 Natalie Burchat1
Natalie Burchat1 Priyanka Sharma1Hong Ye1
Priyanka Sharma1Hong Ye1 Sai Santosh Babu Komakula1,2Agnieszka Dobrzyn1,2Vladimir Vartanian3
Sai Santosh Babu Komakula1,2Agnieszka Dobrzyn1,2Vladimir Vartanian3 R. Stephen Lloyd3,4
R. Stephen Lloyd3,4 Harini Sampath1,5,6*
Harini Sampath1,5,6*- 1Rutgers Center for Lipid Research, New Jersey Institute for Food, Nutrition, and Health, Rutgers University, New Brunswick, NJ, United States
- 2Laboratory of Cell Signaling and Metabolic Disorders, Nencki Institute of Experimental Biology, Warsaw, Poland
- 3Oregon Institute of Occupational Health Sciences, Oregon Health and Science University, Portland, OR, United States
- 4Department of Molecular and Medical Genetics, Oregon Health and Science University, Portland, OR, United States
- 5Department of Nutritional Sciences, Rutgers University, New Brunswick, NJ, United States
- 6Center for Microbiome, Nutrition, and Health, New Jersey Institute for Food, Nutrition, and Health, Rutgers University, New Brunswick, NJ, United States
Obesity and related metabolic disorders are pressing public health concerns, raising the risk for a multitude of chronic diseases. Obesity is multi-factorial disease, with both diet and lifestyle, as well as genetic and developmental factors leading to alterations in energy balance. In this regard, a novel role for DNA repair glycosylases in modulating risk for obesity has been previously established. Global deletion of either of two different glycosylases with varying substrate specificities, Nei-like endonuclease 1 (NEIL1) or 8-oxoguanine DNA glycosylase-1 (OGG1), both predispose mice to diet-induced obesity (DIO). Conversely, enhanced expression of the human OGG1 gene renders mice resistant to obesity and adiposity. This resistance to DIO is mediated through increases in whole body energy expenditure and increased respiration in adipose tissue. Here, we report that hOGG1 expression also confers resistance to genetically-induced obesity. While Agouti obese (Ay/a) mice are hyperphagic and consequently develop obesity on a chow diet, hOGG1 expression in Ay/a mice (Ay/aTg) prevents increased body weight, without reducing food intake. Instead, obesity resistance in Ay/aTg mice is accompanied by increased whole body energy expenditure and tissue mitochondrial content. We also report for the first time that OGG1-mediated obesity resistance in both the Ay/a model and DIO model requires maternal transmission of the hOGG1 transgene. Maternal, but not paternal, transmission of the hOGG1 transgene is associated with obesity resistance and increased mitochondrial content in adipose tissue. These data demonstrate a critical role for OGG1 in modulating energy balance through changes in adipose tissue function. They also demonstrate the importance of OGG1 in modulating developmental programming of mitochondrial content and quality, thereby determining metabolic outcomes in offspring.
Introduction
Oxidatively-induced damage to both nuclear and mitochondrial DNA is repaired via the base-excision repair (BER) pathway, initiated by DNA glycosylases. The most commonly formed oxidative lesion, 8-oxoguanine (8-oxoG), is recognized and cleaved by the enzyme, 8-oxoG DNA glycosylase (OGG1). OGG1 initiates repair of 8-oxoG in both the nuclear and mitochondrial genome and has been shown to play a role in diverse pathologies, including neurodegenerative disease, various cancers, and metabolic dysfunction (Sampath et al., 2012a; Vartanian et al., 2017; Sampath and Lloyd, 2019). We have previously demonstrated that mice lacking endogenous OGG1 (Ogg1–/–) are prone to diet-induced obesity (DIO) and its sequelae, including insulin resistance, ectopic lipid accumulation in liver and skeletal muscle, gut dysbiosis, and chronic inflammation (Sampath et al., 2012b; Vartanian et al., 2017; Simon et al., 2020). Conversely, enhanced expression of human OGG1 downstream of a constitutive mitochondrial targeting sequence protects mice from DIO, insulin resistance, and adipose tissue inflammation (Komakula et al., 2018). This metabolic protection in hOGG1 transgenic mice (Ogg1Tg) was accompanied by increases in whole body energy expenditure and increased mitochondrial content and respiration in white adipose tissue (WAT) (Komakula et al., 2018). Thus, our previous studies reported this novel role for OGG1 in modulating energy balance in the context of a hypercaloric high-fat diet (HFD). In the current study, we sought to explore whether hOGG1 expression could confer protection against genetically-induced obesity. Several models of genetically-induced obesity become obese due to chronic hyperphagia (Robinson et al., 2000; Ellacott and Cone, 2006; Chang et al., 2018). In most of these models, obesity develops spontaneously on a chow diet, without requiring the use of a hypercaloric HFD. We therefore asked the question of whether hOGG1 expression would be protective in the context of genetically-induced obesity. To address this question, we transferred the gene expressing human OGG1 downstream of a constitutive mitochondrial targeting sequence (Ogg1Tg) (Wang et al., 2011; Komakula et al., 2018) into the Ay/a obese mouse (Ay/a). The Ay/a model develops hyperphagia and consequent obesity and insulin resistance due to ectopic overexpression of the Agouti protein, an antagonist of the melanocortin receptor (Klebig et al., 1995; Moussa and Claycombe, 1999; Tschöp and Heiman, 2001). Interestingly, our studies indicate an important function for OGG1 in protecting against genetically-induced obesity. Further, they also uncover a critical role for maternal OGG1 genotype in determining obesity resistance both in the context of genetically-induced as well as DIO.
Experimental Approach
Animals
Age-matched mice on the C57BL6J background were used in all studies. The generation of hOGG1-expressing mice (Ogg1Tg) has been previously described (Wang et al., 2011). Ay/a mice were obtained from Jackson Labs (stock #000021) (Siracusa et al., 1987; Klebig et al., 1995). Ay/a mice expressing the hOGG1 transgene are designated as Ay/aTg and were bred in house by mating Ay/a or Ay/aTg animals with Ogg1Tg animals. Cohort sizes were as follows: males– Ay/a: 10; Ay/aTg: 11; females – Ay/a: 10; Ay/aTg 11; Ay/aTg–dad: 5; all HFD studies—n = 5–9 males per cohort. For studies using Ay/a mice, body weights were measured weekly from week 4 onward, and food intake was measured weekly between weeks 10–22 for males and 18–22 for females. At 50 weeks of age, body composition was measured by NMR (Echo Medical Systems, Houston, TX, United States) and energy expenditure and physical activity were measured via open circuit indirect calorimetry (CLAMS Comprehensive Lab Animal Monitoring System, Columbus Instruments, Columbus, OH, United States). Following an acclimation period of 24 h, oxygen consumption (VO2) and carbon dioxide production (VCO2) were recorded every minute for 48 h, with a room air reference taken following each cycle of measurements. Beam breaks on the X, Y, and Z axes were tallied for voluntary physical activity measurements. Gonadal WAT, subscapular brown adipose tissue (BAT), liver, and gastrocnemius were collected at 52 weeks of age. For HFD feeding studies, 8-week old male mice were individually housed and given ad libitum access to a 60% HFD for 12 weeks, and body weights and food intake were measured weekly. Body composition was assessed, and tissues were collected at 20 weeks of age. All mice were euthanized between 9 and 11 am by isoflurane overdose followed by exsanguination via cardiac puncture. For all in vivo procedures, every effort was made to minimize discomfort and suffering, in accordance with the protocols approved by the Animal Care and Use Committee of Rutgers University, New Brunswick, New Jersey under protocol No. 201900077.
