 Naaziyah Abdulla
Naaziyah Abdulla C. Theresa Vincent2,3
C. Theresa Vincent2,3 Mandeep Kaur
Mandeep Kaur
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 19 November 2021
Sec. Molecular and Cellular Oncology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.728325
This article is part of the Research TopicCholesterol and Cancer drug resistance: molecular, signaling and therapeutic aspectsView all 5 articles
Despite the significant advancements made in targeted anti-cancer therapy, drug resistance constitutes a multifaceted phenomenon leading to therapy failure and ultimately mortality. Emerging experimental evidence highlight a role of cholesterol metabolism in facilitating drug resistance in cancer. This review aims to describe the role of cholesterol in facilitating multi-drug resistance in cancer. We focus on specific signaling pathways that contribute to drug resistance and the link between these pathways and cholesterol. Additionally, we briefly discuss the molecular mechanisms related to the epithelial-mesenchymal transition (EMT), and the documented link between EMT, metastasis and drug resistance. We illustrate this by specifically focusing on hypoxia and the role it plays in influencing cellular cholesterol content following EMT induction. Finally, we provide a proposed model delineating the crucial role of cholesterol in EMT and discuss whether targeting cholesterol could serve as a novel means of combatting drug resistance in cancer progression and metastasis.
Cancer has been predicted to be the leading cause of death in every country in the world in the 21st century (Bray et al., 2018). In 2020, an estimated 10 million deaths were accredited to cancer with approximately 70% of these deaths documented in low- and middle-income countries (Sung et al., 2021). The International Agency for Research on Cancer predicted a worldwide increase in cancer cases from 18,078 957 (2018) to 28,400 00 in the year 2040, while a 78% increase in cancer cases in South Africa has been predicted by the year 2030 (Bray et al., 2018; Sung et al., 2021). Even though scientific advances in cancer research continue to provide renewed hope to cancer patients with major improvements documented in surgery, multidrug resistance (MDR) constitutes a major hurdle leading to therapy failure, and ultimately contributing to over 90% of cancer patient deaths (Bukowski et al., 2020). Intrinsic drug resistance occurs when the tumor cell mass present with resistant mechanisms prior to therapy administration, while acquired resistance describes a condition where cells that were initially sensitive to therapy develop mechanisms that confer them with survival traits during therapy (Haider et al., 2020). Multiple mechanisms implicated in conferring drug resistance to cancer cells have been documented in literature which include but are not limited to: enhanced drug efflux capability, mutations to the drug targets, increased metabolism of xenobiotics leading to drug detoxification and concomitant inactivation, hyperactivation of survival pathways coupled with inactivation of apoptotic pathways as well as the induction of the epithelial-mesenchymal transition (EMT) (Housman et al., 2014; Du and Shim, 2016; Ke and Shen, 2017; Bukowski et al., 2020; Haider et al., 2020). Importantly, the acquisition of resistance is dictated by inherent genetic instability, mutagenicity of tumor cells as well as the influence of cells in the tumor microenvironment (Haider et al., 2020). Increasingly now, emerging experimental evidence highlight the crucial role of cholesterol metabolism in facilitating drug resistance in cancer. This review aims to address the complexities underlying the role of cholesterol in facilitating MDR in cancer cells. Additionally, we document the link between EMT, metastasis and drug resistance, and illustrate this by specifically focusing on hypoxia as an extrinsic factor that influences cholesterol to facilitate EMT-induced drug resistance. This is followed with an accompanying discussion and a proposed hypothesis concerning whether targeting cellular cholesterol may serve as a novel strategy for combating cancer progression.
Cholesterol is a crucial lipid known to maintain cellular homeostasis by regulating the survival and growth of cells (Kuzu et al., 2016). It also serves as the key sterol component in the plasma membrane constituting approximately 30% of the lipid bilayer (Zhang J. et al., 2019). Structural integrity and fluidity of the cell membrane is thus consequently dictated by membrane cholesterol content. As a result, cholesterol is implicated in modulating the homeodynamics of a vast majority of cell surface related proteins (Zhang J. et al., 2019). Additionally, cholesterol serves as a crucial component in the formation of microdomains termed lipid rafts, which contain a multitude of proteins (Murai, 2015), that dictate the functional downstream processes such as signaling, proliferation, differentiation, adhesion and apoptosis (Zalba and Ten Hagen, 2017).
The synthesis of cholesterol is carried out in a highly conserved manner that is regulated by 21 enzymatic reactions (Silvente-Poirot and Poirot, 2014). In the mitochondria, the tricarboxylic acid (TCA) cycle generates a metabolite termed citrate. Cholesterol synthesis is initiated when citrate is converted to acetyl-coenzyme A (Murai, 2015). Subsequently, through the mevalonate pathway acetyl-coenzyme A is converted to lanosterol in a series of enzymatic reactions that take place in the endoplasmic reticulum (ER) (Murai, 2015). 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase (HMGCR) is the pivotal enzyme in this process as it catalyses the conversion of HMG-CoA to mevalonate in a rate-limiting step (Murai, 2015). Cholesterol is subsequently formed when the lanosterol is converted to cholesterol either by the Bloch pathway or the Kandutsch-Russell pathway (Murai, 2015). Mammalian cells are known to possess complex cholesterol homeostatic mechanisms (Figure 1) that are regulated at several levels, namely import, synthesis, export, metabolism and esterification (Das et al., 2014). These processes are facilitated by the presence of several important signaling molecules and enzymes at each stage and are summarized in Figure 1. For a more comprehensive overview concerning cholesterol homeostasis, the reader is referred to our previous publication (Gu et al., 2019).
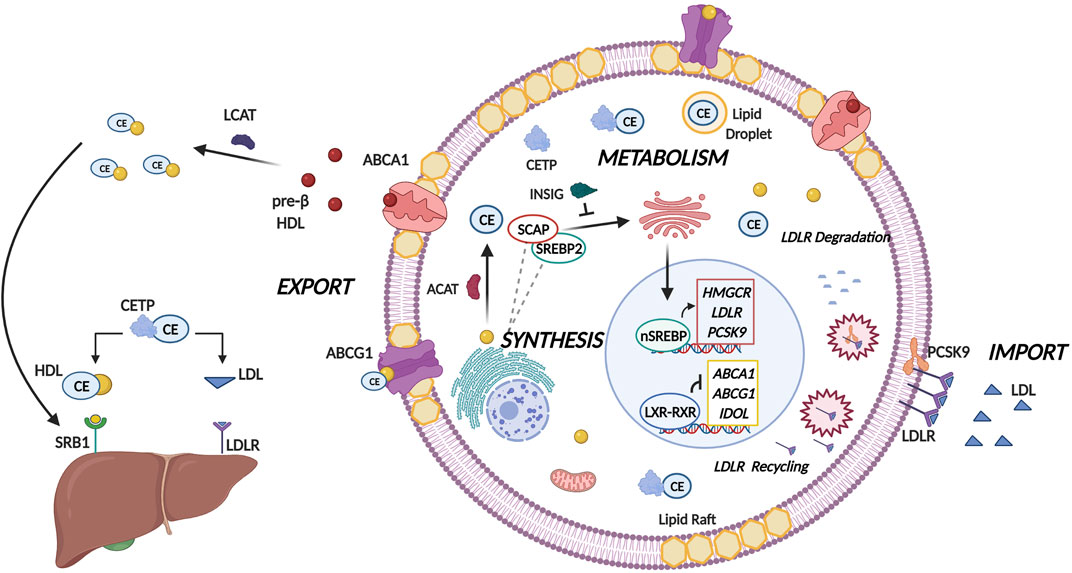
FIGURE 1. Molecular Mechanisms of Cholesterol Homeostasis. Cholesterol homeostasis is maintained by balancing the levels of cholesterol import, synthesis, metabolism, and export. A pivotal group of transcription factors termed the sterol regulatory element binding proteins (SREBPs) regulate lipogenesis and lipid uptake. When intracellular sterol concentrations are low, cholesterol biosynthesis is induced by SREBP2 where following proteolytic cleavage the nuclear isoform of SREBP2 (nSREBP2) binds to sterol response elements to facilitate expression of lipogenesis target genes namely HMGCR, low density lipoprotein receptor (LDLR) and proprotein convertase subtilisin/kexin type-9 (PCSK-9). HMGCR catalyzes the rate limiting step in cholesterol biosynthesis, whereas the LDLR imports cholesterol from the cellular milieu through endocytosis and hydrolysis of low density lipoprotein (LDL) particles. Collectively, this will lead to increased sterol levels within the cell. PCSK-9 regulates LDLR by either degrading LDLR by recruiting it to lysosome, or by prohibiting the endocytic recycling of LDLR by binding to the epidermal like growth factor repeat of LDLR. To prevent the excess accumulation of cholesterol in the cell, acyl-CoA cholesterol acyltransferase ACAT converts free cholesterol to cholesteryl esters (CEs) which are subsequently transported by cholesteryl ester transfer protein (CETP) from the site of synthesis to sites of storage in lipid droplets. Excess cellular cholesterol and cholesterol-derived oxysterols promotes the Liver-X-Receptor (LXR)-Retinoid-X-Receptor (RXR) mediated transcription of the two principal transporters ATP-binding cassette subfamily A member 1 (ABCA1) and subfamily C member 1 (ABCG1). These transporters transfer cholesterol to pre-β high density lipoprotein (HDL) and HDL respectively where cholesterol can either be eliminated by HDL binding to scavenger receptor- B1 (SR-B1). Alternatively, CETP can mediate the shuttling of cholesterol from HDL to LDL which will then result in elimination of cholesterol by binding to LDLR on the liver.
Importantly, cholesterol dyshomeostasis has emerged as a key requirement for cancer initiation and progression (Mandal and Rahman, 2014). Upon carcinogenesis finely tuned mechanisms of cholesterol regulation are altered based on the energetic requirements of the actively dividing cancerous cells (Cruz et al., 2013). Multiple studies conducted validated this notion, highlighting increased expression of sterol regulatory element binding protein (SREBP) target genes including HMGCR responsible for mediating the rate-limiting step in cholesterol synthesis as well as increased production of low density lipoprotein receptor (LDLR) that facilitates cholesterol uptake (Sun et al., 2015; Gallagher et al., 2017; Wen et al., 2018). Interestingly, proprotein convertase subtilisin/kexin type-9 (PCSK-9) (a regulator of LDLR) expression in cancer cells is found to be much lower expressed than their non-cancerous counterparts (Bhat et al., 2015). This allows for the cancer cells to enhance their cholesterol uptake. The decreased expression can be attributed to mutations in PCSK-9 as well as deregulations in cancer signaling pathways (e.g., NF-κβ) that facilitate cholesterol accumulation through decreased PCSK-9 expression (He et al., 2017). In terms of metabolism and efflux, acyl-CoA cholesterol acyltransferase (ACAT) activity in cancer cells has been shown to increase and promotes the conversion of cholesterol to cholesteryl esters (CEs) preventing the accumulation of free cholesterol that is toxic to the cell (Li et al., 2016). As a result of this, cholesterol uptake and synthesis is promoted to meet the metabolic demands of the cancerous cell (Li et al., 2016). Subsequently cholesteryl ester transfer protein (CETP) expression in cancer cells is also increased, which could be explained by the crucial role of CETP in stabilizing the CEs and promoting their storage in lipid droplets (Esau et al., 2016). Contrary to this, LXR modulated genes (such as ABCA1) display decreased expression in multiple cancer types. These deregulations ultimately facilitate accumulation of cholesterol intracellularly, thus contributing to poor prognosis and survival of patients (Kuzu et al., 2016).
From the above it is evident that cholesterol has a multifaceted role in the cell. Cholesterol synthesis is mediated through the highly conserved mevalonate pathway. Mammalian cells are known to possess complex cholesterol homeostatic mechanisms which cancer cells subvert to promote tumor initiation and progression.
Scientific evidence have implicated cholesterol as well as the oxygenated derivatives of cholesterol in the acquisition of drug resistance in cancer. This is facilitated by the influence of cholesterol on cell signaling pathways that contribute to resistance or alternatively through direct effects on the expression and activity of multi-drug transporters.
Kong et al., have documented increased levels of cholesterol due to increased expression of HMGCR in enzalutamide-resistant prostate cancer cell lines relative to enzalutamide-sensitive cell lines, where enzalutamide functions as an androgen receptor inhibitor. Consequently, overexpression of HMGCR in sensitive cell lines can confer resistance whereas shRNA-mediated silencing restores sensitivity (Kong et al., 2018). On this basis, authors documented that treating enzalutamide-resistant cell lines with both simvastatin (a cholesterol synthesis blocker) and enzalutamide efficiently restores sensitivity as evidenced by the decreased half maximal inhibitory concentrations (IC50) of enzalutamide in resistant cell lines (Kong et al., 2018). Similarly, administering a membrane cholesterol depletor, methyl-β-cyclodextrin (MβCD) to gefitinib resistant non-small cell lung cancer (NSCLC) cell lines resulted in a significant decrease in IC50 of gefitinib as a consequence of inhibition of epidermal growth factor receptor (EGFR) activity as well as the activity of downstream MAPK/ERK and PI3K/Akt signaling pathways (Chen et al., 2018). In lung cancer cells, administration of EGFR inhibitors facilitated synthesis of both cellular and mitochondrial cholesterol (Howell et al., 2020). Following exposure to tyrosine kinase inhibitors (TKIs), drug resistant cells showed an increased expression of 7-dehydrocholesterol reductase, 24-dehydrocholesterol reductase, with an approximate 20-fold increase in the expression of SREBF2. Importantly, the administration of ketoconazole (an inhibitor of CYP51A1) together with a TKI acted synergistically to restore TKI sensitivity in resistant cells (Howell et al., 2020). In another study, administering both simvastatin and EGFR TKIs proved effective in combatting T790M (Thr790Met, gatekeeper mutation) mediated EGFR TKI resistance (Hwang et al., 2014). Inhibiting Akt signaling led to reduced β-catenin phosphorylation and further repression of downstream target genes including cyclin-D1 and survivin, where downregulation of survivin has been found to be crucial in mediating apoptosis induction (Hwang et al., 2014).
Cholesterol biosynthesis genes also contribute to cisplatin-resistance in ovarian cancer. This is evidenced by findings that the expression of SREBP2 dependent-genes, including HMGCR, LDLR as well as farnesyl-diphosphate farnesyltransferase 1 (FDFT1) increase following the administration of cisplatin and is dose-dependent (Zheng et al., 2018). The transition from a sensitive to a resistant phenotype correlates with higher levels of plasma membrane and lysosomal cholesterol in resistant cells (Criscuolo et al., 2020). This suggests that along with blocking the endogenous cholesterol synthesis, treating cells with cholesterol depletors to extract cholesterol from the cellular membranes could possibly combat drug resistance. This would be mediated by increasing the expression of genes involved in cholesterol efflux resulting in a concomitant decrease in intracellular cholesterol content (Yokoo et al., 2015; Zimmer et al., 2016). Therefore, further investigations into the field of employing cholesterol depletion as a means to restore drug sensitivity are required.
