
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 11 August 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.716300
This article is part of the Research Topic Hearing Loss: Mechanisms and Prevention View all 35 articles
 Siyu Li1,2†
Siyu Li1,2† Cheng Cheng1,2†
Cheng Cheng1,2† Ling Lu1,2†Xiaofeng Ma1†Xiaoli Zhang1,2†
Ling Lu1,2†Xiaofeng Ma1†Xiaoli Zhang1,2† Ao Li1,2Jie Chen1,2*
Ao Li1,2Jie Chen1,2* Xiaoyun Qian1,2*
Xiaoyun Qian1,2* Xia Gao1,2*
Xia Gao1,2*Sensorineural hearing loss (SNHL) affects approximately 466 million people worldwide, which is projected to reach 900 million by 2050. Its histological characteristics are lesions in cochlear hair cells, supporting cells, and auditory nerve endings. Neurological disorders cover a wide range of diseases affecting the nervous system, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), autism spectrum disorder (ASD), etc. Many studies have revealed that neurological disorders manifest with hearing loss, in addition to typical nervous symptoms. The prevalence, manifestations, and neuropathological mechanisms underlying vary among different diseases. In this review, we discuss the relevant literature, from clinical trials to research mice models, to provide an overview of auditory dysfunctions in the most common neurological disorders, particularly those associated with hearing loss, and to explain their underlying pathological and molecular mechanisms.
Hearing loss is defined by an average pure-tone threshold detection exceeding 20 dB, affecting approximately 466 million people worldwide. According to the value of pure tone thresholds, it can be classified as mild (20–35 dB), moderate (35–50 dB), moderately severe (50–65 dB), severe (65–80 dB), profound (80–95 dB), and total (≥95 dB) hearing loss. Lesions in the cochlea, auditory nerve, and central auditory pathway induce sensorineural hearing loss (SNHL); nearly a third of the population over the age of 65 is suffering from it1. Histological characteristics of age-related hearing loss include degenerative pathology in cochlear hair cells, supporting cells, and auditory nerve endings, resulting in irreversible damage to the sensory epithelium of the cochlea (He et al., 2020; Keithley, 2020; Wu et al., 2020b). Sound is collected and conducted by the external and middle ear, then transformed into electrical signals by cochlear mechanosensory cells: the inner and outer hair cells (OHCs) (Dallos, 1986). OHCs function to enhance sound frequency selectivity and mechanical amplification, and inner hair cells (IHCs) are responsible for subsequent sound detection and transmission. Hair cells are sensitive to aging, acoustic trauma, ototoxic drugs (Fu et al., 2021b), and environmental or genetic influences (Wang et al., 2017; Qian et al., 2020; Fu et al., 2021a; Lv et al., 2021). As damages to either type of hair cells can result in permanent SNHL, many studies have focused on biological treatments for hearing restoration, including gene therapy, hair cell regeneration, etc. (Liu et al., 2016; Li et al., 2018b; Chen et al., 2021; He et al., 2021). These electrical signals are then transduced to the auditory cortex by spiral ganglion neurons (SGNs). SGNs are located in the Rosenthal’s canal of the cochlea and work as the primary sensory neurons to connect the peripheral and central auditory systems, which are susceptible to aging and ototoxic drugs. Hence, preventing the degeneration of SGNs carries critical implications for improving the restoration of hearing (Appler and Goodrich, 2011; Coate and Kelley, 2013; Sun et al., 2016; Liu et al., 2019, 2021; Guo et al., 2021). The pulses ascend into the cochlear nuclei, superior olivary complex, and inferior colliculus for the perception of time and intensity, then target toward the medial geniculate body, and finally, the auditory information is integrated into and further processed by the auditory cortex (Grothe et al., 2010; Profant et al., 2015; Wu et al., 2015). The ascending and reversed descending pathways (originating from the cerebral cortex to the cochlea) form the complete auditory circuitry. Pathology in any portion of the auditory circuitry will lead to auditory dysfunctions, including hearing impairments and central auditory processing disorder, which can be addressed through pure tone audiometry (PTA) and speech tests (such as speech discrimination and speech-in-noise tests). The effects of hearing loss are widespread and profound, resulting in social isolation, psychological illness. And hearing loss is reported to be closely associated with cognitive decline and dementia independently in the elderly population (Lin et al., 2011a, 2013; Jafari et al., 2019).
Neurological disorders include a broad range of diseases that affect the nervous system, of which neurodegenerative diseases and neurodevelopmental disorders have been widely discussed. In the elderly, neurodegenerative diseases are common causes of morbidity and cognitive impairment (Kritsilis et al., 2018; Hou et al., 2019). The progression of these diseases is characterized by the diffusion of protein aggregates, which correlates with clinical severity (Ross and Poirier, 2004; Herrero and Morelli, 2017; Davis et al., 2018). Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by social isolation, stereotypical behaviors, and interests. Genetic and environmental risk factors jointly account for phenotypic variations in ASD (Johnson et al., 2007; Lai et al., 2014a). Recent studies have reported that patients suffering from these neurological disorders are accompanied by hearing impairments and other auditory dysfunctions, especially in Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and ASD. Meanwhile, many mechanisms may account for the complex interplay, including neuropathological changes in the central and peripheral auditory system, social isolation caused by hearing decline, or other potential molecular mechanisms (Fortunato et al., 2016; Shen et al., 2018). It remains unclear whether auditory dysfunction is intrinsic or secondary to these diseases. Here, we discuss the relevant clinical literature to review the most common neurological disorders, particularly those associated with hearing loss, and explain their underlying pathological and molecular mechanisms.
Alzheimer’s disease is a progressive neurodegenerative disorder and the most common form of dementia. One in 10 people aged over 65 years is affected by AD, and the incidence increases with age (Soria Lopez et al., 2019; Alzheimer’s Association, 2020). Late-onset AD (LOAD) is the onset of AD later than 65 years of age, accounting for approximately 94% of all cases. Symptomatic AD exhibits insidious impairments in learning and memory at the initial stage, and then progresses toward impairments in cognition and executive function at the later stage (Long and Holtzman, 2019). AD patients are usually present with deficient perceptual and semantic processing of sounds (Perez et al., 2009; Benarroch, 2010; Ruan et al., 2012; Attems et al., 2014; Albers et al., 2015; van Wijngaarden et al., 2017). Since the 1980s, the association between hearing impairments and AD has been discussed. Evidence has shown that cognitive impairment is often accompanied by hearing loss, and in turn, hearing loss increases the incidence of cognitive decline and AD (Gallacher et al., 2012; Hung et al., 2015; Panza et al., 2015; Fortunato et al., 2016; Ford et al., 2018). Ford et al. (2018) estimated that midlife hearing loss might account for 9.1% of dementia cases globally. Lin et al. (2011a) demonstrated that for every 10 dB increase above the pure tone threshold of 25 dB, the risk of dementia increased by approximately 20%, with risk ratios for mild, moderate, and severe hearing loss of 1.89, 3.00, and 4.94, respectively. Taljaard et al. (2016) conducted a meta-analysis illustrating that hearing impairments coexisted with more inferior cognitive ability in older individuals, and receiving hearing interventions improved cognitive outcomes.
Neuropathological changes in the auditory system of AD have been widely explored, and typical AD pathological changes have been observed in auditory pathways (Uhlmann et al., 1986). Extracellular amyloid-β (Aβ) peptide aggregation and intracellular neurofibrillary tangles (NFTs) are the neuropathological hallmarks of AD (Figure 1; Long and Holtzman, 2019). In the amyloidogenic pathway, amyloid precursor proteins (APP) are membrane proteins that are sequentially cleaved by β-secretase and γ-secretase, resulting in the release of extracellular amyloid-β peptides, where they clump together to form deposits (Aβ plaques) and initiate a cascade of pathogenic processes and neurodegeneration. Tau protein plays a critical role in the development of neurons, and its hyperphosphorylation leads to the production of NFTs. Aβ peptides and NFTs coalesce to induce cellular dysfunctions (inflammation, oxidative stress, etc.), synaptic loss, and neurodegeneration (Guo et al., 2017; Makin, 2018). Genetically, AD is classified into familial (FAD) and sporadic cases. FAD accounts for 5% of AD cases and has an autosomal dominant inheritance pattern. Mutations in APP, PSEN1 (Presenilin 1), and PSEN2 (Presenilin 2) are responsible for the occurrence of FAD, and it was reported that mutations in these genes alter APP processing, induce Aβ formation, and then initiate tau pathology. In contrast, more than 90% of AD patients appear sporadically, which usually presents with late-onset AD (Piaceri et al., 2013). The only confirmed risk gene for sporadic AD is apolipoprotein E (APOE), which encodes an amino acid lipoprotein that can bind to amyloid precursor proteins. The Epsilon4 allele in APOE is strongly associated with an increased risk of AD in either homozygous or heterozygous states. Over 60% of sporadic cases are unrelated to APOE, suggesting that the interplay of genetic and environmental elements contributes to the occurrence of sporadic AD (Verghese et al., 2011). Similar neuropathology is also observed in sporadic AD without such mutations, indicating that Aβ plaques may be the driving force behind tau pathology, but not the sole one (van der Kant et al., 2020).
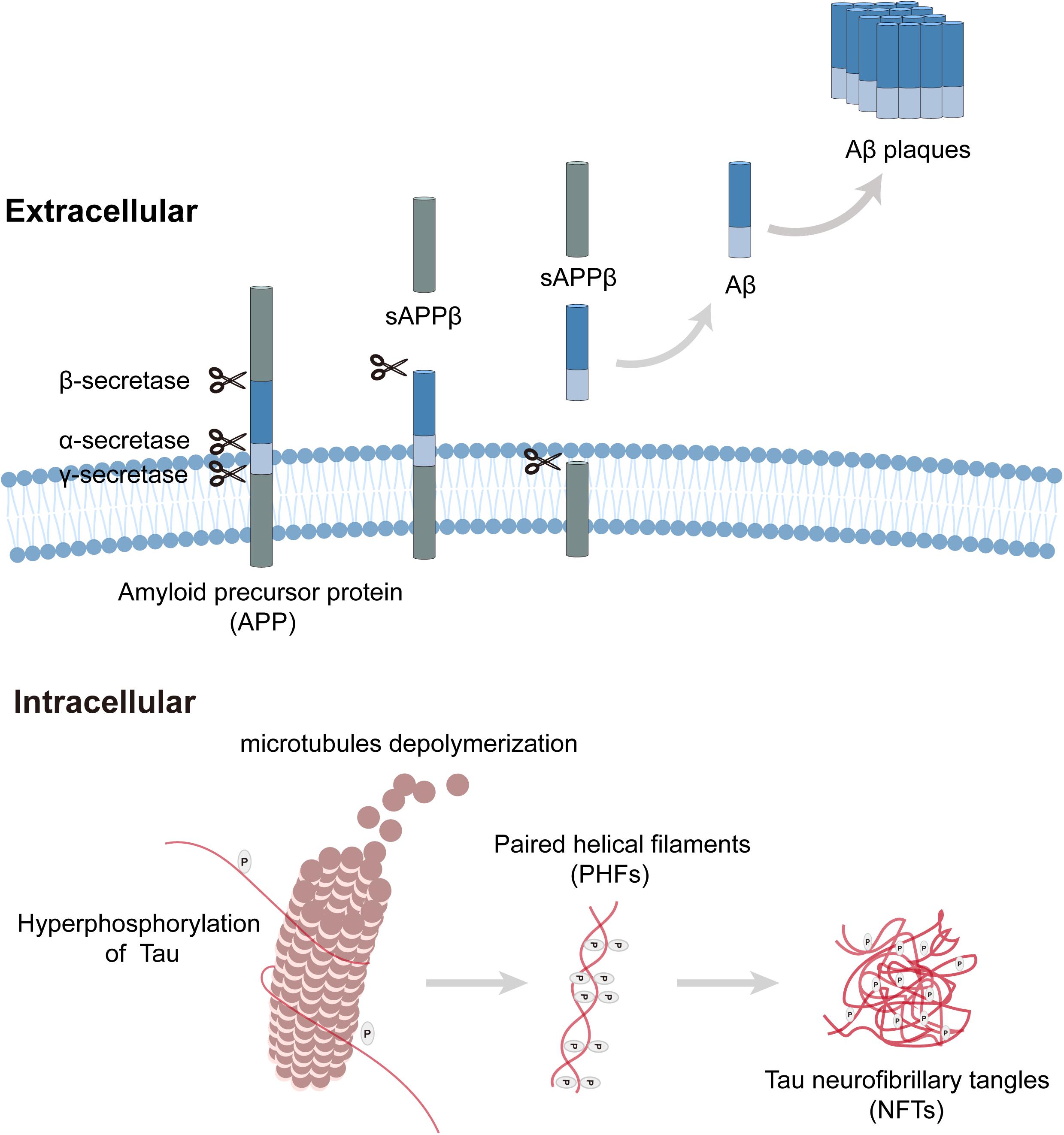
Figure 1. Amyloid plaque formation extracellular and tau pathology intracellular. Amyloid precursor protein (APP) is a transmembrane protein that can be cleaved by three kinds of secretases. In the process of amyloid plaque formation, APP is cleaved by β-secretase and γ-secretase sequentially, then amyloid-β peptides release to extracellular and clump together to form deposits (Aβ plaques). Tau plays a critical role in microtubule assembly and stabilization, hyperphosphorylation of tau leads to microtubules depolymerization, and paired helical filaments (PHF) aggregate to form tau neurofibrillary tangles (NFTs).
