 Tingxuan Wang
Tingxuan Wang Jianjian Li
Jianjian Li Qing Ma
Qing Ma- Shenzhen Key Laboratory of Synthetic Genomics, Guangdong Provincial Key Laboratory of Synthetic Genomics, CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China
Genomic imprinting is a term used for an intergenerational epigenetic inheritance and involves a subset of genes expressed in a parent-of-origin-dependent way. Imprinted genes are expressed preferentially from either the paternally or maternally inherited allele. Long non-coding RNAs play essential roles in regulating this allele-specific expression. In several well-studied imprinting clusters, long non-coding RNAs have been found to be essential in regulating temporal- and spatial-specific establishment and maintenance of imprinting patterns. Furthermore, recent insights into the epigenetic pathological mechanisms underlying human genomic imprinting disorders suggest that allele-specific expressed imprinted long non-coding RNAs serve as an upstream regulator of the expression of other protein-coding or non-coding imprinted genes in the same cluster. Aberrantly expressed long non-coding RNAs result in bi-allelic expression or silencing of neighboring imprinted genes. Here, we review the emerging roles of long non-coding RNAs in regulating the expression of imprinted genes, especially in human imprinting disorders, and discuss three strategies targeting the central long non-coding RNA UBE3A-ATS for the purpose of developing therapies for the imprinting disorders Prader–Willi syndrome and Angelman syndrome. In summary, a better understanding of long non-coding RNA-related mechanisms is key to the development of potential therapeutic targets for human imprinting disorders.
Introduction
In diploid organisms, most genes are transcribed in an unbiased fashion from both alleles. However, in a small subset of genes, genetically identical alleles can be expressed differentially, a process referred to as ASE. In the mammalian genome, common epigenetic examples of ASE include random X-chromosome inactivation in females (Lee, 2011; Deng et al., 2014), genomic imprinting (Peters, 2014), random MAE (Reinius and Sandberg, 2015), allelic expression of antigen receptor (Bergman and Cedar, 2004; Vettermann and Schlissel, 2010), clustered protocadherin (Chen and Maniatis, 2013), and olfactory receptors (Monahan and Lomvardas, 2015). Imprinted genes are expressed strictly or preferentially from either paternally or maternally inherited alleles (referred to as parent-of-origin) (Barlow and Bartolomei, 2014; Huang et al., 2018; Chen and Zhang, 2020). The ASE of imprinted genes depends on differential epigenetic markings during gametogenesis in germline cells, as opposed to gene sequences. After imprinting patterns become established in mature germlines, genomic imprinting in an individual is maintained until genome-wide erasure of epigenetic modification occurs in gamete precursors.
Genomic imprinting has been described in diverse organisms, including marsupials, flowering plants, and insects (Macdonald, 2012). In the human and mouse genome, genomic imprinting has been extensively observed, indicating the conservation and evolutionary significance of this epigenetic regulatory mechanism. While the expression of 1% of human protein-coding genes is estimated to be regulated via genomic imprinting (Im et al., 2005; Patten et al., 2016; Elbracht et al., 2020), many of these imprinted genes are essential for metabolism, development, and the nervous system (Monk et al., 2019; Tucci et al., 2019). Not surprisingly, dysregulated imprinting is closely associated with a broad spectrum of human developmental defects and genetic disorders, including PWS, AS, BWS, SRS, KOS14, and TS14 (Bian and Sun, 2011; Wan et al., 2017). The association between imprinted genes and the clinical features of these human diseases has also been documented in mouse models through the identification of homologous imprinted gene regions corresponding to the imprinted gene regions implicated in human imprinting disorders (Peters, 2014; Tucci et al., 2019).
Long non-coding RNAs are a subgroup of non-coding RNAs defined as having a length longer than 200 nucleotides, and are extensively expressed among the genome (Derrien et al., 2012; Harrow et al., 2012; Knauss and Sun, 2013). The number of lncRNA genes in the human genome has been estimated at 20,000 to 100,000 (Zhao et al., 2016; Fang et al., 2018; Uszczynska-Ratajczak et al., 2018). This number is greater than the canonical protein-coding genes in the human genome (Southan, 2017). lncRNAs are primarily retained in the nucleus, having short half-lives and a rapid turn-over rate compared to mRNAs (Clark et al., 2012; Derrien et al., 2012; Yoon et al., 2015). lncRNAs can regulate gene expression in at least three ways: at the transcription level by modulating gene transcription and chromatin structure, at the post-transcription level by affecting splicing and stability of RNA, and at the translation level by modulating protein translation (referred to review Statello et al., 2021). In the human and mouse genome, imprinted genes often reside together within clusters (2–20 genes), called imprinted clusters or imprinted domains (Ferguson-Smith, 2011). In mammals, lncRNAs are generally located in imprinted clusters that contribute to the establishment and maintenance of monoallelic expression at a genome-scale and long time-range (Andergassen et al., 2019). Here, we summarize the roles of lncRNAs in the regulation of genomic imprinting using several well-established imprinted clusters as examples. We also discuss how the expression pattern of lncRNAs and their epigenetic regulatory functions are affected in imprinting disorders and some cancers. Three potential strategies have been developed to target the central long non-coding RNA Ube3a-ATS for the purpose of therapeutically correcting the PWS/AS locus imprinting disorders. We also discuss the functional mechanisms of imprinted lncRNAs in the regulation of mono-allelic imprinted gene expression and how it could help us understand ASE mechanisms and underlying pathological mechanisms of human imprinting disorders, hopefully inspiring additional efficient therapeutic strategies.
Genomic Basis of Imprinting
Along with more profound analysis of patient samples and well-established mouse reciprocal crossing models using high-throughput sequencing, the monoallelic expression of imprinted genes has been observed extensively in mice and humans (Tucci et al., 2019). Methylomes and transcriptomes derived from human peripheral blood and various adult tissue samples have been combined to identify imprinted methylation and the distribution of imprinted genes across the genome (Baran et al., 2015; Zink et al., 2018). In order to identify mouse imprinted genes, parents from strains with different genetic backgrounds were crossed to obtain heterozygotic individuals, permitting the discrimination of parent-of-origin-dependent transcriptional effects from sequence-dependent allelic expression (Babak et al., 2008; Wang et al., 2008). Imprinted genes in mice are identified based on SNPs specific to paternal or maternal genetic backgrounds, thus permitting the quantitation and comparison of expression levels from both alleles. To date, around 160 imprinted genes have been identified in the human genome, and 200 in the mouse genome (Tucci et al., 2019; Chen and Zhang, 2020). Sixty three of these imprinted genes are shared, suggesting that mouse models could be helpful for understanding imprinting regulation in humans.
In the human and mouse genome, imprinted genes often reside together within imprinted clusters (Ferguson-Smith, 2011). More than 80% of the known imprinted genes in the mouse genome are clustered together in multi-gene ranging in size from less than 100 kb to several megabases (Barlow, 2011). Imprinted lncRNAs located in one imprinted cluster are coordinately controlled by shared regulatory factors, including parent-of-origin-dependent differentially methylated regions (DMRs) and lncRNAs (Peters, 2014). In well-studied imprinted clusters, allele-specific DNA methylation occurs in an independent ICR in the germline, referred to as germline-derived DMRs (gDMRs) or primary DMRs, and persists after fertilization. ICRs in imprinted clusters exhibit parent-of-origin-specific epigenetic modifications, including DNA methylation, governing different expression patterns of parentally inherited alleles (da Rocha and Gendrel, 2019). Around 35 imprinted gDMRs have been identified in the human genome (Monk et al., 2018) and 24 in the mouse genome to date (White et al., 2016). The establishment of gDMRs on paternal or maternal alleles (Figure 1A) is essential for regulating imprinted gene expression in embryonic development (Barlow, 2011; Kelsey and Feil, 2013; Elbracht et al., 2020). In early primordial germ cells, epigenetic marks are extensively erased genome-wide, including DNA methylation and histone modifications. In germline cells, DNA methylation of ICRs is re-established in gametes depending on the parent-of-origin. After fertilization, gDMRs escape secondary global epigenetic reprogramming. DNA methylation information at ICRs of the imprinted regions is retained. In this way, gDMRs of imprinted loci are established robustly during germline development and are resistant to genomic reprogramming after fertilization. Correspondingly, imprinting marks are inherited in a parent-specific manner (Chotalia et al., 2009; Henckel et al., 2012; da Rocha and Gendrel, 2019). gDMRs on the different parent-of-origin alleles are characterized by distinct chromatin configurations, marked with different histone modifications which are corresponding to ‘open chromatin’ and ‘close chromatin’ (Singh et al., 2011; Court et al., 2014; Sanli and Feil, 2015). The allele-specific methylation states of gDMRs are recognized by transcription factors with roles in maintaining parent-of-origin specific expression of the imprinted genes, such as ZFP57 protein (Riso et al., 2016). In total, differential methylation states of gDMRs on parental alleles are essential for the establishment of monoallelic gene expression.
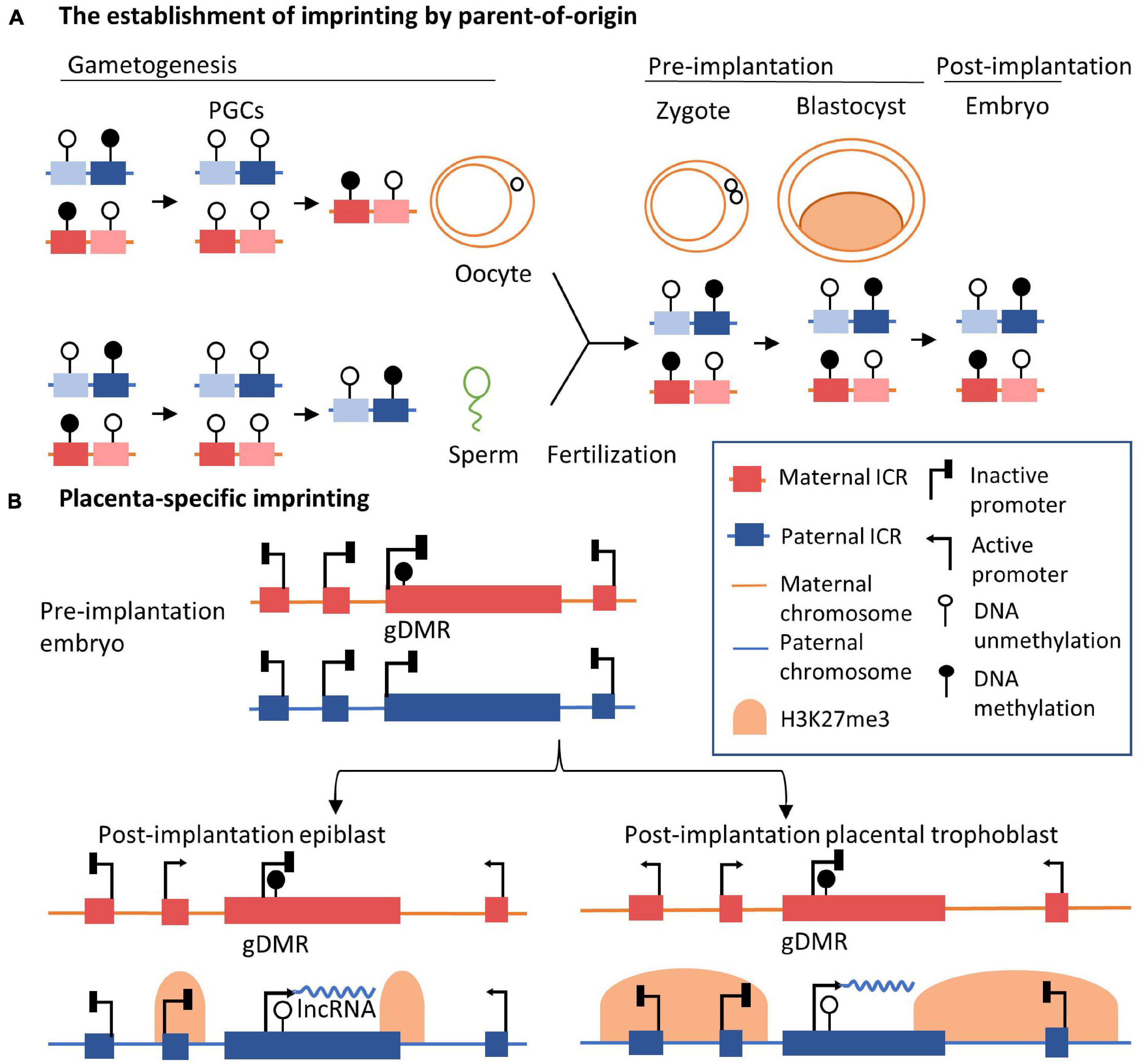
Figure 1. Genomic basis of the regulation of imprinting clusters. (A) The inheritance of allele-specific imprinting epigenetic marks across generations. In the early primordial germ cells, epigenetic modifications are erased at a genomic scale before the formation of germline cells. In the germline, parent-of-origin DNA methylation is established, shown as gDMRs. After fertilization and the formation of the zygote, the gDMRs are further maintained. Established imprinting patterns are maintained in blastocyte and somatic cells in adult tissues. (B) Imprinting in epiblast and placenta in imprinting loci, such as the Kcnq1ot1/Kcnq1 or Airn/Igf2r loci, is shown. In pre-implantation embryos, DNA methylation is inherited in the gDMR on the maternal allele, such as KvDMR1 of the Kcnq1ot1/Kcnq1 imprinting cluster. After implantation, the expression of lncRNA on the maternal allele is repressed by DNA methylation in gDMR, allowing the expression of neighbor genes. On the contrary, lncRNA is expressed from the paternal allele, inducing the spreading of H3K27me3 modifications in adjacent regions in the embryonic lineage (epiblast). In extra-embryonic lineage (placental trophoblast), the extended scale of H3K27me3 marks is longer than that seen in embryonic cells. Adjacent genes further away are also silenced on the paternal allele, indicating placenta-specific imprinting, such as Slc22a18 and Tssc4 genes in the Kcnq1ot1/Kcnq1 imprinting cluster. For simplicity, specific gene names are not shown.
Imprinting control regions govern DNA methylation and chromatin organization in early embryonic and adult lineages, resulting in the persistence of imprinting patterns across generations and their maintenance in adult tissues (Monk et al., 2019). After becoming established at early developmental stages in the germline, gDMRs are maintained in most somatic cells throughout life, resulting in the regulation of allelic expression of imprinted gene clusters. gDMRs also direct the rise of ‘secondary’ DMRs, normally corresponding to repressive chromatin modifications, condensed chromatin structure, and the gene-silencing function of imprinted lncRNAs (Sasaki et al., 1995; Nowak et al., 2011, p. 2; Rao et al., 2014; Tan et al., 2018; Zink et al., 2018). It has been shown that the imprinted expression of some genes is restricted to specific tissues or stages in developmental processes, along with additional allele-specific epigenetic marks further established in somatic cells. The expression patterns of these developmentally expressed imprinted genes are characterized by temporal- and spatial-specific biases (Perez et al., 2015; Andergassen et al., 2017). For example, UBE3A and IGF2 show imprinted expression patterns in specific human brain cell types (Rougeulle et al., 1997; Vu and Hoffman, 1997; Pham et al., 1998; Yamasaki et al., 2003; Li J. et al., 2020). In a study of ASE in diverse tissues from 178 adult post-mortem donors, paternally silenced IGF2 was reported in the human brain, different from the canonical paternal expression observed in other tissues (Baran et al., 2015). In the mouse E6.5 gastrulating epiblast, it has also been observed that Igf2r is expressed from both alleles and further becomes imprinted in the embryonic lineage at the gastrulation stage (Marcho et al., 2015). Besides, the placenta-specific imprinting has been observed, and the underlying mechanism has been well-understood, especially in the potassium voltage-gated channel subfamily Q member 1 (Kcnq1)/Kcnq1 antisense transcript 1 (Kcnq1ot1) cluster and the antisense of Igf2r non-protein coding RNA (Airn)/Igf2r cluster (Figure 1B; Sleutels et al., 2002; Andergassen et al., 2019; Hanna, 2020). The establishment of the placenta-specific imprinting initiates by allelic DNA methylation in pre-implantation embryos. In the placenta, the genomic profile of DNA methylation in imprinted DMRs is different, likely the result of an overall different pattern of placenta compared to other tissues (Schroeder et al., 2013). After implantation, the silencing of imprinted genes on the paternal allele in the post-implantation placental trophoblast expands and tends to be larger than the post-implantation epiblast. This expansion of gene silencing is mediated by the spreading of H3K27me3 marks along the paternal chromosome (Calabrese et al., 2015; Andergassen et al., 2017).
Long Non-Coding RNAs and Their Roles in Regulating the Expression of Imprinted Genes
Two major mechanisms have been described to explain the regulation of the gene expression within an imprinted cluster (Lee and Bartolomei, 2013; Barlow and Bartolomei, 2014; Chen and Zhang, 2020). The first model is the lncRNA model, which may be more common. In this model, imprinted lncRNAs regulate imprinted gene expression. In the lncRNA model, imprinted lncRNAs intimately associate with ICRs. Imprinted lncRNAs are characterized by their capacity to silence imprinted genes in the same cluster (Rao et al., 2014; Kanduri, 2016; Tan et al., 2018; Zink et al., 2018; Tucci et al., 2019). As illustrated by the Kcnq1/Kcnq1ot1 imprinted cluster (Figure 2A), actively expressed imprinted lncRNA Kcnq1ot1 on the paternal allele can silence multiple imprinted genes bidirectionally along their located gene region (Pauler et al., 2012). In contrast, a maternally methylated ICR on the paternal directly inhibits Kcnq1ot1 and its silencing effects, leading to the released expression of imprinted genes from the silencing by Kcnq1ot1. Another model, the insulator model is identified in other imprinted regions, in which parental allele-specific epigenetic differences at ICRs contribute to topological alternations of imprinted gene regions, inducing gene silencing or activation of specific alleles. This model is mainly applied to explain how imprinted genes in the insulin-like growth factor 2 (Igf2)/H19 locus are mechanistically regulated (Figure 2B; Kaffer et al., 2000). H19 is a maternally expressed lncRNA (Bartolomei et al., 1991; DeChiara et al., 1991; Ferguson-Smith et al., 1991). The zinc-finger protein CTCF binds to the unmethylated maternal ICR and creates topologically associating domain boundaries, blocking Igf2 access to the enhancer like an ‘insulator’ (Schoenherr et al., 2003; Gómez-Marín et al., 2015). On the paternal allele, methylated ICR prevents CTCF binding and leads to secondary methylation of the H19 promoter and therefore silencing of lncRNA expression. The enhancers are then accessible to Igf2, permitting paternal-allele expression of Igf2 (Thorvaldsen et al., 1998; Chen and Zhang, 2020). Different from the lncRNA model, imprinted lncRNAs H19 in the insulator model are not the key regulation elements or whether imprinted lncRNAs affect other genes are not clear.
