trace element in mammals. On the one hand, it protects GPX4 from irreversible inactivation; on the other hand, it promotes transcriptional expression of GPX4, protecting cells from OS and inhibiting ferroptosis (Chen et al., 2020b). Nrf2 plays a role in alleviating lipid peroxidation and ferroptosis by regulating the biosynthesis of GSH and promoting the expression of GPX4.
Obviously, ferroptosis is a process regulated by multiple factors (Table 1) and involves many clinical applications. Seeking for valid targets concerning ferroptosis in CCVDs is of great importance.
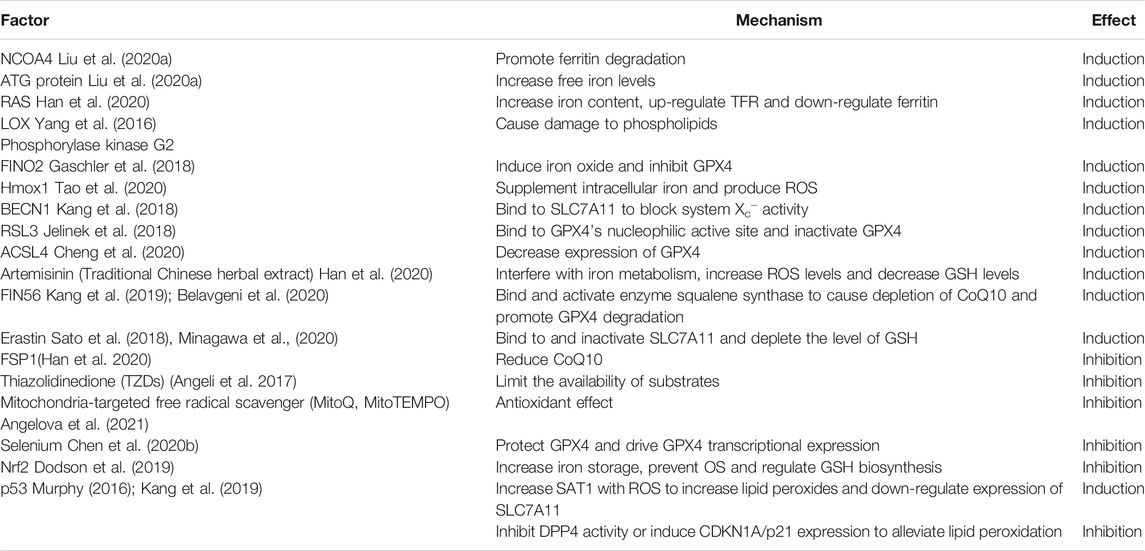
TABLE 1. Initiators and inhibitors of ferroptosis.
3.3 The Down-Stream Regulatory Mechanisms of Ferroptosis
3.3.1 Ferroptosis and Mitochondrion
Mitochondria are the most abundant organelles in cardiomyocytes, which are mainly responsible for energy metabolism (Li N. et al., 2020); meanwhile, cardiomyocyte as an important source of energy, if its mitochondrial function deteriorates, it would lead to myocardial I/R injury. The double membranes structure of mitochondria provides an ideal location for ferroptosis, and some researchers have observed mitochondrial impairment in patients with ferroptosis (Sha et al., 2021). After ferroptosis, excess intracellular free iron can enter the mitochondria, resulting in smaller myocardial mitochondria, increased mitochondrial membrane density, decreased mitochondrial respiration, and mitochondrial membrane potential depolarization (Abdalkader et al., 2018; Sumneang et al., 2020), thus inducing CD and a series of Cardiac Diseases such as arrhythmia (Hu et al., 2021). A recent study shows that if ferroptosis is suppressed, the production of ROS in mitochondria would be reduced, which can save the toxicity caused by inhibition of mitochondrial complex I (Basit et al., 2017) and effectively prevent cardiac ischemic damage. Ferroptosis inhibitor Lip-1 can reduce the level of VDAC1 and mitochondrial ROS in myocardium, to protect the integrity of mitochondrial structure and exert the myocardial protective effect against I/R injury, reducing the myocardial infarct size (Li N. et al., 2021; Wu et al., 2021a). The above findings suggest that ferroptosis plays a vital role in the pathology of CD through mitochondrial function deteriorates and contributes to the development of Cardiac Diseases.
3.3.2 Ferroptosis and Endoplasmic Reticulum Stress
Ferroptosis can also participate in myocardial I/R injury by modulating ERS (Li N. et al., 2020). On the one hand, it has been proved that the occurrence of ferroptosis is accompanied by the production of ERS (Wang et al., 2019), and ferroptosis can also lead to ERS via cystine-glutamate antiporter System Xc− (Sun et al., 2015); on the other hand, ERS induces apoptosis through the activation of three up-stream signaling proteins: inositol requiring protein-1, activating transcription factor-6 and protein kinase RNA (PKR)-like ER kinase. Li et al. (Li W. et al., 2020) found that inhibition of ferroptosis could reduce ERS and mitigate myocardial damage in diabetic rats with I/R injury. It is noteworthy that compared with ERS agonist alone, the combination of ferroptosis inhibitor and ERS agonist can significantly reduce the incidence of ERS and cardiomyocyte injury, while ferroptosis agonist can aggravate cardiac dysfunction. Apparently, ferroptosis promotes ERS and cardiomyocytes injury, and aggravates the progression of Cardiac Diseases.
3.3.3 Ferroptosis and Oxidative Stress
As is well known that OS is induced by the imbalance in the redox state. Accumulating evidence suggests that the regulation of ferroptosis can change the level of OS in cardiomyocytes or vascular cells, resulting in cardiomyocyte injury, cardiac hypertrophy and HF through the interaction between ROS with ERS (Kamoshita et al., 2014; Li W. et al., 2020; Li N. et al., 2021).
In summary, CD is pivotal for internal homeostasis and prevention of CCVDs (Duan et al., 2021). Ferroptosis is accompanied by iron overload, excess lipid peroxidation and a large amount of ROS production, which can accelerate a collection of pathological processes such as inflammation, mitochondrial dysfunction, ERS, and OS that further lead to the increase of CD and damage to the normal function of cardio-cerebral vessels.
3.3.4 Ferroptosis and Inflammation
Inflammation is a basic pathological process including infection and tissue damage, and appears when the organism is stimulated by some specific physiological or pathological factors. Different from other types of RCD, ferroptosis is immunogenic (Sun et al., 2020).
It has been reported that when ferroptosis occurs in neurons, then microglia are activated and release toxic substances including pro-inflammatory factor IL-6, which can cause neuro-inflammation and lead to further brain damage (Tang et al., 2020). In addition, ferroptosis also changes the inflammatory phenotype in macrophages (Ouyang et al., 2021), which promotes macrophage polarization and then aggravates inflammation (Yu et al., 2021). Ferroptosis is capable of directly releasing the pro-inflammatory damaged-associated molecular patterns (DAMPs) that promote the development of inflammation and numerous inflammatory diseases. Similarly, after the death of cells that have experienced ferroptosis, intracellular components are also released in the form of DAMPs upon plasma membrane rupture, triggering the immune system to recruit neutrophils to the injured myocardium (Hu et al., 2021). Once there exists an accumulation of DAMPs, it would trigger the amplification of tissue inflammation, leading to more severe inflammation and CD, forming a vicious circle and disrupting the normal physiological function of tissues and organs (Linkermann et al., 2014; Land et al., 2016). Ferroptosis inhibitor Fer-1 can not only inhibit the infiltration of macrophages (Li N. et al., 2020), but also inhibit the adhesion of neutrophils to coronary vascular endothelial cells (Li N. et al., 2021), effectively reducing inflammation and dysfunction in heart injury.
In a word, as a crucial defensive response in vivo, inflammation is closely associated with CD induced by ferroptosis. As mentioned previously, inflammation can induce myocardial hypertrophy and fibrosis during cardiac injury, destroy BBB and lead to neurological defects in the ischemic region, promoting the progress of CCVDs. To conclude, iron metabolism and lipid peroxidation contribute to the occurrence of ferroptosis, and eventually up-regulate inflammation. Thus, it can be said that inflammation is an important intermediate process that can be regulated by ferroptosis. In the end, ferroptosis-induced inflammation would trigger or even aggravate development of CCVDs.
4 Advances in Drug Application to Improve Cardio-Cerebrovascular by Interfering Ferroptosis
At present, the World Health Organization is committed to the global prevention, management and monitoring of CCVDs, to identify potential mechanisms and effective therapeutic drugs, in order to reduce the incidence, prevalence and mortality of CCVDs (Zhang X. et al., 2020). As one of the results of the imbalance of lipid peroxidation and antioxidant system, ferroptosis has high clinical value in treatment of CCVDs.
For the prevention and control of ferroptosis, we focus on the two main targets in the process of its occurrence and development: iron metabolism and lipid peroxidation. We will concentrate on the drugs or inhibitors acting on the above targets to provide the possibility for the treatment of clinically related CCVDs (Table 2).
