
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 07 October 2021
Sec. Cell Death and Survival
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.729728
This article is part of the Research Topic Programmed Cell Death and Cardiovascular Disease View all 26 articles
 Yingying Li1,2†
Yingying Li1,2† Yingjun Feng2†
Yingjun Feng2† Xiaoyun Ye1†Hanlin Peng1Jiantong Du3Xiaoli Yao2
Xiaoyun Ye1†Hanlin Peng1Jiantong Du3Xiaoli Yao2 Yaqian Huang1*
Yaqian Huang1* Hongfang Jin1*
Hongfang Jin1* Junbao Du1,4*
Junbao Du1,4*SO2, previously known as the product of industrial waste, has recently been proven to be a novel gasotransmitter in the cardiovascular system. It is endogenously produced from the metabolism pathway of sulfur-containing amino acids in mammalians. Endogenous SO2 acts as an important controller in the regulation of many biological processes including cardiovascular physiological and pathophysiological events. Recently, the studies on the regulatory effect of endogenous SO2 on cell apoptosis and its pathophysiological significance have attracted great attention. Endogenous SO2 can regulate the apoptosis of vascular smooth muscle cells, endothelial cells, cardiomyocytes, neuron, alveolar macrophages, polymorphonuclear neutrophils and retinal photoreceptor cells, which might be involved in the pathogenesis of hypertension, pulmonary hypertension, myocardial injury, brain injury, acute lung injury, and retinal disease. Therefore, in the present study, we described the current findings on how endogenous SO2 is generated and metabolized, and we summarized its regulatory effects on cell apoptosis, underlying mechanisms, and pathophysiological relevance.
Previously, SO2 was recognized as a water-soluble, colorless, transparent exhaust gas and air pollutant with a sharp odor. High concentrations of SO2 in the environment could cause various degrees of damage to humans, animals, plants, and even microorganisms. However, it was discovered that SO2 can be endogenously produced in the mammalian metabolism of sulfur-containing amino acids (SAAs) (Stipanuk, 2004; Kimura, 2011). Many studies have shown that endogenous SO2 has the unique characteristics of continuous production, rapid diffusion, low molecular weight, and extensive action (Du et al., 2008a; Huang et al., 2016). More interestingly, it was found to play an important role in regulating cardiovascular function and structure under physiologic and pathophysiologic conditions (Du et al., 2008a; Huang et al., 2016). Therefore, we proposed that SO2 is a new cardiovascular gaseous signaling molecule. However, the molecular mechanisms underlying its biological action require further studies. It has been found recently that endogenous SO2 regulates cell apoptosis, and therefore contributes to the pathogenesis of hypertension, pulmonary hypertension, myocardial injury, brain injury, retinal disease, and acute lung injury (Zhao et al., 2008, 2013, 2019; Wang et al., 2011; Ma et al., 2012; Jin et al., 2013; Han et al., 2014; Du et al., 2018; Liu et al., 2018; Niu et al., 2018; Yang et al., 2018; Zhou et al., 2020). In this article, we described the generation and metabolism of endogenous SO2, and summarized the latest findings of the regulation of endogenous SO2 on cell apoptosis, its mechanisms, and pathophysiological significance.
SO2 can be produced endogenously through the biotransformation of SAAs in various mammal systems (Figure 1), such as the cardiovascular, respiratory, nervous, digestive, urinary, and immune systems. In the body, SAAs, such as methionine and cystine, are primarily metabolized to L-cysteine (L-Cys). L-Cys is a crucial precursor of endogenous SO2 (Singer and Kearney, 1956; Stipanuk, 2004; Du et al., 2008a; Kimura, 2011; Huang et al., 2016). It can be oxidized to produce L-cysteinesulfinate, and this process involves a key enzyme cysteine dioxygenase. Subsequently, the metabolism of L-cysteinesulfinate involves two pathways. In one pathway, L-cysteinesulfinate is converted into β-sulfinylpyruvate via the catalysis of aspartate aminotransferase (AAT), which is followed by spontaneous decomposition of β-sulfinylpyruvate into SO2 and pyruvic acid. In the other pathway, L-cysteinesulfinate is decarboxylated into hypotaurine and CO2 via cysteinesulfinate decarboxylase. Hypotaurine is subsequently oxidized to taurine. There is a competition between these two pathways for the reaction tendency of L-cysteinesulfinate, which is ultimately determined by the activities of AAT and cysteinesulfinate decarboxylase (Singer and Kearney, 1956; Griffith, 1983). Studies have indicated that 3′-phosphoadenosine-5′-phosphosulfate in activated neutrophils functions as an endogenous sulfate donor (Stipanuk, 2004).