Cholesterol uptake due to the increased expression of LDLR facilitated chemoresistance and increased risk of recurrence in pancreatic ductal adenocarcinoma (PDAC) (Guillaumond et al., 2015). Silencing of LDLR led to a disruption in cholesterol distribution, impeding ERK 1/2 signaling, and further sensitized PDAC cells to chemotherapeutic agents both in-vitro and in-vivo (Guillaumond et al., 2015). Similarly, studies conducted by Naito et al. identified the crucial role of LDL in inducing resistance to TKIs in clear cell renal cell carcinoma (ccRCC) as mice fed on a high cholesterol diet (21% milk fat with 1.25% added cholesterol) presented with resistance to sunitinib (TKI) treatment when compared with mice that were on a normal diet, presumably due to LDL mediated activation of the PI3K/Akt signaling pathway (Naito et al., 2017). Consequently, administering a cholesterol depletor, hydroxy-propyl-β-cyclodextrin (HPβCD), resulted in disruption of lipid raft integrity, and abrogated LDL-mediated AKT phosphorylation in the SK-45 RCC cell line as well as the patient-derived PNX0010 ccRCC cell line (Naito et al., 2017).
In addition to LDL, CEs have also been implicated in facilitating chemoresistance. Li et al. identified a significant increase in the fraction of lipid droplet area as well as a modest increase in CEs in the G3K (a resistant PDAC cell line) relative to the sensitive Mia-Pa-Ca2 PDAC cell line (Li et al., 2018). Consequently, administration of an ACAT1 inhibitor avasimibe proved effective in reducing cell viability of both Mia PaCa-2 and G3K cells, whereas administering both avasimibe and gemcitabine enhanced the anti-proliferative effect relative to monotherapy (Li et al., 2018). Authors documented synergism between avasimibe and gemcitabine, and further in-vivo analysis identified that treatment with only gemcitabine resulted in tumor relapse after 20 days, whereas a combination of gemcitabine and avasimibe effectively reduced tumor growth with no observable relapse following 34-days of treatment (Li J. et al., 2018). The combination therapy proved most effective in reducing the levels of CEs as well as the expression of p-AKT highlighting that avasimibe restores chemosensitivity in gemcitabine-resistant cells by impeding Akt signaling (Li et al., 2018). Attempting to demonstrate the link between cholesterol and gallbladder cancer, Zhang and colleagues established that gallbladder cancer patients present with increased expression of genes involved in cholesterol biosynthesis (HMGCR) as well as sterol sulfonation (SULT2B1), reduced expression of genes CYP7B1 and CYP39A1 responsible for cholesterol catabolism as well as decreased expression of genes that facilitate cholesterol efflux (ABCA1, ABCG5, LCAT and CETP) (Zhang Y. et al., 2019). Treating gallbladder cancer cells with both lovastatin and cisplatin proved effective in increasing apoptosis in cells when compared with single treatments. Interestingly cholesterol depletion abrogated activation of checkpoint kinase (CHK) 1, CHK2 as well as γ-H2AX following treatment with cisplatin highlighting reduced DNA damage response (Zhang Y. et al., 2019). This effect was also observed in-vivo indicating reduced growth in the combination-treatment groups (Zhang Y. et al., 2019). Furthermore, Esau et al. by employing both in-vitro and in-vivo assays identified that administration of Acetyl Plumbagin (AP) leads to a decrease in the mRNA expression and protein levels of CETP which results in growth inhibition and contributes to mitochondrial-mediated apoptosis (Esau et al., 2016). On this basis, silencing CETP in MCF-7 cells is shown to enhance the sensitivity of cells to Tamoxifen (TAM) as well as AP as evidenced by the increased caspase 3/7 activity and increased percentage of cells undergoing apoptosis when compared to the non-transfected cells (Esau et al., 2016). Studies conducted by Gu et al. further identify that administering a combination treatment of TAM and AP in CETP knockout MCF-7 cells functions synergistically to enhance mitochondrial-mediated apoptosis relative to monotherapy with either TAM or AP (Gu et al., 2019).
Further developments in this field are beginning to reveal the crucial role cholesterol plays in regulating the expression as well as activity of principal multi-drug transporters contributing to MDR. This is elucidated by the observation that cholesterol serves as a transcriptional regulator of MDR efflux transporters where a positive correlation between LXRα/β protein expression and MDR1 expression has been documented. (Kim S. et al., 2018). In terms of MDR transporter activity, plasma membrane cholesterol content alters membrane fluidity thereby regulating their activity (Kopecka et al., 2020). Interestingly, the lipid environment surrounding these transporters modulate their conformation, maintaining transporters in a low-energy stable conformation (Kopecka et al., 2020). With respect to cholesterol, this molecule surrounds P-gp adopting an asymmetrical distribution together with phospholipids (Alam et al., 2019). The outer leaflet is characterized by an ordered ring of cholesterol whereas the inner leaflet presents with both phospholipids and cholesterol. Importantly, the lipid-transporter interactions mediate structural modifications of P-gp consequently regulating its catalytic activity (Alam et al., 2019). Additionally, studies conducted document that P-gp substrates accumulate in cholesterol-rich regions of the membrane thereby enhancing the activity of the P-gp (Subramanian et al., 2016). Interestingly, evidence exists highlighting the role of cholesterol in altering P-gp substrate KM values thereby implying that cholesterol directly interacts with substrate binding sites (Hegedüs et al., 2015). This is illustrated mechanistically, by studies conducted by Clouser et al. proposing that plasma membrane cholesterol modulates P-gp-transmembrane helix-membrane interactions which consequently promotes long-range conformational changes in the nucleotide-binding domain of P-gp promoting a drastic increase in the rate of ATP hydrolysis (Clouser et al., 2021). This consequently justifies the crucial role cholesterol plays in supporting the activity of P-gp housed within lipid rafts.
The acquisition of chemoresistance following platinum-based therapy is regulated by cholesterol-mediated influence on the expression of ABCG2 (Wu et al., 2015). In this study, individuals who displayed quick chemoresistance presented with increased levels of baseline total cholesterol in tumour tissue relative to the delayed chemoresistance group. Additionally, mRNA and protein expression analysis documented a significant increase in the levels of ABCG2 (Wu et al., 2015). Further, in-vitro studies have shown that culturing the A549 lung cancer cell line in cell culture media supplemented with high cholesterol significantly increased the IC50 of platinum-based chemotherapeutic agents, whereas treating cells with an ABCG2 blocker, Nicardipine or pravastatin combats cholesterol-induced chemoresistance (Wu et al., 2015). Similarly, pre-treatment with cholesterol (cholesterol repletion) resulted in a significant increase in the IC50 to cisplatin and paclitaxel in PA-1 and SCOV-3 ovarian cancer cells which is attributed to cholesterol-mediated increase in the expression of ABCG2 and MDR1 (Kim S. et al., 2018). Interestingly, a positive correlation between LXRα/β protein expression and MDR1 expression has been documented and siRNA-mediated silencing of LXRα/β abrogated cholesterol-mediated induction of MDR1 consequently affecting cholesterol-induced chemoresistance (Kim S. et al., 2018).
Targeting cellular cholesterol is seen to affect lipid raft integrity consequently abolishing the activity of multidrug transporters leading to enhanced retention of chemotherapeutic agents. The treatment with lovastatin and imatinib enhanced cytotoxicity of imatinib in cell lines as well as CD34+ chronic myelogenous leukaemia (CML) cells derived from patients diagnosed with different disease stages (Glodkowska-Mrowka et al., 2014). This is attributed to the inhibition of ABC transporter activity namely ABCB1 and ABCG2 leading to increased retention of imatinib in CML cells (Glodkowska-Mrowka et al., 2014). It is important to note that the expression of these transporters is not affected following treatment with lovastatin. By impeding the activity of these transporters, the accumulation of imatinib in CML cells is enhanced thereby allowing for increased amounts of the active drug to bind to and exert antileukemic activity (Glodkowska-Mrowka et al., 2014). In terms of pancreatic cancer, the principal CD133 marker known to co-localize to lipid rafts display a tendency to co-occur with mevalonate synthesis pathway enzymes (Gupta et al., 2018). Importantly, attempting to eradicate this resistant cancer cell population by the administration of lovastatin alone does not significantly affect cell viability in CD133 + cells, however co-administration of lovastatin and paclitaxel induces cell death in CD133 + cells by disrupting lipid raft integrity which consequently abrogates ABC transporter activity (Gupta et al., 2018).
The above summarised studies emphasize the importance of cholesterol in driving cancer proliferation and therapeutic response by affecting signaling pathways that facilitate resistance or through the direct effects on the expression and activity of MDR transporters. Emerging scientific evidence document that targeting cholesterol through gene silencing, cholesterol synthesis inhibition or cholesterol depletion restores sensitivity to conventional chemotherapeutics.
Seeing that drug resistance is a hallmark of metastatic cancer, there is a gap present in the literature regarding the role of cholesterol in cells undergoing the tumor cell plasticity program of Epithelial-Mesenchymal Transition (EMT). In the following sections, we have addressed this gap at least partly by reviewing the published literature and proposing a potential hypothesis to demonstrate the crucial involvement of cholesterol in EMT and metastasis.
EMT is classified as a tumor cell plasticity program. Initially known to play a role in embryonic development, current scientific evidence supports the role of EMT in various pathological processes including wound healing, fibrosis, inflammation and cancer (Yeung and Yang, 2017). The outline of the EMT program is summarised in Figure 2.
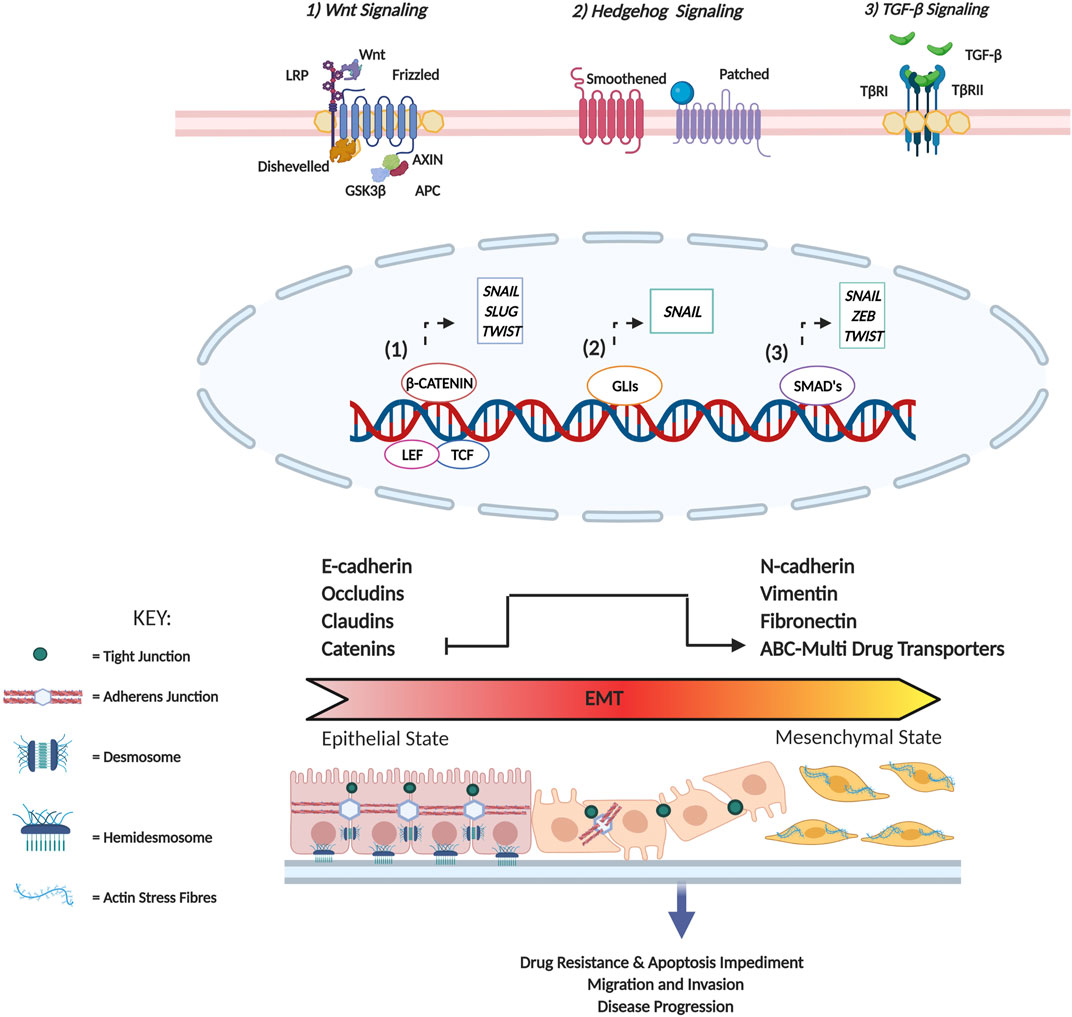
FIGURE 2. Summary of EMT induced changes in cellular physiology. In tissues, epithelial cells are often arranged as either a single layer or exist as multi-layered sheets. These cells are characterized by the presence of specialized contacts called cell junctions that facilitate cell-cell interaction (cadherin-based adherens junctions, tight junctions, and desmosomes) as well as cell-matrix interactions (hemidesmosomes) that maintain structural integrity and apical-basal polarity. In terms of cytoskeletal composition, Type I intermediate filaments such as cytokeratins are expressed, and adherens junctions serve to indirectly connect cortical actin filaments of two adjacent cells. On the contrary, mesenchymal cells are characterized by minimal cell junctions and detached stromal arrangements promoting front-rear polarity. In terms of cytoskeletal composition, vimentin is predominantly expressed to enhance cytoskeletal strength contributing to cell flexibility, and actin fibres arrange to form stress fibres. Additionally, the formation of actin-rich membrane projections facilitates cellular movement as well as sensory reception. Characteristic surface markers utilized to identify epithelial cells include E-cadherin, cytokeratins, occludins as well as claudins, while mesenchymal cells are identified based on the presence of N-cadherin, fibronectin, and vimentin. Cellular stimuli lead to the activation of several signaling pathways (Wnt, Sonic Hedgehog and TGF-β) that promote the expression of master EMT TFs (SNAIL, SLUG, TWIST and ZEB). Following activation of the EMT program these TFs facilitate the loss of several of the aforementioned epithelial characteristics, promote cytoskeletal remodeling and initiate a concomitant gain of mesenchymal traits. This transition consequently facilitates a change in cell morphology from squamous, cuboidal, columnar to a fibroblast/spindle-like morphology conferring upon cancer cells migratory and invasive potential.