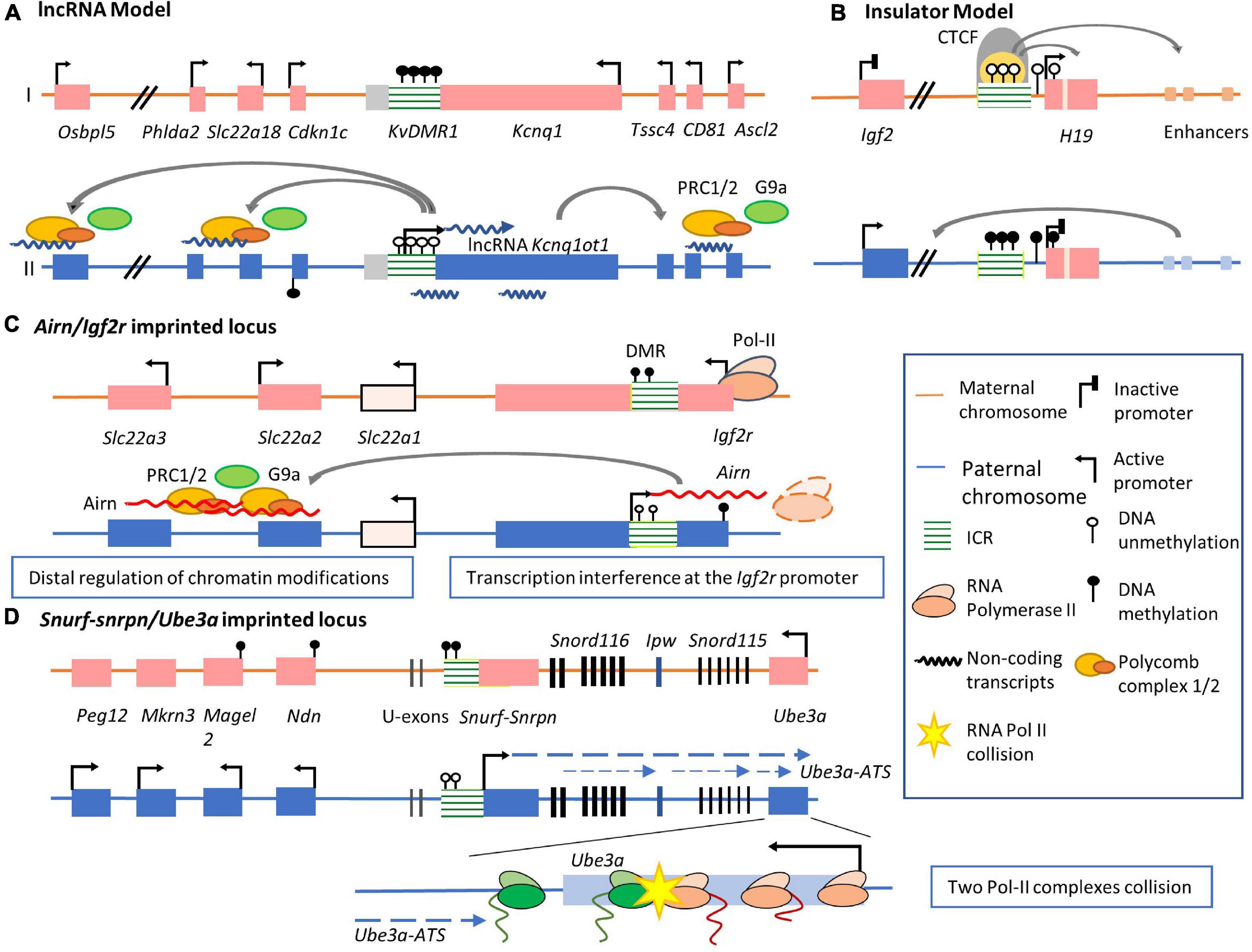
Figure 2. Mechanisms by which imprinted lncRNA regulate allelic expression in imprinted clusters. (A) The lncRNA model of imprinted gene expression regulation. In Kcnq1ot1 imprinted cluster, the ICR is unmethylated on the paternal allele, permitting lncRNA Kcnq1ot1 expression. The expression of this lncRNA recruits the PRC1/2 complex and histone methyltransferase G9a, leading to condensed chromatin and silencing of flanking protein-coding genes. The ICR is methylated on the maternal allele, inhibiting lncRNA expression. The expression of Kcnq1 and several paternal silenced genes are activated. (B) The insulator model of imprinted gene expression regulation. The ICR on the maternal allele is unmethylated. CTCF binds to the maternal ICR and functions as an insulator to block Igf2 access to its distal enhancers. In contrast, the ICR on the paternal allele is methylated, preventing binding of CTCF. The expression of Igf2 is activated via enhancer regulation. (C) lncRNA Airn in Airn/Igf2r locus function in two distinctive mechanisms. On the one hand, methylated DMR on the maternal allele inhibits Airn expression, allowing access of transcription factors to the Igf2r promoter. The paternal DMR is unmethylated, permitting Airn transcription. Airn overlaps with the promoter of Igf2r and inhibits Igf2r expression. On the other hand, Airn transcripts recruit PRC2 complex to distal genes, such as Slc22a3 and Slc22a2, where they silence expression. Slc22a1 is a biallelic expressed protein-coding gene between distal regulated imprinted genes and Igf2r gene loci. (D) In Snurf-Snrpn/Ube3a imprinted cluster, the transcription of Ube3a-ATS starts from the exon upstream of the Snurf-Snrpn gene on the paternal allele. A group of non-coding RNAs are expressed, including Snord116 and Snord115 sno-lncRNAs and SnoRNAs. The elongation of this lncRNA overlaps with the Ube3a protein-coding region. A collision occurs between the converging elongation complexes of Ube3a-ATS and Ube3a resulting in the failure of Ube3a transcription elongation. By contrast, on the maternal allele, the ICR of Snurf-Snrpn/Ube3a cluster is methylated in the brain. G9a is recruited to the methylated DMR. This G9a accumulation leads to condensed chromatin and the silencing of flanking imprinted genes near the Snurf/Snrpn gene region. Consequently, the maternal Ube3a allele is expressed.
Here, we discuss on the role of imprinted lncRNAs in epigenetic regulation in the more common model, lncRNA model (Kopp and Mendell, 2018). lncRNA functions can be characterized based on their specific subcellular locations and interactions with DNA, RNA, and proteins, regulation of chromatin structure, expression of nearby and distal genes, RNA post-transcription modification, or mRNA translation (St Laurent et al., 2015; Kopp and Mendell, 2018; Statello et al., 2021). Imprinted lncRNAs range from 1.9 to 1,000 kb in length (Guenzl and Barlow, 2012) and regulate the expression of adjacent imprinted genes in cis through interacting with promoters and transcription factor binding sites, modifying chromatin status, or affecting higher-order structures (Barlow, 2011). Two major functional mechanisms of imprinted lncRNAs in the regulation of imprinted gene expression are hypothesized: interacting with promoters or enhancers of nearby target genes to affect transcription initiation, or overlapping imprinted gene regions, covering the gene body, and regulating the chromatin state of adjacent gene regions. We will also discuss the mechanisms underlying the regulation of imprinted gene expression by imprinted lncRNAs using well-characterized imprinted clusters as examples.
Transcriptional Interference
Inhibition of Transcriptional Initiation
Transcription of imprinted lncRNAs often overlaps with the promoters or enhancers of imprinted genes and influences their transcription (Lee and Bartolomei, 2013). These imprinted lncRNA transcripts often interfere with the transcription machinery of nearby imprinted genes, influencing the recruitment of transcription factors at their promoters (Latos et al., 2012). Based on an analysis of lncRNA and DNA binding in imprinting clusters from multiple mammalian species, it was suggested that the binding of lncRNAs to promoters of imprinted genes may be common (Liu et al., 2017). The Airn/Igf2r imprinted cluster in the mouse genome is a well-studied example (Figure 2C; Latos et al., 2012). On the paternal allele, the transcription profile of Airn initiates from its promoter embedded within the ICR in a direction antisense to the transcription of the Igf2r gene (Latos and Barlow, 2009). It was noted that intragenic truncations of the endogenous lncRNA Airn in embryonic stem (ES) cells that do not include the overlapping region are unable to silence the Igf2r paternal allele, thus demonstrating that inhibition of RNA polymerase II recruitment to Igf2r promoter region does not depend on the overlap between Airn transcription and the promoter (Sleutels et al., 2002; Latos et al., 2012; Santoro and Pauler, 2013). Furthermore, during ES cells differentiation, Airn expression was also necessary and sufficient to silence Igf2r (Santoro et al., 2013). The overlapping regions between Airn transcription and Igf2r promoter and its gene body instead of Airn lncRNA products themselves lead to silencing of Igf2r expression.
The Disturbance of the Transcriptional Elongation
Another mechanism involves a collision between the converging elongation complexes of imprinted lncRNA and imprinted genes, leading to transcription stalling, premature termination, and subsequent degradation of the imprinted gene transcript (Hao et al., 2017). An example is the UBE3A/UBE3A-ATS imprinted domain on human chromosome 15q11-13, in which imprinted genes, including MAGEL2, NDN, SNRPN, SNORD115, and SNORD116, are silenced on the maternal allele (Horsthemke and Wagstaff, 2008). In contrast, UBE3A, which encodes an E3 ubiquitin ligase, is expressed from the maternal allele, especially in neurons in the brain. The homologous imprinted locus in mice has also been identified and studied, locating at a syntenic loci chromosome 7qC (Yang et al., 1998; Figure 2D). In this imprinted cluster, the ICR embedded within the Snurf-Snrpn gene is unmethylated on the paternal allele. In mouse neurons, Ube3a-ATS lncRNA is expressed specifically from its promoter embedded in the unmethylated ICR (Yin et al., 2012; Meng et al., 2013). Notably, the Ube3a promoter region is not methylated differently like Ube3a-ATS. This, combined with the observation that Ube3a-ATS transcription initiates from an exon region upstream of the Snurf-Snrpn gene and elongates approximately 1,000 kb as far as the intronic region of Ube3a between exons 4 and 5 (Landers et al., 2004; Lewis et al., 2019), it was hypothesized that the two opposing polymerases of Ube3a and Ube3a-ATS collide (Figure 2D). This transcriptional collision may lead to premature termination of Ube3a transcription inside its exon region on the paternal chromosome. In neurons from the monoallelic genetically engineered mouse model with the transcription of paternal Ube3a-ATS allele being terminated, Ube3a allele expression was activated on the paternal allele (Meng et al., 2013), resulting in increased expression comparable to maternal Ube3a (Meng et al., 2012). In cultured AS mouse neurons with biallelic silenced Ube3a expression, Antisense oligonucleotides (ASOs) targeting Ube3a-ATS rescued the expression of Ube3a efficiently (Meng et al., 2015). Consistently, in human induced pluripotent stem cells (iPSC)-derived neuron cells with biallelic silenced UBE3A expression, ASOs targeting UBE3A-ATS lncRNA transcripts lead to transcriptional termination by displacement of RNA Polymerase II, releasing the transcription of UBE3A (Germain et al., 2021). Recently, in human iPSCs, both sufficient expression of UBE3A-ATS lncRNA and two newly identified boundary elements were located inside the IPW gene and the PWAR1 gene (Martins-Taylor et al., 2014; Hsiao et al., 2019). These two genes are located between SNORD115 and SNORD116. In human iPSCs with the boundary elements deleted using gene editing technology, the expression of UBE3A was not silenced by up-regulated UBE3A-ATS expression (Hsiao et al., 2019). Mapping RNAPII density showed that reduced active RNAPII across the 3′ half of UBE3A corresponding to silenced UBE3A. These results together further support the hypothesized collision between UBE3A-ATS and UBE3A transcription complexes, leading to premature termination of the latter. In summary, the overlap between Airn and Igf2r promoter region disrupts the initiation of Igf2r transcription, while Ube3a-ATS silences the expression of Ube3a by disturbing its transcriptional elongation.
Chromatin Modification
Another lncRNA-related imprinting mechanism involves coating the bidirectionally flanking chromosomal region and recruiting repressive chromatin modification factors (Lee and Bartolomei, 2013; Sanli and Feil, 2015; Statello et al., 2021). The interactions between lncRNAs and these chromatin factors facilitate transcriptional silencing of target genes. The repressive chromatin-modification factors methylate DNA and produce histone modifications resulting in condensed chromatin structure and repressed gene expression. Among well-known repressive chromatin-modification factors, PRCs bind and spread across targeted chromatin facilitated by lncRNAs (Kotzin et al., 2016; Marín-Béjar et al., 2017). lncRNAs, genome structures, and CpG islands are essential factors in recruiting these PRCs, which have the capacity to catalyze lysine 119-mono-ubiquitinated histone H2A (H2AK119ub1) and H3K27me3 to repress gene expression through chromatin compaction and antagonization of transcriptional activators (Schwartz and Pirrotta, 2013; Simon and Kingston, 2013; Calabrese et al., 2015; Pintacuda et al., 2017; Colognori et al., 2019; Schertzer et al., 2019; Gil and Ulitsky, 2020; MacDonald and Mann, 2020). In genomic imprinting, some imprinted lncRNAs can bidirectionally direct repression of flanking neighbor imprinted gene region, such as KCNQ1OT1 lncRNA. Some lncRNAs can target distal gene regions in the same imprinted clusters they locate, such as Airn.
Locally Recruiting Condensed Chromatin Structure to Neighbor Gene Region
The Kcnq1/Kcnq1ot1 ICR, also known as KvDMR1 (KvLQT1 differentially methylated region 1), with the embedded lncRNA Kcnq1ot1 promoter, is unmethylated on the paternal allele (Figure 2A; Lee et al., 1999, p. 1; Smilinich et al., 1999; Beatty et al., 2006; Ager et al., 2008). lncRNA Kcnq1ot1 transcripts from the promoter region recruit several epigenetic factors such as the Polycomb group proteins RING1B (Polycomb Repressive Complex 1, PRC1), EZH2 (PRC2), and histone methyltransferase euchromatic histone lysine N-methyltransferase-2 (EHMT2 or G9a) to neighboring gene regions, forming repressive histone modifications such as H3K27me3 and H3K9me2 (Figure 2A II; Umlauf et al., 2004; Pandey et al., 2008). The chromatin state around the flanking regions of this lncRNA becomes condensed and results in silencing of flanking multi-protein coding genes such as Cdkn1c, Slc22a18, and Tssc4. On the maternal allele, DNA methylation of KvDMR1 silences the activation of the Kcnq1ot1 promoter and represses the transcription, releasing the transcription of neighboring genes.
Recruiting Chromatin Modification Factors to Distal Imprinted Genes
A typical example of imprinted lncRNA regulating distal imprinted genes through epigenetic silencing is Airn and recruitment of PRCs in the placenta (Figure 2C; Latos et al., 2012; Lee and Bartolomei, 2013). As mentioned before, the transcription of Airn represses the expression of flanking imprinted gene Igf2r by transcriptional interference of the overlapping Igf2r promoter without repressive chromatin modification involved. In contrast, distal imprinted genes, such as Slc22a2 (about 100 kb to Airn locus) and Slc22a3 (about 300 kb to Airn locus), are also silenced by Airn in the extra-embryonic lineage, where Airn mediates the recruitment of PRC1 and PRC2 to distal targets on the paternal alleles (Terranova et al., 2008; Zhao et al., 2010; Schertzer et al., 2019). Recently, Airn was found to silence Slc22a3 in mouse trophoblast stem cells (Andergassen et al., 2019). Allele-specific chromosome conformation capture studies have suggested that Airn transcription throughout the enhancer of Slc22a3 may silence Slc22a3 expression by disrupting its promoter-enhancer interactions. However, with monoallelic deletion of the entire Airn gene, no essential enhancers for the distal silenced genes were found in the Airn gene region. Nonetheless, it has also been shown that Airn lncRNA is enriched on the Slc22a3 promoter together with an H3K9 dimethylase, G9a (Nagano et al., 2008). These results illustrate that Airn may target the promoters of distal imprinted genes by recruiting PRCs and G9a. The enrichment of these histone modification factors may lead to condensed chromatin in distal imprinted regions and silence imprinted genes.
The Role of Imprinted Long Non-Coding RNAs in Human Imprinting Disorders and Cancer
Long non-coding RNAs play essential roles in many biological processes and are related to various human diseases. Altered expression of imprinted loci has been linked to various neurodevelopmental disorders and cancers (Schaller et al., 2010; Huang et al., 2011; Riordan et al., 2013; Peters, 2014). Since imprinted regions are inherited in a parent-of-origin way, defects in one allele may be sufficient to lead to imprinting disorders (Kopp and Mendell, 2018). More specifically, silencing of parentally expressed imprinted genes can lead to the ultimate loss of its expression. Under abnormal conditions, DNA methylation status, allelic expression, and the biological functions of imprinted lncRNAs may be affected. These alterations may relate to human imprinting disorder-related disease phenotypes (Lee and Bartolomei, 2013). Here, we examine several well-studied imprinting disorders and emphasize the roles of imprinted lncRNAs in pathophysiological processes of imprinting-related diseases and cancers.
Common Molecular Mechanisms of Imprinting Disorders
Appropriate expression patterns of imprinted genes are important to growth and development. Correspondingly, imprinting disorder-related human diseases can be caused by genetic or epigenetic abnormalities on paternally or maternally inherited alleles (Lee and Bartolomei, 2013). Several common molecular mechanisms behind imprinting disorders have been defined, including molecular changes or genetic abnormalities, UPD, and epigenetic alterations (Figures 3A–D; Soellner et al., 2017; Carli et al., 2020). Firstly, genetic alterations, including SNPs and copy number variants on one imprinting allele, can affect imprinting (Figure 3B). Another mechanism is UPD, in which the inheritance of two copies of chromosomes or chromosomal regions are both from either the paternal or maternal allele, resulting in synchronous expression or silencing (Figure 3C; Robinson, 2000). Different from genetic alterations, epigenetic changes known as epimutations in DNA or histone modification without obvious genetic mutations have also been documented in imprinting disorders (Figure 3D; Horsthemke, 2010). Hypermethylation at imprinted DMRs can silence the active allele of the original monoallelic expressed imprinted genes. In contrast, hypomethylation can result in overexpression of the original silenced allele. Epimutations can arise randomly or be driven by their environment during the inheritance of germline epigenetic imprinting marks. DNA methylation in DMRs can thus be abnormally inherited in the absence of genetic sequence alterations (Robertson, 2005). Moreover, as with molecular or genetic alterations, epimutations can be permanently maintained in somatic tissues for life and cause developmental phenotypes (Ioannides et al., 2014; Gillessen-Kaesbach et al., 2018; Monk et al., 2019). Besides the imprinted disorder caused by variations in a single imprinted gene, imprinting disorders with epigenetic alterations at loci across the genome have also been observed in many imprinting diseases, referred to as MLID (Horsthemke, 2010; Fontana et al., 2018). Instead of changes at specific genetic loci, MLID may be caused by a globally disturbed imprinting inheritance process across the genome. However, since current research is mostly limited to a subset of imprinted genes and the mosaic character of MILD (Azzi et al., 2014; Eggermann et al., 2021), the role of MLID in imprinting disorders is still poorly understood.
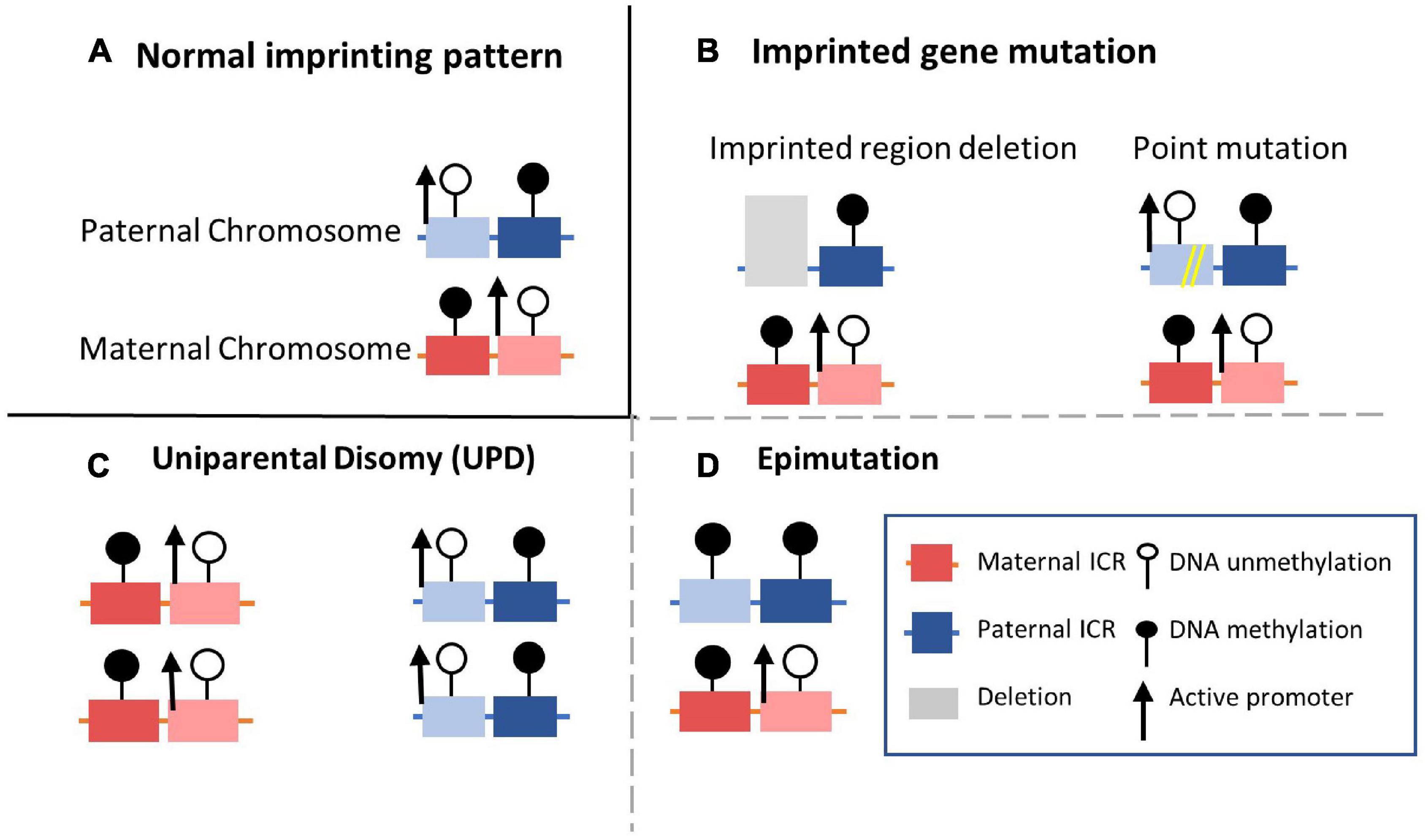
Figure 3. Four common molecular mechanisms of imprinting disorders. (A) The normal state of the established DMR methylation pattern on the maternal and paternal alleles. (B) Imprinting disorder can be caused by copy number variations with imprinted cluster located such as duplication or deletion. Point mutations (single nucleotide polymorphisms) occurring in imprinted genes could also influence normal functions. (C) Both alleles are inherited from same parent-of-origin. In the case shown here, for example, maternally inherited alleles are duplicated without paternal allele participation. (D) Epimutations of DNA modification condition can disturb normal imprinting pattern without alterations in DNA sequences of the imprinted region. For example, DNA methylation of the imprinted gene on paternal allele are hypermethylated and silenced on both alleles.