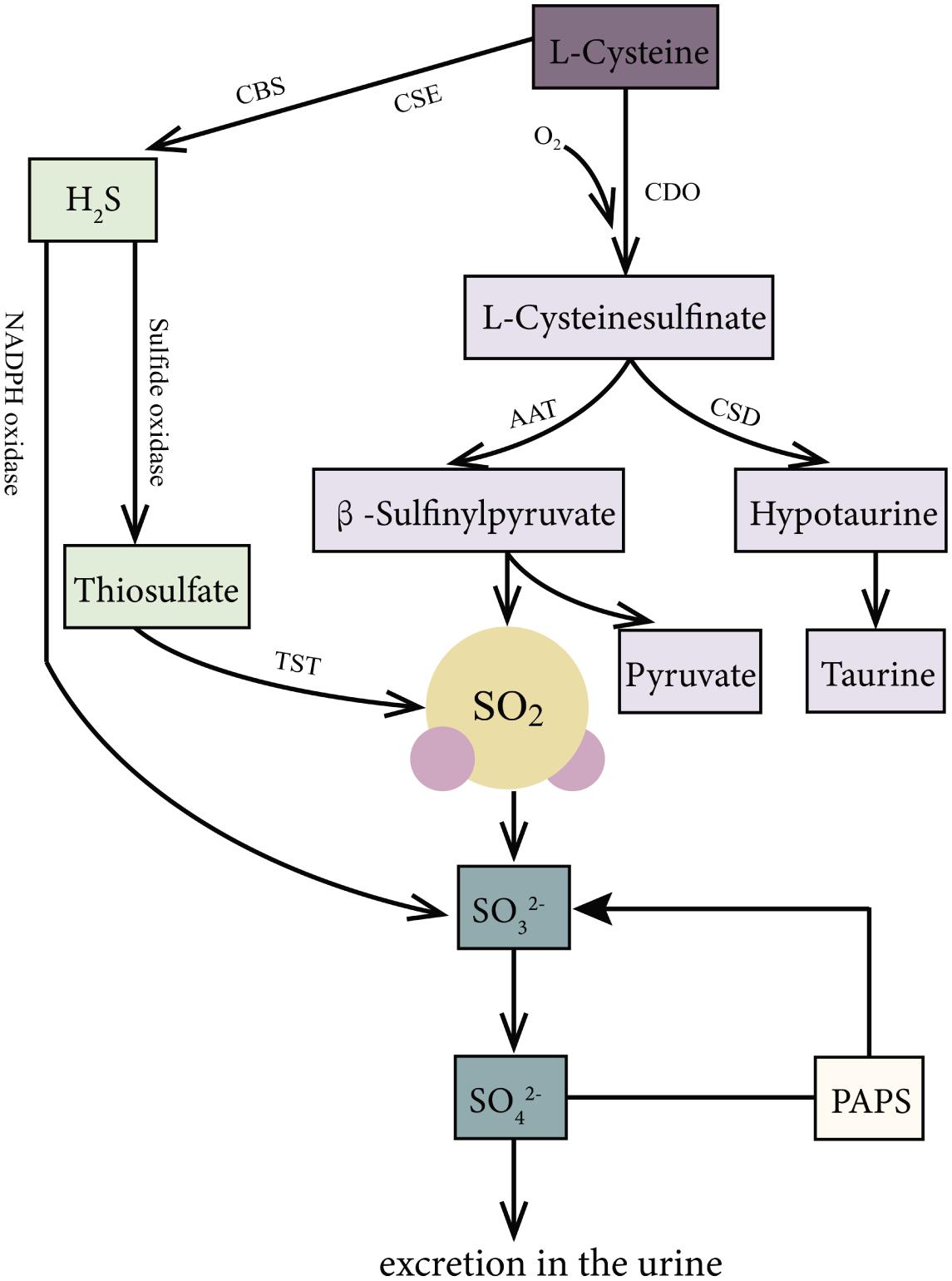
Figure 1. Production and metabolism of endogenous SO2 in mammals. L-Cys is metabolized through important enzymes including CDO, AAT, and CSD to generate endogenous SO2, pyruvic acid, and hypotaurine. Moreover, H2S, a product of the reaction catalyzed by CSE and CBS with L-Cys as substrate, can be metabolized to sulfite or SO2 via the catalyzation of sulfide oxidase, TST, and NADPH oxidase. Thirdly, PAPS in activated neutrophils is also an origin of sulfite. In mammals, SO2 can be hydrated to sulfite, then converted into sulfate by sulfite oxidase, and finally excreted in the urine in the kidney. AAT: aspartate aminotransferase; CBS, cystathionine β-synthase; CDO, cysteine dioxygenase; CSE, cystathionine γ-lyase; L-Cys, L-cysteine; NADPH, nicotinamide dinucleotide phosphate; PAPS, 3′-phosphoadenosine-5′-phosphosulfate; TST, thiosulfate sulfurtransferase.
Besides, hydrogen sulfide (H2S), another gaseous signaling molecule, metabolized from SAAs catalyzed by cystathionine γ-lyase or cystathionine β-synthase (Stipanuk, 2004; Kimura, 2011), can be transformed into SO2 or sulfite through the following two pathways: (1) H2S is oxidized directly to sulfite or SO2 through reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Mitsuhashi et al., 2005). (2) H2S is firstly oxidized by sulfide oxidase into thiosulfate, and then the enzymes such as thiosulfate sulfurtransferase or glutathione-dependent thiosulfate reductase catalyze the transformation of thiosulfate to SO2 (Shapiro, 1977).
In mammals, SO2 can be hydrated to sulfite, and then converted into sulfate by sulfite oxidase, finally excreted in the urine in the kidney (Stipanuk, 2004; Huang et al., 2016). The half-life of SO2 was supposed to be about 5–10 min, represented by the fact that serum SO2 level decreased by 50% about 5–10 min after a bolus intravenous injection of SO2 donor (Du et al., 2008b). It is known that AAT has two isozymes: cytosol AAT1 and mitochondrial AAT2. Huang et al. found that the overexpression of AAT1 or AAT2 increased but the knockdown of AAT1 or AAT2 inhibited the endogenous SO2 production, suggesting that AAT1/2 is the main SO2-generating enzyme (Liu et al., 2014).
SO2 was detected in the headspace gas collected from a closed vial culturing porcine coronary arteries in 2003 by Balazy et al. (2003). Du et al. firstly found the SO2/AAT pathway in the cardiovascular tissues of rats in 2008 (Du et al., 2008a). The SO2 level in rat plasma was 15.54 ± 1.68 μmol/L while the SO2 concentration in the aorta, pulmonary artery, mesenteric artery, tail artery, and renal artery were 3.27 ± 0.21, 2.67 ± 0.17, 2.50 ± 0.20, and 2.23 ± 0.19 μmol/g protein, respectively. To date, SO2/AAT pathway has been found in almost all organs in mammals including vessels, heart, stomach, lung, liver, kidney, brain, retina, pancreas, and spleen, etc. (Luo et al., 2011; Du et al., 2018). It was found that the SO2 level was the highest in the stomach, followed by the ventricle, and the activity of AAT and the expression of AAT1 mRNA are the highest in the left ventricle, whereas the expression of the AAT1 protein is the highest in the right ventricle. Furthermore, the expression of AAT2 mRNA is the highest in the liver, whereas the expression of the AAT2 protein is the highest in the renal medullary membrane (Luo et al., 2011).