Cells exhibit a significant degree of plasticity where instead of transitioning between full epithelial to full mesenchymal phenotype, they traverse through a spectrum of intermediate states along the epithelial-mesenchymal axis expressing varying levels of epithelial and mesenchymal markers (Nieto et al., 2016).
Repression of the epithelial phenotype and activation of the mesenchymal phenotype is dictated by regulatory networks that function to modulate the activity of a group of master transcription factors (TFs) (Fuxe et al., 2010; Craene and Berx, 2013). The master TFs consist of the zinc-finger E-box-binding homeobox factors ZEB1/2 and SNAIL 1/2 as well as the basic helix-loop-helix (bHLH) factors TWIST 1/2 which are also the most comprehensively studied (Nieto et al., 2016). Transcriptional and translational controls of these TFs are governed by the expression of specific non-coding RNAs including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) as well as alternative splicing and protein stability (Craene and Berx, 2013; Nieto et al., 2016). Interestingly, the master EMT-TFs function co-operatively to modulate the expression of target genes as well as each other (Shibue and Weinberg, 2017; Dongre and Weinberg, 2019). In this way, the reciprocal interactions induce comprehensive EMT associated changes even in the event where only one of the TFs are activated (Shibue and Weinberg, 2017). The role of each of these master TFs in facilitating EMT induction is briefly elaborated on below (also see Figure 2).
The SNAIL subfamily of zinc-finger TFs is represented by three members in vertebrates namely Snail (Snail 1), Slug (Snail 2) and Smuc (Snail 3). The transcriptional repression of E-cadherin is achieved through direct binding of Snail and Slug to E-box regions in the proximal promoter region (Batlle et al., 2000; Hajra et al., 2002), while emerging scientific evidence continue to illustrate the recruitment of epigenetic regulators that affect both DNA and chromatin accessibility, and as such E-cadherin expression (Peinado et al., 2004; Herranz et al., 2008; Hou et al., 2008; Lin et al., 2010; Dong et al., 2012; Tong et al., 2012; Wu et al., 2012). Additionally, Snail and Slug are also implicated in repressing the expression of occludins, claudins and CXAR (CAR) which are present in tight junctions, crucial to maintaining cell polarity (Ikenouchi et al., 2003; Martínez-Estrada et al., 2006; Wang et al., 2007; Vincent et al., 2009). Conversely, Snail and Slug can upregulate key mesenchymal markers namely vimentin and fibronectin (Cano et al., 2000; Jethwa et al., 2008).
While several members of this family have been identified, TWIST1 has been documented to play a pivotal role in EMT by repressing the expression of epithelial proteins and by promoting the expression of mesenchymal related proteins. Twist1 binds to E-cadherin promoter regions, recruiting epigenetic regulators that influence DNA and chromatin accessibility as well as modulating expression of ncRNAs that play a role in EMT-induction (Yang et al., 2004; Fu et al., 2011; Yang et al., 2012; Meng et al., 2018).
The ZEB family of TFs in humans comprises ZEB1 and ZEB2 and are classified as zinc finger/homeodomain TFs (Abba et al., 2016; Georgakopoulos-Soares et al., 2020). These TFs possess characteristic protein-binding domains which facilitate the recruitment of co-repressors as well as co-activators leading to gene repression or gene activation, respectively (Zhang et al., 2015). Based on this dual function, they can induce EMT by employing both repression of epithelial proteins and activation of mesenchymal proteins (Bindels et al., 2006).
Interestingly, completion of the EMT program resulting in cells adopting an extreme mesenchymal phenotype has been noted as an infrequent event in both neoplastic and non-neoplastic human tissue (Derynck and Weinberg, 2019). Predominantly, the stable existence of cells in the hybrid state has been linked with increased metastatic potential since the hybrid EMT state facilitates collective cell migration and reinforces ECM attachment (Saitoh, 2018). Furthermore, the plasticity associated with the hybrid phenotype facilitates the acquisition of stemness as well as drug resistance and correlates with poor survival in several cancer types (Pastushenko and Blanpain, 2019; Liao and Yang, 2020). Yet the precise molecular mechanisms governing the process of EMT-mediated stemness is lacking (Dongre and Weinberg, 2019). Additionally, future studies aimed at identifying whether existence in different hybrid states confer differential response to chemotherapeutic agents would be highly informative to elucidate mechanisms governing drug resistance (Thankamony et al., 2020).
Principally, the induction of EMT is associated with an increase in drug resistance potential in cancer cells. This is seen in response to treatment with platinum-based agents, TKI’s as well as taxane-based cancer therapeutics. To sustain resistance to chemotherapeutic agents, cancer cells continuously evolve. In terms of EMT and its link to drug resistance, cancer cells activate several EMT-related signaling pathways that concomitantly induce the expression of EMT master TFs. These TFs consequently regulate key proteins involved in cell survival and apoptosis signaling. This evolutionary process facilitates the acquisition of survival mechanisms that allow for a certain degree of genetic stability to be maintained in the pursuance of cancer cell proliferation and apoptosis induction impediment following DNA damage. Another mechanism employed by EMT to mediate drug resistance is through the presence of several binding sites in the promoter regions of the ATP-binding cassette (ABC) transporters leading to overexpression, which in turn mediates excessive drug efflux. Since altered cellular metabolism has been linked with EMT, we attempt to discuss the emerging role that cholesterol plays in regulating critical signaling pathways associated with EMT. We further elaborate on this concept by discussing hypoxia as an extrinsic factor that modulates cholesterol to mediate EMT-induced drug resistance.
The cellular EMT process is a complex labyrinth dependent on subversion of critical cellular signaling pathways, which crosstalk extensively to mediate the initiation and regulation of the EMT program (Fuxe et al., 2010). Based on the pleotropic role of cholesterol in the cell, it is not surprising that cancer cells have evolved several mechanisms to facilitate cholesterol dyshomeostasis, which in turn influence several EMT associated signaling pathways. In addition to meeting the increased metabolic demands of cancer cells, deregulated cholesterol metabolism also facilitates increased cellular cholesterol availability which is crucial to regulating the activity of protein intermediates in EMT-related signaling pathways. Furthermore, an increase in cholesterol content corresponds to an increase in lipid raft presence. Since lipid rafts contain several cell signaling proteins, alteration to lipid raft domains may therefore influence cell signaling pathways that ultimately augment neoplastic transformation (Zalba and Ten Hagen, 2017). These rafts in turn regulate crucial processes that cancer cells subvert such as survival, proliferation, migration, invasion, and apoptosis. Intra- or extracellular stimuli mediate alterations in lipid raft size and protein composition facilitating receptor-ligand interactions as well as interactions between proteins leading to downstream activation of signal transduction cascades crucial for cancer cell signaling (Mollinedo and Gajate, 2015). The role of cholesterol in regulating some of the most well studied EMT-related signaling pathways (Figure 3) will be further delineated below. It should be noted that this section purely aims to elucidate the role cholesterol plays in the regulation of these EMT-related pathways. The reader is suggested to refer to several existing reviews (Lamouille et al., 2014; Singh et al., 2018; Das et al., 2019; Dongre and Weinberg, 2019) that excellently delineate the precise mechanisms these pathways employ to induce EMT.
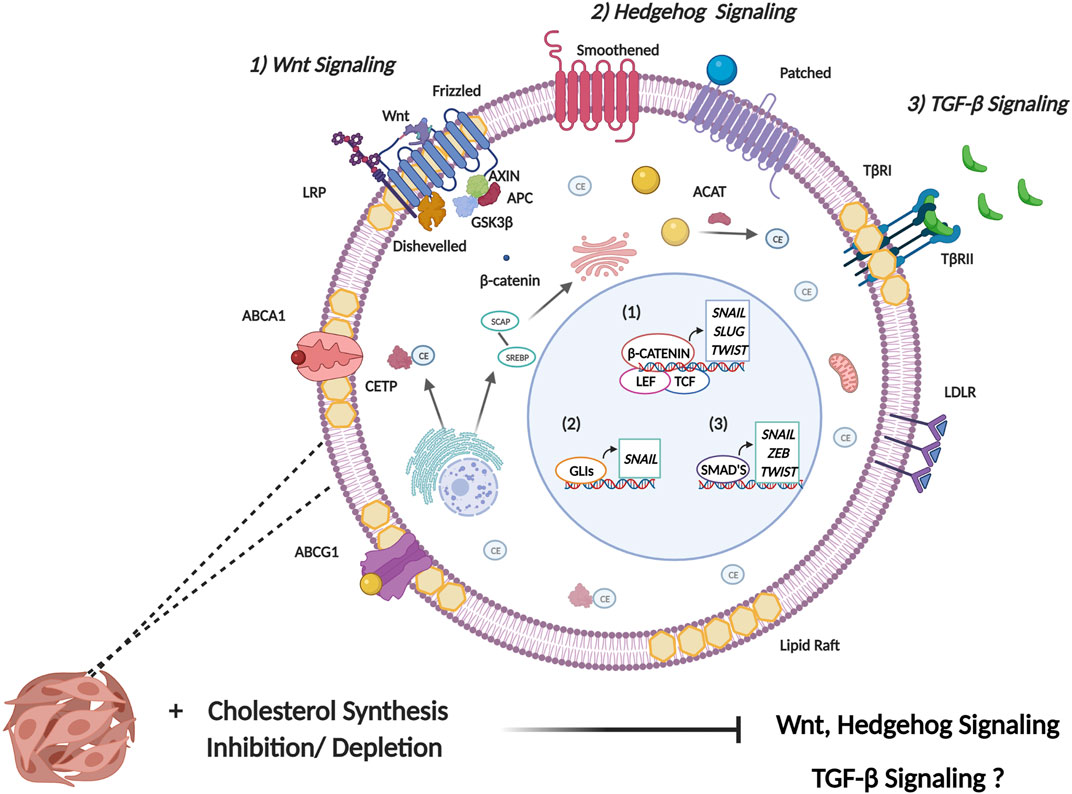
FIGURE 3. Cholesterol modulates signaling pathways involved in EMT induction. EMT is induced by several signaling pathways that facilitate the expression of EMT master transcription factors (SNAIL, SLUG, ZEB, and TWIST). 1) Canonical Wnt signaling is initiated following the binding of a Wnt ligand to Frizzled receptor. This consequently mediates the release of β-catenin from the glycogen synthase kinase 3-β (GSK3β), axis inhibition protein (AXIN), adenomatous polyposis coli (APC) protein destruction complex. This allows for increased stability of β-catenin where it enters the nucleus binding to the transcription factors T-cell factor (TCF) and lymphoid-enhancer factor (LEF) to induce transcription of EMT TFs. Primarily, Wnt binds to receptors in lipid raft domains and cholesterol facilitates recruitment of the Dishevelled to the plasma membrane. Collectively this facilitates activation of the canonical Wnt/β-catenin signaling. 2) Activation of canonical hedgehog signaling occurs through binding of the hedgehog ligand, sonic hedgehog (Shh) to the transmembrane receptor patched which leads to de-repression of Smoothened. Smoothened consequently activates the transcriptional activity of the GLI family of transcription factors. Cholesterol is necessary for post-translational modification of Hh ligands. Furthermore, the activity of the membrane transducer Smo is regulated directly by cholesterol through its cysteine-rich domain and also indirectly through Hh receptor PTCH1 which possesses a sterol-sensing domain. 3) Transmembrane signaling by TGF-β is initiated upon ligand binding to membrane bound serine/threonine kinase receptor, termed TGF-β type II receptor (TβR-II). This leads to recruitment of membrane bound TGF-β type I receptor (TβR-I) resulting in the formation of a heterotetrameric complex. The main mechanism of intracellular TGF-β signaling is mediated through the Smad family of transcription factors which serve to activate or repress TGFβ responsive genes. The role of cholesterol in modulating TGF-β signaling is complex and not fully understood, with some studies implicating cholesterol in TGF-β signaling, and other studies highlighting reduced cholesterol upon TGF-β induced EMT.
In canonical Wnt signalling, binding of Wnt ligands to Frizzled (Fz) receptors in conjunction with low-density lipoprotein receptor-related protein (LRP) facilitates recruitment of Dishevelled (Dvl) protein to the plasma membrane (Zhong and Virshup, 2020). This recruitment is facilitated by cholesterol (Sheng et al., 2014), which in turn supports activation of canonical/β-catenin dependent Wnt signaling (Sezgin et al., 2017). Additionally, Wnt binding to the receptor complex occurs exclusively in the plasma membrane ordered compartments termed lipid rafts (Sezgin et al., 2017). Consequently, administering enzymes/drugs that disrupt key constituents in ordered plasma membrane significantly decreased Wnt/β-catenin signaling when cholesterol was disrupted following the administration of cholesterol oxidase (Sezgin et al., 2017). Furthermore, a class of cholesterol depleting agents termed cyclodextrins have recently been implicated in modulating Wnt signaling through cholesterol depletion from membrane lipid rafts. Lipid rafts play a pivotal role in maintaining the integrity of LRP6 in triple negative breast cancer (TNBC). Treating TNBC cell lines with MβCD resulted in reduced proliferative and migratory potential and also increased apoptosis induction by downregulating several anti-apoptotic proteins as well as decreased expression of LRP6 and β-catenin (Badana et al., 2018). This suggests that lipid raft integrity is crucial in dictating the functionality of the Wnt/β catenin signaling which plays a key role in facilitating TNBC aggressiveness.
Sonic hedgehog (Shh) signaling is essential for embryonic development but has also been documented to play a pivotal role in tumorigenesis and cancer stem cell maintenance (Wu et al., 2017). Hedgehog (Hh) signal transduction can be linked with cholesterol through the post-translational modification on the Hh ligands where covalent modification is initiated at the c-terminal end of the ligand through cholesterolysis (Callahan and Wang, 2015; Blassberg and Jacob, 2017). The translocation of cholesterolysis-defective Hh precursors is impeded from the ER to the Golgi. As they are sequestered in the ER, ER associated protein degradation (ERAD) mediated polyubiquitination and proteasomal degradation is induced which abrogates translocation of the protein out of the ER and hence Hh signaling (Hampton, 2002). In addition to this, the Hh receptor PTCH1 is also documented to possess a sterol-sensing domain (SSD) which is typically present in proteins regulating cholesterol synthesis and transport (Luchetti et al., 2016). A study conducted by Bidet et al. proposes that the SSD of PTCH1 allows for cholesterol efflux from the cell when a Shh ligand is absent, this consequently results in a decrease in the intracellular concentration of cholesterol and impedes the activity of the membrane transducer Smo (Bidet et al., 2011). Conversely, binding of a ligand to PTCH1 facilitates the uptake of PTCH1 and impedes the efflux of cholesterol. The increased intracellular concentration of cholesterol mediates increased abundance of Smo at the membrane (Bidet et al., 2011). Importantly, cholesterol is also seen to directly regulate the activity of Smo by binding to its extracellular cysteine-rich domain (CRD). Mutations found in the CRD impedes cholesterol mediated Smo activation and consequently Shh mediated signaling hence indicating the important role of cholesterol in endogenous Hh signaling (Luchetti et al., 2016).