Congenital Imprinting Disorders and Related Imprinted Long Non-coding RNAs
Molecular disturbances, like loss or gain of methylation at ICRs, and subsequent loss or gain of imprinted gene expression have been described in various congenital human disorders. The frequencies of different molecular abnormalities vary among imprinting disorder-related diseases (Eggermann et al., 2015). More details about typical clinical syndromes and the pathological mechanisms are summarized in Table 1. However, details underlying imprinting disorder mechanisms and how they might impact adult neurobiology and developmental processes remain to be clarified. Fortunately, multiple mouse models of human imprinting disorders have been generated based on genomic conservation in most imprinted clusters. Strong correlates have been shown between these two genomes in imprinting loci, imprinting disorder phenotypes, and underlying molecular mechanisms (Perez et al., 2016). Spatial- and temporal-specific expression of allele-specific genes have been observed in several imprinting clusters in humans and mice. Here, imprinting disorders in three well-studied imprinting clusters are introduced as examples to demonstrate the roles of imprinted lncRNAs in imprinting-related congenital human disorders.
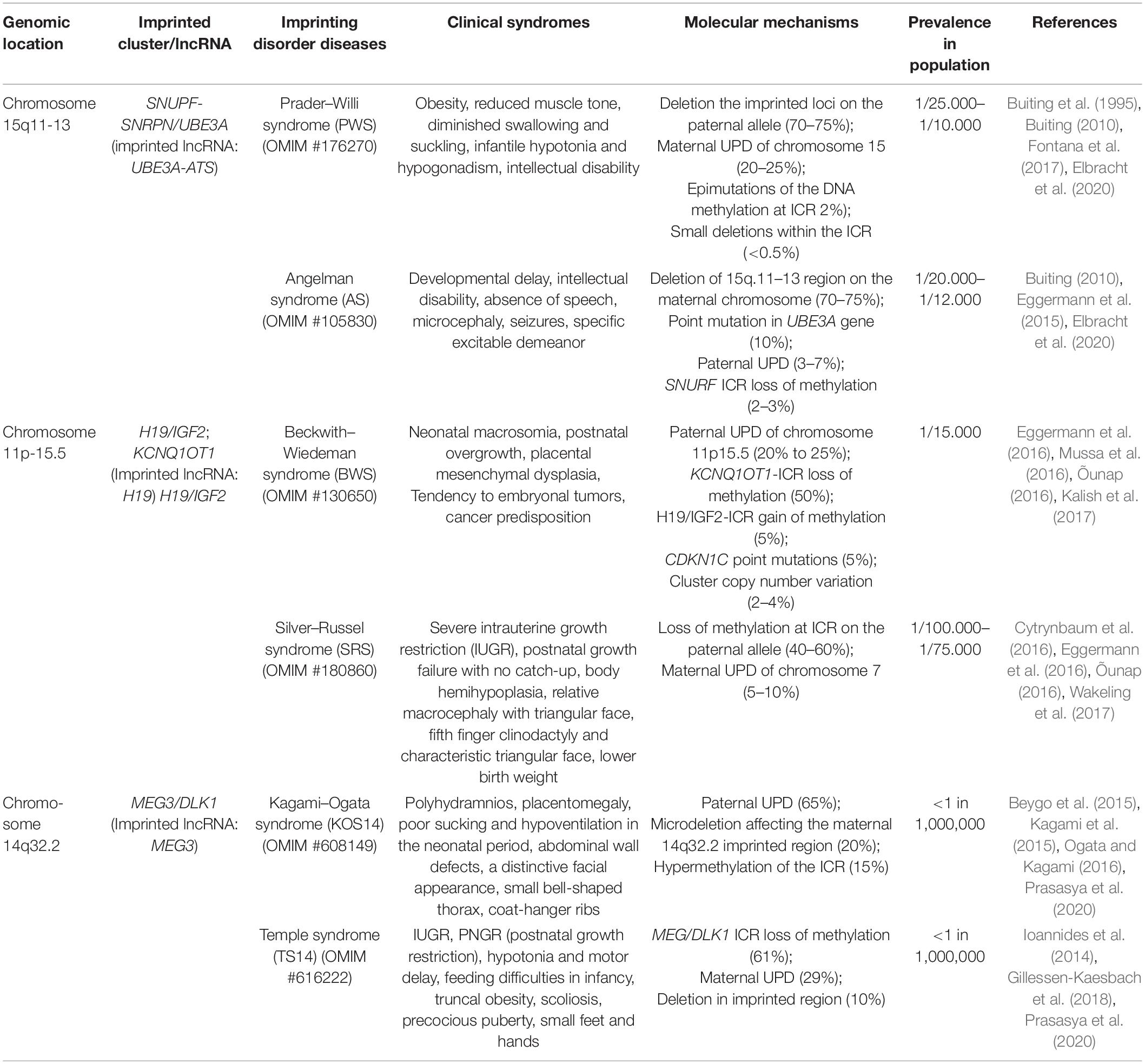
Table 1. Human imprinting disorder-related diseases.
UBE3A-ATS in Prader–Willi Syndrome and Angelman Syndrome
Recent RNA-Seq data revealed strong allele-biased expression in the adult mouse brain, especially in imprinted regions (Perez et al., 2015, 2016), where many of these genes are expressed in cell type-specific manners. Importantly, mutations or disruptions in imprinted genes are linked with extensive neurobehavioral phenotypes, demonstrating that brain-specific imprinted genes may play important roles in neurodevelopmental disorders (Tucci et al., 2019). PWS and AS are two neurodevelopmental disorders caused by oppositely inherited deficiencies occurred in the same imprinted cluster (Peters, 2014; Kalsner and Chamberlain, 2015; Buiting et al., 2016). These two syndromes perform common phenotype characters, including hypotonia at the newborn stage, abnormal sleep patterns, and the deficiency in intellectual development (Buiting, 2010; Kalsner and Chamberlain, 2015). Children affected by PWS exhibit poor suck phenotypes with reduced muscle tone and mental abilities (Buiting et al., 1995; Buiting, 2010; Fontana et al., 2017), while AS is characterized by deficient motor function, intellectual development, and speech abilities (Buiting, 2010; Eggermann et al., 2015). These two disorders are caused by imprinting disorder in the imprinted PWS/AS locus (UBE3A/UBE3A-ATS imprinted cluster) on human chromosome 15q11-13 (Figure 4A). Similar to the mouse homologous locus mentioned previously in Section 3, the E3 ubiquitin ligase-encoding UBE3A gene is specifically imprinted in the brain (Vu and Hoffman, 1997). On the maternal allele, the methylated DMR encompasses the promoter of the SNRPN gene, silencing the SNURF/SNRPN gene and a series of downstream non-coding RNA genes (Rougeulle et al., 1997). In contrast, actively expressed UBE3A-ATS and the non-coding SNORD gene clusters are expressed from the paternal allele.
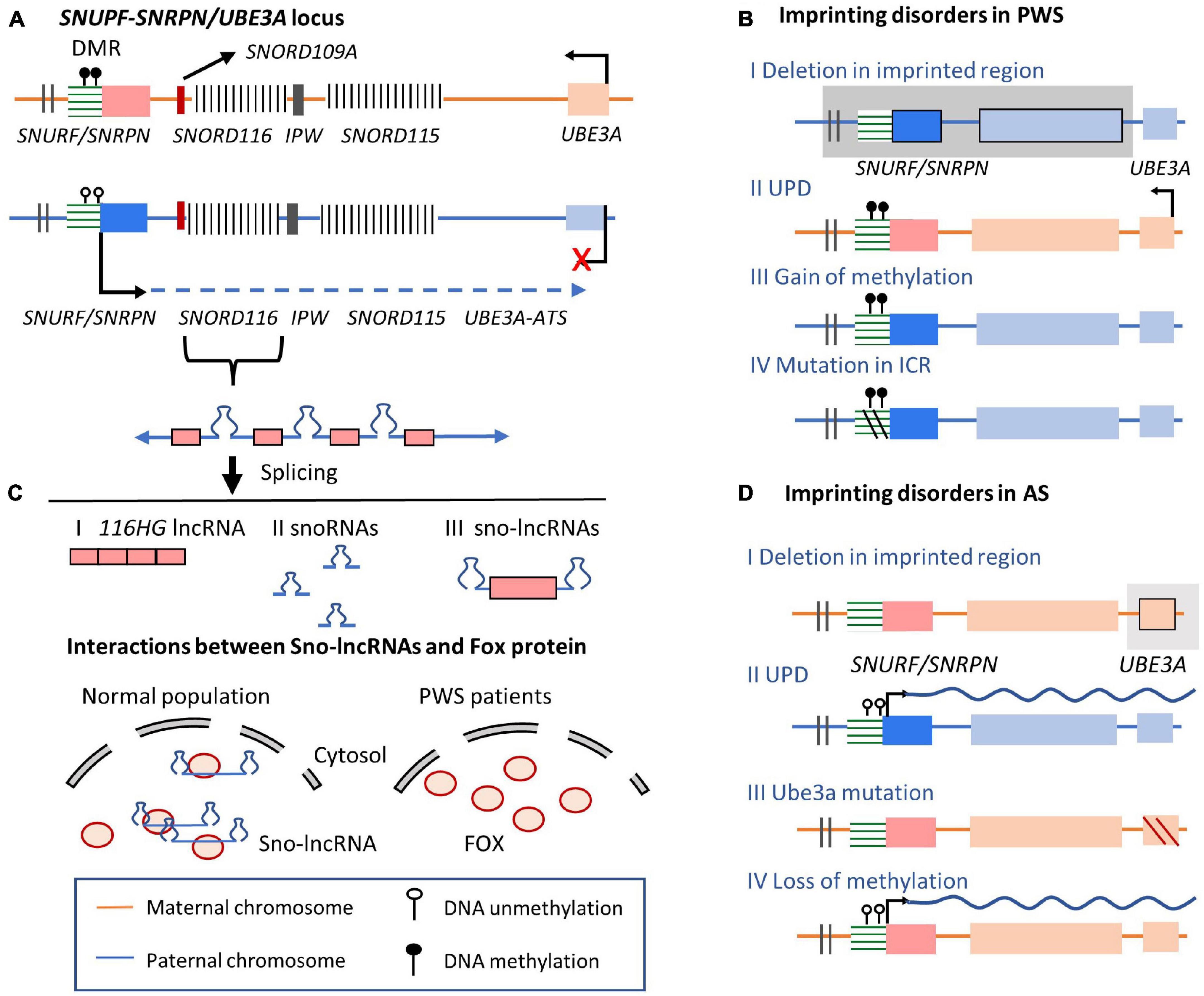
Figure 4. SNUPF-SNRPN/UBE3A imprinted cluster on human chromosome 15 and related imprinting disorders. (A) Allelic expression pattern in SNUPF-SNRPN/UBE3A locus. On the maternal allele, the methylation of ICR silences the expression of UBE3A-ATS, permitting UBE3A expression. On the paternal allele, UBE3A-ATS is expressed from the SNURF gene region, overlapping the exon region of UBE3A gene. On the paternal allele, lncRNA host transcript are processed to give rise to snoRNAs (SNORD115 and SNORD116), lncRNAs (116HG, 115HG, and UBE3A-ATS). Three different spliced non-coding transcripts are produced from SNORD116 gene locus, including 116HG lncRNA, snoRNAs, and sno-lncRNAs. SNORD116 sno-lncRNAs with snoRNAs on two ends are produced after splicing. (B) Imprinting disorders occur in PWS. PWS-related molecular alterations in UBE3A imprinted gene cluster. Line I indicates deletion in the imprinted region; Line II shows double maternal alleles are inherited, losing the paternal copy; Line III shows that the epigenetically mutated DNA methylation in DMR of the ICR leads to the silencing of lncRNA expression. Line IV: small deletion within the ICR. (C) Sno-lncRNAs transcribed from paternal allele can recruit Fox proteins and other related proteins, regulating Fox protein distribution and related alternative splicing functions. However, in PWS patients, loss of the UBE3A-ATS and other noncoding gene expression lead to the accumulation of Fox proteins in the nucleus and global abnormal splicing patterns. (D) Imprinting disorders occur in AS. Line I: deletions of the maternal imprinted regions containing the UBE3A and surrounding genes; Line II: both alleles are inherited from paternal chromosome; Line III: UBE3A mutations lead to transcript loss of function; Line IV: epimutations in the maternal allele lead to lncRNA expression from the maternal allele, preventing normal UBE3A expression.
On the paternal allele of imprinted human PWS/AS locus, the unmethylated PWS-ICR is the region upstream to a protein-coding gene SNRPN and a lncRNA SNHG14 (small nucleolar RNA host gene 14) (Sutcliffe et al., 1994; Buiting et al., 1995; Rougeulle et al., 1997; Runte et al., 2001; Vitali et al., 2010; Chamberlain, 2013; Stanurova et al., 2018; Figure 4A). The neuron-specific non-coding transcript SNHG14 is processed to give rise to a series of non-coding RNA products, such as repeated C/D box small nucleolar RNAs (snoRNAs) and lncRNAs including 116HG, 115HG, and the antisense transcript to UBE3A (Mendiola and LaSalle, 2021; Figure 4A). The most studied RNA product from the host transcript SNHG14 is SNORD116 snoRNA, embedded within intronic regions of SNORD116 gene locus (Cavaillé et al., 2000; de los Santos et al., 2000; Stanurova et al., 2018; Mendiola and LaSalle, 2021). SNORD116 snoRNA present in ribonucleoprotein complexes (snoRNPs) and may participate in splicing, ribosomal RNA maturation, RNA modifications, and regulation of prohormone processing-related gene expression (Bazeley et al., 2008; Burnett et al., 2017). Meanwhile, SNORD116 locus encoded 116HG lncRNA was discovered recently (Vitali et al., 2010). 116HG is stably retained in the nucleus ‘RNA cloud’ at its transcription site (Powell et al., 2013a). 116HG potentially regulates transcript levels of circadian-related genes in the cortex and energy-related metabolism through in a time-of-day-dependent manner (Coulson et al., 2018b). Similarly, SNORD115 locus encodes lncRNA 115HG and SNORD115 snoRNAs. While on the maternal allele, the methylated PWS-ICR occurs the upstream of the SNRPN gene. It silences the expression of the paternally expressed transcripts while allows the expression of UBE3A (Vu and Hoffman, 1997).
Prader–Willi syndrome is the first human disease identified to be caused by the abnormal expression of non-coding RNAs (Sahoo et al., 2008; de Smith et al., 2009; Duker et al., 2010). All cases of PWS in humans involve a deletion in the SNORD116 non-coding gene locus, which regulates the maturation of the central nervous system. The overlap between the phenotype caused by SNORD116 microdeletion and MAGEL2 mutation suggests that transcripts from SNORD116 locus may modify MAGEL2 expression via long-range chromatin interactions (Meziane et al., 2015; Fountain and Schaaf, 2016; Langouët et al., 2018). The loss of the paternal expressed SNORD116 in PWS can be caused by several factors, including large paternal deletions in the imprinted PWS/AS locus (60%), maternal UPD (36%), small microdeletion in SNORD116 locus (<1%), and epigenetic alternations in DNA methylation of the PWS-ICR region (4%) (Sahoo et al., 2008; Duker et al., 2010; Bieth et al., 2015; Rozhdestvensky et al., 2016; Mendiola and LaSalle, 2021; Figure 4B). Rare microdeletions that encompass SNORD116 and its adjacent genes, SNRPN or SNORD115, have been found in PWS patients (Sahoo et al., 2008; de Smith et al., 2009; Duker et al., 2010). Moreover, a small deletion that only covers SNORD116 and its adjacent genes (SNORD109A, and IPW) was identified in a patient with typical PWS syndrome (Bieth et al., 2015; Figure 4A). Since there is no obvious involvement of SNORD109A and IPW genes in PWS, the observations in this PWS case further support that the SNORD116 gene region play key roles in the PWS, independent with SNORD115 or SNRPN deletion. Consistently, SNORD116 is completely silenced in neuron cells derived from PWS patients (Cavaillé et al., 2000; Hsiao et al., 2019). Besides, Snord116 deleted mouse model recapitulates major phenotypes of human PWS patients, including altered metabolism, growth deficiency, memory impairment, hyperphagia and increased anxiety (Skryabin et al., 2007; Ding et al., 2008; Zieba et al., 2015; Qi et al., 2016; Polex-Wolf et al., 2018; Adhikari et al., 2019).
Furthermore, an alternative RNA species (sno-lnRNAs) processed from SNORD116 host non-coding transcript has been described in human (Yin et al., 2012; Powell et al., 2013a; Figure 4A III). The role of SNORD116 sno-lncRNAs in RNA processing and decay of their target mRNAs is not well-understood but may facilitates our understanding of the connection between imprinting disorder and pathological mechanism of PWS (Figure 4C). SNORD116 exon transcript is retained between two snoRNAs, forming sno-lncRNAs with two small nucleolar ribonucleoprotein ends (Yin et al., 2012). These sno-lncRNAs accumulate near the synthesis site together with a type of lncRNAs that are 5′ capped by snoRNAs and 3′ polyadenylated (SPAs) (Wu et al., 2016). These lncRNAs may interact with RNA binding proteins including TDP43 (TAR DNA-binding protein 43), RBFOX2 (RNA Binding Fox-1 Homolog 2), and hnRNP M (Heterogeneous nuclear ribonucleoprotein M). Especially, splicing regulator RBFOX2 are required for the neuron-specific splicing of Snord116 transcript to produce 116HG lncRNA and Snord116 snoRNA (Yeo et al., 2009; Coulson et al., 2018a). Since immunoprecipitation coupled with high-throughput sequencing (CLIP-seq) and RT-PCR assays confirmed that RBFOX2 directly binds to Snord116 snoRNA, it is hypothesized that Snord116 snoRNA may reduce the availability of these splicing-related proteins and regulate alternative splicing in the nucleus (Yin et al., 2012; Wu et al., 2016). Therefore, the disruption of SNORD116 in PWS may lead to more uniform distribution of RBFOX2 protein and global changes in normal alternative splicing patterns, contributing to PWS phenotypes.
In contrast to the paternal-allelic imprinting disorder in PWS, AS, is mainly caused by the lack of maternal UBE3A gene expression (Figure 4D; Buiting et al., 2016). The brain-specific and maternally biased expression of UBE3A has been shown to function in regulating dendritic growth and influencing behavior and neurotransmitters (Avagliano Trezza et al., 2019). In AS patients, the expression of UBE3A or functional UBE3A protein is lost. These alternations can be caused by various imprinting disorder mechanisms including deletions of the maternally imprinted regions containing the UBE3A and surrounding genes. Besides pathological variants in the UBE3A gene, loss of SNURF DMR methylation has also been observed in AS cases (2–3%), in which the expression of UBE3A is silenced by UBE3A-ATS as discussed previously (Dagli et al., 1993).