Apoptosis is the programmed cell death that occurs in multicellular organisms. Increasing evidence implies that endogenous SO2 participates in regulating cell apoptosis (Zhao et al., 2008, 2013, 2019; Wang et al., 2011; Ma et al., 2012; Jin et al., 2013; Han et al., 2014; Du et al., 2018; Liu et al., 2018; Niu et al., 2018; Yang et al., 2018; Zhou et al., 2020, Figure 2). Three main pathways associated with the regulation of SO2 on cell apoptosis have been revealed to date: (1) Extrinsic pathway (Wajant, 2002; Zhao et al., 2008): The combination of Fas ligand (FasL)/Fas causes the binding of the Fas-associated death domain proteins which activate caspase-8 precursor (caspase-10 in humans), initiate the cascade activation of caspase-3, caspase-6, and caspase-7 and then induce cell apoptosis. (2) Intrinsic pathway (Jin et al., 2013): The leakage of cytochrome c (Cytc) from impaired mitochondria to cytosol is the central event of the mitochondria-related intrinsic apoptosis pathway. Its preceding events include the deformation and swelling of mitochondria, the imbalance of apoptotic regulator B-cell lymphoma-2 (bcl-2)/bcl-2-associated X protein (bax), the destroy of mitochondrial membrane potential (MMP), and the opening of mitochondrial permeability transition pore (MPTP). The cytosol Cytc activates caspase-9 and caspase-3, finally induces cell apoptosis. (3) Endoplasmic reticulum stress (ERS)-related apoptosis (Wang et al., 2011): The overactivated ERS induces cell apoptosis via overexpressing C/EBP homologous protein (CHOP) and activating caspase-12.
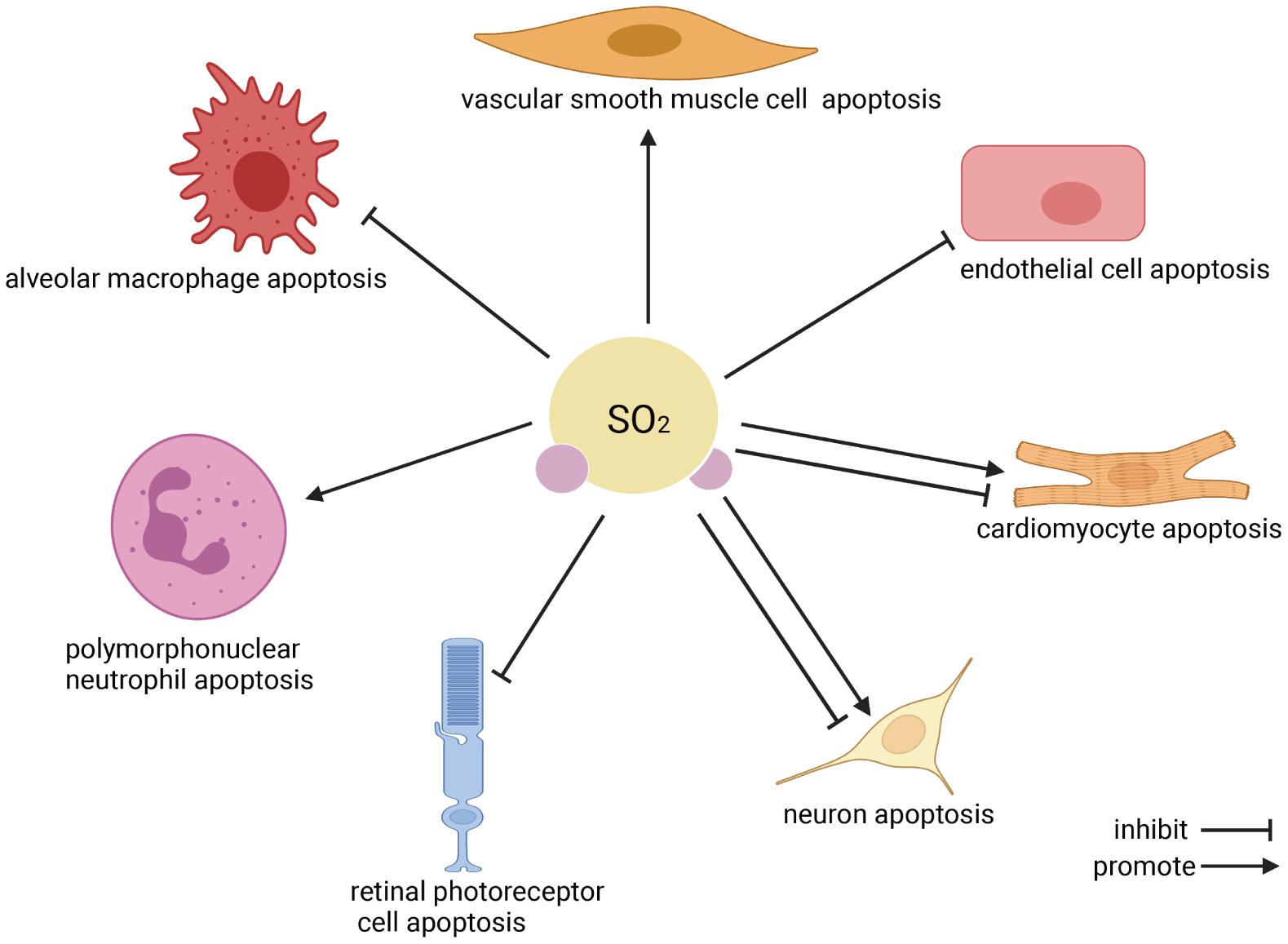
Figure 2. The effect of endogenous SO2 on cell apoptosis. According to the clockwise direction, the SO2 target cells are shown beginning from vascular smooth muscle cells. In the cardiovascular system, endogenous SO2 promotes vascular smooth muscle cell apoptosis, inhibits vascular endothelial cell apoptosis, and inhibits/promotes cardiomyocyte apoptosis. In the nervous system, endogenous SO2 inhibits/promotes neuron apoptosis, and reduces the apoptosis of retinal photoreceptor cells. Regarding immune cells, SO2 promotes polymorphonuclear neutrophil apoptosis but inhibits macrophage apoptosis.