Hepatic Lipids
Hepatic lipids were extracted and separated by thin-layer chromatography, as we have previously described (Komakula et al., 2018).
DNA and RNA Analyses
RNA was isolated using QIAzol Lysis Reagent and the Qiagen RNeasy kit. Superscript III first-strand synthesis system (Invitrogen, Carlsbad, CA, United States) was used to synthesize cDNA from 1 μg of RNA. Quantitative real-time PCR (qPCR) was performed on a QuantStudio 3 Real-Time PCR System (Applied Biosystems, Foster City, CA, United States) with gene-specific primers. Data were normalized to the expression of RNA18SN5 and quantification was done using the 2–ΔΔCt method. Relative copy number was quantified by qRT-PCR amplification of hOGG1 from 10 ng genomic DNA and normalized to expression of GAPDH.
Protein Analyses
Whole cell lysates were prepared from frozen tissue using HEPES homogenization buffer (50 mM pH7.4 HEPES, 150 mM NaCl, 10 mM Na-pyrophosphate, 2 mM EDTA, 1% NP-40, 10% Glycerol) with EDTA-free protease and phosphatase inhibitors. Samples are representative of 3–7 animals, and lysate protein concentrations were determined via Bradford assay. Equal amounts of protein were separated by SDS-PAGE and transferred to nitrocellulose membranes. Ponceau total protein staining was performed to confirm uniform loading and transfer of proteins. Following this, membranes were blocked with 5% non-fat dried milk in Tris-buffered saline (TBS, 150 mM NaCl, 50 mM Tris–HCl, pH 7.4) with 0.1% Tween 20 at room temperature with shaking for an hour. The membranes were then incubated overnight at 4°C with primary antibody. Primary antibodies used were as follows: VDAC (Pierce Biotechnology, Waltham, MA, United States), COXIV (Abcam, Cambridge, United Kingdom), PGC-1α (Novus Biologicals, Littleton, CO, United States), HSP60, SIRT1, and GAPDH (all from Cell Signaling Technology, Danvers, MA, United States). Membranes were incubated with HRP- or Alexa-fluor conjugated secondary antibodies and signal was detected using enhanced chemiluminescence or fluorescence imaging, respectively, on an Azure c600 imaging system (Azure Biosystems, United States).
Statistical Analyses
Data are expressed as mean ± SEM for biological replicates; statistical comparisons were carried out by student’s t-test for 2-group comparisons and by two−way ANOVA for multi−group comparisons, followed by post−hoc analysis (Bonferroni) in Graph Pad Prism (version 8.2.0 for Windows, GraphPad Software, La Jolla, CA, United States).
Results
hOGG1 Expression Attenuates Body Weight in Ay/a Obese Mice
Ay/a yellow obese mice are a genetic model of obesity resulting from antagonism of the melanocortin receptor and consequent hyperphagia (Moussa and Claycombe, 1999; Tschöp and Heiman, 2001). Unlike other common models of obesity such as the leptin-deficient ob/ob or leptin-receptor deficient db/db mice, Ay/a mice express both a functional leptin gene and a leptin receptor. However, these mice develop insulin and leptin resistance, and are hence a valuable model to study genetically-induced obesity in a clinically relevant manner (Klebig et al., 1995; Miltenberger et al., 1997; Tschöp and Heiman, 2001; Rahmouni et al., 2002; Miyazaki et al., 2009). Melanocortin receptor antagonism in Ay/a mice results in chronic hyperphagia and consequent obesity, which is apparent starting at about 16 weeks on a standard rodent chow diet. To determine the potential impact of hOGG1 expression on body weight in this model, we introduced the hOGG1 transgene into Ay/a mice to generate Ay/a; Ogg1Tg (Ay/aTg) mice. Body weights were measured from weaning until 48 weeks of age on a standard chow diet in male and female Ay/a and Ay/aTg littermates. Female Ay/aTg mice had significantly lower body weights than Ay/a counterparts, starting as early as 5 weeks of age. Male Ay/aTg mice also had significantly lower body weights than their Ay/a counterparts, starting at 21 weeks of age (Figures 1A,B).
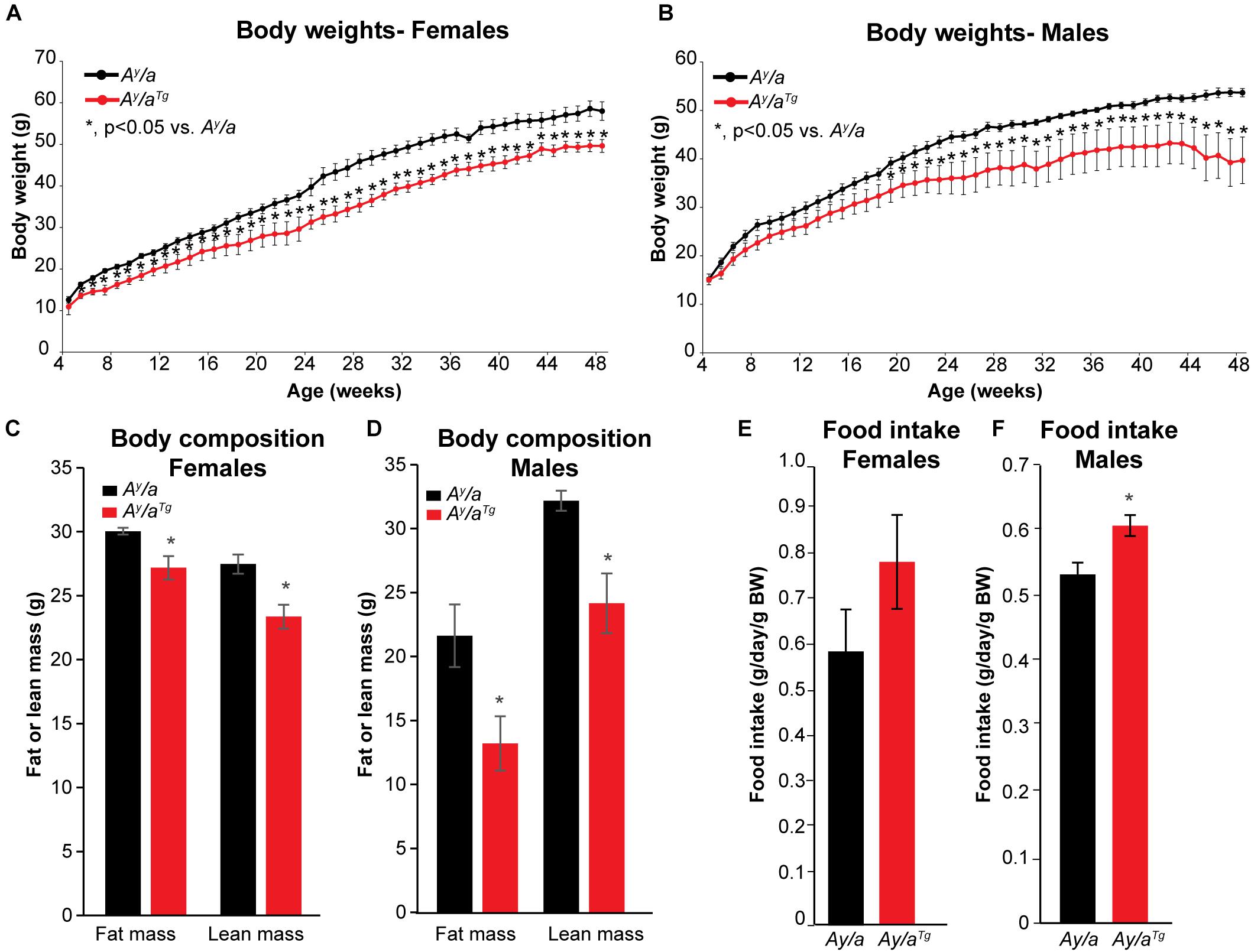
Figure 1. Body weights, body composition, and food intake. (A,B) Body weights were reduced in Ay/aTg mice, relative to Ay/a obese counterparts. (C,D) Reductions in both lean and fat mass were apparent in Ay/aTg mice. (E,F) Food intake was measured weekly between weeks 10–22 for males and 18–22 for females; food intake was slightly increased in Ay/aTg mice (NS in females). Averages ± SEM. *p < 0.05 vs. sex-matched Ay/a.