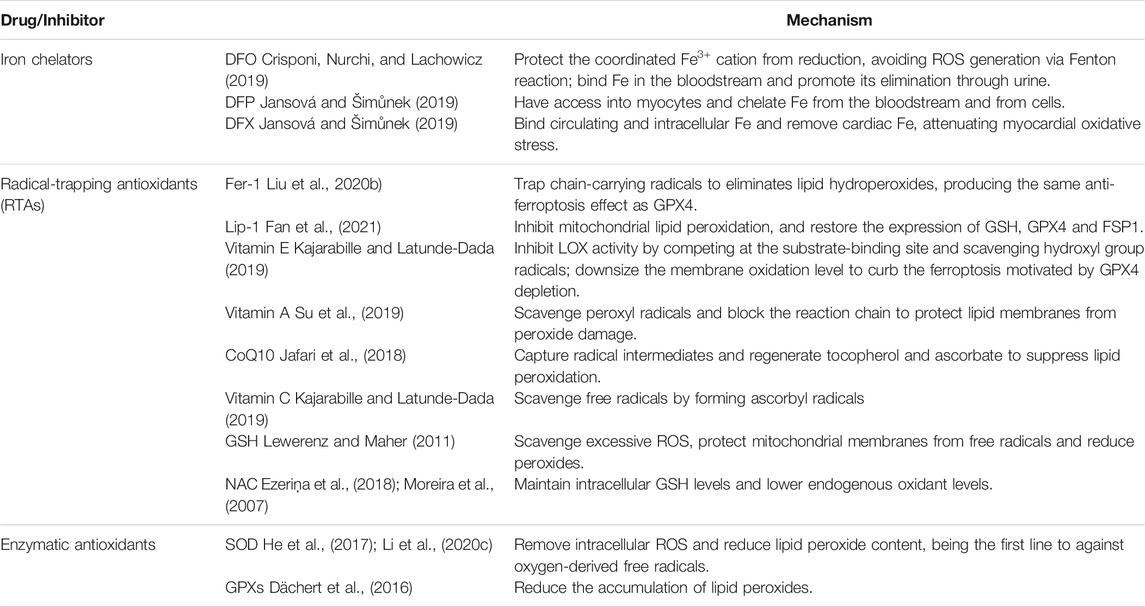
TABLE 2. Treatment strategy of ferroptosis in cardio-cerebrovascular diseases.
4.1 Iron chelators
Iron chelator has the ability to become a stable complex through strong binding with iron ion, thus preventing too much free iron ion from participating in redox reaction in cells to produce ROS, initiate lipid peroxidation, cause cell oxidative damage and trigger ferroptosis. Iron chelators are often used to halt Fenton reaction and related lipid peroxidation, and have been demonstrated to improve some pathological contexts (Conrad et al., 2020).
Currently, there are three specific iron chelators, namely Deferoxamine (DFO), Deferiprone (DFP) and Deferasirox (DFX) (Jansová and Šimůnek 2019).
4.2 Deferoxamine
DFO can be used to treat systemic iron overload and acute iron poisoning (Jansová and Šimůnek 2019). By combining free iron in a complex, DFO protects Fe3+ from reduction and avoids the formation of ROS through Fenton reaction (Crisponi et al., 2019). DFO can effectively reduce the pathological deposition of iron in organs and protect the heart against I/R injury in ex vivo perfused hearts (Kobayashi et al., 2018).
4.3 Deferiprone
DFP is an oral iron chelator with high affinity for iron ions. The lipophilicity and low molecular weight of DFP enable it to cross the cellular membranes and gain access into cardiomyocytes (Crisponi et al., 2019), chelating iron from cells (Jansová and Šimůnek 2019). DFP can improve cardiac contractile function and reduce lipid peroxidation and lactate dehydrogenase release. Two in vitro studies have shown that DFP can protect neonatal rat cardiomyocytes from cardiotoxicity induced by doxorubicin (Jansová and Šimůnek 2019).
4.3 Deferasirox
DFX was the second oral chelator available for clinic use with high oral efficacy and tolerability. It has a high selectivity for Fe3+ (Crisponi et al., 2019). It binds circulating iron and intracellular iron, stabilizing the level of iron in cardiac tissue, also clearing cardiac iron and reducing myocardial oxidative stress in Fe overload in vitro (Jansová and Šimůnek 2019). The administration of DFX on cardiomyocytes in the early phase of reperfusion can effectively reduce the severity of reperfusion injury (Ravingerová et al., 2020). A Study has shown that DFX improved the severity of acute myocardial infarction, with comparative effects (Nishizawa et al., 2020). Also, DFX can alleviate the lipid peroxidation and endothelial dysfunction in MAECs, inhibiting the occurrence of ferroptosis, and then improving AS lesion (Bai et al., 2020).
4.4 Radical-Trapping Antioxidants
RTAs, reacts with chain-carrying peroxyl radicals to produce non-reactive free radicals (Li and Pratt 2015), which destroys the autoxidation of chain-propagating peroxyl radicals (Kajarabille and Latunde-Dada 2019). It is an inhibitor at the initial stage of free radical formation and a chain breaker at the stage of radical breakage and propagation.
RTAs can be divided into lipophilic and hydrophilic free radical scavengers. Ferrostatin-1 (Fer-1), Liproxstatin-1 (Lip-1), and Coenzyme Q10 (CoQ10) are lipophilic; N-acetyl cysteine (NAC) is hydrophilic.
4.5 Ferrostatin-1
Fer-1 acts as a lipophilic free radical scavenger due to its redox cycling activity (Doll and Conrad 2017), and alleviates ferroptosis by inhibiting lipid peroxidation and preventing GPX4 deletion or GSH depletion (Zilka et al., 2017; Liu P. et al., 2020). Zille et al. found that Fer-1 was able to rescue mouse primary cortical neurons from CD induced by hemoglobin chloride and hemoglobin in vitro (Weiland et al., 2019). Ahmad et al. found that Fer-1 prevented reperfusion injury in mice (Cui et al., 2021); similarly, Tuo et al. found that Fer-1 also protected neurons against cerebral ischemic injury in mouse models (Wu et al., 2018). Moreover, Fer-1 could alleviate AS lesion in high-fat diet (HFD)-fed mice model and improve the viability of MAECs treated with ox-LDL (Bai et al., 2020).
4.6 Liproxstatin-1
Lip-1 is a superior antioxidant for capturing free radicals. Lip-1 not only suppresses mitochondrial lipid peroxidation and reduces ROS generation, but also restores GSH, GPX4, and FSP1 levels (Wu et al., 2021a; Fan et al., 2021). Therefore, Lip-1 inhibited ferroptosis with effect in the mice model induced by GPX4 depletion (Doll and Conrad 2017). In addition, a study found that administration of Lip-1 partially rescued neuronal survival and microglia activation, improving neurological function (Cui et al., 2021). There is a study indicated that Lip-1 preserved the expression of GPX4, decreased the expression of ACSL4 and attenuated neuronal injury via inhibition of ferroptosis (Cao et al., 2021).
4.7 Coenzyme Q10
CoQ10 is an endogenous antioxidant, a membrane stabiliser and a cofactor. CoQ10 is able to suppress ferroptosis via inhibition of lipid peroxidation by capturing radical intermediates outside of mitochondria (Stockwell et al., 2020). Numerous studies have investigated that CoQ10 increased energy production by mediating electron transfer in the electron transport chain and stabilized myocardial calcium-dependent ion channels to improve contractility of cardiac muscle (Madmani et al., 2014; Jafari et al., 2018); it prevented low-density lipoproteins (LDL) oxidation to maintain normal cardiovascular and cardiac diastolic function, reducing cardiovascular adverse events (Garrido-Maraver et al., 2014); it also reduced OS to prevent lipid peroxidation on cardiomyocyte membrane and inhibit ferroptosis in cardiomyocytes. In CCVDs, CoQ10 could directly act on ECs to stimulate vasodilation and lower blood pressure; it could reduce left ventricular diastolic dysfunction caused by diabetes; it also slows down the process of ferroptosis in cardiomyocytes during ischemia period to alleviate MI and HF (Zozina et al., 2018); it reduces the release of neurohormonal or diminishes neuro-inflammation to improve the prognosis of IS as well (Yang et al., 2015).
4.8 N-Acetyl Cysteine
NAC is a precursor of GSH synthesis and an antioxidant that scavenges free radicals and oxidants (Yesilbursa et al., 2006; Ezeriņa et al., 2018), protecting cells from free radical injury, inflammation and ferroptosis. It is reported that NAC protected neurons under ischemic stimulation by preserving mitochondrial function and reducing OS (Moreira et al., 2007). Maryam et al. found that the administration of NAC early after acute ischemic stroke improved neurological impairment and prognosis (Sabetghadam et al., 2020); and ischemic animal model pretreated with NAC showed a decrease in the volume of cerebral infarction, a decrease in neuronal cell death and an improvement in neurological function (Zhang et al., 2014). NAC may effectively reduce cardiac iron level, exert antioxidant and anti-inflammatory effects, improving cardiac and cognitive functions (Sumneang et al., 2019). Study by Xiao et al. has demonstrated that in the model of HF, NAC enhanced antioxidant capacity in vivo and reduce cardiomyocyte cell death (Wu et al., 2014). César et al. found that treatment with NAC after MI effectively reduced arrhythmia, cardiac remodeling and infarction expansion (Costa et al., 2020). Furthermore, studies by Dilek and Azita et al. manifested that NAC significantly alleviated OS and I/R injury in patients with MI, improved left ventricular systolic function and attenuated cardiac remodeling (Yesilbursa et al., 2006; Talasaz et al., 2014).
With the deepening of research, ferroptosis has been found in more and more CCVDs. It is of great theoretical significance and practical value to clarify the mechanism of ferroptosis and its role in various diseases, and to develop corresponding inhibitory drugs according to different stages and targets of ferroptosis. This is also the future direction of ferroptosis research (Li J. et al., 2020).
5 Conclusion and Prospect
Globally, CCVDs are the major causes of death and disability. Understanding the pathological process of myocardial and neural cell injury is key to developing protective strategies for CCVDs. Iron overload has been found to play a pathogenic role in cardio-cerebrovascular toxicity both in animal models and in patients with different CCVDs. Ferroptosis, an iron-dependent form of regulated cell death, has received increasing attention and has been associated with the pathology of CCVDs. Many studies have shown that ferroptosis occurs in AS, MI, HF, DCM, hypertensive brain injury, and IS. Inhibitors of ferroptosis may prevent these diseases by inhibiting ferroptosis in cardiac and neuronal cells.