As the main component cell of the vessel, the vascular smooth muscle cell (VSMC) apoptosis markedly affects vascular remodeling. Zhao et al. (2008) found that compared with the WKY rat, the plasma SO2 level and the proportion of apoptotic VSMC in media were downregulated in the spontaneously hypertensive rat (SHR). Correspondingly, the blood pressure and the ratio of aorta media to lumen radius were increased by 53.6 and 28.1% in the SHR rat, respectively. Furthermore, SO2 donor treatment recovered the decreased plasma SO2 content, promoted the VSMC apoptosis, decreased blood pressure, and alleviated the thickening aorta media in the SHR rat. Mechanistically, the expression of antiapoptotic factor bcl-2 was increased but the expression of proapoptotic factor Fas and caspase-3 was decreased in the aorta media in the rats of the SHR + SO2 group compared with those in the rats of the SHR group. These data suggested that SO2 might alleviate the vascular remodeling by promoting VSMC apoptosis, which involved the regulation of bcl-2, Fas, and caspase-3.
However, SO2 did not affect the apoptosis of VSMC with or without fetal bovine serum stimulation in vitro (Liu et al., 2014). The discrepant effects of SO2 on VMSC apoptosis might be associated with the different experimental conditions such as in vivo and in vitro. For example, VSMCs are directly challenged by cyclic mechanical stretch and hydrostatic pressure and indirectly affected by endothelial cell (EC)-transduced shear stress under in vivo circumstances (Qiu et al., 2013; Chen et al., 2021). The abovementioned biomechanical stresses are reported to control the VSMC apoptosis, proliferation, and other behaviors and only are partly mimicked in vivo.
Vascular ECs constitute a huge divider separating the lumen and wall of the vessel, and therefore play a crucial role in maintaining the vascular function and microenvironment homeostasis. Especially, vascular EC apoptosis is a key process involved in the vascular physiological and pathophysiological regulation such as organ development, angiogenesis, vascular injury, and remodeling (Duan et al., 2021; Tisch and Ruiz de Al modóvar, 2021). During the development of hypoxic pulmonary artery structural remodeling, hypoxia-induced vascular EC apoptosis is a critical pathological change. Liu et al. explored the effect of SO2 on the human pulmonary artery endothelial cell (HPAEC) apoptosis based on cobalt chloride (CoCl2)-stimulated cell model (Liu et al., 2018). The endogenous SO2 production and AAT1 expression were down-regulated but the percentage of terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL)-positive nuclei was increased in HPAECs of the vehicle + CoCl2 group as compared with those of the vehicle group. However, there was no difference in the endogenous SO2/AAT1 pathway and apoptosis of HPAECs between the AAT1 overexpression and AAT1 overexpression + CoCl2 groups, suggesting that the sufficient endogenous SO2 was a powerful antagonist to chemical hypoxia-induced HPAEC apoptosis. Furthermore, AAT1 overexpression prevented the downregulation of antiapoptotic factor bcl-2, and the activation of caspase-3 in the CoCl2-treated HPAECs. These data suggested that the endogenous SO2 might inhibit the hypoxia-induced HPAEC apoptosis by increasing bcl-2 expression, and inactivating caspase-3.
Cardiomyocyte apoptosis is a keystone of cardiac pathological changes and is closely correlated with the development of heart failure, myocardial ischemia-reperfusion (I/R) injury and cardiomyopathy, etc. Endogenous SO2 was found to protect against the myocardial I/R, isopropylarterenol (ISO)-induced cardiac injury, sepsis-induced cardiac dysfunction, myocardial infarction, and diabetic cardiomyopathy, suggesting that cardiomyocyte might be an important target cell of endogenous SO2 (Liang et al., 2011; Wang et al., 2011, 2018; Chen et al., 2012; Jin et al., 2013; Zhao et al., 2013; Liu et al., 2017; Yang et al., 2018; Zhou et al., 2020).
SO2 preconditioning significantly reduced the size of the myocardial infarction and decreased the percentage of TUNEL-positive cells in the rats of myocardial I/R (Wang et al., 2011). The mechanisms by which SO2 preconditioning inhibited the cardiomyocyte apoptosis in the myocardial I/R might involve the following (Wang et al., 2011; Zhao et al., 2013): (1) SO2 preconditioning induced a moderate ERS, and then inhibited vigorous ERS-initiated cardiomyocyte apoptosis in the development of myocardial I/R injury. The above process was supported by the fact that SO2 induced early ERS markers glucose-regulated protein 78 (Grp78) expression and eukaryotic initiation factor 2α (eIF2α) phosphorylation, while myocardial I/R induced early and late ERS markers including Grp78 expression, eIF2α phosphorylation, CHOP expression and caspase-12 activation, which could be alleviated by SO2 preconditioning. (2) SO2 preconditioning inhibited the activation of caspase-9 and caspase-3 in the rats of the myocardial I/R group. Considering that caspase-9 activation subsequently follows the leakage of cytosol Cytc from injury mitochondria, the antiapoptotic effect of SO2 preconditioning might involve mitochondrial protection. (3) The inhibitor of PI3K/Akt the pathway blocked the SO2 preconditioning-inhibited caspase-3 activation, suggesting that the activation of the PI3K/Akt pathway might mediate the protective role of SO2 preconditioning.