Unlike in DIO, which is characterized by increases in fat mass, genetically-induced obesity in Ay/a mice results in increases in both lean and fat masses (Miltenberger et al., 1997). Body composition analyses at 52 weeks of age indicated that reductions in body weight in Ay/aTg mice were reflected in decreases in both lean and fat masses (Figures 1C,D). Thus, hOGG1 expression in this model attenuates aberrant increases in both lean and fat compartments of Ay/a mice. Increased body weight in Ay/a mice is a consequence of chronic hyperphagia. Interestingly, both female and male Ay/aTg had higher food intakes than their Ay/a counterparts (Figures 1E,F). This suggested that rather than due to attenuation of food intake, peripheral mechanisms may mediate the observed reductions in body weight in Ay/aTg mice.
hOGG1 Expression Increases Energy Expenditure and Mitochondrial Content in Ay/a Obese Mice
Given the similarities between male and female animals, we focused our downstream studies on female mice due to their larger sample size. Energy expenditure analyses were carried out using open circuit indirect calorimetry. Ay/aTg mice had a trend for elevated O2 consumption and CO2 respiration (Figures 2A,B); however, this was not statistically significant. These data suggested that the reduction in body weights in Ay/aTg mice may be due to physiologically relevant elevations in energy expenditure. Respiratory exchange ratios (RER) were unchanged by genotype (Figure 2C), indicating no differences in whole body substrate utilization. Physical activity was not significantly different between genotypes (Figure 2D).
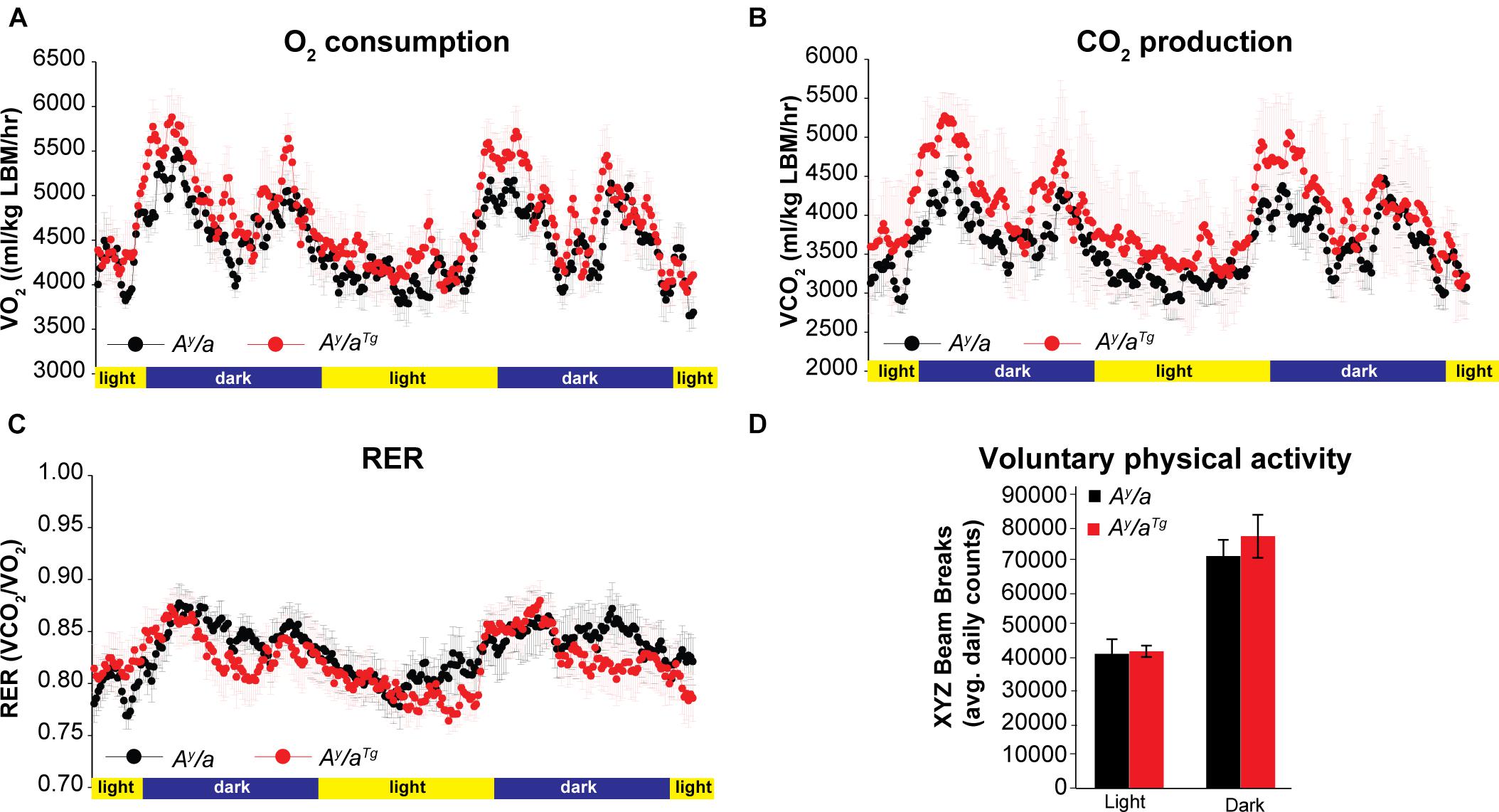
Figure 2. Energy expenditure. (A) O2 consumption and (B) CO2 respiration were measured by indirect calorimetry in female mice. (C) Respiratory exchange ratios (RER) were calculated during indirect calorimetry measurements. (D) Voluntary physical activity was measured during indirect calorimetry. n = 6. Averages ± SEM.