Cardio-cerebrovascular cell death is a fundamental pathological process and ferroptosis occurs during all periods of CCVDs. Although reduced levels of ferroptosis inhibitors (e.g. GSH, GPX4, and Nrf2) and changes in the expression of several other genes are known to occur during CCVDs, current assays are not suitable for routine clinical diagnosis. The molecular mechanisms that lead to ferroptosis in CCVDs are still largely unknown. One of the main features of ferroptosis is impaired mitochondrial structure, but it is not easy to observe changes in mitochondria. In the last few years, an increasing number of targets that induce or inhibit ferroptosis have been uncovered, but which of them provides the best therapeutic effect, which can be used in animal models or patients, and which can be easily handled chemically are still unclear. Although research data on ferroptosis are becoming more detailed and complete, most studies have only revealed the phenomenon of ferroptosis in CCVDs, not its specific mechanisms, location, and stages. More importantly, do other types of RCD have an effect on ferroptosis, and if so, is it synergistic or antagonistic? Moreover, although ferroptosis leads to pathological changes such as inflammation and ERS, in many cases, they actually interact with each other. It is not clear which one is in the absolute up-stream or down-stream. Therefore, it is more difficult to elucidate the complete pathway through which ferroptosis acts. For this reason, we must continue to explore the different roles of ferroptosis in different diseases in order to find efficient and targeted therapeutic approaches. We need to clarify the specific pathways of ferroptosis and figure out whether ferroptosis could act on other cell types. Moreover, when does ferroptosis exert its maximum effect after organism damage? Are there any other factors that could induce ferroptosis except for iron overload and lipid peroxidation? What are the specific effects of different ferroptosis inhibitors? Is combined use of drugs more effective than single use? These are fields that we need to study in the future. Therefore, there is still a long way to go before the findings based on animal or cellular experiments are finally applied in clinical practice.
In summary, ferroptosis is closely associated with the pathogenesis of various CCVDs, and with continued research, inhibition of ferroptosis to prevent cardio-cerebrovascular cell death has the potential to be an effective strategy for the treatment of CCVDs.
Author Contributions
A-PW and YT contributed to the conception of the study; A-PW, YT, S-XG, and NL contributed significantly to the analysis and manuscript preparation; M-YL, J-HS, and A-PW wrote the manuscript; W-QH and WC helped perform the analysis with constructive discussions. All authors have approved it for publication.
Funding
This project was financed by the National Natural Science Foundation of China (grant number 31871169 to YT, 81600040 to A-PW), Innovation guidance project of Hu Nan (grant number 2018SK51606 to S-XG, 2018SK51703 to NL), the province Natural Science Foundation of Hu Nan (grant number 2020JJ8032 to YT, 2021JJ30601 to A-PW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdalkader, M., Lampinen, R., Kanninen, K. M., Malm, T. M., and Liddell, J. R. (2018). Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 12, 466. doi:10.3389/fnins.2018.00466
Abdelwahid, E., Stulpinas, A., and Kalvelyte, A. (2017). Effective Agents Targeting the Mitochondria and Apoptosis to Protect the Heart. Cpd 23 (8), 1153–1166. doi:10.2174/1381612822666161229150120
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 177 (5), 1262–1279. e25. doi:10.1016/j.cell.2019.03.032
Anandhan, A., Dodson, M., Schmidlin, C. J., Liu, P., and Zhang, D. D. (2020). Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cel Chem. Biol. 27 (4), 436–447. doi:10.1016/j.chembiol.2020.03.011
Angeli, J. P. F., Shah, R., Pratt, D. A., and Conrad, M. (2017). Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacological Sciences 38 (5), 489–498. doi:10.1016/j.tips.2017.02.005
Angelova, P. R., Esteras, N., and Abramov, A. Y. (2021). Mitochondria and Lipid Peroxidation in the Mechanism of Neurodegeneration: Finding Ways for Prevention. Med. Res. Rev. 41 (2), 770–784. doi:10.1002/med.21712
Baba, Y., Higa, J. K., Shimada, B. K., Horiuchi, K. M., Suhara, T., Kobayashi, M., et al. (2018). Protective Effects of the Mechanistic Target of Rapamycin against Excess Iron and Ferroptosis in Cardiomyocytes. Am. J. Physiology-Heart Circulatory Physiol. 314 (3), H659–H668. doi:10.1152/ajpheart.00452.2017
Bai, T., Li, M., Liu, Y., Qiao, Z., and Wang, Z. (2020). Inhibition of Ferroptosis Alleviates Atherosclerosis through Attenuating Lipid Peroxidation and Endothelial Dysfunction in Mouse Aortic Endothelial Cell. Free Radic. Biol. Med. 160, 92–102. doi:10.1016/j.freeradbiomed.2020.07.026
Basit, F., van Oppen, L. M., Schöckel, L., Bossenbroek, H. M., van Emst-de Vries, S. E., Hermeling, J. C., et al. (2017). Mitochondrial Complex I Inhibition Triggers a Mitophagy-dependent ROS Increase Leading to Necroptosis and Ferroptosis in Melanoma Cells. Cell Death Dis 8 (3), e2716. doi:10.1038/cddis.2017.133
Bayır, H., Anthonymuthu, T. S., Tyurina, Y. Y., Patel, S. J., Amoscato, A. A., Lamade, A. M., et al. (2020). Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cel Chem. Biol. 27 (4), 387–408. doi:10.1016/j.chembiol.2020.03.014
Belavgeni, A., Meyer, C., Stumpf, J., Hugo, C., and Linkermann, A. (2020). Ferroptosis and Necroptosis in the Kidney. Cel Chem. Biol. 27 (4), 448–462. doi:10.1016/j.chembiol.2020.03.016
Bi, Y., Ajoolabady, A., Demillard, L. J., Yu, W., Hilaire, M. L., Zhang, Y., et al. (2021). Dysregulation of Iron Metabolism in Cardiovascular Diseases: from Iron Deficiency to Iron Overload. Biochem. Pharmacol. 190, 114661. doi:10.1016/j.bcp.2021.114661
Bugger, H., and Abel, E. D. (2014). Molecular Mechanisms of Diabetic Cardiomyopathy. Diabetologia 57 (4), 660–671. doi:10.1007/s00125-014-3171-6
Cai, L., and Kang, Y. J. (2003). Cell Death and Diabetic Cardiomyopathy. Ct 3 (3), 219–228. doi:10.1385/ct:3:3:219
Cao, J. Y., and Dixon, S. J. (2016). Mechanisms of Ferroptosis. Cell. Mol. Life Sci. 73, 2195–2209. doi:10.1007/s00018-016-2194-1
Cao, Y., Li, Y., He, C., Yan, F., Li, J.-R., Xu, H.-Z., et al. (2021). Selective Ferroptosis Inhibitor Liproxstatin-1 Attenuates Neurological Deficits and Neuroinflammation after Subarachnoid Hemorrhage. Neurosci. Bull. 37 (4), 535–549. doi:10.1007/s12264-020-00620-5
Chang, C. F., Cho, S., and Wang, J. (2014). (‐)‐Epicatechin Protects Hemorrhagic Brain via Synergistic Nrf2 Pathways. Ann. Clin. Transl Neurol. 1 (4), 258–271. doi:10.1002/acn3.54
Chen, J., Zhang, Z., and Cai, L. (2014). Diabetic Cardiomyopathy and its Prevention by Nrf2: Current Status. Diabetes Metab. J. 38 (5), 337–345. doi:10.4093/dmj.2014.38.5.337
Chen, X., Xu, S., Zhao, C., and Liu, B. (2019). Role of TLR4/NADPH Oxidase 4 Pathway in Promoting Cell Death through Autophagy and Ferroptosis during Heart Failure. Biochem. biophysical Res. Commun. 516 (1), 37–43. doi:10.1016/j.bbrc.2019.06.015
Chen, Y., Hua, Y., Li, X., Arslan, I. M., Zhang, W., and Meng, G. (2020a). Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front. Pharmacol. 11, 42. doi:10.3389/fphar.2020.00042
Chen, Y., Liu, S., Li, J., Li, Z., Quan, J., Liu, X., et al. (2020b). The Latest View on the Mechanism of Ferroptosis and its Research Progress in Spinal Cord Injury. Oxidative Med. Cell. longevity 2020, 1–11. doi:10.1155/2020/6375938
Cheng, J., Fan, Y. Q., Liu, B. H., Zhou, H., Wang, J. M., and Chen, Q. X. (2020). ACSL4 Suppresses Glioma Cells Proliferation via Activating Ferroptosis. Oncol. Rep. 43 (1), 147–158. doi:10.3892/or.2019.7419