KCNQ1OT1 and H19/IGF in Beckwith–Wiedemann Syndrome and Silver–Russell Syndrome
Beckwith–Wiedemann syndrome and SRS are clinically opposite growth-affecting disorders (Õunap, 2016). The underlying pathological mechanisms involve genetic and epigenetic perturbations of two imprinting clusters on human chromosome 11p15, the KCNQ1/KCNQ1OT1 and H19/IGF loci (Carli et al., 2020; Chang and Bartolomei, 2020; Figures 5A–C). BWS is one of the most common congenital overgrowth conditions (Mussa et al., 2013), with common phenotypes including postnatal overgrowth, placenta mesenchymal dysplasia, and congenital and childhood cancer predisposition. In contrast, SRS patients exhibit postnatal growth failure with body hemihypoplasia, lower birth weight, fetal undergrowth and poor feeding predisposition (Wakeling et al., 2017).
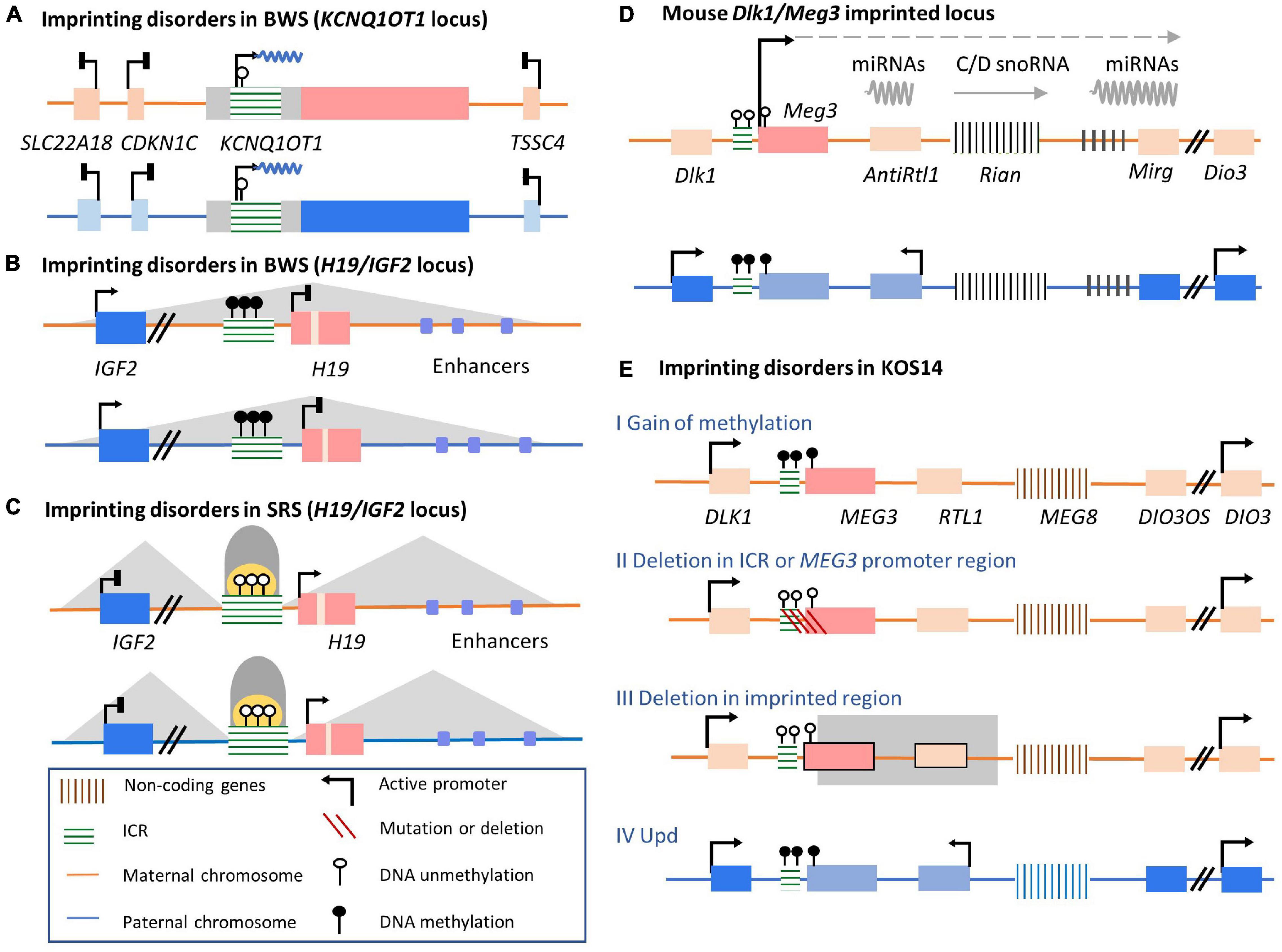
Figure 5. Well-studied imprinting clusters and their conditions in disorder conditions involved in human imprinting disturbance-related diseases. (A,B) BWS and two related imprinted clusters, H19/IGF2 and KCNQ1OT1. (A) The first situation is the hypomethylation of the maternal allele in Kcnq1ot1 ICR leads to lncRNA KCNQ1OT1 overexpression. The expression of neighboring imprinted genes, such as SLC22A18, CDKN1C, and TSSC4, is bi-allelically silenced. (B) The second major imprinting disorder responsible for BWS is hypermethylation of the maternal ICR, resulting in loss of H19 expression and IGF2 overexpression. (C) SRS and alterations in imprinting of H19/IGF2 locus. Hypomethylation of the paternal H19/IGF2 ICR resulting in H19 overexpression and inhibited Igf2 expression. (D) The regulation of mouse Meg3 imprinted cluster. On the paternal allele, the gDMR of Meg3 cluster ICR is methylated, repressing Meg3 lncRNA expression. On the maternal allele, lncRNAs are transcribed from the promoter within the unmethylated ICR. (E) Four cases of MEG3-related imprinting disorders in KOS14 patients are shown. Line I, epimutations in normally activated maternal ICR of MEG3 cluster result in loss of lncRNA transcription, releasing normally silenced adjacent imprinted genes; Line II: maternal deletion in ICR of MEG3 regions; Line III: maternal deletion in the MEG3 gene body; Line IV: both alleles are inherited by silenced paternal allele.
Approximately 50% of BWS patients lose DNA methylation accompanied by loss of H3K9me2 on maternal KvDMR1 (Figure 5A; Robertson, 2005). This epigenetic disturbance results in biallelic expression of the KCNQ1OT1 lncRNA. As a consequence, expression of this lncRNA silences adjacent imprinted genes on both alleles (Soejima and Higashimoto, 2013). Among these silenced genes, CDKN1C is linked to the development of BWS phenotypes (Yan et al., 1997; Zhang et al., 1997; Tunster et al., 2011). These epigenetic mutations in maternal KvDMR1 and biallelic expressed KCNQ1OT1 lncRNAs lead to loss of CDKN1C expression and fetal overgrowth, thus contributing to BWS syndrome (Eggermann et al., 2016; Wakeling et al., 2017). Therefore, after the establishment of DMRs on imprinted alleles, monoallelic expression of KCNQ1OT1 lncRNA is a crucial regulator of adjacent protein-coding genes, which have essential roles in maintaining normal growth processes during early development. Another major abnormal imprinted cluster identified in BWS patients is H19/IGF2 (Figure 5B). Under normal conditions, H19/IGF2 ICR is methylated on the paternal chromosome, controlling the expression of H19. In BWS patients, mutations or hypermethylation of the H19/IGF2 ICR can lead to H19 silencing and subsequent overexpression of IGF2, a circulating hormone and tissue growth factor. The upregulated expression of IGF2 is linked to BWS overgrowth-related phenotypes (Pollak et al., 2004; Brioude et al., 2018a, b; Duffy et al., 2019). As for SRS, loss of H19/IGF2 ICR methylation on the paternal chromosome 11p15 accounts for 40–60% of patients (Wakeling et al., 2017). ICR hypomethylation is bound by the insulator CTCF. The interaction of the IGF2 promoter with its enhancer on both alleles is disrupted, resulting in decreased IGF2 expression and subsequent growth and development delays (Figure 5C; Abi Habib et al., 2017).
Although some BWS and SRS patients can be identified based on clinical features alone, diagnosing imprinting disorders can be complicated by complex molecular alternations (Ibrahim et al., 2014; Wakeling et al., 2017). In addition to the two imprinted loci primarily relevant to BWS and SR phenotypes, MLID has also been observed in an increasingly growing fraction of patients with methylation abnormalities at other imprinted loci (Rossignol et al., 2006; Azzi et al., 2009; Eggermann et al., 2011; Fontana et al., 2018). In addition, symptoms vary widely in patients with imprinting disorders (Wakeling et al., 2017; Brioude et al., 2018b; Mantovani et al., 2018). Therefore, additional insights into the relationship between the epigenetic mechanisms of imprinting disorders and neurological diseases can help clarify more accurate diagnostic guidelines and appropriate clinical therapies.
DLK1/DIO3 in Kagami–Ogata Syndrome and Temple Syndrome
Genetic and epigenetic alterations in delta-like homolog 1 gene/type III iodothyronine deiodinase gene (DLK1/DIO3) imprinted cluster on human chromosome 14q32 are associated with two human imprinting disorder-related diseases, KOS14 and TS14 (Temple et al., 1991; Wang et al., 1991; Ogata and Kagami, 2016). Common KOS14 phenotypes include neonatal respiratory difficulties, a distinctive facial appearance, variable developmental delay, and/or intellectual disability (Ogata and Kagami, 2016; Prasasya et al., 2020). Clinical syndromes observed in TS14 include severe intrauterine growth restriction, postnatal growth restriction, neonatal hypotonia, and feeding difficulties in infancy (Ioannides et al., 2014; Gillessen-Kaesbach et al., 2018; Prasasya et al., 2020).
The distribution of imprinted genes and regulatory mechanisms of DLK1/DIO3 locus are highly conserved between humans and mice. The regulation of this imprinted locus has been revealed in mouse models established with genetic alterations in the Dlk1/Dio3 locus on chromosome 12 (Figure 5D; Paulsen et al., 2001; da Rocha et al., 2008). Three paternally expressed imprinted protein-coding genes are Dlk1, Rtl1, and Dio3. lncRNA Meg3 (also called Gtl2), the Rtl1-antisense Rtl1as, the C/D-box snoRNA cluster Rian, and the microRNA cluster Mirg are transcribed from the maternal allele (da Rocha et al., 2008; Kota et al., 2014). The regulation of imprinted gene expression in this locus relies on an intergenic DMR (IG-DMR). On the maternal allele, AFF3 protein binds to an upstream enhancer of Meg3, activating lncRNA expression. In contrast, on the paternal allele, AFF3 binds instead to the methylated IG-DMR, leading to silencing of Meg3 and other non-coding genes (Luo et al., 2016; Wang et al., 2017). It has also been suggested recently that maternally expressed lncRNA Meg3 is involved in the regulation of the Dlk1/Dio3 imprinted cluster (Sanli et al., 2018). The maternal expression of the Meg3 lncRNA may play a role in preventing maternal Dlk1 activation through interaction with the lysine methyltransferase (KMT) Ezh2 and PRC2 in the maternal Dlk1 gene region (Kaneko et al., 2014; Sanli et al., 2018). Remarkably, Meg3 lncRNA’s regulation of imprinted protein-coding gene Dlk1 is restricted to a developmental window as follows. In embryonic stem cells, the Dlk1 gene is expressed biallelically at a low level. Upon neuronal differentiation, Dlk1 expression is upregulated on the paternal allele. Conversely, the activation of the Dlk1 gene on the maternal allele is prevented by the overlap of Meg3 lncRNA in cis and the recruitment of Ezh2 to the Dlk1 gene region (Sanli et al., 2018). Although the Meg3 lncRNA is necessary for the silencing of Dlk1 expression, the mechanisms underlying the connection between the Meg3 lncRNA and repressed Dlk1 expression on the maternal allele are unknown.
The DLK1/DIO3 locus is predominantly imprinted in the human brain (Davis et al., 2005; Ferrón et al., 2011). Protein-coding genes DLK1, RTL1, and DIO3 are expressed on the paternal allele; lncRNAs (MEG3, MEG8, RTL1as, DIO3OS), snoRNAs, and miRNAs are transcribed on the maternal allele. Importantly, the DLK1 gene plays essential functions in regulating development and metabolism. In KOS14 patients, gain of DNA methylation on the maternal ICR leads to MEG3 silencing (Figure 5E I; Sato et al., 2011). However, maternal micro-deletions of the MEG3 promoter that don’t affect ICR methylation are also observed in some cases (Figure 5E II; Kota et al., 2014). In another case, a maternal micro-deletion has been detected in the MEG3 gene body instead of the IG-DMR or MEG3 promoter (Figure 5E III; van der Werf et al., 2016).
In summary, in these conditions, imprinted lncRNAs play essential roles as upstream regulators of protein-coding genes in the same imprinted cluster. However, the detailed mechanisms are diverse and complicated in different imprinting disorders and remain to be further investigated.
Imprinted Long Non-coding RNAs and Human Cancers
Long non-coding RNAs play important roles in pathways implicated in many cancer types, including prostate (Hua et al., 2018, p. 19), breast (Zhang et al., 2017; Cho et al., 2018), and hepatocellular carcinoma (He et al., 2017; Lecerf et al., 2019; Ye et al., 2020). Long non-coding RNAs can serve as cancer enhancers or repressors in temporal- and spatial-specific manners (Calin et al., 2007; Kanduri, 2016; Quinn and Chang, 2016; Peng et al., 2017). Abnormal functions of lncRNAs have been observed in various tumors and cancer cell lines (Kitagawa et al., 2012; Bhan et al., 2017). Notably, abnormally regulated imprinted gene expression, altered ICR methylation conditions, and altered expression of cancer-related imprinted lncRNAs were observed in cancers such as breast cancer (Kim et al., 2015; Goovaerts et al., 2018). In addition, in imprinting disorders, abnormal silencing of imprinted lncRNAs contributes to congenital and childhood tumors. For instance, susceptibility to Wilm’s tumor and adrenocortical carcinoma is increased in H19-silenced patients (Dao et al., 1999; DeBaun et al., 2000; Weksberg et al., 2010; Brioude et al., 2018a, b).
H19 is one of the most commonly implicated tumorigenesis-promoting lncRNAs (Zheng et al., 2020). The expression of H19 occurs during embryonic development and decreases after birth in most tissues. However, H19 is abnormally upregulated in various cancers, including breast, liver, lung, esophageal, pancreatic, ovarian, and bladder (Vennin et al., 2015; Zhang et al., 2016). H19’s tumor-promoting effects include the inhibition of cell death, promotion of proliferation, downregulation of growth suppressors, and promotion of invasion and metastasis (reviewed in Matouk et al., 2015; Lecerf et al., 2019). Moreover, high H19 expression may be a molecular marker to predict cancers and prognoses after clinical treatment, including the rate of post-therapeutic relapse in hematological cancer patients (Liu et al., 2016). Increased risk of developing congenital and childhood tumors seen in BWS is also associated with aberrant H19. H19 is also associated with growth suppression (Yoshimizu et al., 2008; Lecerf et al., 2019; Zhou et al., 2019). H19’s contribution to tumorigenesis varies by tissue and developmental windows and requires clarification in future investigations.
Another well-studied cancer-related imprinted lncRNA is MEG3, which acts as a cancer repressor. MEG3 is downregulated in breast, neuroblastoma, meningioma, glioma, pituitary adenoma, and hematological malignancies (Benetatos et al., 2011; Cheunsuchon et al., 2011; Zhou et al., 2012; Lyu et al., 2017; Zhu et al., 2019). In pituitary neuroendocrine tumors, hypermethylation of the maternal DLK1/MEG3 locus results in MEG3 downregulation and impaired differentiation (Cheunsuchon et al., 2011; Chen et al., 2020). Hypermethylation of the MEG3 promoter region has also been observed in AML patients (Lyu et al., 2017; Yao et al., 2017; Sellers et al., 2019), while recent studies have begun to reveal the underlying mechanisms in endometrial and breast cancers (Sun et al., 2017; Zhang et al., 2017; Zhu et al., 2019). One such mechanism involves MEG3’s inhibition of the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signaling pathway, a well-known growth-related pathway. Therefore, unraveling the roles of imprinted lncRNAs in cancer may reveal novel biomarkers and therapeutic targets for cancer treatment.
Modulation of the Long Non-Coding RNA Ube3A-ATS to Rescue Abnormal Imprinting in Prader–Willi Syndrome/Angelman Syndrome Imprinted Cluster
Although our understanding of the mechanisms of imprinting disorders has grown, efficient molecular diagnosis and effective treatments are limited to nonexistent (Elbracht et al., 2020). Modulation of imprinted lncRNAs has been proposed as a potential therapeutic strategy to target imprinted genes and rescue imprinting disorders (Peters, 2014; Statello et al., 2021). As the epigenetic regulatory mechanisms of the Ube3a/Ube3a-ATS imprinted cluster are understood best, attempts have been made to rescue Ube3a expression through modulating the collision between the transcriptional machinery of Ube3a and Ube3a-ATS in an allele-specific manner. Herein, three state-of-the art therapeutic strategies by targeting Ube3a-ATS lncRNA, editing Ube3a-ATS gene region, or modulating chromatin transcriptional state by small molecules are discussed along with recent preclinical studies of UBE3A/UBE3A-ATS imprinted cluster-related diseases.
Antisense Oligonucleotides for Imprinted Long Non-coding RNAs
Antisense oligonucleotides are single-stranded DNA oligos designed using sequence homology with their RNA targets that hybridize with the targeted RNA region based on complementary base pairs, and induce subsequent RNA degradation at the ASO-RNA heteroduplex part (Mishra et al., 2019; Li M. et al., 2020). ASOs can be used to alter splicing or gene expression. ASOs have been designed as potential therapies for various diseases, including AS, spinal muscular atrophy (SMA), Duchenne muscular dystrophy, Huntington disease, and hyperlipidemia (Beaudet and Meng, 2016; Dhuri et al., 2020). Several ASO-based therapies, such as Nusinersen (Spinraza) for SMA treatment, have received approval by the United States Food and Drug Administration (FDA) and other regional regulatory agencies (Karaki et al., 2019). Nusinersen is quite effective in rescuing protein deficiency by altering pre-mRNA splicing (Hoy, 2017; Groen et al., 2018; Claborn et al., 2019). The capacities of ASOs to access targeted RNAs through homology base pairing and in inducing RNase H-mediated cleavage at the pairing regions by exonucleases make them suitable to decrease lncRNA levels post-transcriptionally (Chan et al., 2006).
As mentioned before, on the paternal allele of Ube3a/Ube3a-ATS imprinted cluster, Ube3a-ATS represses Ube3a expression by prematurely terminating the elongation of Ube3a transcripts. Therefore, a potential strategy is to rescue the defective Ube3a transcription by targeting Ube3a-ATS transcripts using ASOs (Figure 6A I). To avoid influencing the transcription of sno-lncRNAs essential for neuronal development and PWS, ASOs were designed to be complementary to Ube3a-ATS transcripts downstream of the Snord115 cluster. These ASOs were provided to cultured AS mouse neurons with deficient Ube3a expression (Meng et al., 2015). The treatment achieved sustained ectopic paternal expression of Ube3a, partially rescued UBE3A brain protein levels, and alleviated some cognitive deficits. Remarkably, other splicing products derived from Ube3a-ATS like Snrpn and Snord116 were unaffected. Consistently, ASOs were designed to rescue the expression of UBE3A in AS iPSC-derived neuron cells with a large deletion of maternal 15q11-q13. ASOs targeting UBE3A-ATS transcripts at SNORD115 and SNORD109B, or targeting the snoRNA located between SNORD 115 locus and UBE3A gene region, cleave UBE3A-ATS and release the transcription of UBE3A on the paternal chromosome (Germain et al., 2021). UBE3A-ATS transcription is terminated by displacing RNA Polymerase II several kilobases downstream of the ASO targeting site. Therefore, targeting the lncRNA UBE3A-ATS by ASOs could be a potential strategy for rescuing UBE3A expression and related imprinting disorders. Besides, ASOs have several unique features in treating imprinted disorders, including high in vivo efficacy, broad tissue distribution, low adverse events, and long duration of action (Smith et al., 2006; Kordasiewicz et al., 2012). Considering that several mRNA-targeting ASOs have been approved (Dhuri et al., 2020), targeting lncRNAs using ASOs to treat imprinting diseases could achieve wide application. However, robust delivery systems devoid of associated toxicity should be carefully developed and evaluated.