To investigate metabolic changes that contribute to this lean phenotype and elevated energy expenditure, we evaluated markers of mitochondrial content and energy sensing across multiple metabolically active tissues in Ay/a and Ay/aTg mice (Figure 3). Markers of mitochondrial content, including heat-shock protein 60 (HSP60), voltage-dependent anion channel (VDAC), and cytochrome c oxidase IV (COX4) were significantly elevated in WAT of Ay/aTg mice, relative to Ay/a counterparts. HSP60 and VDAC were increased in BAT and gastrocnemius of Ay/aTg mice (Figures 3A–F). In addition, expression of peroxisome proliferator-activated receptor gamma coactivator-1-alpha (PGC-1α), a master regulator of mitochondrial biogenesis, was elevated across tissues in Ay/aTg mice (NS in gastrocnemius) (Figures 3A–F). Expression of the deacetylase, Sirtuin 1 (SIRT1), which activates PGC-1α, was also significantly increased in WAT of Ay/aTg mice. The elevation in SIRT1 and PGC-1α are consistent with the observed increases in mitochondrial content in these animals. They are also consistent with prior reports from our lab and others indicating a role for OGG1 in altering tissue mitochondrial content and function (Rachek et al., 2002, 2006a,b; Yuzefovych et al., 2013; Vartanian et al., 2017; Komakula et al., 2018). Further, most of the significant changes related to mitochondrial function and content were observed in WAT, rather than in other tissues of Ay/aTg mice. These data are consistent with our prior observations of an important role for WAT remodeling in mediating the metabolic phenotypes of Ogg1Tg mice (Komakula et al., 2018). We thus propose that hOGG1 expression increases mitochondrial content in Ay/aTg mice, particularly in WAT, resulting in elevated energy expenditure and attenuation of body weight, relative to Ay/a littermates.
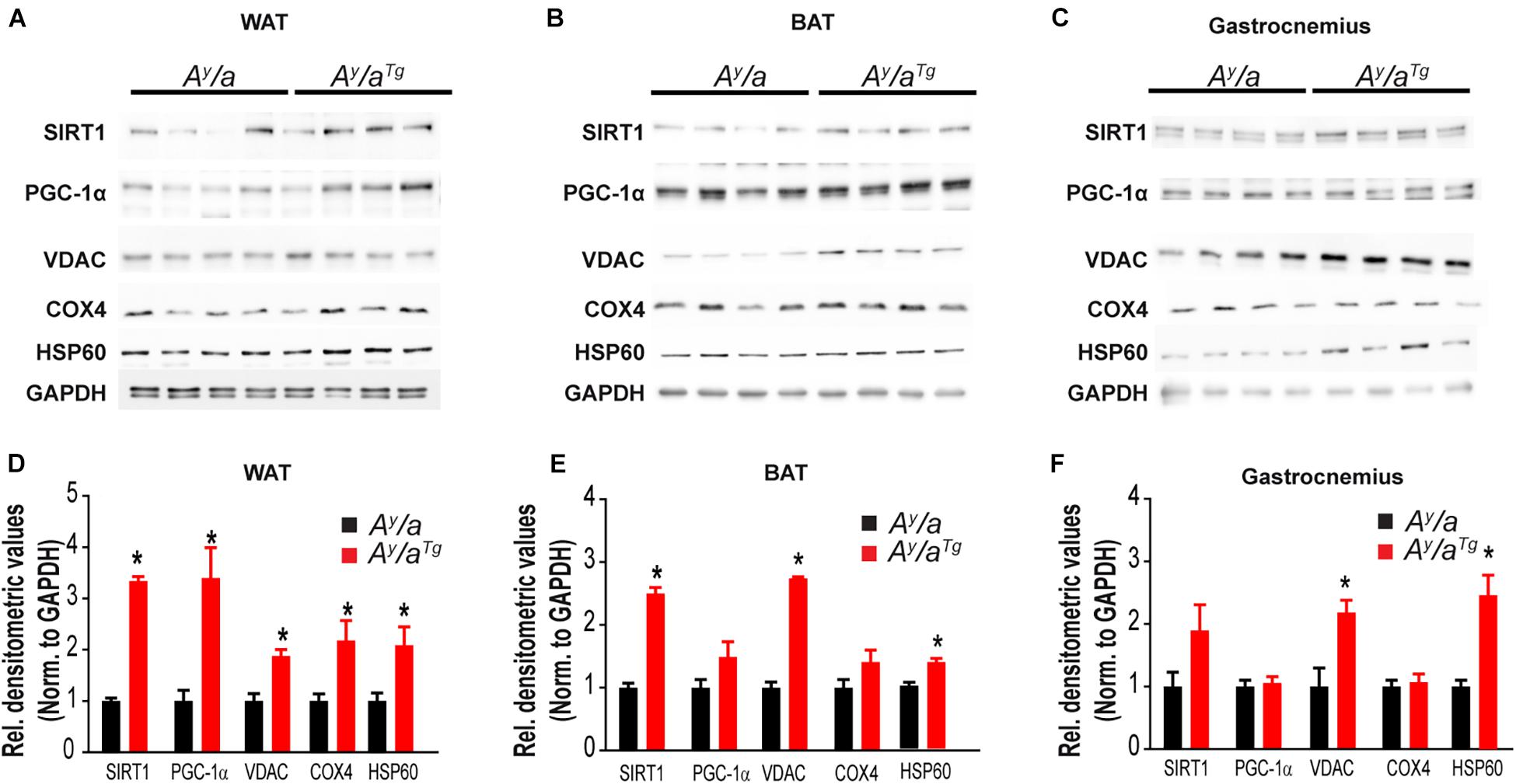
Figure 3. Tissue mitochondrial content. Tissue expression of mitochondrial markers was measured by immunoblotting in (A,D) WAT, (B,E) BAT, and (C,F) Gastrocnemius. BAT, brown adipose tissue, COX4, cytochrome c oxidase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HSP60, heat shock protein-60; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SIRT1, Sirtuin 1; VDAC, voltage-dependent anion channel; WAT, white adipose tissue.
Maternal Transmission of the hOGG1 Transgene Is Necessary for Obesity Resistance
Breeding of Ay/a mice is inherently challenging, as the Ay/a mutation reduces fecundity and is embryonically lethal in offspring of homozygous mutants. Thus, animals used in the studies above were derived from parents where one parent transmitted the Ay/a mutation, with the female parent carrying the hOGG1 transgene. However, during colony expansion, we generated a limited number of offspring where the hOGG1 transgene was transmitted solely by the male parent (Ay/aTg–dad), since the female parent did not carry the hOGG1 transgene. Intriguingly, we noticed that offspring from this mating were not protected from obesity, relative to Ay/a controls. Thus, while offspring of female Ay/aTg mice were protected from agouti-induced obesity, offspring of male Ay/aTg mice were not (Figure 4). Regrettably, these mice were euthanized at 24 weeks, hence longer term body weight curves were not generated for this colony.