Chien, C.-Y., Wen, T.-J., Cheng, Y.-H., Tsai, Y.-T., Chiang, C.-Y., and Chien, C.-T. (2020). Diabetes Upregulates Oxidative Stress and Downregulates Cardiac Protection to Exacerbate Myocardial Ischemia/Reperfusion Injury in Rats. Antioxidants 9 (8), 679. doi:10.3390/antiox9080679
Conrad, M., Lorenz, S. M., and Proneth, B. (2021). Targeting Ferroptosis: New Hope for As-Yet-Incurable Diseases. Trends Molecular Medicine 27, 113–122. doi:10.1016/j.molmed.2020.08.010
Costa, C. R. M., Seara, F. A. C., Peixoto, M. S., Ramos, I. P., Barbosa, R. A. Q., Carvalho, A. B., et al. (2020). Progression of Heart Failure Is Attenuated by Antioxidant Therapy with N-Acetylcysteine in Myocardial Infarcted Female Rats. Mol. Biol. Rep. 47 (11), 8645–8656. doi:10.1007/s11033-020-05907-4
Crisponi, G., Nurchi, V. M., and Lachowicz, J. I. (2019). Iron Chelation for Iron Overload in Thalassemia. Metal ions Life Sci. 19. doi:10.1515/9783110527872-009
Cui, Y., Zhang, Y., Zhao, X., Shao, L., Liu, G., Sun, C., et al. (2021). ACSL4 Exacerbates Ischemic Stroke by Promoting Ferroptosis-Induced Brain Injury and Neuroinflammation. Brain Behav. Immun. 93, 312–321. doi:10.1016/j.bbi.2021.01.003
Dächert, J., Schoeneberger, H., Rohde, K., and Fulda, S. (2016). RSL3 and Erastin Differentially Regulate Redox Signaling to Promote Smac Mimetic-Induced Cell Death. Oncotarget 7 (39), 63779–63792. doi:10.18632/oncotarget.11687
Dalen, J. E., Alpert, J. S., Goldberg, R. J., and Weinstein, R. S. (2014). The Epidemic of the 20th Century: Coronary Heart Disease. Am. J. Med. 127 (9), 807–812. doi:10.1016/j.amjmed.2014.04.015
Dangol, S., Chen, Y., Hwang, B. K., and Jwa, N.-S. (2019). Iron- and Reactive Oxygen Species-dependent Ferroptotic Cell Death in Rice-Magnaporthe Oryzae Interactions. Plant Cell 31 (1), 189–209. doi:10.1105/tpc.18.00535
Datta, A., Sarmah, D., Mounica, L., Kaur, H., Kesharwani, R., Verma, G., et al. (2020). Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 11 (6), 1185–1202. doi:10.1007/s12975-020-00806-z
De Blasio, M. J., Huynh, N., Deo, M., Dubrana, L. E., Walsh, J., Willis, A., et al. (2020). Defining the Progression of Diabetic Cardiomyopathy in a Mouse Model of Type 1 Diabetes. Front. Physiol. 11, 124. doi:10.3389/fphys.2020.00124
DeGregorio-Rocasolano, N., Martí-Sistac, O., and Gasull, T. (2019). Deciphering the Iron Side of Stroke: Neurodegeneration at the Crossroads between Iron Dyshomeostasis, Excitotoxicity, and Ferroptosis. Front. Neurosci. 13, 85. doi:10.3389/fnins.2019.00085
Del Re, D. P., Amgalan, D., Linkermann, A., Liu, Q., and Kitsis, R. N. (2019). Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 99 (4), 1765–1817. doi:10.1152/physrev.00022.2018
DeVon, H. A., Vuckovic, K., Ryan, C. J., Barnason, S., Zerwic, J. J., Pozehl, B., et al. (2017). Systematic Review of Symptom Clusters in Cardiovascular Disease. Eur. J. Cardiovasc. Nurs. 16 (1), 6–17. doi:10.1177/1474515116642594
Dillmann, W. H. (2019). Diabetic Cardiomyopathy. Circ. Res. 124 (8), 1160–1162. doi:10.1161/circresaha.118.314665
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an Iron-dependent Form of Nonapoptotic Cell Death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dodson, M., Castro-Portuguez, R., and Zhang, D. D. (2019). NRF2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol. 23, 101107. doi:10.1016/j.redox.2019.101107
Doll, S., and Conrad, M. (2017). Iron and Ferroptosis: A Still Ill‐defined Liaison. IUBMB life 69 (6), 423–434. doi:10.1002/iub.1616
Dringen, R. (2000). Metabolism and Functions of Glutathione in Brain. Prog. Neurobiol. 62 (6), 649–671. doi:10.1016/s0301-0082(99)00060-x
Duan, J.-Y., Lin, X., Xu, F., Shan, S.-K., Guo, B., Li, F.-X.-Z., et al. (2021). Ferroptosis and its Potential Role in Metabolic Diseases: A Curse or Revitalization? Front. Cel Dev. Biol. 9, 701788. doi:10.3389/fcell.2021.701788
Dumitrescu, L., Popescu-Olaru, I., Cozma, L., Tulbă, D., Hinescu, M. E., Ceafalan, L. C., et al. (20182018). Oxidative Stress and the Microbiota-Gut-Brain Axis. Oxidative Med. Cell. longevity 2018, 1–12. doi:10.1155/2018/2406594
Ezeriņa, D., Takano, Y. K. Hanaoka., Urano, Y., and Dick, T. P. (2018). N-acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular HS and Sulfane Sulfur Production. Cell Chem. Biol. 25 (4), 447–459.e4. doi:10.1016/j.chembiol.2018.01.011
Fang, X., Cai, Z., Wang, H., Han, D., Cheng, Q., Zhang, P., et al. (2020). Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 127 (4), 486–501. doi:10.1161/circresaha.120.316509
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a Target for protection against Cardiomyopathy. Proc. Natl. Acad. Sci. USA 116 (7), 2672–2680. doi:10.1073/pnas.1821022116
Favaloro, B., Allocati, N., Graziano, V., Di Ilio, C., and De Laurenzi, V. (2012). Role of Apoptosis in Disease. Aging 4 (5), 330–349. doi:10.18632/aging.100459
Feigin, V. L., Lawes, C. M., Bennett, D. A., Barker-Collo, S. L., and Parag, V. (2009). Worldwide Stroke Incidence and Early Case Fatality Reported in 56 Population-Based Studies: a Systematic Review. Lancet Neurol. 8 (4), 355–369. doi:10.1016/s1474-4422(09)70025-0
Feng, H., and Stockwell, B. R. (2018). Unsolved Mysteries: How Does Lipid Peroxidation Cause Ferroptosis. Plos Biol. 16 (5), e2006203. doi:10.1371/journal.pbio.2006203
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25 (3), 486–541. doi:10.1038/s41418-017-0012-4
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., et al. (2012). Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cel Death Differ 19 (1), 107–120. doi:10.1038/cdd.2011.96
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cel. 59 (2), 298–308. doi:10.1016/j.molcel.2015.06.011
Gao, S.-Q., Liu, J.-Q., Han, Y.-L., Deji, Q.-Z., Zhaba, W.-D., Deng, H.-J., et al. (2020). Neuroprotective Role of Glutathione Peroxidase 4 in Experimental Subarachnoid Hemorrhage Models. Life Sci. 257, 118050. doi:10.1016/j.lfs.2020.118050
Garrido-Maraver, J., Cordero, M. D., Oropesa-Avila, M., Vega, A. F., Mata, M. D. L., Pavon, A. D., et al. (2014). Clinical Applications of Coenzyme Q₁₀. Front. Biosci. 19, 619–633. doi:10.2741/4231
Gaschler, M. M., Andia, A. A., Liu, H., Csuka, J. M., Hurlocker, B., Vaiana, C. A., et al. (2018). FINO2 Initiates Ferroptosis through GPX4 Inactivation and Iron Oxidation. Nat. Chem. Biol. 14 (5), 507–515. doi:10.1038/s41589-018-0031-6
GBD 2013 Mortality and Causes of Death Collaborators (2015). Global, Regional, and National Age-Sex Specific All-Cause and Cause-specific Mortality for 240 Causes of Death, 1990-2013: a Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 385 (9963), 117–171. doi:10.1016/s0140-6736(14)61682-2
Gimbrone, M. A., and García-Cardeña, G. (2016). Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 118 (4), 620–636. doi:10.1161/circresaha.115.306301
Gorrini, C., Harris, I. S., and Mak, T. W. (2013). Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 12 (12), 931–947. doi:10.1038/nrd4002
Hammad, S. M., Twal, W. O., Barth, J. L., Smith, K. J., Saad, A. F., Virella, G., et al. (2009). Oxidized LDL Immune Complexes and Oxidized LDL Differentially Affect the Expression of Genes Involved with Inflammation and Survival in Human U937 Monocytic Cells. Atherosclerosis 202 (2), 394–404. doi:10.1016/j.atherosclerosis.2008.05.032
Han, C., Liu, Y., Dai, R., Ismail, N., Su, W., and Li, B. (2020). Ferroptosis and its Potential Role in Human Diseases. Front. Pharmacol. 11, 239. doi:10.3389/fphar.2020.00239
Hirschhorn, T., and Stockwell, B. R. (2019). The Development of the Concept of Ferroptosis. Free Radic. Biol. Med. 133, 130–143. doi:10.1016/j.freeradbiomed.2018.09.043
Hu, H., Chen, Y., Jing, L., Zhai, C., and Shen, L. (2021). The Link between Ferroptosis and Cardiovascular Diseases: A Novel Target for Treatment. Front. Cardiovasc. Med. 8, 710963. doi:10.3389/fcvm.2021.710963