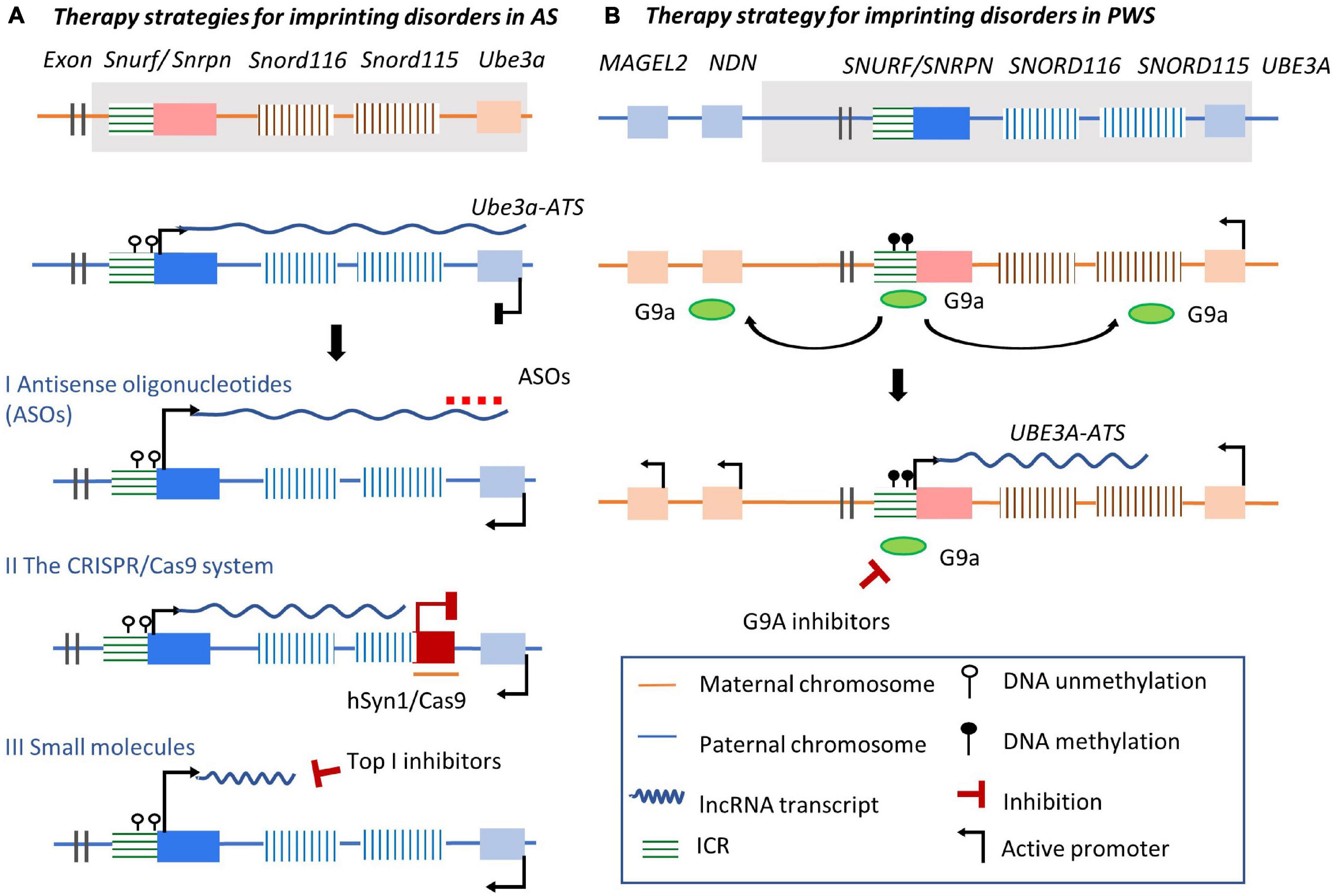
Figure 6. Three state of the art strategies for imprinted disorders via targeting imprinted lncRNAs. (A) Therapeutic strategies for AS. Molecular alterations such as deletions in Ube3a/Ube3a-ATS imprinted cluster can cause the loss of effective Ube3a expression. Line I: ASOs are designed to target the overlapping regions of Ube3a-ATS transcripts and Ube3a, releasing paternal Ube3a expression; Line II: the human synapsin 1 (hSYN1) gene promoter is used drive neuron-specific expression and Cas9 packaged with adeno-associated virus delivering system is inserted into the gene region of the Snord115, leading to disrupted transcription of Ube3a-ATS before extending to the Ube3a gene encoding region. Line III: Top I inhibitors disrupt the elongation of the Ube3a-ATS at the Snord116 region. Ube3a paternal expression is released from transcriptional collision. (B) Therapeutic strategy for PWS. G9A inhibitors prevent G9A recruitment to flanking regions near the ICR, releasing Ube3a-ATS lncRNA expression from its promoter in the ICR.
Modulation of Imprinted Long Non-coding RNA Expression Using the CRISPR/Cas9 System
The CRISPR/Cas9 system permits in vitro and in vivo gene editing tool and is another novel strategy to modulate imprinted lncRNA expression (Deltcheva et al., 2011; Jinek et al., 2012; Konermann et al., 2015; Suzuki et al., 2016; Knott and Doudna, 2018). A series of CRISPR/Cas-engineered systems can be designed to manipulate lncRNAs, including deletion of the lncRNA encoding gene region (pre-transcription level), inhibition or activation of the expression of the lncRNA (transcription level), or direct degradation of the lncRNA transcripts (post-transcriptional level) (Perez-Pinera et al., 2013; Qi et al., 2013; Ran et al., 2013; Abudayyeh et al., 2017). For example, CRISPRi and CRISPRa can modulate lncRNA expression by recruiting transcriptional repressors or activators without inducing genetic mutations (Bester et al., 2018; Kampmann, 2018). At the same time, CRISPR/Cas9 is being studied as a strategy of in vivo genome editing therapy in neurological diseases like schizophrenia and Alzheimer’s disease (Zhuo et al., 2017; Kuruvilla et al., 2018; Park et al., 2019; Sun et al., 2019). It is hoped that effective manipulation of the non-coding regions achieved in human cell lines and animal models could result in novel strategies to eliminate obstacles in developing therapies for lncRNA-related imprinting diseases (Cho et al., 2013; Cong et al., 2013; Jiang et al., 2013; Doudna and Charpentier, 2014, p. 9). Furthermore, when taking into account brain-specific expression of imprinted clusters, CRISPR/Cas9 could be designed to correct abnormal imprinting patterns (Han et al., 2014). Indeed, recently Cas9 gene therapy has shown promise in trapping Ube3a-ATS to activate paternal Ube3a expression (Figure 6A II; Wolter et al., 2020). In addition, a CRISPR/Cas9 system targeting the Snord115 locus in cultured mouse cortical neurons and human neural progenitor-derived neurons was able to successfully increase total Ube3a protein expression while decreasing Snord115 expression. Using a neuron-specific saCas9 and guide RNAs packaged in an adeno-associated virus delivering system and administered to an AS mouse brain during the embryonic and early postnatal stages led to silencing of paternal Snord115 expression with long-lasting effects.
In summary, the CRISPR-Cas9 system offers promising therapeutic strategies with the potential to permanently alter imprinted gene expression with high specificity and low toxicity. Nevertheless, since lncRNAs lack open reading frames and functional protein products, the use of CRISPR-Cas9 system to achieve efficient lncRNA manipulation needs to be further improved (Statello et al., 2021). In addition, an optimal sgRNA design and an effective delivery mechanism to penetrate the blood-brain barrier need further investigation (Zhuo et al., 2017; Hana et al., 2021).
Small Molecules Targeting Histone Modifiers
Small molecules have been screened to target histone modification proteins involved in imprinted lncRNA regulation. As mentioned before, PWS and AS are two imprinting disorders related to the same imprinted cluster. In AS patients, UBE3A expression is decreased. Through high-content screening in mouse-derived primary cortical neurons, about 10 topoisomerase I (Top I) inhibitors have been identified with the capacity to downregulate Ube3a-ATS expression and induce reactivation of UBE3A expression from the paternal allele (Huang et al., 2011; Powell et al., 2013b). The Top I inhibitor topotecan blocks the elongation of the Ube3a-ATS transcription complex in cultured mouse neurons (Powell et al., 2013b). It inhibits sno-lncRNA transcription throughout the Ube3a encoding gene region by stabilizing the formation of R loops between RNA and DNA within paternal Snord116, leading to chromatin decondensation (Liu and Wang, 1987; Belotserkovskii et al., 2010; El Hage et al., 2010; Belotserkovskii and Hanawalt, 2011; French et al., 2011; Skourti-Stathaki et al., 2011; Aguilera and García-Muse, 2012; Ginno et al., 2012; Figure 6A III). The Ube3a-ATS transcription complex stalled before transcription of Sno-lncRNAs completed. Subsequent Ube3a expression was reactivated on the paternal allele. Additional candidates of other Top I inhibitors have also been assessed to identify inhibitors with better pharmacological profiles of Ube3a activation (Lee et al., 2018). Prospective therapeutic safety and central nervous system (CNS) bioavailability studies have also been performed recently in AS mouse neurons (Lee et al., 2018).
A therapeutic strategy for PWS based on the induction of SNORD116 expression has been proposed. SNORD116 is normally silenced on the maternal allele, but its expression can be induced by modulating ‘closed’ chromatin condition into an ‘open’ state (Kim et al., 2017). The methylation of histone H3K9 performs allele-specific pattern in the ICR located upstream of SNRPN gene (PWS-ICR). On the maternal chromosome, histone methyltransferase euchromatic histone lysine N-methyltransferase-2 (G9a) locates at the methylated PWS-ICR and recruits repressive histone modifications (H3K9me2) along the PWS-ICR in a bidirectional manner. This leads to condensed chromatin structure and silencing of PWS-associated genes (Figure 6B). The inactivation of histone H3K9 methyltransferase G9a in mouse embryonic stem (ES) cells leads to reduced DNA methylation in PWS-ICR, and the expression of Snrpn was activated on both chromosome in vitro (Xin et al., 2003). However, in in vivo mouse model, two inhibitors of G9a selected lead to the activation of maternal copy of Snord116 and improve survival of the PWS mouse without effect on the methylation state of the PWS-ICR or Ube3a expression on the maternal allele (Kim et al., 2017). Thus, further studies are needed to clarify the association between DNA methylation of PWS-ICR and allele-specific distribution of G9a. Meanwhile, the reactivation of SNRPN and SNORD116 was recently achieved by preventing the recruitment of H3K9me3 repressive histone modification-related protein factor to SNORD116 locus in PWS-derived iPSCs (Langouët et al., 2020). In summary, small molecules related epigenetic therapy for PWS through modulating the condition of specific chromatin regions could be a potential strategy to be translated in clinical relevance (Crunkhorn, 2017; Chung et al., 2020).
Concluding Remarks
Several imprinting gene clusters have been discovered and studied since the middle of the last century. These studies have shown that lncRNAs play crucial roles in regulating imprinted gene clusters and individual imprinted genes related to human health and diseases. However, from a genomic perspective, the characteristics of gene regulation among imprinting loci remain to be fully elucidated. This is despite the advancement in knowledge of the epigenetic regulatory mechanisms of a subset of genes in imprinted regions. In the three imprinted clusters (Airn/Igfr2, Kcnq1/Kcnq1ot1, and Ube3a/Ube3a-ATS), imprinted lncRNAs which play essential regulatory roles in silencing other imprinted genes are all expressed on the paternal allele. It has been reported that maternal expressed imprinted genes are prominent with protein-coding genes, while paternal expressed genes exhibit consistent distribution between non-coding and protein-coding sequences (Hutter et al., 2010). However, the difference between the establishment of maternal and paternal imprinted genes in lncRNA mechanisms remains unclear. Thus, comprehensive investigations are needed to understand further the mechanisms of imprinted lncRNAs in the epigenetic regulation of imprinted clusters. With technological advancements, studies on lncRNA-associated human imprinting disorders will lead to needed therapies.
Pharmacological treatments for congenital imprinting disorders are limited to symptomatic therapies, which are inefficient in promoting the patients’ quality of life (Chung et al., 2020). Fortunately, the biological role of lncRNAs in the etiology of congenital imprinting disorders has been revealed thanks to the advancement in high-throughput genome-wide sequencing technologies. Therapeutic approaches based on disease-related lncRNAs have been investigated. In a recent study, lncRNA mimics were designed to restore the tissue-specific lncRNA HULC in mice, essential for phenylalanine metabolism (Li et al., 2021). In addition, three strategies mentioned above targeting Ube3a-ATS have efficiently rescued imprinting disorders in PWS/AS imprinted cluster in mouse models and human cell lines. Although the three strategies mentioned here targeting Ube3a-ATS have efficiently rescued imprinting disorders of PWS/AS imprinted cluster in mouse models and human cell lines, therapies for other disease-related clusters have not been investigated. Long non-coding RNA-based and lncRNA-targeting therapies have some unique advantages. For instance, in lncRNA-targeting methods like ASOs, synthesized RNA can be designed with organ-targeting peptides to achieve tissue-specific targeting of endogenous lncRNAs. Besides, synthesized RNA products could be modified to promote in vivo stability. Further translation of these strategies to real clinical tools will require further investigation to overcome related challenges. In vivo delivery of synthesized RNA molecules, cellular permeability, immunogenicity, and potential of organ toxicity also deserve further investigation (Perry and Ulitsky, 2021). Another challenge to extend the lessons learned in PWS and AS into other imprinting disorders is the epigenetic and molecular complexities in different imprinting disorders-related imprinted loci. Considering the complexity of the regulatory network of genomic imprinting, further efforts are needed to reveal underlying pathological mechanisms linked to imprinting disorder phenotypes and support continuous improvement of clinical management and therapeutic strategies.
Author Contributions
TW and QM wrote and edited the manuscript and drew the pictures. JL, LY, and MW assisted in manuscript collation and review. QM provided critical inputs as the corresponding authors and obtained funds. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No: 32070870), Guangdong Province Natural Science Foundation of China (No: 2021A1515010758), Guangdong Provincial Key Laboratory of Synthetic Genomics (No: 2019B030301006), Shenzhen Key Laboratory of Synthetic Genomics (No: ZDSYS201802061806209) and Shenzhen Institute of Synthetic Biology Scientific Research Program (No: ZTXM20200008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ASE, allele-specific gene expression; MAE, monoallelic expression; PWS, Prader–Willi syndrome; AS, Angelman syndrome; BWS, Beckwith–Wiedemann syndrome; SRS, Silver–Russell syndrome; KOS14, Kagami–Ogata syndrome; TS14, Temple syndrome; ICRs, imprinting control centers or imprinting control regions; gDMRs, germline differentially methylated regions; lncRNAs, long non-coding RNAs; ASOs, antisense oligonucleotides; SNPs, single nucleotide polymorphisms; H4R3me2s, histone H4 arginine-3 symmetrical demethylation; H3K9me3, H3 lysine-9 trimethylation; Igf2, the insulin-like growth factor 2; Kcnq1/Kcnq1ot1, potassium voltage-gated channel subfamily Q member 1/Kcnq1 antisense transcript 1; KvDMR1, KvLQT1 differentially methylated region 1; PRC, polycomb repressive complex; EHMT2, histone methyltransferase euchromatic histone lysine N-methyltransferase -2; Airn, the antisense of Igf2r non-protein coding RNA; H2AK119ub1, lysine 119-monoubiquititinated histone H2A; Ube3a-ATS, Ube3a-antisense lncRNA; UPD, uniparental disomy; MLID, multi-locus imprinting disturbance; SNORD116, SnoRNA C/D box cluster 116; RBFOX2, RNA binding protein fox-1 homolog 2; DLK1/DIO3, delta-like homolog 1 gene/type III iodothyronine deiodinase gene; Rtl1as, the Rtl1-antisense; PI3K/Akt, phosphoinositide 3-kinase/protein kinase B; CRISPRi, CRISPR interference; CRISPRa, CRISPR activation; topoisomerase I, (Top I) inhibitors.