The effect of endogenous SO2 on the ISO-induced cardiomyocyte apoptosis was examined targeting the mitochondrion (Liang et al., 2011; Jin et al., 2013). The SO2 level in the myocardial tissue was decreased but the proportion of apoptotic cells was increased in the rats of the ISO group compared with those of the control group. SO2 treatment restored the myocardial SO2 level and inhibited cardiomyocyte apoptosis. Then, the study further focused on the mitochondria. Firstly, the stereological analysis of the mitochondria showed that SO2 alleviated the cardiomyocytic mitochondria swelling and deformation, represented by the fact that SO2 treatment decreased the mitochondrial mean surface area, mean volume and volume density, and increased the mitochondrial numerical density and surface-to-volume ratio in the myocardial tissue of ISO-treated rats. Secondly, SO2 upregulated bcl-2 expression and downregulated bax expression, improved the impaired MMP, and reclosed the MPTP in rat myocardial tissues. Subsequently, SO2 blocked the leakage of Cytc from the mitochondria and inhibited the activation of cytosol caspase-9 and caspase-3. These data suggested that endogenous SO2 prevented ISO-induced cardiomyocytic apoptosis via maintaining the mitochondrial function and structure. Moreover, SO2 decreased the protein and mRNA levels of ERS markers Grp78, CHOP, and caspase-12 in the myocardial tissues of ISO-treated rats, suggesting that the over-activated ERS might also be involved in the inhibitory effect of SO2 on the ISO-induced cardiomyocytic apoptosis (Chen et al., 2012).
Similar to the ISO-induced myocardial injury, SO2 treatment inhibited cardiomyocytic apoptosis in the myocardial tissue of rats with cecal ligation and puncture (CLP), accompanied by an increase in the bcl-2 expression, and a decrease in the bax/bcl-2 ratio and caspase-3 activation (Yang et al., 2018). Moreover, SO2 decreased the level of inflammatory cytokines, inhibited TLR4 and NLRP3 expression, and reduced the inflammatory caspase-1 expression in the myocardial tissue of CLP rats and the LPS-treated primary neonatal rat cardiac ventricular myocytes (Yang et al., 2018), suggesting that NLRP3/caspase-1-mediated cell pyroptosis might also participate in the protective role of SO2 on the sepsis-induced cardiac dysfunction.
In some studies, SO2 promoted cardiomyocytic apoptosis in vitro and in vivo (Li et al., 2019). The data showed that the SO2 increased the proportion of apoptotic cells, activated caspase-9 and caspase-3, destroyed the MMP, reduced bcl-2 expression and the ratio of bcl-2/bax, and upregulated the expression of bax and p53, which could be blocked by antioxidant N-acetylcysteine. We speculated the following explanations for the different effects of SO2 on cardiomyocyte apoptosis. (1) The experimental reagents were different. Li et al. used sodium bisulfite solution and SO2 gas inhalation in vitro and in vivo, respectively, while a mixed solution of sodium sulfite and sodium bisulfite with a molar ratio of 3:1 was used in other studies. (2) The experimental conditions were different. Li et al. used untreated cardiomyocyte H9c2 cells and rats, while other studies were conducted on the different pathological cell and rat models.
The relationship between endogenous SO2 and neuronal apoptosis in the neurological disease was investigated on the kainic acid (KA)-induced epilepsy, febrile seizure (FS), and transient global ischemia/reperfusion models (Han et al., 2014; Niu et al., 2018; Zare Mehrjerdi et al., 2018). The different effects of SO2 on neuronal apoptosis depended on the neurological disorders and SO2 donor concentrations.
In the KA-induced epileptic rats, plasma SO2 level and AAT activity were upregulated compared with those in control rats (Niu et al., 2018). However, an AAT inhibitor HDX treatment retarded the occurrence of hippocampal neuronal apoptosis in the KA-treated rats, represented by the fact that a significant hippocampal neuronal apoptosis was detected in the rats 72 h after treatment with KA, while it was not observed in the rats treated with KA + HDX until 7 days after the intervention. The results suggested that the upregulated endogenous SO2 might promote hippocampal neuronal apoptosis in the development of epilepsy-related brain injury. Furthermore, the KA-upregulated ERS markers Grp78 expression and phosphorylated PERK in the hippocampus could be inhibited by HDX, indicating that ERS might participate in the promotive effect of SO2 on hippocampal neuronal apoptosis.
The endogenous SO2/AAT pathway including SO2 content and AAT1/2 expressions was upregulated in the hippocampus in rats of recurrent FS, accompanied by an increase in the neuronal apoptosis and mossy fiber sprouting (MFS) in the hippocampus (Han et al., 2014). Furthermore, HDX treatment aggravated the increased neuronal apoptosis and MFS in recurrent FS rats, while the supplement of a low concentration of SO2 (1–10 μmol/kg) alleviated the above pathological change, suggesting that the upregulated endogenous SO2 might be a defensive mechanism for antagonizing the neuronal apoptosis in the development of recurrent FS. However, the supplement of a high concentration of SO2 donor (100 μmol/kg) worsened the neuronal apoptosis and MFS in recurrent FS rats, which reminded us to pay attention to the protective dose range of SO2.
A global brain I/R injury increased CA1 neuronal apoptosis in the rats undergoing the occlusion of both common carotid arteries, accompanied by an increase in malondialdehyde level, and a decrease in glutathione level and activity of superoxide dismutase in the CA1 region (Zare Mehrjerdi et al., 2018). Furthermore, SO2 donor treatment improved the learning and memory deficits in the rats with cerebral I/R injury, reduced CA1 neuronal apoptosis, decreased malondialdehyde level, and elevated GSH level and activity of superoxide dismutase in the CA1 region. These findings indicated that SO2 donor prevented brain ischemic injury by decreasing neuronal apoptosis and the underlying mechanisms might involve the anti-oxidative effect of SO2. However, the change of endogenous SO2/AAT pathway in the brain ischemic rats was not detected, which merits further studies for exploring the role of endogenous SO2 in the development of brain ischemic injury.
Oxidative stress can induce apoptosis of retinal photoreceptor cells, which is the main pathological change of blinding retinal diseases (Wenzel et al., 2005). Du et al. found that apoptosis was significantly induced in the 661w mouse photoreceptor cells exposed to H2O2 compared with those without H2O2 exposure, demonstrated by an increase in the percentage of TUNEL positive cells and the ratio of cleaved-caspase3/caspase3 (Du et al., 2018). Simultaneously, endogenous SO2 production and AAT1 expression were decreased in the H2O2-treated 661w cells compared with control cells. HDX treatment mimicked the effects of H2O2 on 661w cells including an increase in the cell apoptosis and the downregulation of endogenous SO2/AAT pathway, indicating that the apoptosis of 661W cells caused by oxidative stress might be mediated by the downregulation of the endogenous SO2 pathway. However, the role of endogenous SO2 in the development of retinal photoreceptor cell apoptosis and its mechanisms require further studies.
