 Kyra Laubach
Kyra Laubach Jin Zhang
Jin Zhang Xinbin Chen
Xinbin Chen- Comparative Oncology Laboratory, Schools of Veterinary Medicine and Medicine, University of California, Davis, Davis, CA, United States
The p53 family of tumor suppressors, which includes p53, p63, and p73, has a critical role in many biological processes, such as cell cycle arrest, apoptosis, and differentiation. In addition to tumor suppression, the p53 family proteins also participate in development, multiciliogenesis, and fertility, indicating these proteins have diverse roles. In this review, we strive to cover the relevant studies that demonstrate the roles of p53, p63, and p73 in lipid and iron metabolism.
Introduction
For over 40 years, p53 has been characterized as a master transcriptional regulator that mediates the expression of various genes to prevent aberrant cell growth (Ko and Prives, 1996). Just before the turn of the century, the TP63 and TP73 genes were discovered due to their significant homology to TP53, particularly in the DNA-binding domain (Kaghad et al., 1997; Schmale and Bamberger, 1997; Trink et al., 1998; Borremans et al., 2001). These three genes constitute the p53 family.
The TP53, TP63, and TP73 genes are expressed as multiple N- and C-terminal isoforms through two promoters and alternative splicing (Figure 1). In TP53, promoter 1 (P1) gives rise to two translation initiation start sites, termed ATG1 and ATG40, which produce full-length p53 (FLp53) and Δ40p53, respectively (Courtois et al., 2002; Yin et al., 2002). Both isoforms possess transactivation function even though Δ40p53 contains a truncated form of the conventional transactivation domain (Zhu et al., 1998; 2000; Harms and Chen, 2005). Similarly driven by P1, TP63/73 express TAp63/73 isoforms, which have a transactivation domain that is comparable to the one found in FLp53 (Arrowsmith, 1999). By using promoter 2 (P2), all family members produce the N-terminally truncated isoforms, termed Δ133p53 and Δ160p53 in TP53, which arise from translation initiation start sites ATG133 and ATG160 (Bourdon et al., 2005), and ΔNp63/73 in TP63/73 (Yang et al., 1998; 2000). Interestingly, despite lacking the conventional transactivation domain, ΔNp63 and ΔNp73 are transcriptionally active and can induce some p53 targets (Liu et al., 2004; Helton et al., 2006). Alternative splicing at the C-terminus of each gene generates additional isoforms. TP53 produces three (α, β, γ) C-terminal isoforms (Bourdon et al., 2005), TP63 produces four (α, β, γ, δ) C-terminal isoforms (Yang et al., 1998; Mangiulli et al., 2009), and TP73 produces at least seven C-terminal isoforms (α, β, γ, δ, ε, ζ, η) (De Laurenzi et al., 1998; 1999). While the N-terminal isoforms of p53, p63, and p73 are well studied, the C-terminal isoforms remain largely uncharacterized.
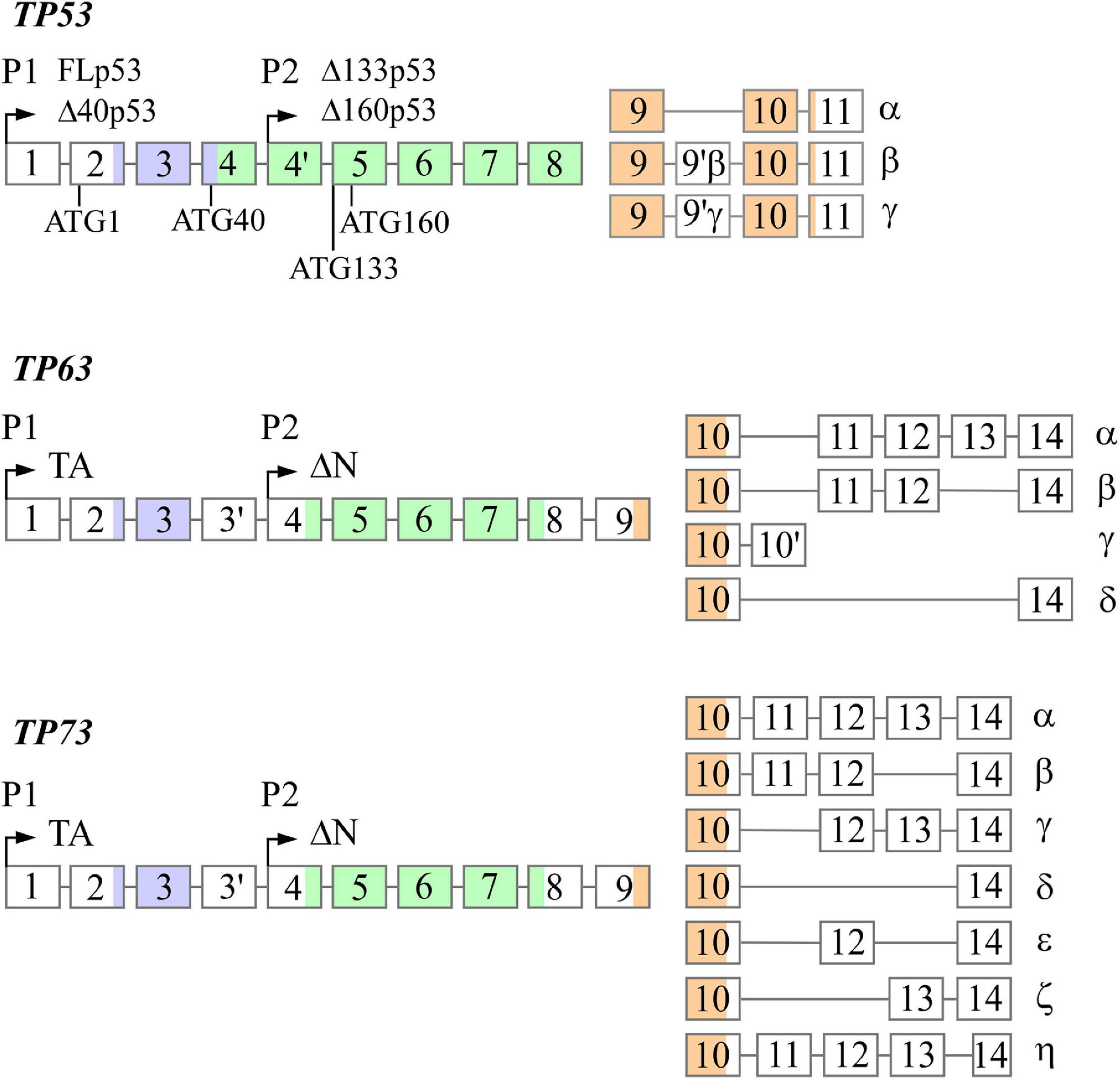
Figure 1. The genomic loci of TP53, TP63, and TP73. All three genes contain two promoters (P1 and P2) through which transcription initiation may occur, resulting in the formation of the N-terminal isoforms. TP53 contains four alternative translation start sites termed ATG1, ATG40, AG133, and ATG160 that give rise to FLp53, Δ40p53, Δ133p53, and Δ160p53, respectively. Alternative splicing at the 3′ end produces the C-terminal isoforms in each gene with the exception of p63γ, which results from transcriptional termination in intron 10. There exists significantly homology between each gene in the transactivation domains (purple), DNA-binding domains (green), and oligomerization domains (orange).
The biological function of the p53 family proteins has been demonstrated through various mouse models. The very first p53-knockout model showed that mice deficient in p53 were prone to spontaneous tumors, but otherwise developed normally (Donehower et al., 1991). Later, it was discovered that p53 dysregulation, predominantly overexpression, can lead to impaired embryogenesis and other developmental defects (Luna et al., 1995; Sah et al., 1995; Parant et al., 2001; Zhang et al., 2012; Nostrand et al., 2014). Unlike p53, total p63-knockout mice exhibit severe epidermal and craniofacial abnormalities and die shortly after birth (Celli et al., 1999; Mills et al., 1999; Yang et al., 1999). Further studies revealed that ΔNp63 is responsible for the observed phenotype (Candi et al., 2006; Koster et al., 2007). In contrast, TAp63-knockout mice did not exhibit birth defects, but were prone to spontaneous tumors, indicating that TAp63 functions as a tumor suppressor to maintain genome stability (Suh et al., 2006; Guo et al., 2009; Su et al., 2009). Similarly, total p73-knockout mice were runty and had severe neurological defects, chronic inflammation, fertility issues (Yang et al., 2000), and impaired multiciliogenesis (Marshall et al., 2016; Nemajerova et al., 2016). It was later found that ΔNp73-knockout mice exhibited neurodegeneration (Wilhelm et al., 2010), whereas TAp73-knockout mice were prone to spontaneous tumors (Tomasini et al., 2008).
Continued research efforts into the more nuanced cancer-associated roles of the p53 family proteins is undeniably valuable. However, emerging evidence suggests that these proteins possess additional important functions that can affect various human diseases, such as diabetes mellitus and liver steatosis. This review will focus specifically on the roles of the p53 family in lipid and iron metabolism.
Lipid Metabolism
Lipids play an important role in various biological processes and serve as an essential building block for many cellular structures. Tight regulation of lipid metabolism is crucial for proper organismal function, and dysregulation has been implicated in numerous diseases, such as Alzheimer’s disease and fatty liver disease (Hooijmans and Kiliaan, 2008; Hasson et al., 2016). There are three main sources of lipids: dietary lipids, fatty acids produced by hepatocytes and adipocytes, and lipoproteins produced by hepatocytes (Giammanco et al., 2015). In the lumen of the gastrointestinal tract, dietary lipids become emulsified by combining with bile salts, which allows for lipid hydrolysis and subsequent import to enterocytes (Hussain, 2014). In enterocytes, lipids are processed by the endoplasmic reticulum and packaged into lipoprotein bundles, called chylomicrons (Hussain, 2014; Giammanco et al., 2015), to allow for transport through the circulation (Alekos et al., 2020). Once chylomicrons arrive at a target cell, lipases break them down to permit cellular import of lipids (Alekos et al., 2020). Hepatocytes are then responsible for recycling chylomicron components to allow for later use (Alekos et al., 2020).
At the cellular level, lipids are categorized into three groups: structural lipids, lipid droplets, and bioactive lipids. Structural lipids are comprised of phospholipids and form cell and organelle membranes (Bohdanowicz and Grinstein, 2013), which are important for cellular compartmentalization. Lipids are also a main source of energy and are stored as modified sterols and fatty acids in specialized organelles called lipid droplets, which are predominantly found in adipocytes (Röhrig and Schulze, 2016; Olzmann and Carvalho, 2019). This modification gives sterols and fatty acids a neutral charge to form sterol esters (Korber et al., 2017) and triglycerides (Alves-Bezerra and Cohen, 2018), respectively. Bioactive lipids are unique in that they are involved in signal transduction and are categorized into multiple classes, including sphingolipids (Hannun and Obeid, 2008), diacylglycerols (Peter-Riesch et al., 1988), and eicosanoids (Levy et al., 2001). Sphingolipids are further categorized into several sub-classes, such as sphingomyelin, galactosylceramide, glucosylceramide, and sphingosine (Hannun and Obeid, 2018). Studies have shown that sphingolipids can modulate cell death and survival pathways, including apoptosis, cell growth/inhibition, and migration (Hannun and Obeid, 2018). Diacylglycerols serve as a secondary messenger in many critical cellular processes, such as neurotransmitter release (Ma et al., 2013) and insulin signaling in islet cells (Peter-Riesch et al., 1988). Eicosanoids have been implicated in mediating the inflammatory response (Levy et al., 2001). Lipids are exceedingly important for many cellular processes, from structure to signaling. In this review, we focus on the role of the p53 family proteins in cholesterol and fatty acid metabolism. Table 1 provides a summary of the p53 family targets that are involved in lipid metabolism, and Figure 2 briefly outlines cholesterol and fatty acid metabolism pathways.
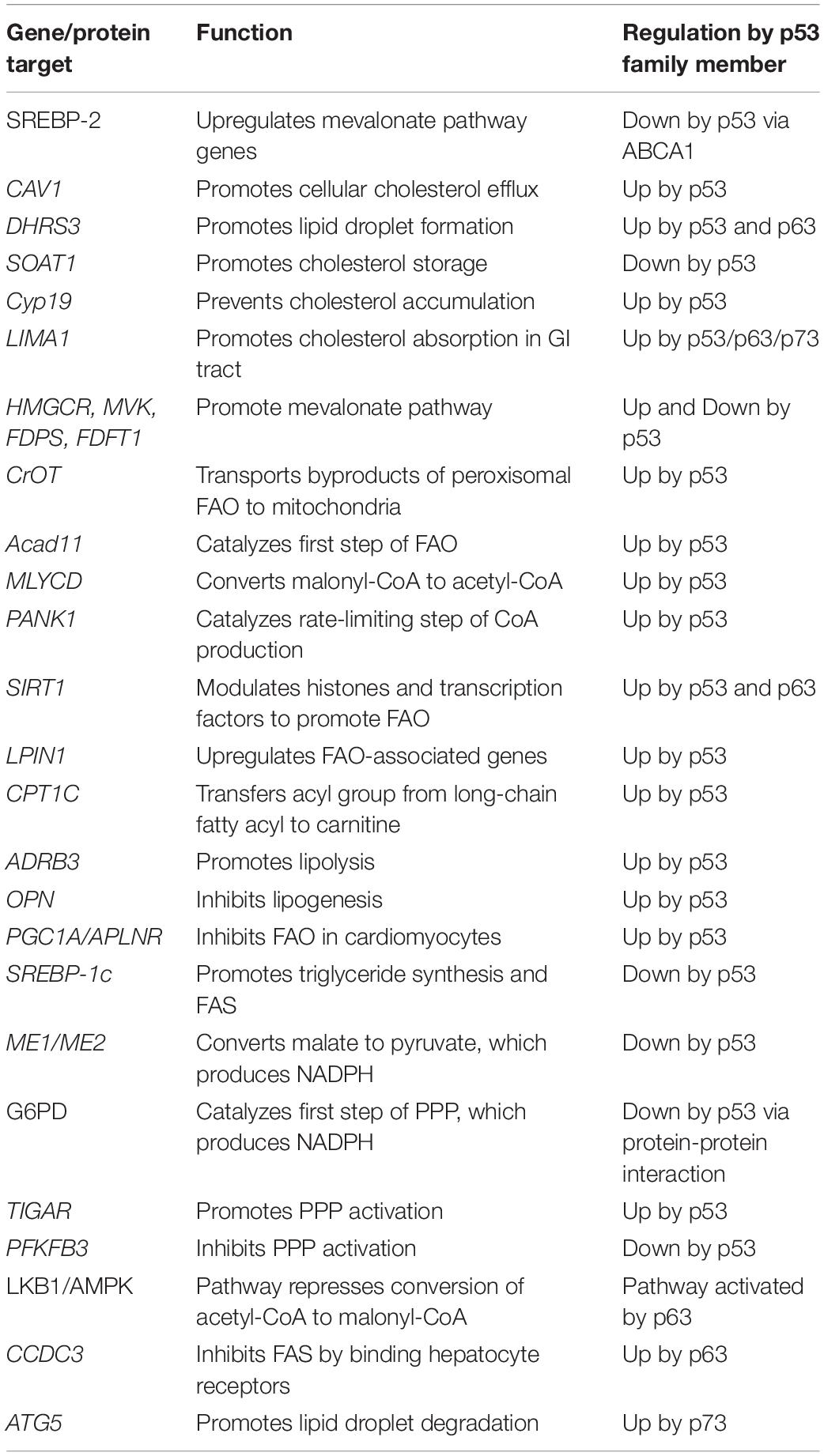
Table 1. Targets of the p53 family that are associated with lipid metabolism.
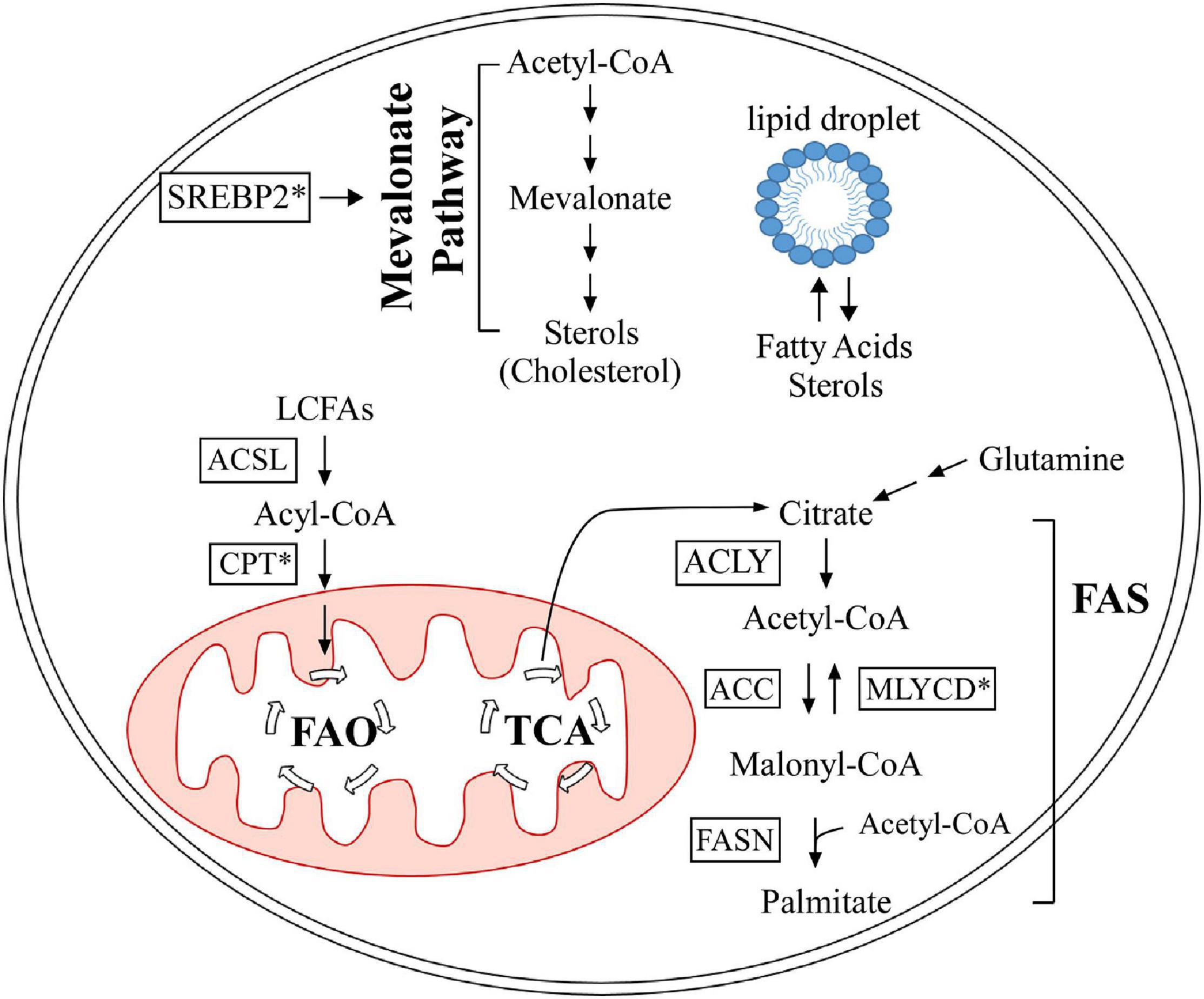
Figure 2. Summary of lipid metabolism, as it pertains to the p53 family. The mevalonate pathway is out lined in the upper half. Fatty acid oxidation and synthesis are portrayed in the lower half. Proteins marked with an asterisk are regulated by the p53 family. FAO, fatty acid oxidation; TCA, tricarboxylic acid cycle; FAS, fatty acid synthesis; ACSL, long-chain acyl-CoA synthetase; CPT, carnitine palmitoyltransferase; ACLY, ATP citrate lyase; MLYCD, malonyl-CoA decarboxylase; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; SREBP2, sterol regulatory element-binding protein 2.
p53
Cholesterol
Multiple studies have shown that p53 is implicated in regulating the levels of intracellular free cholesterol. Sterol Regulatory Element-Binding Protein 2 (SREBP-2) is a master transcriptional regulator of the mevalonate pathway and responds to sterol depletion by promoting cholesterol synthesis (Brown and Goldstein, 1997). It was found that p53 inhibits SREBP-2 maturation through the upregulation of ABCA1 (Moon et al., 2019), an ATP-binding cassette transporter that inhibits cholesterol synthesis and drives cholesterol export when cholesterol stores are high (Yamauchi et al., 2015). Additionally, p53 can promote cholesterol export through the upregulation of CAV1 (Bist et al., 2000), a scaffold protein that binds intracellular free cholesterol and facilitates its efflux (Fielding and Fielding, 1995). To enhance cellular cholesterol storage, p53 transactivates dehydrogenase/reductase member 3 (DHSR3) (Kirschner et al., 2010; Deisenroth et al., 2011), which decreases intracellular free cholesterol by increasing lipid droplet formation (Martin and Parton, 2006). Conversely, p53 has been shown to inhibit cholesterol storage by negatively regulating SOAT1 (Oni et al., 2020). SOAT1 decreases intracellular free cholesterol by increasing cholesterol storage, thus disrupting the negative feedback loop that prevents cholesterol synthesis when free intracellular cholesterol levels are high (Oelkers et al., 1998). Furthermore, Cyp19, an aromatase essential for estrogen synthesis (Thompson and Siiteri, 1974), was found to be upregulated by p53, which prevents intracellular free cholesterol overload and adipocyte formation (Wang et al., 2013). One study revealed a potential link between p53 and LIMA1, also called SREBP3, in mediating cholesterol absorption in the gastrointestinal tract. p53 was shown to upregulate LIMA1 through p53-response elements in its promoter (Ohashi et al., 2017), and LIMA1 promotes cholesterol absorption in the intestine (Zhang et al., 2018). Interestingly, there are some conflicting findings regarding p53 regulation of other mevalonate pathway genes. For example, one group showed that p53 inhibited expression of mevalonate pathway genes HMGCR, MVK, FDPS, and FDFT1 (Moon et al., 2019), but another group showed that p53 enhanced expression of these genes (Laezza et al., 2015), suggesting that p53 may regulate some mevalonate pathway genes in a context-dependent manner. Collectively, these findings suggest that multiple targets are regulated by p53 to prevent intracellular free cholesterol accumulation and to maintain the integrity of the negative feedback loop that regulates cholesterol storage and synthesis.
Fatty Acids
Fatty acid oxidation, also known as β-oxidation and hereafter referred to as FAO, is the process of breaking down long-chain fatty acids (LCFAs), primarily in the mitochondria; FAO can be initiated in peroxisomes, but the byproducts undergo complete oxidation in the mitochondria (Qu et al., 2016). LCFAs are metabolized by long-chain acyl-CoA synthetase to form acyl-CoA (Mashek et al., 2007), which is then transported into the mitochondrial matrix by a series of reactions catalyzed by the carnitine palmitoyltransferase system (Rufer et al., 2009). Acyl-CoA is then used as a substrate to initiate FAO (Qu et al., 2016). Each cycle of FAO in the matrix removes two carbons from the fatty acid, until four carbons remain; these are then used to synthesize acetyl-CoA (Qu et al., 2016).
De novo fatty acid synthesis (FAS) is the process by which cells generate fatty acids that are used in various cellular processes (Röhrig and Schulze, 2016). FAS starts with citrate produced by the tricarboxylic acid (TCA) cycle or glutamine metabolism (Akram, 2014; Röhrig and Schulze, 2016). Citrate is then cleaved by ATP-citrate lyase to form acetyl-CoA, which is the starting substrate for FAS (Zaidi et al., 2012). Acetyl-CoA carboxylases convert acetyl-CoA to malonyl-CoA (Abu-Elheiga et al., 2000), at which point fatty acid synthase (encoded by FASN) catalyzes the reaction between seven malonyl-CoA molecules and one acetyl-CoA molecule to form palmitate, a long-chain fatty acid (Smith et al., 2003). Palmitate is then modified in length (Jakobsson et al., 2006) and degree of saturation (Igal, 2010) to form additional fatty acids.
p53 has been shown to predominantly promote FAO (Parrales and Iwakuma, 2016). RNA-seq analysis revealed that p53 upregulates CrOT (peroxisomal carnitine O-octanoyltransferase) (Goldstein et al., 2012), which is responsible for transporting byproducts of peroxisomal FAO to mitochondria to allow for complete oxidation (Longo et al., 2016). Similarly, another group showed that p53 pathway activation following γ-irradiation led to increased CrOT expression (Hage-Sleiman et al., 2017). In regards to mitochondrial FAO, p53 was found to upregulate Acad11 (Jiang et al., 2015), which encodes acyl-CoA dehydrogenase and catalyzes the first step of FAO in the mitochondrial matrix (He et al., 2011). p53 can additionally promote FAO through upregulation of MLYCD (Liu et al., 2014), which encodes malonyl-CoA decarboxylase and converts the FAO inhibitor malonyl-CoA to acetyl-CoA (Foster, 2004). As evidenced by the name of many FAO intermediates, CoA is a critical molecule in many FAO reactions (Leonardi et al., 2005), and p53 was found to upregulate PANK1, which promotes CoA production (Wang et al., 2013). By complexing with FOXO3a, p53 transactivates SIRT1 (Nemoto et al., 2004), a deacetylase that acts on histones and transcription factors to promote FAO (Rahman and Islam, 2011; Derdak et al., 2013). Activation of p53 in response to DNA damage and glucose starvation results in increased expression of LPIN1, a transcriptional co-activator, to promote FAO (Assaily et al., 2011). Lipin-1 also aids in diacylglycerol formation (Donkor et al., 2007), suggesting a role for p53 in diacylglycerol metabolism. There is evidence that p53 directly upregulates CPT1C, a neuron-specific carnitine palmitoyltransferase that transfers the acyl group from long chain fatty acyl to carnitine to initiate FAO (Lee and Wolfgang, 2012; Sanchez-Macedo et al., 2013). In addition to CPT1C, there are other tissue-specific carnitine palmitoyltransferase family members, such as CPT1a in the liver and CPT1b in muscle (Greenberg et al., 2009). Thus, it is possible that p53 might regulate FAO through CPT1a and CPT1b. p53 was shown to transactivate ADRB3 (Kang et al., 2020), which promotes lipolysis, or the breakdown of triglycerides into fatty acids to allow for FAO (Arner and Langin, 2014). Interestingly, a p53 mutant could induce ADRB3 to a higher degree (Kang et al., 2020). Likewise, studies showed that p53 can prevent lipogenesis through upregulation of OPN, which encodes osteopontin (Gómez-Santos et al., 2020). In vivo analyses in mouse livers showed that OPN levels were increased in response to an increase in p53 (Gómez-Santos et al., 2020). Conversely, a recent report showed that p53 inhibits FAO through PGC1A and APLNR in response to doxorubicin treatment in cardiomyocytes (Saleme et al., 2020). These data lead us to hypothesize that p53 could have tissue/cell-specific effects on FAO.
p53 has been shown to inhibit FAS (Parrales and Iwakuma, 2016). For example, p53 can negatively regulate transcription of SREBP-1c to inhibit FAS (Yahagi et al., 2003). SREBP-1c, a SREBP family member, is involved in triglyceride and fatty acid synthesis predominantly in the liver, which leads to fat accumulation (Shimano et al., 1997; Shimomura et al., 1998). Additionally, p53 has been implicated in inhibiting FAS through repression of NADPH production, a critical energy source utilized during FAS (Brose et al., 2016). p53 can inhibit NADPH production through negative transcriptional regulation of malic enzyme 1 and 2 (ME1 and 2) (Jiang et al., 2013). ME1/2 catalyze the formation of pyruvate from malate, which produces NADPH (Wise and Ball, 1964). Additionally, p53 prevents NADPH production through inhibition of glucose-6-phosphate dehydrogenase (G6PD) via protein-protein interaction, which requires p53’s C-terminus, DNA-binding domain, and tetramerization domain (Jiang et al., 2011). G6PD is an enzyme that catalyzes the first step of the pentose phosphate pathway (PPP), a major source of NADPH production (Ge et al., 2020). While reports show that p53 predominately inhibits NADPH production, some p53 targets have been identified as promoters of NADPH production. For example, TIGAR, a well-defined p53 target, activates PPP to drive NADPH production, which has been shown to prevent ROS formation (Bensaad et al., 2006). Additionally, it was found that p53 promotes NADPH production through suppression of PFKFB3 expression, which favors glycolysis over PPP (Franklin et al., 2016).
p63 and p73
Several studies have unveiled an important role for p63 in lipid metabolism, although the mechanisms are not fully understood. It was shown that TAp63 deficiency in mice increases the incidence of obesity and liver steatosis and impairs FAO function (Su et al., 2012; Liao et al., 2017). It was found that TAp63 promotes FAO through upregulation of SIRT1 (a previously described p53 target) and the LKBI/AMPK pathway, the latter of which prevents the conversion of acetyl-CoA to the FAO inhibitor malonyl-CoA (Li et al., 2020). As previously mentioned, p53 promotes the production of acetyl-CoA from malonyl-CoA (Liu et al., 2014), suggesting that the p53 family can transactivate multiple targets to prevent malonyl-CoA formation. Additionally, TAp63 was shown to inhibit FAS by upregulating CCDC3 (Liao et al., 2017), which encodes a soluble protein that binds to hepatocyte receptors (Kobayashi et al., 2010). While there is limited research on p63 and cholesterol regulation, it has been shown that p63, like p53, inhibits cellular cholesterol accumulation through DHSR3 (Kirschner et al., 2010) and promotes intestinal cholesterol absorption through LIM1A (Zhang et al., 2018).
Phenotypic similarities between p63- and p73-deficient mice suggest that p73 has an analogous role in regulating lipid metabolism. In response to nutrient deprivation, loss of p73 leads to lipid accumulation in mouse livers (He et al., 2013). Mechanistically, TAp73α/β were shown to modulate lipid metabolism through ATG5, a gene that is necessary for autophagy (He et al., 2013). Autophagy is an intracellular process that, among other things, can break down lipid droplets to allow for FAO (Ye et al., 2018; Saito et al., 2019). As such, gene transfer of ATG5 to p73-knockout mice mitigated the accumulation of lipid droplets in the mouse livers (He et al., 2013). As with p53 and p63, p73β can upregulate LIM1A to increase cholesterol absorption (Y. Y. Zhang et al., 2018). These data suggest that p73 prevents lipid accumulation through a mechanism that is quite different from how p53 and p63 regulate this process.
Iron Metabolism
Iron is an essential element for all living entities and plays an important role in many cellular processes, such as oxygen transport and cell proliferation (Kim and Nemeth, 2015; Wallace, 2019). Additionally, iron is a critical cofactor that is required for various metabolic activities, such as DNA synthesis (Puig et al., 2017). An organism’s main source of iron is through dietary intake (Waldvogel-Abramowski et al., 2014). In the gastrointestinal tract, iron exists as non-heme- or heme-iron (Waldvogel-Abramowski et al., 2014); heme is a porphyrin that contains iron (Fiorito et al., 2020). At physiological pH, non-heme iron is present in the ferric (Fe3+) state, but cells can only absorb it in the ferrous (Fe2+) state (Wallace, 2019). Duodenal cytochrome B (Dcytb) reduces Fe3+ to Fe2+ in the lumen and ferrous iron is absorbed by enterocytes through divalent metal cation transporter 1 (DMCT1) (Wallace, 2019). On the other hand, heme-iron is directly imported by haem carrier protein 1 (HCP1). Once in enterocytes, iron is exported by ferroportin 1 (FPN1), whose function is inhibited by hepcidin (Waldvogel-Abramowski et al., 2014). In the plasma, Fe2+ is converted back to Fe3+ by ceruloplasmin and binds to transferrin for transport through the circulation (Attieh et al., 1999). A summary schematic of this process is shown in Figure 3. Once iron has entered the target cell, it binds ferritin until it is needed (Waldvogel-Abramowski et al., 2014). Regulation of iron metabolism is exceedingly important because iron overload, like in hemochromatosis (Bacon et al., 2011), can lead to heart disease and liver cirrhosis, while deficiency can result in anemia and developmental impairments (Abbaspour et al., 2014). The p53 family has been implicated in mediating iron metabolism to prevent iron dysregulation.
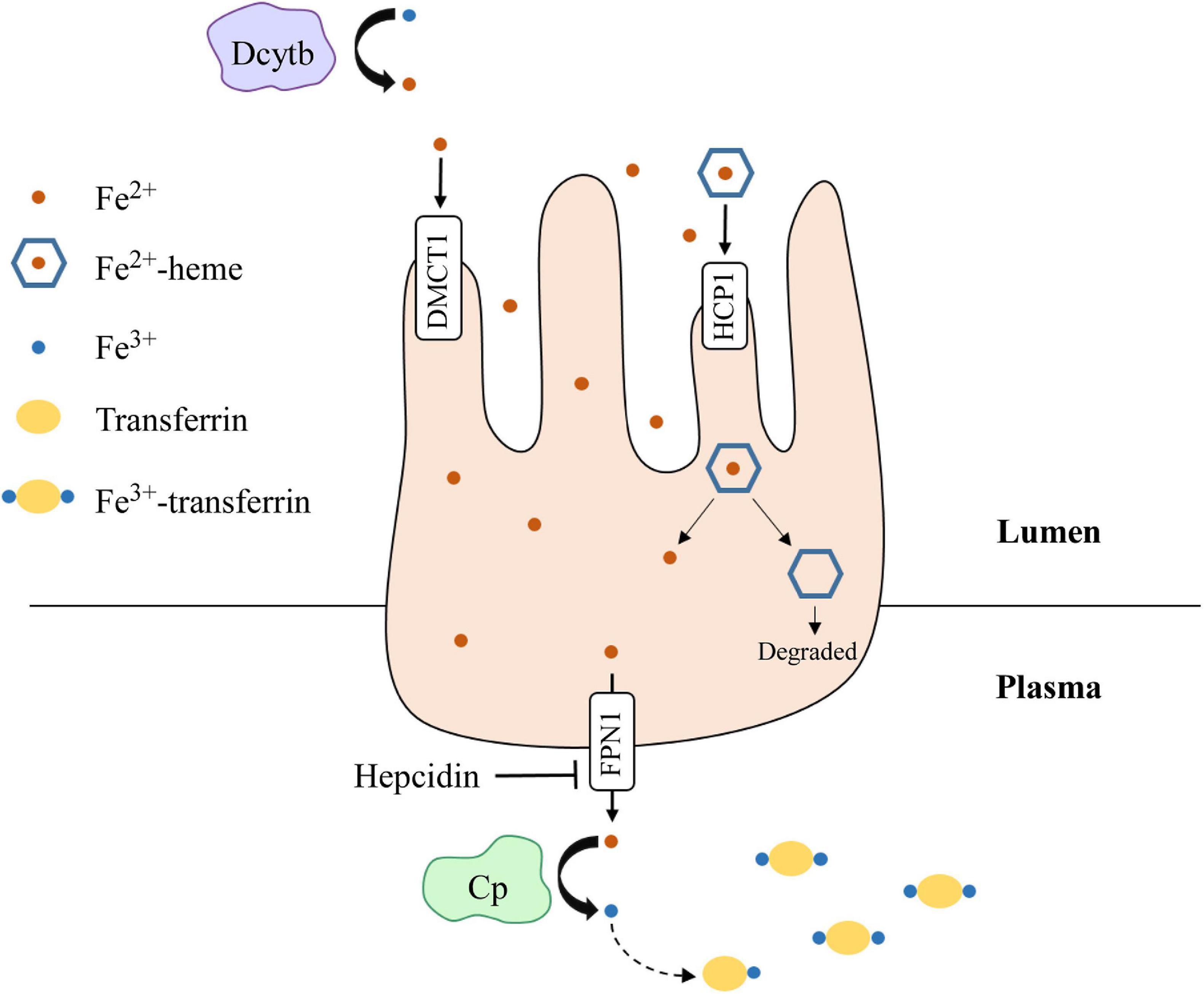
Figure 3. Dietary iron absorption and transport from enterocytes to the plasma. Fe3+ is reduced to Fe2+ via duodenal cytochrome b (Dcytb) in the intestinal lumen. Non-heme-iron is transported into enterocytes via divalent metal cation transporter 1 (DMCT1), while heme-iron is transported through haem carrier protein 1 (HCP1). Heme dissociates from iron and is degraded. Fe2+ is then exported out of enterocytes by ferroportin 1 (FPN1), whose function is inhibited by hepcidin. Once in the plasma, Fe2+ is oxidized to Fe3+ by ceruloplasmin (Cp), which then binds transferrin for transport through the plasma. Iron is transported into target cells for storage or use.
p53
Iron metabolism exerts regulatory functions over p53 and in turn, p53 can regulate iron metabolism. Excess iron leads to decreased p53 expression (Shen et al., 2014), whereas iron depletion leads to p53 accumulation (Liang and Richardson, 2003; Kim et al., 2007). Additionally, direct binding of heme to p53 protein inhibits p53 transcriptional activity and possibly promotes p53 degradation (Shen et al., 2014). As such, a feedback loop between iron and p53 exists wherein iron overload inhibits p53 activity and p53 inhibits iron accumulation. At the systemic level, p53 upregulates HAMP (encoding hepcidin) to inhibit iron efflux from enterocytes (Weizer-Stern et al., 2007) and thus, prevents iron from entering the circulation when it is not needed. To prevent iron overload at the cellular level, p53 directly transactivates several targets, including FXN (frataxin) (Shimizu et al., 2014), FDXR (ferredoxin reductase) (Hwang et al., 2001; Liu and Chen, 2002), and ISCU (iron-sulfur cluster assembly enzyme) (Funauchi et al., 2015). Frataxin is an iron binding protein that regulates mitochondrial iron homeostasis to prevent iron overload (Cavadini et al., 2000) and thus, p53 upregulates frataxin to inhibit mitochondrial iron overload. Additionally, frataxin is necessary for iron-sulfur cluster (ISC) biogenesis (Shimizu et al., 2014) and ISCs are critical for mitochondrial function (Shimizu et al., 2014). In addition to aiding in electron transport during redox reactions (Johnson et al., 2005), ISCs serve as a co-factor for many essential enzymes (Baranovskiy et al., 2012). We also showed that p53 regulates mitochondrial iron metabolism through a FDXR-p53 loop (Liu and Chen, 2002; Zhang et al., 2017). FDXR plays a critical role in ISC biogenesis and steroid hormone synthesis by transferring electrons from NADPH to ferredoxin 1 and 2 (FDX1 and 2) (Brandt and Vickery, 1992; Sheftel et al., 2010). p53 drives the FDXR-p53 loop to upregulate FDXR, which then transfers electrons to FDX2, ultimately preventing iron overload at the cellular level (Zhang et al., 2017). Furthermore, p53 upregulates ISCU, which increases translation of ferritin heavy chain mRNA (FTH1) and destabilizes transferrin receptor mRNA (TFRC) (Funauchi et al., 2015), therefore increasing cellular iron storage and decreasing cellular iron import. p53 can also regulate iron metabolism through post-transcriptional modifications of Iron Regulatory Protein 1 and 2 (IRP1 and IRP2) (Zhang et al., 2008). IRP1/2 alter the expression of proteins associated with iron transport and storage by binding to conserved iron-regulatory elements (IRE) in target mRNAs (Volz, 2008). Interestingly, the binding of IRP1/2 to a target mRNA has context-dependent outcomes, wherein binding can promote both mRNA degradation and mRNA translation (Volz, 2008). Studies showed that overexpression of p53 led to reduced IRP1 and 2 activity, resulting in increased translation of ferritin mRNA and decreased stability of transferrin receptor mRNA (Zhang et al., 2008). This regulation ultimately leads to an increase in cellular iron stores and a decrease in cellular iron import.
Ferroptosis is a specific form of iron-mediated cell death in which oxidative stress from reactive oxygen species (ROS) leads to the formation of lipid peroxides and accumulation of lipid peroxides triggers the ferroptotic response (Dixon et al., 2012; Lu et al., 2018). Iron has a critical role in promoting ROS formation through several mechanisms. First, iron functions as a co-factor for enzymes that catalyze the formation of ROS (Dixon and Stockwell, 2014). In addition, Fe2+ reacts with hydrogen peroxide through the Fenton reaction, resulting in the production of free radicals, a potent form of ROS (Wardman and Candeias, 1996). ROS can then promote lipid peroxidation of cellular membranes, which leads to compromised membrane integrity and cellular damage (Yin et al., 2011). However, several intracellular reducing pathways have been found to block ROS and subsequent accumulation of lipid peroxides (Lu et al., 2018). Import of cystine into the cell via system xc– (encoded by SCL7A11) ultimately results in the synthesis of glutathione, a strong antioxidant (Lu et al., 2018; Sato et al., 2018). GPX4, a member of the glutathione peroxidase family, uses glutathione as a co-activator to reduce lipid peroxides, thus preventing ferroptosis (Lu et al., 2018). Ferroptosis has been implicated in a variety of diseases, such as cell death during ischemia (Gao et al., 2015) and neurodegeneration in Alzheimer’s disease (Masaldan et al., 2019). Interestingly, p53 can promote and inhibit ferroptosis in a context-dependent manner (Liu et al., 2020). For example, p53 is able to inhibit ferroptosis through p21, a primary p53 target that inhibits glutathione degradation (Tarangelo and Dixon, 2018). As such, upregulation of p21 by p53 inhibits glutathione degradation and promotes GPX4 activity (Tarangelo and Dixon, 2018). p53 was also shown to prevent ferroptosis by promoting the nuclear, but inhibiting the plasma membrane, accumulation of dipeptidyl-peptidase 4 (DPP4) (Xie et al., 2017). DPP4 in the nucleus upregulates SLC7A11, leading to increased GPX4 function and subsequent inhibition of ferroptosis (Xie et al., 2017). Interestingly, p53 can promote ferroptosis by directly inhibiting SLC7A11 expression (Jiang et al., 2015). Additionally, p53 promotes ferroptosis through upregulation of SAT1, which facilitates the production of lipid peroxides (Ou et al., 2016). A recent study showed that Mdm2 and Mdm4 can induce ferroptosis (Venkatesh et al., 2020). Since Mdm2 is a target of p53, it is possible that p53 can act through Mdm2/4 to modulate the induction of ferroptosis. The role of both wild-type and mutant p53 in ferroptosis was discussed comprehensively in a recent review (Liu et al., 2020).
p63 and p73
Recent evidence suggests an important role for p63 and p73 in iron metabolism. Like p53, p63, and p73 can be destabilized by an excess of heme (Shen et al., 2014). Conversely, iron depletion was found to stabilize p73, and possibly p63, to promote apoptosis and cell cycle arrest in a p53-independent manner (Calabrese et al., 2020). These data suggest that iron overload inhibits, whereas iron depletion promotes, p63 and p73 activity, which is similar to the effect of iron overload and depletion on p53. Recent studies in our lab revealed a potential mechanism through which iron overload can influence p63/p73 mRNA stability and protein expression. We showed that TAp63 expression can be repressed by IRP2 and likewise, IRP2 deficiency lead to increased expression of TAp63 (Zhang et al., 2020). Additionally, we showed that IRP2 binds to the IRE in p63 mRNA to regulates its stability (Zhang et al., 2020). Similarly, we found that FDXR regulates p73 mRNA stability through IRP2 (Zhang et al., 2020). These observations represent an important step in understanding how iron metabolism regulates p63 and p73.
Several lines of evidence suggest a role for p63 and p73 in mediating ferroptosis. For example, ferroptosis has been shown to promote liver steatosis and inflammation (Tsurusaki et al., 2019). We and others found that p63-deficient mice were prone to liver steatosis (Jiang et al., 2018). Additionally, both p63- and p73-deficient mice exhibited a high degree of liver inflammation (Jiang et al., 2018; Zhang et al., 2020). Moreover, before the term ferroptosis was coined, we found that p63 inhibited cell death caused by oxidative stress through GPX2 (Yan and Chen, 2006), a member of the same phospholipid peroxidase family as GPX4 (Chu, 1994). Aforementioned, ferroptosis ensues when the cell is unable to overcome oxidative stress. Another study revealed that ΔNp63 promoted glutathione metabolism, thus permitting GPX4 function and inhibiting the ferroptotic pathway (G. X. Wang et al., 2017). These findings suggest that p63 regulates ferroptosis through multiple glutathione peroxidase family members. As previously mentioned, p63 activates the LKB1/AMPK pathway and a group recently showed that this pathway inhibits ferroptosis (Li et al., 2020). While the role of p73 in ferroptosis is less studied, one report showed that TAp73-knockout mouse embryonic fibroblasts were particularly prone to oxidative stress (Agostini et al., 2016). Another study showed that TAp73 is able to mitigate the effect of oxidative stress on mitochondrial integrity (Marini et al., 2018). These data suggest a role for TAp73 in suppressing ferroptosis.
Future Directions
There is growing evidence that, in addition to mediating tumor suppression, the p53 family plays an important role in lipid and iron metabolism. However, there is a need for more research on these critical topics. It would be of interest to further explore how p53 is involved in regulating bioactive lipids. Additionally, it would be worthwhile to delve into the mechanisms by which p63/p73 regulate lipid and iron metabolism. While there is evidence that aberrant iron metabolism affects lipid metabolism and ferroptosis, how p53 engages lipid and iron metabolism in ferroptosis needs to be further explored. Moreover, several fundamental questions remain unanswered: Can p63 and p73 function independently of p53 in both lipid and iron metabolism? How does regulation of lipid and iron metabolism differ between the N- and C-terminal isoforms of each protein? Does regulation of lipid and iron metabolism by the p53 family contribute to common diseases associated with these processes, such as diabetes or anemia? Finally, can the p53 family proteins themselves, or the pathways regulated by the p53 family, be manipulated to ameliorate the effect of lipid or iron dysregulation on pathogenesis of diabetes and other diseases? A comprehensive understanding of how the p53 family mediates lipid and iron metabolism will likely provide an insight into the pathways that drive various human diseases.
Author Contributions
KL, JZ, and XC wrote the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the NIH Grants R01 CA081237, CA224433, CA250338, and CA195828 (to XC), T32 HL007013 (to KL), and T31IP1727 (to JZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbaspour, N., Hurrell, R., and Kelishadi, R. (2014). Review on iron and its importance for human health. J. Res. Med. Sci. 19:2.
Abu-Elheiga, L., Brinkley, W. R., Zhong, L., Chirala, S. S., Woldegiorgis, G., and Wakil, S. J. (2000). The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. U. S. A. 97:4. doi: 10.1073/pnas.97.4.1444
Agostini, M., Annicchiarico-Petruzzelli, M., Melino, G., and Rufini, A. (2016). Metabolic pathways regulated by TAp73 in response to oxidative stress. Oncotarget. 7:21. doi: 10.18632/oncotarget.8935
Akram, M. (2014). Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biochem. Biophys. 68:3. doi: 10.1007/s12013-013-9750-1
Alekos, N. S., Moorer, M. C., and Riddle, R. C. (2020). Dual Effects of Lipid Metabolism on Osteoblast Function. Front. Endocrinol. (Lausanne). 11:578194. doi: 10.3389/fendo.2020.578194
Alves-Bezerra, M., and Cohen, D. E. (2018). Triglyceride metabolism in the liver. Compr. Physiol. 8:1. doi: 10.1002/cphy.c170012
Arner, P., and Langin, D. (2014). Lipolysis in lipid turnover, cancer cachexia, and obesity-induced insulin resistance. Trends Endocrinol. Metab. 25:5. doi: 10.1016/j.tem.2014.03.002
Arrowsmith, C. H. (1999). Structure and function in the p53 family. Cell Death Differ. 6:12. doi: 10.1038/sj.cdd.4400619
Assaily, W., Rubinger, D. A., Wheaton, K., Lin, Y., Ma, W., Xuan, W., et al. (2011). ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell. 44:3. doi: 10.1016/j.molcel.2011.08.038
Attieh, Z. K., Mukhopadhyay, C. K., Seshadrit, V., Tripoulas, N. A., and Fox, P. L. (1999). Ceruloplasmin ferroxidase activity stimulates cellular iron uptake by a trivalent cation-specific transport mechanism. J. Biol. Chem. 274:2. doi: 10.1074/jbc.274.2.1116
Bacon, B. R., Adams, P. C., Kowdley, K. V., Powell, L. W., and Tavill, A. S. (2011). Diagnosis and management of hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology. 54:1. doi: 10.1002/hep.24330
Baranovskiy, A. G., Lada, A. G., Siebler, H. M., Zhang, Y., Pavlov, Y. I., and Tahirov, T. H. (2012). DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 287:21. doi: 10.1074/jbc.M112.351122
Bensaad, K., Tsuruta, A., Selak, M. A., Vidal, M. N. C., Nakano, K., Bartrons, R., et al. (2006). TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell. 126:1. doi: 10.1016/j.cell.2006.05.036
Bist, A., Fielding, C. J., and Fielding, P. E. (2000). P53 Regulates Caveolin Gene Transcription, Cell Cholesterol, and Growth By a Novel Mechanism. Biochemistry. 39:8. doi: 10.1021/bi991721h
Bohdanowicz, M., and Grinstein, S. (2013). Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol. Rev. 93:1. doi: 10.1152/physrev.00002.2012
Borremans, B., Hobman, J. L., Provoost, A., and Brown, N. L. (2001). Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat. Med. 4:7.
Bourdon, J.-C., Fernandes, K., Murray-Zmijewski, F., Liu, G., Diot, A., Xirodimas, D. P., et al. (2005). P53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19:18. doi: 10.1101/gad.1339905
Brandt, M. E., and Vickery, L. E. (1992). Expression and characterization of human mitochondrial ferredoxin reductase in Escherichia coli. Arch. Biochem. Biophys. 294:2. doi: 10.1016/0003-9861(92)90749-M
Brose, S. A., Golovko, S. A., and Golovko, M. Y. (2016). Fatty acid biosynthesis inhibition increases reduction potential in neuronal cells under hypoxia. Front. Neurosci. 2016:10. doi: 10.3389/fnins.2016.00546
Brown, M. S., and Goldstein, J. L. (1997). The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 89:3. doi: 10.1016/S0092-8674(00)80213-5
Calabrese, C., Panuzzo, C., Stanga, S., Andreani, G., Ravera, S., Maglione, A., et al. (2020). Deferasirox-dependent iron chelation enhances mitochondrial dysfunction and restores p53 signaling by stabilization of p53 family members in leukemic cells. Int. J. Mol. Sci. 21:20. doi: 10.3390/ijms21207674
Candi, E., Rufini, A., Terrinoni, A., Dinsdale, D., Ranalli, M., Paradisi, A., et al. (2006). Differential roles of p63 isoforms in epidermal development: Selective genetic complementation in p63 null mice. Cell Death Differ. 13:6. doi: 10.1038/sj.cdd.4401926
Cavadini, P., Gellera, C., Patel, P. I., and Isaya, G. (2000). Human frataxin maintains mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum. Mol. Genet. 9:17. doi: 10.1093/hmg/9.17.2523
Celli, J., Duijf, P., Hamel, B. C. J., Bamshad, M., Kramer, B., Smits, A. P. T., et al. (1999). Heterozygous Germline Mutations in the p53 Homolog p63 Are the Cause of EEC Syndrome. Cell 1999:99.
Chu, F. F. (1994). The human glutathione peroxidase genes GPX2, GPX3, and GPX4 map to chromosomes 14, 5, and 19, respectively. Cytogenet. Genome Res. 66:2. doi: 10.1159/000133675
Courtois, S., Verhaegh, G., North, S., Luciani, M. G., Lassus, P., Hibner, U., et al. (2002). ΔN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 21:44. doi: 10.1038/sj.onc.1205874
Deisenroth, C., Itahana, Y., Tollini, L., Jin, A., and Zhang, Y. (2011). P53-inducible DHRS3 is an endoplasmic reticulum protein associated with lipid droplet accumulation. J. Biol. Chem. 286:32. doi: 10.1074/jbc.M111.254227
Derdak, Z., Villegas, K. A., Harb, R., Wu, A. M., Sousa, A., and Wands, J. R. (2013). Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 58:4. doi: 10.1016/j.jhep.2012.11.042
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10:1. doi: 10.1038/nchembio.1416
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 149:5. doi: 10.1016/j.cell.2012.03.042
Donehower, L. A., Harvey, M., Slagle, B. L., Mcarthurt, M. J., Jrt, C. A. M., Butel, J. S., et al. (1991). Mice deficient for p53 are developmentally normal but susceptible to sponatenous tumors. Nature 1991:365.
Donkor, J., Sariahmetoglu, M., Dewald, J., Brindley, D. N., and Reue, K. (2007). Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 282:6. doi: 10.1074/jbc.M610745200
Fielding, P. E., and Fielding, C. J. (1995). Plasma Membrane Caveolae Mediate the Efflux of Cellular Free Cholesterol. Biochemistry 34:44. doi: 10.1021/bi00044a004
Fiorito, V., Chiabrando, D., Petrillo, S., Bertino, F., and Tolosano, E. (2020). The Multifaceted Role of Heme in Cancer. Front. Oncol. 9:1540. doi: 10.3389/fonc.2019.01540
Foster, D. W. (2004). The role of the carnitine system in human metabolism. Ann. N. Y. Acad. Sci. 1033:1. doi: 10.1196/annals.1320.001
Franklin, D. A., He, Y., Leslie, P. L., Tikunov, A. P., Fenger, N., MacDonald, J. M., et al. (2016). P53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci. Rep. 6:38067. doi: 10.1038/srep38067
Funauchi, Y., Tanikawa, C., Yi, Lo, P. H., Mori, J., Daigo, Y., et al. (2015). Regulation of iron homeostasis by the p53-ISCU pathway. Sci. Rep. 5:16497. doi: 10.1038/srep16497
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell. 59:2. doi: 10.1016/j.molcel.2015.06.011
Ge, T., Yang, J., Zhou, S., Wang, Y., Li, Y., and Tong, X. (2020). The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 11:365. doi: 10.3389/fendo.2020.00365
Giammanco, A., Cefalù, A. B., Noto, D., and Averna, M. R. (2015). The pathophysiology of intestinal lipoprotein production. Front. Physiol. 6:61. doi: 10.3389/fphys.2015.00061
Goldstein, I., Ezra, O., Rivlin, N., Molchadsky, A., Madar, S., Goldfinger, N., et al. (2012). P53, a novel regulator of lipid metabolism pathways. J. Hepatol. 56:3. doi: 10.1016/j.jhep.2011.08.022
Gómez-Santos, B., Saenz, de Urturi, D., Nuñez-García, M., Gonzalez-Romero, F., Buque, X., et al. (2020). Liver osteopontin is required to prevent the progression of age-related nonalcoholic fatty liver disease. Aging Cell. 19:8. doi: 10.1111/acel.13183
Greenberg, C. R., Dilling, L. A., Thompson, G. R., Seargeant, L. E., Haworth, J. C., Phillips, S., et al. (2009). The paradox of the carnitine palmitoyltransferase type Ia P479L variant in Canadian Aboriginal populations. Mol. Genet. Metab. 96:4. doi: 10.1016/j.ymgme.2008.12.018
Guo, X., Keyes, W. M., Papazoglu, C., Zuber, J., Li, W., Lowe, S. W., et al. (2009). TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat. Cell Biol. 11:12. doi: 10.1038/ncb1988
Hage-Sleiman, R., Bahmad, H., Kobeissy, H., Dakdouk, Z., Kobeissy, F., and Dbaibo, G. (2017). Genomic alterations during p53-dependent apoptosis induced by γ-irradiation of Molt-4 leukemia cells. PLoS One 12:e0190221. doi: 10.1371/journal.pone.0190221
Hannun, Y. A., and Obeid, L. M. (2008). Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9:2. doi: 10.1038/nrm2329
Hannun, Y. A., and Obeid, L. M. (2018). Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018:107. doi: 10.1038/nrm.2017.107
Harms, K. L., and Chen, X. (2005). The C Terminus of p53 Family Proteins Is a Cell Fate Determinant. Mol. Cell. Biol. 25:5. doi: 10.1128/mcb.25.5.2014-2030.2005
Hasson, D., Feliú, F., and Gormaz, J. G. (2016). Non-alcoholic fatty liver disease and the metabolic syndrome. Nonalcoholic Fat. Liver Dis. 2016, 79–106. doi: 10.2174/138161207781039652
He, M., Pei, Z., Mohsen, A. W., Watkins, P., Murdoch, G., Veldhoven, P. P., et al. (2011). Identification and characterization of new long chain Acyl-CoA dehydrogenases. Mol. Genet. Metab. 102:4. doi: 10.1016/j.ymgme.2010.12.005
He, Z., Liu, H., Agostini, M., Yousefi, S., Perren, A., Tschan, M. P., et al. (2013). P73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell Death Differ. 20:10. doi: 10.1038/cdd.2013.104
Helton, E. S., Zhu, J., and Chen, X. (2006). The unique NH2-terminally deleted (ΔN) residues, the PXXP motif, and the PPXY motif are required for the transcriptional activity of the ΔN variant of p63. J. Biol. Chem. 281:5. doi: 10.1074/jbc.M507964200
Hooijmans, C. R., and Kiliaan, A. J. (2008). Fatty acids, lipid metabolism and Alzheimer pathology. Eur. J. Pharmacol. 585:1. doi: 10.1016/j.ejphar.2007.11.081
Hussain, M. M. (2014). Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 25:3. doi: 10.1097/MOL.0000000000000084
Hwang, P. M., Bunz, F., Yu, J., Rago, C., Chan, T. A., Murphy, M. P., et al. (2001). Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 7:10. doi: 10.1038/nm1001-1111
Igal, R. A. (2010). Stearoyl-coa desaturase-1: A novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis. 31:9. doi: 10.1093/carcin/bgq131
Jakobsson, A., Westerberg, R., and Jacobsson, A. (2006). Fatty acid elongases in mammals: Their regulation and roles in metabolism. Prog. Lipid Res. 45:3. doi: 10.1016/j.plipres.2006.01.004
Jiang, D., LaGory, E. L., KenzelmannBrož, D., Bieging, K. T., Brady, C. A., Link, N., et al. (2015). Analysis of p53 Transactivation Domain Mutants Reveals Acad11 as a Metabolic Target Important for p53 Pro-Survival Function. Cell Rep. 10:7. doi: 10.1016/j.celrep.2015.01.043
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 520:7545. doi: 10.1038/nature14344
Jiang, P., Du, W., Mancuso, A., Wellen, K. E., and Yang, X. (2013). Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 493:7434. doi: 10.1038/nature11776
Jiang, P., Du, W., Wang, X., Mancuso, A., Gao, X., Wu, M., et al. (2011). P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 13:3. doi: 10.1038/ncb2172
Jiang, Y., Xu, E., Zhang, J., Chen, M., Flores, E., and Chen, X. (2018). The Rbm38-p63 feedback loop is critical for tumor suppression and longevity. Oncogene. 37:21. doi: 10.1038/s41388-018-0176-5
Johnson, D. C., Dean, D. R., Smith, A. D., and Johnson, M. K. (2005). Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 74:1. doi: 10.1146/annurev.biochem.74.082803.133518
Kaghad, M., Bonnet, H., Yang, A., Creancier, L., Biscan, J. C., Valent, A., et al. (1997). Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 90:4. doi: 10.1016/S0092-8674(00)80540-1
Kang, J. G., Lago, C. U., Lee, J. E., Park, J. H., Donnelly, M. P., Starost, M. F., et al. (2020). A Mouse Homolog of a Human TP53 Germline Mutation Reveals a Lipolytic Activity of p53. Cell Rep. 30:3. doi: 10.1016/j.celrep.2019.12.074
Kim, A., and Nemeth, E. (2015). New insights into iron regulation and erythropoiesis. Curr. Opin. Hematol. 22:3. doi: 10.1097/MOH.0000000000000132
Kim, B. M., Choi, J. Y., Kim, Y. J., Woo, H. D., and Chung, H. W. (2007). Desferrioxamine (DFX) has genotoxic effects on cultured human lymphocytes and induces the p53-mediated damage response. Toxicology. 229:3. doi: 10.1016/j.tox.2006.10.022
Kirschner, R. D., Rother, K., Müller, G. A., and Engeland, K. (2010). The retinal dehydrogenase/reductase retSDR1/DHRS3 gene is activated by p53 and p63 but not by mutants derived from tumors or EEC/ADULT malformation syndromes. Cell Cycle. 9:11. doi: 10.4161/cc.9.11.11844
Ko, L. J., and Prives, C. (1996). P53: Puzzle and paradigm. Genes Dev. 10:9. doi: 10.1101/gad.10.9.1054
Kobayashi, S., Fukuhara, A., Taguchi, T., Matsuda, M., Tochino, Y., Otsuki, M., et al. (2010). Identification of a new secretory factor, CCDC3/Favine, in adipocytes and endothelial cells. Biochem. Biophys. Res. Commun. 392:1. doi: 10.1016/j.bbrc.2009.12.142
Korber, M., Klein, I., and Daum, G. (2017). Steryl ester synthesis, storage and hydrolysis: A contribution to sterol homeostasis. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 1862:12. doi: 10.1016/j.bbalip.2017.09.002
Koster, M. I., Dai, D., Marinari, B., Sano, Y., Costanzo, A., Karin, M., et al. (2007). P63 Induces Key Target Genes Required for Epidermal Morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 104:9. doi: 10.1073/pnas.0611376104
Laezza, C., D’Alessandro, A., Croce, L., Di, Picardi, P., Ciaglia, E., et al. (2015). P53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis. 6:10. doi: 10.1038/cddis.2015.279
Laurenzi, V., De Catani, M. V., Terrinoni, A., Corazzari, M., Melino, G., Constanzo, A., et al. (1999). Additional complexity in p73: Induction by mitogens in lymphoid cells and identification of two new splicing variants ε and ζ. Cell Death Differ. 6:5. doi: 10.1038/sj.cdd.4400521
Laurenzi, V., De Costanzo, A., Barcaroli, D., Terrinoni, A., Falco, M., Annicchiarico-Petruzzelli, M., et al. (1998). Two new p73 splice variants, γ and δ, with different transcriptional activity. J. Exp. Med. 188:9. doi: 10.1084/jem.188.9.1763
Lee, J., and Wolfgang, M. J. (2012). Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism. BMC Biochem. 13:23. doi: 10.1186/1471-2091-13-23
Leonardi, R., Zhang, Y. M., Rock, C. O., and Jackowski, S. (2005). Coenzyme A: Back in action. Prog. Lipid Res. 44, 2–3. doi: 10.1016/j.plipres.2005.04.001
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, K., and Serhan, C. N. (2001). Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2:7. doi: 10.1038/89759
Li, C., Dong, X., Du, W., Shi, X., Chen, K., Zhang, W., et al. (2020). LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct. Target. Ther. 5:187. doi: 10.1038/s41392-020-00297-2
Liang, S. X., and Richardson, D. R. (2003). The effect of potent iron chelators on the regulation of p53: Examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis. 24:10. doi: 10.1093/carcin/bgg116
Liao, W., Liu, H., Zhang, Y., Jung, J. H., Chen, J., Su, X., et al. (2017). Ccdc3: A new P63 target involved in regulation of liver lipid metabolism. Sci. Rep. 7:1. doi: 10.1038/s41598-017-09228-8
Liu, G., and Chen, X. (2002). The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene. 21:47. doi: 10.1038/sj.onc.1205862
Liu, G., Nozell, S., Xiao, H., and Chen, X. (2004). ΔNp73β Is Active in Transactivation and Growth Suppression. Mol. Cell. Biol. 24:2. doi: 10.1128/mcb.24.2.487-501.2004
Liu, J., Zhang, C., Wang, J., Hu, W., and Feng, Z. (2020). The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int. J. Mol. Sci. 21:21. doi: 10.3390/ijms21218387
Liu, Y., He, Y., Jin, A., Tikunov, A. P., Zhou, L., Tollini, L. A., et al. (2014). Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. U. S. A. 111:23. doi: 10.1073/pnas.1315605111
Longo, N., Frigeni, M., and Pasquali, M. (2016). Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta - Mol. Cell Res. 1863:10. doi: 10.1016/j.bbamcr.2016.01.023
Lu, B., Chen, X. B., Ying, M. D., He, Q. J., Cao, J., and Yang, B. (2018). The role of ferroptosis in cancer development and treatment response. Front. Pharmacol. 8:992. doi: 10.3389/fphar.2017.00992
Luna, R. M. D. O., Wagner, D. S., and Lozano, G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378:6553. doi: 10.1038/378203a0
Ma, C., Liging, S., Seven, A. B., Xu, Y., and Rizo, J. (2013). Reconstitution of the Vital Functions of Munc18 and Munc13 in Neurotransmitter Release. Science 2013:339.
Mangiulli, M., Valletti, A., Caratozzolo, M. F., Tullo, A., Sbisà, E., Pesole, G., et al. (2009). Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 37:18. doi: 10.1093/nar/gkp674
Marini, A., Rotblat, B., Sbarrato, T., Niklison-Chirou, M. V., Knight, J. R. P., Dudek, K., et al. (2018). TAp73 contributes to the oxidative stress response by regulating protein synthesis. Proc. Natl. Acad. Sci. U. S. A. 115:24. doi: 10.1073/pnas.1718531115
Marshall, C. B., Mays, D. J., Beeler, J. S., Rosenbluth, J. M., Boyd, K. L., Santos Guasch, G. L., et al. (2016). P73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 14:10. doi: 10.1016/j.celrep.2016.02.035
Martin, S., and Parton, R. G. (2006). Lipid droplets: a unified view of a dynamic organelle. Mol. Cell 7:912.
Masaldan, S., Belaidi, A. A., Ayton, S., and Bush, A. I. (2019). Cellular senescence and iron dyshomeostasis in alzheimer’s disease. Pharmaceuticals. 12:2. doi: 10.3390/ph12020093
Mashek, D. G., Li, L. O., and Coleman, R. A. (2007). Long-chain acyl-CoA synthetases and fatty acid channeling. Future Lipidol. 2:4. doi: 10.2217/17460875.2.4.465
Mills, A. A., Zheng, B., Wang, X. J., Vogel, H., Roop, D. R., and Bradley, A. (1999). P63 Is a P53 Homologue Required for Limsb and Epidermal Morphogenesis. Nature. 398:6729. doi: 10.1038/19531
Moon, S. H., Huang, C. H., Houlihan, S. L., Regunath, K., Freed-Pastor, W. A., Morris, J. P., et al. (2019). P53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell. 176:3. doi: 10.1016/j.cell.2018.11.011
Nemajerova, A., Kramer, D., Siller, S. S., Herr, C., Shomroni, O., Pena, T., et al. (2016). TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 30:11. doi: 10.1101/gad.279836.116
Nemoto, S., Fergusson, M. M., and Finkel, T. (2004). Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306:5704. doi: 10.1126/science.1101731
Nostrand, J. L., Van, Brady, C. A., Jung, H., Fuentes, D. R., Kozak, M. M., et al. (2014). Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature. 514:7521. doi: 10.1038/nature13585
Oelkers, P., Behari, A., Cromley, D., Billheimer, J. T., and Sturleyt, S. L. (1998). Characterization of two human genes encoding acyl coenzyme A: Cholesterol acyltransferase-related enzymes. J. Biol. Chem. 273:41. doi: 10.1074/jbc.273.41.26765
Ohashi, T., Idogawa, M., Sasaki, Y., and Tokino, T. (2017). P53 mediates the suppression of cancer cell invasion by inducing LIMA1/EPLIN. Cancer Lett. 390:34. doi: 10.1016/j.canlet.2016.12.034
Olzmann, J. A., and Carvalho, P. (2019). Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 20:3. doi: 10.1038/s41580-018-0085-z
Oni, T. E., Biffi, G., Baker, L. A., Hao, Y., Tonelli, C., Somerville, T. D. D., et al. (2020). SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J. Exp. Med. 217:9. doi: 10.1084/jem.20192389
Ou, Y., Wang, S. J., Li, D., Chu, B., and Gu, W. (2016). Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. U. S. A. 113:44. doi: 10.1073/pnas.1607152113
Parant, J., Chavez-Reyes, A., Little, N. A., Yan, W., Reinke, V., Jochemsen, A. G., et al. (2001). Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 29:1. doi: 10.1038/ng714
Parrales, A., and Iwakuma, T. (2016). P53 As a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 17:2074. doi: 10.3390/ijms17122074
Peter-Riesch, B., Fathi, M., Schlegel, W., and Wolheim, C. B. (1988). Glucose and carbachol generate 1,2-diacylglycerols by different mechanisms in pancreatic islets. J. Clin. Invest. 81:4. doi: 10.1172/JCI113430
Puig, S., Ramos-Alonso, L., Romero, A. M., and Martínez-Pastor, M. T. (2017). The elemental role of iron in DNA synthesis and repair. Metallomics. 9:1483. doi: 10.1039/c7mt00116a
Qu, Q., Zeng, F., Liu, X., Wang, Q. J., and Deng, F. (2016). Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 7:5. doi: 10.1038/cddis.2016.132
Rahman, S., and Islam, R. (2011). Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal. 9:11. doi: 10.1186/1478-811X-9-11
Röhrig, F., and Schulze, A. (2016). The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer. 16:11. doi: 10.1038/nrc.2016.89
Rufer, A. C., Thoma, R., and Hennig, M. (2009). Structural insight into function and regulation of carnitine palmitoyltransferase. Cell. Mol. Life Sci. 66:15. doi: 10.1007/s00018-009-0035-1
Sah, V. P., Attardi, L. D., Mulligan, G. J., Williams, B., Bronson, R. T., and Jacks, T. (1995). A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 10, 175–180. doi: 10.1038/ng0695-175
Saito, T., Kuma, A., Sugiura, Y., Ichimura, Y., Obata, M., Kitamura, H., et al. (2019). Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 10:1. doi: 10.1038/s41467-019-08829-3
Saleme, B., Das, S. K., Zhang, Y., Boukouris, A. E., Lorenzana Carrillo, M. A., Jovel, J., et al. (2020). P53-Mediated Repression of the PGC1A (PPARG Coactivator 1α) and APLNR (Apelin Receptor) Signaling Pathways Limits Fatty Acid Oxidation Energetics: Implications for Cardio-oncology. J. Am. Heart Assoc. 9:15. doi: 10.1161/JAHA.120.017247
Sanchez-Macedo, N., Feng, J., Faubert, B., Chang, N., Elia, A., Rushing, E. J., et al. (2013). Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type i tumor model. Cell Death Differ. 20:4. doi: 10.1038/cdd.2012.168
Sato, M., Kusumi, R., Hamashima, S., Kobayashi, S., Sasaki, S., Komiyama, Y., et al. (2018). The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 8:968. doi: 10.1038/s41598-018-19213-4
Schmale, H., and Bamberger, C. (1997). A novel protein with strong homology to the tumor suppressor p53. Oncogene. 15:11. doi: 10.1038/sj.onc.1201500
Sheftel, A. D., Stehling, O., Pierik, A. J., Elsässer, H. P., Mühlenhoff, U., Webert, H., et al. (2010). Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 107:26. doi: 10.1073/pnas.1004250107
Shen, J., Sheng, X., Chang, Z. N., Wu, Q., Wang, S., Xuan, Z., et al. (2014). Iron metabolism regulates p53 signaling through direct Heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 7:1. doi: 10.1016/j.celrep.2014.02.042
Shimano, H., Horton, J. D., Shimomura, I., Hammer, R. E., Brown, M. S., and Goldstein, J. L. (1997). Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J. Clin. Invest. 99:5. doi: 10.1172/JCI119248
Shimizu, R., Lan, N. N., Tai, T. T., Adachi, Y., Kawazoe, A., Mu, A., et al. (2014). P53 directly regulates the transcription of the human frataxin gene and its lack of regulation in tumor cells decreases the utilization of mitochondrial iron. Gene. 551:1. doi: 10.1016/j.gene.2014.08.043
Shimomura, I., Shimano, H., Korn, B. S., Bashmakov, Y., and Horton, J. D. (1998). Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J. Biol. Chem. 273:52. doi: 10.1074/jbc.273.52.35299
Smith, S., Witkowski, A., and Joshi, A. K. (2003). Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 42:4. doi: 10.1016/S0163-7827(02)00067-X
Su, X., Gi, Y. J., Chakravarti, D., Chan, I. L., Zhang, A., Xia, X., et al. (2012). TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 16:4. doi: 10.1016/j.cmet.2012.09.006
Su, X., Paris, M., Gi, Y. J., Tsai, K. Y., Cho, M. S., Lin, Y. L., et al. (2009). TAp63 Prevents Premature Aging by Promoting Adult Stem Cell Maintenance. Cell Stem Cell. 5:1. doi: 10.1016/j.stem.2009.04.003
Suh, E. K., Yang, A., Kettenbach, A., Bamberger, C., Michaelis, A. H., Zhu, Z., et al. (2006). P63 Protects the Female Germ Line During Meiotic Arrest. Nature. 444:7119. doi: 10.1038/nature05337
Tarangelo, A., and Dixon, S. (2018). The p53-p21 pathway inhibits ferroptosis during metabolic stress. Oncotarget. 9:37. doi: 10.18632/oncotarget.25362
Thompson, E. A., and Siiteri, P. K. (1974). The involvement of human placental microsomal cytochrome P 450 in aromatization. J. Biol. Chem. 249:17. doi: 10.1016/s0021-9258(20)79736-x
Tomasini, R., Tsuchihara, K., Wilhelm, M., Fujitani, M., Rufini, A., Cheung, C. C., et al. (2008). TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 22:19. doi: 10.1101/gad.1695308
Trink, B., Okami, K., Wu, L., Sriuranpong, V., Jen, J., and Sidransky, D. (1998). A new human p53 homologue. Nat. Med. 4:7. doi: 10.1038/nm0798-747
Tsurusaki, S., Tsuchiya, Y., Koumura, T., Nakasone, M., Sakamoto, T., Matsuoka, M., et al. (2019). Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 10:6. doi: 10.1038/s41419-019-1678-y
Venkatesh, D., O’Brien, N. A., Zandkarimi, F., Tong, D. R., Stokes, M. E., Dunn, D. E., et al. (2020). MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 34, 7–8. doi: 10.1101/gad.334219.119
Volz, K. (2008). The functional duality of iron regulatory protein 1. Curr. Opin. Struct. Biol. 18:1. doi: 10.1016/j.sbi.2007.12.010
Waldvogel-Abramowski, S., Waeber, G., Gassner, C., Buser, A., Frey, B. M., Favrat, B., et al. (2014). Physiology of iron metabolism. Transfus. Med. Hemotherapy. 41:3. doi: 10.1159/000362888
Wallace, D. F. (2019). Regulation of Folate Homeostasis. Clin. Biochem. 37:2. doi: 10.1515/9783110856262-175
Wang, G. X., Tu, H. C., Dong, Y., Skanderup, A. J., Wang, Y., Takeda, S., et al. (2017). ΔNp63 Inhibits Oxidative Stress-Induced Cell Death, Including Ferroptosis, and Cooperates with the BCL-2 Family to Promote Clonogenic Survival. Cell Rep. 21:10. doi: 10.1016/j.celrep.2017.11.030
Wang, S. J., Yu, G., Jiang, L., Li, T., Lin, Q., Tang, Y., et al. (2013). P53-dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell Cycle. 12:5. doi: 10.4161/cc.23597
Wang, X., Zhao, X., Gao, X., Mei, Y., and Wu, M. (2013). A new role of p53 in regulating lipid metabolism. J. Mol. Cell Biol. 5:2. doi: 10.1093/jmcb/mjs064
Wardman, P., and Candeias, L. P. (1996). Fenton Chemistry: An Introduction. Source Radiat. Res. Vol. 145:3579270.
Weizer-Stern, O., Adamsky, K., Margalit, O., Ashur-Fabian, O., Givol, D., Amariglio, N., et al. (2007). Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br. J. Haematol. 138:2. doi: 10.1111/j.1365-2141.2007.06638.x
Wilhelm, M. T., Rufini, A., Wetzel, M. K., Tsuchihara, K., Inoue, S., Tomasini, R., et al. (2010). Isoform-specific p73 knockout mice reveal a novel role for ΔNp73 in the DNA damage response pathway. Genes Dev. 24:6. doi: 10.1101/gad.1873910
Wise, E. M., and Ball, E. G. (1964). Malic Enzyme and Lipogenesis. Proc. Natl. Acad. Sci. U S. 52:1933. doi: 10.1073/pnas.52.5.1255
Xie, Y., Zhu, S., Song, X., Sun, X., Fan, Y., Liu, J., et al. (2017). The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 20:7. doi: 10.1016/j.celrep.2017.07.055
Yahagi, N., Shimano, H., Matsuzaka, T., Najima, Y., Sekiya, M., Nakagawa, Y., et al. (2003). P53 Activation in Adipocytes of Obese Mice. J. Biol. Chem. 278:28. doi: 10.1074/jbc.M302364200
Yamauchi, Y., Iwamoto, N., Rogers, M. A., Abe-Dohmae, S., Fujimoto, T., Chang, C. C. Y., et al. (2015). Deficiency in the lipid exporter ABCA1 impairs retrograde sterol movement and disrupts sterol sensing at the endoplasmic reticulum. J. Biol. Chem. 290:39. doi: 10.1074/jbc.M115.662668
Yan, W., and Chen, X. (2006). GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J. Biol. Chem. 281:12. doi: 10.1074/jbc.M512655200
Yang, A., Kaghad, M., Wang, Y., Gillett, E., Fleming, M. D., Dötsch, V., et al. (1998). P63, a P53 Homolog At 3Q27-29, Encodes Multiple Products With Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell. 2:3. doi: 10.1016/S1097-2765(00)80275-0
Yang, A., Schweitzer, R., Sun, D., Kaghad, M., Walker, N., Bronson, R. T., et al. (1999). P63 Is Essential for Regenerative Proliferation in Limb, Craniofacial and Epithelial Development. Nature. 398:6729. doi: 10.1038/19539
Yang, A., Walker, N., Bronson, R., Kaghad, M., Oosterwegel, M., Bonnin, J., et al. (2000). P73-Deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 404:6773. doi: 10.1038/35003607
Ye, X., Zhou, X. J., and Zhang, H. (2018). Exploring the role of autophagy-related gene 5 (ATG5ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front. Immunol. 9:2334. doi: 10.3389/fimmu.2018.02334
Yin, H., Xu, L., and Porter, N. A. (2011). Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 111:10. doi: 10.1021/cr200084z
Yin, Y., Stephen, C. W., Luciani, M. G., and Fåhraeus, R. (2002). P53 Stability and Activity Is Regulated By Mdm2-Mediated Induction of Alternative P53 Translation Products. Nat. Cell Biol. 4:6. doi: 10.1038/ncb801
Zaidi, N., Swinnen, J. V., and Smans, K. (2012). ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 72:15. doi: 10.1158/0008-5472.CAN-11-4112
Zhang, F., Wang, W., Tsuji, Y., Torti, S. V., and Torti, F. M. (2008). Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 283:49. doi: 10.1074/jbc.M806432200
Zhang, J., Kong, X., Zhang, Y., Sun, W., Wang, J., Chen, M., et al. (2020). FDXR regulates TP73 tumor suppressor via IRP2 to modulate aging and tumor suppression. J. Pathol. 251:3. doi: 10.1002/path.5451
Zhang, Q., He, X., Chen, L., Zhang, C., Gao, X., Yang, Z., et al. (2012). Synergistic regulation of p53 by Mdm2 and Mdm4 is critical in cardiac endocardial cushion morphogenesis during heart development. J. Pathol. 228:3. doi: 10.1002/path.4077
Zhang, Y. Y., Fu, Z. Y., Wei, J., Qi, W., Baituola, G., Luo, J., et al. (2018). A LIMA1 variant promotes low plasma LDL cholesterol and decreases intestinal cholesterol absorption. Science 360:6393. doi: 10.1126/science.aao6575
Zhang, Y., Feng, X., Zhang, J., and Chen, X. (2020). Iron regulatory protein 2 exerts its oncogenic activities by suppressing TAp63 expression. Mol. Cancer Res. 18:7. doi: 10.1158/1541-7786.MCR-19-1104
Zhang, Y., Qian, Y., Zhang, J., Yan, W., Jung, Y. S., Chen, M., et al. (2017). Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 31:12. doi: 10.1101/gad.299388.117
Zhu, J., Zhang, S., Jiang, J., and Chen, X. (2000). Definition of the p53 functional domains necessary for inducing apoptosis. J. Biol. Chem. 275:51. doi: 10.1074/jbc.M005676200
Keywords: p53, p63, p73, metabolism, lipid, iron
Citation: Laubach K, Zhang J and Chen X (2021) The p53 Family: A Role in Lipid and Iron Metabolism. Front. Cell Dev. Biol. 9:715974. doi: 10.3389/fcell.2021.715974
Received: 27 May 2021; Accepted: 08 July 2021;
Published: 29 July 2021.
Edited by:
Kanaga Sabapathy, National Cancer Centre Singapore, SingaporeReviewed by:
Koji Itahana, Duke-NUS Medical School, SingaporeQing Feng, Nanjing Medical University, China
Copyright © 2021 Laubach, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinbin Chen, eGJjaGVuQHVjZGF2aXMuZWR1