In the early stage of AD, brain atrophy occurs in the central auditory cortex and related functional nuclei; senile plaques (SPs) and NFTs are extensively distributed throughout relay stations in the ascending auditory pathway (Sinha et al., 1993; Parvizi et al., 2001; Rub et al., 2016). Many AD mouse models have been used to explore hearing dysfunction and their underlying mechanisms (summarized in Table 1). Studies have shown that AD mouse models initially exhibit high-frequency hearing loss and finally progress to the entire frequency. 5xFAD and APP/PS1 mice are mainly characterized by β-amyloid plaque deposition and show elevated auditory brainstem response (ABR) thresholds. 5xFAD mice co-express gene mutations in five FAD and can generate Aβ deposits rapidly (Oakley et al., 2006). In 5xFAD mice, amyloid depositions were observed at 2 months of age, while cochlear histopathology revealed a large amount of apical and basal hair cell loss at 13 months of age (O’Leary et al., 2017). The onset of auditory dysfunctions in APP/PS1 mice preceded before neuropathological changes, suggesting that acoustic measurements might be a non-invasive indicator for AD detection (Liu et al., 2020). 3xTg-AD mice express 3 AD-related transgenes, and its neuropathology developments are similar to FAD patients, characterized by Aβ deposition, tau pathology, and neuroinflammation. In 3xTg-AD mice, a reduction of SGN’s relative densities was observed at 9–12 months of age. A transgenic mouse model with overexpression of Aβ peptides in hair cells was established by Omata et al. (2016) and high-frequency hearing impairments were found at 4 months of age. Aligned with the electrophysiological assessment, basal hair cell loss was observed. They further established another model overexpressing tau pathology but found no significant hearing dysfunctions. Nevertheless, double transgenic mice showed an advanced and exaggerated hearing impairments, suggesting that Aβ deposition was a fundamental pathological etiology for hearing defects exhibited by AD and that tau pathology enhanced the dysfunction (Omata et al., 2016). Many previous studies have shown that both oxidative stress and apoptosis play essential roles in the death of hair cells (Yu et al., 2017; Li et al., 2018a), whether hair cell loss observed in the researches can be attributed to Aβ-induced oxidative stress and cell apoptosis need further study.
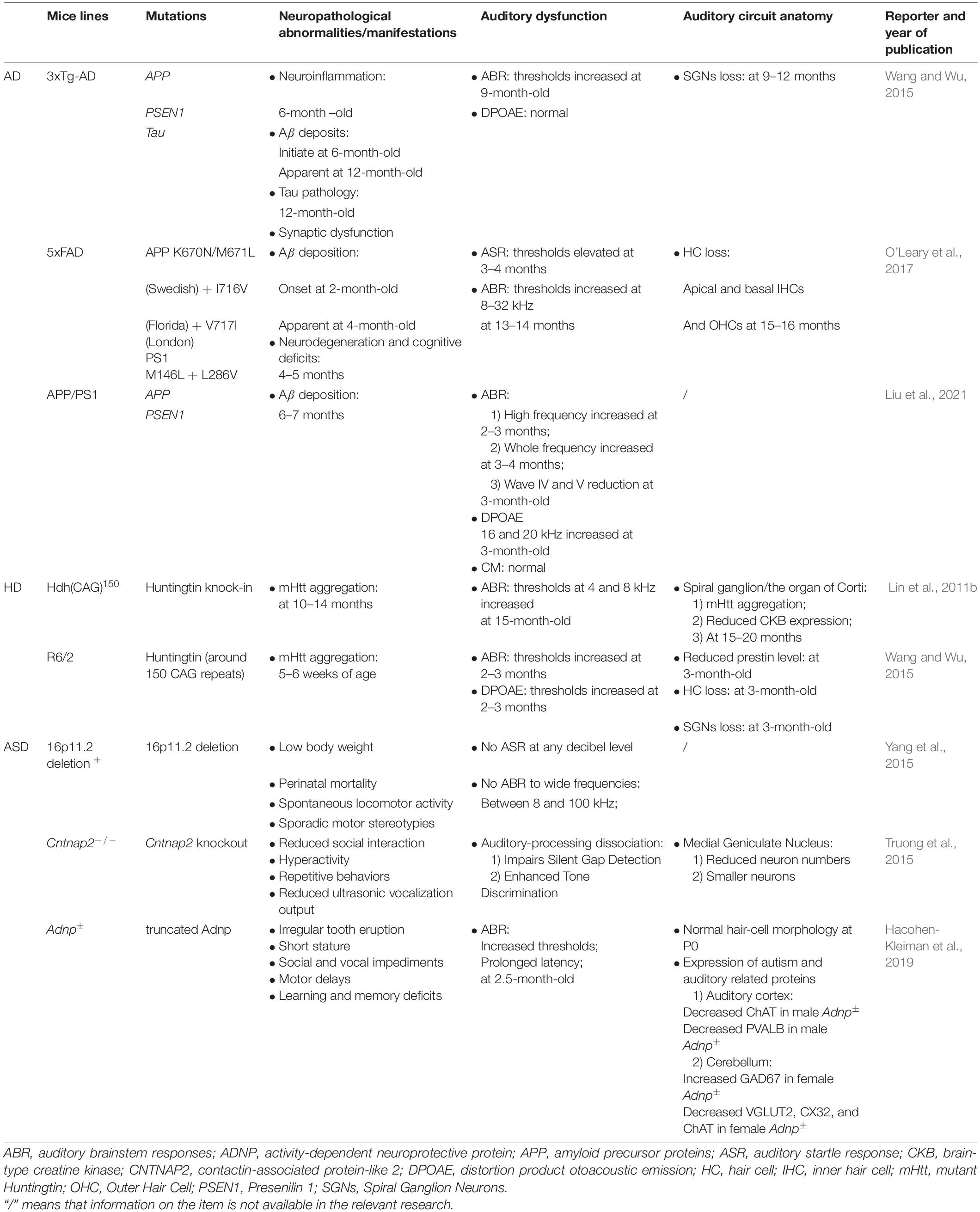
Table 1. List of AD, HD, ASD mice models that have been used for auditory function and anatomy study.
Clinical literature suggests that midlife hearing loss is independently correlated with accelerated progression of sporadic AD and incident dementia. The degree of hearing loss was positively associated with an increased risk of dementia. AD-related neuropathology was found in the central auditory pathway but was not clinically identified in the peripheral auditory pathway. Although multiple clinical studies have been designed to determine the relationship between hearing loss and AD, there are still some flaws in the experimental design. Most of the important information has been neglected, including the extent of hearing loss, measurements of auditory processing, differences between sexes, and auditory condition in different AD classifications, resulting in restricted access to information to determine the relationship between auditory dysfunction and AD. Mouse model studies further illustrated that hearing loss is associated with AD development. In the APP/PS1 mouse model, the shifts of ABR and distortion product otoacoustic emission (DPOAE) preceded the neuropathy observed in the brain. Loss of hair cells and SGNs was observed in AD mouse models, which was likely induced by the spread of AD-related neuropathology (Aβ deposition and tau pathology) in the cochlea. However, the three AD-related mouse models were all designed with mutations in FAD genes, which could not completely mimic the pathogenesis of sporadic AD. The central auditory pathway has not yet been studied in these mouse models. High-frequency hearing loss has been observed in patients with AD and incident dementia. Moreover, both the central and peripheral auditory pathways are affected by AD-related neuropathology, but the concrete cochlear pathology is still debated. The results vary among studies due to different mouse models, sampling times, and hearing measurements. Hence, for a comprehensive understanding of AD-related hearing loss, standard observation criteria should be established in further studies.
Huntington’s disease is an autosomal-dominantly inherited disorder with a mean prevalence of 2.71 per 100,000 individuals worldwide (Pringsheim et al., 2012). HD manifests with midlife cognitive impairment, motor incoordination, and psychiatric symptoms (Martin and Gusella, 1986; Walker, 2007). Late-stage HD patients often present with auditory sensory, processing, and memory problems other than typical dysfunctions. Studies have illustrated that hearing impairment is involved in and is closely correlated with motor deficits in HD (Josiassen et al., 1984; Lin et al., 2011b). Lin et al. (2011b) recruited 19 HD patients and assessed hearing impairments using PTA and ABR. The PTA thresholds showed that an average increase of 15 dB was detected in high frequencies of HD patients, and no significant differences were observed in latency and inter-peak intervals of ABRs, indicating that hearing impairments in HD were more associated with the peripheral auditory pathway than retrocochlear lesions (Lin et al., 2011b). In contrast, other researchers found that HD patients displayed normal sound sensation, but with a significant decrease in speech understanding and sound source lateralization, suggesting that HD-associated neuropathology affects the central auditory system (cortical and subcortical parts) (Beste et al., 2008; Saft et al., 2008; Profant et al., 2017). Wetter et al. (2005) revealed that HD patients had delayed auditory event-related potentials (ERPs), which were also found in individuals at risk for HD. These findings showed HD-related dysfunction during sound processing (Homberg et al., 1986; Josiassen et al., 1988; Wetter et al., 2005).