Lipopolysaccharide (LPS) was intratracheally instilled to induce acute lung injury (ALI) in adult male rats (Huang et al., 2009). The decrease in the apoptosis of polymorphonuclear neutrophils (PMNs) contribute to an increment of the number of PMN and a prolonged inflammatory reaction in the injured lung (Lee and Downey, 2001). LPS treatment reduced the SO2 level in the lung tissue and peripheral blood, while the treatment of SO2 donor alleviated the lung histopathological changes in the rats accompanied by a reduction of PMN number in the bronchoalveolar lavage fluid (Huang et al., 2009; Ma et al., 2012). Furthermore, SO2 promoted the apoptosis of PMN in the bronchoalveolar lavage fluid and peripheral blood in the ALI rats. Mechanistically, SO2 treatment upregulated the expression of bax, downregulated the expression of bcl-2, and elevated the level of activated caspase-3 in the peripheral PMN in the ALI rats. Therefore, it is believed that SO2 may antagonize ALI by promoting PMN apoptosis, in which the bax/bcl-2/caspase-3 pathway is involved.
Many studies focused on the role of alveolar macrophage (AM) in the pathogenesis of ALI (Cheng et al., 2021). It was found that AM apoptosis was significantly increased in the ALI rat model induced by limb I/R injury, resulting in the delayed clearance of inflammatory PMN and the prolongation of ALI (Zhao et al., 2016). Zhao et al. found that SO2 donor prevented limb I/R-induced ALI in rats (Zhao et al., 2016). Furthermore, the primary AM was cultured with the plasma extracted from limb I/R to make the limb I/R-induced AM injury model (Zhao et al., 2019). Compared with the ALI group, the AM apoptosis was reduced in the AMs of the ALI + SO2 group, with an increase in bcl-2 expression, an improvement of the impaired MMP, the reclosing of MPTP and the decrease in caspase-3 expression. These data suggested that the SO2-inhibited AM apoptosis might be involved in the protective effect of SO2 on ALI and mitochondrion was the possible target of SO2.
As a novel gaseous signaling molecule, endogenous SO2 was reported to play a variety of biological effects by regulating cell apoptosis. In summary, endogenous SO2 promoted VSMC apoptosis and inhibited vascular EC apoptosis to alleviate the vascular remodeling in hypertension and pulmonary hypertension. Endogenous SO2 suppressed the apoptosis of cardiomyocytes, contributing to the cardioprotective effect of SO2. In the nervous system, SO2 increased KA-induced hippocampal neuronal apoptosis to aggravate the epileptic brain damage, while SO2 reduced neuronal apoptosis to protect against the neuronal damage in the recurrent FS and global brain I/R injury. Moreover, the downregulation of endogenous SO2 was accompanied by the increase in the 661w retinal photoreceptor cell apoptosis, suggesting that endogenous SO2 might participate in the pathogenesis of blinding retinal disease. In the respiratory system, SO2 facilitated PMN apoptosis and inhibited AM apoptosis to protect against ALI. The abovementioned apoptosis-related studies proved that the abnormal endogenous SO2 was involved in the pathogenesis of many diseases such as cardiovascular disease, neurological disease, and respiratory disease, and then keeping endogenous SO2 in the normal range might be a new and meaningful therapeutic strategy of related diseases in the future.
However, there remain some issues to be taken into account in future studies: (1) The endogenous SO2 generation and its producing enzyme should be firstly detected in the animal and cell models to identify the role of endogenous SO2 in the pathogenesis of diseases. (2) The concentration and type of SO2 donors might affect the effect of SO2, which should be paid attention to in vitro and in vivo. (3) Since mitochondria and their related apoptosis pathways were reported to be involved in almost all the anti- or pro-apoptotic effects of endogenous SO2, the detailed target molecules, and mechanisms by which SO2 affects mitochondria merit expectation in the future. (4) A recent study showed useful findings that plasma SO2 as a practical biomarker could predict the occurrence of acute kidney injury in the patients staying in the surgical intensive care unit (Jiang et al., 2021). Therefore, as a biomarker, the level of SO2 in clinical samples might have translational potential in the diagnosis, treatment decision, and prognosis prediction of the diseases. (5) In the future, the SO2-released prodrug and AAT inhibitor in clinical application for apoptosis-related diseases are expected to further studies (Day et al., 2016; Wang and Wang, 2018; Huang et al., 2021).
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This research was funded by National Natural Science Foundation of China (81770422 to YH, 81770278 to JBD, and 81900872 to JTD), Beijing Natural Science Foundation (7191012 to HJ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Balazy, M., Abu-Yousef, I. A., Harpp, D. N., and Park, J. (2003). Identification of carbonyl sulfide and sulfur dioxide in porcine coronary artery by gas chromatography/mass spectrometry, possible relevance to EDHF. Biochem. Biophys. Res. Commun. 331, 728–734. doi: 10.1016/j.bbrc.2003.10.055
Chen, S., Du, J., Liang, Y., Ochs, T., Liu, D., Zhu, L., et al. (2012). Sulfur dioxide inhibits excessively activated endoplasmic reticulum stress in rats with myocardial injury. Heart Vessels 27, 505–516. doi: 10.1007/s00380-011-0192-7
Chen, Z., Zhang, H., Bai, Y., Cui, C., Li, S., Wang, W., et al. (2021). Single cell transcriptomic analysis identifies novel vascular smooth muscle subsets under high hydrostatic pressure. Sci. China Life Sci. doi: 10.1007/s11427-020-1852-x [Epub ahead of print].