TGF-β is a pleiotropic cytokine known to play an important role during embryonic development as well as regulate homeostasis of adult tissues by modulating processes such as cell proliferation, differentiation, migration, invasion, immunological response and apoptosis (Chruścik et al., 2018; Ahmadi et al., 2019; Derynck and Budi, 2019; Yang et al., 2020). Consequently, aberrant TGF-β signaling often leads to dedifferentiation of several cell types, immunosuppressive effects, enhanced angiogenesis and accommodates an inflammatory milieu which consequently contributes to tumorigenesis (Padua and Massagué, 2009).
The formation of TβRI/TβRII/TGβ-1 complex is dependent on lipid raft integrity and disruption of lipid rafts impede canonical TGF-β signaling (Ma et al., 2007). Importantly, the localization of TGF-β receptors in lipid rafts is crucial to mediating TGF-β induced activation of MAPK signaling and promoting EMT and cell migration. Contrary to the afore-mentioned studies, a study conducted by Zhao et al. suggests that down-regulation of cholesterol may be a consequence of EMT (Zhao et al., 2019). It was recently identified that TGF-β promotes the enrichment of ZEB1 and CtBP at the promoter region of SREBF2 leading to repression of SREBF2 and its downstream target genes. Interestingly the reduction in membrane-bound cholesterol was implicated in enhancing the stability of TGFβRI (Zhao et al., 2019). Additionally, studies conducted by Shapira et al. elucidated that cholesterol depletion facilitates a PKR-dependent phosphorylation of eIF2α which enhances the translation of c-Jun (Shapira et al., 2018). An increase in JNK-mediated c-Jun phosphorylation is also reported which enhances the activation of c-Jun and consequently increases the transcription of Smad 2/3. This ultimately allows for enhanced TGF-β mediated signaling (Shapira et al., 2018). These studies elucidate the complex role that cholesterol plays in regulating the expression and activity of TGF-β signaling module. This consequently necessitates additional investigations into the role of cholesterol in the regulation of TGF-β signaling in a cancer setting to identify whether targeting cellular cholesterol is a feasible means to halt cancer progression.
Based on the principal role of cholesterol in regulating key pathways associated with EMT induction and maintenance, emerging scientific evidence now exist implicating cholesterol-related genes in mediating EMT consequently potentiating an aggressive disease phenotype with increased drug resistance. Studies describing the role of cholesterol-related genes in EMT induction, metastasis and drug resistance are summarized in Table 1 below.
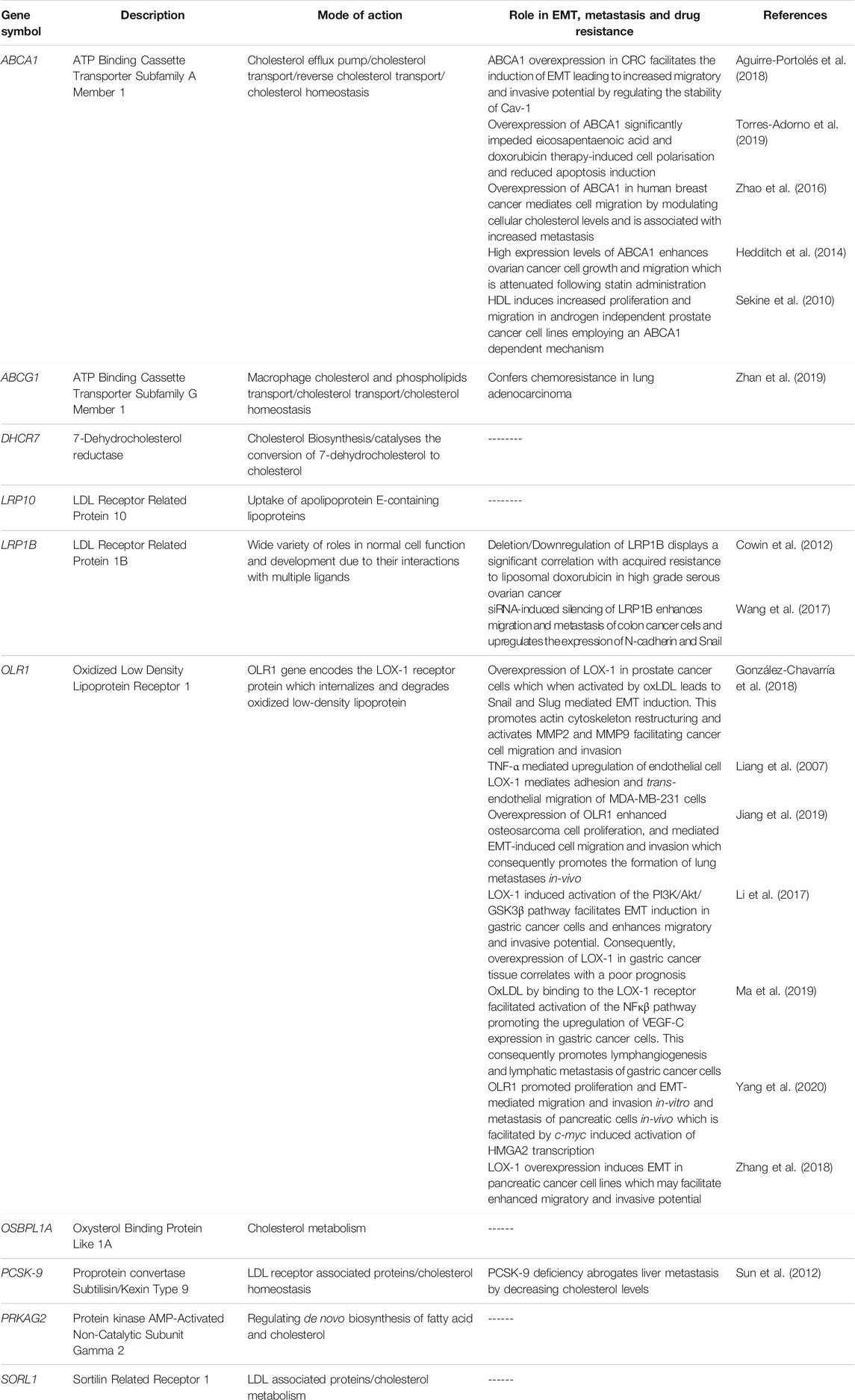
TABLE 1. Summary of cholesterol related genes and their link to EMT, metastasis and drug resistance.
Based on the above documented literature, it can be stated that cholesterol dyshomeostasis influences several EMT associated signaling pathways including Wnt, Shh and TGF-β. This is achieved by directly regulating the activity of EMT-related protein intermediates or through cholesterol’s lipid raft modulating properties. While increased cholesterol is seen to facilitate Wnt and Shh signaling, cholesterol plays a more complex role in the regulation of TGF-β signaling.
Recent scientific evidence also highlight the important role played by the tumor microenvironment in facilitating EMT and drug resistance. The following section of the review aims to discuss the significant contribution of hypoxia to EMT-mediated drug resistance. This is achieved through the direct effect of hypoxia on multidrug transporters and apoptosis impediment as well as indirectly through its immunomodulatory function in the tumour microenvironment. Furthermore, we will also attempt to shed light on mechanisms delineating the effect of hypoxia on cellular cholesterol levels and the resulting effect this has on EMT-mediated drug resistance. This will be completed with the intention of elucidating whether targeting cellular cholesterol could be employed as an efficient novel therapeutic means to combat EMT-mediated drug resistance.
Cancer cells have evolved several mechanisms to synthesize new blood vessels from pre-existing vessels through angiogenesis, and their vasculature often display abnormal characteristics such as distended capillaries, leaky walls, and irregular blood flow (Nurwidya et al., 2012). Additionally, as the cancer progresses, the rate of cell proliferation exceeds the rate of neo-angiogenesis leading to a mismatch between the supply of oxygen and its consumption by the tumor (Challapalli et al., 2017). This in turn results in decreased oxygen levels (≤2%) leading to a condition termed hypoxia (Chee et al., 2019). Hypoxia has been implicated in altering the behavior of cancer cells by manipulating various oxygen-sensitive pathways. The most comprehensively studied, is mediated by the hypoxia-inducible factor (HIF) family of TFs (Nurwidya et al., 2012). Figure 4 briefly describes the molecular mechanisms governing HIF-1α mediated regulation during both normoxic and hypoxic conditions and the resulting changes in cellular physiology that contribute to cancer cell aggressiveness.
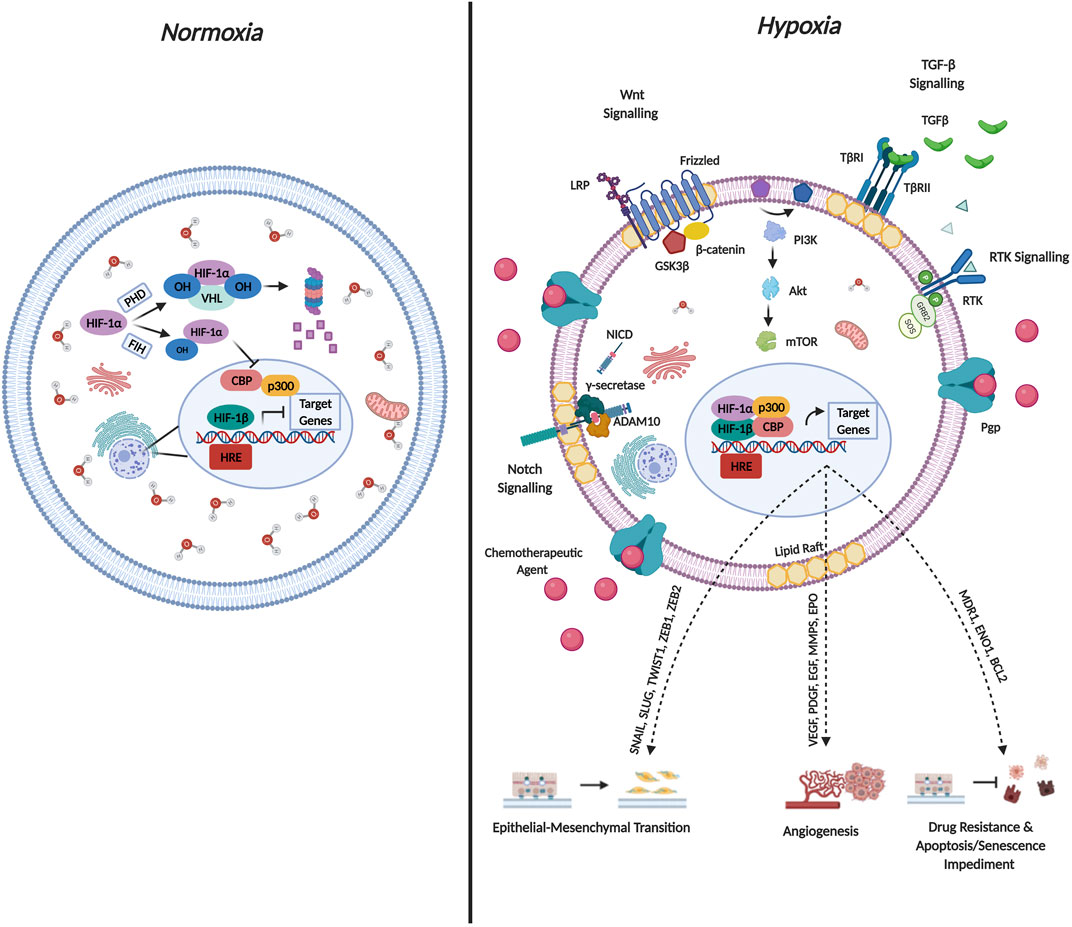
FIGURE 4. Molecular Mechanisms Governing HIF-1α Regulation and Cellular Function. Hypoxia-inducible factors (HIF’s) occur as heterodimers containing an oxygen sensitive subunit (α) and a constitutively expressed β subunit. Both subunits possess a basic helix-loop-helix (bHLH) as well as PER/ARNT/SIM (PAS) domain that is required for DNA binding and dimerization. The oxygen dependent degradation domain (ODD) of HIF-1α mediates oxygen dependent degradation. Under normoxic conditions, prolyl hydroxylase enzymes, hydroxylate HIF-1α on conserved Pro residues within the ODD facilitating von-Hippel-Lindau mediated ubiquitination and subsequent proteasomal degradation. Additionally, Asn-hydroxylation mediated by factor-inhibiting HIF-1α impedes interaction with the co-activators p300/CBP co-activator. Both mechanisms are documented to abrogate HIF transcriptional activation under normoxic conditions. Under conditions of hypoxia, the hydroxylases lose their activity, leading to HIF-1α stabilization. HIF-1α subsequently translocates to the nucleus and binds to HIF-1β as well as the p300/CBP co-activator forming a complex. The activated complex binds to multiple hypoxia-responsive elements (HREs) in the promoter regions to activate transcription of target genes. The concomitant expression of target genes mediates cancer aggressiveness through EMT and angiogenesis induction as well as drug resistance and apoptosis impediment.
In particular, HIF-1α is implicated in contributing to EMT based on its ability to increase the expression of EMT drivers namely SNAIL, ZEB1 and TWIST. Additionally, various signaling pathways described above are activated by hypoxia which are critical to EMT induction including the Notch, Wnt/β-catenin, Sonic Hedgehog as well as the TGF-β pathways (Tirpe et al., 2019). Furthermore, hypoxic conditions increase the production of EMT-induced inflammatory cytokines such as tumor-necrosis factor α (TNF-α), interleukin-6 (IL-6) as well as IL-1β (Bao et al., 2012). Interestingly, hypoxia also contributes to the acquisition of the EMT phenotype by regulating DNA methylation, histone modification as well as the expression of non-coding RNAs required for EMT induction (Choudhry and Harris, 2018).
In terms of hypoxia and drug resistance, early studies conducted by Comerford et al. identified that exposing cells to hypoxic conditions increased the mRNA expression of MDR1 by approximately 7-fold and the MDR1 gene harbored a binding site for HIF-1α (Comerford et al., 2002). Chen et al. documented that both HIF-1α and MDR1 are upregulated following hypoxic exposure (Chen et al., 2014). Interestingly, TNBC cell lines treated with either paclitaxel or gemcitabine demonstrated an increase in the levels of reactive oxygen species which consequently led to the induction of HIF-1α and HIF-2α, and subsequent transcriptional activity (Samanta et al., 2014). Accordingly, co-administration of a HIF-1 inhibitors (digoxin or acriflavine) proved effective in combatting resistance to paclitaxel/gemcitabine by impeding MDR1 expression (Samanta et al., 2014). It is thus plausible to state that a proposed mechanism for hypoxia-induced drug resistance could operate as a positive feedback loop, where exposure to hypoxia increases MDR1 expression and administering chemotherapeutic agents further support the hypoxic environment by increasing the expression of HIF-1α and HIF-2α thereby facilitating maintenance of the drug resistance phenotype.