Hu, X., Rajesh, M., Zhang, J., Zhou, S., Wang, S., Sun, J., et al. (2018). Protection by Dimethyl Fumarate against Diabetic Cardiomyopathy in Type 1 Diabetic Mice Likely via Activation of Nuclear Factor Erythroid-2 Related Factor 2. Toxicol. Lett. 287, 131–141. doi:10.1016/j.toxlet.2018.01.020
Huang, S., and Frangogiannis, N. G. (2018). Anti‐inflammatory Therapies in Myocardial Infarction: Failures, Hopes and Challenges. Br. J. Pharmacol. 175 (9), 1377–1400. doi:10.1111/bph.14155
Iadecola, C., and Anrather, J. (2011). The Immunology of Stroke: from Mechanisms to Translation. Nat. Med. 17 (7), 796–808. doi:10.1038/nm.2399
Jafari, M., Mousavi, S. M., Asgharzadeh, A., and Yazdani, N. (2018). Coenzyme Q10 in the Treatment of Heart Failure: A Systematic Review of Systematic Reviews. Indian Heart J. 70, S111–S117. doi:10.1016/j.ihj.2018.01.031
Jamnongkan, W., Thanan, R., Techasen, A., Namwat, N., Loilome, W., Intarawichian, P., et al. (2017). Upregulation of Transferrin Receptor-1 Induces Cholangiocarcinoma Progression via Induction of Labile Iron Pool. Tumour Biol. 39 (7), 101042831771765. doi:10.1177/1010428317717655
Jansová, H., and Šimůnek, T. (2019). Cardioprotective Potential of Iron Chelators and Prochelators. Cmc 26 (2), 288–301. doi:10.2174/0929867324666170920155439
Jelinek, A., Heyder, L., Daude, M., Plessner, M., Krippner, S., Grosse, R., et al. (2018). Mitochondrial rescue Prevents Glutathione Peroxidase-dependent Ferroptosis. Free Radic. Biol. Med. 117, 45–57. doi:10.1016/j.freeradbiomed.2018.01.019
Jeon, H., Kim, M., Park, W., Lim, J. S., Lee, E., Cha, H., et al. (2021). Upregulation of AQP4 Improves Blood-Brain Barrier Integrity and Perihematomal Edema Following Intracerebral Hemorrhage. Neurotherapeutics. doi:10.1007/s13311-021-01126-2
Jin, R., Liu, L., Zhang, S., Nanda, A., and Li, G. (2013). Role of Inflammation and its Mediators in Acute Ischemic Stroke. J. Cardiovasc. Trans. Res. 6 (5), 834–851. doi:10.1007/s12265-013-9508-6
Kajarabille, N., and Latunde-Dada, G. O. (2019). Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Ijms 20 (19), 4968. doi:10.3390/ijms20194968
Kakhlon, O., and Cabantchik, Z. I. (2002). The Labile Iron Pool: Characterization, Measurement, and Participation in Cellular Processes1 1This Article Is Part of a Series of Reviews on "Iron and Cellular Redox Status." the Full List of Papers May Be Found on the Homepage of the Journal. Free Radic. Biol. Med. 33 (8), 1037–1046. doi:10.1016/s0891-5849(02)01006-7
Kamoshita, M., Ozawa, Y., Kubota, S., Miyake, S., Tsuda, C., Nagai, N., et al. (2014). AMPK-NF-κB Axis in the Photoreceptor Disorder during Retinal Inflammation. PloS one 9 (7), e103013. doi:10.1371/journal.pone.0103013
Kang, R., Kroemer, G., and Tang, D. (2019). The Tumor Suppressor Protein P53 and the Ferroptosis Network. Free Radic. Biol. Med. 133, 162–168. doi:10.1016/j.freeradbiomed.2018.05.074
Kang, R., Zhu, S., Zeh, H. J., Klionsky, D. J., and Tang, D. (2018). BECN1 Is a New Driver of Ferroptosis. Autophagy 14 (12), 2173–2175. doi:10.1080/15548627.2018.1513758
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N-acetylcysteine Targets 5 Lipoxygenase-Derived, Toxic Lipids and Can Synergize with Prostaglandin E2to Inhibit Ferroptosis and Improve Outcomes Following Hemorrhagic Stroke in Mice. Ann. Neurol. 84 (6), 854–872. doi:10.1002/ana.25356
Kenny, E. M., Fidan, E., Yang, Q., Anthonymuthu, T. S., New, L. A., Meyer, E. A., et al. (2019). Ferroptosis Contributes to Neuronal Death and Functional Outcome after Traumatic Brain Injury*. Crit. Care Med. 47 (3), 410–418. doi:10.1097/ccm.0000000000003555
Kerins, M. J., and Ooi, A. (2018). The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signaling 29 (17), 1756–1773. doi:10.1089/ars.2017.7176
Kobayashi, M., Suhara, T., Baba, Y., Kawasaki, N. K., Higa, J. K., and Matsui, T. (2018). Pathological Roles of Iron in Cardiovascular Disease. Cdt 19 (9), 1068–1076. doi:10.2174/1389450119666180605112235
Land, W. G., Agostinis, P., Gasser, S., Garg, A. D., and Linkermann, A. (2016). Transplantation and Damage-Associated Molecular Patterns (DAMPs). Am. J. Transpl. 16 (12), 3338–3361. doi:10.1111/ajt.13963
Lewerenz, J., and Maher, P. (2011). Control of Redox State and Redox Signaling by Neural Antioxidant Systems. Antioxid. Redox Signaling 14 (8), 1449–1465. doi:10.1089/ars.2010.3600
Li, B., and Pratt, D. A. (2015). Methods for Determining the Efficacy of Radical-Trapping Antioxidants. Free Radic. Biol. Med. 82, 187–202. doi:10.1016/j.freeradbiomed.2015.01.020
Li, J., Cao, F., Yin, H.-L., Huang, Z.-J., Lin, Z.-T., Mao, N., et al. (2020a). Ferroptosis: Past, Present and Future. Cel Death Dis 11 (2), 88. doi:10.1038/s41419-020-2298-2
Li, N., Jiang, W., Wang, W., Xiong, R., Wu, X., and Geng, Q. (2021a). Ferroptosis and its Emerging Roles in Cardiovascular Diseases. Pharmacol. Res. 166, 105466. doi:10.1016/j.phrs.2021.105466
Li, N., Wang, W., Zhou, H., Wu, Q., Duan, M., Liu, C., et al. (2020b). Ferritinophagy-mediated Ferroptosis Is Involved in Sepsis-Induced Cardiac Injury. Free Radic. Biol. Med. 160, 303–318. doi:10.1016/j.freeradbiomed.2020.08.009
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017a). Inhibition of Neuronal Ferroptosis Protects Hemorrhagic Brain. JCI insight 2 (7), e90777. doi:10.1172/jci.insight.90777
Li, Q., Weiland, A., Chen, X., Lan, X., Han, X., Durham, F., et al. (2018a). Ultrastructural Characteristics of Neuronal Death and White Matter Injury in Mouse Brain Tissues after Intracerebral Hemorrhage: Coexistence of Ferroptosis, Autophagy, and Necrosis. Front. Neurol. 9, 581. doi:10.3389/fneur.2018.00581
Li, W., Li, W., Leng, Y., Xiong, Y., and Xia, Z. (2020c). Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury through Endoplasmic Reticulum Stress. DNA Cel. Biol. 39 (2), 210–225. doi:10.1089/dna.2019.5097
Li, W., Tang, R., Ouyang, S., Ma, F., Liu, Z., and Wu, J. (2017b). Folic Acid Prevents Cardiac Dysfunction and Reduces Myocardial Fibrosis in a Mouse Model of High-Fat Diet-Induced Obesity. Nutr. Metab. (Lond) 14, 68. doi:10.1186/s12986-017-0224-0
Li, X., Guo, D., Chen, Y., Hu, Y., and Zhang, F. (2021b). Complex Coronary Instent Chronic Total Occlusion Lesions: Oxidative Stress, Inflammation, and Coronary Stent Lengths. Oxidative Med. Cell. longevity 2021, 1–11. doi:10.1155/2021/8815048
Li, Y., Lin, S., Li, L., Tang, Z., Hu, Y., Ban, X., et al. (2018b). PDSS2 Deficiency Induces Hepatocarcinogenesis by Decreasing Mitochondrial Respiration and Reprogramming Glucose Metabolism. Cancer Res. 78 (16), 4471–4481. doi:10.1158/0008-5472.Can-17-2172
Li, Y., Liu, Y., Wu, P., Tian, Y., Liu, B., Wang, J., et al. (2021c). Inhibition of Ferroptosis Alleviates Early Brain Injury after Subarachnoid Hemorrhage In Vitro and In Vivo via Reduction of Lipid Peroxidation. Cell Mol Neurobiol 41 (2), 263–278. doi:10.1007/s10571-020-00850-1
Linkermann, A., Stockwell, B. R., Krautwald, S., and Anders, H.-J. (2014). Regulated Cell Death and Inflammation: an Auto-Amplification Loop Causes Organ Failure. Nat. Rev. Immunol. 14 (11), 759–767. doi:10.1038/nri3743
Liu, B., Zhao, C., Li, H., Chen, X., Ding, Y., and Xu, S. (2018). Puerarin Protects against Heart Failure Induced by Pressure Overload through Mitigation of Ferroptosis. Biochem. biophysical Res. Commun. 497 (1), 233–240. doi:10.1016/j.bbrc.2018.02.061
Liu, J., Kuang, F., Kroemer, G., Klionsky, D. J., Kang, R., and Tang, D. (2020a). Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cel Chem. Biol. 27 (4), 420–435. doi:10.1016/j.chembiol.2020.02.005
Liu, P., Feng, Y., Li, H., Chen, X., Wang, G., Xu, S., et al. (2020b). Ferrostatin-1 Alleviates Lipopolysaccharide-Induced Acute Lung Injury via Inhibiting Ferroptosis. Cell Mol Biol Lett 25, 10. doi:10.1186/s11658-020-00205-0
Liu, Q., Wang, S., and Cai, L. (2014). Diabetic Cardiomyopathy and its Mechanisms: Role of Oxidative Stress and Damage. J. Diabetes Invest. 5 (6), 623–634. doi:10.1111/jdi.12250