The pathophysiological mechanisms underlying HD-related auditory dysfunction are poorly understood; nonetheless, recent studies in transgenic mouse models provide new insight into these mechanisms (Walker, 2007). Aggregated mutant huntingtin (mHtt) is the most classic cellular pathological characteristic of HD; extra amplificated CAG repeats in exon 1 of huntingtin lead to polyglutamine (polyQ) extension at the N-terminal of Htt protein, and mutant Htt accumulates to cause neuronal loss (Figure 2; Macdonald et al., 1993; Mangiarini et al., 1996; Ha and Fung, 2012). Neuronal loss preferentially affects the cortico-striatal circuits, which leads to characteristic chorea, and as HD progresses, mHtt spreads to peripheral tissues, including the inner ear (Vonsattel and DiFiglia, 1998; Cepeda et al., 2007; Snowden, 2017). Mouse models were also used to illustrate auditory dysfunction and pathology in HD (summarized in Table 1). R6/2-HD mice express mHtt and present with HD-related phenotypes at 5–6 weeks of age. Lin et al. (2011b) found that R6/2-HD mice exhibited approximately 10 dB elevation of ABR thresholds at 2 months of age before the presentation of motor deficits. After 3 weeks, the motor defects became apparent, and a 30 dB threshold shift for click stimuli and a 15 dB threshold shift for tone bursts at all frequencies were also observed. In addition, they found no difference in ABR latency and peak intervals between R6/2-HD mice and wild type mice (Lin et al., 2011b). As described in Wang’s research, R6/2-HD mice exhibited increased distortion product otoacoustic emission and ABR thresholds at 2–3 months of age. Furthermore, the relative expression of prestin was reduced in OHCs, which was reported to be responsible for dysfunction in hearing sensitivity and frequency selectivity. Cochlear SGN reduction and hair cell loss (especially OHCs) were observed histologically after that (Wang and Wu, 2015), suggesting that mHtt pathology in the central and peripheral auditory system contributed to the presence of hearing impairments in HD. Hdh(CAG)150 mice are knock-in mice that carry 150 CAG repeats on the Htt locus, of which mHtt aggregation in the nervous system and HD-related characteristics initiate at approximately 10 months of age. In Hdh(CAG)150 mice, thresholds measured by click and tone bursts ABR analysis at 15 months of age revealed that approximately 20 dB thresholds were increased for click and tone bursts at frequencies of 4 and 8 kHz. No differences were observed at frequencies of 16 and 32 kHz because wild type mice developed presbycusis at 15 months of age (Lin et al., 2011b). Moreover, aggregated mHtt and continuing loss of brain-type creatine kinase (CKB), which was previously reported to decline in HD patients, were both obtained in the organ of Corti and the spiral ganglion in both mouse models (Perluigi et al., 2005; Sorolla et al., 2008; Kim et al., 2010; Lin et al., 2011b). Aggregation of mHtt is thought to affect many transcriptional factors and induce mitochondrial dysfunction, which directly leads to the release of cytochrome C and oxidative stress (Kim and Kim, 2014). CKB, a cytosolic enzyme, can regenerate ATP by reversibly transferring high-energy phosphate from phosphocreatine (PCr) to ADP (Jacobus and Lehninger, 1973; Wallimann et al., 1992; Wyss and Kaddurah-Daouk, 2000). CKB also localizes in cochlear hair cells and ligaments and is critical for hearing function (Spicer and Schulte, 1992; Spicer et al., 1997). CKB-knockout mice presented with high-tone hearing loss that can be restored by dietary creatine supplements (Shin et al., 2007; Lin et al., 2011c). These studies suggest that CKB dysregulation may be associated with HD-related auditory dysfunction, synergistic with mHtt (Figure 2).
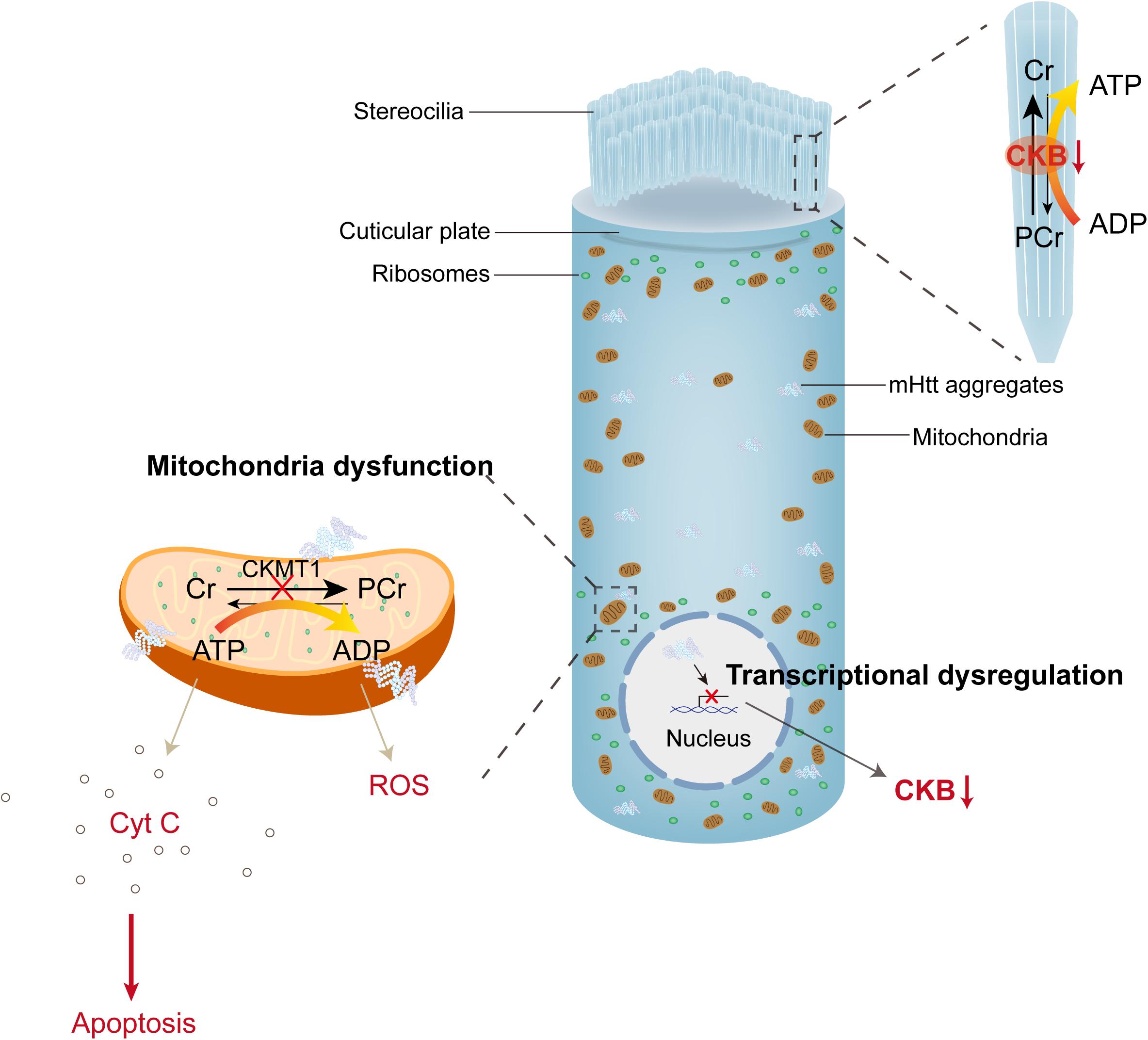
Figure 2. mHtt aggregates induced toxicity and dysregulated PCr-CK system in hair cells. The PCr-CK system plays a critical role in providing ATP in hair bundles of hair cells, mitochondrial creatine kinase (CKMT1) phosphorylates creatine (Cr) to phosphocreatine (PCr). In the stereocilia, brain-type creatine kinase (CKB) regenerates ATP from PCr. Expression of mHtt in hair cells impairs the function of mitochondria, releases cytochrome C and reactive oxygen species (ROS) to the cytoplasm. On the other hand, mHtt aggregates lead to protein sequestration (including many transcriptional factors), then induce transcriptional dysregulation, which reduces the expression of CKB (creatine kinase).
Auditory dysfunction appears to be authentic for HD. Clinical studies have shown that hearing impairments and auditory processing dysfunction are present in HD patients. Delayed ERPs are suggested to be a potential predictor of HD. While there is no consensus that auditory sense, processing, or discrimination is uniparted or jointly present in HD, more research objectives, detailed acoustic measurements, and specified auditory items should be included. Hearing impairment is solid in HD mouse models, and hearing loss precedes the occurrence of motor defects and worsens with the progression of HD in R6/2-HD mice. The loss of hair cells and SGNs was also observed. Hdh(CAG)150 mice exhibited significant low-frequency hearing impairment compared to wild type mice, which was accompanied by presbycusis-related high-frequency hearing loss, suggesting that hearing impairments in HD patients was not merely related to the auditory pathway degeneration caused by natural aging and clarifying that hearing loss was authentic to HD, while the cochlear anatomy has not been assessed in Hdh(CAG)150 mice. Moreover, pathological studies of the central auditory pathway, including cochlear nuclei, superior olivary complex, and auditory cortex, should be performed to determine auditory-related lesions and guide auditory-related tests carried out for HD patients and high-risk groups.
Parkinson’s disease is a neurodegenerative disorder characterized by static tremor, bradykinesia, rigidity, and postural instability, affecting 1–2 per 1,000 individuals (Dorsey et al., 2007; Ascherio and Schwarzschild, 2016). Before the onset of typical motor symptoms, patients with PD often manifest with cognitive impairment, olfactory dysfunction, fatigue, etc. (Postuma et al., 2012; Khoo et al., 2013). Recently, hearing impairment has been considered as another non-motor feature in PD patients. Studies have shown that the incidence of developing PD in the hearing loss group was 1.77 higher than in the control group (Lai et al., 2014b), and high-frequency hearing impairment was observed in PD patients without self-perceiving (Yylmaz et al., 2009; Santos-Garcia et al., 2010; Shetty et al., 2019), which is positively related to PD duration and worsens as it progresses (Scarpa et al., 2020). PTA results showed an average elevation of 10 dB in 4 and 8 kHz in PD patients, and significantly increased latencies in wave V and interpeak were obtained (Yylmaz et al., 2009). PTA performed among the relatively younger (age < 55 years old) population of PD showed that thresholds were elevated at high frequency and low to mid frequencies. This ratio was even higher for low-mid-frequency hearing loss. Concurrently, the brain stem auditory-evoked potentials were comparable to the control group, indicating that hearing loss in PD was independent of aging and that the underlying mechanism appeared to be peripheral according to the study (Shetty et al., 2019). Vitale et al. (2013) calculated the proportion of different degrees of hearing loss in 75 patients with PD and found that 89% of them had mild to moderate hearing loss, and 11% had severe hearing loss. In addition, they revealed that the prevalence of PD with hearing impairments was higher in the male elderly (Vitale et al., 2013). Whole frequencies of distortion product otoacoustic emission thresholds in PD patients also increased. It can be alleviated by dopaminergic treatment (Georgiev et al., 2015; Pisani et al., 2015), which uncovered an undermined dopamine-dependent cochlear dysfunction undermined. Sisto et al. (2020) found that the ipsilateral cochlear dysfunction developed in parallel with asymmetric motor impairment. The abilities of speech discrimination and sound lateralization were also markedly reduced in PD patients (Lewald et al., 2004; Vitale et al., 2016; Folmer et al., 2017), and abnormal auditory evoked potentials were suggested as a measurement of PD duration and severity (Yylmaz et al., 2009; Jafari et al., 2020).