On the other hand, sustained activation of pro-survival pathways is seen to facilitate hypoxia induced drug resistance. In hepatocellular carcinoma, the PI3K/Akt pathway is shown to mediate HIF-1α expression consequently leading to hypoxia-induced EMT and drug resistance, which in turn facilitated increased migratory and invasive potential and impeded apoptotic induction (Jiao and Nan, 2012). Therefore, targeting HIF-1α by siRNA-silencing or treatment with a HIF-1α inhibitor restored the epithelial phenotype. Importantly, treatment with LY294002 a PI3K inhibitor significantly reduced the expression of HIF-1α, and co-administering cisplatin together with a PI3K and HIF-α inhibitor led to a significant reduction in tumor volume and increased median survival time in-vivo (Jiao and Nan, 2012). A different study conducted in hepatocellular carcinoma identified a role for the AMP-activated protein kinase family member 5 (ARK5) in doxorubicin resistance (Xu et al., 2016). Higher expression of ARK5 is shown to decrease doxorubicin sensitivity through EMT induction in Huh7 and Hep3B hepatocellular carcinoma cell lines. (Xu et al., 2016).
Recent studies further elucidate a role for hypoxia in the regulation anti-apoptotic proteins and in this way contributing to drug resistance. Hypoxia has been shown to increase the expression of HIF-1α and HIF-2α as well as the downstream target gene GLUT1 and further induced EMT in malignant mesothelioma, while increasing the expression of the anti-apoptotic Bcl-2 family of proteins. This consequently impeded apoptosis induction in the absence of cisplatin treatment, and also abrogated cisplatin-mediated apoptosis (Kim M.-C. et al., 2018). In colorectal cancer a HIF-1α/miR-338-5p/IL-6 feedback circuit mediates hypoxia induced-drug resistance by increasing the secretion of IL-6 and subsequently activating the IL-6/STAT/Bcl-2 pathway resulting in decreased levels of cleaved-caspase three under hypoxic conditions (Xu et al., 2019a). By impeding the HIF-1α/miR-338-5p/IL-6 feedback loop, sensitivity to oxaliplatin is restored. Interestingly, hypoxia has also been implicated in protecting cells from drug-induced senescence, where pre-exposing MDA-MB-231 cells to hypoxia mediates resistance to various classes of chemotherapeutic agents (Sullivan et al., 2008). Consequently, pre-treating MDA-MB-231 cells with an siRNA targeting HIF-1α abolishes the hypoxia-mediated increase in doxorubicin resistance and restores senescence in transfected cells to that of non-transfected cells (Sullivan et al., 2008). Studies conducted in lung cancer have identified an interesting link between the prolyl-hydroxylase domain 3 (PHD3) protein in mediating EMT, metastasis as well as drug resistance. Authors elucidated that treating lung adenocarcinoma cells with TGF-β induces EMT, increases the expression of HIF-2α and results in a significant reduction in the PHD3 protein expression (Dopeso et al., 2018).
To sum up, the mismatch between the tumor oxygen supply and consumption leads to hypoxia. Hypoxia-mediated activation of the HIF family of TFs leads to pleiotropic changes in cellular physiology thereby promoting EMT and drug resistance. The resistant phenotype is sustained through HIF-mediated regulation of the MDR transporter as well as activation of pro-survival and anti-apoptotic proteins. Based on the above findings it is evident that hypoxia mediated induction of EMT facilitates a more aggressive cancer phenotype.
Although the relationship between hypoxia and EMT has been established, no experimental data is available thus far that sheds light on the dynamics of cholesterol in this relationship. Therefore, to explore this dynamic, we propose a possible hypothesis demonstrating that it is crucial to precisely understand the role of cholesterol in cells growing under hypoxia in an attempt to target cholesterol to combat tumorigenesis.
Acknowledging that cholesterol plays a crucial role in modulating cellular signaling, we propose that cellular cholesterol levels may be directly affected following hypoxia induced EMT to facilitate the acquisition of traits that promote tumor aggressiveness and drug resistance. This hypothesis is based on the evidence implicating hypoxia in the regulation of cholesterol biosynthesis through HIF-1α dependent and independent pathways (Riscal et al., 2019). Indeed, studies document hypoxia mediated translocation of SREBP2 to the nucleus thereby enhancing the expression as well as the activity of HMGCR (Pallottini et al., 2008). This is supported by studies highlighting increased levels of cholesterol in cells exposed to hypoxic conditions (Danza et al., 2013). Moreover, a study implicated the hypoxic tumor microenvironment in promoting SREBP transcriptional activity thereby enhancing the expression of downstream target genes that are crucial to mediating cancer cell survival and tumorigenesis (Lewis et al., 2015). Alternatively, hypoxia induced cholesterol accumulation is also potentiated by decreased cholesterol catabolism and reverse cholesterol transport (Cao et al., 2014).
Based on the fact that cholesterol is crucial to supporting the activity of several EMT-related protein intermediates, together with the observation of increased cholesterol following hypoxic induction, it can be hypothesized, that hypoxia-induced EMT may facilitate increased cellular cholesterol abundance by mediating both cholesterol synthesis and also abrogating cholesterol catabolism and efflux (Figure 5). Importantly, this hypothesis may be supported by studies now showing that exposing cancer cells to hypoxic conditions promotes the coalescence of lipid rafts leading to the formation of large liquid-ordered domains that facilitate compartmentalization of several proteins promoting activation of EMT-related signaling pathways (Danza et al., 2013).
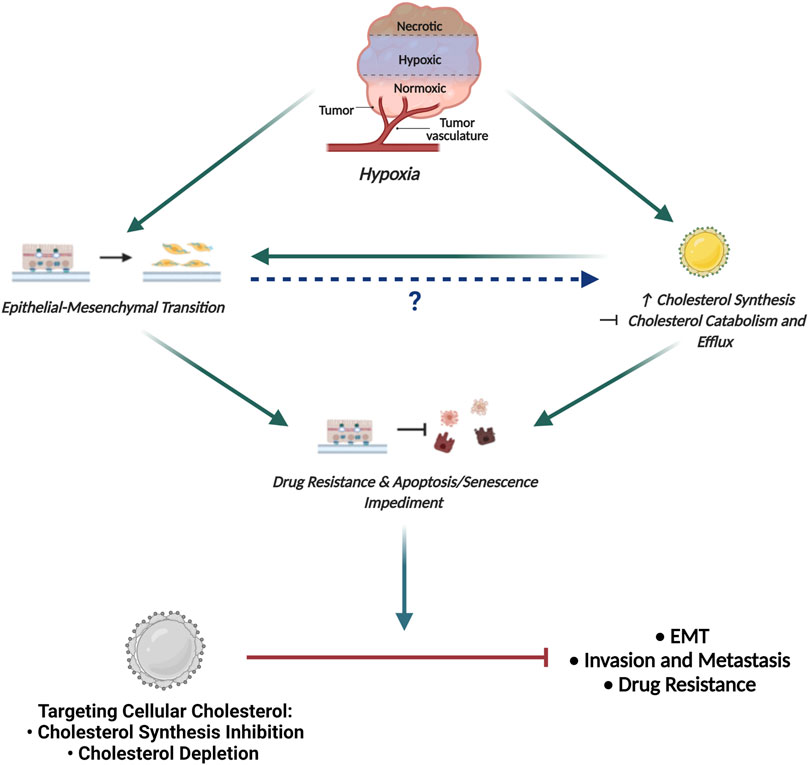
FIGURE 5. Proposed Model Delineating the link between Hypoxia, EMT and Cholesterol in Cancer. The hypoxic tumour microenvironment facilitates EMT induction and promotes increased cellular cholesterol availability by enhancing mechanisms associated with cholesterol synthesis and impeding cholesterol catabolism and efflux. While increased cholesterol content is seen to fuel EMT, how EMT affects the cellular cholesterol profile has not been delineated. Importantly, both EMT and increased cholesterol content is seen to enhance drug resistance and impede apoptotic and senescence induction. We propose that targeting cellular cholesterol, either through cholesterol synthesis inhibition or depletion, will combat EMT, decrease the invasive and metastatic potential of cells and also restore sensitivity to conventional chemotherapeutic agents.
By modulating lipid raft cholesterol content, EMT-associated signaling pathways are consequently affected leading to invasion and metastasis (Morandi et al., 2017). Additionally, the proposed increase in cholesterol content following EMT induction may also be implicated in altering membrane fluidity conferring cancer cells with properties necessary for cancer invasion (Morandi et al., 2017). The increase in lipid raft cholesterol following hypoxia-induced EMT is hence clearly justified, implicating cholesterol in not only directly modulating EMT associated signaling intermediates but also modifying the physical properties of the membrane to support EMT.
Furthermore, exposure to hypoxic conditions is also seen to increase lipid droplet content in cancer cells (Koizume and Miyagi, 2016). Seeing that lipid droplets serve as major storage organelles for CEs, it can be hypothesized that the increased lipid droplet content post hypoxia-induced EMT mediates an increased cellular availability of CEs. Importantly, the increase in CE lipid droplet content is also emerging as a common mechanism employed by advanced cancers to fuel cancer aggressiveness (Yue et al., 2014; Li et al., 2016). Excess of free cholesterol is toxic to cells, accordingly, storing cellular cholesterol as CEs will reduce the energetic burden on cancer cells to synthesize cholesterol de novo. The availability of cholesterol will further prove beneficial to the cell to facilitate membrane biogenesis and alter lipid raft dynamics thus influencing cancer cell signaling (De Gonzalo-Calvo et al., 2015). Consequently, increased levels of CEs have also been associated with increased cancer cell migration and enhanced metastatic potential (Lee et al., 2018).
Accordant with the increased cellular content, membrane lipid raft cholesterol and CEs in cells that possess an aggressive cancer phenotype, it can be hypothesized that cells post-EMT induction may display increased sensitivity to cholesterol targeting agents. This can be corroborated with findings in other studies that elucidate the beneficial outcome associated with employing cholesterol targeting agents to reduce the metastatic potential of cells by decreasing the migratory and invasive potential of cells (Murai et al., 2011; Guerra et al., 2016). This is evidenced by studies reporting that the disruption of lipid raft integrity facilitates the shedding of key cell surface receptor CD44 that cancer cells depend on for conferring an aggressive disease phenotype (Murai et al., 2011). Furthermore, several cancer stem cell and EMT related pathways are also inhibited, which facilitates an increased expression of key epithelial markers while reducing the expression of mesenchymal markers (Guerra et al., 2016; Badana et al., 2018; Sharma et al., 2019; Wu et al., 2020). It can thus be stated that cholesterol targeting agents may effectively combat EMT and metastasis and prevent the acquisition of an aggressive disease phenotype.
Seeing that cholesterol dyshomeostasis is crucial to supporting the activity of several protein intermediates in EMT-related signaling coupled with the observation of increased cellular cholesterol content in cells that have been exposed to hypoxic conditions, it can be proposed that cells post hypoxia-induced EMT should be more susceptible to cholesterol targeting agents. Consequently, targeting cellular cholesterol to combat breast cancer pathogenesis seems like a rational avenue to explore in an attempt to combat tumorigenesis (Figure 5).
To elucidate the crucial role of cholesterol as well as its metabolites in regulating cellular function, and how the deregulation in cholesterol metabolism mediate tumorigenesis and drug resistance is of key importance. Whereas some cancer types are dependent on increased cholesterol uptake to mediate resistance, other cancer types exploit cellular mechanisms of uptake and efflux to meet the increasing metabolic demands.
The abundance of cholesterol in addition to directly regulating protein intermediates, promotes an increase in the presence of lipid rafts which serve as key signaling hubs crucial for EMT induction. Seeing that the induction of EMT is associated with increased drug resistant potential, it can be inferred that cholesterol through its EMT-modulatory effects promotes increased drug resistance potential. Additionally, deregulated cholesterol metabolism is also seen to directly contribute to drug resistance by conferring cells with chemoresistant traits. This is achieved by cholesterol-mediated regulation of pro-survival and anti-apoptotic pathways. Additionally, based on the crucial role cholesterol plays in maintaining lipid raft integrity, evidence exists implicating lipid rafts as key domains for maintaining the activity of MDR transporters. This therefore points to the multifaceted role of cholesterol in promoting EMT and mediating cancer drug resistance.
Consequently, attempts made at targeting cellular cholesterol by employing cholesterol synthesis inhibition to restore sensitivity have been widely exploited in the laboratory setting. This has predominantly been achieved by employing the most widely used lipid lowering agents on the market, i.e. statins (Kuo et al., 2020). While employing cholesterol synthesis inhibition to reduce cellular cholesterol levels and enhance drug sensitivity has proven effective in a laboratory setting, the debilitating side effects associated with statin therapy is seen as a major limitation in a clinical setting. Common side effects include and is not limited to: muscle pains, hepato- and renal-toxicity, neurocognitive decline, myopathy as well as idiopathic polyneuropathy (Gu et al., 2019). Importantly, 30% of patient population display non-responsiveness to statin therapy (Ward et al., 2019). This could be attributed to the long-term impediment of cholesterol synthesis in cells where cholesterol plays an integral role in regulating essential cellular processes. Consequently, the discovery of new cholesterol lowering agents is pivotal to combat drug resistance. Based on the crucial role of altered cholesterol metabolism in drug resistance, screening additional compounds that either as single agents or in combination with anti-cancer therapies will restore drug sensitivity, seems like a promising avenue to pursue. Additionally, further insight is required to delineate the precise role that cholesterol plays in influencing the major components of the tumor microenvironment. This consequently highlights the incipient need to develop models that more closely mimic human immune response and the human tumor microenvironment in general. It is important to emphasize, while most of the afore-mentioned studies target cholesterol synthesis as a means to reduce cellular cholesterol levels, and consequently enhance drug sensitivity, studies conducted employing direct membrane cholesterol depletion intended for drug sensitivity restoration are lacking. Consequently, studies conducted in our laboratory currently seek to delineate the effects of direct membrane cholesterol depletion as a means to target cellular cholesterol and restore drug sensitivity and compare this approach with cholesterol synthesis inhibition strategies. By conducting these studies, a better understanding of cholesterol depletion as a strategy to restore sensitivity will be portrayed, which will further assist in addressing whether membrane cholesterol depletion rather than cholesterol synthesis inhibition could serve as a better approach to enhance drug sensitivity.