Liu, T., Jiang, L., Tavana, O., and Gu, W. (2019). The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 79 (8), 1913–1924. doi:10.1158/0008-5472.Can-18-3037
Lou, J.-S., Zhao, L.-P., Huang, Z.-H., Chen, X.-Y., Xu, J.-T., Tai, W. C.-S., et al. (2021). Ginkgetin Derived from Ginkgo Biloba Leaves Enhances the Therapeutic Effect of Cisplatin via Ferroptosis-Mediated Disruption of the Nrf2/HO-1 axis in EGFR Wild-type Non-small-cell Lung Cancer. Phytomedicine 80, 153370. doi:10.1016/j.phymed.2020.153370
Lu, L.-Q., Tian, J., Luo, X.-J., and Peng, J. (2020). Targeting the Pathways of Regulated Necrosis: a Potential Strategy for Alleviation of Cardio-Cerebrovascular Injury. Cel. Mol. Life Sci. 78, 63–78. doi:10.1007/s00018-020-03587-8
Lu, L.-Q., Tian, J., Luo, X.-J., and Peng, J. (2021). Targeting the Pathways of Regulated Necrosis: a Potential Strategy for Alleviation of Cardio-Cerebrovascular Injury. Cel. Mol. Life Sci. 78 (1), 63–78. doi:10.1007/s00018-020-03587-8
Lv, H., Yang, H., Wang, Z., Feng, H., Deng, X., Cheng, G., et al. (2019). Nrf2 Signaling and Autophagy Are Complementary in Protecting Lipopolysaccharide/d-Galactosamine-Induced Acute Liver Injury by Licochalcone A. Cel Death Dis 10 (4), 313. doi:10.1038/s41419-019-1543-z
Ma, W., Wei, S., Zhang, B., and Li, W. (2020). Molecular Mechanisms of Cardiomyocyte Death in Drug-Induced Cardiotoxicity. Front. Cel Dev. Biol. 8, 434. doi:10.3389/fcell.2020.00434
Madmani, M. E., Yusuf Solaiman, A., Tamr Agha, K., Madmani, Y., Shahrour, Y., Essali, A., et al. (2014). Coenzyme Q10 for Heart Failure. Cochrane database Syst. Rev. (6), CD008684. doi:10.1002/14651858.CD008684.pub2
Magtanong, L., and Dixon, S. J. (2018). Ferroptosis and Brain Injury. Developmental Neurosci. 40, 382–395. doi:10.1159/000496922
Martinet, W., Coornaert, I., Puylaert, P., and De Meyer, G. R. Y. (2019). Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 10, 306. doi:10.3389/fphar.2019.00306
Masaldan, S., Bush, A. I., Devos, D., Rolland, A. S., and Moreau, C. (2019). Striking while the Iron Is Hot: Iron Metabolism and Ferroptosis in Neurodegeneration. Free Radic. Biol. Med. 133, 221–233. doi:10.1016/j.freeradbiomed.2018.09.033
McEvoy, J. W. (2012). Lifetime Risks of Cardiovascular Disease. N. Engl. J. Med. 366 (17), 1641–1643. author reply 1642-3. doi:10.1056/NEJMc1202276
Mendell, J. T. (2008). miRiad Roles for the miR-17-92 Cluster in Development and Disease. Cell 133 (2), 217–222. doi:10.1016/j.cell.2008.04.001
Moore, K. J., and Tabas, I. (2011). Macrophages in the Pathogenesis of Atherosclerosis. Cell 145 (3), 341–355. doi:10.1016/j.cell.2011.04.005
Moreira, P. I., Harris, P. L. R., Zhu, X., Santos, M. S., Oliveira, C. R., Smith, M. A., et al. (2007). Lipoic Acid and N-Acetyl Cysteine Decrease Mitochondrial-Related Oxidative Stress in Alzheimer Disease Patient Fibroblasts. Jad 12 (2), 195–206. doi:10.3233/jad-2007-12210
Morris, G., Walker, A. J., Berk, M., Maes, M., and Puri, B. K. (2018). Cell Death Pathways: a Novel Therapeutic Approach for Neuroscientists. Mol. Neurobiol. 55 (7), 5767–5786. doi:10.1007/s12035-017-0793-y
Murphy, M. E. (2016). Ironing Out How P53 Regulates Ferroptosis. Proc. Natl. Acad. Sci. USA 113 (44), 12350–12352. doi:10.1073/pnas.1615159113
Nishizawa, H., Matsumoto, M., Shindo, T., Saigusa, D., Kato, H., Suzuki, K., et al. (2020). Ferroptosis Is Controlled by the Coordinated Transcriptional Regulation of Glutathione and Labile Iron Metabolism by the Transcription Factor BACH1. J. Biol. Chem. 295 (1), 69–82. doi:10.1074/jbc.RA119.009548
Nunomura, A., Moreira, P. I., Castellani, R. J., Lee, H. G., Zhu, X., Smith, M. A., et al. (2012). Oxidative Damage to RNA in Aging and Neurodegenerative Disorders. Neurotoxicity Res. 22 (3), 231–248. doi:10.1007/s12640-012-9331-x
Orning, P., Ratner, D., Weng, D., Starheim, K., Ratner, D., Best, Z., et al. (2018). Pathogen Blockade of TAK1 Triggers Caspase-8-dependent Cleavage of Gasdermin D and Cell Death. Science 362 (6418), 1064–1069. doi:10.1126/science.aau2818
Ouyang, S., You, J., Zhi, C., Li, P., Lin, X., Tan, X., et al. (2021). Ferroptosis: the Potential Value Target in Atherosclerosis. Cel Death Dis 12 (8), 782. doi:10.1038/s41419-021-04054-3
Parim, B., Sathibabu Uddandrao, V. V., and Saravanan, G. (2019). Diabetic Cardiomyopathy: Molecular Mechanisms, Detrimental Effects of Conventional Treatment, and Beneficial Effects of Natural Therapy. Heart Fail. Rev. 24 (2), 279–299. doi:10.1007/s10741-018-9749-1
Patel, P., and Karch, J. (2020). Regulation of Cell Death in the Cardiovascular System. Int. Rev. Cel. Mol. Biol. 353, 153–209. doi:10.1016/bs.ircmb.2019.11.005
Pistoia, F., Sacco, S., Degan, D., Tiseo, C., Ornello, R., and Carolei, A. (2016). Hypertension and Stroke: Epidemiological Aspects and Clinical Evaluation. High Blood Press. Cardiovasc. Prev. 23 (1), 9–18. doi:10.1007/s40292-015-0115-2
Qiu, Y., Cao, Y., Cao, W., Jia, Y., and Lu, N. (2020). The Application of Ferroptosis in Diseases. Pharmacol. Res. 159, 104919. doi:10.1016/j.phrs.2020.104919
Qu, X. F., Liang, T. Y., Wu, D. G., Lai, N. S., Deng, R. M., Ma, C., et al. (2021). Acyl‐CoA Synthetase Long Chain Family Member 4 Plays Detrimental Role in Early Brain Injury after Subarachnoid Hemorrhage in Rats by Inducing Ferroptosis. CNS Neurosci. Ther. 27 (4), 449–463. doi:10.1111/cns.13548
Rajesh, M., Bátkai, S., Kechrid, M., Mukhopadhyay, P., Lee, W. S., Horváth, B., et al. (2012). Cannabinoid 1 Receptor Promotes Cardiac Dysfunction, Oxidative Stress, Inflammation, and Fibrosis in Diabetic Cardiomyopathy. Diabetes 61 (3), 716–727. doi:10.2337/db11-0477
Ravingerová, T., Kindernay, L., Barteková, M., Ferko, M., Adameová, A., Zohdi, V., et al. (2020). The Molecular Mechanisms of Iron Metabolism and its Role in Cardiac Dysfunction and Cardioprotection. Ijms 21 (21), 7889. doi:10.3390/ijms21217889
Ross, R. (1999). Atherosclerosis - an Inflammatory Disease. N. Engl. J. Med. 340 (2), 115–126. doi:10.1056/nejm199901143400207
Sabetghadam, M., Mazdeh, M., Abolfathi, P., Mohammadi, Y., and Mehrpooya, M. (2020). Evidence for a Beneficial Effect of Oral N-Acetylcysteine on Functional Outcomes and Inflammatory Biomarkers in Patients with Acute Ischemic Stroke. Ndt Vol. 16, 1265–1278. doi:10.2147/ndt.S241497
Sato, M., Kusumi, R., Hamashima, S., Kobayashi, S., Sasaki, S., Komiyama, Y., et al. (2018). The Ferroptosis Inducer Erastin Irreversibly Inhibits System Xc− and Synergizes with Cisplatin to Increase Cisplatin's Cytotoxicity in Cancer Cells. Sci. Rep. 8 (1), 968. doi:10.1038/s41598-018-19213-4
Seibt, T. M., Proneth, B., and Conrad, M. (2019). Role of GPX4 in Ferroptosis and its Pharmacological Implication. Free Radic. Biol. Med. 133, 144–152. doi:10.1016/j.freeradbiomed.2018.09.014
Sha, W., Hu, F., Xi, Y., Chu, Y., and Bu, S. (2021). Mechanism of Ferroptosis and its Role in Type 2 Diabetes Mellitus. J. Diabetes Res. 2021, 1–10. doi:10.1155/2021/9999612
Shen, L., Lin, D., Li, X., Wu, H., Lenahan, C., Pan, Y., et al. (2020). Ferroptosis in Acute Central Nervous System Injuries: The Future Direction. Front. Cel Dev. Biol. 8, 594. doi:10.3389/fcell.2020.00594
Shirazi, L. F., Bissett, J., Romeo, F., and Mehta, J. L. (2017). Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 19 (6), 27. doi:10.1007/s11883-017-0660-3
Shojaie, L., Iorga, A., and Dara, L. (2020). Cell Death in Liver Diseases: A Review. Ijms 21 (24), 9682. doi:10.3390/ijms21249682
Sierra, C., López-Soto, A., and Coca, A. (20112011). Connecting Cerebral white Matter Lesions and Hypertensive Target Organ Damage. J. Aging Res. 2011, 1–7. doi:10.4061/2011/438978
Singh, G. K., Siahpush, M., Azuine, R. E., and Williams, S. D. (2015). Increasing Area Deprivation and Socioeconomic Inequalities in Heart Disease, Stroke, and Cardiovascular Disease Mortality Among Working Age Populations, United States, 1969-2011. Int. J. MCH AIDS 3 (2), 119–133.
