 Joseph Adu-Amankwaah1
Joseph Adu-Amankwaah1 Gabriel Komla Adzika1
Gabriel Komla Adzika1 Adebayo Oluwafemi Adekunle1
Adebayo Oluwafemi Adekunle1 Marie Louise Ndzie Noah1
Marie Louise Ndzie Noah1 Richard Mprah1
Richard Mprah1 Aisha Bushi2
Aisha Bushi2 Nazma Akhter1
Nazma Akhter1 Fei Huang1
Fei Huang1 Yaxin Xu1
Yaxin Xu1 Seyram Yao Adzraku3
Seyram Yao Adzraku3 Iqra Nadeem4
Iqra Nadeem4 Hong Sun1*
Hong Sun1*- 1Department of Physiology, Xuzhou Medical University, Xuzhou, China
- 2Xuzhou Medical University, Xuzhou, China
- 3Key Laboratory of Bone Marrow Stem Cell, Department of Hematology, The Affiliated Hospital of Xuzhou Medical University, Xuzhou, China
- 4Department of Neurobiology and Anatomy, Xuzhou Medical University, Xuzhou, China
Heart failure development is characterized by persistent inflammation and progressive fibrosis owing to chronic catecholamine stress. In a chronic stress state, elevated catecholamines result in the overstimulation of beta-adrenergic receptors (βARs), specifically β2-AR coupling with Gαi protein. Gαi signaling increases the activation of receptor-stimulated p38 mitogen-activated-protein-kinases (p38 MAPKs) and extracellular signal-regulated kinases (ERKs). Phosphorylation by these kinases is a common way to positively regulate the catalytic activity of A Disintegrin and Metalloprotease 17 (ADAM17), a metalloprotease that has grown much attention in recent years and has emerged as a chief regulatory hub in inflammation, fibrosis, and immunity due to its vital proteolytic activity. ADAM17 cleaves and activates proinflammatory cytokines and fibrotic factors that enhance cardiac dysfunction via inflammation and fibrosis. However, there is limited information on the cardiovascular aspect of ADAM17, especially in heart failure. Hence, this concise review provides a comprehensive insight into the structure of ADAM17, how it is activated and regulated during chronic catecholamine stress in heart failure development. This review highlights the inflammatory and fibrotic roles of ADAM17’s substrates; Tumor Necrosis Factor α (TNFα), soluble interleukin-6 receptor (sIL-6R), and amphiregulin (AREG). Finally, how ADAM17-induced chronic inflammation and progressive fibrosis aggravate cardiac dysfunction is discussed.
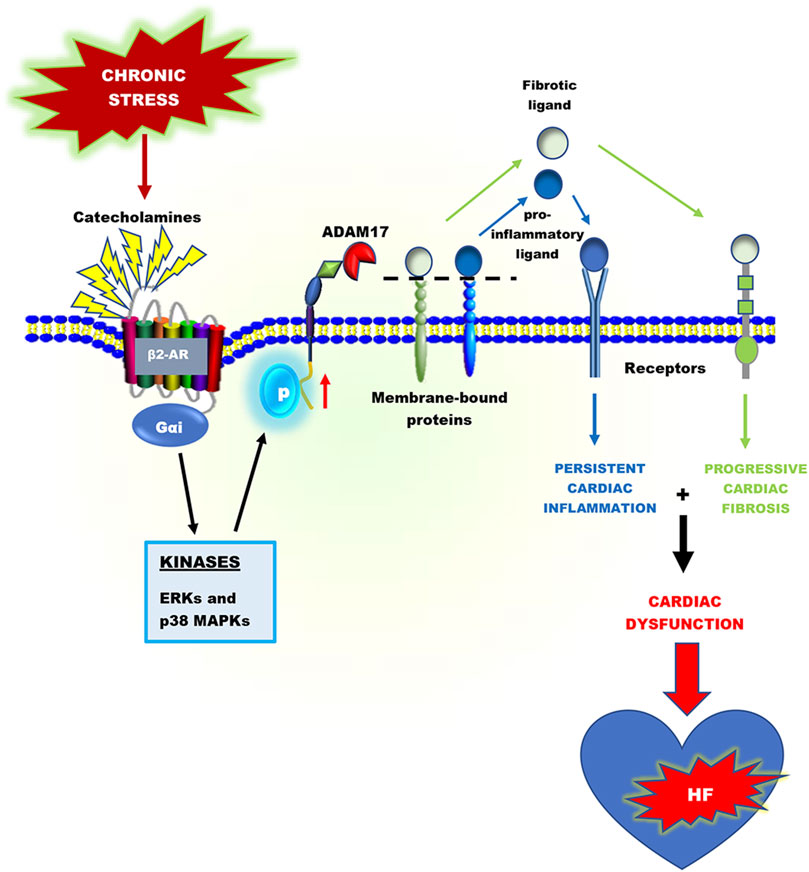
GRAPHICAL ABSTRACT
Introduction
Heart failure (HF) is a serious clinical and public health issue that affects over 23 million people globally, resulting in significant mortality, morbidity, and healthcare expenditures (Ayoub et al., 2017; Orso et al., 2017; Frantz et al., 2018). Despite advances in understanding its pathophysiology and treatment, the prognosis of patients with HF remains poor. Approximately 2–17% of patients die during their first hospital stay, with over 50% of patients dying within 5 years (Ayoub et al., 2017).
Chronic stress-induced adverse cardiac remodeling and HF are generally associated with prolonged activation of proinflammatory responses (Adzika et al., 2021; Huo S. et al., 2021). In an inflammatory driven HF, the inflammatory responses are orchestrated by myosin and troponin (damage-associated molecular patterns (DAMPs)) released from necrotic cardiomyocytes. These cardiac antigens activate and induce the infiltration of neutrophils, macrophages, dendritic cells, as well as T and B cells into the myocardia (Lafuse et al., 2020; Adzika et al., 2021). Following cardiac injury, neutrophils and CD86+ macrophages are rapidly recruited to the injured area, where they initiate inflammatory responses with the goal of cleaning up dead cell debris. However, excessive accumulation and/or delayed switch from these proinflammatory cells infiltration to reparative inflammatory cells (such as CD206+ macrophages) has detrimental effects. By releasing reactive oxygen species, granular components, and proinflammatory mediators such as tumor necrosis factor-alpha (TNFα), soluble interleukin-6 receptor (sIL-6R), and CXC chemokine receptor 2 (CXCR2), neutrophils and macrophages contribute to adverse myocardial injury and remodeling (Adu-Amankwaah et al., 2021a; Ma, 2021). Additionally, the activation of T and B lymphocytes by dendritic cells has been shown to play crucial roles in myocardial inflammation (Santos-Zas et al., 2018; Santos-Zas et al., 2021). Ultimately, without timely resolution of these proinflammatory responses and initiates of reparative functions, genes encoding proinflammatory mediators and fibrotic factors are upregulated excessively (Epelman et al., 2014; Heidt et al., 2014; Adamo et al., 2020).
Proteolytic cleavage of transmembrane proteins is a vital post-translational modification that controls several transmembrane proteins’ biological function, including proinflammatory mediators and growth factors (Lichtenthaler et al., 2018; Düsterhöft et al., 2019). Amid the 560 proteases encoded in the human genome, A Disintegrin and Metalloprotease 17 (ADAM17) has grown much attention in recent years and has emerged as a chief regulatory hub in inflammation, fibrosis, and immunity due to its vital proteolytic activity (Düsterhöft et al., 2019). In immune and non-immune cells, ADAM17 cleaves a number of substrates, including ligands of the epidermal growth factor receptor (EGFR), adhesion molecules, proinflammatory cytokines, and chemokines and their receptors. Some of these substrates include amphiregulin (AREG), epigen, epiregulin, neuregulin, tomoegulin-2, transforming growth factor-alpha (TGF-α), heparin-binding epidermal growth factor (HB-EGF), TNFα, tumor necrosis factor β (TNFβ), the TNF receptors 1 and 2 (TNFR 1 and 2), and interlukin-6 receptor (IL-6R), CXCR2, collagen XVII, desmoglein-2 and nectin-4 (Black et al., 1997; Moss et al., 1997; Tellier et al., 2006; Reddy et al., 2009; Riethmueller et al., 2017; Kawai et al., 2021). According to Cabron et al. ADAM17 is a key regulator of soluble TNFα surface levels in proinflammatory macrophages and dendritic cells (Cabron et al., 2018). Additionally, sIL-6R and CXCR2 on human and mouse neutrophils surfaces are regulated by ADAM17 (Wright et al., 2014; Mishra et al., 2015).
In a physiological state, the expressions of ADAM17 in immune and non-immune cells are regulated by transcriptional and post-transcriptional factors, including nuclear factor kappa B (NF-κB) and Brahma-related gene 1 (BRG1). Furthermore, subcellular localization in the perinuclear region of cells has been shown to regulate ADAM17’s activity (Chemaly et al., 2017; Adu-Amankwaah et al., 2021a). Overexpression and chronic activation of ADAM17 can trigger excessive release of TNFα, sIL-6R, and CXCR2 on the surface of proinflammatory cells, which play crucial roles in the pathogeneses of several inflammatory diseases, including heart failure. Increased levels of TNFα, sIL-6R and CXCR2 have been implicated in immune cells (CD86+ macrophages, neutrophils, and dendritic cells) trafficking, migration, and activation as well as inducing excessive fibrosis, myocardial stiffness, and left ventricular diastolic dysfunction (Cumberbatch and Kimber, 1992; Bozkurt et al., 1998; Satoh et al., 2000; Russo et al., 2009; Jones et al., 2010; Anderson et al., 2013; Arokiasamy et al., 2017).
Several studies have shown that myocardial ADAM17, TNFα, and sIL-6R expressions in both mRNA and protein levels are higher in patients with cardiovascular diseases and complications, although ADAM17’s expression is downregulated in a normal state (Satoh et al., 1999; Damås et al., 2000; Satoh et al., 2000; Satoh et al., 2004; Anderson et al., 2013). Thus, establishing a positive correlation between ADAM17 and heart failure development. The increased expression of ADAM17, TNFα, and sIL-6R has a vital implication in aggravating cardiac dysfunction during heart failure development (Satoh et al., 1999; Satoh et al., 2000; Satoh et al., 2004; Adu-Amankwaah et al., 2021a). Additionally, pro-AREG, a bi-functional growth factor converted to its active form by ADAM17, is crucially involved in enhancing cardiac fibrosis and aggravating cardiac dysfunction (Liu et al., 2018). Besides inducing HF via facilitating hyperactive proinflammatory responses, ADAM17 has been implicated along with HB-EGF and betacellulin (BTC), and angiotensin-converting enzyme 2(ACE2) in causing congenital heart diseases and hypertensive-induced HF, respectively (Jackson et al., 2003; de Queiroz et al., 2015; Xu et al., 2017; Mukerjee et al., 2019). This comprehensive review provides an insight into the structure of ADAM17, how it is activated and regulated during chronic catecholamine stress in heart failure development. This review highlights the inflammatory and fibrotic roles of ADAM17’s substrates; TNFα, sIL-6R, and sAREG. Finally, how ADAM17-induced chronic inflammation and progressive fibrosis aggravate cardiac dysfunction is also discussed.
A Disintegrin and Metalloprotease 17 and Other Related Metalloproteinases
Overview
A disintegrin and metalloproteinases (ADAMs) consist of membrane-bound proteins that belong to a Zn2+-dependent protease superfamily. They are similar to other metalloenzymes, including matrix metalloproteinases (MMPs), meprins, and snake venom metalloproteinases (SVMP) (Calvete et al., 2007; Gooz, 2010). Physiologically, ADAMs and their related metalloenzymes are widely expressed in various body tissues and regulate diverse cellular activities, including cell migration, adhesion, proteolysis, and cellular signaling (Black et al., 1997; Jones et al., 2016). Hence, it is not astonishing that alterations in the expression or function of these proteases are implicated in several pathologies, including cancer, rheumatoid arthritis, kidney fibrosis, diabetes, Alzheimer’s disease, and cardiovascular diseases (Sandgren et al., 1990; Black et al., 1997; Satoh et al., 2000; Umemura et al., 2014; Kefaloyianni et al., 2016; Zhang et al., 2016; Kim et al., 2020; Shalaby et al., 2020; Adu-Amankwaah et al., 2021a). Increasing evidence suggests that various ADAMs and other related metalloenzymes play crucial roles in cardiovascular pathophysiology via the modulation of inflammation, angiogenesis, metabolism, cell proliferation, and cell migration (Adu-Amankwaah et al., 2021a; Kawai et al., 2021). Among the ADAMs identified so far (22 in humans, 34 in mice), ADAM8, 9, 10, 12, 17, 19 and closely related metalloenzymes including MMP2, MMP9, and meprin β are associated with cardiovascular conditions such as hypertension, atherosclerosis, aortic aneurysms, restenosis, acute coronary syndrome, cardiomyopathies and HF (Papazafiropoulou and Tentolouris, 2009; Broder and Becker-Pauly, 2013; Zhang et al., 2016; Adu-Amankwaah et al., 2021a; Kawai et al., 2021). According to Wichert et al., active meprin β is capable of inducing the proteolytic activities of ADAM9, 10, and 17 via specific prodomain cleavage (Wichert et al., 2019). The activation of MMP2 and MMP9 is part of the downstream signaling of ADAM10 and 17, which are closely related in structure and function (Xiao et al., 2012; Jones et al., 2013). While ADAM10’s expression may be important in cancer and neurological disorders, ADAM17 is primarily responsible for coordinating proinflammatory responses during stress. The various substrates of ADAMs have been extensively reviewed elsewhere (Kawai et al., 2021). Remarkably, several members of the ADAM family share the same substrates, and this nonspecific relationship between ADAMs and their substrates complicates and intrigues the physiology of ADAMs. However, the main focus of this review is to elucidate the mechanistic signaling pathways of ADAM17 in HF development during chronic stress.
ADAM17 was discovered in 1997 and named TACE (TNFα converting enzyme), as it was initially known as the protease that converts membrane-bound pro-TNFα (mTNFα) to a soluble form through its cleavage activity (Black et al., 1997; Moss et al., 1997). However, recent studies show that this protease is not only responsible for the liberation of soluble TNFα (sTNFα) but has a relatively broad spectrum of over 90 substrates (Black et al., 1997; Moss et al., 1997; Lammich et al., 1999; Garton et al., 2003; Reddy et al., 2009; Riethmueller et al., 2017). ADAM17 can be activated by intracellular kinases, which include phosphate kinase c (PKC), receptor-stimulated p38 mitogen-activated-protein-kinases (p38 MAPKs), and extracellular signal-regulated kinases (ERKs) (Bell and Gööz, 2010; Xu et al., 2012; Adu-Amankwaah et al., 2021a). These kinases can also phosphorylate and activate rhomboid 1 and 2 (also known as iRhoms or pseudoproteases) (Grieve et al., 2017), which are responsible for trafficking, stabilization as well as activation of ADAM17 (Adrain and Freeman, 2012; McIlwain et al., 2012; Maretzky et al., 2013; Li et al., 2015). Its inhibition is mostly done via tissue inhibitor of metalloproteinase 3 (TIMP3), integrins and protein disulfide isomerases (PDIs) (Düsterhöft et al., 2013; Düsterhöft et al., 2019; Park et al., 2019; Figure 1).
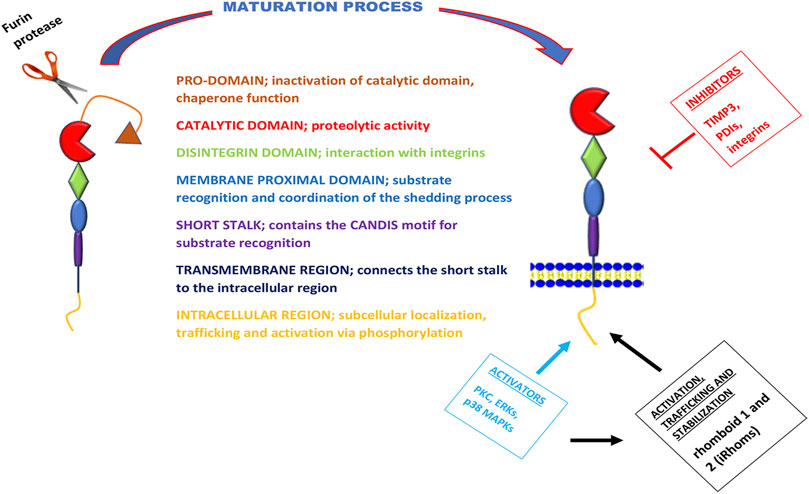
FIGURE 1. Schematic overview of the structure, function, maturation process, and regulation of ADAM17. The metalloprotease ADAM17 can be divided into seven domains with distinct functions, here separated by different colors. During maturation of ADAM17, the pro-domain is cleaved of by furin proteases. The activation of this metalloprotease is via its intracellular region by kinases; PKC, ERKs, and p38 MAPKs. These kinases are also known to phosphorylate and activate iRhoms for trafficking, stabilization, and cell surface expression of ADAM17. However, the inhibition of ADAM17 is mostly carried out by TIMP3, PDIs, and integrins.
A Disintegrin and Metalloprotease 17’s Structure
ADAM17 is a type-I transmembrane protein (Bode et al., 1993; Düsterhöft et al., 2019) with a similar class III snake venom metalloenzymes structure (Bode et al., 1993; Gooz, 2010). It comprises a prodomain, a catalytic domain, a disintegrin-like domain, a membrane-proximal domain (MPD), and a short stalk region, which together forms the extracellular part of the protease and are linked to an intracellular region (ICR) by a transmembrane part (Grötzinger et al., 2017). The catalytic domain possesses this metalloprotease’s proteolytic activity (Bode et al., 1993; Stöcker et al., 1995; Black et al., 1997); however, the preceding prodomain has chaperone-like functions that inhibit this catalytic activity, and it is cleaved off by furin proteases during the maturation of the protease (Schlöndorff et al., 2000). Though this cleavage step was primarily considered a prerequisite for the proteolytic activity of ADAM17, a study by Schwarz et al., revealed that ADAM17 was also active when cleavage by furin proteases was prevented by mutagenesis of the cleavage site (Schwarz et al., 2013). The disintegrin-like domain is needed for the interaction with integrins, a feature that ADAM17 shares with other ADAM family members (Bode et al., 1993; Düsterhöft et al., 2019). However, the membrane-proximal domain is only found in ADAM10 and ADAM17, but not the other family members (Stöcker et al., 1995; Grötzinger et al., 2017), and it is crucially involved in substrate recognition and coordination of the shedding process (Düsterhöft et al., 2013). The membrane-proximal domain is regulated by two disulfide bonds that are vulnerable to isomerization by PDI activity (Düsterhöft et al., 2013). The stalk region of ADAM17 contains the CANDIS motif (Conserved ADAM 17 Dynamic Interaction Sequence), which is located closer to the membrane-proximal domain near the plasma membrane and is vital for substrate recognition (Düsterhöft et al., 2014; Düsterhöft et al., 2019; Figure 1).
Activation of A Disintegrin and Metalloprotease 17 During Chronic Stress
Chronic stress is a renowned risk factor for several cardiovascular diseases (Kivimäki and Steptoe, 2018). One of the central neural pathways activated by stress is the autonomic nervous system. During chronic stress, the sympathetic nervous system can be continuously activated, which results in elevated levels of catecholamines (epinephrine and norepinephrine) (Won and Kim, 2016). Epinephrine and norepinephrine function as hormones and neurotransmitters that maintain homeostasis via adrenergic receptors (ARs), including alpha-adrenergic receptors (α-ARs) and beta-adrenergic receptors (β-ARs). Studies have demonstrated the involvement of ADAM17 with α1-AR (Chen et al., 2006) and β-AR signaling (Zhu and Steinberg, 2021). β-ARs account for the majority of the total ARs in the heart (O’Connell et al., 2014), particularly in apical myocytes (Paur et al., 2012; Machuki et al., 2019) and cardiac non-myocytes such as endothelial and immune cells (Myagmar et al., 2017; Adzika et al., 2021). Hence, a continuous increase in the levels of epinephrine and norepinephrine can result in overstimulation of β-ARs (Machuki et al., 2019). Epinephrine is a more potent ligand for β-ARs compared to norepinephrine (Scanzano and Cosentino, 2015). β-ARs are 7-transmembrane, G-protein coupled receptors which are divided into four subtypes, namely; β1-AR, β2-AR, β3-AR, and β4-AR (Ahlquist, 1948; Bylund et al., 1994; Granneman, 2001). In the heart, the β1-AR, β2-AR, and β3-AR are all broadly expressed, with the β1-AR having the highest expression and the β3-AR having the lowest (Ahlquist, 1948; Madamanchi, 2007). The β4-AR is a low-affinity state of the β1-AR that is yet to be genetically and pharmacologically characterized (Granneman, 2001). The β2-AR and β3-AR can couple with Gαs or Gαi while β1-AR primarily couples with Gαs when activated (Machuki et al., 2019; Schena and Caplan, 2019). In physiological state, the activation of β2-AR and β3-AR couple with Gαs (Adzika et al., 2019; Machuki et al., 2019) and Gαi (Tchivileva et al., 2009; Schena and Caplan, 2019), respectively. Among these βARs, β2-AR is rarely depleted during stress, and it is also the most implicated in mediating signaling cascades in ventricular apical myocytes, cardiac endothelial and immune cells resulting in the initiation and progression of cardiovascular diseases (Paur et al., 2012; Adzika et al., 2019; Adu-Amankwaah et al., 2021a).
In the heart, the overstimulation of β-ARs on ventricular apical myocytes, cardiac endothelial and immune cells due to elevated levels of circulating catecholamine desensitize β1-ARs (Bristow et al., 1990; Paur et al., 2012; Machuki et al., 2019). According to Zhu and Steinberg, the inactivation of β1-ARs and its irresponsiveness to catecholamines in cardiomyocytes during stressful events is via a mechanism involving N-terminal truncation at R31↓L32 by ADAM17 (Zhu and Steinberg, 2021). As such, β2-ARs coupling to Gαi is induced (Bristow et al., 1990; Paur et al., 2012; Machuki et al., 2019; Adzika et al., 2021). In short-terms, Gαi signaling increases via Akt/PI3K/p38 MAPKs/ERKs to prevent cardiac insult (Magocsi et al., 2007; Lajevic et al., 2011; Hou et al., 2018). Recent findings have suggested that the prolonged hyperstimulation of β2-ARs on ventricular apical myocytes and cardiac immune cells induces the bindings of β-arrestin-2 and G protein-coupled receptor kinases (GRKs) to scaffold non-canonical signaling that activates ERKs and p38 MAPKs activities maladaptively, ultimately resulting in HF (Shenoy et al., 2006; Paur et al., 2012; Adzika et al., 2019; Adzika et al., 2021). Intriguingly, ADAM17 initiates its adverse remodeling cascade upon being phosphorylated by these kinases directly and indirectly. For instance, during the maturation of ADAM17, ERK-dependent threonine 735 (Thr735) phosphorylation is vital for it to reach the secretory pathway (Díaz-Rodríguez et al., 2002; Chemaly et al., 2017). Also, indirect ERKs or p38 MAPKs phosphorylation at 14-3-3 binding sites on the N-terminal of iRhoms turns to induce ADAM17’s trafficking, stabilization, and cell surface expressions (Bell and Gööz, 2010; Adrain and Freeman, 2012; McIlwain et al., 2012; Xu et al., 2012; Li et al., 2015; Grieve et al., 2017). On the cell surface, mature ADAM17 proteins exist as inactive homodimers coupled to their inhibitor, TIMP3. However, activation of the ERK or p38 MAPK pathway directly phosphorylates Thr735 on the intracellular domain of ADAM17 and transforms it from a dimer structure into an active monomer structure liberating it from TIMP3 (Xu et al., 2012; Chemaly et al., 2017). Following monomerization, ADAM17 then binds to the phosphatidylserine exposure at the outer leaflet of the cell membrane via its MPD and CANDIS, thereby initiating its cleaving process (Gooz, 2010; Figure 2).
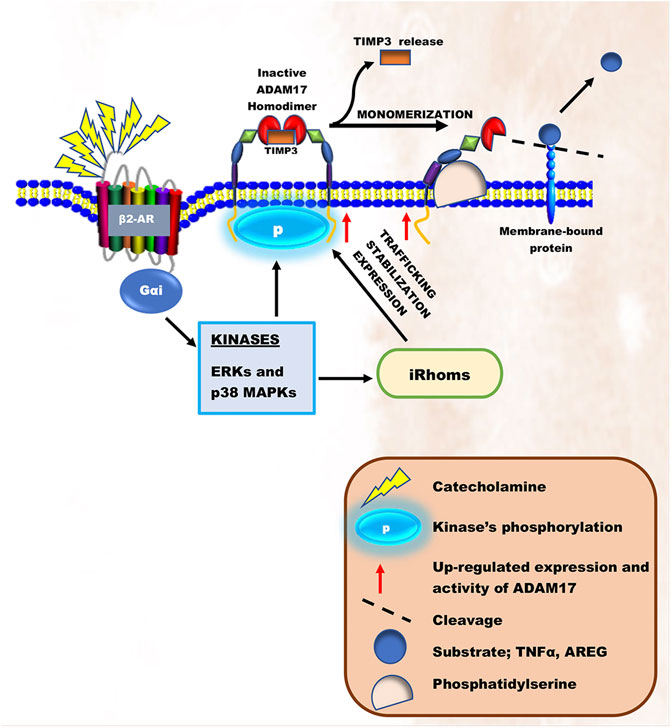
FIGURE 2. Schematic illustration of ADAM17’s activation during chronic catecholamine stress. Elevated catecholamines owning to chronic stress results in overstimulation of β2-ARs coupling with Gαi. Gαi signaling induces the activation of intracellular kinases, ERKs and p38 MAPKs. These kinases are known to either directly phosphorylate and activate ADAM17 or activate iRhoms responsible for trafficking, stabilization, and cell surface expression of ADAM17, thereby initiating its cleaving process.
A Disintegrin and Metalloprotease 17 in Cardiac Inflammation
ADAM17 plays a key role in cardiac inflammation, as it can cleave and activate several proinflammatory cytokines and their receptors. The most prominent examples include the cytokine TNFα, the TNFR 1 and 2, and the IL-6R (Black et al., 1997; Moss et al., 1997; Tellier et al., 2006; Reddy et al., 2009; Riethmueller et al., 2017).
Tumor Necrosis Factor α and its Receptors
The cytokine TNFα is a typical type-II transmembrane protein that belongs to the TNF superfamily (Düsterhöft et al., 2019). It is expressed as a membrane-bound protein, activated by the cleavage process of ADAM17 to release sTNFα (Black et al., 1997; Moss et al., 1997). The majority of the proinflammatory activities of TNFα are attributed to its soluble form. This cytokine activation can signal via two different receptors, TNFR1 and TNFR2 (Defer et al., 2007; Salmeri et al., 2015), expressed on cardiac myocytes (Defer et al., 2007). Interestingly, TNFR1 and 2 can also be cleaved from the surface of cells by ADAM17, and the resulting soluble TNFR (sTNFR) ectodomains retain their ability to bind mTNFα and therefore act as antagonistic decoy receptors (Rego et al., 2013; Figure 3). Besides their function as decoy receptors, sTNFR1 and 2 can also perform a very different biological function by binding to mTNFα on the cell surface and inducing signals within TNFα-expressing cells. This concept is known as “reverse signaling,” which is common among other TNF family members (Juhász et al., 2013). Although both TNFR1/2 can bind to the ligand sTNFα, the intracellular signaling cascades triggered, and the biological responses are markedly different (Düsterhöft et al., 2019). Most importantly, the intracellular region of TNFR1 contains a death domain capable of inducing direct programmed cell death when activated, which is absent in the intracellular region of TNFR2 (Düsterhöft et al., 2019). The binding of sTNFα to TNFR2 can result in the activation of nuclear factor kappa B (NF-κB) (Albensi, 2019; Adu-Amankwaah et al., 2021a), which is also expressed in myocytes, cardiac endothelial, and immune cells (Li et al., 2020). The NF-κB complex exists in an inactive state in the cytoplasm (Ghosh et al., 1998; Albensi, 2019). However, activation of TNFR2 can interact with the IκB kinase (IKK) complex resulting in the phosphorylation of IκB, subsequently causing IκB ubiquitination and degradation, leading to the activation of NF-κB dimer (Li and Karin, 2000; Israël, 2010). When activated, it then migrates into the nucleus (Sen and Smale, 2010) or mitochondria (Bottero et al., 2001; Cogswell et al., 2003). In the nucleus, it encodes genes of proinflammatory cytokines (pro-IL-18 and pro-IL-1β) and NLR family pyrin domain containing 3 (NLRP3) (Sen and Smale, 2010; Albensi, 2019). NLRP3 is an intracellular sensor that identifies a wide range of environmental irritants, microbial motifs, and endogenous danger signals, resulting in the formation and activation of the NLRP3 inflammasome. Activation of the inflammasome triggers caspase 1, which in turn, cleaves pro- IL-1β and pro- IL-18 to release their soluble forms (Bauernfeind et al., 2009; Xing et al., 2017; Swanson et al., 2019), thereby inducing necrosis and inflammation in cardiac cells (Li et al., 2018). Studies show that activated NF-κB can stimulate the intrinsic apoptotic pathway in the mitochondria via releasing cytochrome c, which triggers caspase cascades resulting in programmed cell death (Liu et al., 2004; Albensi, 2019; Adu-Amankwaah et al., 2021a; Figure 3). Irrefutably, increased levels of TNFα in the stress state has been linked to the pathophysiology of heart failure development in various clinical investigations (Ferrari et al., 1995; De Biase et al., 2003; Dunlay et al., 2008) and animal models (Bozkurt et al., 1998; Bryant et al., 1998; Moe et al., 2004; Guggilam et al., 2007; Adzika et al., 2021; Hou H. et al., 2021). For instance, a study carried out by Bryant et al. reveals that cardiac myocytes’ overproduction of TNFα is sufficient to cause heart failure, implying that this cytokine plays a causative role in the development of heart failure (Bryant et al., 1998). In an experimental heart failure model, in vivo TNFα inhibition reduced cardiac mitochondrial dysfunction, oxidative stress, and apoptosis (Moe et al., 2004). Additionally, in heart failure rats, TNF-alpha inhibition reduced chronic catecholamine-induced stress in the paraventricular nucleus and ameliorated cardiac function (Guggilam et al., 2007).
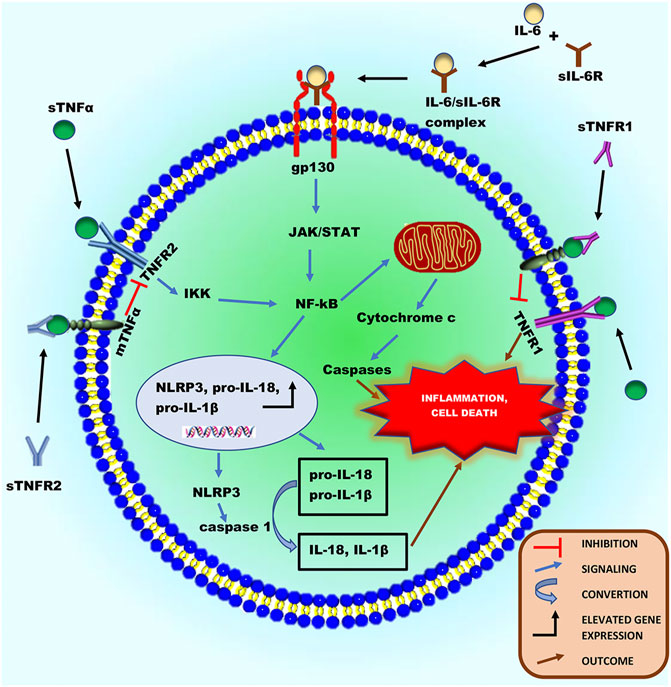
FIGURE 3. Schematic illustration of the inflammatory roles of ADAM17’s substrates, sTNFα and sIL-6R in a cardiac cell during chronic catecholamine stress. Following the proteolytic processing of ADAM17, sTNFα and IL-6 can bind to TNFR1/2 and sIL-6R, respectively activating downstream signaling cascades. Activated TNFR1 can directly induce inflammation and programmed cell death. The activation of TNFR2 can cause it to interact with the IκB kinase (IKK) complex resulting in the phosphorylation of IκB, thereby activating NF-κB. Also, the IL-6/sIL-6R complex formed from IL-6 binding to sIL-6R can directly activate the ubiquitously expressed glycoprotein-130 (gp130), thereby activating NF-κB. Activated NF-κB can either migrates into the nucleus or mitochondria. In the nucleus, it encodes genes of proinflammatory cytokines (pro-IL-18 and pro-IL-1β) and NLRP3, increasing their protein expression. NLRP3 inflammasome can activate caspase 1, which in turn cleaves pro- IL-1β and pro- IL-18 to release their soluble forms, to induce necrosis and inflammation in cardiac cells. Additionally, in the mitochondria, activated NF-κB can stimulate intrinsic apoptotic pathways via releasing cytochrome c, which triggers caspase cascades resulting in programmed cell death and inflammation.
IL-6 and its Receptor
IL-6 is a pleiotropic cytokine released in response to perturbations in homeostasis (Fontes et al., 2015). This cytokine has well-defined pro- and anti-inflammatory properties when activated. Interestingly, its receptor, IL-6R, can be cleaved by ADAM17 (Riethmueller et al., 2017; Garbers et al., 2018). The properties of IL-6 are determined by its stimulation and signaling processes. Thus, acute stimulation of IL-6 is mostly protective, while its chronic response causes long-term signaling leading to inflammation and autoimmunity (Fontes et al., 2015). Signaling via the membrane-bound IL-6 receptor (IL-6R) termed “classic signaling,” can only occur on cell types that express surface IL-6R, including hepatocytes and certain leukocytes’ subpopulations such as neutrophils (Wolf et al., 2014). However, signaling via soluble forms of the IL-6R, called IL-6 trans-signaling, can occur on all body cells since the IL-6/sIL-6R complex can directly bind to and activate the ubiquitously expressed glycoprotein-130 (gp130) without the need of a membrane-bound IL-6R (Wolf et al., 2014; Düsterhöft et al., 2019). IL-6 trans-signaling accounts mainly for the cytokine’s proinflammatory properties (Fontes et al., 2015; Düsterhöft et al., 2019). The glycoprotein-130 (gp130) receptor is widely expressed in mammals, including the developing and adult hearts (Podewski et al., 2003). In physiological state, activation of gp130 in the heart by IL-6 type cytokines induces signaling through three main pathways: 1) the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, 2) the phosphatidylinositol-3-kinase-dependent (PI3K)/Akt pathway and 3) the Ras/mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) signaling pathway (Podewski et al., 2003; Fischer and Hilfiker-Kleiner, 2008). These pathways have been demonstrated to play vital roles in cardiac development and protection (Yajima et al., 2006; Fischer and Hilfiker-Kleiner, 2008). However, in a chronic stress state characterized by elevated IL-6 and sIL-6R, continuous activation of gp130 in the heart can induce cardiac inflammation via gp130/JAK/STAT pathway (Podewski et al., 2003). This pathway can promote NF-κB activation, resulting in the release of proinflammatory cytokines, formation, and activation of inflammasomes, which mediate cell death and cardiac inflammation (Fischer and Hilfiker-Kleiner, 2008; Figure 4). Undeniably, it has been revealed that increased levels of gp130 proteins and IL-6 cytokines are strong predictive markers for morbidity and mortality in patients with HF (Fischer and Hilfiker-Kleiner, 2008). According to Ritschel et al., elevated levels of circulating sIL-6R and IL-6 were linked to future cardiovascular events and mortality in patients, implying that the IL-6 signaling pathway plays an essential role in the development of HF (Ritschel et al., 2016). Studies have also reported that the local gp130 receptor system in myocytes is altered in failing human hearts (Podewski et al., 2003; Fischer and Hilfiker-Kleiner, 2008). Furthermore, many animal studies have demonstrated that in a stress state, upregulated levels of IL-6 in myocardia enhance the development of heart failure while its inhibition improves cardiac function. (Lai et al., 2012; Zhao et al., 2016; Adzika et al., 2021; Hou H. et al., 2021; Huo S. et al., 2021).

FIGURE 4. Schematic illustration of the fibrotic role of sAREG, a substrate of ADAM17. After the proteolytic process of ADAM17, sAREG can activate EGFR, which is widely expressed on cardiac cells. The binding of sAREG to EGFR causes the receptor to undergo a conformational change known as “Dimerization,” resulting in homo- or heterodimers formation. This precedes an intracellular domain activation in its tyrosine residues by phosphorylation, promoting these same residues’ autophosphorylation in their homolog. Autophosphorylation of EGFR can activate it to induce the JAK/STAT pathway, leading to an increase in gene and protein expression of fibroblasts and pro-fibrotic factors such as TGF-β. Elevated fibroblasts can result in fibroblast activation, both mechanically by altered activation patterns and chemically by inflammatory mediators. Activated fibroblasts are transformed into myofibroblasts by TGF-β. Myofibroblasts are not present in normal cardiac tissue unless during cardiac injury and can induce pathological ECM remodeling, which characterizes cardiac fibrosis via the expression of α-SMA, collagen synthesis, and secretion of MMPs.
Currently, ADAM17 has not been directly associated with the regulation of T and B cells functions in the myocardia; however, these immune cells secret TNFα, which is keenly regulated by ADAM17 (Opata et al., 2013; Yang et al., 2013; Adu-Amankwaah et al., 2021a). Also, ADAM17’s targets on T and B cells have been implicated in their migration, differentiation, and effector functions (Link et al., 2017). Typically, Marczynska et al. demonstrated that the costimulatory ligand, ICOS ligand (ICOSL), is preferentially downregulated on the surface of B cells in an ADAM17-dependent way, despite the fact that recombinant ADAM17 does not proteolyze it in vitro (Marczynska et al., 2014). Therefore, it can be speculated that ADAM17 might directly regulate T and B cells’ functions in the myocardia during inflammation.
A Disintegrin and Metalloprotease 17 in Cardiac Fibrosis
ADAM17 is known to activate amphiregulin (AREG) via its proteolytic cleavage activity (Liu et al., 2018). AREG is synthesized as a type-I transmembrane protein (pro-AREG) that can engage in juxtracrine signaling on adjacent cells (Berasain and Avila, 2014). Alternatively, after proteolytic processing, the release of soluble AREG (sAREG) can act as an autocrine or paracrine factor (Berasain and Avila, 2014). sAREG is a ligand of the EGFR (Berasain and Avila, 2014; Liu et al., 2018), widely expressed on cardiac myocytes and fibroblasts (Berasain and Avila, 2014; Liu et al., 2018). In a physiological state, activation of EGFR in the heart induces major intracellular signaling cascades governing fibroblasts proliferation, migration, and collagen synthesis. However, prolonged activation of EGFR in a chronic stress state characterized by continuous elevation of sAREG can enhance cardiac fibroblast activation, proliferation, differentiation to myofibroblast, migration, and collagen synthesis (Liu et al., 2018). The binding of sAREG to EGFR, causes the receptor to undergo a conformational change inducing homo- or heterodimers formation (Dawson et al., 2005; Rayego-Mateos et al., 2018). This precedes an intracellular domain activation in its tyrosine residues by phosphorylation, promoting these same residues’ autophosphorylation in their homolog. Autophosphorylation of EGFR can activate it to induce the JAK/STAT pathway (Dawson et al., 2005; Rayego-Mateos et al., 2018). This signaling pathway plays a vital role in transducing stress and growth signals in the heart during cardiac fibrosis (Wagner and Siddiqui, 2012). The activation of the JAK/STAT pathway can increase the gene expression of fibroblasts and pro-fibrotic factors such as transforming growth factor-beta (TGF-β) (Wang et al., 2002). Physiologically, cardiac fibroblasts are responsible for the homeostasis of the extracellular matrix (ECM), which provides a structural scaffold for cardiomyocytes, distributes mechanical forces through the cardiac tissue, and mediates electrical conduction (Travers et al., 2016). However, elevated fibroblasts can result in fibroblast activation, both mechanically by altered activation patterns and chemically by inflammatory mediators (Wang et al., 2002). Notably, elevated TNFα and IL-6 secretions from macrophages, T and B lymphocytes during chronic inflammation also contributes to the aggravation of cardiac fibrosis as these cytokines stimulate fibroblast proliferation, differentiation to myofibroblast, and their migration (Wang et al., 2002; Adekunle et al., 2021). Also, TGF-β plays a vital role in ECM remodeling, cell mobility, and modulation of immune function. Increased levels of it are crucial in cell differentiation and proliferation of activated fibroblasts to myofibroblasts (Baum and Duffy, 2011). Myofibroblasts are not present in normal cardiac tissue unless during cardiac injury (Manabe et al., 2002). Myofibroblasts can induce pathological ECM remodeling (Manabe et al., 2002) via the expression of smooth muscle alpha-actin (α-SMA) (Sousa et al., 2007), collagen synthesis, and secretion of MMPs (Manabe et al., 2002). MMPs are responsible for the breakdown of the extracellular matrix in many diseases (Liu et al., 2006). Chronic secretion of MMPs in the heart leads to the degradation of collagen and elastin into peptide fragments resulting in elevated collagen deposition in the ECM, leading to scar formation. Although the formation of fibrotic scar tissue is an adaptive way of maintaining the structural integrity and pressure-generating capacity of the heart, myofibroblast persistence due to chronic stress can eventually result in the development of adverse changes in ventricular structure and compliance, which characterizes cardiac fibrosis (Liu et al., 2006; Figure 4). Intriguingly, ADAM17 upregulation does enhance the secretions of the aforementioned cytokine from both innate and adaptive immune cells either via direct or indirect cascade to cause maladaptive interstitial fibrosis.
Synergy of A Disintegrin and Metalloprotease 17-Induced Inflammation and Fibrosis in Heart Failure Development
HF development is characterized by a progressive condition associated with left ventricular (LV) systolic or diastolic dysfunction resulting in insufficient oxygen and nutrient supply to peripheral organs. It can exist in two main forms, namely, HF with preserved ejection fraction (HFpEF) and HF with reduced ejection fraction (HFrEF) (Van Linthout and Tschöpe, 2017; Adu-Amankwaah et al., 2021b). HFpEF is accompanied by diastolic dysfunction characterized by impaired ventricle relaxation and filling, increased ventricle stiffness, and elevated filling pressure to respond to pressure overload (Adekunle et al., 2021). On the flip side, HFrEF is associated with systolic dysfunction characterized by impaired left ventricular contractility, resulting in a reduced ejection fraction (Tanai and Frantz, 2015). Cardiac inflammation and fibrosis play a central role in HF development (Liu et al., 2018). Both can trigger HF development under several conditions, ranging from acute stress to chronic catecholamine stress.
The outcome of inflammation and fibrosis can contribute to the pathogenesis of the two main forms of HF. Although elevated serum concentrations of proinflammatory cytokines and fibrotic factors are common in both forms of HF, the pathomechanisms involved in each are different. For HFpEF, studies reveal that the outcome of chronic inflammation leads to progressive fibrosis, which eventually results in LV hypertrophy (Manabe et al., 2002; Salles et al., 2007; Wynn, 2008). Excessive increase in ECM components and cross-linking during LV hypertrophy can induce myocardial stiffness, thereby triggering HFpEF-specific, characterized by concentric cardiac remodeling and LV diastolic dysfunction (Paulus and Tschöpe, 2013). For example, collagen I which stiffens the myocardia, accounts for about 80% of the total collagen in the myocardia and increases most during LV hypertrophy (Barison et al., 2015). In addition, excessive cross-linking during LV hypertrophy stiffens the collagen matrix, making it more difficult to be broken down by proteinases (Travers et al., 2016). According to Hieda et al., increased myocardial stiffness is frequently observed in patients with HFpEF (Hieda et al., 2020). Regarding HFrEF, excessive cardiomyocyte death preceding necrosis or apoptosis due to persistent cardiac inflammation can result in cardiac atrophy. Continued loss of cardiac tissue can induce systolic dysfunction leading to HFrEF-specific, characterized by eccentric cardiac remodeling and dysfunction (Van Linthout and Tschöpe, 2017). Undeniably, several large studies have reported that patients with HFrEF characterized by systolic dysfunction have elevated serum levels of proinflammatory cytokines such as TNFα, IL-6, and IL-1β (Torre-Amione et al., 1996; Rauchhaus et al., 2000; Deswal et al., 2001).
Conclusion and Future Perspectives
ADAM17 is widely expressed by several mammalian cells. Evidence suggests that the number of identified substrates of this metalloprotease keeps increasing, implying that ADAM17 may play a central role in regulating several physiological and pathophysiological processes. Hence, its implicated in several human diseases as such heart failure is expected. Although, current drug treatments and the subsequent use of recognized medications have reduced mortality and hospitalization rate, particularly in HF patients with reduced ejection fraction (Berliner and Bauersachs, 2017), HF remain a major clinical and public health concern since it affects more than 23 million worldwide (Ayoub et al., 2017; Orso et al., 2017; Frantz et al., 2018); hence there is still a lot to discover about this condition which will serve as a key in establishing specific treatment and management guidelines to significantly reduced the rate of HF. This comprehensive review has provided extensive insight into the mechanisms underlying catecholamine-induced HF. Therefore, therapeutic prospects for the treatment and management of catecholamine-induced HF should also target the inhibition of ADAM17 and/or antagonize its activation and activities.
For decades, ADAM17 has been the subject of intense research. Since its identification as the tumor necrosis factor convertase, it has been an important therapeutic target, particularly in the setting of inflammatory diseases. Nonetheless, developing medications that target ADAM17 has proven more difficult than anticipated. This is owing to ADAM17’s multifunctionality, which includes the release of approximately 90 other substrates aside from tumor necrosis factor (TNF), as well as its structural similarities to other metalloproteinases (Calligaris et al., 2021). The most promising targets of ADAM17 (without any significant physiological consequences) appear to be inhibiting its phosphorylation by ERKs, p38 MAPKs, iRhom1, and iRhom2. These regulators are vital for trafficking, stabilization, and activation of ADAM17 (Bell and Gööz, 2010; Adrain and Freeman, 2012; McIlwain et al., 2012; Xu et al., 2012; Li et al., 2015; Grieve et al., 2017). Usage of pharmacologic agents capable of impeding ADAM17 phosphorylation by ERKs and p38 MAPKs may be an attractive potential target for downregulating ADAM17’s proteolytic activity. Also, it is well-known that iRhom2 is mainly expressed in proinflammatory immune cells, such as macrophages and neutrophils (Adrain and Freeman, 2012), whereas iRhom1 is predominantly expressed in non-immune cells (Issuree et al., 2013; Chemaly et al., 2017). Hence, it is tempting to hypothesize that inhibition of iRhom2 would aid in the downregulation of ADAM17 with no effects on non-immune cells. iRhom1 activities may then compensate for the iRhom2 blockade. Also, the inhibition of ADAM17 could be achieved by injecting its natural inhibitors (TIMP3, PDIs, and integrins). Notably, injection of TIMP3 has been shown to prevent heart failure post-myocardial infarction (Martz, 2014; Takawale et al., 2017; Chintalgattu et al., 2018). PDIs can also interact directly with ADAM17’s MPD, which catalyzes the isomerization of two disulfide bridges, lowering ADAM17’s activity (Willems et al., 2010). Furthermore, the binding of integrin α5β1 to ADAM17 via its disintegrin domain, according to Bax et al., inhibited its activity by altering its mediated cell adhesion and migration (Bax et al., 2004). Besides its natural inhibitors, miRNAs such as miR-124 (Sun et al., 2013), miR-145 (Doberstein et al., 2013), miR-152 (Su et al., 2014), and miR-326 (Cai et al., 2015) have been shown to suppress ADAM17 expression and limit substrate release by binding directly to the ADAM17 3′-UTR.
The modulation of ADAM17 is key in ameliorating cardiac function via attenuation of myocardial inflammation during chronic catecholamine stress. Thus, minimizing the levels of TNFα and other proinflammatory cytokines is necessary for the heart’s normal function; hence, inhibiting ADAM17 which facilitates the activities of these cytokines, might enhance cardiac health or delay the progression of its pathological remodeling into HF.
Author Contributions
The review idea was conceived by JA-A. JA-A drafted and wrote the manuscript. With the supervision of HS; JA-A, GA, AA, MN, RM, AB, NA, FH, YX, SA, and IN revised and proofread the manuscript. All authors read and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant No. 81461138036, No. 81370329), The Natural Science Foundation of the Jiangsu Higher Education Institutes of China (grant No. 17KJB180016), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the help of Prof. Festus Adzaku, Dr. Innocent Afeke, and Miss Mary Nyarko for proofreading the entire manuscript.
References
Adamo, L., Rocha-Resende, C., Prabhu, S. D., and Mann, D. L. (2020). Reappraising the Role of Inflammation in Heart Failure. Nat. Rev. Cardiol. 17, 269–285. doi:10.1038/s41569-019-0315-x
Adekunle, A. O., Adzika, G. K., Mprah, R., Ndzie Noah, M. L., Adu-Amankwaah, J., Rizvi, R., et al. (2021). Predominance of Heart Failure with Preserved Ejection Fraction in Postmenopausal Women: Intra- and Extra-cardiomyocyte Maladaptive Alterations Scaffolded by Estrogen Deficiency. Front. Cell Dev. Biol. 9, 685996. doi:10.3389/fcell.2021.685996
Adrain, C., and Freeman, M. (2012). New Lives for Old: Evolution of Pseudoenzyme Function Illustrated by iRhoms. Nat. Rev. Mol. Cell Biol. 13, 489–498. doi:10.1038/nrm3392
Adu-Amankwaah, J., Adzika, G. K., Adekunle, A. O., Ndzie Noah, M. L., Mprah, R., Bushi, A., et al. (2021a). The Synergy of ADAM17-Induced Myocardial Inflammation and Metabolic Lipids Dysregulation during Acute Stress: New Pathophysiologic Insights into Takotsubo Cardiomyopathy. Front. Cardiovasc. Med. 8, 696413. doi:10.3389/fcvm.2021.696413
Adu-Amankwaah, J., Mprah, R., Adekunle, A. O., Ndzie Noah, M. L., Adzika, G. K., Machuki, J. O. A., et al. (2021b). The Cardiovascular Aspect of COVID-19. Ann. Med. 53, 227–236. doi:10.1080/07853890.2020.1861644
Adzika, G. K., Hou, H., Adekunle, A. O., Rizvi, R., Adzraku, S. Y., Li, K., et al. (2021). Amlexanox and Forskolin Prevents Isoproterenol-Induced Cardiomyopathy by Subduing Cardiomyocyte Hypertrophy and Maladaptive Inflammatory Responses. Front. Cell Dev. Biol. 9, 719351. doi:10.3389/fcell.2021.719351
Adzika, G. K., Machuki, J. O. A., Shang, W., Hou, H., Ma, T., Wu, L., et al. (2019). Pathological Cardiac Hypertrophy: The Synergy of Adenylyl Cyclases Inhibition in Cardiac and Immune Cells during Chronic Catecholamine Stress. J. Mol. Med. 97, 897–907. doi:10.1007/s00109-019-01790-0
Ahlquist, R. P. (1948). A Study of the Adrenotropic Receptors. Am. J. Physiol. Legacy Content 153, 586–600. doi:10.1152/ajplegacy.1948.153.3.586
Albensi, B. C. (2019). What Is Nuclear Factor Kappa B (NF-Κb) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 7, 154. doi:10.3389/fcell.2019.00154
Anderson, D. R., Poterucha, J. T., Mikuls, T. R., Duryee, M. J., Garvin, R. P., Klassen, L. W., et al. (2013). IL-6 and its Receptors in Coronary Artery Disease and Acute Myocardial Infarction. Cytokine 62, 395–400. doi:10.1016/j.cyto.2013.03.020
Arokiasamy, S., Zakian, C., Dilliway, J., Wang, W., Nourshargh, S., and Voisin, M.-B. (2017). Endogenous TNFα Orchestrates the Trafficking of Neutrophils Into and Within Lymphatic Vessels During Acute Inflammation. Sci. Rep. 7, 44189. doi:10.1038/srep44189
Ayoub, K. F., Pothineni, N. V. K., Rutland, J., Ding, Z., and Mehta, J. L. (2017). Immunity, Inflammation, and Oxidative Stress in Heart Failure: Emerging Molecular Targets. Cardiovasc. Drugs Ther. 31, 593–608. doi:10.1007/s10557-017-6752-z
Barison, A., Grigoratos, C., Todiere, G., and Aquaro, G. D. (2015). Myocardial Interstitial Remodelling in Non-ischaemic Dilated Cardiomyopathy: Insights from Cardiovascular Magnetic Resonance. Heart Fail. Rev. 20, 731–749. doi:10.1007/s10741-015-9509-4
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., Macdonald, K., Speert, D., et al. (2009). Cutting Edge: NF-Κb Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 183, 787–791. doi:10.4049/jimmunol.0901363
Baum, J., and Duffy, H. S. (2011). Fibroblasts and Myofibroblasts: What are we Talking About? J. Cardiovasc. Pharmacol. 57, 376–379. doi:10.1097/fjc.0b013e3182116e39
Bax, D. V., Messent, A. J., Tart, J., Van Hoang, M., Kott, J., Maciewicz, R. A., et al. (2004). Integrin α5β1 and ADAM-17 Interact In Vitro and Co-localize in Migrating HeLa Cells. J. Biol. Chem. 279, 22377–22386. doi:10.1074/jbc.m400180200
Bell, H. L., and Gööz, M. (2010). ADAM-17 Is Activated by the Mitogenic Protein Kinase ERK in a Model of Kidney Fibrosis. Am. J. Med. Sci. 339, 105–107. doi:10.1097/MAJ.0b013e3181cb4487
Berasain, C., and Avila, M. A. (2014). Amphiregulin. Semin. Cell Developmental Biol. 28, 31–41. doi:10.1016/j.semcdb.2014.01.005
Berliner, D., and Bauersachs, J. (2017). Current Drug Therapy in Chronic Heart Failure: the New Guidelines of the European Society of Cardiology (ESC). Korean Circ. J. 47, 543–554. doi:10.4070/kcj.2017.0030
Biase, L., Pignatelli, P., Lenti, L., Tocci, G., Piccioni, F., Riondino, S., et al. (2003). Enhanced TNFα and Oxidative Stress in Patients with Heart Failure: Effect of TNFα on Platelet O2 - Production. Thromb. Haemost. 90, 317–325. doi:10.1160/th03-02-0105
Black, R. A., Rauch, C. T., Kozlosky, C. J., Peschon, J. J., Slack, J. L., Wolfson, M. F., et al. (1997). A Metalloproteinase Disintegrin that Releases Tumour-Necrosis Factor-α from Cells. Nature 385, 729–733. doi:10.1038/385729a0
Bode, W., Gomis-Rüth, F.-X., and Stöckler, W. (1993). Astacins, Serralysins, Snake Venom and Matrix Metalloproteinases Exhibit Identical Zinc-Binding Environments (HEXXHXXGXXH and Met-Turn) and Topologies and Should Be Grouped into a Common Family, the ‘metzincins’. FEBS Lett. 331, 134–140. doi:10.1016/0014-5793(93)80312-i
Bottero, V., Busuttil, V., Loubat, A., Magné, N., Fischel, J. L., Milano, G., et al. (2001). Activation of Nuclear Factor kappaB Through the IKK Complex by the Topoisomerase Poisons SN38 and Doxorubicin: A Brake to Apoptosis in HeLa Human Carcinoma Cells. Cancer Res. 61, 7785–7791.
Bozkurt, B., Kribbs, S. B., Clubb, F. J., Michael, L. H., Didenko, V. V., Hornsby, P. J., et al. (1998). Pathophysiologically Relevant Concentrations of Tumor Necrosis Factor-α Promote Progressive Left Ventricular Dysfunction and Remodeling in Rats. Circulation 97, 1382–1391. doi:10.1161/01.cir.97.14.1382
Bristow, M. R., Hershberger, R. E., Port, J. D., Gilbert, E. M., Sandoval, A., Rasmussen, R., et al. (1990). Beta-adrenergic Pathways in Nonfailing and Failing Human Ventricular Myocardium. Circulation 82, I12–I25.
Broder, C., and Becker-Pauly, C. (2013). The Metalloproteases Meprin α and Meprin β: Unique Enzymes in Inflammation, Neurodegeneration, Cancer and Fibrosis. Biochem. J. 450, 253–264. doi:10.1042/bj20121751
Bryant, D., Becker, L., Richardson, J., Shelton, J., Franco, F., Peshock, R., et al. (1998). Cardiac Failure in Transgenic Mice with Myocardial Expression of Tumor Necrosis Factor-α. Circulation 97, 1375–1381. doi:10.1161/01.cir.97.14.1375
Bylund, D. B., Eikenberg, D. C., Hieble, J. P., Langer, S. Z., Lefkowitz, R. J., Minneman, K. P., et al. (1994). International Union of Pharmacology Nomenclature of Adrenoceptors. Pharmacol. Rev. 46, 121–136.
Cabron, A.-S., El Azzouzi, K., Boss, M., Arnold, P., Schwarz, J., Rosas, M., et al. (2018). Structural and Functional Analyses of the Shedding Protease ADAM17 in HoxB8-Immortalized Macrophages and Dendritic-Like Cells. J. Immunol. 201, 3106–3118. doi:10.4049/jimmunol.1701556
Cai, M., Wang, Z., Zhang, J., Zhou, H., Jin, L., Bai, R., et al. (2015). Adam17, a Target of Mir-326, Promotes Emt-Induced Cells Invasion in Lung Adenocarcinoma. Cell Physiol. Biochem. 36, 1175–1185. doi:10.1159/000430288
Calligaris, M., Cuffaro, D., Bonelli, S., Spanò, D. P., Rossello, A., Nuti, E., et al. (2021). Strategies to Target ADAM17 in Disease: From its Discovery to the iRhom Revolution. Molecules 26. doi:10.3390/molecules26040944
Calvete, J., Marcinkiewicz, C., and Sanz, L. (2007). KTS and RTS-Disintegrins: Anti-angiogenic Viper Venom Peptides Specifically Targeting the α1β 1 Integrin. Curr. Pharm. Des. 13, 2853–2859. doi:10.2174/138161207782023766
Chemaly, M., Mcgilligan, V., Gibson, M., Clauss, M., Watterson, S., Alexander, H. D., et al. (2017). Role of Tumour Necrosis Factor Alpha Converting Enzyme (TACE/ADAM17) and Associated Proteins in Coronary Artery Disease and Cardiac Events. Arch. Cardiovasc. Dis. 110, 700–711. doi:10.1016/j.acvd.2017.08.002
Chen, L., Hodges, R. R., Funaki, C., Zoukhri, D., Gaivin, R. J., Perez, D. M., et al. (2006). Effects of α1D-adrenergic Receptors on Shedding of Biologically Active EGF in Freshly Isolated Lacrimal Gland Epithelial Cells. Am. J. Physiol. Cell Physiol. 291, C946–C956. doi:10.1152/ajpcell.00014.2006
Chintalgattu, V., Greenberg, J., Singh, S., Chiueh, V., Gilbert, A., O'neill, J. W., et al. (2018). Utility of Glycosylated TIMP3 Molecules: Inhibition of MMPs and TACE to Improve Cardiac Function in Rat Myocardial Infarct Model. Pharmacol. Res. Perspect. 6, e00442. doi:10.1002/prp2.442
Cogswell, P. C., Kashatus, D. F., Keifer, J. A., Guttridge, D. C., Reuther, J. Y., Bristow, C., et al. (2003). NF-κB and IκBα Are Found in the Mitochondria. J. Biol. Chem. 278, 2963–2968. doi:10.1074/jbc.m209995200
Cumberbatch, M., and Kimber, I. (1992). Dermal Tumour Necrosis Factor-Alpha Induces Dendritic Cell Migration to Draining Lymph Nodes, and Possibly Provides One Stimulus for Langerhans’ Cell Migration. Immunology 75, 257–263.
Damås, J. K., Eiken, H. G., Oie, E., Bjerkeli, V., Yndestad, A., Ueland, T., et al. (2000). Myocardial Expression of CC- and CXC-Chemokines and Their Receptors in Human End-Stage Heart Failure. Cardiovasc. Res. 47, 778–787. doi:10.1016/s0008-6363(00)00142-5
Dawson, J. P., Berger, M. B., Lin, C.-C., Schlessinger, J., Lemmon, M. A., and Ferguson, K. M. (2005). Epidermal Growth Factor Receptor Dimerization and Activation Require Ligand-Induced Conformational Changes in the Dimer Interface. Mol. Cell Biol. 25, 7734–7742. doi:10.1128/mcb.25.17.7734-7742.2005
De Queiroz, T. M., Xia, H., Filipeanu, C. M., Braga, V. A., and Lazartigues, E. (2015). α-Lipoic Acid Reduces Neurogenic Hypertension by Blunting Oxidative Stress-Mediated Increase in ADAM17. Am. J. Physiol. Heart Circ. Physiol. 309, H926–H934. doi:10.1152/ajpheart.00259.2015
Defer, N., Azroyan, A., Pecker, F., and Pavoine, C. (2007). TNFR1 and TNFR2 Signaling Interplay in Cardiac Myocytes. J. Biol. Chem. 282, 35564–35573. doi:10.1074/jbc.m704003200
Deswal, A., Petersen, N. J., Feldman, A. M., Young, J. B., White, B. G., and Mann, D. L. (2001). Cytokines and Cytokine Receptors in Advanced Heart Failure. Circulation 103, 2055–2059. doi:10.1161/01.cir.103.16.2055
Díaz-Rodríguez, E., Montero, J. C., Esparís-Ogando, A., Yuste, L., and Pandiella, A. (2002). Extracellular Signal-Regulated Kinase Phosphorylates Tumor Necrosis Factor Alpha-Converting Enzyme at Threonine 735: A Potential Role in Regulated Shedding. Mol. Biol. Cell 13, 2031–2044.
Doberstein, K., Steinmeyer, N., Hartmetz, A.-K., Eberhardt, W., Mittelbronn, M., Harter, P. N., et al. (2013). MicroRNA-145 Targets the Metalloprotease ADAM17 and Is Suppressed in Renal Cell Carcinoma Patients. Neoplasia 15, 218–IN31. doi:10.1593/neo.121222
Dunlay, S. M., Weston, S. A., Redfield, M. M., Killian, J. M., and Roger, V. L. (2008). Tumor Necrosis Factor-α and Mortality in Heart Failure. Circulation 118, 625–631. doi:10.1161/circulationaha.107.759191
Düsterhöft, S., Jung, S., Hung, C. W., Tholey, A., Sönnichsen, F. D., Grötzinger, J., et al. (2013). Membrane-Proximal Domain of a Disintegrin and Metalloprotease-17 Represents the Putative Molecular Switch of its Shedding Activity Operated by Protein-Disulfide Isomerase. J. Am. Chem. Soc. 135, 5776–5781. doi:10.1021/ja400340u
Düsterhöft, S., Höbel, K., Oldefest, M., Lokau, J., Waetzig, G. H., Chalaris, A., et al. (2014). A Disintegrin and Metalloprotease 17 Dynamic Interaction Sequence, the Sweet Tooth for the Human Interleukin 6 Receptor. J. Biol. Chem. 289, 16336–16348.
Düsterhöft, S., Lokau, J., and Garbers, C. (2019). The Metalloprotease ADAM17 in Inflammation and Cancer. Pathol. Res. Pract. 215, 152410.
Epelman, S., Lavine, K. J., Beaudin, A. E., Sojka, D. K., Carrero, J. A., Calderon, B., et al. (2014). Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity 40, 91–104. doi:10.1016/j.immuni.2013.11.019
Ferrari, R., Bachetti, T., Confortini, R., Opasich, C., Febo, O., Corti, A., et al. (1995). Tumor Necrosis Factor Soluble Receptors in Patients with Various Degrees of Congestive Heart Failure. Circulation 92, 1479–1486. doi:10.1161/01.cir.92.6.1479
Fischer, P., and Hilfiker-Kleiner, D. (2008). Role of Gp130-Mediated Signalling Pathways in the Heart and its Impact on Potential Therapeutic Aspects. Br. J. Pharmacol. 153 (Suppl. 1), S414–S427. doi:10.1038/bjp.2008.1
Fontes, J. A., Rose, N. R., and Čiháková, D. (2015). The Varying Faces of IL-6: From Cardiac protection to Cardiac Failure. Cytokine 74, 62–68. doi:10.1016/j.cyto.2014.12.024
Frantz, S., Falcao-Pires, I., Balligand, J.-L., Bauersachs, J., Brutsaert, D., Ciccarelli, M., et al. (2018). The Innate Immune System in Chronic Cardiomyopathy: a European Society of Cardiology (ESC) Scientific Statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail. 20, 445–459. doi:10.1002/ejhf.1138
Garbers, C., Heink, S., Korn, T., and Rose-John, S. (2018). Interleukin-6: Designing Specific Therapeutics for a Complex Cytokine. Nat. Rev. Drug Discov. 17, 395–412. doi:10.1038/nrd.2018.45
Garton, K. J., Gough, P. J., Philalay, J., Wille, P. T., Blobel, C. P., Whitehead, R. H., et al. (2003). Stimulated Shedding of Vascular Cell Adhesion Molecule 1 (VCAM-1) is Mediated by Tumor Necrosis Factor-α-Converting Enzyme (ADAM 17). J. Biol. Chem. 278, 37459–37464. doi:10.1074/jbc.m305877200
Ghosh, S., May, M. J., and Kopp, E. B. (1998). NF-Κb and REL PROTEINS: Evolutionarily Conserved Mediators of Immune Responses. Annu. Rev. Immunol. 16, 225–260. doi:10.1146/annurev.immunol.16.1.225
Gooz, M. (2010). ADAM-17: The Enzyme that Does it All. Crit. Rev. Biochem. Mol. Biol. 45, 146–169. doi:10.3109/10409231003628015
Granneman, J. G. (2001). The Putative β4-adrenergic Receptor is a Novel State of the β1-adrenergic Receptor. Am. J. Physiol. Endocrinol. Metab. 280, E199–E202. doi:10.1152/ajpendo.2001.280.2.e199
Grieve, A. G., Xu, H., Künzel, U., Bambrough, P., Sieber, B., and Freeman, M. (2017). Phosphorylation of iRhom2 at the Plasma Membrane Controls Mammalian TACE-dependent Inflammatory and Growth Factor Signalling. Elife 6, e23968. doi:10.7554/eLife.23968
Grötzinger, J., Lorenzen, I., and Düsterhöft, S. (2017). Molecular Insights into the Multilayered Regulation of ADAM17: The Role of the Extracellular Region. Biochim. Biophys. Acta Mol. Cell Res. 1864, 2088–2095.
Guggilam, A., Haque, M., Kerut, E. K., Mcilwain, E., Lucchesi, P., Seghal, I., et al. (2007). TNF-α Blockade Decreases Oxidative Stress in the Paraventricular Nucleus and Attenuates Sympathoexcitation in Heart Failure Rats. Am. J. Physiol. Heart Circ. Physiol. 293, H599–H609. doi:10.1152/ajpheart.00286.2007
Heidt, T., Courties, G., Dutta, P., Sager, H. B., Sebas, M., Iwamoto, Y., et al. (2014). Differential Contribution of Monocytes to Heart Macrophages in Steady-State and After Myocardial Infarction. Circ. Res. 115, 284–295. doi:10.1161/circresaha.115.303567
Hieda, M., Sarma, S., Hearon, C. M., Dias, K. A., Martinez, J., Samels, M., et al. (2020). Increased Myocardial Stiffness in Patients with High-Risk Left Ventricular Hypertrophy. Circulation 141, 115–123. doi:10.1161/circulationaha.119.040332
Hou, H., Adzika, G. K., Wu, Q., Ma, T., Ma, Y., Geng, J., et al. (2021). Estrogen Attenuates Chronic Stress-Induced Cardiomyopathy by Adaptively Regulating Macrophage Polarizations via β2-Adrenergic Receptor Modulation. Front. Cell Dev. Biol. 9, 737003. doi:10.3389/fcell.2021.737003
Hou, H., Zhao, Z., Machuki, J. O. a., Zhang, L., Zhang, Y., Fu, L., et al. (2018). Estrogen Deficiency Compromised the β2AR-Gs/Gi Coupling: Implications for Arrhythmia and Cardiac Injury. Pflugers Arch. Eur. J. Physiol. 470, 559–570. doi:10.1007/s00424-017-2098-4
Huo, S., Shi, W., Ma, H., Yan, D., Luo, P., Guo, J., et al. (2021). Alleviation of Inflammation and Oxidative Stress in Pressure Overload-Induced Cardiac Remodeling and Heart Failure via IL-6/STAT3 Inhibition by Raloxifene. Oxid Med. Cell Longev. 2021, 6699054. doi:10.1155/2021/6699054
Israël, A. (2010). The IKK Complex, a Central Regulator of NF-kappaB Activation. Cold Spring Harb Perspect. Biol. 2, a000158. doi:10.1101/cshperspect.a000158
Issuree, P. D., Maretzky, T., Mcilwain, D. R., Monette, S., Qing, X., Lang, P. A., et al. (2013). iRHOM2 is a Critical Pathogenic Mediator of Inflammatory Arthritis. J. Clin. Invest. 123, 928–932. doi:10.1172/JCI66168
Jackson, L. F., Qiu, T. H., Sunnarborg, S. W., Chang, A., Zhang, C., Patterson, C., et al. (2003). Defective Valvulogenesis in HB-EGF and TACE-Null Mice Is Associated with Aberrant BMP Signaling. EMBO J. 22, 2704–2716. doi:10.1093/emboj/cdg264
Jones, A. V., Lambert, D. W., Speight, P. M., and Whawell, S. A. (2013). ADAM 10 is Over Expressed in Oral Squamous Cell Carcinoma and Contributes to Invasive Behaviour through a Functional Association with αvβ6 Integrin. FEBS Lett. 587, 3529–3534. doi:10.1016/j.febslet.2013.09.010
Jones, G. W., Mcloughlin, R. M., Hammond, V. J., Parker, C. R., Williams, J. D., Malhotra, R., et al. (2010). Loss of CD4+ T Cell IL-6R Expression during Inflammation Underlines a Role for IL-6 Trans Signaling in the Local Maintenance of Th17 Cells. J. Immunol. 184, 2130–2139. doi:10.4049/jimmunol.0901528
Jones, J. C., Rustagi, S., and Dempsey, P. J. (2016). ADAM Proteases and Gastrointestinal Function. Annu. Rev. Physiol. 78, 243–276. doi:10.1146/annurev-physiol-021014-071720
Juhász, K., Buzás, K., and Duda, E. (2013). Importance of Reverse Signaling of the TNF Superfamily in Immune Regulation. Expert Rev. Clin. Immunol. 9, 335–348.
Kawai, T., Elliott, K. J., Scalia, R., and Eguchi, S. (2021). Contribution of ADAM17 and Related ADAMs in Cardiovascular Diseases. Cell Mol. Life Sci. 78, 4161–4187. doi:10.1007/s00018-021-03779-w
Kefaloyianni, E., Muthu, M. L., Kaeppler, J., Sun, X., Sabbisetti, V., Chalaris, A., et al. (2016). ADAM17 Substrate Release in Proximal Tubule Drives Kidney Fibrosis. JCI Insight 1. doi:10.1172/jci.insight.87023
Kim, H. J., Trinh, N. T., Choi, Y., Kim, W., Min, K. H., Kang, S. O., et al. (2020). ADAM17 Genetic Variants and the Response of TNF-α Inhibitor in Rheumatoid Arthritis Patients. Pgpm 13, 81–88. doi:10.2147/pgpm.s235035
Kivimäki, M., and Steptoe, A. (2018). Effects of Stress on the Development and Progression of Cardiovascular Disease. Nat. Rev. Cardiol. 15, 215–229.
Lafuse, W. P., Wozniak, D. J., and Rajaram, M. V. S. (2020). Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 10. doi:10.3390/cells10010051
Lai, N. C., Gao, M. H., Tang, E., Tang, R., Guo, T., Dalton, N. D., et al. (2012). Pressure Overload-Induced Cardiac Remodeling and Dysfunction in the Absence of Interleukin 6 in Mice. Lab. Invest. 92, 1518–1526. doi:10.1038/labinvest.2012.97
Lajevic, M. D., Suleiman, S., Cohen, R. L., and Chambers, D. A. (2011). Activation of P38 Mitogen-Activated Protein Kinase by Norepinephrine in T-Lineage Cells. Immunology 132, 197–208. doi:10.1111/j.1365-2567.2010.03354.x
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., et al. (1999). Constitutive and Regulated -Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by a Disintegrin Metalloprotease. Proc. Natl. Acad. Sci. 96, 3922–3927. doi:10.1073/pnas.96.7.3922
Li, H., Xia, Z., Chen, Y., Qi, D., and Zheng, H. (2018). Mechanism and Therapies of Oxidative Stress-Mediated Cell Death in Ischemia Reperfusion Injury. Oxid Med. Cell Longev. 2018, 2910643. doi:10.1155/2018/2910643
Li, N., and Karin, M. (2000). Signaling Pathways Leading to Nuclear Factor-Κb Activation. Methods Enzymol. 319, 273–279. doi:10.1016/s0076-6879(00)19027-5
Li, X., Bian, Y., Pang, P., Yu, S., Wang, X., Gao, Y., et al. (2020). Inhibition of Dectin-1 in Mice Ameliorates Cardiac Remodeling by Suppressing NF-Κb/nlrp3 Signaling after Myocardial Infarction. Int. Immunopharmacol. 80, 106116. doi:10.1016/j.intimp.2019.106116
Li, X., Maretzky, T., Weskamp, G., Monette, S., Qing, X., Issuree, P. D. A., et al. (2015). iRhoms 1 and 2 are Essential Upstream Regulators of ADAM17-Dependent EGFR Signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 6080–6085. doi:10.1073/pnas.1505649112
Lichtenthaler, S. F., Lemberg, M. K., and Fluhrer, R. (2018). Proteolytic Ectodomain Shedding of Membrane Proteins in Mammals-Hardware, Concepts, and Recent Developments. EMBO J. 37, e99456. doi:10.15252/embj.201899456
Link, M. A., Lücke, K., Schmid, J., Schumacher, V., Eden, T., Rose-John, S., et al. (2017). The Role of ADAM17 in the T-Cell Response against Bacterial Pathogens. PLoS One 12, e0184320. doi:10.1371/journal.pone.0184320
Liu, H., Ma, Y., Pagliari, L. J., Perlman, H., Yu, C., Lin, A., et al. (2004). TNF-α-Induced Apoptosis of Macrophages Following Inhibition of NF-Κb: A Central Role for Disruption of Mitochondria. J. Immunol. 172, 1907–1915. doi:10.4049/jimmunol.172.3.1907
Liu, L., Jin, X., Hu, C.-F., Zhang, Y.-P., Zhou, Z. e., Li, R., et al. (2018). Amphiregulin Enhances Cardiac Fibrosis and Aggravates Cardiac Dysfunction in Mice with Experimental Myocardial Infarction Partly through Activating EGFR-dependent Pathway. Basic Res. Cardiol. 113, 12. doi:10.1007/s00395-018-0669-y
Liu, P., Sun, M., and Sader, S. (2006). Matrix Metalloproteinases in Cardiovascular Disease. Can. J. Cardiol. 22 (Suppl. B), 25b–30b. doi:10.1016/s0828-282x(06)70983-7
Ma, Y. (2021). Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 10, 1676. doi:10.3390/cells10071676
Machuki, J. O. a., Zhang, H.-Y., Geng, J., Fu, L., Adzika, G. K., Wu, L., et al. (2019). Estrogen Regulation of Cardiac cAMP-L-type Ca2+ Channel Pathway Modulates Sex Differences in Basal Contraction and Responses to β2AR-mediated Stress in Left Ventricular Apical Myocytes. Cell Commun Signal 17, 34. doi:10.1186/s12964-019-0346-2
Madamanchi, A. (2007). Beta-Adrenergic Receptor Signaling in Cardiac Function and Heart Failure. Mcgill J. Med. 10, 99–104.
Magocsi, M., Vizi, E. S., Selmeczy, Z., Brózik, A., and Szelenyi, J. (2007). Multiple G-Protein-Coupling Specificity of β-adrenoceptor in Macrophages. Immunology 122, 503–513. doi:10.1111/j.1365-2567.2007.02658.x
Manabe, I., Shindo, T., and Nagai, R. (2002). Gene Expression in Fibroblasts and Fibrosis. Circ. Res. 91, 1103–1113. doi:10.1161/01.res.0000046452.67724.b8
Marczynska, J., Ozga, A., Wlodarczyk, A., Majchrzak-Gorecka, M., Kulig, P., Banas, M., et al. (2014). The Role of Metalloproteinase ADAM17 in Regulating ICOS Ligand-Mediated Humoral Immune Responses. J. Immunol. 193, 2753–2763. doi:10.4049/jimmunol.1302893
Maretzky, T., Mcilwain, D. R., Issuree, P. D. A., Li, X., Malapeira, J., Amin, S., et al. (2013). iRhom2 Controls the Substrate Selectivity of Stimulated ADAM17-Dependent Ectodomain Shedding. Proc. Natl. Acad. Sci. 110, 11433–11438. doi:10.1073/pnas.1302553110
Martz, L. (2014). Taking TIMP3 to Heart. Science-Business eXchange 7, 246. doi:10.1038/scibx.2014.246
Mcilwain, D. R., Lang, P. A., Maretzky, T., Hamada, K., Ohishi, K., Maney, S. K., et al. (2012). iRhom2 Regulation of TACE Controls TNF-Mediated Protection Against Listeria and Responses to LPS. Science 335, 229–232. doi:10.1126/science.1214448
Mishra, H. K., Long, C., Bahaie, N. S., and Walcheck, B. (2015). Regulation of CXCR2 Expression and Function by a Disintegrin and Metalloprotease-17 (ADAM17). J. Leukoc. Biol. 97, 447–454. doi:10.1189/jlb.3hi0714-340r
Moe, G. W., Marin-Garcia, J., Konig, A., Goldenthal, M., Lu, X., and Feng, Q. (2004). In Vivo TNF-α Inhibition Ameliorates Cardiac Mitochondrial Dysfunction, Oxidative Stress, and Apoptosis in Experimental Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 287, H1813–H1820. doi:10.1152/ajpheart.00036.2004
Moss, M. L., Jin, S.-L. C., Milla, M. E., Burkhart, W., Carter, H. L., Chen, W.-J., et al. (1997). Cloning of a Disintegrin Metalloproteinase that Processes Precursor Tumour-Necrosis Factor-Alpha. Nature 385, 733–736. doi:10.1038/385733a0
Mukerjee, S., Gao, H., Xu, J., Sato, R., Zsombok, A., and Lazartigues, E. (2019). ACE2 and ADAM17 Interaction Regulates the Activity of Presympathetic Neurons. Hypertension 74, 1181–1191. doi:10.1161/hypertensionaha.119.13133
Myagmar, B. E., Flynn, J. M., Cowley, P. M., Swigart, P. M., Montgomery, M. D., Thai, K., et al. (2017). Adrenergic Receptors in Individual Ventricular Myocytes: The Beta-1 and Alpha-1B are in All Cells, the Alpha-1A is in a Subpopulation, and the Beta-2 and Beta-3 Are Mostly Absent. Circ. Res. 120, 1103–1115. doi:10.1161/circresaha.117.310520
O’connell, T. D., Jensen, B. C., Baker, A. J., and Simpson, P. C. (2014). Cardiac Alpha1-Adrenergic Receptors: Novel Aspects of Expression, Signaling Mechanisms, Physiologic Function, and Clinical Importance. Pharmacol. Rev. 66, 308–333. doi:10.1124/pr.112.007203
Opata, M. M., Ye, Z., Hollifield, M., and Garvy, B. A. (2013). B Cell Production of Tumor Necrosis Factor in Response to Pneumocystis Murina Infection in Mice. Infect. Immun. 81, 4252–4260. doi:10.1128/iai.00744-13
Orso, F., Fabbri, G., and Maggioni, A. P. (2017). Epidemiology of Heart Failure. Handb Exp. Pharmacol. 243, 15–33. doi:10.1007/164_2016_74
Papazafiropoulou, A., and Tentolouris, N. (2009). Matrix Metalloproteinases and Cardiovascular Diseases. Hippokratia 13, 76–82.
Park, S., Lee, E. S., Park, N. H., Hwang, K., and Cho, E. G. (2019). Circadian Expression of TIMP3 Is Disrupted by UVB Irradiation and Recovered by Green Tea Extracts. Int. J. Mol. Sci. 20. doi:10.3390/ijms20040862
Paulus, W. J., and Tschöpe, C. (2013). A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 62, 263–271. doi:10.1016/j.jacc.2013.02.092
Paur, H., Wright, P. T., Sikkel, M. B., Tranter, M. H., Mansfield, C., O'gara, P., et al. (2012). High Levels of Circulating Epinephrine Trigger Apical Cardiodepression in a β2-adrenergic receptor/Gi-dependent Manner: A New Model of Takotsubo Cardiomyopathy. Circulation 126, 697–706. doi:10.1161/circulationaha.112.111591
Podewski, E. K., Hilfiker-Kleiner, D., Hilfiker, A., Morawietz, H., Lichtenberg, A., Wollert, K. C., et al. (2003). Alterations in Janus Kinase (JAK)-signal Transducers and Activators of Transcription (STAT) Signaling in Patients with End-Stage Dilated Cardiomyopathy. Circulation 107, 798–802. doi:10.1161/01.cir.0000057545.82749.ff
Rauchhaus, M., Doehner, W., Francis, D. P., Davos, C., Kemp, M., Liebenthal, C., et al. (2000). Plasma Cytokine Parameters and Mortality in Patients with Chronic Heart Failure. Circulation 102, 3060–3067. doi:10.1161/01.cir.102.25.3060
Rayego-Mateos, S., Rodrigues-Diez, R., Morgado-Pascual, J. L., Valentijn, F., Valdivielso, J. M., Goldschmeding, R., et al. (2018). Role of Epidermal Growth Factor Receptor (EGFR) and its Ligands in Kidney Inflammation and Damage. Mediators Inflamm. 2018, 8739473. doi:10.1155/2018/8739473
Reddy, A. B., Ramana, K. V., Srivastava, S., Bhatnagar, A., and Srivastava, S. K. (2009). Aldose Reductase Regulates High Glucose-Induced Ectodomain Shedding of Tumor Necrosis Factor (TNF)-alpha via Protein Kinase C-delta and TNF-Alpha Converting Enzyme in Vascular Smooth Muscle Cells. Endocrinology 150, 63–74. doi:10.1210/en.2008-0677
Rego, S. L., Swamydas, M., Kidiyoor, A., Helms, R., De Piante, A., Lance, A. L., et al. (2013). Soluble Tumor Necrosis Factor Receptors Shed by Breast Tumor Cells Inhibit Macrophage Chemotaxis. J. Interferon Cytokine Res. 33, 672–681. doi:10.1089/jir.2013.0009
Riethmueller, S., Somasundaram, P., Ehlers, J. C., Hung, C. W., Flynn, C. M., Lokau, J., et al. (2017). Proteolytic Origin of the Soluble Human IL-6R In Vivo and a Decisive Role of N-Glycosylation. PLoS Biol. 15, e2000080. doi:10.1371/journal.pbio.2000080
Ritschel, V. N., Seljeflot, I., Arnesen, H., Halvorsen, S., Eritsland, J., Fagerland, M. W., et al. (2016). Circulating Levels of IL-6 Receptor and Gp130 and Long-Term Clinical Outcomes in ST-Elevation Myocardial Infarction. J. Am. Heart Assoc. 5. doi:10.1161/JAHA.115.003014
Russo, R. C., Guabiraba, R., Garcia, C. C., Barcelos, L. S., Roffê, E., Souza, A. L., et al. (2009). Role of the Chemokine Receptor CXCR2 in Bleomycin-Induced Pulmonary Inflammation and Fibrosis. Am. J. Respir. Cell Mol. Biol. 40, 410–421. doi:10.1165/rcmb.2007-0364oc
Salles, G. F., Fiszman, R., Cardoso, C. R., and Muxfeldt, E. S. (2007). Relation of Left Ventricular Hypertrophy with Systemic Inflammation and Endothelial Damage in Resistant Hypertension. Hypertension 50, 723–728. doi:10.1161/hypertensionaha.107.093120
Salmeri, F. M., Laganà, A. S., Sofo, V., Triolo, O., Sturlese, E., Retto, G., et al. (2015). Behavior of Tumor Necrosis Factor-α and Tumor Necrosis Factor Receptor 1/tumor Necrosis Factor Receptor 2 System in Mononuclear Cells Recovered from Peritoneal Fluid of Women with Endometriosis at Different Stages. Reprod. Sci. 22, 165–172. doi:10.1177/1933719114536472
Sandgren, E. P., Luetteke, N. C., Palmiter, R. D., Brinster, R. L., and Lee, D. C. (1990). Overexpression of TGF Alpha in Transgenic Mice: Induction of Epithelial Hyperplasia, Pancreatic Metaplasia, and Carcinoma of the Breast. Cell 61, 1121–1135. doi:10.1016/0092-8674(90)90075-p
Santos-Zas, I., Lemarié, J., Tedgui, A., and Ait-Oufella, H. (2018). Adaptive Immune Responses Contribute to Post-ischemic Cardiac Remodeling. Front. Cardiovasc. Med. 5, 198. doi:10.3389/fcvm.2018.00198
Santos-Zas, I., Lemarié, J., Zlatanova, I., Cachanado, M., Seghezzi, J. C., Benamer, H., et al. (2021). Cytotoxic CD8(+) T Cells Promote Granzyme B-dependent Adverse post-ischemic Cardiac Remodeling. Nat. Commun. 12, 1483. doi:10.1038/s41467-021-21737-9
Satoh, M., Iwasaka, J., Nakamura, M., Akatsu, T., Shimoda, Y., and Hiramori, K. (2004). Increased Expression of Tumor Necrosis Factor-Alpha Converting Enzyme and Tumor Necrosis Factor-Alpha in Peripheral Blood Mononuclear Cells in Patients with Advanced Congestive Heart Failure. Eur. J. Heart Fail. 6, 869–875. doi:10.1016/j.ejheart.2004.02.007
Satoh, M., Nakamura, M., Saitoh, H., Satoh, H., Maesawa, C., Segawa, I., et al. (1999). Tumor Necrosis Factor-Alpha-Converting Enzyme and Tumor Necrosis Factor-Alpha in Human Dilated Cardiomyopathy. Circulation 99, 3260–3265. doi:10.1161/01.cir.99.25.3260
Satoh, M., Nakamura, M., Satoh, H., Saitoh, H., Segawa, I., and Hiramori, K. (2000). Expression of Tumor Necrosis Factor-Alpha-Cconverting Enzyme and Tumor Necrosis Factor-Alpha in Human Myocarditis. J. Am. Coll. Cardiol. 36, 1288–1294. doi:10.1016/s0735-1097(00)00827-5
Scanzano, A., and Cosentino, M. (2015). Adrenergic Regulation of Innate Immunity: a Review. Front. Pharmacol. 6, 171. doi:10.3389/fphar.2015.00171
Schena, G., and Caplan, M. J. (2019). Everything You Always Wanted to Know about β(3)-AR * (* but Were Afraid to Ask). Cells 8, 357. doi:10.3390/cells8040357
Schlöndorff, J., Becherer, J. D., and Blobel, C. P. (2000). Intracellular Maturation and Localization of the Tumour Necrosis Factor Alpha Convertase (TACE). Biochem. J. 347 (Pt 1), 131–138.
Schwarz, J., Broder, C., Helmstetter, A., Schmidt, S., Yan, I., Müller, M., et al. (2013). Short-term TNFα Shedding Is Independent of Cytoplasmic Phosphorylation or Furin Cleavage of ADAM17. Biochim. Biophys. Acta 1833, 3355–3367. doi:10.1016/j.bbamcr.2013.10.005
Sen, R., and Smale, S. T. (2010). Selectivity of the NF-{kappa}B Response. Cold Spring Harb Perspect. Biol. 2, a000257. doi:10.1101/cshperspect.a000257
Shalaby, L., Thounaojam, M., Tawfik, A., Li, J., Hussein, K., Jahng, W. J., et al. (2020). Role of Endothelial ADAM17 in Early Vascular Changes Associated with Diabetic Retinopathy. J. Clin. Med. 9. doi:10.3390/jcm9020400
Shenoy, S. K., Drake, M. T., Nelson, C. D., Houtz, D. A., Xiao, K., Madabushi, S., et al. (2006). Beta-Arrestin-Dependent, G Protein-independent ERK1/2 Activation by the Beta2 Adrenergic Receptor. J. Biol. Chem. 281, 1261–1273. doi:10.1074/jbc.m506576200
Sousa, A. M., Liu, T., Guevara, O., Stevens, J., Fanburg, B. L., Gaestel, M., et al. (2007). Smooth Muscle Alpha-Actin Expression and Myofibroblast Differentiation by TGFbeta Are Dependent upon MK2. J. Cell Biochem. 100, 1581–1592. doi:10.1002/jcb.21154
Stöcker, W., Grams, F., Baumann, U., Reinemer, P., Gomis-Rüth, F. X., Mckay, D. B., et al. (1995). The Metzincins-Ttopological and Sequential Relations between the Astacins, Adamalysins, Serralysins, and Matrixins (Collagenases) Define a Superfamily of Zinc-Peptidases. Protein Sci. 4, 823–840. doi:10.1002/pro.5560040502
Su, Y., Wang, Y., Zhou, H., Lei, L., and Xu, L. (2014). MicroRNA-152 Targets ADAM17 to Suppress NSCLC Progression. FEBS Lett. 588, 1983–1988. doi:10.1016/j.febslet.2014.04.022
Sun, Y., Li, Q., Gui, H., Xu, D. P., Yang, Y. L., Su, D. F., et al. (2013). MicroRNA-124 Mediates the Cholinergic Anti-Inflammatory Action through Inhibiting the Production of Pro-inflammatory Cytokines. Cell Res. 23, 1270–1283. doi:10.1038/cr.2013.116
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 19, 477–489. doi:10.1038/s41577-019-0165-0
Takawale, A., Zhang, P., Azad, A., Wang, W., Wang, X., Murray, A. G., et al. (2017). Myocardial Overexpression of TIMP3 after Myocardial Infarction Exerts Beneficial Effects by Promoting Angiogenesis and Suppressing Early Proteolysis. Am. J. Physiol. Heart Circ. Physiol. 313, H224–h236. doi:10.1152/ajpheart.00108.2017
Tanai, E., and Frantz, S. (2015). Pathophysiology of Heart Failure. Compr. Physiol. 6, 187–214. doi:10.1002/cphy.c140055
Tchivileva, I. E., Tan, K. S., Gambarian, M., Nackley, A. G., Medvedev, A. V., Romanov, S., et al. (2009). Signaling Pathways Mediating Beta3-Adrenergic Receptor-Induced Production of Interleukin-6 in Adipocytes. Mol. Immunol. 46, 2256–2266. doi:10.1016/j.molimm.2009.04.008
Tellier, E., Canault, M., Rebsomen, L., Bonardo, B., Juhan-Vague, I., Nalbone, G., et al. (2006). The Shedding Activity of ADAM17 is Sequestered in Lipid Rafts. Exp. Cell Res 312, 3969–3980. doi:10.1016/j.yexcr.2006.08.027
Torre-Amione, G., Kapadia, S., Benedict, C., Oral, H., Young, J. B., and Mann, D. L. (1996). Proinflammatory Cytokine Levels in Patients with Depressed Left Ventricular Ejection Fraction: a Report from the Studies of Left Ventricular Dysfunction (SOLVD). J. Am. Coll. Cardiol. 27, 1201–1206. doi:10.1016/0735-1097(95)00589-7
Travers, J. G., Kamal, F. A., Robbins, J., Yutzey, K. E., and Blaxall, B. C. (2016). Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 118, 1021–1040. doi:10.1161/circresaha.115.306565
Umemura, M., Isozaki, T., Ishii, S., Seki, S., Oguro, N., Miura, Y., et al. (2014). Reduction of Serum ADAM17 Level Accompanied with Decreased Cytokines after Abatacept Therapy in Patients with Rheumatoid Arthritis. Int. J. Biomed. Sci. 10, 229–235.
Van Linthout, S., and Tschöpe, C. (2017). Inflammation - Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 14, 251–265. doi:10.1007/s11897-017-0337-9
Wagner, M. A., and Siddiqui, M. A. (2012). The JAK-STAT Pathway in Hypertrophic Stress Signaling and Genomic Stress Response. JAKSTAT 1, 131–141. doi:10.4161/jkst.20702
Wang, X., Shaw, S., Amiri, F., Eaton, D. C., and Marrero, M. B. (2002). Inhibition of the Jak/STAT Signaling Pathway Prevents the High Glucose-Induced Increase in Tgf-Beta and Fibronectin Synthesis in Mesangial Cells. Diabetes 51, 3505–3509. doi:10.2337/diabetes.51.12.3505
Wichert, R., Scharfenberg, F., Colmorgen, C., Koudelka, T., Schwarz, J., Wetzel, S., et al. (2019). Meprin β Induces Activities of A Disintegrin and Metalloproteinases 9, 10, and 17 by Specific Prodomain Cleavage. FASEB J. 33, 11925–11940. doi:10.1096/fj.201801371r
Willems, S. H., Tape, C. J., Stanley, P. L., Taylor, N. A., Mills, I. G., Neal, D. E., et al. (2010). Thiol Isomerases Negatively Regulate the Cellular Shedding Activity of ADAM17. Biochem. J. 428, 439–450. doi:10.1042/bj20100179
Wolf, J., Rose-John, S., and Garbers, C. (2014). Interleukin-6 and its Receptors: A Highly Regulated and Dynamic System. Cytokine 70, 11–20. doi:10.1016/j.cyto.2014.05.024
Won, E., and Kim, Y. K. (2016). Stress, the Autonomic Nervous System, and the Immune-Kynurenine Pathway in the Etiology of Depression. Curr. Neuropharmacol. 14, 665–673. doi:10.2174/1570159x14666151208113006
Wright, H. L., Cross, A. L., Edwards, S. W., and Moots, R. J. (2014). Effects of IL-6 and IL-6 Blockade on Neutrophil Function In Vitro and In Vivo. Rheumatology 53, 1321–1331. doi:10.1093/rheumatology/keu035
Wynn, T. A. (2008). Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 214, 199–210. doi:10.1002/path.2277
Xiao, L. J., Lin, P., Lin, F., Liu, X., Qin, W., Zou, H. F., et al. (2012). ADAM17 Targets MMP-2 and MMP-9 via EGFR-MEK-ERK Pathway Activation to Promote Prostate Cancer Cell Invasion. Int. J. Oncol. 40, 1714–1724. doi:10.3892/ijo.2011.1320
Xing, Y., Yao, X., Li, H., Xue, G., Guo, Q., Yang, G., et al. (2017). Cutting Edge: TRAF6 Mediates TLR/IL-1R Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome. J. Immunol. 199, 1561–1566. doi:10.4049/jimmunol.1700175
Xu, J., Sriramula, S., Xia, H., Moreno-Walton, L., Culicchia, F., Domenig, O., et al. (2017). Clinical Relevance and Role of Neuronal AT(1) Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ. Res. 121, 43–55. doi:10.1161/circresaha.116.310509
Xu, P., Liu, J., Sakaki-Yumoto, M., and Derynck, R. (2012). TACE Activation by MAPK-Mediated Regulation of Cell Surface Dimerization and TIMP3 Association. Sci. Signal. 5, ra34. doi:10.1126/scisignal.2002689
Yajima, T., Yasukawa, H., Jeon, E. S., Xiong, D., Dorner, A., Iwatate, M., et al. (2006). Innate Defense Mechanism against Virus Infection within the Cardiac Myocyte Requiring Gp130-STAT3 Signaling. Circulation 114, 2364–2373. doi:10.1161/circulationaha.106.642454
Yang, H., Li, T., Wei, J., Zhang, H., and He, S. (2013). Induction of Tumor Necrosis Factor (TNF) Release from Subtypes of T Cells by Agonists of Proteinase Activated Receptors. Mediators Inflamm. 2013, 165453. doi:10.1155/2013/165453
Zhang, P., Shen, M., Fernandez-Patron, C., and Kassiri, Z. (2016). ADAMs Family and Relatives in Cardiovascular Physiology and Pathology. J. Mol. Cell Cardiol. 93, 186–199. doi:10.1016/j.yjmcc.2015.10.031
Zhao, L., Cheng, G., Jin, R., Afzal, M. R., Samanta, A., Xuan, Y. T., et al. (2016). Deletion of Interleukin-6 Attenuates Pressure Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Circ. Res. 118, 1918–1929. doi:10.1161/circresaha.116.308688
Keywords: heart failure, cardiac inflammation, cardiac fibrosis, ADAM17, metalloenzymes, pro-inflammatory cytokines, fibrotic factors
Citation: Adu-Amankwaah J, Adzika GK, Adekunle AO, Ndzie Noah ML, Mprah R, Bushi A, Akhter N, Huang F, Xu Y, Adzraku SY, Nadeem I and Sun H (2021) ADAM17, A Key Player of Cardiac Inflammation and Fibrosis in Heart Failure Development During Chronic Catecholamine Stress. Front. Cell Dev. Biol. 9:732952. doi: 10.3389/fcell.2021.732952
Received: 29 June 2021; Accepted: 16 November 2021;
Published: 13 December 2021.
Edited by:
Isotta Chimenti, Sapienza University of Rome, ItalyReviewed by:
Julie Pires Da Silva, University of Colorado Anschutz Medical Campus, United StatesLaurel A. Grisanti, University of Missouri, United States
Copyright © 2021 Adu-Amankwaah, Adzika, Adekunle, Ndzie Noah, Mprah, Bushi, Akhter, Huang, Xu, Adzraku, Nadeem and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Sun, c3VuaEB4emhtdS5lZHUuY24=