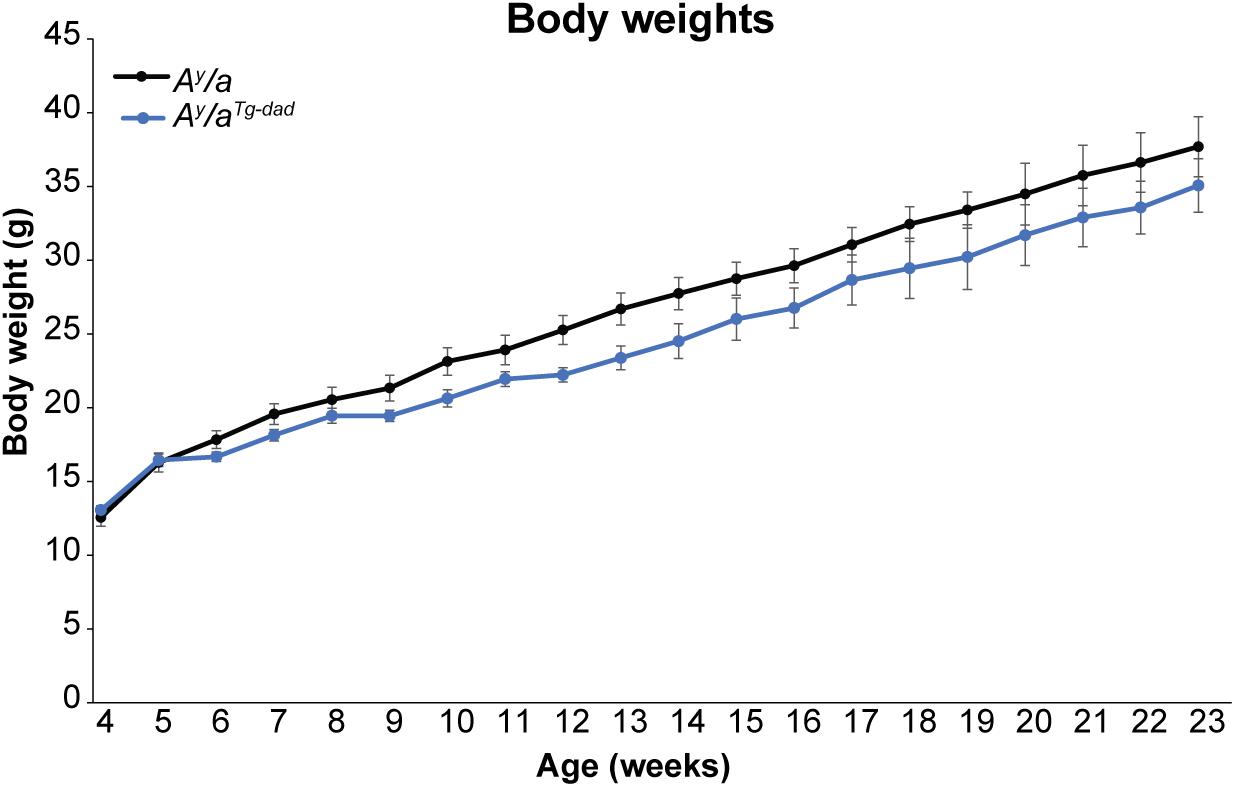
Figure 4. Body weights following paternal transmission of hOGG1. Body weights were measured at weekly intervals in female mice. Averages ± SEM. *p < 0.05 vs. Ay/a.
Since Ay/aTg females were lighter than Ay/a animals at breeding age (8 weeks), we sought to determine if the differences in offspring phenotype were simply a function of maternal body weight during pregnancy. We therefore established breeding colonies in our non-Ay/a colony, such that either the female or male parent was hOGG1 transgenic (Ogg1Tg–mom orOgg1Tg–dad, respectively). Under chow-fed conditions, WT and Ogg1Tg mice do not differ in body weights or litter sizes. WT and Ogg1Tg offspring from all breeding pairs were placed on a 12-week HFD, starting at 8 weeks of age. Mice with maternal transmission of the hOGG1 transgene (Ogg1Tg–mom) were significantly protected from HFD-induced body weight gain, relative to WT littermates, as we have previously reported (Figure 5A). However, mice that received the transgene solely from the male parent (Ogg1Tg–dad) had body weights that were indistinguishable from WT littermates when fed a HFD (Figure 5A). Body composition analyses revealed that fat mass was significantly reduced in Ogg1Tg–mom mice, relative to WT counterparts (Figure 5B). Additionally, lean mass was increased in Ogg1Tg–mom mice, relative to WT controls. In contrast, fat and lean masses in Ogg1Tg–dad mice were indistinguishable from WT controls and significantly higher than Ogg1Tg–mom animals (Figure 5B). Hepatic lipid accumulation is a distinct risk factor for further metabolic dysfunction, and we have previously shown that while OGG1 deficiency increases hepatic lipid storage, OGG1-overexpression significantly reduces hepatic lipids (Sampath et al., 2012b; Komakula et al., 2018). Here we discovered that while stored hepatic lipids such as triglycerides (TG) and cholesterol esters (CE) were significantly reduced in Ogg1Tg–mom mice, no such reduction was apparent in Ogg1Tg–dad livers (Figure 5C). Thus, transgene inheritance from the female parent was both required and sufficient to confer protection against HFD-induced weight gain, whole body adiposity, and hepatic lipid accumulation.
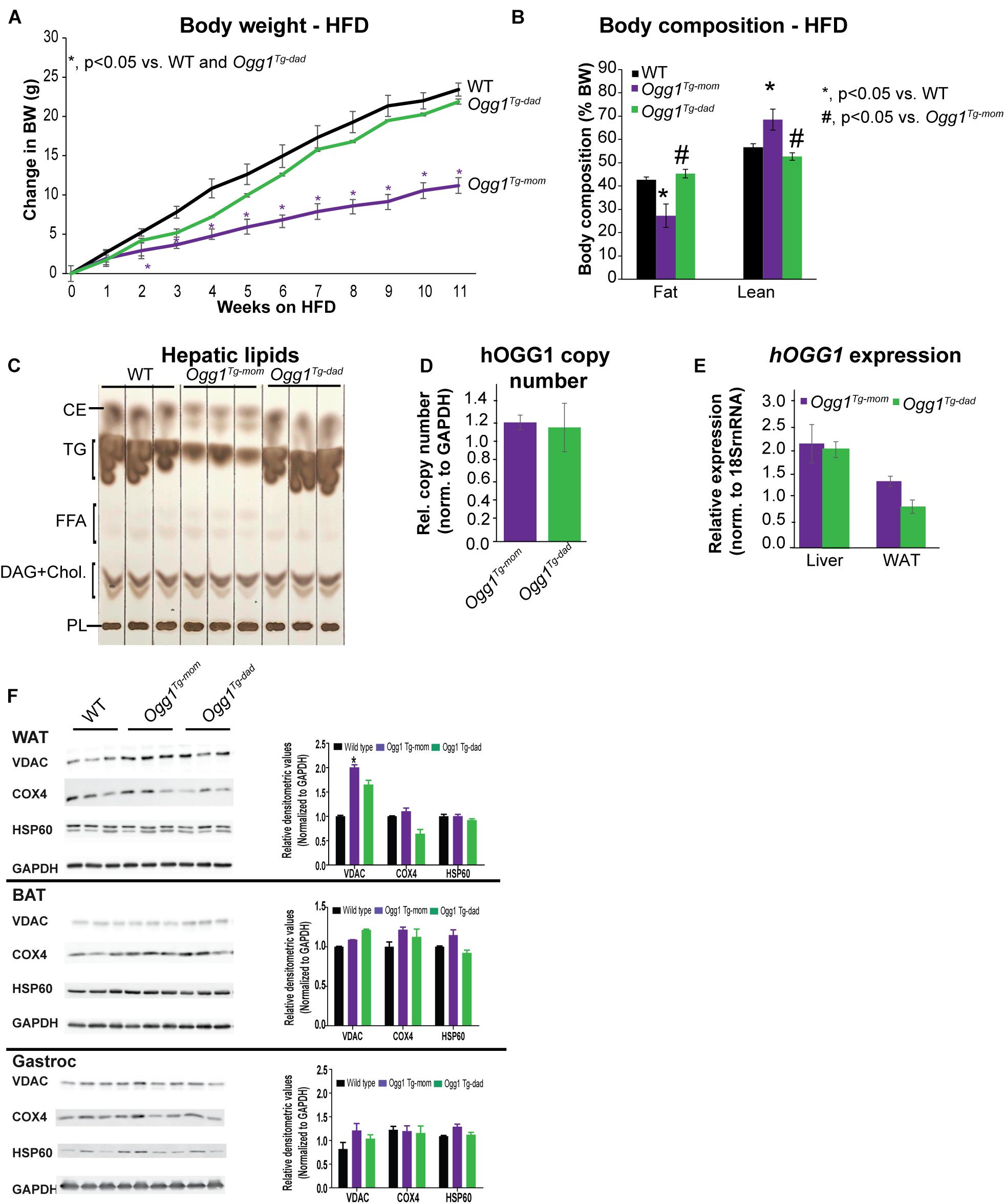
Figure 5. Maternal vs. paternal transmission of hOGG1 influences metabolic phenotype and mitochondrial content of WAT. (A) Ogg1Tg– mom or Ogg1Tg– dad animals were placed on high-fat diets (HFDs) at 8 weeks of age, and body weights were measured weekly. (B) Body composition was measured at the end of 11 weeks of HFD feeding. (C) Hepatic lipids were extracted and separated by thin-layer chromatography. (D) Relative hOGG1 copy number and (E) hOGG1 gene expression were measured by qPCR. (F) Tissue expression of mitochondrial markers was measured by immunoblotting. CE, cholesterol esters; DAG, diacylglycerols; FFA, free fatty acids; PL, phospholipids; TG, triglycerides.