Cheng, P., Li, S., and Chen, H. (2021). Macrophages in lung injury, repair, and fibrosis. Cells 10:436. doi: 10.3390/cells10020436
Day, J. J., Yang, Z., Chen, W., Pacheco, A., and Xian, M. (2016). Benzothiazole sulfinate: a water-soluble and slow-releasing sulfur dioxide donor. ACS Chem. Biol. 11, 1647–1651. doi: 10.1021/acschembio.6b00106
Du, J. T., Huang, Y. Q., Li, K., Yu, X. Q., Jin, H. F., and Yang, L. (2018). Retina-derived endogenous sulfur dioxide might be a novel anti-apoptotic factor. Biochem. Biophys. Res. Commun. 496, 955–960. doi: 10.1016/jbbrc.2018.01.103
Du, S. X., Jin, H. F., Bu, D. F., Zhao, X., Geng, B., Tang, C. S., et al. (2008a). Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacol. Sin. 29, 923–930. doi: 10.1111/j.1745-7254.2008.00845.x
Du, S. X., Jin, H. F., Liang, Y. F., Zhao, X., Wei, H. L., Wang, L., et al. (2008b). Influence of sulfur dioxide and its derivatives on rats’ blood pressure. J. Appl. Clin. Pediatr. 23, 22–24. doi: 10.3969/j.issn.1003-515X.2008.01.007
Duan, H., Zhang, Q., Liu, J., Li, R., Wang, D., Peng, W., et al. (2021). Suppression of apoptosis in vascular endothelial cell, the promising way for natural medicines to treat atherosclerosis. Pharmacol. Res. 168:105599. doi: 10.1016/j.phrs.2021.105599
Griffith, O. W. (1983). Cysteinesulfinate metabolism: altered partitioning between transamination and decarboxylation following administration of beta-methyleneaspartate. J. Biol. Chem. 258, 1591–1598.
Han, Y., Yi, W., Qin, J., Zhao, Y., Zhang, J., and Chang, X. (2014). Dose-dependent effect of sulfur dioxide on brain damage induced by recurrent febrile seizures in rats. Neurosci. lett. 563, 149–154. doi: 10.1016/j.neulet.2013.12.042
Huang, X. L., Zhou, J. L., Zhou, X. H., Xian, X. H., and Ding, C. H. (2009). Ameliorative effects of exogenous sulfur dioxide on lipopolysaccharide-induced acute lung injury in rats. Acta Physiol. Sin. 61, 499–503. doi: 10.3321/j.issn:0371-0874.2009.05.014
Huang, Y. Q., Tang, C. X., Du, J. B., and Jin, H. F. (2016). Endogenous sulfur dioxide: a new member of gasotransmitter family in the cardiovascular system. Oxid. Med. Cell. Longev. 2016:8961951. doi: 10.1155/2016/8961951
Huang, Y. Q., Zhang, H., Lv, B. Y., Tang, C. S., Du, J. B., and Jin, H. F. (2021). Endogenous sulfur dioxide is a new gasotransmitter with promising therapeutic potential in cardiovascular system. Sci. Bull. 66, 1604–1607. doi: 10.1016/j.scib.2021.04.00
Jiang, Y., Wang, J., Zheng, X., and Du, J. (2021). Plasma endogenous sulfur dioxide: a novel biomarker to predict acute kidney injury in critically Ill patients. Int. J. Gen. Med. 14, 2127–2136. doi: 10.2147/IJGM.S312058
Jin, H., Liu, A. D., Holmberg, L., Zhao, M., Chen, S., Yang, J., et al. (2013). The role of sulfur dioxide in the regulation of mitochondrion-related cardiomyocyte apoptosis in rats with isopropylarterenol-induced myocardial injury. Int. J. Mol. Sci. 14, 10465–10482. doi: 10.3390/ijms140510465
Kimura, H. (2011). Hydrogen sulfide: its production, release and functions. Amino Acids 41, 113–121. doi: 10.1007/s00726-010-0510-x
Lee, W. L., and Downey, G. P. (2001). Neutrophil activation and acute lung injury. Curr. Opin. Crit Care. 7, 1–7. doi: 10.1097/00075198-200102000-00001
Li, S., Xu, Z., Jin, X., Qin, G., and Nan, S. (2019). Sulfur dioxide induces apoptosis via reactive oxygen species generation in rat cardiomyocytes. Environ. Sci. Pollut. Res. 26, 8758–8767. doi: 10.1007/s11356-019-04319-7
Liang, Y., Liu, D., Ochs, T., Tang, C., Chen, S., Zhang, S., et al. (2011). Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats. Lab. Invest. 91, 12–23. doi: 10.1038/labinvest.2010.156
Liu, D., Huang, Y., Bu, D., Liu, A. D., Holmberg, L., Jia, Y., et al. (2014). Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the ERK/MAP kinase pathway mediated by cAMP/PKA signaling. Cell Death Dis. 5:e1251. doi: 10.1038/cddis.2014.229
Liu, M., Liu, S., Tan, W., Tang, F., Long, J., Li, Z., et al. (2017). Gaseous signalling molecule SO2 via Hippo-MST pathway to improve myocardial fibrosis of diabetic rats. Mol. Med. Rep. 16, 8953–8963. doi: 10.3892/mmr.2017.7714
Liu, X., Zhang, D., Li, K., Yu, X., Tang, C., Du, J., et al. (2018). Effect of endogenous sulfur dioxide on the apoptosis induced by cobalt chloride in the human pulmonary arterial endothelial cells. Chin. J. Appl. Clin. Pediatr. 33, 999–1003. doi: 10.3760/cma.j.issn.2095-428X.2018.13.009
Luo, L. L., Chen, S., Jin, H. F., Tang, C. S., and Du, J. B. (2011). Endogenous generation of sulfur dioxide in rat tissues. Biochem. Biophys. Res. Commun. 415, 61–67. doi: 10.1016/j.bbrc.2011.10.012
Ma, H. J., Huang, X. L., Liu, Y., and Fan, Y. M. (2012). Sulfur dioxide attenuates LPS-induced acute lung injury via enhancing polymorphonuclear neutrophil apoptosis. Acta Pharmacol. Sin. 33, 983–990. doi: 10.1038/aps.2012.70
Mitsuhashi, H., Yamashita, S., Ikeuchi, H., Kuroiwa, T., Kaneko, Y., Hiromura, K., et al. (2005). Oxidative stress-dependent conversion of hydrogen sulfide to sulfite by activated neutrophils. Shock 24, 529–534. doi: 10.1097/01.shk.0000183393.83272.de
Niu, M., Han, Y., Li, Q., and Zhang, J. (2018). Endogenous sulfur dioxide regulates hippocampal neuron apoptosis in developing epileptic rats and is associated with the PERK signaling pathway. Neurosci. Lett. 665, 22–28. doi: 10.1016/j.neulet.2017.11.036
Qiu, J., Zheng, Y., Hu, J., Liao, D., Gregersen, H., Deng, X., et al. (2013). Biomechanical regulation of vascular smooth muscle cell functions: from in vitro to in vivo understanding. J. R. Soc. Interface 11:20130852. doi: 10.1098/rsif.2013.0852
Shapiro, R. (1977). Genetic effects of bisulfite (sulfur dioxide). Mutat. Res. 39, 149–175. doi: 10.1016/0165-1110(77)90020-3
Singer, T. P., and Kearney, E. B. (1956). Intermediary metabolism of L-cysteinesulfinic acid in animal tissues. Arch. Biochem. Biophys. 61, 397–409. doi: 10.1016/0003-9861(56)90363-0
Stipanuk, M. H. (2004). Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr. 24, 539–577. doi: 10.1146/annurev.nutr.24.012003.132418
Tisch, N., and Ruiz de Al modóvar, C. (2021). Contribution of cell death signaling to blood vessel formation. Cell. Mol. Life Sci. 78, 3247–3264. doi: 10.1007/s00018-020-03738-x
Wajant, H. (2002). The Fas signaling pathway: more than a paradigm. Science 296, 1635–1636. doi: 10.1126/science.1071553
Wang, W. Y., and Wang, B. H. (2018). SO2 donors and prodrugs, and their possible applications: a review. Front. Chem. 6:559. doi: 10.3389/fchem.2018.00559
Wang, X. B., Cui, H., and Du, J. B. (2018). Sulfur dioxide ameliorates rat myocardial fibrosis by inhibiting endoplasmic reticulum stress. Histol. Histopathol. 33, 1089–1097. doi: 10.14670/HH-18-007
Wang, X. B., Huang, X. M., Ochs, T., Li, X. Y., Jin, H. F., Tang, C. S., et al. (2011). Effect of sulfur dioxide preconditioning on rat myocardial ischemia/reperfusion injury by inducing endoplasmic reticulum stress. Basic Res. Cardiol. 106, 865–878. doi: 10.1007/s00395-011-0176-x
Wenzel, A., Grimm, C., Samardzija, M., and Remé, C. E. (2005). Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog. Retin. Eye Res. 24, 275–306. doi: 10.1016/j.preteyeres.2004.08.002
Yang, L., Zhang, H., and Chen, P. (2018). Sulfur dioxide attenuates sepsis-induced cardiac dysfunction via inhibition of NLRP3 inflammasome activation in rats. Nitric Oxide. 81, 11–20. doi: 10.1016/j.niox.2018.09.005
Zare Mehrjerdi, F., Shoshtari, A., Mohseni, F., Khastar, H., Norozi, P., Asadi, Y., et al. (2018). Sulfur dioxide reduces hippocampal cell death and improves learning and memory deficits in a rat model of transient global ischemia/reperfusion. Iran. J. Basic Med. Sci. 21, 998–1003. doi: 10.22038/IJBMS.2018.29404.7106
Zhao, M. M., Yang, J. Y., Wang, X. B., Tang, C. S., Du, J. B., and Jin, H. F. (2013). The PI3K/Akt pathway mediates the protection of SO2 preconditioning against myocardial ischemia/reperfusion injury in rats. Acta Pharmacol. Sin. 34, 501–506. doi: 10.1038/aps.2012.204
Zhao, X., Jin, H. F., Tang, C. S., and Du, J. B. (2008). Effects of sulfur dioxide, on the proliferation and apoptosis of aorta smooth muscle cells in hypertension: experiments with rats. Zhonghua Yi Xue Za Zhi 88, 1279–1283. doi: 10.3321/j.issn:0376-2491.2008.18.011
Zhao, Y. R., Liu, Y., Wang, D., Lv, W. R., and Zhou, J. L. (2019). Effects of sulfur dioxide on alveolar macrophage apoptosis in acute lung injury induced by limb ischemia/reperfusion in rats. J. Peking Univ. 51, 239–244. doi: 10.19723/j.issn.1671-167X.2019.02.007
Zhao, Y. R., Wang, D., Liu, Y., Shan, L., and Zhou, J. L. (2016). The PI3K/Akt, p38MAPK, and JAK2/STAT3 signaling pathways mediate the protection of SO2 against acute lung injury induced by limb ischemia/reperfusion in rats. J. Physiol. Sci. 66, 229–239. doi: 10.1007/s12576-015-0418-z
Keywords: sulfur dioxide, metabolism, apoptosis, mechanism, pathophysiology
Citation: Li Y, Feng Y, Ye X, Peng H, Du J, Yao X, Huang Y, Jin H and Du J (2021) Endogenous SO2 Controls Cell Apoptosis: The State-of-the-Art. Front. Cell Dev. Biol. 9:729728. doi: 10.3389/fcell.2021.729728
Received: 23 June 2021; Accepted: 13 September 2021;
Published: 07 October 2021.
Edited by:
Zhi Qi, Nankai University, ChinaReviewed by:
Linxi Chen, University of South China, ChinaCopyright © 2021 Li, Feng, Ye, Peng, Du, Yao, Huang, Jin and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaqian Huang, eWFxaWFuaHVhbmdAMTI2LmNvbQ==; Hongfang Jin, amluaG9uZ2Zhbmc1MUAxMjYuY29t; Junbao Du, anVuYmFvZHUxQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.