The association between hearing dysfunction and PD suggests a common neuropathological background. Lewy pathology and dopaminergic neuronal degeneration are two primary neuropathological features of PD that spread as PD progresses (Dickson et al., 2009; Dickson, 2012; Kordower et al., 2013). Other protein aggregations, such as Aβ plaques and NFTs, are also present in the nervous system of PD patients (Kalia and Lang, 2015). Lewy pathology consists of insoluble misfolded α-synuclein that can be found in certain regions of the central and peripheral nervous system in PD (Wakabayashi et al., 1989; Spillantini et al., 1997; Beach et al., 2010; Del Tredici et al., 2010; Goedert et al., 2013). In the inner ear, α-synuclein is located predominantly in the efferent neuronal system, especially in the OHC, and contributes to the physiological maintenance of auditory function. Hence, Lewy pathology in the auditory system has been speculated to be associated with PD-related auditory disorders (Akil et al., 2008; Park et al., 2011). On the other hand, common neurotransmitters between the auditory system and basal ganglia were indicated by the curative effect of dopaminergic therapy on auditory responses (Rey et al., 1996; Erro et al., 2015; Georgiev et al., 2015; Pisani et al., 2015). Furthermore, dopamine and glutamate mediate the synaptic interplay oppositely in the basal ganglia. In the auditory system, dopamine also counteracts the excitotoxic effects caused by glutamate to modulate auditory processing and neural plasticity. Since glutamate overdose can induce excitotoxic damage to primary auditory neurons, it was speculated that excessive glutamate caused by the degeneration of dopaminergic neurons might account for PD-related auditory dysfunction (Lendvai et al., 2011). Other common underlying mechanisms, including mitochondrial dysfunction, reduced neurotransmitter levels, perturbed protein homeostasis, and oxidative stress, have also been discussed in previous studies (Simon and Johns, 1999; Raza et al., 2019).
There have been no reports on auditory dysfunction and auditory anatomy in PD mouse model. Although manipulation of specific genes reported in familial PD, including transgenic overexpression for α-synuclein and leucine-rich repeat kinase 2 and knockout models for Parkin, DJ-1, phosphatase, and tensin homolog-induced novel kinase 1, made it possible to establish many mouse models, none of them recapitulate key clinical and neuropathological features of PD entirely, especially in the absence of neurodegeneration of dopaminergic neurons (Dawson et al., 2010). While the objective is to gain insight into the molecular mechanisms underlying auditory dysfunction and PD, studies of auditory function in mouse models with specific gene mutations are still needed. PD is a global neurodegenerative disorder that affects the central and peripheral nervous system, and extensive literature indicated a broad range of auditory dysfunctions from the peripheral auditory system to cortical areas in PD (Pekkonen et al., 1995; Kofler et al., 2001; Putzki et al., 2008; Bronnick et al., 2010; Pisani et al., 2015; Potter-Nerger et al., 2015; Seidel et al., 2015; Liu et al., 2017; Shalash et al., 2017; Guducu et al., 2019), asymptomatic hearing impairments appeared to be a newly non-motor manifestation of both early and late-onset PD, and it can be speculated that the natural aging process combined with PD-related neurodegenerative changes coalesce to induce that. Moreover, the central auditory dysfunctions, including abnormal speech discrimination and sound lateralization, cannot be ignored. The literature suggests hearing measurement as a non-invasive potential biomarker and indicator of disease severity for PD, widespread alpha-synuclein neuropathology, and loss of dopaminergic neurons were suspected of interfering with such auditory dysfunction, and PD mouse models should be applied for precise assessment of hearing function and pathological mechanism exploration.
Autism spectrum disorder is a neurodevelopmental disorder characterized by social isolation, stereotypical behaviors, and interests, with a prevalence of approximately 1% worldwide and has a strong male predominance (Johnson et al., 2007; Lai et al., 2014a). Sense dysfunction, including the feeling of touch, smell, taste, vision, and hearing, is another feature of ASD. The pathogenesis of ASD is not entirely understood, but comorbidities and maternal exposures in placental life may act as risk factors (Arndt et al., 2005). Researchers have suggested that genetic polymorphisms and environmental factors jointly contribute to the phenotypic variation in ASD (Bailey et al., 1995; Veenstra-VanderWeele et al., 2004; Lopez-Rangel and Lewis, 2006). Cerebellar and brainstem hypoplasia was observed in patients with ASD (Courchesne et al., 1988; Hashimoto et al., 1992, 1995), and multiregional neuropathy defects have been identified, including a reduced number of Purkinje cells in the cerebellum, delayed neuron maturation of the forebrain, abnormal development of the frontal and temporal lobes, and sporadic malformation in the brainstem and neocortex (Hardan et al., 2004; Pickett and London, 2005; Lainhart, 2006; Wegiel et al., 2010, 2014; Hampson and Blatt, 2015). These structural abnormalities lead to typical behavioral manifestations and sense dysfunctions in ASD.
Currently, most previous studies identified increased rates of audiological dysfunctions in ASD, including hearing impairments, hyperacusis, difficulty in sound discrimination with background noise and speech sounds encoding (Tomchek and Dunn, 2007; Russo et al., 2009; Stiegler and Davis, 2010). A higher incidence of hearing loss (from unilateral to bilateral) and hyperacusis was demonstrated in the ASD population (Rosenhall et al., 1999; Demopoulos and Lewine, 2016; Do et al., 2017). Fitzpatrick et al. (2014) found that approximately 29.4% of children with ASD had profound hearing loss and that those children with hearing loss benefited from the use of hearing aids. In addition, hearing dysfunction was attributed to ASD-related neuronal degeneration of the auditory pathway (Smith et al., 2019). In contrast, Szymanski et al. (2012) found a high prevalence of ASD among children with hearing loss, supporting that peripheral auditory dysfunction may be associate with functional impairment in ASD (Demopoulos and Lewine, 2016). Previous studies have provided an abundance of evidence supporting both abnormal structure and function in the auditory brainstem of ASD, but there remains a battery of literature showing that the peripheral auditory manifestations of children with ASD were comparable to controls (Gravel et al., 2006; Tharpe et al., 2006). Beers et al. (2014) reviewed 22 articles about peripheral hearing loss in ASD and concluded that there was no solid evidence for an increased risk of peripheral hearing loss among children with ASD. Tas et al. (2007) also evaluated the auditory function of children with ASD through transient evoked otoacoustic emission and ABR. The positive emission and normal hearing level at ABR revealed an insusceptible peripheral auditory system in patients with ASD. Nevertheless, the ABR results showed a prolonged III–V interpeak latencies (IPLs) in children with autism (Rosenhall et al., 2003; Tas et al., 2007).
Approximately 10% of ASD cases have an identifiable genetic background. Many ASD-related genetic and chromosomal disorders have been shown to present with auditory dysfunction (Table 2), underlying a potential common genetic etiology between ASD and auditory dysfunction. Many chromosomal disorders have been reported to manifest with auditory dysfunctions, ASD, developmental retardation, seizures, facial dysmorphism, and multisystem defects. Deletions and duplications range from specific loci to large segments and comprise a considerable number of related genes. Genes with remarkably high risk accounting for these manifestations are listed in Table 2. Most of them are involved in neuron and synaptic development (Smith et al., 2002; Sinajon et al., 2015; Yang et al., 2015; Lahbib et al., 2019; Wu et al., 2020a), among which only two genes are known to be auditory-related: ELMOD3 and FGF2. ELMOD3 is involved in autosomal recessive non-syndromic deafness disability (Lahbib et al., 2019), and FGF2 plays a role in the proliferation and survival of auditory neuroblasts (Wu et al., 2020a). Three monogenic disorders were reported to present with auditory dysfunction and ASD simultaneously, including Fragile X syndrome, MEIS2 syndrome, and ADNP syndrome (Rotschafer et al., 2015; Douglas et al., 2018; Hacohen-Kleiman et al., 2019), related genes all function in brain development, and MEIS2 is responsible for the development of the inner ear in chicken (Douglas et al., 2018). Restricted information about genes and their functions is insufficient to illustrate the genetic association between auditory dysfunction and ASD. Whether these certified ASD-related genes also participate in auditory function is unclear.

Table 2. List of ASD-related chromosomal and monogenic disorders that have been reported co-presented with auditory dysfunction.
ASD-related mouse models have been developed to study auditory dysfunction (Table 1). Chromosomal disorder mice characterized by 16p 11.2 deletions showed whole frequency increased ABR and auditory startle response (ASR) thresholds, indicating that genes located in the area were responsible for auditory dysfunctions, of which KCTD13, SEZ6L2, and MAPK3 were considered to be highly correlated with autism (Konyukh et al., 2011; Golzio et al., 2012; Blumenthal et al., 2014; Yang et al., 2015), while their relationship with the auditory function has not been determined. Monogenic disorder mice were also studied; Fmr1–/–, Cntnap2–/– and Adnp± mice presented with classical characteristics of ASD and showed impaired hearing and auditory process functions. Anatomy of auditory circuits, such as the ventral cochlear nucleus and the medial nucleus of the trapezoid body exhibited reduced neuron size and number. Altered hearing-related protein levels, including VGAT, ChAT, and GAD67, were observed in the auditory cortex and cerebellum (Rotschafer et al., 2015; Ruby et al., 2015; Truong et al., 2015; Hacohen-Kleiman et al., 2019), underlying a central auditory and synaptic pathology.
Increased rates of auditory dysfunction, including hearing impairments, hyperacusis, difficulties in sound discrimination, and speech sounds encoding, were detected in patients with ASD. ASD children with hearing impairments were identified later than those with normal hearing for auditory disorders and related communication delays. Although hearing impairment is an uncommon manifestation of ASD, both diseases affect communication abilities in children, and hearing impairment may contribute to the development of ASD. Comprehensive audiological assessments of confirmed and suspected ASD in children and early hearing interventions are recommended to improve social communication and reduce the aggression of ASD. ASD with chromosomal and monogenetic disorders has been shown to manifest with hearing impairments and auditory process problems, which correspond to auditory dysfunctions in ASD mouse models, and reduced neuron size and number were observed in auditory brainstem nuclei. Studies have also reviewed the genes that might be involved in chromosomal and monogenetic disorders. They concluded that most of them function in neuronal development, suggesting that defective neuropathy in the auditory pathway leads to hearing dysfunction and raises the idea that ASD-related genes may act as potential deafness genes. Hence, specific transgenic mouse models should be applied to clarify their function and influence on the auditory system. For further analysis, next-generation sequencing should be applied to identify more potential ASD-related candidate genes for deafness.
In this review, we presented research on hearing loss in four common neurological disorders (AD, PD, HD, and ASD) and concluded that hearing loss was present in these four disorders. However, the related auditory lesions and underlying mechanisms vary among them.
In AD, high-frequency hearing loss was observed in both the patient and mouse models. Moreover, Aβ deposition appeared to be the initial neurological etiology. Auditory studies on AD mouse models raise the possibility that the auditory pathway is more sensitive to AD-related neuropathology and auditory dysfunction, especially hearing loss, presents before the onset of cognitive impairments. Thus, auditory measurements can provide a reference for preliminary diagnosis and early interventions for patients with AD. Hearing impairments and auditory processing dysfunction have been observed in HD patients. In R6/2-HD mice, hearing loss precedes the characterized presentations of HD, and in the SGNs of Hdh(CAG)150 mice, mHtt aggregation was observed. However, for a comprehensive understanding of auditory dysfunction in AD and HD, more clinical trials involving more subjects and including a variety of detailed auditory measurements should be carried out, and complete studies on auditory circuitry (from the cochlea to the auditory cortex) of mouse models should be conducted in the future.