References
Abi Habib, W., Brioude, F., Azzi, S., Salem, J., Das Neves, C., Personnier, C., et al. (2017). 11p15 ICR1 partial deletions associated with IGF2/H19 DMR hypomethylation and silver-russell syndrome. Hum. Mutat. 38, 105–111. doi: 10.1002/humu.23131
Abudayyeh, O. O., Gootenberg, J. S., Essletzbichler, P., Han, S., Joung, J., Belanto, J. J., et al. (2017). RNA targeting with CRISPR-Cas13. Nature 550, 280–284. doi: 10.1038/nature24049
Adhikari, A., Copping, N. A., Onaga, B., Pride, M. C., Coulson, R. L., Yang, M., et al. (2019). Cognitive deficits in the Snord116 deletion mouse model for Prader-Willi syndrome. Neurobiol. Learn. Mem. 165:106874. doi: 10.1016/j.nlm.2018.05.011
Ager, E. I., Pask, A. J., Gehring, H. M., Shaw, G., and Renfree, M. B. (2008). Evolution of the CDKN1C-KCNQ1 imprinted domain. BMC Evol. Biol. 8:163. doi: 10.1186/1471-2148-8-163
Aguilera, A., and García-Muse, T. (2012). R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 46, 115–124. doi: 10.1016/j.molcel.2012.04.009
Andergassen, D., Dotter, C. P., Wenzel, D., Sigl, V., Bammer, P. C., Muckenhuber, M., et al. (2017). Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. eLife 6:e25125. doi: 10.7554/eLife.25125
Andergassen, D., Muckenhuber, M., Bammer, P. C., Kulinski, T. M., Theussl, H. C., Shimizu, T., et al. (2019). The Airn lncRNA does not require any DNA elements within its locus to silence distant imprinted genes. PLoS Genetics 16:e1009151. doi: 10.1371/journal.pgen.1008268
Avagliano Trezza, R., Sonzogni, M., Bossuyt, S. N. V., Zampeta, F. I., Punt, A. M., van den Berg, M., et al. (2019). Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci. 22, 1235–1247. doi: 10.1038/s41593-019-0425-420
Azzi, S., Blaise, A., Steunou, V., Harbison, M. D., Salem, J., Brioude, F., et al. (2014). Complex tissue-specific epigenotypes in russell-silver syndrome associated with 11p15 ICR1 hypomethylation. Hum. Mutat. 35, 1211–1220. doi: 10.1002/humu.22623
Azzi, S., Rossignol, S., Steunou, V., Sas, T., Thibaud, N., Danton, F., et al. (2009). Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum. Mol. Genet. 18, 4724–4733. doi: 10.1093/hmg/ddp435
Babak, T., Deveale, B., Armour, C., Raymond, C., Cleary, M. A., van der Kooy, D., et al. (2008). Global survey of genomic imprinting by transcriptome sequencing. Curr. Biol. 18, 1735–1741. doi: 10.1016/j.cub.2008.09.044
Baran, Y., Subramaniam, M., Biton, A., Tukiainen, T., Tsang, E. K., Rivas, M. A., et al. (2015). The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 25, 927–936. doi: 10.1101/gr.192278.115
Barlow, D. P. (2011). Genomic imprinting: a mammalian epigenetic discovery model. Annu. Rev. Genet. 45, 379–403. doi: 10.1146/annurev-genet-110410-132459
Barlow, D. P., and Bartolomei, M. S. (2014). Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 6:a018382. doi: 10.1101/cshperspect.a018382
Bartolomei, M. S., Zemel, S., and Tilghman, S. M. (1991). Parental imprinting of the mouse H19 gene. Nature 351, 153–155. doi: 10.1038/351153a0
Bazeley, P. S., Shepelev, V., Talebizadeh, Z., Butler, M. G., Fedorova, L., Filatov, V., et al. (2008). snoTARGET shows that human orphan snoRNA targets locate close to alternative splice junctions. Gene 408, 172–179. doi: 10.1016/j.gene.2007.10.037
Beatty, L., Weksberg, R., and Sadowski, P. D. (2006). Detailed analysis of the methylation patterns of the KvDMR1 imprinting control region of human chromosome 11. Genomics 87, 46–56. doi: 10.1016/j.ygeno.2005.05.015
Beaudet, A. L., and Meng, L. (2016). Gene-targeting pharmaceuticals for single-gene disorders. Hum. Mol. Genet. 25, R18–R26. doi: 10.1093/hmg/ddv476
Belotserkovskii, B. P., and Hanawalt, P. C. (2011). Anchoring nascent RNA to the DNA template could interfere with transcription. Biophys. J. 100, 675–684. doi: 10.1016/j.bpj.2010.12.3709
Belotserkovskii, B. P., Liu, R., Tornaletti, S., Krasilnikova, M. M., Mirkin, S. M., and Hanawalt, P. C. (2010). Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc. Natl. Acad. Sci. U S A. 107, 12816–12821. doi: 10.1073/pnas.1007580107
Benetatos, L., Vartholomatos, G., and Hatzimichael, E. (2011). MEG3 imprinted gene contribution in tumorigenesis. Int. J. Cancer 129, 773–779. doi: 10.1002/ijc.26052
Bergman, Y., and Cedar, H. (2004). A stepwise epigenetic process controls immunoglobulin allelic exclusion. Nat. Rev. Immunol. 4, 753–761. doi: 10.1038/nri1458
Bester, A. C., Lee, J. D., Chavez, A., Lee, Y.-R., Nachmani, D., Vora, S., et al. (2018). An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell 173, 649–664.e20. doi: 10.1016/j.cell.2018.03.052
Beygo, J., Elbracht, M., de Groot, K., Begemann, M., Kanber, D., Platzer, K., et al. (2015). Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur. J. Hum. Genet. 23, 180–188. doi: 10.1038/ejhg.2014.72
Bhan, A., Soleimani, M., and Mandal, S. S. (2017). Long noncoding RNA and cancer: a new paradigm. Cancer Res. 77, 3965–3981. doi: 10.1158/0008-5472.CAN-16-2634
Bian, S., and Sun, T. (2011). Functions of noncoding RNAs in neural development and neurological diseases. Mol. Neurobiol. 44, 359–373. doi: 10.1007/s12035-011-8211-8213
Bieth, E., Eddiry, S., Gaston, V., Lorenzini, F., Buffet, A., Conte Auriol, F., et al. (2015). Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi syndrome. Eur. J. Hum. Genet. 23, 252–255. doi: 10.1038/ejhg.2014.103
Brioude, F., Hennekam, R., Bliek, J., Coze, C., Eggermann, T., Ferrero, G. B., et al. (2018a). Revisiting wilms tumour surveillance in beckwith-wiedemann syndrome with IC2 methylation loss, reply. Eur. J. Hum. Genet. 26, 471–472. doi: 10.1038/s41431-017-0074-72
Brioude, F., Kalish, J. M., Mussa, A., Foster, A. C., Bliek, J., Ferrero, G. B., et al. (2018b). Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat. Rev. Endocrinol. 14, 229–249. doi: 10.1038/nrendo.2017.166
Buiting, K. (2010). Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 365–376. doi: 10.1002/ajmg.c.30273
Buiting, K., Saitoh, S., Gross, S., Dittrich, B., Schwartz, S., Nicholls, R. D., et al. (1995). Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 9, 395–400. doi: 10.1038/ng0495-395
Buiting, K., Williams, C., and Horsthemke, B. (2016). Angelman syndrome - insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 12, 584–593. doi: 10.1038/nrneurol.2016.133
Burnett, L. C., Hubner, G., LeDuc, C. A., Morabito, M. V., Carli, J. F. M., and Leibel, R. L. (2017). Loss of the imprinted, non-coding Snord116 gene cluster in the interval deleted in the Prader Willi syndrome results in murine neuronal and endocrine pancreatic developmental phenotypes. Hum. Mol. Genet. 26, 4606–4616. doi: 10.1093/hmg/ddx342
Calabrese, J. M., Starmer, J., Schertzer, M. D., Yee, D., and Magnuson, T. (2015). A survey of imprinted gene expression in mouse trophoblast stem cells. G3 (Bethesda) 5, 751–759. doi: 10.1534/g3.114.016238
Calin, G. A., Liu, C., Ferracin, M., Hyslop, T., Spizzo, R., Sevignani, C., et al. (2007). Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12, 215–229. doi: 10.1016/j.ccr.2007.07.027
Carli, D., Riberi, E., Ferrero, G. B., and Mussa, A. (2020). Syndromic disorders caused by disturbed human imprinting. J. Clin. Res. Pediatr. Endocrinol. 12, 1–16. doi: 10.4274/jcrpe.galenos.2019.2018.0249
Cavaillé, J., Buiting, K., Kiefmann, M., Lalande, M., Brannan, C. I., Horsthemke, B., et al. (2000). Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. U S A. 97, 14311–14316. doi: 10.1073/pnas.250426397
Chamberlain, S. J. (2013). RNAs of the human chromosome 15q11-q13 imprinted region. Wiley Interdiscip. Rev. RNA 4, 155–166. doi: 10.1002/wrna.1150
Chan, J. H. P., Lim, S., and Wong, W. S. F. (2006). Antisense oligonucleotides: from design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 33, 533–540. doi: 10.1111/j.1440-1681.2006.04403.x
Chang, S., and Bartolomei, M. S. (2020). Modeling human epigenetic disorders in mice: beckwith-Wiedemann syndrome and Silver-Russell syndrome. Dis. Model Mech. 13:dmm044123. doi: 10.1242/dmm.044123
Chen, W. V., and Maniatis, T. (2013). Clustered protocadherins. Development 140, 3297–3302. doi: 10.1242/dev.090621
Chen, Y., Gao, H., Liu, Q., Xie, W., Li, B., Cheng, S., et al. (2020). Functional characterization of DLK1/MEG3 locus on chromosome 14q32.2 reveals the differentiation of pituitary neuroendocrine tumors. Aging (Albany NY) 13, 1422–1439. doi: 10.18632/aging.202376
Chen, Z., and Zhang, Y. (2020). Maternal H3K27me3-dependent autosomal and X chromosome imprinting. Nat. Rev. Genet. 21, 555–571. doi: 10.1038/s41576-020-0245-249
Cheunsuchon, P., Zhou, Y., Zhang, X., Lee, H., Chen, W., Nakayama, Y., et al. (2011). Silencing of the imprinted DLK1-MEG3 locus in human clinically nonfunctioning pituitary adenomas. Am. J. Pathol. 179, 2120–2130. doi: 10.1016/j.ajpath.2011.07.002
Cho, S. W., Kim, S., Kim, J. M., and Kim, J.-S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Cho, S. W., Xu, J., Sun, R., Mumbach, M. R., Carter, A. C., Chen, Y. G., et al. (2018). Promoter of lncRNA Gene PVT1 is a tumor-suppressor DNA boundary element. Cell 173, 1398–1412.e22. doi: 10.1016/j.cell.2018.03.068
Chotalia, M., Smallwood, S. A., Ruf, N., Dawson, C., Lucifero, D., Frontera, M., et al. (2009). Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 23, 105–117. doi: 10.1101/gad.495809
Chung, M. S., Langouët, M., Chamberlain, S. J., and Carmichael, G. G. (2020). Prader-Willi syndrome: reflections on seminal studies and future therapies. Open Biol. 10:200195. doi: 10.1098/rsob.200195
Claborn, M. K., Stevens, D. L., Walker, C. K., and Gildon, B. L. (2019). Nusinersen: a treatment for spinal muscular atrophy. Ann. Pharmacother. 53, 61–69. doi: 10.1177/1060028018789956
Clark, M. B., Johnston, R. L., Inostroza-Ponta, M., Fox, A. H., Fortini, E., Moscato, P., et al. (2012). Genome-wide analysis of long noncoding RNA stability. Genome Res. 22, 885–898. doi: 10.1101/gr.131037.111
Colognori, D., Sunwoo, H., Kriz, A. J., Wang, C.-Y., and Lee, J. T. (2019). Xist deletional analysis reveals an interdependency between xist RNA and polycomb complexes for spreading along the inactive X. Mol. Cell 74, 101–117.e10. doi: 10.1016/j.molcel.2019.01.015
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Coulson, R. L., Powell, W. T., Yasui, D. H., Dileep, G., Resnick, J., and LaSalle, J. M. (2018a). Prader-Willi locus Snord116 RNA processing requires an active endogenous allele and neuron-specific splicing by Rbfox3/NeuN. Hum. Mol. Genet. 27, 4051–4060. doi: 10.1093/hmg/ddy296
Coulson, R. L., Yasui, D. H., Dunaway, K. W., Laufer, B. I., Vogel Ciernia, A., Zhu, Y., et al. (2018b). Snord116-dependent diurnal rhythm of DNA methylation in mouse cortex. Nat. Commun. 9:1616. doi: 10.1038/s41467-018-03676-3670
Court, F., Tayama, C., Romanelli, V., Martin-Trujillo, A., Iglesias-Platas, I., Okamura, K., et al. (2014). Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 24, 554–569. doi: 10.1101/gr.164913.113
Crunkhorn, S. (2017). Genetic disorders: steps towards epigenetic therapy for PWS. Nat. Rev. Drug Discov. 16:85. doi: 10.1038/nrd.2017.3
Cytrynbaum, C., Chong, K., Hannig, V., Choufani, S., Shuman, C., Steele, L., et al. (2016). Genomic imbalance in the centromeric 11p15 imprinting center in three families: further evidence of a role for IC2 as a cause of russell-silver syndrome. Am. J. Med. Genet. A 170, 2731–2739. doi: 10.1002/ajmg.a.37819
da Rocha, S. T., Edwards, C. A., Ito, M., Ogata, T., and Ferguson-Smith, A. C. (2008). Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 24, 306–316. doi: 10.1016/j.tig.2008.03.011
da Rocha, S. T., and Gendrel, A.-V. (2019). The influence of DNA methylation on monoallelic expression. Essays Biochem. 63, 663–676. doi: 10.1042/EBC20190034
Dagli, A. I., Mathews, J., and Williams, C. A. (1993). “Angelman Syndrome,” in GeneReviews, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, G. Mirzaa, et al. (Seattle, WA: University of Washington, Seattle).
Dao, D., Walsh, C. P., Yuan, L., Gorelov, D., Feng, L., Hensle, T., et al. (1999). Multipoint analysis of human chromosome 11p15/mouse distal chromosome 7: inclusion of H19/IGF2 in the minimal WT2 region, gene specificity of H19 silencing in Wilms’ tumorigenesis and methylation hyper-dependence of H19 imprinting. Hum. Mol. Genet. 8, 1337–1352. doi: 10.1093/hmg/8.7.1337
Davis, E., Caiment, F., Tordoir, X., Cavaillé, J., Ferguson-Smith, A., Cockett, N., et al. (2005). RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr. Biol. 15, 743–749. doi: 10.1016/j.cub.2005.02.060
de los Santos, T., Schweizer, J., Rees, C. A., and Francke, U. (2000). Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region, which Is highly expressed in brain. Am. J. Hum. Genet. 67, 1067–1082. doi: 10.1086/303106
de Smith, A. J., Purmann, C., Walters, R. G., Ellis, R. J., Holder, S. E., Van Haelst, M. M., et al. (2009). A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 18, 3257–3265. doi: 10.1093/hmg/ddp263
DeBaun, M. R., King, A. A., and White, N. (2000). Hypoglycemia in Beckwith-Wiedemann syndrome. Semin. Perinatol. 24, 164–171. doi: 10.1053/sp.2000.6366
DeChiara, T. M., Robertson, E. J., and Efstratiadis, A. (1991). Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859. doi: 10.1016/0092-8674(91)90513-x
Deltcheva, E., Chylinski, K., Sharma, C. M., Gonzales, K., Chao, Y., Pirzada, Z. A., et al. (2011). CRISPR RNA maturation by trans -encoded small RNA and host factor RNase III. Nature 471, 602–607. doi: 10.1038/nature09886
Deng, X., Berletch, J. B., Nguyen, D. K., and Disteche, C. M. (2014). X chromosome regulation: diverse patterns in development, tissues and disease. Nat. Rev. Genet. 15, 367–378. doi: 10.1038/nrg3687
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi: 10.1101/gr.132159.111
Dhuri, K., Bechtold, C., Quijano, E., Pham, H., Gupta, A., Vikram, A., et al. (2020). Antisense oligonucleotides: an emerging area in drug discovery and development. J. Clin. Med. 9:2004. doi: 10.3390/jcm9062004
Ding, F., Li, H. H., Zhang, S., Solomon, N. M., Camper, S. A., Cohen, P., et al. (2008). SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS One 3:e1709. doi: 10.1371/journal.pone.0001709
Doudna, J. A., and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096
Duffy, K. A., Cielo, C. M., Cohen, J. L., Gonzalez-Gandolfi, C. X., Griff, J. R., Hathaway, E. R., et al. (2019). Characterization of the beckwith-wiedemann spectrum: diagnosis and management. Am. J. Med. Genet. C Semin. Med. Genet. 181, 693–708. doi: 10.1002/ajmg.c.31740
Duker, A. L., Ballif, B. C., Bawle, E. V., Person, R. E., Mahadevan, S., Alliman, S., et al. (2010). Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur. J. Hum. Genet. 18, 1196–1201. doi: 10.1038/ejhg.2010.102
Eggermann, T., Brioude, F., Russo, S., Lombardi, M. P., Bliek, J., Maher, E. R., et al. (2016). Prenatal molecular testing for Beckwith-Wiedemann and Silver-Russell syndromes: a challenge for molecular analysis and genetic counseling. Eur. J. Hum. Genet. 24, 784–793. doi: 10.1038/ejhg.2015.224
Eggermann, T., Kadgien, G., Begemann, M., and Elbracht, M. (2021). Biallelic PADI6 variants cause multilocus imprinting disturbances and miscarriages in the same family. Eur. J. Hum. Genet. 29, 575–580. doi: 10.1038/s41431-020-00762-760
Eggermann, T., Perez, de Nanclares, G., Maher, E. R., Temple, I. K., Tümer, Z., et al. (2015). Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics 7:123. doi: 10.1186/s13148-015-0143-148
Eggermann, T., Buiting, K., and Temple, I. K. (2011). Clinical utility gene card for: silver-russell syndrome. Eur. J. Hum. Genet. 19. doi: 10.1038/ejhg.2010.202
El Hage, A., French, S. L., Beyer, A. L., and Tollervey, D. (2010). Loss of topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 24, 1546–1558. doi: 10.1101/gad.573310
Elbracht, M., Mackay, D., Begemann, M., Kagan, K. O., and Eggermann, T. (2020). Disturbed genomic imprinting and its relevance for human reproduction: causes and clinical consequences. Hum. Reprod. Update 26, 197–213. doi: 10.1093/humupd/dmz045
Fang, S., Zhang, L., Guo, J., Niu, Y., Wu, Y., Li, H., et al. (2018). NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 46, D308–D314. doi: 10.1093/nar/gkx1107
Ferguson-Smith, A. C. (2011). Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 12, 565–575. doi: 10.1038/nrg3032
Ferguson-Smith, A. C., Cattanach, B. M., Barton, S. C., Beechey, C. V., and Surani, M. A. (1991). Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nature 351, 667–670. doi: 10.1038/351667a0
Ferrón, S. R., Charalambous, M., Radford, E., McEwen, K., Wildner, H., Hind, E., et al. (2011). Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature 475, 381–385. doi: 10.1038/nature10229
Fontana, L., Bedeschi, M. F., Maitz, S., Cereda, A., Faré, C., Motta, S., et al. (2018). Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15.5 related imprinting disorders. Epigenetics 13, 897–909. doi: 10.1080/15592294.2018.1514230
Fontana, P., Grasso, M., Acquaviva, F., Gennaro, E., Galli, M. L., Falco, M., et al. (2017). SNORD116 deletions cause Prader-Willi syndrome with a mild phenotype and macrocephaly. Clin. Genet. 92, 440–443. doi: 10.1111/cge.13005
Fountain, M. D., and Schaaf, C. P. (2016). Prader-Willi syndrome and schaaf-yang syndrome: neurodevelopmental diseases intersecting at the MAGEL2 gene. Diseases 4:E2. doi: 10.3390/diseases4010002
French, S. L., Sikes, M. L., Hontz, R. D., Osheim, Y. N., Lambert, T. E., El Hage, A., et al. (2011). Distinguishing the roles of topoisomerases I and II in relief of transcription-induced torsional stress in yeast rRNA genes. Mol. Cell. Biol. 31, 482–494. doi: 10.1128/MCB.00589-510
Germain, N. D., Gorka, D., Drennan, R., Jafar-nejad, P., Whipple, A., Core, L., et al. (2021). Antisense oligonucleotides targeting UBE3A-ATS restore expression of UBE3A by relieving transcriptional interference. bioRxiv [preprint] doi: 10.1101/2021.07.09.451826
Gil, N., and Ulitsky, I. (2020). Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 21, 102–117. doi: 10.1038/s41576-019-0184-185
Gillessen-Kaesbach, G., Albrecht, B., Eggermann, T., Elbracht, M., Mitter, D., Morlot, S., et al. (2018). Molecular and clinical studies in 8 patients with temple syndrome. Clin. Genet. 93, 1179–1188. doi: 10.1111/cge.13244
Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., and Chédin, F. (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 45, 814–825. doi: 10.1016/j.molcel.2012.01.017
Gómez-Marín, C., Tena, J. J., Acemel, R. D., López-Mayorga, M., Naranjo, S., de la Calle-Mustienes, E., et al. (2015). Evolutionary comparison reveals that diverging CTCF sites are signatures of ancestral topological associating domains borders. Proc. Natl. Acad. Sci. U S A. 112, 7542–7547. doi: 10.1073/pnas.1505463112
Goovaerts, T., Steyaert, S., Vandenbussche, C. A., Galle, J., Thas, O., Van Criekinge, W., et al. (2018). A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat. Commun. 9:4120. doi: 10.1038/s41467-018-06566-6567
Groen, E. J. N., Talbot, K., and Gillingwater, T. H. (2018). Advances in therapy for spinal muscular atrophy: promises and challenges. Nat. Rev. Neurol. 14, 214–224. doi: 10.1038/nrneurol.2018.4
Guenzl, P. M., and Barlow, D. P. (2012). Macro lncRNAs: a new layer of cis-regulatory information in the mammalian genome. RNA Biol. 9, 731–741. doi: 10.4161/rna.19985
Han, J., Zhang, J., Chen, L., Shen, B., Zhou, J., Hu, B., et al. (2014). Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 11, 829–835. doi: 10.4161/rna.29624
Hana, S., Peterson, M., McLaughlin, H., Marshall, E., Fabian, A. J., McKissick, O., et al. (2021). Highly efficient neuronal gene knockout in vivo by CRISPR-Cas9 via neonatal intracerebroventricular injection of AAV in mice. Gene Ther. doi: 10.1038/s41434-021-00224-222 [Epub ahead of print].
Hanna, C. W. (2020). Placental imprinting: emerging mechanisms and functions. PLoS Genet 16:e1008709. doi: 10.1371/journal.pgen.1008709
Hao, N., Palmer, A. C., Dodd, I. B., and Shearwin, K. E. (2017). Directing traffic on DNA-How transcription factors relieve or induce transcriptional interference. Transcription 8, 120–125. doi: 10.1080/21541264.2017.1285851
Harrow, J., Frankish, A., Gonzalez, J. M., Tapanari, E., Diekhans, M., Kokocinski, F., et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. doi: 10.1101/gr.135350.111
He, J.-H., Han, Z.-P., Liu, J.-M., Zhou, J.-B., Zou, M.-X., Lv, Y.-B., et al. (2017). Overexpression of long non-coding RNA MEG3 inhibits proliferation of hepatocellular carcinoma Huh7 cells via negative modulation of miRNA-664. J. Cell. Biochem. 118, 3713–3721. doi: 10.1002/jcb.26018
Henckel, A., Chebli, K., Kota, S. K., Arnaud, P., and Feil, R. (2012). Transcription and histone methylation changes correlate with imprint acquisition in male germ cells. EMBO J. 31, 606–615. doi: 10.1038/emboj.2011.425
Horsthemke, B. (2010). Mechanisms of imprint dysregulation. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 321–328. doi: 10.1002/ajmg.c.30269
Horsthemke, B., and Wagstaff, J. (2008). Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A 146A, 2041–2052. doi: 10.1002/ajmg.a.32364
Hoy, S. M. (2017). Nusinersen: first global approval. Drugs 77, 473–479. doi: 10.1007/s40265-017-0711-717
Hsiao, J. S., Germain, N. D., Wilderman, A., Stoddard, C., Wojenski, L. A., Villafano, G. J., et al. (2019). A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc. Natl. Acad. Sci. U S A. 116, 2181–2186. doi: 10.1073/pnas.1815279116
Hua, J. T., Ahmed, M., Guo, H., Zhang, Y., Chen, S., Soares, F., et al. (2018). Risk SNP-Mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell 174, 564–575.e18. doi: 10.1016/j.cell.2018.06.014
Huang, H.-S., Allen, J. A., Mabb, A. M., King, I. F., Miriyala, J., Taylor-Blake, B., et al. (2011). Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481, 185–189. doi: 10.1038/nature10726
Huang, W. C., Bennett, K., and Gregg, C. (2018). Epigenetic and cellular diversity in the brain through allele-specific effects. Trends Neurosci. 41, 925–937. doi: 10.1016/j.tins.2018.07.005
Hutter, B., Bieg, M., Helms, V., and Paulsen, M. (2010). Imprinted genes show unique patterns of sequence conservation. BMC Genomics 11:649. doi: 10.1186/1471-2164-11-649
Ibrahim, A., Kirby, G., Hardy, C., Dias, R. P., Tee, L., Lim, D., et al. (2014). Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin. Epigenetics 6:11. doi: 10.1186/1868-7083-6-11
Im, M., Jp, R., and Hg, S. (2005). A census of mammalian imprinting. Trends Genet. 21, 457–465. doi: 10.1016/j.tig.2005.06.008
Ioannides, Y., Lokulo-Sodipe, K., Mackay, D. J. G., Davies, J. H., and Temple, I. K. (2014). Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J. Med. Genet. 51, 495–501. doi: 10.1136/jmedgenet-2014-102396
Jiang, W., Bikard, D., Cox, D., Zhang, F., and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239. doi: 10.1038/nbt.2508
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Kaffer, C. R., Srivastava, M., Park, K. Y., Ives, E., Hsieh, S., Batlle, J., et al. (2000). A transcriptional insulator at the imprinted H19/Igf2 locus. Genes Dev. 14, 1908–1919.