Additionally, hypoxia plays an important role in contributing to cancer drug resistance, with the major focus in this review as an inducer of EMT. Following hypoxic exposure, induction of the cellular EMT program is achieved by regulating the expression of master TFs, enhancing the production of EMT-induced inflammatory cytokines, and also regulating epigenetic mechanisms that are linked with EMT induction. It is important to emphasize that cells traverse a spectrum of intermediate states along the E-M axis and may adopt an infinite number of possible states that are dictated by the expression of various EMT markers. Interestingly, the existence in a hybrid state occurs more frequently than expected conferring upon cancer cells dynamic cellular plasticity leading to poor clinical outcome. Further experimental investigation is required to elucidate what conditions dictate whether a cell will undergo a complete EMT or exists in a hybrid state. Importantly, the establishment of molecular markers that can be utilized to distinguish the intermediate states from complete EMT is also required. This will generate a molecular signature that may assist in predicting survival and treatment response in a clinical setting. Correspondingly, studies are required to address whether a possible link exists between a specific hybrid state and drug resistance potential. Importantly, due to inherent cellular plasticity, the occurrence of both inter- and intra-tumoural heterogeneity is inevitable. This heterogeneity facilitates the existence of specific EMT hybrid states within the same niche where different areas of a tumor will be better adapted to deal with environmental pressures as opposed to areas that are not. This will serve as a platform for the selection and outgrowth of the well adapted subtypes consequently promoting metastatic dissemination and relapse. Mouse models designed to study EMT will help in addressing the identification of specific EMT states during tumor development to allow for directed targeting and eradication of resistant cell subtypes.
Furthermore, exposing cells to hypoxic conditions also contributes to drug resistance by influencing the expression and/or the activity of MDR transporters, regulating the expression of anti-apoptotic proteins, protecting cells from drug-induced senescence. Consequently, attempting to understand and delineate the integrated cell signaling biology with hypoxic conditions is crucial to better understand the beneficial outcome associated with targeting hypoxia to improve patient outcome in a clinical setting.
In conclusion, it can be stated that an intricate relationship exists between hypoxia, EMT and cholesterol that governs cancer drug resistance. While studies elucidating the link between EMT, hypoxia and resistance are well-established, studies focusing on the link between cholesterol, EMT and drug resistance are lacking. Given the importance of cholesterol dyshomeostasis in cancer initiation and progression, as well as the observation of increased cellular cholesterol in cells following EMT induction, targeting cellular cholesterol to combat breast cancer pathogenesis may be a promising direction to pursue in the search for novel anticancer therapeutics.
MK and NA conceived, designed, and wrote the review. All authors edited and critically evaluated the manuscript. NA designed figures. MK and CV contributed to the figures and the design of the table.
The authors thank National Research Foundation (NRF), South Africa, and the Swedish Foundation for International Cooperation in Research and Higher Education (STINT), Sweden for providing financial support for the project to MK (UID: 118720) and CV (SA 2018-7661). CV was also supported by a research grant from the Swedish Research Council (VR, 2017-03056) and the Swedish Cancer Society (Cancerfonden, CAN 2018/875). NA was supported with a bursary from NRF, South Africa (Innovations Master’s Scholarship (UID: 121480).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
All figures were created using Biorender.com.
Abba, M., Patil, N., Leupold, J., and Allgayer, H. (2016). MicroRNA Regulation of Epithelial to Mesenchymal Transition. Jcm 5, 8. doi:10.3390/jcm5010008
Aguirre‐Portolés, C., Feliu, J., Reglero, G., and Ramírez de Molina, A. (2018). ABCA1 Overexpression Worsens Colorectal Cancer Prognosis by Facilitating Tumour Growth and Caveolin‐1‐dependent Invasiveness, and These Effects Can Be Ameliorated Using the BET Inhibitor Apabetalone. Mol. Oncol. 12, 1735–1752. doi:10.1002/1878-0261.12367
Ahmadi, A., Najafi, M., Farhood, B., and Mortezaee, K. (2019). Transforming Growth Factor‐β Signaling: Tumorigenesis and Targeting for Cancer Therapy. J. Cel Physiol 234, 12173–12187. doi:10.1002/jcp.27955
Alam, A., Kowal, J., Broude, E., Roninson, I., and Locher, K. P. (2019). Structural Insight into Substrate and Inhibitor Discrimination by Human P-Glycoprotein. Science 363, 753–756. doi:10.1126/Science.Aav7102
Badana, A. K., Chintala, M., Gavara, M. M., Naik, S., Kumari, S., Kappala, V. R., et al. (2018). Lipid Rafts Disruption Induces Apoptosis by Attenuating Expression of LRP6 and Survivin in Triple Negative Breast Cancer. Biomed. Pharmacother. 97, 359–368. doi:10.1016/j.biopha.2017.10.045
Bao, B., Azmi, A. S., Ali, S., Ahmad, A., Li, Y., Banerjee, S., et al. (2012). The Biological Kinship of Hypoxia with CSC and EMT and Their Relationship with Deregulated Expression of miRNAs and Tumor Aggressiveness. Biochim. Biophys. Acta (Bba) - Rev. Cancer 1826, 272–296. doi:10.1016/j.bbcan.2012.04.008
Batlle, E., Sancho, E., Francí, C., Domínguez, D., Monfar, M., Baulida, J., et al. (2000). The Transcription Factor Snail Is a Repressor of E-Cadherin Gene Expression in Epithelial Tumour Cells. Nat. Cel Biol. 2, 84–89. doi:10.1038/35000034
Bhat, M., Skill, N., Marcus, V., Deschenes, M., Tan, X., Bouteaud, J., et al. (2015). Decreased PCSK9 Expression in Human Hepatocellular Carcinoma. BMC Gastroenterol. 15, 176. doi:10.1186/s12876-015-0371-6
Bidet, M., Joubert, O., Lacombe, B., Ciantar, M., Nehmé, R., Mollat, P., et al. (2011). The Hedgehog Receptor Patched Is Involved in Cholesterol Transport. PloS one 6, e23834. doi:10.1371/journal.pone.0023834
Bindels, S., Mestdagt, M., Vandewalle, C., Jacobs, N., Volders, L., Noël, A., et al. (2006). Regulation of Vimentin by SIP1 in Human Epithelial Breast Tumor Cells. Oncogene 25, 4975–4985. doi:10.1038/sj.onc.1209511
Blassberg, R., and Jacob, J. (2017). Lipid Metabolism Fattens up Hedgehog Signaling. BMC Biol. 15, 95. doi:10.1186/s12915-017-0442-y
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer J. Clinicians 68, 394–424. doi:10.3322/caac.21492
Bukowski, K., Kciuk, M., and Kontek, R. (2020). Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Ijms 21, 3233. doi:10.3390/ijms21093233
Callahan, B., and Wang, C. (2015). Hedgehog Cholesterolysis: Specialized Gatekeeper to Oncogenic Signaling. Cancers 7, 2037–2053. doi:10.3390/cancers7040875
Cano, A., Pérez-Moreno, M. A., Rodrigo, I., Locascio, A., Blanco, M. J., Del Barrio, M. G., et al. (2000). The Transcription Factor Snail Controls Epithelial-Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cel Biol. 2, 76–83. doi:10.1038/35000025
Cao, R., Zhao, X., Li, S., Zhou, H., Chen, W., Ren, L., et al. (2014). Hypoxia Induces Dysregulation of Lipid Metabolism in HepG2 Cells via Activation of HIF-2a. Cell. Physiol. Biochem. 34, 1427–1441. doi:10.1159/000366348
Challapalli, A., Carroll, L., and Aboagye, E. O. (2017). Molecular Mechanisms of Hypoxia in Cancer. Clin. Transl Imaging 5, 225–253. doi:10.1007/s40336-017-0231-1
Chee, N. T., Lohse, I., and Brothers, S. P. (2019). mRNA-To-protein Translation in Hypoxia. Mol. Cancer 18, 49. doi:10.1186/s12943-019-0968-4
Chen, J., Ding, Z., Peng, Y., Pan, F., Li, J., Zou, L., et al. (2014). HIF-1α Inhibition Reverses Multidrug Resistance in Colon Cancer Cells via Downregulation of MDR1/P-Glycoprotein. PLoS One 9, e98882. doi:10.1371/journal.pone.0098882
Chen, Q., Pan, Z., Zhao, M., Wang, Q., Qiao, C., Miao, L., et al. (2018). High Cholesterol in Lipid Rafts Reduces the Sensitivity to EGFR‐TKI Therapy in Non‐small Cell Lung Cancer. J. Cel. Physiol. 233, 6722–6732. doi:10.1002/jcp.26351
Choudhry, H., and Harris, A. L. (2018). Advances in Hypoxia-Inducible Factor Biology. Cel Metab. 27, 281–298. doi:10.1016/j.cmet.2017.10.005
Chruścik, A., Gopalan, V., and Lam, A. K.-Y. (2018). The Clinical and Biological Roles of Transforming Growth Factor Beta in colon Cancer Stem Cells: A Systematic Review. Eur. J. Cel Biol. 97, 15–22. doi:10.1016/j.ejcb.2017.11.001
Clouser, A. F., Alam, Y. H., and Atkins, W. M. (2021). Cholesterol Asymmetrically Modulates the Conformational Ensemble of the Nucleotide-Binding Domains of P-Glycoprotein in Lipid Nanodiscs. Biochemistry 60, 85–94. doi:10.1021/acs.biochem.0c00824
Comerford, K. M., Wallace, T. J., Karhausen, J., Louis, N. A., Montalto, M. C., and Colgan, S. P. (2002). Hypoxia-inducible Factor-1-dependent Regulation of the Multidrug Resistance (MDR1) Gene. Cancer Res. 62, 3387–3394.
Cowin, P. A., George, J., Fereday, S., Loehrer, E., Van Loo, P., Cullinane, C., et al. (2012). LRP1B Deletion in High-Grade Serous Ovarian Cancers Is Associated with Acquired Chemotherapy Resistance to Liposomal Doxorubicin. Cancer Res. 72, 4060–4073. doi:10.1158/0008-5472.CAN-12-0203
Craene, B. D., and Berx, G. (2013). Regulatory Networks Defining EMT during Cancer Initiation and Progression. Nat. Rev. Cancer 13, 97–110. doi:10.1038/nrc3447
Criscuolo, D., Avolio, R., Calice, G., Laezza, C., Paladino, S., Navarra, G., et al. (2020). Cholesterol Homeostasis Modulates Platinum Sensitivity in Human Ovarian Cancer. Cells 9, 828. doi:10.3390/cells9040828
Cruz, P. M. R., Mo, H., Mcconathy, W. J., Sabnis, N., and Lacko, A. G. (2013). The Role of Cholesterol Metabolism and Cholesterol Transport in Carcinogenesis: a Review of Scientific Findings, Relevant to Future Cancer Therapeutics. Front. Pharmacol. 4, 119. doi:10.3389/fphar.2013.00119
Danza, G., Di Serio, C., Ambrosio, M. R., Sturli, N., Lonetto, G., Rosati, F., et al. (2013). Notch3 Is Activated by Chronic Hypoxia and Contributes to the Progression of Human Prostate Cancer. Int. J. Cancer 133, 2577–2586. doi:10.1002/ijc.28293
Das, A., Brown, M. S., Anderson, D. D., Goldstein, J. L., and Radhakrishnan, A. (2014). Three Pools of Plasma Membrane Cholesterol and Their Relation to Cholesterol Homeostasis. eLife 3, e02882. doi:10.7554/eLife.02882
Das, V., Bhattacharya, S., Chikkaputtaiah, C., Hazra, S., and Pal, M. (2019). The Basics of Epithelial-Mesenchymal Transition (EMT): A Study from a Structure, Dynamics, and Functional Perspective. J. Cel Physiol 234, 14535–14555. doi:10.1002/jcp.28160
De Gonzalo-Calvo, D., López-Vilaró, L., Nasarre, L., Perez-Olabarria, M., Vázquez, T., Escuin, D., et al. (2015). Intratumor Cholesteryl Ester Accumulation Is Associated with Human Breast Cancer Proliferation and Aggressive Potential: a Molecular and Clinicopathological Study. BMC Cancer 15, 460. doi:10.1186/s12885-015-1469-5
Derynck, R., and Budi, E. H. (2019). Specificity, Versatility, and Control of TGF-β Family Signaling. Sci. Sig. 12, eaav5183. doi:10.1126/scisignal.aav5183
Derynck, R., and Weinberg, R. A. (2019). EMT and Cancer: More Than Meets the Eye. Develop. Cel 49, 313–316. doi:10.1016/j.devcel.2019.04.026
Dong, C., Wu, Y., Yao, J., Wang, Y., Yu, Y., Rychahou, P. G., et al. (2012). G9a Interacts with Snail and Is Critical for Snail-Mediated E-Cadherin Repression in Human Breast Cancer. J. Clin. Invest. 122, 1469–1486. doi:10.1172/JCI57349
Dongre, A., and Weinberg, R. A. (2019). New Insights into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cel Biol. 20, 69–84. doi:10.1038/s41580-018-0080-4
Dopeso, H., Jiao, H.-K., Cuesta, A. M., Henze, A.-T., Jurida, L., Kracht, M., et al. (2018). PHD3 Controls Lung Cancer Metastasis and Resistance to EGFR Inhibitors through TGFα. Cancer Res. 78, 1805–1819. doi:10.1158/0008-5472.CAN-17-1346
Du, B., and Shim, J. (2016). Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 21, 965. doi:10.3390/molecules21070965
Esau, L., Sagar, S., Bangarusamy, D., and Kaur, M. (2016). Identification of CETP as a Molecular Target for Estrogen Positive Breast Cancer Cell Death by Cholesterol Depleting Agents. Genes Cancer 7, 309–322. doi:10.18632/genesandcancer.122
Fu, J., Qin, L., He, T., Qin, J., Hong, J., Wong, J., et al. (2011). The TWIST/Mi2/NuRD Protein Complex and its Essential Role in Cancer Metastasis. Cell Res 21, 275–289. doi:10.1038/cr.2010.118
Fuxe, J., Vincent, T., and Garcia De Herreros, A. (2010). Transcriptional Crosstalk between TGFβ and Stem Cell Pathways in Tumor Cell Invasion: Role of EMT Promoting Smad Complexes. Cell Cycle 9, 2363–2374. doi:10.4161/cc.9.12.12050
Gallagher, E. J., Zelenko, Z., Neel, B. A., Antoniou, I. M., Rajan, L., Kase, N., et al. (2017). Elevated Tumor LDLR Expression Accelerates LDL Cholesterol-Mediated Breast Cancer Growth in Mouse Models of Hyperlipidemia. Oncogene 36, 6462–6471. doi:10.1038/onc.2017.247
Georgakopoulos-Soares, I., Chartoumpekis, D. V., Kyriazopoulou, V., and Zaravinos, A. (2020). EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 10, 499. doi:10.3389/fonc.2020.00499
Glodkowska-Mrowka, E., Mrowka, P., Basak, G. W., Niesiobedzka-Krezel, J., Seferynska, I., Wlodarski, P. K., et al. (2014). Statins Inhibit ABCB1 and ABCG2 Drug Transporter Activity in Chronic Myeloid Leukemia Cells and Potentiate Antileukemic Effects of Imatinib. Exp. Hematol. 42, 439–447. doi:10.1016/j.exphem.2014.02.006
González-Chavarría, I., Fernandez, E., Gutierrez, N., González-Horta, E. E., Sandoval, F., Cifuentes, P., et al. (2018). LOX-1 Activation by oxLDL Triggers an Epithelial Mesenchymal Transition and Promotes Tumorigenic Potential in Prostate Cancer Cells. Cancer Lett. 414, 34–43. doi:10.1016/j.canlet.2017.10.035
Gu, L., Saha, S. T., Thomas, J., and Kaur, M. (2019). Targeting Cellular Cholesterol for Anticancer Therapy. FEBS J. 286, 4192–4208. doi:10.1111/febs.15018
Guerra, F. S., Sampaio, L. D. S., Konig, S., Bonamino, M., Rossi, M. I. D., Costa, M. L., et al. (2016). Membrane Cholesterol Depletion Reduces Breast Tumor Cell Migration by a Mechanism that Involves Non-canonical Wnt Signaling and IL-10 Secretion. Transl. Med. Commun. 1, 3. doi:10.1186/s41231-016-0002-4
Guillaumond, F., Bidaut, G., Ouaissi, M., Servais, S., Gouirand, V., Olivares, O., et al. (2015). Cholesterol Uptake Disruption, in Association with Chemotherapy, Is a Promising Combined Metabolic Therapy for Pancreatic Adenocarcinoma. Proc. Natl. Acad. Sci. USA 112, 2473–2478. doi:10.1073/pnas.1421601112
Gupta, V. K., Sharma, N. S., Kesh, K., Dauer, P., Nomura, A., Giri, B., et al. (2018). Metastasis and Chemoresistance in CD133 Expressing Pancreatic Cancer Cells Are Dependent on Their Lipid Raft Integrity. Cancer Lett. 439, 101–112. doi:10.1016/j.canlet.2018.09.028
Haider, T., Pandey, V., Banjare, N., Gupta, P. N., and Soni, V. (2020). Drug Resistance in Cancer: Mechanisms and Tackling Strategies. Pharmacol. Rep. 72, 1125–1151. doi:10.1007/s43440-020-00138-7
Hajra, K. M., Chen, D. Y., and Fearon, E. R. (2002). The SLUG Zinc-Finger Protein Represses E-Cadherin in Breast Cancer. Cancer Res. 62, 1613–1618.