Increased Mitochondrial Content in WAT Requires Maternal Transmission of the hOGG1 Transgene
Inheritance of the hOGG1 transgene from the male or female parent did not alter the relative transgene copy number nor gene expression of hOGG1 (Figures 5D,E). This led us to conclude that differences in obesity resistance were not a function of unexpected differences in copy number or hOgg1 expression levels in the offspring. We have previously shown that in the context of HFD-induced obesity, changes in mitochondrial content and function in WAT may mediate obesity resistance in Ogg1Tg mice. Consistent with these previous reports, we discovered that Ogg1Tg–mom mice had increased mitochondrial content, reflected in significantly increased content of VDAC in WAT. However, Ogg1Tg–dad animals did not have a similar increase in mitochondrial content, consistent with the lack of obesity resistance in these mice (Figure 5F). Similar increases in mitochondrial content were not observed in BAT or gastroc of Ogg1Tg mice (Figure 5F). These findings are consistent with our previous reports (Komakula et al., 2018) and suggest that differences in metabolic phenotypes in Ogg1Tg mice may stem from alterations in WAT mitochondrial content and function. Overall, our results indicate that maternal transmission of the hOGG1 transgene confers resistance not only to genetically-induced obesity (Figure 4), but also to HFD-induced obesity.
Discussion
We demonstrate for the first time that enhanced expression of the human OGG1 gene confers protection against genetically-induced obesity. The Agouti yellow mouse obesity syndrome is a result of dominant mutations at the Agouti locus (Moussa and Claycombe, 1999). Ectopic expression of Agouti in multiple tissues in these mice results in a yellow coat color, chronic hyperphagia, obesity, increased linear growth, leptin and insulin resistance, and hyperglycemia. Mechanisms mediating hyperphagia and obesity in this model involve antagonism of the melanocortin receptor, and similar phenotypes have been described in mice overexpressing Agouti-Related protein (AGRP), a potent antagonist of the melanocortin receptors-3 and -4 (Moussa and Claycombe, 1999; Small et al., 2001). Human obesity is also frequently associated with resistance to both leptin and insulin, and genome-wide association studies have implicated mutations near the melanocortin receptor-4 in the development of obesity and insulin resistance (Chambers et al., 2008; Loos et al., 2008). Thus, our results indicating a role for OGG1 in attenuating body weights in the Ay/a obese model (Figure 1) are particularly relevant from a translational standpoint. As hOGG1 expression lowered body weight without lowering food intake (Figure 1), body weight reduction in Ay/aTg mice likely results from alterations in peripheral tissues. Consistently, we observed increases in whole body energy expenditure (Figure 2) and mitochondrial content in several metabolically important tissues, particularly in WAT of Ay/aTg mice (Figure 3).
An intriguing discovery of these studies is that of obesity resistance being determined by the maternal OGG1 genotype. Male Ogg1Tg parents were unable to transmit obesity resistance to their Ogg1Tg offspring. Female Ogg1Tg mice, conversely, could transmit this metabolically beneficial phenotype to both male and female (not shown) offspring. These data are strongly suggestive of the mitochondrial genome, which is inherited solely from the female parent, as being a critical determinant of the obesity resistance phenotype in Ogg1Tg mice.
Studies examining the developmental origins of disease have established a strong link between maternal obesity and adverse metabolic outcomes in offspring (Das et al., 2021). For instance, in both human and rodent studies, maternal obesity is associated not only with increased risk of pregnancy complications such as gestational diabetes, but also with adverse outcomes in the offspring, including increased risk of obesity and diabetes. The mechanisms underlying these effects are not completely known but likely involve hormonal regulation, epigenetic changes, oxidative stress in the uterine environment, and dysregulated gut microbiota, among other factors (Saben et al., 2016; Das et al., 2021). While these links between maternal obesity and fetal outcomes have been extensively studied in animal models, relatively few studies have examined a link between maternal obesity resistance and metabolic outcomes in the offspring. Our studies indicate that maternal, but not paternal, OGG1 genotype influences tissue mitochondrial content and energy balance in both male and female Ogg1Tg offspring. These findings clearly implicate a role for mitochondrial quality in determining obesity resistance phenotypes in offspring. They also raise intriguing questions about interactions between the intrauterine environment, which is influenced by maternal genotype, and offspring genotype. Prior studies have reported roles for increased maternal oxidative stress and DNA damage in impacting comparable stress in the fetus and offspring (Luo et al., 2006; Simmons, 2006; Raicević et al., 2010; Rodríguez-Rodríguez et al., 2018). Further, epigenetic modifications during development have been shown to be influenced by an adverse fetal environment and to, in turn, impair metabolic outcomes in adult offspring (Seki et al., 2012; Houde et al., 2013; Ruchat et al., 2013; Marchlewicz et al., 2016; Zhu et al., 2019). In this regard, 8-oxoG itself may serve as an epigenetic mark, thereby altering promoter transcription rates via the recruitment of OGG1 to sites of oxidation (Perillo et al., 2008; Ruchko et al., 2009; Zarakowska et al., 2014; Allgayer et al., 2016; Fleming et al., 2017a, b; Hao et al., 2020). In addition to being recruited to 8-oxoG sites, OGG1 activity influences DNA methylation, as oxidized guanines in CpG sequences are resistant to the action of DNA methyltransferases (Weitzman et al., 1994; Valinluck et al., 2004; Maltseva et al., 2009; Ba and Boldogh, 2018). Thus, oxidative stress and OGG1 activity are both critical determinants of epigenetic programming, although their roles in the uterine environment have not been investigated. We observed that protection from DIO required both the female parent and offspring to carry the hOGG1 transgene. Thus, it is possible that reduced uterine oxidative stress in an Ogg1Tg female may result in altered metabolic programming in Ogg1Tg offspring. The mechanisms mediating these interactions between maternal or in utero genotype with offspring genotype require further investigation.