Kagami, M., Mizuno, S., Matsubara, K., Nakabayashi, K., Sano, S., Fuke, T., et al. (2015). Epimutations of the IG-DMR and the MEG3-DMR at the 14q32.2 imprinted region in two patients with Silver-Russell Syndrome-compatible phenotype. Eur. J. Hum. Genet. 23, 1062–1067. doi: 10.1038/ejhg.2014.234
Kalish, J. M., Biesecker, L. G., Brioude, F., Deardorff, M. A., Di Cesare-Merlone, A., Druley, T., et al. (2017). Nomenclature and definition in asymmetric regional body overgrowth. Am. J. Med. Genet. A 173, 1735–1738. doi: 10.1002/ajmg.a.38266
Kalsner, L., and Chamberlain, S. J. (2015). Prader-Willi, angelman, and 15q11-q13 duplication syndromes. Pediatr. Clin. North Am. 62, 587–606. doi: 10.1016/j.pcl.2015.03.004
Kampmann, M. (2018). CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem. Biol. 13, 406–416. doi: 10.1021/acschembio.7b00657
Kanduri, C. (2016). Long noncoding RNAs: lessons from genomic imprinting. Biochim. Biophys. Acta (BBA) - Gene Regulatory Mechan. 1859, 102–111. doi: 10.1016/j.bbagrm.2015.05.006
Kaneko, S., Bonasio, R., Saldaña-Meyer, R., Yoshida, T., Son, J., Nishino, K., et al. (2014). Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell. 53, 290–300. doi: 10.1016/j.molcel.2013.11.012
Karaki, S., Paris, C., and Rocchi, P. (2019). “Antisense oligonucleotides, a novel developing targeting therapy,” in Antisense Therapy, eds S. Sharad and S. Kapur (London: IntechOpen), doi: 10.5772/intechopen.82105
Kelsey, G., and Feil, R. (2013). New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20110336. doi: 10.1098/rstb.2011.0336
Kim, J., Bretz, C. L., and Lee, S. (2015). Epigenetic instability of imprinted genes in human cancers. Nucleic Acids Res. 43, 10689–10699. doi: 10.1093/nar/gkv867
Kim, Y., Lee, H.-M., Xiong, Y., Sciaky, N., Hulbert, S. W., Cao, X., et al. (2017). Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat. Med. 23, 213–222. doi: 10.1038/nm.4257
Kitagawa, M., Kotake, Y., and Ohhata, T. (2012). Long non-coding RNAs involved in cancer development and cell fate determination. Curr. Drug Targets 13, 1616–1621. doi: 10.2174/138945012803530026
Knauss, J. L., and Sun, T. (2013). Regulatory mechanisms of long noncoding RNAs in vertebrate central nervous system development and function. Neuroscience 235, 200–214. doi: 10.1016/j.neuroscience.2013.01.022
Knott, G. J., and Doudna, J. A. (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. doi: 10.1126/science.aat5011
Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. doi: 10.1038/nature14136
Kopp, F., and Mendell, J. T. (2018). Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407. doi: 10.1016/j.cell.2018.01.011
Kordasiewicz, H. B., Stanek, L. M., Wancewicz, E. V., Mazur, C., McAlonis, M. M., Pytel, K. A., et al. (2012). Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044. doi: 10.1016/j.neuron.2012.05.009
Kota, S. K., Llères, D., Bouschet, T., Hirasawa, R., Marchand, A., Begon-Pescia, C., et al. (2014). ICR noncoding RNA expression controls imprinting and DNA replication at the Dlk1-Dio3 domain. Dev. Cell 31, 19–33. doi: 10.1016/j.devcel.2014.08.009
Kotzin, J. J., Spencer, S. P., McCright, S. J., Kumar, D. B. U., Collet, M. A., Mowel, W. K., et al. (2016). The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature 537, 239–243. doi: 10.1038/nature19346
Kuruvilla, J., Sasmita, A. O., and Ling, A. P. K. (2018). Therapeutic potential of combined viral transduction and CRISPR/Cas9 gene editing in treating neurodegenerative diseases. Neurol. Sci. 39, 1827–1835. doi: 10.1007/s10072-018-3521-3520
Landers, M., Bancescu, D. L., Le Meur, E., Rougeulle, C., Glatt-Deeley, H., Brannan, C., et al. (2004). Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res. 32, 3480–3492. doi: 10.1093/nar/gkh670
Langouët, M., Glatt-Deeley, H. R., Chung, M. S., Dupont-Thibert, C. M., Mathieux, E., Banda, E. C., et al. (2018). Zinc finger protein 274 regulates imprinted expression of transcripts in Prader-Willi syndrome neurons. Hum. Mol. Genet. 27, 505–515. doi: 10.1093/hmg/ddx420
Langouët, M., Gorka, D., Orniacki, C., Dupont-Thibert, C. M., Chung, M. S., Glatt-Deeley, H. R., et al. (2020). Specific ZNF274 binding interference at SNORD116 activates the maternal transcripts in Prader-Willi syndrome neurons. Hum. Mol. Genet. 29, 3285–3295. doi: 10.1093/hmg/ddaa210
Latos, P. A., and Barlow, D. P. (2009). Regulation of imprinted expression by macro non-coding RNAs. RNA Biol. 6, 100–106. doi: 10.4161/rna.6.2.7854
Latos, P. A., Pauler, F. M., Koerner, M. V., Şenergin, H. B., Hudson, Q. J., Stocsits, R. R., et al. (2012). Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338, 1469–1472. doi: 10.1126/science.1228110
Lecerf, C., Le Bourhis, X., and Adriaenssens, E. (2019). The long non-coding RNA H19: an active player with multiple facets to sustain the hallmarks of cancer. Cell Mol. Life. Sci. 76, 4673–4687. doi: 10.1007/s00018-019-03240-z
Lee, H.-M., Clark, E. P., Kuijer, M. B., Cushman, M., Pommier, Y., and Philpot, B. D. (2018). Characterization and structure-activity relationships of indenoisoquinoline-derived topoisomerase I inhibitors in unsilencing the dormant Ube3a gene associated with Angelman syndrome. Mol. Autism 9:45. doi: 10.1186/s13229-018-0228-222
Lee, J. T. (2011). Gracefully ageing at 50, X-chromosome inactivation becomes a paradigm for RNA and chromatin control. Nat. Rev. Mol. Cell Biol. 12, 815–826. doi: 10.1038/nrm3231
Lee, J. T., and Bartolomei, M. S. (2013). X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152, 1308–1323. doi: 10.1016/j.cell.2013.02.016
Lee, M. P., DeBaun, M. R., Mitsuya, K., Galonek, H. L., Brandenburg, S., Oshimura, M., et al. (1999). Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. U S A. 96, 5203–5208. doi: 10.1073/pnas.96.9.5203
Lewis, M. W., Vargas-Franco, D., Morse, D. A., and Resnick, J. L. (2019). A mouse model of Angelman syndrome imprinting defects. Hum. Mol. Genet. 28, 220–229. doi: 10.1093/hmg/ddy345
Li, J., Lin, X., Wang, M., Hu, Y., Xue, K., Gu, S., et al. (2020). Potential role of genomic imprinted genes and brain developmental related genes in autism. BMC Med. Genomics 13:54. doi: 10.1186/s12920-020-0693-692
Li, M., Ding, X., Zhang, Y., Li, X., Zhou, H., Yang, L., et al. (2020). Antisense oligonucleotides targeting lncRNA AC104041.1 induces antitumor activity through Wnt2B/β-catenin pathway in head and neck squamous cell carcinomas. Cell Death Dis. 11:672. doi: 10.1038/s41419-020-02820-2823
Li, Y., Tan, Z., Zhang, Y., Zhang, Z., Hu, Q., Liang, K., et al. (2021). A noncoding RNA modulator potentiates phenylalanine metabolism in mice. Science 373, 662–673. doi: 10.1126/science.aba4991
Liu, F., Pan, H., Xia, G., Qiu, C., and Zhu, Z. (2016). Prognostic and clinicopathological significance of long noncoding RNA H19 overexpression in human solid tumors: evidence from a meta-analysis. Oncotarget 7, 83177–83186. doi: 10.18632/oncotarget.13076
Liu, H., Shang, X., and Zhu, H. (2017). LncRNA/DNA binding analysis reveals losses and gains and lineage specificity of genomic imprinting in mammals. Bioinformatics. 33, 1431–1436. doi: 10.1093/bioinformatics/btw818
Liu, L. F., and Wang, J. C. (1987). Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. U S A. 84, 7024–7027. doi: 10.1073/pnas.84.20.7024
Luo, Z., Lin, C., Woodfin, A. R., Bartom, E. T., Gao, X., Smith, E. R., et al. (2016). Regulation of the imprinted Dlk1-Dio3 locus by allele-specific enhancer activity. Genes Dev. 30, 92–101. doi: 10.1101/gad.270413.115
Lyu, Y., Lou, J., Yang, Y., Feng, J., Hao, Y., Huang, S., et al. (2017). Dysfunction of the WT1-MEG3 signaling promotes AML leukemogenesis via p53-dependent and -independent pathways. Leukemia 31, 2543–2551. doi: 10.1038/leu.2017.116
Macdonald, W. A. (2012). Epigenetic mechanisms of genomic imprinting: common themes in the regulation of imprinted regions in mammals, plants, and insects. Genet Res. Int. 2012:585024. doi: 10.1155/2012/585024
MacDonald, W. A., and Mann, M. R. W. (2020). Long noncoding RNA functionality in imprinted domain regulation. PLoS Genet 16:e1008930. doi: 10.1371/journal.pgen.1008930
Mantovani, G., Bastepe, M., Monk, D., de Sanctis, L., Thiele, S., Usardi, A., et al. (2018). Diagnosis and management of pseudohypoparathyroidism and related disorders: first international consensus statement. Nat. Rev. Endocrinol. 14, 476–500. doi: 10.1038/s41574-018-0042-40
Marcho, C., Bevilacqua, A., Tremblay, K. D., and Mager, J. (2015). Tissue-specific regulation of Igf2r/Airn imprinting during gastrulation. Epigenet. Chromatin 8:10. doi: 10.1186/s13072-015-0003-y
Marín-Béjar, O., Mas, A. M., González, J., Martinez, D., Athie, A., Morales, X., et al. (2017). The human lncRNA LINC-PINT inhibits tumor cell invasion through a highly conserved sequence element. Genome Biol. 18:202. doi: 10.1186/s13059-017-1331-y
Martins-Taylor, K., Hsiao, J. S., Chen, P.-F., Glatt-Deeley, H., De Smith, A. J., Blakemore, A. I. F., et al. (2014). Imprinted expression of UBE3A in non-neuronal cells from a Prader-Willi syndrome patient with an atypical deletion. Hum. Mol. Genet. 23, 2364–2373. doi: 10.1093/hmg/ddt628
Matouk, I. J., Halle, D., Gilon, M., and Hochberg, A. (2015). The non-coding RNAs of the H19-IGF2 imprinted loci: a focus on biological roles and therapeutic potential in Lung Cancer. J. Trans. Med. 13:113. doi: 10.1186/s12967-015-0467-463
Mendiola, A. J. P., and LaSalle, J. M. (2021). Epigenetics in Prader-Willi syndrome. Front. Genet. 12:624581. doi: 10.3389/fgene.2021.624581
Meng, L., Person, R. E., and Beaudet, A. L. (2012). Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 21, 3001–3012. doi: 10.1093/hmg/dds130
Meng, L., Person, R. E., Huang, W., Zhu, P. J., Costa-Mattioli, M., and Beaudet, A. L. (2013). Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 9:e1004039. doi: 10.1371/journal.pgen.1004039
Meng, L., Ward, A. J., Chun, S., Bennett, C. F., Beaudet, A. L., and Rigo, F. (2015). Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518, 409–412. doi: 10.1038/nature13975
Meziane, H., Schaller, F., Bauer, S., Villard, C., Matarazzo, V., Riet, F., et al. (2015). An early postnatal oxytocin treatment prevents social and learning deficits in adult mice deficient for Magel2, a gene involved in Prader-willi syndrome and autism. Biol. Psychiatry 78, 85–94. doi: 10.1016/j.biopsych.2014.11.010
Mishra, S., Verma, S. S., Rai, V., Awasthee, N., Chava, S., Hui, K. M., et al. (2019). Long non-coding RNAs are emerging targets of phytochemicals for cancer and other chronic diseases. Cell Mol. Life. Sci. 76, 1947–1966. doi: 10.1007/s00018-019-03053-3050
Monahan, K., and Lomvardas, S. (2015). Monoallelic expression of olfactory receptors. Annu. Rev. Cell Dev. Biol. 31, 721–740. doi: 10.1146/annurev-cellbio-100814-125308
Monk, D., Mackay, D. J. G., Eggermann, T., Maher, E. R., and Riccio, A. (2019). Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 20, 235–248. doi: 10.1038/s41576-018-0092-90
Monk, D., Morales, J., den Dunnen, J. T., Russo, S., Court, F., Prawitt, D., et al. (2018). Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics 13, 117–121. doi: 10.1080/15592294.2016.1264561
Mussa, A., Russo, S., De Crescenzo, A., Chiesa, N., Molinatto, C., Selicorni, A., et al. (2013). Prevalence of beckwith-wiedemann syndrome in north West of Italy. Am. J. Med. Genet. A 161A, 2481–2486. doi: 10.1002/ajmg.a.36080
Mussa, A., Russo, S., de Crescenzo, A., Freschi, A., Calzari, L., Maitz, S., et al. (2016). Fetal growth patterns in Beckwith-Wiedemann syndrome. Clin. Genet. 90, 21–27. doi: 10.1111/cge.12759
Nagano, T., Mitchell, J. A., Sanz, L. A., Pauler, F. M., Ferguson-Smith, A. C., Feil, R., et al. (2008). The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720. doi: 10.1126/science.1163802
Nowak, K., Stein, G., Powell, E., He, L. M., Naik, S., Morris, J., et al. (2011). Establishment of paternal allele-specific DNA methylation at the imprinted mouse Gtl2 locus. Epigenetics 6, 1012–1020. doi: 10.4161/epi.6.8.16075
Ogata, T., and Kagami, M. (2016). Kagami-Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet. 61, 87–94. doi: 10.1038/jhg.2015.113
Õunap, K. (2016). Silver-Russell syndrome and beckwith-wiedemann syndrome: opposite phenotypes with heterogeneous molecular etiology. Mol. Syndromol. 7, 110–121. doi: 10.1159/000447413
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J., et al. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 32, 232–246. doi: 10.1016/j.molcel.2008.08.022
Park, H., Oh, J., Shim, G., Cho, B., Chang, Y., Kim, S., et al. (2019). In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer’s disease. Nat. Neurosci. 22, 524–528. doi: 10.1038/s41593-019-0352-0
Patten, M. M., Cowley, M., Oakey, R. J., and Feil, R. (2016). Regulatory links between imprinted genes: evolutionary predictions and consequences. Proc. Biol. Sci. 283:20152760. doi: 10.1098/rspb.2015.2760
Pauler, F. M., Barlow, D. P., and Hudson, Q. J. (2012). Mechanisms of long range silencing by imprinted macro non-coding RNAs. Curr. Opin. Genet. Dev 22, 283–289. doi: 10.1016/j.gde.2012.02.005
Paulsen, M., Takada, S., Youngson, N. A., Benchaib, M., Charlier, C., Segers, K., et al. (2001). Comparative sequence analysis of the imprinted Dlk1-Gtl2 locus in three mammalian species reveals highly conserved genomic elements and refines comparison with the Igf2-H19 region. Genome Res. 11, 2085–2094. doi: 10.1101/gr.206901
Peng, W.-X., Koirala, P., and Mo, Y.-Y. (2017). LncRNA-mediated regulation of cell signaling in cancer. Oncogene 36, 5661–5667. doi: 10.1038/onc.2017.184
Perez, J. D., Rubinstein, N. D., and Dulac, C. (2016). New perspectives on genomic imprinting, an essential and multifaceted mode of epigenetic control in the developing and adult brain. Annu. Rev. Neurosci. 39, 347–384. doi: 10.1146/annurev-neuro-061010-113708
Perez, J. D., Rubinstein, N. D., Fernandez, D. E., Santoro, S. W., Needleman, L. A., Ho-Shing, O., et al. (2015). Quantitative and functional interrogation of parent-of-origin allelic expression biases in the brain. eLife 4:e07860. doi: 10.7554/eLife.07860
Perez-Pinera, P., Kocak, D. D., Vockley, C. M., Adler, A. F., Kabadi, A. M., Polstein, L. R., et al. (2013). RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 10, 973–976. doi: 10.1038/nmeth.2600
Perry, R. B.-T., and Ulitsky, I. (2021). Therapy based on functional RNA elements. Science 373, 623–624. doi: 10.1126/science.abj7969
Peters, J. (2014). The role of genomic imprinting in biology and disease: an expanding view. Nat. Rev. Genet. 15, 517–530. doi: 10.1038/nrg3766
Pham, N. V., Nguyen, M. T., Hu, J. F., Vu, T. H., and Hoffman, A. R. (1998). Dissociation of IGF2 and H19 imprinting in human brain. Brain Res. 810, 1–8. doi: 10.1016/s0006-8993(98)00783-785
Pintacuda, G., Wei, G., Roustan, C., Kirmizitas, B. A., Solcan, N., Cerase, A., et al. (2017). hnRNPK Recruits PCGF3/5-PRC1 to the Xist RNA B-repeat to establish polycomb-mediated chromosomal silencing. Mol. Cell. 68, 955–969.e10. doi: 10.1016/j.molcel.2017.11.013
Polex-Wolf, J., Lam, B. Y., Larder, R., Tadross, J., Rimmington, D., Bosch, F., et al. (2018). Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome. J. Clin. Invest. 128, 960–969. doi: 10.1172/JCI97007
Pollak, M. N., Schernhammer, E. S., and Hankinson, S. E. (2004). Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 4, 505–518. doi: 10.1038/nrc1387
Powell, W. T., Coulson, R. L., Crary, F. K., Wong, S. S., Ach, R. A., Tsang, P., et al. (2013a). A Prader-Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum. Mol. Genet. 22, 4318–4328. doi: 10.1093/hmg/ddt281
Powell, W. T., Coulson, R. L., Gonzales, M. L., Crary, F. K., Wong, S. S., Adams, S., et al. (2013b). R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. U S A. 110, 13938–13943. doi: 10.1073/pnas.1305426110
Prasasya, R., Grotheer, K. V., Siracusa, L. D., and Bartolomei, M. S. (2020). Temple syndrome and Kagami-Ogata syndrome: clinical presentations, genotypes, models and mechanisms. Hum. Mol. Genet. 29, R107–R116. doi: 10.1093/hmg/ddaa133
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi: 10.1016/j.cell.2013.02.022
Qi, Y., Purtell, L., Fu, M., Lee, N. J., Aepler, J., Zhang, L., et al. (2016). Snord116 is critical in the regulation of food intake and body weight. Sci. Rep. 6:18614. doi: 10.1038/srep18614
Quinn, J. J., and Chang, H. Y. (2016). Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 17, 47–62. doi: 10.1038/nrg.2015.10
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. doi: 10.1038/nprot.2013.143
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Reinius, B., and Sandberg, R. (2015). Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat. Rev. Genet. 16, 653–664. doi: 10.1038/nrg3888
Riordan, J. D., Keng, V. W., Tschida, B. R., Scheetz, T. E., Bell, J. B., Podetz-Pedersen, K. M., et al. (2013). Identification of rtl1, a retrotransposon-derived imprinted gene, as a novel driver of hepatocarcinogenesis. PLoS Genet 9:e1003441. doi: 10.1371/journal.pgen.1003441
Riso, V., Cammisa, M., Kukreja, H., Anvar, Z., Verde, G., Sparago, A., et al. (2016). ZFP57 maintains the parent-of-origin-specific expression of the imprinted genes and differentially affects non-imprinted targets in mouse embryonic stem cells. Nucleic Acids Res. 44, 8165–8178. doi: 10.1093/nar/gkw505
Robertson, K. D. (2005). DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610. doi: 10.1038/nrg1655
Robinson, W. P. (2000). Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 22, 452–459.