Hampton, R. Y. (2002). ER-associated Degradation in Protein Quality Control and Cellular Regulation. Curr. Opin. Cel Biol. 14, 476–482. doi:10.1016/s0955-0674(02)00358-7
He, M., Zhang, W., Dong, Y., Wang, L., Fang, T., Tang, W., et al. (2017). Pro-inflammation NF-Κb Signaling Triggers a Positive Feedback via Enhancing Cholesterol Accumulation in Liver Cancer Cells. J. Exp. Clin. Cancer Res. 36, 15. doi:10.1186/s13046-017-0490-8
Hedditch, E. L., Gao, B., Russell, A. J., Lu, Y., Emmanuel, C., Beesley, J., et al. (2014). ABCA Transporter Gene Expression and Poor Outcome in Epithelial Ovarian Cancer. JNCI 106, dju149. doi:10.1093/jnci/dju149
Hegedüs, C., Telbisz, Á., Hegedűs, T., Sarkadi, B., and Özvegy-Laczka, C. (2015). “Lipid Regulation of the ABCB1 and ABCG2 Multidrug Transporters,” in Adv Cancer Res. Editors J. D. Schuetz,, and T. Ishikawa (Academic Press), 97–137. doi:10.1016/bs.acr.2014.10.004
Herranz, N., Pasini, D., Díaz, V. M., Francí, C., Gutierrez, A., Dave, N., et al. (2008). Polycomb Complex 2 Is Required for E-Cadherin Repression by the Snail1 Transcription Factor. Mol. Cel. Biol. 28, 4772–4781. doi:10.1128/MCB.00323-08
Hou, Z., Peng, H., Ayyanathan, K., Yan, K.-P., Langer, E. M., Longmore, G. D., et al. (2008). The LIM Protein AJUBA Recruits Protein Arginine Methyltransferase 5 to Mediate SNAIL-dependent Transcriptional Repression. Mol. Cel. Biol. 28, 3198–3207. doi:10.1128/MCB.01435-07
Housman, G., Byler, S., Heerboth, S., Lapinska, K., Longacre, M., Snyder, N., et al. (2014). Drug Resistance in Cancer: an Overview. Cancers 6, 1769–1792. doi:10.3390/cancers6031769
Howell, M. C., Green, R., Khalil, R., Foran, E., Quarni, W., Nair, R., et al. (2020). Lung Cancer Cells Survive Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Exposure through Upregulation of Cholesterol Synthesis. FASEB BioAdvances 2, 90–105. doi:10.1096/fba.2019-00081
Hwang, K.-E., Kwon, S.-J., Kim, Y.-S., Park, D.-S., Kim, B.-R., Yoon, K.-H., et al. (2014). Effect of Simvastatin on the Resistance to EGFR Tyrosine Kinase Inhibitors in a Non-small Cell Lung Cancer with the T790M Mutation of EGFR. Exp. Cel Res. 323, 288–296. doi:10.1016/j.yexcr.2014.02.026
Ikenouchi, J., Matsuda, M., Furuse, M., and Tsukita, S. (2003). Regulation of Tight Junctions during the Epithelium-Mesenchyme Transition:direct Repression of the Gene Expression of Claudins/occludin by Snail. J. Cel Sci 116, 1959–1967. doi:10.1242/jcs.00389
Jethwa, P., Naqvi, M., Hardy, R. G., Hotchin, N. A., Roberts, S., Spychal, R., et al. (2008). Overexpression of Slug Is Associated with Malignant Progression of Esophageal Adenocarcinoma. Wjg 14, 1044–1052. doi:10.3748/wjg.14.1044
Jiang, L., Jiang, S., Zhou, W., Huang, J., Lin, Y., Long, H., et al. (2019). Oxidized Low Density Lipoprotein Receptor 1 Promotes Lung Metastases of Osteosarcomas through Regulating the Epithelial-Mesenchymal Transition. J. Transl Med. 17, 369. doi:10.1186/s12967-019-2107-9
Ke, X., and Shen, L. (2017). Molecular Targeted Therapy of Cancer: The Progress and Future prospect. Front. Lab. Med. 1, 69–75. doi:10.1016/j.flm.2017.06.001
Kim, M.-C., Hwang, S.-H., Kim, N.-Y., Lee, H.-S., Ji, S., Yang, Y., et al. (2018a). Hypoxia Promotes Acquisition of Aggressive Phenotypes in Human Malignant Mesothelioma. BMC Cancer 18, 819. doi:10.1186/s12885-018-4720-z
Kim, S., Lee, M., Dhanasekaran, D. N., and Song, Y. S. (2018b). Activation of LXRɑ/β by Cholesterol in Malignant Ascites Promotes Chemoresistance in Ovarian Cancer. BMC cancer 18, 1232. doi:10.1186/s12885-018-5152-5
Koizume, S., and Miyagi, Y. (2016). Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Ijms 17, 1430. doi:10.3390/ijms17091430
Kong, Y., Cheng, L., Mao, F., Zhang, Z., Zhang, Y., Farah, E., et al. (2018). Inhibition of Cholesterol Biosynthesis Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer (CRPC). J. Biol. Chem. 293, 14328–14341. doi:10.1074/jbc.RA118.004442
Kopecka, J., Trouillas, P., Gašparović, A. Č., Gazzano, E., Assaraf, Y. G., and Riganti, C. (2020). Phospholipids and Cholesterol: Inducers of Cancer Multidrug Resistance and Therapeutic Targets. Drug Resist. Updates 49, 100670. doi:10.1016/j.drup.2019.100670
Kuo, T.-T., Huang, Y.-B., and Hsieh, C.-J. (2020). Consumption and Market Share of Cholesterol-Lowering Drugs in High-Risk Patients before and after the Release of the 2013 ACC/AHA Cholesterol Guidelines: a Retrospective Observational Study. BMJ Open 10, e036769. doi:10.1136/bmjopen-2020-036769
Kuzu, O. F., Noory, M. A., and Robertson, G. P. (2016). The Role of Cholesterol in Cancer. Cancer Res. 76, 2063–2070. doi:10.1158/0008-5472.CAN-15-2613
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cel Biol. 15, 178–196. doi:10.1038/nrm3758
Lee, H. J., Li, J., Vickman, R. E., Li, J., Liu, R., Durkes, A. C., et al. (2018). Cholesterol Esterification Inhibition Suppresses Prostate Cancer Metastasis by Impairing the Wnt/β-Catenin Pathway. Mol. Cancer Res. 16, 974–985. doi:10.1158/1541-7786.MCR-17-0665
Lewis, C. A., Brault, C., Peck, B., Bensaad, K., Griffiths, B., Mitter, R., et al. (2015). SREBP Maintains Lipid Biosynthesis and Viability of Cancer Cells under Lipid- and Oxygen-Deprived Conditions and Defines a Gene Signature Associated with Poor Survival in Glioblastoma Multiforme. Oncogene 34, 5128–5140. doi:10.1038/onc.2014.439
Li, C., Zhang, J., Wu, H., Li, L., Yang, C., Song, S., et al. (2017). Lectin-like Oxidized Low-Density Lipoprotein Receptor-1 Facilitates Metastasis of Gastric Cancer through Driving Epithelial-Mesenchymal Transition and PI3K/Akt/GSK3β Activation. Sci. Rep. 7, 45275. doi:10.1038/srep45275
Li, J., Gu, D., Lee, S. S.-Y., Song, B., Bandyopadhyay, S., Chen, S., et al. (2016). Abrogating Cholesterol Esterification Suppresses Growth and Metastasis of Pancreatic Cancer. Oncogene 35, 6378–6388. doi:10.1038/onc.2016.168
Li, J., Qu, X., Tian, J., Zhang, J.-T., and Cheng, J.-X. (2018). Cholesterol Esterification Inhibition and Gemcitabine Synergistically Suppress Pancreatic Ductal Adenocarcinoma Proliferation. PLoS One 13, e0193318. doi:10.1371/journal.pone.0193318
Liang, M., Zhang, P., and Fu, J. (2007). Up-regulation of LOX-1 Expression by TNF-α Promotes Trans-endothelial Migration of MDA-MB-231 Breast Cancer Cells. Cancer Lett. 258, 31–37. doi:10.1016/j.canlet.2007.08.003
Liao, T.-T., and Yang, M.-H. (2020). Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 9, 623. doi:10.3390/cells9030623
Lin, Y., Wu, Y., Li, J., Dong, C., Ye, X., Chi, Y.-I., et al. (2010). The SNAG Domain of Snail1 Functions as a Molecular Hook for Recruiting Lysine-specific Demethylase 1. Embo J. 29, 1803–1816. doi:10.1038/emboj.2010.63
Luchetti, G., Sircar, R., Kong, J. H., Nachtergaele, S., Sagner, A., Byrne, E. F., et al. (2016). Cholesterol Activates the G-Protein Coupled Receptor Smoothened to Promote Hedgehog Signaling. eLife 5, e20304. doi:10.7554/eLife.20304
Ma, C., Xie, J., Luo, C., Yin, H., Li, R., Wang, X., et al. (2019). OxLDL Promotes Lymphangiogenesis and Lymphatic Metastasis in Gastric Cancer by Upregulating VEGF-C E-xpression and S-ecretion. Int. J. Oncol. 54, 572–584. doi:10.3892/ijo.2018.4648
Ma, X., Wang, Q., Jiang, Y., Xiao, Z., Fang, X., and Chen, Y.-G. (2007). Lateral Diffusion of TGF-β Type I Receptor Studied by Single-Molecule Imaging. Biochem. Biophysical Res. Commun. 356, 67–71. doi:10.1016/j.bbrc.2007.02.080
Mandal, C. C., and Rahman, M. M. (2014). Targeting Intracellular Cholesterol Is a Novel Therapeutic Strategy for Cancer Treatment. J. Cancer Sci. Ther. 6, 510–513. doi:10.4172/1948-5956.1000316
Martínez-Estrada, O. M., Cullerés, A., Soriano, F. X., Peinado, H., Bolós, V., Martínez, F. O., et al. (2006). The Transcription Factors Slug and Snail Act as Repressors of Claudin-1 Expression in Epithelial Cells1. Biochem. J. 394, 449–457. doi:10.1042/BJ20050591
Meng, J., Chen, S., Han, J.-X., Qian, B., Wang, X.-R., Zhong, W.-L., et al. (2018). Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 78, 4150–4162. doi:10.1158/0008-5472.CAN-17-3009
Mollinedo, F., and Gajate, C. (2015). Lipid Rafts as Major Platforms for Signaling Regulation in Cancer. Adv. Biol. Regul. 57, 130–146. doi:10.1016/j.jbior.2014.10.003
Morandi, A., Taddei, M. L., Chiarugi, P., and Giannoni, E. (2017). Targeting the Metabolic Reprogramming that Controls Epithelial-To-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 7, 40. doi:10.3389/fonc.2017.00040
Murai, T. (2015). Cholesterol Lowering: Role in Cancer Prevention and Treatment. Biol. Chem. 396, 1–11. doi:10.1515/hsz-2014-0194
Murai, T., Maruyama, Y., Mio, K., Nishiyama, H., Suga, M., and Sato, C. (2011). Low Cholesterol Triggers Membrane Microdomain-dependent CD44 Shedding and Suppresses Tumor Cell Migration. J. Biol. Chem. 286, 1999–2007. doi:10.1074/jbc.M110.184010
Naito, S., Makhov, P., Astsaturov, I., Golovine, K., Tulin, A., Kutikov, A., et al. (2017). LDL Cholesterol Counteracts the Antitumour Effect of Tyrosine Kinase Inhibitors against Renal Cell Carcinoma. Br. J. Cancer 116, 1203–1207. doi:10.1038/bjc.2017.77
Nan, K.-J., and Nan, K.-J. (2012). Activation of PI3 kinase/Akt/HIF-1α Pathway Contributes to Hypoxia-Induced Epithelial-Mesenchymal Transition and Chemoresistance in Hepatocellular Carcinoma. Int. J. Oncol. 40, 461–468. doi:10.3892/ijo.2011.1197
Nieto, M. A., Huang, R. Y.-J., Jackson, R. A., and Thiery, J. P. (2016). EMT: 2016. Cell 166, 21–45. doi:10.1016/j.cell.2016.06.028
Nurwidya, F., Takahashi, F., Minakata, K., Murakami, A., and Takahashi, K. (2012). From Tumor Hypoxia to Cancer Progression: the Implications of Hypoxia-Inducible Factor-1 Expression in Cancers. Anat. Cel Biol. 45, 73–78. doi:10.5115/acb.2012.45.2.73
Padua, D., and Massagué, J. (2009). Roles of TGFβ in Metastasis. Cel Res 19, 89–102. doi:10.1038/cr.2008.316
Pallottini, V., Guantario, B., Martini, C., Totta, P., Filippi, I., Carraro, F., et al. (2008). Regulation of HMG-CoA Reductase Expression by Hypoxia. J. Cel. Biochem. 104, 701–709. doi:10.1002/jcb.21757
Pastushenko, I., and Blanpain, C. (2019). EMT Transition States during Tumor Progression and Metastasis. Trends Cel Biol. 29, 212–226. doi:10.1016/j.tcb.2018.12.001
Peinado, H., Ballestar, E., Esteller, M., and Cano, A. (2004). Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol. Cel. Biol. 24, 306–319. doi:10.1128/MCB.24.1.306-319.2004
Riscal, R., Skuli, N., and Simon, M. C. (2019). Even Cancer Cells Watch Their Cholesterol!. Mol. Cel 76, 220–231. doi:10.1016/j.molcel.2019.09.008
Saitoh, M. (2018). Involvement of Partial EMT in Cancer Progression. J. Biochem. 164, 257–264. doi:10.1093/jb/mvy047