Singh, V. P., Le, B., Khode, R., Baker, K. M., and Kumar, R. (2008). Intracellular Angiotensin II Production in Diabetic Rats Is Correlated with Cardiomyocyte Apoptosis, Oxidative Stress, and Cardiac Fibrosis. Diabetes 57 (12), 3297–3306. doi:10.2337/db08-0805
Song, X., Zhu, S., Chen, P., Hou, W., Wen, Q., Liu, J., et al. (2018). AMPK-mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc- Activity. Curr. Biol. 28 (15), 2388–2399. e5. doi:10.1016/j.cub.2018.05.094
Song, Y., Wang, B., Zhu, X., Hu, J., Sun, J., Xuan, J., et al. (2021). Human Umbilical Cord Blood-Derived MSCs Exosome Attenuate Myocardial Injury by Inhibiting Ferroptosis in Acute Myocardial Infarction Mice. Cell Biol Toxicol 37 (1), 51–64. doi:10.1007/s10565-020-09530-8
Speer, R. E., Karuppagounder, S. S., Basso, M., Sleiman, S. F., Kumar, A., Brand, D., et al. (2013). Hypoxia-inducible Factor Prolyl Hydroxylases as Targets for Neuroprotection by "antioxidant" Metal Chelators: From Ferroptosis to Stroke. Free Radic. Biol. Med. 62, 26–36. doi:10.1016/j.freeradbiomed.2013.01.026
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171 (2), 273–285. doi:10.1016/j.cell.2017.09.021
Stockwell, B. R., Jiang, X., and Gu, W. (2020). Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biology 30 (6), 478–490. doi:10.1016/j.tcb.2020.02.009
Stoyanovsky, D. A., Tyurina, Y. Y., Shrivastava, I., Bahar, I., Tyurin, V. A., Protchenko, O., et al. (2019). Iron Catalysis of Lipid Peroxidation in Ferroptosis: Regulated Enzymatic or Random Free Radical Reaction. Free Radic. Biol. Med. 133, 153–161. doi:10.1016/j.freeradbiomed.2018.09.008
Su, G., Yang, W., Wang, S., Geng, C., and Guan, X. (2021). SIRT1-autophagy axis Inhibits Excess Iron-Induced Ferroptosis of Foam Cells and Subsequently Increases IL-1Β and IL-18. Biochem. biophysical Res. Commun. 561, 33–39. doi:10.1016/j.bbrc.2021.05.011
Su, L.-J., Zhang, J.-H., Gomez, H., Murugan, R., Hong, X., Xu, D., et al. (20192019). Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. longevity 2019, 1–13. doi:10.1155/2019/5080843
Sullivan, J. L. (2009). Iron in Arterial Plaque: A Modifiable Risk Factor for Atherosclerosis. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1790 (7), 718–723. doi:10.1016/j.bbagen.2008.06.005
Sumneang, N., Kumfu, S., Khamseekaew, J., Siri-Angkul, N., Fucharoen, S., Chattipakorn, S. C., et al. (2019). Combined Iron Chelator with N-Acetylcysteine Exerts the Greatest Effect on Improving Cardiac Calcium Homeostasis in Iron-Overloaded Thalassemic Mice. Toxicology 427, 152289. doi:10.1016/j.tox.2019.152289
Sumneang, N., Siri-Angkul, N., Kumfu, S., Chattipakorn, S. C., and Chattipakorn, N. (2020). The Effects of Iron Overload on Mitochondrial Function, Mitochondrial Dynamics, and Ferroptosis in Cardiomyocytes. Arch. Biochem. Biophys. 680, 108241. doi:10.1016/j.abb.2019.108241
Sun, X., Ou, Z., Xie, M., Kang, R., Fan, Y., Niu, X., et al. (2015). HSPB1 as a Novel Regulator of Ferroptotic Cancer Cell Death. Oncogene 34 (45), 5617–5625. doi:10.1038/onc.2015.32
Sun, Y., Chen, P., Zhai, B., Zhang, M., Xiang, Y., Fang, J., et al. (2020). The Emerging Role of Ferroptosis in Inflammation. Biomed. Pharmacother. 127, 110108. doi:10.1016/j.biopha.2020.110108
Talasaz, A. H., Khalili, H., Fahimi, F., Jenab, Y., Ma, Broumand., Salarifar, M., et al. (2014). Effects of N-Acetylcysteine on the Cardiac Remodeling Biomarkers and Major Adverse Events Following Acute Myocardial Infarction: a Randomized Clinical Trial. Am. J. Cardiovasc. Drugs : Drugs devices, other interventions 14 (1), 51–61. doi:10.1007/s40256-013-0048-x
Tang, S., Gao, P., Chen, H., Zhou, X., Ou, Y., and He, Y. (2020). The Role of Iron, its Metabolism and Ferroptosis in Traumatic Brain Injury. Front. Cel. Neurosci. 14, 590789. doi:10.3389/fncel.2020.590789
Tao, N., Li, K., and Liu, J. (2020). Molecular Mechanisms of Ferroptosis and its Role in Pulmonary Disease. Oxidative Med. Cell. longevity 2020, 1–12. doi:10.1155/2020/9547127
Thackeray, J. T., Hupe, H. C., Wang, Y., Bankstahl, J. P., Berding, G., Ross, T. L., et al. (2018). Myocardial Inflammation Predicts Remodeling and Neuroinflammation after Myocardial Infarction. J. Am. Coll. Cardiol. 71 (3), 263–275. doi:10.1016/j.jacc.2017.11.024
Thong, P. S., Selley, M., and Watt, F. (1996). Elemental Changes in Atherosclerotic Lesions Using Nuclear Microscopy. Cel Mol Biol (Noisy-le-grand) 42 (1), 103–110.
Tuo, Q.-Z., Lei, P., Jackman, K. A., Li, X.-L., Xiong, H., Li, X.-L., et al. (2017). Tau-mediated Iron export Prevents Ferroptotic Damage after Ischemic Stroke. Mol. Psychiatry 22 (11), 1520–1530. doi:10.1038/mp.2017.171
Ursini, F., and Maiorino, M. (2020). Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 152, 175–185. doi:10.1016/j.freeradbiomed.2020.02.027
van Asch, C. J., Luitse, M. J., Rinkel, G. J., van der Tweel, I., Algra, A., and Klijn, C. J. (2010). Incidence, Case Fatality, and Functional Outcome of Intracerebral Haemorrhage over Time, According to Age, Sex, and Ethnic Origin: a Systematic Review and Meta-Analysis. Lancet Neurol. 9 (2), 167–176. doi:10.1016/s1474-4422(09)70340-0
Vasan, R. S., Larson, M. G., Leip, E. P., Evans, J. C., O'Donnell, C. J., Kannel, W. B., et al. (2001). Impact of High-normal Blood Pressure on the Risk of Cardiovascular Disease. N. Engl. J. Med. 345 (18), 1291–1297. doi:10.1056/NEJMoa003417
Veseli, B. E., Perrotta, P., De Meyer, G. R. A., Roth, L., Donckt, C. V. D., Martinet, W., et al. (2017). Animal Models of Atherosclerosis. Eur. J. Pharmacol. 816, 3–13. doi:10.1016/j.ejphar.2017.05.010
Wang, G., Song, X., Zhao, L., Li, Z., and Liu, B. (2018). Resveratrol Prevents Diabetic Cardiomyopathy by Increasing Nrf2 Expression and Transcriptional Activity. Biomed. Research International, 2150218. doi:10.1155/2018/2150218
Wang, H., Liu, C., Zhao, Y., and Gao, G. (2020a). Mitochondria Regulation in Ferroptosis. Eur. J. Cel. Biol. 99 (1), 151058. doi:10.1016/j.ejcb.2019.151058
Wang, J. (2010). Preclinical and Clinical Research on Inflammation after Intracerebral Hemorrhage. Prog. Neurobiol. 92 (4), 463–477. doi:10.1016/j.pneurobio.2010.08.001
Wang, N., Zeng, G.-Z., Yin, J.-L., and Bian, Z.-X. (2019). Artesunate Activates the ATF4-CHOP-CHAC1 Pathway and Affects Ferroptosis in Burkitt's Lymphoma. Biochem. biophysical Res. Commun. 519 (3), 533–539. doi:10.1016/j.bbrc.2019.09.023
Wang, Y., Peng, X., Zhang, M., Jia, Y., Yu, B., and Tian, J. (2020b). Revisiting Tumors and the Cardiovascular System: Mechanistic Intersections and Divergences in Ferroptosis. Oxidative Med. Cell. longevity 2020, 1–13. doi:10.1155/2020/9738143
Weber, C., and Noels, H. (2011). Atherosclerosis: Current Pathogenesis and Therapeutic Options. Nat. Med. 17 (11), 1410–1422. doi:10.1038/nm.2538
Weiland, A., Wang, Y., Wu, W., Lan, X., Han, X., Li, Q., et al. (2019). Ferroptosis and its Role in Diverse Brain Diseases. Mol. Neurobiol. 56 (7), 4880–4893. doi:10.1007/s12035-018-1403-3
Westermann, D., Van Linthout, S., Dhayat, S., Dhayat, N., Escher, F., Bücker-Gärtner, C., et al. (2007). Cardioprotective and Anti-inflammatory Effects of Interleukin Converting Enzyme Inhibition in Experimental Diabetic Cardiomyopathy. Diabetes 56 (7), 1834–1841. doi:10.2337/db06-1662