Rossignol, S., Steunou, V., Chalas, C., Kerjean, A., Rigolet, M., Viegas-Pequignot, E., et al. (2006). The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J. Med. Genet. 43, 902–907. doi: 10.1136/jmg.2006.042135
Rougeulle, C., Glatt, H., and Lalande, M. (1997). The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 17, 14–15. doi: 10.1038/ng0997-14
Rozhdestvensky, T. S., Robeck, T., Galiveti, C. R., Raabe, C. A., Seeger, B., Wolters, A., et al. (2016). Maternal transcription of non-protein coding RNAs from the PWS-critical region rescues growth retardation in mice. Sci. Rep. 6:20398. doi: 10.1038/srep20398
Runte, M., Hüttenhofer, A., Gross, S., Kiefmann, M., Horsthemke, B., and Buiting, K. (2001). The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 10, 2687–2700. doi: 10.1093/hmg/10.23.2687
Sahoo, T., del Gaudio, D., German, J. R., Shinawi, M., Peters, S. U., Person, R. E., et al. (2008). Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 40, 719–721. doi: 10.1038/ng.158
Sanli, I., and Feil, R. (2015). Chromatin mechanisms in the developmental control of imprinted gene expression. Int. J. Biochem. Cell Biol. 67, 139–147. doi: 10.1016/j.biocel.2015.04.004
Sanli, I., Lalevée, S., Cammisa, M., Perrin, A., Rage, F., Llères, D., et al. (2018). Meg3 Non-coding RNA expression controls imprinting by preventing transcriptional upregulation in cis. Cell Rep. 23, 337–348. doi: 10.1016/j.celrep.2018.03.044
Santoro, F., Mayer, D., Klement, R. M., Warczok, K. E., Stukalov, A., Barlow, D. P., et al. (2013). Imprinted Igf2r silencing depends on continuous Airn lncRNA expression and is not restricted to a developmental window. Development 140, 1184–1195. doi: 10.1242/dev.088849
Santoro, F., and Pauler, F. M. (2013). Silencing by the imprinted Airn macro lncRNA: transcription is the answer. Cell Cycle 12, 711–712. doi: 10.4161/cc.23860
Sasaki, H., Ferguson-Smith, A. C., Shum, A. S., Barton, S. C., and Surani, M. A. (1995). Temporal and spatial regulation of H19 imprinting in normal and uniparental mouse embryos. Development 121, 4195–4202.
Sato, S., Yoshida, W., Soejima, H., Nakabayashi, K., and Hata, K. (2011). Methylation dynamics of IG-DMR and Gtl2-DMR during murine embryonic and placental development. Genomics 98, 120–127. doi: 10.1016/j.ygeno.2011.05.003
Schaller, F., Watrin, F., Sturny, R., Massacrier, A., Szepetowski, P., and Muscatelli, F. (2010). A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 19, 4895–4905. doi: 10.1093/hmg/ddq424
Schertzer, M. D., Braceros, K. C. A., Starmer, J., Cherney, R. E., Lee, D. M., Salazar, G., et al. (2019). lncRNA-Induced spread of polycomb controlled by genome architecture, RNA abundance, and CpG Island DNA. Mol. Cell. 75, 523–537.e10. doi: 10.1016/j.molcel.2019.05.028
Schoenherr, C. J., Levorse, J. M., and Tilghman, S. M. (2003). CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 33, 66–69. doi: 10.1038/ng1057
Schroeder, D. I., Blair, J. D., Lott, P., Yu, H. O. K., Hong, D., Crary, F., et al. (2013). The human placenta methylome. Proc. Natl. Acad. Sci. U S A. 110, 6037–6042. doi: 10.1073/pnas.1215145110
Schwartz, Y. B., and Pirrotta, V. (2013). A new world of Polycombs: unexpected partnerships and emerging functions. Nat. Rev. Genet. 14, 853–864. doi: 10.1038/nrg3603
Sellers, Z. P., Bolkun, L., Kloczko, J., Wojtaszewska, M. L., Lewandowski, K., Moniuszko, M., et al. (2019). Increased methylation upstream of the MEG3 promotor is observed in acute myeloid leukemia patients with better overall survival. Clin. Epigenetics 11:50. doi: 10.1186/s13148-019-0643-z
Simon, J. A., and Kingston, R. E. (2013). Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell. 49, 808–824. doi: 10.1016/j.molcel.2013.02.013
Singh, P., Wu, X., Lee, D.-H., Li, A. X., Rauch, T. A., Pfeifer, G. P., et al. (2011). Chromosome-wide analysis of parental allele-specific chromatin and DNA methylation. Mol. Cell. Biol. 31, 1757–1770. doi: 10.1128/MCB.00961-910
Skourti-Stathaki, K., Proudfoot, N. J., and Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 42, 794–805. doi: 10.1016/j.molcel.2011.04.026
Skryabin, B. V., Gubar, L. V., Seeger, B., Pfeiffer, J., Handel, S., Robeck, T., et al. (2007). Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet. 3:e235. doi: 10.1371/journal.pgen.0030235
Sleutels, F., Zwart, R., and Barlow, D. P. (2002). The non-coding air RNA is required for silencing autosomal imprinted genes. Nature 415, 810–813. doi: 10.1038/415810a
Smilinich, N. J., Day, C. D., Fitzpatrick, G. V., Caldwell, G. M., Lossie, A. C., Cooper, P. R., et al. (1999). A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl. Acad. Sci. U S A. 96, 8064–8069. doi: 10.1073/pnas.96.14.8064
Smith, R. A., Miller, T. M., Yamanaka, K., Monia, B. P., Condon, T. P., Hung, G., et al. (2006). Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 116, 2290–2296. doi: 10.1172/JCI25424
Soejima, H., and Higashimoto, K. (2013). Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J. Hum. Genet. 58, 402–409. doi: 10.1038/jhg.2013.51
Soellner, L., Begemann, M., Mackay, D. J. G., Grønskov, K., Tümer, Z., Maher, E. R., et al. (2017). Recent advances in imprinting disorders. Clin. Genet. 91, 3–13. doi: 10.1111/cge.12827
Southan, C. (2017). Last rolls of the yoyo: assessing the human canonical protein count. F1000Res 6:448. doi: 10.12688/f1000research.11119.1
St Laurent, G., Wahlestedt, C., and Kapranov, P. (2015). The landscape of long noncoding RNA classification. Trends Genet. 31, 239–251. doi: 10.1016/j.tig.2015.03.007
Stanurova, J., Neureiter, A., Hiber, M., de Oliveira Kessler, H., Stolp, K., Goetzke, R., et al. (2018). Corrigendum: angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci. Rep. 8:46952. doi: 10.1038/srep46952
Statello, L., Guo, C.-J., Chen, L.-L., and Huarte, M. (2021). Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118. doi: 10.1038/s41580-020-00315-319
Sun, J., Carlson-Stevermer, J., Das, U., Shen, M., Delenclos, M., Snead, A. M., et al. (2019). CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat. Commun. 10:53. doi: 10.1038/s41467-018-07971-7978
Sun, K.-X., Wu, D.-D., Chen, S., Zhao, Y., and Zong, Z.-H. (2017). LncRNA MEG3 inhibit endometrial carcinoma tumorigenesis and progression through PI3K pathway. Apoptosis 22, 1543–1552. doi: 10.1007/s10495-017-1426-1427
Sutcliffe, J. S., Nakao, M., Christian, S., Orstavik, K. H., Tommerup, N., Ledbetter, D. H., et al. (1994). Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat. Genet. 8, 52–58. doi: 10.1038/ng0994-52
Suzuki, K., Tsunekawa, Y., Hernandez-Benitez, R., Wu, J., Zhu, J., Kim, E. J., et al. (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149. doi: 10.1038/nature20565
Tan, L., Xing, D., Chang, C.-H., Li, H., and Xie, X. S. (2018). Three-dimensional genome structures of single diploid human cells. Science 361, 924–928. doi: 10.1126/science.aat5641
Temple, I. K., Cockwell, A., Hassold, T., Pettay, D., and Jacobs, P. (1991). Maternal uniparental disomy for chromosome 14. J. Med. Genet. 28, 511–514. doi: 10.1136/jmg.28.8.511
Terranova, R., Yokobayashi, S., Stadler, M. B., Otte, A. P., van Lohuizen, M., Orkin, S. H., et al. (2008). Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev. Cell 15, 668–679. doi: 10.1016/j.devcel.2008.08.015
Thorvaldsen, J. L., Duran, K. L., and Bartolomei, M. S. (1998). Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 12, 3693–3702. doi: 10.1101/gad.12.23.3693
Tucci, V., Isles, A. R., Kelsey, G., and Ferguson-Smith, A. C. Erice Imprinting and Group. (2019). Genomic imprinting and physiological processes in mammals. Cell 176, 952–965. doi: 10.1016/j.cell.2019.01.043
Tunster, S. J., Van de Pette, M., and John, R. M. (2011). Fetal overgrowth in the Cdkn1c mouse model of Beckwith-Wiedemann syndrome. Dis. Model. Mech. 4, 814–821. doi: 10.1242/dmm.007328
Umlauf, D., Goto, Y., Cao, R., Cerqueira, F., Wagschal, A., Zhang, Y., et al. (2004). Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat. Genet. 36, 1296–1300. doi: 10.1038/ng1467
Uszczynska-Ratajczak, B., Lagarde, J., Frankish, A., Guigó, R., and Johnson, R. (2018). Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 19, 535–548. doi: 10.1038/s41576-018-0017-y
van der Werf, I. M., Buiting, K., Czeschik, C., Reyniers, E., Vandeweyer, G., Vanhaesebrouck, P., et al. (2016). Novel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami-Ogata syndrome. Eur. J. Hum. Genet. 24, 1724–1729. doi: 10.1038/ejhg.2016.82
Vennin, C., Spruyt, N., Dahmani, F., Julien, S., Bertucci, F., Finetti, P., et al. (2015). H19 non coding RNA-derived miR-675 enhances tumorigenesis and metastasis of breast cancer cells by downregulating c-Cbl and Cbl-b. Oncotarget 6, 29209–29223. doi: 10.18632/oncotarget.4976
Vettermann, C., and Schlissel, M. S. (2010). Allelic exclusion of immunoglobulin genes: models and mechanisms. Immunol. Rev. 237, 22–42. doi: 10.1111/j.1600-065X.2010.00935.x
Vitali, P., Royo, H., Marty, V., Bortolin-Cavaillé, M.-L., and Cavaillé, J. (2010). Long nuclear-retained non-coding RNAs and allele-specific higher-order chromatin organization at imprinted snoRNA gene arrays. J. Cell Sci. 123, 70–83. doi: 10.1242/jcs.054957
Vu, T. H., and Hoffman, A. R. (1997). Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 17, 12–13. doi: 10.1038/ng0997-12
Wakeling, E. L., Brioude, F., Lokulo-Sodipe, O., O’Connell, S. M., Salem, J., Bliek, J., et al. (2017). Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat. Rev. Endocrinol. 13, 105–124. doi: 10.1038/nrendo.2016.138
Wan, P., Su, W., and Zhuo, Y. (2017). The role of long noncoding RNAs in neurodegenerative diseases. Mol. Neurobiol. 54, 2012–2021. doi: 10.1007/s12035-016-9793-9796
Wang, J. C., Passage, M. B., Yen, P. H., Shapiro, L. J., and Mohandas, T. K. (1991). Uniparental heterodisomy for chromosome 14 in a phenotypically abnormal familial balanced 13/14 Robertsonian translocation carrier. Am. J. Hum. Genet. 48, 1069–1074.
Wang, X., Sun, Q., McGrath, S. D., Mardis, E. R., Soloway, P. D., and Clark, A. G. (2008). Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One 3:e3839. doi: 10.1371/journal.pone.0003839
Wang, Y., Shen, Y., Dai, Q., Yang, Q., Zhang, Y., Wang, X., et al. (2017). A permissive chromatin state regulated by ZFP281-AFF3 in controlling the imprinted Meg3 polycistron. Nucleic Acids Res. 45, 1177–1185. doi: 10.1093/nar/gkw1051
Weksberg, R., Shuman, C., and Beckwith, J. B. (2010). Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 18, 8–14. doi: 10.1038/ejhg.2009.106
White, C. R., MacDonald, W. A., and Mann, M. R. W. (2016). Conservation of DNA methylation programming between mouse and human gametes and preimplantation embryos. Biol. Reprod. 95:61. doi: 10.1095/biolreprod.116.140319
Wolter, J. M., Mao, H., Fragola, G., Simon, J. M., Krantz, J. L., Bazick, H. O., et al. (2020). Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 587, 281–284. doi: 10.1038/s41586-020-2835-2832
Wu, H., Yin, Q.-F., Luo, Z., Yao, R.-W., Zheng, C.-C., Zhang, J., et al. (2016). Unusual processing generates SPA LncRNAs that sequester multiple RNA binding proteins. Mol. Cell. 64, 534–548. doi: 10.1016/j.molcel.2016.10.007
Xin, Z., Tachibana, M., Guggiari, M., Heard, E., Shinkai, Y., and Wagstaff, J. (2003). Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J. Biol. Chem. 278, 14996–15000. doi: 10.1074/jbc.M211753200
Yamasaki, K., Joh, K., Ohta, T., Masuzaki, H., Ishimaru, T., Mukai, T., et al. (2003). Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 12, 837–847. doi: 10.1093/hmg/ddg106
Yan, Y., Frisén, J., Lee, M. H., Massagué, J., and Barbacid, M. (1997). Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 11, 973–983. doi: 10.1101/gad.11.8.973
Yang, T., Adamson, T. E., Resnick, J. L., Leff, S., Wevrick, R., Francke, U., et al. (1998). A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat. Genet. 19, 25–31. doi: 10.1038/ng0598-25
Yao, H., Duan, M., Lin, L., Wu, C., Fu, X., Wang, H., et al. (2017). TET2 and MEG3 promoter methylation is associated with acute myeloid leukemia in a Hainan population. Oncotarget 8, 18337–18347. doi: 10.18632/oncotarget.15440
Ye, Y., Guo, J., Xiao, P., Ning, J., Zhang, R., Liu, P., et al. (2020). Macrophages-induced long noncoding RNA H19 up-regulation triggers and activates the miR-193b/MAPK1 axis and promotes cell aggressiveness in hepatocellular carcinoma. Cancer Lett. 469, 310–322. doi: 10.1016/j.canlet.2019.11.001
Yeo, G. W., Coufal, N. G., Liang, T. Y., Peng, G. E., Fu, X.-D., and Gage, F. H. (2009). An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 16, 130–137. doi: 10.1038/nsmb.1545
Yin, Q.-F., Yang, L., Zhang, Y., Xiang, J.-F., Wu, Y.-W., Carmichael, G. G., et al. (2012). Long noncoding RNAs with snoRNA ends. Mol. Cell. 48, 219–230. doi: 10.1016/j.molcel.2012.07.033
Yoon, J.-H., Kim, J., and Gorospe, M. (2015). Long noncoding RNA turnover. Biochimie 117, 15–21. doi: 10.1016/j.biochi.2015.03.001
Yoshimizu, T., Miroglio, A., Ripoche, M.-A., Gabory, A., Vernucci, M., Riccio, A., et al. (2008). The H19 locus acts in vivo as a tumor suppressor. Proc. Natl. Acad. Sci. U S A. 105, 12417–12422. doi: 10.1073/pnas.0801540105
Zhang, C.-Y., Yu, M.-S., Li, X., Zhang, Z., Han, C.-R., and Yan, B. (2017). Overexpression of long non-coding RNA MEG3 suppresses breast cancer cell proliferation, invasion, and angiogenesis through AKT pathway. Tumour Biol, 39:1010428317701311. doi: 10.1177/1010428317701311
Zhang, K., Luo, Z., Zhang, Y., Zhang, L., Wu, L., Liu, L., et al. (2016). Circulating lncRNA H19 in plasma as a novel biomarker for breast cancer. Cancer Biomark. 17, 187–194. doi: 10.3233/CBM-160630
Zhang, P., Liégeois, N. J., Wong, C., Finegold, M., Hou, H., Thompson, J. C., et al. (1997). Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 387, 151–158. doi: 10.1038/387151a0
Zhao, J., Ohsumi, T. K., Kung, J. T., Ogawa, Y., Grau, D. J., Sarma, K., et al. (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell. 40, 939–953. doi: 10.1016/j.molcel.2010.12.011
Zhao, Y., Li, H., Fang, S., Kang, Y., Wu, W., Hao, Y., et al. (2016). NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 44, D203–D208. doi: 10.1093/nar/gkv1252
Zheng, J.-F., Guo, N.-H., Zi, F.-M., and Cheng, J. (2020). Long noncoding RNA H19 promotes tumorigenesis of multiple myeloma by activating BRD4 signaling by targeting MicroRNA 152-3p. Mol. Cell. Biol. 40:e00382-19. doi: 10.1128/MCB.00382-319
Zhou, H., Wang, B., Yang, Y.-X., Jia, Q.-J., Zhang, A., Qi, Z.-W., et al. (2019). Long Noncoding RNAs in pathological cardiac remodeling: a review of the update literature. Biomed. Res. Int. 2019:7159592. doi: 10.1155/2019/7159592
Zhou, Y., Zhang, X., and Klibanski, A. (2012). MEG3 noncoding RNA: a tumor suppressor. J. Mol. Endocrinol. 48, R45–R53. doi: 10.1530/JME-12-18
Zhu, M., Wang, X., Gu, Y., Wang, F., Li, L., and Qiu, X. (2019). MEG3 overexpression inhibits the tumorigenesis of breast cancer by downregulating miR-21 through the PI3K/Akt pathway. Arch. Biochem. Biophys. 661, 22–30. doi: 10.1016/j.abb.2018.10.021
Zhuo, C., Hou, W., Hu, L., Lin, C., Chen, C., and Lin, X. (2017). Genomic editing of Non-Coding RNA genes with CRISPR/Cas9 ushers in a potential novel approach to study and treat schizophrenia. Front. Mol. Neurosci. 10:28. doi: 10.3389/fnmol.2017.00028
Zieba, J., Low, J. K., Purtell, L., Qi, Y., Campbell, L., Herzog, H., et al. (2015). Behavioural characteristics of the Prader-Willi syndrome related biallelic Snord116 mouse model. Neuropeptides 53, 71–77. doi: 10.1016/j.npep.2015.06.009
Keywords: genomic imprinting, lncRNA, epigenetic regulation, imprinting disorders, UBE3A-ATS, ASO, CRISPR-Cas9
Citation: Wang T, Li J, Yang L, Wu M and Ma Q (2021) The Role of Long Non-coding RNAs in Human Imprinting Disorders: Prospective Therapeutic Targets. Front. Cell Dev. Biol. 9:730014. doi: 10.3389/fcell.2021.730014
Received: 24 June 2021; Accepted: 23 September 2021;
Published: 25 October 2021.
Edited by:
Lan Jiang, Key Laboratory of Genome Sciences & Information, Beijing Institute of Genomics (CAS), ChinaReviewed by:
Joseph Mauro Calabrese, University of North Carolina at Chapel Hill, United StatesJanine M. LaSalle, University of California, Davis, United States
Copyright © 2021 Wang, Li, Yang, Wu and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Ma, cWluZy5tYUBzaWF0LmFjLmNu