Samanta, D., Gilkes, D. M., Chaturvedi, P., Xiang, L., and Semenza, G. L. (2014). Hypoxia-inducible Factors Are Required for Chemotherapy Resistance of Breast Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 111, E5429–E5438. doi:10.1073/pnas.1421438111
Sekine, Y., Demosky, S. J., Stonik, J. A., Furuya, Y., Koike, H., Suzuki, K., et al. (2010). High-Density Lipoprotein Induces Proliferation and Migration of Human Prostate Androgen-independent Cancer Cells by an ABCA1-dependent Mechanism. Mol. Cancer Res. 8, 1284–1294. doi:10.1158/1541-7786.MCR-10-0008
Sezgin, E., Azbazdar, Y., Ng, X. W., Teh, C., Simons, K., Weidinger, G., et al. (2017). Binding of Canonical Wnt Ligands to Their Receptor Complexes Occurs in Ordered Plasma Membrane Environments. FEBS J. 284, 2513–2526. doi:10.1111/febs.14139
Shapira, K. E., Ehrlich, M., and Henis, Y. I. (2018). Cholesterol Depletion Enhances TGF-β Smad Signaling by Increasing C-Jun Expression through a PKR-dependent Mechanism. MBoC 29, 2494–2507. doi:10.1091/mbc.E18-03-0175
Sharma, A., Bandyopadhayaya, S., Chowdhury, K., Sharma, T., Maheshwari, R., Das, A., et al. (2019). Metformin Exhibited Anticancer Activity by Lowering Cellular Cholesterol Content in Breast Cancer Cells. PLoS One 14, e0209435. doi:10.1371/journal.pone.0209435
Sheng, R., Kim, H., Lee, H., Xin, Y., Chen, Y., Tian, W., et al. (2014). Cholesterol Selectively Activates Canonical Wnt Signalling over Non-canonical Wnt Signalling. Nat. Commun. 5, 4393. doi:10.1038/ncomms5393
Shibue, T., and Weinberg, R. A. (2017). EMT, CSCs, and Drug Resistance: the Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 14, 611–629. doi:10.1038/nrclinonc.2017.44
Silvente-Poirot, S., and Poirot, M. (2014). Cholesterol and Cancer, in the Balance. Science 343, 1445–1446. doi:10.1126/science.1252787
Singh, M., Yelle, N., Venugopal, C., and Singh, S. K. (2018). EMT: Mechanisms and Therapeutic Implications. Pharmacol. Ther. 182, 80–94. doi:10.1016/j.pharmthera.2017.08.009
Subramanian, N., Schumann-Gillett, A., Mark, A. E., and O’mara, M. L. (2016). Understanding the Accumulation of P-Glycoprotein Substrates Within Cells: The Effect of Cholesterol on Membrane Partitioning. Biochim. Biophys. Acta Biomembr. 1858, 776–782. doi:10.1016/j.bbamem.2015.12.025
Sullivan, R., Paré, G. C., Frederiksen, L. J., Semenza, G. L., and Graham, C. H. (2008). Hypoxia-induced Resistance to Anticancer Drugs Is Associated with Decreased Senescence and Requires Hypoxia-Inducible Factor-1 Activity. Mol. Cancer Ther. 7, 1961–1973. doi:10.1158/1535-7163.MCT-08-0198
Sun, X., Essalmani, R., Day, R., Khatib, A. M., Seidah, N. G., and Prat, A. (2012). Proprotein Convertase Subtilisin/kexin Type 9 Deficiency Reduces Melanoma Metastasis in Liver. Neoplasia 14, 1122–1131. doi:10.1593/neo.121252
Sun, Y., He, W., Luo, M., Zhou, Y., Chang, G., Ren, W., et al. (2015). SREBP1 Regulates Tumorigenesis and Prognosis of Pancreatic Cancer through Targeting Lipid Metabolism. Tumor Biol. 36, 4133–4141. doi:10.1007/s13277-015-3047-5
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A. Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Thankamony, A. P., Saxena, K., Murali, R., Jolly, M. K., and Nair, R. (2020). Cancer Stem Cell Plasticity - A Deadly Deal. Front. Mol. Biosci. 7, 79. doi:10.3389/fmolb.2020.00079
Tirpe, A. A., Gulei, D., Ciortea, S. M., Crivii, C., and Berindan-Neagoe, I. (2019). Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Ijms 20, 6140. doi:10.3390/ijms20246140
Tong, Z.-T., Cai, M.-Y., Wang, X.-G., Kong, L.-L., Mai, S.-J., Liu, Y.-H., et al. (2012). EZH2 Supports Nasopharyngeal Carcinoma Cell Aggressiveness by Forming a Co-repressor Complex with HDAC1/HDAC2 and Snail to Inhibit E-Cadherin. Oncogene 31, 583–594. doi:10.1038/onc.2011.254
Torres-Adorno, A. M., Vitrac, H., Qi, Y., Tan, L., Levental, K. R., Fan, Y.-Y., et al. (2019). Eicosapentaenoic Acid in Combination with EPHA2 Inhibition Shows Efficacy in Preclinical Models of Triple-Negative Breast Cancer by Disrupting Cellular Cholesterol Efflux. Oncogene 38, 2135–2150. doi:10.1038/s41388-018-0569-5
Vincent, T., Neve, E. P. A., Johnson, J. R., Kukalev, A., Rojo, F., Albanell, J., et al. (2009). A SNAIL1‐SMAD3/4 Transcriptional Repressor Complex Promotes TGF‐β Mediated Epithelial‐Mesenchymal Transition. Nat. Cell Biol. 11, 943–950. doi:10.1038/ncb1905
Wang, Z., Sun, P., Gao, C., Chen, J., Li, J., Chen, Z., et al. (2017). Down-regulation of LRP1B in colon Cancer Promoted the Growth and Migration of Cancer Cells. Exp. Cel Res. 357, 1–8. doi:10.1016/j.yexcr.2017.04.010
Wang, Z., Wade, P., Mandell, K. J., Akyildiz, A., Parkos, C. A., Mrsny, R. J., et al. (2007). Raf 1 Represses Expression of the Tight junction Protein Occludin via Activation of the Zinc-finger Transcription Factor Slug. Oncogene 26, 1222–1230. doi:10.1038/sj.onc.1209902
Wen, Y.-A., Xiong, X., Zaytseva, Y. Y., Napier, D. L., Vallee, E., Li, A. T., et al. (2018). Downregulation of SREBP Inhibits Tumor Growth and Initiation by Altering Cellular Metabolism in colon Cancer. Cel Death Dis 9, 265. doi:10.1038/s41419-018-0330-6
Wu, F., Zhang, Y., Sun, B., Mcmahon, A. P., and Wang, Y. (2017). Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cel Chem. Biol. 24, 252–280. doi:10.1016/j.chembiol.2017.02.010
Wu, Y., Si, R., Tang, H., He, Z., Zhu, H., Wang, L., et al. (2015). Cholesterol Reduces the Sensitivity to Platinum-Based Chemotherapy via Upregulating ABCG2 in Lung Adenocarcinoma. Biochem. Biophysical Res. Commun. 457, 614–620. doi:10.1016/j.bbrc.2015.01.035
Wu, Y., Zhao, Y., He, X., He, Z., Wang, T., Wan, L., et al. (2020). Hydroxypropyl-β-cyclodextrin A-ttenuates the E-pithelial-to-mesenchymal T-ransition via E-ndoplasmic R-eticulum S-tress in MDA-MB-231 B-reast C-ancer C-ells. Mol. Med. Rep. 21, 249–257. doi:10.3892/mmr.2019.10802
Wu, Z.-Q., Li, X.-Y., Hu, C. Y., Ford, M., Kleer, C. G., and Weiss, S. J. (2012). Canonical Wnt Signaling Regulates Slug Activity and Links Epithelial-Mesenchymal Transition with Epigenetic Breast Cancer 1, Early Onset (BRCA1) Repression. Proc. Natl. Acad. Sci. 109, 16654–16659. doi:10.1073/pnas.1205822109
Xu, K., Zhan, Y., Yuan, Z., Qiu, Y., Wang, H., Fan, G., et al. (2019a). Hypoxia Induces Drug Resistance in Colorectal Cancer through the HIF-1α/miR-338-5p/IL-6 Feedback Loop. Mol. Ther. 27, 1810–1824. doi:10.1016/j.ymthe.2019.05.017
Xu, T., Zhang, J., Chen, W., Pan, S., Zhi, X., Wen, L., et al. (2016). ARK5 Promotes Doxorubicin Resistance in Hepatocellular Carcinoma via Epithelial-Mesenchymal Transition. Cancer Lett. 377, 140–148. doi:10.1016/j.canlet.2016.04.026
Yang, F., Sun, L., Li, Q., Han, X., Lei, L., Zhang, H., et al. (2012). SET8 Promotes Epithelial-Mesenchymal Transition and Confers TWIST Dual Transcriptional Activities. EMBO J. 31, 110–123. doi:10.1038/emboj.2011.364
Yang, G., Xiong, G., Feng, M., Zhao, F., Qiu, J., Liu, Y., et al. (2020). OLR1 Promotes Pancreatic Cancer Metastasis via Increased C-Myc Expression and Transcription of HMGA2. Mol. Cancer Res. 18, 685–697. doi:10.1158/1541-7786.MCR-19-0718
Yang, J., Mani, S. A., Donaher, J. L., Ramaswamy, S., Itzykson, R. A., Come, C., et al. (2004). Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 117, 927–939. doi:10.1016/j.cell.2004.06.006
Yeung, K. T., and Yang, J. (2017). Epithelial-mesenchymal Transition in Tumor Metastasis. Mol. Oncol. 11, 28–39. doi:10.1002/1878-0261.12017
Yokoo, M., Kubota, Y., Motoyama, K., Higashi, T., Taniyoshi, M., Tokumaru, H., et al. (2015). 2-Hydroxypropyl-β-Cyclodextrin Acts as a Novel Anticancer Agent. PloS one 10, e0141946. doi:10.1371/journal.pone.0141946
Yue, S., Li, J., Lee, S.-Y., Lee, H. J., Shao, T., Song, B., et al. (2014). Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cel Metab. 19, 393–406. doi:10.1016/j.cmet.2014.01.019
Zalba, S., and Ten Hagen, T. L. M. (2017). Cell Membrane Modulation as Adjuvant in Cancer Therapy. Cancer Treat. Rev. 52, 48–57. doi:10.1016/j.ctrv.2016.10.008
Zhan, J., Wang, P., Li, S., Song, J., He, H., Wang, Y., et al. (2019). HOXB13 Networking with ABCG1/EZH2/Slug Mediates Metastasis and Confers Resistance to Cisplatin in Lung Adenocarcinoma Patients. Theranostics 9, 2084–2099. doi:10.7150/thno.29463
Zhang, J., Li, Q., Wu, Y., Wang, D., Xu, L., Zhang, Y., et al. (2019a). Cholesterol Content in Cell Membrane Maintains Surface Levels of ErbB2 and Confers a Therapeutic Vulnerability in ErbB2-Positive Breast Cancer. Cell Commun. Signal 17, 15. doi:10.1186/s12964-019-0328-4
Zhang, J., Zhang, L., Li, C., Yang, C., Li, L., Song, S., et al. (2018). LOX-1 Is a Poor Prognostic Indicator and Induces Epithelial-Mesenchymal Transition and Metastasis in Pancreatic Cancer Patients. Cell Oncol. 41, 73–84. doi:10.1007/s13402-017-0360-6
Zhang, P., Sun, Y., and Ma, L. (2015). ZEB1: at the Crossroads of Epithelial-Mesenchymal Transition, Metastasis and Therapy Resistance. Cell cycle 14, 481–487. doi:10.1080/15384101.2015.1006048
Zhang, Y., Liu, Y., Duan, J., Wang, H., Zhang, Y., Qiao, K., et al. (2019b). Cholesterol Depletion Sensitizes Gallbladder Cancer to Cisplatin by Impairing DNA Damage Response. Cell cycle 18, 3337–3350. doi:10.1080/15384101.2019.1676581
Zhao, N., Sun, H., Sun, B., Zhu, D., Zhao, X., Wang, Y., et al. (2016). miR-27a-3p Suppresses Tumor Metastasis and VM by Down-Regulating VE-Cadherin Expression and Inhibiting EMT: an Essential Role for Twist-1 in HCC. Sci. Rep. 6, 23091. doi:10.1038/srep23091
Zhao, Z., Hao, D., Wang, L., Li, J., Meng, Y., Li, P., et al. (2019). CtBP Promotes Metastasis of Breast Cancer through Repressing Cholesterol and Activating TGF-β Signaling. Oncogene 38, 2076–2091. doi:10.1038/s41388-018-0570-z
Zheng, L., Li, L., Lu, Y., Jiang, F., and Yang, X.-A. (2018). SREBP2 Contributes to Cisplatin Resistance in Ovarian Cancer Cells. Exp. Biol. Med. (Maywood) 243, 655–662. doi:10.1177/1535370218760283
Zhong, Z., and Virshup, D. M. (2020). Wnt Signaling and Drug Resistance in Cancer. Mol. Pharmacol. 97, 72–89. doi:10.1124/mol.119.117978
Keywords: cholesterol, drug resistance, hypoxia, EMT, cancer
Citation: Abdulla N, Vincent CT and Kaur M (2021) Mechanistic Insights Delineating the Role of Cholesterol in Epithelial Mesenchymal Transition and Drug Resistance in Cancer. Front. Cell Dev. Biol. 9:728325. doi: 10.3389/fcell.2021.728325
Received: 15 July 2021; Accepted: 29 October 2021;
Published: 19 November 2021.
Edited by:
Laura Rosanò, Italian National Research Council, ItalyReviewed by:
Rosanna Sestito, Regina Elena National Cancer Institute (IRCCS), ItalyCopyright © 2021 Abdulla, Vincent and Kaur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mandeep Kaur, bWFuZGVlcC5rYXVyQHdpdHMuYWMuemE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.