A broad range of auditory dysfunctions, including hearing loss, abnormal speech discrimination, and sound lateralization, have been reported in PD, and asymptomatic hearing impairments appear to be a new non-motor symptom of PD patients, and hearing measurements may act as a non-invasive potential biomarker and indicator of disease severity. There are no transgenic mouse models that can completely mimic the important neuropathological features of PD, especially the neurodegeneration of dopaminergic neurons. Hence, better PD mouse models should be established, and to gain insight into the underlying molecular mechanisms, auditory studies in existing mouse models still worth exploring.
Many auditory dysfunctions, including hearing impairments, hyperacusis, difficulty in sound discrimination, and speech sound encoding, have been detected in patients with ASD. Concomitant hearing loss makes the diagnosis of ASD more challenging. As both disorders affect communication abilities in children and early hearing interventions have been reported to improve social communication and reduce aggression in ASD, comprehensive audiological assessments should be carried out in confirmed and suspected ASD in children. Moreover, approximately 10% of ASD cases have an identifiable genetic background. Clinical and transgenic mouse model studies revealed the involvement of hearing impairments, raising the possibility that associated genes may act as potential deafness genes. Hence, more potential ASD-related candidate genes should be identified, and specific transgenic mouse models should be applied to explore the function of autism-related genes in the auditory system.
Sensorineural hearing loss affects a large population of people worldwide, and the impact of hearing loss is broad and profound, including delayed language development in children, social isolation, and psychological illness. Hearing loss is not only present in neurological disorders mentioned above but can also affect the prognosis of these diseases to some extent. Hence, exploring hearing loss in neurological disorders is beneficial for understanding the pathogenesis and improving the prognosis of these diseases.
SL and CC wrote the manuscript. LL, XM, XZ, and AL collated the resource. JC, XQ, and XG wrote and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (82071059, 82071044, 81970884, 81900941, 81970885, 81900944, 81870721, and 81771019), the Natural Science Foundation of Jiangsu Province (BK20190121), the Jiangsu Provincial Key Research and Development Fund (BE2018605), and the China Postdoctoral Science Foundation (2020M681555).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alzheimer’s Association (2020). 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 16, 391–460. doi: 10.1002/alz.12068
Akil, O., Weber, C. M., Park, S. N., Ninkina, N., Buchman, V., and Lustig, L. R. (2008). Localization of synucleins in the mammalian cochlea. J. Assoc. Res. Otolaryngol. 9, 452–463. doi: 10.1007/s10162-008-0134-y
Albers, M. W., Gilmore, G. C., Kaye, J., Murphy, C., Wingfield, A., Bennett, D. A., et al. (2015). At the interface of sensory and motor dysfunctions and Alzheimer’s disease. Alzheimers Dement. 11, 70–98. doi: 10.1016/j.jalz.2014.04.514
Appler, J. M., and Goodrich, L. V. (2011). Connecting the ear to the brain: molecular mechanisms of auditory circuit assembly. Prog. Neurobiol. 93, 488–508. doi: 10.1016/j.pneurobio.2011.01.004
Arndt, T. L., Stodgell, C. J., and Rodier, P. M. (2005). The teratology of autism. Int. J. Dev. Neurosci. 23, 189–199. doi: 10.1016/j.ijdevneu.2004.11.001
Ascherio, A., and Schwarzschild, M. A. (2016). The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol 15, 1257–1272. doi: 10.1016/s1474-4422(16)30230-7
Attems, J., Walker, L., and Jellinger, K. A. (2014). Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol. 127, 459–475. doi: 10.1007/s00401-014-1261-7
Bailey, A., Lecouteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., et al. (1995). Autism as a strongly genetic disorder - evidence from a British twin study. Psychol. Med. 25, 63–77. doi: 10.1017/S0033291700028099
Beach, T. G., Adler, C. H., Sue, L. I., Vedders, L., Lue, L., White Iii, C. L., et al. (2010). Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 119, 689–702. doi: 10.1007/s00401-010-0664-3
Beers, A. N., McBoyle, M., Kakande, E., Dar Santos, R. C., and Kozak, F. K. (2014). Autism and peripheral hearing loss: a systematic review. Int. J. Pediatr. Otorhinolaryngol. 78, 96–101. doi: 10.1016/j.ijporl.2013.10.063
Benarroch, E. E. (2010). Olfactory system: functional organization and involvement in neurodegenerative disease. Neurology 75, 1104–1109. doi: 10.1212/WNL.0b013e3181f3db84
Beste, C., Saft, C., Gunturkun, O., and Falkenstein, M. (2008). Increased cognitive functioning in symptomatic Huntington’s disease as revealed by behavioral and event-related potential indices of auditory sensory memory and attention. J. Neurosci. 28, 11695–11702. doi: 10.1523/JNEUROSCI.2659-08.2008
Blumenthal, I., Ragavendran, A., Erdin, S., Klei, L., Sugathan, A., Guide, J. R., et al. (2014). Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet. 94, 870–883. doi: 10.1016/j.ajhg.2014.05.004
Bronnick, K. S., Nordby, H., Larsen, J. P., and Aarsland, D. (2010). Disturbance of automatic auditory change detection in dementia associated with Parkinson’s disease: a mismatch negativity study. Neurobiol. Aging 31, 104–113. doi: 10.1016/j.neurobiolaging.2008.02.021
Cepeda, C., Wu, N., Andre, V. M., Cummings, D. M., and Levine, M. S. (2007). The corticostriatal pathway in Huntington’s disease. Prog. Neurobiol. 81, 253–271. doi: 10.1016/j.pneurobio.2006.11.001
Chen, Y., Gu, Y., Li, Y., Li, G. L., Chai, R., Li, W., et al. (2021). Generation of mature and functional hair cells by co-expression of Gfi1, Pou4f3, and Atoh1 in the postnatal mouse cochlea. Cell Rep. 35:109016. doi: 10.1016/j.celrep.2021.109016
Coate, T. M., and Kelley, M. W. (2013). Making connections in the inner ear: recent insights into the development of spiral ganglion neurons and their connectivity with sensory hair cells. Semin. Cell Dev. Biol. 24, 460–469. doi: 10.1016/j.semcdb.2013.04.003
Courchesne, E., Yeungcourchesne, R., Press, G. A., Hesselink, J. R., and Jernigan, T. L. (1988). Hypoplasia of cerebellar vermal Lobule-Vi and Lobule-Vii in autism. N. Engl. J. Med. 318, 1349–1354. doi: 10.1056/Nejm198805263182102
Dallos, P. (1986). Neurobiology of cochlear inner and outer hair cells: intracellular recordings. Hear. Res. 22, 185–198. doi: 10.1016/0378-5955(86)90095-x
Davis, A. A., Leyns, C. E. G., and Holtzman, D. M. (2018). Intercellular spread of protein aggregates in neurodegenerative disease. Annu. Rev. Cell Dev. Biol. 34, 545–568. doi: 10.1146/annurev-cellbio-100617-062636
Dawson, T. M., Ko, H. S., and Dawson, V. L. (2010). Genetic animal models of Parkinson’s disease. Neuron 66, 646–661. doi: 10.1016/j.neuron.2010.04.034
Del Tredici, K., Hawkes, C. H., Ghebremedhin, E., and Braak, H. (2010). Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 119, 703–713. doi: 10.1007/s00401-010-0665-2
Demopoulos, C., and Lewine, J. D. (2016). Audiometric profiles in autism spectrum disorders: does subclinical hearing loss impact communication? Autism Res. 9, 107–120. doi: 10.1002/aur.1495
Dickson, D. W. (2012). Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2:a009258. doi: 10.1101/cshperspect.a009258
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., et al. (2009). Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 8, 1150–1157. doi: 10.1016/S1474-4422(09)70238-8
Do, B., Lynch, P., Macris, E. M., Smyth, B., Stavrinakis, S., Quinn, S., et al. (2017). Systematic review and meta-analysis of the association of Autism spectrum disorder in visually or hearing impaired children. Ophthalmic Physiol. Opt. 37, 212–224. doi: 10.1111/opo.12350
Dorsey, E. R., Constantinescu, R., Thompson, J. P., Biglan, K. M., Holloway, R. G., Kieburtz, K., et al. (2007). Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68, 384–386. doi: 10.1212/01.wnl.0000247740.47667.03
Douglas, G., Cho, M. T., Telegrafi, A., Winter, S., Carmichael, J., Zackai, E. H., et al. (2018). De novo missense variants in MEIS2 recapitulate the microdeletion phenotype of cardiac and palate abnormalities, developmental delay, intellectual disability and dysmorphic features. Am. J. Med. Genet. A 176, 1845–1851. doi: 10.1002/ajmg.a.40368
Erro, R., Picillo, M., Amboni, M., Moccia, M., Vitale, C., Longo, K., et al. (2015). Nonmotor predictors for levodopa requirement in de novo patients with Parkinson’s disease. Mov. Disord. 30, 373–378. doi: 10.1002/mds.26076
Fitzpatrick, E. M., Lambert, L., Whittingham, J., and Leblanc, E. (2014). Examination of characteristics and management of children with hearing loss and autism spectrum disorders. Int. J. Audiol. 53, 577–586. doi: 10.3109/14992027.2014.903338
Folmer, R. L., Vachhani, J. J., Theodoroff, S. M., Ellinger, R., and Riggins, A. (2017). Auditory processing abilities of Parkinson’s disease patients. Biomed. Res. Int. 2017:2618587. doi: 10.1155/2017/2618587
Ford, A. H., Hankey, G. J., Yeap, B. B., Golledge, J., Flicker, L., and Almeida, O. P. (2018). Hearing loss and the risk of dementia in later life. Maturitas 112, 1–11. doi: 10.1016/j.maturitas.2018.03.004
Fortunato, S., Forli, F., Guglielmi, V., De Corso, E., Paludetti, G., Berrettini, S., et al. (2016). A review of new insights on the association between hearing loss and cognitive decline in ageing. Acta Otorhinolaryngol. Ital. 36, 155–166. doi: 10.14639/0392-100X-993
Fu, X., An, Y., Wang, H., Li, P., Lin, J., Yuan, J., et al. (2021a). Deficiency of Klc2 induces low-frequency sensorineural hearing loss in C57BL/6 J mice and human. Mol. Neurobiol. doi: 10.1007/s12035-021-02422-w
Fu, X., Wan, P., Li, P., Wang, J., Guo, S., Zhang, Y., et al. (2021b). Mechanism and prevention of ototoxicity induced by aminoglycosides. Front. Cell Neurosci. 15:692762. doi: 10.3389/fncel.2021.692762
Gallacher, J., Ilubaera, V., Ben-Shlomo, Y., Bayer, A., Fish, M., Babisch, W., et al. (2012). Auditory threshold, phonologic demand, and incident dementia. Neurology 79, 1583–1590. doi: 10.1212/WNL.0b013e31826e263d
Georgiev, D., Jahanshahi, M., Dreo, J., Cus, A., Pirtosek, Z., and Repovs, G. (2015). Dopaminergic medication alters auditory distractor processing in Parkinson’s disease. Acta Psychol. 156, 45–56. doi: 10.1016/j.actpsy.2015.02.001
Goedert, M., Spillantini, M. G., Del Tredici, K., and Braak, H. (2013). 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24. doi: 10.1038/nrneurol.2012.242
Golzio, C., Willer, J., Talkowski, M. E., Oh, E. C., Taniguchi, Y., Jacquemont, S., et al. (2012). KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485, 363–367. doi: 10.1038/nature11091
Gravel, J. S., Dunn, M., Lee, W. W., and Ellis, M. A. (2006). Peripheral audition of children on the autistic spectrum. Ear Hear. 27, 299–312. doi: 10.1097/01.aud.0000215979.65645.22
Grothe, B., Pecka, M., and McAlpine, D. (2010). Mechanisms of sound localization in mammals. Physiol. Rev. 90, 983–1012. doi: 10.1152/physrev.00026.2009
Guducu, C., Eskicioglu, E., Oz, D., Oniz, A., Cakmur, R., and Ozgoren, M. (2019). Auditory brain oscillatory responses in drug-naive patients with Parkinson’s disease. Neurosci. Lett. 701, 170–174. doi: 10.1016/j.neulet.2019.02.039
Guo, R. R., Li, J., Chen, C. T., Xiao, M., Liao, M. H., Hu, Y. N., et al. (2021). Biomimetic 3D bacterial cellulose-graphene foam hybrid scaffold regulates neural stem cell proliferation and differentiation. Colloids Surf. B-Biointerfaces 200:111590. doi: 10.1016/j.colsurfb.2021.111590
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. doi: 10.1007/s00401-017-1707-9
Ha, A. D., and Fung, V. S. (2012). Huntington’s disease. Curr. Opin. Neurol. 25, 491–498. doi: 10.1097/WCO.0b013e3283550c97
Hacohen-Kleiman, G., Yizhar-Barnea, O., Touloumi, O., Lagoudaki, R., Avraham, K. B., Grigoriadis, N., et al. (2019). Atypical auditory brainstem response and protein expression aberrations related to asd and hearing loss in the adnp haploinsufficient mouse brain. Neurochem. Res. 44, 1494–1507. doi: 10.1007/s11064-019-02723-6
Hampson, D. R., and Blatt, G. J. (2015). Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 9:420. doi: 10.3339/fnins.2075.00420
Hardan, A. Y., Jou, R. J., Keshavan, M. S., Varma, R., and Minshew, N. J. (2004). Increased frontal cortical folding in autism: a preliminary MRI study. Psychiatry Res. 131, 263–268. doi: 10.1016/j.pscychresns.2004.06.001
Hashimoto, T., Tayama, M., Miyazaki, M., Sakurama, N., Yoshimoto, T., Murakawa, K., et al. (1992). Reduced brainstem size in children with autism. Brain Dev. 14, 94–97. doi: 10.1016/s0387-7604(12)80093-3
Hashimoto, T., Tayama, M., Murakawa, K., Yoshimoto, T., Miyazaki, M., Harada, M., et al. (1995). Development of the brainstem and cerebellum in autistic patients. J. Autism Dev. Disord. 25, 1–18. doi: 10.1007/BF02178163
He, Z. H., Li, M., Fang, Q. J., Liao, F. L., Zou, S. Y., Wu, X., et al. (2021). FOXG1 promotes aging inner ear hair cell survival through activation of the autophagy pathway. Autophagy 1–22. doi: 10.1080/15548627.2021.1916194
He, Z. H., Zou, S. Y., Li, M., Liao, F. L., Wu, X., Sun, H. Y., et al. (2020). The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biol. 28:101364. doi: 10.1016/j.redox.2019.101364
Herrero, M. T., and Morelli, M. (2017). Multiple mechanisms of neurodegeneration and progression. Prog. Neurobiol. 155:1. doi: 10.1016/j.pneurobio.2017.06.001
Homberg, V., Hefter, H., Granseyer, G., Strauss, W., Lange, H., and Hennerici, M. (1986). Event-related potentials in patients with Huntington’s disease and relatives at risk in relation to detailed psychometry. Electroencephalogr. Clin. Neurophysiol. 63, 552–569. doi: 10.1016/0013-4694(86)90143-4
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Hung, S. C., Liao, K. F., Muo, C. H., Lai, S. W., Chang, C. W., and Hung, H. C. (2015). Hearing loss is associated with risk of Alzheimer’s disease: a case-control study in older people. J. Epidemiol. 25, 517–521. doi: 10.2188/jea.JE20140147
Jacobus, W. E., and Lehninger, A. L. (1973). Creatine kinase of rat heart mitochondria. coupling of creatine phosphorylation to electron transport. J. Biol. Chem. 248, 4803–4810.
Jafari, Z., Kolb, B. E., and Mohajerani, M. H. (2019). Age-related hearing loss and tinnitus, dementia risk, and auditory amplification outcomes. Ageing Res. Rev. 56:100963. doi: 10.1016/j.arr.2019.100963
Jafari, Z., Kolb, B. E., and Mohajerani, M. H. (2020). Auditory dysfunction in Parkinson’s disease. Mov. Disord. 35, 537–550. doi: 10.1002/mds.28000
Johnson, C. P., Myers, S. M., and American Academy of Pediatrics Council on Children With Disabilities (2007). Identification and evaluation of children with autism spectrum disorders. Pediatrics 120, 1183–1215. doi: 10.1542/peds.2007-2361
Josiassen, R. C., Shagass, C., Mancall, E. L., and Roemer, R. A. (1984). Auditory and visual evoked potentials in Huntington’s disease. Electroencephalogr. Clin. Neurophysiol. 57, 113–118. doi: 10.1016/0013-4694(84)90169-x
Josiassen, R. C., Shagass, C., Roemer, R. A., and Mancall, E. (1988). A sensory evoked potential comparison of persons ‘at risk’ for Huntington’s disease and hospitalized neurotic patients. Int. J. Psychophysiol. 6, 281–289. doi: 10.1016/0167-8760(88)90015-3
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/s0140-6736(14)61393-3
Keithley, E. M. (2020). Pathology and mechanisms of cochlear aging. J. Neurosci. Res. 98, 1674–1684. doi: 10.1002/jnr.24439
Khoo, T. K., Yarnall, A. J., Duncan, G. W., Coleman, S., O’Brien, J. T., Brooks, D. J., et al. (2013). The spectrum of nonmotor symptoms in early Parkinson disease. Neurology 80, 276–281. doi: 10.1212/WNL.0b013e31827deb74
Kim, J., Amante, D. J., Moody, J. P., Edgerly, C. K., Bordiuk, O. L., Smith, K., et al. (2010). Reduced creatine kinase as a central and peripheral biomarker in Huntington’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1802, 673–681. doi: 10.1016/j.bbadis.2010.05.001
Kim, S., and Kim, K. T. (2014). Therapeutic approaches for inhibition of protein aggregation in Huntington’s disease. Exp. Neurobiol. 23, 36–44. doi: 10.5607/en.2014.23.1.36
Kofler, M., Muller, J., Wenning, G. K., Reggiani, L., Hollosi, P., Bosch, S., et al. (2001). The auditory startle reaction in parkinsonian disorders. Mov. Disord. 16, 62–71. doi: 10.1002/1531-8257(200101)16:1<62::aid-mds1002<3.0.co;2-v
Konyukh, M., Delorme, R., Chaste, P., Leblond, C., Lemiere, N., Nygren, G., et al. (2011). Variations of the candidate SEZ6L2 gene on Chromosome 16p11.2 in patients with autism spectrum disorders and in human populations. PLoS One 6:e17289. doi: 10.1371/journal.pone.0017289
Kordower, J. H., Olanow, C. W., Dodiya, H. B., Chu, Y., Beach, T. G., Adler, C. H., et al. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136(Pt 8), 2419–2431. doi: 10.1093/brain/awt192
Kritsilis, M., Rizou, S. V., Koutsoudaki, P. N., Evangelou, K., Gorgoulis, V. G., and Papadopoulos, D. (2018). Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 19:2937. doi: 10.3390/ijms19102937
Lahbib, S., Leblond, C. S., Hamza, M., Regnault, B., Lemee, L., Mathieu, A., et al. (2019). Homozygous 2p11.2 deletion supports the implication of ELMOD3 in hearing loss and reveals the potential association of CAPG with ASD/ID etiology. J. Appl. Genet. 60, 49–56. doi: 10.1007/s13353-018-0472-3
Lai, M.-C., Lombardo, M. V., and Baron-Cohen, S. (2014a). Autism. Lancet 383, 896–910. doi: 10.1016/s0140-6736(13)61539-1
Lai, S. W., Liao, K. F., Lin, C. L., Lin, C. C., and Sung, F. C. (2014b). Hearing loss may be a non-motor feature of Parkinson’s disease in older people in Taiwan. Eur. J. Neurol. 21, 752–757. doi: 10.1111/ene.12378
Lainhart, J. E. (2006). Advances in autism neuroimaging research for the clinician and geneticist. Am. J. Med. Genet. C Semin. Med. Genet. 142C, 33–39. doi: 10.1002/ajmg.c.30080
Lendvai, B., Halmos, G. B., Polony, G., Kapocsi, J., Horvath, T., Aller, M., et al. (2011). Chemical neuroprotection in the cochlea: the modulation of dopamine release from lateral olivocochlear efferents. Neurochem. Int. 59, 150–158. doi: 10.1016/j.neuint.2011.05.015
Lewald, J., Schirm, S. N., and Schwarz, M. (2004). Sound lateralization in Parkinson’s disease. Brain Res. Cogn. Brain Res. 21, 335–341. doi: 10.1016/j.cogbrainres.2004.06.008
Li, A., You, D., Li, W. Y., Cui, Y. J., He, Y. Z., Li, W., et al. (2018a). Novel compounds protect auditory hair cells against gentamycin-induced apoptosis by maintaining the expression level of H3K4me2. Drug Deliv. 25, 1033–1043. doi: 10.1080/10717544.2018.1461277
Li, H., Song, Y. D., He, Z. H., Chen, X. Y., Wu, X. M., Li, X. F., et al. (2018b). Meclofenamic acid reduces reactive oxygen species accumulation and apoptosis, inhibits excessive autophagy, and protects hair cell-like HEI-OC1 cells from Cisplatin-induced damage. Front. Cell. Neurosci. 12:139. doi: 10.3389/fncel.2018.00139
Lin, F. R., Metter, E. J., O’Brien, R. J., Resnick, S. M., Zonderman, A. B., and Ferrucci, L. (2011a). Hearing loss and incident dementia. Arch. Neurol. 68, 214–220. doi: 10.1001/archneurol.2010.362
Lin, F. R., Yaffe, K., Xia, J., Xue, Q. L., Harris, T. B., Purchase-Helzner, E., et al. (2013). Hearing loss and cognitive decline in older adults. JAMA Intern. Med. 173, 293–299. doi: 10.1001/jamainternmed.2013.1868
Lin, Y. S., Chen, C. M., Soong, B. W., Wu, Y. R., Chen, H. M., Yeh, W. Y., et al. (2011b). Dysregulated brain creatine kinase is associated with hearing impairment in mouse models of Huntington disease. J. Clin. Invest. 121, 1519–1523. doi: 10.1172/JCI43220
Lin, Y. S., Wang, C. H., and Chern, Y. (2011c). Besides Huntington’s disease, does brain-type creatine kinase play a role in other forms of hearing impairment resulting from a common pathological cause? Aging 3, 657–662. doi: 10.18632/aging.100338
Liu, C., Zhang, Y., Tang, W., Wang, B., Wang, B., and He, S. (2017). Evoked potential changes in patients with Parkinson’s9;s disease. Brain Behav. 7:e00703. doi: 10.1002/brb3.703
Liu, L., Chen, Y., Qi, J., Zhang, Y., He, Y., Ni, W., et al. (2016). Wnt activation protects against neomycin-induced hair cell damage in the mouse cochlea. Cell Death Dis. 7:e2136. doi: 10.1038/cddis.2016.35
Liu, W. W., Xu, L., Wang, X., Zhang, D. G., Sun, G. Y., Wang, M., et al. (2021). PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy 1–23. doi: 10.1080/15548627.2021.1905466
Liu, W. W., Xu, X. C., Fan, Z. M., Sun, G. Y., Han, Y. C., Zhang, D. G., et al. (2019). Wnt signaling activates TP53-induced glycolysis and apoptosis regulator and protects against cisplatin-induced spiral ganglion neuron damage in the mouse cochlea. Antioxid. Redox Signal. 30, 1389–1410. doi: 10.1089/ars.2017.7288
Liu, Y., Fang, S., Liu, L. M., Zhu, Y., Li, C. R., Chen, K., et al. (2020). Hearing loss is an early biomarker in APP/PS1 Alzheimer’s disease mice. Neurosci. Lett. 717:134705. doi: 10.1016/j.neulet.2019.134705
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Lopez-Rangel, E., and Lewis, M. E. (2006). Loud and clear evidence for gene silencing by epigenetic mechanisms in autism spectrum and related neurodevelopmental disorders. Clin. Genet. 69, 21–22. doi: 10.1111/j.1399-0004.2006.00543a.x
Lv, J., Fu, X., Li, Y., Hong, G., Li, P., Lin, J., et al. (2021). Deletion of Kcnj16 in mice does not alter auditory function. Front. Cell. Dev. Biol. 9:630361. doi: 10.3389/fcell.2021.630361
Macdonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntingtons-disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Makin, S. (2018). The amyloid hypothesis on trial. Nature 559, S4–S7. doi: 10.1038/d41586-018-05719-4
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/S0092-8674(00)81369-0
Martin, J. B., and Gusella, J. F. (1986). Huntington’s disease. pathogenesis and mannagement. N. Engl. J. Med. 315, 1267–1276. doi: 10.1056/NEJM198611133152006
O’Leary, T. P., Shin, S., Fertan, E., Dingle, R. N., Almuklass, A., Gunn, R. K., et al. (2017). Reduced acoustic startle response and peripheral hearing loss in the 5xFAD mouse model of Alzheimer’s disease. Genes Brain Behav. 16, 554–563. doi: 10.1111/gbb.12370
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Omata, Y., Tharasegaran, S., Lim, Y. M., Yamasaki, Y., Ishigaki, Y., Tatsuno, T., et al. (2016). Expression of amyloid-beta in mouse cochlear hair cells causes an early-onset auditory defect in high-frequency sound perception. Aging 8, 427–439. doi: 10.18632/aging.100899
Panza, F., Solfrizzi, V., and Logroscino, G. (2015). Age-related hearing impairment-a risk factor and frailty marker for dementia and AD. Nat. Rev. Neurol. 11, 166–175. doi: 10.1038/nrneurol.2015.12
Park, S. N., Back, S. A., Choung, Y. H., Kim, H. L., Akil, O., Lustig, L. R., et al. (2011). alpha-Synuclein deficiency and efferent nerve degeneration in the mouse cochlea: a possible cause of early-onset presbycusis. Neurosci. Res. 71, 303–310. doi: 10.1016/j.neures.2011.07.1835
Parvizi, J., Van Hoesen, G. W., and Damasio, A. (2001). The selective vulnerability of brainstem nuclei to Alzheimer’s disease. Ann. Neurol. 49, 53–66. doi: 10.1002/1531-8249(200101)49:1<53::aid-ana30<3.0.co;2-q
Pekkonen, E., Jousmaki, V., Reinikainen, K., and Partanen, J. (1995). Automatic auditory discrimination is impaired in Parkinson’s disease. Electroencephalogr. Clin. Neurophysiol. 95, 47–52. doi: 10.1016/0013-4694(94)00304-4
Perez, S. E., Lumayag, S., Kovacs, B., Mufson, E. J., and Xu, S. (2009). Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer’s disease. Invest. Ophthalmol. Vis. Sci. 50, 793–800. doi: 10.1167/iovs.08-2384
Perluigi, M., Poon, H. F., Maragos, W., Pierce, W. M., Klein, J. B., Calabrese, V., et al. (2005). Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol. Cell Proteomics 4, 1849–1861. doi: 10.1074/mcp.M500090-MCP200
Piaceri, I., Nacmias, B., and Sorbi, S. (2013). Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. 5:167–177. doi: 10.2741/e605
Pickett, J., and London, E. (2005). The neuropathology of autism: a review. J. Neuropathol. Exp. Neurol. 64, 925–935. doi: 10.1097/01.jnen.0000186921.42592.6c
Pisani, V., Sisto, R., Moleti, A., Di Mauro, R., Pisani, A., Brusa, L., et al. (2015). An investigation of hearing impairment in de-novo Parkinson’s disease patients: a preliminary study. Park. Relat. Disord. 21, 987–991. doi: 10.1016/j.parkreldis.2015.06.007
Postuma, R. B., Aarsland, D., Barone, P., Burn, D. J., Hawkes, C. H., Oertel, W., et al. (2012). Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease. Mov. Disord. 27, 617–626. doi: 10.1002/mds.24996
Potter-Nerger, M., Govender, S., Deuschl, G., Volkmann, J., and Colebatch, J. G. (2015). Selective changes of ocular vestibular myogenic potentials in Parkinson’s disease. Mov. Disord. 30, 584–589. doi: 10.1002/mds.26114
Pringsheim, T., Wiltshire, K., Day, L., Dykeman, J., Steeves, T., and Jette, N. (2012). The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov. Disord. 27, 1083–1091. doi: 10.1002/mds.25075
Profant, O., Roth, J., Bures, Z., Balogova, Z., Liskova, I., Betka, J., et al. (2017). Auditory dysfunction in patients with Huntington’s disease. Clin. Neurophysiol. 128, 1946–1953. doi: 10.1016/j.clinph.2017.07.403
Profant, O., Tintera, J., Balogova, Z., Ibrahim, I., Jilek, M., and Syka, J. (2015). Functional changes in the human auditory cortex in ageing. PLoS One 10:e0116692. doi: 10.1371/journal.pone.0116692
Putzki, N., Graf, K., Stude, P., Diener, H. C., and Maschke, M. (2008). Habituation of the auditory startle response in cervical dystonia and Parkinson’s disease. Eur. Neurol. 59, 172–178. doi: 10.1159/000114038
Qian, F. P., Wang, X., Yin, Z. H., Xie, G. C., Yuan, H. J., Liu, D., et al. (2020). The slc4a2b gene is required for hair cell development in zebrafish. Aging 12, 18804–18821. doi: 10.18632/aging.103840
Raza, C., Anjum, R., and Shakeel, N. U. A. (2019). Parkinson’s disease: mechanisms, translational models and management strategies. Life Sci. 226, 77–90. doi: 10.1016/j.lfs.2019.03.057
Rey, R. D., Garretto, N. S., Bueri, J. A., Simonetti, D. D., Sanz, O. P., and Sica, R. E. (1996). The effect of levodopa on the habituation of the acoustic-palpebral reflex in Parkinson’s disease. Electromyogr. Clin. Neurophysiol. 36, 357–360.
Rosenhall, U., Nordin, V., Brantberg, K., and Gillberg, C. (2003). Autism and auditory brain stem responses. Ear Hear. 24, 206–214. doi: 10.1097/01.AUD.0000069326.11466.7E
Rosenhall, U., Nordin, V., Sandstrom, M., Ahlsen, G., and Gillberg, C. (1999). Autism and hearing loss. J. Autism Dev. Disord. 29, 349–357. doi: 10.1023/a:1023022709710
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl.) S10–S17. doi: 10.1038/nm1066
Rotschafer, S. E., Marshak, S., and Cramer, K. S. (2015). Deletion of Fmr1 alters function and synaptic inputs in the auditory brainstem. PLoS One 10:e0117266. doi: 10.1371/journal.pone.0117266
Ruan, Y., Zheng, X. Y., Zhang, H. L., Zhu, W., and Zhu, J. (2012). Olfactory dysfunctions in neurodegenerative disorders. J. Neurosci. Res. 90, 1693–1700. doi: 10.1002/jnr.23054
Rub, U., Stratmann, K., Heinsen, H., Turco, D. D., Seidel, K., Dunnen, W., et al. (2016). The brainstem tau cytoskeletal pathology of Alzheimer’s disease: a brief historical overview and description of its anatomical distribution pattern, evolutional features, pathogenetic and clinical relevance. Curr. Alzheimer Res. 13, 1178–1197. doi: 10.2174/1567205013666160606100509
Ruby, K., Falvey, K., and Kulesza, R. J. (2015). Abnormal neuronal morphology and neurochemistry in the auditory brainstem of Fmr1 knockout rats. Neuroscience 303, 285–298. doi: 10.1016/j.neuroscience.2015.06.061
Russo, N., Nicol, T., Trommer, B., Zecker, S., and Kraus, N. (2009). Brainstem transcription of speech is disrupted in children with autism spectrum disorders. Dev. Sci. 12, 557–567. doi: 10.1111/j.1467-7687.2008.00790.x
Saft, C., Schuttke, A., Beste, C., Andrich, J., Heindel, W., and Pfleiderer, B. (2008). fMRI reveals altered auditory processing in manifest and premanifest Huntington’s disease. Neuropsychologia 46, 1279–1289. doi: 10.1016/j.neuropsychologia.2007.12.002
Santos-Garcia, D., Aneiros-Diaz, A., Macias-Arribi, M., Llaneza-Gonzalez, M. A., Abella-Corral, J., and Santos-Canelles, H. (2010). Sensory symptoms in Parkinson’s disease. Rev. Neurol. 50(Suppl. 2) S65–S74.
Scarpa, A., Cassandro, C., Vitale, C., Ralli, M., Policastro, A., Barone, P., et al. (2020). A comparison of auditory and vestibular dysfunction in Parkinson’s disease and multiple system atrophy. Park. Relat. Disord. 71, 51–57. doi: 10.1016/j.parkreldis.2020.01.018
Seidel, K., Mahlke, J., Siswanto, S., Kruger, R., Heinsen, H., Auburger, G., et al. (2015). The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol. 25, 121–135. doi: 10.1111/bpa.12168
Shalash, A. S., Hassan, D. M., Elrassas, H. H., Salama, M. M., Mendez-Hernandez, E., Salas-Pacheco, J. M., et al. (2017). Auditory- and vestibular-evoked potentials correlate with motor and non-motor features of Parkinson’s disease. Front. Neurol. 8:55. doi: 10.3389/fneur.2017.00055
Shen, Y., Ye, B., Chen, P., Wang, Q., Fan, C., Shu, Y., et al. (2018). cognitive decline, dementia, Alzheimer’s disease and presbycusis: examination of the possible molecular mechanism. Front. Neurosci. 12:394. doi: 10.3389/fnins.2018.00394
Shetty, K., Krishnan, S., Thulaseedharan, J. V., Mohan, M., and Kishore, A. (2019). Asymptomatic hearing impairment frequently occurs in early-onset Parkinson’s disease. J. Mov. Disord. 12, 84–90. doi: 10.14802/jmd.18048
Shin, J. B., Streijger, F., Beynon, A., Peters, T., Gadzala, L., McMillen, D., et al. (2007). Hair bundles are specialized for ATP delivery via creatine kinase. Neuron 53, 371–386. doi: 10.1016/j.neuron.2006.12.021
Simon, D. K., and Johns, D. R. (1999). Mitochondrial disorders: clinical and genetic features. Annu. Rev. Med. 50, 111–127.
Sinajon, P., Gofine, T., Ingram, J., and So, J. (2015). Microdeletion 8q22.2-q22.3 in a 40-year-old male. Eur. J. Med. Genet. 58, 569–572. doi: 10.1016/j.ejmg.2015.10.004
Sinha, U. K., Hollen, K. M., Rodriguez, R., and Miller, C. A. (1993). Auditory system degeneration in Alzheimer’s disease. Neurology 43, 779–785. doi: 10.1212/wnl.43.4.779
Sisto, R., Viziano, A., Stefani, A., Moleti, A., Cerroni, R., Liguori, C., et al. (2020). Lateralization of cochlear dysfunction as a specific biomarker of Parkinson’s disease. Brain Commun. 2:fcaa144. doi: 10.1093/braincomms/fcaa144
Smith, A., Storti, S., Lukose, R., and Kulesza, R. J. Jr. (2019). Structural and functional aberrations of the auditory brainstem in autism spectrum disorder. J. Am. Osteopath. Assoc. 119, 41–50. doi: 10.7556/jaoa.2019.007
Smith, M., Woodroffe, A., Smith, R., Holguin, S., Martinez, J., Filipek, P. A., et al. (2002). Molecular genetic delineation of a deletion of chromosome 13q12–>q13 in a patient with autism and auditory processing deficits. Cytogenet. Genome Res. 98, 233–239. doi: 10.1159/000071040
Snowden, J. S. (2017). The neuropsychology of Huntington’s disease. Arch. Clin. Neuropsychol. 32, 876–887. doi: 10.1093/arclin/acx086
Soria Lopez, J. A., Gonzalez, H. M., and Leger, G. C. (2019). Alzheimer’s disease. Handb. Clin. Neurol. 167, 231–255. doi: 10.1016/B978-0-12-804766-8.00013-3
Sorolla, M. A., Reverter-Branchat, G., Tamarit, J., Ferrer, I., Ros, J., and Cabiscol, E. (2008). Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 45, 667–678. doi: 10.1016/j.freeradbiomed.2008.05.014
Spicer, S. S., Gratton, M. A., and Schulte, B. A. (1997). Expression patterns of ion transport enzymes in spiral ligament fibrocytes change in relation to strial atrophy in the aged gerbil cochlea. Hear. Res. 111, 93–102. doi: 10.1016/s0378-5955(97)00097-x
Spicer, S. S., and Schulte, B. A. (1992). Creatine kinase in epithelium of the inner ear. J. Histochem. Cytochem. 40, 185–192. doi: 10.1177/40.2.1313059
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stiegler, L. N., and Davis, R. (2010). Understanding sound sensitivity in individuals with autism spectrum disorders. Focus Autism Other Dev. Disabil. 25, 67–75. doi: 10.1177/1088357610364530
Sun, G., Liu, W., Fan, Z., Zhang, D., Han, Y., Xu, L., et al. (2016). The three-dimensional culture system with matrigel and neurotrophic factors preserves the structure and function of spiral ganglion neuron in vitro. Neural Plast. 2016:4280407. doi: 10.1155/2016/4280407
Szymanski, C. A., Brice, P. J., Lam, K. H., and Hotto, S. A. (2012). Deaf children with autism spectrum disorders. J. Autism Dev. Disord. 42, 2027–2037. doi: 10.1007/s10803-012-1452-9
Taljaard, D. S., Olaithe, M., Brennan-Jones, C. G., Eikelboom, R. H., and Bucks, R. S. (2016). The relationship between hearing impairment and cognitive function: a meta-analysis in adults. Clin. Otolaryngol. 41, 718–729. doi: 10.1111/coa.12607
Tas, A., Yagiz, R., Tas, M., Esme, M., Uzun, C., and Karasalihoglu, A. R. (2007). Evaluation of hearing in children with autism by using TEOAE and ABR. Autism 11, 73–79. doi: 10.1177/1362361307070908
Tharpe, A. M., Bess, F. H., Sladen, D. P., Schissel, H., Couch, S., and Schery, T. (2006). Auditory characteristics of children with autism. Ear Hear. 27, 430–441. doi: 10.1097/01.aud.0000224981.60575.d8
Tomchek, S. D., and Dunn, W. (2007). Sensory processing in children with and without autism: a comparative study using the short sensory profile. Am. J. Occup. Ther. 61, 190–200. doi: 10.5014/ajot.61.2.190
Truong, D. T., Rendall, A. R., Castelluccio, B. C., Eigsti, I. M., and Fitch, R. H. (2015). Auditory processing and morphological anomalies in medial geniculate nucleus of Cntnap2 mutant mice. Behav. Neurosci. 129, 731–743. doi: 10.1037/bne0000096
Uhlmann, R. F., Larson, E. B., and Koepsell, T. D. (1986). Hearing impairment and cognitive decline in senile dementia of the Alzheimer’s type. J. Am. Geriatr. Soc. 34, 207–210. doi: 10.1111/j.1532-5415.1986.tb04204.x
van der Kant, R., Goldstein, L. S. B., and Ossenkoppele, R. (2020). Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21, 21–35. doi: 10.1038/s41583-019-0240-3
van Wijngaarden, P., Hadoux, X., Alwan, M., Keel, S., and Dirani, M. (2017). Emerging ocular biomarkers of Alzheimer disease. Clin. Exp. Ophthalmol. 45, 54–61. doi: 10.1111/ceo.12872
Veenstra-VanderWeele, J., Christian, S. L., and Cook, E. H. (2004). Autism as a paradigmatic complex genetic disorder. Annu. Rev. Genomics Hum. Genet. 5, 379–405. doi: 10.1146/annurev.genom.5.061903.180050
Verghese, P. B., Castellano, J. M., and Holtzman, D. M. (2011). Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 10, 241–252. doi: 10.1016/S1474-4422(10)70325-2
Vitale, C., Marcelli, V., Abate, T., Pianese, A., Allocca, R., Moccia, M., et al. (2016). Speech discrimination is impaired in parkinsonian patients: expanding the audiologic findings of Parkinson’s disease. Park. Relat. Disord. 22(Suppl. 1) S138–S143. doi: 10.1016/j.parkreldis.2015.09.040
Vitale, C., Marcelli, V., Allocca, R., Santangelo, G., Riccardi, P., Erro, R., et al. (2013). Hearing impairment in Pa19;s disease: expanding the nonmotor phenotype (vol 27, pg 1530, 2012). Mov. Disord. 28, 410–410. doi: 10.1002/mds.25406
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Wakabayashi, K., Takahashi, H., Takeda, S., Ohama, E., and Ikuta, F. (1989). Lewy bodies in the enteric nervous system in Parkinson’s disease. Arch. Histol. Cytol. 52(Suppl.) 191–194. doi: 10.1679/aohc.52.suppl_191
Wallimann, T., Wyss, M., Brdiczka, D., Nicolay, K., and Eppenberger, H. M. (1992). Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 281(Pt 1), 21–40. doi: 10.1042/bj2810021
Wang, S. E., and Wu, C. H. (2015). Physiological and histological evaluations of the cochlea between 3xTg-AD mouse model of Alzheimer’s diseases and R6/2 mouse model of Huntington’s diseases. Chin. J. Physiol. 58, 359–366. doi: 10.4077/CJP.2015.BAD334
Wang, Y., Li, J., Yao, X., Li, W., Du, H., Tang, M., et al. (2017). Loss of CIB2 causes profound hearing loss and abolishes mechanoelectrical transduction in mice. Front. Mol. Neurosci. 10:401. doi: 10.3389/fnmol.2017.00401
Wegiel, J., Flory, M., Kuchna, I., Nowicki, K., Ma, S. Y., Imaki, H., et al. (2014). Brain-region-specific alterations of the trajectories of neuronal volume growth throughout the lifespan in autism. Acta Neuropathol. Commun. 2:28. doi: 10.1186/2051-5960-2-28
Wegiel, J., Kuchna, I., Nowicki, K., Imaki, H., Wegiel, J., Marchi, E., et al. (2010). The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 119, 755–770. doi: 10.1007/s00401-010-0655-4
Wetter, S., Peavy, G., Jacobson, M., Hamilton, J., Salmon, D., and Murphy, C. (2005). Olfactory and auditory event-related potentials in Huntington’s disease. Neuropsychology 19, 428–436. doi: 10.1037/0894-4105.19.4.428
Wu, C., Stefanescu, R. A., Martel, D. T., and Shore, S. E. (2015). Listening to another sense: somatosensory integration in the auditory system. Cell Tissue Res. 361, 233–250. doi: 10.1007/s00441-014-2074-7
Wu, M., Zheng, X., Wang, X., Zhang, G., and Kuang, J. (2020a). 4q27 deletion and 7q36.1 microduplication in a patient with multiple malformations and hearing loss: a case report. BMC Med. Genomics 13:31. doi: 10.1186/s12920-020-0697-y
Wu, P. Z., O’Malley, J. T., de Gruttola, V., and Liberman, M. C. (2020b). Age-related hearing loss is dominated by damage to inner ear sensory cells, not the cellular battery that powers them. J. Neurosci. 40, 6357–6366. doi: 10.1523/JNEUROSCI.0937-20.2020
Wyss, M., and Kaddurah-Daouk, R. (2000). Creatine and creatinine metabolism. Physiol. Rev. 80, 1107–1213. doi: 10.1152/physrev.2000.80.3.1107
Yang, M., Mahrt, E. J., Lewis, F., Foley, G., Portmann, T., Dolmetsch, R. E., et al. (2015). 16p11.2 deletion syndrome mice display sensory and ultrasonic vocalization deficits during social interactions. Autism Res. 8, 507–521. doi: 10.1002/aur.1465
Yu, X. Y., Liu, W. W., Fan, Z. M., Qian, F. P., Zhang, D. G., Han, Y. C., et al. (2017). c-Myb knockdown increases the neomycin-induced damage to hair-cell-like HEI-OC1 cells in vitro. Sci. Rep. 7:41094. doi: 10.1038/srep41094
Keywords: hearing loss, neurodegenerative diseases, autism spectrum disorder, pathological mechanisms, molecular mechanisms
Citation: Li S, Cheng C, Lu L, Ma X, Zhang X, Li A, Chen J, Qian X and Gao X (2021) Hearing Loss in Neurological Disorders. Front. Cell Dev. Biol. 9:716300. doi: 10.3389/fcell.2021.716300
Received: 28 May 2021; Accepted: 23 July 2021;
Published: 11 August 2021.
Edited by:
Renjie Chai, Southeast University, ChinaReviewed by:
Lukas D. Landegger, Medizinische Universität Wien, AustriaCopyright © 2021 Li, Cheng, Lu, Ma, Zhang, Li, Chen, Qian and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Chen, bmpqaWVjaGVuQDE2My5jb20=; Xiaoyun Qian, cXh5NTIyQDE2My5jb20=; Xia Gao, eGlhZ2FvZ2FvQGhvdG1haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.