These studies also suggested that among markers of mitochondrial content, which can directly influence energy expenditure, increased mitochondrial content in WAT was consistent with obesity resistance in Ogg1Tg–mom but not Ogg1Tg–dad mice (Figure 5F). These data further support our working hypothesis that the improved metabolic phenotype in Ogg1Tg mice is a consequence of increased mitochondrial content and improved function in WAT (Komakula et al., 2018). In further support of a role for OGG1 in modulating adipocyte behavior, we recently showed that OGG1 genotype corresponds with adipocyte differentiation capacity (Komakula et al., 2021). Preadipocytes lacking OGG1 differentiated faster and accumulated more lipids than WT cells, while hOGG1 expression significantly blunted adipocyte differentiation and lipid accretion. These phenotypes in isolated preadipocytes and 3T3 cells correspond with obesity predisposition or resistance in Ogg1–/– and Ogg1Tg animals, respectively (Komakula et al., 2021). They also indicate an important cell-intrinsic role for OGG1 in the adipocyte, as supported by our current studies.
In summary, we show here for the first time that hOGG1 expression is protective against obesity resulting not only from HFD consumption, but also from genetically-induced obesity. In both models, hOGG1 expression alters tissue mitochondrial content, particularly in WAT, and enhances overall energy expenditure. Importantly, maternal transmission of the transgene is both necessary and sufficient to confer resistance to obesity. These data have important implications to our understanding of the etiology of obesity and the role that DNA damage and repair may play in the process. They also establish an important role for interactions between the in utero environment, shaped by maternal genotype, with offspring genotype in impacting developmental programming and influencing metabolic outcomes in adult animals.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Animal Care and Use Committee of Rutgers University, New Brunswick, NJ, United States.
Author Contributions
NB, RL, and HS contributed to conceptualization and design of the study. NB, PS, HY, SK, VV, and HS carried out experiments and analyzed data. NB, PS, and HS prepared figures and wrote the first draft of the manuscript. AD, RL, and HS edited the manuscript and obtained funding. All authors have read and approved the submitted version.
Funding
This work was supported by NIH grants DK100640 (HS) and DK075974 (RL), a grant from European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska Curie COFUND grant agreement no. 665735 (Bio4Med) to AD and HS, by funding from the Polish Ministry of Science and Higher Education as part of 2016–2020 funds for the implementation of international projects (agreement no. 548/H2020/COFUND/2016/2) to AD and HS, funds from the Oregon Institute of Occupational Health Sciences at Oregon Health and Science University via funds from the Division of Consumer and Business Services of the State of Oregon (ORS 656.630) to RL, and funds from Rutgers University to HS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Dr. Lars Eide (University of Oslo) for the gift of Ogg1Tg animals.
References
Allgayer, J., Kitsera, N., Bartelt, S., Epe, B., and Khobta, A. (2016). Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 44, 7267–7280.
Ba, X., and Boldogh, I. (2018). 8-Oxoguanine DNA glycosylase 1: beyond repair of the oxidatively modified base lesions. Redox Biol. 14, 669–678. doi: 10.1016/j.redox.2017.11.008
Chambers, J. C., Elliott, P., Zabaneh, D., Zhang, W., Li, Y., Froguel, P., et al. (2008). Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat. Genet. 40, 716–718. doi: 10.1038/ng.156
Chang, J. Y., Park, J. H., Park, S. E., Shon, J., and Park, Y. J. (2018). The fat mass-and obesity-associated (FTO) gene to obesity: lessons from mouse models. Obesity 26, 1674–1686. doi: 10.1002/oby.22301
Das, M., Sauceda, C., and Webster, N. J. G. (2021). Mitochondrial dysfunction in obesity and reproduction. Endocrinology 162:bqaa158.
Ellacott, K. L., and Cone, R. D. (2006). The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1265–1274. doi: 10.1098/rstb.2006.1861
Fleming, A. M., Ding, Y., and Burrows, C. J. (2017a). Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. 114, 2604–2609. doi: 10.1073/pnas.1619809114
Fleming, A. M., Zhu, J., Ding, Y., and Burrows, C. J. (2017b). 8-Oxo-7,8-dihydroguanine in the context of a gene promoter G-Quadruplex is an on-off switch for transcription. ACS Chem. Biol. 12, 2417–2426. doi: 10.1021/acschembio.7b00636
Hao, W., Wang, J., Zhang, Y., Wang, C., Xia, L., Zhang, W., et al. (2020). Enzymatically inactive OGG1 binds to DNA and steers base excision repair toward gene transcription. FASEB J. 34, 7427–7441. doi: 10.1096/fj.201902243r
Houde, A.-A., Hivert, M.-F., and Bouchard, L. (2013). Fetal epigenetic programming of adipokines. Adipocyte 2, 41–46. doi: 10.4161/adip.22055
Klebig, M. L., Wilkinson, J. E., Geisler, J. G., and Woychik, R. P. (1995). Ectopic expression of the agouti gene in transgenic mice causes obesity, features of type II diabetes, and yellow fur. Proc. Natl. Acad. Sci. U.S.A. 92:4728. doi: 10.1073/pnas.92.11.4728
Komakula, S. S. B., Tumova, J., Kumaraswamy, D., Burchat, N., Vartanian, V., Ye, H., et al. (2018). The DNA repair protein OGG1 protects against obesity by altering mitochondrial energetics in white adipose tissue. Sci. Rep. 8:14886.
Komakula, S. S., Blaze, B., Ye, H., Dobrzyn, A., and Sampath, H. A. (2021). Novel role for the DNA repair enzyme 8-Oxoguanine DNA glycosylase in adipogenesis. Int. J. Mol. Sci. 22:1152. doi: 10.3390/ijms22031152
Loos, R. J., Lindgren, C. M., Li, S., Wheeler, E., Zhao, J. H., Prokopenko, I., et al. (2008). Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 40, 768–775.
Luo, Z. C., Fraser, W. D., Julien, P., Deal, C. L., Audibert, F., Smith, G. N., et al. (2006). Tracing the origins of “fetal origins” of adult diseases: programming by oxidative stress? Med. Hypotheses 66, 38–44. doi: 10.1016/j.mehy.2005.08.020
Maltseva, D. V., Baykov, A. A., Jeltsch, A., and Gromova, E. S. (2009). Impact of 7,8-dihydro-8-oxoguanine on methylation of the CpG site by Dnmt3a. Biochemistry 48, 1361–1368. doi: 10.1021/bi801947f
Marchlewicz, E. H., Dolinoy, D. C., Tang, L., Milewski, S., Jones, T. R., Goodrich, J. M., et al. (2016). Lipid metabolism is associated with developmental epigenetic programming. Sci. Rep. 6:34857.
Miltenberger, R. J., Mynatt, R. L., Wilkinson, J. E., and Woychik, R. P. (1997). The role of the agouti gene in the yellow obese syndrome. J. Nutr. 127, 1902S–1907S.