Wu, H., Wu, T., Xu, X., Wang, J., and Wang, J. (2011). Iron Toxicity in Mice with Collagenase-Induced Intracerebral Hemorrhage. J. Cereb. Blood Flow Metab. 31 (5), 1243–1250. doi:10.1038/jcbfm.2010.209
Wu, J.-r., Tuo, Q.-z., and Lei, P. (2018). Ferroptosis, a Recent Defined Form of Critical Cell Death in Neurological Disorders. J. Mol. Neurosci. 66 (2), 197–206. doi:10.1007/s12031-018-1155-6
Wu, X.-Y., Luo, A.-Y., Zhou, Y.-R., and Ren, J.-H. (2014). N-acetylcysteine Reduces Oxidative Stress, Nuclear Factor-Κb Activity and Cardiomyocyte Apoptosis in Heart Failure. Mol. Med. Rep. 10 (2), 615–624. doi:10.3892/mmr.2014.2292
Wu, X., Li, Y., Zhang, S., and Zhou, X. (2021a). Ferroptosis as a Novel Therapeutic Target for Cardiovascular Disease. Theranostics 11 (7), 3052–3059. doi:10.7150/thno.54113
Wu, X., Liu, Z., Yu, X. Y., Xu, S., and Luo, J. (2021b). Autophagy and Cardiac Diseases: Therapeutic Potential of Natural Products. Med. Res. Rev. 41 (1), 314–341. doi:10.1002/med.21733
Xiao, F.-J., Zhang, D., Wu, Y., Jia, Q.-H., Zhang, L., Li, Y.-X., et al. (2019). miRNA-17-92 Protects Endothelial Cells from Erastin-Induced Ferroptosis through Targeting the A20-ACSL4 axis. Biochem. biophysical Res. Commun. 515 (3), 448–454. doi:10.1016/j.bbrc.2019.05.147
Xie, X., Ma, X., Zeng, S., Tang, W., Xiao, L., Zhu, C., et al. (2020). Mechanisms of Berberine for the Treatment of Atherosclerosis Based on Network Pharmacology. Evidence-Based Complement. Altern. Med. 2020, 1–11. doi:10.1155/2020/3568756
Xu, S. (2019). Iron and Atherosclerosis: The Link Revisited. Trends Molecular Medicine 25 (8), 659–661. doi:10.1016/j.molmed.2019.05.012
Yan, H., Zhang, D., Hao, S., Li, K., and Hang, C.-H. (2015). Role of Mitochondrial Calcium Uniporter in Early Brain Injury after Experimental Subarachnoid Hemorrhage. Mol. Neurobiol. 52 (3), 1637–1647. doi:10.1007/s12035-014-8942-z
Yang, J., Wang, M., Wang, S., Li, G., and Gao, Y. (20201993). Study on Ferroptosis Pathway that Operates in Hypertensive Brain Damage. Clin. Exp. Hypertens. 42 (8), 748–752. doi:10.1080/10641963.2020.1783545
Yang, K., Song, H., and Yin, D. (2021). PDSS2 Inhibits the Ferroptosis of Vascular Endothelial Cells in Atherosclerosis by Activating Nrf2. J. Cardiovasc. Pharmacol. 77 (6), 767–776. doi:10.1097/fjc.0000000000001030
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 113 (34), E4966–E4975. doi:10.1073/pnas.1603244113
Yang, Y.-K., Wang, L.-P., Chen, L., Yao, X.-P., Yang, K.-Q., Gao, L.-G., et al. (2015). Coenzyme Q10 Treatment of Cardiovascular Disorders of Ageing Including Heart Failure, Hypertension and Endothelial Dysfunction. Clinica Chim. Acta 450, 83–89. doi:10.1016/j.cca.2015.08.002
Yao, X., Feng, S.-Q., Fan, B.-Y., Pang, Y.-L., Li, W.-X., Zhao, C.-X., et al. (2021). Liproxstatin-1 Is an Effective Inhibitor of Oligodendrocyte Ferroptosis Induced by Inhibition of Glutathione Peroxidase 4. Neural Regen. Res. 16 (3), 561–566. doi:10.4103/1673-5374.293157
Yesilbursa, D., Serdar, A., Senturk, T., Serdar, Z., Sağ, S., and Cordan, J. (2006). Effect of N-Acetylcysteine on Oxidative Stress and Ventricular Function in Patients with Myocardial Infarction. Heart and vessels 21 (1), 33–37. doi:10.1007/s00380-005-0854-4
Yesilbursa, D., Serdar, A., Senturk, T., Serdar, Z., Sağ, S., and Cordan, J. (2006). Effect of N-Acetylcysteine on Oxidative Stress andVentricular Function in Patients with Myocardial Infarction. Heart and vessels 21 (1), 33–37. doi:10.1007/s00380-005-0854-4
Yu, Y., Yan, Y., Niu, F., Wang, Y., Chen, X., Su, G., et al. (2021). Ferroptosis: a Cell Death Connecting Oxidative Stress, Inflammation and Cardiovascular Diseases. Cell Death Discov. 7 (1), 193. doi:10.1038/s41420-021-00579-w
Zang, H., Wu, W., Qi, L., Tan, W., Nagarkatti, P., Nagarkatti, M., et al. (2020). Autophagy Inhibition Enables Nrf2 to Exaggerate the Progression of Diabetic Cardiomyopathy in Mice. Diabetes 69 (12), 2720–2734. doi:10.2337/db19-1176
Zecca, L., Youdim, M. B. H., Riederer, P., Connor, J. R., and Crichton, R. R. (2004). Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 5 (11), 863–873. doi:10.1038/nrn1537
Zhang, J., Zhang, M., Yang, Z., Huang, S., Wu, X., Cao, L., et al. (2020a). PDCD4 Deficiency Ameliorates Left Ventricular Remodeling and Insulin Resistance in a Rat Model of Type 2 Diabetic Cardiomyopathy. BMJ Open Diab Res. Care 8 (1), e001081. doi:10.1136/bmjdrc-2019-001081
Zhang, P., Dong, G., Sun, B., Zhang, L., Chen, X., Ma, N., et al. (2011). Long-term Exposure to Ambient Air Pollution and Mortality Due to Cardiovascular Disease and Cerebrovascular Disease in Shenyang, China. PloS one 6 (6), e20827. doi:10.1371/journal.pone.0020827
Zhang, W.-J., Wei, H., and Frei, B. (2010). The Iron Chelator, Desferrioxamine, Reduces Inflammation and Atherosclerotic Lesion Development in Experimental Mice. Exp. Biol. Med. (Maywood) 235 (5), 633–641. doi:10.1258/ebm.2009.009229
Zhang, X., Yu, Y., Lei, H., Cai, Y., Shen, J., Zhu, P., et al. (2020b). The Nrf-2/HO-1 Signaling Axis: A Ray of Hope in Cardiovascular Diseases. Cardiol. Res. Pract. 2020, 1–9. doi:10.1155/2020/5695723
Zhang, Y., Wu, H., Xu, Y., Qin, H., Lan, C., and Wang, W. (2020c). The Correlation between Neck Circumference and Risk Factors in Patients with Hypertension. Medicine 99 (47), e22998. doi:10.1097/md.0000000000022998
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione Peroxidase 4 Participates in Secondary Brain Injury through Mediating Ferroptosis in a Rat Model of Intracerebral Hemorrhage. Brain Res. 1701, 112–125. doi:10.1016/j.brainres.2018.09.012
Zhang, Z., Yan, J., Taheri, S., Liu, K. J., and Shi, H. (2014). Hypoxia-inducible Factor 1 Contributes to N-Acetylcysteine's protection in Stroke. Free Radic. Biol. Med. 68, 8–21. doi:10.1016/j.freeradbiomed.2013.11.007
Zhaolin, Z., Guohua, L., Shiyuan, W., and Zuo, W. (2019). Role of Pyroptosis in Cardiovascular Disease. Cell Prolif 52 (2), e12563. doi:10.1111/cpr.12563
Zilka, O, Shah, R., Li, B, Friedmann Angeli, J. P., Conrad, M., et al. (2017). On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 3 (3), 232–243. doi:10.1021/acscentsci.7b00028
Zille, M., Karuppagounder, S. S., Chen, Y., Gough, P. J., Bertin, J., Finger, J., et al. (2017). Neuronal Death after Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 48 (4), 1033–1043. doi:10.1161/strokeaha.116.015609
Keywords: ferroptosis, cardio-cerebrovascular disease, inflammation, iron, lipid peroxidation
Citation: Luo M-Y, Su J-H, Gong S-X, Liang N, Huang W-Q, Chen W, Wang A-P and Tian Y (2021) Ferroptosis: New Dawn for Overcoming the Cardio-Cerebrovascular Diseases. Front. Cell Dev. Biol. 9:733908. doi: 10.3389/fcell.2021.733908
Received: 06 July 2021; Accepted: 25 October 2021;
Published: 11 November 2021.
Edited by:
Yi Zhang, First Affiliated Hospital of Zhengzhou University, ChinaReviewed by:
Qianyi He, First Affiliated Hospital of Zhengzhou University, ChinaNao Yan, Wuhan University, China
Copyright © 2021 Luo, Su, Gong, Liang, Huang, Chen, Wang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ai-Ping Wang, d2FpcGluZzIwMTFAMTYzLmNvbQ==; Ying Tian, dXNjdHlAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship