 Gabriel E. Matos-Rodrigues
Gabriel E. Matos-Rodrigues Rodrigo A. P. Martins
Rodrigo A. P. Martins- Programa de Biologia Celular e do Desenvolvimento, Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
Several inherited human syndromes that severely affect organogenesis and other developmental processes are caused by mutations in replication stress response (RSR) genes. Although the molecular machinery of RSR is conserved, disease-causing mutations in RSR-genes may have distinct tissue-specific outcomes, indicating that progenitor cells may differ in their responses to RSR inactivation. Therefore, understanding how different cell types respond to replication stress is crucial to uncover the mechanisms of RSR-related human syndromes. Here, we review the ocular manifestations in RSR-related human syndromes and summarize recent findings investigating the mechanisms of RSR during eye development in vivo. We highlight a remarkable heterogeneity of progenitor cells responses to RSR inactivation and discuss its implications for RSR-related human syndromes.
Introduction
Maintenance of genome stability is essential for development and homeostasis, and failures in processes required for genomic stability are associated with various human syndromes (Ciccia and Elledge, 2010; Negrini et al., 2010; O’Driscoll, 2012). DNA replication, transcriptional regulation and chromatin modifications must be precisely coordinated to ensure faithful transmission of genetic information to stem/progenitor cell pools that expand during development (Prioleau and MacAlpine, 2016). During DNA synthesis, many sources of genotoxic stress may slow or stall the progression of replication forks, a condition defined as replication stress. As a consequence, cells trigger the replication stress response (RSR). Activation of the RSR signaling pathways may slow DNA replication and allow extra time for DNA repair, preventing DNA mutations, chromosomal rearrangements and, therefore, genomic instability (Zeman and Cimprich, 2014; Techer et al., 2017; Tubbs and Nussenzweig, 2017). Due to its essential role during replication and development, mutations in genes that code proteins required for RSR are associated with several developmental syndromes (Zeman and Cimprich, 2014; Munoz and Mendez, 2017). Here, we review the ocular manifestations in RSR-related human syndromes and discuss recent findings investigating tissue-specific RSR in the developing eye that may contribute to understanding how defective-RSR drives developmental malformations.
Replication Stress Response
Single-stranded DNA (ssDNA) breaks are proposed to be the most frequent DNA lesion (∼75%) and those are normally generated during DNA replication (Lindahl and Barnes, 2000; Tubbs and Nussenzweig, 2017). The formation ssDNA stretches and aberrant replication fork structures lead to the activation of the ATR kinase, the master regulator of the RSR (Figure 1A). When exposed, long ssDNA stretches are coated by the replication protein A (RPA) complex. ATR-interacting protein (ATRIP), a mutually dependent partner of ATR, directly binds to RPA and recruits ATR to the RPA-ssDNA sites (Hekmat-Nejad et al., 2000; Cortez et al., 2001; Zou and Elledge, 2003; Dart et al., 2004; Ball et al., 2005) (Figure 1A). ATR recruitment is not sufficient for its full activation and many regulatory partners are necessary (Saldivar et al., 2017). In double-stranded DNA-ssDNA (dsDNA-ssDNA) junctions, such as the ones found in stalled replication forks, ATR activation requires DNA topoisomerase II-binding protein 1 (TOPBP1) (Kumagai et al., 2006). TOPBP1 recruitment to dsDNA-ssDNA junctions depends on its interaction with RAD9, member of the 9-1-1 clamp complex (RAD9-RAD1-HUS1) that is recruited by the clamp load factor RAD17 (Bermudez et al., 2003; Kumagai et al., 2006; Delacroix et al., 2007) (Figure 1A). TOPBP1 recruitment depends on other proteins, including the MRE11-RAD50-NBS1 (MRN) complex and RHINO (Cotta-Ramusino et al., 2011; Duursma et al., 2013). Importantly, NBS1 and the MRN complex are directly involved in ATR activation and cells from patients with inactivating mutations in NBS1 exhibit defective RSR (Stiff et al., 2005; Duursma et al., 2013; Shiotani et al., 2013). In ssDNA regions without ssDNA-dsDNA junctions, RSR activation can be mediated by ETAA1, that directly interacts with RPA and activates ATR through its ATR-activating domain (AAD) domain (Figure 1A; Bass et al., 2016; Haahr et al., 2016; Lee et al., 2016; Thada and Cortez, 2019). Studies in human cell lines suggested that ATR activation by TOPBP1 and ETAA1 may occur in different contexts. TOPBP1 would activate ATR upon induced replication stress and ETAA1 would trigger ATR activation in unchallenged replication to avoid under-replicated DNA during the S-M transition (Saldivar et al., 2018). In addition, ATR can be directly activated by NBS1, although the mechanisms are not clear since NBS1 does not have an AAD domain (Kobayashi et al., 2013).
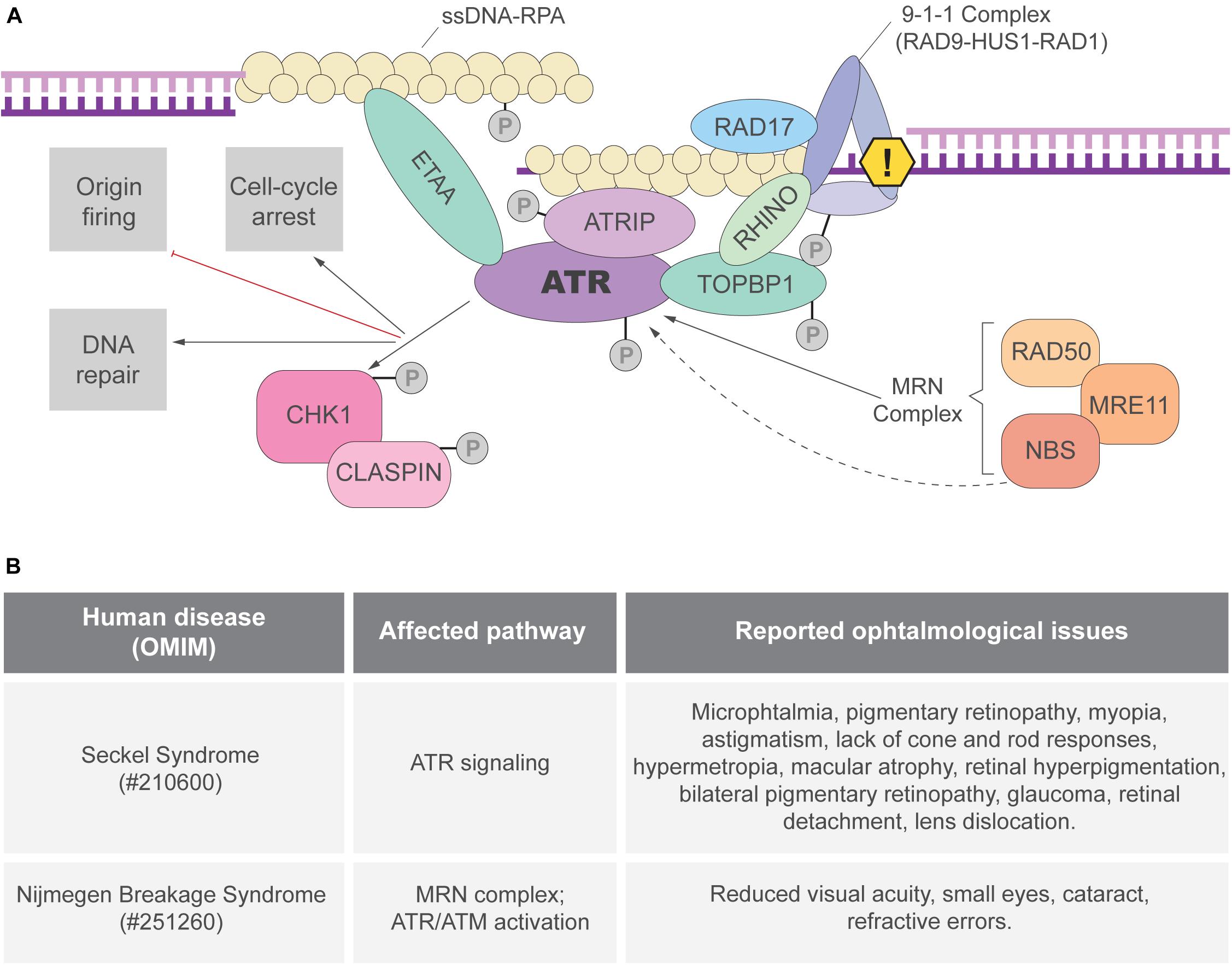
Figure 1. ATR activation and the replication stress response (RSR). (A) The ATRIP-ATR complex is recruited to RPA coated ssDNA. ATR can be directly activated by TOPBP1 or ETAA1 via their AAD domain. In regions of dsDNA-ssDNA junctions, the 9-1-1 complex is responsible for TOPBP1 recruitment and ATR activation. ETAA1-mediated ATR activation is not dependent on ssDNA-dsDNA junctions as ETAA1 directly binds to RPA-coated ssDNA. RHINO and the MRN complex are also important for ATR activation that phosphorylates different targets, including the CHK1 kinase. Once the RSR is activated, ATR and its downstream targets can modify different aspects of cell metabolism to prevent genome instability. (B) Ocular manifestations reported in patients of the RSR-related syndromes: Seckel (Lim and Wong, 1973; Guirgis et al., 2001; Reddy and Starr, 2007; Aktas et al., 2013; Krzyzanowska-Berkowska et al., 2014) and Nijmegen breakage syndromes (Varon et al., 1998; Gralek et al., 2011).
RSR depends not only on ATR-mediated signal transduction but also on its downstream effectors, specially the checkpoint protein 1 (CHK1) (Saldivar et al., 2017). ATR phosphorylates CHK1 in multiple sites and CHK1 activation depends on its partner CLASPIN (Kumagai and Dunphy, 2000; Liu et al., 2000, 2006; Zhao and Piwnica-Worms, 2001; Figure 1A). Once activated, the ATR-CHK1 signaling triggers local (e.g., dormant replication fork firing) and global (e.g., cell cycle arrest) responses to ensure the faithful duplication of the genome (Saldivar et al., 2017).
Inactivation of the Replication Stress Response in vivo
Highlighting the importance of ATR activation for unchallenged cell proliferation during development in vivo, inactivation of various “RSR genes” (here defined as genes necessary for full activation of ATR-CHK1 signaling following replication stress) is embryonic lethal in mice (Luo et al., 1999; Brown and Baltimore, 2000; de Klein et al., 2000; Liu et al., 2000; Weiss et al., 2000; Zhu et al., 2001; Dumon-Jones et al., 2003; Budzowska et al., 2004; Hopkins et al., 2004; Wang et al., 2005; Han et al., 2010; Jeon et al., 2011; Yang et al., 2016; Miosge et al., 2017). Although RSR has been extensively studied in various models, the mechanisms of the ATR activation and, therefore, the exact roles of ATR regulators in unchallenged replication in vivo are still not completely understood. For example, it was clear that ATR protein stability and function depend on its interaction with ATRIP in human cells (Cortez et al., 2001), however, prior to our recent work (Matos-Rodrigues et al., 2020a,b) ATRIP function had not been investigated in vivo. Moreover, while ETAA1 plays an essential role in an ATR-regulated S-G2 checkpoint in immortalized cells (Saldivar et al., 2018), ETAA1 null mice show a mild phenotype of partial embryonic lethality (Miosge et al., 2017). In contrast, ATR activation by TOPBP1 has an essential role in unchallenged replication in vivo, since disruption of ATR activation by TOPBP1 leads to embryonic lethality in mice (Zhou et al., 2013). These data indicate that ATR activation by TOPBP1, but not ETAA1, is essential for unchallenged replication in mice. The reason behind these distinct requirements in cultured human cells and in mouse development remains unclear.
Replication Stress Response in vivo: Focus on the Eye
The eye is the sensory organ responsible for vision and is composed of three main tissues: cornea, lens and retina (Figure 2A). The anterior segment of the eye comprises the cornea, the iris and the lens, a transparent structure that focus the light to the back of the eye. The main tissue of its posterior segment is the retina, the neural part of the eye responsible for detection and preprocessing of the visual stimuli before transmission to the visual centers of the brain through the optic nerve (Dowling, 1987). The development of these ocular tissues is extremely interdependent. In mice, on the ninth day of embryonic development (E9), a projection of the diencephalon, the optic vesicle, encounters the surface ectoderm of the head and starts eye organogenesis by triggering the invagination of both structures. While the invagination of the surface ectoderm gives rise to the lens, the retina originates from the invaginating optic vesicle (Miesfeld and Brown, 2019).
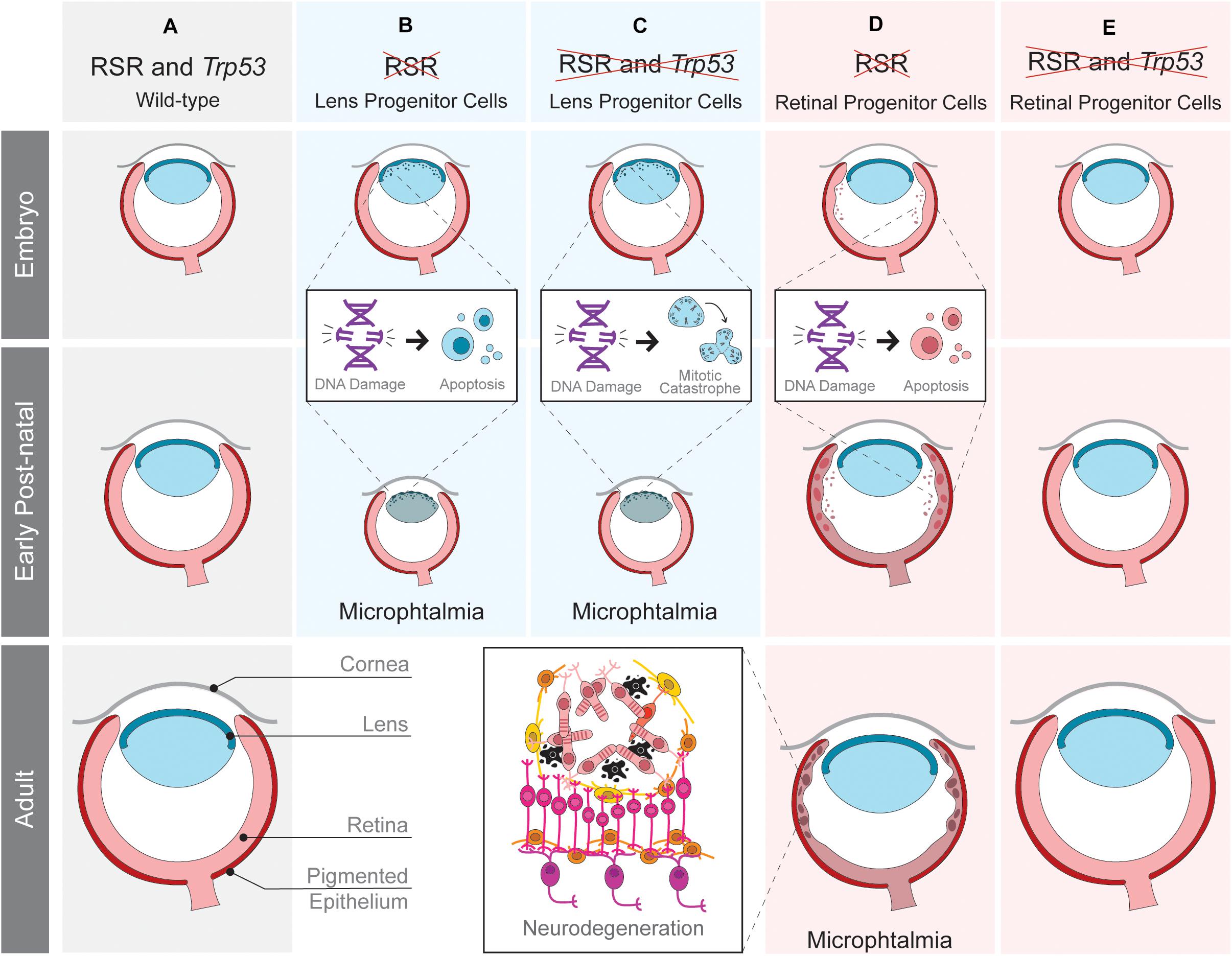
Figure 2. Tissue-specific effects of replication stress response (RSR) inactivation in developing mouse eye. Schematic representation of ocular development in wild-type mice (A) and the consequences of RSR inactivation in lens (B,C) or retinal progenitor cells (D,E) in Trp53-proficient (B,D) and Trp53-deficient scenarios (C,E). RSR inactivation by the loss of ATRIP leads to progenitor cell apoptosis in both the lens and retina. Only in the retina, Trp53-deficiency rescued embryonic apoptosis and the consequent secondary phenotypes.
Importantly, the eye represents a unique model to study the impact of defective RSR to organogenesis because: (1) of the vast knowledge about its development in mammals; (2) it is a non-essential organ, therefore a powerful model to analyze genetic interactions, and evaluate the long term consequences of essential genes inactivation; (3) there is a substantial amount of genetic tools available; (4) it is composed of tissues derived from distinct developmental lineages, making it ideal to study progenitor cells of different lineages. In addition, although clinical studies have shown ophthalmological manifestations in RSR-related syndromes (Figure 1B), the origins of these manifestations in these syndromes have been underexplored and raising awareness to this topic may bring important contributions to patients.
Loss-of-function mutations in ATR/ATRIP or in NBS1 are among the known causes of Seckel or Nijmegen breakage syndrome, respectively. These syndromes are characterized by moderate to severe tissue-growth impairments, neurodevelopmental defects and a series of ocular manifestations that have been reported in patients (Lim and Wong, 1973; Varon et al., 1998; Guirgis et al., 2001; Reddy and Starr, 2007; Gralek et al., 2011; Aktas et al., 2013; Krzyzanowska-Berkowska et al., 2014). Due to the recent advances on the understanding of these genes in eye development, we focus on their functions and its related human syndromes.
Ocular Manifestations in Replication Stress-Related Human Syndromes
Microphthalmia
Microphthalmia is a disorder characterized by abnormally small eyes that display high genetic heterogeneity and may occur as part of a syndrome. Disproportional ocular growth may contribute to microphthalmia, since microphthalmic eyes are more affected in the posterior segment than the anterior (Verma and Fitzpatrick, 2007). Microphthalmia has been reported in both Seckel and Nijmegen breakage syndromes (Figure 1B). Studies in animal models (discussed in the next sections) suggested that defective cell proliferation and increased cell death may be the cause of microphthalmia following the inactivation of RSR genes (Yang et al., 2006; Rodrigues et al., 2013; Matos-Rodrigues et al., 2020a,b). However, the mechanisms driving eye growth defects in syndromes caused by mutations in RSR-genes are far from being completely understood.
Cataract
Although treatable, cataracts are the most common cause of blindness. Congenital cataracts, the ones in which the opacification of the lens is detected at birth, are a clinical feature of almost 200 syndromic genetic diseases (Liu et al., 2017; Berry et al., 2020). Many evidences directly associates cataractogenesis and DNA damage. Increased DNA oxidation has been found in cataract patients and is thought to trigger cataractogenesis (Osnes-Ringen et al., 2013; Zhang et al., 2014; Erol Tinaztepe et al., 2017; Uwineza et al., 2019). DNA repair genes are known risk factors for cataract (Su et al., 2013; Cui et al., 2017; Yang et al., 2018) and cataracts have been reported in Seckel syndrome patients (Rao et al., 2011) (Figure 1B). Human lens progenitor cells from cataract patients display increased levels of DNA single strand breaks, a hallmark of replication stress (Kleiman and Spector, 1993). Finally, sources of replication stress, such as oxidative damage, UV-light and ionizing radiation cause cataract (Liu et al., 2017). As expected, induced DNA damage disturbs the proliferation and differentiation of lens progenitor cells, which is proposed to be an underlying cause of ionizing radiation induced cataract (Uwineza et al., 2019). The molecular mechanisms driving these processes are still to be determined.
Retinal Neurodegeneration
Glaucoma is characterized by structural damage to the optic nerve and retinal ganglion cell degeneration, leading to loss of vision due to the interruption of the transmission of information from the eye to the brain (Quigley, 2011; Calkins, 2012; Gemenetzi et al., 2012). Other retinopathies leading to neurodegeneration and vision loss include macular degeneration, retinopathy diabetic and retinitis pigmentosa (Massengill et al., 2018). Glaucoma, photoreceptors degeneration and lack of photoreceptor electrical responses were reported in patients with Seckel syndrome (Figure 1B). Importantly, replication stress has also been associated with the activation of pro-inflammatory pathways, which might fuel retinal neurodegeneration (Charlier and Martins, 2020; Ragu et al., 2020).
Lessons From Mouse Models
Genetic inactivation of NBS1 in mice was key to understanding the etiology of Nijmegen breakage syndrome (Frappart and McKinnon, 2008). While NBS1 knockout in mice led to early embryonic lethality (Zhu et al., 2001), neural tissue-specific inactivation of NBS1 resulted in abnormalities similar to patients including microcephaly, growth retardation, cerebellar defects and ataxia (Frappart et al., 2005). Importantly, NBS1 loss in the developing brain led to distinct outcomes depending on the progenitor cell affected. For example, NBS1 deficiency in progenitor cells of the neocortex induced cell cycle arrest. In the cerebellum, growth defects are driven by progenitor cell death (Frappart et al., 2005; Li et al., 2012; Rodrigues et al., 2013).
In the developing eye, NBS1-deficiency in the lens leads to cell death, proliferation defects and microphthalmia (Yang et al., 2006; Baranes et al., 2009; Rodrigues et al., 2013). During retinogenesis, NBS1 is also required for retinal progenitor cell survival, but its inactivation does not affect eye growth (Rodrigues et al., 2013), most likely due to a minor contribution of retinal growth to eye size. Finding that NBS1 loss led to microphthalmia only when inactivated in lens progenitor cells provided a first hint of how RSR inactivation could affect eye development in a tissue-specific manner (Yang et al., 2006; Rodrigues et al., 2013). Interestingly, NBS1-deficient mature retinas undergo degeneration of the optic nerve and loss of retinal function (Baranes et al., 2009), but the molecular and cellular mechanisms underlying this neurodegeneration remain unclear.
Interestingly, a specific synergy between NBS1 loss and TRP53 was also revealed in lens progenitor cells. In the developing brain, TRP53 inactivation rescues cell death and proliferation defects and brain growth defects caused by NBS1 loss (Frappart et al., 2005; Li et al., 2012). In the lens, however, Trp53 inactivation rescued progenitor cell death caused by NBS1 loss, but it did not rescue the defects in eye growth or cataract (Yang et al., 2006). Therefore, in NBS1-deficient lens progenitors, cell proliferation is blocked even when TRP53 is not functional, but the underlying mechanisms are still unknown. Importantly, in addition to its roles in RSR, NBS1 also participates in double-strand break signaling (Lee and Paull, 2005; Syed and Tainer, 2018), which could also factor in the diversity of outcomes observed.
Recently, we explored the function of another RSR gene by analyzing the function of ATRIP following tissue-specific inactivation in mice (Figure 2). As shown in transformed human cells (Cortez et al., 2001), ATR protein stability also depends on ATRIP in embryonic neural progenitor cells (Matos-Rodrigues et al., 2020a). Nestin-Cre-mediated inactivation of ATRIP in the developing central nervous system and in the eye leads to tissue growth defects (microphthalmia and microcephaly) that mirror the ones observed upon Atr inactivation (Lee et al., 2012). To understand the mechanisms underlying microphthalmia caused by ATRIP loss, we evaluated its contribution to cell cycle progression in Trp53-proficient and Trp53-deficient lens progenitor cells. In the presence of Trp53, ATRIP loss increases DNA damage and cell death, while in Trp53-deficient progenitors, ATRIP loss does not increase cell death, but leads to mitotic DNA damage and mitotic defects (Matos-Rodrigues et al., 2020a). These data suggest that inactivation of both genes might confer the ability to bypass the TRP53-mediated checkpoint and avoid cell death in S-phase, but ultimately culminating in mitotic catastrophe. Finally, as observed for NBS1, TRP53 deficiency does not rescue the microphthalmia caused by Atrip inactivation in lens progenitor cells.
We have also evaluated the effects of RSR inactivation in the mouse retina. ATRIP loss in embryonic retinal progenitor cells induces DNA damage accumulation and cell death, leading to lamination defects, photoreceptor degeneration and loss of vision (Matos-Rodrigues et al., 2020b). A previous study revealed photoreceptor degeneration in mice carrying an Atr hypomorphic mutation (Valdes-Sanchez et al., 2013). A role of ATR in the photoreceptor cilia was suggested to explain the observed neurodegeneration. Importantly, we found no evidence for a role of ATRIP in photoreceptors, since inactivation of Atrip specifically in these post-mitotic neurons did not affect retinal morphology or function. Because ATRIP is essential for ATR stability and all of its known functions are interdependent, further research is required to define the possible roles of the ATR-ATRIP complex in post-mitotic photoreceptor neurons.
In contrast to the lens, inactivation of Trp53 rescues the cell death of retinal progenitor cells, neurodegeneration and visual impairment caused by ATRIP loss, indicating that TRP53-dependent apoptosis is the driver of retinal malformations caused by Atrip inactivation (Matos-Rodrigues et al., 2020b). These findings reinforced the existence of tissue-specific effects of RSR inactivation in the developing eye. An intact RSR is essential for lens progenitor cell proliferation since Atrip inactivation in the lens either abolishes lens formation (aphakia) or causes microphthalmia (Matos-Rodrigues et al., 2020a). In retinal progenitor cells, Atrip inactivation also leads to DNA damage accumulation and cell death. However, retinal development is not completely impaired by the slight modifications in proliferation and differentiation caused by defective RSR (Matos-Rodrigues et al., 2020b). These results suggest that lens progenitor cells are more sensitive to RSR inactivation than retinal ones and point to a different synergy between Atrip and Trp53 when comparing retinal and lens progenitors. Trp53 inactivation rescues lens progenitor cells apoptosis, but does not rescue eye growth defects, which were likely caused by enhanced mitotic DNA damage and mitotic defects (Matos-Rodrigues et al., 2020a). In opposition, Trp53 inactivation completely rescues the developmental defects and the consequent neurodegeneration of the Atrip-deficient retinas (Figure 2). These observations are in agreement with previous data on the effects of NBS1 inactivation during mouse eye development.
Discussion
Based on the above-described studies we propose that the eye growth defects observed in replication-stress related syndrome patients are caused by the essential function of the affected genes in RSR in progenitor cells during embryogenesis. For example, tissue dysplasia and photoreceptor degeneration observed in Atrip-deficient retinas are a secondary consequence of progenitor apoptosis caused by the defective RSR in progenitor cells during embryonic development (Matos-Rodrigues et al., 2020b). Reports of retinal malformations and degeneration have been found in Seckel and Nijmegen breakage syndrome (Figure 1B). However, possible non-canonical functions of RSR genes in post-mitotic cells should not be overlooked, as it has been recently shown that ATR-CHK1 pathway can have a direct function on post-mitotic neurons activity and regeneration in model organisms (Kirtay et al., 2021; Li et al., 2021). Clinical investigations performing follow up in RSR-related syndromes patients associated with molecular diagnosis can bring important insights on the eye manifestations of these disorders.
The DDR is an evolutionarily conserved process that is often believed to operate by universal uniform principles. However, given that different progenitor cells have distinct transcriptional programs, metabolism, microenvironment and face different DNA-damaging insults, the DDR presents cell type- and developmental stage-specific adaptations (Blanpain et al., 2011; Rodrigues et al., 2013; Kafer and Cesare, 2020). The heterogeneous cellular outcomes of RSR inactivation in retinal and lens progenitor cells leads to the question of why progenitor cells show different sensitivity to RSR inactivation. Future studies in this field might bring exciting new contributions to the understanding of the RSR and its implications for developmental syndromes.
Author Contributions
Both authors wrote the manuscript and read and agreed to the published version of the manuscript.
Funding
This work was supported by the Brazilian National Council of Scientific and Technological Development (CNPq) (439031/2018-4 and 313064/2017-2 to RM), the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (E-26/210.500/2019 to RM), and The International Retinal Research Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We apologize to the authors whose work could not be cited due to space limitations. We would like to thank Clara F. Charlier for reading and helpful discussion.
References
Aktas, Z., Yuksel, N., Kula, S., Akman, A., and Hasanreisoglu, B. (2013). Childhood glaucoma as an ophthalmic manifestation of Seckel syndrome. J. Glaucoma 22, e3–e4. doi: 10.1097/IJG.0b013e318237cadf
Ball, H. L., Myers, J. S., and Cortez, D. (2005). ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell 16, 2372–2381. doi: 10.1091/mbc.e04-11-1006
Baranes, K., Raz-Prag, D., Nitzan, A., Galron, R., Ashery-Padan, R., Rotenstreich, Y., et al. (2009). Conditional inactivation of the NBS1 gene in the mouse central nervous system leads to neurodegeneration and disorganization of the visual system. Exp. Neurol. 218, 24–32. doi: 10.1016/j.expneurol.2009.03.026
Bass, T. E., Luzwick, J. W., Kavanaugh, G., Carroll, C., Dungrawala, H., Glick, G. G., et al. (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 18, 1185–1195. doi: 10.1038/ncb3415
Bermudez, V. P., Lindsey-Boltz, L. A., Cesare, A. J., Maniwa, Y., Griffith, J. D., Hurwitz, J., et al. (2003). Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 1633–1638. doi: 10.1073/pnas.0437927100
Berry, V., Georgiou, M., Fujinami, K., Quinlan, R., Moore, A., and Michaelides, M. (2020). Inherited cataracts: molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br. J. Ophthalmol. 104, 1331–1337. doi: 10.1136/bjophthalmol-2019-315282
Blanpain, C., Mohrin, M., Sotiropoulou, P. A., and Passegue, E. (2011). DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 8, 16–29. doi: 10.1016/j.stem.2010.12.012
Brown, E. J., and Baltimore, D. (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 14, 397–402.
Budzowska, M., Jaspers, I., Essers, J., de Waard, H., van Drunen, E., Hanada, K., et al. (2004). Mutation of the mouse Rad17 gene leads to embryonic lethality and reveals a role in DNA damage-dependent recombination. EMBO J. 23, 3548–3558. doi: 10.1038/sj.emboj.7600353
Calkins, D. J. (2012). Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog. Retin. Eye Res. 31, 702–719. doi: 10.1016/j.preteyeres.2012.07.001
Charlier, C. F., and Martins, R. A. P. (2020). Protective mechanisms against DNA replication stress in the nervous system. Genes (Basel) 11:730. doi: 10.3390/genes11070730
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Cortez, D., Guntuku, S., Qin, J., and Elledge, S. J. (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. doi: 10.1126/science.1065521
Cotta-Ramusino, C., McDonald, E. R. III, Hurov, K., Sowa, M. E., Harper, J. W., and Elledge, S. J. (2011). A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317. doi: 10.1126/science.1203430
Cui, N. H., Qiao, C., Chang, X. K., and Wei, L. (2017). Associations of PARP-1 variant rs1136410 with PARP activities, oxidative DNA damage, and the risk of age-related cataract in a Chinese Han population: A two-stage case-control analysis. Gene 600, 70–76. doi: 10.1016/j.gene.2016.11.019
Dart, D. A., Adams, K. E., Akerman, I., and Lakin, N. D. (2004). Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J. Biol. Chem. 279, 16433–16440. doi: 10.1074/jbc.M314212200
de Klein, A., Muijtjens, M., van Os, R., Verhoeven, Y., Smit, B., Carr, A. M., et al. (2000). Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 10, 479–482. doi: 10.1016/s0960-9822(00)00447-4
Delacroix, S., Wagner, J. M., Kobayashi, M., Yamamoto, K., and Karnitz, L. M. (2007). The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 21, 1472–1477. doi: 10.1101/gad.1547007
Dowling, J. E. (1987). The Retina: An Approachable Part of the Brain. Cambridge, MA: Belknap Press of Harvard University Press.
Dumon-Jones, V., Frappart, P. O., Tong, W. M., Sajithlal, G., Hulla, W., Schmid, G., et al. (2003). Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res. 63, 7263–7269.
Duursma, A. M., Driscoll, R., Elias, J. E., and Cimprich, K. A. (2013). A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol. Cell 50, 116–122. doi: 10.1016/j.molcel.2013.03.006
Erol Tinaztepe, O., Ay, M., and Eser, E. (2017). Nuclear and mitochondrial DNA of age-related cataract patients are susceptible to oxidative damage. Curr. Eye Res. 42, 583–588. doi: 10.1080/02713683.2016.1200100
Frappart, P. O., and McKinnon, P. J. (2008). Mouse models of DNA double-strand break repair and neurological disease. DNA Repair (Amst.) 7, 1051–1060. doi: 10.1016/j.dnarep.2008.03.007
Frappart, P. O., Tong, W. M., Demuth, I., Radovanovic, I., Herceg, Z., Aguzzi, A., et al. (2005). An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat. Med. 11, 538–544. doi: 10.1038/nm1228
Gemenetzi, M., Yang, Y., and Lotery, A. J. (2012). Current concepts on primary open-angle glaucoma genetics: a contribution to disease pathophysiology and future treatment. Eye (Lond.) 26, 355–369. doi: 10.1038/eye.2011.309
Gralek, M., Chrzanowska, K. H., Kanigowska, K., and Kocyla-Karczmarewicz, B. (2011). [Ocular findings in Nijmegen breakage syndrome]. Klin. Oczna 113, 153–155.
Guirgis, M. F., Lam, B. L., and Howard, C. W. (2001). Ocular manifestations of Seckel syndrome. Am. J. Ophthalmol. 132, 596–597. doi: 10.1016/s0002-9394(01)01046-7
Haahr, P., Hoffmann, S., Tollenaere, M. A., Ho, T., Toledo, L. I., Mann, M., et al. (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 18, 1196–1207. doi: 10.1038/ncb3422
Han, L., Hu, Z., Liu, Y., Wang, X., Hopkins, K. M., Lieberman, H. B., et al. (2010). Mouse Rad1 deletion enhances susceptibility for skin tumor development. Mol. Cancer 9:67. doi: 10.1186/1476-4598-9-67
Hekmat-Nejad, M., You, Z., Yee, M. C., Newport, J. W., and Cimprich, K. A. (2000). Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr. Biol. 10, 1565–1573. doi: 10.1016/s0960-9822(00)00855-1
Hopkins, K. M., Auerbach, W., Wang, X. Y., Hande, M. P., Hang, H., Wolgemuth, D. J., et al. (2004). Deletion of mouse rad9 causes abnormal cellular responses to DNA damage, genomic instability, and embryonic lethality. Mol. Cell Biol. 24, 7235–7248. doi: 10.1128/MCB.24.16.7235-7248.2004
Jeon, Y., Ko, E., Lee, K. Y., Ko, M. J., Park, S. Y., Kang, J., et al. (2011). TopBP1 deficiency causes an early embryonic lethality and induces cellular senescence in primary cells. J. Biol. Chem. 286, 5414–5422. doi: 10.1074/jbc.M110.189704
Kafer, G. R., and Cesare, A. J. (2020). A survey of essential genome stability genes reveals that replication stress mitigation is critical for peri-implantation embryogenesis. Front. Cell Dev. Biol. 8:416. doi: 10.3389/fcell.2020.00416
Kirtay, M., Sell, J., Marx, C., Haselmann, H., Ceanga, M., Zhou, Z. W., et al. (2021). ATR regulates neuronal activity by modulating presynaptic firing. Nat. Commun. 12:4067. doi: 10.1038/s41467-021-24217-2
Kleiman, N. J., and Spector, A. (1993). DNA single strand breaks in human lens epithelial cells from patients with cataract. Curr. Eye Res. 12, 423–431. doi: 10.3109/02713689309024624
Kobayashi, M., Hayashi, N., Takata, M., and Yamamoto, K. (2013). NBS1 directly activates ATR independently of MRE11 and TOPBP1. Genes Cells 18, 238–246. doi: 10.1111/gtc.12031
Krzyzanowska-Berkowska, P., Szumny, D., Mlynczak, T., Kisza, K., and Oficjalska, J. (2014). Bilateral retinal detachment in Seckel syndrome. Can. J. Ophthalmol. 49, e130–e131. doi: 10.1016/j.jcjo.2014.07.013
Kumagai, A., and Dunphy, W. G. (2000). Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 6, 839–849. doi: 10.1016/s1097-2765(05)00092-4
Kumagai, A., Lee, J., Yoo, H. Y., and Dunphy, W. G. (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955. doi: 10.1016/j.cell.2005.12.041
Lee, J. H., and Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554. doi: 10.1126/science.1108297
Lee, Y., Shull, E. R., Frappart, P. O., Katyal, S., Enriquez-Rios, V., Zhao, J., et al. (2012). ATR maintains select progenitors during nervous system development. EMBO J. 31, 1177–1189. doi: 10.1038/emboj.2011.493
Lee, Y. C., Zhou, Q., Chen, J., and Yuan, J. (2016). RPA-binding protein ETAA1 is an ATR activator involved in DNA replication stress response. Curr. Biol. 26, 3257–3268. doi: 10.1016/j.cub.2016.10.030
Li, F., Lo, T. Y., Miles, L., Wang, Q., Noristani, H. N., Li, D., et al. (2021). The Atr-Chek1 pathway inhibits axon regeneration in response to Piezo-dependent mechanosensation. Nat. Commun. 12:3845. doi: 10.1038/s41467-021-24131-7
Li, R., Yang, Y. G., Gao, Y., Wang, Z. Q., and Tong, W. M. (2012). A distinct response to endogenous DNA damage in the development of Nbs1-deficient cortical neurons. Cell Res. 22, 859–872. doi: 10.1038/cr.2012.3
Lim, K. H., and Wong, H. B. (1973). Ocular anomalies in Seckel’s syndrome. Aust. N. Z. J. Med. 3, 520–522. doi: 10.1111/j.1445-5994.1973.tb03132.x
Lindahl, T., and Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 65, 127–133. doi: 10.1101/sqb.2000.65.127
Liu, Q., Guntuku, S., Cui, X. S., Matsuoka, S., Cortez, D., Tamai, K., et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459.
Liu, S., Bekker-Jensen, S., Mailand, N., Lukas, C., Bartek, J., and Lukas, J. (2006). Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol. Cell Biol. 26, 6056–6064. doi: 10.1128/MCB.00492-06
Liu, Y. C., Wilkins, M., Kim, T., Malyugin, B., and Mehta, J. S. (2017). Cataracts. Lancet 390, 600–612. doi: 10.1016/S0140-6736(17)30544-5
Luo, G., Yao, M. S., Bender, C. F., Mills, M., Bladl, A. R., Bradley, A., et al. (1999). Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. U.S.A. 96, 7376–7381. doi: 10.1073/pnas.96.13.7376
Massengill, M. T., Ahmed, C. M., Lewin, A. S., and Ildefonso, C. J. (2018). Neuroinflammation in retinitis pigmentosa, diabetic retinopathy, and age-related macular degeneration: a minireview. Adv. Exp. Med. Biol. 1074, 185–191. doi: 10.1007/978-3-319-75402-4_23
Matos-Rodrigues, G. E., Grigaravicius, P., Lopez, B. S., Hofmann, T. G., Frappart, P. O., and Martins, R. A. P. (2020a). ATRIP protects progenitor cells against DNA damage in vivo. Cell Death Dis. 11:923. doi: 10.1038/s41419-020-03090-9
Matos-Rodrigues, G. E., Tan, P. B., Rocha-Martins, M., Charlier, C. F., Gomes, A. L., Cabral-Miranda, F., et al. (2020b). Progenitor death drives retinal dysplasia and neuronal degeneration in a mouse model of ATRIP-Seckel syndrome. Dis. Model. Mech. 13:dmm045807. doi: 10.1242/dmm.045807
Miesfeld, J. B., and Brown, N. L. (2019). Eye organogenesis: a hierarchical view of ocular development. Curr. Top. Dev. Biol. 132, 351–393. doi: 10.1016/bs.ctdb.2018.12.008
Miosge, L. A., Sontani, Y., Chuah, A., Horikawa, K., Russell, T. A., Mei, Y., et al. (2017). Systems-guided forward genetic screen reveals a critical role of the replication stress response protein ETAA1 in T cell clonal expansion. Proc. Natl. Acad. Sci. U.S.A. 114, E5216–E5225. doi: 10.1073/pnas.1705795114
Munoz, S., and Mendez, J. (2017). DNA replication stress: from molecular mechanisms to human disease. Chromosoma 126, 1–15. doi: 10.1007/s00412-016-0573-x
Negrini, S., Gorgoulis, V. G., and Halazonetis, T. D. (2010). Genomic instability–an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228. doi: 10.1038/nrm2858
O’Driscoll, M. (2012). Diseases associated with defective responses to DNA damage. Cold Spring Harb. Perspect. Biol. 4:a012773. doi: 10.1101/cshperspect.a012773
Osnes-Ringen, O., Azqueta, A. O., Moe, M. C., Zetterstrom, C., Roger, M., Nicolaissen, B., et al. (2013). DNA damage in lens epithelium of cataract patients in vivo and ex vivo. Acta Ophthalmol. 91, 652–656. doi: 10.1111/j.1755-3768.2012.02500.x
Prioleau, M. N., and MacAlpine, D. M. (2016). DNA replication origins-where do we begin? Genes Dev. 30, 1683–1697. doi: 10.1101/gad.285114.116
Ragu, S., Matos-Rodrigues, G., and Lopez, B. S. (2020). Replication stress, DNA damage, inflammatory cytokines and innate immune response. Genes (Basel) 11:409. doi: 10.3390/genes11040409
Rao, V., Deshpande, G., Rao, G., and Rehman, P. (2011). Cataract in Seckel Syndrome. Asian J. Ophthalmol. 13, 12–15. doi: 10.35119/asjoo.v13i1.19
Reddy, S., and Starr, C. (2007). Seckel syndrome and spontaneously dislocated lenses. J. Cataract Refract. Surg. 33, 910–912. doi: 10.1016/j.jcrs.2006.12.027
Rodrigues, P. M., Grigaravicius, P., Remus, M., Cavalheiro, G. R., Gomes, A. L., Rocha-Martins, M., et al. (2013). Nbn and atm cooperate in a tissue and developmental stage-specific manner to prevent double strand breaks and apoptosis in developing brain and eye. PLoS One 8:e69209. doi: 10.1371/journal.pone.0069209
Saldivar, J. C., Cortez, D., and Cimprich, K. A. (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 18, 622–636. doi: 10.1038/nrm.2017.67
Saldivar, J. C., Hamperl, S., Bocek, M. J., Chung, M., Bass, T. E., Cisneros-Soberanis, F., et al. (2018). An intrinsic S/G2 checkpoint enforced by ATR. Science 361, 806–810. doi: 10.1126/science.aap9346
Shiotani, B., Nguyen, H. D., Hakansson, P., Marechal, A., Tse, A., Tahara, H., et al. (2013). Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 3, 1651–1662. doi: 10.1016/j.celrep.2013.04.018
Stiff, T., Reis, C., Alderton, G. K., Woodbine, L., O’Driscoll, M., and Jeggo, P. A. (2005). Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 24, 199–208. doi: 10.1038/sj.emboj.7600504
Su, S., Yao, Y., Zhu, R., Liang, C., Jiang, S., Hu, N., et al. (2013). The associations between single nucleotide polymorphisms of DNA repair genes, DNA damage, and age-related cataract: Jiangsu Eye Study. Invest. Ophthalmol. Vis. Sci. 54, 1201–1207. doi: 10.1167/iovs.12-10940
Syed, A., and Tainer, J. A. (2018). The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 87, 263–294. doi: 10.1146/annurev-biochem-062917-012415
Techer, H., Koundrioukoff, S., Nicolas, A., and Debatisse, M. (2017). The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 18, 535–550. doi: 10.1038/nrg.2017.46
Thada, V., and Cortez, D. (2019). Common motifs in ETAA1 and TOPBP1 required for ATR kinase activation. J. Biol. Chem. 294, 8395–8402. doi: 10.1074/jbc.RA119.008154
Tubbs, A., and Nussenzweig, A. (2017). Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656. doi: 10.1016/j.cell.2017.01.002
Uwineza, A., Kalligeraki, A. A., Hamada, N., Jarrin, M., and Quinlan, R. A. (2019). Cataractogenic load–a concept to study the contribution of ionizing radiation to accelerated aging in the eye lens. Mutat. Res. Rev. Mutat. Res. 779, 68–81. doi: 10.1016/j.mrrev.2019.02.004
Valdes-Sanchez, L., De la Cerda, B., Diaz-Corrales, F. J., Massalini, S., Chakarova, C. F., Wright, A. F., et al. (2013). ATR localizes to the photoreceptor connecting cilium and deficiency leads to severe photoreceptor degeneration in mice. Hum. Mol. Genet. 22, 1507–1515. doi: 10.1093/hmg/dds563
Varon, R., Vissinga, C., Platzer, M., Cerosaletti, K. M., Chrzanowska, K. H., Saar, K., et al. (1998). Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93, 467–476. doi: 10.1016/s0092-8674(00)81174-5
Verma, A. S., and Fitzpatrick, D. R. (2007). Anophthalmia and microphthalmia. Orphanet. J. Rare Dis. 2:47. doi: 10.1186/1750-1172-2-47
Wang, Y., Putnam, C. D., Kane, M. F., Zhang, W., Edelmann, L., Russell, R., et al. (2005). Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 37, 750–755. doi: 10.1038/ng1587
Weiss, R. S., Enoch, T., and Leder, P. (2000). Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev. 14, 1886–1898.
Yang, C. C., Suzuki, M., Yamakawa, S., Uno, S., Ishii, A., Yamazaki, S., et al. (2016). Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat. Commun. 7:12135. doi: 10.1038/ncomms12135
Yang, M., Zhang, J., Su, S., Qin, B., Kang, L., Zhu, R., et al. (2018). Allelic interaction effects of DNA damage and repair genes on the predisposition to age-related cataract. PLoS One 13:e0184478. doi: 10.1371/journal.pone.0184478
Yang, Y. G., Frappart, P. O., Frappart, L., Wang, Z. Q., and Tong, W. M. (2006). A novel function of DNA repair molecule Nbs1 in terminal differentiation of the lens fibre cells and cataractogenesis. DNA Repair (Amst.) 5, 885–893. doi: 10.1016/j.dnarep.2006.05.004
Zeman, M. K., and Cimprich, K. A. (2014). Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9. doi: 10.1038/ncb2897
Zhang, J., Wu, J., Yang, L., Zhu, R., Yang, M., Qin, B., et al. (2014). DNA damage in lens epithelial cells and peripheral lymphocytes from age-related cataract patients. Ophthalmic Res. 51, 124–128. doi: 10.1159/000356399
Zhao, H., and Piwnica-Worms, H. (2001). ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 21, 4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001
Zhou, Z. W., Liu, C., Li, T. L., Bruhn, C., Krueger, A., Min, W., et al. (2013). An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet. 9:e1003702. doi: 10.1371/journal.pgen.1003702
Zhu, J., Petersen, S., Tessarollo, L., and Nussenzweig, A. (2001). Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 11, 105–109. doi: 10.1016/s0960-9822(01)00019-7
Keywords: genome stability, cell cycle, DNA damage, chekcpoint, ATR, organogenesis, retina, lens
Citation: Matos-Rodrigues GE and Martins RAP (2021) An Eye in the Replication Stress Response: Lessons From Tissue-Specific Studies in vivo. Front. Cell Dev. Biol. 9:731308. doi: 10.3389/fcell.2021.731308
Received: 26 June 2021; Accepted: 21 September 2021;
Published: 04 November 2021.
Edited by:
Nicolas Hoch, University of São Paulo, BrazilReviewed by:
Chunsik Lee, Sun Yat-sen University, ChinaVanesa Gottifredi, IIBBA-CONICET Leloir Institute Foundation, Argentina
Copyright © 2021 Matos-Rodrigues and Martins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel E. Matos-Rodrigues, Z2FicmllbC5yb2RyaWd1ZXNAbmloLmdvdg==; Rodrigo A. P. Martins, cm9kcmlnby5tYXJ0aW5zQGljYi51ZnJqLmJy
†Present address: Gabriel E. Matos-Rodrigues, Laboratory of Genome Integrity, National Cancer Institute, National Institutes of Health (NIH), Bethesda, MD, United States