Miyazaki, M., Sampath, H., Liu, X., Flowers, M. T., Chu, K., Dobrzyn, A., et al. (2009). Stearoyl-CoA desaturase-1 deficiency attenuates obesity and insulin resistance in leptin-resistant obese mice. Biochem. Biophys. Res. Commun. 380, 818–822. doi: 10.1016/j.bbrc.2009.01.183
Moussa, N. M., and Claycombe, K. J. (1999). The yellow mouse obesity syndrome and mechanisms of agouti-induced obesity. Obes. Res. 7, 506–514. doi: 10.1002/j.1550-8528.1999.tb00440.x
Perillo, B., Ombra, M. N., Bertoni, A., Cuozzo, C., Sacchetti, S., Sasso, A., et al. (2008). DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206. doi: 10.1126/science.1147674
Rachek, L. I., Grishko, V. I., Ledoux, S. P., and Wilson, G. L. (2006a). Role of nitric oxide-induced mtDNA damage in mitochondrial dysfunction and apoptosis. Free Radic. Biol. Med. 40, 754–762. doi: 10.1016/j.freeradbiomed.2005.09.028
Rachek, L. I., Grishko, V. I., Musiyenko, S. I., Kelley, M. R., Ledoux, S. P., and Wilson, G. L. (2002). Conditional targeting of the DNA repair enzyme hOGG1 into mitochondria. J. Biol. Chem. 277, 44932–44937. doi: 10.1074/jbc.m208770200
Rachek, L. I., Thornley, N. P., Grishko, V. I., Ledoux, S. P., and Wilson, G. L. (2006b). Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes 55, 1022–1028. doi: 10.2337/diabetes.55.04.06.db05-0865
Rahmouni, K., Haynes, W. G., Morgan, D. A., and Mark, A. L. (2002). Selective resistance to central neural administration of leptin in agouti obese mice. Hypertension 39, 486–490. doi: 10.1161/hy0202.102836
Raicević, S., Cubrilo, D., Arsenijević, S., Vukcević, G., Zivković, V., Vuletić, M., et al. (2010). Oxidative stress in fetal distress: potential prospects for diagnosis. Oxid. Med. Cell. Longev. 3, 214–218. doi: 10.4161/oxim.3.3.12070
Robinson, S. W., Dinulescu, D. M., and Cone, R. D. (2000). Genetic models of obesity and energy balance in the mouse. Annu. Rev. Genet. 34, 687–745. doi: 10.1146/annurev.genet.34.1.687
Rodríguez-Rodríguez, P., Ramiro-Cortijo, D., Reyes-Hernández, C. G., López de Pablo, A. L., González, M. C., and Arribas, S. M. (2018). Implication of oxidative stress in fetal programming of cardiovascular disease. Front. Physiol. 9:602. doi: 10.3389/fphys.2018.00602
Ruchat, S.-M., Hivert, M.-F., and Bouchard, L. (2013). Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr. Rev. 71(suppl_1), S88–S94.
Ruchko, M. V., Gorodnya, O. M., Pastukh, V. M., Swiger, B. M., Middleton, N. S., Wilson, G. L., et al. (2009). Hypoxia-induced oxidative base modifications in the VEGF hypoxia-response element are associated with transcriptionally active nucleosomes. Free Radic. Biol. Med. 46, 352–359. doi: 10.1016/j.freeradbiomed.2008.09.038
Saben, J. L., Boudoures, A. L., Asghar, Z., Thompson, A., Drury, A., Zhang, W., et al. (2016). Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep. 16, 1–8. doi: 10.1016/j.celrep.2016.05.065
Sampath, H., and Lloyd, R. S. (2019). Roles of OGG1 in transcriptional regulation and maintenance of metabolic homeostasis. DNA Repair 81:102667. doi: 10.1016/j.dnarep.2019.102667
Sampath, H., McCullough, A. K., and Lloyd, R. S. (2012a). Regulation of DNA glycosylases and their role in limiting disease. Free Radic. Res. 46, 460–478. doi: 10.3109/10715762.2012.655730
Sampath, H., Vartanian, V., Rollins, M. R., Sakumi, K., Nakabeppu, Y., and Lloyd, R. S. (2012b). 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS One 7:17. doi: 10.1371/journal.pone.0051697
Seki, Y., Williams, L., Vuguin, P. M., and Charron, M. J. (2012). Minireview: epigenetic programming of diabetes and obesity: animal models. Endocrinology 153, 1031–1038. doi: 10.1210/en.2011-1805
Simmons, R. A. (2006). Developmental origins of diabetes: the role of oxidative stress. Free Radic. Biol. Med. 40, 917–922. doi: 10.1016/j.freeradbiomed.2005.12.018
Simon, H., Vartanian, V., Wong, M. H., Nakabeppu, Y., Sharma, P., Lloyd, R. S., et al. (2020). OGG1 deficiency alters the intestinal microbiome and increases intestinal inflammation in a mouse model. PLoS One 15:e0227501. doi: 10.1371/journal.pone.0227501
Siracusa, L. D., Russell, L. B., Eicher, E. M., Corrow, D. J., Copeland, N. G., and Jenkins, N. A. (1987). Genetic organization of the agouti region of the mouse. Genetics 117, 93–100. doi: 10.1093/genetics/117.1.93
Small, C. J., Kim, M. S., Stanley, S. A., Mitchell, J. R. D., Murphy, K., Morgan, D. G. A., et al. (2001). Effects of chronic central nervous system administration of agouti-related protein in pair-fed animals. Diabetes 50:248. doi: 10.2337/diabetes.50.2.248
Tschöp, M., and Heiman, M. L. (2001). Rodent obesity models: an overview. Exp. Clin. Endocrinol. Diabetes 109, 307–319. doi: 10.1055/s-2001-17297
Valinluck, V., Tsai, H. H., Rogstad, D. K., Burdzy, A., Bird, A., and Sowers, L. C. (2004). Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 32, 4100–4108. doi: 10.1093/nar/gkh739
Vartanian, V., Tumova, J., Dobrzyn, P., Dobrzyn, A., Nakabeppu, Y., Lloyd, R. S., et al. (2017). 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in lipid and mitochondrial metabolism in muscle. PLoS One 12:e0181687. doi: 10.1371/journal.pone.0181687
Wang, W., Esbensen, Y., Kunke, D., Suganthan, R., Rachek, L., Bjørås, M., et al. (2011). Mitochondrial DNA damage level determines neural stem cell differentiation fate. J. Neurosci. 31, 9746–9751. doi: 10.1523/jneurosci.0852-11.2011
Weitzman, S. A., Turk, P. W., Milkowski, D. H., and Kozlowski, K. (1994). Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. 91, 1261–1264. doi: 10.1073/pnas.91.4.1261
Yuzefovych, L., Schuler, A., Chen, J., Alvarez, D., Eide, L., Ledoux, S., et al. (2013). Alteration of mitochondrial function and insulin sensitivity in primary mouse skeletal muscle cells isolated from transgenic and knockout mice: role of OGG1. Endocrinology 154, 2640–2649. doi: 10.1210/en.2013-1076
Zarakowska, E., Gackowski, D., Foksinski, M., and Olinski, R. (2014). Are 8-oxoguanine (8-oxoGua) and 5-hydroxymethyluracil (5-hmUra) oxidatively damaged DNA bases or transcription (epigenetic) marks? Mutat. Res. Genet. Toxicol. Environ. Mutagen. 76, 58–63. doi: 10.1016/j.mrgentox.2013.09.002
Keywords: DNA repair, metabolic syndrome (in offspring), obesity, developmental origins of disease, mitochondrial function
Citation: Burchat N, Sharma P, Ye H, Komakula SSB, Dobrzyn A, Vartanian V, Lloyd RS and Sampath H (2021) Maternal Transmission of Human OGG1 Protects Mice Against Genetically- and Diet-Induced Obesity Through Increased Tissue Mitochondrial Content. Front. Cell Dev. Biol. 9:718962. doi: 10.3389/fcell.2021.718962
Received: 01 June 2021; Accepted: 09 August 2021;
Published: 15 September 2021.
Edited by:
Jianjun Zhao, Lerner Research Institute, United StatesReviewed by:
Youliang Wang, Beijing Institute of Technology, ChinaRahul N. Kanadia, University of Connecticut, United States
Copyright © 2021 Burchat, Sharma, Ye, Komakula, Dobrzyn, Vartanian, Lloyd and Sampath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harini Sampath, aGFyaW5pLnNhbXBhdGhAcnV0Z2Vycy5lZHU=